WO2017126602A1 - 多孔質金属酸化物の製造方法 - Google Patents
多孔質金属酸化物の製造方法 Download PDFInfo
- Publication number
- WO2017126602A1 WO2017126602A1 PCT/JP2017/001729 JP2017001729W WO2017126602A1 WO 2017126602 A1 WO2017126602 A1 WO 2017126602A1 JP 2017001729 W JP2017001729 W JP 2017001729W WO 2017126602 A1 WO2017126602 A1 WO 2017126602A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- metal oxide
- porous
- firing
- slurry
- pore
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F7/00—Compounds of aluminium
- C01F7/02—Aluminium oxide; Aluminium hydroxide; Aluminates
- C01F7/34—Preparation of aluminium hydroxide by precipitation from solutions containing aluminium salts
- C01F7/36—Preparation of aluminium hydroxide by precipitation from solutions containing aluminium salts from organic aluminium salts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/04—Oxides; Hydroxides
- C01G23/047—Titanium dioxide
- C01G23/053—Producing by wet processes, e.g. hydrolysing titanium salts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G25/00—Compounds of zirconium
- C01G25/02—Oxides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/14—Pore volume
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/16—Pore diameter
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/80—Compositional purity
Definitions
- the present invention relates to a method for producing a porous metal oxide.
- Porous metal oxides such as porous alumina and porous zirconia are widely used as catalyst carriers and adsorbents.
- Patent documents 1 to 3 can be cited as technical documents related to porous alumina or its production.
- Patent Document 4 is cited as a technical document relating to the use of a metal oxide having a mesoporous structure as a catalyst carrier.
- the porous metal oxide is as a Mo adsorbent in a technetium 99m ( 99m Tc) generator.
- the 99m Tc generator is an apparatus in which molybdenum 99 ( 99 Mo) as a parent nuclide is adsorbed on a column packed with Mo adsorbent, and 99m Tc generated from 99 Mo by passing physiological saline through the column. It is comprised so that it can elute. 99m Tc extracted by the elution is combined with an appropriate drug and used as a so-called radiopharmaceutical (technetium preparation) for nuclear medical diagnosis and the like.
- Non-patent document 1 is cited as a technical document related to porous alumina for a Mo adsorbent of a 99m Tc generator.
- Non-Patent Document 1 describes the production of porous alumina using glucose as a template for pore formation.
- porous metal oxide is as a catalyst carrier.
- a three-way catalyst containing a metal such as platinum as a catalyst component is typically widely used in a form supported on a catalyst carrier.
- a metal oxide powder can be preferably used.
- the catalyst carrier having a porous structure can be advantageous from the viewpoint of the contact efficiency between the catalyst component and the target component.
- a suitable example of the catalyst carrier is a porous metal oxide such as porous alumina or porous zirconia.
- Patent Documents 1 to 3 describe porous alumina as a catalyst carrier, an adsorbent and the like, there is no description of using porous alumina as a Mo adsorbent for a 99m Tc generator.
- the method for producing mesoporous alumina described in Patent Document 1 includes a step of preparing a slurry in which an aqueous solution of aluminum salt such as aluminum nitrate and an aqueous solution of ammonium carbonate are mixed, but the step of preparing the slurry is unstable.
- Patent Document 2 proposes a method for producing a porous alumina product comprising hydrolyzing aluminum alcoholate in an aqueous solution containing 8 to 20% by weight of a compound capable of decomposing NH 3 and / or CO 2 .
- the above compound is used in an amount of 0.5 to 3.0 parts by weight per 10 parts by weight of aluminum alcoholate, which is clearly different from the production method provided by this specification.
- Patent Document 3 relates to a method for producing alumina suitable for use as a catalyst carrier for treating exhaust products from an internal combustion engine, and is a technology that preferentially takes into account the heat resistance required in the above-mentioned use, Relevance to the disclosed invention is low.
- Patent Document 4 also does not describe the use of a porous metal oxide as the Mo adsorbent of the 99m Tc generator.
- hexadecyltrimethylammonium bromide is dissolved in an aqueous hydrochloric acid solution, and tetrapropoxide zirconium is further added and dissolved.
- a separately prepared aqueous ammonium sulfate solution is mixed, stirred, cooled, centrifuged, and the like. Manufacturing porous zirconia is described, but this manufacturing method is complicated and there is room for improvement.
- the present invention has been made in view of such circumstances, and an object of the present invention is to provide a highly versatile production method for a porous metal oxide that can be suitably used in various applications.
- the method for producing a porous metal oxide includes mixing a metal source, a pore forming agent, and an aqueous solvent to prepare a slurry.
- the method includes drying the slurry to obtain a metal oxide precursor.
- it includes firing the metal oxide precursor to produce a porous metal oxide.
- the metal source an organometallic compound containing a metal constituting the porous metal oxide or a hydrolyzate thereof is used.
- the pore forming agent an inorganic compound that decomposes at a temperature equal to or lower than the temperature at which the metal oxide precursor is calcined to generate a gas is used.
- the slurry is prepared using about 50 parts by weight or more of the pore forming agent with respect to 100 parts by weight of the metal source.
- an organometallic compound or a hydrolyzate thereof is used as a metal source, a slurry with good homogeneity can be easily prepared. Since this slurry is prepared using about 50 parts by weight or more of a pore-forming agent with respect to 100 parts by weight of the metal source, the slurry is dried and fired for various applications (for example, Mo adsorbent, catalyst, etc.). It is possible to accurately produce a porous metal oxide having a structure suitable for use such as a carrier.
- the technique disclosed herein can be preferably implemented in a mode in which the organometallic compound is a metal alkoxide.
- a metal isopropoxide is mentioned as a suitable example of a metal alkoxide.
- the pore forming agent at least one compound selected from ammonium salts, carbonates and hydrogen carbonates can be preferably used.
- a pore forming agent pores suitable for various applications (for example, adsorption of Mo) tend to be efficiently formed.
- the slurry is prepared using about 1 part by weight or more and about 50 parts by weight or less of the metal source (for example, metal alkoxide) with respect to 100 parts by weight of the aqueous solvent. Can be done. According to such an embodiment, a slurry with good homogeneity tends to be easily prepared.
- the metal source for example, metal alkoxide
- the slurry is prepared using about 2 parts by weight or more and about 30 parts by weight or less of the pore forming agent with respect to 100 parts by weight of the aqueous solvent. be able to. According to such an embodiment, a slurry with good homogeneity tends to be easily prepared.
- the firing of the metal oxide precursor is preferably performed at a firing temperature of about 500 ° C. or higher and about 700 ° C. or lower.
- the metal oxide precursor is fired in N stages (N is an integer of 2 or more) from the first stage to the Nth stage. Can be included in order. According to this aspect, the homogeneity of the porous metal oxide can be improved. From the viewpoint of obtaining a higher effect, the fired product of the firing stage in the n-1st stage (n is an integer of 2 or more and N or less) among the N stages of firing is once heated to a temperature of less than 300 ° C. After cooling, it may be subjected to the next (that is, n-th) firing stage.
- the firing of the metal oxide precursor is performed at a temperature equal to or higher than a temperature at which the pore forming agent decomposes to generate gas (for example, approximately 500 ° C. or more and approximately 700 ° C. or less).
- a first firing stage and a second firing stage performed at a firing temperature).
- the fired product obtained in the first firing stage can be once cooled to a temperature of less than 300 ° C. and then subjected to the second firing stage. According to such an embodiment, a porous metal oxide with better homogeneity can be produced.
- the porous metal oxide having an average pore diameter of 9 nm or more is generated by firing the metal oxide precursor.
- a porous metal oxide having such an average pore diameter can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- the porous metal oxide having a pore volume of 0.3 cm 3 / g or more is generated by firing the metal oxide precursor.
- the porous metal oxide having such a pore volume can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- the porous metal oxide having a specific surface area of 180 m 2 / g or more is generated by firing the metal oxide precursor.
- the porous metal oxide having such a specific surface area can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- porous metal oxides in the technology disclosed herein include porous alumina and porous zirconia. Such a porous metal oxide can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- a preferable example of the porous alumina is porous alumina substantially composed of ⁇ -alumina.
- a method for manufacturing a 99m Tc generator includes producing a porous metal oxide by any of the methods disclosed herein and incorporating the porous metal oxide into a 99m Tc generator shield. According to such a method, a high-performance 99m Tc generator can be manufactured by providing a porous metal oxide (for example, porous alumina) with good Mo adsorption performance.
- a porous metal oxide for example, porous alumina
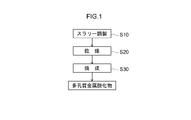
- FIG. 1 is a flowchart showing a method for producing a porous metal oxide according to the present invention.
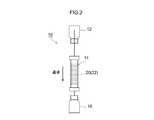
- FIG. 2 is a schematic diagram showing a schematic configuration of a 99m Tc generator.
- FIG. 3 is a characteristic diagram showing the Mo adsorption amount of the porous alumina produced in Examples 1 to 4 and 6.
- FIG. 4 is a TEM image of porous zirconia produced according to Example 7.
- FIG. 5 is a TEM image of porous zirconia produced according to Example 8.
- the porous metal oxide production method provided by this specification can be preferably applied to the production of porous alumina, for example. That is, according to this specification, an aluminum source (Al source), a pore forming agent, and an aqueous solvent are mixed to prepare a slurry; the slurry is dried to obtain an alumina precursor; and the alumina A method for producing porous alumina is provided that includes calcining a precursor to produce porous alumina.
- the Al source is an organometallic compound containing aluminum which is a metal constituting the porous alumina (that is, an organoaluminum compound) or a hydrolyzate thereof.
- the pore forming agent is an inorganic compound that decomposes at a temperature equal to or lower than the temperature at which the alumina precursor is calcined to generate gas.
- the slurry is prepared using 50 parts by weight or more of the pore forming agent with respect to 100 parts by weight of the Al source.
- porous metal oxide other than porous alumina and a method for producing the same can be provided by using another metal source, for example, another metal alkoxide, instead of the Al source. Therefore, according to this specification, porous metal oxides (including porous alumina and other porous metal oxides) and methods for producing the same are included. Examples of porous metal oxides other than porous alumina include porous zirconia. According to this specification, a slurry is prepared by mixing a zirconium source (Zr source), a pore forming agent and an aqueous solvent; drying the slurry to obtain a zirconia precursor; and the zirconia precursor To produce porous zirconia.
- Zr source zirconium source
- the Zr source may be an organic zirconium compound or a hydrolyzate thereof.
- the pore-forming agent may be an inorganic compound that decomposes at a temperature lower than the temperature for firing the zirconia precursor to generate a gas.
- the slurry may be prepared using 50 parts by weight or more of the pore forming agent with respect to 100 parts by weight of the Zr source.
- Another example of the porous metal oxide other than porous alumina is porous titania.
- a porous titania production method can be provided, wherein a titanium source (for example, titanium alkoxide) is used in place of the Al source in the porous alumina production method.
- a typical embodiment of a method for producing a porous metal oxide (for example, porous alumina such as ⁇ -alumina or porous zirconia) disclosed herein will be described with reference to FIG.
- a metal source, a pore forming agent, and an aqueous solvent are mixed to prepare a slurry (step S10).
- the slurry prepared in step S10 is dried to obtain a metal oxide precursor (step S20).
- the metal oxide precursor obtained by step S20 is baked, and a porous metal oxide is produced
- a porous metal oxide for example, porous alumina such as ⁇ -alumina or porous zirconia
- an organometallic compound containing a metal corresponding to the target porous metal oxide or a hydrolyzate thereof is used as a metal source.
- the metal is not particularly limited as long as it can form a porous oxide.
- the metal can be, for example, a metal belonging to any one of groups 2 to 14 (typically groups 3 to 13) of the periodic table.
- the metal may be a typical metal element or a transition metal element. Specific examples of the metal include aluminum and zirconium.
- an organoaluminum compound or a hydrolyzate thereof is used.
- an organic zirconium compound or a hydrolyzate thereof is used. It can preferably be employed as a metal source. By using such a metal source, a slurry with good homogeneity can be easily prepared. This is preferable from the viewpoint of productivity and quality stability of the porous metal oxide.
- an organoaluminum compound refers to a compound containing Al and an organic group. Accordingly, non-limiting examples of the organoaluminum compounds mentioned here include compounds having a direct bond between an organic group carbon (C) and Al, such as alkylaluminum, for example, and the organic group C is another element. And compounds having a structure bonded to Al via An organoaluminum compound can be used individually by 1 type or in combination of 2 or more types. The same applies to organic zirconium compounds and other organometallic compounds.
- a water-insoluble compound can be preferably used as the organoaluminum compound.
- a nonionic organoaluminum compound can be preferably employed from the viewpoint of ease of preparation of the slurry. The same applies to organic zirconium compounds and other organometallic compounds.
- the organometallic compound in the technology disclosed herein can be, for example, a compound having a structure in which C of the organic group is bonded to the metal via oxygen (O).
- a typical example of such an organometallic compound is a metal alkoxide.
- the technique disclosed here can be preferably implemented in an embodiment using a metal alkoxide as the organometallic compound.
- a metal alkoxide as the organometallic compound.
- the process of preparing the said slurry can be grasped
- the metal alkoxide may be used for preparing the slurry in a form in which at least a part thereof is hydrolyzed.
- the number of carbon atoms of the alkoxy group in the metal alkoxide is not particularly limited.
- the number of carbon atoms of the alkoxy group may be, for example, about 1 or more and 12 or less.
- a metal alkoxide in which the number of carbon atoms of the alkoxy group is 6 or less, more preferably 5 or less, for example 4 or less can be preferably used.
- a metal alkoxide having 2 or more carbon atoms in the alkoxy group can be preferably used.
- Preferable examples of the metal alkoxide that can be used in the technology disclosed herein include ethoxide, sec-butoxide and tert-butoxide of the metal.
- aluminum ethoxide, aluminum isopropoxide, aluminum sec-butoxide, and aluminum tert-butoxide are preferred examples of aluminum alkoxides that can be used to produce the porous alumina. Is mentioned. Of these, aluminum isopropoxide is preferred.
- the technique disclosed here can be preferably implemented in an embodiment in which aluminum isopropoxide is used alone as the organoaluminum compound.
- zirconium alkoxide that can be used for the production of porous zirconia
- zirconium ethoxide zirconium isopropoxide
- zirconium sec-butoxide zirconium sec-butoxide
- zirconium tert-butoxide zirconium tert-butoxide
- zirconium isopropoxide is preferable.
- the technique disclosed here can be preferably implemented in an embodiment in which zirconium isopropoxide is used alone as the organic zirconium compound.
- metal alkoxides that can be used in the production of other porous metal oxides (for example, titanium alkoxides that can be used in the production of porous titania).
- a slurry is prepared by mixing the metal source, the pore forming agent and an aqueous solvent, and the slurry is dried and fired to obtain a porous metal oxide.
- the pore-forming agent an inorganic compound that decomposes at a temperature not higher than the temperature at which the firing is performed to generate a gas is used.
- a pore forming agent can be used individually by 1 type or in combination of 2 or more types. Without wishing to be bound by theory, the inventor believes that the gas generated from the pore former (eg, one or both of CO 2 and NH 3 ) during at least one of the drying and firing steps.
- the metal may be, for example, aluminum or zirconium, but is not limited thereto.
- a compound that generates gas at a temperature of about 500 ° C. or lower, more preferably about 400 ° C. or lower, typically about 300 ° C. or lower can be preferably used.
- a compound that generates gas at a temperature of about 200 ° C. or lower, or about 100 ° C. or lower may be used.
- a compound that generates gas at a temperature of about 30 ° C. or higher, more preferably about 40 ° C. or higher, typically about 50 ° C. or higher can be preferably used.
- the technology disclosed herein can be preferably implemented in an embodiment in which a compound that generates a gas at a temperature of about 40 ° C. or higher and about 300 ° C. or lower (eg, about 50 ° C. or higher and about 150 ° C. or lower) is used as the pore forming agent. .
- the pore forming agent is not particularly limited, and for example, ammonium salts, carbonates, bicarbonates and the like can be used.
- a compound belonging to at least one of ammonium salt and carbonate can be preferably used.
- Specific examples of the compound that can be used as the pore forming agent include ammonium carbonate, ammonium hydrogen carbonate, ammonium carbamate, sodium hydrogen carbonate and the like.
- the pore-forming agent substantially not containing a metal element other than the metal constituting the target porous metal oxide. can be preferably employed.
- a water-soluble compound can be preferably employed as the pore forming agent from the viewpoint of ease of slurry preparation.
- the technique disclosed herein can be preferably implemented in an embodiment using at least ammonium carbonate as a pore forming agent.
- a pore forming agent for example, it is preferable that about 70% or more (more preferably about 85% or more, for example, about 95% or more) of the pore forming agent used is ammonium carbonate on a weight basis.
- substantially only ammonium carbonate may be used as the pore forming agent.
- substantially means that a pore forming agent other than ammonium carbonate is not intentionally used, and the presence of unavoidable impurities and denatured products can be tolerated.
- the procedure for preparing the slurry is not particularly limited.
- a preparation procedure may be employed in which a metal source is added to and mixed with an aqueous solvent containing a pore former (eg, an aqueous solution of a pore former).
- the metal may be, for example, aluminum or zirconium, but is not limited thereto.
- the aqueous solvent is a concept including water and a mixed solvent containing water as a main component (typically a component that is contained most on a weight basis).
- the components other than water constituting the mixed solvent include solvents that can be uniformly mixed with water, such as lower alcohols (typically monoalcohols having about 1 to 4 carbon atoms such as methanol, ethanol, etc.) and lower ketones.
- lower alcohols typically monoalcohols having about 1 to 4 carbon atoms such as methanol, ethanol, etc.
- One type or two or more types of solvents selected from the above may be used.
- the aqueous solvent in the technology disclosed herein has a water ratio of typically more than 50% and 100% or less by weight.
- the proportion of water in the aqueous solvent is typically 50% by weight. In general, 70% or more is appropriate.
- the ratio of the water may be 80% or more, 90% or more, 95% or more, 99% or more, and substantially 100% or more. May be.
- substantially means that a solvent other than water is not used at least intentionally.
- the technique disclosed here can be implemented, for example, in a mode in which a metal source is added to an aqueous solution of a pore forming agent and mixed.
- the metal source may be added as it is, or may be added together with an appropriate solvent, for example, in a form dissolved or dispersed in the solvent.
- the technique disclosed herein is a method for forming a pore-forming agent by using a metal source that is solid at normal temperature (for example, 25 ° C.) as in aluminum isopropoxide in the solid state (that is, without being dissolved in a solvent). It can preferably be carried out in an embodiment in which it is added to an aqueous solvent containing
- the solid metal source is preferably added in the form of a powder.
- the average particle size of the powder is usually about 300 ⁇ m or less, preferably about 100 ⁇ m or less. The lower limit of the average particle size of the powder is not particularly limited.
- the average particle size of the powder can be, for example, about 100 nm or more from the viewpoint of the handleability of the powder.
- the average particle diameter can also be applied to the average particle diameter of the powder in an embodiment in which the metal source powder is added in a form dispersed in a solvent.
- the “average particle diameter” means a particle diameter (50% volume average particle) at an integrated value of 50% in a particle size distribution measured based on a particle size distribution measuring apparatus based on a laser diffraction scattering method. Diameter; hereinafter, it may be abbreviated as D50).
- the solvent used for dissolving or dispersing the metal source is water, and uniformly mixed with water.
- Usable solvents, mixed solvents thereof and the like can be used.
- the solvent that can be uniformly mixed with water include one or more selected from lower alcohols (typically monoalcohols having about 1 to 4 carbon atoms, such as methanol and ethanol), lower ketones, and the like. Can be used.
- the amount of the solvent used for dissolving or dispersing the metal source can be, for example, about 500 g or less with respect to 100 g of the metal source, or about 300 g or less, about 200 g or less, May be about 150 g or less.
- the amount of the solvent used for 100 g of the metal source can be, for example, about 10 g or more, about 50 g or more, about 80 g or more, and further about 100 g or more.
- the proportion of water in the total aqueous solvent used to prepare the slurry is typically greater than 50% by weight. It is preferably 100% or less, and usually 70% or more and 100% or less.
- the ratio of the water may be 80% or more, 90% or more, 95% or more, and further 99% or more. In one embodiment, the percentage of water may be substantially 100%. That is, the aqueous solvent used for preparing the slurry may be only water.
- the ratio of the water to the aqueous solvent used for preparation of a slurry may be less than 95%, for example, and may be less than 90%.
- the technique disclosed here can be implemented in a mode in which the ratio of water in the aqueous solvent used for preparing the slurry is, for example, 80% or more and 100% or less.
- the amount of the pore forming agent used for the preparation of the slurry is suitably about 50 parts by weight or more, preferably about 100 parts by weight or more, more preferably about 150 parts by weight with respect to 100 parts by weight of the metal source. More than a part. Increasing the amount of pore-forming agent used for the metal source tends to increase the amount of metal (for example, Mo, platinum group metals, and other catalytic metals) that can be retained per weight of the porous metal oxide. is there. In one embodiment, the use amount of the pore forming agent with respect to 100 parts by weight of the metal source may be about 200 parts by weight or more, and further about 250 parts by weight or more.
- the upper limit of the amount of the pore-forming agent used is not particularly limited, but usually it is suitably about 1000 parts by weight or less with respect to 100 parts by weight of the metal source, preferably about 500 parts by weight or less, For example, it can be about 350 parts by weight or less.
- the amount of the aqueous solvent used for the preparation of the slurry is such that the amount of the pore-forming agent used is about 30 parts by weight or less, preferably about 20 parts by weight or less, more preferably about 10 parts by weight or less with respect to 100 parts by weight of the aqueous solvent.
- the lower limit of the amount of pore-forming agent used with respect to 100 parts by weight of the aqueous solvent is not particularly limited, but it is usually appropriate to be about 2 parts by weight or more with respect to 100 parts by weight of the aqueous solvent, and about 4 parts by weight or more. For example, it may be about 5 parts by weight or more.
- the amount of the aqueous solvent used for the preparation of the slurry is such that the amount of the metal source used is about 50 parts by weight or less, preferably about 30 parts by weight or less, more preferably about 20 parts by weight or less, for example, 10 parts by weight based on 100 parts by weight of the aqueous solvent. It can set so that it may become below a weight part.
- a slurry that provides a porous metal oxide capable of efficiently supporting a metal (for example, Mo, catalytic metal, etc.) by adding a metal source to a liquid containing such an amount of an aqueous solvent and a pore-forming agent and mixing them is suitable. Tend to be prepared.
- the gelation of the slurry can be suppressed to maintain a good stirring state, and the hydrolysis reaction of the metal alkoxide can proceed more uniformly. This tends to form a porous metal oxide with better structural homogeneity.
- the minimum of the usage-amount of a metal source with respect to 100 weight part of aqueous solvents is not specifically limited. From the standpoint of slurry drying efficiency, the amount of metal source used per 100 parts by weight of water is usually about 1 part by weight or more, preferably about 2 parts by weight or more, for example, about 3 parts. It can be more than parts by weight.
- the time for mixing the metal source, the pore forming agent and the aqueous solvent is not particularly limited, and the metal used Depending on the type of the source and the like, it can be set so that a slurry that provides a suitable porous metal oxide is prepared. From the viewpoint of the homogeneity of the slurry, it is usually appropriate that the mixing time is about 1 hour or longer, preferably 3 hours or longer, more preferably 6 hours or longer, for example, 12 hours or longer.
- the mixing time is suitably about 12 hours or more, preferably about 24 hours or more.
- the mixing time is usually suitably 72 hours or less, preferably 48 hours or less, and more preferably 36 hours or less.
- the slurry can be prepared, for example, at a temperature of less than about 60 ° C., and at a temperature of less than about 50 ° C., or even less than about 40 ° C. (eg, less than about 35 ° C.). Also good. According to the said temperature, a more homogeneous slurry can be prepared stably.
- the slurry is usually prepared at a temperature of about 10 ° C. or higher, preferably from about 15 ° C. or higher, from the viewpoint of productivity.
- the slurry preparation temperature mentioned above can be preferably applied, for example in the aspect using a metal alkoxide (for example, metal isopropoxide) as a metal source.
- the porous metal oxide production method disclosed herein typically includes a step of drying the slurry to obtain a metal oxide precursor.
- the metal may be, for example, aluminum or zirconium, but is not limited thereto.
- a metal alkoxide for example, metal isopropoxide
- the method disclosed herein can be preferably carried out in an embodiment in which no additional organic solvent is used in the process of obtaining the metal oxide precursor from the slurry.
- it can carry out without performing operation which wash
- the method disclosed herein includes an operation of separating (separating) the solid content contained in the slurry from the solvent by filtration, centrifugation, etc. Can be carried out in such a manner that the operation of washing (in particular, washing with a solvent capable of dissolving the pore forming agent) is not performed. By this, the function of the said pore formation agent can be exhibited effectively at the time of baking of the said metal oxide precursor.
- the fact that separation and washing of solid content is not required can be advantageous from the viewpoint of simplification of manufacturing equipment, reduction of environmental burden due to reduction of waste liquid, and improvement of productivity.
- the drying temperature of the slurry is not particularly limited.
- the slurry is dried at room temperature (typically about 15 ° C. or more, eg, about 20 ° C. or more, typically less than about 40 ° C., preferably about 35 ° C. or less, eg, about 30 ° C. or less). It can be carried out.
- the drying temperature of the slurry may be about 40 ° C. or higher (preferably about 60 ° C. or higher, more preferably about 70 ° C. or higher). Further, the drying temperature can be, for example, about 150 ° C.
- the drying time of the slurry is not particularly limited, and can be set so that an appropriate drying state can be obtained. From the viewpoint of productivity, it is usually appropriate to set the drying time to 120 hours or shorter (for example, 72 hours or shorter).
- a porous metal oxide is obtained by firing the metal oxide precursor.
- the calcined porous metal oxide can be prepared to an appropriate particle size by a known method such as sieving after crushing as necessary.
- the firing conditions of the metal oxide precursor can be set so as to obtain a porous metal oxide having desired properties, and is not particularly limited. For example, in order to obtain a porous metal oxide having desired properties, only one-stage (one time) firing may be performed, or two or more N-stage (N times) firing may be performed. . That is, the firing of the metal oxide precursor may include N stages (N is an integer of 2 or more) of firing stages from the first stage to the Nth stage in this order.
- the firing temperature of the metal oxide precursor can be, for example, about 150 ° C. or higher, and is usually about 250 ° C. or higher, preferably about 300 ° C. or higher, about 350 ° C. or higher, and further about 400 ° C. or higher. It is good. Moreover, the said calcination temperature can be made into about 1200 degreeC or less, for example, about 1000 degreeC or less is suitable normally, and about 800 degreeC or less is preferable. In one embodiment of the technology disclosed herein, the baking can be performed at approximately 300 ° C. or higher and approximately 1000 ° C. or lower (eg, approximately 400 ° C. or higher and 800 ° C. or lower).
- Calcination time is not particularly limited, and can be set so that a porous metal oxide having desired properties is generated.
- the firing time is usually suitably 1 hour or longer, 2 hours or longer, 3 hours or longer, for example 5 hours or longer.
- the firing temperature is usually 72 hours or less, preferably 48 hours or less, more preferably 50 hours or less.
- the said baking time means the time which maintains a baking target object in the said baking temperature, and the aspect maintained in the said baking temperature in each baking stage in the aspect which performs a 2 or more times baking process so that it may mention later. The total time.
- the firing temperature and firing time described above can be applied to firing conditions in the production of porous alumina, porous zirconia and other porous metal oxides, for example.
- porous alumina In the production of porous alumina, it is easy to increase the amount of metal (for example, Mo, catalytic metal, etc.) retained per weight of alumina, so that the porous alumina is sometimes referred to as ⁇ -alumina (activated alumina). Is preferably fired so as to form. Whether the porous alumina is ⁇ -alumina can be confirmed by, for example, X-ray diffraction.
- metal for example, Mo, catalytic metal, etc.
- the firing temperature of the alumina precursor can be, for example, about 400 ° C. or higher, and is usually about 450 ° C. or higher, preferably about 500 ° C. or higher, more preferably about 550 ° C. or higher.
- the firing temperature is suitably about 800 ° C. or lower, preferably about 700 ° C. or lower, and more preferably about 650 ° C. or lower.
- the technique disclosed herein can be preferably implemented, for example, in an embodiment in which the firing is performed at about 500 ° C. or higher and about 700 ° C. or lower (more preferably, about 550 ° C. or higher and about 650 ° C. or lower).
- Calcination time is not particularly limited, and can be set so that porous alumina having desired properties is generated.
- the firing time is usually suitably 1 hour or longer, preferably 2 hours or longer, for example 3 hours or longer.
- the firing time may be 6 hours or longer, and may be 12 hours or longer, for example, 24 hours or longer.
- the firing temperature is usually 72 hours or less, preferably 48 hours or less, more preferably 50 hours or less.
- the number of times of firing in the case where the metal precursor is fired in a plurality of times can be, for example, 2 to 5 times.
- a more homogeneous porous metal oxide can be obtained.
- firing at the firing temperature for example, firing at about 300 ° C. or higher, preferably about 400 ° C. or higher, more preferably about 500 ° C. or higher
- firing at the firing temperature for example, firing at about 300 ° C. or higher, preferably about 400 ° C. or higher, more preferably about 500 ° C. or higher
- N stages for example, firing at about 300 ° C. or higher, preferably about 400 ° C. or higher, more preferably about 500 ° C. or higher
- the firing object (the previous firing step) After cooling to a temperature of less than about 300 ° C. (typically about 100 ° C. or less, preferably about 50 ° C. or less, more preferably about 40 ° C. or less). ) Can be subjected to a firing stage. It is preferable that the cooled object to be fired is subjected to an operation of crushing (unwinding) as necessary, and then subjected to the next firing step. This tends to provide a more homogeneous porous metal oxide.
- an operation of passing the firing object through a sieve having an appropriate size can be mentioned.
- a sieve having a mesh size of approximately 200 mesh or more and approximately 30 mesh or less typically having a mesh opening of approximately 77 ⁇ m or more and approximately 600 ⁇ m or less
- the above operation is performed between the first stage firing (first firing stage) and the second stage firing (second firing stage). It can be carried out. You may perform the said operation after each baking step.
- this manufacturing method is a metal source, pore, for example. It can be carried out in a mode in which materials other than the forming agent and the aqueous solvent are not substantially used. This can be advantageous from the viewpoints of ease of setting conditions in each process, ease of procurement of materials used for manufacturing, and improvement of productivity.
- the porous metal oxide production method according to the above aspect can be carried out in an aspect using only two kinds of materials other than the aqueous solvent. .
- a porous metal in which the amount of Mo retained (the weight (mg) of Mo atoms retained per gram of porous metal oxide) measured by the following method is approximately 200 mg / g or more.
- An oxide can be provided.
- a porous metal oxide having a Mo holding amount of about 300 mg / g or more typically about 400 mg / g or more, for example, about 500 mg / g or more, further about 600 mg / g or more).
- the upper limit of the Mo holding amount is not particularly limited, and may be, for example, about 2000 mg / g or less (typically about 1000 mg / g or less).
- the porous metal oxide can be, for example, porous alumina (preferably porous ⁇ -alumina), porous zirconia, or the like.
- the amount of Mo retained (supported) on the porous metal oxide is determined from the change in the amount of Mo.
- Mo retention amount [mg / g] is calculated by converting this Mo amount into a numerical value per 1 g of the porous metal oxide used. The same method is adopted for the embodiments described later.
- the specific surface area of the porous metal oxide provided by this specification is not particularly limited.
- the specific surface area of the porous metal oxide can be, for example, 4.0 m 2 / g or more, and is usually 5.0 m 2 / g or more (typically 6.0 m 2 / g or more).
- the specific surface area of the porous metal oxide eg, porous alumina, preferably porous ⁇ -alumina
- the specific surface area of the porous metal oxide may be about 280 m 2 / g or more (eg, about 300 m 2 / g or more).
- the upper limit of the specific surface area of the porous metal oxide is not particularly limited.
- the specific surface area of the porous metal oxide can be measured by a nitrogen gas adsorption amount measurement method (BET method). The same method is adopted for the embodiments described later.
- the average pore diameter of the porous metal oxide provided by this specification is not particularly limited.
- the average pore diameter of the porous metal oxide can be, for example, about 5 nm or more, typically about 7 nm or more, usually about 9 nm or more, preferably about 10 nm or more (for example, more than 10 nm), more preferably It is about 12 nm or more, more preferably about 15 nm or more.
- the retention performance of Mo tends to be improved by increasing the average pore diameter.
- the average pore size of the porous metal oxide may be, for example, about 18 nm or more.
- the average pore diameter of the porous metal oxide can be, for example, about 500 nm or less, typically about 300 nm or less, usually about 100 nm or less, preferably about 50 nm or less, more preferably about 30 nm or less ( For example, approximately 25 nm or less).
- the technology disclosed herein can be preferably applied to, for example, a porous metal oxide having an average pore diameter in the range of about 5 nm to about 50 nm (for example, about 10 nm to about 25 nm) and the production thereof.
- the porous metal oxide having such an average pore diameter tends to show a good holding performance for a metal such as Mo, it can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- the average pore diameter of the porous metal oxide can be measured by a nitrogen gas adsorption amount measurement method. The same method is adopted for the embodiments described later.
- the porous metal oxide can be, for example, porous alumina (preferably porous ⁇ -alumina), porous zirconia, or the like.
- the pore volume of the porous metal oxide is not particularly limited.
- the pore volume of the porous metal oxide for example, be a 0.010 cm 3 / g or more, is generally suitable 0.020 cm 3 / g or more.
- the pore volume of the porous metal oxide eg, porous alumina, preferably porous ⁇ -alumina
- the pore volume of the porous metal oxide may be approximately 1.2 cm 3 / g or more. Further, the pore volume of the porous metal oxide may be about 3.0 cm 3 / g or less, typically about 2.5 cm 3 / g or less, for example, about 2.0 cm 3 / g or less. In the technique disclosed herein, for example, the pore volume is about 0.5 cm 3 / g or more and about 2.5 cm 3 / g or less (preferably about 0.6 cm 3 / g or more and about 2.0 cm 3 / g or less). It can preferably be applied to porous metal oxides in the range and their production.
- the porous metal oxide having such a pore volume tends to show a good holding performance for a metal such as Mo, it can be preferably used for various applications such as a Mo adsorbent and a catalyst carrier.
- the pore volume of the porous metal oxide can be measured by a nitrogen gas adsorption amount measurement method. The same method is adopted for the embodiments described later.
- a porous metal oxide for example, porous alumina having a large amount of Mo retained per surface area (weight of Mo atoms (mg) retained per 1 m 2 of surface area of the porous metal oxide). ) May be provided.
- a porous metal oxide having a Mo retention amount per surface area of about 0.9 mg / m 2 or more can be provided.
- the upper limit of the Mo retention amount per surface area is not particularly limited, and may be, for example, about 10 mg / m 2 or less.
- a porous metal oxide for example, porous alumina having a pore volume of 0.3 cm 3 / g or more and an average pore diameter of 9 nm or more is provided.
- a porous metal oxide satisfying the above pore volume and average pore diameter and having a specific surface area of 180 m 2 / g or more is particularly preferred.
- the porous metal oxide having such a structure tends to exhibit good holding performance with respect to metals (including a simple substance of metal, an alloy, and a metal compound). Therefore, it can be preferably used for various applications such as a catalyst carrier and a metal adsorbent.
- porous alumina having such a structure tends to exhibit excellent retention performance with respect to Mo, and thus is suitable for applications such as Mo adsorbents.
- the porous metal oxide production method disclosed herein is a method for producing a porous metal oxide satisfying the pore volume and the average pore diameter (preferably a porous metal oxide satisfying the specific surface area). It can be suitably employed.
- the outer shape of the porous metal oxide is not particularly limited, and can be prepared in an appropriate shape according to the application and usage.
- the porous metal oxide can be prepared, for example, in the form of particles having an average particle diameter of about 1 ⁇ m to about 400 ⁇ m (more preferably about 10 ⁇ m to about 300 ⁇ m).
- it can be prepared so as not to substantially contain particles having a diameter of more than about 500 ⁇ m.
- the metal oxide provided by the technique disclosed herein can be preferably used in various fields.
- the porous alumina provided by the technology disclosed herein can be preferably applied to a Mo adsorbent in a 99m Tc generator.
- the technology disclosed herein can be preferably applied to the production of porous alumina for such applications.
- the matter provided by this specification includes porous alumina produced by any of the methods disclosed herein, any porous alumina disclosed herein (any of those disclosed herein) 99m Tc generator comprising Mo as an adsorbent, and porous alumina for Mo adsorbent, and any porous alumina disclosed herein.
- a method of manufacturing a 99m Tc generator is included.
- the 99m Tc generator can be configured in the same manner as the conventional 99m Tc generator except that any porous alumina disclosed herein is used as the Mo adsorbent. An outline of a configuration example of such a 99m Tc generator will be described with reference to FIG.
- the 99m Tc generator 10 shown in FIG. 2 includes a column 14 containing 99 Mo adsorption alumina 20, a physiological saline container 12 connected to the upstream side of the column 14, and a 99m Tc solution connected to the downstream side of the column 14.
- a recovery container 16 At least the periphery of the column 14 of the 99m Tc generator 10 is covered with a shield (not typically shown) (typically a lead shield).
- a shield typically a lead shield.
- Column 99 Mo adsorbed alumina 20 in 14, to one of the porous alumina 22 disclosed herein, after receiving prior to or accommodated in the column 14 is obtained by adsorbing 99 Mo.
- 99 Mo can be adsorbed on the porous alumina 22 by supplying an aqueous solution containing ( 99 Mo) O 4 2 ⁇ to the column 14.
- the adsorbed 99 Mo decays by emitting ⁇ -rays to generate 99m Tc.
- the produced 99m Tc is eluted as 99m TcO 4 ⁇ by passing the physiological saline supplied from the physiological saline container 12 through the column 14 and is recovered in the 99m Tc solution recovery container 16, and the radiopharmaceutical (Technetium) Used in the manufacture of pharmaceutical preparations).
- porous alumina provided by the technology disclosed herein is not limited to the Mo adsorbent as described above, but as a general porous metal oxide, it can be used as another adsorbent or as a catalyst carrier. It can be preferably used for applications and the like.
- porous metal oxides other than porous alumina provided by the technology disclosed herein are not limited to metal adsorbents (for example, Mo adsorbents), but other adsorbents as well as general porous metal oxides. It can also be preferably used for applications as a material or a catalyst carrier.
- Example 1 ⁇ Manufacture of porous alumina> (Example 1) At room temperature, 970 g of distilled water was charged into a glass beaker, and 30 g of ammonium carbonate as a pore-forming agent was charged and dissolved while stirring with a magnetic stirrer. To this aqueous ammonium carbonate solution, 32 g of powdered aluminum isopropoxide was added, and stirring was continued at room temperature for 24 hours to prepare a slurry. As the aluminum isopropoxide, a commercially available product was crushed with a mortar and prepared to have an average particle size of about 100 ⁇ m.
- the slurry (the contents of the beaker) was transferred to a heat-resistant tray made of silicone resin, left in an oven at 80 ° C. and dried for 48 hours. Thereafter, the alumina precursor (dried product of the slurry) in the heat-resistant tray was transferred to a mortar and fired in a firing furnace in an atmospheric atmosphere.
- the first firing step of heating at 600 ° C. for 6 hours is first performed, once taken out of the firing furnace, cooled to room temperature, passed through a 50-mesh wire mesh, the particle size is made uniform to some extent, and then fired.
- the second baking step of returning to the furnace and further heating at 600 ° C. for 24 hours was performed, allowed to cool to room temperature, and again passed through a 50-mesh wire mesh to obtain porous alumina according to Example 1.
- Example 2 and 3 Porous alumina according to Examples 2 and 3 was obtained in the same manner as in Example 1 except that the amount of ammonium carbonate used was changed to 60 g (Example 2) and 90 g (Example 3), respectively.
- Example 4 In Example 1, 30 g of glucose was used as the pore forming agent instead of 30 g of ammonium carbonate as the pore forming agent. In addition, aluminum isopropoxide was added to an aqueous ammonium carbonate solution, and then the pH was adjusted to 5.0 using HNO 3 , and then stirred at room temperature for 24 hours to prepare a slurry. Otherwise in the same manner as in Example 1, porous alumina according to Example 4 was obtained.
- Example 5 In Example 2, an attempt was made to produce porous alumina using aluminum nitrate instead of aluminum isopropoxide. However, when the aluminum nitrate aqueous solution was started to be added to the ammonium carbonate aqueous solution, gelation rapidly progressed in the middle and the stirring was stopped. Therefore, it was judged that it was difficult to obtain a homogeneous slurry, and the subsequent experiments were stopped.
- Example 6 Porous alumina (Aluminum Oxide, model number 1999966) available from Sigma Aldrich USA was used.
- Example 6 As shown in Table 1, the porous materials of Examples 1 to 3 manufactured using ammonium carbonate as compared to the porous alumina of Example 4 manufactured using glucose and the commercially available porous alumina (Example 6). Alumina had a higher molybdenum retention per alumina weight. Among them, the porous aluminas of Examples 2 and 3 showed excellent Mo adsorptivity 4.5 times or more as compared with Example 6.
- a porous alumina was produced in the same manner as in Example 2 except that sodium hydrogen carbonate was used instead of ammonium carbonate, and the measurement and evaluation were conducted in the same manner.
- the specific surface area was 3.9 g / m 2
- the pore volume was 0.06 cm 3 / g
- the average pore diameter was 62.9 nm
- the amount of Mo retained per weight was 412 mg / g.
- Example 7 ⁇ Manufacture of porous zirconia> (Example 7) At room temperature, 300 g of distilled water was charged into a glass beaker, and 60 g of ammonium carbonate as a pore-forming agent was charged and dissolved while stirring with a magnetic stirrer. A solution B containing 32 g of zirconium tetraisopropoxide in 50 mL of methanol was added to this aqueous ammonium carbonate solution (solution A), and stirring was continued at room temperature for 24 hours to prepare a slurry. Next, the slurry (the contents of the beaker) was transferred to a heat-resistant tray made of silicone resin and dried at room temperature.
- the zirconia precursor (the dried product of the slurry) in the heat-resistant tray was transferred to a mortar and fired at 600 ° C. for 6 hours in a firing furnace in an air atmosphere.
- the fired product was allowed to cool to room temperature and passed through a 50-mesh wire mesh to obtain porous zirconia according to Example 7.
- a TEM (transmission electron microscope) image of the obtained porous zirconia is shown in FIG.
- the specific surface area was 8.5 m 3 / g
- the pore volume was 0.027 cm 3 / g
- the average pore diameter was 11.1 nm. It was.
- Example 8 A porous zirconia according to Example 8 was obtained in the same manner as in Example 7 except that the firing conditions of the zirconia precursor were changed to 200 ° C. for 6 hours.
- a TEM image of the obtained porous zirconia is shown in FIG.
- the specific surface area of this porous zirconia was 66.9 m 3 / g, the pore volume was 0.062 cm 3 / g, and the average pore diameter was 2.3 nm.
- porous zirconia in obtaining porous zirconia by firing a zirconia precursor containing a pore-forming agent, the properties of porous zirconia obtained by varying the firing conditions (here, firing temperature) of the precursor are different. It has been shown that (eg average pore size) can be adjusted.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Environmental & Geological Engineering (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Catalysts (AREA)
- Compounds Of Alkaline-Earth Elements, Aluminum Or Rare-Earth Metals (AREA)
Abstract
Description
本出願は、2016年1月21日出願の米国仮特許出願第62/281,361号および2016年5月19日出願の米国仮特許出願第62/338,756号に基づく優先権を主張しており、それらの出願の全内容は本明細書中に参照として組み入れられている。
99mTcジェネレータのMo吸着材として好ましく用いられる得る多孔質金属酸化物の一例として、多孔質アルミナが挙げられる。また、99mTcジェネレータのMo吸着材用の多孔質アルミナに係る技術文献として、非特許文献1が挙げられる。この非特許文献1には、細孔形成のテンプレートとしてグルコースを用いて多孔質アルミナを製造することが記載されている。
以下の図面において、同じ作用を奏する部材・部位には同じ符号を付して説明することがあり、重複する説明は省略または簡略化することがある。また、図面に記載の実施形態は、本発明を明瞭に説明するために模式化されており、実際に提供される製品のサイズや縮尺を必ずしも正確に表したものではない。
ここに開示される多孔質金属酸化物製造方法では、金属源として、目的とする多孔質金属酸化物に対応する金属を含む有機金属化合物またはその加水分解物を使用する。上記金属は、多孔質の酸化物を形成し得るものであればよく、特に限定されない。上記金属は、例えば、周期表2族~14族(典型的には3族~13族)のいずれかに属する金属であり得る。上記金属は、典型金属元素であってもよく、遷移金属元素であってもよい。上記金属の具体例としては、アルミニウム、ジルコニウム等が挙げられる。例えば、上記方法を多孔質アルミナの製造に適用する場合には有機アルミニウム化合物またはその加水分解物を、該方法を多孔質ジルコニアの製造に適用する場合には有機ジルコニウム化合物またはその加水分解物を、金属源として好ましく採用し得る。かかる金属源を用いることにより、均質性のよいスラリーを容易に調製することができる。このことは、多孔質金属酸化物の生産性や品質安定性の観点から好ましい。
ここに開示される多孔質金属酸化物製造方法では、上記金属源と細孔形成剤と水性溶媒とを混合してスラリーを調製し、該スラリーを乾燥および焼成して多孔質金属酸化物を得る。上記細孔形成剤としては、上記焼成を行う温度以下の温度で分解してガスを生じる無機化合物が用いられる。細孔形成剤は、1種を単独でまたは2種以上を組み合わせて使用することができる。理論により拘束されることを望むものではないが、本発明者は、上記乾燥および焼成の少なくともいずれかの過程で上記細孔形成剤から生じるガス(例えば、CO2およびNH3の一方または両方)が、種々の機能(例えばMo等の金属の保持、触媒担体としての機能等)を好適に発揮するために適した多孔質構造の形成に効果的に寄与するものと考えている。上記金属は、例えばアルミニウム、ジルコニウム等であり得るが、これらに限定されない。
ここに開示される多孔質金属酸化物製造方法において、スラリーを調製する手順は特に限定されない。一態様において、細孔形成剤を含む水性溶媒(例えば、細孔形成剤の水溶液)に金属源を加えて混合するという調製手順を採用し得る。上記金属は、例えばアルミニウム、ジルコニウム等であり得るが、これらに限定されない。
ここに開示される多孔質金属酸化物製造方法は、典型的には、上記スラリーを乾燥させて金属酸化物前駆体を得る工程を含む。上記金属は、例えばアルミニウム、ジルコニウム等であり得るが、これらに限定されない。金属源として金属アルコキシド(例えば金属イソプロポキシド)を使用する態様では、上記乾燥により、上記スラリーの調製に用いた水性溶媒のほか、上記金属アルコキシドから生じたアルコールを揮発させて除去することが好ましい。
ここに開示される方法では、上記金属酸化物前駆体を焼成することにより多孔質金属酸化物を得る。焼成後の多孔質金属酸化物は、必要に応じて解砕を行った後、篩分け等の公知の手法によって適当な粒子サイズに調製することができる。
金属酸化物前駆体の焼成条件は、所望の性質を有する多孔質金属酸化物が得られるように設定することができ、特に限定されない。例えば、所望の性質を有する多孔質金属酸化物を得るために、1段階(1回)の焼成のみを行ってもよいし、2段階以上のN段(N回)の焼成を行ってもよい。すなわち、上記金属酸化物前駆体の焼成は、第1段目から第N段目までのN段(Nは2以上の整数)の焼成段階をこの順に含んでもよい。
上記複数回の焼成は、例えば、上記焼成温度での焼成(例えば凡そ300℃以上、好ましくは凡そ400℃以上、より好ましくは凡そ500℃以上での焼成)を2段以上のN段に分けて行う態様で実施することができる。
ここに開示される技術によると、下記の方法により測定されるMo保持量(多孔質金属酸化物1g当たりに保持されるMo原子の重量(mg))が凡そ200mg/g以上である多孔質金属酸化物が提供され得る。より好適な態様によると、Mo保持量が凡そ300mg/g以上(典型的には凡そ400mg/g以上、例えば凡そ500mg/g以上、さらには凡そ600mg/g以上)である多孔質金属酸化物が提供され得る。Mo保持量の上限は特に制限されず、例えば凡そ2000mg/g以下(典型的には凡そ1000mg/g以下)であり得る。上記多孔質金属酸化物は、例えば、多孔質アルミナ(好適には多孔質γ-アルミナ)、多孔質ジルコニア等であり得る。
内径2mmのガラス管の下端を塞ぎ、各例に係る粉末状の多孔質金属酸化物0.5gを充填する。濃度30重量%のモリブデン酸ナトリウム水溶液を用意し、上記ガラス管内に多孔質金属酸化物が浸るまで注ぐ。この状態で5分間放置した後、上記ガラス管の下端からモリブデン酸ナトリウム水溶液を抜き出す。高周波誘導結合プラズマ発光分析(ICP-OES;Inductively Coupled Plasma-Optical Emission Spectrometry)により、ガラス管に供給したモリブデン酸ナトリウム水溶液に含まれるMo量と、ガラス管から抜き出したモリブデン酸ナトリウム水溶液に含まれるMo量とを決定し、Mo量の変化から、多孔質金属酸化物に保持(担持)されたMo量を求める。このMo量を、使用した多孔質金属酸化物1g当たりの数値に換算することにより、Mo保持量[mg/g]を算出する。後述の実施例についても同様の方法が採用される。
多孔質金属酸化物の比表面積は、窒素ガス吸着量測定法(BET法)により測定することができる。後述の実施例についても同様の方法が採用される。
このような構造を有する多孔質金属酸化物は、金属(金属の単体、合金および金属化合物を包含する意味である。)に対して良好な保持性能を示す傾向にある。したがって触媒担体や金属吸着材等の種々の用途に好ましく用いられ得る。例えば、このような構造を有する多孔質アルミナは、Moに対して優れた保持性能を示す傾向にあることから、Mo吸着材等の用途に好適である。ここに開示される多孔質金属酸化物製造方法は、上記細孔容積および平均細孔径を満たす多孔質金属酸化物(好ましくは、さらに上記比表面積を満たす多孔質金属酸化物)を製造する方法として好適に採用され得る。
ここに開示される技術により提供される金属酸化物は、種々の分野において好ましく用いられ得る。
例えば、ここに開示される技術により提供される多孔質アルミナは、99mTcジェネレータにおいてMo吸着材に好ましく適用され得る。また、ここに開示される技術は、かかる用途向けの多孔質アルミナの製造に好ましく適用され得る。したがって、この明細書により提供される事項には、ここに開示されるいずれかの方法により製造された多孔質アルミナ、ここに開示されるいずれかの多孔質アルミナ(ここに開示されるいずれかの方法により製造された多孔質アルミナであり得る。以下同じ。)をMo吸着材として備える99mTcジェネレータ、該Mo吸着材用多孔質アルミナ、および、ここに開示されるいずれかの多孔質アルミナを用いて99mTcジェネレータを製造する方法が包含される。
カラム14内の99Mo吸着アルミナ20は、ここに開示されるいずれかの多孔質アルミナ22に、カラム14への収容前または収容後に99Moを吸着させたものである。例えば、カラム14に多孔質アルミナ22を充填した後、このカラム14に(99Mo)O4 2-を含む水溶液を供給することにより、多孔質アルミナ22に99Moを吸着させることができる。吸着された99Moは、β線を放出して崩壊し、99mTcを生じる。
生成した99mTcは、生理食塩水容器12から供給される生理食塩水をカラム14に通液することにより、99mTcO4 -として溶出して99mTc溶液回収容器16に回収され、放射性医薬品(テクネチウム製剤)の製造等に利用される。
また、ここに開示される技術により提供される多孔質アルミナ以外の多孔質金属酸化物も、金属吸着材(例えばMo吸着材)のほか、一般的な多孔質金属酸化物と同様、その他の吸着材としての用途や触媒担体としての用途等にも好ましく利用され得る。
(例1)
室温において、ガラス製のビーカーに蒸留水970gを投入し、マグネチックスターラーで攪拌しながら、細孔形成剤としての炭酸アンモニウム30gを投入して溶解させた。この炭酸アンモニウム水溶液に粉末状のアルミニウムイソプロポキシド32gを投入し、室温で24時間攪拌を継続してスラリーを調製した。上記アルミニウムイソプロポキシドとしては、市販品を乳鉢ですり潰して平均粒径100μm程度に調製したものを使用した。
次いで、上記スラリー(ビーカーの内容物)をシリコーン樹脂製の耐熱トレーに移し、80℃の乾燥器内に静置して48時間乾燥させた。
その後、上記耐熱トレー内のアルミナ前駆体(上記スラリーの乾燥物)を匣鉢に移し、大気雰囲気の焼成炉にて焼成した。具体的には、まず600℃で6時間加熱する第1焼成段階を行い、いったん焼成炉から取り出して室温まで冷却し、50メッシュの金網を通過させることにより粒子サイズをある程度均一化した後、焼成炉に戻し、さらに600℃で24時間加熱する第2焼成段階を行い、室温まで放冷し、再度50メッシュの金網を通過させて、例1に係る多孔質アルミナを得た。
炭酸アンモニウムの使用量を60g(例2)および90g(例3)にそれぞれ変更した他は例1と同様にして、例2,3に係る多孔質アルミナを得た。
例1において、細孔形成剤としての炭酸アンモニウム30gに代えて、細孔形成剤としてグルコース30gを使用した。また、炭酸アンモニウム水溶液にアルミニウムイソプロポキシドを投入した後、HNO3を用いてpHを5.0に調整し、その後、室温で24時間攪拌してスラリーを調製した。その他の点は例1と同様にして、例4に係る多孔質アルミナを得た。
例2において、アルミニウムイソプロポキシドに代えて硝酸アルミニウムを用いて多孔質アルミナを製造することを試みた。しかし、炭酸アンモニウム水溶液に硝酸アルミニウム水溶液を加え始めたところ、途中で急激にゲル化が進行して攪拌が停止したため、均質なスラリーを得ることは困難と判断し、それ以降の実験を中止した。
Sigma Aldrich USA社から入手可能な多孔質アルミナ(Aluminum Oxide、型番199966)を使用した。
例1~4,6に係る多孔質アルミナの比表面積、細孔容積および平均細孔径を上述した方法により測定した。結果を表1に示す。また、上述した方法により、これらの多孔質アルミナの重量当たりのモリブデン保持量を測定した。結果を表1および図3に示す。

(例7)
室温において、ガラス製のビーカーに蒸留水300gを投入し、マグネチックスターラーで攪拌しながら、細孔形成剤としての炭酸アンモニウム60gを投入して溶解させた。この炭酸アンモニウム水溶液(溶液A)に、メタノール50mL中にジルコニウムテトライソプロポキシド32gを含む溶液Bを加え、室温で24時間攪拌を継続してスラリーを調製した。次いで、上記スラリー(ビーカーの内容物)をシリコーン樹脂製の耐熱トレーに移し、室温で乾燥させた。その後、上記耐熱トレー内のジルコニア前駆体(上記スラリーの乾燥物)を匣鉢に移し、大気雰囲気の焼成炉にて、600℃で6時間焼成した。その焼成物を室温まで放冷し、50メッシュの金網を通過させて、例7に係る多孔質ジルコニアを得た。得られた多孔質ジルコニアのTEM(透過型電子顕微鏡)像を図4に示す。この多孔質ジルコニアの性状を上述した多孔質アルミナと同様にして評価したところ、比表面積は8.5m3/g、細孔容積は0.027cm3/g、平均細孔径は11.1nmであった。
ジルコニア前駆体の焼成条件を200℃で6時間に変更した他は例7と同様にして、例8に係る多孔質ジルコニアを得た。得られた多孔質ジルコニアのTEM像を図5に示す。この多孔質ジルコニアの比表面積は66.9m3/g、細孔容積は0.062cm3/g、平均細孔径は2.3nmであった。
12 生理食塩水容器
14 カラム
16 99mTc溶液回収容器
20 99Mo吸着金属酸化物
22 多孔質アルミナ(Mo吸着材)
Claims (16)
- 多孔質金属酸化物を製造する方法であって、
金属源と細孔形成剤と水性溶媒とを混合してスラリーを調製すること;
前記スラリーを乾燥させて金属酸化物前駆体を得ること;および
前記金属酸化物前駆体を焼成して多孔質金属酸化物を生成すること;
を包含し、
ここで、前記金属源は、前記多孔質金属酸化物を構成する金属を含む有機金属化合物またはその加水分解物であり、
前記細孔形成剤は、前記金属酸化物前駆体を焼成する温度以下の温度で分解してガスを生じる無機化合物であり、
前記スラリーの調製は、前記金属源100重量部に対して前記細孔形成剤50重量部以上を用いて行われる、多孔質金属酸化物の製造方法。 - 前記有機金属化合物が金属アルコキシドである、請求項1に記載の方法。
- 前記金属アルコキシドは、アルミニウムアルコキシド、ジルコニウムアルコキシドおよびチタニウムアルコキシドからなる群から選択される少なくとも1種を含む、請求項2に記載の方法。
- 前記細孔形成剤は、アンモニウム塩、炭酸塩および炭酸水素塩からなる群から選択される少なくとも1種の化合物を含む、請求項1から3のいずれか一項に記載の方法。
- 前記スラリーの調製は、前記水性溶媒100重量部に対して1重量部以上50重量部以下の前記金属源を用いて行われる、請求項1から4のいずれか一項に記載の方法。
- 前記スラリーの調製は、前記水性溶媒100重量部に対して2重量部以上30重量部以下の前記細孔形成剤を用いて行われる、請求項1から5のいずれか一項に記載の方法。
- 前記金属酸化物前駆体の焼成を500℃以上700℃以下の焼成温度で行う、請求項1から6のいずれか一項に記載の方法。
- 前記金属酸化物前駆体の焼成は、第1段目から第N段目までのN段の焼成段階をこの順に含み、ここでNは2以上の整数であり、
前記N段の焼成段階のうち第n-1段目(ここで、nは2以上N以下の整数である。)の焼成段階による焼成物は、いったん300℃未満の温度まで冷却した後に第n段目の焼成段階に供される、請求項1から7のいずれか一項に記載の方法。 - 前記金属酸化物前駆体の焼成は、平均細孔径が9nm以上の前記多孔質金属酸化物を生成するように行われる、請求項1から8のいずれか一項に記載の方法。
- 前記金属酸化物前駆体の焼成は、細孔容積が0.3cm3/g以上の前記多孔質金属酸化物を生成するように行われる、請求項1から9のいずれか一項に記載の方法。
- 前記金属酸化物前駆体の焼成は、比表面積が180m2/g以上の前記多孔質金属酸化物を生成するように行われる、請求項1から10のいずれか一項に記載の方法。
- 前記多孔質金属酸化物は多孔質アルミナである、請求項1から11のいずれか一項に記載の方法。
- 前記多孔質金属酸化物は多孔質ジルコニアである、請求項1から11のいずれか一項に記載の方法。
- 請求項1から13のいずれかに記載の方法により製造された多孔質金属酸化物。
- 細孔容積が0.3cm3/g以上であり、かつ平均細孔径が9nm以上である、多孔質金属酸化物。
- 比表面積が180m2/g以上である、請求項15に記載の多孔質金属酸化物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17741479.4A EP3409640B1 (en) | 2016-01-21 | 2017-01-19 | Method for producing porous metal oxide |
| JP2017562883A JPWO2017126602A1 (ja) | 2016-01-21 | 2017-01-19 | 多孔質金属酸化物の製造方法 |
| US16/071,214 US11554967B2 (en) | 2016-01-21 | 2017-01-19 | Method for producing porous metal oxide |
| PL17741479T PL3409640T3 (pl) | 2016-01-21 | 2017-01-19 | Sposób wytwarzania porowatego tlenku metalu |
| US18/094,542 US12012337B2 (en) | 2016-01-21 | 2023-01-09 | Method for producing porous metal oxide |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662281361P | 2016-01-21 | 2016-01-21 | |
| US62/281,361 | 2016-01-21 | ||
| US201662338756P | 2016-05-19 | 2016-05-19 | |
| US62/338,756 | 2016-05-19 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/071,214 A-371-Of-International US11554967B2 (en) | 2016-01-21 | 2017-01-19 | Method for producing porous metal oxide |
| US18/094,542 Continuation US12012337B2 (en) | 2016-01-21 | 2023-01-09 | Method for producing porous metal oxide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017126602A1 true WO2017126602A1 (ja) | 2017-07-27 |
Family
ID=59362429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/001729 Ceased WO2017126602A1 (ja) | 2016-01-21 | 2017-01-19 | 多孔質金属酸化物の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US11554967B2 (ja) |
| EP (1) | EP3409640B1 (ja) |
| JP (1) | JPWO2017126602A1 (ja) |
| PL (1) | PL3409640T3 (ja) |
| TW (1) | TW201736265A (ja) |
| WO (1) | WO2017126602A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102316874B1 (ko) * | 2021-01-28 | 2021-10-25 | 영남대학교 산학협력단 | 다공성 중공 구조의 은 나노입자의 제조방법, 그로부터 제조된 다공성 중공 구조의 은 나노입자 및 이를 포함하는 폴리머 겔 |
| WO2022030593A1 (ja) | 2020-08-06 | 2022-02-10 | 株式会社フジミインコーポレーテッド | 吸着方法およびその方法に用いられるメソポーラスアルミナ |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5275698A (en) * | 1975-12-17 | 1977-06-24 | Condea Petrochemie Gmbh | Process for preparing porous alumina product |
| US4175118A (en) | 1975-12-17 | 1979-11-20 | Condea Chemie Gmbh | Method for manufacturing a high porous alumina having a large surface area and a low bulk density |
| JP2002320850A (ja) | 2001-04-25 | 2002-11-05 | Toyota Motor Corp | 触媒及びその触媒を用いた排気ガス浄化装置 |
| WO2007021037A1 (ja) * | 2005-08-19 | 2007-02-22 | Kyoto University | 無機系多孔質体及びその製造方法 |
| JP2007522066A (ja) * | 2004-01-26 | 2007-08-09 | エイビービー ラマス グローバル インコーポレイテッド | メソ細孔性またはメソ細孔性およびミクロ細孔性の組み合わせの無機酸化物を製造する方法 |
| US20120122671A1 (en) | 2010-11-16 | 2012-05-17 | Rhodia Operations | Alumina catalyst support |
| JP2013522164A (ja) * | 2010-03-22 | 2013-06-13 | ブリガム・ヤング・ユニバーシティ | 細孔構造が制御された、高度に多孔性の、安定な金属酸化物を製造する方法 |
| JP2013129574A (ja) | 2011-12-22 | 2013-07-04 | Taki Chem Co Ltd | メソポーラスアルミナの製造方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1258854B (de) * | 1963-05-11 | 1968-01-18 | Deutsche Erdoel Ag | Verfahren zur kontinuierlichen Herstellung von Alkoholen durch Hydrolyse von Aluminiumalkoxiden in einem homogenen System aus n-Butanol und Wasser |
| US3419352A (en) | 1964-12-02 | 1968-12-31 | Continental Oil Co | Process for producing alpha alumina monohydrate |
| US3987155A (en) * | 1971-10-20 | 1976-10-19 | Continental Oil Company | High-porosity, high-surface area, low-bulk density alumina |
| US4543341A (en) * | 1983-12-23 | 1985-09-24 | Massachusetts Institute Of Technology | Synthesis and processing of monosized oxide powders |
| JPH10324580A (ja) | 1997-03-21 | 1998-12-08 | Mitsubishi Heavy Ind Ltd | 金属酸化物の多孔質化方法 |
| US6399540B1 (en) * | 1999-08-12 | 2002-06-04 | Sumitomo Chemical Co., Ltd. | Porous titania, catalyst comprising the porous titania |
| WO2010114561A1 (en) | 2009-04-03 | 2010-10-07 | Carrier Corporation | Production of tailored metal oxide materials using a reaction sol-gel approach |
| US20110003085A1 (en) | 2008-04-04 | 2011-01-06 | Carrier Corporation | Production Of Tailored Metal Oxide Materials Using A Reaction Sol-Gel Approach |
| US10464811B2 (en) | 2009-04-06 | 2019-11-05 | Nanyang Technological University | Method of forming a particulate porous metal oxide or metalloid oxide |
-
2017
- 2017-01-19 PL PL17741479T patent/PL3409640T3/pl unknown
- 2017-01-19 JP JP2017562883A patent/JPWO2017126602A1/ja active Pending
- 2017-01-19 US US16/071,214 patent/US11554967B2/en active Active
- 2017-01-19 TW TW106101886A patent/TW201736265A/zh unknown
- 2017-01-19 EP EP17741479.4A patent/EP3409640B1/en active Active
- 2017-01-19 WO PCT/JP2017/001729 patent/WO2017126602A1/ja not_active Ceased
-
2023
- 2023-01-09 US US18/094,542 patent/US12012337B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5275698A (en) * | 1975-12-17 | 1977-06-24 | Condea Petrochemie Gmbh | Process for preparing porous alumina product |
| US4175118A (en) | 1975-12-17 | 1979-11-20 | Condea Chemie Gmbh | Method for manufacturing a high porous alumina having a large surface area and a low bulk density |
| JP2002320850A (ja) | 2001-04-25 | 2002-11-05 | Toyota Motor Corp | 触媒及びその触媒を用いた排気ガス浄化装置 |
| JP2007522066A (ja) * | 2004-01-26 | 2007-08-09 | エイビービー ラマス グローバル インコーポレイテッド | メソ細孔性またはメソ細孔性およびミクロ細孔性の組み合わせの無機酸化物を製造する方法 |
| WO2007021037A1 (ja) * | 2005-08-19 | 2007-02-22 | Kyoto University | 無機系多孔質体及びその製造方法 |
| JP2013522164A (ja) * | 2010-03-22 | 2013-06-13 | ブリガム・ヤング・ユニバーシティ | 細孔構造が制御された、高度に多孔性の、安定な金属酸化物を製造する方法 |
| US20120122671A1 (en) | 2010-11-16 | 2012-05-17 | Rhodia Operations | Alumina catalyst support |
| JP2014500217A (ja) * | 2010-11-16 | 2014-01-09 | ロディア オペレーションズ | アルミナ触媒担体 |
| JP2013129574A (ja) | 2011-12-22 | 2013-07-04 | Taki Chem Co Ltd | メソポーラスアルミナの製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| INDUSTRIAL & ENGINEERING CHEMISTRY RESEARCH (IND. ENG. CHEM. RES., vol. 52, 2013, pages 11673 - 11684 |
| See also references of EP3409640A4 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022030593A1 (ja) | 2020-08-06 | 2022-02-10 | 株式会社フジミインコーポレーテッド | 吸着方法およびその方法に用いられるメソポーラスアルミナ |
| KR20230048519A (ko) | 2020-08-06 | 2023-04-11 | 가부시키가이샤 후지미인코퍼레이티드 | 흡착 방법 및 그 방법에 사용되는 메조포러스 알루미나 |
| KR102316874B1 (ko) * | 2021-01-28 | 2021-10-25 | 영남대학교 산학협력단 | 다공성 중공 구조의 은 나노입자의 제조방법, 그로부터 제조된 다공성 중공 구조의 은 나노입자 및 이를 포함하는 폴리머 겔 |
Also Published As
| Publication number | Publication date |
|---|---|
| US11554967B2 (en) | 2023-01-17 |
| US12012337B2 (en) | 2024-06-18 |
| EP3409640B1 (en) | 2021-05-19 |
| EP3409640A4 (en) | 2019-12-04 |
| JPWO2017126602A1 (ja) | 2018-11-08 |
| US20210139342A1 (en) | 2021-05-13 |
| EP3409640A1 (en) | 2018-12-05 |
| US20230159347A1 (en) | 2023-05-25 |
| PL3409640T3 (pl) | 2021-11-02 |
| TW201736265A (zh) | 2017-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020143450A1 (zh) | 阶梯式梯度升温煅烧法制备臭氧催化剂的方法及应用 | |
| CN105772048B (zh) | 一种碳化钼与二氧化钛复合光催化分解水产氢催化剂及其制备方法 | |
| CN109794241B (zh) | 一种氧化铈选择性包覆负载型钯催化剂及其制备方法 | |
| CN102302933A (zh) | 磁性氧化物空心微球/二氧化钛复合光催化剂的制备方法 | |
| JP2009091238A (ja) | 酸化セリウムウオッシュコート | |
| CN103894194B (zh) | 一种室温去除甲醛的负载型催化剂 | |
| CN101973590A (zh) | 小尺寸介孔金属氧化物的制备方法 | |
| CN105948098B (zh) | 一种球形氧化镧 | |
| Gaikwad et al. | Photocatalytic performance of magnetically separable Fe, N co-doped TiO2-cobalt ferrite nanocomposite | |
| CN106458593A (zh) | 大直径、低密度碳纳米管以及制备该碳纳米管的方法 | |
| Fu et al. | Exceptionally thermal-stable Al2O3/TiO2 nanofibers by depressing surface-initiated grain growth as new supports for anti-sintering Pt nanoparticles | |
| US20230159347A1 (en) | Method for producing porous metal oxide | |
| CN103339061A (zh) | 多孔质氧化铝材料及其制造方法、以及催化剂 | |
| JP5533783B2 (ja) | 内燃機関の排気浄化用触媒 | |
| JP2016123932A (ja) | 排ガス浄化触媒及びその製造方法 | |
| WO2013038674A1 (ja) | 実質的に面心立方構造を有するルテニウム微粒子およびその製造方法 | |
| CN113617331A (zh) | 一种双层金属有机骨架材料衍生石墨碳包裹纳米铁的制备方法与应用 | |
| CN106103343A (zh) | 高密度捆绑形碳纳米管及其制备方法 | |
| CN105585035B (zh) | 一种氧化铝空心微球的制备方法 | |
| CN1736603A (zh) | 一种耐高温磁性载体及其制备方法及应用 | |
| CN111530461A (zh) | 一种低负载量、高分散单活性位点Cu催化剂及其制备方法和应用 | |
| CN101791578A (zh) | 有序双孔Al2O3-TiO2及其制备方法和应用 | |
| CN108620064B (zh) | 一种高比表面积贵金属基硅铝酸盐催化剂及其制备方法 | |
| JP4883241B2 (ja) | 触媒材料の製造方法およびそれによって製造される触媒材料並びに触媒体 | |
| CN107456982B (zh) | 一种介孔铬基气相氟化催化剂的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17741479 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017562883 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017741479 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017741479 Country of ref document: EP Effective date: 20180821 |