WO2017190218A1 - Liquid-biopsy signatures for prostate cancer - Google Patents
Liquid-biopsy signatures for prostate cancer Download PDFInfo
- Publication number
- WO2017190218A1 WO2017190218A1 PCT/CA2017/000114 CA2017000114W WO2017190218A1 WO 2017190218 A1 WO2017190218 A1 WO 2017190218A1 CA 2017000114 W CA2017000114 W CA 2017000114W WO 2017190218 A1 WO2017190218 A1 WO 2017190218A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- peptide
- peptides
- subject
- expression
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/10—Gene or protein expression profiling; Expression-ratio estimation or normalisation
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57555—Immunoassay; Biospecific binding assay; Materials therefor for cancer of the prostate
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/20—Supervised data analysis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Definitions
- This application relates to methods, compositions and systems for the diagnosis or classification of prostate cancer.
- prostate cancer The worldwide incidence of prostate cancer has been steadily increasing, but many patients harbor tumors of an indolent nature. These indolent tumors grow slowly and pose minimal threat to the life of the patient, in the absence of treatment (i.e. are clinically insignificant). However, once prostate cancer begins to grow aggressively, it metastasizes quickly with lethal consequences. The management of prostate cancer has become an urgent clinical dilemma with significant over-diagnosis and challenges in predicting patient survival Prostate cancers are uniquely heterogeneous with major spatial 3 ⁇ 4 3 and temporal 4 variability in their genomes. Therefore, once cancer has been confirmed, the optimal course of action is tailored to spare patients with indolent disease from unnecessary procedures, while identifying and treating those who would benefit from treatment intensification.
- a fluid-based biomarker would be ideal.
- Liquid biopsies such as circulating tumor cells and cell-free DNA 16 have been proposed as promising non-invasive prostate cancer biomarkers, but their detection and enrichment remains technically challenging.
- Cataloguing the secreted and soluble factors released into the interstitial fluid that bathes the organ of interest may provide a novel inventory of putative disease biomarkers.
- EPS prostatic secretions
- Reproducible detection and Quantificatioii of multiple proteins in complex biological matrices is an essential requirement for any potential disease biomarker, but verification of these candidates is a major bottleneck in the pipeline from discovery to clinical implementation.
- immunoaffmily based assays namely enzyme-linked immunosorbent assays (ELISAs) are used to validate protein biomarkers, but this approach is time-coiisuming, costly, and relies on the existence of validated antibody pairs for every target protein.
- ELISAs enzyme-linked immunosorbent assays
- MS Targeted mass spectrometry
- SRM-MS Selected reaction momtcrirtg mass spectrometry
- SRM-MS Selected reaction momtcrirtg mass spectrometry
- a method of diagnosing a subject with prostate cancer comprising: (a) deteennining an expression level of at least 1 gene in a teat sample from the subject selected from the group consisting of the genes identified in Fig.4b; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
- a method of classifying a subject with prostate cancer between having a pT2 stage (organ confined) tumor and having a pT3 stage (extracapsular) tumor comprising: (a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig.
- a composition comprising a plurality of antibodies capable of specifically binding to a plurality of peptides corresponding to a plurality of the genes in Fig. 4b and Fig. 4d.
- the plurality of peptides are a plurality of the peptides identified in Fig. 4b and Fig. 4d.
- the plurality of peptides are the 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig.4d.
- a computer program product for use in conjunction with a computer having a processor and a memory connected to the processor, the computer program product comprising a computer readable storage medium having a computer mechanism encoded thereon, wherein the computer program mechanism may be loaded into the memory of the computer and cause the computer to carry out the method described herein.
- a computer implemented product for diagnosing or classifying a subject with prostate cancer comprising: (a) a means for receiving values corresponding to a subject expression profile in a subject sample; (b) a database comprising a reference expression profile representing a control, wherein the subject expression profile and the reference profile each have at least one value representing the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4&, wherein the computer implemented product compares the reference expression profile to the subject biomarker expression profile, wherein a statistically significant difference or similarity in the expression profiles is used to diagnose or classify the subject with prostate cancer.
- the computer implemented product carries out the method described herein.
- a computer readable medium having stored thereon a data structure for storing the computer implemented product described herein.
- a computer system comprising (a) a database including records comprising a reference expression profile of the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a user interface capable of receiving a selection of expression levels of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d, for use in comparing to the reference expression profile in the database; (c) an output that displays a prediction of diagnosis or classification wherein a statistically significant difference or similarity in the expression levels is used to diagnose or classify the subject with prostate cancer.
- kits comprising reagents for detecting the level of at least 1 gene identified in Fig, 4b and Fig. 4d, preferably at least 1 peptide in Fig.4b and Fig.4d, in a sample.
- Figure 1 shows systematic development of targeted proteomics assays in EPS-urines
- Discovery proteomics data from direct-EPS derived from patients with extracapsular (EC) or organ-confined (OC) prostatic tumors was used to select putative candidates. Proteotypic peptides from these candidates were carefully selected and evaluated by SRM-MS in an EPS- urine background
- All peptides that passed the above selection criteria were analyzed in clinically stratified EPS-urines (Cohort A). Peptide quantification by SRM-MS was performed and 34 candidates with diagnostic and prognostic potential were identified based on relative abundance changes
- Figure 2 shows absolute peptide quantification in an independent patient cohort
- Right panel correlation plot
- Figure 3 shows univariate analyses to distinguish patient risk groups, (a) Heafmap representation of absolute peptide expression levels for all candidate peptides within Cohort B samples (represented as fmol EPS-urine protein), Peptide expression heatmap is clustered using
- SPSA Serum PSA
- Figure 4 shows maohinc-lcaming model to identify biomarker signatures
- (a) Schematic overview of the macbinc-lcaining approach used to develop multi-feature biomarker signatures
- (d) The predictive importance of individual peptides to distinguish pathological stage pT3 from stage pT2.
- Figure 5 shows Pearson correlation for replicate analyses (all sample types analyzed in duplicate), Representation of the reproducibility (Pearson's correlation) for replicate analyses of all peptides analyzed in Cohort B (n-207). Dotted red line represents samples with high correlation (RX),7).
- Figure 6 shows Pearson correlation for replicate analyses (individual sample types; risk groups). Representation of the reproducibility (Pearson's correlation) for replicate analyses of all peptides analyzed in Cohort B stratified by patient risk group (normal, BPH, pT2, pT3). Dolled red line represents samples with high correlation (RX).7).
- Figure 7 shows Pearson correlation compared by peptide abundance. Comparison of peptide abundance plotted as a function of Pearson's correlation. Peptides with a high concordance between replicate analyses (RXJ.7) are significantly mote abundant based on SRM quantification.
- Figure 8 shows chromatographic retention time of individual peptides, (a) Representative chromatogram of the 34 peptides quantified in cohort B. (b) The 34 peptides quantified by SRM- MS in all cohort B samples (207 samples analyzed in duplicates; n-414 SRM-MS analyses) demonstrate highly reproducible retention times.
- Figure 9 shows peptide abundance in the two separate patient cohorts, (a) Average fold change correlation of cancer vs. normal samples for the 34 peptides quantified in cohorts A and B, left side: avenge fold change (log2) in both cohorts-, rights side: correlation blot (b) Average fold change correlation of pT2 vs. pT3 samples for the 34 peptides quantified in cohorts A and B. left side: average fold change (l°g2) in both cohorts; rights side: correlation blot.
- Figure 10 shows patient characteristics for all cohort B samples (n-207).
- Age distribution (a) Age distribution; (b) Ethnicity; (c) Serum PSA distribution (SPSA).
- Figure 11 shows ROC curves for test set analysis, (a) Diagnostic signature: the performance for the selected peptide signature (pink), serum PSA alone (blue) and randomly selected peptides (grey) are compared, (b) Prognostic signature: the performance for the selected peptide signature (pink), serum PSA alone (blue) and randomly selected peptides (grey) are compered.
- ROC curves axe generated from test set
- Figure 12 shows area under the ROC for randomly generated signatures. Distribution of AUCs for randomly selected peptides to generate predictive models for cancer vs. normal (top panel) or for pT3 vs. pT2 (bottom panel). Pink line indicates AUC for our predictive models based on identified peptides. AUCs are measured from test set.
- Figure 13 shows power analyses for all analyzed samples to distinguish indicated patient risk groups.
- Figure 14 shows inter-correlation between peptides, (a) Correlation matrix of peptide-peptide expression. Expression profiles between all 34 peptides quantified in cohort B are compared using Pearson's correlation coefficient (R). Peptide-peptide matrix comparing the quantitative expression profiles of (b) the 6 diagnostic signature peptides and (c) the 7 prognostic signature peptides.
- Figure 15 shows suitable configured computer device, and associated communications networks, devices, software and firmware to provide a platform for enabling one or more embodiments as described herein.
- a method of diagnosing a subject with prostate cancer comprising: (a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig. 4b; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
- classifying means predicting or identifying the particular state of a disease. For example, with respect to prostate cancer, patients may be classified as having a pT2 stage (organ confined) tumor or a pT3 stage (extracapsular) tumor.
- diagnosis is tine identification of the nature and cause of a certain phenomenon, such as, the identification of disease state in a patient
- the methods described herein are useful for determining whether a subject has prostate cancer.
- subject refers to any member of the animal kingdom, preferably a human being and most preferably a human being that has prostate cancer or that is suspected of having prostate cancer.
- test sample refers to any fluid, cell or tissue sample from a subject which can be assayed for biomarker expression products and/or a reference expression profile, e.g. peptides differentially present in a liquid biopsy.
- RNA includes mRNA transcripts, and/or specific spliced or other alternative variants of mRNA, including anti-sense products.
- protein or “peptides”, it refers to proteins expressed by genes are measurable in a sample.
- level of expression or “expression level” as used herein refers to a measurable level of expression of the products of biomarlcers, such as, without limitation, micro-RNA, or messenger RNA transcript expressed or of a specific exon or other portion of a transcript, the level of proteins, peptides or portions thereof expressed of the biomarkers, the number or presence of DNA polymorphisms of the biomarkers, the enzymatic or other activities of the biomarkers, and the level of specific metabolites.
- control refers to a specific value or dataset that can be used to prognose or classify the value e.g, expression level or reference expression profile obtained from the test sample associated with an outcome class.
- a dataset may be obtained from samples from a group of subjects known to have prostate cancer having different tumor states or healthy individuals.
- the expression data of the biomarkers in the dataset can be used to create a control value that is used in testing samples from new patients.
- differential expression refers to a difference in the level of expression of the biomarkers that can be assayed by measuring the level of expression of the products of the biomarkers, such as the difference in level of peptide or protein expressed. In a preferred embodiment, the difference is statistically significant.
- difference in the level of expression refers to an increase or decrease in the measurable expression level of a given biomarker, for example as measured by me amount of peptide as compared with the measurable expression level of a given peptide in a control.
- expression profile refers to a dataset representing the expression level(s) of one or more biomarkers.
- An expression profile may represent one subject, or alternatively a consolidated dataset of a cohort of subjects, for example to establish a reference expression profile as a control.
- the at least 1 gene is at least 2, 3, 4, 5, or 6 genes associated with the top- ranked peptides in Fig.4b.
- the at least 1 gene comprises 6 genes associated with the 6 top-ranked peptides in Fig.4b.
- a method of classifying a subject with prostate cancer between having a pT2 stage (organ confined) tumor and having a pT3 stage (extracapsular) tumor comprising: (a) detemnning an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig.
- the at least 1 gene is at least 2, 3, 4, 5, 6, or 7 genes associated with the top-ranked peptides in Fig. 4d.
- the at least 1 gene comprises 7 genes associated with the 7 top-ranked peptides in Fig.4d.
- the method further comprises producing gene expression profiles comprising a subject gene expression profile and a gene reference expression profile, each having values representing the expression level of the at least 1 gene corresponding the test and control samples respectively.
- the test sample comprises at least one of prostate-proximal fluid and/or expressed prostatic secretions.
- test sample is collected directly from the prostate, preferably prior to radical prostatectomy, and/or from urine, preferably post-digital rectal examination urine.
- determining the expression level of the at least one gene in the test sample comprises measuring in the test sample, the level of at least one peptide corresponding to the protein product of the at least one genc.
- the method further comprises producing peptide presence profiles comprising t subject peptide expression profile and a reference peptide expression profile, each having values representing the peptide levels of the at least 1 peptide corresponding the test and control samples respectively.
- the at least one peptide comprises 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig. 4d.
- the level of the at least 1 peptide is measured using mass spectrometry, Preferably, the mass spectrometry is targeted mass spectrometry using selected reaction monitoring mass spectrometry.
- composition comprising a plurality of antibodies capable of specifically binding to a plurality of peptides corresponding to a plurality of the genes in Fig. 4b and Fig. 4d
- the plurality of peptides are a plurality of the peptides identified in Fig. 4b and Fig. 4d.
- the plurality of peptides are the 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig.4d
- Figure IS shows a generic computer device 100 that may include a central processing unit (“CPU") 102 connected to a storage unit 104 and to a random access memory 106.
- the CPU 102 may process an operating system 101, application program 103, and data 123.
- the operating system 101, application program 103, and data 123 may be stored in storage unit 104 and loaded into memory 106, as may be required.
- Computer device 100 may further include a graphics processing unit (GPU) 122 which is operatively connected to CPU 102 and to memory 106 to offload intensive image processing calculations from CPU 102 and run these calculations in parallel with CPU 102.
- An operator 107 may interact with the computer device 100 using a video display 108 connected by a video interface 105, and various input/output devices such as a keyboard 1 IS, mouse 112, and disk drive or solid state drive 114 connected by an I/O interface 109.
- the mouse 112 may be configured to control movement of a cursor in the video display 108, and to operate various graphical user interface (GUI) controls appearing in the video display 108 with a mouse button.
- GUI graphical user interface
- the disk drive or solid state drive 114 may be configured to accept computer readable media 116.
- the computer device 100 may form part of a network via a network interface 111, allowing the computer device 100 to communicate >with other suitably configured data processing systems (not shown).
- One or more different types of sensors 135 may
- the present system and method may be practiced on virtually any manner of computer device including a desktop computer, laptop computer, tablet computer or wireless handheld.
- the present system and method may also be implemented as a cor ⁇ uter-raadableAiseable medium that includes computer program code to enable one or more computer devices to implement each of the various process steps in a method in accordance with the present invention.
- the computer devices are networked to distribute the various steps of the operation.
- the terms computer-readable medium or computer useable medium comprises one or more of any type of physical embodiment of the program code.
- me ccniputer-readable/useable medium can comprise program code embodied on one or more portable storage articles of manufacture (e.g. an optical disc, a magnetic disk, a tape, etc.), on one or more data storage portioned of a computing device, such as memory associated with a computer and/or a storage system.
- a computer program product for use in conjunction with a computer having a processor and a memory connected to the processor, the computer program product comprising a computer readable storage medium having a computer mechanism encoded thereon, wherein roe computer program mechanism may be loaded into the memory of the computer and cause the computer to carry out the method described herein.
- a computer implemented product for diagnosing or classifying a subject with prostate cancer comprising: (a) a means for receiving values corresponding to a subject expression profile in a subject sample; (b) a database comprising a reference expression profile representing a control, wherein the subject expression profile and the reference profile each have at least one value representing the level of at least 1 gene identified in Fig. 4b and Fig.
- the computer implemented product compares the reference expression profile to the subject biomarker expression profile, wherein a statistically significant difference or similarity in the expression profiles is used to diagnose or classify the subject with prostate cancer.
- the computer implemented product carries out the method described herein.
- a computer readable medium having stored thereon a data structure for storing the computer implemented product described herein.
- the data structure is capable of configuring a computer to respond to queries based on records belonging to the data structure, each of the records comprising: (a) a value that identifies a reference expression profile of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a value that identifies the probability of a diagnosis or classification associated with the reference expression profile.
- a computer system comprising (a) a database including records comprising a reference expression profile of the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a user interface capable of receiving a selection of expression levels of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d, for use m comparing to the reference expression profile in the database; (c) an output that displays a prediction of diagnosis or classification wherein a statistically significant difference or similarity in the expression levels is used to diagnose or classify the subject with prostate cancer.
- kits comprising reagents for detecting the level of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig.4b and Fig.4d, in a sample.
- reagents for detecting the level of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig.4b and Fig.4d in a sample.
- Prostate cancer patients were selected on the basis of organ confinement or pathological stage. Non-cancer individuals had biopsy confirmed BPH or were considered as individuals with no indication of prostatic disease based on biopsy results. Sample preparation for mass spectrometry
- Ultrapurc-grade 2,2,2-trifluoroethanoI (TFB), trifluoroacetic add (TFA), iodoacetamide (IAA), and dithiotreitol (DTT) were from Sigma-AldricL HPLC-grade solvents (methanol, acctonitrilc, and water) and formic acid were fiom Fisher Scientific. Mass spcctrometry-grade trypshVLys-C was fiom Promega (Madison, WI). Amicon spin filters, O.S ml, 3 kDa MWCO, were fiom Millipore.
- Solid phase extraction C18 tips were fiom Agilent Four ml of EPS-urine were concentrated to approximately 500 ⁇ by using a spin filter with a molecular weight cutoff of 3 kDa, and proteins were precipitated overnight by the addition of ice-cold 100% methanol. Protein pellets were washed twice with 100% methanol and air-dried. Protein resolubilization was performed by the addition of 50% TFE at 60 °C for 2 hours. Following reduction with DTT and alkylation with IAA, proteins were digested overnight at 37 °C using mass 2 ⁇ g trypsin/Lys- C. The reaction was quenched by the addition of TFA. Desalting was performed by solid phase extraction using CI 8 tips.
- Phase 1 peptides were selected and purchased as bulk heavy-isotope labeled peptide standards (JPT Peptide Technologies), also containing 8 peptides that were deemed potentially interesting from our additional EPS proteomics studies
- 250 finol of each heavy peptide standard was spiked into 1 ug of EPS- urine-digest, with 4-6 transitions monitored over a 40-mmute chromatographic gradient Of the 232 Phase J peptides, 147 (Phase 2 peptides) were reproducibly detectable with a minimum of three transitions in the complex EPS-urine background.
- a cohort of individual EPS-urines (n - 74) from a heterogeneous population of patients with EC, OC and control (BPH, normal) (Cohort A) was used to analyze all Phase 2 peptides.
- a total of 1 ⁇ g of peptide from each sample was spiked with 200 fmol of heavy peptide standards that were combined into six batches (batch A-F), consisting of -20 peptides per batch. Visualization and inspection of peaks was performed in Skyline.
- Each peptide was quantified in a sample by integrating the quantifier ion (most intense ion) of the light peptide with its co-eluting heavy peptide ion, in order to derive a light-to-heavy peptide ratio.
- the Student's t-test was used to compare the ratios between cancer and controls, as well as EC and OC prostate cancers.
- a K- fold cross-validation was performed to investigate the diagnostic and prognostic power of the peptides at different p-vahie cutoffs.
- p- value cut-offs were used, respectively. Further refinements to this list were made by including additional peptides that did not meet the p-value cut-offs, but were potentially promising.
- peptides SSEDPNEDIVER from protein IGJ and TPAQFDADELR from protein ANXA1 were added to the list of putative prognostic candidates because they were the only candidates that were elevated in the EC tumor group (p-value - 0,25), Two KLK3 peptides (HSQPWQVLVASR and LSEPAELTDAVK) were also added in order to monitor PSA levels in EPS-urine.
- a multiplexed SRM-MS assay was developed by scheduling all 34 candidates in a single 40-mimrte chromatographic gradient A total of 3 transitions were monitored for the light and heavy versions of each peptide for a total of 204 transitions per analysis. A 2-mimitc acquisition time window was scheduled around the expected peptide elutiontime.
- MStem approach A recently published approach termed MStern (Berger et al. Mol Cell Proteomics. 2015 Oct;14(10):2814-23) that is based on the high protein binding capabilities of porous PVDF membranes (similar to a Western Blot). This approach can be performed in a 96-well format and is rapidly automatable.
- the Generalized Linear Model was used to classify samples into two classes: cancer vs. normal (diagnosis) and pT3 vs. pT2 (prognosis).
- OLM is a widely used machine-learning algorithm that has been applied in various types of biomarker identification ( «.£.
- OLM outperformed eight other machine learning algorithms, which look for different types of patterns and data properties; random forest (rf), stochastic gradient boosting (gbm), Naive Bayes (nb), boosted generalized linear model (glmboost), lasso and elastic-net-regularized generalized linear model (ghnnet), support vector machine with linear kernel (svrnLinear) and radial basis function (RBF) kernel (svmRadial).
- rf random forest
- gbm stochastic gradient boosting
- nb Naive Bayes
- glmboost boosted generalized linear model
- lasso and elastic-net-regularized generalized linear model ghnnet
- support vector machine with linear kernel svrnLinear
- RBF radial basis function kernel
- Z is the normalized peptide expression.
- X is the peptide expression in each sample, ⁇ is the mean of the normal controls, ⁇ is the standard deviation of the normal controls.
- ⁇ is the mean of the normal controls.
- ⁇ is the standard deviation of the normal controls.
- top-ranked peptides from top 3 to top IS
- AUQ receiver operating characteristic curve
- Phase 1 peptides were first evaluated for reproducible detection by SRM-MS in EPS-urine samples.
- Each Phase 1 peptide was purchased as a crude heavy isotope labeled synthetic peptide and spiked into pooled EPS-urines to evaluate their suitability for targeted proteomics assays, directly within the biomarker matrix.
- Light (endogenous) and heavy (synthetic) peptides were monitored in SRM mode, and data were manually inspected to select peptides that had at least 3 fragments ions aligned at the expected peptide elution time, had co-eluting light and heavy peptides, had minimal interference and were reproducible.
- Phase 2 peptides we performed relative quantification in a medium-sized cohort of EPS-urines (n*74; Cohort A; Table 1), using the crude heavy isotope labeled synthetic peptides as internal standards (see Methods). The goal of this initial quantification was to evaluate peptide performance in relevant clinical samples, while reducing the rwmber of peptides to be moved forward to the next development steps. Briefly, a Student's t-test was performed to compare the ' ratios of peptide abundance between cancer and non-cancer groups (termed: diagnostic), as well as EC and OC prostate cancers (termed: prognostic).
- the first criteria used to select candidates as potential diagnostic and prognostic biomarkers were p- value cutoffs of 0.05 and 0.1, respectively.
- a higher p- value cut-off was used to select prognostic candidates in order to avoid removing putative candidates at mis early stage for distinguishing cancer comparisons (EC vs. OC cancer groups), Refinements to the candidate list were made by adding peptides representing the proteins IGJ and ANXAJ, because these peptides were the only candidates even trend up- regulated in the EC tumor group (p ⁇ 0.25).
- two KLK3 peptides were added to monitor PSA levels.
- each peptide that met the above criteria was manually inspected for SRM-MS trace quality.
- Fig. 4a To identify subsets of peptides that can serve as liquid-biopsy signatures and integrate them into unified predictors mat accurately discriminate among our distinct patient risk groups, we employed a machine-learning analysis (Fig. 4a). Briefly, we evaluated quantified peptides as input features for machine learning and identified the most relevant ones using Generalized Linear Models (GLMs) 22 . The selection of reliable features reduces the dimensionality of the feature space and leads to better performance in machine learning To select relevant peptides, we measured the importance of each peptide to discriminate two classes of patient risk groups (cancer vs. normal controls or pT2 vs. pT3 prostate cancers) and subsequently systematically selected the top-ranked peptides as features to build predictive models.
- GLMs Generalized Linear Models
- Nelder JA Wedderburn RWM. Generalized Linear Models. Journal of the Royal Statistical Society 135, 370-384 (1972).
- Prostate-specific antigen (PSA) isoform p2PSA in combination with total PSA and free PSA improves diagnostic accuracy in prostate cancer detection. Eur Urol 57, 921-927 (2010).
- Ju H Brasier AR. Variable selection methods for developing a biomarker panel for prediction of dengue hemorrhagic fever. BMC research notes 6, 365 (2013).
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Medical Informatics (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Analytical Chemistry (AREA)
- Evolutionary Biology (AREA)
- Theoretical Computer Science (AREA)
- Bioinformatics & Computational Biology (AREA)
- Pathology (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Zoology (AREA)
- Hematology (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Data Mining & Analysis (AREA)
- Cell Biology (AREA)
- Hospice & Palliative Care (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Databases & Information Systems (AREA)
- Epidemiology (AREA)
- Evolutionary Computation (AREA)
- Public Health (AREA)
- Software Systems (AREA)
- Bioethics (AREA)
- Oncology (AREA)
- Artificial Intelligence (AREA)
Abstract
There is described herein, a method of diagnosing a subject with prostate cancer comprising: (a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig. 4b; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
Description
LIQUID-BIOPSY SIGNATURES FOR PROSTATE CANCER
FIELD OF THE INVENTION
This application relates to methods, compositions and systems for the diagnosis or classification of prostate cancer.
BACKGROUND
The worldwide incidence of prostate cancer has been steadily increasing, but many patients harbor tumors of an indolent nature. These indolent tumors grow slowly and pose minimal threat to the life of the patient, in the absence of treatment (i.e. are clinically insignificant). However, once prostate cancer begins to grow aggressively, it metastasizes quickly with lethal consequences. The management of prostate cancer has become an urgent clinical dilemma with significant over-diagnosis and challenges in predicting patient survival Prostate cancers are uniquely heterogeneous with major spatial ¾ 3 and temporal 4 variability in their genomes. Therefore, once cancer has been confirmed, the optimal course of action is tailored to spare patients with indolent disease from unnecessary procedures, while identifying and treating those who would benefit from treatment intensification. Current clinical stratification employs the Gleason Score (OS), estimates of tumour size and pre-treatoent PSA levels. However, generating the OS requires obtaining biopsy specimens, a procedure that increases the risk of hospitalizations from post-biopsy complications, posing a significant burden on health care and risking serious complications
 In addition, biopsies under-sample the prostate, and significant lesions are frequently missed
In addition, biopsies under-sample the prostate, and significant lesions are frequently missed
 Localized prostate cancer has excellent prognosis, with almost 100% 5-year survival (http-J/seer.cancer.gov/stat^ but nearly 70% of these men receive early intervention in the form of surgery or radiotherapy
Localized prostate cancer has excellent prognosis, with almost 100% 5-year survival (http-J/seer.cancer.gov/stat^ but nearly 70% of these men receive early intervention in the form of surgery or radiotherapy
 These therapies carry significant morbidities and healthcare costs, and surveillance protocols rely on repeat prostate specific antigen (PSA) testing, digital rectal examination (DRE) and multiple biopsies 13' A major gap that currently exists in prostate cancer diagnostics is that no accurate biomarkers are available mat could overcome these potential complications (i.e. invasive, heterogeneity). A fluid-based biomarker would be ideal.
Liquid biopsies, such as circulating tumor cells and cell-free DNA 16 have been proposed as promising non-invasive prostate cancer biomarkers, but their detection and enrichment remains technically challenging. Cataloguing the secreted and soluble factors released into the interstitial fluid that bathes the organ of interest may provide a novel inventory of putative disease biomarkers. We have interrogated the collection of proteins comprising a prostate-proximal fluid, expressed prostatic secretions (EPS) 11 ' 18, 19, that is collected either directly from the prostate prior to radical prostatectomy (termed: direct-EPS), or from post-digital rectal examination urine (termed: post-DRE urine or EPS-urine). Reproducible detection and Quantificatioii of multiple proteins in complex biological matrices is an essential requirement for any potential disease biomarker, but verification of these candidates is a major bottleneck in the pipeline from discovery to clinical implementation. Traditionally, immunoaffmily based assays, namely enzyme-linked immunosorbent assays (ELISAs) are used to validate protein biomarkers, but this approach is time-coiisuming, costly, and relies on the existence of validated antibody pairs for every target protein. Targeted mass spectrometry (MS) offers an alternative approach for the rapid verification of candidate biomarkers. Selected reaction momtcrirtg mass spectrometry (SRM-MS) is currently the leading method for targeted quantification of proteins via MS. It offers excellent selectivity, sensitivity, and throughput, without the need for validated antibodies.
These therapies carry significant morbidities and healthcare costs, and surveillance protocols rely on repeat prostate specific antigen (PSA) testing, digital rectal examination (DRE) and multiple biopsies 13' A major gap that currently exists in prostate cancer diagnostics is that no accurate biomarkers are available mat could overcome these potential complications (i.e. invasive, heterogeneity). A fluid-based biomarker would be ideal.
Liquid biopsies, such as circulating tumor cells and cell-free DNA 16 have been proposed as promising non-invasive prostate cancer biomarkers, but their detection and enrichment remains technically challenging. Cataloguing the secreted and soluble factors released into the interstitial fluid that bathes the organ of interest may provide a novel inventory of putative disease biomarkers. We have interrogated the collection of proteins comprising a prostate-proximal fluid, expressed prostatic secretions (EPS) 11 ' 18, 19, that is collected either directly from the prostate prior to radical prostatectomy (termed: direct-EPS), or from post-digital rectal examination urine (termed: post-DRE urine or EPS-urine). Reproducible detection and Quantificatioii of multiple proteins in complex biological matrices is an essential requirement for any potential disease biomarker, but verification of these candidates is a major bottleneck in the pipeline from discovery to clinical implementation. Traditionally, immunoaffmily based assays, namely enzyme-linked immunosorbent assays (ELISAs) are used to validate protein biomarkers, but this approach is time-coiisuming, costly, and relies on the existence of validated antibody pairs for every target protein. Targeted mass spectrometry (MS) offers an alternative approach for the rapid verification of candidate biomarkers. Selected reaction momtcrirtg mass spectrometry (SRM-MS) is currently the leading method for targeted quantification of proteins via MS. It offers excellent selectivity, sensitivity, and throughput, without the need for validated antibodies.
SUMMARY
In an aspect, there is provided a method of diagnosing a subject with prostate cancer comprising: (a) deteennining an expression level of at least 1 gene in a teat sample from the subject selected from the group consisting of the genes identified in Fig.4b; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
In an aspect, there is provided a method of classifying a subject with prostate cancer between having a pT2 stage (organ confined) tumor and having a pT3 stage (extracapsular) tumor, the method comprising: (a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig. 4d; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of subjects with pT2 stage (organ confined) tumors and/or a pT3 stage (extracapsular) tumors; wherein a statistically significant difference or similarity in the expression of the at least 1 gene in the test sample compared to the corresponding reference expression level is an indication that the subject correspondingly has a pT2 stage (organ confined) tumor or a pT3 stage (extracapsular) tumor.
In an aspect, there is provided a composition comprising a plurality of antibodies capable of specifically binding to a plurality of peptides corresponding to a plurality of the genes in Fig. 4b and Fig. 4d. Preferably, the plurality of peptides are a plurality of the peptides identified in Fig. 4b and Fig. 4d. In some embodiments, the plurality of peptides are the 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig.4d.
In an aspect, there is provided a computer program product for use in conjunction with a computer having a processor and a memory connected to the processor, the computer program product comprising a computer readable storage medium having a computer mechanism encoded thereon, wherein the computer program mechanism may be loaded into the memory of the computer and cause the computer to carry out the method described herein.
In an aspect, there is provided a computer implemented product for diagnosing or classifying a subject with prostate cancer comprising: (a) a means for receiving values corresponding to a subject expression profile in a subject sample; (b) a database comprising a reference expression profile representing a control, wherein the subject expression profile and the reference profile each have at least one value representing the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4&, wherein the computer implemented product compares the reference expression profile to the subject biomarker expression profile, wherein a statistically significant difference or similarity in the expression profiles is used to
diagnose or classify the subject with prostate cancer. In some embodiments, the computer implemented product carries out the method described herein.
In an aspect, there is provided a computer readable medium having stored thereon a data structure for storing the computer implemented product described herein.
In an aspect, there is provided a computer system comprising (a) a database including records comprising a reference expression profile of the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a user interface capable of receiving a selection of expression levels of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d, for use in comparing to the reference expression profile in the database; (c) an output that displays a prediction of diagnosis or classification wherein a statistically significant difference or similarity in the expression levels is used to diagnose or classify the subject with prostate cancer.
In an aspect, there is provided a kit comprising reagents for detecting the level of at least 1 gene identified in Fig, 4b and Fig. 4d, preferably at least 1 peptide in Fig.4b and Fig.4d, in a sample.
DRAWINGS
These and other features of the preferred embodiments of the invention will become more apparent in the following detailed description in which reference is made to the appended drawings and tables, which form a part of the specification.
Figure 1 shows systematic development of targeted proteomics assays in EPS-urines, (a) Discovery proteomics data from direct-EPS derived from patients with extracapsular (EC) or organ-confined (OC) prostatic tumors was used to select putative candidates. Proteotypic peptides from these candidates were carefully selected and evaluated by SRM-MS in an EPS- urine background, (b) All peptides that passed the above selection criteria were analyzed in clinically stratified EPS-urines (Cohort A). Peptide quantification by SRM-MS was performed and 34 candidates with diagnostic and prognostic potential were identified based on relative
abundance changes, (c) Venn diagram depicting the distribution of peptides with diagnostic potential {i.e. differential expression in cancer vs. controls) and the number of peptides with prognostic potential (ie. differential expression in EC VJ. OC tumors), (d) Bar charts depict directional expression of the 34 peptide candidates; diagnostic - left panel, prognostic - right panel. (SpC = spectral counts; PTP - proteotypic peptides)
Figure 2 shows absolute peptide quantification in an independent patient cohort (a) All 34 peptide candidates were accurately quantified in an independent cohort of EPS-urines (Cohort B). (b) Peptide abundance correlation between both replicates analyzed by SRM-MS. (c) Correlation of peptide expression for all 34 peptides in EPS-urines from Cohort A and B (prognostic comparison shown). Left panel: directionality of peptide expression; Right panel: correlation plot (d) Absolute peptide quantification in all EPS-urines from Cohort B. Box plots represent the median and interquartile range. Whiskers represent the 1-99 percentile. Outliers are represented by red dots and the mean is represented by '+'.
Figure 3 shows univariate analyses to distinguish patient risk groups, (a) Heafmap representation of absolute peptide expression levels for all candidate peptides within Cohort B samples (represented as fmol EPS-urine protein), Peptide expression heatmap is clustered using
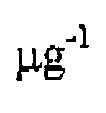
consensus clustering. Pearson's correlation was used as the similarity metric to generate clusters and k-means method was used as a clustering algorithm. Serum PSA (SPSA) levels,

ethnicity and patient risk group status are shown. On the right-hand side peptide expression levels for "cancer vs. normal" and "
 " prostate cancers are represented as box plots (shown as logz ratios of endogenous divided by spike-in standard "H" peak ratios), (b)
" prostate cancers are represented as box plots (shown as logz ratios of endogenous divided by spike-in standard "H" peak ratios), (b)

Quantification of individual peptides in normal versus all cancer patient EPS-urines. Peptides passing indicated statistical cut-off criteria are color-coded in red. Peptide sequences and gene names are indicated, (c) The area under the ROC curve (AUC) was used to evaluate the ability of individual peptides to distinguish between cancer patients and normal controls. SPSA values, available for these patients, were used as a positive control (indicated as blue bar), (d/e) Same analyses performed for prostate cancer risk groups (pT3 vs. pT2).
Figure 4 shows maohinc-lcaming model to identify biomarker signatures, (a) Schematic overview of the macbinc-lcaining approach used to develop multi-feature biomarker signatures, (b) The predictive importance of individual peptides to distinguish prostate cancer from normal controls. Pink bars represent the selected relevant peptides to build the predictor. Blue bar represents the predictive importance of serum PSA (SPSA). (c) ROC curves for diagnosis. The performance for the selected peptide signature (pink), serum PSA alone (blue) and randomly selected peptides (grey) are compared. ROC curves are generated from 10-fold cross validation. ROC curves generated from test set are in Fig. 11. (d) The predictive importance of individual peptides to distinguish pathological stage pT3 from stage pT2. (e) ROC curve analyses for prognosis.
Figure 5 shows Pearson correlation for replicate analyses (all sample types analyzed in duplicate), Representation of the reproducibility (Pearson's correlation) for replicate analyses of all peptides analyzed in Cohort B (n-207). Dotted red line represents samples with high correlation (RX),7).
Figure 6 shows Pearson correlation for replicate analyses (individual sample types; risk groups). Representation of the reproducibility (Pearson's correlation) for replicate analyses of all peptides analyzed in Cohort B stratified by patient risk group (normal, BPH, pT2, pT3). Dolled red line represents samples with high correlation (RX).7).
Figure 7 shows Pearson correlation compared by peptide abundance. Comparison of peptide abundance plotted as a function of Pearson's correlation. Peptides with a high concordance between replicate analyses (RXJ.7) are significantly mote abundant based on SRM quantification.
Figure 8 shows chromatographic retention time of individual peptides, (a) Representative chromatogram of the 34 peptides quantified in cohort B. (b) The 34 peptides quantified by SRM- MS in all cohort B samples (207 samples analyzed in duplicates; n-414 SRM-MS analyses) demonstrate highly reproducible retention times.
Figure 9 shows peptide abundance in the two separate patient cohorts, (a) Average fold change correlation of cancer vs. normal samples for the 34 peptides quantified in cohorts A and B, left
side: avenge fold change (log2) in both cohorts-, rights side: correlation blot (b) Average fold change correlation of pT2 vs. pT3 samples for the 34 peptides quantified in cohorts A and B. left side: average fold change (l°g2) in both cohorts; rights side: correlation blot.
Figure 10 shows patient characteristics for all cohort B samples (n-207). (a) Age distribution; (b) Ethnicity; (c) Serum PSA distribution (SPSA).
Figure 11 shows ROC curves for test set analysis, (a) Diagnostic signature: the performance for the selected peptide signature (pink), serum PSA alone (blue) and randomly selected peptides (grey) are compared, (b) Prognostic signature: the performance for the selected peptide signature (pink), serum PSA alone (blue) and randomly selected peptides (grey) are compered. ROC curves axe generated from test set
Figure 12 shows area under the ROC for randomly generated signatures. Distribution of AUCs for randomly selected peptides to generate predictive models for cancer vs. normal (top panel) or for pT3 vs. pT2 (bottom panel). Pink line indicates AUC for our predictive models based on identified peptides. AUCs are measured from test set.
Figure 13 shows power analyses for all analyzed samples to distinguish indicated patient risk groups.
Figure 14 shows inter-correlation between peptides, (a) Correlation matrix of peptide-peptide expression. Expression profiles between all 34 peptides quantified in cohort B are compared using Pearson's correlation coefficient (R). Peptide-peptide matrix comparing the quantitative expression profiles of (b) the 6 diagnostic signature peptides and (c) the 7 prognostic signature peptides.
Figure 15 shows suitable configured computer device, and associated communications networks, devices, software and firmware to provide a platform for enabling one or more embodiments as described herein.
DESCRIPTION
Biomarkers are rapidly gaining importance in personalized medicine. Although numerous molecular signatures have been developed over the past decade, mere is a lack of overlap and many biomarkers fail to validate in independent patient cohorts and hence are not useful for clinical application. For these reasons, identification of novel and robust biomarkers remains a formidable challenge. We combine targeted pxoteomics with computational biology to discover robust proteomic signatures for prostate cancer. Quantitative pxoteomics conducted in expressed prostatic secretions from men with extrapro static and organ-coniined prostate cancers identified 133 differentially expressed proteins. Using synthetic peptides, we evaluate them by targeted proteomics in a 74-patient cohort of expressed prostatic secretions in urine. We quantify a panel of 34 candidates in an independent 207-oatient cohort We apply machine-learning approaches to develop clinical predictive models for prostate cancer diagnosis and prognosis. Our results demonstrate that computationally guided proteomics can discover highly accurate non-invasive biomarkers.
In an aspect, there is provided a method of diagnosing a subject with prostate cancer comprising: (a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig. 4b; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
The term "classifying" as used herein means predicting or identifying the particular state of a disease. For example, with respect to prostate cancer, patients may be classified as having a pT2 stage (organ confined) tumor or a pT3 stage (extracapsular) tumor.
As used herein "diagnosis" is tine identification of the nature and cause of a certain phenomenon, such as, the identification of disease state in a patient For example, the methods described herein are useful for determining whether a subject has prostate cancer.
The term "subject" as used herein refers to any member of the animal kingdom, preferably a human being and most preferably a human being that has prostate cancer or that is suspected of having prostate cancer.
The term "test sample" as used herein refers to any fluid, cell or tissue sample from a subject which can be assayed for biomarker expression products and/or a reference expression profile, e.g. peptides differentially present in a liquid biopsy.
The phrase "dctcrrnimng the expression" as used herein refers to determining or quantifying RNA or proteins or protein activities or protein-related metabolites expressed by the biomarkers. The term "RNA" includes mRNA transcripts, and/or specific spliced or other alternative variants of mRNA, including anti-sense products. In the case of "protein" or "peptides", it refers to proteins expressed by genes are measurable in a sample.
The term "level of expression" or "expression level" as used herein refers to a measurable level of expression of the products of biomarlcers, such as, without limitation, micro-RNA, or messenger RNA transcript expressed or of a specific exon or other portion of a transcript, the level of proteins, peptides or portions thereof expressed of the biomarkers, the number or presence of DNA polymorphisms of the biomarkers, the enzymatic or other activities of the biomarkers, and the level of specific metabolites.
As used herein, the term "control" refers to a specific value or dataset that can be used to prognose or classify the value e.g, expression level or reference expression profile obtained from the test sample associated with an outcome class. In one embodiment, a dataset may be obtained from samples from a group of subjects known to have prostate cancer having different tumor states or healthy individuals. The expression data of the biomarkers in the dataset can be used to create a control value that is used in testing samples from new patients.
The term "differentially expressed'' or "differential expression" as used herein refers to a difference in the level of expression of the biomarkers that can be assayed by measuring the level of expression of the products of the biomarkers, such as the difference in level of peptide or protein expressed. In a preferred embodiment, the difference is statistically significant The term "difference in the level of expression" refers to an increase or decrease in the measurable
expression level of a given biomarker, for example as measured by me amount of peptide as compared with the measurable expression level of a given peptide in a control.
The tarn "expression profile" as used herein refers to a dataset representing the expression level(s) of one or more biomarkers. An expression profile may represent one subject, or alternatively a consolidated dataset of a cohort of subjects, for example to establish a reference expression profile as a control.
In some embodiments, the at least 1 gene is at least 2, 3, 4, 5, or 6 genes associated with the top- ranked peptides in Fig.4b. Preferably, the at least 1 gene comprises 6 genes associated with the 6 top-ranked peptides in Fig.4b.
In an aspect, there is provided a method of classifying a subject with prostate cancer between having a pT2 stage (organ confined) tumor and having a pT3 stage (extracapsular) tumor, the method comprising: (a) detemnning an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig. 4d; and (b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of subjects with pT2 stage (organ confined) tumors and/or a pT3 stage (extracapsular) tumors; wherein a statistically significant difference or similarity in the expression of the at least 1 gene in the test sample compared to the corresponding reference expression level is an indication that the subject correspondingly has a pT2 stage (organ confined) tumor or a pT3 stage (extracapsular) tumor.
In some embodiments, the at least 1 gene is at least 2, 3, 4, 5, 6, or 7 genes associated with the top-ranked peptides in Fig. 4d. Preferably the at least 1 gene comprises 7 genes associated with the 7 top-ranked peptides in Fig.4d.
In some embodiments, the method further comprises producing gene expression profiles comprising a subject gene expression profile and a gene reference expression profile, each having values representing the expression level of the at least 1 gene corresponding the test and control samples respectively.
In some embodiments, the test sample comprises at least one of prostate-proximal fluid and/or expressed prostatic secretions.
In some embodiments, the test sample is collected directly from the prostate, preferably prior to radical prostatectomy, and/or from urine, preferably post-digital rectal examination urine.
In some embodiments, determining the expression level of the at least one gene in the test sample comprises measuring in the test sample, the level of at least one peptide corresponding to the protein product of the at least one genc.
In some embodiments, the method further comprises producing peptide presence profiles comprising t subject peptide expression profile and a reference peptide expression profile, each having values representing the peptide levels of the at least 1 peptide corresponding the test and control samples respectively. Preferably, the at least one peptide comprises 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig. 4d.
In some embodiments, the level of the at least 1 peptide is measured using mass spectrometry, Preferably, the mass spectrometry is targeted mass spectrometry using selected reaction monitoring mass spectrometry.
In an aspect, mere is provided a composition comprising a plurality of antibodies capable of specifically binding to a plurality of peptides corresponding to a plurality of the genes in Fig. 4b and Fig. 4d, Preferably, the plurality of peptides are a plurality of the peptides identified in Fig. 4b and Fig. 4d. In some embodiments, the plurality of peptides are the 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig.4d
The present system and method may be practiced in various embodiments. A suitably configured computer device, and associated communications networks, devices, software and firmware may provide a platform for enabling one or more embodiments as described above. By way of example, Figure IS shows a generic computer device 100 that may include a central processing unit ("CPU") 102 connected to a storage unit 104 and to a random access memory 106. The CPU 102 may process an operating system 101, application program 103, and data 123. The operating system 101, application program 103, and data 123 may be stored in storage unit 104 and loaded
into memory 106, as may be required. Computer device 100 may further include a graphics processing unit (GPU) 122 which is operatively connected to CPU 102 and to memory 106 to offload intensive image processing calculations from CPU 102 and run these calculations in parallel with CPU 102. An operator 107 may interact with the computer device 100 using a video display 108 connected by a video interface 105, and various input/output devices such as a keyboard 1 IS, mouse 112, and disk drive or solid state drive 114 connected by an I/O interface 109. In known manner, the mouse 112 may be configured to control movement of a cursor in the video display 108, and to operate various graphical user interface (GUI) controls appearing in the video display 108 with a mouse button. The disk drive or solid state drive 114 may be configured to accept computer readable media 116. The computer device 100 may form part of a network via a network interface 111, allowing the computer device 100 to communicate >with other suitably configured data processing systems (not shown). One or more different types of sensors 135 may be used to receive input from various sources.
The present system and method may be practiced on virtually any manner of computer device including a desktop computer, laptop computer, tablet computer or wireless handheld. The present system and method may also be implemented as a cor^uter-raadableAiseable medium that includes computer program code to enable one or more computer devices to implement each of the various process steps in a method in accordance with the present invention. In case of more than computer devices rjerrorrning the entire operation, the computer devices are networked to distribute the various steps of the operation. It is understood that the terms computer-readable medium or computer useable medium comprises one or more of any type of physical embodiment of the program code. In particular, me ccniputer-readable/useable medium can comprise program code embodied on one or more portable storage articles of manufacture (e.g. an optical disc, a magnetic disk, a tape, etc.), on one or more data storage portioned of a computing device, such as memory associated with a computer and/or a storage system.
In an aspect, there is provided a computer program product for use in conjunction with a computer having a processor and a memory connected to the processor, the computer program product comprising a computer readable storage medium having a computer mechanism encoded thereon, wherein roe computer program mechanism may be loaded into the memory of the computer and cause the computer to carry out the method described herein.
In an aspect, there is provided a computer implemented product for diagnosing or classifying a subject with prostate cancer comprising: (a) a means for receiving values corresponding to a subject expression profile in a subject sample; (b) a database comprising a reference expression profile representing a control, wherein the subject expression profile and the reference profile each have at least one value representing the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4cL, wherein the computer implemented product compares the reference expression profile to the subject biomarker expression profile, wherein a statistically significant difference or similarity in the expression profiles is used to diagnose or classify the subject with prostate cancer. In some embodiments, the computer implemented product carries out the method described herein.
In an aspect, there is provided a computer readable medium having stored thereon a data structure for storing the computer implemented product described herein.
In some embodiments, the data structure is capable of configuring a computer to respond to queries based on records belonging to the data structure, each of the records comprising: (a) a value that identifies a reference expression profile of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a value that identifies the probability of a diagnosis or classification associated with the reference expression profile.
In an aspect, there is provided a computer system comprising (a) a database including records comprising a reference expression profile of the level of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d; (b) a user interface capable of receiving a selection of expression levels of at least 1 gene identified in Fig. 4b and Fig. 4d, preferably at least 1 peptide in Fig. 4b and Fig. 4d, for use m comparing to the reference expression profile in the database; (c) an output that displays a prediction of diagnosis or classification wherein a statistically significant difference or similarity in the expression levels is used to diagnose or classify the subject with prostate cancer.
In an aspect, there is provided a kit comprising reagents for detecting the level of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig.4b and Fig.4d, in a sample.
We build on our proteomics studies of EPS and systematically develop SRM-MS assays in post- DRE urines. Step-wise applications of these assays to two independent, richly annotated patient cohorts in conjunction with computational modeling identify liquid biopsy signatures that accurately distinguish patients with organ-confined stage pT2 and extracapsular stage pT3 prostate cancers, prior to radical prostatectomy. These data highlight the value of readily accessible tissue proximal fluids and multiplexed quantitative proteomic signatures to identify extracapsular disease prior to invasive surgery, potentially nxxlifying treatment options for these patients.
The advantages of the present invention are further illustrated by the following example, The example and its particular details set forth herein are presented for illustration only and should not be construed as a limitation on the claims of the present invention.
EXAMPLES
METHODS/MATERIALS Sample collection and annotation
Samples were obtained from patients following informed consent and use of Institutional Review Board approved protocols at Urology of Virginia and Eastern Virginia Medical School (# 06-12- FB-0343) and the Research Ethics Review Board at the University Health Network (lO-0159-Τ). Twenty ml of EPS-urine was centrifuged at 1100 g for 15 minutes at 4 °C to pellet debris. The resulting EPS-urinfi supernatant was aliquoted in volumes of 3.5 ml and diluted with 2,5 ml of PBS (pH 7.4). The mixture was vortexed, combined, and centrifuged at 1100 g for 15 minutes. The resulting supematants were stored at -40 °C. Detailed clinical information for all patients enrolled in mis study are available but not shown. Prostate cancer patients were selected on the basis of organ confinement or pathological stage. Non-cancer individuals had biopsy confirmed BPH or were considered as individuals with no indication of prostatic disease based on biopsy results.
Sample preparation for mass spectrometry
Ultrapurc-grade 2,2,2-trifluoroethanoI (TFB), trifluoroacetic add (TFA), iodoacetamide (IAA), and dithiotreitol (DTT) were from Sigma-AldricL HPLC-grade solvents (methanol, acctonitrilc, and water) and formic acid were fiom Fisher Scientific. Mass spcctrometry-grade trypshVLys-C was fiom Promega (Madison, WI). Amicon spin filters, O.S ml, 3 kDa MWCO, were fiom Millipore. Solid phase extraction C18 tips were fiom Agilent Four ml of EPS-urine were concentrated to approximately 500 μΐ by using a spin filter with a molecular weight cutoff of 3 kDa, and proteins were precipitated overnight by the addition of ice-cold 100% methanol. Protein pellets were washed twice with 100% methanol and air-dried. Protein resolubilization was performed by the addition of 50% TFE at 60 °C for 2 hours. Following reduction with DTT and alkylation with IAA, proteins were digested overnight at 37 °C using mass 2 μg trypsin/Lys- C. The reaction was quenched by the addition of TFA. Desalting was performed by solid phase extraction using CI 8 tips. Solvents were removed by vacuum centrifugation and peptides were resolubilized in 5% acetonitrile, 0.1 % formic acid. Peptide concentrations were determined by the micro BCA assay kit (Thermo Fisher Scientific).
Selected reaction monitoring mass spectrometry
Samples were analyzed on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific) equipped with an EASY-Spray (Thermo Fisher Scientific) electrospray ion source, Separations were performed on EASY-Spray columns (15 cm x 75 um ID packed with 3μπι CIS particles, Thermo Fisher Scientific) heated to 50 °C- Peptides were kept at 4° C and loaded onto the column fiom an EASY-nLC (Thermo Fisher Scientific) autosampler. Chromatographic conditions were as follows: 40 minute gradient at a flow rate of 300 nl min*1 starting with 100% A (water), stepping up to 5% B (ACN) in 5 minutes, followed by 25% B at 35 minutes, followed by a steep increase to 50% B at 38 minutes and 100% B at 40 minutes. Targeted acquisition of eluting ions was performed by the mass spectrometer operated in SRM-MS mode with Ql and Q3 set to 0.7 m/z fwhm resolution and a cycle time of 1 second. For all SRM-MS runs, with the exception of the measurement of Phase 3 peptides fiom Cohort B, multiple unscheduled injections were used, each targeting approximately 200 transitions. For Cohort B, a single scheduled method was utilized with a 2-minute elution window.
Selection of Phase 2 peptides
In a previous study , 133 differentially expressed proteins were identified when comparing the proteomc profiles of 16 direct-EPS samples from individuals with EC and OC prostatic tumors. A spectral library was built from the resulting LTQ-Orbitrap XL data using the Skyline software tool (version 2.1.0) 41. All spectra had scores that passed a stringent peptide score as determined by an X! Tandem target decoy search (~ 0.5% FDR). Protein sequences were converted to FASTA format and uploaded into Skyline for the prediction of proteotypic peptides. Peptides were chosen based on previously reported specifications
 A total of 232 proteotypic peptides {Phase 1 peptides) were selected and purchased as bulk heavy-isotope labeled peptide standards (JPT Peptide Technologies), also containing 8 peptides that were deemed potentially interesting from our additional EPS proteomics studies
A total of 232 proteotypic peptides {Phase 1 peptides) were selected and purchased as bulk heavy-isotope labeled peptide standards (JPT Peptide Technologies), also containing 8 peptides that were deemed potentially interesting from our additional EPS proteomics studies
 In order to assess the suitability of the Phase 1 peptides for SRM-MS, 250 finol of each heavy peptide standard was spiked into 1 ug of EPS- urine-digest, with 4-6 transitions monitored over a 40-mmute chromatographic gradient Of the 232 Phase J peptides, 147 (Phase 2 peptides) were reproducibly detectable with a minimum of three transitions in the complex EPS-urine background.
In order to assess the suitability of the Phase 1 peptides for SRM-MS, 250 finol of each heavy peptide standard was spiked into 1 ug of EPS- urine-digest, with 4-6 transitions monitored over a 40-mmute chromatographic gradient Of the 232 Phase J peptides, 147 (Phase 2 peptides) were reproducibly detectable with a minimum of three transitions in the complex EPS-urine background.
Quantification of Phase 2 and selection at Phase 3 peptides
A cohort of individual EPS-urines (n - 74) from a heterogeneous population of patients with EC, OC and control (BPH, normal) (Cohort A) was used to analyze all Phase 2 peptides. A total of 1 μg of peptide from each sample was spiked with 200 fmol of heavy peptide standards that were combined into six batches (batch A-F), consisting of -20 peptides per batch. Visualization and inspection of peaks was performed in Skyline. Each peptide was quantified in a sample by integrating the quantifier ion (most intense ion) of the light peptide with its co-eluting heavy peptide ion, in order to derive a light-to-heavy peptide ratio. The Student's t-test was used to compare the ratios between cancer and controls, as well as EC and OC prostate cancers. A K- fold cross-validation was performed to investigate the diagnostic and prognostic power of the peptides at different p-vahie cutoffs, For the diagnostic and prognostic peptide candidates, p- value cut-offs of 0.05 and 0.1 were used, respectively. Further refinements to this list were made by including additional peptides that did not meet the p-value cut-offs, but were potentially promising. For instance, peptides SSEDPNEDIVER from protein IGJ and TPAQFDADELR
from protein ANXA1 were added to the list of putative prognostic candidates because they were the only candidates that were elevated in the EC tumor group (p-value - 0,25), Two KLK3 peptides (HSQPWQVLVASR and LSEPAELTDAVK) were also added in order to monitor PSA levels in EPS-urine. A total of 34 peptides, comprising the Phase 3 candidates (24 diagnostic and 14 prognostic peptides, of which 4 overlapped), were taken forward for verification.
Multiplexed selected reaction monitoring mass spectrometry
In order to increase throughput, a multiplexed SRM-MS assay was developed by scheduling all 34 candidates in a single 40-mimrte chromatographic gradient A total of 3 transitions were monitored for the light and heavy versions of each peptide for a total of 204 transitions per analysis. A 2-mimitc acquisition time window was scheduled around the expected peptide elutiontime.
Verification of Phase 3 peptides in EPS urines
For verification, a heterogeneous population of patients with pathological stage pT3 and pT2 prostate tumors, BPH, and normal individuals (n " 207) were enrolled (Cohort B). One ug of total peptide from each sample was spiked with 100 rmol of heavy peptide and 10 frnol of corresponding light peptide for all candidates with the exception of the KLK3 peptide, HSQPWQVLVASR, which was spiked in at 500 frnol of heavy peptide. Visualization and inspection of peaks was performed on Skyline. Each sample was analyzed in two technical replicates using the same instrument parameters as described above.
Quantitative and statistical analyses
Each of the light and heavy peptides were checked for quality of data by observing co-elution of all three transitions, alignment of light and heavy peptide elution times, and reproducibility between technical replicates. Relative ratios of the area under the curve of the most predorninant ion (quantifier) of the light peptide versus the corresponding heavy quantifier were calculated. To evaluate the reproducibility of proteomics data, we compared peptide abundance in two replicates using Pearson's correlation coefficient (R). On average, peptides show the R of 0.97 across all samples. Of quantified peptides, 30 (88.24%), 28 (82.35%), 31 (91.18%), and 31
(91.18%) peptides show strong reproducibility (R > 0.7) in normal controls, BPH, pT2, and pT3 samples, respectively (Fig. 6). Peptides were comparatively tested for abundance differences between cancer vs. normal and pathological stage pT3 vs. pathological stage pT2. All statistical analyses were performed in R environment (v3-2.1).
Generating predictors for prostate cancer
To generate a predictor that distinguishes cancer patients from control individuals (diagnosis), peptide expressions of 90 cancer patients (positive set) and 48 normal controls (negative set) were used. For the prediction of EC and OC cancers (prognosis), 29 pT3 samples (positive set) and 61 pT2 samples (negative set) were used (Table 3). All datasets (positive and negative sets) were divided into two groups to build predictive models: a training set (90% of data set) and a testing set (10% of data set). When 90% of the dataset was used as a training set, the predictive model was able to capture the properties of context classification associated with patient risk groups and, thus, showed the maximirm performance on the testing set compared to other sized training sets (60% of data set and 80% of data set) (Table 4). To examine the reliability of sample size for the training, power analyses were performed (Fig. 13). Power analyses allow for the determination of the appropriate sample size for statistical analysis. In general, there is a large difference effect between the two groups when effect size is bigger than 0.S at a power of 0.8 43. In our study, at a power of 0.8, the effect sizes of the training sets are 0.79 (diagnosis, cancer vs. normal control, p = 0.001) and 1.01 (prognosis, pT3 vs. pT2, p ·» 0.0Q1), Power analysis was performed using 'pwr' package (1.1-3) in R (version 3.2.1).
Additional Optimizations - Targeted quantification via Parallel Reaction Monitoring Mass Spectrometry (PRM-MS)
A number of optimizations are contemplated in respect to our recently published assays (Kim et al. Nature Cornmumcations; 2016). In mis manuscript we used nano-flow liquid chromatography for peptide separation. Eluting peptides were quantified by a multiplexed Selected Reaction Monitoring Mass Spectrometry (SRM-MS) assay using a Thermo TSQ Vantage mass spectrometer. Recent advances in LC-MS technologies, both at the level of separation and detection are taken into account for mis patent application. For example, we will use long
column (50cm) nano-flow ultra-performance liquid chromatography (nUPLC) for peptide separation. La our hand this technology provides more robust separation (i.e. minimal changes in peptide elution times), improved peptide separation and better signal-to-noise ratios. The advances are mainly due to the longer columns and the significantly higher pressure (~1000psi vs. ISOpsi), compared to the older set-up, Improved separation and sharper peaks will reduce the possibility for interference from co-cluting peaks, hence improving the assay specificity. Further, recent technical advances in mass analyzers, especially the introduction of hybrid Orbitrap mass analyzers such as the Q Executive HF, now enable targeted quantification using a quasi SRM approach (termed PRM-MS). While the concept for these assays is identical, there are several advantages. This Included acquisition of a full tandem mass spectrum in the Orbitrap mass analyzer following peak selection in the quadrupole. This improves the simplicity of assay development, since fragment ions for quantification are selected post-acquisition, as compared to monitoring of defined transitions by the classic SRM-MS assays. La addition, the fragment ions are recorded in the high-resolution, accurate mass (HR/AM) Orbitrap analyzer, providing an additional means for reducing mass interference, hence improving selectivity of the assay.
Additional Optimisations - Sample processing
It is further contemplated that one can evaluate the methods for a possibly more rapid, automatable way of processing post-DRE urines that could also reduce required sample amounts (--0,2 ml from the currently used 4ml): MStem approach: A recently published approach termed MStern (Berger et al. Mol Cell Proteomics. 2015 Oct;14(10):2814-23) that is based on the high protein binding capabilities of porous PVDF membranes (similar to a Western Blot). This approach can be performed in a 96-well format and is rapidly automatable.
Machine learning and feature selection
The Generalized Linear Model (GLM) was used to classify samples into two classes: cancer vs. normal (diagnosis) and pT3 vs. pT2 (prognosis). OLM is a widely used machine-learning algorithm that has been applied in various types of biomarker identification («.£. cancer, HTV/AlDS and infection diseases) with reliable performance 45' 46 , In addition, we demonstrate that OLM outperformed eight other machine learning algorithms, which look for different types
of patterns and data properties; random forest (rf), stochastic gradient boosting (gbm), Naive Bayes (nb), boosted generalized linear model (glmboost), lasso and elastic-net-regularized generalized linear model (ghnnet), support vector machine with linear kernel (svrnLinear) and radial basis function (RBF) kernel (svmRadial). We generated predictive models using all peptides, tested them by 100 bootstrap samples and measured AUC, accuracy, sensitivity and specificity. As shown in Table 5, OLM shows relatively higher performance in all performance metrics.
After collecting data for all 34 peptides, we identified the best peptide biomarkers to build predictive models. To do this, firstly, all peptide expressions (abundances) were normalized against peptide expressions of normal controls:

Z is the normalized peptide expression. X is the peptide expression in each sample, μ is the mean of the normal controls, σ is the standard deviation of the normal controls. We then examined inter-correlation between peptides. To do mis, we generated a feature-feature matrix by comparing expression profiles between peptides (Fig. 14a). There is low expression similarity among all tested peptides (median Pearson's R is 0.15) suggesting that there is no inter- dependency between tested peptides and they have their own prediction importance. Next, we calculated the importance of each peptide to discriminate two classes (e.g. cancer and normal controls) and subsequently selected the top-ranked peptides (from top 3 to top IS), We used these top-ranked peptides as features to build predictive models. To avoid overfitting, 10-fold cross-validation was conducted on a training set We then examined their prediction performance on a testing set To obtain stable predictions, predictive models were tested by 100-fold bootstrapping, The resulting average area under the receiver operating characteristic curve (AUQ was calculated and used as a performance measure. Sets of top-ranked peptides that show the highest AUC were chosen as the most relevant features. As a result, we selected 6 peptides for diagnosis and 7 peptides for prognosis as the most relevant features (Table 6). Peptides that were used as predictors also showed low expression similarity. Median Pearson's R is 0.32 (cancer vs. normal controls, Fig. 14b) and 0.18 (pT3 vs. pT2, Fig. 14c).
Data availability
The raw mass spectrometry data associated with this manuscript have been submitted to a public repository (the Mass Spectrometry Interactive Virtual Environment, http://inassive.ucsd.edu) for others to download. These data are associated with the identifier MassIVE ID MSV000079401 at the FTP site fh;:/Anassive.ucsd.edu/MSV000079401. Cohort B SRM traces for the 34 measured peptides are available through Panorama at the following link (¾t^s://panoramaweb.org/labkey/projcct/EPS_SRM/
des%29/begm, view?). Skyline exported data for all quantified peptides of Cohort B was done but not shown. The authors declare that all other data supporting the findings of this study are available within the article and its supplementary information files.
RESULTS
Candidate selection and assay evaluation
Our previously generated discovery proteomic profiles from direct-EPS derived from extracapsular (EC) or organ-confined (OC) prostate cancers laid the foundation for the current project 18. Relative quantification revealed 133 proteins that were significantly differentially expressed between both patient groups. These proteins were identified by 1-346 distinct peptide sequences. Data mining of all peptides based on sequence and biophysical properties led to 232 proteotypio peptides (Phase 1 peptides) suitable for evaluation by SRM-MS in EPS-urines (Fig. la). To abrogate potential confounding influences due to the differing modes of data acquisition (discovery proteomics vs. targeted proteomics), instruments (ion-trap vs. triple quadrupole) and sample types (direct-EPS vs. EPS-urine), all Phase 1 peptides were first evaluated for reproducible detection by SRM-MS in EPS-urine samples. Each Phase 1 peptide was purchased as a crude heavy isotope labeled synthetic peptide and spiked into pooled EPS-urines to evaluate their suitability for targeted proteomics assays, directly within the biomarker matrix. Light (endogenous) and heavy (synthetic) peptides were monitored in SRM mode, and data were manually inspected to select peptides that had at least 3 fragments ions aligned at the expected peptide elution time, had co-eluting light and heavy peptides, had minimal interference and were reproducible. In total, 147 peptides (63%) met these quality criteria (Phase 2 peptides), and were
taken forward to an independent cohort of EPS-urine samples (Fig. la). These results demonstrate that systematic SRM-MS assay development based on previous discovery proteomics data is rapid and feasible in clinically-useful EPS-urines.
Peptide quantification In EPS urine Cohort A
To evaluate Phase 2 peptides, we performed relative quantification in a medium-sized cohort of EPS-urines (n*74; Cohort A; Table 1), using the crude heavy isotope labeled synthetic peptides as internal standards (see Methods). The goal of this initial quantification was to evaluate peptide performance in relevant clinical samples, while reducing the rwmber of peptides to be moved forward to the next development steps. Briefly, a Student's t-test was performed to compare the ' ratios of peptide abundance between cancer and non-cancer groups (termed: diagnostic), as well as EC and OC prostate cancers (termed: prognostic). The first criteria used to select candidates as potential diagnostic and prognostic biomarkers were p- value cutoffs of 0.05 and 0.1, respectively. A higher p- value cut-off was used to select prognostic candidates in order to avoid removing putative candidates at mis early stage for distinguishing cancer comparisons (EC vs. OC cancer groups), Refinements to the candidate list were made by adding peptides representing the proteins IGJ and ANXAJ, because these peptides were the only candidates even trend up- regulated in the EC tumor group (p < 0.25). Furthermore, two KLK3 peptides were added to monitor PSA levels. Finally, each peptide that met the above criteria was manually inspected for SRM-MS trace quality. Overall, 34 candidates {Phase 3 peptides) demonstrated a potential for classifying individuals based on prostatic disease status (Fig. lb). Overall 24 diagnostic candidates (21 over-expressed in cancers) and 14 prognostic ones (2 over-expressed in invasive tumours) were identified (Figs, lc, d). Our data suggest mat SRM-MS assays developed in EPS- urines are capable of separating patients into defined categories using retrospective cohorts, while quickly triaging candidates affected by large patient variability.
Absolute quantification in EPS urine Cohort B
To generate optimized SRM-MS assays for absolute quantification, highly purified f>97% purity) heavy stable isotope labeled standards (AQUA peptides) were purchased for all 34 Phase 3 peptides. Prior to quantification in independent samples (n=207; Cohort B, Table 1), a multiplexed, scheduled SRM-MS method was developed, enabling quantification of all candidate peptides in a single chromatographic gradient (Fig. 2a; see Methods). Next, all Phase 3 peptides were quantified, using single-point quantification based on 100 finol addition of each AQUA peptide to standardized amounts of total EPS-urine (1 μg total protein on column), in an independent cohort of 207 EPS-urines (Table 1; Methods). Each EPS-urine was measured in two technical replicates. The duplicate analyses demonstrated strong reproducibility with a Pearson's correlation coefficients (R) of 0.97 (p < 2,2 x 10-16, Fig.2b). Of the 34 peptides, the majority (30 peptides, 8824%) showed strong correlations between two replicates (R > 0.7, Fig. 5). Furthermore, we examined the reproducibility depending on sample types (Normal, BPH, pT2 and pT3 samples) and found that 31 peptides (91.18%) show strong reproducibility (R > 0.7) in at least one type of sample (Fig. 6). Peptides with R < 0.7 were generally of lower abundance (mean difference of peptide abundance [log. L:H ratio] between the groups is 4.96, p = 0.01, Student's t-test; Fig. 7). The analysis also demonstrated good chromatographic reproducibility (coefficient of variation < 1.5%) with most peptides eluting between 20 and 30 minutes over the 40-minute chromatographic gradient (Fig.8a, b).
To compare the correlation of our quantitative results in both EPS-urine cohorts, fold-change correlation plots were generated for the various group comparisons. While different EPS-urine cohorts, spike-in standards and SRM-MS methods were used, we were able to observe a generally positive correlation. On average, 73 % of our peptides showed concordant expression in both cohorts (Fig.2c and Fig. 9).
Next, the absolute concentration of each peptide across all 207 samples in Cohort B was determined. The 34 peptides quantified spanned approximately 5 orders of magnitude, from the low rmol to nmol range per μg total EPS-urines protein (Fig.2d; 21). As expected, the two most abundant peptides represented KLK3, and in fact the KLK3 peptide HSQPWQVLVASR demonstrated such a strong detector response that it was ultimately removed from further
analyses, as it would requite sample dilution to achieve accurate quantification via AQUA spike- in. The remaining peptides spanned approximately 3 orders of magnitude and significant inter- patient variation in peptide expression was observed (Fig. 3a). These data demonstrate that selected biomarker candidates can be rapidly quantified in large numbers of EPS-urines spanning approximately five orders of concentration.
Univariate analyses to o^urtfnguish patient groups
We investigated the abundance of peptides in different patient risk groups (Normal, BPH, pT2 and pT3). All risk groups had similar age distributions (average age of 60 years) and ethnic compositions (White
 and Asian American - 0.5%; Fig.3a, Fig. 10a and b). As we expected, normal controls had the lowest concentration of serum
and Asian American - 0.5%; Fig.3a, Fig. 10a and b). As we expected, normal controls had the lowest concentration of serum
 and pT3 stage had the highest concentration of SPSA
and pT3 stage had the highest concentration of SPSA
 Student's t-test; Fig. 10c). Next, we evaluated the ability of individual peptides to distinguish among distinct patient groups based on absolute quantification of individual peptides. Most (27/33) are more abundant in cancer patients (middle panel in Fig. 3a) compared to normal controls (1.03 - 4.82 times more expressed, Fig.3b). Among them, ten peptides had, on average, 2.2-fold change in expression (1.4 - 3.8 fold changes) with p < 0.1 (Student's t-test, red dots in Fig.3b). Further, of all quantified peptides, 28 (84.85%) were under- expressed in pT3 stage (right panel in Fig.3a) compared to pT2 stage (1.03 - 2.37 times under- expressed, Fig.3d). Nine peptides were significantly down-regulated in pT3 stage (0.5 - 0.8 fold changes with p < 0.1, Student's t-test; red dots in Fig.3d).
Student's t-test; Fig. 10c). Next, we evaluated the ability of individual peptides to distinguish among distinct patient groups based on absolute quantification of individual peptides. Most (27/33) are more abundant in cancer patients (middle panel in Fig. 3a) compared to normal controls (1.03 - 4.82 times more expressed, Fig.3b). Among them, ten peptides had, on average, 2.2-fold change in expression (1.4 - 3.8 fold changes) with p < 0.1 (Student's t-test, red dots in Fig.3b). Further, of all quantified peptides, 28 (84.85%) were under- expressed in pT3 stage (right panel in Fig.3a) compared to pT2 stage (1.03 - 2.37 times under- expressed, Fig.3d). Nine peptides were significantly down-regulated in pT3 stage (0.5 - 0.8 fold changes with p < 0.1, Student's t-test; red dots in Fig.3d).
To evaluate the power of each peptide to distinguish individual patient risk groups, we measured the area under the receiver operating characteristic curve (AUG) of each peptide. For diagnosis (cancer vs. normal), one peptide (VEITYTPSDGTQK) showed higher AUC (AUC - 0.69) than SPSA (AUC = 0.67), the currently used biomarker (Fig.3c). For prognosis (pT3 vs. pT2), SPSA showed the highest prediction ability (AUC = 0.66, Fig. 3e). Neither SPSA nor any of our individually selected peptides were able to accurately predict patient risk groups with high confidence (AUC > 0.7). These findings are in line with the general belief that signatures, rather than individual biomarkers, are required to accurately distinguish patient groups.
Signatures that distinguish patient groups
To identify subsets of peptides that can serve as liquid-biopsy signatures and integrate them into unified predictors mat accurately discriminate among our distinct patient risk groups, we employed a machine-learning analysis (Fig. 4a). Briefly, we evaluated quantified peptides as input features for machine learning and identified the most relevant ones using Generalized Linear Models (GLMs) 22. The selection of reliable features reduces the dimensionality of the feature space and leads to better performance in machine learning To select relevant peptides, we measured the importance of each peptide to discriminate two classes of patient risk groups (cancer vs. normal controls or pT2 vs. pT3 prostate cancers) and subsequently systematically selected the top-ranked peptides as features to build predictive models. To obtain stable predictions, predictive models were tested by 100-fold bootstrapping (see Methods). We evaluated the prediction performance of selected peptides using AUC. A set of peptides that showed the highest AUC was selected as the most relevant features. Using these peptides as features, the best predictive model was generated.
We found that the six top-ranked features, mapping to five distinct gene products (IDHC, SERA., IGJ, EF2 and KCRB; pink bars in Fig. 4b) showed the best performance to discriminate all prostate cancer patients (pT2 and pT3) from biopsy-verified normal controls. From the 10-fold cross validation, these features predicted 70% of cases correctly (82% sensitivity, 47% specificity). We found that combination of these peptides outperforms the traditional FDA- approved biomarker, PSA. The predictive model showed an AUC of 0.77 (95% confidence interval, 0.68 - 0.87, pink line in Fig. 4c). In the test set (10% of entire dataset), our predictive model showed an AUC of 0.79 (95% of confidence interval, 0.76 - 0.81), 82% sensitivity and 49% specificity (Fig. 11a). This compares favorably with the performance of SPSA alone in which the AUC is 0.67 (blue line in Fig. 4c), similar to previous reports * Furthermore, we compared our six-peptide biomarkers to the null distribution in this dataset by performing a large-scale re-sampling study 1( 25. We generated 100 random sets of 6 peptides, trained them using the same OLM approach used above and measured the performance using AUC. A random model achieved an AUC of 0.64 from the 10-fold cross validation (grey line in Fig. 4c) and 0.61 from test set (Fig. 11a), Indeed, our selected peptide biomarkers showed a significant improvement with respect to randomly selected signatures (bootstrap
 Fig. 12).
Taken together, targeted proteomics quantification in poat-DRE urines is capable of identifying peptide signatures that are superior to the current gold Standard biomarker for prostate cancer screening.
Fig. 12).
Taken together, targeted proteomics quantification in poat-DRE urines is capable of identifying peptide signatures that are superior to the current gold Standard biomarker for prostate cancer screening.
We next sought to examine "whether our approach can be used to address an important clinical challenge: to distinguish pT2 stage (organ-confined) tumors from pT3 stage (extracapsular) tumors, prior to radical prostatectomy, Of note, our retrospective patient Cohort B had detailed clinical annotation, both at the stage of needle biopsy and following surgical resection of the prostate (Table 2). It is hence important to note that all patients' tumors were staged as T2 via diagnostic needle biopsy, but up-staged to pT3 following precise pathological examination of the surgical specimen. From 100-fold bootstrapping, we selected seven top-ranked peptides (6PQL, SERA, GELS, PEDF, PARK7, 1433S and AGV7; pink bars in Kg. 4d) as the most relevant biomarker signatures to predict pT3 samples prior to radical prostatectomy (i.e. EPS-urines were collected prior to surgery). From the 10-fbld cross validation, the combination of these seven features showed AUC of 0.74 (95% confidence interval, 0.62 - 0.85), 69% accuracy, 39% sensitivity and 85% specificity (Fig. 4e). In the test set, our predictive model achieved 74% accuracy and an AUC of 0.77 (95% confidence interval, 0.74 - 0.80; Fig. lib). Meanwhile, the single SPSA-based approach achieved an AUC of 0.66 and a random model achieved an AUC of 0.70 (10-fold cross validation, Fig. 4E) and 0.69 (test set, Fig. lib). Our predictive model significantly outperformed 100 random sets of 7 peptide signatures (bootstrap p - 4.57 x 10'7, Fig. 12). These results suggest that targeted proteomics analyses in clinically relevant tissue proximal fluids are capable of detecting peptide signatures that predict prostate cancer stage independent of needle biopsy and prior to surgical removal of the prostate.
Discussion
The timely verification of extensive lists of candidate disease biomarkers generated by high- resolution proteomic technologies is becoming increasingly feasible and necessary to identify putative candidates. Despite this, the implementation of biomarkers into clinical practice is largely lagging behind, This is, in part, attributed to the lack of validated methods of candidate testing and further evaluation. Targeted proteomics by SRM-MS has emerged as the method of choice for candidate protein quantification and verification, due to its relatively low cost and
amenability to robust high-throughput assay development workflows
 The aim of the current study was to systematically evaluate targeted proteomic assays in a clinically applicable, yet to date rarely utilized tissue proximal fluid - EPS-urine, and develop liquid-biopsy signatures for accurate patient classification.
The aim of the current study was to systematically evaluate targeted proteomic assays in a clinically applicable, yet to date rarely utilized tissue proximal fluid - EPS-urine, and develop liquid-biopsy signatures for accurate patient classification.
This work is an extension of previously published proteoxnics data in direct-EPS from patients with EC and OC tumors The feasibility of obtaining coordinatcs (peptide clution times and the most intense fragment ions) from different modes of data acquisition and instrumentation was demonstrated by extracting relevant information from shotgun proteomics in order to lay the foundation for targeted assay development using a triple quadrupole mass spectrometer. By performing multiple rounds of assay refinement and statistical evaluation, we narrowed over 200 promising candidates, to 34 peptides with high biomarker potential By using heavy isotope- labeled peptide standards, we estimated and later absolutely quantified these candidates in richly annotated cohorts of EPS-urines.
Interestingly, a small number of peptides in EPS-urine overlapped with those that were previously explored in urine 29. The cancer-associated proteins that were evaluated in that study were empirically gathered from reports based on protein and nucleic acid changes in human plasma and tissue 20130 , In contrast, we directly compared prostate-proximal fluids from EC and OC tumor groups to derive biomarker candidates, We previously demonstrated mat post-DRE urines (EPS-urines) contain a unique subset of proteins, when compared to matched urines from the same patient, supporting our hypothesis that prostate-enriched proteins are released as a result of the digital rectal examination 19 , Interestingly, applying a subset of our SRM-MS assays to an independent cohort of prostate cancer urines (non post-DRE urines) demonstrated that the majority of peptides were not quantifiable and likely below our detection limits.
A notable finding from the work presented here is the trend towards lower abundance of the majority of candidates with advancing disease. For instance, elevated serum levels of PSA are indicative of prostate cancer; however, EPS levels of PSA have been consistently lower in disease involving extracapsular extension (EC and/or pT3) Similarly, a trend of decreased PSA in EPS-urine from cancer patients compared to controls has been noted by Drake et at . This may be indicative of PSA leakage out of the prostate gland and into the circulation 32 or by
diminished secretory functions that have been observed for high GS tumors, a histology hallmark of which are smaller, rounder glands. Similarly, it can be conceptualized that other proteins are escaping the prostate and entering the circulation due to the deteriorating structural mtegrity of advanced prostate cancers. It would be beneficial to measure both EPS and serum levels of such proteins from the same patient in order to further test this observation. Although the majority of candidate expression levels were in agreement between Cohorts A and B, some differences were noted. For instance, the protein ΛΝΧΑ3 has one peptide (LTFDEYR) that is up-regulated in the EC group of EPS-urines in Cohort A, while the other peptide (SEIDLLDIR) is down-regulated in the EC group. This could be due to inaccurate quantification, the presence of multiple proteoforms post-translational modifications, or variations in proteolytic digestion efficiencies of endogenous proteins
 and warrant further investigation using additional peptides.
and warrant further investigation using additional peptides.
To evaluate the effect of extra peptides from the same protein on risk assessment, we examined the performance of our predictors by adding these additional peptides. In Cohort B, there are six proteins (ΛΝΧΑ3, IDHC, PEDF, PRDX6, SERA and TGM4) that each have two unique peptides used in our SRM-MS assays. Our original predictive models contained both IDHC peptides and one SERA peptide (diagnostic model) and one SERA peptide (prognostic model). We hence evaluated if adding the second SERA peptide would change the performance of our predictors. As it turns out, there is no significant performance change between our original predictors (AUC of 0.79 for diagnosis and 0,77 for prognosis) and the modified prediction models using both peptides (AUC of 0.S1 for diagnosis with p-value = 0.18, AUC of 0.77 for prognosis with p- value = 0,97, Student's t-test; Table 2). Indeed, the additional SERA peptide has a low variable importance score, For example, for the diagnostic analysis, the additional peptide "TLGILGLGR" from SERA has a variable importance score of 17.04. Meanwhile the selected peptide "GGIVDEGALLR" from SERA has a variable importance score of 90.30 (average variable importance score is 37.21, Fig. 4b).
For the comparison between BPH and normal individuals from Cohort B, only the protein IDHC (peptide VEITYTPSDGTQK) was found to demonstrate a significant increase in BPH. This may be explained by the fact that the original discovery cohort
 lacked a non-cancer group consisting of BPH and normal individuals. Furthermore, candidates that were selected from Cohort A for verification were not specifically aimed at verifying their differential expression
between BPH and normal EPS-urines. Future studies could be aimed at identification of peptide signatures that accurately distinguish normal from BPH, since mis is a major limitation of SPSA measurements.
lacked a non-cancer group consisting of BPH and normal individuals. Furthermore, candidates that were selected from Cohort A for verification were not specifically aimed at verifying their differential expression
between BPH and normal EPS-urines. Future studies could be aimed at identification of peptide signatures that accurately distinguish normal from BPH, since mis is a major limitation of SPSA measurements.
Although not obtained for the current study, additional parameters such as tumor microenvironment measurements may also provide information about patient outcome. Indeed, in a recent study, intra-prostatic hypoxia was combined with DNA indices (copy number alterations) to robustly predict 5-year biochemical recurrence (BCR) Such multi-feature signatures may provide a cornrmhensive, powerful predictor of patient outcome, Patients enrolled in Cohort B of this study have clinical information regarding BCR. In the pT2 group, 11 individuals out of 61 developed BCR within 2 years post-RP; in the pT3 group, 10 individuals out of 29 developed BCR. Although not yet explored in this study, one approach to the analysis of the data may be to comparatively analyze the protein expression changes between individuals who developed recurrence and those that did not
Our study is currently the largest investigation of prostate-proximal fluids in the context of biomarker discovery utilizing discovery proteomic data to design targeted proteomic assays. While developed assays are subsequently evaluated across multiple independent cohorts, substantial work is still required prior to possible clinical application. This will include additional validation in independent patient cohorts, preferably using longitudinally collected samples, and additional assay optirnizations
 . Furthermore, alternative approaches such as analyses of extracellular vesicles
. Furthermore, alternative approaches such as analyses of extracellular vesicles
 or glycosylated proteins
or glycosylated proteins
 should be applied for the discovery of prostate cancer biomarkers, ideally in parallel to analyses of matching tissue specimens applying carefully designed multi-omic/protcogenoiiric workflows Here, we provide the first work that
should be applied for the discovery of prostate cancer biomarkers, ideally in parallel to analyses of matching tissue specimens applying carefully designed multi-omic/protcogenoiiric workflows Here, we provide the first work that

utilizes SRM-MS for the systematic identification of novel biomarker signatures for distinguishing prostate cancer patient risk groups in a medium sized cohort of biofluid.
Although preferred embodiments of the invention have been described herein, it will be understood by those skilled in the art that variations may be made thereto without departing from the spirit of the invention or the scope of the appended claims. All documents disclosed herein, including those in the following reference list, are incorporated by reference.




References
1. Lalondc E, et al. Tumour genomic and microcmvironmental heterogeneity for integrated prediction of S-year biochemical recurrence of prostate cancer a retrospective cohort study. Lancet Oncol IS, 1521-1532 (2014).
2. Cooper CS, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet 47, 367-372 (2015).
3. Boutros PC et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet 47, 736-745 (2015).
4. Oundem O, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353-357 (2015).
5. Grover SA, Zowall H, Coupal L, Krahn MD. Prostate cancer 12. The economic burden.
CMAJ ; Canadian Medical Association journal - Journal de I'Associatlon medicale canadtenne 160, 685-690 (1999).
6. Loeb S, et al Systematic review of complications of prostate biopsy. Eur Urol 64, 876- 892 (2013).
7. Klein EA, et al A 17-gene assay to predict prostate cancer aggressiveness in the context of Oleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur Urol 66, 550-560 (2014).
8. Cooperberg MR, Broering JM, Carroll PR. Time trends and local variation in primary treatment of localized prostate cancer. J Clin Oncol 28, 1117-1123 (2010).
9. Welch HO, Albertsen PC. Prostate cancer diagnosis and treatment after the introduction of prostate-specific antigen screening: 1986-2005. J Natl Cancer bat 101, 1325-1329 (2009).
Thompson IM, Klotz L. Active surveillance for prostate cancer. JAMA 304, 2411-2412 (2010).
Ganz PA, et al National Institutes of Health State-of-the-Science Conference; role of active surveillance in the management of men with localized prostate cancer. Annals of interna medicine 156, 591-595 (2012).
Sieh W, et al. Treatment and Mortality in Men with Localized Prostate Cancer: A Population-Based Study in California. The open prostate cancer journal 6, 1-9 (2013).
Klotz L, et al. Long-term follow-up of a large active surveillance cohort of patients with prostate cancer, J Gin Oncol 33, 272-277 (2015).
Jain S, et al. Oleason Upgrading with Time in a Large Prostate Cancer Active Surveillance Cohort J Urol 194, 79-84 (2015).
Scher HI et al, Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J Clin Oncol 33, 1348-1355 (2015).
Cortese R, et al Epigenetic markers of prostate cancer in plasma circulating DNA. Human molecular genetics 21, 3619-3631 (2012).
Principe S, et al. In-depth proteomic analyses of exosomes isolated from expressed prostatic secretions in urine. Proteomics 13, 1667-1671 (2013).
Kim Y, et al Identification of differentially expressed proteins in direct expressed prostatic secretions of men with organ-confined versus extracapsular prostate cancer. Mot Cell Proteomics 11, 1870-1884 (2012).
19. Principe S, et al Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. JProteome Res 11, 2386-2396 (2012).
20. Drake RR, et al. In-depth proteomic analyses of direct expressed prostatic secretions. J Proteotne Res 9, 2109-2116 (2010).
21. Chen YT, et al Multiplexed quantification of 63 proteins in human urine by multiple reaction niomtoring-based mass spectrometry for discovery of potential bladder cancer biomarkers. JProteomics 75, 3529-3545 (2012).
22. Nelder JA, Wedderburn RWM. Generalized Linear Models. Journal of the Royal Statistical Society 135, 370-384 (1972).
23. Ouyon I, Elisseeff A. An Introduction to Variable and Feature Selection. Journal of Machine Learning Research 3, 11S7-1182 (2003).
24. Jansen FH, et al. Prostate-specific antigen (PSA) isoform p2PSA in combination with total PSA and free PSA improves diagnostic accuracy in prostate cancer detection. Eur Urol 57, 921-927 (2010).
25. Boutros PC, et al. Prognostic gene signatures for non-small-cell lung cancer. Proc Natl AcadSci USA 106, 2824-2828 (2009).
26. Surinova S, et al Non-invasive prognostic protein biomarker signatures associated with colorectal cancer. EMBO molecular medicine 7, 1153-1165 (2015).
27. Kennedy JJ, et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify hitman proteins. Nat Methods 11, 149-155 (2014).
28. Abbatiello SE, et al. Large-Scale Interlaboratory Study to Develop, Analytically Validate and Apply Highly Multiplexed, Quantitative Peptide Assays to Measure Cancer-Relevant Proteins in Plasma. Mol Cell Proteomics 14, 2357-2374 (2015).
29. Huttenhain R, et at. Reproducible quantification of cancer-associated proteins in body fluids using targeted protcomics. Sci Transl Med 4, 142ral94 (2012).
30. Polansld M, Anderson NL. A list of candidate cancer biomarkers for targeted proteomics.
Biomarker insights 1, M8 (2007).
31. Drake RR, et al. Clinical collection and protein properties of expressed prostatic secretions as a source for biomarkers of prostatic disease. J Proteomics 72, 907-917 (2009).
32. Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nature clinical practice Oncology 5, 588- 599 (2008).
33. Smith LM, Kelleher NL, Pioteomics CTD. Proteoform: a single term describing protein complexity. Nat Methods 10, 186-187 (2013).
34. Schrciner D, Simicevic J, Ahrne E, Schmidt A, Scheiffele P. Quantitative isofonn- profiling of highly diversified recognition molecules. eUfe 4, (2015).
35. Arsene CO, et al. Protein quantification by isotope dilution mass spectrometry of proteolytic fragments: cleavage rate and accuracy. Anal Chem 80, 4154-4160 (2008).
36. Rifai N, Gillette MA, Can SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 24, 971-983 (2006).
37. Nawaz M, et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat Rev Urol 11, 688-701 (2014).
38. Liu Y, et al Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol Cell Proteomics 13, 1753-1768 (2014).
39. Cerciello F, et al. Identification of a seven glycopeptide signature for malignant pleural mesothelioma in human serum by selected reaction monitoring. Clin Proteomics 10, 16 (2013).
40. Alfaro JA, Sinha A, Kislmger T, Boutros PC. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods 11, 1107-1113 (2014).
41. MacLean B, et al Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformattcs 26, 966-968 (2010).
42. Elschenbroich S, et al. In-depth proteomics of ovarian cancer ascites: combining shotgun proteomics and selected reaction monitoring mass spectrometry. J Proteome Res 10, 2286-2299 (2011).
43. Cohen J. Statistical power analysis for the behavioral sciences, 2nd edn. L. Erlbaum Associates (1988).
44. Wason J, Marshall A, Dunn J, Stein RC, Stallard N, Adaptive designs for clinical trials assessing biomarker-guided treatment strategies. Br J Cancer 110, 1950-1957 (2014).
45. Ju H, Brasier AR. Variable selection methods for developing a biomarker panel for prediction of dengue hemorrhagic fever. BMC research notes 6, 365 (2013).
46. Foulkes AS, et al. Prediction based classification for longitudinal biomarkers. The annals of applied statistics 4, 1476-1497 (2010).
Claims
1. A method of diagnosing a subject with prostate cancer comprising:
(a) detenniiiing an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig.4b; and
(b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of healthy subjects; wherein a statistically significant difference in the expression of the at least 1 gene in the test sample compared to the reference expression level is an indication that the subject has prostate cancer.
2. The method of claim 1, wherein the at least 1 gene is at least 2, 3, 4, 5, or 6 genes associated with the top-ranked peptides in Fig. 4b.
3. The method of claim 2, wherein the at least 1 gene comprises 6 genes associated with the 6 top-ranked peptides in Fig.4b.
4. A method of classifying a subject with prostate cancer between having a pT2 stage (organ confined) tumor and having a pT3 stage (extracapsular) tumor, the method comprising:
(a) determining an expression level of at least 1 gene in a test sample from the subject selected from the group consisting of the genes identified in Fig.4d; and
(b) comparing the expression level of the at least 1 gene in the test sample with a reference expression level of the at least 1 gene from control samples of subjects with pT2 stage (organ confined) tumors and/or a pT3 stage (extracapsular) tumors;
wherein a statistically significant difference or similarity in the expression of the at least 1 gene in the test sample compared to the corresponding reference expression level is an indication that the subject correspondingly has a pT2 stage (organ confined) tumor or a pT3 stage (extracapsular) tumor.
5. The method of claim 1, wherein the at least 1 gene is at least 2, 3, 4, 5, 6, or 7 genes associated 'with the top-ranked peptides in Fig.4d.
6. The method of claim 2, wherein the at least 1 gene comprises 7 genes associated with the 7 top-ranked peptides in Fig.4d.
7. The method of any one of claims 1-6, further comprising producing gene expression profiles comprising a subject gene expression profile and a gene reference expression profile, each having values representing the expression level of the at least 1 gene corresponding the test and control samples respectively.
8. The method of any one of claims 1-7, wherein the test sample comprises at least one of prostate-proximal fluid and/or expressed prostatic secretions.
9. The method of any one of claims 1-8, wherein the test sample is collected directly from the prostate, preferably prior to radical prostatectomy, and/or from urine, preferably post- digital rectal examination urine.
10. The method of any one of claims 1-9, wherein determining the expression level of the at least one gene in the test sample comprises measuring in the test sample, the level of at least one peptide corresponding to the protein product of the at least one gene.
11. The method of any one of claims 1-10, further comprising producing peptide presence profiles comprising a subject peptide expression profile and a reference peptide
expression profile, each having values representing the peptide levels of the at least 1 peptide corresponding foe test and control samples respectively.
12. The method of claim 11, wherein the at least one peptide comprises 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig. 4d.
13. The method of any one of claims 10-12, wherein the level of the at least 1 peptide is measured using mass spectrometry.
14. The method of claim 13, wherein the mass spectrometry is targeted mass spectrometry using selected reaction monitoring mass spectrometry.
15. A composition compriaing a plurality of antibodies capable of specifically binding to a plurality of peptides cone-bonding to a plurality of the genes in Fig, 4b and Fig.4d.
16. The composition of claim 15, wherein the plurality of peptides are a plurality of the peptides identified in Fig.4b and Fig.4d
17. The composition of claim 16, wherein the plurality of peptides are the 2, 3, 4, 5, 6, or 7 of the top-ranked peptides in Fig.4b and Fig.4&
18. A computer program product for use in conjunction with a computer having a processor and a memory connected to the processor, the computer program product comprising a computer readable storage medium having a computer mechanism encoded thereon, wherein the computer program mechanism may be loaded into the memory of the computer and cause the computer to carry out the method of any one of claims 1-14.
19. A computer implemented product for diagnosing or classifying a subject with prostate cancer comprising;
(a) a means for receiving values corresponding to a subject expression profile in a subject sample;
(b) a database comprising a reference expression profile representing a control, wherein the subject expression profile and the reference profile each have at least one value representing the level of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig. 4b and Fig.44 wherein the computer implemented product compares the reference expression profile to the subject biomarker expression profile, wherein a statistically significant difference or similarity in the expression profiles is used to diagnose or or classify the subject with prostate cancer.
20. The computer implemented product of claim 19 carrying out the method of any one of claims
1-14.
21. A computer readable medium having stored thereon a data structure for storing the computer implemented product of any one of claims 19 and 20.
22. The computer readable medium according to claim 21, wherein the data structure is capable of configuring a computer to respond to queries based on records belonging to the data structure, each of the records comprising:
(a) a value that identifies a reference expression profile of at least 1 gene identified in Fig. 4b and Fig.4d, preferably at least 1 peptide in Fig. 4b and Fig.4d;
(b) a value that identifies the probability of a diagnosis or classification associated with the reference expression profile.
23. A computer system comprising
(a) a database including records comprising a reference expression profile of the level of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig.4b end Fig.4d;
(b) a user interface capable of receiving a selection of expression levels of at least 1 gene identified in Fig.4b and Fig.4d, preferably at least 1 peptide in Fig.4b and Fig.4d, for use in comparing to the reference expression profile in the database;
(c) an output that displays a prediction of diagnosis or classification wherein a statistically significant difference or similarity in the expression levels is used to diagnose or classify the subject with prostate cancer.
24. A kit comprising reagents for detecting the level of at least 1 gene identified in Fig. 4b and Fig.4d, preferably at least 1 peptide in Fig.4b and Fig.4d, in a sample.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662332742P | 2016-05-06 | 2016-05-06 | |
| US62/332,742 | 2016-05-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017190218A1 true WO2017190218A1 (en) | 2017-11-09 |
Family
ID=60202659
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2017/000114 Ceased WO2017190218A1 (en) | 2016-05-06 | 2017-05-05 | Liquid-biopsy signatures for prostate cancer |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017190218A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10837970B2 (en) | 2017-09-01 | 2020-11-17 | Venn Biosciences Corporation | Identification and use of glycopeptides as biomarkers for diagnosis and treatment monitoring |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5952177A (en) * | 1997-12-03 | 1999-09-14 | Incyte Pharmaceuticals, Inc. | Human cytosolic isocitrate dehydrogenase |
| US20140106981A1 (en) * | 2006-08-09 | 2014-04-17 | Integrated Diagnostics, Inc. | Organ-Specific Proteins and Methods of Their Use |
| US20150086570A1 (en) * | 2012-03-29 | 2015-03-26 | Biogen Idec Ma Inc. | Biomarkers for use in integrin therapy applications |
| WO2016187508A2 (en) * | 2015-05-20 | 2016-11-24 | The Broad Institute Inc. | Shared neoantigens |
-
2017
- 2017-05-05 WO PCT/CA2017/000114 patent/WO2017190218A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5952177A (en) * | 1997-12-03 | 1999-09-14 | Incyte Pharmaceuticals, Inc. | Human cytosolic isocitrate dehydrogenase |
| US20140106981A1 (en) * | 2006-08-09 | 2014-04-17 | Integrated Diagnostics, Inc. | Organ-Specific Proteins and Methods of Their Use |
| US20150086570A1 (en) * | 2012-03-29 | 2015-03-26 | Biogen Idec Ma Inc. | Biomarkers for use in integrin therapy applications |
| WO2016187508A2 (en) * | 2015-05-20 | 2016-11-24 | The Broad Institute Inc. | Shared neoantigens |
Non-Patent Citations (5)
| Title |
|---|
| CHONDROS K ET AL.: "Validation of revised Epstein's criteria for insignificant prostate cancer prediction in a Greek subpopulation", HIPPOKRATIA, vol. 19, no. l, January 2015 (2015-01-01), pages 30 - 33, XP055436296 * |
| IDE H ET AL.: "Circulating pigment epithelium-derived factor (PEDF) is associated with pathological grade of prostate cancer", ANTICANCER RES ., vol. 35, no. 3, March 2015 (2015-03-01), pages 1703 - 1708, XP055436294 * |
| KIM Y ET AL.: "Identification of differentially expressed proteins in direct expressed prostatic secretions of men with organ-confined versus extracapsular prostate cancer", MOL CELL PROTEOMICS, vol. ll, no. 12, 17 September 2012 (2012-09-17), pages 1870 - 1884, XP055349206 * |
| LEIBOVICI D ET AL.: "How accurate is our clinical prediction of ''minimal prostate cancer''?", ISR MED ASSOC J., vol. 15, July 2013 (2013-07-01), pages 359 - 363, XP055436298 * |
| MHAWECH P ET AL.: "Immunohistochemical expression of 14-3-3 sigma protein in human urological and gynecological tumors using a multi-tumor microarray analysis", PATHOL INT., vol. 55, no. 2, February 2005 (2005-02-01), pages 77 - 82, XP055436300 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10837970B2 (en) | 2017-09-01 | 2020-11-17 | Venn Biosciences Corporation | Identification and use of glycopeptides as biomarkers for diagnosis and treatment monitoring |
| US11624750B2 (en) | 2017-09-01 | 2023-04-11 | Venn Biosciences Corporation | Identification and use of glycopeptides as biomarkers for diagnosis and treatment monitoring |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Kim et al. | Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer | |
| Issaq et al. | Cancer biomarker discovery: Opportunities and pitfalls in analytical methods | |
| Zhang et al. | Metabolomics for biomarker discovery: moving to the clinic | |
| Frantzi et al. | Clinical proteomic biomarkers: relevant issues on study design & technical considerations in biomarker development | |
| Huang et al. | Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application | |
| Diamandis | Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations | |
| Drabovich et al. | Toward an integrated pipeline for protein biomarker development | |
| CN102027373B (en) | Methods for discovery of biomarkers and drug targets for prostate cancer diagnosis and treatment and assays for established biomarkers | |
| Neuhaus et al. | Seminal plasma as a source of prostate cancer peptide biomarker candidates for detection of indolent and advanced disease | |
| Qin et al. | SRM targeted proteomics in search for biomarkers of HCV‐induced progression of fibrosis to cirrhosis in HALT‐C patients | |
| Kikuchi et al. | Proteomics analysis in lung cancer: challenges and opportunities | |
| Domon et al. | Implications of new proteomics strategies for biology and medicine | |
| Li et al. | High-throughput metabolomics to identify metabolites to serve as diagnostic biomarkers of prostate cancer | |
| CN106461647A (en) | Protein biomarker profiles for detecting colorectal tumors | |
| Wessels et al. | Plasma glycoproteomics delivers high-specificity disease biomarkers by detecting site-specific glycosylation abnormalities | |
| Kočevar et al. | The progress of proteomic approaches in searching for cancer biomarkers | |
| Patel et al. | Unique and novel urinary metabolomic features in malignant versus benign adrenal neoplasms | |
| Nedelkov | Population proteomics: addressing protein diversity in humans | |
| Cai et al. | Diagnosis and prognosis prediction of gastric cancer by high-performance serum lipidome fingerprints | |
| Song et al. | MALDI‐TOF‐MS analysis in low molecular weight serum peptidome biomarkers for NSCLC | |
| Piga et al. | Paving the path toward multi-omics approaches in the diagnostic challenges faced in thyroid pathology | |
| Huang et al. | Liquid chromatography–mass spectrometry based serum peptidomic approach for renal clear cell carcinoma diagnosis | |
| Gromov et al. | Proteomic analysis of tissue samples in translational breast cancer research | |
| Wouters | Proteomics: methodologies and applications in oncology | |
| Fan et al. | Serum peptidome patterns for early screening of esophageal squamous cell carcinoma |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17792309 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17792309 Country of ref document: EP Kind code of ref document: A1 |