WO2017169599A1 - アモルファス酸化物系正極活物質、その製造方法及びその用途 - Google Patents
アモルファス酸化物系正極活物質、その製造方法及びその用途 Download PDFInfo
- Publication number
- WO2017169599A1 WO2017169599A1 PCT/JP2017/009294 JP2017009294W WO2017169599A1 WO 2017169599 A1 WO2017169599 A1 WO 2017169599A1 JP 2017009294 W JP2017009294 W JP 2017009294W WO 2017169599 A1 WO2017169599 A1 WO 2017169599A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- active material
- electrode active
- amorphous oxide
- ions
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






















Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0561—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of inorganic materials only
- H01M10/0562—Solid materials
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/131—Electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/136—Electrodes based on inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/139—Processes of manufacture
- H01M4/1391—Processes of manufacture of electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/139—Processes of manufacture
- H01M4/1397—Processes of manufacture of electrodes based on inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/362—Composites
- H01M4/364—Composites as mixtures
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/485—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of mixed oxides or hydroxides for inserting or intercalating light metals, e.g. LiTi2O4 or LiTi2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/582—Halogenides
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/5825—Oxygenated metallic salts or polyanionic structures, e.g. borates, phosphates, silicates, olivines
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/026—Electrodes composed of, or comprising, active material characterised by the polarity
- H01M2004/028—Positive electrodes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0065—Solid electrolytes
- H01M2300/0068—Solid electrolytes inorganic
- H01M2300/0071—Oxides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to an amorphous oxide-based positive electrode active material, a production method thereof, and an application thereof. More specifically, the present invention relates to an amorphous oxide-based positive electrode active material that can exhibit high conductivity even with a relatively thick film positive electrode, a method for producing the same, a positive electrode including the same, and an all-solid-state secondary battery.
- Lithium ion secondary batteries have high voltage and high capacity, and are therefore widely used as power sources for mobile phones, digital cameras, video cameras, notebook computers, electric vehicles, and the like.
- lithium secondary batteries in circulation use a liquid electrolyte in which an electrolytic salt is dissolved in a non-aqueous solvent as an electrolyte. Since non-aqueous solvents contain a lot of flammable solvents, it is desired to ensure safety.
- an all-solid secondary battery using a so-called solid electrolyte in which an electrolyte is formed from a solid material without using a non-aqueous solvent has been proposed. As this solid material, many sulfide-based solid electrolytes represented by Li 2 S—P 2 S 5 have been reported.
- the sulfide-based solid electrolyte has a problem from the viewpoint of workability because sulfide is used as a raw material for the production thereof. Therefore, the applicant of the present application has proposed an oxide-based solid electrolyte as a solid electrolyte that is not sulfide-based (Japanese Patent Laid-Open No. 2015-76854: Patent Document 1).
- Non-Patent Document 1 proposes a technique for forming a positive electrode containing a positive electrode active material on a solid electrolyte layer by a sputtering method, and heat applied to the solid electrolyte layer during the formation can be suppressed. It is said that formation is suppressed.
- an amorphous oxide-based positive electrode active material that is a raw material for producing a positive electrode for an all-solid-state secondary battery
- the amorphous oxide-based positive electrode active material is (i) an alkali metal selected from Li and Na, Co, Ni, Mn, Fe, Cr, V, Cu, Ti, Zn, Zr, Nb, Mo, Ru, and Sn.
- a second metal selected from phosphate ions, sulfate ions, borate ions, silicate ions, aluminate ions, germanate ions, nitrate ions, carbonate ions and halide ions, and oxygen atoms (Excluding oxygen atoms constituting ionic species),
- An amorphous oxide-based positive electrode active material that is a raw material for producing a positive electrode having at least an amorphous phase and (iii) a thickness of 20 ⁇ m or more is provided.
- a method for producing the amorphous oxide-based positive electrode active material wherein the amorphous oxide-based positive electrode active material is obtained by mixing the raw materials by mechanical milling.
- a method for producing an active material is provided.
- the positive electrode which contains the said amorphous oxide type positive electrode active material and has a thickness of 20 micrometers or more is provided.
- the all-solid-state secondary battery provided with at least the said positive electrode, a negative electrode, and the solid electrolyte layer located between the said positive electrode and the said negative electrode is provided.
- an amorphous oxide-based positive electrode active material that can exhibit high conductivity even with a relatively thick positive electrode.
- an amorphous oxide-based positive electrode active material that can exhibit higher conductivity even with a relatively thick positive electrode can be provided.
- an alkali metal selected from Li and Na; a second metal selected from Co, Ni, Mn, Fe, Cr, V, Cu, Ti, Zn, Zr, Nb, Mo, Ru, and Sn; Ion species selected from phosphate ion, sulfate ion, borate ion, silicate ion, aluminate ion, germanate ion, nitrate ion, carbonate ion and halide ion, and oxygen atom (however, oxygen constituting the ion species) Is a raw material for producing a positive electrode for an all-solid-state secondary battery having an amorphous state and a thickness of 20 ⁇ m or more.
- the amorphous oxide-based positive electrode active material exhibits an amorphous state in which the peak at the minimum 2 ⁇ in the XRD pattern has a half width of 0.5 or more.
- the amorphous oxide-based positive electrode active material includes an amorphous phase and a crystalline phase.
- the amorphous oxide-based positive electrode active material includes a component derived from an alkali metal oxide and an alkali metal salt, and the alkali metal oxide is LiCoO 2 , LiNiO 2 , LiMnO 2 , Li 2 MnO 3 , Li ( Ni, Co, Mn) O 2 , Li 2 TiO 3 , LiFeO 2 , LiCrO 2 , Li 2 CuO 2 , LiCuO 2 , LiMoO 2 , Li 2 RuO 3 , Li 3 NbO 4 , Li 3 V 2 (PO 4 ) 3 LiMn 2 O 4 and Li (Ni, Mn) O 4 lithium compounds, NaCoO 2 , NaNiO 2 , NaMnO 2 , Na 2 MnO 3 , Na (Ni, Co, Mn) O 2 , NaFeO 2 , Na 2 TiO 3, NaCrO 2, Na 2 CuO 2, NaCuO 2, NaMoO 2, Na 2 RuO 3, Na 3 NbO 4, Na
- the component derived from the alkali metal oxide and the component derived from the alkali metal salt are present in the amorphous oxide-based positive electrode active material in a molar ratio of 1: 9 to 9: 1.
- the amorphous oxide-based positive electrode active material includes a component derived from LiCoO 2 and Li 3 PO 4 and / or Li 2 SO 4 .
- FIG. 2 is an XRD pattern of a positive electrode active material of Example 1.
- FIG. 2 is a mapping diagram of the positive electrode active material of Example 1 by SEM and EDX.
- FIG. 3 is an impedance plot of the positive electrode active material of Example 1. It is discharge current reduction for every elapsed time of the positive electrode active material of Example 1.
- FIG. 3 is a cross-sectional photograph of a cell of Example 2. It is a graph showing the result which attached
- 4 is a SEM photograph of a cross section of the positive electrode of the cell of Example 2. It is an XRD pattern of the pellet of the positive electrode before charge of the cell of Example 2, after charge, and after discharge.
- Example 7 is an XRD pattern of a positive electrode active material of Example 8. It is a graph showing the result of attaching
- 10 is an XRD pattern of a positive electrode active material of Example 9. It is a graph showing the result of attaching
- the amorphous oxide-based positive electrode active material includes an alkali metal selected from Li and Na, Co, Ni, Mn, Fe, Cr, V, Cu, Ti, Zn, Zr, A second metal selected from Nb, Mo, Ru and Sn and selected from phosphate ion, sulfate ion, borate ion, silicate ion, aluminate ion, germanate ion, nitrate ion, carbonate ion and halide ion Ion species and oxygen atoms (however, excluding oxygen atoms constituting the ion species).
- alkali metal selected from Li and Na, Co, Ni, Mn, Fe, Cr, V, Cu, Ti, Zn, Zr
- a second metal selected from Nb, Mo, Ru and Sn and selected from phosphate ion, sulfate ion, borate ion, silicate ion, aluminate ion, germanate ion, nitrate
- the positive electrode active material includes components derived from alkali metal oxides and alkali metal salts, and the alkali metal oxides are LiCoO 2 , LiNiO 2 , LiMnO 2 , Li 2 MnO 3 , Li (Ni, Co, Mn) O 2.
- the alkali metal salt is selected from the group consisting of: AxByOz (wherein A is Li or Na, B is selected from P, S, B, C, Si, Al, Ge and
- alkali metal oxides and alkali metal salts may be selected one by one or a plurality of types may be selected. More specific alkali metal salts include Li 3 PO 4 , Li 4 P 2 O 7 , LiPO 3 , Li 2 SO 4 , Li 3 BO 3 , Li 4 BO 5 , LiBO 2 , Li 2 CO 3 , Li 4.
- the positive electrode active material preferably includes a component derived from LiCoO 2 and Li 3 PO 4 and / or Li 2 SO 4 .
- an alkali metal salt having a relatively low melting point such as LiNO 3 can easily follow the increase / decrease in the positive electrode volume due to the charge / discharge reaction, and thus can suppress the generation of irreversible capacity.
- LiNO 3 can be contained in the positive electrode active material in an amount of 5 to 20 mol%.
- an oxide of a metal selected from Co, Ni, Mn, Fe, Cr, V, Cu, Ti, Zn, Zr, Nb, Mo, Ru, and Sn may be included.
- the capacity of the battery may be improved.
- TiO 2 and Mn 2 O 3 are considered to have an effect of improving the capacity of the battery by having a function of improving the oxidation-reduction reactivity.
- These metal oxides can be used at a molar ratio of 0.01 to 3 with respect to the alkali metal oxide 1.
- the component derived from the alkali metal oxide and the component derived from the alkali metal salt are preferably present in the positive electrode active material in a molar ratio of 1: 9 to 9: 1.
- the molar ratio of the component derived from the alkali metal salt is less than 1, it may be difficult to form an amorphous material.
- the electrode function may not be exhibited.
- the molar ratio can be 1: 9, 2: 8, 3: 7, 4: 6, 5: 5, 6: 4, 7: 3, 8: 2, 9: 1.
- the molar ratio is more preferably 3: 7 to 9: 1, and further preferably 5: 5 to 8: 2.
- the positive electrode active material may contain a crystalline phase as long as it does not inhibit the effect of the present invention as long as it contains an amorphous phase.
- a crystalline state in which this amorphous phase is an essential contained phase and a crystalline phase is arbitrarily contained is referred to as an amorphous state.
- the crystallinity increases, interface formation becomes difficult and the resistance tends to increase. Therefore, it is desired to provide a positive electrode active material having low crystallinity (amorphous state).
- the amorphous state has a lower density than the crystalline state, the positive electrode active material has high followability with respect to a volume change during charge and discharge.
- the minimum 2 ⁇ peak in the XRD pattern indicates a state in which the half-value width is 0.5 or more. means.
- the half width is 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1. 6, 1.7, 1.8, 1.9, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0.
- the inventors have found that it may be appropriate that the positive electrode active material contains a small amount of crystalline phase.
- a thick positive electrode is usually formed by pressing a particulate raw material (raw material particle) such as a positive electrode active material.
- raw material particle such as a positive electrode active material.
- the raw material particles composed of the positive electrode active material containing a small amount of the crystalline phase are easily crushed by pressing, the adhesion of the raw material particles can be improved.
- the crystalline phase should just be contained in the positive electrode active material in the grade which shows the half value width of 5.0 or less, 4.0 or less, 3.0 or less, 2.0 or less, and 1.0 or less.
- this positive electrode active material a positive electrode having a thickness of 20 ⁇ m or more can be easily formed by pressing, so this positive electrode active material is used as a material for forming a positive electrode for a high-capacity bulk type all-solid-state secondary battery. Is suitable.
- This thickness of 20 ⁇ m or more is difficult to realize by a vapor phase growth method such as sputtering.
- the thickness can be 20 ⁇ m, 25 ⁇ m, 30 ⁇ m, 35 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 80 ⁇ m, 100 ⁇ m, 120 ⁇ m, 140 ⁇ m, 150 ⁇ m.
- the positive electrode active material can be produced by mixing the raw materials by mechanical milling.
- Li 2 SO 4 is expected to be decomposed by the sputtering method as described in Non-Patent Document 1, and it is difficult to form a positive electrode as a positive electrode active material. Therefore, Li 2 SO 4 has many advantages for manufacturing by mechanical milling.
- the treatment by mechanical milling is not particularly limited to the treatment apparatus and treatment conditions as long as the raw materials are sufficiently mixed to make the positive electrode active material amorphous.
- a ball mill can be used normally.
- a ball mill is preferable because large mechanical energy can be obtained.
- the planetary ball mill is preferable because the pot rotates and revolves around the base plate so that high impact energy can be efficiently generated.
- the processing conditions can be appropriately set according to the processing apparatus to be used.
- the raw materials can be mixed more uniformly as the rotational speed is higher and / or the processing time is longer.
- the conditions are 50 to 600 rotations / minute, 0.1 to 200 hours of processing time, 1 to 100 kWh / raw material mixture of 1 kg.
- the treatment atmosphere is preferably an inert atmosphere such as argon.
- the treatment time is more preferably 10 to 70 hours in order to realize a more appropriate amorphous state.
- a raw material is an anhydrous state from a viewpoint of avoiding the hydrolysis reaction at the time of a mechanical milling process.
- the raw material contains crystal water
- the temperature of heat processing can be suitably determined according to the kind of raw material.
- the alkali metal salt may be obtained by reacting a hydroxide of lithium and / or sodium with a corresponding acid.
- the positive electrode includes at least the positive electrode active material. Moreover, it has a thickness of 20 ⁇ m or more. A positive electrode having a thickness of 20 ⁇ m or more is difficult to form by sputtering. Moreover, the oxide type solid electrolyte may be included as needed. The proportion of the oxide-based solid electrolyte in the positive electrode is preferably 30% by weight or less, and more preferably 10% by weight or less. Examples of the oxide-based solid electrolyte include Li 3 PO 4 , Li 2 SO 4 , Li 3 BO 3 and Li 2 CO 3 lithium salts, Na 3 PO 4 , Na 2 SO 4 , Na 3 BO 3 and Na 2. Examples thereof include a sodium salt of CO 3 . This oxide-based solid electrolyte can impart sufficient conductivity to the positive electrode even by heating at about 300 ° C. or less.
- the oxide solid electrolyte may be in the form of glass ceramics or glass.
- a glass-ceramic electrolyte has a lower conductivity than b-like electrolyte if b is smaller. It is high, and when b is large, the conductivity tends to be low.
- the glass-ceramic electrolyte is usually obtained by heating the glass-like electrolyte at a temperature equal to or higher than the crystallization temperature. Therefore, the glass ceramic electrolyte is more costly than the glass electrolyte.
- a glass ceramic form is a state in which the glass transition point which existed in the corresponding glass form does not exist. Further, the glass ceramic form may be in a state in which a crystalline part is dispersed in an amorphous glass component.
- the ratio of the crystalline part is preferably 50% by weight or more, and more preferably 80% by weight or more based on the whole. The proportion of the crystalline part can be measured by solid state NMR.
- a glassy oxide-based solid electrolyte can be obtained by mixing an alkali metal salt. For mixing, it is preferable to use a mechanical milling process under the same conditions as the positive electrode active material from the viewpoint of more uniformly mixing the salt.
- the glass ceramic oxide-based solid electrolyte can be obtained by subjecting the glass-like solid electrolyte to a heat treatment. This heat treatment is preferably performed at a temperature equal to or higher than the crystallization temperature of the glassy solid electrolyte.
- the glass transition point (T g ) varies depending on the constituent elements of the solid electrolyte, but is, for example, in the range of 190 to 250 ° C. in the case of the Li 3 BO 3 —Li 2 SO 4 system.
- the first crystallization temperature (T c ) is in the range of 210 to 270 ° C. Although the upper limit of heat processing temperature is not specifically limited, Usually, it is 1st crystallization temperature +100 degreeC.
- the heat treatment time is a time during which glass can be converted into glass ceramics, and is short when the heat treatment temperature is high and long when the heat treatment temperature is low. The heat treatment time is usually in the range of 0.1 to 10 hours.
- the positive electrode may further contain a binder, a conductive agent, and the like.
- the binder include polyvinylidene fluoride, polytetrafluoroethylene, polyvinyl alcohol, polyvinyl acetate, polymethyl methacrylate, and polyethylene.
- the conductive agent include natural graphite, artificial graphite, acetylene black, ketjen black, Denka black, carbon black, vapor grown carbon fiber (VGCF) and the like.
- the positive electrode may be formed on a current collector such as SUS, aluminum, or copper.
- the positive electrode can be obtained in the form of a pellet, for example, by mixing a positive electrode active material and optionally an oxide solid electrolyte, a binder, a conductive agent and the like, and pressing the obtained mixture.
- the pressing can be performed at a temperature of about 300 ° C. or lower and a pressure of 100 to 800 MPa for 0.1 to 5 hours.
- the positive electrode active material of the present invention can impart high conductivity to the positive electrode even when the press temperature is lower than conventional ones.
- this press may serve as the press at the time of formation of a solid electrolyte layer. By serving also, the adhesiveness in the interface of a positive electrode and a solid electrolyte layer can be improved more.
- the all-solid-state secondary battery includes at least a positive electrode, a negative electrode, and a solid electrolyte layer positioned between the positive electrode and the negative electrode.
- a positive electrode a negative electrode
- a solid electrolyte layer positioned between the positive electrode and the negative electrode.
- the above-mentioned thing can be used for a positive electrode.
- the solid electrolyte demonstrated in the column of the said positive electrode can be used for a solid electrolyte layer as it is.
- the negative electrode is not particularly limited.
- the negative electrode may be composed of only the negative electrode active material, and may be mixed with a binder, a conductive agent, an electrolyte, and the like.
- the negative electrode active material examples include metals such as Li, Na, In, and Sn, Li alloys, Na alloys, graphite, hard carbon, Li 4/3 Ti 5/3 O 4 , Na 3 V 2 (PO 4 ) 3 , SnO. And various transition metal oxides.
- the binder and the conductive agent any of those mentioned in the column of the positive electrode can be used.
- the electrolyte both oxide-based and sulfide-based solid electrolytes can be used.
- the negative electrode can be obtained in the form of pellets by, for example, mixing a negative electrode active material and optionally a binder, a conductive agent, an electrolyte, and the like, and pressing the obtained mixture.
- the metal sheet (foil) which consists of a metal or its alloy as a negative electrode active material, it can be used as it is.
- the negative electrode may be formed on a current collector such as SUS, aluminum, or copper.
- the all solid state secondary battery can be obtained, for example, by laminating and pressing a positive electrode, an electrolyte layer, and a negative electrode.
- Example 1 Two kinds of positive electrode active materials using LiCoO 2 and Li 3 PO 4 or Li 2 SO 4 as raw materials were produced by the following procedure.
- the two types of positive electrode active materials are hereinafter referred to as LiCoO 2 —Li 3 PO 4 and LiCoO 2 —Li 2 SO 4 .
- LiCoO 2 manufactured by Nippon Kagaku Kogyo Co., Ltd.
- Li 3 PO 4 manufactured by Wako Pure Chemical Industries, Ltd.
- Li 2 SO 4 .H 2 O manufactured by Wako Pure Chemical Industries, Ltd.
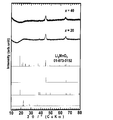
- FIG. 1 shows XRD patterns of cubic LiCoO 2 , hexagonal LiCoO 2 , Li 3 PO 4, and Li 2 SO 4 .
- the peaks corresponding to LiCoO 2 are broadened, and the positive electrode active material is in an amorphous state. In particular, it can be seen that all the observed 2 ⁇ peaks have a half width of 0.5 or more.
- FIG. 2A a scanning electron microscope (SEM) photograph of a LiCoO 2 —Li 2 SO 4 positive electrode active material is shown in FIG. 2A, and energy dispersive X-ray spectroscopy of Co and S atoms corresponding to FIG. 2A. Mapping diagrams by (EDX) are shown in FIGS. 2 (b) and 2 (c). 2 (a) to 2 (c), it can be seen that in the obtained positive electrode active material, Co and S are uniformly dispersed in the particles. This indicates that the original LiCoO 2 and Li 2 SO 4 particles reacted to form new LiCoO 2 —Li 2 SO 4 particles.
- FIG. 2A scanning electron microscope
- FIG. 3 shows a Nyquist plot of the positive electrode active material of LiCoO 2 —Li 2 SO 4 by the AC impedance method
- FIG. 4 shows the current behavior for each elapsed time when DC polarization measurement is performed at an applied voltage of 0.1 V.
- FIG. 3 shows the results of measurement with the positive electrode active material sandwiched between SUS plates
- FIG. 4 shows the results of measurement with the positive electrode active material sandwiched between solid electrolyte layers (Li 3 PS 4 ) and further sandwiched between Li plates.
- LiCoO 2 —Li 2 SO 4 has an electronic conductivity of 3.7 ⁇ 10 ⁇ 5 Scm ⁇ 1 and a lithium ion conductivity of 1.2 ⁇ 10 ⁇ 6 Scm ⁇ 1 .
- LiCoO 2 —Li 2 PO 4 has an electron conductivity of 2.5 ⁇ 10 ⁇ 5 Scm ⁇ 1 and a lithium ion conductivity of 5.3 ⁇ 10 ⁇ 7 Scm ⁇ 1 . Since it has an electron conductivity and an ionic conductivity of 10 ⁇ 7 Scm ⁇ 1 or more at room temperature, it satisfies the conditions necessary for a positive electrode of an all-solid battery.
- the solid electrolyte layer was produced by the following procedure. First, LiOH.H 2 O (manufactured by Wako Pure Chemical Industries, Ltd.) and H 3 BO 3 (manufactured by Wako Pure Chemical Industries, Ltd.) are mixed, heated at 500 ° C. for 1 hour, and then fired at 600 ° C. for 2 hours. Li 3 BO 3 was synthesized. Li 2 SO 4 .H 2 O (manufactured by Wako Pure Chemical Industries, Ltd.) was dehydrated by heating at 300 ° C. for 3 hours under an Ar atmosphere to obtain Li 2 SO 4 .
- Li 3 BO 3 , Li 2 SO 4 and Li 2 CO 3 are weighed to a molar ratio of 1: 1: 1, mixed in a mortar, and then the mixture is put into a planetary ball mill. I put it in. After the charging, mechanical milling was performed to obtain a glassy solid electrolyte (33Li 3 BO 3 .33Li 2 SO 4 .33Li 2 CO 3 ) having a particle size of several ⁇ m.
- Pulverisette P-7 manufactured by Fritsch was used as the planetary ball mill.
- the pot and balls were made of zirconium oxide, and a mill containing 160 balls having a diameter of 5 mm in a 45 ml pot was used.
- the mechanical milling process was performed for 90 hours in a glove box with a rotational speed of 370 rpm, room temperature, and a dry Ar atmosphere.
- the glassy solid electrolyte was heated at 260 ° C., which is equal to or higher than the crystallization temperature, to obtain a glass ceramic, thereby obtaining a glass ceramic solid electrolyte.
- 40 mg of the obtained solid electrolyte was pressed at a pressure of 720 MPa using a pellet molding machine having a molding part having an area of 0.785 cm 2 to obtain a pellet-shaped solid electrolyte layer (thickness: about 1 mm).
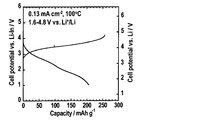
- Example 2 A positive electrode (LiCoO 2 —Li 2 SO 4 ), the solid electrolyte layer, and an indium foil as a counter electrode are stacked in this order, and the stacked body is sandwiched between stainless steel (SUS) current collectors. A solid lithium secondary battery) was obtained. A cross-sectional photograph of the cell is shown in FIG. This cell was subjected to a charge / discharge test at 100 ° C. and a current density of 0.13 mAcm ⁇ 2 . The test results are shown in the graph of FIG.
- FIG. 5 shows a SEM photograph of the cross section of the positive electrode, and FIGS.
- FIG. 7B to 7D show mapping diagrams of S, Co, and O atoms corresponding to FIG. 7A by EDX.
- 7A to 7D show that Co and S are uniformly dispersed in the obtained positive electrode active material.
- the XRD pattern of the pellet of the positive electrode before charge, after charge, and after discharge is shown in FIG.
- FIG. 8 also shows the XRD pattern of hexagonal LiCoO 2 and Li 2 SO 4 . Note that the peak with Si in FIG. 8 means the peak of the reference material.
- FIG. 8 shows that the positive electrode active material is in an amorphous state after charging and after discharging.
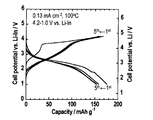
- Example 3 A battery cell was produced in the same manner as in Example 2 except that LiCoO 2 —Li 2 SO 4 was changed to LiCoO 2 —Li 2 PO 4 , and charge / discharge was performed at 100 ° C. and a current density of 0.13 mAcm ⁇ 2. Tested. The test results are shown in the graph of FIG. FIG. 9 shows that charging and discharging can be performed even with a so-called bulk-type all-solid secondary battery formed by pressing.
- Example 4 Except that LiNiO 2 (manufactured by Toshima Seisakusho) and Li 2 SO 4 .H 2 O (manufactured by Wako Pure Chemical Industries, Ltd.) are used, and the processing time of the mechanical milling process is changed to 50 hours, the same as in Example 1. Thus, LiNiO 2 —Li 2 SO 4 as a positive electrode active material was obtained. A battery cell obtained in the same manner as in Example 2 except that the positive electrode active material was used was subjected to a charge / discharge test. The test results are shown in the graph of FIG. FIG. 10 shows that LiNiO 2 —Li 2 SO 4 can also be charged and discharged.
- Example 5 Using LiNi 1/3 Mn 1/3 Co 1/3 O 2 (manufactured by Toda Kogyo Co., Ltd.) and Li 2 SO 4 .H 2 O (manufactured by Wako Pure Chemical Industries, Ltd.), the processing time for mechanical milling treatment is 50 hours. Except for changing, LiNi 1/3 Mn 1/3 Co 1/3 O 2 —Li 2 SO 4 as a positive electrode active material was obtained in the same manner as in Example 1. A battery cell obtained in the same manner as in Example 2 except that the positive electrode active material was used was subjected to a charge / discharge test. The test results are shown in the graph of FIG. FIG. 11 shows that LiNi 1/3 Mn 1/3 Co 1/3 O 2 —Li 2 SO 4 can also be charged and discharged.
- Example 6 The molar ratio of LiCoO 2 to Li 2 SO 4 (x: 1-x) was changed to 100: 0, 90:10, 80:20, 70:30, 60:40, and the processing time of mechanical milling treatment was 50 Except for changing the time, in the same manner as in Example 1, five types of LiCoO 2 —Li 2 SO 4 as positive electrode active materials were obtained.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG. 12 also shows XRD patterns of cubic LiCoO 2 , hexagonal LiCoO 2 , Li 3 PO 4, and Li 2 SO 4 . From FIG.
- Example 7 In the same manner as in Example 1, except that the molar ratio of LiCoO 2 and Li 2 SO 4 was changed to 80:20 and the processing time of the mechanical milling process was changed to 1 hour, 10 hours and 50 hours. Three types of LiCoO 2 —Li 2 SO 4 as materials were obtained.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG. 14 also shows the XRD pattern of the mixture before mechanical milling, cubic LiCoO 2 , hexagonal LiCoO 2 , Li 3 PO 4 and Li 2 SO 4 .
- FIG. 14 shows that the peak corresponding to LiCoO 2 becomes broader as the treatment time is extended, and the positive electrode active material becomes in an amorphous state.
- a battery cell obtained in the same manner as in Example 2 except that the positive electrode active material was used was subjected to a charge / discharge test. The test results are shown in the graph of FIG. FIG. 15 shows that the charge / discharge capacity increases as the processing time increases.
- Example 8 The molar ratio of LiCoO 2 (manufactured by Nippon Kagaku Kogyo Co., Ltd.) and Li 2 MnO 3 (manufactured by Toshima Seisakusho Co., Ltd.) is set to 40:40 or 60:20, and the total of LiCoO 2 and Li 2 MnO 3 and Li LiCoO 2 —Li 2 MnO 3 as the positive electrode active material was changed in the same manner as in Example 1 except that the molar ratio with 2 SO 4 was changed to 80:20 and the processing time for mechanical milling was changed to 50 hours. -Li 2 SO 4 was obtained.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG.
- FIG. 16 also shows XRD patterns of Li 2 MnO 3 , cubic LiCoO 2 , hexagonal LiCoO 2, and Li 2 SO 4 . From FIG. 16, it can be seen that the peaks corresponding to LiCoO 2 are all broad, and the positive electrode active material is in an amorphous state. In particular, it can be seen that all the observed 2 ⁇ peaks have a half width of 0.5 or more.
- Example 9 The molar ratio of LiCoO 2 (manufactured by Nippon Kagaku Kogyo Co., Ltd.) and Li 2 TiO 3 (manufactured by Wako Pure Chemical Industries, Ltd.) is set to 40:40 or 60:20, and the total of LiCoO 2 and Li 2 TiO 3 the molar ratio between Li 2 SO 4 was changed to 80:20 and, except for changing the processing time of the mechanical milling for 50 hours in the same manner as in example 1, LiCoO 2 -Li 2 as the positive electrode active material TiO 3 —Li 2 SO 4 was obtained.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG.
- FIG. 19 also shows XRD patterns of Li 2 TiO 3 , cubic LiCoO 2 , hexagonal LiCoO 2, and Li 2 SO 4 . From FIG. 19, it can be seen that the peaks corresponding to LiCoO 2 are all broad, and the positive electrode active material is in an amorphous state. In particular, it can be seen that all the observed 2 ⁇ peaks have a half width of 0.5 or more.
- FIG. 20 shows that LiCoO 2 —Li 2 TiO 3 —Li 2 SO 4 can also be charged and discharged.
- Example 10 Li 2 CO 3 (manufactured by Nippon Kagaku Kogyo Co., Ltd.), TiO 2 (manufactured by Wako Pure Chemical Industries, Ltd.) and Mn 2 O 3 (manufactured by Kojun Chemical Co., Ltd.) were mixed, and the resulting mixture was heated to 700 ° C. in the air. For 2 hours. The obtained fired product was pelletized. The pellet was sintered in air at 950 ° C. for 96 hours to obtain crystalline Li 1.2 Ti 0.4 Mn 0.4 O 2 .
- the obtained mixture was subjected to the same mechanical milling treatment as in Example 1 to obtain a positive electrode active material.
- the number of balls was changed to 50 and the processing time was changed to 50 hours.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG.
- FIG. 21 also shows an XRD pattern with literature values of LiTiMnO 2 . From FIG. 21, it can be seen that the peaks corresponding to LiTiMnO 2 are all broad and the positive electrode active material is in an amorphous state. In particular, it can be seen that all the observed 2 ⁇ peaks have a half width of 0.5 or more.
- Li 1.2 Ti 0.4 Mn 0.4 O 2 —Li 2 SO 4 can also be charged and discharged. Moreover, it turns out that the capacity
- the solid electrolyte was 90Li 3 BO 3 ⁇ obtained in the same manner as in Example 1 except that Li 2 CO 3 was not used and Li 3 BO 3 and Li 2 SO 4 had a molar ratio of 9: 1. 10Li 2 SO 4 was used.
- Example 11 Using LiCoO 2 (manufactured by Nippon Kagaku Kogyo Co., Ltd.), Li 2 SO 4 .H 2 O (manufactured by Wako Pure Chemical Industries, Ltd.) and LiNO 3 (manufactured by Wako Pure Chemical Industries, Ltd.) (LiCoO 2 and Li 2 SO 4 .H
- the molar ratio of 2 O to LiNO 3 is 80: 15: 5 and 70:20:10), the processing time of mechanical milling is 50 hours, and the number of balls is changed to 67, the same as in Example 1.
- LiCoO 2 —Li 2 SO 4 —LiNO 3 as a positive electrode active material was obtained.
- the XRD pattern of the positive electrode active material was measured in the same manner as in Example 1, and is shown in FIG. FIG. 24 also shows the XRD patterns of hexagonal LiCoO 2 , LiNO 3 and Li 2 SO 4 .
- FIG. 24 shows that peaks corresponding to LiCoO 2 are all broad, and the positive electrode active material is in an amorphous state. In particular, it can be seen that all the observed 2 ⁇ peaks have a half width of 0.5 or more.
- the battery cell obtained in the same manner as in Example 2 was charged except that the positive electrode active material was used, the solid electrolyte layer was changed to a solid electrolyte layer in Example 10, and the current collector on the solid electrolyte layer side was changed to Cu. Subjected to a discharge test. The test results are shown in the graph of FIG. 25 (molar ratio 80: 15: 5).
- FIG. 25 shows that LiCoO 2 —Li 2 SO 4 —LiNO 3 can also be charged and discharged.
Landscapes
- Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Composite Materials (AREA)
- Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- General Physics & Mathematics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Secondary Cells (AREA)
Abstract
Description
安全性を確保するために、非水系溶媒を使用せずに、電解質を固体材料から形成する、いわゆる固体電解質を使用した全固体二次電池が提案されている。この固体材料としては、Li2S-P2S5で表される硫化物系固体電解質が多く報告されている。しかし、硫化物系固体電解質は、その製造原料に硫化物を使用するため、作業性の観点からの課題があった。そこで、硫化物系ではない固体電解質として、本願出願人は、酸化物系の固体電解質を提案している(特開2015-76854号公報:特許文献1)。
一方、全固体二次電池は、固体電荷質層以外にも、正極及び負極が構成要件として必要とされる。特許文献1の実施例では、正極を構成する正極活物質としてLiCoO2が使用されている。上記した界面での高抵抗相の形成を抑制し、電子伝導性を有する正極活物質としてのLixMyPOzが提案されている(Sabi等、Journal of Power Soureces,258(2014),p.54-60:非特許文献1)。非特許文献1では、正極活物質を含む正極を、固体電解質層上にスパッタ法により形成する技術が提案されており、その形成時に固体電解質層に加えられる熱を抑制できるので、高抵抗相の形成が抑制される、とされている。
前記アモルファス酸化物系正極活物質が
(i)Li及びNaから選択されるアルカリ金属と、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される第2金属と、リン酸イオン、硫酸イオン、硼酸イオン、ケイ酸イオン、アルミン酸イオン、ゲルマン酸イオン、硝酸イオン、炭酸イオン及びハロゲン化物イオンから選択されるイオン種と、酸素原子(但し、イオン種を構成する酸素原子を除く)とを含み、
(ii)少なくともアモルファス相を含み、かつ
(iii)20μm以上の厚さの正極の作製原料である
アモルファス酸化物系正極活物質が提供される。
また、本発明によれば、上記アモルファス酸化物系正極活物質を含み、20μm以上の厚さを有する正極が提供される。
更に、本発明によれば、上記正極、負極、及び前記正極と前記負極間に位置する固体電解質層とを少なくとも備える全固体二次電池が提供される。
(1)Li及びNaから選択されるアルカリ金属と、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される第2金属と、リン酸イオン、硫酸イオン、硼酸イオン、ケイ酸イオン、アルミン酸イオン、ゲルマン酸イオン、硝酸イオン、炭酸イオン及びハロゲン化物イオンから選択されるイオン種と、酸素原子(但し、イオン種を構成する酸素原子を除く)とを含み、アモルファス状態であり、20μm以上の厚さの全固体二次電池用の正極の作製原料である。
(2)アモルファス酸化物系正極活物質は、そのXRDパターンにおける最小2θでのピークが、0.5以上の半値幅となるアモルファス状態を示す。
(3)アモルファス酸化物系正極活物質が、アモルファス相と結晶質相とを含む。
(4)アモルファス酸化物系正極活物質が、アルカリ金属酸化物及びアルカリ金属塩に由来する成分を含み、前記アルカリ金属酸化物が、LiCoO2、LiNiO2、LiMnO2、Li2MnO3、Li(Ni,Co,Mn)O2、Li2TiO3、LiFeO2、LiCrO2、Li2CuO2、LiCuO2、LiMoO2、Li2RuO3、Li3NbO4、Li3V2(PO4)3、LiMn2O4及びLi(Ni,Mn)O4のリチウム系化合物と、NaCoO2、NaNiO2、NaMnO2、Na2MnO3、Na(Ni,Co,Mn)O2、NaFeO2、Na2TiO3、NaCrO2、Na2CuO2、NaCuO2、NaMoO2、Na2RuO3、Na3NbO4、Na3V2(PO4)3、NaMn2O4及びNa(Ni,Mn)O4のナトリウム系化合物とからなる群から選択され、前記アルカリ金属塩が、AxByOz(式中、AはLi又はNaであり、BはP,S,B,C,Si,Al,Ge及びNから選択され、xは1以上、yは1以上、zは1以上であり、かつx、y及びzは化学量論的に可能な値である)及びAX(AはLi又はNaであり、XはF,Cl,Br及びIから選択される)から選択される。
(5)アルカリ金属酸化物に由来する成分とアルカリ金属塩に由来する成分とが、アモルファス酸化物系正極活物質中に、1:9~9:1のモル比で存在する。
(6)アモルファス酸化物系正極活物質が、LiCoO2と、Li3PO4及び/又はLi2SO4とに由来する成分を含む。
アモルファス酸化物系正極活物質(以下、単に正極活物質と称する)は、Li及びNaから選択されるアルカリ金属と、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される第2金属と、リン酸イオン、硫酸イオン、硼酸イオン、ケイ酸イオン、アルミン酸イオン、ゲルマン酸イオン、硝酸イオン、炭酸イオン及びハロゲン化物イオンから選択されるイオン種と、酸素原子(但し、イオン種を構成する酸素原子を除く)とを含む。
正極活物質は、アルカリ金属酸化物及びアルカリ金属塩に由来する成分を含み、アルカリ金属酸化物が、LiCoO2、LiNiO2、LiMnO2、Li2MnO3、Li(Ni,Co,Mn)O2、Li2TiO3、LiFeO2、LiCrO2、Li2CuO2、LiCuO2、LiMoO2、Li2RuO3、Li3NbO4、Li3V2(PO4)3、LiMn2O4及びLi(Ni,Mn)O4のリチウム系化合物と、NaCoO2、NaNiO2、NaMnO2、Na2MnO3、Na(Ni,Co,Mn)O2、Na2TiO3、NaFeO2、NaCrO2、Na2CuO2、NaCuO2、NaMoO2、Na2RuO3、Na3NbO4、Na3V2(PO4)3、NaMn2O4及びNa(Ni,Mn)O4のナトリウム系化合物とからなる群から選択され、アルカリ金属塩が、AxByOz(式中、AはLi又はNaであり、BはP,S,B,C,Si,Al,Ge及びNから選択され、xは1以上、yは1以上、zは1以上であり、かつx、y及びzは化学量論的に可能な値である)及びAX(AはLi又はNaであり、XはF,Cl,Br及びIから選択される)から選択されることが好ましい。これらアルカリ金属酸化物及びアルカリ金属塩は、それぞれ、1種ずつ選択されていてもよく、複数種ずつ選択されていてもよい。
より具体的なアルカリ金属塩としては、Li3PO4、Li4P2O7、LiPO3、Li2SO4、Li3BO3、Li4BO5、LiBO2、Li2CO3、Li4SiO4、Li6Si2O7、Li2SiO3、Li3AlO3、Li4Al2O5、LiAlO2、Li4GeO4、Li6Ge2O7、Li2GeO3、LiNO3及びLiX(X=F,Cl,Br,I)のリチウム塩と、Na3PO4、Na2SO4、Na3BO3、Na2CO3、Na4SiO4、Na3AlO3、Na4GeO4、NaNO3及びNaX(X=F,Cl,Br,I)のナトリウム塩とからなる群から選択されることが好ましい。
上記の具体例の中でも、正極活物質は、LiCoO2と、Li3PO4及び/又はLi2SO4とに由来する成分を含むことが好ましい。また、LiNO3のような比較的融点の低いアルカリ金属塩は、充放電反応による正極体積の増減に追随し易いため、不可逆容量の発生を抑制できると考えられる。LiNO3をLi3PO4及び/又はLi2SO4と併用する場合、LiNO3は、正極活物質中に、5~20モル%含ませ得る。
更に、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される金属の酸化物を含んでいてもよい。金属の酸化物を含むことで、電池の容量を向上できる場合がある。例えば、TiO2やMn2O3は、酸化還元反応性を向上させる働きを有していることにより、電池の容量を向上効果を奏すると考えられる。これら金属の酸化物は、アルカリ金属酸化物1に対して、0.01~3のモル比で使用し得る。
また、正極活物質は、アモルファス相を含んでいさえすれば、本発明による効果を阻害しない範囲内で結晶質相を含んでいてもよい。このアモルファス相を必須の含有相とし、結晶質相を任意に含有相とする結晶状態をアモルファス状態と称する。結晶性が高くなると、界面形成が困難となり抵抗が高くなる傾向があるため、結晶性(アモルファス状態)の低い正極活物質の提供が望まれている。また、アモルファス状態は、結晶状態に比べて、密度が低いため、充放電の際の体積変化に対する正極活物質の追従性が高い。その結果、粉末化による未導電部のような不可逆容量の発生を抑制することができる。ここで、アモルファス状態であることは、不可逆容量の発生を抑制し得る程度の状態であり、例えば、そのXRDパターンにおける最小2θのピークが、0.5以上の半値幅となる状態を示すことを意味する。半値幅は、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.5、3.0、3.5、4.0、4.5、5.0をとり得る。
ここで、発明者等は、正極活物質が少量の結晶質相を含んでいることが適切な場合があることを見い出している。厚い正極は、通常、正極活物質等の粒子状の原料(原料粒子)をプレスすることにより形成される。厚い正極では、原料粒子間の隙間(空間)をできるだけ減らし、原料粒子の密着性を向上させることが、電池特性を向上させる観点から強く望まれている。少量の結晶質相を含む正極活物質から構成される原料粒子は、プレスによりつぶれ易いため、原料粒子の密着性を向上させることができる。なお、結晶質相は、5.0以下、4.0以下、3.0以下、2.0以下、1.0以下の半値幅を示す程度で正極活物質内に含まれていればよい。
更に、この正極活物質を用いれば、プレスによって20μm以上の厚さの正極も容易に形成できるので、この正極活物質は、高容量のバルク型全固体二次電池用の正極を形成する材料として適している。この20μm以上の厚さは、スパッタ法のような気相成長法では、実現することが困難である。厚さは、20μm、25μm、30μm、35μm、40μm、50μm、60μm、80μm、100μm、120μm、140μm、150μmをとり得る。
正極活物質が、その原料をメカニカルミリングにより混合することで製造できる。特に、Li2SO4は、非特許文献1のようなスパッタ法では分解することが予想され、正極活物質として正極を構成することは困難である。従って、Li2SO4は、メカニカルミリング処理による製造に利点が多い。
メカニカルミリングでの処理は、原料を十分混合することで、正極活物質をアモルファス状態とすることができさえすれば、処理装置及び処理条件には特に限定されない。
処理装置としては、通常ボールミルが使用できる。ボールミルは、大きな機械的エネルギーが得られるため好ましい。ボールミルの中でも、遊星型ボールミルは、ポットが、自転回転すると共に、台盤により公転回転するため、高い衝撃エネルギーを効率よく発生させることができるので、好ましい。
なお、原料は、メカニカルミリング処理時の加水分解反応を回避するという観点から、無水状態であることが好ましい。原料が結晶水を含む場合、予め空気中で2時間程度熱処理しておくことが好ましい。熱処理の温度は、原料の種類に応じて適宜決定できる。更に、アルカリ金属塩は、リチウム及び/又はナトリウムの水酸化物と対応する酸とを反応させることで得てもよい。
正極は、上記正極活物質を少なくとも含む。また、20μm以上の厚さを有する。20μm以上の厚さを有する正極は、スパッタ法では形成困難である。
また、必要に応じて、酸化物系固体電解質を含んでいてもよい。正極中、酸化物系固体電解質の占める割合は、30重量%以下であることが好ましく、10重量%以下であることがより好ましい。
酸化物系固体電解質としては、例えば、Li3PO4、Li2SO4、Li3BO3及びLi2CO3のリチウム塩、Na3PO4、Na2SO4、Na3BO3及びNa2CO3のナトリウム塩等が挙げられる。この酸化物系固体電解質は、300℃程度以下の加熱でも、十分な導電性を正極に付与できる。
なお、ガラスセラミックス状は、対応するガラス状に存在していたガラス転移点が存在しない状態であることが好ましい。また、ガラスセラミックス状は、非晶質状態のガラス成分中に、結晶質部が分散した状態であってもよい。結晶質部の割合は、全体に対して、50重量%以上であることが好ましく、80重量%以上であることがより好ましい。なお、結晶質部の割合は固体NMRにより測定可能である。
(2)ガラスセラミックス状の酸化物系固体電解質は、上記ガラス状の固体電解質を、熱処理に付すことにより得ることができる。この熱処理は、ガラス状の固体電解質の結晶化温度以上の温度で行うことが好ましい。
ガラス転移点(Tg)は、固体電解質の構成元素に応じて相違するが、例えば、Li3BO3-Li2SO4系の場合、190~250℃の範囲にある。また第一結晶化温度(Tc)は210~270℃の範囲にある。熱処理温度の上限は、特に限定されないが、通常、第一結晶化温度+100℃である。
熱処理時間は、ガラス状をガラスセラミックス状に変換し得る時間であり、熱処理温度が高いと短く、低いと長くなる。熱処理時間は、通常、0.1~10時間の範囲である。
結着剤としては、例えば、ポリフッ化ビニリデン、ポリテトラフルオロエチレン、ポリビニルアルコール、ポリ酢酸ビニル、ポリメチルメタクリレート、ポリエチレン等が挙げられる。
導電剤としては、天然黒鉛、人工黒鉛、アセチレンブラック、ケッチェンブラック、デンカブラック、カーボンブラック、気相成長カーボンファィバ(VGCF)等が挙げられる。
正極は、例えば、正極活物質と、任意に酸化物系固体電解質、結着剤、導電剤等を混合し、得られた混合物をプレスすることで、ペレット状として得ることができる。プレスは、300℃程度以下の温度、100~800MPaの圧力で、0.1~5時間行うことができる。本発明の正極活物質は、プレス温度が従来より低温であっても、高い導電性を正極に付与できる。
また、このプレスは、固体電解質層の形成時のプレスと兼ねていてもよい。兼ねることで、正極と固体電解質層の界面での密着性をより向上できる。
全固体二次電池は、正極、負極、及び前記正極と前記負極間に位置する固体電解質層とを少なくとも備えている。この内、正極は、上記の物を使用できる。また、固体電解質層には、上記正極の欄で説明した固体電解質をそのまま使用できる。
負極は、特に限定されない。負極は、負極活物質のみからなっていてもよく、結着剤、導電剤、電解質等と混合されていてもよい。
負極活物質としては、Li、Na、In、Sn等の金属、Li合金、Na合金、グラファイト、ハードカーボン、Li4/3Ti5/3O4、Na3V2(PO4)3、SnO等の種々の遷移金属酸化物等が挙げられる。
結着剤及び導電剤は、上記正極の欄で挙げた物をいずれも使用できる。電解質は、酸化物系及び硫化物系の固体電解質をいずれも使用できる。
負極は、例えば、負極活物質及び、任意に結着剤、導電剤、電解質等を混合し、得られた混合物をプレスすることで、ペレット状として得ることができる。また、負極活物質として金属又はその合金からなる金属シート(箔)を使用する場合、それをそのまま使用可能である。
負極は、SUS、アルミニウム又は銅等の集電体の上に形成されていてもよい。
全固体二次電池は、例えば、正極と、電解質層と、負極とを積層し、プレスすることにより得ることができる。
実施例1
LiCoO2とLi3PO4又はLi2SO4とを原料とする2種類の正極活物質を以下の手順で製造した。なお、2種類の正極活物質は、以下では、LiCoO2-Li3PO4とLiCoO2-Li2SO4と称する。
LiCoO2(日本化学工業社製)とLi3PO4(和光純薬工業社製)又はLi2SO4・H2O(和光純薬工業社製)を、Ar雰囲気下、300℃で3時間加熱することによって脱水して得たLi2SO4とを、7:3のモル比となるように計り取り、乳鉢で混合した。得られた混合物を、メカニカルミリング処理に付すことで2種類の約3μmの粒子径の正極活物質を得た。この処理には、遊星型ボールミルである、Fritsch社製Pulverisette P-7を使用し、ポット及びボールはZrO2製であり、45mlのポット内に直径5mmのボール(40g)が500個入っているミルを使用した。メカニカルミリング処理は、370rpmの回転速度、室温、アルゴン雰囲気中で20時間行った。
なお、上記製造法は、Akitoshi Hayashi et al., Journal of Non-Crystalline Solids 356 (2010),p.2670-2673のExperimentalの記載に準じている。
得られた2種類の正極活物質のペレットのXRDパターンを図1に示す。図1には、立方晶(cubic)のLiCoO2、六方晶(hexagonal)のLiCoO2、Li3PO4及びLi2SO4のXRDパターンも示す。図1から、LiCoO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
また、LiCoO2-Li2SO4の正極活物質の走査型電子顕微鏡(SEM)写真を図2(a)に、図2(a)に対応するCo及びS原子のエネルギー分散型X線分光法(EDX)によるマッピング図を図2(b)及び(c)に示す。図2(a)~(c)から、得られた正極活物質においては、粒子内においてCoとSが均一に分散されていることがわかる。これは、もとのLiCoO2とLi2SO4の粒子が反応して、新たなLiCoO2-Li2SO4の粒子が形成されていることを示す。
LiCoO2-Li2SO4の正極活物質の交流インピーダンス法によるナイキストプロットを図3に、印加電圧0.1Vで直流分極測定した際の経過時間毎の電流挙動を図4に、それぞれ示す。図3は、正極活物質をSUS板で挟んで測定した結果を、図4は、正極活物質を固体電解質層(Li3PS4)で挟み、更にLi板で挟んで測定した結果を、それぞれ意味する。図3及び4から、LiCoO2-Li2SO4は3.7×10-5Scm-1の電子伝導度と1.2×10-6Scm-1のリチウムイオン伝導度を有していることが分かる。同様に、LiCoO2-Li2PO4は2.5×10-5Scm-1の電子伝導度と5.3×10-7Scm-1のリチウムイオン伝導度を有していることが分かる。室温において10-7Scm-1以上の電子伝導度とイオン伝導度を有していることから、全固体電池の正極として必要な条件を満たしている。
まず、LiOH・H2O(和光純薬工業社製)とH3BO3(和光純薬工業社製)を混合して、500℃で1時間加熱後、600℃で2時間焼成することによってLi3BO3を合成した。Li2SO4・H2O(和光純薬工業社製)をAr雰囲気下で300℃で3時間加熱することによって脱水し、Li2SO4を得た。Li3BO3とLi2SO4及びLi2CO3(和光純薬工業社製)を1:1:1のモル比になるように計り取り、乳鉢で混合した後、混合物を遊星型ボールミルに投入した。投入後、メカニカルミリング処理することで、数μmの粒径のガラス状固体電解質(33Li3BO3・33Li2SO4・33Li2CO3)を得た。遊星型ボールミルは、Fritsch社製Pulverisette P-7を使用し、ポット及びボールは酸化ジルコニウム製であり、45mlのポット内に直径5mmのボールが160個入っているミルを使用した。メカニカルミリング処理は、370rpmの回転速度、室温、乾燥Ar雰囲気のグローブボックス内で90時間行った。上記ガラス状固体電解質を、結晶化温度以上の260℃で加熱し、ガラスセラミックス化することによりガラスセラミック状固体電解質を得た。得られた固体電解質40mgを面積0.785cm2の成形部を有するペレット成形機を用いて、720MPaの圧力でプレスすることで、ペレット状の固体電解質層(厚さ約1mm)を得た。
正極(LiCoO2-Li2SO4)、上記固体電解質層、及び対極としてのインジウム箔をこの順で積層し、積層体をステンレススチール(SUS)製の集電体で挟むことで電池セル(全固体リチウム二次電池)を得た。セルの断面写真を図5に示す。このセルを、100℃、0.13mAcm-2の電流密度での、充放電試験に付した。試験結果を図6のグラフに示す。図の左の縦軸にはLi-In対極に対する電位を、右の縦軸にはLi-InとLiの電位差である0.62Vを考慮して算出したLi基準の電位をそれぞれ示す。
図5から、正極と固体電解質層との界面は隙間なく密着しており、界面における副反応層(高抵抗層)の存在も確認できなかった。また正極層の厚みは約100μmであることが分かる。図6から、プレスにより形成した所謂バルク型の全固体二次電池であっても充放電を行うことができることが分かる。
正極の断面のSEM写真を図7(a)に、図7(a)に対応するS、Co及びO原子のEDXによるマッピング図を図7(b)~(d)に示す。図7(a)~(d)から、得られた正極活物質において、CoとSとが均一に分散されていることが分かる。
更に、充電前、充電後及び放電後の正極のペレットのXRDパターンを図8に示す。図8には、六方晶(hexagonal)のLiCoO2及びLi2SO4のXRDパターンも示す。なお、図8中のSiが付されたピークは、基準材料のピークを意味する。図8から、正極活物質が充電後及び放電後においてもアモルファス状態であることが分かる。
LiCoO2-Li2SO4をLiCoO2-Li2PO4に変更すること以外は実施例2と同様にして電池セルを作製し、100℃、0.13mAcm-2の電流密度での、充放電試験に付した。試験結果を図9のグラフに示す。図9から、プレスにより形成した所謂バルク型の全固体二次電池であっても充放電を行うことができることが分かる。
LiNiO2(豊島製作所社製)とLi2SO4・H2O(和光純薬工業社製)を用い、メカニカルミリング処理の処理時間を50時間に、変更すること以外は実施例1と同様にして、正極活物質としてのLiNiO2-Li2SO4を得た。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図10のグラフに示す。図10から、LiNiO2-Li2SO4も充放電を行うことができることが分かる。
実施例5
LiNi1/3Mn1/3Co1/3O2(戸田工業社製)とLi2SO4・H2O(和光純薬工業社製)を用い、メカニカルミリング処理の処理時間を50時間に変更すること以外は実施例1と同様にして、正極活物質としてのLiNi1/3Mn1/3Co1/3O2-Li2SO4を得た。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図11のグラフに示す。図11から、LiNi1/3Mn1/3Co1/3O2-Li2SO4も充放電を行うことができることが分かる。
LiCoO2とLi2SO4とのモル比(x:1-x)を100:0、90:10、80:20、70:30、60:40に変更し、メカニカルミリング処理の処理時間を50時間に変更すること以外は実施例1と同様にして、正極活物質としての5種のLiCoO2-Li2SO4を得た。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図12に示す。図12には、立方晶(cubic)のLiCoO2、六方晶(hexagonal)のLiCoO2、Li3PO4及びLi2SO4のXRDパターンも示す。図12から、LiCoO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図13のグラフに示す。図13から、LiCoO2とLi2SO4の割合を変更しても充放電を行うことができることが分かる。
LiCoO2とLi2SO4とのモル比を80:20に変更し、メカニカルミリング処理の処理時間を1時間、10時間及び50時間に変更すること以外は実施例1と同様にして、正極活物質としての3種のLiCoO2-Li2SO4を得た。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図14に示す。図14には、メカニカルミリング処理前の混合物、立方晶(cubic)のLiCoO2、六方晶(hexagonal)のLiCoO2、Li3PO4及びLi2SO4のXRDパターンも示す。図14から、処理時間が延びる程、LiCoO2に対応するピークがブロード化しており、正極活物質がアモルファス状態となることが分かる。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図15のグラフに示す。図15から、処理時間が延びる程、充放電容量が増えることが分かる。
LiCoO2(日本化学工業社製)とLi2MnO3(豊島製作所社製)とのモル比80-x:xを40:40又は60:20とし、LiCoO2とLi2MnO3の合計とLi2SO4とのモル比を80:20に変更し、メカニカルミリング処理の処理時間を50時間に変更すること以外は実施例1と同様にして、正極活物質としてのLiCoO2-Li2MnO3-Li2SO4を得た。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図16に示す。図16には、Li2MnO3、立方晶(cubic)のLiCoO2、六方晶(hexagonal)のLiCoO2及びLi2SO4のXRDパターンも示す。図16から、LiCoO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図17(x=20)及び図18(x=40)のグラフに示す。図17及び18から、LiCoO2-Li2MnO3-Li2SO4も充放電を行うことができることが分かる。
LiCoO2(日本化学工業社製)とLi2TiO3(和光純薬工業社製)とのモル比80-x:xを40:40又は60:20とし、LiCoO2とLi2TiO3の合計とLi2SO4とのモル比を80:20に変更し、メカニカルミリング処理の処理時間を50時間に変更すること以外は実施例1と同様にして、正極活物質としてのLiCoO2-Li2TiO3-Li2SO4を得た。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図19に示す。図19には、Li2TiO3、立方晶(cubic)のLiCoO2、六方晶(hexagonal)のLiCoO2及びLi2SO4のXRDパターンも示す。図19から、LiCoO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
上記正極活物質を使用すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図20(x=20)のグラフに示す。図20から、LiCoO2-Li2TiO3-Li2SO4も充放電を行うことができることが分かる。
Li2CO3(日本化学工業社製)、TiO2(和光純薬工業社製)とMn2O3(高純度化学社製)とを混合し、得られた混合物を、空気中、700℃で2時間焼成した。得られた焼成物をペレット化した。ペレットを、空気中、950℃で96時間焼結することにより、結晶性のLi1.2Ti0.4Mn0.4O2を得た。Li1.2Ti0.4Mn0.4O2と、Li2SO4・H2O(和光純薬工業社製)を、Ar雰囲気下、300℃で3時間加熱することによって脱水して得たLi2SO4とを、7:3及び8:2のモル比となるように計り取り、乳鉢で混合した。得られた混合物を、実施例1と同様のメカニカルミリング処理に付すことで正極活物質を得た。但し、この実施例での処理は、ボール数を50個に、処理時間を50時間に変更した。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図21に示す。図21には、LiTiMnO2の文献値でのXRDパターンも示す。図21から、LiTiMnO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
上記正極活物質を使用し、固体電解質層を以下の固体電解質層、固体電解質層側の集電体をCu製に変更すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図22(モル比7:3)及び図23(モル比8:2)のグラフに示す。図22及び23から、Li1.2Ti0.4Mn0.4O2-Li2SO4も充放電を行うことができることが分かる。また、充放電の繰り返しによる容量低下が比較的小さいことが分かる。
固体電解質は、Li2CO3を使用せず、Li3BO3とLi2SO4を9:1のモル比とすること以外は、実施例1と同様にして得られた90Li3BO3・10Li2SO4を使用した。
LiCoO2(日本化学工業社製)、Li2SO4・H2O(和光純薬工業社製)とLiNO3(和光純薬工業社製)とを用い(LiCoO2とLi2SO4・H2OとLiNO3のモル比は80:15:5及び70:20:10)、メカニカルミリング処理の処理時間を50時間に、ボール数を67個に変更すること以外は実施例1と同様にして、正極活物質としてのLiCoO2-Li2SO4-LiNO3を得た。
上記正極活物質のXRDパターンを実施例1と同様にして測定し、図24に示す。図24には、六方晶(hexagonal)のLiCoO2、LiNO3及びLi2SO4のXRDパターンも示す。図24から、LiCoO2に対応するピークがいずれもブロード化しており、正極活物質がアモルファス状態であることが分かる。特に、観測された全ての2θのピークが、0.5以上の半値幅となっていることが分かる。
上記正極活物質を使用し、固体電解質層を実施例10の固体電解質層、固体電解質層側の集電体をCu製に変更すること以外は実施例2と同様にして得た電池セルを充放電試験に付した。試験結果を図25(モル比80:15:5)のグラフに示す。図25から、LiCoO2-Li2SO4-LiNO3も充放電を行うことができることが分かる。
Claims (10)
- 全固体二次電池用の正極の作製原料であるアモルファス酸化物系正極活物質であり、
前記アモルファス酸化物系正極活物質が
(i)Li及びNaから選択されるアルカリ金属と、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される第2金属と、リン酸イオン、硫酸イオン、硼酸イオン、ケイ酸イオン、アルミン酸イオン、ゲルマン酸イオン、硝酸イオン、炭酸イオン及びハロゲン化物イオンから選択されるイオン種と、酸素原子(但し、イオン種を構成する酸素原子を除く)とを含み、
(ii)少なくともアモルファス相を含み、かつ
(iii)20μm以上の厚さの正極の作製原料である
アモルファス酸化物系正極活物質。 - Li及びNaから選択されるアルカリ金属と、Co、Ni、Mn、Fe、Cr、V、Cu、Ti、Zn、Zr、Nb、Mo、Ru及びSnから選択される第2金属と、リン酸イオン、硫酸イオン、硼酸イオン、ケイ酸イオン、アルミン酸イオン、ゲルマン酸イオン、硝酸イオン、炭酸イオン及びハロゲン化物イオンから選択されるイオン種と、酸素原子(但し、イオン種を構成する酸素原子を除く)とを含み、アモルファス状態であり、20μm以上の厚さの全固体二次電池用の正極の作製原料である請求項1に記載のアモルファス酸化物系正極活物質。
- 前記アモルファス酸化物系正極活物質は、そのXRDパターンにおける最小2θでのピークが、0.5以上の半値幅となるアモルファス状態を示す請求項1に記載のアモルファス酸化物系正極活物質。
- 前記アモルファス酸化物系正極活物質が、アモルファス相と結晶質相とを含む請求項3に記載のアモルファス酸化物系正極活物質。
- 前記アモルファス酸化物系正極活物質が、アルカリ金属酸化物及びアルカリ金属塩に由来する成分を含み、前記アルカリ金属酸化物が、LiCoO2、LiNiO2、LiMnO2、Li2MnO3、Li(Ni,Co,Mn)O2、Li2TiO3、LiFeO2、LiCrO2、Li2CuO2、LiCuO2、LiMoO2、Li2RuO3、Li3NbO4、Li3V2(PO4)3、LiMn2O4及びLi(Ni,Mn)O4のリチウム系化合物と、NaCoO2、NaNiO2、NaMnO2、Na2MnO3、Na(Ni,Co,Mn)O2、NaFeO2、Na2TiO3、NaCrO2、Na2CuO2、NaCuO2、NaMoO2、Na2RuO3、Na3NbO4、Na3V2(PO4)3、NaMn2O4及びNa(Ni,Mn)O4のナトリウム系化合物とからなる群から選択され、前記アルカリ金属塩が、AxByOz(式中、AはLi又はNaであり、BはP,S,B,C,Si,Al,Ge及びNから選択され、xは1以上、yは1以上、zは1以上であり、かつx、y及びzは化学量論的に可能な値である)及びAX(AはLi又はNaであり、XはF,Cl,Br及びIから選択される)から選択される請求項1に記載のアモルファス酸化物系正極活物質。
- 前記アルカリ金属酸化物に由来する成分とアルカリ金属塩に由来する成分とが、前記アモルファス酸化物系正極活物質中に、1:9~9:1のモル比で存在する請求項3に記載のアモルファス酸化物系正極活物質。
- 前記アモルファス酸化物系正極活物質が、LiCoO2と、Li3PO4及び/又はLi2SO4とに由来する成分を含む請求項1に記載のアモルファス酸化物系正極活物質。
- 請求項1に記載のアモルファス酸化物系正極活物質の製造方法であって、前記アモルファス酸化物系正極活物質が、その原料をメカニカルミリングにより混合することで得られるアモルファス酸化物系正極活物質の製造方法。
- 請求項1に記載のアモルファス酸化物系正極活物質を含み、20μm以上の厚さを有する正極。
- 請求項9に記載の正極、負極、及び前記正極と前記負極間に位置する固体電解質層とを少なくとも備える全固体二次電池。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/089,275 US11349123B2 (en) | 2016-03-31 | 2017-03-08 | Amorphous oxide-based positive electrode active material, method for producing same and use of same |
| CN201780021201.1A CN108886145B (zh) | 2016-03-31 | 2017-03-08 | 非晶相氧化物基正极活性材料及其制备方法和应用 |
| JP2018508899A JP6935926B2 (ja) | 2016-03-31 | 2017-03-08 | アモルファス酸化物系正極活物質、その製造方法及びその用途 |
| EP17774144.4A EP3439083B1 (en) | 2016-03-31 | 2017-03-08 | Amorphous oxide-based positive electrode active material, method for producing same and use of same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016070298 | 2016-03-31 | ||
| JP2016-070298 | 2016-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017169599A1 true WO2017169599A1 (ja) | 2017-10-05 |
Family
ID=59963063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/009294 Ceased WO2017169599A1 (ja) | 2016-03-31 | 2017-03-08 | アモルファス酸化物系正極活物質、その製造方法及びその用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11349123B2 (ja) |
| EP (1) | EP3439083B1 (ja) |
| JP (1) | JP6935926B2 (ja) |
| CN (1) | CN108886145B (ja) |
| WO (1) | WO2017169599A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019087717A1 (ja) * | 2017-10-31 | 2019-05-09 | 国立大学法人横浜国立大学 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極、リチウムイオン二次電池、電子機器及び車両 |
| WO2020085015A1 (ja) * | 2018-10-26 | 2020-04-30 | 株式会社豊田自動織機 | 電極及び固体型リチウムイオン二次電池 |
| US20210104748A1 (en) * | 2018-10-31 | 2021-04-08 | Lg Chem, Ltd. | Lithium secondary battery |
| US20210399296A1 (en) * | 2018-11-28 | 2021-12-23 | Hyundai Motor Company | Lithium secondary battery and manufacturing method thereof |
| WO2023095889A1 (ja) | 2021-11-26 | 2023-06-01 | 公立大学法人大阪 | 酸化物系正極活物質及びその利用 |
| JP2023131550A (ja) * | 2022-03-09 | 2023-09-22 | 株式会社豊田中央研究所 | リチウム複合酸化物、蓄電デバイス及びリチウム複合酸化物の製造方法 |
| JP2024025946A (ja) * | 2022-08-15 | 2024-02-28 | トヨタ自動車株式会社 | 正極活物質、正極活物質層、全固体リチウムイオン電池、正極活物質の製造方法、及び全固体リチウムイオン電池の製造方法 |
| KR20250016111A (ko) | 2022-05-30 | 2025-02-03 | 스미또모 가가꾸 가부시키가이샤 | 리튬 함유 산화물, 전극 및 전지 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE112019001591B4 (de) * | 2018-03-28 | 2024-07-25 | Tdk Corporation | Festkörperbatterie |
| CN110400931B (zh) * | 2019-07-31 | 2022-09-02 | 陕西师范大学 | 一种具有超晶格有序结构的锰基储钠型正极材料及其制备方法 |
| CN113078299B (zh) * | 2020-01-06 | 2023-09-29 | 中国科学院物理研究所 | 钠锂铁锰基层状氧化物材料、制备方法和用途 |
| GB2613896A (en) * | 2021-12-20 | 2023-06-21 | Dyson Technology Ltd | A cathode composition |
| CN115632117A (zh) * | 2022-10-25 | 2023-01-20 | 湖北亿纬动力有限公司 | 一种锰基固溶体正极材料及制备方法与用途 |
| CN118117063B (zh) * | 2024-01-24 | 2025-09-26 | 中南大学 | NaaFebXcYd(BO3)e正极活性材料及其制备和在钠离子电池中的应用 |
| WO2025178830A1 (en) * | 2024-02-21 | 2025-08-28 | Wildcat Discovery Technologies, Inc. | Anionic redox high energy cathodes |
| CN119419244B (zh) * | 2024-10-31 | 2025-09-23 | 蜂巢能源科技股份有限公司 | 一种复合正极材料及其制备方法和应用 |
| CN119038635B (zh) * | 2024-11-01 | 2025-04-08 | 宁波维科电池有限公司 | 一种非晶金属氧化物正极材料的制备方法及其应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013161646A (ja) * | 2012-02-03 | 2013-08-19 | Sumitomo Electric Ind Ltd | 非水電解質電池及びその製造方法、並びにこの電池を備える電動車両 |
| WO2015087734A1 (ja) * | 2013-12-09 | 2015-06-18 | 日本電気硝子株式会社 | ナトリウムイオン電池用電極合材、及びその製造方法並びにナトリウム全固体電池 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1392623A (zh) * | 2001-06-15 | 2003-01-22 | 未来科技研究院有限公司 | 锂磷氧氮硫化合物电解质薄膜 |
| KR101065307B1 (ko) * | 2004-01-19 | 2011-09-16 | 삼성에스디아이 주식회사 | 리튬이차전지용 캐소드 활물질 및 이를 이용한 리튬이차전지 |
| JP5029540B2 (ja) * | 2008-09-01 | 2012-09-19 | ソニー株式会社 | 正極活物質、これを用いた正極および非水電解質二次電池 |
| JP5157781B2 (ja) * | 2008-09-25 | 2013-03-06 | トヨタ自動車株式会社 | 全固体リチウム二次電池 |
| JP2010177024A (ja) * | 2009-01-29 | 2010-08-12 | Sumitomo Electric Ind Ltd | 非水電解質電池用正極と非水電解質電池および非水電解質電池用正極の製造方法 |
| JP5549192B2 (ja) * | 2009-11-18 | 2014-07-16 | ソニー株式会社 | 固体電解質電池および正極活物質 |
| JP6051514B2 (ja) | 2010-12-02 | 2016-12-27 | ソニー株式会社 | 固体電解質電池および正極活物質 |
| KR101623720B1 (ko) * | 2013-03-15 | 2016-05-26 | 주식회사 엘지화학 | 고용량 음극 활물질 및 이를 포함하는 리튬 이차전지 |
| JP6362371B2 (ja) | 2014-03-18 | 2018-07-25 | 公立大学法人大阪府立大学 | 酸化物系固体電解質及びその用途 |
| JP6083406B2 (ja) * | 2014-03-19 | 2017-02-22 | トヨタ自動車株式会社 | 活物質粉体及びその製造方法 |
| JP2016035912A (ja) * | 2014-07-31 | 2016-03-17 | 富士フイルム株式会社 | 全固体二次電池、固体電解質組成物、これを用いた電池用電極シート、電池用電極シートの製造方法および全固体二次電池の製造方法 |
-
2017
- 2017-03-08 US US16/089,275 patent/US11349123B2/en active Active
- 2017-03-08 CN CN201780021201.1A patent/CN108886145B/zh active Active
- 2017-03-08 JP JP2018508899A patent/JP6935926B2/ja active Active
- 2017-03-08 WO PCT/JP2017/009294 patent/WO2017169599A1/ja not_active Ceased
- 2017-03-08 EP EP17774144.4A patent/EP3439083B1/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013161646A (ja) * | 2012-02-03 | 2013-08-19 | Sumitomo Electric Ind Ltd | 非水電解質電池及びその製造方法、並びにこの電池を備える電動車両 |
| WO2015087734A1 (ja) * | 2013-12-09 | 2015-06-18 | 日本電気硝子株式会社 | ナトリウムイオン電池用電極合材、及びその製造方法並びにナトリウム全固体電池 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3439083A4 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019087717A1 (ja) * | 2017-10-31 | 2019-05-09 | 国立大学法人横浜国立大学 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極、リチウムイオン二次電池、電子機器及び車両 |
| JPWO2019087717A1 (ja) * | 2017-10-31 | 2020-11-26 | 国立大学法人横浜国立大学 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極、リチウムイオン二次電池、電子機器及び車両 |
| JP7094570B2 (ja) | 2017-10-31 | 2022-07-04 | 国立大学法人横浜国立大学 | リチウムイオン二次電池、電子機器及び車両 |
| WO2020085015A1 (ja) * | 2018-10-26 | 2020-04-30 | 株式会社豊田自動織機 | 電極及び固体型リチウムイオン二次電池 |
| US20210104748A1 (en) * | 2018-10-31 | 2021-04-08 | Lg Chem, Ltd. | Lithium secondary battery |
| US12191498B2 (en) * | 2018-10-31 | 2025-01-07 | Lg Energy Solution, Ltd. | Lithium secondary battery |
| US12046749B2 (en) * | 2018-11-28 | 2024-07-23 | Hyundai Motor Company | Lithium secondary battery and manufacturing method thereof |
| US20210399296A1 (en) * | 2018-11-28 | 2021-12-23 | Hyundai Motor Company | Lithium secondary battery and manufacturing method thereof |
| WO2023095889A1 (ja) | 2021-11-26 | 2023-06-01 | 公立大学法人大阪 | 酸化物系正極活物質及びその利用 |
| JP2023131550A (ja) * | 2022-03-09 | 2023-09-22 | 株式会社豊田中央研究所 | リチウム複合酸化物、蓄電デバイス及びリチウム複合酸化物の製造方法 |
| JP7797924B2 (ja) | 2022-03-09 | 2026-01-14 | 株式会社豊田中央研究所 | リチウム複合酸化物、蓄電デバイス及びリチウム複合酸化物の製造方法 |
| KR20250016111A (ko) | 2022-05-30 | 2025-02-03 | 스미또모 가가꾸 가부시키가이샤 | 리튬 함유 산화물, 전극 및 전지 |
| JP2024025946A (ja) * | 2022-08-15 | 2024-02-28 | トヨタ自動車株式会社 | 正極活物質、正極活物質層、全固体リチウムイオン電池、正極活物質の製造方法、及び全固体リチウムイオン電池の製造方法 |
| JP7619341B2 (ja) | 2022-08-15 | 2025-01-22 | トヨタ自動車株式会社 | 正極活物質、正極活物質層、全固体リチウムイオン電池、正極活物質の製造方法、及び全固体リチウムイオン電池の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190181446A1 (en) | 2019-06-13 |
| EP3439083A4 (en) | 2019-08-21 |
| CN108886145A (zh) | 2018-11-23 |
| JP6935926B2 (ja) | 2021-09-15 |
| CN108886145B (zh) | 2022-03-15 |
| JPWO2017169599A1 (ja) | 2019-02-14 |
| US11349123B2 (en) | 2022-05-31 |
| EP3439083A1 (en) | 2019-02-06 |
| EP3439083B1 (en) | 2021-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6935926B2 (ja) | アモルファス酸化物系正極活物質、その製造方法及びその用途 | |
| JP6362371B2 (ja) | 酸化物系固体電解質及びその用途 | |
| JP7218725B2 (ja) | バイポーラ型全固体ナトリウムイオン二次電池 | |
| JP5817657B2 (ja) | 電池システム、電池システムの製造方法、電池の制御装置 | |
| JPWO2018225494A1 (ja) | 全固体ナトリウムイオン二次電池 | |
| JP5682318B2 (ja) | 全固体電池 | |
| JP6937009B2 (ja) | 全固体アルカリ金属二次電池用の固体電解質層及び全固体アルカリ金属二次電池 | |
| CN114946049B (zh) | 固体电池 | |
| JP7810291B2 (ja) | 固体電池 | |
| JP2018018578A (ja) | 固体電解質粉末、並びにそれを用いてなる電極合材及び全固体ナトリウムイオン二次電池 | |
| WO2015079509A1 (ja) | リチウムイオン伝導性酸化物および蓄電デバイス | |
| JP2016225089A (ja) | 電極、電極の製造方法及び電池 | |
| JPWO2018131627A1 (ja) | ナトリウムイオン二次電池用電極合材及びその製造方法 | |
| JP6578743B2 (ja) | 電極の製造方法 | |
| JP2013120700A (ja) | 電池 | |
| WO2013100002A1 (ja) | 全固体電池およびその製造方法 | |
| JP7476867B2 (ja) | 硫化物固体電解質、電池および硫化物固体電解質の製造方法 | |
| JP6285317B2 (ja) | 全固体電池システム | |
| JP2012104280A (ja) | 電池用焼結体、全固体リチウム電池および電池用焼結体の製造方法 | |
| JP2022129560A (ja) | 積層型固体電池 | |
| JP2022111714A (ja) | 積層型固体電池 | |
| KR20220112160A (ko) | 전고체 이차전지 | |
| CN120693696A (zh) | 二次电池用电极及其制造方法和全固态二次电池 | |
| CN104364956B (zh) | 电池系统、电池系统的制造方法、电池的控制装置 | |
| JP2022129559A (ja) | 積層型固体電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018508899 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017774144 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017774144 Country of ref document: EP Effective date: 20181031 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17774144 Country of ref document: EP Kind code of ref document: A1 |