WO2016194983A1 - 共役ジエンの製造方法 - Google Patents
共役ジエンの製造方法 Download PDFInfo
- Publication number
- WO2016194983A1 WO2016194983A1 PCT/JP2016/066297 JP2016066297W WO2016194983A1 WO 2016194983 A1 WO2016194983 A1 WO 2016194983A1 JP 2016066297 W JP2016066297 W JP 2016066297W WO 2016194983 A1 WO2016194983 A1 WO 2016194983A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- unsaturated alcohol
- alcohol
- formaldehyde
- conjugated diene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/20—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon starting from organic compounds containing only oxygen atoms as heteroatoms
- C07C1/24—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon starting from organic compounds containing only oxygen atoms as heteroatoms by elimination of water
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/14—Phosphorus; Compounds thereof
- B01J27/16—Phosphorus; Compounds thereof containing oxygen, i.e. acids, anhydrides and their derivates with N, S, B or halogens without carriers or on carriers based on C, Si, Al or Zr; also salts of Si, Al and Zr
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/12—Alkadienes
- C07C11/173—Alkadienes with five carbon atoms
- C07C11/18—Isoprene
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/36—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring increasing the number of carbon atoms by reactions with formation of hydroxy groups, which may occur via intermediates being derivatives of hydroxy, e.g. O-metal
- C07C29/38—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring increasing the number of carbon atoms by reactions with formation of hydroxy groups, which may occur via intermediates being derivatives of hydroxy, e.g. O-metal by reaction with aldehydes or ketones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/44—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring increasing the number of carbon atoms by addition reactions, i.e. reactions involving at least one carbon-to-carbon double or triple bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2527/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- C07C2527/14—Phosphorus; Compounds thereof
- C07C2527/16—Phosphorus; Compounds thereof containing oxygen
- C07C2527/167—Phosphates or other compounds comprising the anion (PnO3n+1)(n+2)-
- C07C2527/173—Phosphoric acid or other acids with the formula Hn+2PnO3n+1
Definitions
- the present invention relates to a method for producing a conjugated diene by producing a ⁇ , ⁇ -unsaturated alcohol from an ⁇ -olefin and formaldehyde and subjecting the obtained ⁇ , ⁇ -unsaturated alcohol to a dehydration reaction.
- the methods for producing conjugated dienes there is a method of synthesizing from ⁇ -olefin and formaldehyde by a one-stage reaction.
- a method for producing isoprene is known in which isobutene, formaldehyde, and water are continuously or intermittently supplied into an acidic aqueous solution and reacted while distilling the produced isoprene out of the reaction system (see Patent Document 1). .
- this production method has a problem that the selectivity of isoprene is as low as about 73% and the amount of high-boiling by-products generated is large.
- dividing the reaction into multiple stages each having a high selectivity may improve the total selectivity compared to synthesis in a single stage reaction.
- a ⁇ , ⁇ -unsaturated alcohol is produced from an ⁇ -olefin and formaldehyde, and the resulting ⁇ , ⁇ -unsaturated alcohol is subjected to a dehydration reaction to produce a conjugated diene. If the selectivity of each reaction is high, there is a possibility that it exceeds the selectivity when produced in a single-stage reaction.
- the reaction between ⁇ -olefin and formaldehyde is carried out in the absence of a catalyst using an alcohol having 3 to 10 carbon atoms as a solvent.
- a method is disclosed that is carried out at a temperature of 30 to 500 atmospheres (see Patent Document 2). The process yields 3-methyl-3-buten-1-ol with a selectivity of up to about 91%.
- a dehydration reaction can be performed in a liquid phase system having a lower reaction temperature.
- 1-ol is described (see Patent Document 5).
- the document does not describe any industrial and continuous reaction method, and describes a method for starting the reaction by breaking the glass tube sealed with the catalyst together with the start of stirring. Not immediately applicable.
- an object of the present invention is to provide a method for industrially and stably producing a conjugated diene with high selectivity.
- the ⁇ , ⁇ -unsaturated alcohol obtained by reacting ⁇ -olefin with formaldehyde is subjected to a dehydration reaction at a high temperature in the presence of an acidic catalyst, thereby selecting a conjugated diene.
- the inventors have found that the rate can be stably increased and have completed the present invention.
- the present invention provides the following [1] to [8].
- Step A in which ⁇ -olefin and formaldehyde are reacted to produce ⁇ , ⁇ -unsaturated alcohol, and ⁇ , ⁇ -unsaturated alcohol is dehydrated at 135-210 ° C in the presence of an aqueous acidic catalyst.
- the manufacturing method of the conjugated diene which has the process B to be made.
- [2] The production method of [1], wherein a solvent is allowed to coexist in the step A.
- [3] The production method of [2], wherein the solvent is alcohol.
- Step B both ⁇ , ⁇ -unsaturated alcohol and water are continuously or intermittently supplied to the reactor, and the produced conjugated diene and water are continuously or intermittently removed from the reaction system.
- the method according to any one of [1] to [4], wherein [6] The process according to [5], wherein ⁇ water supply rate (mol / hour) / ⁇ , ⁇ -unsaturated alcohol supply rate (mol / hour) ⁇ is 0.5 to 12.
- a method for producing a conjugated diene with high selectivity can be provided industrially and stably.
- the method for producing a conjugated diene of the present invention comprises a step of producing a ⁇ , ⁇ -unsaturated alcohol from an ⁇ -olefin and formaldehyde (hereinafter referred to as “step A”), and the ⁇ , ⁇ -obtained in step A. And a step of dehydrating the unsaturated alcohol in the presence of an acidic catalyst (hereinafter referred to as “step B”).
- Step A includes the following general formula (I)
- R 1 , R 2 and R 3 each independently represents a hydrogen atom or an alkyl group having 1 to 10 carbon atoms. R 1 and R 3 may be linked together to form a ring
- ⁇ -olefin (I) An ⁇ -olefin represented by the following formula (hereinafter referred to as ⁇ -olefin (I)) and formaldehyde,
- alkyl group having 1 to 10 carbon atoms independently represented by R 1 , R 2 and R 3 examples include a methyl group, an ethyl group, and various propyl groups (“various” means linear and all branched chain groups). The same applies hereinafter.) Examples include various butyl groups, various hexyl groups, various octyl groups, and various decyl groups. Of these, an alkyl group having 1 to 5 carbon atoms is preferable, an alkyl group having 1 to 3 carbon atoms is more preferable, and a methyl group is more preferable.
- the ring in the case where R 1 and R 3 are connected to each other to form a ring is preferably a saturated aliphatic ring having 5 to 10 carbon atoms such as a cyclopentane ring, a cyclohexane ring or a cyclooctane ring. More preferred.
- the R 1, R 2 and R 3, at least one of R 1 and R 2 are hydrogen atoms, and more preferably R 3 is an alkyl group having 1 to 10 carbon atoms, R 1 and R It is particularly preferred that 2 is a hydrogen atom and R 3 is an alkyl group having 1 to 10 carbon atoms. More preferable alkyl groups are as described above.
- ⁇ -olefin (I) examples include (1) propylene, wherein R 1 , R 2 and R 3 are all hydrogen atoms, (2) isobutene as an example in which R 1 and R 2 are a hydrogen atom and R 3 is an alkyl group having 1 to 10 carbon atoms, (3) 2-methyl-1-butene, 2-methyl-1-pentene, 2 as examples in which one of R 1 and R 2 is a hydrogen atom and R 3 is an alkyl group having 1 to 10 carbon atoms -Methyl-1-hexene, 2-methyl-1-heptene, 2-methyl-1-octene, (4) 2,3-dimethyl-1-butene as an example where R 1 , R 2 and R 3 are all alkyl groups having 1 to 10 carbon atoms, (5) Methylenecyclohexane as an example in which R 2 is a hydrogen atom and R 1 and R 3 are connected to each other to form a ring.
- the amount of ⁇ -olefin (I) used in Step A is preferably 1 to 50 moles, more preferably 3 to 30 moles, and even more preferably 3 to 20 moles per mole of formaldehyde. If the amount of ⁇ -olefin (I) used is 1 mol or more with respect to 1 mol of formaldehyde, the selectivity of the target ⁇ , ⁇ -unsaturated alcohol is improved, and if it is 50 mol or less, ⁇ -olefin is used. The equipment required for the recovery of (I) is reduced, the industrial value is improved, the volumetric efficiency is improved, and the productivity is improved.
- formaldehyde may be used as it is, or dissolved in a solvent and used.
- the solvent for dissolving formaldehyde is not particularly limited, but water is preferable from the viewpoint of availability, that is, it is preferable to use an aqueous formaldehyde solution.
- the concentration of formaldehyde is high from the viewpoint of volume efficiency. However, if the concentration is too high, a problem of precipitation occurs and handling becomes difficult. Therefore, the concentration of formaldehyde in the formaldehyde solution is usually 10 to 70% by mass. Preferably, it is 30 to 60% by mass.
- the reaction of Step A can be carried out in the presence or absence of a solvent, but is preferably carried out in the presence of a solvent.
- the solvent used is not particularly limited as long as it does not adversely affect the reaction, and aliphatic hydrocarbons such as pentane, hexane, heptane, octane, nonane, decane, cyclohexane, and cyclooctane; aromatic carbons such as benzene, toluene, xylene, and mesitylene Hydrogen; alcohols such as methanol, ethanol and tert-butyl alcohol; and organic solvents such as ethers such as diethyl ether, diisopropyl ether and tetrahydrofuran.
- the solvent is preferably an alcohol, more preferably an alcohol having 3 to 10 carbon atoms.
- the alcohol having 3 to 10 carbon atoms include n-propanol, isopropanol, n-butanol, tert-butyl alcohol, isobutanol, sec-butyl alcohol, n-amyl alcohol, isoamyl alcohol, tert-amyl alcohol, hexanol, Aliphatic alcohols such as 2-methyl-2-butanol, 3-methyl-3-pentanol, 2-ethylhexanol, heptanol, octanol, and nonanol; alicyclic alcohols such as cyclohexanol, methylcyclohexanol, and cyclopentanol; Aromatic alcohols such as benzyl alcohol can be mentioned, but are not limited thereto.
- isopropanol, isobutanol, sec-butyl alcohol, tert-butyl alcohol, isoamyl alcohol, and tert-amyl alcohol are preferable, and tert-butyl alcohol is preferable from the viewpoint of uniformly dissolving ⁇ -olefin (I) and formaldehyde. More preferred.
- a solvent may be used individually by 1 type and may use 2 or more types together. In addition, other solvents may be used in combination as long as the reaction is not adversely affected.
- the amount of the solvent used is preferably 0.5 to 20 moles, more preferably 1 to 10 moles per mole of formaldehyde. If the amount of solvent used is 0.5 mol or more with respect to 1 mol of formaldehyde, by-product formation of alkyl-m-dioxane can be suppressed, and if it is 20 mol or less, the scale of distillation equipment for separation and recovery In addition, the industrial value is improved because the amount of steam and electric power used as a heat source can be simplified.
- the reaction temperature in step A is preferably 150 to 350 ° C., more preferably 200 to 330 ° C., and further preferably 240 to 310 ° C. If it is 150 ° C or higher, the reaction rate is high and the reaction time can be shortened. If it is 350 ° C or lower, the decomposition reaction of formaldehyde and the produced ⁇ , ⁇ -unsaturated alcohol is suppressed, and the intended ⁇ , ⁇ -unsaturation is achieved. The yield of saturated alcohol is improved.
- the reaction time is appropriately determined depending on the reaction temperature, but the reaction is usually completed in 1 minute to 30 minutes. Therefore, even when the reaction is performed continuously as described later, the residence time in the reaction tube may be 1 to 30 minutes.
- the reaction pressure in step A may be usually equal to or higher than the vapor pressure at the reaction temperature of ⁇ -olefin (I), but when ⁇ -olefin (I) exceeding the critical condition at a predetermined temperature is used, the pressure may be increased as necessary. It is recommended to control.
- the reaction pressure is preferably 3 to 50 MPa, more preferably 3 to 30 MPa, still more preferably 5 to 30 MPa, and particularly preferably 10 to 30 MPa. If the reaction pressure is equal to or higher than the vapor pressure of ⁇ -olefin (I) at a predetermined temperature, the concentration of ⁇ -olefin (I) in the reaction solution increases, and the reaction rate and selectivity of ⁇ , ⁇ -unsaturated alcohol are increased. Bring improvement. In addition, by controlling the reaction pressure to 50 MPa or less, the construction cost of the pressure-resistant equipment can be suppressed, and the risk of the reactor bursting is also reduced.
- step A it is preferable to use a reactor capable of controlling the reaction temperature, reaction time and reaction pressure.
- the reaction in step A can be carried out by any method such as batch, semi-batch and continuous methods. Among them, it is preferable to carry out the reaction in a continuous manner in which the conversion rate of formaldehyde, the selectivity of ⁇ , ⁇ -unsaturated alcohol and the yield are increased.
- a specific and preferred embodiment in the case of carrying out the reaction in a continuous manner is as follows. A mixed solution containing ⁇ -olefin (I), an aqueous formaldehyde solution and a solvent in a predetermined ratio is sent to a reaction tube heated to a predetermined temperature at a desired flow rate.
- the reaction pressure is adjusted so that the outlet of the cooling pipe connected to the outlet of the reaction tube is kept at a predetermined pressure, the mixed solution is allowed to stay in the reaction tube for a predetermined time, and the reaction solution is allowed to flow out from the outlet of the reaction tube.
- ⁇ -olefin (I) remains in the obtained reaction liquid, it is preferable to fractionate ⁇ -olefin (I) and use it again as a raw material.
- Step A ′ cleaning
- cleaning method in process A ' is not specifically limited, It is preferable to perform the alkali washing
- the method of bringing the reaction solution into contact with the alkaline aqueous solution there is no particular limitation on the method of bringing the reaction solution into contact with the alkaline aqueous solution.
- a method of introducing the reaction solution and the alkaline aqueous solution into a container equipped with a stirrer and stirring them (batch type)
- a method (continuous method) in which the reaction solution and the aqueous alkali solution are continuously brought into contact with each other in the column by a countercurrent method preferably a complete countercurrent method
- alkali metal hydroxide, alkali metal carbonate, alkali metal acetate, alkali metal phosphate, alkaline earth metal hydroxide, alkaline earth metal carbonate, alkaline earth metal It is preferable to use at least one selected from acetates of the following and alkaline earth metal phosphates.
- sodium hydroxide, potassium hydroxide, sodium carbonate, and potassium carbonate are preferred from the viewpoints of availability, removal efficiency of formic acid and formate esters, and selectivity and yield of ⁇ , ⁇ -unsaturated alcohol.
- Sodium is more preferred.
- An alkali may be used individually by 1 type and may use 2 or more types together.
- an “aqueous solution” of alkali is used from the viewpoints of removal efficiency of formic acid and formic acid ester and selectivity and yield of ⁇ , ⁇ -unsaturated alcohol.
- concentration of alkali in the aqueous alkali solution is not particularly limited, but it is 0.01 from the viewpoint of ease of handling, removal efficiency of formic acid and formate, and selectivity and yield of ⁇ , ⁇ -unsaturated alcohol.
- 0.1 to 20 mol / L is more preferable, 0.1 to 10 mol / L is further preferable, and 0.1 to 5 mol / L is particularly preferable.
- the pH of the aqueous solution in the resulting solution is adjusted to 9 to 13, preferably 10 to 13, more preferably 11 to 13, and more preferably 12 to 13. Adjust to.
- the pH of the aqueous solution in the solution obtained by bringing the alkaline aqueous solution into contact with the reaction solution is from the viewpoint of the removal efficiency of formic acid and formic acid ester, and the selectivity and yield of ⁇ , ⁇ -unsaturated alcohol. If it is less than pH 9, it is inadequate and needs to show stronger alkalinity.
- the temperature at which the reaction solution is brought into contact with the aqueous alkali solution is not particularly limited, but is 10 to 90 ° C. from the viewpoint of the removal efficiency of formic acid and formate esters and the selectivity and yield of ⁇ , ⁇ -unsaturated alcohol.
- 20 to 90 ° C is more preferable, 35 to 85 ° C is more preferable, and 50 to 80 ° C is particularly preferable.
- the contact time between the reaction solution and the aqueous alkali solution may be any length as long as it can sufficiently remove the formic acid and the ester derived from the formic acid in the raw material solution, but preferably 2 minutes to 600 minutes. Preferably it is 5 minutes to 500 minutes.
- the time in the column is adjusted to 2 to 600 minutes (preferably 5 to 500 minutes, more preferably 30 to 500 minutes). It is preferable to do this.
- the reaction solution that has undergone alkali cleaning has a very low content of formic acid as an impurity, so there is no risk of corrosion of the apparatus due to formic acid.
- formic acid formic acid
- ⁇ , ⁇ -unsaturated alcohols have a high boiling point due to formic acid. The yield can be maintained high without fear of compounding.
- step A After passing through step A and step A ′ as necessary, purification yields a higher purity ⁇ , ⁇ -unsaturated alcohol.
- the purification method is not particularly limited, and may be purified by column chromatography after separating the organic layer, but distillation purification is preferred when it is carried out industrially continuously.
- the theoretical number of distillation columns is preferably 10 to 60, more preferably 10 to 40, and still more preferably 10 to 30.
- the reflux ratio is preferably 0.5 to 1.5, more preferably 0.7 to 1.2.
- distillation purification at 100 to 180 ° C. and 3 to 10 kPa is preferable, and distillation purification at 120 to 160 ° C. and 3 to 7 kPa is preferable. More preferred.
- high-purity ⁇ , ⁇ -unsaturated alcohol can be obtained by a single distillation operation using one distillation column, or a plurality of distillation operations using two or more distillation columns. Impurities may be separated stepwise in two or more steps, and the purity of the ⁇ , ⁇ -unsaturated alcohol may be gradually increased.
- step B the ⁇ , ⁇ -unsaturated alcohol obtained in step A and optionally step A ′ is dehydrated in the presence of an acidic catalyst to give the following general formula (III) (Wherein R 1 , R 2 and R 3 are as defined above.) Is a step of producing a conjugated diene represented by
- Acid catalysts include phosphoric acid, phosphorous acid, nitric acid, sulfuric acid, hydrogen chloride, hydrogen bromide, hydrogen fluoride, hypochlorous acid, chlorous acid, chloric acid, perchloric acid, boric acid, fluorosulfonic acid, benzenesulfone Examples thereof include acid, p-toluenesulfonic acid, methanesulfonic acid, trifluoromethanesulfonic acid, trifluoroacetic acid, and oxalic acid.
- phosphoric acid sulfuric acid, boric acid, p-toluenesulfonic acid, and methanesulfonic acid are preferable, and phosphoric acid is more preferable from the viewpoint of selectivity of the conjugated diene.
- the acidic catalyst is preferably used as an aqueous solution from the viewpoint of suppressing the generation of high-boiling byproducts in the reaction system.
- the concentration of the acidic catalyst in the reaction system is preferably 0.2 to 10% by mass, more preferably 0.5 to 8% by mass, more preferably 1 to 5% by mass, and further preferably 1 to 4% by mass. . If the concentration of the acidic catalyst in the reaction system is within this range, the formation of high-boiling byproducts can be suppressed, the selectivity of the conjugated diene can be increased, and the conversion rate of ⁇ , ⁇ -unsaturated alcohol can be increased. Can be kept high.
- the acidic catalyst aqueous solution is usually charged into the reactor before the reaction starts, and the acidic catalyst concentration of the acidic catalyst aqueous solution at that time is preferably in the above range.
- the water supply rate to the reactor is preferably adjusted so that the ratio is within a certain range with respect to the supply rate of ⁇ , ⁇ -unsaturated alcohol.
- the ⁇ water supply rate (mol / hour) / ⁇ , ⁇ -unsaturated alcohol supply rate (mol / hour) ⁇ is usually 0.8 to 12, preferably 0.8 to 10.
- the reaction temperature in Step B is 135 to 210 ° C. If it is 135 ° C. or higher, the reaction rate is high, and conjugated dienes can be obtained with high selectivity. If it is 210 ° C. or less, side reactions of the produced conjugated dienes can be suppressed and yield reduction can be prevented. Since the amount used can be suppressed, the industrial value is improved.
- the reaction temperature is preferably 135 to 200 ° C, more preferably 135 to 195 ° C.
- the reaction pressure is usually 0.35 to 1.6 MPa.
- the reaction pressure is preferably 0.5 to 1.4 MPa, more preferably 0.5 to 1.2 MPa, and still more preferably 0.5 to 1.0 MPa. If the reaction pressure is less than 0.35 MPa, it becomes difficult to set the reaction temperature to 135 ° C. or higher.
- the reaction in step B is preferably carried out in an inert gas atmosphere such as nitrogen or argon, and the inside of the reaction system is preferably pressurized to some extent with an inert gas.
- an inert gas atmosphere such as nitrogen or argon
- the pressure in the reactor continues to increase due to the effect of the conjugated diene produced and water. Accordingly, by appropriately releasing the gas, the reaction pressure is adjusted to be within the above range.
- Step B the ⁇ , ⁇ -unsaturated alcohol and water are continuously supplied to the reactor at the predetermined ratio from the viewpoint of stabilizing the reaction pressure and reaction results, and the produced conjugated diene and water are continuously added. It is preferable to distill out of the reaction system.
- water include (i) water of an acidic catalyst aqueous solution, (ii) water generated by a dehydration reaction, (iii) water supplied to the reactor together with ⁇ , ⁇ -unsaturated alcohol, and (iv) distilled from the reactor.
- water to be discharged it is preferable to adjust various reaction conditions so that the amount of water in the reaction system is always almost constant from the viewpoint of stabilizing the reaction results.
- the reaction system is sealed until the pressure in the reactor reaches a predetermined value, and when the target pressure is reached, gas (nitrogen, conjugated diene and water vapor are added so as to maintain the pressure.
- gas nitrogen, conjugated diene and water vapor are added so as to maintain the pressure.
- the pressure in the reactor can be kept at a predetermined value by adjusting the gas contained) to the outside of the reaction system.
- the total distillate rate of conjugated diene and water distilled from the reactor (however, converted to a liquid form obtained by cooling after distillation) is the ⁇ , ⁇ -unsaturated alcohol fed to the reactor.
- the mass is preferably 0.8 to 1.2 times the mass of the total supply rate of water, and more preferably 0.9 to 1.1 times the mass.
- Example 1 [Step A] A stainless steel reaction tube heated to 280 ° C. and having an inner diameter of 2 mm and a length of 3180 mm (internal volume: 10 mL) was charged with 2.4% by mass of formaldehyde, 2.4% by mass of water, 66.1% by mass of isobutene and tert-butyl alcohol (organic). Solvent) A mixed solution consisting of 29.1% by mass was fed at 1 mL / min. Here, the isobutene: tert-butyl alcohol: formaldehyde (molar ratio) in the mixed solution is 15: 5: 1, and the residence time of the mixed solution is 10 minutes.
- the outlet of the reaction tube was connected to a cooling tube having an inner diameter of 2 mm and a length of 2,000 mm, the outlet pressure of the cooling tube was kept at 20 MPa, and the reaction solution was allowed to flow out.
- the conversion of formaldehyde was 81.6%, and the selectivity for 3-methyl-3-buten-1-ol was 90.5%.
- by-product formic acid was 1.4%, and formate was 0.8%.
- Step A ′ To 100 g of the reaction solution obtained in step A, 12 mL of a 1 mol / L sodium hydroxide aqueous solution (corresponding to 12 mmol of sodium hydroxide) was added and stirred at 70 ° C. for 5 minutes.
- the organic layer (upper layer) was subjected to gas chromatography analysis, neither formic acid nor 3-methyl-3-buten-1-ol formate remained in the organic layer.
- the organic layer (upper layer) after alkali washing was purified by distillation under the conditions of 20 theoretical plates, reflux ratio of 1, bath temperature of 140 ° C. and pressure of 5.3 kPa. As a result, 3-methyl-3 having a purity of 99.4% by mass was obtained.
- -Buten-1-ol was obtained with a distillation yield of 95.0%.
- Step B After putting 300 g of 1.8 mass% phosphoric acid aqueous solution into a 500 mL autoclave made of Hastelloy and replacing the inside of the autoclave with nitrogen, the pressure was increased to 0.7 MPa with nitrogen, and heating and stirring (1000 times / min) were started.
- the internal pressure reached 0.85 MPa, gas release in the autoclave was started so as to maintain the pressure.
- the released gas was continuously cooled by a condenser attached to the autoclave to agglomerate the reaction product, and the reaction product was received in a tank and operated continuously for 4 hours.
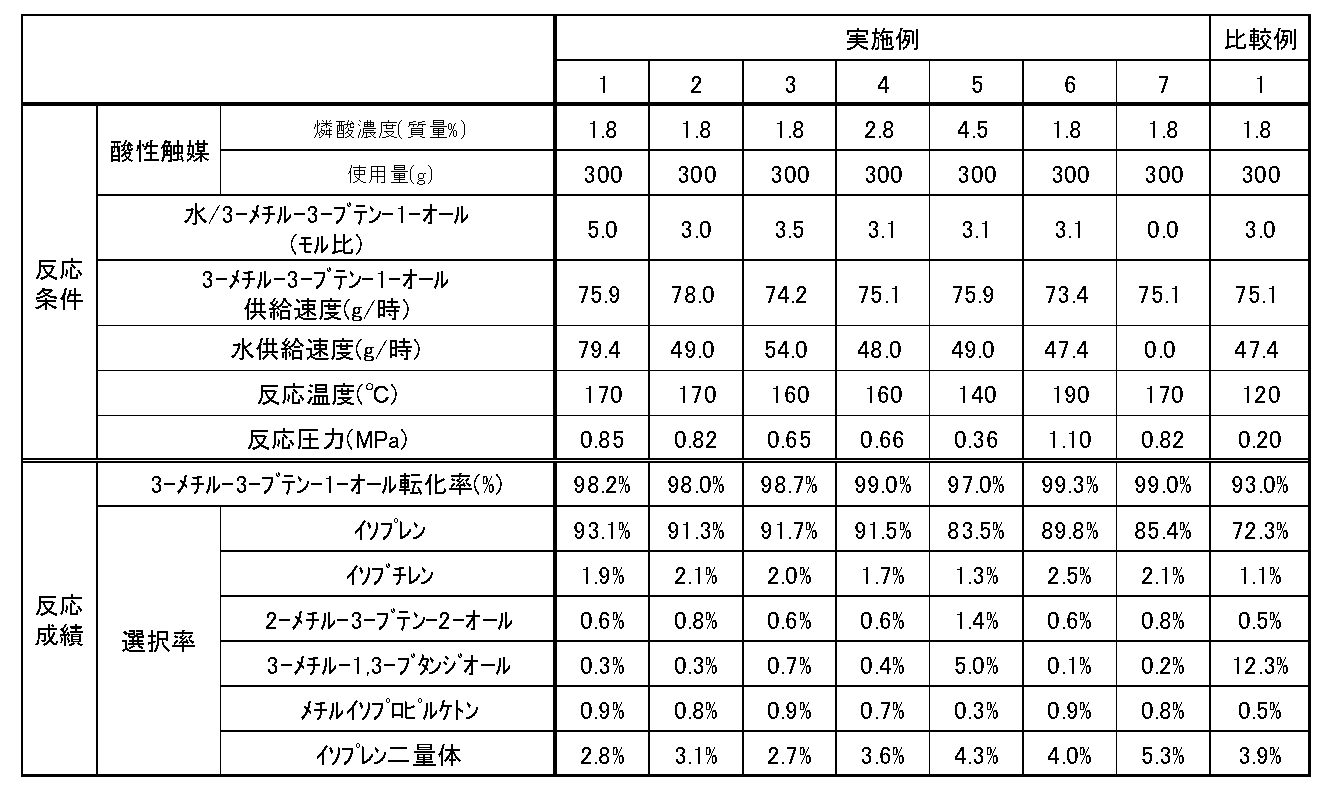
- the conversion of 3-methyl-3-buten-1-ol was 98.2%, and the selectivity for isoprene was 93.1%. Met.
- the selectivity for isobutene as a by-product is 1.9%
- the selectivity for 2-methyl-3-buten-2-ol is 0.6%
- methyl isopropyl ketone selectivity was 0.9%
- isoprene dimer selectivity was 2.8%.
- Table 1 The results are shown in Table 1.
- Step B of Example 1 the phosphoric acid concentration, the reaction temperature, the reaction pressure, the ratio of water to 3-methyl-3-buten-1-ol, and the feed rates of 3-methyl-3-buten-1-ol and water were determined. The same operation was performed except that the changes were made as shown in Table 1. The results are shown in Table 1.
- step B of Example 1 the same operation was performed except that the type of acidic catalyst was changed as shown in Table 2. The results are shown in Table 2.
- ⁇ , ⁇ -unsaturated alcohol can be obtained from ⁇ -olefin and formaldehyde with a selectivity of 90.5%, and purified with a distillation yield of 95.0%.
- a conjugated diene was obtained with a selectivity of 93.1%.
- the total selectivity was 80.0%, and it was found that the desired conjugated diene can be produced with high selectivity.
- the conjugated diene obtained by the production method of the present invention is useful as various chemicals and polymer raw materials.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
[1]α-オレフィンとホルムアルデヒドとを反応させてγ,δ-不飽和アルコールを製造する工程Aと、酸性触媒水溶液の存在下、γ,δ-不飽和アルコールを135~210℃にて脱水反応させる工程Bを有する、共役ジエンの製造方法。
[2]前記工程Aにおいて、溶媒を共存させる、[1]の製造方法。
[3]前記溶媒がアルコールである、[2]の製造方法。
[4]前記α-オレフィンがイソブテンであり、かつ前記γ,δ-不飽和アルコールが3-メチル-3-ブテン-1-オールである、[1]~[3]のいずれかの製造方法。
[5]前記工程Bにおいて、γ,δ-不飽和アルコールと水を共に反応器へ連続的または断続的に供給し、かつ生成した共役ジエンおよび水を連続的または断続的に反応系外へ取り出すことを特徴とする、[1]~[4]のいずれかの製造方法。
[6]{水の供給速度(モル/時)/γ,δ-不飽和アルコールの供給速度(モル/時)}が0.5~12である、[5]の製造方法。
[7]前記酸性触媒が燐酸である、[1]~[6]のいずれかの製造方法。
[8]前記脱水反応における反応圧力が0.35~1.6MPaである、[1]~[7]のいずれかの製造方法。
工程Aは、下記一般式(I)

で示されるα-オレフィン(以下、α-オレフィン(I)と称する)とホルムアルデヒドとを反応させ、下記一般式(II)
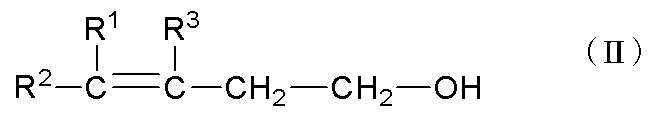
で示されるγ,δ-不飽和アルコールを製造する工程である。
R1、R2およびR3がそれぞれ独立して表す炭素数1~10のアルキル基としては、例えばメチル基、エチル基、各種プロピル基(「各種」とは、直鎖状およびあらゆる分岐鎖状を含むことを示し、以下同様である。)各種ブチル基、各種ヘキシル基、各種オクチル基、各種デシル基などが挙げられる。中でも炭素数1~5のアルキル基が好ましく、炭素数1~3のアルキル基がより好ましく、メチル基がさらに好ましい。R1とR3が互いに連結して環を形成している場合の環としては、シクロペンタン環、シクロヘキサン環、シクロオクタン環などの炭素数5~10の飽和脂肪族環が好ましく、シクロヘキサン環がより好ましい。
R1、R2およびR3としては、R1およびR2のうちの少なくとも一方が水素原子であり、且つR3が炭素数1~10のアルキル基であることがさらに好ましく、R1およびR2がいずれも水素原子であり、且つR3が炭素数1~10のアルキル基であることが特に好ましい。アルキル基としてより好ましいものは前述の通りである。
(1)R1、R2およびR3がいずれも水素原子であるプロピレン、
(2)R1およびR2が水素原子、且つR3が炭素数1~10のアルキル基である例としてのイソブテン、
(3)R1およびR2のうちの一方が水素原子、且つR3が炭素数1~10のアルキル基である例としての2-メチル-1-ブテン、2-メチル-1-ペンテン、2-メチル-1-ヘキセン、2-メチル-1-ヘプテン、2-メチル-1-オクテン、
(4)R1、R2およびR3がいずれも炭素数1~10のアルキル基である例としての2,3-ジメチル-1-ブテン、
(5)R2が水素原子、且つR1とR3とが互いに連結して環を形成している例としてのメチレンシクロヘキサンなどが挙げられる。
工程Aにおいて、ホルムアルデヒドはそのまま用いてもよく、溶媒に溶解して使用してもよい。ホルムアルデヒドを溶解させる溶媒は特に限定されないが、入手容易性から水であること、つまりホルムアルデヒド水溶液を用いることが好ましい。この場合、容積効率の観点からホルムアルデヒドの濃度は高いことが好ましいが、濃度が高すぎると析出の問題が生じて取り扱いが難しくなることから、ホルムアルデヒド溶液のホルムアルデヒドの濃度は、通常10~70質量%、好ましくは30~60質量%である。
工程Aの反応は、溶媒の存在下または非存在下に実施することができるが、溶媒の存在下に実施するのが好ましい。反応に悪影響を与えない限り用いる溶媒に特に制限はなく、ペンタン、ヘキサン、ヘプタン、オクタン、ノナン、デカン、シクロヘキサン、シクロオクタン等の脂肪族炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;メタノール、エタノール、tert-ブチルアルコール等のアルコール;ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン等のエーテルなどの有機溶媒が挙げられる。
前述したホルムアルデヒドを水溶液として用いる場合、溶媒としてはアルコールが好ましく、炭素数3~10のアルコールがより好ましい。炭素数3~10のアルコールとしては、例えば、n-プロパノール、イソプロパノール、n-ブタノール、tert-ブチルアルコール、イソブタノール、sec-ブチルアルコール、n-アミルアルコール、イソアミルアルコール、tert-アミルアルコール、ヘキサノール、2-メチル-2-ブタノール、3-メチル-3-ペンタノール、2-エチルヘキサノール、ヘプタノール、オクタノール、ノナノール等の脂肪族アルコール;シクロヘキサノール、メチルシクロヘキサノール、シクロペンタノール等の脂環式アルコール;ベンジルアルコール等の芳香族アルコールなどが挙げられるが、これらに限定されない。α-オレフィン(I)とホルムアルデヒドとを均一に溶解する観点から、前記の中でもイソプロパノール、イソブタノール、sec-ブチルアルコール、tert-ブチルアルコール、イソアミルアルコール、tert-アミルアルコールが好ましく、tert-ブチルアルコールがより好ましい。
溶媒は1種を単独で使用してもよいし、2種以上を併用してもよい。また、本反応に悪影響を及ぼさない限りその他の溶媒を併用してもよい。
工程Aにおける反応温度は150~350℃が好ましく、200~330℃がより好ましく、240~310℃がさらに好ましい。150℃以上であれば反応速度が大きく、反応時間が短縮でき、350℃以下であれば、ホルムアルデヒドおよび生成したγ,δ-不飽和アルコールの分解反応が抑制され、目的とするγ,δ-不飽和アルコールの収率が向上する。
反応時間は反応温度により適宜決定されるが、通常1分~30分で反応は完結する。よって、後述するように連続式にて反応を行なう場合でも、反応管内の滞留時間は1分~30分でよい。
連続式で反応を行なう場合の具体的且つ好ましい実施形態は次の通りである。所定温度に加熱した反応管に、α-オレフィン(I)、ホルムアルデヒド水溶液および溶媒を所定比率で含有する混合溶液を所望の流速で送液する。反応管の出口につないだ冷却管の出口を所定圧力に保つように反応圧力を調整しておき、前記混合溶液を反応管内に所定時間滞留させ、反応管の出口から反応液を流出させながら反応を行なう。得られた反応液中にα-オレフィン(I)が残存している場合には、α-オレフィン(I)を分取し、再度、原料として使用することが好ましい。
本発明の製造方法では、工程Aの後、得られた反応液を洗浄する工程(工程A’)を有してもよい。工程A’における洗浄方法は特に限定されないが、アルカリ水溶液と接触させる工程を有するアルカリ洗浄を行うことが好ましい。得られた反応液とアルカリ水溶液を接触させた後に分離することにより、副生成物として発生するギ酸およびギ酸エステルの除去とγ,δ-不飽和アルコールの収率向上を同時に達成できる。これは、得られた反応液中のギ酸およびギ酸エステルがいずれもギ酸塩となって除去されてγ,δ-不飽和アルコールの純度が高まること、また、反応液中のギ酸エステルはギ酸とγ,δ-不飽和アルコールとの縮合物であるため、分解により目的物であるγ,δ-不飽和アルコールが形成されることによるものと推測される。ギ酸は、蒸留精製の際にその存在によってγ,δ-不飽和アルコールが高沸点化合物化し易くなること、およびギ酸エステルは、蒸留によってγ,δ-不飽和アルコールから分離することが困難であることが判明しており、本工程にてギ酸およびギ酸エステルの混入量を十分に低減できることの意義は大きい。
反応液をアルカリ水溶液と接触させる方法に特に制限はなく、例えば、(i)攪拌装置を備えた容器に反応液とアルカリ水溶液とを導入し、両者を攪拌する方法(バッチ式)、(ii)塔内で反応液とアルカリ水溶液とを向流方式(好ましくは完全向流方式)にて連続的に接触させる方法(連続式)などを採用できる。
アルカリは、1種を単独で用いてもよいし、2種以上を併用してもよい。
反応液とアルカリ水溶液との接触時間は、十分に原料液中のギ酸とそのギ酸由来のエステルを取り除くことができればどのような長さであってもよいが、好ましくは2分~600分、より好ましくは5分~500分である。なお、前述の完全向流方式を採用する場合には、塔内に存在する時間が2分~600分(好ましくは5分~500分、より好ましくは30分~500分)となるように調整するのが好ましい。
工程Aおよび必要に応じて工程A’を経た後、精製によってより高純度のγ,δ-不飽和アルコールが得られる。
工程Bは、工程Aおよび場合により工程A’により得られたγ,δ-不飽和アルコールを、酸性触媒の存在下で脱水させて、下記一般式(III)
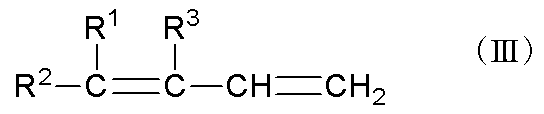
で示される共役ジエンを製造する工程である。
酸性触媒としては、燐酸、亜燐酸、硝酸、硫酸、塩化水素、臭化水素、フッ化水素、次亜塩素酸、亜塩素酸、塩素酸、過塩素酸、ホウ酸、フルオロスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、メタンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸、シュウ酸などが挙げられる。これらの中でも、共役ジエンの選択率の観点から、燐酸、硫酸、ホウ酸、p-トルエンスルホン酸、メタンスルホン酸が好ましく、燐酸がより好ましい。
工程Bにおける反応では、γ,δ-不飽和アルコールと共に水を反応器へ供給することが好ましい。これによって、反応系内の酸性触媒の濃度の変動を抑制し、同時に、反応系内の圧力を一定値に維持し易くすることができる。反応器への水の供給速度は、工業的に安定して生産する観点から、γ,δ-不飽和アルコールの供給速度に対して一定範囲の比率になるよう調整することが好ましい。この観点から、{水の供給速度(モル/時)/γ,δ-不飽和アルコールの供給速度(モル/時)}は通常0.8~12であり、好ましくは0.8~10であり、より好ましくは0.9~9であり、さらに好ましくは1~8であり、特に好ましくは2~7であり、最も好ましくは4~6である。
使用する水としては特に制限は無く、例えばイオン交換水や蒸留水を好適に用いることができる。また、反応で生成する水を回収して用いても良い。
工程Bにおける反応温度は、135~210℃である。135℃以上であれば反応速度が大きく、共役ジエンを高い選択率で得ることができ、210℃以下であれば、生成した共役ジエンの副反応が抑制され収率低下を防げること、また熱源の使用量を抑制できることから工業的価値が向上する。反応温度は135~200℃が好ましく、135~195℃がより好ましい。
なお、各例において、ガスクロマトグラフィー分析は以下の条件にて実施した。
[ガスクロマトグラフィー分析条件]
分析機器:GC14A(株式会社島津製作所製)
検出器:FID(水素炎イオン化型検出器)
使用カラム:DB-1(30m,膜厚5μm)(J&W Scientific社製)
分析条件:注入口温度280℃、検出器温度280℃
昇温条件:〈γ,δ-不飽和アルコール〉70℃(0分保持)→(5℃/分で昇温)→250℃(4分保持)、〈共役ジエン〉40℃(10分保持)→(5℃/分で昇温)→250℃(4分保持)
[工程A]
280℃に加熱した内径2mm、長さ3180mm(内容量10mL)のステンレス製反応管に、ホルムアルデヒド2.4質量%、水2.4質量%、イソブテン66.1質量%およびtert-ブチルアルコール(有機溶媒)29.1質量%からなる混合溶液を1mL/分で送液した。ここで、該混合溶液中のイソブテン:tert-ブチルアルコール:ホルムアルデヒド(モル比)は、15:5:1であり、混合溶液の滞留時間は10分間である。反応管の出口を内径2mmおよび長さ2,000mmの冷却管につなぎ、冷却管の出口圧力を20MPaに保ち、反応液を流出させた。
得られた反応液をガスクロマトグラフィーにより分析した結果、ホルムアルデヒドの転化率は81.6%、3-メチル-3-ブテン-1-オールの選択率は90.5%であった。また、副生成物であるギ酸は1.4%、ギ酸エステルは0.8%であった。
工程Aで得られた反応液100gに1mol/Lの水酸化ナトリウム水溶液を12mL(水酸化ナトリウム12mmol相当)加えて、70℃で5分撹拌した。有機層(上層)についてガスクロマトグラフィー分析を行なったところ、ギ酸、および3-メチル-3-ブテン-1-オールのギ酸エステルは、いずれも有機層には残存していなかった。
アルカリ洗浄後の有機層(上層)を、理論段数20、還流比1、バス温度140℃および圧力5.3kPaの条件で蒸留精製を行なったところ、純度99.4質量%の3-メチル-3-ブテン-1-オールが蒸留収率95.0%で得られた。
ハステロイ製の500mLオートクレーブに1.8質量%燐酸水溶液300gを入れ、オートクレーブ内を窒素置換した後に、窒素にて0.7MPaに加圧し、加熱と撹拌(1000回/分)を開始した。内温が170℃になったところで、工程A’で得られた3-メチル-3-ブテン-1-オールを75.9g/時、水を79.4g/時[水/3-メチル-3-ブテン-1-オール=5.0/1.0(モル比)]にてオートクレーブ内へ供給した。
内圧が0.85MPaになったところで、当該圧力を保つようにオートクレーブ内のガスの放出を開始した。放出したガスを、オートクレーブに付属しているコンデンサで連続的に冷却して反応生成物を凝集し、該反応生成物をタンクに受け入れながら、4時間連続で運転を行った。
タンク内の有機層および反応器内の反応液をガスクロマトグラフィーにより分析した結果、3-メチル-3-ブテン-1-オールの転化率は98.2%、イソプレンの選択率は93.1%であった。また、副生成物であるイソブテンの選択率は1.9%、2-メチル-3-ブテン-2-オールの選択率は0.6%、3-メチル-1,3-ブタンジオールの選択率は0.3%、メチルイソプロピルケトンの選択率は0.9%、イソプレン二量体の選択率は2.8%であった。結果を表1に示す。
実施例1の工程Bにおいて、燐酸濃度、反応温度、反応圧力、水と3-メチル-3-ブテン-1-オールの比率、3-メチル-3-ブテン-1-オールおよび水の供給速度を表1に示すとおりに変更したこと以外は同様に操作を行なった。結果を表1に示す。
実施例1の工程Bにおいて、酸性触媒の種類を表2に示すとおりに変更したこと以外は同様に操作を行なった。結果を表2に示す。

Claims (8)
- α-オレフィンとホルムアルデヒドとを反応させてγ,δ-不飽和アルコールを製造する工程Aと、酸性触媒水溶液の存在下、γ,δ-不飽和アルコールを135~210℃にて脱水反応させる工程Bを有する、共役ジエンの製造方法。
- 前記工程Aにおいて、溶媒を共存させる、請求項1に記載の製造方法。
- 前記溶媒がアルコールである、請求項2に記載の製造方法。
- 前記α-オレフィンがイソブテンであり、かつ前記γ,δ-不飽和アルコールが3-メチル-3-ブテン-1-オールである、請求項1~3のいずれかに記載の製造方法。
- 前記工程Bにおいて、γ,δ-不飽和アルコールと水を共に反応器へ連続的または断続的に供給し、かつ生成した共役ジエンおよび水を連続的または断続的に反応系外へ取り出すことを特徴とする、請求項1~4のいずれかに記載の製造方法。
- {水の供給速度(モル/時)/γ,δ-不飽和アルコールの供給速度(モル/時)}が0.5~12である、請求項5に記載の製造方法。
- 前記酸性触媒が燐酸である、請求項1~6のいずれかに記載の製造方法。
- 前記脱水反応における反応圧力が0.35~1.6MPaである、請求項1~7のいずれかに記載の製造方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MYPI2017704587A MY183026A (en) | 2015-06-03 | 2016-06-01 | Method for producing conjugated diene |
| SG11201709802XA SG11201709802XA (en) | 2015-06-03 | 2016-06-01 | Method for producing conjugated diene |
| EP16803417.1A EP3305747B1 (en) | 2015-06-03 | 2016-06-01 | Method for producing conjugated diene |
| CN201680031521.0A CN107614464A (zh) | 2015-06-03 | 2016-06-01 | 共轭二烯的制造方法 |
| US15/577,988 US11198657B2 (en) | 2015-06-03 | 2016-06-01 | Method for producing conjugated diene |
| RU2017146189A RU2723241C2 (ru) | 2015-06-03 | 2016-06-01 | Способ производства сопряженных диенов |
| KR1020177034292A KR102486605B1 (ko) | 2015-06-03 | 2016-06-01 | 공액 디엔의 제조 방법 |
| JP2017522225A JP6687281B2 (ja) | 2015-06-03 | 2016-06-01 | 共役ジエンの製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015113006 | 2015-06-03 | ||
| JP2015-113006 | 2015-06-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016194983A1 true WO2016194983A1 (ja) | 2016-12-08 |
Family
ID=57440365
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/066297 Ceased WO2016194983A1 (ja) | 2015-06-03 | 2016-06-01 | 共役ジエンの製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US11198657B2 (ja) |
| EP (1) | EP3305747B1 (ja) |
| JP (1) | JP6687281B2 (ja) |
| KR (1) | KR102486605B1 (ja) |
| CN (1) | CN107614464A (ja) |
| MY (1) | MY183026A (ja) |
| RU (1) | RU2723241C2 (ja) |
| SA (1) | SA517390417B1 (ja) |
| SG (1) | SG11201709802XA (ja) |
| WO (1) | WO2016194983A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017191794A1 (ja) * | 2016-05-06 | 2017-11-09 | 株式会社クラレ | 共役ジエンの製造方法 |
| JP2020530468A (ja) * | 2017-08-11 | 2020-10-22 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 3−メチルブタ−3−エン−1−オールの回収方法 |
| RU2828416C1 (ru) * | 2023-11-23 | 2024-10-11 | Общество с ограниченной ответственностью "Оргнефтехим-Холдинг" | Способ производства изопрена из изобутилена и формальдегида без выделения промежуточных продуктов |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020058119A1 (en) * | 2018-09-18 | 2020-03-26 | Basf Se | Process to recover high quality 3-methyl-but-3-en-1-ol |
| CN110963882A (zh) * | 2018-09-29 | 2020-04-07 | 万华化学集团股份有限公司 | 一种制备2-甲基-1,3-戊二烯的方法 |
| CN112430178B (zh) * | 2021-01-26 | 2021-06-22 | 天津安德胜科技服务有限公司 | 一种工业化异戊烯醇的生产工艺 |
| CN115007181B (zh) * | 2022-07-18 | 2023-12-19 | 中国科学院长春应用化学研究所 | 一种催化合成异戊二烯的催化剂及其制备方法,以及异戊二烯的制备方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59184137A (ja) * | 1983-04-04 | 1984-10-19 | Kuraray Co Ltd | イソプレンの製造方法 |
| JPH071714A (ja) * | 1993-02-01 | 1995-01-06 | Stork Contiweb Bv | 短縮された再スタートのドライヤー |
| JPH0714105A (ja) * | 1993-06-22 | 1995-01-17 | Matsushita Electric Ind Co Ltd | デジタル磁気記録再生装置 |
| JPH07285899A (ja) * | 1994-04-18 | 1995-10-31 | Kuraray Co Ltd | γ,δ−不飽和アルコールの製造方法 |
| WO2004087625A1 (ja) * | 2003-03-31 | 2004-10-14 | Kuraray Co., Ltd. | イソプレンの製造方法 |
| JP2013075877A (ja) * | 2011-09-30 | 2013-04-25 | Kuraray Co Ltd | イソプレンの製造方法 |
| WO2015186699A1 (ja) * | 2014-06-02 | 2015-12-10 | 株式会社クラレ | γ,δ-不飽和アルコールの製造方法 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2626284A (en) * | 1949-09-17 | 1953-01-20 | Standard Oil Dev Co | Aqueous caustic treat of iso-octyl alcohol |
| DE1275049B (de) * | 1967-02-25 | 1968-08-14 | Basf Ag | Verfahren zur Herstellung von 3-Methylbuten-(3)-ol-(1) |
| US3544603A (en) * | 1967-02-27 | 1970-12-01 | Pilot Chem Co | Alkyl-tetrahydropyranols and ethoxylated alkyl-tetrahydropyranols |
| GB1205397A (en) | 1967-05-26 | 1970-09-16 | British Petroleum Co | Production of isoprene |
| DE1928632A1 (de) | 1969-06-06 | 1970-12-10 | Basf Ag | Verfahren zur Herstellung von Isopren |
| DE2031900C3 (de) | 1970-06-27 | 1979-02-01 | Basf Ag, 6700 Ludwigshafen | Verfahren zur Herstellung von Isopren |
| DE2061803C3 (de) | 1970-12-16 | 1979-03-22 | Basf Ag, 6700 Ludwigshafen | Verfahren zur Herstellung von Isopren |
| JPS5242763B2 (ja) * | 1971-01-12 | 1977-10-26 | ||
| DE2116948A1 (en) | 1971-04-07 | 1972-10-19 | Badische Anilin- & Soda-Fabrik Ag, 6700 Ludwigshafen | Isoprene prepn - by dehydrating 3-methyl-3-buten-1-ol with acids giving quantitative yields of highly pure prods |
| JPS471571A (ja) | 1971-06-25 | 1972-01-26 | ||
| JPS5236725B2 (ja) | 1973-05-07 | 1977-09-17 | ||
| US4028424A (en) * | 1974-11-15 | 1977-06-07 | Japan Synthetic Rubber Co., Ltd. | Process for preparing unsaturated alcohols |
| DE2504151A1 (de) | 1975-02-01 | 1976-08-05 | Basf Ag | Verfahren zur herstellung von isopren durch dehydratisierung von 3-methyl-3-buten-1-ol |
| US3993702A (en) | 1975-09-29 | 1976-11-23 | Phillips Petroleum Company | Production of unsaturated alcohols |
| JPS6059886B2 (ja) * | 1979-12-04 | 1985-12-27 | 株式会社クラレ | イソプレンの製造方法 |
| US4511751A (en) | 1982-10-14 | 1985-04-16 | Kuraray Company, Ltd. | Process for producing isoprene |
| CN102206136A (zh) | 2011-03-23 | 2011-10-05 | 中国科学院山西煤炭化学研究所 | 一种无催化剂合成3-甲基-3-丁烯-1-醇的制备方法 |
| CN102516009B (zh) | 2011-11-16 | 2014-05-21 | 万华化学集团股份有限公司 | 一种液相法制备异戊二烯的方法 |
| CN103224444A (zh) | 2013-05-23 | 2013-07-31 | 南京工业大学 | 两步法合成3-甲基-3-丁烯-1-醇的方法 |
-
2016
- 2016-06-01 SG SG11201709802XA patent/SG11201709802XA/en unknown
- 2016-06-01 EP EP16803417.1A patent/EP3305747B1/en active Active
- 2016-06-01 US US15/577,988 patent/US11198657B2/en active Active
- 2016-06-01 MY MYPI2017704587A patent/MY183026A/en unknown
- 2016-06-01 KR KR1020177034292A patent/KR102486605B1/ko active Active
- 2016-06-01 RU RU2017146189A patent/RU2723241C2/ru active
- 2016-06-01 JP JP2017522225A patent/JP6687281B2/ja active Active
- 2016-06-01 CN CN201680031521.0A patent/CN107614464A/zh active Pending
- 2016-06-01 WO PCT/JP2016/066297 patent/WO2016194983A1/ja not_active Ceased
-
2017
- 2017-11-27 SA SA517390417A patent/SA517390417B1/ar unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59184137A (ja) * | 1983-04-04 | 1984-10-19 | Kuraray Co Ltd | イソプレンの製造方法 |
| JPH071714A (ja) * | 1993-02-01 | 1995-01-06 | Stork Contiweb Bv | 短縮された再スタートのドライヤー |
| JPH0714105A (ja) * | 1993-06-22 | 1995-01-17 | Matsushita Electric Ind Co Ltd | デジタル磁気記録再生装置 |
| JPH07285899A (ja) * | 1994-04-18 | 1995-10-31 | Kuraray Co Ltd | γ,δ−不飽和アルコールの製造方法 |
| WO2004087625A1 (ja) * | 2003-03-31 | 2004-10-14 | Kuraray Co., Ltd. | イソプレンの製造方法 |
| JP2013075877A (ja) * | 2011-09-30 | 2013-04-25 | Kuraray Co Ltd | イソプレンの製造方法 |
| WO2015186699A1 (ja) * | 2014-06-02 | 2015-12-10 | 株式会社クラレ | γ,δ-不飽和アルコールの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3305747A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017191794A1 (ja) * | 2016-05-06 | 2017-11-09 | 株式会社クラレ | 共役ジエンの製造方法 |
| JP2020530468A (ja) * | 2017-08-11 | 2020-10-22 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 3−メチルブタ−3−エン−1−オールの回収方法 |
| JP7332583B2 (ja) | 2017-08-11 | 2023-08-23 | ビーエーエスエフ ソシエタス・ヨーロピア | 3-メチルブタ-3-エン-1-オールの回収方法 |
| RU2828416C1 (ru) * | 2023-11-23 | 2024-10-11 | Общество с ограниченной ответственностью "Оргнефтехим-Холдинг" | Способ производства изопрена из изобутилена и формальдегида без выделения промежуточных продуктов |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2016194983A1 (ja) | 2018-03-22 |
| EP3305747B1 (en) | 2020-10-14 |
| US11198657B2 (en) | 2021-12-14 |
| EP3305747A1 (en) | 2018-04-11 |
| RU2723241C2 (ru) | 2020-06-09 |
| SA517390417B1 (ar) | 2021-07-04 |
| CN107614464A (zh) | 2018-01-19 |
| RU2017146189A (ru) | 2019-07-11 |
| SG11201709802XA (en) | 2017-12-28 |
| KR102486605B1 (ko) | 2023-01-09 |
| MY183026A (en) | 2021-02-08 |
| EP3305747A4 (en) | 2019-01-09 |
| JP6687281B2 (ja) | 2020-04-22 |
| RU2017146189A3 (ja) | 2019-10-01 |
| US20180290947A1 (en) | 2018-10-11 |
| KR20180015632A (ko) | 2018-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6687281B2 (ja) | 共役ジエンの製造方法 | |
| JP6485456B2 (ja) | γ,δ−不飽和アルコールの製造方法 | |
| JP6935399B2 (ja) | イソプロピルアルコールの製造方法 | |
| CN104640836A (zh) | 使用反应精馏制备乙二醇酯的方法 | |
| KR102224243B1 (ko) | 트리메틸올프로판의 제조장치 및 이를 이용한 제조방법 | |
| JP5101495B2 (ja) | 3−メチル−1,5−ペンタンジオールの製造方法 | |
| JP7043427B2 (ja) | γ,δ-不飽和アルコールの製造方法 | |
| WO2012033055A1 (ja) | ジトリメチロールプロパンの製造方法 | |
| CN116924909A (zh) | 3-甲氧基丙酸甲酯、3-甲氧基-n,n-二甲基丙酰胺的制备方法 | |
| CN102341360A (zh) | 催化卤化二醇的方法 | |
| JP4471078B2 (ja) | アルキルベンズアルデヒド類の製造方法 | |
| JP4890107B2 (ja) | 2−ヒドロキシ−4−メチルテトラヒドロピランの製造方法 | |
| JPWO2018193749A1 (ja) | インダンカルボアルデヒドの製造方法 | |
| CN107074711A (zh) | 改进的甲醛回收方法 | |
| RU2560156C1 (ru) | Способ получения этриола | |
| JP2009035522A (ja) | シクロペンチルアルコール化合物の製造方法 | |
| JP4779294B2 (ja) | アルコールの製造方法 | |
| CN119912305A (zh) | 一种用于mtbe分解制备异丁烯的方法 | |
| WO2015010928A1 (en) | Continuous process for the production of purified cyclohexanone | |
| JPWO2013005636A1 (ja) | ジトリメチロールプロパンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16803417 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017522225 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20177034292 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11201709802X Country of ref document: SG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15577988 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016803417 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017146189 Country of ref document: RU |