WO2017009578A1 - Procédé de préparation de liquides ioniques biosources pour la catalyse - Google Patents
Procédé de préparation de liquides ioniques biosources pour la catalyse Download PDFInfo
- Publication number
- WO2017009578A1 WO2017009578A1 PCT/FR2016/051806 FR2016051806W WO2017009578A1 WO 2017009578 A1 WO2017009578 A1 WO 2017009578A1 FR 2016051806 W FR2016051806 W FR 2016051806W WO 2017009578 A1 WO2017009578 A1 WO 2017009578A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- alkyl
- alkene
- heteroalkyl
- alkyne
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)C*1C=*N(C)C1 Chemical compound CC(*)C*1C=*N(C)C1 0.000 description 2
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0277—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides comprising ionic liquids, as components in catalyst systems or catalysts per se, the ionic liquid compounds being used in the molten state at the respective reaction temperature
- B01J31/0298—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides comprising ionic liquids, as components in catalyst systems or catalysts per se, the ionic liquid compounds being used in the molten state at the respective reaction temperature the ionic liquids being characterised by the counter-anions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/40—Substitution reactions at carbon centres, e.g. C-C or C-X, i.e. carbon-hetero atom, cross-coupling, C-H activation or ring-opening reactions
- B01J2231/49—Esterification or transesterification
Definitions
- the present invention relates to the field of ionic liquids.
- the invention relates to recyclable ionic liquids, their manufacturing process and their use as esterification catalysts for the manufacture of cosmetic agents, flavorings, biofuels, pesticides or low volatile solvents.
- Esters are chemical compounds widely used in the industry; in particular in cosmetics, perfumery, agri-food and bio-refinery. These products are obtained by an esterification reaction of contacting an alcohol function with a carboxylic acid function in the presence of an acid catalyst. Generally, it is chosen from mineral acids (for example, sulfuric acid (H 2 SO 4 ) or phosphoric acid (H 3 PO 4 ) (WO 2014/149669)) or organic acids (for example acetic acid or methacrylic acid (DE 10246869)).
- mineral acids for example, sulfuric acid (H 2 SO 4 ) or phosphoric acid (H 3 PO 4 ) (WO 2014/149669)
- organic acids for example acetic acid or methacrylic acid (DE 10246869)
- Ionic liquids are defined as a subset of molten salts having a melting temperature below 100 ° C under normal pressure conditions.
- the organic cation is bulky and asymmetric aromatic nitrogen type (alkylpyrrolidiniums, alkylpyridiniums and alkylimidazolium).
- anion it is often chosen from halogen (F ", Cl", Br “or ⁇ ) or from inorganic molecular anions such as tetrafluoroborate, [BF 4 ⁇ ], hexafluorophosphate [PFO], or fluorinated anions and / or sulfonic such as [CF3CO2], [CF3SO3], [HS0 4 ⁇ ], for example.
- ionic liquids having an imidazolium unit cation as the cation have been used as a new class of non-volatile solvents, alternative to conventional organic solvents (Plechkova, NV, Seddon, KR Chem, Soc Rev. 2008, 37, 123).
- organic solvents Plechkova, NV, Seddon, KR Chem, Soc Rev. 2008, 37, 123.
- Messadi, A. et al. have postponed the use of ionic liquids as complexing agents of heavy metals for the depollution of liquid effluents (Sep. Purif Technol 2013, 107, 172).
- Ionic liquids used as solvents have also improved the separation of conventional metal catalyst products.
- Ionic liquids have also been studied as catalysts for chemical reactions such as the esterification of cellulose derivatives (EP 2 692 738, Kano et al.), The preparation of biofuel in the presence of enzymes (EP 2 189 535, De Diego et al.) or as a catalyst for curing epoxy resin (US 2012/0157572).
- the use of these catalysts did not fully solve the problems of contamination of the end products.
- the preparation of these ionic liquids, used as catalysts has many implementation constraints.
- ionic liquids imidazolium unit can be carried out in one step, in the presence of mineral acids without halides; for example, (1) in the presence of acetic acid, (WO 2011/056924), or (2) under an inert atmosphere in the presence of HBF 4 , (JP 2008074740).
- this synthesis uses in all cases, reagents both classified (i) "CMR” (Carcinogenic, Mutagenic and Reprotoxic) such as formaldehyde, and (ii) toxic by ingestion, corrosive and flammable like amines.
- CMR Carcinogenic, Mutagenic and Reprotoxic
- the yields obtained by these processes are variable depending on the nature of the acid and are between 50 to 70%.
- the invention therefore relates to a process for preparing ionic liquids of formula
- R 1 and R 2 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, cycloalkyl, cycloalkenyl, heteroalkyl, heteroaryl or heterocycloalkyl; optionally substituted with at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl;
- R3 and R4 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, alkoxy or heteroalkyl; optionally substituted with at least one group selected from hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne or heteroalkyl; preferably R3 and R4 are the same; more preferably, R3 and R4 are identical and represent an H atom;
- a " represents an anion selected from alkali metal anions or conjugate bases of an organic acid or mineral acid having a pKa of less than 14, comprising:
- the reagents of step (i-0) comprise oxalic acid and an amine.
- the amine is an amino acid.
- step (i) comprises at least one ammonium source, a source of formaldehyde and a compound comprising an oxalyl function.
- the source of formaldehyde is chosen from organic compounds having at least one aldehyde function or organic compounds which can release under the reaction conditions a compound having at least one aldehyde function; preferably, the organic compound comprising an aldehyde function is formed in situ; more preferably, the organic compound comprising an aldehyde function is urotropin.
- alkylimidazolium hydrogen oxalate is chosen from the group of imidazolium hydrogen oxalate of formula (IV):
- R 1 and R 2 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, cycloalkyl, cycloalkenyl, heteroalkyl, heteroaryl or heterocycloalkyl; optionally substituted with at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl;
- R3 and R4 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, alkoxy or heteroalkyl; optionally substituted with at least one group selected from hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne or heteroalkyl; preferably R3 and R4
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in step (i) and an acid having a pKa of less than 2; preferably, less than 1.2; more preferably with an acid selected from H2SO4, HBF 4, HPF 6 or HNTf 2.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in step (i) and an alkaline salt.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in step (i) and an acid having a pKa greater than 1.2, in an electrochemical cell subjected to an intensity. electric.
- the electrical intensity applied in the electrochemical cell is in a range from 40 mA to 900 mA, preferably from 50 mA to 80 mA; more preferably, the electrical intensity is equal to about 60 mA.
- the electrochemical cell comprises a solvent chosen from polar solvents; preferably, acetonitrile.
- the electrochemical cell does not comprise an electrolyte.
- the present invention also relates to an ionic liquid obtainable by the process of the invention, of formula (I):
- R 1 and R 2 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, cycloalkyl, cycloalkenyl, heteroalkyl, heteroaryl or heterocycloalkyl; optionally substituted with at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl;
- R3 and R4 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, alkoxy or heteroalkyl; optionally substituted with at least one group selected from hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne or heteroalkyl; preferably R3 and R4 are the same; more preferably, R3 and R4 are identical and represent an H atom; A " represents an anion selected from alkali metal anions or conjugate bases of an organic acid or mineral acid having a pKa of less than 14.
- the present invention also relates to the use of an ionic liquid as an esterification reaction catalyst in which the product (ester) formed is an aroma or a cosmetic agent.
- the aroma formed is butyl levulinate.
- the cosmetic agent formed is isopropyl myristate.
- Ionic liquid relates to a salt consisting of an organic cation and an organic or inorganic anion having a melting temperature below 100 ° C under normal pressure conditions.
- the organic cation is an imidazolium or a derivative thereof;
- Recyclable relates to a compound that can be re-engaged in several successive chemical reactions while maintaining good efficiency
- Standard acid means an acid derived from an inorganic mineral
- Conjugate base of an acid relates to a chemical species obtained by the deprotonation of the corresponding acid; this species forms with the corresponding acid an acid / base pair characterized by its pKa;
- Alkyl relates to a hydrocarbon chain, linear or branched, having from 1 to 20 carbon atoms; preferably, from 1 to 15 carbon atoms; preferably, methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl;
- Aryl relates to a mono- or polycyclic system of 5 to 20, preferably 6 to 12, carbon atoms having one or more aromatic rings (when there are two rings, it is referred to as a biaryl) among which mention may be made of the phenyl group, the biphenyl group, the 1-naphthyl group, the 2-naphthyl
- Alkene relates to an unsaturated hydrocarbon chain, linear or branched, comprising at least 2 carbon atoms, characterized by the presence of at least one covalent double bond between two carbon atoms; preferably having from 2 to 20 carbon atoms; more preferably, from 2 to 15 carbon atoms;
- Alkyne relates to a monovalent unsaturated hydrocarbyl group, wherein the unsaturation results from the presence of one or more carbon-carbon triple bonds.
- the alkynyl groups in general, and preferably, have the same number of carbon atoms as described above with respect to the alkyl groups.
- Non-limiting examples of alkynyl groups are ethynyl, 2-propynyl, 2-butynyl, 3-butynyl, 2-pentynyl and isomers thereof;
- amino acid relates to an organic compound carrying both a carboxylic acid function and an amine function.
- the amino acids are preferably the 22 proteinogenic acids; preferably, the amino acids are chosen from alanine, arginine, asparagine, aspartate, cysteine, glutamate, glutamine, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, pyrrolysine, selenocysteine, serine, threonine, tryptophan tyrosine, valine;
- Aldehyde relates to a group -CHO
- Tetrafluoroboric Acid relates to a compound of formula HBF 4 ;
- Halophosphoric ide relates to a compound of formula HPF 6 ;
- Catalyst relates to a chemical compound capable of accelerating the rate of a chemical reaction, preferably an esterification reaction
- Cycloalkyl relates to a cyclic or polycyclic alkyl group, optionally branched, substituted or unsubstituted; preferably a cyclopropyl, cyclopentyl or cyclohexyl group;
- Cycloalkenyl relates to a cyclic or polycyclic alkene group, optionally branched, substituted or unsubstituted; preferably a cyclopropenyl, cyclopentenyl or cyclohexenyl group;
- Heteroatom refers to an atom of an organic molecule having at least one electron pair that differs from carbon or hydrogen.
- a heteroatom is chosen from O, S or N;
- Heteroalkyl relates to an alkyl group as described above, comprising one or more heteroatoms selected from O, S or N;
- Heteroalkene relates to an alkene group as described above, comprising one or more heteroatoms chosen from O, S or N;
- Heterocycloalkyl relates to a cycloalkyl group as described above, comprising one or more heteroatoms chosen from O, S or N;
- Heteroaryl relates to an aryl group as described above, comprising one or more heteroatoms chosen from O, S or N;
- Haldrogenoxalate relates to an ion of formula C2HO4 " ;
- Niro relates to a function -NO2
- Alkoxy relates to an O-alkyl group
- NTf 2 represents the triflimide compound (or bis (trifluoromethylsulfonyl) imide) of empirical formula C2F6NO4S2;
- pKa is less than 14; preferably, the pKa is less than or equal to 2; more preferably, the pKa is less than or equal to 1.2.
- the pKa is from more than 0 to 14; preferably from more than 0 to 2; even more preferably, from more than 0 to 1.2.
- the pKa of the acid is greater than 1.2; preferably from 1.2 to 14; more preferably; from 2 to 14;
- Esterification relates to an organic chemistry reaction between an alcohol function and a carboxylic acid function leading to the formation of an ester function (-COO-);
- “Flavor” means any product or substance intended to be added to a foodstuff to give it an odor, ie nasal or retro-nasal perception, and / or taste, that is, Lingual perception, which belongs to one of the categories of flavoring agents as defined by European Directive 88-388;
- Cosmetic agent any substance intended to be placed in contact with various superficial parts of the human body (such as, for example, the epidemic, hair and hair systems, nails, etc.), with the teeth and / or with oral mucosa, exclusively or principally, to cleanse, perfume, modify the appearance, protect, maintain or repair body odor.
- the cosmetic agent satisfies the requirements defined by the Regulation on Registration, Evaluation, Authorization and Restriction of Chemicals (REACH: Registration, Evaluation, Authorization and Restriction of CHemicals);
- Aliphatic relates to a non-aromatic organic compound
- Alkaline salt relates to a salt consisting of a cation which is an alkali metal, and an anion.
- the alkaline salt is chosen from NaBF 4 ,
- the subject of the present invention is an ionic liquid and / or catalyst of general formula (I)
- R 1 and R 2 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, cycloalkyl, cycloalkenyl, heteroalkyl, heteroaryl or heterocycloalkyl; optionally substituted with at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl; preferably Ri and R 2 are identical; more preferably, R 1 and R 2 are identical and represent an unsubstituted alkyl chain or an alkyl chain substituted by an alkyl group or an aryl group;
- R3 and R4 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, alkoxy or heteroalkyl; optionally substituted with at least one group selected from hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne or heteroalkyl; preferably R3 and R4 are the same; more preferably, R3 and R4 are identical and represent an H atom;
- a " represents an anion chosen from anions of alkaline salts or conjugate bases of an organic acid or a mineral acid having a pKa of less than 14, preferably having a pKa of less than or equal to 2, more preferably, having a pKa less than or equal to 1.2
- said anion is obtained by an anion exchange step on an alkylimidazolium hydrogen oxalate.
- the catalyst of general formula (I) is a recyclable ionic liquid. In one embodiment, the catalyst of general formula (I) is a bio-sourced recyclable ionic liquid.
- an ionic liquid as described above does not include any of the following ionic liquids:
- a " is an anion which is a conjugate base of an inorganic acid selected from H2SO4, HBF 4, HPF 6, HNTf 2.
- A represents a halogen anion.
- a " is an anion which is not aromatic.
- a " is an anion which is an anion of an alkaline salt, preferably A " is an anion which is an anion of an alkaline salt selected from NaBF 4 , KPF 6 or LiNTf 2 .
- a " is not a halide, because the presence of halide inhibits the catalytic properties of the compounds
- a " is not Cl " .
- a " is not Br “
- a " is not F.
- a " is not ⁇ .
- a " does not comprise halogen,
- a " does not include a brominated atom.
- a " does not include a fluorine atom,
- a " does not include an iodine atom.
- a " does not include a chlorine atom.
- the cation of the general formula (I) is symmetrical. In a second embodiment, the cation of the general formula (I) is not symmetrical.
- the catalyst (ionic liquid) of general formula (I) is liquid. According to one embodiment, the catalyst (ionic liquid) of general formula (I) is not a gel. According to one embodiment, the catalyst (ionic liquid) of general formula (I) is not a polymer. According to one embodiment, R 1, R 2 , R 3 and R 4 do not comprise polymer chains. In one embodiment, R 1, R 2 , R 3 and R 4 do not include an acid function; preferably, R 1, R 2 , R 3 and R 4 do not include a sulfonic acid function. According to one embodiment, R1 does not include a sulfonic acid function. According to one embodiment, R 2 does not comprise a sulphonic acid function. According to one embodiment, R3 does not include a sulphonic acid function. According to one embodiment, R4 does not include a sulfonic acid function.
- R 1 and R 2 do not represent H. In one embodiment, R 1 and R 2 do not represent CH 3.
- the catalyst (ionic liquid) of general formula (I) is an ionic liquid that is not polyionic. In one embodiment, the catalyst of general formula (I) is not a phase transfer catalyst.
- the invention relates to the use of the compound (I) as an esterification reaction catalyst.
- the invention relates to the use of the compound (I), as an esterification reaction catalyst for the manufacture of cosmetic agents, flavorings, biofuels, pesticides or low volatile solvents; preferably for the synthesis and manufacture of cosmetic agents or flavorings.
- the invention relates to the use of an ionic liquid as described above as an esterification reaction catalyst for the manufacture of cosmetic agents, flavorings, biofuels, pesticides or weakly volatile solvents, very preferably for the synthesis and manufacture of cosmetic agents or flavors.
- the esterification reaction leads to esters useful in the agrifood field.
- said esterification reaction is a reaction in which the reagents of the esterification reaction comprise at least one acyclic aliphatic organic compound having an alcohol function.
- said esterification reaction is a reaction in which the reagents of the esterification reaction comprise at least one acyclic aliphatic organic compound having a carboxylic acid function.
- the use of an ionic liquid as described above does not include the ionic liquids of formula (I) in which R 1 or R 2 represents a hydrogen atom (H).
- the catalyst of general formula (I) is a recyclable ionic liquid which is separated from the reaction medium by decantation at the end of the reaction, in particular of the esterification reaction.
- the catalyst of general formula (I) is separated from the reaction medium by decantation at the end of the reaction, in particular the esterification reaction.
- the esterification reaction is not conducted in microwaves.
- the esterification reaction is conducted at atmospheric pressure. According to one embodiment, the esterification reaction is not carried out in an autoclave. According to one embodiment, the esterification reaction is not conducted under reduced pressure.
- the esterification reaction is conducted at a temperature of from 20 to 400 ° C; preferably, ranging from 40 to 150 ° C; more preferably, the esterification reaction is conducted at about 70 ° C, 80 ° C or 100 ° C.
- the esterification reaction is conducted for less than 72 hours; preferably, less than 48 hours; more preferably, less than 24 hours. According to one embodiment, the esterification reaction is conducted for about 50h. According to one embodiment, the esterification reaction is conducted for about 16 hours. According to one embodiment, the esterification reaction is conducted for about 1 h.
- the catalyst is added in the medium at a concentration of greater than 0 to 10 mol%; preferably at a concentration of from 1 to 6 mol%. ; more preferably, the catalyst is added in the medium at a concentration of about 4 mol%.
- the reagent which is a non-aromatic organic compound having at least one alcohol function, comprises methanol, ethanol, propanol, isopropanol, butanol.
- the non-aromatic organic compound having at least one alcohol function is chosen from methanol, butanol, ethanol or isopropanol.
- the nonaromatic organic compound having at least one alcohol function is methanol.
- the non-aromatic organic compound having at least one alcohol function is ethanol. According to one embodiment, the non-aromatic organic compound having at least one alcohol function is isopropanol. In one embodiment, the nonaromatic organic compound having at least one alcohol function does not include an alkene function.
- the reagent which is a non-aromatic organic compound having at least one carboxylic acid function, comprises methanoic acid, ethanoic acid, propanoic acid, butanoic acid, pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, nonanoic acid, decanoic acid, undecanoic acid, dodecanoic acid, tridecanoic acid, tetradecanoic acid (or myristic acid), 3-oxopentanoic acid, 4-oxopentanoic acid (or levulinic acid).
- the nonaromatic organic compound having at least one carboxylic acid function is tetradecanoic acid or 4-oxopentanoic acid. According to one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is myristic acid or levulinic acid. According to one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is myristic acid. According to one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is levulinic acid. According to one embodiment, the nonaromatic organic compound having at least one carboxylic acid function comprises several carboxylic acid functions.
- the nonaromatic organic compound having at least one carboxylic acid function is not pivalic acid. In one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is not myristic acid. In one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is not palmitic acid. In one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is not acetic acid. In one embodiment, the nonaromatic organic compound having at least one carboxylic acid function does not include an alkene function. According to one embodiment, the nonaromatic organic compound having at least one carboxylic acid function is not linoleic acid.
- the nonaromatic organic compound having several carboxylic acid functions is not sebacic acid.
- the reaction medium is not heterogeneous.
- the esterification reaction is conducted without the addition of a phase transfer catalyst.
- the esterification reaction leads to an ester useful as a flavor.
- the flavor is butyl levulinate.
- the esterification reaction leads to an ester that is useful as a cosmetic agent; such as, but not limited to, an emollient, depigmenting, healing, moisturizing, scenting, deodorant, antiperspirant, cleanser, colorant, preservative, volumizer, plumping agent and / or tensor.
- the cosmetic agent is isopropyl myristate.
- the cosmetic agent is butyl levulinate.
- the esterification reaction leads to an ester which is not a polymer.
- the conversion of the limiting reagent is greater than 80 mol%. ; preferably greater than 90 mol%. ; more preferably, the conversion is total.
- the preferred compounds of formula (I) are the compounds of general formula (II):
- the preferred ionic liquids of formula (II) are the ionic liquids of formula (IIa):
- the preferred compounds of formula (I) are the compounds of general formula (III):
- Ri and R 2 are identical and represent an unsubstituted alkyl chain or an alkyl chain substituted by an alkyl group or an aryl group;
- a " is defined as previously.
- the preferred ionic liquids of formula (III) are the ionic liquids of formula (IIIa):
- the recyclable ionic liquid is chosen from 1,3-diisobutylimidazolium hydrogen sulphate or butyl methylimidazolium hydrogen sulphate.
- the preferred ionic liquids of formula (I) are the compounds shown in Table 1 below. Table 1
- the invention also relates to a process for preparing ionic liquids and / or catalysts of general formula (I)
- R 1, R 2, R 3, R 4 and A " are defined as above, comprising:
- step (i) further comprises a preliminary step of synthesis of an ammonium hydrogen oxalate (denoted (i-0)).
- the present invention relates to a process for preparing ionic liquids and / or catalysts of general formula (I)
- R 1, R 2, R 3, R 4 and A " are defined as above, comprising:
- the method of the invention does not include an exothermic step.
- R 1, R 2 , R 3 , R 4 and A " are defined as above, in the form of a product which is not brown or black, preferably in the form of a colorless product.
- the process of the invention provides a compound of formula (I) which is not an oil.
- the process of the invention provides a compound of formula (I) which does not require heavy purification steps.
- the method of the invention comprises a purification step in an oxygenated solvent; preferably in an oxygenated solvent which is not hydrogen peroxide or sodium hydroxide.
- the method of the invention comprises a purification step which is a recrystallization.
- the method of the invention comprises a purification step which comprises washing and / or filtration.
- the method of the invention does not comprise a purification step comprising hydrogen peroxide. According to one embodiment, the method of the invention does not comprise a purification step comprising the use of sodium hydroxide. According to one embodiment, the method of the invention does not include a purification step at a temperature above 60 ° C. According to one embodiment, the method of the invention does not include a purification step at a temperature of 60 ° C to 100 ° C. According to one embodiment, the method of the invention comprises a purification step conducted at room temperature.
- the reagents of step (i-0) comprise an acid, preferably a diacid; and an amine.
- the molar ratio of diacid / amine is in a range from 0.5 to 10; preferably from 1 to 2; more preferably, is equal to about 1. According to one embodiment, in step (i-0), the molar ratio diacid / amine is in a range from 1 to 10. According to one embodiment the diacid and the amine are introduced in stoichiometric amount in step (i-0).
- the diacid used as reagent in step (i-0) is oxalic acid.
- the diacid used as reagent in step (i-0) is an aqueous solution of oxalic acid.
- the diacid used as reagent in step (i-0) is an aqueous solution of oxalic acid preheated to about 50 ° C.
- the aqueous solution of oxalic acid is at a concentration in a range of 1 to 3 mol / L; preferably from 1.5 to 2.5 mol / L; more preferably, the concentration of the oxalic acid solution is about 1.8 mol / L.
- the acid is not a monoacid, ie an organic acid having a carboxylic function.
- the acid is not the acid acetic acid, formic acid, ethanoic acid, proanoic acid, pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, nonanoic acid or decanoic acid.
- the acid is not benzoic acid.
- the amine used as reagent in step (i-0) is from commercial source.
- the amine used as reagent in step (i-0) is an organic compound, linear or branched, having at least one amine function.
- the amine used as reagent in step (i-0) is an organic compound having at least one amine function and comprising from 1 to 10 carbon atoms; preferably comprising from 1 to 6 carbon atoms; more preferably, the amine is selected from methylamine, ethylamine, propylamine, butylamine, pentylamine and hexylamine.
- the amine used as reagent in step (i-0) is isobutylamine.
- the amine used as reagent in step (i-0) is propylamine.
- the primary amine is phenylethylamine.
- the amine used as reagent in step (i-0) is of natural source (i.e. biosourced); preferably, the amine is selected from amino acids.
- the amino acid used as reagent in step (i-0) is chosen from alanine, arginine, asparagine, aspartate, cysteine, glutamate, glutamine, glycine, histidine, isoleucine, leucine, lysine. , methionine, phenylalanine, proline, pyrrolysine, selenocysteine, serine, threonine, tryptophan, tyrosine, valine.
- the amino acid is of L-form.
- the amine used as reagent in step (i-0) is L-Valine.
- the amino acids as amine source in step (i-0) are preferred over conventional amines because of their non-toxicity in the reaction medium.
- the solvent of step (i-0) is chosen from organic solvents; preferably aromatic ketones; more preferably, acetophenone.
- step (i-0) is conducted at a temperature ranging from 100 ° C to 150 ° C; preferably from 120 ° C to 140 ° C; more preferably, the temperature used in step (i-0) is equal to about 140 ° C.
- step (i-0) is carried out at ambient pressure.
- step (i-0) further comprises a purification step at the end of the reaction.
- the purification step of (i-0) comprises the implementation of at least one of the techniques known to those skilled in the art; preferably, said purification step comprises washing cycles; more preferably, cycles of washing in polar solvent such as acetone and / or ethyl acetate.
- step (i-O) leads to the formation of alkylammonium hydrogen oxalate; preferably alkylammonium hydrogen oxalate wherein the alkyl group may be linear or branched, and is selected from compounds having from 1 to 6 carbon atoms; preferably, methyl, ethyl, propyl, isopropyl, sec-butyl, butyl, iso-butyl, sec-butyl, tert-butyl, pentyl or hexyl.
- step (i-0) leads to the formation of isobutylammonium hydrogen oxalate.
- step (i-0) leads to the formation of propylammonium hydrogen oxalate.
- Step (i) the synthesis of alkylimidazolium hydrogen oxalate according to step (i) comprises at least one ammonium source, a source of formaldehyde and a compound comprising an oxalyl function.
- the ammonium source is chosen from organic compounds having at least one ammonium function or compounds which can release, under the reaction conditions, a compound having at least one ammonium function.
- the ammonium source is formed in situ by chemical reaction between at least one amine and an acid; preferably, by chemical reaction between at least one amine and a diacid; even more preferentially, between at least one amine and oxalic acid.
- the ammonium source is chosen from ammonium hydrogen oxalate salts; preferably, the ammonium hydrogenoxalate salts obtained according to step (i-0).
- the ammonium source is isobutylammonium hydrogen oxalate.
- the aldehyde source is chosen from organic compounds having at least one aldehyde function or compounds which can release, under the reaction conditions, a compound having at least one aldehyde function.
- the source of aldehyde is paraformaldehyde.
- the compound comprising an aldehyde function formed in situ is obtained from urotropin (also called hexamethylenetetramine or methenamine).
- the compound comprising an aldehyde function can be a commercial compound directly involved in the mixture or a compound formed in situ during the mixing step.
- the compound comprising an oxalyl functional group is glyoxal.
- glyoxal and oxalic acid are bio-sourced.
- the source of formaldehyde and the compound comprising an oxalyl function are introduced into the reaction mixture in stoichiometric amounts.
- the molar ratio of the alkylammonium hydrogen oxalate: source of formaldehyde is in a range from 1 to 3; preferably is about 2.
- the synthesis of alkylimidazolium hydrogen oxalate according to step (i) is carried out at a temperature ranging from 0 ° C. to 180 ° C .; preferably, the temperature is in a range of 25 ° C to 100 ° C.
- the synthesis of alkylimidazolium hydrogen oxalate according to step (i) is carried out at room temperature.
- the synthesis of alkylimidazolium hydrogen oxalate according to step (i) comprises azeotropic distillation of the water at a temperature of about 85 ° C.
- the azeotropic distillation is carried out using a Dean-Stark device.
- the synthesis of alkylimidazolam hydrogenoxalate according to step (i) is carried out from 1 to 10 h; preferably from 2 to 6h. According to one embodiment, the synthesis of alkylimidazolam hydrogenoxalate is carried out for 5 hours.
- the efficiency of step (i) is greater than 70%. According to one embodiment, the yield obtained during step (i) is equal to about 76%.
- step (i) leads to an intermediate product which is an imidazolium hydrogen oxalate.
- alkylimidazolam hydrogenoxalate is selected from the group of imidazolium hydrogen oxalate of formula (IV):
- R 1 and R 2 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, cycloalkyl, cycloalkenyl, heteroalkyl, heteroaryl or heterocycloalkyl; optionally substituted with at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl; preferably Ri and R 2 are identical; more preferably, R 1 and R 2 are identical and represent an unsubstituted alkyl chain or an alkyl chain substituted by an alkyl group or an aryl group; R3 and R4 are the same or different, and each represents a group selected from H, alkyl, alkene, alkyne, alkoxy or heteroalkyl; optionally substituted
- R 1 and R 2 do not represent H.
- Ri, R 2, R3 and R4 do not include acid functional groups; preferably, R 1, R 2 , R 3 and R 4 do not include carboxylic acid functions or sulphonic acid functional groups.
- Scheme 1 The method for the preparation of alkylimidazolium hydrogen oxalate of general formula (IV) is shown below (Scheme 1):
- the method for the preparation of alkylimidazolium hydrogenoxalate is more preferably carried out from an ammonium salt according to the scheme Ibis below:
- alkylimidazolium hydrogenoxalate of general formula (IV) can also be carried out according to the method described below (Scheme Iter):
- the preferred compounds of formula (IV) are symmetrical.
- the invention also relates to an imidazolium hydrogen oxalate of formula (IV):
- the preferred compounds of formula (IV) are the compounds of formula (V):
- the preferred compounds of formula (IV) are the compounds of formula (Vbis) (called 1,3-dialkylimidazolium hydrogenoxalate):
- R 1 and R 2 are the same or different, and each represents a group selected from alkyl or heteroalkyl; optionally substituted by at least one group selected from aryl, hydroxyl, oxo, nitro, amido, amino, cyano, alkoxy, alkyl, alkene, alkyne, cycloalkyl, cycloalkene, heteroalkyl, heteroaryl or heterocycloalkyl.
- the preferred compounds of formula (V) are the compounds of formula (VI):
- R 1 and R 2 are the same and represent an unsubstituted alkyl chain or an alkyl chain substituted by an alkyl group or an aryl group.
- the preferred compounds of formula (V) are the compounds chosen from: dipropylimidazolium hydrogen oxalate
- the preferred compounds of formula (V) are chosen from dipropylimidazolium hydrogen oxalate or diisobutylimidazolium hydrogen oxalate.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an acid having a pKa of less than 14; preferably, an acid having a pKa of less than or equal to 2; more preferably, an acid having a pKa less than or equal to 1.2.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an acid having a pKa ranging from 2 to 14. According to one embodiment, the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an acid having a pKa in a range from 1.2 to 14. According to one embodiment, the acid having a pKa ranging from 1.2 to 14 is acetic acid. In one embodiment, the acid having a pKa in a range of from 1.2 to 14 is formic acid.
- the acid is not acetic acid. According to one embodiment, the acid is not formic acid.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an acid having a pKa of less than 2; preferably less than 1.2.
- the acid is selected from H2SO4, HBF 4, HPF 6 or HNTf 2; preferably, H 2 SO 4 .
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an alkaline salt.
- the anion exchange is anionic metathesis.
- the alkali salt is selected from NaBF 4, KPF 6 or LiNTf 2.
- the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and an acid having a pKa greater than 1.2 in an electrochemical cell; preferably, greater than 2.
- the molar ratio between the alkylimidazolium hydrogen oxalate obtained in (i) and the acid is in a range from 0.6 to 1.2; preferably, is about 0.8.
- the exchange of anions takes place in an electrochemical cell; preferably in an electrochemical cell with a compartment.
- the electrochemical cell comprises at least two electrodes.
- the electrochemical cell comprises a working electrode, a reference electrode and / or an auxiliary electrode.
- the electrochemical cell comprises at least one electrode chosen from Al, Au, Ag, Boron-doped diamond, C, Cd, Co, Cr, Cu, Ga, Hg, In, Ir, Mg, Mo, Nb.
- the electrode is selected from C, Pt or stainless steel.
- the electrode is chosen from stainless steel, carbon felt or vitreous carbon.
- the electrochemical cell comprises a reference electrode chosen from saturated calomel electrode (SCE), silver-silver chloride electrode (Ag / AgCl) or Ag / AgNO 3 .
- the electrochemical cell is coupled to a potentiostat and an integrator.
- the applied electrical intensity is in a range from 40 mA to 900 mA; preferably from 50 mA to 80 mA; more preferably, the applied electrical intensity is equal to about 60 mA.
- the solvent is chosen from organic solvents; preferably, the polar solvents; more preferably, acetonitrile.
- step (ii) further comprises a reflux heating step.
- the yield obtained in step (ii) is greater than 80%; preferably, greater than or equal to 85%. According to one embodiment, the yield obtained in step (ii) is equal to about 92%.
- the anion exchange is conducted at a temperature ranging from 0 ° C to 180 ° C; preferably, the temperature is in the range of 10 ° C to 150 ° C; more preferably, the anion exchange is conducted at a temperature ranging from 50 ° C to 90 ° C. According to one embodiment, the anion exchange is conducted at room temperature. According to one embodiment, the anion exchange is conducted at a temperature ranging from 0 ° C. to 30 ° C. when the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and a acid having a pKa of less than or equal to 2; preferably, less than or equal to 1.2.
- the anion exchange is conducted at a temperature ranging from 50 ° C. to 90 ° C. when the anion exchange is carried out between the alkylimidazolium hydrogen oxalate obtained in (i) and a acid having a pKa less than 14.
- the anion exchange is conducted under reduced pressure.
- the anion exchange is conducted at a pressure ranging from 500 to 2 mbar; preferably at a pressure of about 10 mbar.
- the anion exchange is not carried out in an autoclave.
- the exchange of anions is not carried out under reduced pressure.
- the anion exchange step leads to the formation of a recyclable ionic liquid of general formula (I) as described. previously, and oxalic acid.
- the anion exchange step is carried out in acetone.
- the anion exchange step is carried out in an ether.
- the anion exchange step is carried out in diethyl ether.
- the anion exchange step is carried out in tert-butyl methyl ether (MTBE).
- the invention also relates to a compound of formula (I) as described above, obtainable by the process of the invention, and its use as an esterification reaction catalyst; preferably as a catalyst in the esterification reaction leading to butyl levulinate or isopropyl myristate.
- the invention also relates to an ionic liquid of formula (I) as described above, obtainable by the method of the invention, and its use as an esterification reaction catalyst; preferably as a catalyst in the esterification reaction leading to butyl levulinate or isopropyl myristate.
- Figure 1 is a photograph showing the phase separation between the catalyst and isopropyl myristate at the end of the reaction.
- Solvents, reagents and starting material were purchased from suppliers of known chemicals (eg Sigma Aldrich, Acros Organics, VWR Int., Sopachem, Carlo Erba, Alfa-Aesar or Avocado) and were used as received unless otherwise indicated.
- CO2 carbon dioxide
- H2SO4 sulfuric acid
- L liter (s)
- NTf 2 bis (trifluoromethylsulfonyl) amide
- Step 2 was carried out with various sources of aldehyde: either from formaldehyde (method 2a); either from urotropin (method 2b).
- Propylamine (7.20 g, 121.8 mmol, M 59, 11 g / mol) is added to a suspension of paraformaldehyde (3.66 g) in toluene (100 ml) cooled by a cold water bath. The reaction mixture is stirred for 30 minutes and then cooled to 0 ° C. A second equivalent of propylamine (7.200 g, 121.8 mmol) and oxalic acid (10.99 g, 122.1 mmol) are added. The solution is stirred for 2 hours at room temperature. Then glyoxal (40% w / w in water, 13.9 mL, 121.7 mmol) is added.
- the anion exchange reaction from the hydrogen oxalate salt can be carried out:
- Method A either by the addition of an acid having a pKa of less than about 1.2;
- Method B either by the addition of an acid having a pKa greater than about 1.2 under electrolysis;
- the diisobutylimidazolium hydrogen oxalate (0.500 g, 1.850 mmol) is suspended in 50 mL of diethyl ether at room temperature.
- HBF 4 Et 2 O
- 0.300 g, 1.851 mmol is added and the solution is stirred for 1 hour.
- the ether phase is removed and the residue is washed with diethyl ether (5 times 10 ml) and then dried under vacuum.
- the 1,3-dipropylimidazolium hydrogen oxalate (2.189 g, 8.998 mmol) is suspended in 20 mL of acetone at room temperature. H2SO4 (0.915 g, 9.330 mmol) is added and the solution is stirred for 1 hour. The solution is filtered and the filtrate is evaporated under vacuum. The residue is washed with diethyl ether (3 ⁇ 10 mL), cyclohexane (2 ⁇ 10 mL) and then dried under vacuum.
- Example 4 Anionic metathesis from 1,3-diisobutylimidazolium hydrogenoxalate (product B1)
- 1,3-diisobutylimidazolium salts as described are ionic liquids.
- Example 7 esterification of levulinic acid catalyzed by diisobutylimidazolium hydrogen sulfate
- the esterification of levulinic acid is obtained with a conversion of less than 60% after 24 hours;
- the time and the conversion of the esterification reaction are comparable to those obtained in the presence of H 2 SO 4 .
- the imidazolium salts having the character of an ionic liquid and having the hydrogen sulfate as a counterion, make it possible to catalyze the esterification of levulinic acid with butanol as effectively as a conventional catalyst such as the acid sulfuric.
- the esters synthesized by this process can be easily separated from the catalyst and are obtained with a purity greater than 99%. 7.2. with ethanol
- the catalyst (compound C3) was used for several cycles according to the reaction conditions described above. The results are shown in Table 4 below.
- Isobutylimidazolium acetate was synthesized according to the protocol described in Example 1 of application US 2010/0249432 substituting ethylamine by isobutylamine. A red-brown product was obtained showing a large number of impurities.
- the Applicant purified the product according to the protocol described in US 2010/0249432, that is to say by reacting the crude reaction with hydrogen peroxide (30%) at 80 ° C for 5 hours; followed by treatment with sodium hydroxide (40%) at a temperature of 65 ° C to 95 ° C for 2h. A straw yellow liquid was obtained. Characterization of the liquid by I MN 1 !! shows that the purification conditions led to a high degradation of the product (> 20%).
- the Applicant has also prepared, according to the protocol of Example 1, isobutylammonium acetate substituting oxalic acid with acetic acid.
- acetic acid is not suitable for the synthesis of alkylammonium salts in contrast to the oxalic acid used in the present invention.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La présente invention concerne le domaine des liquides ioniques. En particulier, la présente invention concerne des liquides ioniques recyclables et leur procédé de fabrication. D'autre part, la présente invention concerne l'utilisation d'un liquide ionique recyclable comme catalyseur de réaction d'estérification pour la synthèse d'arômes et/ou d'agents cosmétiques, dans laquelle le liquide ionique recyclable est de formule générale (I) : dans laquelle, R1, R2, R3, R4 et A- sont tels que définis dans les revendications.
Description
PROCÉDÉ DE PRÉPARATION DE LIQUIDES IONIQUES BIOSOURCES
POUR LA CATALYSE
DOMAINE DE L'INVENTION La présente invention concerne le domaine des liquides ioniques. En particulier, l'invention a pour objet des liquides ioniques recyclables, leur procédé de fabrication et leur utilisation comme catalyseurs d'estérification pour la fabrication d'agents cosmétiques, d'arômes, de biocarburants, de pesticides ou de solvants faiblement volatils.
ÉTAT DE LA TECHNIQUE
Les esters sont des composés chimiques largement employés dans l'industrie ; notamment en cosmétique, parfumerie, agroalimentaire et bio-raffinerie. Ces produits sont obtenus par une réaction d'estérification consistant à mettre en contact une fonction alcool avec une fonction acide carboxylique en présence d'un catalyseur acide. Généralement, celui-ci est choisi parmi les acides minéraux (par exemple, l'acide sulfurique (H2S04) ou l'acide phosphorique (H3P04) (WO 2014/149669)) ou les acides organiques (par exemple l'acide acétique ou l'acide méthacrylique (DE 10246869)).
Cependant, l'utilisation de tels composés induit une corrosion des équipements et une contamination des produits esters finaux. En effet, ces acides minéraux ou organiques sont difficilement éliminables du milieu en fin de réaction et les méthodes de synthèse conventionnelles nécessitant la plupart du temps une distillation à hautes températures et sous très basses pressions, restent inefficaces.
De plus, ces synthèses chimiques sont majoritairement menées en solvants organiques souvent volatils, inflammables et toxiques. Or, l'actuel essor de la chimie verte cherche à minimiser l'impact environnemental de ces procédés de synthèse en réduisant le flux de ces déchets pour les procédés industriels considérés comme trop polluants.
Π existe donc un besoin pour développer des composés non-corrosifs pouvant être utilisés à la fois comme co-solvants et comme catalyseurs d'estérification. Il existe également un besoin pour développer des procédés d'estérification à la fois plus efficaces et plus respectueux des contraintes économiques et environnementales actuelles. Une des alternatives aux solvants et/ou catalyseurs conventionnels est de substituer le solvant et/ou le catalyseur conventionnel par un liquide ionique.
Les liquides ioniques sont définis comme étant un sous-ensemble des sels fondus ayant une température de fusion inférieure à 100°C dans des conditions normales de pression.
Le plus souvent, ils sont constitués d'un cation organique et d'un anion organique ou inorganique. En particulier, le cation organique est volumineux et dissymétrique de type aromatiques azotés (alkylpyrrolidiniums, alkylpyridiniums et alkylimidazolium). Quant à anion, il est souvent choisi parmi les halogènes (F", Cl", Br" ou Γ) ou parmi les anions moléculaires inorganiques tels que le tétrafluoroborate [BF4 ~], l'hexafluorophosphate [PFÔ ], ou les anions fluorés et/ou sulfoniques comme [CF3CO2 ], [CF3SO3 ], [HS04 ~], par exemple.
Depuis la synthèse officielle du premier liquide ionique en 1914 par Walden (Bull. Acad. Sci. Petersburg 21, 1914, 405-422), ces composés ont suscité beaucoup d'intérêt à la fois en raison de leur sûreté d'emploi (non-inflammabilité, non-miscibilité avec la plupart des solvants organiques, bonne conductivité électrique) et de par les propriétés remarquables qu'ils apportent en synthèse comme très bons solvants.
Tout d'abord, les liquides ioniques ayant comme cation un motif imidazolium ont été utilisés comme nouvelle classe de solvants non volatils, alternatifs aux solvants organiques conventionnels (Plechkova, N. V.; Seddon, K. R. Chem. Soc. Rev. 2008, 37, 123). Par exemple, Messadi, A. et al. ont reporté l'utilisation de liquides ioniques comme complexants de métaux lourds pour la dépollution d'effluents liquides (Sep. Purif. Technol. 2013, 107, 172). Les liquides ioniques employés comme solvants, ont également permis d'améliorer la séparation des catalyseurs métalliques conventionnels de produits
finaux, lors de la dimérisation d'oléfines dans des procédés continus biphasiques sans solvant (Olivier-Bourbigou, H. et al. In Handbook of Green Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: 2010).
Les liquides ioniques ont également été étudiés comme catalyseurs de réactions chimiques telles que l'estérification de dérivés de cellulose (EP 2 692 738, Kano et al.), la préparation de biocarburant en présence d'enzymes (EP 2 189 535, De Diego et al.) ou encore comme catalyseur de durcissement de résine époxy (US 2012/0157572). Cependant, l'utilisation de ces catalyseurs n'a pas permis de résoudre pleinement les problèmes de contamination des produits finaux. Par ailleurs, la préparation de ces liquides ioniques, utilisés comme catalyseurs, connaît de nombreuses contraintes de mise en œuvre.
En effet, en 1991, Arduengo a décrit une synthèse de liquides ioniques à motif imidazolium portant des anions halogénures (WO 91/14678), qui pourraient ensuite être remplacés par une métathèse d' anion. Néanmoins, la réaction d'échange de cet anion par un autre dérivant d'un acide ayant un pKa supérieur à 4 peut s'avérer problématique pour des raisons thermodynamiques, engendrant des rendements très variables (Green Chem. 2005, 7, 39-42).
De plus, un tel procédé de transformation reste très énergivore (WO 00/16902 ; J. Taïwan Institute of Chem. Eng., 2014, 45, page 432 and Green Chem. 2013, 15, page 138) et conduit à une réaction difficilement quantitative : la présence d'halogénures, même à faible teneur (1%), diminue drastiquement les performances de ces liquides ioniques (Green Chem. 2005, 7, 39-42, J.A.C.S. 2002, 124, 14247-14254) et/ou conduit à l'inhibition complète de leur activité de catalyseurs organométalliques.
La synthèse de liquides ioniques à motif imidazolium peut être réalisée en une étape, en présence d'acide minéraux sans halogénures ; par exemple, (1) en présence d'acide acétique, (WO 2011/056924), ou (2) sous atmosphère inerte en présence d'HBF4, (JP 2008074740). Cependant, cette synthèse utilise dans tous les cas, des réactifs à la fois classés (i) « CMR » (Cancérogène, Mutagène et Reprotoxique) comme le formaldéhyde,
et (ii) toxique par ingestion, corrosif et inflammable comme les aminés. Les rendements obtenus par ces procédés sont variables selon la nature de l'acide et sont compris entre 50 à 70%.
Les procédés de synthèse classiques ne permettent pas de fournir des liquides ioniques ayant une pureté élevée. Les méthodes existantes à ce jour ne requièrent pas l'utilisation de sels alcalins mais utilisent des réactifs très alkylants, tel que le diméthylsulfonate par exemple, conduisant la plupart du temps au dégagement de gaz toxiques et corrosifs (US 2007/0255064). Ceci limite donc fortement leur utilisation dans des domaines à forte valeur ajoutée tels que celui de la cosmétologie, de la parfumerie ou de agroalimentaire (arômes).
Par ailleurs, les procédés de synthèse de liquides ioniques sous forme de sels d'imidazoliums conduisent le plus souvent à des produits fortement contaminés par les sous-produits de réaction (coloration brun/noir) et impliquent des étapes lourdes de purification (US 2010/0249432). II existe donc un besoin de développer des procédés de préparation de liquides ioniques plus purs et exempts d'halogénures permettant d'une part, d'accroître la sélectivité et les rendements des synthèses dans lesquelles ils sont employés ; et d'autre part, permettant d'utiliser des réactifs présentant une dangerosité amoindrie, plus favorable à l'application de la réglementation REACh (« Registration, Evaluation and Autorisation of Chemicals »). En particulier, il existe un besoin de fournir des procédés de préparation de liquides ioniques qui permettent la production à grande échelle de ces composés dans des conditions de sécurité optimales pour l'opérateur tout en limitant le coût et la complexité des étapes de purification en fin de réaction. Il existe également un besoin de développer des procédés de synthèse de liquides ioniques plus respectueux de l'environnement ; en particulier, permettant de facilement recycler les liquides ioniques après utilisation.
Un autre défi majeur est de pouvoir développer des catalyseurs renouvelables à partir des ressources issues de la biomasse. A ce jour, dans le cas des liquides ioniques, les cations les plus utilisés sont issus d'ammoniums naturels tels que la choline, l'éphédrine, la
nicotine, la bétaïne ou les aminoalcools. Peu d'exemples de liquides ioniques bio-sourcés ayant un cation imidazolium ont été reportés dans la littérature (Villa et al., Green Chemistry, 2003, 5, 623-626 ; Kirchhecker et al., Green Chemistry, 2014, 16, 3705-3709 ; Esposito et al., Chem. Eur. ]., 2013, 19, 15097-15100). La Demanderesse a mis en évidence un nouveau procédé de synthèse de liquides ioniques qui permet de répondre aux besoins de l'art antérieur. En premier lieu, tous les liquides ioniques de l'invention sont bio-sourcés et le procédé est mis en œuvre à partir d'hydrogénoxalate d'alkylimidazolium qui est lui-même obtenu à partir de réactifs non CMR et non toxique. De plus, le Demanderesse a également mis en évidence que l'utilisation de ces composés comme catalyseurs d'estérification permet d'une part d'obtenir des rendements élevés (supérieurs à 99%) tout en évitant la dégradation du matériel de synthèse ; et d'autre part, permet de séparer plus facilement le catalyseur du milieu en fin de réaction et de le recycler.
RÉSUMÉ
L'invention concerne donc un procédé de préparation de liquides ioniques de formule
(I) :

(I)
dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle,
hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle ;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H ;
A" représente un anion choisi parmi les anions de sels alcalins ou les bases conjuguées d'un acide organique ou d'un acide minéral ayant un pKa inférieur à 14 ; comprenant :
(i-0) une étape de synthèse d'un hydrogénoxalate d'alkylammonium ;
(i) une étape de synthèse d'un hydrogénoxalate d' alkylimidazolium ; puis
(ii) une étape d'échange d'anions.
Selon un mode de réalisation, les réactifs de l'étape (i-0) comprennent de l'acide oxalique et une aminé.
Selon un mode de réalisation, l'aminé est un acide aminé.
Selon un mode de réalisation, l'étape (i) comprend au moins une source d'ammonium, une source de formaldéhyde et un composé comprenant une fonction oxalyle.
Selon un mode de réalisation, la source de formaldéhyde est choisie parmi les composés organiques ayant au moins une fonction aldéhyde ou les composés organiques pouvant libérer dans les conditions de réaction un composé ayant au moins une fonction aldéhyde ; de préférence, le composé organique comprenant une fonction aldéhyde est formé in situ ; plus préférentiellement, le composé organique comprenant une fonction aldéhyde est l'urotropine. Selon un mode de réalisation, Γ hydrogénoxalate d' alkylimidazolium est choisi parmi le groupe des hydrogénoxalate d'imidazolium de formule (IV) :

(IV) dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H.
Selon un mode de réalisation, l'échange d' anions s'effectue entre l'hydrogénoxalate d'alkylimidazolium obtenu à l'étape (i) et un acide ayant un pKa inférieur à 2; de préférence, inférieur à 1,2 ; plus préférentiellement, avec un acide choisi parmi H2SO4, HBF4, HPF6 ou HNTf2.
Selon un mode de réalisation, l'échange d' anions s'effectue entre l'hydrogénoxalate d'alkylimidazolium obtenu à l'étape (i) et un sel alcalin.
Selon un mode de réalisation, l'échange d' anions s'effectue entre l'hydrogénoxalate d'alkylimidazolium obtenu à l'étape (i) et un acide ayant un pKa supérieur à 1,2, dans une cellule électrochimique soumise à une intensité électrique.
Selon un mode de réalisation, l'intensité électrique appliquée dans la cellule électrochimique est comprise dans une gamme allant de 40 mA à 900 mA, de préférence de 50 mA à 80 mA ; plus préférentiellement, l'intensité électrique est égale à environ 60 mA. Selon un mode de réalisation, la cellule électrochimique comprend un solvant choisi parmi les solvants polaires ; de préférence, l'acétonitrile.
Selon un mode de réalisation, la cellule électrochimique ne comprend pas d'électrolyte.
La présente invention concerne également un liquide ionique susceptible d'être obtenu par le procédé de l'invention, de formule (I) :

(D dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle ;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H ;
A" représente un anion choisi parmi les anions de sels alcalins ou les bases conjuguées d'un acide organique ou d'un acide minéral ayant un pKa inférieur à 14.
La présente invention concerne également l'utilisation d'un liquide ionique comme catalyseur de réaction d'estérification dans laquelle le produit (ester) formé est un arôme ou un agent cosmétique.
Selon un mode de réalisation, l'arôme formé est le lévulinate de butyle.
Selon un mode de réalisation, l'agent cosmétique formé est le myristate d'isopropyle.
DÉFINITIONS
Dans la présente invention, les termes ci-dessous sont définis de la manière suivante :
« Liquide ionique » : concerne un sel constitué d'un cation organique et d'un anion organique ou inorganique ayant une température de fusion inférieure à 100°C dans des conditions normales de pression. Dans la présente invention, le cation organique est un imidazolium ou l'un de ses dérivés ;
« Recyclable » : est relatif à un composé pouvant être réengagé dans plusieurs réactions chimiques successives tout en conservant une bonne efficacité ;
« Acide minéral » : concerne un acide dérivant d'un minéral inorganique ;
« Base conjuguée d'un acide » : concerne une espèce chimique obtenue par la déprotonation de l'acide correspondant ; cette espèce forme avec l'acide correspondant un couple acide/base caractérisé par son pKa ;
« Alkyle » : concerne une chaîne hydrocarbonée, linéaire ou ramifiée, comportant de 1 à 20 atomes de carbone ; préférentiellement, de 1 à 15 atomes de carbone ; préférentiellement, méthyle, éthyle, propyle, isopropyle, w-butyle, sec-butyle, isobutyle, iert-butyle, pentyle, hexyle, heptyle, octyle, nonyle, décyle, undécyle, dodécyle, tridécyle, tétradécyle, pentadécyle ;
« Aryle » : concerne un système mono- ou polycyclique de 5 à 20, de préférence de 6 à 12, atomes de carbone possédant un ou plusieurs noyaux aromatiques (quand il y a deux noyaux, il est fait référence à un biaryle) parmi lesquels on peut citer le groupe phényle, le groupe biphényle, le groupe 1-naphtyle, le groupe 2-naphtyle, le groupe tétrahydronaphtyle, le groupe indanyle, et le groupe binaphtyle ;
« Alcène » : concerne une chaîne hydrocarbonée insaturée, linéaire ou ramifiée, comportant au moins 2 atomes de carbone, caractérisée par la présence d'au moins une double liaison covalente entre deux atomes de carbone ; de préférence, comportant de 2 à 20 atomes de carbone ; plus préférentiellement, de 2 à 15 atomes de carbone ;
« Alcyne » : concerne un groupe hydrocarbyle insaturé monovalent, dans lequel l'insaturation provient de la présence d'une ou plusieurs liaisons carbone-carbone triple. Les groupes alcynyles en général, et de préférence, ont le même nombre d'atomes de carbone tel que décrit ci-dessus relativement aux groupes alkyles. Des exemples non-limitatifs de groupes alcynyles sont les groupes éthynyles, 2- propynyle, 2-butynyle, 3-butynyle, 2-pentynyle et ses isomères ;
« Acide aminé » : concerne un composé organique portant à la fois une fonction acide carboxylique et une fonction aminé. Au sens de la présente invention, les acides aminés sont préférentiellement les 22 acides protéinogènes ; de préférence, les acides aminés sont choisis parmi l'alanine, arginine, asparagine, aspartate, cystéine, glutamate, glutamine, glycine, histidine, isoleucine, leucine, lysine, méthionine, phénylalanine, proline, pyrrolysine, sélénocystéine, sérine, thréonine, tryptophane, tyrosine, valine ;
« Aldéhyde » : concerne un groupement -CHO ;
« Acide sulfurique » : concerne un composé de formule H2SO4 ;
« Acide tétrafluoroborique » : concerne un composé de formule HBF4 ;
« ide hexafluorophosphorique » : concerne un composé de formule HPF6 ;
« Catalyseur » : concerne un composé chimique capable d'accélérer la vitesse d'une réaction chimique, de préférence, d'une réaction d'estérification ;
« Cycloalkyle » : concerne un groupement alkyle cyclique ou polycyclique, optionnellement ramifié, substitué ou non substitué ; de préférence un groupement cyclopropyle, cyclopentyle ou cyclohexyle ;
« Cycloalcényle » : concerne un groupement alcène cyclique ou polycyclique, optionnellement ramifié, substitué ou non substitué ; de préférence un groupement cyclopropényle, cyclopentényle ou cyclohexényle ;
« Hétéroatome » : concerne un atome d'une molécule organique possédant au moins un doublet électronique qui diffère du carbone ou de l'hydrogène. Au sens de la présente invention, un hétéroatome est choisi parmi O, S ou N ;
« Hétéroalkyle » : concerne un groupement alkyle tel que décrit ci-dessus, comprenant un ou plusieurs hétéroatomes choisis parmi O, S ou N ;
« Hétéroalcène » : concerne un groupement alcène tel que décrit ci-dessus, comprenant un ou plusieurs hétéroatomes choisis parmi O, S ou N ;
« Hétérocycloalkyle » : concerne un groupement cycloalkyle tel que décrit ci- dessus, comprenant un ou plusieurs hétéroatomes choisis parmi O, S ou N ;
« Hétéroaryle » : concerne un groupement aryle tel que décrit ci-dessus, comprenant un ou plusieurs hétéroatomes choisis parmi O, S ou N ;
« Hydrogénoxalate » : concerne un ion de formule C2HO4" ;
« Oxo » : concerne une fonction C=0 ;
« Oxalyle » : concerne un composé ayant un groupe dioxo vicinal; Le. deux fonctions C=0 côte-à-côte ;
« Nitro » : concerne une fonction -NO2 ;
« Amido » : concerne une fonction -NR-CO- dans laquelle R représente H ou un groupe alkyl tel que défini ci-dessus ;
« Cyano » : concerne une fonction -C≡N ;
« Alcoxy » : concerne un groupe O-alkyle ;
« NTf2 » : représente le composé triflimide (ou bis(trifluorométhylsulfonyl)imide) de formule brute C2F6NO4S2 ;
« pKa » : concerne une indication de la constante d'acidité Ka (pKa= -log Ka) caractérisant l'équilibre d'un couple acide/base. Dans la présente invention, le pKa est inférieur à 14 ; de préférence, le pKa est inférieur ou égal à 2 ; plus préférentiellement, le pKa est inférieur ou égal à 1,2. Selon la présente invention, le pKa est compris de plus de 0 à 14 ; de préférence, de plus de 0 à 2 ; encore plus préférentiellement, de plus de 0 à 1,2. Selon la présente invention, lorsqu'une cellule électrochimique est utilisée, le pKa de l'acide est supérieur à 1,2 ; de préférence, compris de 1,2 à 14 ; plus préférentiellement ; compris de 2 à 14 ;
« estérification » : concerne une réaction de chimie organique entre une fonction alcool et une fonction acide carboxylique conduisant à la formation d'une fonction ester (-COO-) ;
« arôme » : concerne tout produit ou substance destiné à être ajouté à une denrée alimentaire pour lui conférer une odeur, c'est-à-dire une perception par voie nasale ou rétro-nasale, et/ou un goût, c'est-à-dire une perception par voie linguale, et qui appartient à l'une des catégories d'agents d'aromatisations tels que définis par la directive européenne 88-388 ;
« agent cosmétique » : concerne toute substance destinée à être mise en contact avec diverses parties superficielles du corps humain (telles que par exemple, l'épidémie, les systèmes pileux et capillaire, ongles, etc .), avec les dents et/ou avec les muqueuses buccales, en vue exclusivement ou principalement, de les nettoyer, de les parfumer, d'en modifier l'aspect, de les protéger, de les maintenir en bon état ou de corriger les odeurs corporelles. En particulier, l'agent cosmétique satisfait les exigences définies par le règlement sur l'enregistrement, l'évaluation, l'autorisation et les restrictions des substances chimiques (REACH : Registration, Evaluation, Authorization and Restriction of CHemicals) ;
« aliphatique » : concerne un composé organique non aromatique ;
« environ » : placé devant un nombre, signifie plus ou moins 10% de la valeur nominale de ce nombre ;
« bio-sourcé » : caractérise un produit issu de la biomasse d'origine végétale ou animale ;
« sel alcalin » : concerne un sel constitué d'un cation qui est un métal alcalin, et d'un anion. De préférence, dans la présente invention, le sel alcalin est choisi parmi NaBF4,
KPF6 ou LiNTf2.
DESCRIPTION DÉTAILLÉE
Dans un premier aspect, la présente invention a pour objet un liquide ionique et/ou catalyseur de formule générale (I)

(I) dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; préférablement Ri et R2 sont identiques; plus préférentiellement, Ri et R2 sont identiques et représentent une chaîne alkyle non substituée ou une chaîne alkyle substituée par un groupe alkyle ou un groupe aryle ;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H ;
A" représente un anion choisi parmi les anions de sels alcalins ou les bases conjuguées d'un acide organique ou d'un acide minéral ayant un pKa inférieur à 14 ; de préférence, ayant un pKa inférieur ou égal à 2 ; plus préférentiellement, ayant un pKa inférieur ou égal à 1,2. Avantageusement, ledit anion est obtenu par une étape d'échange d' anion sur un hydrogénoxalate d'alkylimidazolium.
Dans un mode de réalisation, le catalyseur de formule générale (I) est un liquide ionique recyclable. Dans un mode de réalisation, le catalyseur de formule générale (I) est un liquide ionique recyclable bio-sourcé.
Dans un mode de réalisation, l'utilisation d'un liquide ionique telle que décrite ci-dessus ne comprend pas l'un des liquides ioniques suivantes :
- hydrogénosulfate de l-butyl-3-méthylimidazolium ;
- hydrogénosulfate de l-méthyl-3-éthylimidazolium ;
- hydrogénosulfate de 1-méthylimidazolium ;
- hydrogénosulfate de l-sulfobutyl-3-méthylimidazolium ;
- sulfate de 1-méthylimidazolium :
- dihydrogénophosphate de l-éthyl-3-méthylimidazolium ;
- dihydrogénophosphate de l-hexyl-3-méthylimidazolium ;
- dihydrogénophosphate de 1-méthylimidazolium ;
- nitrate de 1-méthylimidazolium ;
- chlorure de 1-méthylimidazolium ;
- perchlorate de l-butyl-3-méthylimidazolium ;
- tétrafluoroborate de 1-méthylimidazolium.
Selon un mode de réalisation, A" est un anion qui est une base conjuguée d'un acide minéral choisi parmi H2SO4, HBF4, HPF6, HNTf2. Selon un second mode de réalisation, A" représente un anion non halogéné. Selon un mode de réalisation, A" est un anion qui n'est pas aromatique.
Selon un mode de réalisation, A" est un anion qui est un anion d'un sel alcalin ; de préférence, A" est un anion qui est un anion d'un sel alcalin choisi parmi NaBF4, KPF6 ou LiNTf2.
Selon un mode de réalisation, A" n'est pas un halogénure. En effet, la présence d'halogénure inhibe les propriétés catalytiques des composés. Selon un mode de réalisation, A" n'est pas Cl". Selon un mode de réalisation, A" n'est pas Br". Selon un mode de réalisation, A" n'est pas F". Selon un mode de réalisation, A" n'est pas Γ. Selon un mode de réalisation, A" ne comprend pas d'halogène. Selon un mode de réalisation, A" ne comprend pas d'atome brome. Selon un mode de réalisation, A" ne comprend pas d'atome fluor. Selon un mode de réalisation, A" ne comprend pas d'atome d'iode. Selon un mode de réalisation, A" ne comprend pas d'atome chlore.
Selon un mode de réalisation, le cation de la formule générale (I) est symétrique. Dans un second mode de réalisation, le cation de la formule générale (I) n'est pas symétrique.
Selon un mode de réalisation, le catalyseur (liquide ionique) de formule générale (I) est liquide. Selon un mode de réalisation, le catalyseur (liquide ionique) de formule générale (I) n'est pas un gel. Selon un mode de réalisation, le catalyseur (liquide ionique) de formule générale (I) n'est pas un polymère. Selon un mode de réalisation, Ri, R2, R3 et R4 ne comprennent pas de chaînes polymères. Dans un mode de réalisation, Ri, R2, R3 et R4 ne comprennent pas de fonction acide ; de préférence, Ri, R2, R3 et R4 ne comprennent pas de fonction acide sulfonique. Selon un mode de réalisation, Ri ne comprend pas de fonction acide sulfonique. Selon un mode de réalisation, R2 ne comprend pas de fonction acide sulfonique. Selon un mode de réalisation, R3 ne comprend pas de fonction acide sulfonique. Selon un mode de réalisation, R4 ne comprend pas de fonction acide sulfonique.
Dans un mode de réalisation, Ri et R2 ne représentent pas H. Dans un mode de réalisation, Ri et R2 ne représentent pas CH3.
Dans un mode de réalisation, le catalyseur (liquide ionique) de formule générale (I) est un liquide ionique qui n'est pas polyionique. Dans un mode de réalisation, le catalyseur de formule générale (I) n'est pas un catalyseur de transfert de phase.
Dans un second aspect, l'invention concerne l'utilisation du composé (I), comme catalyseur de réaction d'estérification.
Dans un troisième aspect, l'invention concerne l'utilisation du composé (I), comme catalyseur de réaction d'estérification pour la fabrication d'agents cosmétiques, d'arômes, de biocarburants, de pesticides ou de solvants faiblement volatils ; de préférence, pour la synthèse et la fabrication d'agents cosmétiques ou d'arômes. Selon un mode de réalisation, l'invention concerne l'utilisation d'un liquide ionique tel que décrit ci-dessus comme catalyseur de réaction d'estérification pour la fabrication d'agents cosmétiques, d'arômes, de biocarburants, de pesticides ou de solvants faiblement volatils, très préférentiellement pour la synthèse et la fabrication d'agents cosmétiques ou d'arômes. Dans un mode de réalisation, la réaction d'estérification conduit à des esters utiles dans le domaine agroalimentaire.
Dans un mode de réalisation, ladite réaction d'estérification est une réaction dans laquelle les réactifs de la réaction d'estérification comprennent au moins un composé organique aliphatique acyclique ayant une fonction alcool.
Dans un autre mode de réalisation, ladite réaction d'estérification est une réaction dans laquelle les réactifs de la réaction d'estérification comprennent au moins un composé organique aliphatique acyclique ayant une fonction acide carboxylique.
Selon un mode de réalisation, l'utilisation d'un liquide ionique telle que décrite ci-dessus ne comprend pas les liquides ioniques de formule (I) dans laquelle Ri ou R2 représente un atome d'hydrogène (H). Selon un mode de réalisation, le catalyseur de formule générale (I) est un liquide ionique recyclable qui est séparé du milieu réactionnel par décantation à la fin de la réaction, en particulier de la réaction d'estérification. Selon un mode de réalisation, le catalyseur de formule générale (I) est séparé du milieu réactionnel par décantation à la fin de la réaction, en particulier de la réaction d'estérification. Dans un mode de réalisation, la réaction d'estérification n'est pas menée en micro-ondes.
Dans un mode de réalisation, la réaction d'estérification est menée à pression atmosphérique. Selon un mode de réalisation, la réaction d'estérification n'est pas menée
en autoclave. Selon un mode de réalisation, la réaction d'estérification n'est pas menée à pression réduite.
Dans un mode de réalisation, la réaction d'estérification est menée à une température allant de 20 à 400°C ; de préférence, allant de 40 à 150°C ; plus préférentiellement, la réaction d'estérification est menée à environ 70°C, 80°C ou à 100°C.
Dans un mode de réalisation, la réaction d'estérification est menée pendant moins de 72h ; de préférence, moins de 48h ; plus préférentiellement, moins de 24h. Selon un mode de réalisation, la réaction d'estérification est menée pendant environ 50h. Selon un mode de réalisation, la réaction d'estérification est menée pendant environ 16h. Selon un mode de réalisation, la réaction d'estérification est menée pendant environ lh.
Dans un mode de réalisation, le catalyseur est ajouté dans le milieu à une concentration allant de plus de 0 à 10% mol ; de préférence, à une concentration allant de 1 à 6%mol. ; plus préférentiellement, le catalyseur est ajouté dans le milieu à une concentration d'environ 4% mol. Dans un mode de réalisation, le réactif qui est un composé organique non aromatique ayant au moins une fonction alcool, comprend le méthanol, l'éthanol, le propanol, Γ isopropanol, le butanol. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction alcool est choisi parmi le méthanol, le butanol, l'éthanol ou l'isopropanol. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction alcool est le méthanol. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction alcool est l'éthanol. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction alcool est l'isopropanol. Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction alcool ne comprend pas de fonction alcène.
Dans un mode de réalisation, le réactif qui est un composé organique non aromatique ayant au moins une fonction acide carboxylique, comprend l'acide méthanoïque, l'acide éthanoïque, l'acide propanoïque, l'acide butanoïque, l'acide pentanoïque, l'acide hexanoïque, l'acide heptanoïque, l'acide octanoïque, l'acide nonanoïque, l'acide
décanoïque, l'acide undécanoïque, l'acide dodécanoïque, l'acide tridécanoïque, l'acide tétradécanoïque (ou acide myristique), l'acide 3-oxopentanoïque, l'acide 4- oxopentanoïque (ou acide lévulinique). Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique est l'acide tétradécanoïque ou l'acide 4-oxopentanoïque. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique est l'acide myristique ou l'acide lévulinique. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique est l'acide myristique. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique est l'acide lévulinique. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique comprend plusieurs fonctions acides carboxyliques.
Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique n'est pas l'acide pivalique. Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique n'est pas l'acide myristique. Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique n'est pas l'acide palmitique. Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique n'est pas l'acide acétique. Dans un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique ne comprend pas de fonction alcène. Selon un mode de réalisation, le composé organique non aromatique ayant au moins une fonction acide carboxylique n'est pas l'acide linoléique.
Dans un mode de réalisation, le composé organique non aromatique ayant plusieurs fonctions acides carboxyliques n'est pas l'acide sébacique. Dans un mode de réalisation, le milieu réactionnel n'est pas hétérogène. Dans un mode de réalisation, la réaction d'estérification est menée sans ajout de catalyseur de transfert de phase.
Selon un mode de réalisation, la réaction d'estérification conduit à un ester utile comme arôme. Selon un mode de réalisation préféré, l'arôme est le lévulinate de butyle.
Selon un mode de réalisation, la réaction d'estérification conduit à un ester utile comme agent cosmétique ; tel que, mais non limité à, un agent émollient, dépigmentant, cicatrisant, hydratant, parfumant, désodorisant, anti- transpirant, nettoyant, colorant, conservateur, volumateur, repulpant et/ou tenseur. Selon un mode de réalisation préféré, l'agent cosmétique est le myristate d'isopropyle. Selon un mode de réalisation préféré, l'agent cosmétique est le lévulinate de butyle.
Selon un mode de réalisation, la réaction d'estérification conduit à un ester qui n'est pas un polymère.
Dans un mode de réalisation, la conversion du réactif limitant est supérieure à 80% mol. ; de préférence, supérieure à 90% mol. ; plus préférentiellement, la conversion est totale.
Dans un mode de réalisation, les composés préférés de formule (I) sont les composés de formule générale (II) :

(Π) dans laquelle, Ri, R2 et A" sont définis tels que précédemment.
Selon un mode de réalisation, les liquides ioniques préférés de formule (II) sont les liquides ioniques de formule (Ha) :

(lia) dans laquelle, Ri et R2 sont définis tels que précédemment.
Dans un mode de réalisation, les composés préférés de formule (I) sont les composés de formule générale (III) :

(III) dans laquelle,
Ri et R2 sont identiques et représentent une chaîne alkyle non substituée ou une chaîne alkyle substituée par un groupe alkyle ou un groupe aryle ; et
A" est défini tel que précédemment.
Selon un mode de réalisation, les liquides ioniques préférés de formule (III) sont les liquides ioniques de formule (Illa) :

(Ma) dans laquelle, Ri et R2 sont définis tels que précédemment.
Selon un mode de réalisation préféré, le liquide ionique recyclable est choisi l'hydrogénosulfate de 1,3-diisobutylimidazolium ou l'hydrogénosulfate de butylméthylimidazolium. Dans un mode de réalisation, les liquides ioniques préférés de formule (I) sont les composés présentés dans le Tableau 1 ci-dessous.
Tableau 1


Bis(trifluorométhylsulfonyl)amidure de 1 ,3-diisobutyl-2,3-dihydro- iH-imidazolium
^ /— \ ^ Θ -^ ^ N\^ N ^^^ BF4
Tétrafluoroborate de l,3-dipropyl-2,3-dihydro-7H-imidazolium
^ /— \ _ 0 ^Ν^ Νχ^Ν -^^^ PF6
Hexafluorophosphate de l,3-dipropyl-2,3-dihydro-7H-imidazolium
^Ν^ Ν ^ Ν ^.^^^ NTf2
Bis(trifluorométhylsulfonyl)amidure de l,3-dipentyl-2,3-dihydro- iH-imidazolium
Tétrafluoroborate de l,3-diphényléthyl-2,3-dihydro-7H- imidazolium
Hexafluorophosphate de l,3-diphényléthyl-2,3-dihydro-7H- imidazolium
Bis(trifluorométhylsulfonyl)amidure de l,3-diphényléthyl-2,3- dihydro-iH-imidazolium
/— \ , Θ
^^ Ν ^Ν ^/ HS04
Hydrogénosulfate de l,3-diéthyl-2,3-dihydro-7H-imidazolium
/— \ Θ
N^ N^N -^/ BF4
Tétrafluoroborate de l,3-diéthyl-2,3-dihydro-7H-imidazolium
/— \ „ Θ
Ν^ Νχ^Ν ~^/ PF6
Hexafluorophosphate de l,3-diéthyl-2,3-dihydro-7H-imidazolium N^N NTf2
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-diéthyl-2,3-dihydro-7H- imidazolium
/— \ Θ
^ Ν ^Ν ^ HS04
Hydrogénosulfate de l,3-diméthyl-2,3-dihydro-7H-imidazolium
/— \ Θ
^ N^N ^ BF4
Tétrafluoroborate de l,3-diméthyl-2,3-dihydro-7H-imidazolium
/— \ Θ
^Ν ^Ν ^ PF6
Hexafluorophosphate de l,3-diméthyl-2,3-dihydro-7H-imidazolium
/— \ Θ
^ Νχά^Ν^, NTf2
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-diméthyl-2,3-dihydro-7H- imidazolium
Hydrogénosulfate de l,3-dibutyl-2,3-dihydro-7H-imidazolium N^N ^7 BF4 0
Tétrafluoroborate de l,3-dibutyl-2,3-dihydro-7H-imidazolium
Hexafluorophosphate de l,3-dibutyl-2,3-dihydro-7H-imidazolium
Ns^'\^ Nv^; N ¾Tf2
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-dibutyl-2,3-dihydro-7H- imidazolium
Hydrogénosulfate de l,3-dipentyl-2,3-dihydro-7H-imidazolium
Tétrafluoroborate de l,3-dipentyl-2,3-dihydro-7H-imidazolium
Hexafluorophosphate de l,3-dipentyl-2,3-dihydro-7H-imidazolium

Bis(trifluorométhylsulfonyl)amidure de 1 ,3-dipentyl-2,3-dihydro-7H- imidazolium

Hydrogénosulfate de l,3-dihexyl-2,3-dihydro-7H-imidazolium
^^^ - N■ N ^/^^0
Tétrafluoroborate de l,3-dihexyl-2,3-dihydro-7H-imidazolium
"^^^^-^V^ N N -^ ^^ΐ^ Θ
Hexafluorophosphate de l,3-dihexyl-2,3-dihydro-7H-imidazolium
^^^^- N ^/^^^ 0 NTf2
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-dihexyl-2,3-dihydro-7H- imidazolium
Hydrogéno sulfate de l,3-butylméthyl-2,3-dihydro-7H-imidazolium
^N^N ^^/ BF4
Tétrafluoroborate de l,3-butylméthyl-2,3-dihydro-7H-imidazolium
Hexafluorophosphate de 1,3- butylméthyl -2,3-dihydro-iH-imidazolium i— \ ^ / 0
^N^N ^7 NTf2
Bis(trifluorométhylsulfonyl)amidure de 1,3- butylméthyl-2,3-dihydro- iH-imidazolium
/— \ / Q
^ Nv^N ^/ HS04
Hydrogéno sulfate de l,3-éthylméthyl-2,3-dihydro-7H-imidazolium

Tétrafluoroborate de 1,3-éthylméthyl -2,3-dihydro-iH-imidazolium
^N^N^ PF6 U
Hexafluorophosphate de 1,3-éthylméthyl -2,3-dihydro-iH-imidazolium
/_\ / Θ
^Νχ^Ν^ ¾Tf2
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-éthylméthyl-2,3-dihydro- iH-imidazolium
Hydrogénosulfate de l,3-propylméthyl-2,3-dihydro-7H-imidazolium
Tétrafluoroborate de l,3-propylméthyl-2,3-dihydro-7H-imidazolium
^N^N^ PF6 U
Hexafluorophosphate de 1,3-propylméthyl -2,3-dihydro-iH- imidazolium
/— \ ^ Θ
^ Νχ^Ν ^^^ NTf2
44
Bis(trifluorométhylsulfonyl)amidure de 1 ,3-propylméthyl-2,3-dihydro- iH-imidazolium
L'invention concerne également un procédé de préparation de liquides ioniques et /ou catalyseurs de formule générale (I)

(I) dans laquelle,
Ri, R2, R3, R4 et A" sont définis tels que précédemment, comprenant :
(i) une étape de synthèse d'un hydrogénoxalate d'alkylimidazolium ; puis
(ii) une étape d'échange d'anions.
Selon un mode de réalisation, l'étape (i) comprend en outre une étape préliminaire de synthèse d'un hydrogénoxalate d'ammonium (notée (i-0)).
En particulier, la présente invention concerne un procédé de préparation de liquides ioniques et/ou catalyseurs de formule générale (I)

(I)
dans laquelle,
Ri, R2, R3, R4 et A" sont définis tels que précédemment ; comprenant :
une étape de synthèse d'un hydrogénoxalate d'alkylammonium ; une étape de synthèse d'un hydrogénoxalate d'alkylimidazolium ; puis
(ϋ) une étape d'échange d'anions.
Selon un mode de réalisation, le procédé de l'invention ne comprend pas d'étape exothermique.
Avantageusement, le procédé de l'invention fournit un composé de formule générale (I)

(I) dans laquelle,
Ri, R2, R3, R4 et A" sont définis tels que précédemment, sous la forme d'un produit qui n'est pas brun ou noir ; de préférence sous la forme d'un produit incolore.
Avantageusement, le procédé de l'invention fournit un composée de formule (I) qui n'est pas une huile.
Avantageusement, le procédé de l'invention fournit un composée de formule (I) qui ne nécessite pas d'étapes de purification lourdes. Selon un mode de réalisation, le procédé de l'invention comprend une étape de purification dans un solvant oxygéné ; de préférence, dans un solvant oxygéné qui n'est pas du peroxyde d'hydrogène ou de l'hydroxyde de sodium. Selon un mode de réalisation, le procédé de l'invention comprend une étape de purification qui est une recristallisation. Selon un mode de réalisation, le
procédé de l'invention comprend une étape de purification qui comprend des lavages et/ou une filtration.
Selon un mode de réalisation, le procédé de l'invention ne comprend pas d'étape de purification comprenant du peroxyde d'hydrogène. Selon un mode de réalisation, le procédé de l'invention ne comprend pas d'étape de purification comprenant l'utilisation d'hydroxyde de sodium. Selon un mode de réalisation, le procédé de l'invention ne comprend pas d'étape de purification à une température supérieure à 60°C. Selon un mode de réalisation, le procédé de l'invention ne comprend pas d'étape de purification à une température comprise de 60°C à 100°C. Selon un mode de réalisation, le procédé de l'invention comprend une étape de purification menée à température ambiante.
Etape (i-0)
Selon un mode de réalisation, les réactifs de l'étape (i-0) comprennent un acide, de préférence, un diacide ; et une aminé.
Selon un mode de réalisation, dans l'étape (i-0), le ratio molaire diacide/amine est compris dans une gamme allant de 0,5 à 10 ; de préférence, de 1 à 2 ; plus préférentiellement, est égale à environ 1. Selon un mode de réalisation, dans l'étape (i-0), le ratio molaire diacide/amine est compris dans une gamme allant de 1 à 10. Selon un mode de réalisation le diacide et l'aminé sont introduits en quantité stœchiométrique dans l'étape (i-0).
Selon un mode de réalisation, le diacide utilisé comme réactif dans l'étape (i-0) est l'acide oxalique. Selon un mode de réalisation, le diacide utilisé comme réactif dans l'étape (i-0) est une solution aqueuse d'acide oxalique. Selon un mode de réalisation, le diacide utilisé comme réactif dans l'étape (i-0) est une solution aqueuse d'acide oxalique préalablement chauffée à environ 50°C. Selon un mode de réalisation, la solution aqueuse d'acide oxalique est à une concentration comprise dans une gamme de 1 à 3 mol/L ; de préférence, de 1,5 à 2,5 mol/L ; plus préférentiellement, la concentration de la solution d'acide oxalique est égale à environ 1,8 mol/L.
Selon un mode de réalisation, l'acide n'est pas un monoacide, i.e. un acide organique ayant une fonction carboxylique. Selon un mode de réalisation, l'acide n'est pas l'acide
acétique, l'acide formique, l'acide éthanoïque, l'acide proanoique, l'acide pentanoïque, l'acide hexanoïque, l'acide heptanoique, l'acide octanoïque, l'acide nonanoïque ou l'acide décanoique. Selon un mode de réalisation, l'acide n'est pas l'acide benzoïque.
Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est de source commerciale. Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est un composé organique, linéaire ou ramifiée, ayant au moins une fonction amine.
Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est un composé organique ayant au moins une fonction amine et comprenant de 1 à 10 atomes de carbone; de préférence, comprenant de 1 à 6 atomes de carbones ; plus préférentiellement, l'aminé est choisie parmi méthylamine, éthylamine, propylamine, butylamine, pentylamine et hexylamine. Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est l'isobutylamine. Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est la propylamine. Selon un mode de réalisation, l'aminé primaire est la phényléthylamine.
Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est de source naturelle (i.e. biosourcée); de préférence, l'aminé est choisie parmi les acides aminés. Selon un mode de réalisation, l'acide aminé utilisé comme réactif dans l'étape (i-0) est choisi parmi l'alanine, arginine, asparagine, aspartate, cystéine, glutamate, glutamine, glycine, histidine, isoleucine, leucine, lysine, méthionine, phénylalanine, proline, pyrrolysine, sélénocystéine, sérine, thréonine, tryptophane, tyrosine, valine. Selon un mode de réalisation, l'acide aminé est de forme L. Selon un mode de réalisation, l'aminé utilisée comme réactif dans l'étape (i-0) est la L- Valine.
Selon la présente invention, les acides aminés comme source d'amine dans l'étape (i-0) sont privilégiés par rapport aux aminés conventionnelles en raison de leur non-toxicité dans le milieu réactionnel.
Selon un mode de réalisation, le solvant de l'étape (i-0) est choisi parmi les solvants organiques ; de préférence, les cétones aromatiques ; plus préférentiellement, l'acétophénone.
Selon un mode de réalisation, l'étape (i-0) est menée à une température comprise allant de 100°C à 150°C ; de préférence, de 120°C à 140°C ; plus préférentiellement, la température mise en œuvre de l'étape (i-0) est égale à environ 140°C.
Selon un mode de réalisation, l'étape (i-0) est menée à pression ambiante. Selon un mode de réalisation, l'étape (i-0) comprend en outre une étape de purification en fin de réaction. Selon un mode de réalisation, l'étape de purification de (i-0) comprend la mise en œuvre d'au moins une des techniques connues de l'homme du métier ; de préférence, ladite étape de purification comprend des cycles de lavages ; plus préférentiellement, des cycles de lavages en solvant polaire tel que l'acétone et/ou l'acétate d'éthyle.
Selon un mode de réalisation, l'étape (i-0) conduit à la formation d'hydrogénoxalate d'alkylammonium ; de préférence, d'hydrogénoxalate d'alkylammonium dans lequel le groupe alkyle peut être linéaire ou ramifié, et est choisi parmi les composés ayant de 1 à 6 atomes de carbones ; de préférence, méthyle, éthyle, propyle, isopropyle, sec-butyle, butyle, iso-butyle, sec-butyle, tert-butyle, pentyle ou hexyle. Selon un mode de réalisation, l'étape (i-0) conduit à la formation d'hydrogénoxalate d'isobutylammonium. Selon un mode de réalisation, l'étape (i-0) conduit à la formation d'hydrogénoxalate de propylammonium.
Etape (i) Selon un mode de réalisation, la synthèse d'hydrogénoxalate d'alkylimidazolium selon l'étape (i) comprend au moins une source d'ammonium, une source de formaldéhyde et un composé comprenant une fonction oxalyle.
Selon un mode de réalisation préféré, la source d'ammonium est choisie parmi les composés organiques ayant au moins une fonction ammonium ou les composés pouvant libérer dans les conditions de réaction, un composé ayant au moins une fonction ammonium. Selon un mode de réalisation préféré, la source d'ammonium est formée in situ par réaction chimique entre au moins une aminé et un acide ; de préférence, par réaction chimique entre au moins une aminé et un diacide ; encore plus
préférentiellement, entre au moins une aminé et l'acide oxalique. Selon un mode de réalisation préféré, la source d'ammonium est choisie parmi les sels d'hydrogénoxalate d'ammonium ; de préférence, les sels d'hydrogénoxalate d'ammonium obtenus selon l'étape (i-0). Selon un mode de réalisation préféré, la source d'ammonium est l'hydrogénoxalate d'isobutylammonium.
Selon un mode de réalisation préféré, la source d'aldéhyde est choisie parmi les composés organiques ayant au moins une fonction aldéhyde ou les composés pouvant libérer dans les conditions de réaction, un composé ayant au moins une fonction aldéhyde. Selon un mode de réalisation préféré, la source d'aldéhyde est du paraformaldéhyde. Selon un mode de réalisation, le composé comprenant une fonction aldéhyde formé in situ est obtenu à partir de l'urotropine (aussi nommée hexaméthylènetétramine ou méthenamine).
Selon un mode de réalisation, le composé comprenant une fonction aldéhyde peut être un composé commercial directement engagé dans le mélange ou un composé formé in situ lors de l'étape de mélange. Selon un mode de réalisation, le composé comprenant une fonction oxalyle est le glyoxal.
Selon un mode de réalisation, le glyoxal et l'acide oxalique sont bio-sourcés.
Selon un mode de réalisation, la source de formaldéhyde et le composé comprenant une fonction oxalyle sont introduits dans le mélange réactionnel en quantités stœchiométriques . Selon un mode de réalisation, le ratio molaire de l'hydrogénoxalate d'alkylammonium : source de formaldéhyde est compris dans une gamme allant de 1 à 3 ; de préférence est égal à environ 2.
Selon un mode de réalisation, la synthèse d'hydrogénoxalate d'alkylimidazolium selon l'étape (i) est menée à une température allant de 0°C à 180°C ; de préférence, la température est comprise dans une gamme allant de 25°C à 100°C. Selon un mode de réalisation, la synthèse d'hydrogénoxalate d'alkylimidazolium selon l'étape (i) est menée à température ambiante. Selon un mode de réalisation, la synthèse d'hydrogénoxalate d'alkylimidazolium selon l'étape (i) comprend une distillation azéotropique de l'eau à
une température égale à environ 85°C. Selon un mode de réalisation, la distillation azéotropique est effectuée à l'aide d'un dispositif Dean-Stark.
Selon un mode de réalisation, la synthèse d'hydrogénoxalate d' alkylimidazolmm selon l'étape (i) est menée de 1 à lOh ; de préférence, de 2 à 6h. Selon un mode de réalisation, la synthèse d'hydrogénoxalate d' alkylimidazolmm est menée pendant 5h.
Selon un mode de réalisation, le rendement de l'étape (i) est supérieur à 70%. Selon un mode de réalisation, le rendement obtenu lors de l'étape (i) est égal à environ 76%.
Selon le procédé de l'invention, l'étape (i) conduit à un produit intermédiaire qui est un hydrogénoxalate d'imidazolium.
Dans un mode de réalisation, Γ hydrogénoxalate d' alkylimidazolmm est choisi parmi le groupe des hydrogénoxalate d'iimidazolium de formule (IV) :
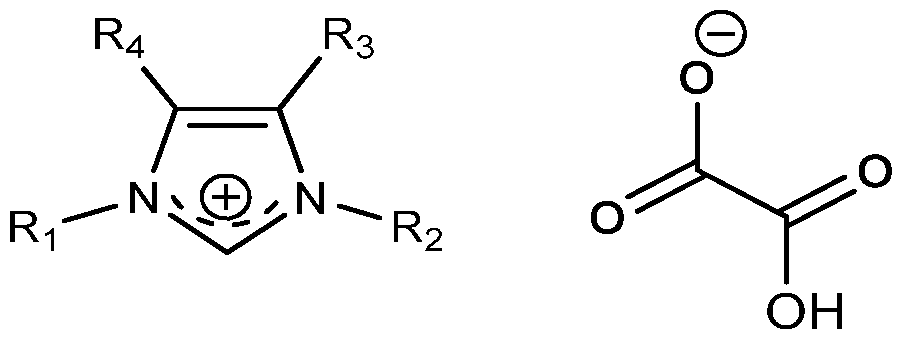
(IV) dans laquelle, Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; préférablement Ri et R2 sont identiques; plus préférentiellement, Ri et R2 sont identiques et représentent une chaîne alkyle non substituée ou une chaîne alkyle substituée par un groupe alkyle ou un groupe aryle ;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H.
Dans un mode de réalisation, Ri et R2 ne représentent pas H.
Dans un mode de réalisation, Ri, R2, R3 et R4, ne comprennent pas de fonctions acides ; de préférence, Ri, R2, R3 et R4, ne comprennent pas de fonctions acides carboxyliques ou de fonctions acides sulfoniques. La méthode de préparation d'hydrogénoxalate d' alkylimidazolium de formule générale (IV) est présentée ci-dessous (schéma 1) :

Selon la présente invention, la méthode de préparation d'hydrogénoxalate d' alkylimidazolium est plus préférentiellement menée à partir d'un sel d'ammonium selon le schéma Ibis ci-dessous :

La préparation d'hydrogénoxalate d' alkylimidazolium de formule générale (IV) peut également être menée selon la méthode décrite ci-dessous (schéma lter) :

Avantageusement, l'invention concerne également un hydrogénoxalate d'imidazolium de formule (IV) :

(IV) dans laquelle Ri, R2, R3 et R4 sont définis tels que précédemment.
Selon un mode de réalisation, les composés préférés de formule (IV) sont les composés de formule (V) :

(V) dans laquelle, Ri et R2 sont définis tels que précédemment.
Selon un mode de réalisation, les composés préférés de formule (IV) sont les compo de formule (Vbis) (appelés hydrogénoxalate de 1,3-dialkylimidazolium) :

(V) dans laquelle, Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi alkyle ou hétéroalkyle; optionnellement substitué par au moins un groupe
choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle.
Selon un mode de réalisation, les composés préférés de formule (V) sont les composés de formule (VI) :

(VI) dans laquelle, Ri et R2 sont identiques et représentent une chaîne alkyle non substituée ou une chaîne alkyle substituée par un groupe alkyle ou un groupe aryle.
Selon un mode de réalisation, les composés préférés de formule (V) sont les composés choisis parmi : l'hydrogénoxalate de dipropylimidazolium

l'hydrogénoxalate de diisobutylimidazolium

Etape (ii) Suivant un mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa inférieur à 14 ; de préférence, un acide ayant un pKa inférieur ou égal à 2 ; plus préférentiellement, un acide ayant un pKa inférieur ou égal à 1,2.
Selon un mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa compris dans une gamme allant de 2 à 14. Selon un mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa compris dans une gamme allant de 1,2 à 14. Selon un mode de réalisation, l'acide ayant un pKa compris dans une gamme allant de 1,2 à 14 est l'acide acétique. Selon un mode de réalisation, l'acide ayant un pKa compris dans une gamme allant de 1,2 à 14 est l'acide formique.
Selon un mode de réalisation, l'acide n'est pas l'acide acétique. Selon un mode de réalisation, l'acide n'est pas l'acide formique.
Selon la présente invention, l'échange d'anions peut être mené selon trois méthodes. Méthode A
Suivant un premier mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa inférieur à 2; de préférence, inférieur à 1,2. Selon un mode de réalisation, l'acide est choisi parmi H2SO4, HBF4, HPF6 ou HNTf2 ; de préférence, H2S04.
Méthode B
Suivant un deuxième mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un sel alcalin. Dans ce mode de réalisation, de préférence, l'échange d'anions est une métathèse anionique. Selon un mode de réalisation, le sel alcalin est choisi parmi NaBF4, KPF6 ou LiNTf2.
Méthode C
Suivant un troisième mode de réalisation, l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa supérieur à 1,2 dans une cellule électrochimique ; de préférence, supérieure à 2. Selon un mode de réalisation, le ratio molaire entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et l'acide est compris dans une gamme allant de 0, 6 à 1,2 ; de préférence, est égale à environ 0,8.
Selon un mode de réalisation, l'échange d'anions s'effectue dans une cellule électrochimique ; de préférence, dans une cellule électrochimique à un compartiment. Selon un mode de réalisation, la cellule électrochimique comprend au moins deux électrodes. Selon un mode de réalisation, la cellule électrochimique comprend une électrode de travail, une électrode de référence et/ou une électrode auxiliaire. Selon un mode de réalisation, la cellule électrochimique comprend au moins une électrode choisie parmi Al, Au, Ag, diamant dopé au Bore, C, Cd, Co, Cr, Cu, Ga, Hg, In, Ir, Mg, Mo, Nb, Ni, oxyde de fer, alliage Li-Fe, Os, Pb, Pd, Pt, Rh, Ru, Sn, alliage Sn, Ti, V, W, Zn, alliage Co-Cr-Ni, acier austénitique, acier duplex, acier ferrique, acier martensitique, acier inoxydable, Si- dopé et Si:As dopé; de préférence, l'électrode est choisi parmi C, Pt ou acier inoxydable. Selon un mode de réalisation, l'électrode est choisi parmi l'acier inoxydable, le feutre de carbone ou le carbone vitreux. Selon un mode de réalisation, la cellule électrochimique comprend une électrode de référence choisie parmi électrode au calomel saturé (SCE), électrode d'argent-chlorure d'argent (Ag/AgCl) ou Ag/AgN03.
Selon un mode de réalisation, la cellule électrochimique est couplée à un potentiostat et à un intégrateur.
Selon un mode de réalisation, l'intensité électrique appliquée est comprise dans une gamme allant de 40 mA à 900 mA ; de préférence, de 50 mA à 80 mA ; plus préférentiellement, l'intensité électrique appliquée est égale à environ 60 mA.
Selon un mode de réalisation, le solvant est choisi parmi les solvants organiques ; de préférence, les solvants polaires ; plus préférentiellement, l'acétonitrile.
Selon un mode de réalisation, l'étape (ii) comprend en outre une étape de chauffage à reflux. Selon un mode de réalisation, le rendement obtenu à l'étape (ii) est supérieur à 80% ; de préférence, supérieur ou égal à 85%. Selon un mode de réalisation, le rendement obtenu à l'étape (ii) est égal à environ 92%.
Selon un mode de réalisation, l'échange d'anions est mené à une température allant de 0°C à 180°C ; de préférence, la température est comprise dans une gamme allant de 10°C à 150°C ; plus préférentiellement, l'échange d'anions est mené à une température allant de 50°C à 90°C. Selon un mode de réalisation, l'échange d'anions est mené à température ambiante. Selon un mode de réalisation, l'échange d'anions est mené à une température allant de 0°C à 30°C quand l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa inférieur ou égal à 2 ; de préférence, inférieur ou égal à 1,2. Selon un mode de réalisation, l'échange d'anions est mené à une température allant de 50°C à 90°C quand l'échange d'anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu en (i) et un acide ayant un pKa inférieur à 14. Selon un mode de réalisation, l'échange d'anions est mené à pression réduite. Selon un mode de réalisation, l'échange d'anions est mené à une pression allant de 500 à 2 mbar ; de préférence, à une pression d'environ 10 mbar. Selon un mode de réalisation, l'échange d'anions n'est pas mené en autoclave. Selon un mode de réalisation, l'échange d'anions n'est pas mené à pression réduite.
Dans un mode de réalisation, l'étape d'échange d' anions selon l'une des méthodes A, B ou C telles que décrites précédemment, conduit à la formation d'un liquide ionique recyclable de formule général (I) tel que décrit précédemment, et d'acide oxalique. Selon un mode de réalisation, l'étape d'échange d' anions est réalisée dans l'acétone. Selon un mode de réalisation, l'étape d'échange d' anions est réalisée dans un éther. Selon un mode de réalisation, l'étape d'échange d' anions est réalisée dans le diéthyléther. Selon un mode de réalisation, l'étape d'échange d' anions est réalisée dans le iert-butylméthyléther (MTBE).
Sans vouloir être lié par une théorie, la Demanderesse a mis en évidence que l'emploi d'un éther comme solvant lors de l'étape d'échange d' anions solubilise le sous-produit de réaction (l'acide oxalique) dans le milieu. Ainsi, le liquide ionique de formule générale (I) obtenu par ce procédé est plus efficacement purifié.
L'invention concerne également un composé de formule (I) tel que décrit précédemment, susceptible d'être obtenu par le procédé de l'invention, et son utilisation comme catalyseur de réaction d'estérification ; de préférence, comme catalyseur dans la réaction d'estérification conduisant au lévulinate de butyle ou au myristate d'isopropyle.
En particulier, l'invention concerne également un liquide ionique de formule (I) tel que décrit précédemment, susceptible d'être obtenu par le procédé de l'invention, et son utilisation comme catalyseur de réaction d'estérification ; de préférence, comme catalyseur dans la réaction d'estérification conduisant au lévulinate de butyle ou au myristate d'isopropyle.
BRÈVE DESCRIPTION DES FIGURES
Figure 1 est une photographie montrant la séparation de phases entre le catalyseur et le myristate d'isopropyle en fin de réaction.
EXEMPLES
La présente invention se comprendra mieux à la lecture des exemples suivants qui illustrent non-limitativement l'invention.
Matériel et Méthodes Les solvants, les réactifs et le matériel de départ ont été achetés chez des fournisseurs de produits chimiques connus (par exemple : Sigma Aldrich, Acros Organics, VWR Int., Sopachem, Carlo Erba, Alfa-Aesar ou Avocado) et ont été utilisés tels que reçus, sauf indication contraire.
Toutes les températures sont données en degrés Celsius (°C) ; toutes les réactions ont été réalisées à température ambiante sauf indication contraire.
Les spectres RMN1H et RMN13C ont été enregistrés via un spectrophotomètre Bruker Avance III 300 MHz (Bruker France, Wissembourg, France). Les déplacements chimiques sont exprimés en parties par million (ppm, δ). Les constantes de couplage sont exprimées en Hertz (Hz). Les abréviations concernant les multiplicités observées sont les suivantes : s (singulet), d (doublet), t (triplet), q (quadruplet), sext (sextuplet), hept (heptuplet), m (multiplet), m (massif).
Abréviations
CDC : chloroforme deutéré ;
CO2 : dioxyde de carbone ;
d: densité ;
eq.: équivalent(s) ;
Et20: diéthyléther ;
EtOH: ethanol ;
g: gramme(s) ;
h: heure(s) ;
HA: acide fort ;
H2SO4: acide sulfurique ;
L: litre(s) ;
M: masse molaire (en g/mol) ;
mg: milligramme(s) ;
mL: millilitre(s) ;
mmol: millimole(s) ;
min: minute(s) ;
NTf2 : bis(trifluorométhylsulfonyl)amidure ;
RMN: Résonnance Magnétique Nucléaire ;
Rdt : rendement ;
TA: température ambiante ;
THF: tétrahydrofurane.
Les intermédiaires et composes décrits ci-dessous ont été nommés en utilisant le logiciel ChemBioDraw® Ultra version 12.0 (Cambridgesoft).
I. Synthèse des composés de l'invention Hydrogénoxalate d'alkylimidazolÎum
Exemple 1 : synthèse de hydrogénoxalate d'isobutylammonium à partir d' aminés bio- sourcées
Etape 1

Produit A La L-Valine (300 g ; 2,56 mol ; M=l 17,15 g/mol) est mise à chauffer à 140°C dans 1,135 L d' acétophénone (1102 g, 9,17 mol ; M=120,15 g/mol) pendant 1 jour. Un dégagement de C02 et de vapeur d'eau est observé. La solution est filtrée puis une solution d'acide oxalique à 50°C (324 g ; 2,56 mol ; M=126,7 g/mol, dans 1,4 L d'eau) est ajoutée dans le milieu réactionnel. Le mélange est refroidi à température ambiante. Une fois la phase acétophénone solidifiée, elle est séparée de la phase aqueuse par filtration et elle
est mélangée avec 1,35 L d'acétone. Le précipité en suspension est filtré et lavé avec de l'acétone. La phase aqueuse est évaporée, puis le solide obtenu est lavé sous agitation avec 400 mL d'acétone. Il est filtré et lavé avec 200 mL d'acétone. Les deux solides sont lavés sous agitation dans 800 mL d'acétate d'éthyle, puis filtrés et lavés à nouveau avec 3 x 200 mL d'acétate d'éthyle. Le produit A (hydrogénoxalate d'isobutylammonium) est obtenu comme un poudre blanche (279 g ; 1,71 mol ; M=163,17 g/mol, Rdt = 67%).
Caractérisation du produit A
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 2.89 (CH2, d, 2H), 1.99 (CH, hept, 1H), 1.03 (CH3, d, 6H) ; 13C NMR (D20, 75 MHz, 298 K) δ (ppm) 165.4 (COO), 46.4 (CH2), 26.3 (CH), 18.8 (CH3).
Etape 2
L'étape 2 a été réalisée avec des sources variées d'aldéhyde : soit à partir de formaldéhyde (méthode 2a) ; soit à partir d'urotropine (méthode 2b).
Méthode 2a

Produit A Produit B1
Dans 300 mL de toluène, sont introduits de Γ hydrogénoxalate d'isobutylammonium (Produit A, 50 g, 307 mmol), du paraformaldéhyde (4,6 g ; 153 mmol ; M=30,03 g/mol) et du glyoxal (17,5 mL ; 153 mmol ; M= 58,04 g/mol, solution à 40%, d=l,271). Après agitation quelques minutes, l'eau est éliminée par distillation azéotropique à 84°C à l'aide d'un Dean-Stark. Le solvant est évaporé, et l'addition de 120 mL d'acétone sur le résidu conduit à la formation d'un précipité qui est filtré puis recristallisé dans un mélange EtOH/MeTHF. Le produit B l (hydrogénoxalate de diisobutylimidazolium) est obtenu avec un rendement de 76% (31,4 g ; 11,6 mmol ; M=270,33 g/mol).
Caractérisations du produit B l
1H NMR (CDCb, 300 MHz, 298 K) δ (ppm) 10.65 (ΗΠ, s, 1H 8.77 (H2, s, 1H 7.30 (H4, d, 2H), 4.14 (H5, d, 4H), 2.13 (H6, hept, 2H), 0.91 (H7, d, 12H) ; 13C NMR (CDCb, 75 MHz, 298 K) δ (ppm) 165.16 (Ci l), 139.86 (C2), 122.08 (C4), 56.78 (C5), 29.61 (C6), 19.50 (C7). HRMS (ESI-MS) m/z calculée pour C11H21N2 [M]+: 181.16993, trouvée: 181.16998. Analyse élémentaire: calculée pour C13H23N2O4: C, 57.55; H, 8.54; N, 10.32; trouvée: C, 57.41 ; H, 8.58; N,10.61.
Méthode 2b)

Produit B3 Dans 75 mL de toluène, sont mis à réagir l'hydrogénoxalate d'isobutylammonium (Produit A, 10 g, 61,35 mmol), l'urotropine (2,15g, 15,3 mmol) et le glyoxal (7 mL, 61,35 mmol, 40% m/m dans l'eau). Après élimination de l'eau par distillation en Dean- Stark, le milieu réactionnel est évaporé. Le résidu est lavé avec un solvant oxygéné, préférentiellement de l'acétone. Le produit B3 est isolé par lavage avec un solvant oxygéné, préférentiellement de l'éthanol et purifié avec un solvant oxygéné, préférentiellement de l'eau et de l'acétone (m = 1,72 g).
Caractérisations du produit B l :
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 8.83 (H2, s, 1H), 7.52 (H4, d, 2H 4.05 (H5, d, 4H), 2.17 (H6, hept, 2H), 0.94 (H7, d, 12H) ; 13C NMR (D20, 75 MHz, 298 K) δ (ppm) 165.28 (Ci l), 135.52 (C2), 122.68 (C4), 56.37 (C5), 2.81 (C6), 18.50 (C7).
Caractérisations du produit B2 :
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 8.75 (H2, s, 1H), 7.51 (H4, m, 1H 7.50 (Η4', m, 1H), 4.09 (H5, d, 2H), 2.19 (H6, hept, 1H), 0.95 (H7, d, 6H) ; 13C NMR (D20, 75 MHz, 298 K) δ (ppm) 165.28 (Ci l), 134.47 (C2), 122.12 (C4'), 119.53 (C4), 56.18 (C5), 28.84 (C5), 18.52 (C7).
Caractérisations du produit B3 :
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 8.74 (H2, s, 1H), 7.52 (H4, s, 2H) ; 13C NMR (D20, 75 MHz, 298 K) δ (ppm) 165.60 (Ci l), 133.29 (C2), 118.82 (C4).
Le mélange B l et B2 est obtenu après évaporation de l'éthanol (masse totale = 8,95 g ; rapport molaire B 1 :B2 = 52:48).
Exemple 2 : synthèse de l'hydrogénoxalate de dipropylimidazolium

Produit B4
La propylamine (7,20 g, 121,8 mmol, M=59, l l g/mol) est ajoutée à une suspension de paraformaldéhyde (3,66 g) dans le toluène (100 mL) refroidi par un bain d'eau froide. Le mélange réactionnel est agité pendant 30 min puis refroidi à 0°C. Un deuxième équivalent de propylamine (7,200 g, 121,8 mmol) et l'acide oxalique (10,99 g, 122,1 mmol) sont ajoutés. La solution est agitée 2h à température ambiante. Puis le glyoxal (40% m/m dans l'eau, 13,9 mL, 121,7 mmol) est ajouté. La solution est agitée pendant 2h puis l'eau est éliminée grâce à un Dean-Stark. Le solvant est évaporé sous vide, le résidu dissout dans un minimum d'acétonitrile, filtré et précipité à l'éther diéthylique. Une poudre blanche correspondant à l'hydrogénoxalate de dipropylimidazolium est obtenue après recristallisation dans un mélange chloroforme/THF (18,3 g, 75,2 mmol, rdt = 62%).
RMN 1H (DMSO-de, 300 MHz, 298 K) δ (ppm) 9,46 (s, 1H), 7,84 (br s, 1H), 7,83 (d, 2H), 4,15 (ί, 4H), 1,81 (sext, 4H), 0,83 (ί, 6H) ; RMN 13C (DMSO-d6, 75 MHz, 298 K) δ (ppm) 164,8 ; 136,4 ; 122,5 ; 50,3 ; 22,8 ; 10,3. HRMS (ESI-MS) m/z calculée pour
C9H17N2 [M-C204H]+: 153,13863, trouvée: 153,13792. Analyse élémentaire pour CnHi8N204: calculée % : C, 54,53; H, 7,49; N, 11,56; trouvée: C, 54,45; H, 7,54; N, 11,31.
Sels d'alkylimidazolium - liquides ioniques
Selon la présente invention, la réaction d'échanges d' anions à partir du sel d'hydrogénoxalate peut être mise en œuvre :
(i) méthode A : soit par l'addition d'un acide ayant un pKa inférieur à environ 1,2 ;
(ii) méthode B : soit par l'addition d'un acide ayant un pKa supérieur à environ 1,2 sous électrolyse ;
(iii) méthode C : soit par l'addition d'un sel alcalin.
Exemple 3 : Méthode A - réactions d'échanges d' anions à partir d'un sel hydrogénoxalate d'imidazolium
3.1. A partir d'hydrogénoxalate de 1,3-diisobutylimidazolium (produit Bl)

Produit B1 Produit C1
L'hydrogénoxalate de diisobutylimidazolium (0,500 g ; 1,850 mmol) est mis en suspension dans 50 mL de diéthyléther, à température ambiante. HBF4(Et20) (0,300 g ; 1,851 mmol) est ajouté et la solution est agitée pendant 1 heure. La phase éthérée est éliminée et le résidu est lavé avec de l'éther diéthylique (5 fois 10 mL) puis séché sous vide. Le tétrafluoroborate de 1,3-diisobutylimidazolium est obtenu sous forme d'une poudre (0,472 g ; 1,754 mmol ; rdt = 95%).

Produit B1 Produit C2
L'hydrogénoxalate de diisobutylimidazolium (10,02 g ; 37,07 mmol) est mis en suspension dans 50 mL de diéthyléther, à température ambiante. HPF6 (65% m/m dans l'eau, 8,354 g ; 37,20 mmol) est ajouté et la solution est agitée pendant 10 heures. La phase éthérée est éliminée et le résidu est lavé à l'eau (3 fois 10 mL). L'hexafluorophosphate de diisobutylimidazolium est séché sous vide et est obtenu sous forme d'un liquide visqueux (10,581 g ; 32,330 mmol ; rdt = 87 %).

Produit B1 Produit C3
L'hydrogénoxalate de diisobutylimidazolium (5,012 g, 18,54 mmol) est mis en suspension dans 50 mL de diéthyléther ou de MTBE, à température ambiante. H2SO4 (1,000 g. 18,66 mmol) est ajouté et la solution est agitée pendant 1 heure. La phase éthérée est éliminée et le résidu est lavé avec de l'éther diéthylique (5 fois 10 mL), du cyclohexane (2 fois 10 mL) puis séché sous vide. L'hydrogénosulfate de diisobutylimidazolium est obtenu sous forme d'un liquide visqueux (5,040 g ; 18,04 mmol ; rdt = 97%). RMN 1H (DMSO-de, 300 MHz, 298 K): 9,95 (HS04, br s, 1H), 9,20 (s, 1H 7,80 (d, 2H), 4,03 (d, 4H), 2, 10 iept, 2H), 0,86 {d, 12H) ; RMN 13C (DMSO-d6, 75 MHz, 298 K) δ (ppm) 136,4 ; 122,8 ; 55,5 ; 28,7 ; 19,0 ; Tf. < 25 °C ; Analyse élémentaire pour C11H22N2O4S : calculée % : C, 47,46; H, 7,97; N, 10,06; trouvée: C, 47,09; H, 8,24; N, 10,41. 3.2. A partir d'hvdrogénoxalate de L3-dipropylimidazolium (produit B4)

Produit B4 Produit G
L'hydrogénoxalate de 1 ,3-dipropylimidazolium (2,189 g, 8,998 mmol) est mis en suspension dans 20 mL d'acétone à température ambiante. H2SO4 (0,915 g ; 9,330 mmol) est ajouté et la solution est agitée pendant 1 heure. La solution est filtrée et le filtrat évaporé sous vide. Le résidu est lavé avec de l'éther diéthylique (3 fois 10 mL), du
cyclohexane (2 fois 10 mL) puis séché sous vide. L'hydrogénosulfate de 1.3- dipropylimidazolium est obtenu sous forme d'un liquide visqueux (Produit G ; 2,216 g ; 8,817 mmol ; rdt = 98%).
3.3. A partir d'hvdrogénoxalate de 3-diphényléthylimidazolium (produit B5)
Ph^N^N^Ph + H2S°4 ► Ph^N^N^Ph + ° P"
HO O HO O
Produit B5 Produit E
L'hydrogénoxalate de 1,3-diphényléthylimidazolium (Produit B5 ; 6,000 g ; 16,37 mmol) (6,000 g, 16,38 mmol) est mis en suspension dans 50 mL de diéthyléther à température ambiante. H2SO4 (1,647 g, 16,79 mmol) est ajouté et la solution est agitée pendant 2 heures. La phase éthérée est éliminée et le résidu est lavé avec de l'éther diéthylique (5 fois 10 mL), du cyclohexane (2 fois 10 mL) puis séché sous vide. Le produit E est obtenu sous forme d'une poudre blanche (6,119 g, 16,30 mmol, rdt = 99%).
RMN 1H (DMSO-de, 300 MHz, 298 K) δ (ppm) 8,81 (s, 1H), 7,73 (d, 2H), 7,55-7,38 {Hméta-para, m, 6H), 7,26 (Hortho, m, 4H , 4,41 (ί, 4H), 3,08 (i, 4H); RMN 13C (DMSO-d6, 75 MHz, 298 K) δ (ppm) 138,7 ; 129,8 ; 128,7 ; 127,8 ; 125,7 ; 121,6 ; 50,6 ; 36,2. Tf. 142 °C ; Analyse élémentaire pour C19H22N2O4S: calculée % : C, 60,94; H, 5,92; N, 7,48; S, 8,56; trouvée: C, 61,10; H, 6,40; N, 7,82; S, 8,09.
Exemple 4 : Méthode B - Métathèse anionique à partir d'hydrogénoxalate de 1,3 - diisobutylimidazolium (produit Bl)
Produit B1 Produit C1

Produit Β1 Produit C2
O 0 <3 Θ ©
/==\ acétone, TA, 1 h f=\
^ + LiNTf2 Θ O. O Li
Νχ©-Ν
HO O
Produit B1 Produit C6
L'hydrogénoxalate de diisobutylimidazolium (2,010 g, 7,436 mmol) est mis en suspension dans 20 mL de diéthyléther à température ambiante. LiNTf2 (2,139 g, 7,451 mmol), ou un tout autre sel (NaBF4, KPF6,„), est ajouté et la solution est agitée pendant 4 jours. La solution est filtrée, le filtrat mis à -18°C puis refiltré. Après évaporation du solvant, le produit est séché sous vide et est obtenu sous forme d'un liquide visqueux (2,622 g, 6,091 mmol, Rdt = 82%).
Exemple 5 : Méthode C - Echange d'anions par électrolyse à partir d'hydrogénoxalate de diisobutylimidazolium (Produit Bl)
5.1. Réaction d'échange avec l'acide formique

i = 60 mA Produit C6 Le produit Bl (1,0 g ; 3,7 mmol ; M=270,33 g/mol) est mis en suspension dans 175 mL d'acétonitrile et de l'acide formique (241 mg ; 4,4 mmol ; M=46,03 g/mol, solution à 85%) est ajouté. Le mélange est électrolysé sous 60 mA (minimum 2 éq. d'électrons/mol de Bl). 10 mL d'eau sont ajoutés à la solution, qui est ensuite portée à reflux quelques minutes. Après évaporation des solvants, le composé C6 est obtenu sous forme d'un liquide marron (775 mg, 92%).
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 8.80 (Hl l, s, 1H) ; 8.33 (H2, s, 1H) ; 7.49 (H4, s, 2H), 4.02 (H5, d, 4H) ; 2.14 (H6, hept, 2H) ; 0.91 (H7, d, 72H).13C NMR (D20, 75 MHz, 298 K) δ (ppm) 167.9 (Ci l) ; 135.5 (C2) ; 122.7 (C4) ; 56.4 (C5) ; 28.8 (C6) ; 18.5 (C7). 5.2. Réaction d'échange avec l'acide acétique

i = 60 mA ro u t
Le produit B l (1,0 g ; 3,7 mmol ; M=270,33 g/mol) est mis en suspension dans 175 mL d'acétonitrile et de l'acide acétique (266 mg ; 4,4 mmol ; M=60,05 g/mol) est ajouté. Le mélange est électrolysé sous 60 mA (minimum 2 éq. d'électrons/mol de B l). 10 mL d'eau sont ajoutés à la solution, qui est ensuite portée à reflux quelques minutes. Après évaporation des solvants, le composé C7 est obtenu sous forme d'un liquide visqueux jaune (757 mg, 85%).
1H NMR (D20, 300 MHz, 298 K) δ (ppm) 8.80 (H2, s, 1H, échange avec D20) ; 7.48 (H4, s, 2H), 4.02 (H5, d, 4H) ; 2.14 (H6, hept, 2H) ; 1.93 (H12, s, 3H) ; 0.91 (H7, d, 12H). 13C NMR (D20, 75 MHz, 298 K) δ (ppm) 180.4 (Cl 1) ; 135.6 (t, C2) ; 122.7 (C4) ; 56.4 (C5) ; 28.8 (C6) ; 22.8 (C12) ; 18.5 (C7).
Exemple 6 : Caractérisation des sels de 1,3-diisobutyllimidazolium comme liquides ioniques
Afin de savoir si les sels d'imidazolium présentent un caractère de type liquide ionique, la température de fusion de divers sels de 1,3-diisobutylimidazolium a été mesurée (Cf. tableau 2).
Tableau 2
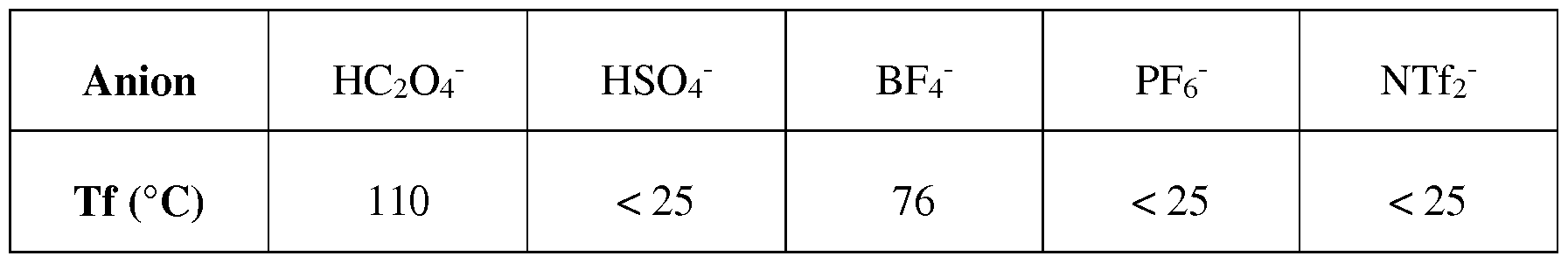
Les résultats montrent que les sels de 1,3-diisobutylimidazolium ayant un contre-ion choisi parmi HS04 ", BF4", PFÔ" et NTf2 " ont une température de fusion inférieure à 100°C.
Par conséquent, les sels de 1,3-diisobutylimidazolium tels que décrits, sont des liquides ioniques.
IL Utilisation des composés de l'invention comme catalyseurs
Exemple 7 : estérification de l'acide lévulinique catalysée par l'hydrogénosulfate de diisobutylimidazolium
7.1. avec du butanol

Dans un ballon sont introduits de l'acide lévulinique (lg ; 9 mmol ; M= 116,12 g/mol) et le catalyseur (4% mol.). Le milieu réactionnel est placé à 100°C sous agitation magnétique. Puis du butanol (5 eq., 4 mL ; d= 0,81 ; 44 mmol, M=74,12 g/mol) est ajouté. La réaction est suivie par
 dans CDCI3. Puis une extraction de l'ester est réalisée en solvant apolaire.
dans CDCI3. Puis une extraction de l'ester est réalisée en solvant apolaire.
Les résultats de la synthèse sont présentés dans le Tableau 3 ci-dessous.
Tableau 3
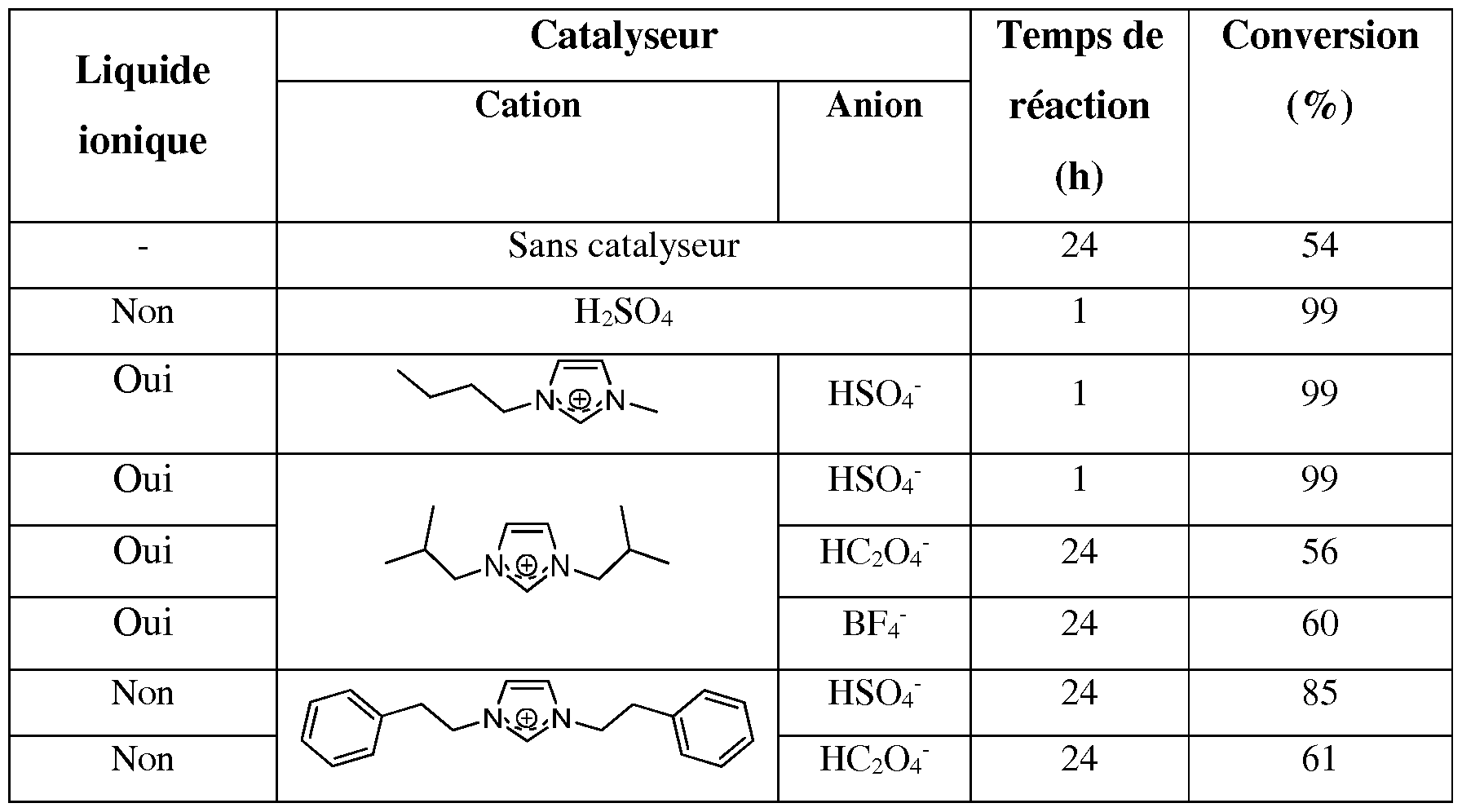
Les résultats montrent que :
- sans catalyseur, l'estérification de l'acide lévulinique est obtenu avec une conversion inférieure à 60% après 24h ;
- en présence d'H2S04 (catalyseur acide conventionnel), le temps de réaction est fortement diminué (lh) et la conversion est de 99% ;
- en présence d'un catalyseur n'ayant pas un caractère de liquide ionique, pour un même temps de réaction sans catalyseur, la conversion est plus élevée ;
- en présence d'un catalyseur ayant un caractère de liquide ionique, le temps et la conversion de la réaction d'estérification sont comparables à ceux obtenus en présence d'H2S04.
En conclusion, les sels d'imidazolium présentant un caractère de liquide ionique et ayant pour contre-ion l'hydrogénosulfate, permettent de catalyser l'estérification de l'acide lévulinique par le butanol aussi efficacement qu'un catalyseur conventionnel tel que l'acide sulfurique. Les esters synthétisés par ce procédé peuvent être facilement séparés du catalyseur et sont obtenus avec une pureté supérieure à 99%.
7.2. avec de l'éthanol

Dans un ballon sont introduits de l'acide lévulinique (10,4g ; 90 mmol ; M= 116,12 g/mol) et le catalyseur (composé C3, 4% mol.). Le milieu réactionnel est placé à 100°C sous agitation magnétique. Puis du butanol (5 eq., 26,3 mL ; d= 0,789; 450 mmol, M=46,07 g/mol) est ajouté. La réaction est suivie par I MN1!! dans CDCI3. La conversion est totale après lh de réaction. L'ester formé est obtenu avec une pureté supérieure à 99%.
7.3. avec du méthanol
 L'acide lévulinique (10,4 g, 89,8 mmol) et le catalyseur (composé C3 ; 1 g, 3,6 mmol,
L'acide lévulinique (10,4 g, 89,8 mmol) et le catalyseur (composé C3 ; 1 g, 3,6 mmol,
4 mol %) sont agités dans un bain d'huile à 72 °C. Le méthanol (14,4 g, 449 mmol,
5 équivalents) est ajouté et la réaction est suivie par ΡνΜΝ1!! dans CDCI3. La conversion est totale après 9h. Le méthanol est ensuite évaporé. L'ester formé est obtenu avec une pureté supérieure à 99%. Exemple 8 : Catalyse de l'estérification de l'acide myristique et recyclage du catalyseur
Estérification

L'acide myristique (20,5 g, 89,8 mmol) et l'hydrogénosulfate de diisobutylimidazolium (1 g, 3,6 mmol, 4 mol %) sont agités dans un bain d'huile à 87 °C. L'isopropanol (3 éq.) est ajouté et la réaction est suivie par RMN proton dans le chloroforme deutéré. Après 16h, la conversion est totale et l'isopropanol est évaporé.
Le myristate d' isopropyle est facilement séparé du catalyseur par décantation et contient moins de 1% de catalyseur. La Figure 1 est une photographie du milieu en fin de réaction, montrant la séparation de phases entre le catalyseur et le milieu réactionnel.
Recyclage
Le catalyseur (composé C3) a été utilisé lors de plusieurs cycles selon les conditions réactionnelles décrites précédemment. Les résultats sont présentés dans le Tableau 4 ci- dessous.
Tableau 4

III. Tests comparatifs
Exemple 9
Le but de cette expérience est de comparer le procédé de l'invention avec celui décrit dans la demande US 2010/0249432 relatif à la synthèse d'acétate de diéthylimidazolium.
L'acétate d'isobutylimidazolium a été synthétisé selon le protocole décrit dans l'exemple 1 de la demande US 2010/0249432 en substituant l'éthylamine par l'isobutylamine. Un produit rouge brun a été obtenu mettant en évidence un grand nombre d'impuretés.
Puis, la Demanderesse a purifié le produit selon le protocole décrit dans US 2010/0249432, c'est-à-dire en faisant réagir le brut de réaction avec du peroxyde d'hydrogène (30%) à 80°C pendant 5h ; suivi d'un traitement par l'hydroxyde de soude
(40%) à une température comprise de 65°C à 95°C pendant 2h. Un liquide jaune paille a été obtenu. La caractérisation du liquide par I MN1!! montre que les conditions de purification ont conduit à une forte dégradation du produit (>20%).
En conclusion, le procédé de l'art antérieur mettant en jeu un monoacide ; en particulier, de l'acide acétique ne permet pas de fournir directement des produits purs (non colorés) et requière des conditions de purification sévères engendrant une forte dégradation du produit final.
Exemple 10 : préparation d'acétate d'isobutylammonium
La Demanderesse a également préparé, selon le protocole de l'exemple 1, de l'acétate d'isobutylammonium en substituant l'acide oxalique par l'acide acétique.
Les résultats ont montré que le produit obtenu est difficile à purifier et n'est pas thermodynamiquement stable. En effet, le produit se décompose dès 65°C.
En conclusion, l'acide acétique n'est pas adapté à la synthèse de sels d'alkylammonium contrairement à l'acide oxalique utilisé dans la présente invention.
Claims
REVENDICATIONS
1. Procédé de préparation de liquides ioniques formule (I)

(I)
dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle ;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H ;
A" représente un anion choisi parmi les anions de sels alcalins ou les bases conjuguées d'un acide organique ou d'un acide minéral ayant un pKa inférieur à 14 ; comprenant : (i-0) une étape de synthèse d'un hydrogénoxalate d'alkylammonium ;
(i) une étape de synthèse d'un hydrogénoxalate d'alkylimidazolium ; puis
(ii) une étape d'échange d'anions.
2. Procédé selon la revendication 1, dans lequel les réactifs de l'étape (i-0) comprennent de l'acide oxalique et une aminé.
Procédé selon la revendication 2, dans lequel l'aminé est un acide aminé.
Procédé selon l'une quelconque des revendications 1 à 3, dans lequel l'étape (i) comprend au moins une source d'ammonium, une source de formaldéhyde et un composé comprenant une fonction oxalyle.
Procédé selon la revendication 4, dans lequel la source de formaldéhyde est choisie parmi les composés organiques ayant au moins une fonction aldéhyde ou les composés organiques pouvant libérer dans les conditions de réaction un composé ayant au moins une fonction aldéhyde ; de préférence, le composé organique comprenant une fonction aldéhyde est formée in situ ; plus préférentiellement, le composé organique comprenant une fonction aldéhyde est l'urotropine.
6. Procédé selon l'une quelconque des revendications 1 à 5, dans lequel l'hydrogénoxalate d'alkylimidazolium est choisi parmi le groupe des hydrogénoxalate de 1,3-dialkylimidazolium de formule (IV) :

(IV) dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano,
alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H.
7. Procédé selon l'une quelconque des revendications 1 à 6, dans lequel l'échange d' anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu à l'étape (i) et un acide ayant un pKa inférieur à 2; de préférence, inférieur à 1,2 ; plus préférentiellement, avec un acide choisi parmi H2SO4, HBF4, HPF6 ou HNTf2.
8. Procédé selon l'une quelconque des revendications 1 à 6, dans lequel l'échange d' anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu à l'étape (i) et un sel alcalin.
9. Procédé selon l'une quelconque des revendications 1 à 6, dans lequel l'échange d' anions s'effectue entre l'hydrogénoxalate d' alkylimidazolium obtenu à l'étape (i) et un acide ayant un pKa supérieur à 1,2, dans une cellule électrochimique soumise à une intensité électrique. 10. Procédé selon la revendication 9, dans lequel l'intensité électrique appliquée dans la cellule électrochimique est comprise dans une gamme allant de 40 mA à 900 mA ; de préférence de 50 mA à 80 mA ; plus préférentiellement, l'intensité électrique est égale à environ 60 mA.
Procédé selon la revendication 9 ou la revendication 10, dans lequel la cellule électrochimique comprend un solvant choisi parmi les solvants polaires ; de préférence, l'acétonitrile.
12. Liquide ionique susceptible d'être obtenu par le procédé selon l'une quelconque des revendications 1 à 11, de formule (I) :

(D
dans laquelle,
Ri et R2 sont identiques ou différents, et représentent chacun un groupe choisi parmi H, alkyle, alcène, alcyne, cycloalkyle, cycloalcényle, hétéroalkyle, hétéroaryle ou hétérocycloalkyle; optionnellement substitué par au moins un groupe choisi parmi aryle, hydroxyle, oxo, nitro, amido, amino, cyano, alcoxy, alkyle, alcène, alcyne, cycloalkyle, cycloalcène, hétéroalkyle, hétéroaryle ou hétérocycloalkyle;
R3 et R4 sont identiques ou différents, et représentent chacun un groupe choisi parmi un atome H, alkyle, alcène, alcyne, alcoxy ou hétéroalkyle; optionnellement substitué par au moins un groupe choisi parmi hydroxyle, oxo, nitro, amido, amino, cyano, alkoxy, alkyle, alcène, alcyne ou hétéroalkyle; préférablement R3 et R4 sont identiques; plus préférentiellement, R3 et R4 sont identiques et représentent un atome H ;
A" représente un anion choisi parmi les anions de sels alcalins ou les bases conjuguées d'un acide organique ou d'un acide minéral ayant un pKa inférieur à 14.
Utilisation d'un liquide ionique selon la revendication 12 comme catalyseur de réaction d'estérification dans laquelle le produit (ester) formé est un arôme ou un agent cosmétique.
Utilisation d'un liquide ionique selon la revendication 13 comme catalyseur de réaction d'estérification dans laquelle l'arôme formé est le lévulinate de butyle.
Utilisation d'un liquide ionique selon la revendication 13 comme catalyseur de réaction d'estérification dans laquelle l'agent cosmétique formé est le myristate d'isopropyle.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16750462.0A EP3322527A1 (fr) | 2015-07-16 | 2016-07-13 | Procédé de préparation de liquides ioniques biosources pour la catalyse |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1556711A FR3038853B1 (fr) | 2015-07-16 | 2015-07-16 | Utilisation de liquides ioniques recyclables comme catalyseurs d'esterification |
| FR1556711 | 2015-07-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017009578A1 true WO2017009578A1 (fr) | 2017-01-19 |
Family
ID=54186147
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2016/051806 Ceased WO2017009578A1 (fr) | 2015-07-16 | 2016-07-13 | Procédé de préparation de liquides ioniques biosources pour la catalyse |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP3322527A1 (fr) |
| FR (1) | FR3038853B1 (fr) |
| WO (1) | WO2017009578A1 (fr) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108558662A (zh) * | 2018-05-16 | 2018-09-21 | 浙江工业大学 | 一种多磺酸基功能化离子液体催化合成棕榈酸异丙酯的方法 |
| CN110878018A (zh) * | 2019-11-19 | 2020-03-13 | 陕西科技大学 | 一种制备尼泊金酯的方法 |
| CN120864987A (zh) * | 2025-07-30 | 2025-10-31 | 广东邦固化学科技有限公司 | 一种异冰片酯单体的制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0521870A1 (fr) * | 1990-03-29 | 1993-01-13 | E.I. Du Pont De Nemours And Company | Preparation de sels d'imidazolium 1,3-disubstitue |
| WO2000016902A1 (fr) * | 1998-09-24 | 2000-03-30 | Bp Chemicals Limited | Liquides ioniques |
| CN101070282A (zh) * | 2006-05-09 | 2007-11-14 | 黑龙江大学 | 室温离子液体催化制备亚油酸乙酯的方法 |
| US20090235574A1 (en) * | 2005-03-11 | 2009-09-24 | Earle Martyn J | Production of Bio-Diesel |
| US20100249432A1 (en) * | 2007-12-12 | 2010-09-30 | Basf Se | Method for the production of disubstituted imidazolium salts |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2948671B1 (fr) * | 2009-07-31 | 2011-08-19 | Univ Paris Curie | Procede de synthese de polyesters en milieu liquide ionique acide |
-
2015
- 2015-07-16 FR FR1556711A patent/FR3038853B1/fr not_active Expired - Fee Related
-
2016
- 2016-07-13 EP EP16750462.0A patent/EP3322527A1/fr not_active Withdrawn
- 2016-07-13 WO PCT/FR2016/051806 patent/WO2017009578A1/fr not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0521870A1 (fr) * | 1990-03-29 | 1993-01-13 | E.I. Du Pont De Nemours And Company | Preparation de sels d'imidazolium 1,3-disubstitue |
| WO2000016902A1 (fr) * | 1998-09-24 | 2000-03-30 | Bp Chemicals Limited | Liquides ioniques |
| US20090235574A1 (en) * | 2005-03-11 | 2009-09-24 | Earle Martyn J | Production of Bio-Diesel |
| CN101070282A (zh) * | 2006-05-09 | 2007-11-14 | 黑龙江大学 | 室温离子液体催化制备亚油酸乙酯的方法 |
| US20100249432A1 (en) * | 2007-12-12 | 2010-09-30 | Basf Se | Method for the production of disubstituted imidazolium salts |
Non-Patent Citations (6)
| Title |
|---|
| BEHZAD AGHABARARI ET AL: "Esterification of fatty acids by new ionic liquids as acid catalysts", JOURNAL OF THE TAIWAN INSTITUTE OF CHEMICAL ENGINEERS, vol. 45, no. 2, 1 March 2014 (2014-03-01), AMSTERDAM, NL, pages 431 - 435, XP055270719, ISSN: 1876-1070, DOI: 10.1016/j.jtice.2013.08.003 * |
| BEILEI ZHOU ET AL: "Ionic liquid mediated esterification of alcohol with acetic acid", FRONTIERS OF CHEMICAL ENGINEERING IN CHINA, vol. 3, no. 2, 27 January 2009 (2009-01-27), CN, pages 211 - 214, XP055270709, ISSN: 1673-7369, DOI: 10.1007/s11705-009-0054-3 * |
| CINZIA CHIAPPE ET AL: "A dramatic effect of the ionic liquid structure in esterification reactions in protic ionic media", GREEN CHEMISTRY, vol. 15, no. 1, 1 January 2013 (2013-01-01), GB, pages 137 - 143, XP055270712, ISSN: 1463-9262, DOI: 10.1039/C2GC35941C * |
| COLE A C ET AL: "Novel Bronsted Acidic Ionic Liquids and Their Use as Dual Solvent-Catalysts", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, US, vol. 124, 7 May 2002 (2002-05-07), pages 5962 - 5963, XP002447834, ISSN: 0002-7863, DOI: 10.1021/JA026290W * |
| DATABASE WPI Week 200830, 2008 Derwent World Patents Index; AN 2008-E27971, XP002757389 * |
| YING LI ET AL: "Acidic ionic liquid-catalyzed esterification of oleic acid for biodiesel synthesis", CHINESE JOURNAL OF CATALYSIS / DALIAN INSTITUTE OF CHEMICAL PHYSICS, vol. 35, no. 3, 1 March 2014 (2014-03-01), AMSTERDAM, NL, pages 396 - 406, XP055270706, ISSN: 1872-2067, DOI: 10.1016/S1872-2067(14)60005-X * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108558662A (zh) * | 2018-05-16 | 2018-09-21 | 浙江工业大学 | 一种多磺酸基功能化离子液体催化合成棕榈酸异丙酯的方法 |
| CN108558662B (zh) * | 2018-05-16 | 2021-05-07 | 浙江工业大学 | 一种多磺酸基功能化离子液体催化合成棕榈酸异丙酯的方法 |
| CN110878018A (zh) * | 2019-11-19 | 2020-03-13 | 陕西科技大学 | 一种制备尼泊金酯的方法 |
| CN120864987A (zh) * | 2025-07-30 | 2025-10-31 | 广东邦固化学科技有限公司 | 一种异冰片酯单体的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3322527A1 (fr) | 2018-05-23 |
| FR3038853A1 (fr) | 2017-01-20 |
| FR3038853B1 (fr) | 2019-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017009578A1 (fr) | Procédé de préparation de liquides ioniques biosources pour la catalyse | |
| BE1023898B1 (fr) | Composition comprenant du silicium et son procédé de préparation | |
| EP3007822B1 (fr) | Procédé de synthèse d'acide glycolique | |
| EP2794543B1 (fr) | Procédé de scission moléculaire oxydative d'un composé gras | |
| EP3416944B1 (fr) | Procédé de préparation de composés aminothiolester et leurs sels | |
| CA2795741C (fr) | Nouveau procede de synthese de l'ivabradine et de ses sels d'addition a un acide pharmaceutiquement acceptable | |
| FR2939790A1 (fr) | Procede catalytique de fabrication de composes de type diol | |
| EP4100388A1 (fr) | Nouvelle voie de synthese du sinapoyl malate, de la sinapine et de leurs analogues, et leur utilisation en tant que molecules anti-microbiennes | |
| EP3066069A1 (fr) | Procédé de synthèse d'esters et catalyseur de ladite synthèse | |
| FR2957076A1 (fr) | Procede de production d'acide pyrrolidonecarboxylique ou d'un sel de celui-ci, acide pyrrolidonecarboxylique ainsi obtenu et cosmetique le contenant | |
| FR3071495B1 (fr) | Nouveau procede de fabrication du resorcinol | |
| EP3233779B1 (fr) | Nouveau procede de preparation d'intermediaires de synthese utilisant des produits d'origine naturelle et application des intermedaires obtenus | |
| WO2010037786A1 (fr) | Procede de reduction de la concentration en ruthenium residuel dans des solutions, notamment dans des solutions contenant des produits issus de metathese d'olefines | |
| EP2614046A1 (fr) | Nouveaux dérivés guanidines en série cinnamique | |
| EP3066066B1 (fr) | Procédé de synthèse d'esters | |
| EP0200621B1 (fr) | Procédé de synthèse du trifluoro-2,2,2 éthanol | |
| WO2014068256A1 (fr) | Procede de synthese de l'h exanitrohexaazaisowurtzitane a partir de l'hexaallylhexaazaisowurtzitane; intermediaires. | |
| WO2016120574A1 (fr) | Adduit hydrosilane / acide de lewis, notamment l'aluminium, le fer et le zinc, son procede de preparation et son utilisation dans des reactions de reduction de derives carbonyles | |
| WO2006082200A1 (fr) | Procédé direct de préparation d'amino-alcools ou d'alcools utilisant le borohydrure d'halogénozinc | |
| WO2006097594A1 (fr) | Procede de preparation d'amides tertiaires aliphatiques n-alkyl substitues | |
| BE819380A (fr) | Procede de racemisation d'amines optiquement actives | |
| FR2930551A1 (fr) | Procedes de synthese asymetrique de la 6-fluoro-l-dopa et de ses analogues | |
| FR2853652A1 (fr) | Diphosphines chirales, leur preparation et leurs utilisations comme ligands dans la synthese de complexes destines a la catalyse asymetrique | |
| FR2849037A1 (fr) | Diphosphines chirales, leur preparation et leurs utilisations coome ligands dans la synthese de complexes destines a la catalyse asymetrique | |
| FR3073844A1 (fr) | Un compose organique, sa methode de fabrication et ses utilisations |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16750462 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016750462 Country of ref document: EP |