WO2017111137A1 - オリゴヌクレオチドの製造方法 - Google Patents
オリゴヌクレオチドの製造方法 Download PDFInfo
- Publication number
- WO2017111137A1 WO2017111137A1 PCT/JP2016/088580 JP2016088580W WO2017111137A1 WO 2017111137 A1 WO2017111137 A1 WO 2017111137A1 JP 2016088580 W JP2016088580 W JP 2016088580W WO 2017111137 A1 WO2017111137 A1 WO 2017111137A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- protected
- hydroxyl
- atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a method for producing an oligonucleotide. More specifically, the present invention relates to a method for producing an oligonucleotide using a silyl protecting group, a method for producing an oligonucleotide using a phosphitylating agent, a method for producing an oligonucleotide by fragment condensation, and the like.
- Oligonucleotide synthesis methods include the phosphate triester method, the H-phosphonate method, and the phosphoramidite method.
- solid phase synthesis solid phase method using the phosphoramidite method is most widely used.
- Non-Patent Document 1 Oligonucleotides consisting of several nucleotides can be synthesized by sequentially linking nucleotides as raw materials. However, when synthesizing oligonucleotides of about 20 mer or more, a building block group of 2 to 3 nucleotides is used.
- a block-mer synthesis method to obtain a product with a desired chain length by repeating this ligation in advance and unit coupling (fragment condensation) synthesis to obtain an oligonucleotide by ligation between oligonucleotides of about 10 bases or more Laws are also used.
- This fragment condensation method requires special care for ensuring solubility and selectively deprotecting and activating the reaction site.
- a levulinyl ester is used as a protective group capable of selectively deprotecting the 3′-hydroxy group of an oligonucleic acid fragment, and extension is performed by a phosphoramidite method.
- Non-Patent Document 2 states that removal of the base protecting group can proceed under deprotection conditions.
- Non-Patent Document 3 describes a blockmer synthesis method in which an oligonucleic acid is synthesized using a 3-mer amidite.
- Patent Document 1 uses a protecting group that is difficult to leave, but this protecting group is special and conventional reaction conditions and the like cannot be used as they are.
- Non-Patent Document 4 describes a method for synthesizing oligonucleotides by fragment condensation using the triester method, but the triester method is hardly practical at present.
- a technique for synthesizing a longer-chain oligonucleotide using a 3′-terminal phosphoramididized oligonucleotide prepared using a phosphitylating agent is known.
- 2-Cyanoethyl-N, N-diisopropylchlorophosphoramidite has been commonly used for phosphitylation of nucleosides.
- Patent Document 2 this is combined with N-methylimidazole to phosphitylate the deprotected 3′-end of the oligonucleic acid.
- 2-cyanoethyl-N, N-diisopropylchlorophosphoramidite is inferior in storage stability and has a high market value.
- 2-Cyanoethyl-N, N, N ′, N′-tetraisopropyl phosphorodiamidite is used as a cheaper and more stable alternative to the above-mentioned reagents (Non-patent Document 5).
- the agent itself has poor reactivity, and an activating agent is required for phosphitylation of nucleosides.
- Non-patent Documents 6 and 6 when a strong acid and base salt such as trifluoromethanesulfonic acid is used as an activator, it is reported that phosphite and a pentavalent phosphorus compound are by-produced due to excessive activity (Non-patent Documents 6 and 6). 7).
- a silyl protecting group is used as a protecting group for the hydroxyl group at the 2′-position in the oligonucleotide synthesis process.
- 3HF-TEA is an acidic salt, and 4,4′-dimethoxytriethylamine, which is commonly used as a 5′-terminal protecting group for oligonucleic acid synthesis, is acidic and promotes elimination.
- a molar ratio of salt is used.
- the object of the present invention is to use a phosphitylating agent having superior storage stability, suppress excessive activation of the phosphitylating agent, and suppress detachment of the protecting group on phosphoric acid, thereby reducing the 3 ′ terminal phospho
- a method for producing a rhamididized oligonucleotide, a method for producing an oligonucleotide by allowing desilylation without removal of a cyanoethyl protecting group, and a method for producing an oligonucleotide more efficiently using these methods It is intended to provide a method for producing an oligonucleotide having various functional groups linked to the 3 ′ end.
- the present inventors have used diamidite as a phosphitylating agent, implemented a specific phosphitylating agent activator and activation method, and carried out phosphitylation in the presence of a specific base. It has been found that the above problems can be solved. Furthermore, as a result of intensive studies, the present inventors have found a specific silyl protecting group capable of suppressing decyanoethylation and its deprotection conditions.
- the present invention includes the following.
- n-polymerized oligonucleotide in which a 5′-position hydroxyl group or a 5′-position phosphate group is protected in a solvent and a 3′-position hydroxyl group or a 3′-position amino group is protected with a silyl protecting group (n is 2 And the 5′-position hydroxyl group or 5′-position phosphate group is protected, including the step of allowing a fluoride ion source to act in the presence of one or more organic bases.
- s + 1 pieces of A 1 is independently an oxygen atom or -NH-, s + 1 Bases each independently represents a nucleobase that may be protected; P 1 represents a protecting group for a hydroxyl group, or one of the hydroxyl groups is replaced with —OL n1 —OH (wherein L n1 represents an organic group and the hydroxyl group is protected) as —O—P 1.
- the phosphate group s number of P 2 each independently represent a protecting group of a phosphate group
- s R 40 s independently represent an oxygen atom or a sulfur atom
- s + 1 Y's each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom or an organic group that crosslinks to the 4-position carbon atom
- P 3 represents a silyl protecting group
- s represents an arbitrary integer of 1 or more.
- DABCO octane
- organic base pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, piperazine, piperidine, imidazole, N-methylimidazole, N-methylmorpholine, N-ethylmorpholine, aniline, toluidine, The method according to any one of [1] to [7] above, wherein at least one selected from dimethylaniline, ethylaniline, diethylaniline, ethylmethylaniline, and anisidine is used. [9] The method according to any one of [1] to [8] above, wherein a mixture of triethylamine and pyridine is used as the organic base.
- fluoride ion source is at least one selected from triethylamine pentahydrofluoride, triethylamine trihydrofluoride, and pyridine hydrofluoride The method described in 1.
- the silyl protecting group is such that three substituents on silicon are selected from an alkyl group, an aryl group, an aralkyl group, an alkoxy group, an aryloxy group, an aralkyloxy group, an alkylthio group, an arylthio group, and at least one of them
- the method according to any one of [1] to [10] above, wherein one is a silyl protecting group selected from an aryl group, an alkoxy group, an aryloxy group, and an aralkyloxy group.
- At least one of the s + 1 Bases has the formula: -LY- L- Z [Where: L represents the formula (a1):
- R 4 represents a hydrogen atom or, when R b is a group represented by the following formula (a3), together with R 6 represents a single bond or —O—, And may form a fluorenyl group or a xanthenyl group
- k R 5 's each independently represents an organic group having an aliphatic hydrocarbon group having 10 or more carbon atoms
- k represents an integer of 1 to 4
- Ring A includes, in addition to k OR 5 , a halogen atom, a C 1-6 alkyl group which may be substituted with a halogen atom, and a C 1-6 alkoxy group which may be substituted with a halogen atom. May have a substituent selected from the group consisting of; R a represents a hydrogen atom or a phenyl group optionally substituted by a halogen atom; and R b represents a hydrogen atom or the formula (a3):
- the base of the 3 ′ terminal nucleoside is a protecting group represented by the formula: —LY— L— Z (where each symbol has the same definition as in the above [12]), or the formula: —Z ′ ( The method according to any one of [1] to [15] above, wherein each symbol is defined as defined in [12] above).
- a method for producing a phosphoramidite-ized oligonucleotide comprising the following steps (1) and (2). (1) In a solvent, the following formula (1):
- X represents an oxygen atom or a sulfur atom
- R 10 represents an aromatic ring, a hydroxyl protecting group or a thiol protecting group
- R 20 and R 30 each independently represents an alkyl group, and the alkyl group may form a ring together with the adjacent nitrogen atom.
- An activating agent is allowed to act on a phosphitylating agent precursor represented by the following formula (2):
- q + 1 A 1 independently represent an oxygen atom or —NH—
- Base A represents a nucleobase which may be independently protected
- P 1a represents a hydroxyl-protecting group, or one of the hydroxyl groups is replaced with —OL n1 —OH (wherein L n1 represents an organic group and the hydroxyl group is protected) as —OP 1
- the phosphate group q P 2a each independently represents a protecting group for a phosphate group
- q R 40a each independently represents an oxygen atom or a sulfur atom
- q + 1 Y a s each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to a 4-position carbon atom
- q represents an arbitrary integer of 1 or more.
- q + 1 A 1 s independently represent an oxygen atom or —NH—
- Base A represents a nucleobase which may be independently protected
- P 1a ′ represents a hydroxyl group or a protecting group for an amino group, or one of the hydroxyl groups as —A 1 —P 1a ′ represents —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected).
- the phosphoric acid group replaced by q P 2a each independently represents a protecting group for a phosphate group
- q R 40a each independently represents an oxygen atom or a sulfur atom
- q + 1 Y a s each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to a 4-position carbon atom
- q represents an arbitrary integer of 1 or more.
- At least one of q + 1 Base A is a protecting group represented by the formula: -LY L -Z (the definitions of each symbol are as defined in the above [12]), or the formula: -Z ' The method according to any one of [17] to [19] above, which is protected with a protecting group represented by the definition of each symbol as defined in [12] above.
- step (1) The method according to any one of [17] to [20] above, wherein the activator used in step (1) is a weakly acidic activator having a pKa of 5 or more.
- the activator used in step (1) is an azole compound.
- the activator used in step (1) is dicyanoimidazole or dichloroimidazole.
- the activator used in step (1) is used in an amount of 1.5 to 20 molar equivalents relative to the phosphitylating agent precursor.
- the solvent used in the step (1) dissolves the phosphitylating agent precursor, and the activator becomes hardly soluble, and has no acidic or basic functional group.
- the solvent used in step (1) is toluene, benzene, o-xylene, m-xylene, p-xylene, ethylbenzene, pentane, hexane, heptane, octane, cyclopentane, cyclohexane, diethyl ether, diisopropyl ether, tert
- q + 1 A 1 independently represent an oxygen atom or —NH—
- Base A represents a nucleobase which may be independently protected
- P 1a is either a protecting group for hydroxyl group, or one hydroxyl group -OL n1 -OH as -O-P 1a (wherein, L n1 represents an organic group, a hydroxyl group is protected) replaced the
- the phosphate group q P 2a each independently represents a protecting group for a phosphate group
- q R 40a each independently represents an oxygen atom or a sulfur atom
- q + 1 Y a s each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to a 4-position carbon atom
- X represents an oxygen atom or a sulfur atom
- R 10 represents an aromatic ring, a hydroxyl protecting group or a thiol protecting group
- R 30 represents an alkyl group independently
- q represents
- P 1a ′ represents a hydroxyl group or an amino-protecting group, or one of the hydroxyl groups as —A 1 —P 1a ′ represents —OL n1 —OH (wherein L n1 represents an organic group, The hydroxyl group is protected), and the other symbols are as defined in formula (Ia-1)]
- P 1a ′ represents a hydroxyl group or an amino-protecting group, or one of the hydroxyl groups as —A 1 —P 1a ′ represents —OL n1 —OH (wherein L n1 represents an organic group, The hydroxyl group is protected), and the other symbols are as defined in formula (Ia-2)]
- At least one of q + 1 Base A is a protecting group represented by the formula: -L Y L -Z (the definitions of each symbol are the same as defined in [12] above), or the formula: -Z ' (The definition of each symbol is synonymous with the above [12])
- the method in any one of.
- n′-polymerized oligonucleotide having a functional group protected at the 3′-position hydroxyl group, the 3′-position amino group or the 3′-position phosphate group and not protected at the 5′-position hydroxyl group (n ′ is 2 The method according to [31] above, wherein any integer above is shown.
- n′-polymerized oligonucleotide in which the functional group is protected at the 5′-position hydroxyl group and the 3′-position hydroxyl group or the 3′-position amino group is not protected (n ′ represents an integer of 2 or more)
- n ′ represents an integer of 2 or more
- r + 1 A 1 each independently represents an oxygen atom or NH; r + 1 Base Bs each independently represent a nucleobase that may be protected; r P 2b represents a protecting group for a phosphate group; r R 40b represents an oxygen atom or a sulfur atom, P 3b represents a hydroxyl group or a protecting group for an amino group, or one of the hydroxyl groups as —A 1 -P 3b is —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected) ) Indicates the phosphate group replaced, r + 1 Yb represents a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to the 4-position carbon atom; r represents an arbitrary integer of 1 or more.
- n ′ polymerized oligonucleotide has the following structure (I
- P 3b ′ represents a protecting group for a hydroxyl group, or one of the hydroxyl groups as —OP 3b ′ represents —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected)
- the other phosphates have the same definitions as in formula (Ib)]
- At least one of r + 1 Base Bs is a protecting group represented by the formula: -L Y L -Z (the definitions of each symbol are the same as defined in [12] above), or the formula: -Z ' (The definition of each symbol is synonymous with the above [12])
- the protecting group for the hydroxyl group in P 3b or P 3b ′ is represented by the formula: -LY L -Z (the definitions of each symbol are the same as defined in [12] above), or the formula: -Z ′ (each symbol The definition
- q + 1 A 1 each independently represents an oxygen atom or NH; q + 1 pieces of Base A ′ are each independently represented by the formula: —L′—Y L —Z (where L ′ is a succinyl group, Y L , Z is the same as defined in [12] above).
- P 1a is either a protecting group for hydroxyl group, or one hydroxyl group -OL n1 -OH as -O-P 1a (wherein, L n1 represents an organic group, a hydroxyl group is protected) replaced the
- the phosphate group q P 2a each independently represents a protecting group for a phosphate group;
- q R 40a each independently represents an oxygen atom or a sulfur atom;
- q + 1 Y a s each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to a 4-position carbon atom;
- X represents an oxygen atom or a sulfur atom,
- R 10 represents an aromatic ring, a hydroxyl protecting group or a thiol protecting group
- r + 1 pieces of A 1 each independently represent an oxygen atom or NH; r + 1 Base Bs each independently represent a nucleobase that may be protected; r P 2b represents a protecting group for a phosphate group; r R 40b represents an oxygen atom or a sulfur atom, P 3b represents a hydroxyl group or a protecting group for an amino group, or one of the hydroxyl groups as —A 1 -P 3b is —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected) ) Indicates the phosphate group replaced, r + 1 Yb represents a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to the 4-position carbon atom; r represents an arbitrary integer of 1 or more.
- At least one of r + 1 Base Bs is a protecting group represented by the formula: -L Y L -Z (the definitions of each symbol are the same as defined in [12] above), or the formula: -Z ' (The definition of each symbol is synonymous with said [12].)
- the hydroxyl protecting group for P 3b ′ is represented by the formula: -LY L -Z (the definitions of each symbol are the same as defined in [12] above), or the formula: -Z ′ (the definition of each symbol is The method according to [46] or [47] above, which is synonymous with [12] above.
- q + 1 A 1 each independently represents an oxygen atom or NH; q + 1 Bases each independently represents a nucleobase that may be protected; P 1 represents a protecting group for a hydroxyl group, or one of the hydroxyl groups is replaced with —OL n1 —OH (wherein L n1 represents an organic group and the hydroxyl group is protected) as —O—P 1.
- the phosphate group q pieces of P 2 each independently represent a protecting group for a phosphate group; q R 40 s each independently represent an oxygen atom or a sulfur atom; q + 1 Y's each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom or an organic group that crosslinks to the 4-position carbon atom; P 3 represents a silyl protecting group, q represents an arbitrary integer of 1 or more.
- a compound represented by [51] At least one of q + 1 Bases is a protecting group represented by the formula: -LY L -Z (the definitions of each symbol are as defined in the above [12]), or the formula: -Z '( The definition of each symbol is the same as defined in [12] above, and is protected by a protecting group represented by [50].
- the method of the present invention it is possible to prepare a phosphitylating agent from a stable diamidite during storage, and it is possible to suppress the removal of the protecting group on phosphoric acid during the phosphitylation reaction. Therefore, phosphoramididized oligonucleotides can be produced more stably and efficiently. Furthermore, according to the method of the present invention, the silyl protecting group can be deprotected without removing the cyanoethyl protecting group on the internucleotide phosphate. Therefore, by using these methods, more efficient oligonucleotide production and fragment condensation using it are possible.
- nucleoside which is a structural unit of an oligonucleotide means that a nucleobase is linked to a sugar (for example, 2-deoxyribose, ribose, 2-position carbon atom and 4-position carbon atom by a divalent organic group.
- a sugar for example, 2-deoxyribose, ribose, 2-position carbon atom and 4-position carbon atom by a divalent organic group.
- the “sugar” includes an amino sugar in which a hydroxyl group is replaced with an amino group, and a ribose in which a hydroxyl group at the 2-position is replaced with a halogen atom.
- nucleotide means a compound in which a phosphate group is bonded to a nucleoside
- oligonucleotide means a compound in which one or more nucleotides are linked to a nucleoside
- oligonucleotide refers to a phosphorothioate-type oligonucleotide in which the oxygen atom of the phosphate group is replaced with a sulfur atom, an oligonucleotide in which —O— of the phosphate group is replaced with —NH—, phosphor Oligonucleotides in which the hydroxyl group (—OH) in the acid group is replaced by —OR p (wherein R p represents an organic group) are also included.
- the number of nucleosides of the oligonucleotide in the present invention is not particularly limited, but is preferably 3 to 50, more preferably 5 to 30.
- the “3′-position amino group” means an amino group bonded to the 3′-position carbon atom of a nucleoside, nucleotide or oligonucleotide.
- the “5′-position amino group” means an amino group bonded to the 5′-position carbon atom of a nucleoside, nucleotide or oligonucleotide.
- the “3′-position phosphate group” means a phosphate group bonded to the 3′-position carbon atom of a nucleotide or oligonucleotide.
- the “5′-position phosphate group” means a phosphate group bonded to the 5′-position carbon atom of a nucleotide or oligonucleotide.
- the “phosphate group” includes not only —O—P (O) (OH) 2 but also a group in which an oxygen atom is replaced with a sulfur atom or NH (for example, —O—P (S) (OH ) 2, -NH-P (O ) (OH) 2, -NH-P (S) (OH) 2) also encompasses.
- a group for example, a protected phosphate group in which a hydroxyl group (—OH) in the phosphate group is replaced by —OR p (wherein R p represents an organic group such as a protecting group for a phosphate group). ) Is also included in the “phosphate group”.
- nucleobase is not particularly limited as long as it is used for nucleic acid synthesis, and examples thereof include pyrimidine bases such as cytosyl group, uracil group and thyminyl group, adenyl group, guanyl group and the like. Mention may be made of purine bases.
- nucleobase includes a nucleobase having an arbitrary substituent (eg, halogen atom, alkyl group, aralkyl group, alkoxy group, acyl group, alkoxyalkyl group, hydroxy group, amino group).
- Modified nucleobases eg, 8-bromoadenyl group, 8-bromoguanyl group, 5-bromocytosyl group
- Modified nucleobases substituted at any position by a group, monoalkylamino, dialkylamino, carboxy, cyano, nitro, etc.
- halogen atom is a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom.
- examples of the “alkyl (group)” include linear or branched alkyl groups having 1 or more carbon atoms.
- C 1- 10 alkyl group more preferably C 1-6 alkyl group.
- Preferable specific examples in the case where the carbon number range is not limited include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl and the like, and methyl and ethyl are particularly preferable. .
- examples of the “aralkyl (group)” include a C 7-20 aralkyl group, preferably a C 7-16 aralkyl group (C 6-10 aryl-C 1-6 alkyl group). Specific examples include benzyl, 1-phenylethyl, 2-phenylethyl, 1-phenylpropyl, naphthylmethyl, 1-naphthylethyl, 1-naphthylpropyl and the like, and benzyl is particularly preferable.
- examples of the “alkoxy (group)” include an alkoxy group having 1 or more carbon atoms.
- the carbon number range it is preferably a C 1-10 alkoxy group, more preferably Is a C 1-6 alkoxy group.
- the carbon number range is not limited include methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, pentyloxy, hexyloxy, etc. Ethoxy is preferred.
- examples of the “acyl (group)” include a linear or branched C 1-6 alkanoyl group, a C 7-13 aroyl group, and the like. Specific examples include formyl, acetyl, n-propionyl, isopropionyl, n-butyryl, isobutyryl, pivaloyl, valeryl, hexanoyl, benzoyl, naphthoyl, levulinyl and the like, which may each be substituted.
- alkenyl (group) is preferably a linear or branched C 2-6 alkenyl group, such as vinyl, 1-propenyl, allyl, isopropenyl, butenyl, isobutenyl and the like. Can be mentioned. Of these, a C 2 -C 4 alkenyl group is preferable.
- alkynyl (group) is preferably a C 2-6 alkynyl group, for example, ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1- Examples include pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, 5-hexynyl and the like. Among them, C 2 -C 4 alkynyl group.
- cycloalkyl (group) means a cyclic alkyl group, and examples thereof include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like. Among them, C 3 -C 6 cycloalkyl groups such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like are preferable, and cyclohexyl is particularly preferable.
- aryl (group) or “aromatic ring (group)” means a monocyclic or polycyclic (fused) hydrocarbon group exhibiting aromaticity, specifically, for example, And C 6-14 aryl groups such as phenyl, 1-naphthyl, 2-naphthyl, biphenylyl, 2-anthryl and the like. Among them, a C 6-10 aryl group is more preferable, and phenyl is particularly preferable.
- aryloxy (group) is preferably a C 6-14 aryloxy group, and preferably includes phenoxy, tolyloxy, xylyloxy, naphthoxy, dimethylnaphthoxy and the like.
- aralkyloxy (group) means the above “alkyloxy (group)” substituted with the above “aryl group”, and specifically includes benzyloxy, phenethyloxy, 2-phenylpropane. -2-yloxy, diphenylmethyloxy and the like.
- examples of the “hydrocarbon group” include an aliphatic hydrocarbon group, an araliphatic hydrocarbon group, a monocyclic saturated hydrocarbon group, an aromatic hydrocarbon group, and the like.
- a monovalent group such as an alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, an aryl group, an aralkyl group, and a divalent group derived therefrom.
- the “organic group having a hydrocarbon group” means a group having the “hydrocarbon group”, and the portion other than the “hydrocarbon group” in the “organic group having a hydrocarbon group” It can be set arbitrarily.
- the linker may have sites such as —O—, —S—, —COO—, —OCONH—, and —CONH—.
- Step 2 [Method for Deprotecting Oligonucleotide with 3′-position Hydroxyl or 3′-position Amino Group Protected by Silyl-protecting Group (Step) —Desilylation]
- n-polymerized oligos in which the 5′-position hydroxyl group or the 5′-position phosphate group is protected and the 3′-position hydroxyl group or the 3′-position amino group is protected with a silyl protecting group are used.
- the silyl protecting group of the nucleotide (n represents an arbitrary integer of 2 or more) is deprotected (also referred to as desilylation) to protect the “5′-position hydroxyl group or 5′-position phosphate group”.
- the protecting group for the 5′-position hydroxyl group is not particularly limited as long as it can be deprotected under acidic conditions and can be used as a protecting group for a hydroxyl group, but is not limited to a trityl group or a 9- (9-phenyl) xanthenyl group.
- Di (C 1-6 alkoxy) trityl groups such as 9-phenylthioxanthenyl group, 1,1-bis (4-methoxyphenyl) -1-phenylmethyl group (dimethoxytrityl group), 1- (4-methoxy And mono (C 1-18 alkoxy) trityl group such as phenyl) -1,1-diphenylmethyl group (monomethoxytrityl group).
- a monomethoxytrityl group and a dimethoxytrityl group are preferable, and a dimethoxytrityl group is more preferable.
- the 5 ′ end of the n-polymerized oligonucleotide to be desilylated may be a phosphate group that may be protected.
- the protected phosphate group at the 5′-position include, for example, one of the hydroxyl groups in the phosphate group is —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group can be deprotected under acidic conditions) And a phosphate group that is replaced by a protecting group).
- the organic group of L n1 means a hydrocarbon group or a group in which a carbon atom in the hydrocarbon group is replaced with a hetero atom.
- the hetero atom include an oxygen atom, a nitrogen atom, and a sulfur atom.
- the organic group may have a substituent such as a hydroxyl group, an amino group, or an oxo group ( ⁇ O).
- the hydroxyl group and amino group that the organic group may have are preferably protected with a protecting group.
- the shape of the organic group may be any of a chain (linear or branched), a ring, or a combination thereof.
- the organic group may have a group having functionality for cells.
- the group having functionality with respect to the cells is preferably bonded to the end of the main chain or side chain of the organic group. Examples of groups having functionality with respect to cells include, for example, “groups that improve the cell membrane permeability of compounds by improving the lipid solubility of compounds”, “incorporation of compounds into cells via cell membrane receptors” And the like ".
- Examples of the “group that improves the cell membrane permeability of the compound by improving the fat solubility of the compound” include cholesterol residues and tocopherol residues.
- Examples of the “group that improves the uptake of a compound into a cell via a cell membrane receptor” include an N-acetylgalactosamine residue and the like.
- Specific examples of —OL n1 —OH include the following (in the following formula, * represents a bonding position with a phosphorus atom, and Ac represents an acetyl group).
- the substituent on silicon is selected from an alkyl group, an aryl group, an aralkyl group, an alkoxy group, an aryloxy group, an aralkyloxy group, an alkylthio group, and an arylthio group
- a silyl group in which at least one of the three substituents is selected from an aryl group, an alkoxy group, an aryloxy group, and an aralkyloxy group.
- TDMS tert-butyldimethylsilyl
- DIPPS diisopropylphenylsilyl
- TBODPS tert-butoxydiphenylsilyl
- IPIPS isopropoxydiisopropylsilyl
- n-polymerized oligonucleotide in which the 5′-position hydroxyl group or the 5′-position phosphate group is protected and the 3′-position hydroxyl group or the 3′-position amino group is protected with a silyl protecting group (n is 2 or more).
- n is 2 or more.
- An arbitrary integer is shown.
- I a compound having the following structure (I) (hereinafter referred to as compound (I)).
- s + 1 pieces of A 1 is independently an oxygen atom or -NH-, s + 1 Bases each independently represents a nucleobase that may be protected; P 1 represents a protecting group for a hydroxyl group, or one of the hydroxyl groups is replaced with —OL n1 —OH (wherein L n1 represents an organic group and the hydroxyl group is protected) as —O—P 1.
- the phosphate group s number of P 2 each independently represent a protecting group of a phosphate group
- s R 40 s independently represent an oxygen atom or a sulfur atom
- s + 1 Y's each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom or an organic group that crosslinks to the 4-position carbon atom
- P 3 represents a silyl protecting group
- s represents an arbitrary integer of 1 or more.
- the hydroxyl protecting group for P 1 the same hydroxyl group protecting groups as those exemplified above for the 5′-position hydroxyl protecting group can be used. A dimethoxytrityl group is preferred.
- a phosphate group in which one of the hydroxyl groups in P 1 is replaced with —OL n1 —OH is a protected group at the 5′-end.
- L n1 represents an organic group and the hydroxyl group is protected
- the protecting group for the phosphate group in P 2 is not particularly limited as long as it can be deprotected under basic conditions and can be used as a protecting group for the phosphate group, but is not limited to —CH 2 CH 2 WG ( WG represents an electron-withdrawing group.), And WG is preferably a cyano group.
- R 40 is preferably the same and is an oxygen atom or a sulfur atom.
- the protecting group for the hydroxyl group which may be protected in Y is not particularly limited.
- An arbitrary protecting group described in, for example, JOHN WILLY & SONS publication (2006) can be mentioned.
- a triethylsilyl group, a triisopropylsilyl group, or a tert-butyldimethylsilyl group is preferable, and a tert-butyldimethylsilyl group is particularly preferable from the viewpoints of economy and availability.
- the “organic group capable of cross-linking to the 4-position carbon atom” in Y is not particularly limited as long as the 2-position and 4-position of the nucleoside are cross-linked, and examples thereof include a C 2-7 alkylene group.
- the alkylene group includes, for example, —O—, —NR 37 — (R 37 represents a hydrogen atom or a C 1-6 alkyl group), —S—, —CO—, —COO—, —OCONR 38 — (R 38 represents a hydrogen atom or a C 1-6 alkyl group), —CONR 39 — (R 39 represents a hydrogen atom or a C 1-6 alkyl group) or the like, and is one or more positions (preferably 1 or 2 locations) may be interrupted.
- the “organic group that bridges to the 4-position carbon atom” is preferably, for example, —ORi (Ri represents a C 1-6 alkylene group that bridges to the 4-position), —O—NR 37 —Rj (Rj is in the 4-position)
- R 37 is as defined above
- —O—Rk—O—Rl Rk is a C 1-6 alkylene group, Rl is a C 1 bridge at the 4-position
- -6 represents an alkylene group).
- a methylene group or an ethylene group is preferable independently.
- Examples of the “organic group bridging to the 4-position carbon atom” include —O—CH 2 —, —O—CH 2 —CH 2 —, —O—NR 37 —CH 2 — (R 37 is as defined above). , —O—CH 2 —O—CH 2 — and the like are preferable, —O—CH 2 —, —O—CH 2 —CH 2 —, —O—NH—CH 2 —, —O—NMe—CH 2 — , —O—CH 2 —O—CH 2 — (each bonded to the 2-position on the left side and bonded to the 4-position on the right side) is more preferable.
- the silyl protecting group for P 3 those exemplified as the silyl protecting group for the 3′-position hydroxyl group can be used.
- the “optionally protected nucleobase” of Base means, for example, that an amino group may be protected in an adenyl group, a guanyl group, or a cytosyl group, which is a nucleobase having an amino group, Nucleobases in which the amino group of the nucleobase is protected by a protecting group that can withstand deprotection conditions at the 5 ′ position are preferred.
- amino-protecting group is not particularly limited. For example, Green's PROTECTIVE GROUPS IN ORGANIC SYNTHESIS, 4th edition, John Willy and -The protecting group described in Sands (John WILLY & SONS) publication (2006) etc. can be mentioned.
- amino-protecting group examples include, for example, pivaloyl group, pivaloyloxymethyl group, trifluoroacetyl group, phenoxyacetyl group, 4-isopropylphenoxyacetyl group, 4-tert-butylphenoxyacetyl group, Examples thereof include an acetyl group, a benzoyl group, an isobutyryl group, a dimethylformamidinyl group, and a 9-fluorenylmethyloxycarbonyl group.
- phenoxyacetyl group 4-isopropylphenoxyacetyl group, acetyl group, benzoyl group, isobutyryl group, and dimethylformamidinyl group are preferable.
- the carbonyl group of the nucleobase may be protected, for example, phenol, 2,5-dichlorophenol, 3-chlorophenol, 3,5-dichlorophenol, 2-formylphenol, 2-naphthol, 4-methoxy Phenol, 4-chlorophenol, 2-nitrophenol, 4-nitrophenol, 4-acetylaminophenol, pentafluorophenol, 4-pivaloyloxybenzyl alcohol, 4-nitrophenethyl alcohol, 2- (methylsulfonyl) ethanol, 2 -(Phenylsulfonyl) ethanol, 2-cyanoethanol, 2- (trimethylsilyl) ethanol, dimethylcarbamic acid chloride, diethylcarbamic acid chloride, ethylphenyl
- a protective group for the carbonyl group may not be particularly introduced.
- protecting groups for nucleic bases it can also be used protecting groups described (C 5-30 straight-chain or branched-chain alkyl group and / or a C 5-30 group having a straight chain or branched chain alkenyl) in WO2013 / 122236 .
- protecting groups for nucleic bases the following formula: -L-Y L -Z represented by protecting groups, or the formula: can also be used a protective group represented by -Z '.
- the protecting group is solubilized in a nonpolar solvent when the group is bonded to a reaction substrate, can be reacted in a liquid phase, and precipitated by the addition of a polar solvent, allowing solid-liquid separation. It is a protecting group that has both reactivity and ease of post-treatment, and is a stable group under acidic conditions that can remove the protecting group at the 5′-terminal hydroxyl group. Accordingly, at least one of the s + 1 Bases is protected with a protecting group represented by the formula (1): -LY L -Z, or (2) a protecting group represented by the formula: -Z '. Preferably it is. (1)
- R 4 represents a hydrogen atom or, when R b is a group represented by the following formula (a3), together with R 6 represents a single bond or —O—, And may form a fluorenyl group or a xanthenyl group
- k R 5 's each independently represents an organic group having an aliphatic hydrocarbon group having 10 or more carbon atoms
- k represents an integer of 1 to 4
- Ring A includes, in addition to k OR 5 , a halogen atom, a C 1-6 alkyl group which may be substituted with a halogen atom, and a C 1-6 alkoxy group which may be substituted with a halogen atom. May have a substituent selected from the group consisting of; R a represents a hydrogen atom or a phenyl group optionally substituted by a halogen atom; and R b represents a hydrogen atom or the formula (a3):
- L 1 represents a C 1-6 alkylene group
- L 2 represents a single bond, or the formula: * C (R 3a ) (R 3b ) —O—R 1 ** , formula: * C ( ⁇ O) N (R 2 ) —R 1 —N ( R 3a ) ** or formula: * C ( ⁇ O) N (R 2 ) —R 1 —C (R 3a ) (R 3b ) ** (wherein * represents the position of bonding with L 1 , ** represents a bonding position to C ⁇ O, R 1 represents a C 1-6 alkylene group, and R 2 , R 3a and R 3b are each independently a hydrogen atom or substituted A good C 1-6 alkyl group, or R 2 and R 3a may be combined to form an optionally substituted C 1-6 alkylene bond.) There; and R c and R d are each independently hydrogen atoms or taken
- L 1 represents an ethylene group
- L 2 represents a single bond
- R c and R d together form a single carbonyl group.
- linker L of formula (a1) wherein (a1), L 1 is, represents an ethylene group; and in L 2 in N (R 2) -R 1 -N ( The R 3a ) moiety represents a piperazinylene group; and R c and R d are taken together to form a single carbonyl group.
- L 1 represents an ethylene group
- R c and R d together form a single carbonyl group.
- L 1 represents an ethylene group
- the N (R 2 ) —R 1 —C (R 3a ) (R 3b ) moiety in L 2 represents a piperidinylene group
- R c and R d together form a single carbonyl group It is a group.
- L 1 represents a methylene group or an ethylene group
- L 2 represents a single bond
- R c and R d are a hydrogen atom.
- L 1 represents a butylene group
- L 2 represents a single bond
- R c and R d together form a single carbonyl group.
- L 1 represents a methylene group
- the C (R 3a ) (R 3b ) —O—R 1 moiety in L 2 represents —CH 2 —O—CH 2 —; and R c and R d together form a single carbonyl group The group that is forming.
- linker L is a succinyl group that is easily available and inexpensive.
- a preferable embodiment of Z is a group represented by the formula (a2), the formula (a2 ′) or the formula (a2 ′′).
- R a and R b both represent a hydrogen atom;
- R 4 represents a hydrogen atom,
- k R 5 's each independently represents an organic group having an aliphatic hydrocarbon group having 10 or more carbon atoms (eg, a C 10-40 alkyl group); and
- k represents an integer of 1 to 3. It is a group. Also preferred is an embodiment in which R a and R b together form a single carbonyl group.
- k represents an integer of 1 to 3; R a and R b both represent a hydrogen atom; R 4 represents a hydrogen atom; k R 5 's each independently represent a benzyl group having 1 to 3 aliphatic hydrocarbon groups having 10 or more carbon atoms, or a cyclohexyl group having 1 to 3 aliphatic hydrocarbon groups having 10 or more carbon atoms.
- ring a in addition to the k oR 5, further halogen atom, a C 1-6 alkyl group optionally substituted by a halogen atom, and may be substituted by a halogen atom C 1-6 It is a group optionally having a substituent selected from the group consisting of alkoxy groups.
- R a represents a hydrogen atom
- R b represents the above formula (a3) (wherein * represents a bonding position; j represents an integer of 0 to 3; j R 7 each independently represents a C 10-40 alkyl group)
- R 4 and R 6 both represent a hydrogen atom. It is a group that is a group represented by
- R a represents a hydrogen atom
- R b represents the above formula (a3) (wherein * represents a bonding position; j represents an integer of 0 to 3; j R 7 each independently represents a C 10-40 alkyl group)
- R 6 together with R 4 of ring A forms a single bond or —O—, whereby ring A and ring B together represent a fluorenyl group or a xanthenyl group. It is a group that is a group represented by
- k R 5 s are each independently an organic group having an aliphatic hydrocarbon group having 10 or more carbon atoms ( For example, a C 10-40 alkyl group; and k is a group that represents an integer of 1 to 3.
- —Z ′, k R 5 's each independently represents an organic group having an aliphatic hydrocarbon group having 10 or more carbon atoms (eg, a C 10-40 alkyl group); A group representing an integer of 1 to 3;
- the protecting group represented by the above formula: -LY L -Z or the protecting group represented by the formula: -Z ' is not easily cleaved under acidic conditions that can remove the protecting group of the nucleotide terminal hydroxyl group, Groups that are cleaved under basic conditions are preferred.
- L in the formula: -L Y L -Z is a group represented by the above formula (a1) (preferably a succinyl group or the like), and Y L -Z is the following group; A group represented by the formula: -Z 'is the following group.
- the solvent used in this method (step) is not particularly limited as long as the reaction proceeds, as long as it dissolves the substrate and is not an acid, but halogens such as dichloromethane, 1,2-dichloroethane, chloroform, carbon tetrachloride, etc.
- Hydrocarbon solvents aromatic hydrocarbon solvents such as benzene, toluene and xylene, or aliphatic hydrocarbon solvents such as pentane, hexane, heptane and octane, or ether solvents such as diethyl ether, tetrahydrofuran and cyclopentyl methyl ether Or a mixed solvent thereof.
- a mixed solvent of tetrahydrofuran and dichloromethane is preferable, and a mixed solvent of 2: 1 is particularly preferable.
- Desilylation is carried out by applying a fluoride ion source in the presence of an organic base.
- the organic base one or a mixture of two or more types is used, and a salt of at least one of these bases and hydrogen fluoride is used as the fluoride ion source.
- the organic base may be a mixture of strong and weak bases, a mixture of two or more weak bases, or a single weak base.
- the strong base is preferably one having pKa ⁇ 8, specifically, methylamine, dimethylamine, trimethylamine, ethylamine, diethylamine, triethylamine, propylamine, isopropylamine, diisopropylamine, diisopropylethylamine, butylamine, isobutylamine , Tert-butylamine, 1,4-diazabicyclo [2.2.2] octane (DABCO), morpholine amines, and preferably triethylamine.
- DABCO 1,4-diazabicyclo [2.2.2] octane
- the weak base is preferably 4 ⁇ pKa ⁇ 8, specifically, pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, piperazine, piperidine, imidazole, N-methylimidazole And heterocyclic compounds such as N-methylmorpholine and N-ethylmorpholine, and anilines such as aniline, toluidine, dimethylaniline, diethylaniline, ethylaniline, ethylmethylaniline and anisidine, preferably pyridine, N-methylmorpholine More preferably, it is pyridine.
- the organic base is preferably used at 1 molar equivalent or more with respect to fluoride ions.
- the organic base is a mixture of a strong base and a weak base
- the amount of each used can be appropriately set within a range that does not adversely affect the desired reaction, but decyanoethylation on thiophosphoric acid may occur. Therefore, it is preferable to avoid the use of excess strong base.
- the strong base is used at 1/3 molar equivalent or less with respect to fluoride ions.
- the weak base is used in an amount of at least 1 molar equivalent together with the strong base. Even if the strong base is used at 1/3 molar equivalent or less, the 5′-terminal protecting group may be removed unless the weak base is used at 1 molar equivalent or more together with the strong base.
- the amount of the weak base used is not particularly limited as long as the substrate dissolves.
- the fluoride ion source is a salt of hydrogen fluoride with at least one of the above bases, specifically, triethylamine pentahydrofluoride (5HF-TEA), triethylamine trihydrofluoride (3HF). -TEA), pyridine hydrofluoride (HF-pyridine) and the like, and 3HF-TEA is preferred.
- This method (step) is a mixture obtained by adding an organic base to the fluoride ion source, preferably a mixture obtained by adding a weak base to a strong base hydrogen fluoride salt, more preferably, A mixture of pyridine added to 5HF-TEA or 3HF-TEA, particularly preferably a mixture of pyridine added to 3HF-TEA (3HF-TEA-pyridine).
- the reaction temperature and reaction time of this method are not particularly limited as long as the substrate / product is not precipitated, but each is usually ⁇ 10 to 80 ° C., preferably 0 to 40 ° C., more preferably 10 to 20 ° C. In general, it is 0.5 to 96 hours, preferably 1 to 48 hours, and more preferably 1 to 24 hours.
- a phosphitylating agent precursor having two nitrogen substituents on trivalent phosphorus is monoselectively activated to form a phosphitylating agent, which is used to free oligonucleotide in the presence of a base. It includes a reaction of phosphitylating the 3 ′ end or 5 ′ end. That is, this method is a method for producing a phosphoramididized oligonucleotide comprising the following steps (1) and (2). (1) In a solvent, the following formula (1):
- X represents an oxygen atom or a sulfur atom
- R 10 represents an aromatic ring, a hydroxyl protecting group or a thiol protecting group
- R 20 and R 30 each independently represents an alkyl group, and the alkyl group may form a ring together with the adjacent nitrogen atom.
- An activating agent is allowed to act on a phosphitylating agent precursor represented by the following formula (2):
- Za represents a group derived from an activator; Other symbols are as defined above.
- N represents an arbitrary integer of 2 or more
- the phosphitylating agent obtained in step (1) is allowed to act in the presence of a base to phosphitylate the hydroxyl group at the terminal of the oligonucleotide.
- the aromatic ring of R 10 includes phenyl, 4-nitrophenyl, 2,4-dinitrophenyl, pentafluorophenyl, pentachlorophenyl, 2-bromophenyl, 4-bromophenyl, 2-methylphenyl, 2,6-dimethylphenyl and the like can be mentioned, and 4-nitrophenyl is preferable.
- the hydroxyl protecting group or thiol protecting group for R 10 is specifically a C 1-6 alkyl group (eg, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl).
- Cyanated C 1-6 alkyl group eg, 2-cyanoethyl, 2-cyano-1,1-dimethylethyl
- Ethyl group substituted with substituted silyl group Eg, 2-methyldiphenylsilylethyl, 2-trimethylsilylethyl, 2-triphenylsilylethyl
- halogenated C 1-6 alkyl group eg, 2,2,2-trichloroethyl, 2,2,2-trimethyl) bromoethyl, 2,2,2-trifluoroethyl
- C 2-6 alkenyl group e.g., ethenyl, 1-propenyl, 2-propenyl, 1-methyl-2-propenyl Le, 1-methyl-1-propenyl
- C 3-6 cycloalkyl group e.g., cyclopropyl, cyclobutyl, cyclopent
- R 20 and R 30 each independently represent an alkyl group, and the alkyl group may be combined with an adjacent nitrogen atom to form a ring (eg, pyrrolidine).
- R 20 and R 30 are preferably both isopropyl groups.
- the phosphitylating agent precursor the following compounds are particularly preferable.
- a phosphitylating agent is obtained by making an activator act on a phosphitylating agent precursor.
- An activator is an acid that can displace an amine on the phosphoramidite to give a reactive substituent with a hydroxyl group or the like. Specifically, it is at least one selected from a weakly acidic activator having a pKa of 5 or more, more preferably an azole compound having a pKa of 5 or more and a C-substituted product thereof. Examples of the azole compound include tetrazole, triazole, imidazole, and the like.
- a compound disubstituted with a halogen atom such as dicyanoimidazole, bis (trifluoromethyl) imidazole, dichloroimidazole, or the like is used. Particularly preferred are dicyanoimidazole and dichloroimidazole.
- Za is a group derived from an activator, for example, a group obtained by removing one hydrogen atom from the activator. When dicyanoimidazole is used as the activator, Za is dicyanoimidazolyl. Yes, when dichloroimidazole is used as the activator, Za is dichloroimidazolyl.
- the solvent used in the step (1) is not particularly limited as long as it dissolves the phosphitylation precursor and the activator becomes hardly soluble, but usually does not have an acidic or basic functional group.
- “slightly soluble” refers to the fact that the concentration of the activator in the solvent is 6 ⁇ M or less.
- Examples include ether, carbon tetrachloride, etc., preferably toluene and cyclohexane, particularly preferably toluene.
- reaction temperature and reaction time of this method are not particularly limited as long as the substrate / product does not precipitate, but each is usually 40 ° C. or less, preferably 0 to 30 ° C., more preferably 5 to 15 ° C., particularly
- the temperature is preferably about 10 ° C., and is usually 0.5 to 24 hours, preferably 1 to 12 hours, and more preferably 1 to 6 hours.
- the amount of the activator and the phosphitylating agent precursor used is not particularly limited as long as the phosphitylating precursor is activated, but it is usually an excess amount relative to the phosphitylating precursor, preferably 1.5. Used in ⁇ 10 molar equivalents.
- the phosphitylation agent precursor is activated, and at the same time, diisopropylamine produced as a by-product upon phosphitylation is precipitated as a salt with the activator. Therefore, if necessary, a step of separating insoluble matters such as precipitates can be performed between the step (1) and the following step (2), and is preferably performed.
- Step (2) In this step, n-polymerized oligos in which either the 5′-position hydroxyl group or the 5′-position phosphate group or the 3′-position hydroxyl group or the 3′-position amino group is protected and the other is not protected in the solvent.
- N-polymerized oligonucleotide in which either one of the 5′-position hydroxyl group or the 5′-position phosphate group or the 3′-position hydroxyl group or the 3′-position amino group is protected and the other is not protected n is 2 or more
- q + 1 A 1 independently represent an oxygen atom or —NH—
- Base A represents a nucleobase which may be independently protected
- P 1a is either a protecting group for hydroxyl group, or one hydroxyl group -OL n1 -OH as -O-P 1a (wherein, L n1 represents an organic group, a hydroxyl group is protected) replaced the
- the phosphate group q P 2a each independently represents a protecting group for a phosphate group
- q R 40a each independently represents an oxygen atom or a sulfur atom
- q + 1 Y a s each independently represent a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to a 4-position carbon atom
- q represents an arbitrary integer of 1 or more.
- hydroxyl protecting group for P 1a those exemplified as the hydroxyl protecting group for P 1 can be used.
- a dimethoxytrityl group is preferred.
- the protecting group for the phosphate group in P 2a the same groups as those exemplified as the protecting group for the phosphate group in P 2 can be used.
- a group represented by —CH 2 CH 2 WG (WG represents an electron-withdrawing group) is preferable, and WG is preferable as WG.
- R 40a is preferably the same and is an oxygen atom or a sulfur atom.
- Y As the protecting group of hydroxyl group which may be protected in a, it can be the same as those exemplified as the protecting group of the hydroxyl group which may be protected in the Y, preferably tert- butyldimethylsilyl It is a group.
- the “organic group bridging to the 4-position carbon atom” in Y a those exemplified as the “organic group bridging to the 4-position carbon atom” in Y can be used.
- the Base can be the same as the “optionally protected nucleic acid base which may", at least one of Base A present one q + is It is preferably protected with a protecting group represented by the formula -LY L -Z (as defined above) or a protecting group represented by the formula -Z '(as defined above). The following is a scheme when this step (2) is carried out using compound (Ia) as a raw material.
- N “N” -polymerized oligonucleotides used in this step, wherein the “5′-position hydroxyl group or 5′-position phosphate group” is protected and the 3′-position hydroxyl group or the 3′-position amino group is not protected, exemplified by the compound (Ia) (N represents an arbitrary integer of 2 or more.) ”Is obtained by desilylation of the silyl protecting group at the 3 ′ end of the compound (I), preferably by the desilylation of the present invention described above. It may be.
- N-polymerized oligonucleotide in which either one of the 5′-position hydroxyl group or the 5′-position phosphate group or the 3′-position hydroxyl group or the 3′-position amino group is protected and the other is not protected Is a compound having the following structure (Ia ′) in which the 3′-position hydroxyl group or the 3′-position amino group is protected, and the 5′-position hydroxyl group is not protected. (Hereinafter, compound (Ia ′)).
- P 1a ′ represents a hydroxyl group or an amino-protecting group, or one of the hydroxyl groups as —A 1 —P 1a ′ represents —OL n1 —OH (wherein L n1 represents an organic group, The hydroxyl group is protected), and the other symbols are as defined in formula (Ia).
- the hydroxyl protecting group for P 1a ′ those exemplified as the hydroxyl protecting group for P 1 can be used.
- the protecting group for the amino group in P 1a ′ is not particularly limited, and examples thereof include pivaloyl group, pivaloyloxymethyl group, acetyl group, trifluoroacetyl group, phenoxyacetyl group, 4-isopropylphenoxyacetyl group, 4-tert -Butylphenoxyacetyl group, benzoyl group, isobutyryl group, (2-hexyl) decanoyl group, dimethylformamidinyl group, 1- (dimethylamino) ethylidene group, 9-fluorenylmethyloxycarbonyl group.
- the following is a scheme when this step (2) is carried out using compound (Ia ′) as a raw material.
- the same solvents as those used in the step (1) can be used.
- toluene, dichloromethane, chloroform and the like can be mentioned, preferably toluene and dichloromethane.
- It is a mixed solvent.
- the base used in step (2) has sufficient basicity for neutralization of the acid (activator) generated by the reaction, and does not cause decyanoethyl on phosphoric acid and does not form a PN bond. Is selected.
- Specific examples of such a base include pKa 5-8 bases, preferably collidine, N-methylmorpholine, diethylaniline, and the like.
- the phosphitylated precursor may or may not be added, but it is preferable to add it.
- the method includes linking a functional group directly or via a linker to the end of an oligonucleotide having a terminal hydroxyl group phosphorylated. More specifically, a functional group is linked directly or via a linker to the 3 ′ end of the 3 ′ end phosphoramididized oligonucleotide, or directly or directly to the 5 ′ end of the 5 ′ end phosphoramididized oligonucleotide or Linking functional groups via a linker.
- the “3′-terminal phosphoramididized oligonucleotide” and “5′-terminal phosphoramididized oligonucleotide” may be synthesized by any method, but preferably the “terminal phosphoramidated oligonucleotide” of the present invention described above.
- a functional group is linked directly or via a linker to the 3 ′ end of compound (Ia-1) to give the following compound (Ia-2) directly to the 5 ′ end of compound (Ia′-1) or via a linker
- the following compound (Ia′-2) is produced by linking functional groups.
- Lx represents a single bond or a linker.
- the functional group is directly linked to compound (Ia-1) or compound (Ia′-1).
- the functional group is directly linked to the 3 ′ end of the compound (Ia-1) or the 5 ′ end of (Ia′-1) with a hydroxyl group, thiol group or amino group.
- Lx is a linker
- the compound is formed via a linker (eg, —O—, —S—, —COO—, —OCONH—, and —CONH—) having a hydroxyl group, a thiol group, and an amino group as a reaction site.
- G is a functional group, and examples thereof include those derived from at least one selected from the group consisting of oligonucleotides, mononucleosides, cholesterol, GalNac3, PEG, low molecular weight drugs, biotin, peptides and labeled compounds. . Preferred are oligonucleotides and mononucleosides, and more preferred are oligonucleotides.
- the functional group is a 3′-position hydroxyl group or a 3′-position amino group or a 3′-phosphate group protected, and the 5′-position hydroxyl group is not protected, the following structure (Ib):
- r + 1 pieces of A 1 each independently represent an oxygen atom or NH; r + 1 Base Bs each independently represent a nucleobase that may be protected; r P 2b represents a protecting group for a phosphate group; r R 40b represents an oxygen atom or a sulfur atom, P 3b represents a hydroxyl group or a protecting group for an amino group, or one of the hydroxyl groups as —A 1 -P 3b is —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected) ) Indicates the phosphate group replaced, r + 1 Yb represents a hydrogen atom, an optionally protected hydroxyl group, a halogen atom, or an organic group that crosslinks to the 4-position carbon atom; r represents an arbitrary integer of 1 or more.
- n ′ polymerized oligonucleotide having n ′ (n ′ represents any integer of 2 or more
- the functional group has the following structure (Ib ') in which the 5'-position hydroxyl group or the 5'-position phosphate group is protected and the 3'-position hydroxyl group or the 3'-position amino group is not protected:
- P 3b represents a protecting group for a hydroxyl group, or one of the hydroxyl groups as —OP 3b ′ is —OL n1 —OH (wherein L n1 represents an organic group, and the hydroxyl group is protected) N'-polymerized oligonucleotides (where n 'represents any integer of 2 or more) having a phosphate group replaced with the formula (Ib).
- the method corresponds to fragment condensation of oligonucleotides and specifically comprises the following steps.
- the protecting group of a phosphate group at P 2b is not particularly limited, -CH 2 CH 2 WG ( WG represents an electron-withdrawing group.), And WG is preferably a cyano group.
- R 40b is preferably the same and is an oxygen atom or a sulfur atom.
- the protecting group for the hydroxyl group which may be protected in Y b is not particularly limited, and those exemplified in the definition of Y can be used similarly. As "organic group bridging the 4-position carbon atoms" in the Y b are is used similarly to that illustrated in the definition of Y b.
- the protecting group for the hydroxyl group in P 3b ′ is a protecting group for the 5′-position hydroxyl group of the oligonucleotide obtained after ligation (in the case of the compound (Ib)) or a protecting group for the 3′-position hydroxyl group (in the case of the compound (Ib ′)).
- a protecting group represented by the formula -LY L -Z (the definition of each symbol is as described above) or a protecting group represented by the formula -Z '(as defined above) is used.
- L is a succinyl group.
- the hydroxyl-protecting group for P 3b ′ those described in the above-mentioned desilylation section can be used, and in this case, a dimethoxytrityl group is preferable.
- P 3b pieces polymerization oligonucleotide 'n having a -L-Y L -Z as'(1b') is 5 'has a -L-Y L -Z in position, the mononucleoside 3' position is a hydroxyl group
- a monomer nucleoside having a hydroxyl group protected at the 3′-position and a phosphoramidite at the 5′-position can be prepared by repeating ligation by the phosphoramidite method in the same liquid phase as described in WO2012157723A1.
- the monomer nucleoside can be prepared based on known literature (eg, Wagner, T .; Pfleiderer, W. Nucleoside Nucleotides 1997, 16, 1657-1660 .; US8541569B2).
- One preferred embodiment of the present invention is the following formula (Ia-1 ′):
- Base A ′ are each independently a protecting group represented by the formula —L′—Y L —Z (wherein L ′ is a succinyl group and Y L and Z are as defined above). Or a nucleobase which may be protected with a protecting group represented by the formula: -Z '(as defined above), The definitions of the other symbols are as defined above.]
- a functional group is linked directly or via a linker to the 3 ′ end of the 3 ′ end phosphoramidite-modified oligonucleotide represented by the following formula (Ia-2 ′):
- the present invention is a method for producing an oligonucleotide comprising the following steps (1) and (2).
- n-polymerized oligonucleotide in which the 3′-position hydroxyl group is converted to a phosphoramidite and the 5′-position hydroxyl group is protected, and the 3′-position hydroxyl group is protected and the 5′-position hydroxyl group is not protected.
- Step (1) (Condensation step)
- n oligonucleotides in which the 3′-position hydroxyl group is converted to phosphoramidite and the 5′-position hydroxyl group is protected are converted into n ′, the 3′-position hydroxyl group is protected and the 5′-position hydroxyl group is not protected.
- This is a step of condensing the individual polymerized oligonucleotide with a phosphite triester bond via its 5′-position hydroxyl group.
- the condensation step with the nucleotide is usually performed in the presence of a condensation activator.
- the condensation activator used in this step is not particularly limited, but 5- (benzylthio) -1H-tetrazole (BTT), 5- (ethylthio) -1H-tetrazole (ETT), 4,5-dicyanoimidazole ( DCI), tetrazole, 5- [3,5-bis (trifluoromethyl) phenyl] -1H-tetrazole (Activator 42 (registered trademark)), benzimidazole triflate (BIT), pyridine trifluoroacetate, etc. are used. It is preferable. BTT, ETT, and DCI are more preferable, and BTT is particularly preferable.
- the amount of the condensation activator used in this step is usually 0.5 to 10 mol, preferably 1.0 to 2.0 mol, particularly preferably 1.0 mol, relative to 1 mol of the phosphoramidite-ized oligonucleotide. is there.
- the reaction temperature in this step is not particularly limited as long as the reaction proceeds, but is preferably 0 to 100 ° C, more preferably 20 to 50 ° C, and particularly preferably 20 to 30 ° C.
- the reaction time varies depending on the type of oligonucleotide used, the type of solvent, the reaction temperature, etc., but is 30 minutes to 24 hours.
- This step is performed in a solvent that does not affect the reaction.
- a solvent that does not affect the reaction.
- Specific examples include pyridine, THF, dichloromethane, chloroform, toluene, acetonitrile, or any mixed solvent thereof.
- the amount used depends on which substrate is intended to approach more complete consumption, but the former is preferably 0.1 to 10 moles and 0.5 to 3 moles relative to 1 mole of the latter. More preferred is 1.0 to 2.0 mol.
- Process (2) (oxidation process or sulfurization process) By reacting the n + n ′ polymerized oligonucleotide obtained in step (1) with an oxidizing agent or a sulfurizing agent, the phosphite triester bond of the n + n ′ polymerized oligonucleotide is converted into a phosphate triester bond or a thiophosphate triester. It is a process of converting into a bond.
- This step can be performed by directly adding an oxidizing agent or a sulfurizing agent to the reaction solution after step (2) without isolating the n + n′-polymerized oligonucleotide obtained in step (1). it can.
- the “oxidant” used in this step is not particularly limited as long as it has an ability to oxidize a phosphite triester bond to a phosphate triester bond without oxidizing other sites.
- iodine, (1S)-(+)-(10-camphanylsulfonyl) oxaziridine, tert-butyl hydroperoxide, 2-butanone peroxide, 1,1-dihydroperoxycyclododecane More preferably, iodine, (1S)-(+)-(10-camphanylsulfonyl) oxaziridine, tert-butyl hydroperoxide and 2-butanone peroxide are more preferable, iodine and tert-butyl hydroperoxide are still more preferable, and iodine is preferable. Particularly preferred.
- Such an oxidizing agent can be used by diluting with an appropriate solvent so as to have a concentration of 0.05 to 2M.
- a diluting solvent is not particularly limited as long as it is an inert solvent for the reaction, and examples thereof include pyridine, THF, dichloromethane, water, or any mixed solvent thereof. Among them, for example, iodine / water / pyridine-THF or iodine / pyridine-acetic acid or a peroxide (TBHP) / dichloromethane is preferably used.
- the “sulfurizing agent” used in this step is not particularly limited as long as it has an ability to convert a phosphite triester bond into a thiophosphate triester bond, but 3-((N, N-dimethylamino) is not limited.
- Methylidene) amino) -3H-1,2,4-dithiazol-5-thione DDTT
- 3H-1,2-benzodithiol-3-one-1,1-dioxide Beaucage reagent
- 3H-1, 2-Benzodithiol-3-one phenylacetyl disulfide (PADS), tetraethylthiuram disulfide (TETD), 3-amino-1,2,4-dithiazol-5-thione (ADTT), and sulfur are preferably used.
- PADS phenylacetyl disulfide
- TETD tetraethylthiuram disulfide
- ADTT 3-amino-1,2,4-dithiazol-5-thione
- Such a sulfurizing agent can be used by diluting with a suitable solvent so as to have a concentration of 0.05 to 2M.
- a diluting solvent is not particularly limited as long as it is an inert solvent for the reaction, and examples thereof include dichloromethane, acetonitrile, pyridine, and any mixed solvent thereof.
- the amount of the oxidizing agent or sulfurizing agent used can be 1 to 50 mol, preferably 1 to 5 mol, per 1 mol of the n + n ′ polymerized oligonucleotide (iv) obtained in step (1). .
- the reaction temperature is not particularly limited as long as the reaction proceeds, but is preferably 0 ° C to 100 ° C, more preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of n + n′-polymerized oligonucleotide (iv), the type of oxidizing agent or sulfurizing agent used, the reaction temperature, etc., but is 1 minute to 3 hours.
- step (3) it is possible to easily and effectively remove excess raw materials and by-products and purify n + n′-polymerized oligonucleotides.
- step (3) A step of adding a polar solvent to the reaction solution obtained in step (2) to precipitate n + n′-polymerized oligonucleotides and obtaining them by solid-liquid separation.
- the oligonucleotide can be isolated and produced.
- Step (3) (precipitation and solid-liquid separation step)
- the n + n′-polymerized oligonucleotide is obtained by adding a polar solvent to the reaction solution containing the n + n′-polymerized oligonucleotide having the phosphate triester bond or thiophosphate triester bond obtained in step (2). It is a step of precipitating and obtaining by solid-liquid separation.
- Examples of the polar solvent for precipitating the target product (n + n ′ polymerized oligonucleotide) in this step include alcohol solvents such as methanol, ethanol and isopropanol, nitrile solvents such as acetonitrile and propionitrile, dimethylformamide, Examples thereof include amide solvents such as dimethylacetamide, dimethyl sulfoxide, water, and a mixed solvent of two or more of these. Among these, alcohol solvents and nitrile solvents are preferable, and methanol or acetonitrile is more preferably used. As the polar solvent in the present invention, acetonitrile is particularly preferable from a practical viewpoint.
- coloring by iodine can be removed by using a solution in which sodium thiosulfate (hypo) is saturated in methanol as the precipitation solvent, It is possible to isolate the n + n′-polymerized oligonucleotide in which the 5′-position hydroxyl group is protected with high purity.
- trivalent phosphorus reagent for example, trimethyl phosphite, triethyl phosphite, tris (2-carboxyethyl) phosphine, etc.
- hypo etc.
- the desired oligonucleotide can be obtained with high purity and high yield by repeating the above steps (1) to (3) a desired number of times.
- Step (4) (Deprotection / Oligonucleotide Isolation Step)
- the oligonucleotide can be isolated by performing deprotection according to the type and nature of the protecting group.
- Deprotection methods include, for example, Green's PROTECTIVE GROUPS IN ORGANIC SYNTHESIS, 4th edition, published by John WILLY & SONS (2006) ) Etc., the process of removing all the protecting groups of oligonucleotide can be performed.
- phenoxyacetyl groups, acetyl groups, and the like which are nucleobase protecting groups in the present invention, are all removed by treatment with an aqueous ammonia / ethanol solution, such as cyanoethyl groups bonded to a phosphate group. be able to. Since an oligonucleotide having no protecting group is easily degraded by an enzyme, it is preferable to isolate the oligonucleotide under air cleanliness control.
- the confirmation of the progress of the reaction in each of the above steps can be performed by the same method as a general liquid phase organic synthesis reaction. That is, the reaction can be followed using thin layer silica gel chromatography, high performance liquid chromatography or the like.
- the oligonucleotide obtained from the step (3) or the step (4) can be led to a desired oligonucleotide derivative by further performing an organic synthesis reaction.
- the oligonucleotide obtained from the step (4) can provide RNA, DNA, oligonucleic acid drug.
- the functional group is an n ′ polymer having the following structure (Ib ′) in which the 5′-position hydroxyl group or the 5′-position phosphate group is protected and the 3′-position hydroxyl group or the 3′-position amino group is not protected.
- n ′ represents an arbitrary integer of 2 or more.
- tetramethylsilane was used as an internal standard, and CDCl 3 was used as a measurement solvent.
- NMR spectra were measured using a Bruker AVANCE 400 (400 MHZ) nuclear magnetic resonance apparatus.
- Electrospray ionization liquid chromatography / mass spectrometry (hereinafter abbreviated as LC / MS) was measured using Agilent Technologies 1290 Infinity. Abbreviations used in the following preparation examples and examples are as follows. If the nucleoside nucleobase is protected, the protecting group shall be indicated with a superscript after each nucleoside.
- Example 1 N 4 - [3,4,5- tris (octadecyl) benzoyl] -3'-O- (tert- butyldimethylsilyl)-5'-O-(4,4'-dimethoxytrityl) deoxy Synthesis of cytidine (DMTr-dC CO-TOP -TBDMS) (1) Synthesis of 5′-O- (4,4′-dimethoxytrityl) -3′-O- (tert-butyldimethylsilyl) deoxycytidine
- the white precipitate on the filter paper was dispersed in acetonitrile (40 mL), then filtered and washed again with a Kiriyama funnel, and after vacuum drying, the target DMTr-dC CO-TOP -TBDMS (1.51 g, 0.973 mmol, 97.3%).
- Example 2 5'-O- (4,4'-dimethoxytrityl) -deoxythymidine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxyguanosine 3'-[O- ( 2-cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyloxy) benzoyl]-3'-tert-butyldimethylsilyl-deoxycytidine (DMTr-d [TG iBu C CO-TOP] -TBDMS) Synthesis of
- Trifluoroacetic acid (55.7 ⁇ L, 0.75 mL) was added dropwise thereto and stirred at room temperature for 30 minutes, and completion of the reaction was confirmed by UPLC.
- the reaction mixture was neutralized with 2,4,6, -trimethylpyridine and then 5′-O- (4,4′-dimethoxytrityl) -N 2 -isobutyryldeoxyguanosine-3 ′-[O— ( 2-Cyanoethyl) (N, N-diisopropyl)] phosphoramidite (1.68 g, 2.00 mmol), 5- (benzylthio) -1H-tetrazole (0.384 g, 2.00 mmol) in acetonitrile (3.00 mL) The solution was added and stirred at room temperature for 2 hours.
- Example 3 31 P ⁇ 1 H ⁇ NMR analysis of cyanoethyl protected product About 0.003 g of DMTr-d [TG iBu C CO-TOP ] -TBDMS obtained in the same manner as in Example 2 was added to chloroform-d (0.7 mL). Dissolved in. When 31 P ⁇ 1 H ⁇ NMR was measured, all peaks were observed at 66.4 to 67.2 ppm.
- Example 4 Obtaining a decyanoethylated preparation using DBU DMTr-d [TG iBu C CO-TOP ] -TBDMS (0.0043 g, 0.00179 mmol) obtained in the same manner as in Example 2 was added to tetrahydrofuran (0 15 mL). To this, 1,8-diazabiccyclo [5.4.0] undec-7-ene (hereinafter DBU) (2.0 ⁇ L, 0.0133 mmol) was added and mixed. Decyanoethylation was allowed to proceed for 5 minutes at 23 ° C. Acetonitrile (1.0 mL) was added to the reaction solution and centrifuged at 10,000 G, 4 ° C. for 5 minutes.
- DBU 1,8-diazabiccyclo [5.4.0] undec-7-ene
- Example 5 Study of the desilylated using various fluoride ion source (1) 3HF-TEA was obtained in the same manner as Study Example 2 using (DMTr-d [TG iBu C CO-TOP] -TBDMS ( 0.0239 g, 0.00998 mmol) was dissolved in dichloromethane (0.3 mL) and cooled to 15 ° C. A solution of 3HF-TEA (29.0 mg, 0.180 mmol) in THF (0.60 mL) was added thereto. The desilylation was allowed to proceed for 24 hours at 15 ° C. The reaction solution was divided into approximately 200 ⁇ L, and acetonitrile (1.0 mL) was added to each to add 10,000 G, 4 ° C.
- the reaction solution was divided into approximately 200 ⁇ L in 4 portions, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the entire precipitate was vacuum-dried, the weight was 0.0169 g. When 31 P ⁇ 1 H ⁇ NMR of the product was measured, all peaks were observed at 65.6 to 68.3 ppm corresponding to the cyanoethyl protected product, and no signal of 60 ppm or less corresponding to the decyanoethylated product was observed. . In addition, the completion of desilylation was confirmed by measurement with LC-TOF MS.
- the reaction solution was divided into four portions of approximately 200 ⁇ L, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the whole precipitate was vacuum-dried, the weight was 0.01953 g. When 31 P ⁇ 1 H ⁇ NMR of the product was measured, all peaks were observed at 65.7 to 68.2 ppm corresponding to the cyanoethyl protected product, and no signal of 60 ppm or less corresponding to the decyanoethylated product was observed. . In addition, the completion of desilylation was confirmed by measurement with LC-TOF MS.
- the reaction solution was divided into four portions of approximately 200 ⁇ L, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the entire precipitate was vacuum-dried, the weight was 0.02133 g. When 31 P ⁇ 1 H ⁇ NMR of the product is measured, all peaks are observed at 65.8 to 68.1 ppm corresponding to the cyanoethyl protected product, and no signal below 60 ppm corresponding to the decyanoethylated product is observed. It was. In addition, the completion of desilylation was confirmed by measurement with LC-TOF MS.
- the reaction solution was divided into four portions of approximately 200 ⁇ L, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the whole precipitate was vacuum-dried, the weight was 0.0188 g.
- 31 P ⁇ 1 H ⁇ NMR of the product was measured, a peak of 60 ppm or higher corresponding to the cyanoethyl protected product was not observed, and only a peak of 60 ppm or lower corresponding to the decyanoethylated product was observed. It was revealed that the upper cyanoethyl group was completely removed. The completion of desilylation was also confirmed by analysis with LC-TOF MS.
- the reaction solution was divided into approximately 200 ⁇ L in 4 portions, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the entire precipitate was vacuum-dried, the weight was 0.0169 g.
- the integration ratio of the peak of 60 ppm or more corresponding to the cyanoethyl protected product and the peak of 60 ppm or less corresponding to the decyanoethylated product is as follows.
- the reaction solution was divided into four portions of approximately 200 ⁇ L, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the entire precipitate was vacuum-dried, the weight was 0.0208 g. When 31 P ⁇ 1 H ⁇ NMR of the product was measured, all peaks were observed at 65.8 to 68.2 ppm corresponding to the cyanoethyl protected product, and a signal of 60 ppm or less corresponding to the decyanoethylated product was not observed. . In addition, the completion of desilylation was confirmed by measurement with LC-TOF MS.
- the reaction solution was divided into approximately 200 ⁇ L in 4 portions, acetonitrile (1.0 mL) was added to each, and the mixture was centrifuged at 10,000 G, 4 ° C. for 5 minutes. After removing the supernatant, it was dispersed in acetonitrile (1.0 mL) and centrifuged again in the same manner. When the entire precipitate was vacuum-dried, the weight was 0.0168 g.
- 31 P ⁇ 1 H ⁇ NMR of the product was measured, a peak of 60 ppm or higher corresponding to the cyanoethyl protected product was not observed, and only a peak of 60 ppm or lower corresponding to the decyanoethylated product was observed. It was revealed that the upper cyanoethyl group was completely removed. The completion of desilylation was also confirmed by analysis with LC-TOF MS.
- Example 6 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxyguanosine 3 ′-[O— (2 - cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyloxy) benzoyl] deoxycytidine-3'-yl - [O-(2-cyanoethyl)] - N, N-diisopropyl phosphoramidite ( Synthesis of DMTr-d [TG iBu C CO-TOP ] -PA) (1) 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl- N 2 - is
- 2-Cyanoethyl-N, N, N ′, N′-tetraisopropyl phosphorodiamidite (960 ⁇ L, 3.02 mmol) was dissolved in dehydrated toluene (6.6 mL), and molecular sieve 3A powder (0.10 g) was added for 30 minutes. After dehydration, it was filtered through a 0.45 ⁇ m membrane filter, and 5.25 mL was dispensed into a dry screw mouth bottle. To this toluene solution, 4,5-dicyanoimidazole (0.616 g, 5.21 mmol) was added and stirred for 30 minutes, and then filtered through a 0.45 ⁇ m membrane filter to fractionate 1.5 mL of the solution.
- the suspension was added at 0 ° C.
- Dehydrated dichloromethane (3.0 mL) was added at the same temperature to obtain a homogeneous solution, and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by LC / MS, and the reaction was stopped with 2,4,6-trimethylpyridine.
- the reaction was cooled to 0 ° C. and acetonitrile (110 mL) was added to precipitate the product.
- Example 7 DMTr-d [TG iBu C CO-TOP ] -suc-TOB-condensation of DMTr-d [TG iBu C CO-TOP A Bz TT] -suc- by condensation of PA and HO-d [A Bz TT] -suc-TOB Synthesis of TOB
- Example 8 5 ′-(4,4′-dimethoxytrityl) -3′-diisopropylphenylsilyl-N 3- [3,4,5-tris (octadecyloxy) benzoyl] deoxythymidine (DMTr-dT CO-TOP Synthesis of -DIPPS (1) Preparation of diisopropylphenylsilane Phenyllithium (1.6 M dibutyl ether solution, 20.5 mL, 32.8 mmol) was dissolved in dehydrated tetrahydrofuran (60 mL) cooled to -78 ° C.
- the aqueous layer was extracted with dichloromethane (20 mL), and the combined organic layer was washed with water and then with a saturated saline solution, dehydrated with sodium sulfate, and the solvent was distilled off under reduced pressure to obtain the desired diisopropylphenylsilane. 14.9 g of a light brown oily liquid was obtained. This was used in the next step without purification.
- the oily liquid containing diisopropylphenylsilane obtained in (1) was dissolved in dry dichloromethane (30 mL), molecular sieve 3A (0.5 g) activated under heating was added thereto for dehydration, and the solution was filtered. Additional dichloromethane (282 mL) was added. To the solution was added 1,3-dichloro-5,5-dimethylhydantoin (9.54 g, 48.4 mmol), and the mixture was stirred at room temperature for 30 minutes.
- Example 9 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxy Guanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl-
- the reaction solution was neutralized by adding 1-methylimidazole (0.112 mL, 1.42 mmol).
- 1-methylimidazole 0.112 mL, 1.42 mmol.
- Acetonitrile 200 mL was added to the reaction solution to precipitate a mixture of oligonucleic acid trimer and 2,4,6-tris (octadecyloxy) benzyl acetate. After filtering and washing the suspension, 3.56 g of a mixture of the target nucleic acid trimer and 3,4,6-tris (octadecyloxy) benzyl acetate (of which 2.21 g of nucleic acid, 0.887 mmol, 95.7% yield) Rate). After dissolving the sample in tetrahydrofuran, the cyanoethyl protecting group on phosphoric acid was removed with DBU, and the target product was identified by LC / MS. LC / MS m / z: calcd 1188.14, found 1188.14 [M-2H] 2-
- Example 10 5′-O- (4,4′-Dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl deoxyguanosine 3 '- [O- (2- cyanoethyl)] Hosuhorochioniru -N 4 - benzoyl deoxycytidine 3' - [O- (2- cyanoethyl)] Ho
- Example 11 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- Acetonitrile 200 mL was added to the reaction solution to precipitate a mixture of oligonucleic acid dimer and 2,4,6-tris (octadecyloxy) benzyl acetate. After filtering and washing the suspension, 3.24 g of a mixture of the target nucleic acid dimer and 3,4,6-tris (octadecyloxy) benzyl acetate (of which 1.80 g of nucleic acid, 0.943 mmol, 96.9% yield) Rate). After dissolving the sample in tetrahydrofuran, the cyanoethyl protecting group on phosphoric acid was removed with DBU, and the target product was identified by LC / MS. LC / ESI TOF MS: calcd 1858.13, found 1858.13 [MH] -
- Acetonitrile 200 mL was added to the reaction solution to precipitate a mixture of oligonucleic acid trimer and 2,4,6-tris (octadecyloxy) benzyl acetate. After filtering and washing the suspension, 3.59 g of a mixture of the desired nucleic acid trimer and 3,4,6-tris (octadecyloxy) benzyl acetate (of which 1.20 g of nucleic acid, 0.916 mmol, 97.1% yield) Rate). After dissolving the sample in tetrahydrofuran, the cyanoethyl protecting group on phosphoric acid was removed with DBU, and the target product was identified by LC / MS. LC / MS m / z: calcd 1145.09, found 1145.09 [M-2H] 2-
- DMTr-d [A Bz TT] -suc-TOB (2.49 g, 1.00 mol) and 5-methoxyindole obtained in (2) were dissolved in dehydrated dichloromethane (50 mL). To this was added trifluoroacetic acid (76.5 ⁇ L, 1.00 mmol), and the mixture was stirred at room temperature for 2 hours. Completion of dedimethoxytritylation was confirmed by LC / TOF MS and 2,4,6-trimethylpyridine (265 ⁇ L, 2.00 mmol) was added. Acetonitrile (250 mL) was added to precipitate the target product.
- nucleoside amidites were sequentially extended. The following were used as nucleoside amidites.
- Example 12 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- Example 13 5 ′-(4,4′-dimethoxytrityl) -3′-tert-butyldimethylsilyl-4- [3,4,5-tris (octadecyloxy) benzoyl] deoxythymidine (DMTr-dT CO— Synthesis of TOP- TBDMS) (1) Synthesis of 5 ′-(4,4′-dimethoxytrityl) -3′-tert-butyldimethylsilyldeoxythymidine (DMTr-dT-TBDMS)
- DMTr-dT-TBMDS (3.28 g, 4.98 mmol) obtained in (1) was dissolved in dehydrated pyridine (45 mL).
- 3,4,5-Tris (octadecyloxy) benzoic acid chloride (6.91 g, 7.3 mmol) and diisopropylethylamine (3.9 mL, 22.4 mmol) were added thereto and stirred at 65 ° C. for 6 hours. .
- dichloromethane 100 mL was added, washed with a saturated aqueous sodium hydrogen carbonate solution (100 mL), water (50 mL), and brine (50 mL), and dehydrated over anhydrous sodium sulfate.
- Example 14 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- Example 15 DMTr-d [TC Bz C Bz C Bz G iBu C Bz C Bz TG iBu T CO-TOP ] -TBDMS desilylation (1) Under the same fluoride ion concentration and temperature rising conditions as in Example 10 Desilylation of
- the solid was obtained by filtration through a Kiriyama funnel, washed with acetonitrile, and then vacuum dried to obtain 0.216 g of a white solid (of which 0.168 g, 0.310 mmol, 62.8% yield of oligonucleic acid).
- the cyanoethyl protecting group on phosphoric acid was removed with DBU, and the target product was identified by LC / MS.
- a short-chain peak was observed in the subsequent retention time of the target compound, and the area ratio of the target compound peak was 62.0%.
- Example 16 5'-O- (4,4'- dimethoxytrityl) -N 4 - [3,4,5-tris (octadecyloxy) benzyl] succinyl-3'-O-(tert-butyldimethylsilyl) Synthesis of deoxycytidine (DMTr-dC suc-TOB- TBDMS)
- Example 17 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl-deoxyguanosine 3 ′-[O- (2 - cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyloxy) benzyl] succinyl-3'-tert-butyldimethylsilyl-deoxycytidine (DMTr-d [TG iBu C suc-TOB] -TBDMS) synthesis (1) 5'-O- (4,4'- dimethoxytrityl) -N 2 - isobutyryl - deoxyguanosine 3 '- [O- (2- cyanoethyl)] Hosuhorochioniru -N 4 -
- Example 16 Under an argon atmosphere, DMTr-dC suc-TOB- TBDMS (820 mg, 0.500 mmol) obtained in Example 16 was dissolved in dehydrated dichloromethane (23 mL), 5-methoxyindole (1.47 g, 10.0 mmol), Fluoroacetic acid (55.7 ⁇ L, 0.750 mmol) was added and stirred at room temperature for 30 minutes, and the completion of the reaction was confirmed by UPLC.
- reaction mixture was neutralized with 2,4,6, -trimethylpyridine and then 5′-O- (4,4′-dimethoxytrityl) -N 2 -isobutyryl-deoxyguanosine-3 ′-[O— (2 -Cyanoethyl) (N, N-diisopropyl)] phosphoramidite (1.88 g, 2.24 mmol), 5- (benzylthio) -1H-tetrazole (0.432 g, 2.25 mmol) in acetonitrile (3.00 mL) was added and stirred at room temperature for 2 hours.
- reaction mixture was neutralized by adding 2,4,6, -trimethylpyridine (325 ⁇ L, 2.45 mmol), and then 5′-O- (4,4′-dimethoxytrityl) -thymidine-3 ′-[ O- (2-cyanoethyl) (N, N-diisopropyl)] phosphoramidite (1.02 g, 1.37 mmol), 5- (benzylthio) -1H-tetrazole 0.264 g (1.37 mmol) in acetonitrile (3. 00 mL) solution was added and stirred at room temperature for 2 hours.
- the reaction solution was filtered using a Kiriyama funnel, then washed with acetonitrile, and the target DMTr-d [TG iBu C suc-TOB ] -TBDMS (1.05 g, 0.419 mmol, 92%) was obtained as a white solid by vacuum drying. Got as. After dissolving the sample in tetrahydrofuran, the cyanoethyl protecting group on phosphoric acid was removed with DBU, and the target product was identified by LC / MS. LC / MS m / z: calcd 1185.64, found 1185.64 [M-2H] 2-
- Example 18-1 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl-deoxyguanosine 3 ′-[O— (2-cyanoethyl)] Hosuhorochioniru -N 4 - synthesis of [3,4,5-tris (octadecyloxy) benzyl] succinyl deoxycytidine (DMTr-d [TG iBu C suc-TOB] -H) ( desilylation)
- Example 18-2 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxyguanosine-3 ′-[ O-(2-cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyloxy) benzyl] succinyl deoxycytidine-3'-yl - [O-(2-cyanoethyl)] - N, N- Similarly diisopropyl phosphoramidite (DMTr-d [TG iBu C suc-TOB] -PA) DMTr-d [TG iBu C suc-TOB] example 6- (5) obtained in synthesis example 18-1 To obtain DMTr-d [TG iBu C suc-TOB ] -PA.
- Example 19 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxyguanosine-3 ′-[O— (2-cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyloxy) benzyl] succinyl deoxycytidine-3'-yl - [O-(2-cyanoethyl)] - N, N-diisopropyl phosphonate Loamidite (DMTr-d [TG iBu C suc-TOB ] -PA) and N 4 -benzoyldeoxyadenosine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-deoxythymidine 3 ′-[O- (2
- Example 20 6-mer oligonucleic acid DMTr-d [TG iBu C suc-TOB A Bz TT] -suc-TOB and DMTr-d [TG iBu C CO-TOP A Bz TT] -suc- produced by fragment condensation Comparison of deprotection of TOB in ammonia water
- Example 21 Diamidite activation and phosphitylation with 4,5-dichloroimidazole (1) pKa determination by titration of 4,5-dichloroimidazole 4,5-dichloroimidazole (0.2396 g, 1.749 mmol) It melt
- Example 22 5′-O- (4,4′-dimethoxytrityl) -deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryldeoxyguanosine 3 ′-[O— ( 2-cyanoethyl)] Hosuhorochioniru -N 4 - [3,4,5-tris (octadecyl) benzoyl] -3'-isopropoxyphenyl diisopropylsilyl deoxycytidine (DMTr-d [TG iBu C CO-TOP] -IPODIPS) Synthesis (1) Synthesis of Isopropoxydiisopropylsilane Imidazole (3.27 g, 48.1 mmol) was dissolved in dry N, N-dimethylformamide (40 mL) under a dry argon atmosphere.
- Example 23 Comparison of deprotection efficiency of 3′-silyl group (1) Desilylation of DMTr-d [TG iBu C CO-TOP ] -IPODIPS DMTr-d [TG iBu C CO— obtained in Example 22 TOP ] -IPODIPS (0.4915 g, 0.200 mmol) was dissolved in 6.0 ml of dichloromethane. To this was added a solution of 3HF-TEA (146 mg, 0.906 mmol) and pyridine (146 ⁇ L, 1.80 mmol) in THF (12 mL), and the mixture was kept at 15 ° C. The reaction solution was sampled and analyzed by HPLC, and the progress of 3′-desilylation was followed. The half-life of the 3′-silyl compound was 30 minutes, and the 3′-silyl compound was below the detection limit in 8 hours. became.
- 3HF-TEA 146 mg, 0.906 mmol
- pyridine 146 ⁇ L, 1.80 mmol
- Example 24 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- the title compound was obtained as a white solid by subjecting 3,4,5-tris (octadecyloxy) benzoic acid to the same condensation conditions as in Example 1- (2) without purification of the product (3,4,5). (Yield 43% based on the amount of 5-tris (octadecyloxy) benzoic acid used).
- Example 25 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- Example 26 5′-O- (4,4′-dimethoxytrityl) deoxythymidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl) )] Phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3 ′-[O- (2-cyanoethyl)] phosphorothionyl-N 2 -isobutyryl Deoxyguanosine 3 '-[O- (2-cyanoethyl)] phosphorothionyl-N 4 -benzoyldeoxycytidine 3'-[O- (2-cyanoethyl)] phosphorothionyl
- dA dma 5 ′-(4,4′-dimethoxytrityl) -N 4 -dimethylaminoethylidene-2′-deoxyadenosine-3 ′-[(2-cyanoethyl) -N, N-diisopropyl] phosphoramidite dC HD : 5 '- (4,4'-dimethoxytrityl) -N 4 - (2-hexyldecanoyl) -2'-deoxycytidine-3' - [(2-cyanoethyl) -N, N-diisopropyl] phosphoramidite dG iBu : 5 ′-(4,4′-dimethoxytrityl) -N 2 -isobutyryl-2′-deoxyguanosine-3 ′-[(2-cyanoethyl) -N, N-diisopropyl] phosphoramidite dT
- Example 27 5'-O- (4,4'-dimethoxytrityl) deoxythymidine 3'-phosphorothionyldeoxycytidine 3'-phosphorothionyldeoxycytidine 3'-phosphorothionyl by 10 + 10 fragment condensation and deprotection Deoxycytidine 3'-phosphorothionyldeoxyguanosine 3'-phosphorothionyldeoxycytidine 3'-phosphorothionyldeoxycytidine 3'-phosphorothionyldeoxythymidine 3'-phosphorothionyldeoxyguanosine 3'-phosphorothionyldeoxycytidine 3'-phosphorothionyldeoxyguanosine 3 '-phosphorothionyldeoxycytidine 3'-phosphorothionyldeoxyguanosine 3 '-[phosphorothionyldeoxyadenosine 3'-[
- H-d [G iBu A dma C HD A dma TG iBu C HD A dma TT] the -suc-TOB at a content 82 wt% 3,4,5-tris (octadecyloxy) benzoic acid was 10.6mg accurately weighed unreacted coupling mixture in sample solid similarly deprotected, diluted to admixture with methyl as a standard H-d [G iBu a dma C HD a dma TG iBu C HD a dma TT] -suc-TOB quantitative determination was actually used, a mixture of crystallization aid (82 w / w%) as 137mg equivalent of H-d [G iBu a dma C HD a dma TG iBu C HD a dma TT] -suc and it contains -TOB, H-d by condensation reactions [G iBu a
- Example 6 a phosphitylating agent solution prepared from 2-cyanoethyl-N, N, N ′, N′-tetraisopropyl phosphorodiamidite and 4,5-dicyanoimidazole in dehydrated toluene (1.5 mL as the filtrate, 0.60 mmol) was added N-methylmorpholine (0.66 mL, 6.0 mmol). Hd [C Ac A Bz TT] -suc-TOB (0.496 g, 0.20 mmol) was added thereto at 10 ° C. to obtain a suspension. Dichloromethane (10.0 mL) was added at the same temperature to make a homogeneous solution, and then stirred for 2 hours.
- Hd [C Ac A Bz TT] -suc-TOB is obtained by condensing d [A Bz TT] -suc-TOB obtained in the same manner as in Preparation Example 1- (3) with a nucleoside phosphoramidite and further removing it. Prepared by DMTr.
- the nucleoside phosphoramidites used are as follows.
- the method of the present invention it is possible to prepare a phosphitylating agent from a stable diamidite during storage, and it is possible to suppress the removal of the protecting group on phosphoric acid during the phosphitylation reaction. Therefore, the 3′-terminal phosphoramidite-modified oligonucleotide can be produced more stably and efficiently. Furthermore, according to the method of the present invention, the silyl protecting group at the 3 ′ end can be removed without removing the cyanoethyl protecting group on the internucleotide phosphate. Therefore, by using these methods, more efficient oligonucleotide production and fragment condensation using it are possible.
- This application is based on a patent application No. 2015-250665 filed in Japan (filing date: December 22, 2015), the contents of which are incorporated in full herein.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
Abstract
本発明はより安定で、効率的なオリゴヌクレオチド、特に、3'末端に種々の官能基を連結してなるオリゴヌクレオチドを製造する方法等を提供することを課題とする。3'末端がシリル保護基によって保護されたオリゴ核酸を、シリル基以外の保護基に影響を与えない脱シリル条件にて、3'末端選択的に脱保護する。これをホスホロジアミダイト試薬を用いて、オリゴ核酸上の保護基に影響を与えないホスフィチル化条件に付し、式(Ia-1)(各記号の定義は明細書中に記載の通り)で表される3'末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3'末端ホスホロアミダイト化オリゴヌクレオチドの3'末端に、直接又はリンカーを介して官能基を連結させる工程を含む、式(Ia-2)(各記号の定義は明細書中に記載の通り)で表される3'末端に官能基を有するオリゴヌクレオチドの製造方法、等により効率的なオリゴヌクレオチドの製造が可能となる。
Description
本発明は、オリゴヌクレオチドの製造方法に関する。より詳細には、シリル保護基を利用したオリゴヌクレオチドの製造方法及びホスフィチル化剤を利用したオリゴヌクレオチドの製造方法、並びにフラグメント縮合によるオリゴヌクレオチドの製造方法等に関する。
オリゴヌクレオチドの合成方法には、リン酸トリエステル法、H-ホスホネート法、ホスホロアミダイト法などがあり、現在ではホスホロアミダイト法を用いた固相合成(固相法)が最も汎用されている(非特許文献1)。数個のヌクレオチドからなるオリゴヌクレオチドは、原料となるヌクレオチドを逐次連結していくことで合成可能であるが、20mer程度以上のオリゴヌクレオチドを合成する場合には、2~3ヌクレオチドのビルディングブロック群を予め調製しておき、これの連結を繰り返すことで所望の鎖長の生成物を得るブロックマー合成法や10塩基程度以上のオリゴヌクレオチド間の連結でオリゴヌクレオチドを得るユニットカップリング(フラグメント縮合)合成法等も利用されている。このフラグメント縮合の手法は、溶解性の確保や、反応部位の選択的な脱保護・活性化等に特段のケアを必要とする。
特許文献2の実施例、非特許文献2においては、オリゴ核酸フラグメントの3’-水酸基の選択的脱保護可能な保護基としてレブリニルエステルを採用して、ホスホロアミダイト法にて伸長を行い、インターヌクレオチドのリン酸エステルがシアノエチル基で保護されたオリゴマーを取得している。この脱保護には、安全性に問題があるヒドラジンを用いる必要がある。また、脱保護条件において塩基部保護基の脱落が進行しうることが非特許文献2で述べられている。
非特許文献3には、3-merアミダイトを用いてオリゴ核酸を合成するブロックマー合成法が記載されているが、反応過程でリン酸エステルの開裂や保護基であるシアノエチルの脱離という問題があった。特許文献1は、脱離しにくい保護基が使用されているが、該保護基は特殊なものであり、従来の反応条件等をそのまま利用することができない。
非特許文献4にはトリエステル法を用いたフラグメント縮合によるオリゴヌクレオチドの合成法が記載されているが、トリエステル法は現在殆ど実用されていない。
一方、ホスフィチル化剤を用いて調製した3’末端ホスホロアミダイト化オリゴヌクレオチドを用いたより長鎖のオリゴヌクレオチドを合成する手法が知られている。
ヌクレオシドのホスフィチル化には、2-シアノエチル-N,N-ジイソプロピルクロロホスホロアミダイトが常用されてきた。特許文献2においてはこれと、N-メチルイミダゾールとを組み合わせることで、オリゴ核酸の脱保護された3’-末端のホスフィチル化を実施している。しかし、2-シアノエチル-N,N-ジイソプロピルクロロホスホロアミダイトは保存安定性に劣る上、市価も高い。
上述の試剤より安価かつ安定な代替として、2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトが用いられる(非特許文献5)。同剤それ自体の反応性は乏しく、ヌクレオシドのホスフィチル化に際しては活性化剤が必要となる。脆弱な保護基を持たないヌクレオシドモノマーのホスフィチル化においては、常法としてジイソプロピルアンモニウムテトラゾリドが用いられる。しかしながら、オリゴ核酸フラグメントの3’-水酸基のシアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトによるホスフィチル化において上述のアンモニウム塩を用いるとインターヌクレオチドのリン酸部上の、塩基に対して脆弱なシアノエチル基が顕著に脱落することが知られている(非特許文献3)。また、トリフルオロメタンスルホン酸を始めとする強酸と塩基の塩を活性化剤とした場合、過剰活性によりホスファイトや五価のリン化合物が副生することが報告されている(非特許文献6及び7)。また、オリゴヌクレオチドの合成過程で、2’-位の水酸基の保護基としてシリル保護基が用いられるが、その脱保護、即ち脱シリル化には、フッ化水素:トリエチルアミン=3:1の塩(3HF-TEA)が一般に用いられている(非特許文献8)。3HF-TEAは酸性の塩であり、オリゴ核酸合成の5’末端保護基として常用される4,4’-ジメトキシトリエチルアミンは酸性で脱離が亢進される。ジメトキシトリチル基の好ましくない脱落を防止するため、非特許文献9では、3’末端のシリル保護基(乃至リンカー)の開裂に3HF-TEAにさらにトリエチルアミンを追加して酸:アミン=1:1のモル比の塩が使用されている。また、リン酸部位がシアノエチル保護されたオリゴ核酸の3’末端の脱シリル化にトリエチルアミンを用いた場合、シアノエチル保護基が脱落する等の問題点があった。
特許文献2の実施例、非特許文献2においては、オリゴ核酸フラグメントの3’-水酸基の選択的脱保護可能な保護基としてレブリニルエステルを採用して、ホスホロアミダイト法にて伸長を行い、インターヌクレオチドのリン酸エステルがシアノエチル基で保護されたオリゴマーを取得している。この脱保護には、安全性に問題があるヒドラジンを用いる必要がある。また、脱保護条件において塩基部保護基の脱落が進行しうることが非特許文献2で述べられている。
非特許文献3には、3-merアミダイトを用いてオリゴ核酸を合成するブロックマー合成法が記載されているが、反応過程でリン酸エステルの開裂や保護基であるシアノエチルの脱離という問題があった。特許文献1は、脱離しにくい保護基が使用されているが、該保護基は特殊なものであり、従来の反応条件等をそのまま利用することができない。
非特許文献4にはトリエステル法を用いたフラグメント縮合によるオリゴヌクレオチドの合成法が記載されているが、トリエステル法は現在殆ど実用されていない。
一方、ホスフィチル化剤を用いて調製した3’末端ホスホロアミダイト化オリゴヌクレオチドを用いたより長鎖のオリゴヌクレオチドを合成する手法が知られている。
ヌクレオシドのホスフィチル化には、2-シアノエチル-N,N-ジイソプロピルクロロホスホロアミダイトが常用されてきた。特許文献2においてはこれと、N-メチルイミダゾールとを組み合わせることで、オリゴ核酸の脱保護された3’-末端のホスフィチル化を実施している。しかし、2-シアノエチル-N,N-ジイソプロピルクロロホスホロアミダイトは保存安定性に劣る上、市価も高い。
上述の試剤より安価かつ安定な代替として、2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトが用いられる(非特許文献5)。同剤それ自体の反応性は乏しく、ヌクレオシドのホスフィチル化に際しては活性化剤が必要となる。脆弱な保護基を持たないヌクレオシドモノマーのホスフィチル化においては、常法としてジイソプロピルアンモニウムテトラゾリドが用いられる。しかしながら、オリゴ核酸フラグメントの3’-水酸基のシアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトによるホスフィチル化において上述のアンモニウム塩を用いるとインターヌクレオチドのリン酸部上の、塩基に対して脆弱なシアノエチル基が顕著に脱落することが知られている(非特許文献3)。また、トリフルオロメタンスルホン酸を始めとする強酸と塩基の塩を活性化剤とした場合、過剰活性によりホスファイトや五価のリン化合物が副生することが報告されている(非特許文献6及び7)。また、オリゴヌクレオチドの合成過程で、2’-位の水酸基の保護基としてシリル保護基が用いられるが、その脱保護、即ち脱シリル化には、フッ化水素:トリエチルアミン=3:1の塩(3HF-TEA)が一般に用いられている(非特許文献8)。3HF-TEAは酸性の塩であり、オリゴ核酸合成の5’末端保護基として常用される4,4’-ジメトキシトリエチルアミンは酸性で脱離が亢進される。ジメトキシトリチル基の好ましくない脱落を防止するため、非特許文献9では、3’末端のシリル保護基(乃至リンカー)の開裂に3HF-TEAにさらにトリエチルアミンを追加して酸:アミン=1:1のモル比の塩が使用されている。また、リン酸部位がシアノエチル保護されたオリゴ核酸の3’末端の脱シリル化にトリエチルアミンを用いた場合、シアノエチル保護基が脱落する等の問題点があった。
S. L. Beaucage et al., Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons (2000)
Chen, C.-H. et al., Aust. J. Chem. 2010, 63, 227-235
Muller, S. et al., Org. Biomol. Chem. 2012, 10, 1510-1513
Chekhmakhcheva, O.G. et al., A Nucleic Acid Res. 1983, 11, 8369-8387
Gukathasan, R. et al., J. Organomet. Chem. 2005, 690, 2603-2607
Yogesh S. Sanghvi et al., Org. Proc. Res. Dev. 2000, 4, 175-181
Xie, C. et al., Org. Proc. Res. Dev. 2005, 9, 730-737
Gasparutto D. et al., Nucleic Acid Res. 1992, 20(19), 5159-5166
Ohkubo, A. et al., Bioorg. Med. Chem. 2008, 16, 5345-5351
本発明の課題は、より保存安定性に優れたホスフィチル化剤を用い、ホスフィチル化剤の過剰活性化を抑え、且つリン酸上の保護基の脱落が抑えられたホスフィチル化反応により3’末端ホスホロアミダイト化オリゴヌレオチドを製造する方法、シアノエチル保護基の脱落を伴わずに脱シリル化を可能にしてオリゴヌクレオチドを製造する方法、及びそれらの方法を用いて、より効率よくオリゴヌクレオチドを製造する方法、3’末端に種々の官能基を連結してなるオリゴヌクレオチドを製造する方法等を提供することである。
本発明者らは鋭意検討の結果、ホスフィチル化剤として、ジアミダイトを利用し、特定のホスフィチル化剤の活性化剤及び活性化方法を実施すること、特定の塩基の存在下でホスフィチル化を実施することにより、上記課題が解決できることを見出した。さらに、本発明者らは、鋭意検討の結果、脱シアノエチル化を抑制し得る特定のシリル保護基とその脱保護条件を見出した。
本発明は以下を含む。
[1]溶媒中で5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)に1又は2種類以上の有機塩基の存在下、フッ化物イオン源を作用させる工程を含む、5’位水酸基又は5’位リン酸基が保護され、3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチドを製造する方法であって、該1又は2種類以上の有機塩基のうち少なくとも1種とフッ化水素との塩を該フッ化物イオン源とすることを特徴とする、方法。
[2]5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチドが、下記構造(I)を有する、上記[1]記載の方法。
本発明は以下を含む。
[1]溶媒中で5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)に1又は2種類以上の有機塩基の存在下、フッ化物イオン源を作用させる工程を含む、5’位水酸基又は5’位リン酸基が保護され、3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチドを製造する方法であって、該1又は2種類以上の有機塩基のうち少なくとも1種とフッ化水素との塩を該フッ化物イオン源とすることを特徴とする、方法。
[2]5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチドが、下記構造(I)を有する、上記[1]記載の方法。
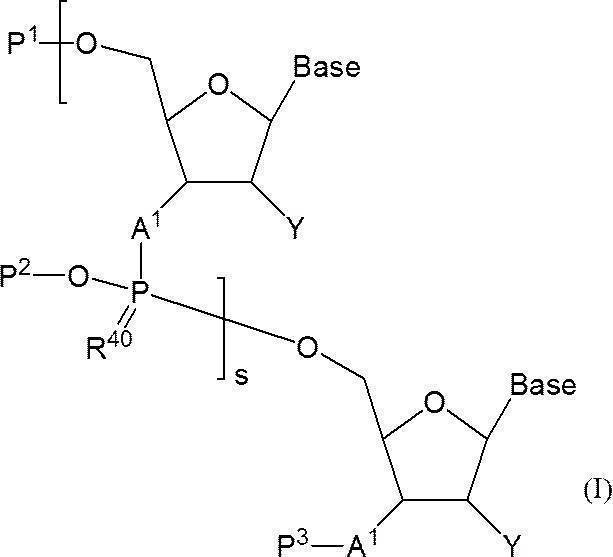
[式中、
s+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
s+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
s個のP2は、それぞれ独立してリン酸基の保護基を示し、
s個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
s+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
sは、1以上の任意の整数を示す。]
[3]フッ化物イオン源に対し1モル当量以上の有機塩基を使用する、上記[1]又は[2]に記載の方法。
[4]有機塩基が強塩基と弱塩基との混合物、2種類以上の弱塩基の混合物又は単一の弱塩基である、上記[1]~[3]のいずれかに記載の方法。
[5]強塩基がpKa≧8であり、弱塩基が4≦pKa<8である、上記[4]に記載の方法。
[6]有機塩基として強塩基を1/3モル当量以下で使用する、上記[1]~[5]のいずれかに記載の方法。
[7]有機塩基として、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、プロピルアミン、イソプロピルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、ブチルアミン、イソブチルアミン、tert-ブチルアミン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、モルホリンから選択される1種以上を使用する、上記[1]~[6]のいずれかに記載の方法。
[8]有機塩基として、ピリジン、2,6-ジメチルピリジン、2,4,6-トリメチルピリジン、ピペラジン、ピペリジン、イミダゾール、N-メチルイミダゾール、N-メチルモルホリン、N-エチルモルホリン、アニリン、トルイジン、ジメチルアニリン、エチルアニリン、ジエチルアニリン、エチルメチルアニリン、アニシジンから選択される1種以上を使用する、上記[1]~[7]のいずれかに記載の方法。
[9]有機塩基として、トリエチルアミンとピリジンとの混合物を使用する、上記[1]~[8]のいずれかに記載の方法。
[10]フッ化物イオン源が、トリエチルアミン五フッ化水素酸塩、トリエチルアミン三フッ化水素酸塩及びピリジンフッ化水素酸塩から選択される1種以上である上記[1]~[9]のいずれかに記載の方法。
s+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
s+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
s個のP2は、それぞれ独立してリン酸基の保護基を示し、
s個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
s+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
sは、1以上の任意の整数を示す。]
[3]フッ化物イオン源に対し1モル当量以上の有機塩基を使用する、上記[1]又は[2]に記載の方法。
[4]有機塩基が強塩基と弱塩基との混合物、2種類以上の弱塩基の混合物又は単一の弱塩基である、上記[1]~[3]のいずれかに記載の方法。
[5]強塩基がpKa≧8であり、弱塩基が4≦pKa<8である、上記[4]に記載の方法。
[6]有機塩基として強塩基を1/3モル当量以下で使用する、上記[1]~[5]のいずれかに記載の方法。
[7]有機塩基として、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、プロピルアミン、イソプロピルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、ブチルアミン、イソブチルアミン、tert-ブチルアミン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、モルホリンから選択される1種以上を使用する、上記[1]~[6]のいずれかに記載の方法。
[8]有機塩基として、ピリジン、2,6-ジメチルピリジン、2,4,6-トリメチルピリジン、ピペラジン、ピペリジン、イミダゾール、N-メチルイミダゾール、N-メチルモルホリン、N-エチルモルホリン、アニリン、トルイジン、ジメチルアニリン、エチルアニリン、ジエチルアニリン、エチルメチルアニリン、アニシジンから選択される1種以上を使用する、上記[1]~[7]のいずれかに記載の方法。
[9]有機塩基として、トリエチルアミンとピリジンとの混合物を使用する、上記[1]~[8]のいずれかに記載の方法。
[10]フッ化物イオン源が、トリエチルアミン五フッ化水素酸塩、トリエチルアミン三フッ化水素酸塩及びピリジンフッ化水素酸塩から選択される1種以上である上記[1]~[9]のいずれかに記載の方法。
[11]シリル保護基が、ケイ素上の3つの置換基がアルキル基、アリール基、アラルキル基、アルコキシ基、アリールオキシ基、アラルキルオキシ基、アルキルチオ基、アリールチオ基から選択され、且つそのうちの少なくとも1つがアリール基、アルコキシ基、アリールオキシ基、アラルキルオキシ基から選択されるシリル保護基である、上記[1]~[10]のいずれかに記載の方法。
[12]s+1個のBaseの少なくとも一つが、式:-L-YL-Z
[式中、
Lは、式(a1):
[12]s+1個のBaseの少なくとも一つが、式:-L-YL-Z
[式中、
Lは、式(a1):

(式中、**は、核酸との結合位置を示し;
*は、YLとの結合位置を示し;
L1は、置換されていてもよい2価のC1-22炭化水素基、酸素原子、-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)を示し;かつ
L2は、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3a)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、CRcRdとの結合位置を示し、R1は、置換されていてもよいC1-22アルキレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくは置換されていてもよいC1-22アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-22アルキレン結合を形成していてもよく、R3bは水素原子若しくは置換されていても良いC1-22アルキル基を示す)で示される基を示し;
RcおよびRdはそれぞれ独立して水素原子、置換されていてもよいC1-22アルキル基もしくはRcおよびRdが一緒になって、単一のカルボニル基を形成していてもよい)で表される基(リンカー)を示し;
YLは、単結合、酸素原子、または-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)、硫黄原子を示し;ならびに
Zは式(a2):
*は、YLとの結合位置を示し;
L1は、置換されていてもよい2価のC1-22炭化水素基、酸素原子、-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)を示し;かつ
L2は、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3a)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、CRcRdとの結合位置を示し、R1は、置換されていてもよいC1-22アルキレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくは置換されていてもよいC1-22アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-22アルキレン結合を形成していてもよく、R3bは水素原子若しくは置換されていても良いC1-22アルキル基を示す)で示される基を示し;
RcおよびRdはそれぞれ独立して水素原子、置換されていてもよいC1-22アルキル基もしくはRcおよびRdが一緒になって、単一のカルボニル基を形成していてもよい)で表される基(リンカー)を示し;
YLは、単結合、酸素原子、または-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)、硫黄原子を示し;ならびに
Zは式(a2):

(式中、*は、YLとの結合位置を示し;
R4は、水素原子を示すか、あるいはRbが、下記式(a3)で表される基である場合には、R6と一緒になって単結合または-O-を示して、環Bと共にフルオレニル基またはキサンテニル基を形成していてもよく;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
kは、1~4の整数を示し;
環Aは、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
Raは、水素原子、またはハロゲン原子により置換されていてもよいフェニル基を示し;ならびに
Rbは、水素原子、または式(a3):
R4は、水素原子を示すか、あるいはRbが、下記式(a3)で表される基である場合には、R6と一緒になって単結合または-O-を示して、環Bと共にフルオレニル基またはキサンテニル基を形成していてもよく;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
kは、1~4の整数を示し;
環Aは、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
Raは、水素原子、またはハロゲン原子により置換されていてもよいフェニル基を示し;ならびに
Rbは、水素原子、または式(a3):

(式中、*は結合位置を示し;
jは、0~4の整数を示し;
j個のR7は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
R6は、水素原子を示すか、またはR4と一緒になって単結合または-O-を示して、環Aと共にフルオレニル基またはキサンテニル基を形成していてもよく;かつ
環Bは、j個のOR7に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
RaおよびRbが一緒になって、単一のカルボニル基を形成していてもよい。)で表される基);
式(a2’):
jは、0~4の整数を示し;
j個のR7は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
R6は、水素原子を示すか、またはR4と一緒になって単結合または-O-を示して、環Aと共にフルオレニル基またはキサンテニル基を形成していてもよく;かつ
環Bは、j個のOR7に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
RaおよびRbが一緒になって、単一のカルボニル基を形成していてもよい。)で表される基);
式(a2’):
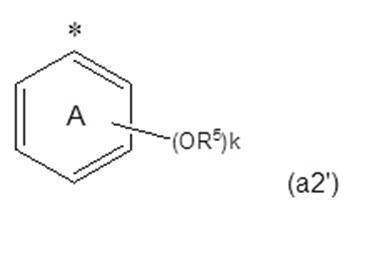
(式中、*は、YLとの結合位置を示し;
それ以外の各記号の定義は式(a2)と同義である。)で示される基
または、
式(a2’’):
それ以外の各記号の定義は式(a2)と同義である。)で示される基
または、
式(a2’’):
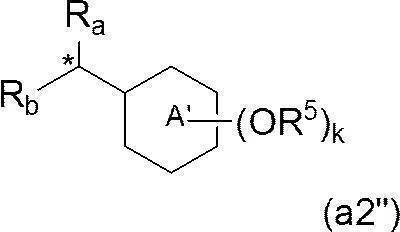
(式中、*は、YLとの結合位置を示し;
環A’は、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
各記号の定義は式(a2)と同義である。)
で表される基を示す]で表される保護基、若しくは式:-Z’
環A’は、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
各記号の定義は式(a2)と同義である。)
で表される基を示す]で表される保護基、若しくは式:-Z’

[式中、**は、核酸との結合位置を示し;
各記号の定義は式(a2’’)と同義である。]
で表される保護基で保護されている、上記[1]~[11]のいずれかに記載の方法。
[13]Lがスクシニル基である、上記[12]に記載の方法。
[14]R5及び/又はR7が炭素原子数10~40のアルキル基である、上記[12]又は[13]に記載の方法。
[15]R5がオクタデシル基である、上記[12]又は[13]に記載の方法。
[16]3’末端のヌクレオシドのBaseが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[1]~[15]のいずれか1項に記載の方法。
[17]以下の工程(1)及び(2)を含有する、ホスホロアミダイト化オリゴヌクレオチドの製造方法。
(1)溶媒中で、下記式(1):
各記号の定義は式(a2’’)と同義である。]
で表される保護基で保護されている、上記[1]~[11]のいずれかに記載の方法。
[13]Lがスクシニル基である、上記[12]に記載の方法。
[14]R5及び/又はR7が炭素原子数10~40のアルキル基である、上記[12]又は[13]に記載の方法。
[15]R5がオクタデシル基である、上記[12]又は[13]に記載の方法。
[16]3’末端のヌクレオシドのBaseが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[1]~[15]のいずれか1項に記載の方法。
[17]以下の工程(1)及び(2)を含有する、ホスホロアミダイト化オリゴヌクレオチドの製造方法。
(1)溶媒中で、下記式(1):

[式中、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R20およびR30はそれぞれ独立してアルキル基を示し、該アルキル基は隣接する窒素原子と一緒になって環を形成してもよい。]
で表されるホスフィチル化剤前駆体に活性化剤を作用させて下記式(2):
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R20およびR30はそれぞれ独立してアルキル基を示し、該アルキル基は隣接する窒素原子と一緒になって環を形成してもよい。]
で表されるホスフィチル化剤前駆体に活性化剤を作用させて下記式(2):

[式中、
Zaは活性化剤に由来する基を示し、
それ以外の各記号は、前記と同義である。]
で表されるホスフィチル化剤を調製する工程、および
(2)溶媒中で、5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程
[18]該n個重合オリゴヌクレオチドが、下記構造(Ia)を有する、上記[17]記載の方法。
Zaは活性化剤に由来する基を示し、
それ以外の各記号は、前記と同義である。]
で表されるホスフィチル化剤を調製する工程、および
(2)溶媒中で、5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程
[18]該n個重合オリゴヌクレオチドが、下記構造(Ia)を有する、上記[17]記載の方法。
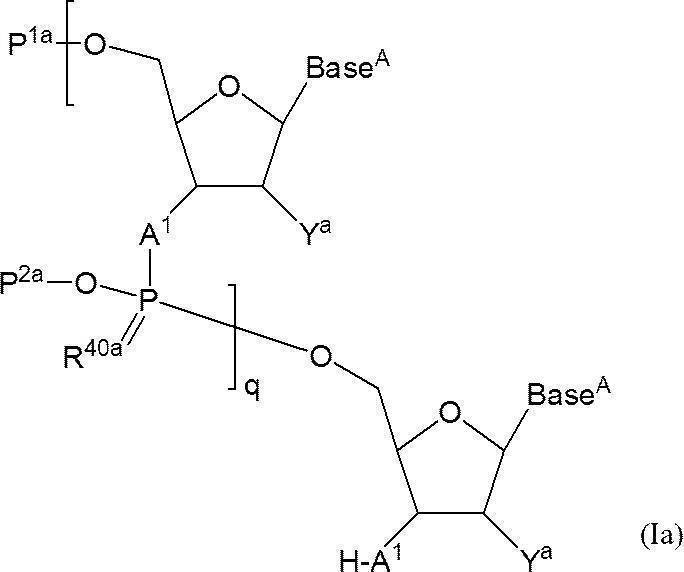
[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
[19]該n個重合オリゴヌクレオチドが、下記構造(Ia’)を有する、上記[17]記載の方法。
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
[19]該n個重合オリゴヌクレオチドが、下記構造(Ia’)を有する、上記[17]記載の方法。

[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
[20]q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[17]~[19]のいずれかに記載の方法。
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
[20]q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[17]~[19]のいずれかに記載の方法。
[21]工程(1)で用いる活性化剤が、pKa5以上の弱酸性活性化剤である、上記[17]~[20]のいずれかに記載の方法。
[22]工程(1)で用いる活性化剤が、アゾール系化合物である、上記[17]~[21]のいずれかに記載の方法。
[23]工程(1)で用いる活性化剤が、ジシアノイミダゾール又はジクロロイミダゾールである、上記[17]~[22]のいずれかに記載の方法。
[24]工程(1)で用いる活性化剤が、ホスフィチル化剤前駆体に対して1.5~20モル当量で用いられる、上記[17]~[23]のいずれかに記載の方法。
[25]工程(1)で用いる溶媒が、ホスフィチル化剤前駆体を溶解し、かつ活性化剤が難溶性となるものであって、酸性または塩基性官能基を有さないことを特徴とする、上記[17]~[24]のいずれかに記載の方法。
[26]工程(1)で用いる溶媒が、トルエン、ベンゼン、o-キシレン、m-キシレン、p-キシレン、エチルベンゼン、ペンタン、ヘキサン、ヘプタン、オクタン、シクロペンタン、シクロヘキサン、ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、シクロペンチルメチルエーテル、四塩化炭素から選択される1種以上の溶媒である、上記[17]~[25]のいずれかに記載の方法。
[27]工程(1)で用いる溶媒が、トルエンである、上記[17]~[26]のいずれかに記載の方法。
[28]工程(2)で用いる塩基が、pKa5~8の塩基である、上記[17]~[27]のいずれかに記載の方法。
[29]工程(2)で用いる塩基が、コリジン、N-メチルモルホリン及びジエチルアニリンから選択される1種以上の塩基である、上記[17]~[28]のいずれかに記載の方法。
[30]工程(1)と工程(2)の間に、不溶物を分離する工程を含む、上記[17]~[29]のいずれかに記載の方法。
[22]工程(1)で用いる活性化剤が、アゾール系化合物である、上記[17]~[21]のいずれかに記載の方法。
[23]工程(1)で用いる活性化剤が、ジシアノイミダゾール又はジクロロイミダゾールである、上記[17]~[22]のいずれかに記載の方法。
[24]工程(1)で用いる活性化剤が、ホスフィチル化剤前駆体に対して1.5~20モル当量で用いられる、上記[17]~[23]のいずれかに記載の方法。
[25]工程(1)で用いる溶媒が、ホスフィチル化剤前駆体を溶解し、かつ活性化剤が難溶性となるものであって、酸性または塩基性官能基を有さないことを特徴とする、上記[17]~[24]のいずれかに記載の方法。
[26]工程(1)で用いる溶媒が、トルエン、ベンゼン、o-キシレン、m-キシレン、p-キシレン、エチルベンゼン、ペンタン、ヘキサン、ヘプタン、オクタン、シクロペンタン、シクロヘキサン、ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、シクロペンチルメチルエーテル、四塩化炭素から選択される1種以上の溶媒である、上記[17]~[25]のいずれかに記載の方法。
[27]工程(1)で用いる溶媒が、トルエンである、上記[17]~[26]のいずれかに記載の方法。
[28]工程(2)で用いる塩基が、pKa5~8の塩基である、上記[17]~[27]のいずれかに記載の方法。
[29]工程(2)で用いる塩基が、コリジン、N-メチルモルホリン及びジエチルアニリンから選択される1種以上の塩基である、上記[17]~[28]のいずれかに記載の方法。
[30]工程(1)と工程(2)の間に、不溶物を分離する工程を含む、上記[17]~[29]のいずれかに記載の方法。
[31]上記[18]に記載の方法により下記式(Ia-1):

[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia-2):
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia-2):

[式中、
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。
[32]上記[19]に記載の方法により下記式(Ia’-1):
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。
[32]上記[19]に記載の方法により下記式(Ia’-1):

[式中、P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ia-1)と同義である]
で表される5’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該5’末端ホスホロアミダイト化オリゴヌクレオチドの5’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia’-2):
で表される5’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該5’末端ホスホロアミダイト化オリゴヌクレオチドの5’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia’-2):
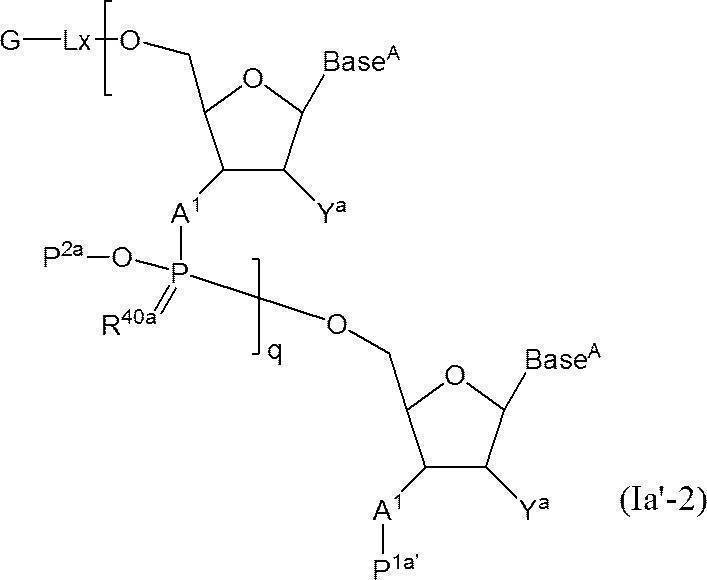
[式中、P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ia-2)と同義である]
で表される5’末端に官能基を有するオリゴヌクレオチドの製造方法。
[33]q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[31]又は[32]に記載の方法。
[34]官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、上記[31]~[33]のいずれかに記載の方法。
[35]官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[31]記載の方法。
[36]官能基が、5’位水酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[31]記載の方法。
[37]n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、上記[35]記載の方法。
で表される5’末端に官能基を有するオリゴヌクレオチドの製造方法。
[33]q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[31]又は[32]に記載の方法。
[34]官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、上記[31]~[33]のいずれかに記載の方法。
[35]官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[31]記載の方法。
[36]官能基が、5’位水酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[31]記載の方法。
[37]n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、上記[35]記載の方法。

[式中、r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
[38]n’個重合オリゴヌクレオチドが、下記構造(Ib’)を有する、上記[36]記載の方法。
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
[38]n’個重合オリゴヌクレオチドが、下記構造(Ib’)を有する、上記[36]記載の方法。

[式中、P3b’は、水酸基の保護基を示すか、又は-O-P3b’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ib)と同義である]
[39]r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[37]又は[38]に記載の方法。
[40]P3b又はP3b’における水酸基の保護基が、式:-L-YL-Z(各記号の定義は上記[12]と同義である)、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される、上記[37]~[39]のいずれかに記載の方法。
[39]r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[37]又は[38]に記載の方法。
[40]P3b又はP3b’における水酸基の保護基が、式:-L-YL-Z(各記号の定義は上記[12]と同義である)、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される、上記[37]~[39]のいずれかに記載の方法。
[41]Lがスクシニル基である、上記[40]に記載の方法。
[42]下記式(Ia-1’):
[42]下記式(Ia-1’):
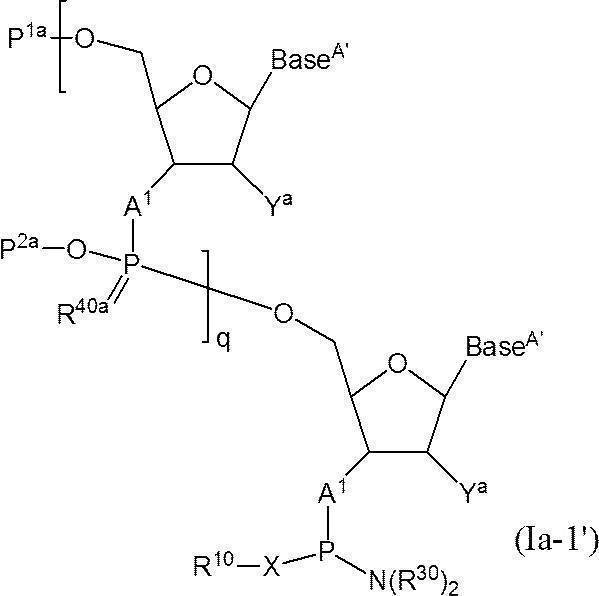
[式中、
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseA’は、それぞれ独立して式:-L’-YL-Z(式中、L’はスクシニル基、YL、Zは上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させて下記式(Ia-2’):
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseA’は、それぞれ独立して式:-L’-YL-Z(式中、L’はスクシニル基、YL、Zは上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させて下記式(Ia-2’):

[式中、
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドを得た後、保護基を除去する工程を含む、下記式(II):
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドを得た後、保護基を除去する工程を含む、下記式(II):
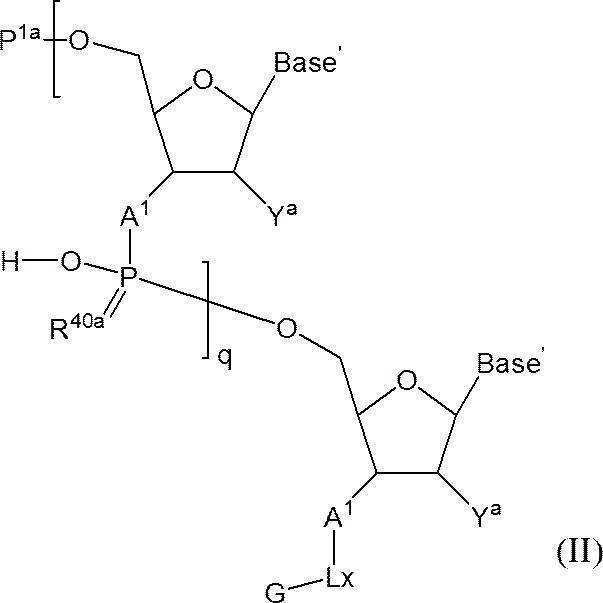
[式中、q+1個のBase’は無保護の核酸塩基を示し、それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。
[43]上記[17]~[41]のいずれかに記載の方法により3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドを使用する、上記[42]記載の方法。
[44]官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、上記[42]又は[43]記載の方法。
[45]官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[42]~[44]のいずれかに記載の方法。
[46]n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、上記[45]記載の方法。
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。
[43]上記[17]~[41]のいずれかに記載の方法により3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドを使用する、上記[42]記載の方法。
[44]官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、上記[42]又は[43]記載の方法。
[45]官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、上記[42]~[44]のいずれかに記載の方法。
[46]n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、上記[45]記載の方法。
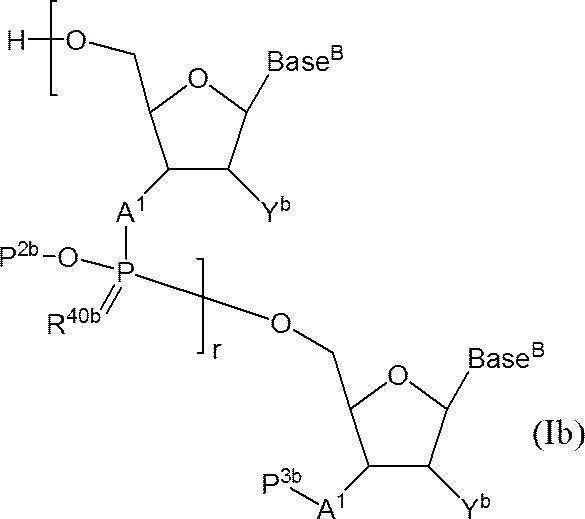
[式中、
r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
[47]r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[46]に記載の方法。
[48]P3b’における水酸基の保護基が、式:-L-YL-Z(各記号の定義は上記[12]と同義である)、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される、上記[46]又は[47]記載の方法。
[49]Lがスクシニル基である、上記[48]に記載の方法。
[50]下記式:
r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
[47]r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[46]に記載の方法。
[48]P3b’における水酸基の保護基が、式:-L-YL-Z(各記号の定義は上記[12]と同義である)、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される、上記[46]又は[47]記載の方法。
[49]Lがスクシニル基である、上記[48]に記載の方法。
[50]下記式:

[式中、
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2は、それぞれ独立してリン酸基の保護基を示し、
q個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
qは、1以上の任意の整数を示す。]
で表される化合物。
[51]q+1個のBaseの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[50]記載の化合物。
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2は、それぞれ独立してリン酸基の保護基を示し、
q個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
qは、1以上の任意の整数を示す。]
で表される化合物。
[51]q+1個のBaseの少なくとも一つが、式:-L-YL-Z(各記号の定義は上記[12]と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は上記[12]と同義である)で表される保護基で保護されている、上記[50]記載の化合物。
本発明の方法によれば、保管中安定なジアミダイトからホスフィチル化剤を調製することが可能であり、ホスフィチル化反応の際のリン酸上の保護基が脱落することを抑制することができる。従って、より安定、且つ効率よくホスホロアミダイト化オリゴヌレオチドを製造することができる。さらに本発明の方法によれば、ヌクレオチド間リン酸上のシアノエチル保護基を脱落することなく、シリル保護基を脱保護することが可能となる。従って、これらの方法を用いることで、より効率的なオリゴヌクレオチドの製造及びそれを用いたフラグメント縮合が可能となる。
文中で特に断らない限り、本明細書で用いるすべての技術用語および科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様または同等の任意の方法および材料は、本発明の実施または試験において使用することができるが、好ましい方法および材料を以下に記載する。本明細書で言及したすべての刊行物および特許は、例えば、記載された発明に関連して使用されうる刊行物に記載されている、構築物および方法論を記載および開示する目的で、参照として本明細書に組み入れられる。
本明細書において、オリゴヌクレオチドの構成単位となる「ヌクレオシド」とは、核酸塩基が糖(例えば、2-デオキシリボース、リボース、2位炭素原子および4位炭素原子が2価の有機基により結合された2-デオキシリボースまたはリボースなど)の1位にN-グリコシド化により結合された化合物を意味する。
本明細書における「糖」は、水酸基がアミノ基に置き換わったアミノ糖、および2位水酸基がハロゲン原子に置き換わったリボースも包含する。
本明細書における「糖」は、水酸基がアミノ基に置き換わったアミノ糖、および2位水酸基がハロゲン原子に置き換わったリボースも包含する。
本明細書中、「ヌクレオチド」とは、ヌクレオシドにリン酸基が結合した化合物を意味し、「オリゴヌクレオチド」とは、ヌクレオシドにヌクレオチドが1個以上連結した化合物を意味する。本明細書中、なお、「オリゴヌクレオチド」には、リン酸基の酸素原子が硫黄原子に置き換わったホスホロチオエート型のオリゴヌクレオチド、リン酸基の-O-が-NH-に置き換わったオリゴヌクレオチド、リン酸基中の水酸基(-OH)が-ORp(式中、Rpは有機基を示す)に置き換わったオリゴヌクレオチドも包含される。本発明におけるオリゴヌクレオチドのヌクレオシドの数は特に限定されないが、好ましくは3~50、より好ましくは5~30である。
「3’位アミノ基」とは、ヌクレオシド、ヌクレオチドまたはオリゴヌクレオチドの3’位の炭素原子に結合したアミノ基を意味する。
「5’位アミノ基」とは、ヌクレオシド、ヌクレオチドまたはオリゴヌクレオチドの5’位の炭素原子に結合したアミノ基を意味する。
「3’位リン酸基」とは、ヌクレオチドまたはオリゴヌクレオチドの3’位の炭素原子に結合したリン酸基を意味する。
本明細書中、「5’位リン酸基」とは、ヌクレオチドまたはオリゴヌクレオチドの5’位の炭素原子に結合したリン酸基を意味する。
「5’位アミノ基」とは、ヌクレオシド、ヌクレオチドまたはオリゴヌクレオチドの5’位の炭素原子に結合したアミノ基を意味する。
「3’位リン酸基」とは、ヌクレオチドまたはオリゴヌクレオチドの3’位の炭素原子に結合したリン酸基を意味する。
本明細書中、「5’位リン酸基」とは、ヌクレオチドまたはオリゴヌクレオチドの5’位の炭素原子に結合したリン酸基を意味する。
本明細書中、「リン酸基」は、-O-P(O)(OH)2だけでなく、酸素原子が硫黄原子またはNHに置き換わった基(例えば、-O-P(S)(OH)2、-NH-P(O)(OH)2、-NH-P(S)(OH)2)も包含する。また、リン酸基中の水酸基(-OH)が-ORp(式中、Rpは、リン酸基の保護基などの有機基を示す)に置き換わった基(例えば、保護されたリン酸基)も、「リン酸基」に包含される。
本明細書において、「核酸塩基」とは、核酸の合成に使用されるものであれば特に制限されず、例えば、シトシル基、ウラシル基、チミニル基等のピリミジン塩基、アデニル基、グアニル基等のプリン塩基を挙げることができる。また、該「核酸塩基」には、上記した基の他に、核酸塩基が任意の置換基(例えば、ハロゲン原子、アルキル基、アラルキル基、アルコキシ基、アシル基、アルコキシアルキル基、ヒドロキシ基、アミノ基、モノアルキルアミノ、ジアルキルアミノ、カルボキシ、シアノ、ニトロ等)により任意の位置に1~3個置換されている修飾核酸塩基(例えば、8-ブロモアデニル基、8-ブロモグアニル基、5-ブロモシトシル基、5-ヨードシトシル基、5-ブロモウラシル基、5-ヨードウラシル基、5-フルオロウラシル基、5-メチルシトシル基、8-オキソグアニル基、ヒポキサンチニル基等)も包含される。
本明細書中、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子、またはヨウ素原子である。
本明細書中、「アルキル(基)」としては、直鎖状または分岐鎖状の炭素数1以上のアルキル基が挙げられ、特に炭素数範囲の限定がない場合には、好ましくはC1-10アルキル基であり、より好ましくはC1-6アルキル基である。炭素数範囲の限定がない場合の好適な具体例としては、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル等が挙げられ、特にメチル、エチルが好ましい。
本明細書中、「アラルキル(基)」としては、C7-20アラルキル基が挙げられ、好ましくはC7-16アラルキル基(C6-10アリール-C1-6アルキル基)である。好適な具体例としては、ベンジル、1-フェニルエチル、2-フェニルエチル、1-フェニルプロピル、ナフチルメチル、1-ナフチルエチル、1-ナフチルプロピル等が挙げられ、特にベンジルが好ましい。
本明細書中、「アルコキシ(基)」としては、炭素数1以上のアルキコキシ基が挙げられ、特に炭素数範囲の限定がない場合には、好ましくはC1-10アルコキシ基であり、より好ましくはC1-6アルコキシ基である。炭素数範囲の限定がない場合の好適な具体例としては、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、sec-ブトキシ、tert-ブトキシ、ペンチルオキシ、ヘキシルオキシ等が挙げられ、特にメトキシ、エトキシが好ましい。
本明細書中、「アシル(基)」としては、例えば、直鎖状または分岐鎖状のC1-6アルカノイル基、C7-13アロイル基等が挙げられる。具体的には、例えば、ホルミル、アセチル、n-プロピオニル、イソプロピオニル、n-ブチリル、イソブチリル、ピバロイル、バレリル、ヘキサノイル、ベンゾイル、ナフトイル、レブリニル等が挙げられ、これらはそれぞれ置換されていてもよい。
本明細書中、「アルケニル(基)」としては、直鎖状または分岐鎖状のC2-6アルケニル基等が好ましく、例えば、ビニル、1-プロペニル、アリル、イソプロペニル、ブテニル、イソブテニル等が挙げられる。中でも、C2-C4アルケニル基が好ましい。
本明細書中、「アルキニル(基)」としては、C2-6アルキニル基等が好ましく、例えば、エチニル、1-プロピニル、2-プロピニル、1-ブチニル、2-ブチニル、3-ブチニル、1-ペンチニル、2-ペンチニル、3-ペンチニル、4-ペンチニル、1-ヘキシニル、2-ヘキシニル、3-ヘキシニル、4-ヘキシニル、5-ヘキシニル等が挙げられる。中でも、C2-C4アルキニル基が好ましい。
本明細書中、「シクロアルキル(基)」は、環状アルキル基を意味し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル等が挙げられる。中でも、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル等のC3-C6シクロアルキル基が好ましく、シクロヘキシルが特に好ましい。
本明細書中、「アリール(基)」又は「芳香環(基)」は、芳香族性を示す単環式あるいは多環式(縮合)の炭化水素基を意味し、具体的には、例えば、フェニル、1-ナフチル、2-ナフチル、ビフェニリル、2-アンスリル等のC6-14アリール基等が挙げられる。中でもC6-10アリール基がより好ましく、フェニルが特に好ましい。
本明細書中、「アリールオキシ(基)」としては、C6-14アリールオキシ基が好ましくは、フェノキシ、トリルオキシ、キシリルオキシ、ナフトキシ、ジメチルナフトキシ等が挙げられる。
本明細書中、「アラルキルオキシ(基)」は、上記「アリール基」で置換された上記「アルキルオキシ(基)」を意味し、具体的には、ベンジルオキシ、フェネチルオキシ、2-フェニルプロパン-2-イルオキシ、ジフェニルメチルオキシ等が挙げられる。
本明細書中、「炭化水素基」としては、例えば、脂肪族炭化水素基、芳香脂肪族炭化水素基、単環式飽和炭化水素基および芳香族炭化水素基等が挙げられ、具体的には、例えば、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アリール基、アラルキル基等の1価基およびそれらから誘導される2価基である。
本明細書中、「炭化水素基を有する有機基」とは、前記「炭化水素基」を有する基を意味し、「炭化水素基を有する有機基」中の「炭化水素基」以外の部位は任意に設定することができる。例えば、リンカーとして-O-、-S-、-COO-、-OCONH-、および-CONH-等の部位を有していてもよい。
[3’位水酸基又は3’位アミノ基がシリル保護基で保護されているオリゴヌクレオチドを脱保護する方法(工程)-脱シリル化]
本方法(工程)は、溶媒中、「5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)」のシリル保護基を脱保護(脱シリル化ともいう)して、「5’位水酸基又は5’位リン酸基が保護され、3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)」を製造する方法(工程)である。
5’位水酸基の保護基としては、酸性条件下で脱保護可能であり、水酸基の保護基として用いられるものであれば、特に限定はされないが、トリチル基、9-(9-フェニル)キサンテニル基、9-フェニルチオキサンテニル基、1,1-ビス(4-メトキシフェニル)-1-フェニルメチル基(ジメトキシトリチル基)等のジ(C1-6アルコキシ)トリチル基、1-(4-メトキシフェニル)-1,1-ジフェニルメチル基(モノメトキシトリチル基)等のモノ(C1-18アルコキシ)トリチル基等を挙げることができる。これらの中でも、脱保護のしやすさ、入手の容易さの観点から、モノメトキシトリチル基、ジメトキシトリチル基であることが好ましく、より好ましくは、ジメトキシトリチル基である。
本方法(工程)は、溶媒中、「5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)」のシリル保護基を脱保護(脱シリル化ともいう)して、「5’位水酸基又は5’位リン酸基が保護され、3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)」を製造する方法(工程)である。
5’位水酸基の保護基としては、酸性条件下で脱保護可能であり、水酸基の保護基として用いられるものであれば、特に限定はされないが、トリチル基、9-(9-フェニル)キサンテニル基、9-フェニルチオキサンテニル基、1,1-ビス(4-メトキシフェニル)-1-フェニルメチル基(ジメトキシトリチル基)等のジ(C1-6アルコキシ)トリチル基、1-(4-メトキシフェニル)-1,1-ジフェニルメチル基(モノメトキシトリチル基)等のモノ(C1-18アルコキシ)トリチル基等を挙げることができる。これらの中でも、脱保護のしやすさ、入手の容易さの観点から、モノメトキシトリチル基、ジメトキシトリチル基であることが好ましく、より好ましくは、ジメトキシトリチル基である。
脱シリル化されるn個重合オリゴヌクレオチドの5’位末端は保護されていてもよいリン酸基であり得る。5’位末端における保護されたリン酸基としては、例えばリン酸基中の水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基が酸性条件下で脱保護可能な保護基で保護されている)に置き換わっているリン酸基が挙げられる。
Ln1の有機基とは、炭化水素基、または炭化水素基中の炭素原子がヘテロ原子で置き換わった基を意味する。ヘテロ原子としては、例えば、酸素原子、窒素原子、硫黄原子等が挙げられる。また、有機基は、水酸基、アミノ基、オキソ基(=O)等の置換基を有していてもよい。有機基が有し得る水酸基およびアミノ基は、保護基で保護されていることが好ましい。有機基の形状は、鎖状(直鎖状または分岐鎖状)、環状またはこれらの組合せのいずれでもよい。
有機基は、細胞に対して機能性を有する基を有していてもよい。細胞に対して機能性を有する基は、有機基の主鎖または側鎖の末端に結合していることが好ましい。細胞に対して機能性を有する基としては、例えば、「化合物の脂溶性を向上させることによって、化合物の細胞膜透過性を向上させる基」、「細胞膜受容体を介して細胞内への化合物の取込みを向上させる基」等が挙げられる。「化合物の脂溶性を向上させることによって、化合物の細胞膜透過性を向上させる基」としては、例えば、コレステロール残基、トコフェロール残基等が挙げられる。「細胞膜受容体を介して細胞内への化合物の取込みを向上させる基」としては、例えば、N-アセチルガラクトサミン残基等が挙げられる。
-OLn1-OHの具体例としては、以下のものが挙げられる(下記式中の*は、リン原子との結合位置を示し、Acはアセチル基を示す。)。
Ln1の有機基とは、炭化水素基、または炭化水素基中の炭素原子がヘテロ原子で置き換わった基を意味する。ヘテロ原子としては、例えば、酸素原子、窒素原子、硫黄原子等が挙げられる。また、有機基は、水酸基、アミノ基、オキソ基(=O)等の置換基を有していてもよい。有機基が有し得る水酸基およびアミノ基は、保護基で保護されていることが好ましい。有機基の形状は、鎖状(直鎖状または分岐鎖状)、環状またはこれらの組合せのいずれでもよい。
有機基は、細胞に対して機能性を有する基を有していてもよい。細胞に対して機能性を有する基は、有機基の主鎖または側鎖の末端に結合していることが好ましい。細胞に対して機能性を有する基としては、例えば、「化合物の脂溶性を向上させることによって、化合物の細胞膜透過性を向上させる基」、「細胞膜受容体を介して細胞内への化合物の取込みを向上させる基」等が挙げられる。「化合物の脂溶性を向上させることによって、化合物の細胞膜透過性を向上させる基」としては、例えば、コレステロール残基、トコフェロール残基等が挙げられる。「細胞膜受容体を介して細胞内への化合物の取込みを向上させる基」としては、例えば、N-アセチルガラクトサミン残基等が挙げられる。
-OLn1-OHの具体例としては、以下のものが挙げられる(下記式中の*は、リン原子との結合位置を示し、Acはアセチル基を示す。)。

3’位水酸基又は3’位アミノ基におけるシリル保護基としては、ケイ素上の置換基がアルキル基、アリール基、アラルキル基、アルコキシ基、アリールオキシ基、アラルキルオキシ基、アルキルチオ基、アリールチオ基から選択されるシリル基であって、3つの置換基のうち少なくとも1つがアリール基、アルコキシ基、アリールオキシ基、アラルキルオキシ基から選択されるシリル基が挙げられる。具体的には、tert-ブチルジメチルシリル(TBDMS)、ジイソプロピルフェニルシリル(DIPPS)、tert-ブトキシジフェニルシリル(TBODPS)、イソプロポキシジイソプロピルシリル(IPODIPS)等が挙げられ、好ましくはDIPPSである。
好ましくは、「5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)」は、下記構造(I)を有する化合物である(以下、化合物(I))。

[式中、
s+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
s+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
s個のP2は、それぞれ独立してリン酸基の保護基を示し、
s個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
s+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
sは、1以上の任意の整数を示す。]
P1における水酸基の保護基としては、上記5’位水酸基の保護基として例示したものと同様のものを用いることができる。好ましくはジメトキシトリチル基である。P1における、水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基としては、上記5’位末端における保護されたリン酸基として記載されたものと同様のものを用いることができる。
P2におけるリン酸基の保護基としては、塩基性条件下で脱保護可能であり、リン酸基の保護基として用いられるものであれば、特に限定はされないが、-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基が好ましく、WGとしてはシアノ基が好ましい。
R40は、好ましくは同一で酸素原子又は硫黄原子である。
Yにおける保護されていてもよい水酸基の保護基としては、特に限定されず、例えば、グリーンズ・プロテクティブ・グループス・イン・オーガニック・シンセシス(Greene’s PROTECTIVE GROUPS IN ORGANIC SYNTHESIS)、第4版、ジョン・ウィリー・アンド・サンズ(JOHN WILLY&SONS)出版(2006年)等に記載されている任意の保護基を挙げることができる。具体的には、メチル、ベンジル、p-メトキシベンジル、tert-ブチル、メトキシメチル、メトキシエチル、2-テトラヒドロピラニル、エトキシエチル、シアノエチル、シアノエトキシメチル、フェニルカルバモイル、1,1-ジオキソチオモルホリン-4-チオカルバモイル、アセチル、ピバロイル、ベンゾイル、トリメチルシリル、トリエチルシリル、トリイソプロピルシリル、tert-ブチルジメチルシリル、[(トリイソプロピルシリル)オキシ]メチル(Tom)、1-(4-クロロフェニル)-4-エトキシピペリジン-4-イル(Cpep)基等を挙げることができる。これらの中でも、トリエチルシリル基、トリイソプロピルシリル基、またはtert-ブチルジメチルシリル基であることが好ましく、経済性及び入手の容易さの観点から、tert-ブチルジメチルシリル基であることが特に好ましい。
Yにおける「4位炭素原子に架橋する有機基」としては、ヌクレオシド2位と4位を架橋する限り特に限定はないが、例えば、C2-7アルキレン基が挙げられる。当該アルキレン基は、例えば、-O-、-NR37-(R37は水素原子またはC1-6アルキル基を示す)、-S-、-CO-、-COO-、-OCONR38-(R38は水素原子またはC1-6アルキル基を示す)、-CONR39-(R39は水素原子またはC1-6アルキル基を示す)等から選ばれるリンカーで1箇所以上(好ましくは、1又は2箇所)中断されていてもよい。
「4位炭素原子に架橋する有機基」として好ましくは、例えば、-ORi(Riは4位に架橋するC1-6アルキレン基を示す)、-O-NR37-Rj(Rjは4位に架橋するC1-6アルキレン基を示し、R37は前記と同義を示す)、-O-Rk-O-Rl(RkはC1-6アルキレン基を示し、Rlは4位に架橋するC1-6アルキレン基を示す)等が挙げられる。Ri、Rj、Rk及びRlで示されるC1-6アルキレン基としては、それぞれ独立して、メチレン基またはエチレン基が好ましい。
「4位炭素原子に架橋する有機基」としては、-O-CH2-、-O-CH2-CH2-、-O-NR37-CH2-(R37は前記と同義を示す)、-O-CH2-O-CH2-等が好ましく、-O-CH2-、-O-CH2-CH2-、-O-NH-CH2-、-O-NMe-CH2-、-O-CH2-O-CH2-(それぞれ、左側が2位に結合し、右側が4位に結合する。)等がより好ましい。
P3におけるシリル保護基としては、上記3’位水酸基におけるシリル保護基として例示したものと同様のものを用いることができる。
s+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
s+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
s個のP2は、それぞれ独立してリン酸基の保護基を示し、
s個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
s+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
sは、1以上の任意の整数を示す。]
P1における水酸基の保護基としては、上記5’位水酸基の保護基として例示したものと同様のものを用いることができる。好ましくはジメトキシトリチル基である。P1における、水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基としては、上記5’位末端における保護されたリン酸基として記載されたものと同様のものを用いることができる。
P2におけるリン酸基の保護基としては、塩基性条件下で脱保護可能であり、リン酸基の保護基として用いられるものであれば、特に限定はされないが、-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基が好ましく、WGとしてはシアノ基が好ましい。
R40は、好ましくは同一で酸素原子又は硫黄原子である。
Yにおける保護されていてもよい水酸基の保護基としては、特に限定されず、例えば、グリーンズ・プロテクティブ・グループス・イン・オーガニック・シンセシス(Greene’s PROTECTIVE GROUPS IN ORGANIC SYNTHESIS)、第4版、ジョン・ウィリー・アンド・サンズ(JOHN WILLY&SONS)出版(2006年)等に記載されている任意の保護基を挙げることができる。具体的には、メチル、ベンジル、p-メトキシベンジル、tert-ブチル、メトキシメチル、メトキシエチル、2-テトラヒドロピラニル、エトキシエチル、シアノエチル、シアノエトキシメチル、フェニルカルバモイル、1,1-ジオキソチオモルホリン-4-チオカルバモイル、アセチル、ピバロイル、ベンゾイル、トリメチルシリル、トリエチルシリル、トリイソプロピルシリル、tert-ブチルジメチルシリル、[(トリイソプロピルシリル)オキシ]メチル(Tom)、1-(4-クロロフェニル)-4-エトキシピペリジン-4-イル(Cpep)基等を挙げることができる。これらの中でも、トリエチルシリル基、トリイソプロピルシリル基、またはtert-ブチルジメチルシリル基であることが好ましく、経済性及び入手の容易さの観点から、tert-ブチルジメチルシリル基であることが特に好ましい。
Yにおける「4位炭素原子に架橋する有機基」としては、ヌクレオシド2位と4位を架橋する限り特に限定はないが、例えば、C2-7アルキレン基が挙げられる。当該アルキレン基は、例えば、-O-、-NR37-(R37は水素原子またはC1-6アルキル基を示す)、-S-、-CO-、-COO-、-OCONR38-(R38は水素原子またはC1-6アルキル基を示す)、-CONR39-(R39は水素原子またはC1-6アルキル基を示す)等から選ばれるリンカーで1箇所以上(好ましくは、1又は2箇所)中断されていてもよい。
「4位炭素原子に架橋する有機基」として好ましくは、例えば、-ORi(Riは4位に架橋するC1-6アルキレン基を示す)、-O-NR37-Rj(Rjは4位に架橋するC1-6アルキレン基を示し、R37は前記と同義を示す)、-O-Rk-O-Rl(RkはC1-6アルキレン基を示し、Rlは4位に架橋するC1-6アルキレン基を示す)等が挙げられる。Ri、Rj、Rk及びRlで示されるC1-6アルキレン基としては、それぞれ独立して、メチレン基またはエチレン基が好ましい。
「4位炭素原子に架橋する有機基」としては、-O-CH2-、-O-CH2-CH2-、-O-NR37-CH2-(R37は前記と同義を示す)、-O-CH2-O-CH2-等が好ましく、-O-CH2-、-O-CH2-CH2-、-O-NH-CH2-、-O-NMe-CH2-、-O-CH2-O-CH2-(それぞれ、左側が2位に結合し、右側が4位に結合する。)等がより好ましい。
P3におけるシリル保護基としては、上記3’位水酸基におけるシリル保護基として例示したものと同様のものを用いることができる。
Baseの「保護されていてもよい核酸塩基」とは、例えば、アミノ基を有する核酸塩基であるアデニル基、グアニル基、またはシトシル基において、アミノ基が保護されていてもよいことを意味し、核酸塩基のアミノ基が5’位の脱保護条件に耐え得る保護基により保護されている核酸塩基が好ましい。かかる「アミノ基の保護基」としては、特に限定されず、例えば、グリーンズ・プロテクティブ・グループス・イン・オーガニック・シンセシス(Greene’s PROTECTIVE GROUPS IN ORGANIC SYNTHESIS)、第4版、ジョン・ウィリー・アンド・サンズ(JOHN WILLY&SONS)出版(2006年)等に記載されている保護基を挙げることができる。かかる「アミノ基の保護基」の具体例としては、例えば、ピバロイル基、ピバロイロキシメチル基、トリフルオロアセチル基、フェノキシアセチル基、4-イソプロピルフェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、アセチル基、ベンゾイル基、イソブチリル基、ジメチルホルムアミジニル基、9-フルオレニルメチルオキシカルボニル基等を挙げることができる。これらの中でも、フェノキシアセチル基、4-イソプロピルフェノキシアセチル基、アセチル基、ベンゾイル基、イソブチリル基、及びジメチルホルムアミジニル基が好ましい。また、核酸塩基のカルボニル基が保護されていてもよく、例えば、フェノール、2,5-ジクロロフェノール、3-クロロフェノール、3,5-ジクロロフェノール、2-ホルミルフェノール、2-ナフトール、4-メトキシフェノール、4-クロロフェノール、2-ニトロフェノール、4-ニトロフェノール、4-アセチルアミノフェノール、ペンタフルオロフェノール、4-ピバロイロキシベンジルアルコール、4-ニトロフェネチルアルコール、2-(メチルスルフォニル)エタノール、2-(フェニルスルフォニル)エタノール、2-シアノエタノール、2-(トリメチルシリル)エタノール、ジメチルカルバミン酸クロライド、ジエチルカルバミン酸クロライド、エチルフェニルカルバミン酸クロライド、1-ピロリジンカルボン酸クロライド、4-モルホリンカルボン酸クロライド、ジフェニルカルバミン酸クロライド等を反応させて、カルボニル基を保護することが出来る。ここで、カルボニル基の保護基については、特に導入しなくてもよい場合がある。
核酸塩基の保護基として、WO2013/122236に記載の保護基(C5-30直鎖又は分岐鎖アルキル基及び/又はC5-30直鎖又は分岐鎖アルケニルを有する基)を使用することもできる。
核酸塩基の保護基として、下記式:-L-YL-Zで表される保護基、若しくは式:-Z’で表される保護基を使用することもできる。当該保護基は、該基が反応基質に結合することにより非極性溶媒に可溶化し、液相中の反応が可能であると共に、極性溶媒の添加により沈殿し、固液分離が可能となる、反応性と後処理の簡便性を兼ね備えた保護基であって、5’末端水酸基の保護基を除去し得る酸性条件で安定な基である。従って、s+1個存在するBaseの少なくとも1つは(1)式:-L-YL-Zで表される保護基、若しくは(2)式:-Z’で表される保護基で保護されていることが好ましい。
(1)
核酸塩基の保護基として、WO2013/122236に記載の保護基(C5-30直鎖又は分岐鎖アルキル基及び/又はC5-30直鎖又は分岐鎖アルケニルを有する基)を使用することもできる。
核酸塩基の保護基として、下記式:-L-YL-Zで表される保護基、若しくは式:-Z’で表される保護基を使用することもできる。当該保護基は、該基が反応基質に結合することにより非極性溶媒に可溶化し、液相中の反応が可能であると共に、極性溶媒の添加により沈殿し、固液分離が可能となる、反応性と後処理の簡便性を兼ね備えた保護基であって、5’末端水酸基の保護基を除去し得る酸性条件で安定な基である。従って、s+1個存在するBaseの少なくとも1つは(1)式:-L-YL-Zで表される保護基、若しくは(2)式:-Z’で表される保護基で保護されていることが好ましい。
(1)
[式中、
Lは、式(a1):
Lは、式(a1):

(式中、**は、核酸との結合位置を示し;
*は、YLとの結合位置を示し;
L1は、置換されていてもよい2価のC1-22炭化水素基、酸素原子、-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)を示し;かつ
L2は、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、CRcRdとの結合位置を示し、R1は、置換されていてもよいC1-22アルキレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくは置換されていてもよいC1-22アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-22アルキレン結合を形成していてもよく、R3bは水素原子若しくは置換されていても良いC1-22アルキル基を示す)で示される基を示し;
RcおよびRdはそれぞれ独立して水素原子、置換されていてもよいC1-22アルキル基もしくはRcおよびRdが一緒になって、単一のカルボニル基を形成していてもよい)で表される基(リンカー)を示し;
YLは、単結合、酸素原子、または-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)、硫黄原子を示し;ならびに
Zは式(a2):
*は、YLとの結合位置を示し;
L1は、置換されていてもよい2価のC1-22炭化水素基、酸素原子、-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)を示し;かつ
L2は、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、CRcRdとの結合位置を示し、R1は、置換されていてもよいC1-22アルキレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくは置換されていてもよいC1-22アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-22アルキレン結合を形成していてもよく、R3bは水素原子若しくは置換されていても良いC1-22アルキル基を示す)で示される基を示し;
RcおよびRdはそれぞれ独立して水素原子、置換されていてもよいC1-22アルキル基もしくはRcおよびRdが一緒になって、単一のカルボニル基を形成していてもよい)で表される基(リンカー)を示し;
YLは、単結合、酸素原子、または-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)、硫黄原子を示し;ならびに
Zは式(a2):

(式中、*は、YLとの結合位置を示し;
R4は、水素原子を示すか、あるいはRbが、下記式(a3)で表される基である場合には、R6と一緒になって単結合または-O-を示して、環Bと共にフルオレニル基またはキサンテニル基を形成していてもよく;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
kは、1~4の整数を示し;
環Aは、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
Raは、水素原子、またはハロゲン原子により置換されていてもよいフェニル基を示し;ならびに
Rbは、水素原子、または式(a3):
R4は、水素原子を示すか、あるいはRbが、下記式(a3)で表される基である場合には、R6と一緒になって単結合または-O-を示して、環Bと共にフルオレニル基またはキサンテニル基を形成していてもよく;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
kは、1~4の整数を示し;
環Aは、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
Raは、水素原子、またはハロゲン原子により置換されていてもよいフェニル基を示し;ならびに
Rbは、水素原子、または式(a3):

(式中、*は、YLとの結合位置を示し;
jは、0~4の整数を示し;
j個のR7は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
R6は、水素原子を示すか、またはR4と一緒になって単結合または-O-を示して、環Aと共にフルオレニル基またはキサンテニル基を形成していてもよく;かつ
環Bは、j個のOR7に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
RaおよびRbが一緒になって、単一のカルボニル基を形成していてもよい。)で表される基);
式(a2’):
jは、0~4の整数を示し;
j個のR7は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
R6は、水素原子を示すか、またはR4と一緒になって単結合または-O-を示して、環Aと共にフルオレニル基またはキサンテニル基を形成していてもよく;かつ
環Bは、j個のOR7に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
RaおよびRbが一緒になって、単一のカルボニル基を形成していてもよい。)で表される基);
式(a2’):

(式中、*は、YLとの結合位置を示し;それ以外の各記号の定義は式(a2)と同義である。)で示される基
または、
式(a2’’):
または、
式(a2’’):

(式中、*は、YLとの結合位置を示し;
環A’は、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
各記号の定義は式(a2)と同義である。)
で表される基を示す]
環A’は、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
各記号の定義は式(a2)と同義である。)
で表される基を示す]
(2)
式:-Z’
式:-Z’

[式中、**は、核酸との結合位置を示し;
各記号の定義は式(a2’’)と同義である。]
各記号の定義は式(a2’’)と同義である。]
上記式(a1)で表されるリンカーLの好ましい態様としては、式(a1)中、L1が、C1-6アルキレン基を示し;
L2が、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3a)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、C=Oとの結合位置を示し、R1は、C1-6アルキレン基を示し、R2、R3aおよびR3bは、独立してそれぞれ水素原子、もしくは置換されていてもよいC1-6アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-6アルキレン結合を形成していてもよい。)で表される基であり;かつ
RcおよびRdがそれぞれ独立して水素原子、またはRcおよびRdが一緒になって、単一のカルボニル基を形成している、基である。
L2が、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3a)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、C=Oとの結合位置を示し、R1は、C1-6アルキレン基を示し、R2、R3aおよびR3bは、独立してそれぞれ水素原子、もしくは置換されていてもよいC1-6アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-6アルキレン結合を形成していてもよい。)で表される基であり;かつ
RcおよびRdがそれぞれ独立して水素原子、またはRcおよびRdが一緒になって、単一のカルボニル基を形成している、基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、エチレン基を示し;
L2が、単結合を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している基である。
L2が、単結合を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、エチレン基を示し;かつ
L2中のN(R2)-R1-N(R3a)部分が、ピペラジニレン基を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
L2中のN(R2)-R1-N(R3a)部分が、ピペラジニレン基を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
上記式(a1)で表されるリンカーLの更に別の好ましい態様としては、式(a1)中、L1が、エチレン基を示し;
L2が、式:*C(=O)N(R2)-R1-N(R3a)**(式中、*は、L1との結合位置を示し、**は、C=Oとの結合位置を示し、R1は、ペンチレン基、またはヘキシレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくはメチル基を示す。)で表される基であり;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
L2が、式:*C(=O)N(R2)-R1-N(R3a)**(式中、*は、L1との結合位置を示し、**は、C=Oとの結合位置を示し、R1は、ペンチレン基、またはヘキシレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくはメチル基を示す。)で表される基であり;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、エチレン基を示し;
L2中のN(R2)-R1-C(R3a)(R3b)部分が、ピペリジニレン基を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
L2中のN(R2)-R1-C(R3a)(R3b)部分が、ピペリジニレン基を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、メチレン基またはエチレン基を示し;
L2が、単結合を示し;かつ
RcおよびRdが水素原子である、基である。
L2が、単結合を示し;かつ
RcおよびRdが水素原子である、基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、ブチレン基を示し;
L2が、単結合を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
L2が、単結合を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
上記式(a1)で表されるリンカーLの別の好ましい態様としては、式(a1)中、L1が、メチレン基を示し;
L2中のC(R3a)(R3b)-O-R1部分が、-CH2-O-CH2-を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
L2中のC(R3a)(R3b)-O-R1部分が、-CH2-O-CH2-を示し;かつ
RcおよびRdが一緒になって単一のカルボニル基を形成している、基である。
上記リンカーLの特に好ましい例は、入手が容易で安価なスクシニル基である。
Zの好ましい態様としては、式(a2)、式(a2’)または式(a2’’)で表される基である。
上記式(a2)または式(a2’’)で表されるZの好ましい態様としては、式(a2)または式(a2’’)中、RaおよびRbは、共に水素原子を示し;
R4は、水素原子を示し、
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基(例えば、C10-40アルキル基)を示し;かつ
kは、1~3の整数を示す、基である。
また、RaおよびRbが一緒になって単一のカルボニル基を形成する態様も又好ましい。
R4は、水素原子を示し、
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基(例えば、C10-40アルキル基)を示し;かつ
kは、1~3の整数を示す、基である。
また、RaおよびRbが一緒になって単一のカルボニル基を形成する態様も又好ましい。
上記式(a2)または式(a2’’)で表されるZの別の好ましい態様としては、式(a2)または式(a2’’)中、
kは、1~3の整数を示し;
RaおよびRbは、共に水素原子を示し;
R4は、水素原子を示し;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を1~3個有するベンジル基、または炭素数10以上の脂肪族炭化水素基を1~3個有するシクロヘキシル基を示し;かつ
環Aが、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよい、基である。
kは、1~3の整数を示し;
RaおよびRbは、共に水素原子を示し;
R4は、水素原子を示し;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を1~3個有するベンジル基、または炭素数10以上の脂肪族炭化水素基を1~3個有するシクロヘキシル基を示し;かつ
環Aが、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよい、基である。
上記式(a2)または式(a2’’)で表されるZの別の好ましい態様としては、式(a2)または式(a2’’)中、
Raは、水素原子を示し;
Rbは、上記式(a3)(式中、*は結合位置を示し;jは、0~3の整数を示し;j個のR7は、独立してそれぞれC10-40アルキル基を示し;
R4およびR6は、共に水素原子を示す。)で表される基である、基である。
Raは、水素原子を示し;
Rbは、上記式(a3)(式中、*は結合位置を示し;jは、0~3の整数を示し;j個のR7は、独立してそれぞれC10-40アルキル基を示し;
R4およびR6は、共に水素原子を示す。)で表される基である、基である。
上記式(a2)または式(a2’’)で表されるZの更に別の好ましい態様としては、式(a2)または式(a2’’)中、
Raは、水素原子を示し;
Rbは、上記式(a3)(式中、*は結合位置を示し;jは、0~3の整数を示し;j個のR7は、独立してそれぞれC10-40アルキル基を示し;
R6は、環AのR4と一緒になって単結合または-O-を形成し、それにより環Aと環Bは一緒になってフルオレニル基またはキサンテニル基を示す。)で表される基である、基である。
Raは、水素原子を示し;
Rbは、上記式(a3)(式中、*は結合位置を示し;jは、0~3の整数を示し;j個のR7は、独立してそれぞれC10-40アルキル基を示し;
R6は、環AのR4と一緒になって単結合または-O-を形成し、それにより環Aと環Bは一緒になってフルオレニル基またはキサンテニル基を示す。)で表される基である、基である。
上記式(a2’)で表されるZの好ましい態様としては、式(a2’)中、k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基(例えば、C10-40アルキル基)を示し;かつ
kは、1~3の整数を示す、基である。
kは、1~3の整数を示す、基である。
上記式:-Z’中、k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基(例えば、C10-40アルキル基)を示し;かつ
kは、1~3の整数を示す、基である。
kは、1~3の整数を示す、基である。
上記式:-L-YL-Zで表される保護基、若しくは式:-Z’で表される保護基としては、ヌクレオチド末端水酸基の保護基を除去し得る酸性条件下では切断されにくく、塩基性条件下で切断される基が好ましい。かかる保護基の代表的な例としては、例えば、式:-L-YL-Z中のLが、上記式(a1)で表される基(好ましくは、スクシニル基等)であり、かつYL-Zが、以下の基であるもの;
式:-Z’で表される基が以下の基であるものが挙げられる。
3,4,5-トリス(オクタデシルオキシ)ベンジルオキシ基、
3,5-ビス(ドコシルオキシ)ベンジルオキシ基、
3,5-ビス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルオキシ基、
3,4,5-トリス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、
3,4,5-トリス(オクタデシルオキシ)ベンジルアミノ基、
2,4-ビス(ドコシルオキシ)ベンジルアミノ基、
3,5-ビス(ドコシルオキシ)ベンジルアミノ基、
ビス(4-ドコシルオキシフェニル)メチルアミノ基、
4-メトキシ-2-[3',4',5'-トリス(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、
4-メトキシ-2-[3',4',5'-トリス(オクタデシルオキシ)シクロヘキシルメチルオキシ]ベンジルアミノ基、
2,4-ビス(ドデシルオキシ)ベンジルアミノ基、
フェニル(2,3,4-トリス(オクタデシルオキシ)フェニル)メチルアミノ基、
ジ[4-(12-ドコシルオキシドデシルオキシ)フェニル]メチルアミノ基、
3,5-ビス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、または
3,4,5-トリス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基。
式:-Z’で表される基が以下の基であるものが挙げられる。
3,4,5-トリス(オクタデシルオキシ)ベンジルオキシ基、
3,5-ビス(ドコシルオキシ)ベンジルオキシ基、
3,5-ビス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルオキシ基、
3,4,5-トリス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、
3,4,5-トリス(オクタデシルオキシ)ベンジルアミノ基、
2,4-ビス(ドコシルオキシ)ベンジルアミノ基、
3,5-ビス(ドコシルオキシ)ベンジルアミノ基、
ビス(4-ドコシルオキシフェニル)メチルアミノ基、
4-メトキシ-2-[3',4',5'-トリス(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、
4-メトキシ-2-[3',4',5'-トリス(オクタデシルオキシ)シクロヘキシルメチルオキシ]ベンジルアミノ基、
2,4-ビス(ドデシルオキシ)ベンジルアミノ基、
フェニル(2,3,4-トリス(オクタデシルオキシ)フェニル)メチルアミノ基、
ジ[4-(12-ドコシルオキシドデシルオキシ)フェニル]メチルアミノ基、
3,5-ビス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基、または
3,4,5-トリス[3',4',5'-トリ(オクタデシルオキシ)ベンジルオキシ]ベンジルアミノ基。
下記は化合物(I)を原料として本方法(工程)を実施した場合のスキームである。
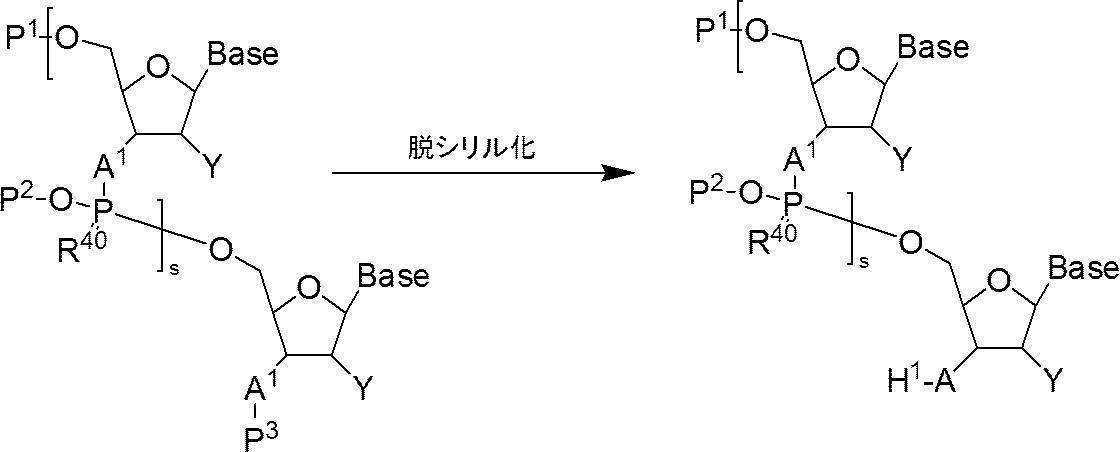
[式中、各記号の定義は上述の通りである]
本方法(工程)で用いられる溶媒としては、基質を溶解し、酸でないものであれば、反応が進行する限り特に限定されないが、ジクロロメタン、1,2-ジクロロエタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素溶媒、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒、またはペンタン、ヘキサン、ヘプタン、オクタン等の脂肪族炭化水素系溶媒、またはジエチルエーテル、テトラヒドロフラン、シクロペンチルメチルエーテル等のエーテル系溶媒、あるいはそれらの混合溶媒が好ましい。中でもテトラヒドロフランとジクロロメタンの混合溶媒が好ましく、特に2:1の混合溶媒が好ましい。
脱シリル化は、有機塩基の存在下、フッ化物イオン源を作用させることで実施される。該有機塩基は1又は2種類以上の混合物が用いられ、それらの塩基のうち少なくとも1種とフッ化水素との塩を該フッ化物イオン源とすることを特徴とする。
有機塩基は強塩基と弱塩基との混合物、2種類以上の弱塩基の混合物又は単一の弱塩基であってもよい。
強塩基としては、好ましくはpKa≧8のものであり、具体的には、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、プロピルアミン、イソプロピルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、ブチルアミン、イソブチルアミン、tert-ブチルアミン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、モルホリンのアミン類が挙げられ、好ましくはトリエチルアミンである。弱塩基としては、好ましくは4≦pKa<8のものであり、具体的には、ピリジン、2,6-ジメチルピリジン、2,4,6-トリメチルピリジン、ピペラジン、ピペリジン、イミダゾール、N-メチルイミダゾール、N-メチルモルホリン、N-エチルモルホリン等のヘテロ環状化合物、アニリン、トルイジン、ジメチルアニリン、ジエチルアニリン、エチルアニリン、エチルメチルアニリン、アニシジン等のアニリン類が挙げられ、好ましくはピリジン、N-メチルモルホリンであり、より好ましくはピリジンである。
有機塩基は、フッ化物イオンに対し1モル当量以上で用いることが好ましい。
有機塩基が強塩基と弱塩基との混合物の場合、それぞれの使用量は所望する反応に悪影響を及ぼさない範囲で適宜設定することができるが、チオリン酸上の脱シアノエチル化が起こる可能性があるので、過剰な強塩基の使用は避けることが好ましい。通常、強塩基はフッ化物イオンに対し1/3モル当量以下で使用する。弱塩基は強塩基とあわせて少なくとも1モル当量以上となる量で使用する。強塩基を1/3モル当量以下で使用しても、弱塩基を強塩基とあわせて1モル当量以上で使用しないと5’末端保護基の脱落が起こる。弱塩基の使用量は基質が溶解する限り特に限定されない。
フッ化物イオン源は、上記塩基のうち少なくとも1種とフッ化水素との塩であって、具体的にはトリエチルアミン五フッ化水素酸塩(5HF-TEA)、トリエチルアミン三フッ化水素酸塩(3HF-TEA)、ピリジンフッ化水素酸塩(HF-ピリジン)等が挙げられ、好ましくは3HF-TEAである。
本方法(工程)は上記フッ化物イオン源に有機塩基を添加した混合物、好ましくは強塩基のフッ化水素塩に弱塩基を添加した混合物、より好ましくは、
5HF-TEA又は3HF-TEAにピリジンを添加した混合物、特に好ましくは3HF-TEAにピリジンを添加した混合物(3HF-TEA-ピリジン)である。弱塩基のフッ化水素塩に弱塩基を添加してもよい。例えば、HF-ピリジンにN-メチルモルホリンを添加してもよい。
本方法(工程)の反応温度及び反応時間は、基質・生成物が析出しない限り特に限定されないが、それぞれ、通常、-10~80℃、好ましくは0~40℃、より好ましくは10~20℃、通常0.5~96時間、好ましくは1~48時間、より好ましくは1~24時間である。
本方法(工程)で用いられる溶媒としては、基質を溶解し、酸でないものであれば、反応が進行する限り特に限定されないが、ジクロロメタン、1,2-ジクロロエタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素溶媒、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒、またはペンタン、ヘキサン、ヘプタン、オクタン等の脂肪族炭化水素系溶媒、またはジエチルエーテル、テトラヒドロフラン、シクロペンチルメチルエーテル等のエーテル系溶媒、あるいはそれらの混合溶媒が好ましい。中でもテトラヒドロフランとジクロロメタンの混合溶媒が好ましく、特に2:1の混合溶媒が好ましい。
脱シリル化は、有機塩基の存在下、フッ化物イオン源を作用させることで実施される。該有機塩基は1又は2種類以上の混合物が用いられ、それらの塩基のうち少なくとも1種とフッ化水素との塩を該フッ化物イオン源とすることを特徴とする。
有機塩基は強塩基と弱塩基との混合物、2種類以上の弱塩基の混合物又は単一の弱塩基であってもよい。
強塩基としては、好ましくはpKa≧8のものであり、具体的には、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、プロピルアミン、イソプロピルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、ブチルアミン、イソブチルアミン、tert-ブチルアミン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、モルホリンのアミン類が挙げられ、好ましくはトリエチルアミンである。弱塩基としては、好ましくは4≦pKa<8のものであり、具体的には、ピリジン、2,6-ジメチルピリジン、2,4,6-トリメチルピリジン、ピペラジン、ピペリジン、イミダゾール、N-メチルイミダゾール、N-メチルモルホリン、N-エチルモルホリン等のヘテロ環状化合物、アニリン、トルイジン、ジメチルアニリン、ジエチルアニリン、エチルアニリン、エチルメチルアニリン、アニシジン等のアニリン類が挙げられ、好ましくはピリジン、N-メチルモルホリンであり、より好ましくはピリジンである。
有機塩基は、フッ化物イオンに対し1モル当量以上で用いることが好ましい。
有機塩基が強塩基と弱塩基との混合物の場合、それぞれの使用量は所望する反応に悪影響を及ぼさない範囲で適宜設定することができるが、チオリン酸上の脱シアノエチル化が起こる可能性があるので、過剰な強塩基の使用は避けることが好ましい。通常、強塩基はフッ化物イオンに対し1/3モル当量以下で使用する。弱塩基は強塩基とあわせて少なくとも1モル当量以上となる量で使用する。強塩基を1/3モル当量以下で使用しても、弱塩基を強塩基とあわせて1モル当量以上で使用しないと5’末端保護基の脱落が起こる。弱塩基の使用量は基質が溶解する限り特に限定されない。
フッ化物イオン源は、上記塩基のうち少なくとも1種とフッ化水素との塩であって、具体的にはトリエチルアミン五フッ化水素酸塩(5HF-TEA)、トリエチルアミン三フッ化水素酸塩(3HF-TEA)、ピリジンフッ化水素酸塩(HF-ピリジン)等が挙げられ、好ましくは3HF-TEAである。
本方法(工程)は上記フッ化物イオン源に有機塩基を添加した混合物、好ましくは強塩基のフッ化水素塩に弱塩基を添加した混合物、より好ましくは、
5HF-TEA又は3HF-TEAにピリジンを添加した混合物、特に好ましくは3HF-TEAにピリジンを添加した混合物(3HF-TEA-ピリジン)である。弱塩基のフッ化水素塩に弱塩基を添加してもよい。例えば、HF-ピリジンにN-メチルモルホリンを添加してもよい。
本方法(工程)の反応温度及び反応時間は、基質・生成物が析出しない限り特に限定されないが、それぞれ、通常、-10~80℃、好ましくは0~40℃、より好ましくは10~20℃、通常0.5~96時間、好ましくは1~48時間、より好ましくは1~24時間である。
[ホスホロアミダイト化オリゴヌクレオチドの製造方法]
本方法は、三価のリン上に2つの窒素置換基を有するホスフィチル化剤前駆体をモノ選択的に活性化してホスフィチル化剤とし、該剤を用いて、塩基存在下、オリゴヌクレオチドのフリーな3’末端あるいは5’末端をホスフィチル化する反応を含む。
即ち、本方法は、以下の工程(1)及び(2)を含有する、ホスホロアミダイト化オリゴヌクレオチドの製造方法である。
(1)溶媒中で、下記式(1):
本方法は、三価のリン上に2つの窒素置換基を有するホスフィチル化剤前駆体をモノ選択的に活性化してホスフィチル化剤とし、該剤を用いて、塩基存在下、オリゴヌクレオチドのフリーな3’末端あるいは5’末端をホスフィチル化する反応を含む。
即ち、本方法は、以下の工程(1)及び(2)を含有する、ホスホロアミダイト化オリゴヌクレオチドの製造方法である。
(1)溶媒中で、下記式(1):
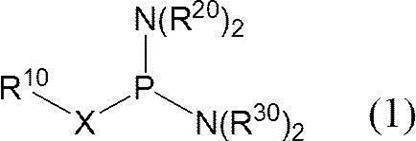
[式中、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R20およびR30はそれぞれ独立してアルキル基を示し、該アルキル基は隣接する窒素原子と一緒になって環を形成してもよい。]
で表されるホスフィチル化剤前駆体に活性化剤を作用させて下記式(2):
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R20およびR30はそれぞれ独立してアルキル基を示し、該アルキル基は隣接する窒素原子と一緒になって環を形成してもよい。]
で表されるホスフィチル化剤前駆体に活性化剤を作用させて下記式(2):

[式中、
Zaは活性化剤に由来する基を示し、
それ以外の各記号は、前記と同義である。]
で表されるホスフィチル化剤を調製する工程、および
(2)溶媒中で、5’位水酸基又は3’位水酸基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程
Zaは活性化剤に由来する基を示し、
それ以外の各記号は、前記と同義である。]
で表されるホスフィチル化剤を調製する工程、および
(2)溶媒中で、5’位水酸基又は3’位水酸基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程
工程(1)
式(1)中、R10の芳香環としては、フェニル、4-ニトロフェニル、2,4-ジニトロフェニル、ペンタフルオロフェニル、ペンタクロロフェニル、2-ブロモフェニル、4-ブロモフェニル、2-メチルフェニル、2,6-ジメチルフェニル等が挙げられ、好ましくは4-ニトロフェニルである。
式(1)中、R10における水酸基の保護基又はチオール基の保護基としては、具体的にはC1-6アルキル基(例、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、s-ブチル、t-ブチル、ペンチル、イソペンチル);シアノ化C1-6アルキル基(例、2-シアノエチル、2-シアノ-1,1-ジメチルエチル);置換シリル基で置換されたエチル基(例、2-メチルジフェニルシリルエチル、2-トリメチルシリルエチル、2-トリフェニルシリルエチル);ハロゲン化C1-6アルキル基(例、2,2,2-トリクロロエチル、2,2,2-トリブロモエチル、2,2,2-トリフルオロエチル);C2-6アルケニル基(例、エテニル、1-プロペニル、2-プロペニル、1-メチル-2-プロペニル、1-メチル-1-プロペニル);C3-6シクロアルキル基(例、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル);シアノ化C1-6アルケニル基(例、2-シアノブテニル);C7-11アラルキル基(例、ベンジル、α-ナフチルメチル、β-ナフチルメチル);C6-10アリール基(例、フェニル、インデニル、ナフチル)が挙げられ、より好ましくはシアノ化C1-6アルキル基であり、特に好ましくは2-シアノエチルである。
式(1)中、R20およびR30はそれぞれ独立してアルキル基を示すが、該アルキル基は隣接する窒素原子と一緒になって環(例、ピロリジン)を形成してもよい。R20およびR30は好ましくはいずれもイソプロピル基である。
ホスフィチル化剤前駆体としては、下記化合物が特に好ましい。
式(1)中、R10の芳香環としては、フェニル、4-ニトロフェニル、2,4-ジニトロフェニル、ペンタフルオロフェニル、ペンタクロロフェニル、2-ブロモフェニル、4-ブロモフェニル、2-メチルフェニル、2,6-ジメチルフェニル等が挙げられ、好ましくは4-ニトロフェニルである。
式(1)中、R10における水酸基の保護基又はチオール基の保護基としては、具体的にはC1-6アルキル基(例、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、s-ブチル、t-ブチル、ペンチル、イソペンチル);シアノ化C1-6アルキル基(例、2-シアノエチル、2-シアノ-1,1-ジメチルエチル);置換シリル基で置換されたエチル基(例、2-メチルジフェニルシリルエチル、2-トリメチルシリルエチル、2-トリフェニルシリルエチル);ハロゲン化C1-6アルキル基(例、2,2,2-トリクロロエチル、2,2,2-トリブロモエチル、2,2,2-トリフルオロエチル);C2-6アルケニル基(例、エテニル、1-プロペニル、2-プロペニル、1-メチル-2-プロペニル、1-メチル-1-プロペニル);C3-6シクロアルキル基(例、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル);シアノ化C1-6アルケニル基(例、2-シアノブテニル);C7-11アラルキル基(例、ベンジル、α-ナフチルメチル、β-ナフチルメチル);C6-10アリール基(例、フェニル、インデニル、ナフチル)が挙げられ、より好ましくはシアノ化C1-6アルキル基であり、特に好ましくは2-シアノエチルである。
式(1)中、R20およびR30はそれぞれ独立してアルキル基を示すが、該アルキル基は隣接する窒素原子と一緒になって環(例、ピロリジン)を形成してもよい。R20およびR30は好ましくはいずれもイソプロピル基である。
ホスフィチル化剤前駆体としては、下記化合物が特に好ましい。
活性化剤をホスフィチル化剤前駆体に作用させることで、ホスフィチル化剤が得られる。
活性化剤は、ホスホロアミダイト上のアミンを置換して、水酸基等との反応性置換基を付与し得る酸である。具体的には、pKa5以上の弱酸性活性化剤、より好ましくはpKa5以上のアゾール系化合物及びそのC上置換体から選ばれる少なくとも1種である。アゾール系化合物としては、テトラゾール、トリアゾール、イミダゾール等が挙げられ、C上置換体としては、ジシアノイミダゾール、ビス(トリフルオロメチル)イミダゾール、ジクロロイミダゾール等のハロゲン原子でジ置換された化合物が用いられる。特に好ましくはジシアノイミダゾール及びジクロロイミダゾールである。
式(2)において、Zaは活性化剤に由来する基であり、例えば活性化剤から水素原子を1つ除いた基であり、活性化剤としてジシアノイミダゾールを用いた場合、Zaはジシアノイミダゾリルであり、活性剤としてジクロロイミダゾールを用いた場合、Zaはジクロロイミダゾリルである。
活性化剤は、ホスホロアミダイト上のアミンを置換して、水酸基等との反応性置換基を付与し得る酸である。具体的には、pKa5以上の弱酸性活性化剤、より好ましくはpKa5以上のアゾール系化合物及びそのC上置換体から選ばれる少なくとも1種である。アゾール系化合物としては、テトラゾール、トリアゾール、イミダゾール等が挙げられ、C上置換体としては、ジシアノイミダゾール、ビス(トリフルオロメチル)イミダゾール、ジクロロイミダゾール等のハロゲン原子でジ置換された化合物が用いられる。特に好ましくはジシアノイミダゾール及びジクロロイミダゾールである。
式(2)において、Zaは活性化剤に由来する基であり、例えば活性化剤から水素原子を1つ除いた基であり、活性化剤としてジシアノイミダゾールを用いた場合、Zaはジシアノイミダゾリルであり、活性剤としてジクロロイミダゾールを用いた場合、Zaはジクロロイミダゾリルである。
工程(1)で使用する溶媒は、ホスフィチル化前駆体を溶解し、活性化剤が難溶性となるものであれば特に限定されないが、酸性または塩基性の官能基を通常有さない。ここで「難溶性」とは、溶媒中の活性化剤の濃度が6μM以下であることを目安とする。具体的には、トルエン、ベンゼン、o-キシレン、m-キシレン、p-キシレン、エチルベンゼン、ペンタン、ヘキサン、ヘプタン、オクタン、シクロペンタン、シクロヘキサン、ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、シクロペンチルメチルエーテル、四塩化炭素等が挙げられ、好ましくはトルエン、シクロヘキサン、特に好ましくはトルエンである。
本方法(工程)の反応温度及び反応時間は、基質・生成物が析出しない限り特に限定されないが、それぞれ、通常、40℃以下、好ましくは0~30℃、より好ましくは5~15℃、特に好ましくは10℃程度であり、通常0.5~24時間、好ましくは1~12時間、より好ましくは1~6時間である。
活性化剤とホスフィチル化剤前駆体との使用量は、ホスフィチル化前駆体が活性化される限り特に限定されないが、通常、ホスフィチル化前駆体に対して、通常過剰量で、好ましくは1.5~10モル当量で用いられる。過剰量の活性化剤をホスフィチル化前駆体に溶媒中で作用させることで、ホスフィチル化剤前駆体を活性化すると同時に、ホスフィチル化時に副生するジイソプロピルアミンを活性化剤との塩として沈殿させる。従って、必要に応じて、工程(1)と下記工程(2)との間に、沈殿等の不溶物を分離する工程を実施することができ、また、実施することが好ましい。
工程(2)
本工程は、溶媒中で、5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程を含む。
本工程は、溶媒中で、5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程を含む。
「5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)」の好ましい一態様は、5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていない下記構造(Ia)を有する化合物である(以下、化合物(Ia))。
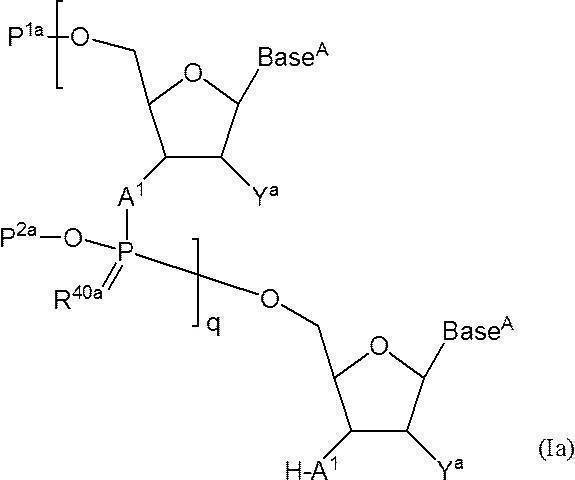
[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
P1aにおける水酸基の保護基としては、上記P1における水酸基の保護基として例示したものと同様のものを用いることができる。好ましくはジメトキシトリチル基である。
P2aにおけるリン酸基の保護基としては、上記P2におけるリン酸基の保護基として例示したものと同様のものを用いることができる。好ましくは-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基であり、WGとしてはシアノ基が好ましい。
R40aは、好ましくは同一で酸素原子又は硫黄原子である。
Yaにおける保護されていてもよい水酸基の保護基としては、上記Yにおける保護されていてもよい水酸基の保護基として例示したものと同様のものを用いることができ、好ましくはtert-ブチルジメチルシリル基である。
Yaにおける「4位炭素原子に架橋する有機基」としては、上記Yにおける「4位炭素原子に架橋する有機基」として例示したものと同様のものを用いることができる。
BaseAの「保護されていてもよい核酸塩基」としては、上記Baseの「保護されていてもよい核酸塩基」と同様のものを用いることができ、q+1個存在するBaseAの少なくとも1つは式-L-YL-Z(上述と同義)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基で保護されていることが好ましい。
下記は化合物(Ia)を原料として本工程(2)を実施した場合のスキームである。
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。]
P1aにおける水酸基の保護基としては、上記P1における水酸基の保護基として例示したものと同様のものを用いることができる。好ましくはジメトキシトリチル基である。
P2aにおけるリン酸基の保護基としては、上記P2におけるリン酸基の保護基として例示したものと同様のものを用いることができる。好ましくは-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基であり、WGとしてはシアノ基が好ましい。
R40aは、好ましくは同一で酸素原子又は硫黄原子である。
Yaにおける保護されていてもよい水酸基の保護基としては、上記Yにおける保護されていてもよい水酸基の保護基として例示したものと同様のものを用いることができ、好ましくはtert-ブチルジメチルシリル基である。
Yaにおける「4位炭素原子に架橋する有機基」としては、上記Yにおける「4位炭素原子に架橋する有機基」として例示したものと同様のものを用いることができる。
BaseAの「保護されていてもよい核酸塩基」としては、上記Baseの「保護されていてもよい核酸塩基」と同様のものを用いることができ、q+1個存在するBaseAの少なくとも1つは式-L-YL-Z(上述と同義)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基で保護されていることが好ましい。
下記は化合物(Ia)を原料として本工程(2)を実施した場合のスキームである。

[式中、各記号の定義は上述の通りである]
本工程で用いる、化合物(Ia)に例示される「5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)」は、上記化合物(I)の3’末端のシリル保護基を脱シリル化、好ましくは上記した本発明の脱シリル化によって得られたものであってもよい。
「5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)」の好ましい一態様は、3’位水酸基又は3’位アミノ基が保護され、かつ5’位水酸基が保護されていない下記構造(Ia’)を有する化合物である(以下、化合物(Ia’))。

[式中、P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ia)と同義である]
P1a’における水酸基の保護基としては、上記P1における水酸基の保護基として例示したものと同様のものを用いることができる。
P1a’におけるアミノ基の保護基としては、特に限定されず、例えば、ピバロイル基、ピバロイロキシメチル基、アセチル基、トリフルオロアセチル基、フェノキシアセチル基、4-イソプロピルフェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、ベンゾイル基、イソブチリル基、(2-ヘキシル)デカノイル基、ジメチルホルムアミジニル基、1-(ジメチルアミノ)エチリデン基、9-フルオレニルメチルオキシカルボニル基を挙げることができる。
下記は化合物(Ia’)を原料として本工程(2)を実施した場合のスキームである。
P1a’における水酸基の保護基としては、上記P1における水酸基の保護基として例示したものと同様のものを用いることができる。
P1a’におけるアミノ基の保護基としては、特に限定されず、例えば、ピバロイル基、ピバロイロキシメチル基、アセチル基、トリフルオロアセチル基、フェノキシアセチル基、4-イソプロピルフェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、ベンゾイル基、イソブチリル基、(2-ヘキシル)デカノイル基、ジメチルホルムアミジニル基、1-(ジメチルアミノ)エチリデン基、9-フルオレニルメチルオキシカルボニル基を挙げることができる。
下記は化合物(Ia’)を原料として本工程(2)を実施した場合のスキームである。
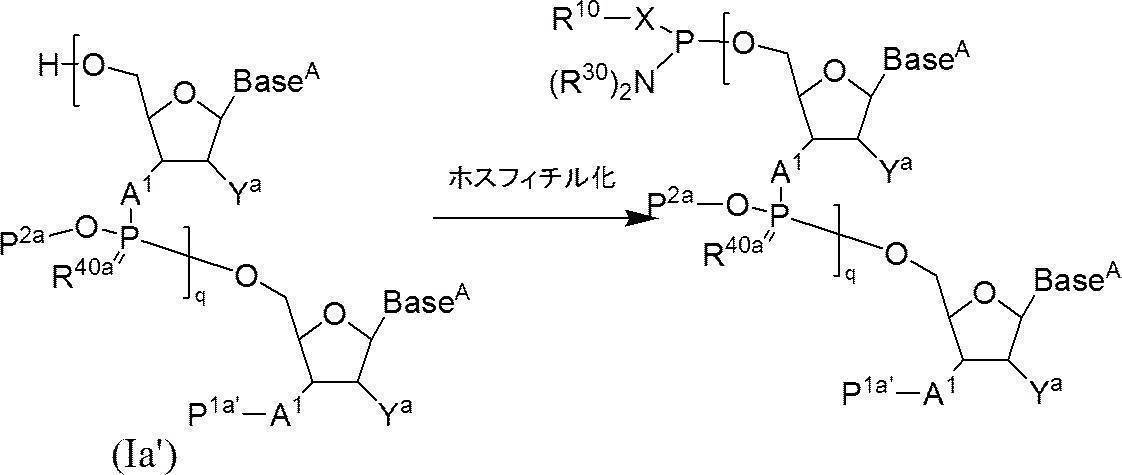
[式中、各記号の定義は上述の通りである]
工程(2)で使用する溶媒としては、工程(1)で使用した溶媒と同様のものを用いることができ、具体的には、トルエン、ジクロロメタン、クロロホルム等が挙げられ、好ましくはトルエンとジクロロメタンの混合溶媒である。
工程(2)で使用する塩基は、反応により生じた酸(活性化剤)の中和に十分な塩基性を有し、且つリン酸上の脱シアノエチルを引き起こさずP-N結合を形成しない塩基が選択される。そのような塩基としては、具体的には、pKa5~8の塩基、好ましくは、コリジン、N-メチルモルホリン、ジエチルアニリン等が用いられる。pKa8より高い塩基を用いると脱シアノ化が顕著となり、pKa5未満の塩基を用いると反応の進行に伴い再生した活性化剤の捕捉が不十分となり副生物が生じる。
工程(2)では、ホスフィチル化前駆体は添加してもしなくてもよいが、添加することが好ましい。
工程(2)で使用する塩基は、反応により生じた酸(活性化剤)の中和に十分な塩基性を有し、且つリン酸上の脱シアノエチルを引き起こさずP-N結合を形成しない塩基が選択される。そのような塩基としては、具体的には、pKa5~8の塩基、好ましくは、コリジン、N-メチルモルホリン、ジエチルアニリン等が用いられる。pKa8より高い塩基を用いると脱シアノ化が顕著となり、pKa5未満の塩基を用いると反応の進行に伴い再生した活性化剤の捕捉が不十分となり副生物が生じる。
工程(2)では、ホスフィチル化前駆体は添加してもしなくてもよいが、添加することが好ましい。
[末端に官能基を有するオリゴヌクレオチドの製造方法]
本方法は、末端の水酸基がホスホロアミダイト化されたオリゴヌクレオチドの該末端に直接又はリンカーを介して官能基を連結させることを含む。より具体的には、3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に直接又はリンカーを介して官能基を連結させること、あるいは5’末端ホスホロアミダイト化オリゴヌクレオチドの5’末端に直接又はリンカーを介して官能基を連結させることを含む。「3’末端ホスホロアミダイト化オリゴヌクレオチド」及び「5’末端ホスホロアミダイト化オリゴヌクレオチド」は任意の方法で合成されるものであってもよいが、好ましくは上記した本発明の「末端ホスホロアミダイト化オリゴヌクレオチドの製造方法」によって得られる下記構造を有する化合物(Ia-1)及び(Ia’-1)である。
本方法は、末端の水酸基がホスホロアミダイト化されたオリゴヌクレオチドの該末端に直接又はリンカーを介して官能基を連結させることを含む。より具体的には、3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に直接又はリンカーを介して官能基を連結させること、あるいは5’末端ホスホロアミダイト化オリゴヌクレオチドの5’末端に直接又はリンカーを介して官能基を連結させることを含む。「3’末端ホスホロアミダイト化オリゴヌクレオチド」及び「5’末端ホスホロアミダイト化オリゴヌクレオチド」は任意の方法で合成されるものであってもよいが、好ましくは上記した本発明の「末端ホスホロアミダイト化オリゴヌクレオチドの製造方法」によって得られる下記構造を有する化合物(Ia-1)及び(Ia’-1)である。

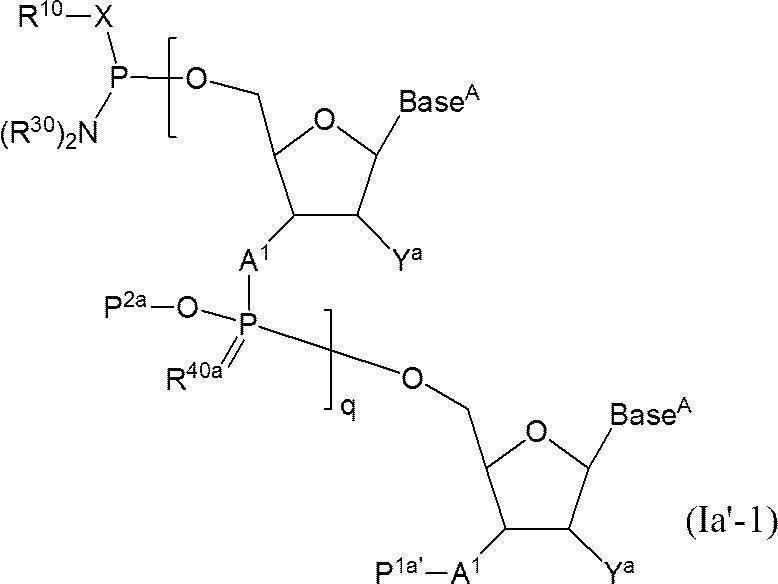
[式中、各記号の定義は上述の通りである]
化合物(Ia-1)の3’末端に直接又はリンカーを介して官能基を連結して下記化合物(Ia-2)を、化合物(Ia’-1)の5’末端に直接又はリンカーを介して官能基を連結して下記化合物(Ia’-2)を製造する。
化合物(Ia-1)の3’末端に直接又はリンカーを介して官能基を連結して下記化合物(Ia-2)を、化合物(Ia’-1)の5’末端に直接又はリンカーを介して官能基を連結して下記化合物(Ia’-2)を製造する。


式中、Lxは単結合又はリンカーを示す。Lxが単結合の場合、官能基は化合物(Ia-1)あるいは化合物(Ia’-1)に直接連結している。官能基の水酸基、チオール基若しくはアミノ基で直接化合物(Ia-1)の3’末端に、あるいは(Ia’-1)の5’末端に連結する。Lxがリンカーである場合には、水酸基、チオール基、アミノ基を反応点として有するリンカー(例、-O-、-S-、-COO-、-OCONH-、および-CONH-)を介して化合物(Ia-1)の3’末端に、あるいは(Ia’-1)の5’末端に連結する。
式中、Gは官能基であり、例えばオリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来するものが挙げられる。好ましくはオリゴヌクレオチド及びモノヌクレオシドであり、より好ましくはオリゴヌクレオチドである。
式中、Gは官能基であり、例えばオリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来するものが挙げられる。好ましくはオリゴヌクレオチド及びモノヌクレオシドであり、より好ましくはオリゴヌクレオチドである。
例えば、官能基が、3’位水酸基又は3’位アミノ基あるいは3’リン酸基が保護され、かつ5’位水酸基が保護されていない、下記構造(Ib):

[式中、
r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
を有するn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である場合、本方法は、オリゴヌクレオチドのフラグメント縮合に相当し、具体的には以下の工程を含む。
r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。]
を有するn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である場合、本方法は、オリゴヌクレオチドのフラグメント縮合に相当し、具体的には以下の工程を含む。

例えば、官能基が、5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていない、下記構造(Ib’):

[式中、P3bは、水酸基の保護基を示すか、又は-O-P3b’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、各記号の定義は式(Ib)と同義である]を有するn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である場合、本方法は、オリゴヌクレオチドのフラグメント縮合に相当し、具体的には以下の工程を含む。

[式中、各記号の定義は上述の通りである。]
BaseBの「保護されていてもよい核酸塩基」としては、上記Baseの「保護されていてもよい核酸塩基」と同様のものを用いることができ、r+1個存在するBaseBの少なくとも1つは式-L-YL-Z(上述と同義)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基で保護されていることが好ましい。
P2bにおけるリン酸基の保護基としては、塩基性条件下で脱保護可能であり、リン酸基の保護基として用いられるものであれば、特に限定はされないが、-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基が好ましく、WGとしてはシアノ基が好ましい。
R40bは、好ましくは同一で酸素原子又は硫黄原子である。
Ybにおける保護されていてもよい水酸基の保護基としては、特に限定されず、Yの定義において例示されたものが同様に用いられる。
Ybにおける「4位炭素原子に架橋する有機基」としては、Ybの定義において例示されたものが同様に用いられる。
P3b’における水酸基の保護基としては、連結後に得られるオリゴヌクレオチドの5’位水酸基の保護基(化合物(Ib)の場合)あるいは3’位水酸基の保護基(化合物(Ib’)の場合)を除去し得る酸性条件で安定な基であり、縮合反応において反応が進行するように反応溶媒にn’個重合オリゴヌクレオチド(化合物(Ib)または化合物(Ib’))を溶解させうるものであれば特に限定はないが、式-L-YL-Z(各記号の定義は上述の通り)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基を使用することができる。好ましくは、Lはスクシニル基である。また、P3b’における水酸基の保護基としては、上記脱シリル化の項で記載したものと同様のものを用いることもでき、その場合、好ましくはジメトキシトリチル基である。
BaseBの「保護されていてもよい核酸塩基」としては、上記Baseの「保護されていてもよい核酸塩基」と同様のものを用いることができ、r+1個存在するBaseBの少なくとも1つは式-L-YL-Z(上述と同義)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基で保護されていることが好ましい。
P2bにおけるリン酸基の保護基としては、塩基性条件下で脱保護可能であり、リン酸基の保護基として用いられるものであれば、特に限定はされないが、-CH2CH2WG(WGは、電子吸引性基を示す。)で表される基が好ましく、WGとしてはシアノ基が好ましい。
R40bは、好ましくは同一で酸素原子又は硫黄原子である。
Ybにおける保護されていてもよい水酸基の保護基としては、特に限定されず、Yの定義において例示されたものが同様に用いられる。
Ybにおける「4位炭素原子に架橋する有機基」としては、Ybの定義において例示されたものが同様に用いられる。
P3b’における水酸基の保護基としては、連結後に得られるオリゴヌクレオチドの5’位水酸基の保護基(化合物(Ib)の場合)あるいは3’位水酸基の保護基(化合物(Ib’)の場合)を除去し得る酸性条件で安定な基であり、縮合反応において反応が進行するように反応溶媒にn’個重合オリゴヌクレオチド(化合物(Ib)または化合物(Ib’))を溶解させうるものであれば特に限定はないが、式-L-YL-Z(各記号の定義は上述の通り)で表される保護基、若しくは式-Z’(上述と同義)で表される保護基を使用することができる。好ましくは、Lはスクシニル基である。また、P3b’における水酸基の保護基としては、上記脱シリル化の項で記載したものと同様のものを用いることもでき、その場合、好ましくはジメトキシトリチル基である。
P3b’として-L-YL-Zを有するn’個重合オリゴヌクレオチド(1b’)は5’位に-L-YL-Zを有し、3’位が水酸基であるモノヌクレオシドに、3’位が保護された水酸基、5’位がホスホロアミダイトであるモノマーヌクレオシドをWO2012157723A1記載と同様の液相中でのホスホロアミダイト法により連結を繰り返すことで調製可能である。ここで、該モノマーヌクレオシドは、既知文献(例、Wagner, T.; Pfleiderer, W. Nucleoside Nucleotides 1997, 16, 1657-1660.;US8541569B2)に基づいて調製することができる。
本発明の好ましい一実施態様は、下記式(Ia-1’):

[式中、
q+1個のBaseA’は、それぞれ独立して式-L’-YL-Z(式中、L’はスクシニル基であり、YL、Zは上述と同義である)で表される保護基、若しくは式:-Z’(上述と同義)で表される保護基で保護されていてもよい核酸塩基を示し、
それ以外の各記号の定義は上述と同義である]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させて下記式(Ia-2’):
q+1個のBaseA’は、それぞれ独立して式-L’-YL-Z(式中、L’はスクシニル基であり、YL、Zは上述と同義である)で表される保護基、若しくは式:-Z’(上述と同義)で表される保護基で保護されていてもよい核酸塩基を示し、
それ以外の各記号の定義は上述と同義である]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させて下記式(Ia-2’):

[式中、
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドを得た後、保護基を除去する工程を含む、式(II):
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドを得た後、保護基を除去する工程を含む、式(II):

[式中、q+1個のBase’は無保護の核酸塩基を示し、それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法である。
〔本発明の製造方法〕
次に、本発明の方法を用いたオリゴヌクレオチドの製造方法(以下、「本発明の製造方法」ともいう。)について説明する。
本発明は、以下の工程(1)及び(2)を含有するオリゴヌクレオチドの製造方法である。
(1)3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個重合オリゴヌクレオチドを、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドと、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程、ならびに
(2)工程(1)の反応液に、酸化剤、または硫化剤を添加して、工程(1)で得られたn+n’個重合オリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合へと変換する工程。
以下に、各工程について詳細に説明する。
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法である。
〔本発明の製造方法〕
次に、本発明の方法を用いたオリゴヌクレオチドの製造方法(以下、「本発明の製造方法」ともいう。)について説明する。
本発明は、以下の工程(1)及び(2)を含有するオリゴヌクレオチドの製造方法である。
(1)3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個重合オリゴヌクレオチドを、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドと、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程、ならびに
(2)工程(1)の反応液に、酸化剤、または硫化剤を添加して、工程(1)で得られたn+n’個重合オリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合へと変換する工程。
以下に、各工程について詳細に説明する。
工程(1)(縮合工程)
本工程は、3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個オリゴヌクレオチドを、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドと、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程である。
3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個重合オリゴヌクレオチドと、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドとの縮合工程は、通常、縮合活性化剤の存在下で実施される。
本工程は、3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個オリゴヌクレオチドを、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドと、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程である。
3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個重合オリゴヌクレオチドと、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドとの縮合工程は、通常、縮合活性化剤の存在下で実施される。
本工程に使用する縮合活性化剤としては、特に限定されないが、5-(ベンジルチオ)-1H-テトラゾール(BTT)、5-(エチルチオ)-1H-テトラゾール(ETT)、4,5-ジシアノイミダゾール(DCI)、テトラゾール、5-[3,5-ビス(トリフルオロメチル)フェニル]-1H-テトラゾール(Activator 42(登録商標))、ベンゾイミダゾールトリフラート(BIT)、ピリジン・トリフルオロ酢酸塩等を使用することが好ましい。BTT、ETT、DCIがより好ましく、BTTが特に好ましい。
本工程における縮合活性化剤の使用量は、ホスホロアミダイト化オリゴヌクレオチド1モルに対し、通常0.5~10モル、好ましくは1.0~2.0モル、特に好ましくは1.0モルである。
本工程の反応温度は、反応が進行しさえすれば特に限定されないが、0~100℃が好ましく、20~50℃がより好ましく、20~30℃が特に好ましい。反応時間は、使用するオリゴヌクレオチドの種類、溶媒の種類、反応温度等により異なるが、30分~24時間である。
本工程は、反応に影響を及ぼさない溶媒中で行われる。具体的には、ピリジン、THF、ジクロロメタン、クロロホルム、トルエン、アセトニトリルまたはこれら任意の混合溶媒を挙げることができる。
3’位水酸基がホスホロアミダイト化され、かつ5’位水酸基が保護されたn個オリゴヌクレオチド及び、3’位水酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチドとの使用量は、いずれの基質をより完全な消費に近付ける事を意図するかに依存するが、後者1モルに対して前者が0.1~10モルが好ましく、0.5~3モルがより好ましく、1.0~2.0モルが特に好ましい。
工程(2)(酸化工程、または硫化工程)
工程(1)で得られたn+n’個重合オリゴヌクレオチドに酸化剤または硫化剤を反応させることにより、該n+n’個重合オリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合へと変換する工程である。
工程(1)で得られたn+n’個重合オリゴヌクレオチドに酸化剤または硫化剤を反応させることにより、該n+n’個重合オリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合へと変換する工程である。
本工程は、工程(1)で得られたn+n’個重合オリゴヌクレオチドを単離することなく、工程(2)後の反応液に、酸化剤または硫化剤を、直接添加するだけで行うことができる。
本工程に使用する「酸化剤」としては、他の部位を酸化することなく、ホスファイトトリエステル結合を、ホスフェートトリエステル結合に酸化する能力がありさえすれば、特に限定されないが、ヨウ素、(1S)-(+)-(10-カンファニルスルフォニル)オキサジリジン、tert-ブチルヒドロペルオキシド(TBHP)、2-ブタノンペルオキシド、1,1-ジヒドロペルオキシシクロドデカン、ビス(トリメチルシリル)ペルオキシド、m-クロロ過安息香酸を使用することが好ましい。良好な酸化反応が達成できるという観点で、ヨウ素、(1S)-(+)-(10-カンファニルスルフォニル)オキサジリジン、tert-ブチルヒドロペルオキシド、2-ブタノンペルオキシド、1,1-ジヒドロペルオキシシクロドデカンがより好ましく、ヨウ素、(1S)-(+)-(10-カンファニルスルフォニル)オキサジリジン、tert-ブチルヒドロペルオキシド、2-ブタノンペルオキシドが更に好ましく、ヨウ素、tert-ブチルヒドロペルオキシドが更に一層好ましく、ヨウ素が特に好ましい。かかる酸化剤は、0.05~2Mの濃度になるように適当な溶媒で希釈して使用することができる。かかる希釈溶媒としては、反応に不活性な溶媒であれば特に限定されないが、ピリジン、THF、ジクロロメタン、水、またはこれら任意の混合溶媒を挙げることができる。中でも、例えば、ヨウ素/水/ピリジン―THFあるいはヨウ素/ピリジン―酢酸や過酸化剤(TBHP)/ジクロロメタンを用いるのが好ましい。
本工程に使用する「硫化剤」としては、ホスファイトトリエステル結合を、チオホスフェートトリエステル結合に変換しうる能力がありさえすれば、特に限定されないが、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン(DDTT)、3H-1,2-ベンゾジチオール-3-オン-1,1-ジオキシド(Beaucage試薬)、3H-1,2-ベンゾジチオール-3-オン、フェニルアセチルジスルフィド(PADS)、テトラエチルチウラムジスルフィド(TETD)、3-アミノ-1,2,4-ジチアゾール-5-チオン(ADTT)、硫黄を使用することが好ましい。良好な反応が進行しうるという観点で、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン(DDTT)、3H-1,2-ベンゾジチオール-3-オン-1,1-ジオキシド(Beaucage試薬)、3H-1,2-ベンゾジチオール-3-オン、フェニルアセチルジスルフィド(PADS)がより好ましく、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン、3H-1,2-ベンゾジチオール-3-オン-1,1-ジオキシドが更に好ましく、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオンが特に好ましい。かかる硫化剤は、0.05~2Mの濃度になるように適当な溶媒で希釈して使用することができる。かかる希釈溶媒としては、反応に不活性な溶媒であれば特に限定されないが、例えば、ジクロロメタン、アセトニトリル、ピリジン又はこれら任意の混合溶媒が挙げられる。
酸化剤または硫化剤の使用量は、工程(1)で得られたn+n’個重合オリゴヌクレオチド(iv)1モルに対し、1~50モル使用することができ、好ましくは1~5モルである。
反応温度は、反応が進行しさえすれば特に限定されないが、0℃~100℃が好ましく、20℃~50℃がより好ましい。反応時間は、n+n’個重合オリゴヌクレオチド(iv)の種類、使用する酸化剤または硫化剤の種類、反応温度等によって異なるが、1分~3時間である。
更に、所望により下記工程(3)を含有させることにより、簡便かつ効果的に過剰原料や副生物を除去して、n+n’個重合オリゴヌクレオチドを精製することができる。
(3)工程(2)で得られた反応液に極性溶媒を添加して、n+n’個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程。
(3)工程(2)で得られた反応液に極性溶媒を添加して、n+n’個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程。
本発明の製造方法には、更に、工程(4)を含有させることにより、オリゴヌクレオチドを単離・製造することができる。
(4)工程(3)で得られたn+n’個重合オリゴヌクレオチドの保護基を全て除去する工程。
(4)工程(3)で得られたn+n’個重合オリゴヌクレオチドの保護基を全て除去する工程。
工程(3)(沈殿化および固液分離工程)
本工程は、工程(2)で得られたホスフェートトリエステル結合またはチオホスフェートトリエステル結合を有するn+n’個重合オリゴヌクレオチドを含む反応液に極性溶媒を添加することにより該n+n’個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程である。
本工程は、工程(2)で得られたホスフェートトリエステル結合またはチオホスフェートトリエステル結合を有するn+n’個重合オリゴヌクレオチドを含む反応液に極性溶媒を添加することにより該n+n’個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程である。
本工程における目的物(n+n’個重合オリゴヌクレオチド)を沈殿させるための極性溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のアルコール系溶媒、アセトニトリル、プロピオニトリル等のニトリル系溶媒、ジメチルホルムアミド、ジメチルアセトアミド等のアミド系溶媒、ジメチルスルホキシド、水等、ならびにこれら2種以上の混合溶媒が挙げられる。中でも、アルコール系溶媒、ニトリル系溶媒が好ましく、メタノールまたはアセトニトリルがより好適に使用される。本発明における極性溶媒としては、特に実用的観点からアセトニトリルが好ましい。
酸化剤としてヨウ素を使用した場合の沈殿化の際には、沈殿化溶媒であるメタノールにチオ硫酸ナトリウム(ハイポ)を飽和させた溶液を使用することにより、ヨウ素による着色を除去することができ、5’位水酸基が保護されたn+n’個重合オリゴヌクレオチドを純度良く単離することが可能である。
硫化剤を使用した場合の沈殿化の際には、沈殿化溶媒であるメタノールに3価のリン試薬(例えば、トリメチルホスファイト、トリエチルホスファイト、トリス(2-カルボキシエチル)ホスフィン等)、ハイポ等の還元剤を飽和させた溶液を使用することにより、5’位水酸基が保護されたn+n’個重合オリゴヌクレオチドを純度良く単離することが可能である。
本発明のオリゴヌクレオチドの製造方法は、上記工程(1)~(3)を所望の回数繰返すことで高純度かつ高収率で目的のオリゴヌクレオチドを得ることができる。
工程(4)(脱保護・オリゴヌクレオチド単離工程)
本発明のオリゴヌクレオチドの製造方法においては、工程(3)の後に、保護基の種類・性質に応じて、脱保護を行い、オリゴヌクレオチドを単離することができる。脱保護の方法としては、例えば、グリーンズ プロテクティブ・グループス・イン・オーガニック・シンセシス(Greene’s PROTECTIVE GROUPS IN ORGANIC SYNTHESIS)、第4版、ジョン・ウィリー・アンド・サンズ(JOHN WILLY&SONS)出版(2006年)等に記載されている脱保護方法に従い、オリゴヌクレオチドの全ての保護基を除去する工程を行うことができる。具体的には、本発明における核酸塩基の保護基であるフェノキシアセチル基、アセチル基等、リン酸基に結合しているシアノエチル基等は、アンモニア水/エタノール溶液で処理することにより、全て除去することができる。
保護基を有しないオリゴヌクレオチドは酵素により容易に分解されやすいため、空気清浄度管理下でオリゴヌクレオチドを単離することが好ましい。
本発明のオリゴヌクレオチドの製造方法においては、工程(3)の後に、保護基の種類・性質に応じて、脱保護を行い、オリゴヌクレオチドを単離することができる。脱保護の方法としては、例えば、グリーンズ プロテクティブ・グループス・イン・オーガニック・シンセシス(Greene’s PROTECTIVE GROUPS IN ORGANIC SYNTHESIS)、第4版、ジョン・ウィリー・アンド・サンズ(JOHN WILLY&SONS)出版(2006年)等に記載されている脱保護方法に従い、オリゴヌクレオチドの全ての保護基を除去する工程を行うことができる。具体的には、本発明における核酸塩基の保護基であるフェノキシアセチル基、アセチル基等、リン酸基に結合しているシアノエチル基等は、アンモニア水/エタノール溶液で処理することにより、全て除去することができる。
保護基を有しないオリゴヌクレオチドは酵素により容易に分解されやすいため、空気清浄度管理下でオリゴヌクレオチドを単離することが好ましい。
上記各工程における反応の進行の確認は、いずれも一般的な液相有機合成反応と同様の方法を適用できる。すなわち、薄層シリカゲルクロマトグラフィー、高速液体クロマトグラフィー等を用いて反応を追跡することができる。
工程(3)、あるいは工程(4)より得られたオリゴヌクレオチドは、更に有機合成反応を施すことにより、所望のオリゴヌクレオチド誘導体へと導くこともできる。
工程(4)より得られたオリゴヌクレオチドにより、RNA、DNA、オリゴ核酸医薬を提供することができる。
工程(4)より得られたオリゴヌクレオチドにより、RNA、DNA、オリゴ核酸医薬を提供することができる。
また、官能基が、5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていない、下記構造(Ib’)を有するn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である場合も同様に実施することができる。

[式中、各記号の定義は上述の通りである]
以下、実施例に沿って本発明を詳細に説明するが、これら調製例、実施例は本発明の範囲をなんら限定するものではない。また、本発明において使用する試薬や装置、材料はとくに言及されない限り、商業的に入手可能である。また、本明細書において、略号で表示する場合、各表示はIUPAC-IUB Commission on Biochemical Nomenclatureにより略号あるいは当該分野における慣用名に基づくものである。
以下の調製例及び、実施例中の収率はmol/mol%を示す。特段の定義が無い限り、本明細書中における「%」は「質量%」を表す。また、以下の調製例および実施例中の溶媒の比率は、体積比を示す。1H-NMRスペクトルは内部標準としてテトラメチルシランを用い、測定溶媒としてCDCl3を用いた。NMRスペクトルはBruker AVANCE 400 (400MHZ)核磁気共鳴装置を用いて測定した。
エレクトロスプレーイオン化液体クロマトグラフィー/質量分析(以下LC/MSと略す)はAgilent Technologies 1290 Infinityを用いて測定した。
下記調製例、実施例中で使用される略号は以下の通りである。ヌクレオシドの核酸塩基が保護されている場合は、保護基は各ヌクレオシドの後に上付きで表示するものとする。
dT:2’-デオキシチミジン
dC:2’-デオキシシチジン
dG:2’-デオキシグアノシン
dA:2’-デオキシアデノシン
DMTr:4,4’-ジメトキシトリチル
Ac:アセチル
Bz:ベンゾイル
dma:ジメチルアミノエチリデン
iBu:イソブチリル
PA:(2-シアノエチル)-N,N-ジイソプロピルホスホロアミダイト
TBDMS:tert-ブチルジメチルシリル
DIPPS:ジイソプロピルフェニルシリル
HD:ヘキシルデカノイル
IPODIPS:イソプロポキシジイソプロピルシリル
TOB:3,4,5-トリス(オクタデシルオキシ)ベンジル
CO-TOP:3,4,5-トリス(オクタデシルオキシ)ベンゾイル
suc:スクシネート
TEA:トリエチルアミン
以下の調製例及び、実施例中の収率はmol/mol%を示す。特段の定義が無い限り、本明細書中における「%」は「質量%」を表す。また、以下の調製例および実施例中の溶媒の比率は、体積比を示す。1H-NMRスペクトルは内部標準としてテトラメチルシランを用い、測定溶媒としてCDCl3を用いた。NMRスペクトルはBruker AVANCE 400 (400MHZ)核磁気共鳴装置を用いて測定した。
エレクトロスプレーイオン化液体クロマトグラフィー/質量分析(以下LC/MSと略す)はAgilent Technologies 1290 Infinityを用いて測定した。
下記調製例、実施例中で使用される略号は以下の通りである。ヌクレオシドの核酸塩基が保護されている場合は、保護基は各ヌクレオシドの後に上付きで表示するものとする。
dT:2’-デオキシチミジン
dC:2’-デオキシシチジン
dG:2’-デオキシグアノシン
dA:2’-デオキシアデノシン
DMTr:4,4’-ジメトキシトリチル
Ac:アセチル
Bz:ベンゾイル
dma:ジメチルアミノエチリデン
iBu:イソブチリル
PA:(2-シアノエチル)-N,N-ジイソプロピルホスホロアミダイト
TBDMS:tert-ブチルジメチルシリル
DIPPS:ジイソプロピルフェニルシリル
HD:ヘキシルデカノイル
IPODIPS:イソプロポキシジイソプロピルシリル
TOB:3,4,5-トリス(オクタデシルオキシ)ベンジル
CO-TOP:3,4,5-トリス(オクタデシルオキシ)ベンゾイル
suc:スクシネート
TEA:トリエチルアミン
実施例1:N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-O-(tert-ブチルジメチルシリル)-5’-O-(4,4’-ジメトキシトリチル)デオキシシチジン(DMTr-dCCO-TOP-TBDMS)の合成
(1)5’-O-(4,4’-ジメトキシトリチル)-3’-O-(tert-ブチルジメチルシリル)デオキシシチジンの合成
(1)5’-O-(4,4’-ジメトキシトリチル)-3’-O-(tert-ブチルジメチルシリル)デオキシシチジンの合成

アルゴン雰囲気下、5’-O-(4,4’-ジメトキシトリチル)デオキシシチジン(6.32g,11.9mmol)を乾燥N,N-ジメチルホルムアミド(24.0mL)に溶解し、イミダゾール(2.47g,36.4mmol)、tert-ブチルジメチルシリルクロライド(2.71g,18.0mmol)を加えて、35℃で12時間撹拌した。反応の完結を薄層クロマトグラフィーにより確認後の反応液にジクロロメタン(20mL)及び5%炭酸水素ナトリウム水溶液(10mL)を加えて抽出した後、水(20mL)、続いて飽和食塩水(20mL)で洗浄した。有機層を硫酸ナトリウムにより乾燥させた後、溶媒を減圧下留去して得られた粗生成物をシリカゲルカラムクロマトグラフィー精製(溶出液:1%トリエチルアミン含有のn-ヘキサン:酢酸エチル=1:1→0:1続いて、1%トリエチルアミン含有のジクロロメタン:メタノール=10:1→5:1)し、表題化合物(6.81g,10.5mmol,88.9%)を得た。
1H NMR (400 MHz, CDCl3) δ: -0.07 (s, 3H, SiCH3), -0.01 (s, 3H, SiCH3), 0.80 (s, 9H, tBu), 1.81 (m, 2H, 4-NH2), 2.19 (ddd, J = 4.8, 6.4, 13.2 Hz, 1H, 2'-CHH ), 2.47 (ddd, J = 6.4, 6.8, 13.2 Hz, 2'-CHH), 3.29 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.53 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.80 (s, 6H, OCH3 of DMTr), 3.94 (ddd, J = 2.8, 2.8, 6.4 Hz, 1H, 4'-CH), 4.60 (dd, J = 6.4, 6.4, 6.4 Hz, 1H, 3'-H), 5.38 (d, J = 7.2 Hz, 5-H), 6.26 (dd, J = 6.8, 4.8 Hz, 1H, 1'-H), 6.84 (d, J = 8.8 Hz, 4H, 2, 2', 6, 6'-H of DMTr), 7.2-7.4 (m, 8H, Ar and 5-H of cytidine), 7.40 (d, J = 8.4 Hz, 2H, 2, 6-H of Ph), 8.0-8.2 (m, 2H, -NH2).
(2)N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-O-(tert-ブチルジメチルシリル)-5’-O-(4,4’-ジメトキシトリチル)デオキシシチジン(DMTr-dCCO-TOP-TBDMS)の合成
1H NMR (400 MHz, CDCl3) δ: -0.07 (s, 3H, SiCH3), -0.01 (s, 3H, SiCH3), 0.80 (s, 9H, tBu), 1.81 (m, 2H, 4-NH2), 2.19 (ddd, J = 4.8, 6.4, 13.2 Hz, 1H, 2'-CHH ), 2.47 (ddd, J = 6.4, 6.8, 13.2 Hz, 2'-CHH), 3.29 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.53 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.80 (s, 6H, OCH3 of DMTr), 3.94 (ddd, J = 2.8, 2.8, 6.4 Hz, 1H, 4'-CH), 4.60 (dd, J = 6.4, 6.4, 6.4 Hz, 1H, 3'-H), 5.38 (d, J = 7.2 Hz, 5-H), 6.26 (dd, J = 6.8, 4.8 Hz, 1H, 1'-H), 6.84 (d, J = 8.8 Hz, 4H, 2, 2', 6, 6'-H of DMTr), 7.2-7.4 (m, 8H, Ar and 5-H of cytidine), 7.40 (d, J = 8.4 Hz, 2H, 2, 6-H of Ph), 8.0-8.2 (m, 2H, -NH2).
(2)N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-O-(tert-ブチルジメチルシリル)-5’-O-(4,4’-ジメトキシトリチル)デオキシシチジン(DMTr-dCCO-TOP-TBDMS)の合成
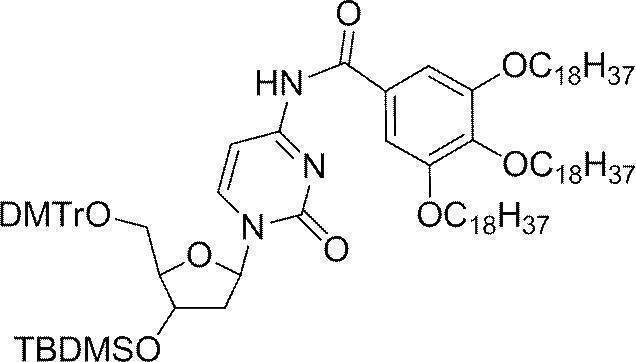
3,4,5-トリス(オクタデシルオキシ)安息香酸(0.927g,1.00mmol)、ジイソプロピルエチルアミン(0.520mL,2.04mmol)をクロロホルム(10.0mL)に溶解し、ここに0℃で2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)(0.573g,1.51mmol)を添加した後、室温に昇温し40分間撹拌した。反応液に(1)で得た5’-O-(4,4’-ジメトキシトリチル)-3’-O-(tert-ブチルジメチルシリル)デオキシシチジン(0.969g,1.50mmol)を添加して40℃にて21時間撹拌した。薄層クロマトグラフィーで反応の完結を確認し、室温に冷却した後、アセトニトリル(50mL)を添加して目的物を析出させ、桐山ロートでろ過、洗浄した。ろ紙上の白色沈殿をアセトニトリル(40mL)中に分散させた後、再び桐山ロートでろ過・洗浄し、真空乾燥の後、目的のDMTr-dCCO-TOP-TBDMS(1.51g,0.973mmol,97.3%)を取得した。
1H NMR (400 MHz, CDCl3) δ: -0.04 (s, 3H, Si(CH3)(CH3)), 0.02 (s, 3H, Si(CH3)(CH3)), 0.82 (s, 9H, tBu), 0.89 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 1.20-1.50 (m, 90H, OCH2CH2(CH2)15CH3), 1.75 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.83 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.22 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2'-CHH), 2.585 (ddd, J = 13.6, 6.4, 6.4 Hz, 1H, 2'-CHH), 3.34 (dd, J =10.8, 3.6 Hz, 1H, 5'-CHH), 3.53 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.806 (s, 3H, DMTr 4-OCH3), 3.809 (s, 3H, DMTr 4'-OCH3), 3.99-4.04(m, 7H, 3'-H and OCH2(CH2)16CH3), 4.46 (ddd, J = 6.4, 6.0, 6.0, 1H, 3'-H), 6.27 (dd, J = 6.4, 4.8 Hz, 1H, 1'-H), 6.68 (d, J =8.8 Hz, 4H,3,3',4,4'-H of p-methoxyphenyl), 7.05 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.27-7.33 (m, 8H, 2,2',6,6'-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.40 (d, J =7.2 Hz, 2H, 2,6-H of phenyl), 8.40 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.53 (s, 1H, cytosine N-H).
1H NMR (400 MHz, CDCl3) δ: -0.04 (s, 3H, Si(CH3)(CH3)), 0.02 (s, 3H, Si(CH3)(CH3)), 0.82 (s, 9H, tBu), 0.89 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 1.20-1.50 (m, 90H, OCH2CH2(CH2)15CH3), 1.75 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.83 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.22 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2'-CHH), 2.585 (ddd, J = 13.6, 6.4, 6.4 Hz, 1H, 2'-CHH), 3.34 (dd, J =10.8, 3.6 Hz, 1H, 5'-CHH), 3.53 (dd, J = 10.8, 2.8 Hz, 1H, 5'-CHH), 3.806 (s, 3H, DMTr 4-OCH3), 3.809 (s, 3H, DMTr 4'-OCH3), 3.99-4.04(m, 7H, 3'-H and OCH2(CH2)16CH3), 4.46 (ddd, J = 6.4, 6.0, 6.0, 1H, 3'-H), 6.27 (dd, J = 6.4, 4.8 Hz, 1H, 1'-H), 6.68 (d, J =8.8 Hz, 4H,3,3',4,4'-H of p-methoxyphenyl), 7.05 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.27-7.33 (m, 8H, 2,2',6,6'-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.40 (d, J =7.2 Hz, 2H, 2,6-H of phenyl), 8.40 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.53 (s, 1H, cytosine N-H).
実施例2:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[TGiBuCCO-TOP]-TBDMS)の合成

(1)5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[GiBuCCO-TOP]-TBDMS)の合成
実施例1で得たDMTr-dCCO-TOP-TBDMS(0.777g,0.500mmol)、5-メトキシインドール(10.0mmol)を脱水ジクロロメタン(23.0mL)に溶解した。ここにトリフルオロ酢酸(55.7μL,0.75mL)を滴下して、室温で30分撹拌し、反応の完結をUPLCにより確認した。反応混合液を2,4,6,-トリメチルピリジンで中和した後、5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.68g,2.00mmol)、5-(ベンジルチオ)-1H-テトラゾール(0.384g,2.00mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.452g,2.20mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[GiBuCCO-TOP]-TBDMS(0.970g,0.480mmol,96.0%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1966.23,found 1966.24[M-H]-
実施例1で得たDMTr-dCCO-TOP-TBDMS(0.777g,0.500mmol)、5-メトキシインドール(10.0mmol)を脱水ジクロロメタン(23.0mL)に溶解した。ここにトリフルオロ酢酸(55.7μL,0.75mL)を滴下して、室温で30分撹拌し、反応の完結をUPLCにより確認した。反応混合液を2,4,6,-トリメチルピリジンで中和した後、5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.68g,2.00mmol)、5-(ベンジルチオ)-1H-テトラゾール(0.384g,2.00mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.452g,2.20mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[GiBuCCO-TOP]-TBDMS(0.970g,0.480mmol,96.0%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1966.23,found 1966.24[M-H]-
(2)5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[TGiBuCCO-TOP]-TBDMS)の合成
アルゴン雰囲気下、(1)で得たDMTr-d[GiBuCCO-TOP]-TBDMS(950mg,0.470mmol)を脱水ジクロロメタン(23mL)に溶解し、5-メトキシインドール(1.38g,9.40mmol)、トリフルオロ酢酸(41.9μL,0.564mmol)を添加し、室温で30分撹拌し、反応の完結をLC/MSにより確認した。反応混合液に2,4,6,-トリメチルピリジン(1.49μL,1.13mmol)を添加して中和した後、5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.42g,1.91mmol)、5-(ベンジルチオ)-1H-テトラゾール(0.367g,1.91mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。反応の完結をLC/MSにより確認した後、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.431g,2.01mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[TGiBuCCO-TOP]-TBDMS(1.08g,0.453mmol,96.4%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1142.62,found 1142.63 [M-2H]2-
アルゴン雰囲気下、(1)で得たDMTr-d[GiBuCCO-TOP]-TBDMS(950mg,0.470mmol)を脱水ジクロロメタン(23mL)に溶解し、5-メトキシインドール(1.38g,9.40mmol)、トリフルオロ酢酸(41.9μL,0.564mmol)を添加し、室温で30分撹拌し、反応の完結をLC/MSにより確認した。反応混合液に2,4,6,-トリメチルピリジン(1.49μL,1.13mmol)を添加して中和した後、5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.42g,1.91mmol)、5-(ベンジルチオ)-1H-テトラゾール(0.367g,1.91mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。反応の完結をLC/MSにより確認した後、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.431g,2.01mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[TGiBuCCO-TOP]-TBDMS(1.08g,0.453mmol,96.4%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1142.62,found 1142.63 [M-2H]2-
実施例3:シアノエチル保護体の31P{1H}NMR分析
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMSおよそ0.003gをクロロホルム-d(0.7mL)に溶解した。31P{1H}NMRを測定した所、全てのピークは66.4~67.2ppmに観測された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMSおよそ0.003gをクロロホルム-d(0.7mL)に溶解した。31P{1H}NMRを測定した所、全てのピークは66.4~67.2ppmに観測された。
実施例4:DBUを用いた脱シアノエチル化体標品の取得
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0043g,0.00179mmol)をテトラヒドロフラン(0.15mL)に溶解した。ここに1,8-diazabicyclo[5.4.0]undec-7-ene(以後DBU)(2.0μL,0.0133mmol)を添加・混合した。23℃にて5分間、脱シアノエチル化を進行させた。反応液にアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0017gであった。生成物の31P{1H}NMR(CDCl3)を測定すると全てのピークは56.5~56.8ppmに出現し、脱シアノエチル化体はシアノエチル保護体と顕著に異なる領域に31P{1H}NMRのシグナルを与える事を確認した。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0043g,0.00179mmol)をテトラヒドロフラン(0.15mL)に溶解した。ここに1,8-diazabicyclo[5.4.0]undec-7-ene(以後DBU)(2.0μL,0.0133mmol)を添加・混合した。23℃にて5分間、脱シアノエチル化を進行させた。反応液にアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0017gであった。生成物の31P{1H}NMR(CDCl3)を測定すると全てのピークは56.5~56.8ppmに出現し、脱シアノエチル化体はシアノエチル保護体と顕著に異なる領域に31P{1H}NMRのシグナルを与える事を確認した。
実施例5:各種フッ化物イオン源を用いた脱シリル化の検討
(1)3HF-TEAを用いた検討
実施例2と同様に得た(DMTr-d[TGiBuCCO-TOP]-TBDMS(0.0239g,0.00998mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.0mg,0.180mmol)のTHF(0.60mL)溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0230gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.1ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認され、同時に4,4’-ジメトキシトリチル基の0.4%が脱落していた。
(1)3HF-TEAを用いた検討
実施例2と同様に得た(DMTr-d[TGiBuCCO-TOP]-TBDMS(0.0239g,0.00998mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.0mg,0.180mmol)のTHF(0.60mL)溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0230gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.1ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認され、同時に4,4’-ジメトキシトリチル基の0.4%が脱落していた。
(2)3HF-TEA-2ピリジンを用いた脱シリル化の検討
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0204g,0.00835mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.4mg,0.182mmol)、ピリジン(0.065mL,0.360mL)をTHF(0.60mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0169gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.6~68.3ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0204g,0.00835mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.4mg,0.182mmol)、ピリジン(0.065mL,0.360mL)をTHF(0.60mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0169gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.6~68.3ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(3)3HF-TEA-4ピリジンを用いた脱シリル化の検討
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.719g,0.300mmol)をジクロロメタン(9.0mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(870mg,5.44mmol)、ピリジン(1.75mL,21.6mmol)をTHF(18mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液にアセトニトリル(150mL)を添加して目的物を析出させた。桐山ロートにより濾過し、アセトニトリルで洗浄した。全沈殿を真空乾燥させ、0.652g(0.258mmol,95.2%)の白色固体を得た。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.6~68.3ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.719g,0.300mmol)をジクロロメタン(9.0mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(870mg,5.44mmol)、ピリジン(1.75mL,21.6mmol)をTHF(18mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液にアセトニトリル(150mL)を添加して目的物を析出させた。桐山ロートにより濾過し、アセトニトリルで洗浄した。全沈殿を真空乾燥させ、0.652g(0.258mmol,95.2%)の白色固体を得た。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.6~68.3ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(4)3HF-TEA-2N-メチルモルホリンを用いた脱シリル化の検討
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0241g,0.0101mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.1mg,0.181mmol)、N-メチルモルホリン(39.6μL,0.360mmol)をTHF(0.6mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.01953gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.7~68.2ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0241g,0.0101mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.1mg,0.181mmol)、N-メチルモルホリン(39.6μL,0.360mmol)をTHF(0.6mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.01953gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.7~68.2ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(5)3HF-TEA-2コリジンを用いた脱シリル化の検討
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0240g,0.0100mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.1mg,0.181mmol)、コリジン(2,4,6-トリメチルピリジン)(47.7μL,0.360mmol)をTHF(0.6mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.02133gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.1 ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0240g,0.0100mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.1mg,0.181mmol)、コリジン(2,4,6-トリメチルピリジン)(47.7μL,0.360mmol)をTHF(0.6mL)に溶解した混合溶液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.02133gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.1 ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(6)3HF-TEA-2モルホリンを用いた脱シリル化の検討
3HF-TEA(29.4mg,0.182mmol)、モルホリン(31.3μL,0.360mmol)をTHF(0.6mL)で混合し沈殿を含む溶液としこれを15℃に冷却した。ここに、実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0236g,0.0099mmol)のジクロロメタン(0.3mL)溶液を添加した。
15℃にて72時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0188gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークは見られず、脱シアノエチル化体に対応する60ppm以下のピークのみが観測され、本条件では、チオリン酸上のシアノエチル基が完全に脱落することが明らかとなった。また、LC-TOF MSでの分析により、脱シリルの完結も確認された。
3HF-TEA(29.4mg,0.182mmol)、モルホリン(31.3μL,0.360mmol)をTHF(0.6mL)で混合し沈殿を含む溶液としこれを15℃に冷却した。ここに、実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0236g,0.0099mmol)のジクロロメタン(0.3mL)溶液を添加した。
15℃にて72時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0188gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークは見られず、脱シアノエチル化体に対応する60ppm以下のピークのみが観測され、本条件では、チオリン酸上のシアノエチル基が完全に脱落することが明らかとなった。また、LC-TOF MSでの分析により、脱シリルの完結も確認された。
(7)HF-TEAを用いた検討
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0204g,0.00851mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.4mg,0.182mmol)、トリエチルアミン(0.0501mL,0.359mL)をTHF(0.60mL)中で混合した懸濁液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0169gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークと、脱シアノエチル化体に対応する60ppm以下のピークの積分比は以下の通りとなり、本条件では顕著な脱シアノエチル化が進行することが明らかとなった。
(シアノエチル保護チオリン酸):(脱保護チオリン酸)=1.00:1.66
LC-TOF MSによる測定では、脱シリル化の完結が確認された。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0204g,0.00851mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに3HF-TEA(29.4mg,0.182mmol)、トリエチルアミン(0.0501mL,0.359mL)をTHF(0.60mL)中で混合した懸濁液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0169gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークと、脱シアノエチル化体に対応する60ppm以下のピークの積分比は以下の通りとなり、本条件では顕著な脱シアノエチル化が進行することが明らかとなった。
(シアノエチル保護チオリン酸):(脱保護チオリン酸)=1.00:1.66
LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(8)HF-0.12ピリジン-0.88NMMを用いた検討
HF-ピリジン(0.0162mg;HF含量67.2%,HF0.544mmol,ピリジン0.0672mmol)、N-メチルモルホリン(52.1μL,0.474mmol)をTHF(0.6mL)で混合し沈殿を含む溶液としこれを15℃に冷却した。ここに、実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0240g,0.0100mmol)のジクロロメタン(0.3mL)溶液を添加した。
15℃にて96時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0208gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.2ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
HF-ピリジン(0.0162mg;HF含量67.2%,HF0.544mmol,ピリジン0.0672mmol)、N-メチルモルホリン(52.1μL,0.474mmol)をTHF(0.6mL)で混合し沈殿を含む溶液としこれを15℃に冷却した。ここに、実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMS(0.0240g,0.0100mmol)のジクロロメタン(0.3mL)溶液を添加した。
15℃にて96時間、脱シリル化を進行させた。反応液をおよそ200μLずつに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0208gであった。生成物の31P{1H}NMRを測定すると全てのピークはシアノエチル保護体に対応する65.8~68.2ppmに観測され、脱シアノエチル化体に対応する60ppm以下のシグナルは観測されなかった。また、LC-TOF MSによる測定では、脱シリル化の完結が確認された。
(9)TBAF-AcOHを用いた検討
実施例2と同様に得たDMTr-d[TGiBuTCO-TOP]-TBDMS(0.0204g,0.00851mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに1.0Mテトラブチルアンモニウムフロリドのテトラヒドロフラン溶液(0.54mL,0.54mmol)、酢酸(0.0309mL,0.536mmol)、テトラヒドロフラン(0.06mL)の混合液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0168gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークは見られず、脱シアノエチル化体に対応する60ppm以下のピークのみが観測され、本条件では、チオリン酸上のシアノエチル基が完全に脱落することが明らかとなった。また、LC-TOF MSでの分析により、脱シリルの完結も確認された。
実施例2と同様に得たDMTr-d[TGiBuTCO-TOP]-TBDMS(0.0204g,0.00851mmol)をジクロロメタン(0.3mL)に溶解し、これを15℃まで冷却した。ここに1.0Mテトラブチルアンモニウムフロリドのテトラヒドロフラン溶液(0.54mL,0.54mmol)、酢酸(0.0309mL,0.536mmol)、テトラヒドロフラン(0.06mL)の混合液を添加・混合した。15℃にて24時間、脱シリル化を進行させた。反応液をおよそ200μLに4分割し、それぞれにアセトニトリル(1.0mL)を添加して、10,000G,4℃にて5分間遠心分離した。上澄みを除いた後、アセトニトリル(1.0mL)に分散させ、再度同様に遠心分離した。全沈殿を真空乾燥させると重量は0.0168gであった。生成物の31P{1H}NMRを測定するとシアノエチル保護体に対応する60ppm以上のピークは見られず、脱シアノエチル化体に対応する60ppm以下のピークのみが観測され、本条件では、チオリン酸上のシアノエチル基が完全に脱落することが明らかとなった。また、LC-TOF MSでの分析により、脱シリルの完結も確認された。
実施例6:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシシチジン-3’-イル-[O-(2-シアノエチル)]-N,N-ジイソプロピルホスホロアミダイト(DMTr-d[TGiBuCCO-TOP]-PA)の合成
(1)5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシシチジン(DMTr-d[TGiBuCCO-TOP]-H)の合成
(1)5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシシチジン(DMTr-d[TGiBuCCO-TOP]-H)の合成

実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMSと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物に実施例18と同様の脱シリル化操作を施して、DMTr-d[TGiBuCCO-TOP]-Hと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1085.58, found 1085.59 [M-2H]2-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1085.58, found 1085.59 [M-2H]2-
(2)モノイミダゾリダイトとN-メチルモルホリンを用いたホスフィチル化

2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(960μL,3.02mmol)を脱水トルエン(6.6mL)に溶解し、モレキュラシーブ3Aの粉末(0.10g)により30分脱水した後、0.45μmのメンブレンフィルターによりろ過し5.25mLを乾燥したねじ口瓶に分取した。このトルエン溶液に、4,5-ジシアノイミダゾール(0.616g,5.21mmol)を添加して30分撹拌した後に、0.45μmのメンブレンフィルターにより濾過して、溶液1.5mLを分取した。N-メチルモルホリン(0.66mL,6.0mmol,pKa=7.38)及び(1)で得たDMTr-d[TGiBuCCO-TOP]-H(0.455g,0.200mmol)を10℃で添加して懸濁液とした。同温度で脱水ジクロロメタン(3.0mL)を添加して均一溶液とした後、2時間撹拌した。反応の終了をLC/MSにより確認し、2,4,6-トリメチルピリジンで反応を停止した。反応液を0℃に冷却して、アセトニトリル(110mL)を添加し、生成物を析出させた。桐山ロートでろ過・洗浄し、真空乾燥によりDMTr-d[TGiBuCCO-TOP]-PA(0.465g,0.187mmol,93.8%)を得た。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかったが、反応中に副生したジシアノイミダゾールによって生成物が活性化を受けたことに起因すると考えられるH-ホスホネートジエステルが目的物に対して5%検出された。生成物のTHF溶液をDBUにて脱シアノエチル化したサンプルのLC/MS分析では、アミダイト部位の酸化体として検出された。
LC/MS m/z:calcd 1167.12,found 1167.12 [M-2H]2-
LC/MS m/z:calcd 1167.12,found 1167.12 [M-2H]2-
(3)モノイミダゾリダイトとコリジンを用いたホスフィチル化
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数の2,4,6-トリメチルピリジンに変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率92.8%。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかったが、反応中に副生したジシアノイミダゾールによって生成物が活性化を受けたことに起因すると考えられるH-ホスホネートジエステルが目的物に対して6%検出された。
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数の2,4,6-トリメチルピリジンに変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率92.8%。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかったが、反応中に副生したジシアノイミダゾールによって生成物が活性化を受けたことに起因すると考えられるH-ホスホネートジエステルが目的物に対して6%検出された。
(4)モノイミダゾリダイトとピリジンを用いたホスフィチル化
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数のピリジン(pKa=5.17)に変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率92.8%。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかったが、反応中に副生したジシアノイミダゾールによって生成物が活性化を受けたことに起因すると考えられるH-ホスホネートジエステルが顕著に生成し、生成比は
目的物:H-ホスホネートジエステル=1:1.05
であった。
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数のピリジン(pKa=5.17)に変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率92.8%。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかったが、反応中に副生したジシアノイミダゾールによって生成物が活性化を受けたことに起因すると考えられるH-ホスホネートジエステルが顕著に生成し、生成比は
目的物:H-ホスホネートジエステル=1:1.05
であった。
(5)モノイミダゾリダイトと1,4-ジアザビシクロ[2.2.2]オクタンを用いたホスフィチル化
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数の1,4-ジアザビシクロ[2.2.2]オクタン(pKa=8.82)に変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率96.7%。31P NMR分析の結果、チオリン酸上のシアノエチル保護基の86%が脱落していることが明らかとなった。また、目的のアミダイトに対して加水分解体であるH-ホスホネートジエステルが9%生成していた。
ホスフィチル化時に添加する塩基をN-メチルモルホリンから同じ当量数の1,4-ジアザビシクロ[2.2.2]オクタン(pKa=8.82)に変更した以外は(2)と同じ混合比・手順にて0.067mmolの基質に対してホスフィチル化を行った。収率96.7%。31P NMR分析の結果、チオリン酸上のシアノエチル保護基の86%が脱落していることが明らかとなった。また、目的のアミダイトに対して加水分解体であるH-ホスホネートジエステルが9%生成していた。
(6)モノイミダゾリダイトとジアミダイトとN-メチルモルホリンを用いたホスフィチル化
2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(1.43mL,4.5mmol)を脱水トルエン(9.8mL)に溶解し、モレキュラシーブ3Aの粉末(0.5g)により30分脱水した後、0.45μmのメンブレンフィルターによりろ過した。このトルエン溶液に、4,5-ジシアノイミダゾール(0.886g,7.5mmol)を添加して30分撹拌した後に、0.45μmのメンブレンフィルターにより濾過して固体を除いた。この反応液に2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.238mL,0.750mmol)、N-メチルモルホリン(0.826mL,7.50mmol)及び(1)で取得したDMTr-d[TGiBuCCO-TOP]-H(0.532g,0.233mmol)を10℃で添加して懸濁液とした。同温度で脱水ジクロロメタン(4.0mL)を添加して均一溶液とした後、2時間撹拌した。反応の終了をLC/MSにより確認し、2,4,6-トリメチルピリジンで反応を停止した。反応液を0℃に冷却して、アセトニトリル(110mL)を添加し、生成物を析出させた。桐山ロートでろ過・洗浄し、真空乾燥によりDMTr-d[TGiBuCCO-TOP]-PA(0.525g,0.204mmol,94.4%)を得た。収量0.530g(2.14mmol,91.6%収率)。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかった。生成物のTHF溶液をDBUにて脱シアノエチル化したサンプルのLC/MS分析では、アミダイト部位の酸化体として検出された。
LC/MS m/z:calcd 1167.12,found 1167.12 [M-2H]2-
2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(1.43mL,4.5mmol)を脱水トルエン(9.8mL)に溶解し、モレキュラシーブ3Aの粉末(0.5g)により30分脱水した後、0.45μmのメンブレンフィルターによりろ過した。このトルエン溶液に、4,5-ジシアノイミダゾール(0.886g,7.5mmol)を添加して30分撹拌した後に、0.45μmのメンブレンフィルターにより濾過して固体を除いた。この反応液に2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.238mL,0.750mmol)、N-メチルモルホリン(0.826mL,7.50mmol)及び(1)で取得したDMTr-d[TGiBuCCO-TOP]-H(0.532g,0.233mmol)を10℃で添加して懸濁液とした。同温度で脱水ジクロロメタン(4.0mL)を添加して均一溶液とした後、2時間撹拌した。反応の終了をLC/MSにより確認し、2,4,6-トリメチルピリジンで反応を停止した。反応液を0℃に冷却して、アセトニトリル(110mL)を添加し、生成物を析出させた。桐山ロートでろ過・洗浄し、真空乾燥によりDMTr-d[TGiBuCCO-TOP]-PA(0.525g,0.204mmol,94.4%)を得た。収量0.530g(2.14mmol,91.6%収率)。31P NMR分析の結果、チオリン酸上の脱シアノエチル化は確認されなかった。生成物のTHF溶液をDBUにて脱シアノエチル化したサンプルのLC/MS分析では、アミダイト部位の酸化体として検出された。
LC/MS m/z:calcd 1167.12,found 1167.12 [M-2H]2-
実施例7:DMTr-d[TGiBuCCO-TOP]-PAとHO-d[ABzTT]-suc-TOBの縮合によるDMTr-d[TGiBuCCO-TOPABzTT]-suc-TOBの合成

実施例6-(5)と同様に得たDMTr-d[TGiBuCCO-TOP]-PA(0.149g,0.0601mmol)と調製例1-(3)と同様に得たd[ABzTT]-suc-TOB(0.104g,0.0497mmol)をジクロロメタン(5.0mL)に溶解し、モレキュラシーブ3A(0.229g)で脱水した。この溶液4.5mLを0.45μmのメンブレンフィルターによりろ過した。ろ過した溶液に5-(ベンジルチオ)-1H-テトラゾール(21.8mg,0.113mmol)を添加して室温で1時間30分撹拌した。縮合の完結をLC/MSにより確認した後、ピリジン(48.5μL,0.600mmol)、次いで3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(16.4mg,0.0799mmol)を添加し、室温で30分撹拌してホスファイト中間体を硫化した。反応液にメタノール(20mL)を添加して目的物を析出させ、桐山ロートでろ過・洗浄し、目的物を含む固体(0.222g)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1412.72,found 1412.71 [M-2H]2-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1412.72,found 1412.71 [M-2H]2-
実施例8:5’-(4,4’-ジメトキシトリチル)-3’-ジイソプロピルフェニルシリル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシチミジン(DMTr-dTCO-TOP-DIPPS)の合成
(1)ジイソプロピルフェニルシランの調製
-78℃に冷却した脱水テトラヒドロフラン(60mL)にフェニルリチウム(1.6Mジブチルエーテル溶液,20.5mL,32.8mmol)を溶解した。溶液を同温度で撹拌しながら、ここにジイソプロピルクロロシラン(5.20mL,30.4mmol)を6分間かけて滴下すると濃褐色となった。反応液を同温度で30分間撹拌すると、淡褐色となった。反応液に飽和塩化アンモニウム水溶液(50mL)を添加した後、室温まで昇温し、水層と有機層を分離した。水層をジクロロメタン(20mL)で抽出し、合わせた有機層を水、続いて飽和食塩水で洗浄した後、硫酸ナトリウムで脱水し、溶媒を減圧下、留去することで目的のジイソプロピルフェニルシランを含む淡褐色の油状液体14.9gを得た。これを精製すること無く次のステップに用いた。
(1)ジイソプロピルフェニルシランの調製
-78℃に冷却した脱水テトラヒドロフラン(60mL)にフェニルリチウム(1.6Mジブチルエーテル溶液,20.5mL,32.8mmol)を溶解した。溶液を同温度で撹拌しながら、ここにジイソプロピルクロロシラン(5.20mL,30.4mmol)を6分間かけて滴下すると濃褐色となった。反応液を同温度で30分間撹拌すると、淡褐色となった。反応液に飽和塩化アンモニウム水溶液(50mL)を添加した後、室温まで昇温し、水層と有機層を分離した。水層をジクロロメタン(20mL)で抽出し、合わせた有機層を水、続いて飽和食塩水で洗浄した後、硫酸ナトリウムで脱水し、溶媒を減圧下、留去することで目的のジイソプロピルフェニルシランを含む淡褐色の油状液体14.9gを得た。これを精製すること無く次のステップに用いた。
(2)5’-(4,4’-ジメトキシトリチル)-3’-ジイソプロピルフェニルシリルデオキシチミジンの合成

(1)で得たジイソプロピルフェニルシランを含む油状液体を乾燥ジクロロメタン(30mL)に溶解し、ここに加熱下で活性化したモレキュラシーブ3A(0.5g)を添加して脱水後、溶液をろ過した。更にジクロロメタン(282mL)を添加した。溶液に1,3-ジクロロ-5,5-ジメチルヒダントイン(9.54g,48.4mmol)を添加し、室温で30分撹拌した。ここにイミダゾール(7.20g,106mmol)、続いて、5’-(4,4’-ジメトキシトリチル)デオキシチミジン(13.0g,23.8mmol)を添加して50分間、室温で撹拌した。反応液に飽和炭酸水素ナトリウム水溶液(50mL)を添加してクエンチした後、分液した。有機層を水、続いて飽和食塩水で洗浄後、更に炭酸水素ナトリウム水溶液で洗浄した。有機層を硫酸ナトリウムで脱水の後、減圧下、溶媒を留去して、褐色の固体を得た。これをシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=10:1~1:1)で精製し、目的の5’-(4,4’-ジメトキシトリチル)-3’-ジイソプロピルフェニルシリルデオキシチミジン(9.80g,13.3mmol,55.9%)を白色固体として得た。
1H NMR (400 MHz, CDCl3) δ: 0.95-1.02 (m, 12 H, 2CH(CH3)2 ), 1.18-1.24 (m, 2H, 2CH(CH3)2), 1.46 (d, J = 0.8 Hz, 3H, CH3(thymidine)), 2.23 (ddd, J = 13.4, 8.0, 5.6 Hz, 1H, 2’-CHH), 2.44 (ddd, J = 13.4, 6.0, 2.0 Hz, 1H, 2’-CHH), 3.24 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.44 (dd, J = 10.8, 2.8 Hz, 2H, 5’-CHH), 3.78 (s, 6H, 2(-OCH3)), 4.09-4.15 (m, 2H), 4.64 (m, 1H, 3’-CH), 6.47 (dd, J = 8.0, 5.6 Hz, 1H , 1’-CH), 6.80 (dd, J = 8.8, 4.4 Hz, 4H, 3,3’,5,5’-CH of DMTr), 71.5-7.40 (m, 11H), 7.46 (d, J =6.8 Hz, 2H, 2,6-CH of Si-Ph), 7.63 (s, 1H, 6-CH (thymidine)), 8.40(brs, 1H, 3-NH).
1H NMR (400 MHz, CDCl3) δ: 0.95-1.02 (m, 12 H, 2CH(CH3)2 ), 1.18-1.24 (m, 2H, 2CH(CH3)2), 1.46 (d, J = 0.8 Hz, 3H, CH3(thymidine)), 2.23 (ddd, J = 13.4, 8.0, 5.6 Hz, 1H, 2’-CHH), 2.44 (ddd, J = 13.4, 6.0, 2.0 Hz, 1H, 2’-CHH), 3.24 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.44 (dd, J = 10.8, 2.8 Hz, 2H, 5’-CHH), 3.78 (s, 6H, 2(-OCH3)), 4.09-4.15 (m, 2H), 4.64 (m, 1H, 3’-CH), 6.47 (dd, J = 8.0, 5.6 Hz, 1H , 1’-CH), 6.80 (dd, J = 8.8, 4.4 Hz, 4H, 3,3’,5,5’-CH of DMTr), 71.5-7.40 (m, 11H), 7.46 (d, J =6.8 Hz, 2H, 2,6-CH of Si-Ph), 7.63 (s, 1H, 6-CH (thymidine)), 8.40(brs, 1H, 3-NH).
(3)3,4,5-トリスオクタデシルオキシ安息香酸塩化物の調製

3,4,5-トリス(オクタデシルオキシ)安息香酸(14.4g,15.5mmol)をクロロホルム(54.2mL)中に懸濁しスラリーとした。ここに、N,N-ジメチルホルムアミドを2滴と塩化チオニル2.25mL(31.0mmol)を添加し35℃で2時間撹拌した。溶媒を減圧下留去した。得られた淡黄色の固体を脱水トルエンに溶解、過剰の塩化チオニルと共に減圧下留去する操作を2回繰り返した後、固体を減圧下乾燥して目的の3,4,5-トリス(オクタデシルオキシ)安息香酸塩化物を定量的に得た。
1H NMR (400 MHz, CDCl3) δ: 0.879 (t, J = 6.6 Hz, 9H, CH3), 1.2-1.9 (m, 96 H, CH2), 4.01 (t, J = 6.4 ppm, 4H, 3,5-OCH2), 4.07 (t, J = 6.4 Hz, 2H, 4-OCH2), 7.32 (s, 2H, Ar).
1H NMR (400 MHz, CDCl3) δ: 0.879 (t, J = 6.6 Hz, 9H, CH3), 1.2-1.9 (m, 96 H, CH2), 4.01 (t, J = 6.4 ppm, 4H, 3,5-OCH2), 4.07 (t, J = 6.4 Hz, 2H, 4-OCH2), 7.32 (s, 2H, Ar).
(4)5’-(4,4’-ジメトキシトリチル)-3’-ジイソプロピルフェニルシリル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-デオキシチミジン(DMTr-dTCO-TOP-DIPPS)の合成

(2)で得た5’-(4,4’-ジメトキシトリチル)-3’-ジイソプロピルフェニルシリルデオキシチミジン(5.14g,6.99mmol)、(3)で得た3,4,5-トリス(オクタデシルオキシ)安息香酸塩化物(9.97g,10.5mmol)、ジイソプロピルエチルアミン(3.62g,14.0mmol)を脱水ピリジン(65.0mL)に溶解して、65℃で4時間撹拌した。反応の終了をTLCにより確認した後、ピリジンを留去し、固体をアセトニトリル中スラリー洗浄・ろ過した。固体をジクロロメタン中に懸濁し、濾過した。回収したジクロロメタン溶液を濃縮し、淡褐色の固体を得た。これをシリカゲルカラムクロマトグラフィーにより精製し、目的のDMTr-dTCO-TOP-DIPPS(9.52g,5.79mmol,82.7%)を得た。
1H NMR (400 MHz, CDCl3) δ: 0.878 (t, J = 7.2 Hz, 9H, octadecyl-CH3), 0.94-1.00 (m, 12 H, SiCH(CH3)2), 1.19-1.85 (m, 101 H, octadecyl-CH2 , thymine-CH3 and SiCH(CH3)2), 2.22-2.29 (m, 1H, 2’-CHH), 2.46-2.53 (m, 1H, 2’-CHH), 3.27 (dd, J = 10.8, 2.4 Hz, 1H, 5’-CHH), 3.47 (dd, J = 10.8, 2.4 Hz, 1H, 5’-CHH), 3.79 (s, 6H, OCH3 of DMTr), 3.98 (t, J = 6.4 Hz, 4H, 3,5-OCH2(CH2)16CH3), 4.03 (t, J = 6.4 Hz, 2H, 4- OCH2(CH2)16CH3), 4.17 (m, 1H, 4’-H), 4.64 (m, 1H, 3’-H ), 6.46 (dd, J = 5.6, 8.0 Hz, 1H, 1’-H), 6.79-6.83 (m, 4H, 3,3’,5,5’-H of DMTr), 7.14 (s, 2H, 2,6-H of substituted benzoate), 7.2-7.5 (m, 14 H, Ar), 7.75 (s, 1H, 6-H of thymidine).
1H NMR (400 MHz, CDCl3) δ: 0.878 (t, J = 7.2 Hz, 9H, octadecyl-CH3), 0.94-1.00 (m, 12 H, SiCH(CH3)2), 1.19-1.85 (m, 101 H, octadecyl-CH2 , thymine-CH3 and SiCH(CH3)2), 2.22-2.29 (m, 1H, 2’-CHH), 2.46-2.53 (m, 1H, 2’-CHH), 3.27 (dd, J = 10.8, 2.4 Hz, 1H, 5’-CHH), 3.47 (dd, J = 10.8, 2.4 Hz, 1H, 5’-CHH), 3.79 (s, 6H, OCH3 of DMTr), 3.98 (t, J = 6.4 Hz, 4H, 3,5-OCH2(CH2)16CH3), 4.03 (t, J = 6.4 Hz, 2H, 4- OCH2(CH2)16CH3), 4.17 (m, 1H, 4’-H), 4.64 (m, 1H, 3’-H ), 6.46 (dd, J = 5.6, 8.0 Hz, 1H, 1’-H), 6.79-6.83 (m, 4H, 3,3’,5,5’-H of DMTr), 7.14 (s, 2H, 2,6-H of substituted benzoate), 7.2-7.5 (m, 14 H, Ar), 7.75 (s, 1H, 6-H of thymidine).
実施例9:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチミジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-DIPPS)の合成
(1):5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチミジン(DMTr-d[GiBuTCO-TOP]-DIPPS)の合成
(1):5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチミジン(DMTr-d[GiBuTCO-TOP]-DIPPS)の合成
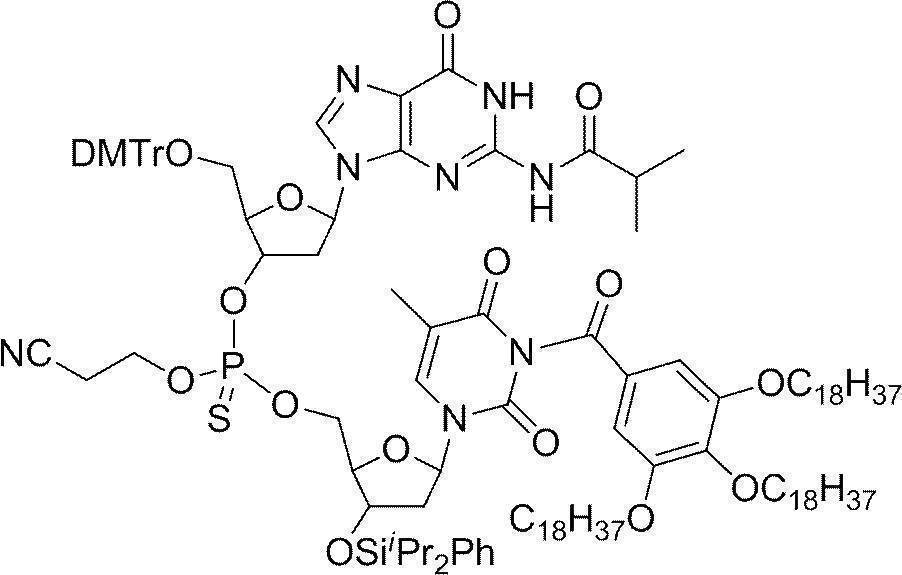
実施例8と同様に得たDMTr-dTCO-TOP-DIPPS(1.49g,0.903mmol)と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテート(1.38g,1.44mmol)及びインドール(1.37g,1.17mmol)を脱水ジクロロメタン(50mL)に溶解し、ここにトリフルオロ酢酸(0.103mL,1.34mmol)を滴下した。反応液を室温で1時間撹拌し、薄層クロマトグラフィーにより脱ジメトキシトリチル化を確認した。反応液に1-メチルイミダゾール(0.112mL,1.42mmol)を添加して中和した。ここに、5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(2.04g,2.43mmol)と4,5-ジシアノイミダゾール(0.422g,3.57mmol)の脱水アセトニトリル溶液(12.5mL)を添加して室温で1時間30分撹拌し、カップリング反応の完結を薄層クロマトグラフィーにより確認した。3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.540g,2.63mmol)を添加して室温で10分間撹拌してホスファイト中間体を硫化した。続いて、無水酢酸(80.1μL)、2,4,6-トリメチルピリジン(119μL)、1-メチルイミダゾール(71.2μL)を添加して室温で30分撹拌した。反応液に、室温でアセトニトリル(200mL)を添加して目的化合物と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートを析出させた後、桐山ロートを用いてろ過・洗浄(アセトニトリル)した。得られた固体を乾燥して、DMTr-d[GiBuTCO-TOP]-DIPPSを定量的に得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 2057.26,found 1185.64 [M-2H]2-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 2057.26,found 1185.64 [M-2H]2-
(2)5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチミジン(DMTr-d[TGiBuTCO-TOP]-DIPPS)の合成
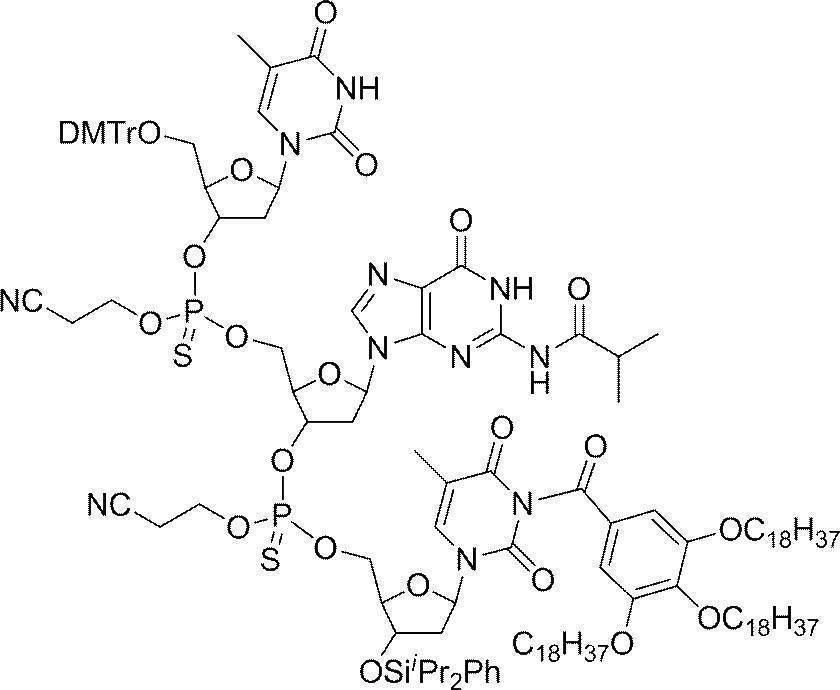
(1)で得た、DMTr-d[GiBuTCO-TOP]-DIPPS(0.903mmol)と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物およびインドール(1.42g,12.1mmol)を乾燥ジクロロメタン(50.0mL)に溶解した。ここにトリフルオロ酢酸(106μL,1.39mmol)を滴下し、室温で1時間撹拌した。脱ジメトキシトリチル化の完了をLC/MSにより確認し、1-メチルイミダゾール(116μL,1.46mmol)で中和した。反応液に、5’-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト(2.00g,2.69mmol)と4,5-ジシアノイミダゾール(0.479g,4.05mmol)の脱水アセトニトリル(12.5mL)溶液を添加して室温で1時間30分撹拌し、カップリング反応完結をLC/MSにより確認した。続いて、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.612g,2.98mmol)を添加して10分撹拌してホスファイト中間体を硫化した。反応液に無水酢酸(87.6μL,0.927mmol)、2,4,6-トリメチルピリジン(123μL,0.927 mmol)、1-メチルイミダゾール(73.3μL,0.927mmol)を添加して10分間撹拌した。反応液にアセトニトリル(200mL)を添加してオリゴ核酸トリマーと2,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。懸濁液をろ過・洗浄した後、目的の核酸トリマーと3,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物3.56g(うち、核酸2.21g,0.887mmol,95.7%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1188.14,found 1188.14 [M-2H]2-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1188.14,found 1188.14 [M-2H]2-
(3)5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチミジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP](配列番号1)-DIPPS)の合成

(2)で取得したDMTr-d[TGiBuTCO-TOP]-DIPPSから、(2)と同様に、所望の配列に対応するヌクレオシドアミダイトを順次用いて伸長を実施した。ヌクレオシドアミダイトとしては以下を用いた。
dCBz:5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
各ステップの収率を以下に示す。
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
各ステップの収率を以下に示す。
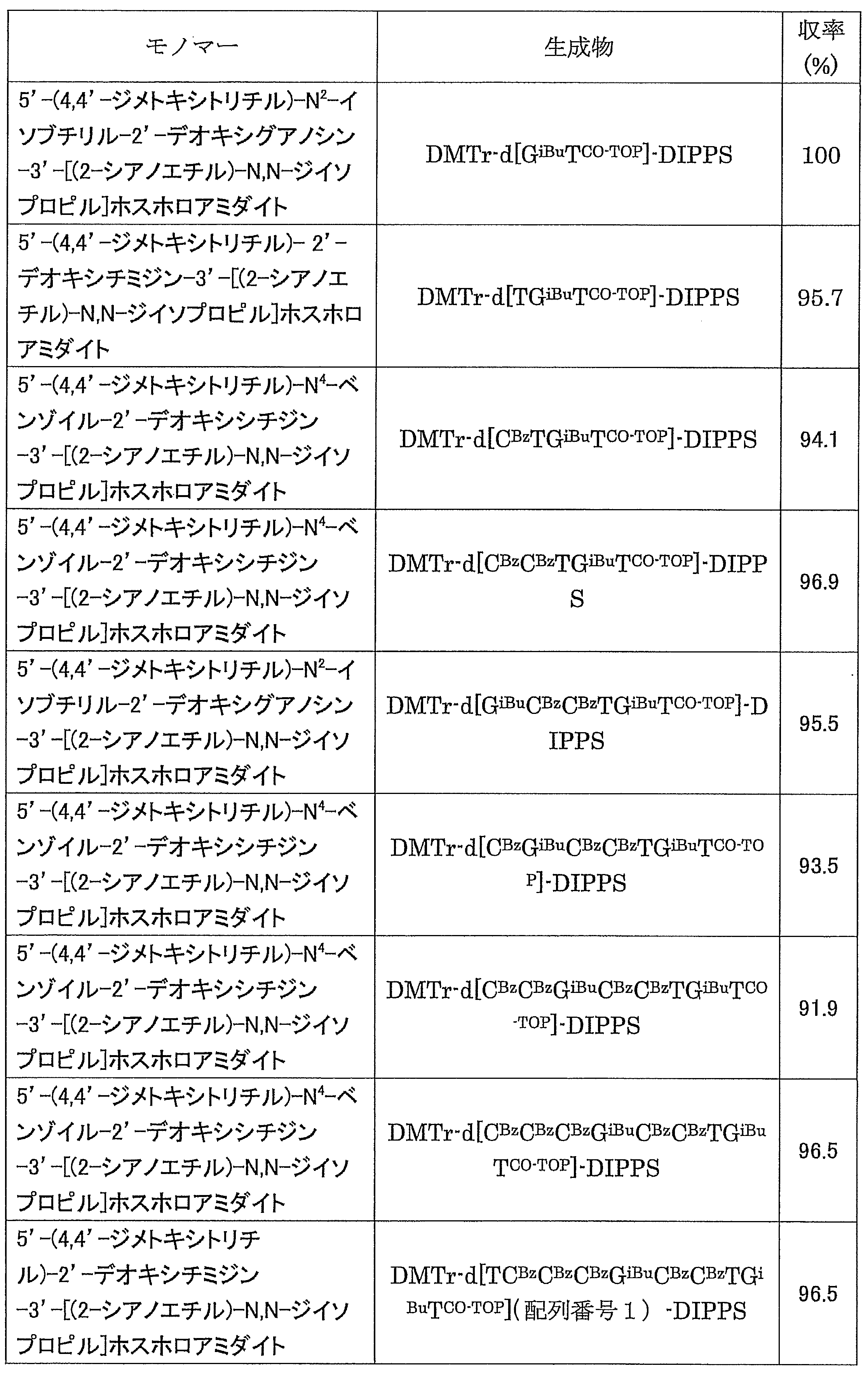
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1030.72,found 1030.72 [M-5H]5-
LC/MS m/z:calcd 1030.72,found 1030.72 [M-5H]5-
実施例10: 5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニルデオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシチミジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP](配列番号1)-H)の合成(脱シリル化)

実施例9で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-DIPPSと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(1.69g、0.235mmol)をジクロロメタン(7.20mL)に溶解し10℃に冷却した。ここに、トリエチルアミン三フッ化水素酸塩(0.174g,1.08mmol)、ピリジン(175μL,2.16mmol)のテトラヒドロフラン(14.4mL)溶液を添加した。反応液を10℃で24時間撹拌し、脱シリル化の完結をLC/MSにより確認した。反応液にアセトニトリル(100mL)を添加して、目的のDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-OHと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。固体を桐山ロートによるろ過で取得し、アセトニトリルによる洗浄の後、真空乾燥し白色固体1.46g(うち、オリゴ核酸は1.14g,0.209mmol,87.5%収率)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。HPLCによる分析では、目的化合物の以降の保持時間に短鎖のピークが見られ、目的化合物ピークの面積比は94.0%であった。
LC/MS m/z:calcd 1655.16,found 1655.14 [M-3H]3-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。HPLCによる分析では、目的化合物の以降の保持時間に短鎖のピークが見られ、目的化合物ピークの面積比は94.0%であった。
LC/MS m/z:calcd 1655.16,found 1655.14 [M-3H]3-
実施例11:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]チミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP](配列番号1)-PA)の合成

2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.971mL,3.06mmol)を脱水トルエン(6.68mL)に溶解し、モレキュラシーブ3Aの粉末により30分脱水し、0.45μmのメンブレンフィルターにより溶液3.18mLをろ別した。このトルエン溶液に、4,5-ジシアノイミダゾール(0.886g,7.5mmol)を添加して30分撹拌した後に、0.45μmのメンブレンフィルターにより濾過して溶液1.28mLを取得した。この反応液に2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.162mL,0.510mmol)、N-メチルモルホリン(0.572mL,5.10mmol)及び実施例10で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-Hと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(1.18g)を15℃で添加して懸濁液とした。同温度で脱水ジクロロメタン(16.0mL)を添加して均一溶液とした後、6時間撹拌した。反応の終了をLC/MSにより確認し、2,4,6-トリメチルピリジンで反応を停止した。反応液を0℃に冷却して、アセトニトリル(110mL)を添加し、生成物と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートを析出させた。桐山ロートでろ過・洗浄し、真空乾燥によりDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-PAと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物1.13g(うち、オリゴ核酸は0.888g,0.1571mmol, 92.4%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1709.52,found 1709.52 [M-5H]5-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1709.52,found 1709.52 [M-5H]5-
調製例1:N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニルデオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニルデオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-イル-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシネート(H-d[GiBuABzCBzABzTGiBuCBzABzTT](配列番号2)-suc-TOB)の合成
(1)5’-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(DMTr-d[TT]-suc-TOB)の合成
(1)5’-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(DMTr-d[TT]-suc-TOB)の合成

5’-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-イル[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシネート(1.51g,0.977mmol)、3,4,5-トリス(オクタデシルオキシ)ベンジルアセテート(1.49g,1.56mmol)およびインドール(1.50g,12.7mmol)を乾燥ジクロロメタン(38.9mL)に溶解した。ここにトリフルオロ酢酸(112μL,1.46mmol)を滴下し、室温で1時間撹拌した。脱ジメトキシトリチル基の完了を薄層クロマトグラフィーにより確認し、1-メチルイミダゾール(122μL,1.54mmol)で中和した。反応液に、5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト(2.00g,2.69mmol)と4,5-ジシアノイミダゾール(0.476g,4.03mmol)の脱水アセトニトリル(9.7mL)溶液を添加して室温で2時間撹拌した。続いて、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.582g,2.83mmol)を添加して10分撹拌してホスファイト中間体を硫化した。反応液に無水酢酸(92.1μL,0.974mmol)、2,4,6-トリメチルピリジン(129μL,0.974mmol)、1-メチルイミダゾール(77.1μL,0.974mmol)を添加して10分間撹拌した。反応液にアセトニトリル(200mL)を添加してオリゴ核酸ダイマーと2,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。懸濁液をろ過・洗浄した後、目的の核酸ダイマーと3,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物3.24g(うち、核酸1.80g,0.943mmol,96.9%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/ESI TOF MS:calcd 1858.13,found 1858.13 [M-H]-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/ESI TOF MS:calcd 1858.13,found 1858.13 [M-H]-
(2)5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(DMTr-d[ABzTT]-suc-TOB)の合成

(1)で得た、DMTr-d[TT]-suc-TOB(0.977mmol)と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物およびインドール(1.44g,12.2mmol)を乾燥ジクロロメタン(38.9mL)に溶解した。ここにトリフルオロ酢酸(108μL,1.42mmol)を滴下し、室温で1時間撹拌した。脱ジメトキシトリチル基の完了をLC/MSにより確認し、1-メチルイミダゾール(118μL,1.49mmol)で中和した。反応液に、5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシアデノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト(2.53g,2.95mmol)と4,5-ジシアノイミダゾール(0.526g,4.03mmol)の脱水アセトニトリル(10.0mL)溶液を添加して室温で2時間撹拌した。続いて、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.638g,3.11mmol)を添加して10分撹拌してホスファイト中間体を硫化した。反応液に無水酢酸(89.2μL,0.943mmol)、2,4,6-トリメチルピリジン(125μL,0.943mmol)、1-メチルイミダゾール(74.7μL,0.943mmol)を添加して10分間撹拌した。反応液にアセトニトリル(200mL)を添加してオリゴ核酸トリマーと2,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。懸濁液をろ過・洗浄した後、目的の核酸トリマーと3,4,6-トリス(オクタデシルオキシ)ベンジルアセテートの混合物3.59g(うち、核酸1.20g,0.916mmol,97.1%収率)を得た。サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1145.09,found 1145.09[M-2H]2-
LC/MS m/z:calcd 1145.09,found 1145.09[M-2H]2-
(3)N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン-3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(d[ABzTT]-suc-TOB)の合成(脱DMTr化)

(2)で得たDMTr-d[ABzTT]-suc-TOB(2.49g,1.00mol)と5-メトキシインドールを脱水ジクロロメタン(50mL)に溶解した。ここに、トリフルオロ酢酸(76.5μL,1.00mmol)を添加し、室温にて2時間撹拌した。脱ジメトキシトリチル化の完結をLC/TOF MSにより確認し、2,4,6-トリメチルピリジン(265μL,2.00mmol)を添加した。アセトニトリル(250mL)を添加して、目的物を沈殿させた。スラリーを桐山ロートにてろ過・洗浄の後、真空乾燥により目的化合物(2.10g,0.999mmol,99.5%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/ MS: calcd 1989.06, found 1989.09 [M-H]-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/ MS: calcd 1989.06, found 1989.09 [M-H]-
(4)5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン-3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(DMTr-d[GiBuABzCBzABzTGiBuCBzABzTT](配列番号2)-suc-TOB)の合成
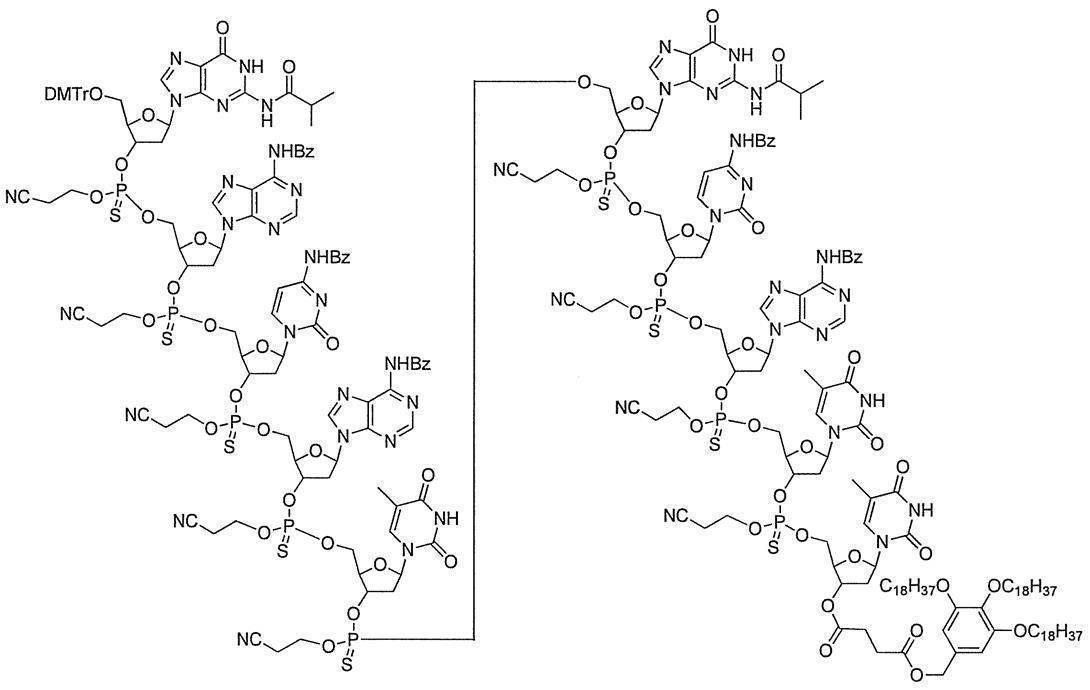
(3)で取得したd[ABzTT]-suc-TOBと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物から、(1)と同様の操作を繰り返して、所望の配列に対応するヌクレオシドアミダイトを順次用いて伸長を実施した。ヌクレオシドアミダイトとしては以下を用いた。
dABz:5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシアデノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dCBz:5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
各ステップの収率を以下に示す。
dABz:5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシアデノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dCBz:5’-(4,4’-ジメトキシトリチル)-N4-ベンゾイル-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
各ステップの収率を以下に示す。
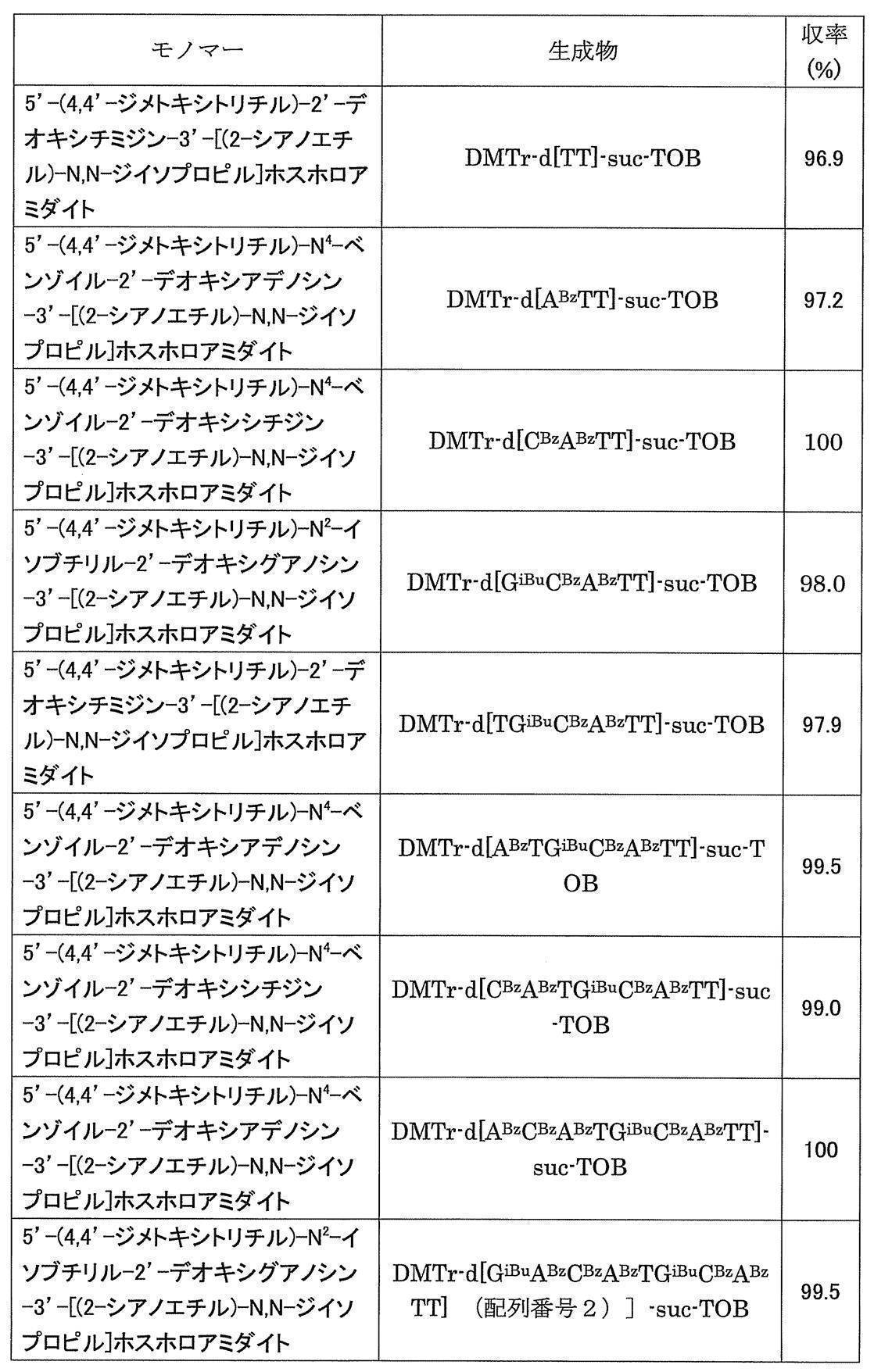
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1280.64,found 1280.66[M-4H]4-
LC/MS m/z:calcd 1280.64,found 1280.66[M-4H]4-
(5)N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-デオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(H-d[GiBuABzCBzABzTGiBuCBzABzTT](配列番号2)-suc-TOB)の合成(脱DMTr化)

(4)で得た、DMTr-d[GiBuABzCBzABzTGiBuCBzABzTT](配列番号2)-suc-TOBと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物4.51g(うちオリゴ核酸3.53g,0.631mmol)、インドール(0.971g,8.29mmol)を脱水ジクロロメタン(31.2mL)に溶解し、トリフルオロ酢酸(217μL,2.84mmol)を添加した。室温で2時間撹拌した後、LC/TOF MSにより脱ジメトキシトリチル化の完結を確認した。1-メチルイミダゾール(236μL,2.98mmol)により中和の後、アセトニトリル(200mL)を添加して、目的化合物と3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートを析出させた。桐山ロートによりろ過、洗浄した後、真空乾燥で白色固体4.19g(うちオリゴ核酸3.25g,0.614mmol,97.5% yield)を取得した。
サンプルをテトラヒドロフランに溶解の後、1,8-diazabicyclo[5.4.0]undec-7-ene (以下DBU)でリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1205.11,found 1205.13 [M-4H]4-
サンプルをテトラヒドロフランに溶解の後、1,8-diazabicyclo[5.4.0]undec-7-ene (以下DBU)でリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1205.11,found 1205.13 [M-4H]4-
実施例12:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]チミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン-3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート (DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOPGiBuABzCBzABzTGiBuCBzABzTT](配列番号3)-suc-TOB)の合成

実施例11で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-PAと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(0.0862g,0.121mmol)と調製例1で取得したH-d[GiBuABzCBzABzTGiBuCBzABzTT]-suc-TOBと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.61の混合物(0.0678g,0.0100mmol)を脱水ジクロロメタン(1.0mL)に溶解し、モレキュラシーブ3A(0.0588g)で脱水した。この溶液、0.6mLを0.45μmのメンブレンフィルターによりろ過し、乾燥した5φ NMRチューブに取得した。溶液に5-(ベンジルチオ)-1H-テトラゾール(2.88mg,0.0149mmol)を添加して室温で3時間撹拌した。縮合の完結を31P{1H}NMRにより確認した後、ピリジン(9.7μL,0.12mmol)、次いで3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(2.82mg,0.0137mmol)を添加し、室温で30分撹拌してホスファイト中間体を硫化した。反応液にメタノール(4mL)を添加して目的物を析出させ、桐山ロートでろ過・洗浄し、真空乾燥させ、カップリング生成物の白色固体(0.0931g)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1095.76,found 1095.66 [M-5H]5-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 1095.76,found 1095.66 [M-5H]5-
実施例13: 5’-(4,4’-ジメトキシトリチル)-3’-tert-ブチルジメチルシリル-4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシチミジン(DMTr-dTCO-TOP-TBDMS)の合成
(1)5’-(4,4’-ジメトキシトリチル)-3’-tert-ブチルジメチルシリルデオキシチミジン(DMTr-dT-TBDMS)の合成
(1)5’-(4,4’-ジメトキシトリチル)-3’-tert-ブチルジメチルシリルデオキシチミジン(DMTr-dT-TBDMS)の合成

5’-(4,4’-ジメトキシトリチル)デオキシチミジン(3.24g,5.95mmol)、イミダゾール(1.03g,15.1mmol)を脱水N,N-ジメチルホルムアミド(12mL)に溶解した。ここにtert-ブチルジメチルシリルクロリド(1.35g,8.95mmol)を添加して、アルゴン下、45℃にて1.5時間撹拌した。反応液を室温まで冷却した後、ジクロロメタン(10mL)、5%炭酸水素ナトリウム水溶液(10mL)を添加して撹拌した。分液の後、水層を10mLのジクロロメタンで抽出し、合わせた有機層を水10mL×2回、続いて飽和食塩水10mLで洗浄した。ジクロロメタン溶液を無水硫酸ナトリウムにより脱水の後、ろ過し、減圧下で濃縮下。得られた微黄色透明のオイルをシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1~3:2)にて精製し、目的のDMTr-dT-TBDMSを白色固体3.29g(4.9mmol,84.0%)として得た。
(2)5’-(4,4’-ジメトキシトリチル)-3’-tert-ブチルジメチルシリル-4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]チミジン(DMTr-dTCO-TOP-TBDMS)の合成

(1)にて取得したDMTr-dT-TBMDS(3.28g,4.98mmol)を脱水ピリジン(45mL)に溶解した。ここに3,4,5-トリス(オクタデシルオキシ)安息香酸塩化物(6.91g,7.3mmol)、ジイソプロピルエチルアミン(3.9mL,22.4mmol)を添加し、65℃にて6時間撹拌した。反応液を室温まで冷却後、ジクロロメタン(100mL)を添加し飽和炭酸水素ナトリウム水溶液(100mL)、水(50mL)、食塩水(50mL)で洗浄し、無水硫酸ナトリウム上で脱水した。ジクロロメタン-ピリジン溶液を減圧下濃縮した。得られた褐色の固体にジクロロメタン(30mL)を添加し、ろ過した。溶分を再度濃縮し、シリカゲルカラムクロマトグラフィー(ヘキサン:テトラヒドロフラン=14:1~7:1)にて精製し目的のDMTr-dTCO-TOP-TBDMS(2.21g,1.41mmol,28.3%)を白色固体として得た。
1H NMR (400 MHz, CDCl3) δ: -0.03 (s, 3H, SitBu(CH3)(CH3)), 0.02 (s, 3H, SitBu(CH3)(CH3)), 0.82 (s, 9H, SitBu(CH3)(CH3)), 0.85-0.91(t, J = 6.8 Hz, 9H, octadecyl-CH3), 1.18-1.47 (m, 90H, OCH2CH2(CH2)15CH3), 1.52 (d, J = 0.6 Hz, 3H, thymine 5-CH3), 1.72 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH2)15CH3), 1.78 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH2)15CH3), 2,24 (ddd, J = 13.6, 6.8, 6.8 Hz, 1H, 2’-CHH), 2.36 (ddd, J = 13.6, 5.8, 3.2 Hz, 1H, 2’-CHH), 3.30 (dd, J = 10.7, 3.6 Hz, 1H, 5’-CHH); 3.49 (dd, J = 10.7, 2.6 Hz, 1H), 3.80 (two singlets, 6H, 4,4’-OMe), 3.98 (t, J
= 6.5 Hz, 4H, 3,5-OCH2CH2(CH2)15CH3), 4.04 (t, J = 6.5 Hz, 2H, 3,5-OCH2CH2(CH2)15CH3), 3.95-4.05 (probably m, 1H), 4.51 (m, 1H), 6.33 (m, 1H), 6.84 (d, 1.2 Hz, 2H), 6.84-6.87 (m,4H), 7.13 (s, 2H); 7.24-7.271 (m, 1H), 7.28-7.33(m, 6H), 7.41 (d, J = 7.2 Hz, 2H); 7.76 (s, 1H).
1H NMR (400 MHz, CDCl3) δ: -0.03 (s, 3H, SitBu(CH3)(CH3)), 0.02 (s, 3H, SitBu(CH3)(CH3)), 0.82 (s, 9H, SitBu(CH3)(CH3)), 0.85-0.91(t, J = 6.8 Hz, 9H, octadecyl-CH3), 1.18-1.47 (m, 90H, OCH2CH2(CH2)15CH3), 1.52 (d, J = 0.6 Hz, 3H, thymine 5-CH3), 1.72 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH2)15CH3), 1.78 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH2)15CH3), 2,24 (ddd, J = 13.6, 6.8, 6.8 Hz, 1H, 2’-CHH), 2.36 (ddd, J = 13.6, 5.8, 3.2 Hz, 1H, 2’-CHH), 3.30 (dd, J = 10.7, 3.6 Hz, 1H, 5’-CHH); 3.49 (dd, J = 10.7, 2.6 Hz, 1H), 3.80 (two singlets, 6H, 4,4’-OMe), 3.98 (t, J
= 6.5 Hz, 4H, 3,5-OCH2CH2(CH2)15CH3), 4.04 (t, J = 6.5 Hz, 2H, 3,5-OCH2CH2(CH2)15CH3), 3.95-4.05 (probably m, 1H), 4.51 (m, 1H), 6.33 (m, 1H), 6.84 (d, 1.2 Hz, 2H), 6.84-6.87 (m,4H), 7.13 (s, 2H); 7.24-7.271 (m, 1H), 7.28-7.33(m, 6H), 7.41 (d, J = 7.2 Hz, 2H); 7.76 (s, 1H).
実施例14:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N3-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-tert-ブチルジメチルシリルデオキシチミジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP](配列番号1)-TBDMS)の合成
実施例13で得たDMTr-dTCO-TOP-TBDMSを実施例9と同様の伸長に付すことで目的の10merを得た。
各ステップの収率を以下に示す。
実施例13で得たDMTr-dTCO-TOP-TBDMSを実施例9と同様の伸長に付すことで目的の10merを得た。
各ステップの収率を以下に示す。

実施例15:DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-TBDMSの脱シリル化
(1)実施例10と同一フッ化物イオン濃度、昇温条件での脱シリル化
(1)実施例10と同一フッ化物イオン濃度、昇温条件での脱シリル化

実施例14で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-TBDMSと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(0.141g,0.0199mmol)をジクロロメタン(0.6mL)に溶解し20℃に冷却した。ここに、トリエチルアミン三フッ化水素酸塩(16.7μL,0.100mmol)、ピリジン(32.3μL,0.40mmol)のテトラヒドロフラン(1.2mL)溶液を添加・混合した。反応液を20℃で24時間進行させた所、脱シリル化は未完であった。
(2)フッ化物イオン濃度を向上させた条件
実施例14で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-TBDMSと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(0.350g,0.0493mmol)をジクロロメタン(1.5mL)に溶解し20℃に冷却した。ここに、トリエチルアミン三フッ化水素酸塩(0.167mL,1.00mmol)、ピリジン(0.324mL,4.01mmol)のテトラヒドロフラン(3.0mL)溶液を添加した。反応液を20℃で24時間撹拌した、脱シリル化の完結をLC/MSにより確認した。反応液にアセトニトリル(18mL)を添加して、目的のDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-OHと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。固体を桐山ロートによるろ過で取得し、アセトニトリルによる洗浄の後、真空乾燥し白色固体0.216g(うち、オリゴ核酸は0.168g,0.310mmol,62.8%収率)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。HPLCによる分析では、目的化合物の以降の保持時間に短鎖のピークが見られ、目的化合物ピークの面積比は62.0%であった。
実施例14で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-TBDMSと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートのモル比1.00:1.60の混合物(0.350g,0.0493mmol)をジクロロメタン(1.5mL)に溶解し20℃に冷却した。ここに、トリエチルアミン三フッ化水素酸塩(0.167mL,1.00mmol)、ピリジン(0.324mL,4.01mmol)のテトラヒドロフラン(3.0mL)溶液を添加した。反応液を20℃で24時間撹拌した、脱シリル化の完結をLC/MSにより確認した。反応液にアセトニトリル(18mL)を添加して、目的のDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuTCO-TOP]-OHと3,4,5-トリス(オクタデシルオキシ)ベンジルアセテートの混合物を析出させた。固体を桐山ロートによるろ過で取得し、アセトニトリルによる洗浄の後、真空乾燥し白色固体0.216g(うち、オリゴ核酸は0.168g,0.310mmol,62.8%収率)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。HPLCによる分析では、目的化合物の以降の保持時間に短鎖のピークが見られ、目的化合物ピークの面積比は62.0%であった。
実施例16:5’-O-(4,4’-ジメトキシトリチル)-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニル-3’-O-(tert-ブチルジメチルシリル)デオキシシチジン(DMTr-dCsuc-TOB-TBDMS)の合成

3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート・トリエチルアンモニウム塩(1.11g,1.0mmol)を脱水クロロホルム3.3mLに溶解した。ここに、ジイソプロピルエチルアミン(0.512mL,3.00mmol)を添加した後、0℃で2-(1Hベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)(0.759g,2.00mmol)を加えた。反応液を室温にて30分撹拌した後、3’-O-(tert-ブチルジメチルシリル)-5’-O-(4,4’-ジメトキシトリチル)-2’-デオキシシチジン(0.966g,1.50mmol)を転化して、40℃にて17時間撹拌した。反応の終了を薄層クロマトグラフィーで確認した後、室温に冷却した。反応液にアセトニトリル(5mL)を添加して目的物を析出させて濾過した。得られたケーキを5mLのアセトニトリルで洗浄したのち真空乾燥して1.64g(1.00mmol,100%)の目的物を取得した。
1H NMR (400 MHz, CDCl3) δ: -0.06 (s, 3H, Si-CH3), 0.00 (s, 3H, Si-CH3), 0.81 (s, 9H, tBu), 0.88 (t, J = 6.8 Hz, 9H , OCH2CH2(CH2)15CH3), 1.2-1.5 (m, 90H, OCH2CH2(CH2)15CH3), 1.73 (quint, J = 8.0 Hz, 2H, 4- OCH2CH2(CH2)15CH3), 1.78 (quint, J = 8.0 Hz, 4H, 2,5- OCH2CH2(CH2)15CH3), 2.19 (ddd, J = 13.6, 6.0, 4.4 Hz, 2’-CHH), 2.55 (ddd, J = 13.6, 6.4, 6.4, 2’-CHH), 2.68-2.75 (m, 4H, -COCH2CH2CO-), 3.32 (dd, J = 10.8, 3.2 Hz, 1 H, 5’-CHH), 3.52 (dd, J =10.8, 2.8 Hz, 1H, 5’-CHH), 3.79 (s, 6H, OCH3 of DMTr), 3.91-3.97 (m, 4H,-OCH2(CH2)16CH3), 4.00 (ddd, J =5.6, 3.2, 2.8 Hz, 1H, 4’-H) ,4.42 (ddd, J = 6.4, 6.0, 5.6 Hz, 1H, 3’-H), 5.02 (s, 2H, suc-OCH2Ar), 6.22 (dd, J =6.4, 4.4 Hz, 1H, 1’-H), 6.54 (s, 2H, 2,6-H of tri(octadecyloxy)phenyl), 6.84 (d, J = 8.8 Hz, 4H, 3,5-H of methoxyphenyl), 7.06 (d, 7.2 Hz, 1H, 6H of cytosine), 7.20-7.30 (m, 7H, Ar), 7.38 (d, J =7.2 Hz, 2H, 2,6-of phenyl), 8.19 (brs, 1H, N-H), 8.37 (d, J = 7.2 Hz, 5-H of cytosine).
1H NMR (400 MHz, CDCl3) δ: -0.06 (s, 3H, Si-CH3), 0.00 (s, 3H, Si-CH3), 0.81 (s, 9H, tBu), 0.88 (t, J = 6.8 Hz, 9H , OCH2CH2(CH2)15CH3), 1.2-1.5 (m, 90H, OCH2CH2(CH2)15CH3), 1.73 (quint, J = 8.0 Hz, 2H, 4- OCH2CH2(CH2)15CH3), 1.78 (quint, J = 8.0 Hz, 4H, 2,5- OCH2CH2(CH2)15CH3), 2.19 (ddd, J = 13.6, 6.0, 4.4 Hz, 2’-CHH), 2.55 (ddd, J = 13.6, 6.4, 6.4, 2’-CHH), 2.68-2.75 (m, 4H, -COCH2CH2CO-), 3.32 (dd, J = 10.8, 3.2 Hz, 1 H, 5’-CHH), 3.52 (dd, J =10.8, 2.8 Hz, 1H, 5’-CHH), 3.79 (s, 6H, OCH3 of DMTr), 3.91-3.97 (m, 4H,-OCH2(CH2)16CH3), 4.00 (ddd, J =5.6, 3.2, 2.8 Hz, 1H, 4’-H) ,4.42 (ddd, J = 6.4, 6.0, 5.6 Hz, 1H, 3’-H), 5.02 (s, 2H, suc-OCH2Ar), 6.22 (dd, J =6.4, 4.4 Hz, 1H, 1’-H), 6.54 (s, 2H, 2,6-H of tri(octadecyloxy)phenyl), 6.84 (d, J = 8.8 Hz, 4H, 3,5-H of methoxyphenyl), 7.06 (d, 7.2 Hz, 1H, 6H of cytosine), 7.20-7.30 (m, 7H, Ar), 7.38 (d, J =7.2 Hz, 2H, 2,6-of phenyl), 8.19 (brs, 1H, N-H), 8.37 (d, J = 7.2 Hz, 5-H of cytosine).
実施例17:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニル-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[TGiBuCsuc-TOB]-TBDMS)の合成
(1)5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニル-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[GiBuCsuc-TOB]-TBMDS)の合成
(1)5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニル-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[GiBuCsuc-TOB]-TBMDS)の合成

アルゴン雰囲気下、実施例16で取得したDMTr-dCsuc-TOB-TBDMS(820mg,0.500mmol)を脱水ジクロロメタン(23mL)に溶解し、5-メトキシインドール(1.47g,10.0mmol)、トリフルオロ酢酸(55.7μL,0.750mmol)を添加し、室温で30分撹拌し、反応の完結をUPLCにより確認した。反応混合液を2,4,6,-トリメチルピリジンで中和した後、5’-O-(4,4’-ジメトキシトリチル)-N2-イソブチリル-デオキシグアノシン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.88g,2.24mmol)、5-(ベンジルチオ)-1H-テトラゾール(0.432g,2.25mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.620g,3.02mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[GiBuCsuc-TOB]-TBMDS(1.00g,0.471mmol,94%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 2052.26, found 2052.26 [M-H]-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 2052.26, found 2052.26 [M-H]-
(2)5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニル-3’-tert-ブチルジメチルシリルデオキシシチジン(DMTr-d[TGiBuCsuc-TOB]-TBDMS)の合成

アルゴン雰囲気下、(1)で得たDMTr-d[GiBuCsuc-TOB]-TBMDS(974mg,0.458mmol)を脱水ジクロロメタン23mLに溶解し、5-メトキシインドール(1.47g,10.0mmol)、トリフルオロ酢酸(47.6μL,0.750mmol)を添加し、室温で30分撹拌し、反応の完結をLC/MSにより確認した。反応混合液に2,4,6,-トリメチルピリジン(325μL,2.45mmol)を添加して中和した後、5’-O-(4,4’-ジメトキシトリチル)-チミジン-3’-[O-(2-シアノエチル)(N,N-ジイソプロピル)]ホスホロアミダイト(1.02g,1.37mmol)、5-(ベンジルチオ)-1H-テトラゾール0.264g(1.37mmol)のアセトニトリル(3.00mL)溶液を添加して、室温で2時間撹拌した。反応の完結をLC/MSにより確認した後、3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン0.311g(1.51mmol)を添加し、室温で45分撹拌してホスファイト中間体を硫化した。室温でアセトニトリル(120mL)を添加して目的化合物を析出させた。反応液を桐山ロートを用いてろ過した後、アセトニトリルで洗浄し、真空乾燥により目的のDMTr-d[TGiBuCsuc-TOB]-TBDMS(1.05g,0.419mmol,92%)を白色固体として得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1185.64, found 1185.64 [M-2H]2-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1185.64, found 1185.64 [M-2H]2-
実施例18-1:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリル-デオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニルデオキシシチジン(DMTr-d[TGiBuCsuc-TOB]-H)の合成(脱シリル化)
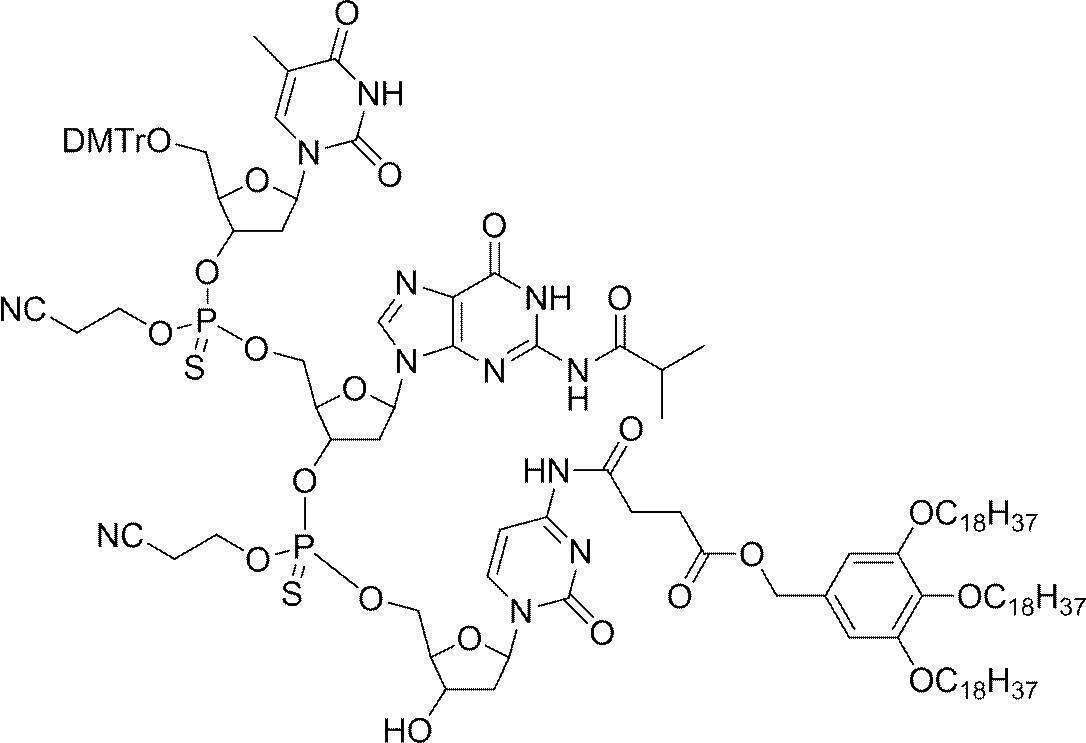
実施例17で取得したDMTr-d[TGiBuCsuc-TOB]-TBDMS(0.748g,0.301mmol)をジクロロメタン9.0mLに溶解し15℃に冷却した。ここに、トリエチルアミン三フッ化水素塩(875mg,5.42mmol)、ピリジン(1.75mL,22.1mmol)のテトラヒドロフラン(18.0mL)溶液を添加し、15℃で30時間撹拌した。反応液にアセトニトリル(108mL)を添加して析出させた後、桐山ロートにてろ過・洗浄し、真空乾燥により目的のDMTr-d[TGiBuCsuc-TOB]-H(0.686g,2.29mmol,96.6%)を取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 2258.20, found 2258.21 [M-H]-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 2258.20, found 2258.21 [M-H]-
実施例18-2:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン-3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニルデオキシシチジン-3’-イル-[O-(2-シアノエチル)]-N,N-ジイソプロピルホスホロアミダイト(DMTr-d[TGiBuCsuc-TOB]-PA)の合成
実施例18-1で取得したDMTr-d[TGiBuCsuc-TOB]を実施例6-(5)と同様にしてホスフィチル化して、DMTr-d[TGiBuCsuc-TOB]-PAを得た。
実施例18-1で取得したDMTr-d[TGiBuCsuc-TOB]を実施例6-(5)と同様にしてホスフィチル化して、DMTr-d[TGiBuCsuc-TOB]-PAを得た。
実施例19:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン-3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニルデオキシシチジン-3’-イル-[O-(2-シアノエチル)]-N,N-ジイソプロピルホスホロアミダイト(DMTr-d[TGiBuCsuc-TOB]-PA)とN4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン-3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネートの縮合による5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン-3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシニルデオキシシチジン-3’-イル-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン-3’-イル-3,4,5-トリス(オクタデシルオキシ)ベンジルスクシネート(DMTr-d[TGiBuCsuc-TOBABzTT]-suc-TOB)の合成

実施例18-2で得たDMTr-d[TGiBuCsuc-TOB]-PA(0.156g,0.0606mmol)と調製例1-(3)で得たd[ABzTT]-suc-TOB(0.103g,0.492mmol)をジクロロメタン5.0mLに溶解し、モレキュラシーブ3A,0.229gで脱水した。この溶液、4.5mLを0.45μmのメンブレンフィルターによりろ過した。ろ過した溶液に5-(ベンジルチオ)-1H-テトラゾール(20.5mg,0.107mmol)を添加して室温で1時間30分撹拌した。縮合の完結をLC/MSにより確認した後、ピリジン(48.5μL,0.600mmol)、次いで3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(0.311g,1.51mmol)を添加し、室温で30分撹拌してホスファイト中間体を硫化した。反応液にメタノール(20mL)を添加して目的物を析出させ、桐山ロートでろ過・洗浄し、目的物を含む固体0.221gを取得した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1441.40, found 1441.40 [M-3H]3-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z: calcd 1441.40, found 1441.40 [M-3H]3-
実施例20:フラグメント縮合により生成した6-merオリゴ核酸DMTr-d[TGiBuCsuc-TOBABzTT]-suc-TOB及びDMTr-d[TGiBuCCO-TOPABzTT]-suc-TOBのアンモニア水中での脱保護の比較

(1)DMTr-d[TGiBuCsuc-TOBABzTT]-suc-TOBの脱保護
実施例19の生成物6.05mgを1.5mLのアンモニア水中に分散させた。分散液を23℃でマグネチックスターラーで撹拌し、12時間までは2時間おきに、それ以降は12時間ごとに反応液をサンプリングし、LC/MSにより分析した。
実施例19の生成物は2時間以内に、シアノエチル基、アセチル基、ベンゾイル基、スクシニル基の脱離が完結しており、イソブチリル基の一部が残存していた。イソブチリル基の脱保護は36時間で完結した。
実施例19の生成物6.05mgを1.5mLのアンモニア水中に分散させた。分散液を23℃でマグネチックスターラーで撹拌し、12時間までは2時間おきに、それ以降は12時間ごとに反応液をサンプリングし、LC/MSにより分析した。
実施例19の生成物は2時間以内に、シアノエチル基、アセチル基、ベンゾイル基、スクシニル基の脱離が完結しており、イソブチリル基の一部が残存していた。イソブチリル基の脱保護は36時間で完結した。
(2)DMTr-d[TGiBuCCO-TOPABzTT]-suc-TOBの脱保護
実施例7の生成物6.01mgを1.5mLのアンモニア水中に分散させた。分散液を23℃でマグネチックスターラーで撹拌し、12時間までは2時間おきに、それ以降は12時間ごとに反応液をサンプリングし、LC/MSにより分析した。
実施例7の生成物は2時間の時点でシアノエチル基、アセチル基、ベンゾイル基、スクシニル基の脱離が完結しており、3,4,5-トリス(オクタデシルオキシ)ベンゾイル基、イソブチリル基の一部が残存していた。3,4,5-トリス(オクタデシルオキシ)ベンゾイル基の一部は12時間の時点でも残存しており、24時間において完全な脱離が確認された。イソブチリル基の脱保護は36時間で完結した。
実施例7の生成物6.01mgを1.5mLのアンモニア水中に分散させた。分散液を23℃でマグネチックスターラーで撹拌し、12時間までは2時間おきに、それ以降は12時間ごとに反応液をサンプリングし、LC/MSにより分析した。
実施例7の生成物は2時間の時点でシアノエチル基、アセチル基、ベンゾイル基、スクシニル基の脱離が完結しており、3,4,5-トリス(オクタデシルオキシ)ベンゾイル基、イソブチリル基の一部が残存していた。3,4,5-トリス(オクタデシルオキシ)ベンゾイル基の一部は12時間の時点でも残存しており、24時間において完全な脱離が確認された。イソブチリル基の脱保護は36時間で完結した。
実施例21:4,5-ジクロロイミダゾールを用いたジアミダイト活性化とホスフィチル化
(1)4,5-ジクロロイミダゾールの滴定によるpKa決定
4,5-ジクロロイミダゾール(0.2396g,1.749mmol)を、煮沸により脱気したイオン交換水に溶解し、50.0mL(0.0350M)とした。この溶液20mLを25℃で、0.10M NaOHにより滴定した。
滴定結果の非対数直線化プロット(Benet, L. Z.; Goyan, J. E. J. Pharm. Sci. 1967, 56, 665-680.)によりジクロロイミダゾールの25℃の水中におけるpKaは9.09と求められた。
(1)4,5-ジクロロイミダゾールの滴定によるpKa決定
4,5-ジクロロイミダゾール(0.2396g,1.749mmol)を、煮沸により脱気したイオン交換水に溶解し、50.0mL(0.0350M)とした。この溶液20mLを25℃で、0.10M NaOHにより滴定した。
滴定結果の非対数直線化プロット(Benet, L. Z.; Goyan, J. E. J. Pharm. Sci. 1967, 56, 665-680.)によりジクロロイミダゾールの25℃の水中におけるpKaは9.09と求められた。
(2)4,5-ジクロロイミダゾールを用いたジアミダイト活性化とホスフィチル化
2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.120g,0.40mmol)を乾燥したトルエン:シクロヘキサン=1:1(v/v)溶媒(1.5mL)に溶解した。ここに4,5-ジクロロイミダゾール(0.138g,1.00mmol)を加えて乾燥アルゴン下、17時間撹拌し、ホスフィチル化剤を含む溶液を調製した。この溶液全量を0.45μmメンブレンフィルターによりろ過した。ろ液にN-メチルモルホリン(0.881mL,8.0mmol)を添加したのち、実施例6-(1)と同様に得たDMTr-d[TGiBuCCO-TOP]-H(0.229g,0.100mmol)とMeOCOC6H2(OC18H37)3(0.162g)からなる混合物およびN-メチルイミダゾール(0.040mL,0.50mmol)を15℃で加えて懸濁液とした。同温度でジクロロメタン(10.0mL)を添加して均一溶液とした後43時間撹拌した。0℃に冷却の上、2,4,6-トリメチルピリジン(0.53mL,4.9mmol)を添加し、アセトニトリル(60mL)を添加して生成物を析出させた。桐山ロートによりろ過・洗浄し、真空乾燥によりDMTr-d[TGiBuCCO-TOP]-P(NiPr2)(OCH2CH2CN)を含む白色固体(0.386g)を得た。31P{1H}NMRによる分析の結果、3’-末端へのアミダイト導入率は50%であった。インターヌクレオチドのリン酸上の脱シアノエチル化体は観測されなかった。
2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.120g,0.40mmol)を乾燥したトルエン:シクロヘキサン=1:1(v/v)溶媒(1.5mL)に溶解した。ここに4,5-ジクロロイミダゾール(0.138g,1.00mmol)を加えて乾燥アルゴン下、17時間撹拌し、ホスフィチル化剤を含む溶液を調製した。この溶液全量を0.45μmメンブレンフィルターによりろ過した。ろ液にN-メチルモルホリン(0.881mL,8.0mmol)を添加したのち、実施例6-(1)と同様に得たDMTr-d[TGiBuCCO-TOP]-H(0.229g,0.100mmol)とMeOCOC6H2(OC18H37)3(0.162g)からなる混合物およびN-メチルイミダゾール(0.040mL,0.50mmol)を15℃で加えて懸濁液とした。同温度でジクロロメタン(10.0mL)を添加して均一溶液とした後43時間撹拌した。0℃に冷却の上、2,4,6-トリメチルピリジン(0.53mL,4.9mmol)を添加し、アセトニトリル(60mL)を添加して生成物を析出させた。桐山ロートによりろ過・洗浄し、真空乾燥によりDMTr-d[TGiBuCCO-TOP]-P(NiPr2)(OCH2CH2CN)を含む白色固体(0.386g)を得た。31P{1H}NMRによる分析の結果、3’-末端へのアミダイト導入率は50%であった。インターヌクレオチドのリン酸上の脱シアノエチル化体は観測されなかった。
実施例22:5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-イソプロポキシジイソプロピルシリルデオキシシチジン(DMTr-d[TGiBuCCO-TOP]-IPODIPS)の合成
(1)イソプロポキシジイソプロピルシランの合成
乾燥アルゴン雰囲気下、イミダゾール(3.27g,48.1mmol)を乾燥N,N-ジメチルホルムアミド(40mL)に溶解した。ここに2-プロパノール(3.70mL,52.2mmol)及びジイソプロピルクロロシラン(4.00mL,23.4mL)を滴下した。この混合液を40℃で11時間撹拌した。反応液を室温まで冷却した後に、ヘキサン(40mL)を添加し、続いて、飽和炭酸水素ナトリウム水溶液(20mL)、イオン交換水(20mL)を順次添加後、気泡の発生が収まるまで撹拌した。ヘキサン相を分取し、水(40mL)、飽和食塩水(40mL)で順次洗浄し、無水硫酸ナトリウム上で乾燥したのち、ロータリーエバポレーター(100mmHg,水浴35℃)にて溶媒を留去しイソプロポキシジイソプロピルシラン(2.69g)を無色透明の液体として得た。生成物の精製は行わずに次の工程に用いた。
(1)イソプロポキシジイソプロピルシランの合成
乾燥アルゴン雰囲気下、イミダゾール(3.27g,48.1mmol)を乾燥N,N-ジメチルホルムアミド(40mL)に溶解した。ここに2-プロパノール(3.70mL,52.2mmol)及びジイソプロピルクロロシラン(4.00mL,23.4mL)を滴下した。この混合液を40℃で11時間撹拌した。反応液を室温まで冷却した後に、ヘキサン(40mL)を添加し、続いて、飽和炭酸水素ナトリウム水溶液(20mL)、イオン交換水(20mL)を順次添加後、気泡の発生が収まるまで撹拌した。ヘキサン相を分取し、水(40mL)、飽和食塩水(40mL)で順次洗浄し、無水硫酸ナトリウム上で乾燥したのち、ロータリーエバポレーター(100mmHg,水浴35℃)にて溶媒を留去しイソプロポキシジイソプロピルシラン(2.69g)を無色透明の液体として得た。生成物の精製は行わずに次の工程に用いた。
(2)5’-O-(4,4’-ジメトキシトリチル)-3’-O-(イソプロポキシジイソプロピルシリル)デオキシシチジンの合成
(1)で得たイソプロポキシジイソプロピルシラン(1.40g,8.01mmol)を乾燥ジクロロメタン(8.0mL)に溶解し、ここに加熱下で活性化したモレキュラシーブ3A(0.1g)を添加して脱水後、溶液をろ過した。更にジクロロメタン(20mL)を添加した。溶液に1,3-ジクロロ-5,5-ジメチルヒダントイン(1.58g,8.00mmol)を添加し、室温で1時間撹拌した。ピリジン(0.65mL,8.03mmol)、5’-(4,4’-ジメトキシトリチル)チミジン(2.18g,4.00mmol)、イミダゾール(2.74g,40.2mmol)を添加して5時間、室温で撹拌した。反応液にチオ硫酸ナトリウム飽和水溶液(40mL)を添加してクエンチした後、分液した。有機層を水、続いて飽和食塩水で洗浄後、更に炭酸水素ナトリウム水溶液で洗浄した。有機層を硫酸ナトリウムで脱水の後、減圧下、溶媒を留去して表題化合物を含む黄色の固体3.45gを得た。精製は行わずに次工程に用いた。
(1)で得たイソプロポキシジイソプロピルシラン(1.40g,8.01mmol)を乾燥ジクロロメタン(8.0mL)に溶解し、ここに加熱下で活性化したモレキュラシーブ3A(0.1g)を添加して脱水後、溶液をろ過した。更にジクロロメタン(20mL)を添加した。溶液に1,3-ジクロロ-5,5-ジメチルヒダントイン(1.58g,8.00mmol)を添加し、室温で1時間撹拌した。ピリジン(0.65mL,8.03mmol)、5’-(4,4’-ジメトキシトリチル)チミジン(2.18g,4.00mmol)、イミダゾール(2.74g,40.2mmol)を添加して5時間、室温で撹拌した。反応液にチオ硫酸ナトリウム飽和水溶液(40mL)を添加してクエンチした後、分液した。有機層を水、続いて飽和食塩水で洗浄後、更に炭酸水素ナトリウム水溶液で洗浄した。有機層を硫酸ナトリウムで脱水の後、減圧下、溶媒を留去して表題化合物を含む黄色の固体3.45gを得た。精製は行わずに次工程に用いた。
(3)N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-5’-O-(4,4’-ジメトキシトリチル)-3’-O-(イソプロポキシジイソプロピルシリル)デオキシシチジンの合成
(2)で得た5’-O-(4,4’-ジメトキシトリチル)-3’-O-(イソプロポキシジイソプロピルシリル)デオキシシチジンを3,4,5-トリス(オクタデシルオキシ)安息香酸(1.85g,2.00mmol)と共に実施例1-(2)同様の縮合条件に付して表題化合物を含む淡黄色固体2.57gを取得した。これをシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1~3:1)により精製して表題の化合物1.20g(0.744mmol,37%収率)を得た。
1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 0.94-1.00 (m, 12H, SiCH(CH3)2), 1.11 (d, J = 6.4 Hz, 2H, -OCH(CH3)(CH3)), 1.13 (d, J = 6.4 Hz, 2H, -OCH(CH3)(CH3)), 1.20-1.50 (m, 92H, OCH2CH2(CH2)15CH3 and SiCH(CH3)2), 1.74 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.85 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.22 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2’-CHH), 2.70 (ddd, J = 13.6, 6.4, 6.4 Hz, 1H, 2’-CHH), 3.35 (dd, J =10.8, 3.6 Hz, 1H, 5’-CHH), 3.51 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.798 (s, 3H, DMTr 4-OCH3), 3.803 (s, 3H, DMTr 4’-OCH3), 3.99-4.04(m, 6H, 3’-H and OCH2(CH2)16CH3), 4.09 (sep, J = 6.0 Hz, 1H, OCHMe2), 4.65 (ddd, J = 6.4, 6.0, 6.0, 1H, 4’-H), 6.32 (dd, J = 6.0, 6.0 Hz, 1H, 1’-H), 6.68 (d, J =9.2 Hz, 4H, 3, 3’, 5, 5’-H of p-methoxyphenyl), 7.04 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.26-7.31(m, 8H, 2,2’,6,6’-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.40(d, J =7.2 Hz, 2H, 2,6-H of phenyl), 8.31 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.50 (s, 1H, cytosine N-H).
(2)で得た5’-O-(4,4’-ジメトキシトリチル)-3’-O-(イソプロポキシジイソプロピルシリル)デオキシシチジンを3,4,5-トリス(オクタデシルオキシ)安息香酸(1.85g,2.00mmol)と共に実施例1-(2)同様の縮合条件に付して表題化合物を含む淡黄色固体2.57gを取得した。これをシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1~3:1)により精製して表題の化合物1.20g(0.744mmol,37%収率)を得た。
1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 0.94-1.00 (m, 12H, SiCH(CH3)2), 1.11 (d, J = 6.4 Hz, 2H, -OCH(CH3)(CH3)), 1.13 (d, J = 6.4 Hz, 2H, -OCH(CH3)(CH3)), 1.20-1.50 (m, 92H, OCH2CH2(CH2)15CH3 and SiCH(CH3)2), 1.74 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.85 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.22 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2’-CHH), 2.70 (ddd, J = 13.6, 6.4, 6.4 Hz, 1H, 2’-CHH), 3.35 (dd, J =10.8, 3.6 Hz, 1H, 5’-CHH), 3.51 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.798 (s, 3H, DMTr 4-OCH3), 3.803 (s, 3H, DMTr 4’-OCH3), 3.99-4.04(m, 6H, 3’-H and OCH2(CH2)16CH3), 4.09 (sep, J = 6.0 Hz, 1H, OCHMe2), 4.65 (ddd, J = 6.4, 6.0, 6.0, 1H, 4’-H), 6.32 (dd, J = 6.0, 6.0 Hz, 1H, 1’-H), 6.68 (d, J =9.2 Hz, 4H, 3, 3’, 5, 5’-H of p-methoxyphenyl), 7.04 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.26-7.31(m, 8H, 2,2’,6,6’-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.40(d, J =7.2 Hz, 2H, 2,6-H of phenyl), 8.31 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.50 (s, 1H, cytosine N-H).
(4)5’-O-(4,4’-ジメトキシトリチル)-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-イソプロポキシジイソプロピルシリルデオキシシチジン(DMTr-d[TGiBuCCO-TOP]-IPODIPS)の合成

(3)で得たN4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-5’-O-(4,4’-ジメトキシトリチル)-3’-O-(イソプロポキシジイソプロピルシリル)デオキシシチジンを原料として実施例2と同様に伸長した。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 2346.31, found 2346.30[M-H]-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 2346.31, found 2346.30[M-H]-
実施例23:3’-シリル基の脱保護効率の比較
(1)DMTr-d[TGiBuCCO-TOP]-IPODIPSの脱シリル化
実施例22で得たDMTr-d[TGiBuCCO-TOP]-IPODIPS(0.4915g,0.200mmol)をジクロロメタン6.0 mlに溶解した。ここに3HF-TEA(146mg,0.906mmol)及びピリジン(146μL,1.80mmol)のTHF(12mL)溶液を添加し15℃に保冷した。反応液をサンプリング、HPLCにより分析し3’-脱シリル化の進捗を追跡した所、3’-シリル体の半減期は30分であり、8時間にて3’-シリル体は検出限界以下となった。
(1)DMTr-d[TGiBuCCO-TOP]-IPODIPSの脱シリル化
実施例22で得たDMTr-d[TGiBuCCO-TOP]-IPODIPS(0.4915g,0.200mmol)をジクロロメタン6.0 mlに溶解した。ここに3HF-TEA(146mg,0.906mmol)及びピリジン(146μL,1.80mmol)のTHF(12mL)溶液を添加し15℃に保冷した。反応液をサンプリング、HPLCにより分析し3’-脱シリル化の進捗を追跡した所、3’-シリル体の半減期は30分であり、8時間にて3’-シリル体は検出限界以下となった。
(2)DMTr-d[TGiBuCCO-TOP]-TBDMSの脱シリル化
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMSを(1)と同一の脱シリル化条件に付した所、8時間における脱シリル化の進行度は53%であった。
実施例2と同様に得たDMTr-d[TGiBuCCO-TOP]-TBDMSを(1)と同一の脱シリル化条件に付した所、8時間における脱シリル化の進行度は53%であった。
実施例24:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-3’-ジイソプロピルフェニルシリルデオキシチジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP](配列番号4)-DIPPS)の合成
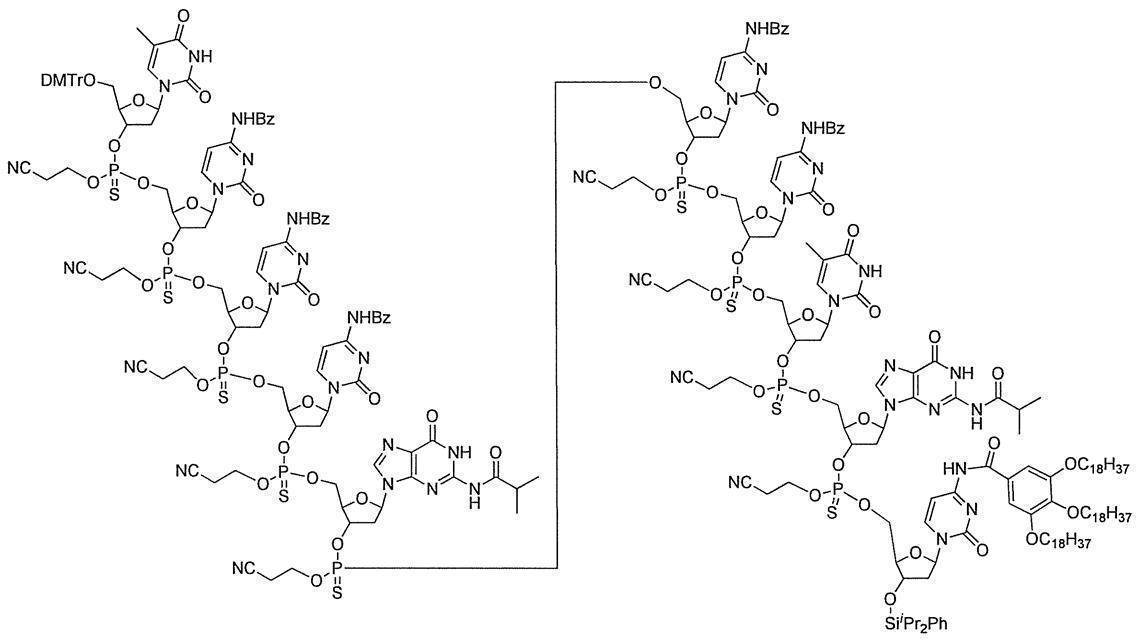
(1)4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]-5’-O-(4,4’-ジメトキシトリチル)-3’-O-ジイソプロピルフェニルシリルデオキシシチジン(DMTr-dCCO-TOP-DIPPS)の合成
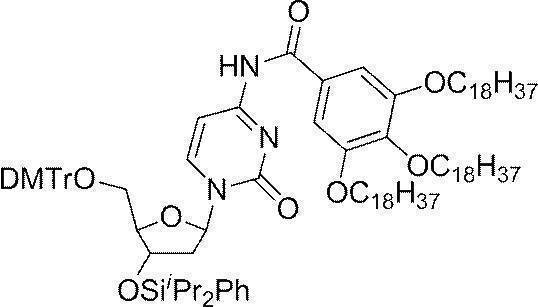
実施例8-(1)、(2)同様にジイソプロピルクロロシランと5’-O-(4,4’-ジメトキシトリチル)デオキシシチジンから5’-O-(4,4’-ジメトキシトリチル)-3’-O-ジイソプロピルフェニルシリルデオキシシチジンを調製した。生成物を精製すること無く3,4,5-トリス(オクタデシルオキシ)安息香酸と実施例1-(2)同様の縮合条件に付すことで表題の化合物を白色固体として得た(3,4,5-トリス(オクタデシルオキシ)安息香酸使用量に基づく収率43%)。
1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 0.95-1.01 (m, 12H, SiCH(CH3)2), 1.20-1.50 (m, 92H, OCH2CH2(CH2)15CH3 and SiCH(CH3)2), 1.82 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.85 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.18 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2’-CHH), 2.76 (m, 1H, 2’-CHH), 3.31 (dd, J =10.8, 3.6 Hz, 1H, 5’-CHH), 3.50 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.790 (s, 3H, DMTr 4-OCH3), 3.794 (s, 3H, DMTr 4’-OCH3), 3.99-4.07(m, 7H, 3’-H and OCH2(CH2)16CH3), 4.59 (ddd, J = 6.4, 6.0, 6.0, 1H, 4’-H), 6.32 (dd, J = 6.0, 6.0 Hz, 1H, 1’-H), 6.80-6.83 (d, J =9.2 Hz, 4H, 3, 3’, 5, 5’-H of DMTr), 7.04 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.22-7.40 (m, 8H, 2,2’,6,6’-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.46 (dd, J = 6.4, 2.0 Hz, 2H, 2,6-H of C-phenyl), 8.29 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.48 (s, 1H, cytosine N-H).
1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J =6.8 Hz, 9H, O(CH2)17CH3), 0.95-1.01 (m, 12H, SiCH(CH3)2), 1.20-1.50 (m, 92H, OCH2CH2(CH2)15CH3 and SiCH(CH3)2), 1.82 (quint, J =6.8 Hz, 2H, 4-OCH2CH2(CH)2CH3), 1.85 (quint, J =6.8 Hz, 4H, 3,5-OCH2CH2(CH)15CH3), 2.18 (ddd, J = 13.6, 6.0, 4.8Hz, 1H, 2’-CHH), 2.76 (m, 1H, 2’-CHH), 3.31 (dd, J =10.8, 3.6 Hz, 1H, 5’-CHH), 3.50 (dd, J = 10.8, 2.8 Hz, 1H, 5’-CHH), 3.790 (s, 3H, DMTr 4-OCH3), 3.794 (s, 3H, DMTr 4’-OCH3), 3.99-4.07(m, 7H, 3’-H and OCH2(CH2)16CH3), 4.59 (ddd, J = 6.4, 6.0, 6.0, 1H, 4’-H), 6.32 (dd, J = 6.0, 6.0 Hz, 1H, 1’-H), 6.80-6.83 (d, J =9.2 Hz, 4H, 3, 3’, 5, 5’-H of DMTr), 7.04 (brs, 2H, 2,6-H of 3,4,5-tris(octadecyloxy)benzoyl), 7.22-7.40 (m, 8H, 2,2’,6,6’-H of p-methoxyphenyl, 3,4,5-H of phenyl and 6-H of cytosine), 7.46 (dd, J = 6.4, 2.0 Hz, 2H, 2,6-H of C-phenyl), 8.29 (d, J = 7.2 Hz, 1H, 5-H of cytosine), 8.48 (s, 1H, cytosine N-H).
(2)(1)で得たDMTr-dCCO-TOP-DIPPS(1.63g,1.00 mmol)に3,4,5-トリス(オクタデシルオキシ)安息香酸メチル(1.65g,1.75mmol)を添加した上で、実施例9同様の伸長を施すことで表題の10残基オリゴ核酸を取得した。

サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 965.70,found 965.79 [M-5H]5-
LC/MS m/z:calcd 965.70,found 965.79 [M-5H]5-
実施例25:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]デオキシチジン(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP](配列番号4)-H)の合成(脱シリル化)
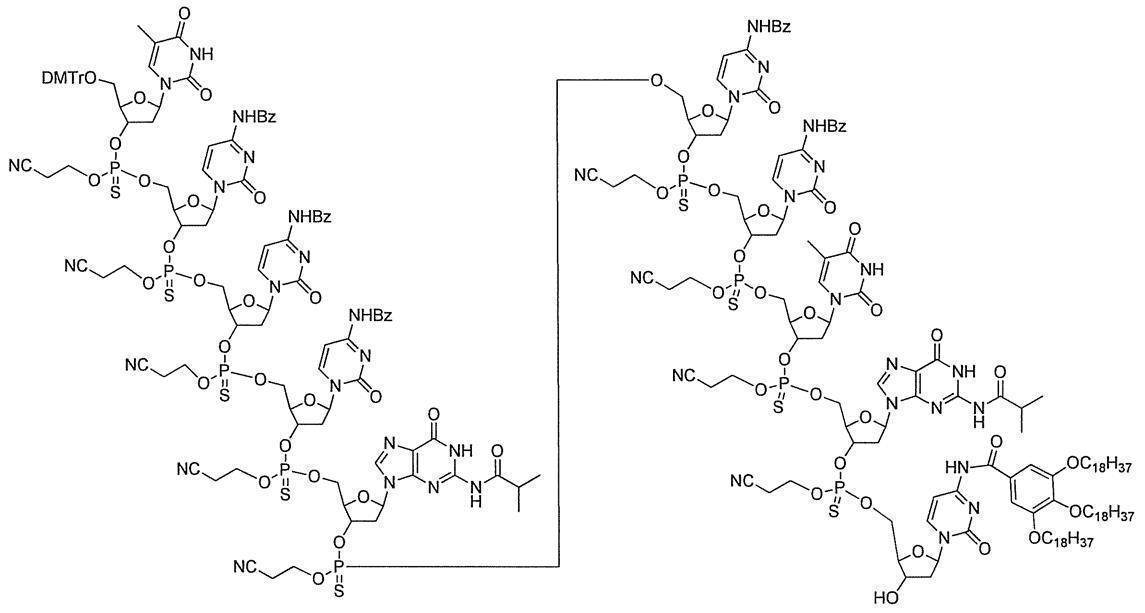
実施例24で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP]-DIPPSを76wt%の含量で含有する3,4,5-トリス(オクタデシルオキシ)安息香酸メチルとの混合物4.88g(0.70mmol)を実施例10と同一の脱シリル化条件に26時間付し、表題の化合物を75wt%の含量で含有する3,4,5-トリス(オクタデシルオキシ)安息香酸メチルとの混合物4.40g(0.650mmol,93%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 927.68,found 927.68 [M-5H]5-
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 927.68,found 927.68 [M-5H]5-
実施例26:5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ベンゾイルデオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-[3,4,5-トリス(オクタデシルオキシ)ベンゾイル]シチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト(DMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP]-PA)の合成
実施例25で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP]-H(4.09g)(含量75.71wt%,0.606mmol)を実施例11と同様のホスフィチル化条件に付し、3,4,5-トリス(オクタデシルオキシ)安息香酸メチルと共に表題化合物を含量56wt%で含有する白色固体(4.10g)(0.430mmol,71%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。目的物のオリゴヌクレオチド間リン酸上のみで脱シアノエチル化が進行し、3’-末端のホスホロアミダイト上のシアノエチル基は保持され同リンがイオン化条件にて酸化された化学種が主ピークとして観測された。
LC/MS m/z:calcd 970.90,found 970.85 [M-5H]5-
実施例25で取得したDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP]-H(4.09g)(含量75.71wt%,0.606mmol)を実施例11と同様のホスフィチル化条件に付し、3,4,5-トリス(オクタデシルオキシ)安息香酸メチルと共に表題化合物を含量56wt%で含有する白色固体(4.10g)(0.430mmol,71%収率)を得た。
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。目的物のオリゴヌクレオチド間リン酸上のみで脱シアノエチル化が進行し、3’-末端のホスホロアミダイト上のシアノエチル基は保持され同リンがイオン化条件にて酸化された化学種が主ピークとして観測された。
LC/MS m/z:calcd 970.90,found 970.85 [M-5H]5-
調製例2 N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ジメチルアミノエチリデンデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-(2-ヘキシルデカノイル)デオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ジメチルアミノエチリデンデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N2-イソブチリルデオキシグアノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-(2-ヘキシルデカノイル)デオキシシチジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-N4-ジメチルアミノエチリデンデオキシアデノシン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-[O-(2-シアノエチル)]ホスホロチオニル-デオキシチミジン 3’-イル-[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシネート(H-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT](配列番号5)-suc-TOB)の合成

5’-(4,4’-ジメトキシトリチル)-デオキシチミジン-3’-イル[3,4,5-トリス(オクタデシルオキシ)ベンジル]スクシネート(1.24g,0.80mmol)、3,4,5-トリス(オクタデシルオキシ)安息香酸メチル(1.23g,1.31mmol)の混合物から調製例1と同様の伸長・脱DMTr化を行い、表題の化合物を得た。
ヌクレオシドアミダイトとしては以下を用いた。
dAdma:5’-(4,4’-ジメトキシトリチル)-N4-ジメチルアミノエチリデン-2’-デオキシアデノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dCHD:5’-(4,4’-ジメトキシトリチル)-N4-(2-ヘキシルデカノイル)-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 996.58,found 996.54 [M-5H]5-
ヌクレオシドアミダイトとしては以下を用いた。
dAdma:5’-(4,4’-ジメトキシトリチル)-N4-ジメチルアミノエチリデン-2’-デオキシアデノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dCHD:5’-(4,4’-ジメトキシトリチル)-N4-(2-ヘキシルデカノイル)-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dGiBu:5’-(4,4’-ジメトキシトリチル)-N2-イソブチリル-2’-デオキシグアノシン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
dT:5’-(4,4’-ジメトキシトリチル)-2’-デオキシチミジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
サンプルをテトラヒドロフランに溶解の後、DBUでリン酸上のシアノエチル保護基を脱落させ、LC/MSにより目的物を同定した。
LC/MS m/z:calcd 996.58,found 996.54 [M-5H]5-
実施例27:10+10フラグメント縮合と脱保護によるによる5’-O-(4,4’-ジメトキシトリチル)デオキシチミジン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシグアノシン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシチミジン 3’-ホスホロチオニルデオキシグアノシン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシグアノシン 3’-[ホスホロチオニルデオキシアデノシン 3’-[ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシアデノシン 3’-ホスホロチオニルデオキシチミジン 3’-ホスホロチオニルデオキシグアノシン 3’-ホスホロチオニルデオキシシチジン 3’-ホスホロチオニルデオキシアデノシン 3’-ホスホロチオニルデオキシチミジン 3’-ホスホロチオニルデオキシチミジン (DMTr-d[TCCCGCCTGCGACATGCATT](配列番号6)-H)の合成
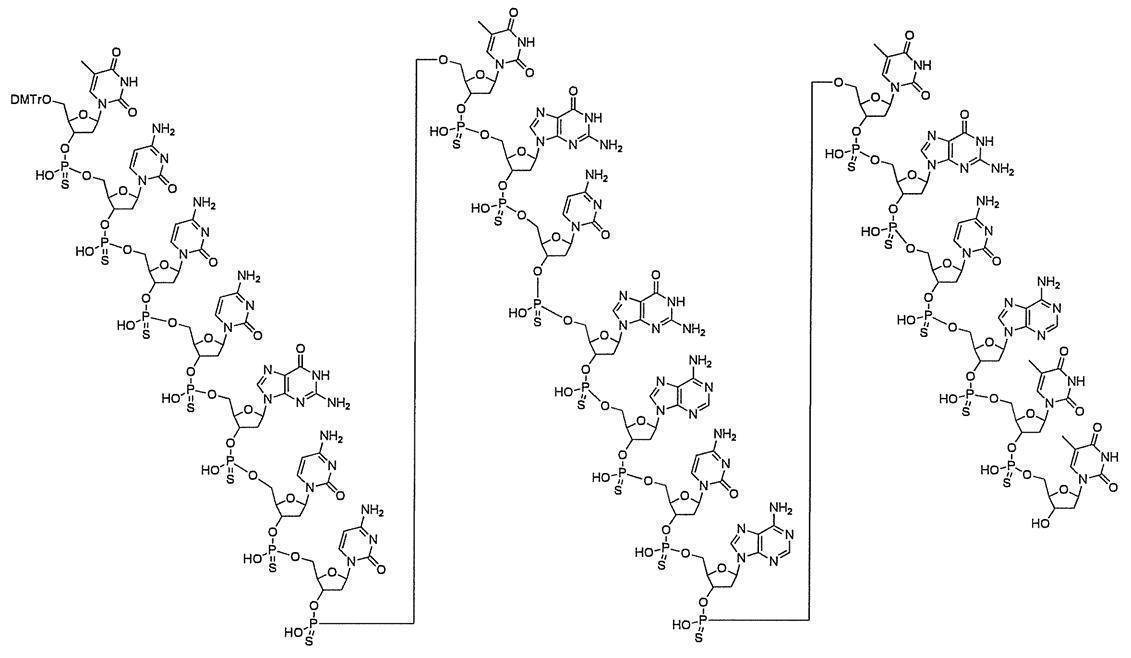
実施例26で得たDMTr-d[TCBzCBzCBzGiBuCBzCBzTGiBuCCO-TOP]-PAを含量56wt%で3,4,5-トリス(オクタデシルオキシ)安息香酸メチルと共に含有する固体(1.00g,0.100mmol)、調製例2で得たH-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT]-suc-TOBを含量82wt%で3,4,5-トリス(オクタデシルオキシ)安息香酸メチルと共に含有する固体(0.556g,0.083mmol)を、予め強熱脱水したモレキュラシーブ3Aの粉末(0.544g)と共に脱水ジクロロメタン(7.5mL)中、1時間撹拌することで脱水溶液とした。この懸濁液に脱水アセトニトリル(0.75mL)を添加し0.45μmのメンブレンフィルターにより全量を濾過して20mLシュレンクチューブに取得した。ここに5-(ベンジルチオ)-1H-テトラゾール(47.2mg,0.245mmol)を添加して乾燥アルゴン下で3時間撹拌した。反応液にピリジン(80μL,1.0mmol)及びエタノール(5.84μL)を添加して系をクエンチした。ここに3-[(N,N-ジメチルアミノメチリデン)アミノ]-3H-1,2,4-ジチアゾール-5-チオン(33.7mg,0.164mmol)を添加し30分撹拌することで、ホスファイト中間体を硫化した。アセトニトリル(42mL)を添加してアンカーを有する化合物群を沈殿させた。沈殿をろ過により取得し、アセトニトリル(42mL)で洗浄した。これを真空乾燥することで、1.34gの固体を取得した。サンプルの内、10.6mgをスターラーチップを入れた2mLのガラスバイアル中で、メタノール(250μL)および28%アンモニア水(1.0mL)と共に密封して45℃のオイルバス中18時間撹拌し、脱保護を行った。この水溶液を室温まで冷却の後、遠心エバポレータ中35℃で10分間アンモニアの除去を行った。2Mトリエチルアミン-酢酸バッファーで5.0mLまでメスアップし、0.45μmのメンブレンフィルターにより濾過し不溶分を除去し、液体クロマトグラフィーにより分析した。別途、10.6mg精秤したH-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT]-suc-TOBを含量82wt%で3,4,5-トリス(オクタデシルオキシ)安息香酸メチルと共に含有する固体を同様に脱保護・希釈したサンプルを標品としてカップリング混合物中の未反応のH-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT]-suc-TOBを定量した所、実際に用いた、晶析補助剤との混合物(82 w/w%)として137mg相当のH-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT]-suc-TOBを含有しており、縮合反応によるH-d[GiBuAdmaCHDAdmaTGiBuCHDAdmaTT]-suc-TOBの転化率は76%と求まった。
前述の脱保護・希釈を行ったサンプルをLC/MSにより分析し、表題の化合物を同定した。
LC/MS m/z:calcd 2209.57,found 2209.55 [M-3H]3-
前述の脱保護・希釈を行ったサンプルをLC/MSにより分析し、表題の化合物を同定した。
LC/MS m/z:calcd 2209.57,found 2209.55 [M-3H]3-
実施例28:5’-末端のホスフィチル化
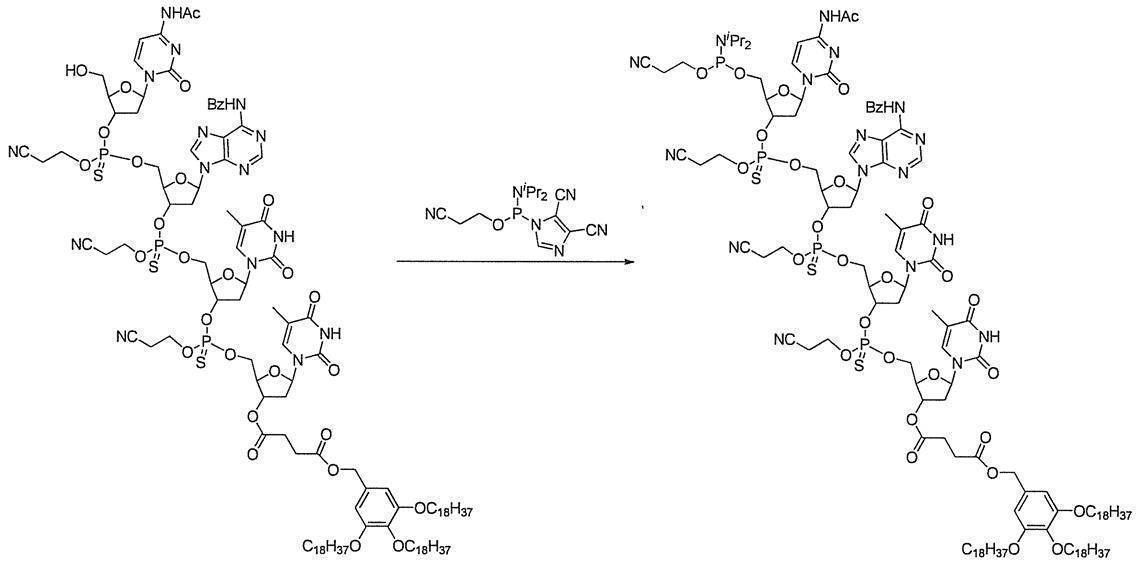
実施例6同様に、脱水トルエン中,2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトと4,5-ジシアノイミダゾールから調製したホスフィチル化剤溶液(濾液として1.5mL,0.60mmol)にN-メチルモルホリン(0.66mL,6.0mmol)を添加した。ここにH-d[CAcABzTT]-suc-TOB(0.496g,0.20mmol)を10℃で加えて懸濁液とした。同温度でジクロロメタン(10.0mL)を添加して均一溶液とした後、2時間、撹拌した。原料の完全転化をLC/MSにより確認した後2,4,6-トリメチルピリジン(0.79mL,6.0mmol)で反応を停止した。反応液を0℃に冷却してアセトニトリルを添加して生成物を析出させた。桐山ロートによりろ過・洗浄し、真空乾燥により(iPr2N)(NCCH2CH2O)P-d[CAcABzTT]-suc-TOB(0.4227g,0.16mmol,79%収率)を白色固体として得た。H-d[CAcABzTT]-suc-TOBは、調製例1-(3)と同様にして得たd[ABzTT]-suc-TOBをヌクレオシドホスホロアミダイトと縮合し、さらに脱DMTr化することで調製した。用いたヌクレオシドホスホロアミダイトは以下の通り。
dCAc:5’-(4,4’-ジメトキシトリチル)-N4-アセチル-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
生成物を31P{1H}NMRにより分析した所、H-ホスホネートジエステルが目的物に対して9%検出されたが、インターヌクレオチドリン酸上の脱シアノエチル化体は検出されず、本反応で導入したリンを介して2つのオリゴ核酸5’-末端同士が連結したホスファイトは検出限界以下であった。
5’末端がホスフィチル化されたホスホロアミダイトを用いることで5’末端側に直接又はリンカーを介して官能基が連結されたオリゴヌクレオチドを得ることができる。
dCAc:5’-(4,4’-ジメトキシトリチル)-N4-アセチル-2’-デオキシシチジン-3’-[(2-シアノエチル)-N,N-ジイソプロピル]ホスホロアミダイト
生成物を31P{1H}NMRにより分析した所、H-ホスホネートジエステルが目的物に対して9%検出されたが、インターヌクレオチドリン酸上の脱シアノエチル化体は検出されず、本反応で導入したリンを介して2つのオリゴ核酸5’-末端同士が連結したホスファイトは検出限界以下であった。
5’末端がホスフィチル化されたホスホロアミダイトを用いることで5’末端側に直接又はリンカーを介して官能基が連結されたオリゴヌクレオチドを得ることができる。
本発明の方法によれば、保管中安定なジアミダイトからホスフィチル化剤を調製することが可能であり、ホスフィチル化反応の際のリン酸上の保護基が脱落することを抑制することができる。従って、より安定、且つ効率よく3’末端ホスホロアミダイト化オリゴヌレオチドを製造することができる。さらに本発明の方法によれば、ヌクレオチド間リン酸上のシアノエチル保護基を脱落することなく、3’末端のシリル保護基を脱保護することが可能となる。従って、これらの方法を用いることで、より効率的なオリゴヌクレオチドの製造及びそれを用いたフラグメント縮合が可能となる。
本出願は、日本で出願された特願2015-250665(出願日:2015年12月22日)を基礎としており、その内容は本明細書にすべて包含されるものである。
本出願は、日本で出願された特願2015-250665(出願日:2015年12月22日)を基礎としており、その内容は本明細書にすべて包含されるものである。
Claims (51)
- 溶媒中で5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチド(nは2以上の任意の整数を示す。)に1又は2種類以上の有機塩基の存在下、フッ化物イオン源を作用させる工程を含む、5’位水酸基又は5’位リン酸基が保護され、3’位水酸基又は3’位アミノ基が保護されていないn個重合オリゴヌクレオチドを製造する方法であって、該1又は2種類以上の有機塩基のうち少なくとも1種とフッ化水素との塩を該フッ化物イオン源とすることを特徴とする、方法。
- 5’位水酸基又は5’位リン酸基が保護され、かつ3’位水酸基又は3’位アミノ基がシリル保護基で保護されているn個重合オリゴヌクレオチドが、下記構造(I)を有する、請求項1記載の方法。

[式中、
s+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
s+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
s個のP2は、それぞれ独立してリン酸基の保護基を示し、
s個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
s+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
sは、1以上の任意の整数を示す。] - フッ化物イオン源に対し1モル当量以上の有機塩基を使用する、請求項1又は2に記載の方法。
- 有機塩基が強塩基と弱塩基との混合物、2種類以上の弱塩基の混合物又は単一の弱塩基である、請求項1~3のいずれか1項に記載の方法。
- 強塩基がpKa≧8であり、弱塩基が4≦pKa<8である、請求項4に記載の方法。
- 有機塩基として強塩基を1/3モル当量以下で使用する、請求項1~5のいずれか1項に記載の方法。
- 有機塩基として、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、プロピルアミン、イソプロピルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、ブチルアミン、イソブチルアミン、tert-ブチルアミン、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、モルホリンから選択される1種以上を使用する、請求項1~6のいずれか1項に記載の方法。
- 有機塩基として、ピリジン、2,6-ジメチルピリジン、2,4,6-トリメチルピリジン、ピペラジン、ピペリジン、イミダゾール、N-メチルイミダゾール、N-メチルモルホリン、N-エチルモルホリン、アニリン、トルイジン、ジメチルアニリン、エチルアニリン、ジエチルアニリン、エチルメチルアニリン、アニシジンから選択される1種以上を使用する、請求項1~7のいずれか1項に記載の方法。
- 有機塩基として、トリエチルアミンとピリジンとの混合物を使用する、請求項1~8のいずれか1項に記載の方法。
- フッ化物イオン源が、トリエチルアミン五フッ化水素酸塩、トリエチルアミン三フッ化水素酸塩及びピリジンフッ化水素酸塩から選択される1種以上である請求項1~9のいずれか1項に記載の方法。
- シリル保護基が、ケイ素上の3つの置換基がアルキル基、アリール基、アラルキル基、アルコキシ基、アリールオキシ基、アラルキルオキシ基、アルキルチオ基、アリールチオ基から選択され、且つそのうちの少なくとも1つがアリール基、アルコキシ基、アリールオキシ基、アラルキルオキシ基から選択されるシリル保護基である、請求項1~10のいずれか1項に記載の方法。
- s+1個のBaseの少なくとも一つが、式:-L-YL-Z
[式中、
Lは、式(a1):
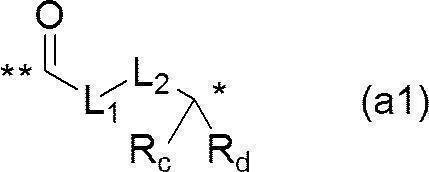
(式中、**は、核酸との結合位置を示し;
*は、YLとの結合位置を示し;
L1は、置換されていてもよい2価のC1-22炭化水素基、酸素原子、-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)を示し;かつ
L2は、単結合を示すか、または式:*C(R3a)(R3b)-O-R1**、式:*C(=O)N(R2)-R1-N(R3a)**あるいは式:*C(=O)N(R2)-R1-C(R3a)(R3b)**(式中、*は、L1との結合位置を示し、**は、CRcRdとの結合位置を示し、R1は、置換されていてもよいC1-22アルキレン基を示し、R2およびR3aは、それぞれ独立して、水素原子もしくは置換されていてもよいC1-22アルキル基を示すか、またはR2およびR3aが一緒になって、置換されていてもよいC1-22アルキレン結合を形成していてもよく、R3bは水素原子若しくは置換されていても良いC1-22アルキル基を示す)で示される基を示し;
RcおよびRdはそれぞれ独立して水素原子、置換されていてもよいC1-22アルキル基もしくはRcおよびRdが一緒になって、単一のカルボニル基を形成していてもよい)で表される基(リンカー)を示し;
YLは、単結合、酸素原子、または-NR-(Rは、水素原子、アルキル基またはアラルキル基を示す。)、硫黄原子を示し;ならびに
Zは式(a2):

(式中、*は、YLとの結合位置を示し;
R4は、水素原子を示すか、あるいはRbが、下記式(a3)で表される基である場合には、R6と一緒になって単結合または-O-を示して、環Bと共にフルオレニル基またはキサンテニル基を形成していてもよく;
k個のR5は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
kは、1~4の整数を示し;
環Aは、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
Raは、水素原子、またはハロゲン原子により置換されていてもよいフェニル基を示し;ならびに
Rbは、水素原子、または式(a3):
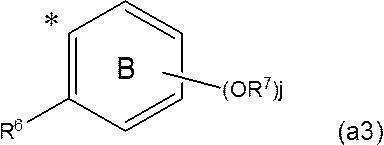
(式中、*は結合位置を示し;
jは、0~4の整数を示し;
j個のR7は、独立してそれぞれ炭素数10以上の脂肪族炭化水素基を有する有機基を示し;
R6は、水素原子を示すか、またはR4と一緒になって単結合または-O-を示して、環Aと共にフルオレニル基またはキサンテニル基を形成していてもよく;かつ
環Bは、j個のOR7に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
RaおよびRbが一緒になって、単一のカルボニル基を形成していてもよい。)で表される基);
式(a2’):

(式中、*は、YLとの結合位置を示し;
それ以外の各記号の定義は式(a2)と同義である。)で示される基
または、
式(a2’’):
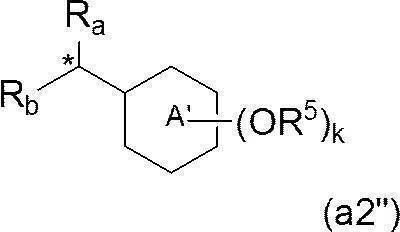
(式中、*は、YLとの結合位置を示し;
環A’は、k個のOR5に加えて、更にハロゲン原子、ハロゲン原子で置換されていてもよいC1-6アルキル基、およびハロゲン原子で置換されていてもよいC1-6アルコキシ基からなる群から選択される置換基を有していてもよく;
各記号の定義は式(a2)と同義である。)
で表される基を示す]で表される保護基、若しくは式:-Z’

[式中、**は、核酸との結合位置を示し;
各記号の定義は式(a2’’)と同義である。]
で表される保護基で保護されている、請求項1~11のいずれか1項に記載の方法。 - Lがスクシニル基である、請求項12に記載の方法。
- R5及び/又はR7が炭素原子数10~40のアルキル基である、請求項12又は13に記載の方法。
- R5がオクタデシル基である、請求項12又は13に記載の方法。
- 3’末端のヌクレオシドのBaseが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項1~15のいずれか1項に記載の方法。
- 以下の工程(1)及び(2)を含有する、ホスホロアミダイト化オリゴヌクレオチドの製造方法。
(1)溶媒中で、下記式(1):

[式中、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R20およびR30はそれぞれ独立してアルキル基を示し、該アルキル基は隣接する窒素原子と一緒になって環を形成してもよい。]
で表されるホスフィチル化剤前駆体に活性化剤を作用させて下記式(2):
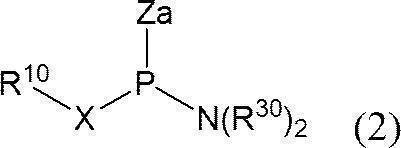
[式中、
Zaは活性化剤に由来する基を示し、
それ以外の各記号は、前記と同義である。]
で表されるホスフィチル化剤を調製する工程、および
(2)溶媒中で、5’位水酸基若しくは5’位リン酸基又は3’位水酸基若しくは又は3’位アミノ基のいずれか一方が保護され、かつもう一方が保護されていないn個重合オリゴヌクレオチド(nは、2以上の任意の整数を示す。)に、塩基の存在下、工程(1)で得られたホスフィチル化剤を作用させて、該オリゴヌクレオチドの末端の水酸基をホスフィチル化する工程 - 該n個重合オリゴヌクレオチドが、下記構造(Ia)を有する、請求項17記載の方法。

[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。] - 該n個重合オリゴヌクレオチドが、下記構造(Ia’)を有する、請求項17記載の方法。

[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
qは、1以上の任意の整数を示す。] - q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項17~19のいずれか1項に記載の方法。
- 工程(1)で用いる活性化剤が、pKa5以上の弱酸性活性化剤である、請求項17~20のいずれか1項に記載の方法。
- 工程(1)で用いる活性化剤が、アゾール系化合物である、請求項17~21のいずれか1項に記載の方法。
- 工程(1)で用いる活性化剤が、ジシアノイミダゾール又はジクロロイミダゾールである、請求項17~22のいずれか1項に記載の方法。
- 工程(1)で用いる活性化剤が、ホスフィチル化剤前駆体に対して1.5~20モル当量で用いられる、請求項17~23のいずれか1項に記載の方法。
- 工程(1)で用いる溶媒が、ホスフィチル化剤前駆体を溶解し、かつ活性化剤が難溶性となるものであって、酸性または塩基性官能基を有さないことを特徴とする、請求項17~24のいずれか1項に記載の方法。
- 工程(1)で用いる溶媒が、トルエン、ベンゼン、o-キシレン、m-キシレン、p-キシレン、エチルベンゼン、ペンタン、ヘキサン、ヘプタン、オクタン、シクロペンタン、シクロヘキサン、ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、シクロペンチルメチルエーテル、四塩化炭素から選択される1種以上の溶媒である、請求項17~25のいずれか1項に記載の方法。
- 工程(1)で用いる溶媒が、トルエンである、請求項17~26のいずれか1項に記載の方法。
- 工程(2)で用いる塩基が、pKa5~8の塩基である、請求項17~27のいずれか1項に記載の方法。
- 工程(2)で用いる塩基が、コリジン、N-メチルモルホリン及びジエチルアニリンから選択される1種以上の塩基である、請求項17~28のいずれか1項に記載の方法。
- 工程(1)と工程(2)の間に、不溶物を分離する工程を含む、請求項17~29のいずれか1項に記載の方法。
- 請求項18に記載の方法により下記式(Ia-1):
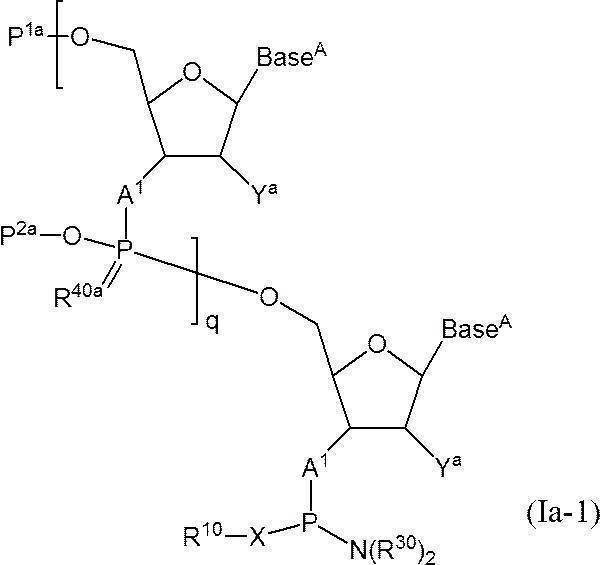
[式中、
q+1個のA1は、それぞれ独立して酸素原子又は-NH-を示し、
q+1個のBaseAは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia-2):

[式中、
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。 - 請求項19に記載の方法により下記式(Ia’-1):

[式中、P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ia-1)と同義である]
で表される5’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該5’末端ホスホロアミダイト化オリゴヌクレオチドの5’末端に、直接又はリンカーを介して官能基を連結させる工程を含む、下記式(Ia’-2):

[式中、P1a’は、水酸基又はアミノ基の保護基を示すか、又は-A1-P1a’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ia-2)と同義である]
で表される5’末端に官能基を有するオリゴヌクレオチドの製造方法。 - q+1個のBaseAの少なくとも一つが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項31又は32に記載の方法。
- 官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、請求項31~33のいずれか1項に記載の方法。
- 官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、請求項31記載の方法。
- 官能基が、5’位水酸基が保護され、かつ3’位水酸基又は3’位アミノ基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、請求項31記載の方法。
- n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、請求項35記載の方法。

[式中、r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。] - n’個重合オリゴヌクレオチドが、下記構造(Ib’)を有する、請求項36記載の方法。
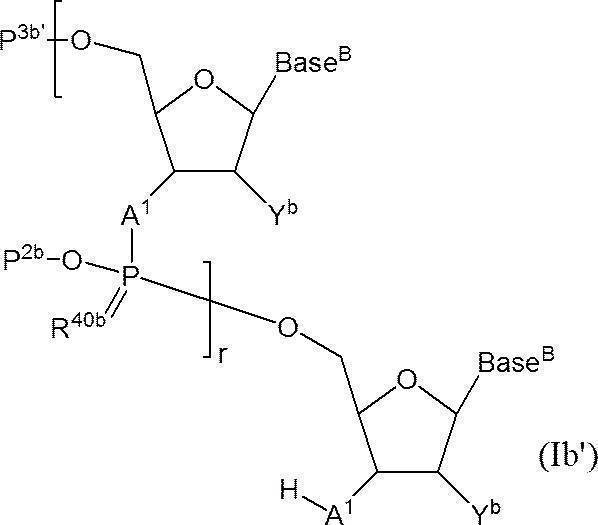
[式中、P3b’は、水酸基の保護基を示すか、又は-O-P3b’として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、それ以外の各記号の定義は式(Ib)と同義である] - r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項37又は38に記載の方法。
- P3b又はP3b’における水酸基の保護基が、式:-L-YL-Z(各記号の定義は請求項12と同義である)、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される、請求項37~39のいずれか1項に記載の方法。
- Lがスクシニル基である、請求項40に記載の方法。
- 下記式(Ia-1’):
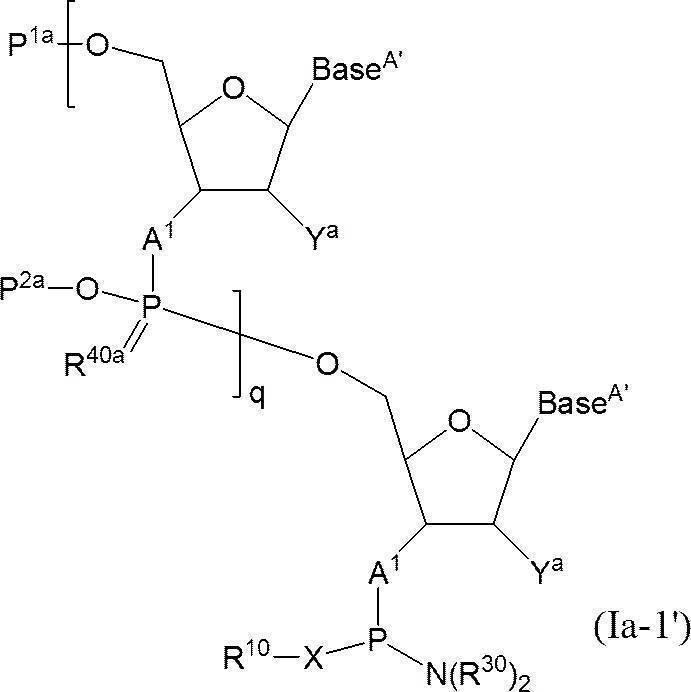
[式中、
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseA’は、それぞれ独立して式:-L’-YL-Z(式中、L’はスクシニル基、YL、Zは請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されていてもよい核酸塩基を示し、
P1aは、水酸基の保護基を示すか、又は-O-P1aとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2aは、それぞれ独立してリン酸基の保護基を示し、
q個のR40aは、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYaは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
Xは、酸素原子又は硫黄原子を示し、
R10は、芳香環、水酸基の保護基又はチオール基の保護基を示し、
R30はそれぞれ独立してアルキル基を示し、
qは、1以上の任意の整数を示す。]
で表される3’末端ホスホロアミダイト化オリゴヌクレオチドの3’末端に、直接又はリンカーを介して官能基を連結させて下記式(Ia-2’):

[式中、
Lxは単結合又はリンカーを示し、
Gは官能基を示し、
それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドを得た後、保護基を除去する工程を含む、下記式(II):
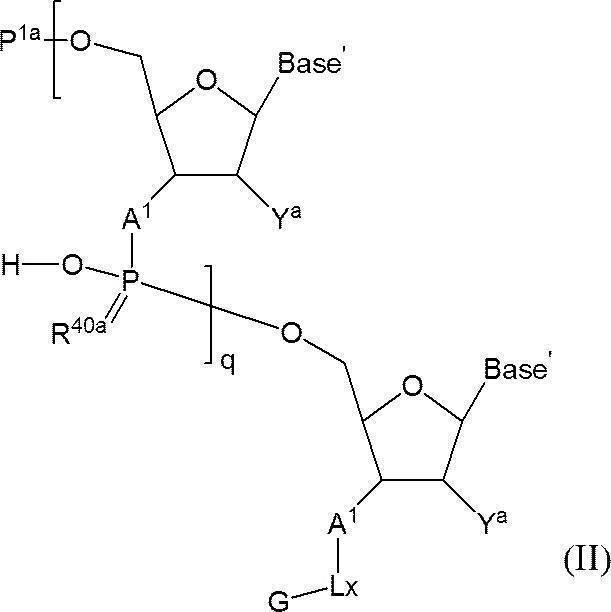
[式中、q+1個のBase’は無保護の核酸塩基を示し、それ以外の各記号の定義は上述の通りである]
で表される3’末端に官能基を有するオリゴヌクレオチドの製造方法。 - 請求項17~41のいずれか1項に記載の方法により3’末端ホスホロアミダイト化オリゴヌクレオチドを得た後、該3’末端ホスホロアミダイト化オリゴヌクレオチドを使用する、請求項42記載の方法。
- 官能基が、オリゴヌクレオチド、モノヌクレオシド、コレステロール、GalNac3、PEG、低分子医薬、ビオチン、ペプチド及び標識化合物からなる群より選択される少なくとも1種に由来する、請求項42又は43記載の方法。
- 官能基が、3’位水酸基又は3’位アミノ基あるいは3’位リン酸基が保護され、かつ5’位水酸基が保護されていないn’個重合オリゴヌクレオチド(n’は、2以上の任意の整数を示す。)である、請求項42~44のいずれか1項に記載の方法。
- n’個重合オリゴヌクレオチドが、下記構造(Ib)を有する、請求項45記載の方法。

[式中、
r+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
r+1個のBaseBは、それぞれ独立して保護されていてもよい核酸塩基を示し、
r個のP2bは、リン酸基の保護基を示し、
r個のR40bは、酸素原子又は硫黄原子を示し、
P3bは、水酸基又はアミノ基の保護基を示すか、又は-A1-P3bとして水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
r+1個のYbは、水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
rは、1以上の任意の整数を示す。] - r+1個のBaseBの少なくとも一つが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項46に記載の方法。
- P3bおける水酸基の保護基が、式:-L-YL-Z(各記号の定義は請求項12と同義である)、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される、請求項46又は47記載の方法。
- Lがスクシニル基である、請求項48に記載の方法。
- 下記式:

[式中、
q+1個のA1は、それぞれ独立して酸素原子又はNHを示し、
q+1個のBaseは、それぞれ独立して保護されていてもよい核酸塩基を示し、
P1は、水酸基の保護基を示すか、又は-O-P1として水酸基の1つが-OLn1-OH(式中、Ln1は有機基を示し、水酸基は保護されている)に置き換わっているリン酸基を示し、
q個のP2は、それぞれ独立してリン酸基の保護基を示し、
q個のR40は、それぞれ独立して酸素原子又は硫黄原子を示し、
q+1個のYは、それぞれ独立して水素原子、保護されていてもよい水酸基、ハロゲン原子又は4位炭素原子に架橋する有機基を示し、
P3は、シリル保護基を示し、
qは、1以上の任意の整数を示す。]
で表される化合物。 - q+1個のBaseの少なくとも一つが、式:-L-YL-Z(各記号の定義は請求項12と同義である)で表される保護基、若しくは式:-Z’(各記号の定義は請求項12と同義である)で表される保護基で保護されている、請求項50記載の化合物。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017558314A JPWO2017111137A1 (ja) | 2015-12-22 | 2016-12-22 | オリゴヌクレオチドの製造方法 |
| US16/014,536 US20180291056A1 (en) | 2015-12-22 | 2018-06-21 | Oligonucleotide manufacturing method |
| US17/654,700 US12559517B2 (en) | 2015-12-22 | 2022-03-14 | Oligonucleotide manufacturing method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015250665 | 2015-12-22 | ||
| JP2015-250665 | 2015-12-22 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/014,536 Continuation US20180291056A1 (en) | 2015-12-22 | 2018-06-21 | Oligonucleotide manufacturing method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017111137A1 true WO2017111137A1 (ja) | 2017-06-29 |
Family
ID=59090655
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/088580 Ceased WO2017111137A1 (ja) | 2015-12-22 | 2016-12-22 | オリゴヌクレオチドの製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US20180291056A1 (ja) |
| JP (2) | JPWO2017111137A1 (ja) |
| WO (1) | WO2017111137A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019212061A1 (ja) * | 2018-05-02 | 2019-11-07 | 株式会社四国核酸化学 | オリゴヌクレオチド合成用セグメントおよびその製造方法、ならびにそれを用いたオリゴヌクレオチドの合成方法 |
| WO2019212063A1 (ja) * | 2018-05-02 | 2019-11-07 | 株式会社四国核酸化学 | 立体制御オリゴヌクレオチド合成用光学活性セグメントおよびその製造方法、ならびにそれを用いた立体制御オリゴヌクレオチドの合成方法 |
| US10919928B2 (en) | 2015-12-16 | 2021-02-16 | Ajinomoto Co., Inc. | Oligonucleotide production method, and nucleoside, nucleotide, or oligonucleotide |
| WO2021141072A1 (ja) * | 2020-01-08 | 2021-07-15 | 日東電工株式会社 | セグメント型アミダイトを用いた核酸合成法 |
| JP2022527569A (ja) * | 2019-03-21 | 2022-06-02 | ミトセラピューティクス・エルエルシー | 治療剤の標的化送達のための多価リガンドクラスター |
| JP7220439B1 (ja) | 2022-03-14 | 2023-02-10 | 国立大学法人東北大学 | キメラ分子の製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL287798B2 (en) | 2019-05-08 | 2025-09-01 | Biogen Ma Inc | Convergent Syntheses of Oligonucleotides in Liquid Phase |
| CN115843299A (zh) | 2020-03-31 | 2023-03-24 | 詹森生物制药有限公司 | 寡核苷酸的合成和相关化合物 |
| JP2025515631A (ja) * | 2022-05-02 | 2025-05-20 | アムジェン インコーポレイテッド | 炭水化物ターゲティング部分であるnag-25、及びその中間体を製造する方法 |
| WO2024083746A1 (en) | 2022-10-17 | 2024-04-25 | Bachem Holding Ag | Method and composition for oligonucleotide synthesis |
| EP4695265A1 (en) | 2023-10-16 | 2026-02-18 | Bachem Holding AG | Method and composition for oligonucleotide synthesis |
Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09505307A (ja) * | 1993-11-16 | 1997-05-27 | ジンタ・インコーポレイテッド | 多キラル合成フォスホネートオリゴマー類 |
| JPH09505306A (ja) * | 1993-11-16 | 1997-05-27 | ジンタ・インコーポレイテッド | 非ホスホネートヌクレオシド間結合と混合している不確定キラリティーのホスホネートヌクレオシド間結合を有する合成オリゴマー |
| JPH11322783A (ja) * | 1998-05-06 | 1999-11-24 | Tosoh Corp | 光学活性なホスホン酸ジエステル結合を持つdnaプローブ |
| WO2000000499A1 (en) * | 1998-06-30 | 2000-01-06 | Isis Pharmaceuticals, Inc. | Method for preparing oligonucleotides with boranophosphate linkages |
| JP2001089496A (ja) * | 1999-07-22 | 2001-04-03 | Sankyo Co Ltd | 新規ビシクロヌクレオシド類縁体 |
| JP2002255990A (ja) * | 2000-11-21 | 2002-09-11 | Sankyo Co Ltd | N3’−p5’結合を有する2’,4’−bnaオリゴヌクレオチド |
| WO2005070859A1 (ja) * | 2004-01-27 | 2005-08-04 | Takeshi Wada | フルオラス担体およびそれを用いたオリゴヌクレオチド誘導体の製造方法 |
| JP2008163024A (ja) * | 2007-12-20 | 2008-07-17 | Geron Corp | オリゴヌクレオチドn3’→p5’ホスホルアミデートの固相合成 |
| JP2009516521A (ja) * | 2005-11-23 | 2009-04-23 | エフ.ホフマン−ラ ロシュ アーゲー | リン酸塩模倣物を含むポリヌクレオチド |
| JP2009524695A (ja) * | 2006-01-27 | 2009-07-02 | アイシス ファーマシューティカルズ, インコーポレーテッド | 6−修飾された二環式核酸類似体 |
| JP2010275254A (ja) * | 2009-05-29 | 2010-12-09 | Tokyo Univ Of Agriculture & Technology | 疎水性基結合ヌクレオシド、疎水性基結合ヌクレオシド溶液、及び疎水性基結合オリゴヌクレオチド合成方法 |
| WO2011005861A1 (en) * | 2009-07-07 | 2011-01-13 | Alnylam Pharmaceuticals, Inc. | Oligonucleotide end caps |
| WO2011061114A1 (de) * | 2009-11-23 | 2011-05-26 | Ernst-Moritz-Arndt-Universität Greifswald | Verfahren zur herstellung von trinukleotiden |
| WO2011156202A1 (en) * | 2010-06-08 | 2011-12-15 | Isis Pharmaceuticals, Inc. | Substituted 2 '-amino and 2 '-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2012024776A1 (en) * | 2010-08-23 | 2012-03-01 | The Royal Institution For The Advancement Of Learning/Mcgill University | Block synthesis of oligoribonucleotides |
| JP2012506701A (ja) * | 2008-10-24 | 2012-03-22 | アイシス ファーマシューティカルズ, インコーポレーテッド | 5’及び2’ビス置換ヌクレオシド及びそれから製造されるオリゴマー化合物 |
| JP2012510460A (ja) * | 2008-12-02 | 2012-05-10 | 株式会社キラルジェン | リン原子修飾核酸の合成方法 |
| JP2012111728A (ja) * | 2010-11-26 | 2012-06-14 | Nokodai Tlo Kk | 高分散性液相支持体を用いたオリゴヌクレオチド合成法 |
| WO2012157723A1 (ja) * | 2011-05-17 | 2012-11-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
| WO2013022966A1 (en) * | 2011-08-11 | 2013-02-14 | Isis Pharmaceuticals, Inc. | Linkage modified gapped oligomeric compounds and uses thereof |
| WO2013122236A1 (ja) * | 2012-02-17 | 2013-08-22 | 味の素株式会社 | 塩基部保護オリゴヌクレオチド |
| US20130267697A1 (en) * | 2012-02-17 | 2013-10-10 | Ajinomoto Co., Inc. | Oligonucleotide with protected base |
| WO2013179412A1 (ja) * | 2012-05-30 | 2013-12-05 | 北海道システム・サイエンス株式会社 | 高分散性液相支持体を用いたオリゴヌクレオチド合成法 |
| WO2014077292A1 (ja) * | 2012-11-14 | 2014-05-22 | 武田薬品工業株式会社 | 核酸の液相合成方法 |
| WO2015168172A1 (en) * | 2014-04-28 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US5571902A (en) | 1993-07-29 | 1996-11-05 | Isis Pharmaceuticals, Inc. | Synthesis of oligonucleotides |
| US5955597A (en) | 1993-11-16 | 1999-09-21 | Genta, Incorporated | Chirally enriched synthetic phosphate oligomers |
| US6060456A (en) | 1993-11-16 | 2000-05-09 | Genta Incorporated | Chimeric oligonucleoside compounds |
| AU678085B2 (en) | 1993-11-16 | 1997-05-15 | Genta Incorporated | Synthetic oligomers having chirally pure phosphonate internucleosidyl linkages mixed with non-phosphonate internucleosidyl linkages |
| US6340749B1 (en) | 1995-10-06 | 2002-01-22 | Avecia Biotechnology Inc. | Preparation of nucleoside phosphoramidites and oligonucleotide synthesis |
| US6476216B1 (en) | 1995-10-20 | 2002-11-05 | Mcgill University | Preparation of phosphorothioate oligomers |
| US5734041A (en) | 1995-10-20 | 1998-03-31 | Mcgill University | Preparation of chiral phosphorothioate oligomers |
| US6090937A (en) | 1998-03-17 | 2000-07-18 | Ajinomoto Co., Inc. | Methods for producing nucleoside derivatives and intermediates therefor |
| US6211354B1 (en) | 1998-05-06 | 2001-04-03 | Tosch Corporation | Optically active DNA probe having phosphonic diester linkage |
| ES2386709T3 (es) | 2004-08-26 | 2012-08-27 | Nippon Shinyaku Co., Ltd. | Compuesto de fosforamidita y método para producir un oligo-ARN |
| CA2671873C (en) | 2006-12-12 | 2018-10-09 | Brian Stephen Sproat | Oligonucleotides containing high concentrations of guanine monomers |
| US20080206851A1 (en) * | 2007-02-28 | 2008-08-28 | Dellinger Douglas J | Methods and compositions for RNA synthesis |
| EP2358398A2 (en) | 2008-10-24 | 2011-08-24 | Isis Pharmaceuticals, Inc. | Oligomeric compounds and methods |
| US10017764B2 (en) | 2011-02-08 | 2018-07-10 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| KR102080743B1 (ko) | 2013-03-08 | 2020-02-24 | 엘지전자 주식회사 | 이동 단말기 및 그것의 제어 방법 |
| KR102143576B1 (ko) | 2015-01-19 | 2020-09-09 | 엘에스엠트론 주식회사 | 내부 터미널의 설치 구조가 개선된 전기에너지 저장장치 |
| EP3263579B1 (en) | 2015-01-21 | 2022-11-30 | Ajinomoto Co., Inc. | Precipitation promoter and precipitation method in which same is used |
| CN108350018A (zh) | 2015-11-17 | 2018-07-31 | 日产化学工业株式会社 | 寡核苷酸的制造方法 |
| WO2017104836A1 (ja) | 2015-12-16 | 2017-06-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法、およびヌクレオシド、ヌクレオチドまたはオリゴヌクレオチド |
-
2016
- 2016-12-22 JP JP2017558314A patent/JPWO2017111137A1/ja active Pending
- 2016-12-22 WO PCT/JP2016/088580 patent/WO2017111137A1/ja not_active Ceased
-
2018
- 2018-06-21 US US16/014,536 patent/US20180291056A1/en not_active Abandoned
-
2022
- 2022-02-25 JP JP2022028524A patent/JP7484951B2/ja active Active
- 2022-03-14 US US17/654,700 patent/US12559517B2/en active Active
Patent Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09505307A (ja) * | 1993-11-16 | 1997-05-27 | ジンタ・インコーポレイテッド | 多キラル合成フォスホネートオリゴマー類 |
| JPH09505306A (ja) * | 1993-11-16 | 1997-05-27 | ジンタ・インコーポレイテッド | 非ホスホネートヌクレオシド間結合と混合している不確定キラリティーのホスホネートヌクレオシド間結合を有する合成オリゴマー |
| JPH11322783A (ja) * | 1998-05-06 | 1999-11-24 | Tosoh Corp | 光学活性なホスホン酸ジエステル結合を持つdnaプローブ |
| WO2000000499A1 (en) * | 1998-06-30 | 2000-01-06 | Isis Pharmaceuticals, Inc. | Method for preparing oligonucleotides with boranophosphate linkages |
| JP2001089496A (ja) * | 1999-07-22 | 2001-04-03 | Sankyo Co Ltd | 新規ビシクロヌクレオシド類縁体 |
| JP2002255990A (ja) * | 2000-11-21 | 2002-09-11 | Sankyo Co Ltd | N3’−p5’結合を有する2’,4’−bnaオリゴヌクレオチド |
| WO2005070859A1 (ja) * | 2004-01-27 | 2005-08-04 | Takeshi Wada | フルオラス担体およびそれを用いたオリゴヌクレオチド誘導体の製造方法 |
| JP2009516521A (ja) * | 2005-11-23 | 2009-04-23 | エフ.ホフマン−ラ ロシュ アーゲー | リン酸塩模倣物を含むポリヌクレオチド |
| JP2009524695A (ja) * | 2006-01-27 | 2009-07-02 | アイシス ファーマシューティカルズ, インコーポレーテッド | 6−修飾された二環式核酸類似体 |
| JP2008163024A (ja) * | 2007-12-20 | 2008-07-17 | Geron Corp | オリゴヌクレオチドn3’→p5’ホスホルアミデートの固相合成 |
| JP2012506701A (ja) * | 2008-10-24 | 2012-03-22 | アイシス ファーマシューティカルズ, インコーポレーテッド | 5’及び2’ビス置換ヌクレオシド及びそれから製造されるオリゴマー化合物 |
| JP2012510460A (ja) * | 2008-12-02 | 2012-05-10 | 株式会社キラルジェン | リン原子修飾核酸の合成方法 |
| JP2010275254A (ja) * | 2009-05-29 | 2010-12-09 | Tokyo Univ Of Agriculture & Technology | 疎水性基結合ヌクレオシド、疎水性基結合ヌクレオシド溶液、及び疎水性基結合オリゴヌクレオチド合成方法 |
| WO2011005861A1 (en) * | 2009-07-07 | 2011-01-13 | Alnylam Pharmaceuticals, Inc. | Oligonucleotide end caps |
| WO2011061114A1 (de) * | 2009-11-23 | 2011-05-26 | Ernst-Moritz-Arndt-Universität Greifswald | Verfahren zur herstellung von trinukleotiden |
| WO2011156202A1 (en) * | 2010-06-08 | 2011-12-15 | Isis Pharmaceuticals, Inc. | Substituted 2 '-amino and 2 '-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2012024776A1 (en) * | 2010-08-23 | 2012-03-01 | The Royal Institution For The Advancement Of Learning/Mcgill University | Block synthesis of oligoribonucleotides |
| JP2012111728A (ja) * | 2010-11-26 | 2012-06-14 | Nokodai Tlo Kk | 高分散性液相支持体を用いたオリゴヌクレオチド合成法 |
| WO2012157723A1 (ja) * | 2011-05-17 | 2012-11-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
| WO2013022966A1 (en) * | 2011-08-11 | 2013-02-14 | Isis Pharmaceuticals, Inc. | Linkage modified gapped oligomeric compounds and uses thereof |
| WO2013122236A1 (ja) * | 2012-02-17 | 2013-08-22 | 味の素株式会社 | 塩基部保護オリゴヌクレオチド |
| US20130267697A1 (en) * | 2012-02-17 | 2013-10-10 | Ajinomoto Co., Inc. | Oligonucleotide with protected base |
| WO2013179412A1 (ja) * | 2012-05-30 | 2013-12-05 | 北海道システム・サイエンス株式会社 | 高分散性液相支持体を用いたオリゴヌクレオチド合成法 |
| WO2014077292A1 (ja) * | 2012-11-14 | 2014-05-22 | 武田薬品工業株式会社 | 核酸の液相合成方法 |
| WO2015168172A1 (en) * | 2014-04-28 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
Non-Patent Citations (4)
| Title |
|---|
| FETTES KEVIN J. ET AL.: "Synthesis and nucleic- acid-binding properties of sulfamide- and 3'-N- sulfamate-modified DNA", JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, vol. 4, 2002, pages 485 - 495, XP055395813 * |
| GREENE THEODORA W ET AL.: "PROTECTIVE GROUPS IN ORGANIC SYNTHESIS", pages 77 - 86 * |
| KIM SHOKAKU ET AL.: "Liquid-Phase RNA Synthesis by Using Alkyl-Chain-Soluble Support", CHEMISTRY - A EUROPEAN JOURNAL, vol. 19, no. 26, 2013, pages 8615 - 8620, XP055211240 * |
| SHOJI TAKAO ET AL.: "Synthesis of conjugated oligonucleotide in solution phase using alkyl- chain-soluble support", CHEMISTRY LETTERS, vol. 43, no. 8, 2014, pages 1251 - 1253, XP055393419 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10919928B2 (en) | 2015-12-16 | 2021-02-16 | Ajinomoto Co., Inc. | Oligonucleotide production method, and nucleoside, nucleotide, or oligonucleotide |
| US12139507B2 (en) | 2018-05-02 | 2024-11-12 | Natias Inc. | Segment for use in synthesis of oligonucleotide, method for producing the same, and method for synthesizing oligonucleotide using the same |
| WO2019212063A1 (ja) * | 2018-05-02 | 2019-11-07 | 株式会社四国核酸化学 | 立体制御オリゴヌクレオチド合成用光学活性セグメントおよびその製造方法、ならびにそれを用いた立体制御オリゴヌクレオチドの合成方法 |
| JP7803520B2 (ja) | 2018-05-02 | 2026-01-21 | 株式会社ナティアス | オリゴヌクレオチド合成用セグメントおよびその製造方法、ならびにそれを用いたオリゴヌクレオチドの合成方法 |
| WO2019212061A1 (ja) * | 2018-05-02 | 2019-11-07 | 株式会社四国核酸化学 | オリゴヌクレオチド合成用セグメントおよびその製造方法、ならびにそれを用いたオリゴヌクレオチドの合成方法 |
| JPWO2019212063A1 (ja) * | 2018-05-02 | 2021-07-29 | 株式会社ナティアス | 立体制御オリゴヌクレオチド合成用光学活性セグメントおよびその製造方法、ならびにそれを用いた立体制御オリゴヌクレオチドの合成方法 |
| CN114269764A (zh) * | 2018-05-02 | 2022-04-01 | 株式会社纳蒂亚斯 | 立体控制寡核苷酸合成用光学活性片段及其制造方法、以及使用其的立体控制寡核苷酸的合成方法 |
| JP2022065127A (ja) * | 2018-05-02 | 2022-04-26 | 株式会社ナティアス | オリゴヌクレオチド合成用セグメントおよびその製造方法、ならびにそれを用いたオリゴヌクレオチドの合成方法 |
| JP7075681B2 (ja) | 2018-05-02 | 2022-05-26 | 株式会社ナティアス | 立体制御オリゴヌクレオチド合成用光学活性セグメントおよびその製造方法、ならびにそれを用いた立体制御オリゴヌクレオチドの合成方法 |
| JP2022527569A (ja) * | 2019-03-21 | 2022-06-02 | ミトセラピューティクス・エルエルシー | 治療剤の標的化送達のための多価リガンドクラスター |
| JPWO2021141072A1 (ja) * | 2020-01-08 | 2021-07-15 | ||
| JP7657736B2 (ja) | 2020-01-08 | 2025-04-07 | 日東電工株式会社 | セグメント型アミダイトを用いた核酸合成法 |
| JP2025089484A (ja) * | 2020-01-08 | 2025-06-12 | 日東電工株式会社 | セグメント型アミダイトを用いた核酸合成法 |
| WO2021141072A1 (ja) * | 2020-01-08 | 2021-07-15 | 日東電工株式会社 | セグメント型アミダイトを用いた核酸合成法 |
| JP7848377B2 (ja) | 2020-01-08 | 2026-04-20 | 日東電工株式会社 | セグメント型アミダイトを用いた核酸合成法 |
| WO2023176773A1 (ja) * | 2022-03-14 | 2023-09-21 | 国立大学法人東北大学 | キメラ分子の製造方法 |
| JP2023133992A (ja) * | 2022-03-14 | 2023-09-27 | 国立大学法人東北大学 | キメラ分子の製造方法 |
| JP7220439B1 (ja) | 2022-03-14 | 2023-02-10 | 国立大学法人東北大学 | キメラ分子の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US12559517B2 (en) | 2026-02-24 |
| JP2022068360A (ja) | 2022-05-09 |
| JPWO2017111137A1 (ja) | 2018-10-18 |
| US20220235090A1 (en) | 2022-07-28 |
| JP7484951B2 (ja) | 2024-05-16 |
| US20180291056A1 (en) | 2018-10-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7484951B2 (ja) | オリゴヌクレオチドの製造方法 | |
| JP6673373B2 (ja) | 擬似固相保護基およびヌクレオチド | |
| CN112533892B (zh) | 烷氧基苯基衍生物、核苷保护体和核苷酸保护体、寡核苷酸制造方法以及取代基除去方法 | |
| JP6237237B2 (ja) | 塩基部保護オリゴヌクレオチド | |
| USRE34069E (en) | Process for the preparation of oligonucleotides | |
| JPS6250479B2 (ja) | ||
| JPWO2017104836A1 (ja) | オリゴヌクレオチドの製造方法、およびヌクレオシド、ヌクレオチドまたはオリゴヌクレオチド | |
| CN112424213A (zh) | 寡核苷酸合成用片段及其制造方法、以及使用其的寡核苷酸的合成方法 | |
| WO2016060135A1 (ja) | モルフォリノオリゴヌクレオチドの製造方法 | |
| JP2025028988A (ja) | ホスホロチオエート化部位を有するオリゴヌクレオチドの製造方法 | |
| EP3660021B1 (en) | Photoresponsive nucleotide analog capable of photocrosslinking in visible light region | |
| KR20030081303A (ko) | 올리고뉴클레오티드 합성용 신톤 | |
| US12269841B2 (en) | Production method for oligonucleotides | |
| JP7075681B2 (ja) | 立体制御オリゴヌクレオチド合成用光学活性セグメントおよびその製造方法、ならびにそれを用いた立体制御オリゴヌクレオチドの合成方法 | |
| JPH0588240B2 (ja) | ||
| JP2022177332A (ja) | オリゴヌクレオチドを製造する方法 | |
| CN103936805B (zh) | 一种核苷酸和/或寡核苷酸及其制备方法 | |
| JP7423533B2 (ja) | 配糖体化合物の製造方法 | |
| Dabkowski et al. | Synthesis of phosphorofluoridates and phosphorofluoridothioates via the phosphoramidite approach | |
| JP2003088374A (ja) | チミジン誘導体およびオリゴヌクレオチド |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16879023 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017558314 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16879023 Country of ref document: EP Kind code of ref document: A1 |