WO2018012572A1 - シーラントフィルム及びその製造方法 - Google Patents
シーラントフィルム及びその製造方法 Download PDFInfo
- Publication number
- WO2018012572A1 WO2018012572A1 PCT/JP2017/025497 JP2017025497W WO2018012572A1 WO 2018012572 A1 WO2018012572 A1 WO 2018012572A1 JP 2017025497 W JP2017025497 W JP 2017025497W WO 2018012572 A1 WO2018012572 A1 WO 2018012572A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyester
- film
- units
- sealant film
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/66—Polyesters containing oxygen in the form of ether groups
- C08G63/668—Polyesters containing oxygen in the form of ether groups derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/676—Polyesters containing oxygen in the form of ether groups derived from polycarboxylic acids and polyhydroxy compounds in which at least one of the two components contains aliphatic unsaturation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B15/08—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
- B32B15/09—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin comprising polyesters
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/001—Combinations of extrusion moulding with other shaping operations
- B29C48/0018—Combinations of extrusion moulding with other shaping operations combined with shaping by orienting, stretching or shrinking, e.g. film blowing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/022—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the choice of material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C55/00—Shaping by stretching, e.g. drawing through a die; Apparatus therefor
- B29C55/005—Shaping by stretching, e.g. drawing through a die; Apparatus therefor characterised by the choice of materials
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C55/00—Shaping by stretching, e.g. drawing through a die; Apparatus therefor
- B29C55/02—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/10—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of paper or cardboard
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/20—Layered products comprising a layer of synthetic resin characterised by the use of special additives using fillers, pigments, thixotroping agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/22—Layered products comprising a layer of synthetic resin characterised by the use of special additives using plasticisers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/36—Layered products comprising a layer of synthetic resin comprising polyesters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K3/00—Materials not provided for elsewhere
- C09K3/10—Materials in mouldable or extrudable form for sealing or packing joints or covers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2023/00—Use of polyalkenes or derivatives thereof as moulding material
- B29K2023/04—Polymers of ethylene
- B29K2023/06—PE, i.e. polyethylene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29L—INDEXING SCHEME ASSOCIATED WITH SUBCLASS B29C, RELATING TO PARTICULAR ARTICLES
- B29L2007/00—Flat articles, e.g. films or sheets
- B29L2007/008—Wide strips, e.g. films, webs
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/20—Properties of the layers or laminate having particular electrical or magnetic properties, e.g. piezoelectric
- B32B2307/21—Anti-static
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/306—Resistant to heat
- B32B2307/3065—Flame resistant or retardant, fire resistant or retardant
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/31—Heat sealable
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/40—Properties of the layers or laminate having particular optical properties
- B32B2307/402—Coloured
- B32B2307/4026—Coloured within the layer by addition of a colorant, e.g. pigments, dyes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/71—Resistive to light or to UV
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/724—Permeability to gases, adsorption
- B32B2307/7242—Non-permeable
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/732—Dimensional properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/732—Dimensional properties
- B32B2307/734—Dimensional stability
- B32B2307/736—Shrinkable
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2439/00—Containers; Receptacles
- B32B2439/70—Food packaging
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2553/00—Packaging equipment or accessories not otherwise provided for
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2190/00—Compositions for sealing or packing joints
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2250/00—Compositions for preparing crystalline polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/06—Unsaturated polyesters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2200/00—Chemical nature of materials in mouldable or extrudable form for sealing or packing joints or covers
- C09K2200/06—Macromolecular organic compounds, e.g. prepolymers
- C09K2200/0645—Macromolecular organic compounds, e.g. prepolymers obtained otherwise than by reactions involving carbon-to-carbon unsaturated bonds
- C09K2200/0655—Polyesters
Definitions
- the present invention mainly comprises at least one diol unit selected from the group consisting of 2,5-furandicarboxylic acid units and ethylene glycol units, 1,3-propanediol units and 1,4-butanediol units.
- the present invention relates to a sealant film made of polyester. Moreover, it is related with the manufacturing method.
- Sealant films are widely used in foods, beverages, medicines, cosmetics, packaging materials for medical equipment, and the like.
- the sealant film can be placed in the innermost layer of the packaging material and heat sealed to seal the package.
- resins used as such sealant films polyolefins such as polyethylene and polypropylene, ionomers, ethylene-methyl methacrylate copolymers, and the like are known.
- organic compounds are easily adsorbed to these resins, there are cases where odor components and contents are adsorbed on a sealant film made of the resin, which may be a problem.
- polyesters such as polyethylene terephthalate (hereinafter may be abbreviated as PET) are resins that are difficult to adsorb organic compounds and have excellent non-adsorption properties. Polyesters are also excellent in properties such as mechanical properties, gas barrier properties, flavor barrier properties, and transparency. Furthermore, polyester has less concern about residual monomers and harmful additives when formed into molded articles, and is excellent in hygiene and safety.
- PET polyethylene terephthalate
- Patent Document 1 describes a polymer compound having a structural unit represented by the following formula. According to this, it is said that it is a polymer compound excellent in moldability and mechanical strength (bending strength), and has physical properties that can sufficiently withstand the specifications of optical equipment, bottles, and housing materials. However, Patent Document 1 does not describe use as a sealant film, and the tensile elongation is not always good, and improvement has been demanded.
- A represents an aromatic hydrocarbon group which may be substituted, an aliphatic hydrocarbon group which may be substituted or an alicyclic hydrocarbon group which may be unsubstituted, and n
- the degree of polymerization is 185 or more and 600 or less.
- the present invention has been made to solve the above-described problems, and an object thereof is to provide a sealant film having excellent mechanical properties, particularly tensile elongation, and excellent heat sealability, non-adsorption properties, and gas barrier properties.
- the above-mentioned problem is a sealant film having at least an outermost layer made of polyester, the polyester comprising 20 to 50 mol% of 2,5-furandicarboxylic acid units, ethylene glycol units, and 1,3-propanediol units. And at least one diol unit selected from the group consisting of 1,4-butanediol units, 18 to 49.5 mol%, and diethylene glycol units 0.5 to 2.5 mol%,
- a sealant film having a crystallinity of less than 14%, and the sealant film is a stretched film having a shrinkage ratio in a maximum shrinkage direction of 6% or more when left at 125 ° C. for 20 seconds. It is solved by providing.
- the polyester preferably further contains 0.01 to 30 mol% of another comonomer unit having 5 or more carbon atoms, and the uniaxial stretching ratio is preferably 1.1 to 4.5 times. It is preferable that the surface draw ratio is 1.1 to 9.0 times.
- a sealant film having a tensile elongation measured by conducting a tensile test under the conditions of a temperature of 23 ° C., a relative humidity of 65% and a tensile speed of 50 mm / min is 130% or more is a preferred embodiment of the present invention.
- the above problem can also be solved by providing a method for producing the sealant film in which the film is obtained by extruding the polyester and then the film is uniaxially stretched.
- the above problem can also be solved by providing a method for producing the sealant film, in which a film is obtained by extruding the polyester and then the film is biaxially stretched.
- the sealant film of the present invention is excellent in mechanical properties, particularly tensile elongation, and also excellent in heat sealability, non-adsorption property and gas barrier property. Therefore, such a sealant film is suitably used as the innermost layer of various packaging materials. Moreover, according to the manufacturing method of this invention, such a sealant film can be obtained simply.
- the sealant film of the present invention is a sealant film having at least a layer made of polyester as the outermost layer, wherein the polyester contains 20 to 50 mol% of 2,5-furandicarboxylic acid units, ethylene glycol units, 1,3- 18 to 49.5 mol% of at least one diol unit selected from the group consisting of propanediol units and 1,4-butanediol units, and 0.5 to 2.5 mol% of diethylene glycol units,
- the degree of crystallinity of the sealant film is less than 14%
- the sealant film is a stretched film having a shrinkage rate in the maximum shrinkage direction of 6% or more when allowed to stand at 125 ° C. for 20 seconds. Is. It has been clarified by the present inventors that a sealant film satisfying such a configuration is excellent in mechanical properties, particularly tensile elongation, while maintaining excellent heat sealability and gas barrier properties.
- the content of 2,5-furandicarboxylic acid unit in the polyester is 20 to 50 mol%, and if it is less than this, the oxygen barrier property and the water vapor barrier property are lowered.
- the content of 2,5-furandicarboxylic acid units is preferably 25 mol% or more, more preferably 30 mol% or more, and even more preferably 35 mol% or more.
- the diol unit in the polyester is at least one selected from the group consisting of an ethylene glycol unit, a 1,3-propanediol unit and a 1,4-butanediol unit.
- the content of the diol unit in the polyester is 18 to 49.5 mol%, and if it is less than this, there will be a problem of a decrease in heat resistance due to a decrease in glass transition temperature and a decrease in gas barrier properties.
- the content of the diol unit is preferably 25 mol% or more, more preferably 28 mol% or more, and further preferably 30 mol% or more.
- the content of diethylene glycol units in the polyester is 0.5 to 2.5 mol%. Usually, a unit by-produced by dimerization of ethylene glycol during the polycondensation reaction is contained in the polyester. The content of diethylene glycol units is preferably 2 mol% or less.
- the polyester includes at least one diol unit selected from the group consisting of 2,5-furandicarboxylic acid units, ethylene glycol units, 1,3-propanediol units, and 1,4-butanediol units, other than diethylene glycol units. It is preferable to contain 0.01 to 30 mol% of other comonomer units. By containing 0.01 mol% or more of such other comonomer units, the crystallinity and melting point of the polyester can be lowered, heat sealability can be improved, and extrusion film forming properties can be improved. Or you can.
- the content of other comonomer units is preferably 0.03 mol% or more.
- the total content of 2,5-furandicarboxylic acid units and other comonomer units is preferably 30 to 80 mol%.
- the total content is more preferably 75 mol% or less.
- the other comonomer unit preferably has 5 or more carbon atoms. If the number of carbon atoms is less than 5, the boiling point of the comonomer of the raw material is lowered and volatilizes during the condensation polymerization reaction, so there is a possibility that it is difficult to recover ethylene glycol or the like. Moreover, when the carbon number is 5 or more, the crystallinity of the polyester can be effectively reduced.
- the upper limit of the carbon number is not particularly limited, but is usually 50 or less.
- One type of other comonomer units contained in the polyester may be used, or two or more types may be used in combination.
- Other comonomer units include dicarboxylic acid units other than 2,5-furandicarboxylic acid units, ethylene glycol units, 1,3-propanediol units, diol units other than 1,4-butanediol units and diethylene glycol units, and hydroxy units.
- Bifunctional compound units such as carboxylic acid units are mainly used.
- Other difunctional compound units used as comonomer units include dicarboxylic acid units other than 2,5-furandicarboxylic acid units, ethylene glycol units, 1,3-propanediol units, 1,4-butanediol units, and diethylene glycol units. Diol units other than units are preferred.
- the polyfunctional compound unit which has 3 or more of carboxyl groups or hydroxyl groups, and the monofunctional compound unit which is a monocarboxylic acid unit or a monoalcohol unit can also be used together with the said bifunctional compound unit.
- dicarboxylic acid units used as comonomer units include aliphatic dicarboxylic acids such as glutaric acid, adipic acid, azelaic acid, sebacic acid, and dimer acid, and ester-forming derivatives thereof; cyclohexanedicarboxylic acid, norbornene dicarboxylic acid, tricyclohexane Alicyclic dicarboxylic acids such as decanedicarboxylic acid and ester-forming derivatives thereof; terephthalic acid, isophthalic acid, phthalic acid, biphenyldicarboxylic acid, diphenyletherdicarboxylic acid, diphenylsulfonedicarboxylic acid, diphenylketonedicarboxylic acid, sodium sulfoisophthalate, Derived from aromatic dicarboxylic acids such as 2,6-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, 2,7-na
- the dimer acid is a dicarboxylic acid obtained by dimerizing an unsaturated fatty acid.
- unsaturated fatty acid include unsaturated fatty acids having 18 carbon atoms such as linoleic acid, linolenic acid, and oleic acid.
- the dimer acid may be non-hydrogenated or hydrogenated, but the latter is preferable because it is less colored during polymerization.
- the dicarboxylic acid unit used as another comonomer unit is preferably at least one selected from the group consisting of a terephthalic acid unit, an isophthalic acid unit, a sebacic acid unit, a dimer acid unit and an adipic acid unit, a terephthalic acid unit, More preferably, it is at least one selected from the group consisting of an isophthalic acid unit, a sebacic acid unit and a dimer acid unit, and more preferably at least one selected from the group consisting of a terephthalic acid unit and an isophthalic acid unit.
- Diol units used as other comonomer units include aliphatic diols such as 1,5-pentanediol, neopentyl glycol, 1,6-hexanediol, and methylpentanediol; cyclohexanedimethanol, cyclooctanedimethanol, norbornenediol Examples are alicyclic diols such as methanol and tricyclodecane dimethanol; those derived from isosorbide. Examples of the methylpentanediol include 3-methyl-1,5-pentanediol and 2-methyl-1,5-pentanediol.
- cyclohexanedimethanol examples include 1,2-cyclohexanedimethanol, 1,3-cyclohexanedimethanol and 1,4-cyclohexanedimethanol.
- a unit derived from a diol in which one or more molecules of ethylene oxide are added to two hydroxyl groups of an aromatic diol can also be used.
- a unit derived from a bisphenol A ethylene oxide adduct in which 1 to 8 molecules of ethylene oxide are added to two phenolic hydroxyl groups of bisphenol A is exemplified.
- diol units used as other comonomer units are 3-methyl-1,5-pentanediol units, units derived from bisphenol A ethylene oxide adducts, 1,4-cyclohexanedimethanol units, cyclooctane dimers.
- It is preferably at least one selected from the group consisting of methanol units and isosorbide units, 3-methyl-1,5-pentanediol units, units derived from bisphenol A ethylene oxide adducts and 1,4-cyclohexanedi More preferred is at least one selected from the group consisting of methanol units, and more preferred are units derived from 3-methyl-1,5-pentanediol units or bisphenol A ethylene oxide adducts.
- hydroxycarboxylic acid units used as comonomer units include aliphatic hydroxycarboxylic acids such as 10-hydroxyoctadecanoic acid or ester-forming derivatives thereof; hydroxymethylcyclohexanecarboxylic acid, hydroxymethylnorbornenecarboxylic acid, hydroxymethyltricyclohexane Alicyclic hydroxycarboxylic acids such as decanecarboxylic acid or ester-forming derivatives thereof; hydroxybenzoic acid, hydroxytoluic acid, hydroxynaphthoic acid, 3- (hydroxyphenyl) propionic acid, hydroxyphenylacetic acid, 3-hydroxy-3- Illustrative are those derived from aromatic hydroxycarboxylic acids such as phenylpropionic acid and their ester-forming derivatives.
- the bifunctional compound unit used as the other comonomer unit is derived from a terephthalic acid unit, an isophthalic acid unit, a 3-methyl-1,5-pentanediol unit, a sebacic acid unit, and a bisphenol A ethylene oxide adduct.
- Polyfunctional compound units having 3 or more carboxyl groups or hydroxyl groups used as other comonomer units are derived from trimellitic acid, pyromellitic acid, trimesic acid, trimethylolpropane, pentaerythritol and their ester-forming derivatives. Are illustrated. These can be added in a small amount to increase the melt tension and can be used to adjust melt moldability.
- the content of the polyfunctional compound unit in the polyester is preferably 1 mol% or less, and more preferably 0.5 mol% or less. If the content of the polyfunctional compound unit exceeds 1 mol%, gelation tends to occur, which is not preferable.
- the monofunctional compound unit having only one carboxyl group or hydroxyl group used as another comonomer unit is derived from benzoic acid, 2,4,6-trimethoxybenzoic acid, 2-naphthoic acid, stearic acid and stearyl alcohol. Are illustrated. These function as sealing monomer units, seal molecular chain end groups in the polyester, and may be blended to prevent excessive crosslinking and gel formation in the polyester.
- the content of the monofunctional compound unit in the polyester is preferably 1 mol% or less, and more preferably 0.5 mol% or less. When the ratio of the monofunctional compound unit exceeds 1 mol%, the polymerization rate in producing the polyester is slowed, and the productivity tends to be lowered.
- the polyester production method includes 2,5-furandicarboxylic acid or an ester-forming derivative thereof, and at least one selected from the group consisting of ethylene glycol, 1,3-propanediol and 1,4-butanediol.
- a method of melt polycondensation by adding another comonomer having 5 or more carbon atoms, if necessary, or a method of solid phase polymerization after the melt polycondensation is preferred.
- esterification is performed by heating 2,5-furandicarboxylic acid or an ester-forming derivative thereof and at least one selected from the group consisting of ethylene glycol, 1,3-propanediol and 1,4-butanediol. Reaction or transesterification proceeds to obtain an oligomer. At this time, another comonomer having 5 or more carbon atoms may be added in advance and an esterification reaction or transesterification reaction may be allowed to proceed simultaneously to obtain an oligomer, or another comonomer having 5 or more carbon atoms may be obtained after obtaining the oligomer. May be added to the melt polycondensation reaction.
- the esterification reaction or transesterification reaction is preferably carried out at a temperature of 180 to 300 ° C. while distilling off the produced water or alcohol at an absolute pressure of about 0.3 MPa or less or at normal pressure.
- the ratio of raw materials in the esterification reaction or transesterification reaction is preferably such that the molar ratio (diol component / dicarboxylic acid component) is in the range of 1.1 to 2.5.
- the melt polycondensation reaction following the esterification reaction or transesterification reaction is carried out by adding additives such as the above-mentioned raw materials, polycondensation catalyst and coloring inhibitor to the obtained polyester oligomer as necessary. It is preferable to carry out under reduced pressure at a temperature of 200 to 300 ° C. until a polyester having a desired viscosity is obtained.
- the melt polycondensation reaction can be performed using, for example, a tank-type batch polycondensation apparatus or a continuous polycondensation apparatus including a biaxial rotating horizontal reactor.
- a compound containing a germanium element, an antimony element, or a titanium element is preferable.
- antimony element antimony trioxide, antimony chloride, antimony acetate, etc. are used.
- germanium dioxide, germanium tetrachloride, germanium tetraethoxide, etc. are used.
- the compound to be used include organic titanium compounds such as tetraisopropyl titanate and tetrabutyl titanate, and inorganic titanium compounds such as titanium oxide and composite particles of hydrotalcite and titanium dioxide.
- the amount added is preferably in the range of 0.002 to 0.8% by mass based on the mass of the dicarboxylic acid component.
- an anti-coloring agent for example, phosphorous acid, phosphoric acid, trimethyl phosphite, triphenyl phosphite, tridecyl phosphite, trimethyl phosphate, tridecyl phosphate, triphenyl phosphite Phosphorus compounds such as fate can be used. These phosphorus compounds may be used alone or in combination of two or more.
- an anti-coloring agent comprising the above-described phosphorus compound it is preferably in the range of 0.001 to 0.5 mass% based on the mass of the dicarboxylic acid component. Further, in order to suppress coloring due to thermal decomposition of the polyester, 0.001 to 0.5 mass% of a cobalt compound such as cobalt acetate can be added based on the mass of the dicarboxylic acid component.
- the polyester thus obtained may be used for the production of a film, or the obtained polyester may be further solid-phase polymerized as follows.
- a polyester having a higher degree of polymerization can be obtained.
- the intrinsic viscosity of the polyester subjected to solid phase polymerization is preferably in the range of 0.4 to 0.85 dl / g.
- Polyester obtained by melt polycondensation is extruded into strands, sheets, etc., cooled, and then cut with a strand cutter, sheet cutter, etc., in the shape of cylinders, elliptic cylinders, disks, dies, etc. Intermediate pellets are produced.
- the above-described cooling after extrusion can be performed by, for example, a water cooling method using a water tank, a method using a cooling drum, an air cooling method, or the like.
- the intermediate pellet thus obtained is solid-phase polymerized, but it is preferable to crystallize by heating before solid-phase polymerization. By doing so, it is possible to prevent the pellets from sticking during solid phase polymerization.
- the crystallization temperature is preferably 100 to 180 ° C.
- crystallization may be performed in a vacuum tumbler, or crystallization may be performed by heating in an air circulation type heating apparatus.
- the time required for crystallization is usually about 30 minutes to 24 hours.
- the temperature of solid phase polymerization is preferably 170 to 250 ° C., and the time of solid phase polymerization is usually about 5 to 70 hours. Moreover, you may coexist the catalyst used by melt polycondensation at the time of solid-phase polymerization.
- the solid phase polymerization is preferably performed under reduced pressure or in an inert gas such as nitrogen gas. Further, it is preferable to perform solid-state polymerization while moving the pellets by an appropriate method such as a rolling method or a gas fluidized bed method so that no sticking occurs between the pellets.
- the pressure when solid-state polymerization is performed under reduced pressure is preferably 1 kPa or less.
- the intrinsic viscosity of the polyester after solid phase polymerization is preferably in the range of 0.8 to 1.3 dL / g.
- the sealant film of the present invention may contain additives as long as the effects of the present invention are not impaired.
- colorants such as dyes and pigments, stabilizers such as ultraviolet absorbers, antistatic agents, Examples include flame retardants, flame retardant aids, lubricants, plasticizers, inorganic fillers, inorganic layered compounds, and organically treated inorganic layered compounds. These can be added to the polyester when forming a film.
- the content of these additives in the sealant film is preferably 10% by mass or less, and more preferably 2% by mass or less.
- the polyester used in the present invention preferably has a glass transition temperature (Tg) of 60 to 90 ° C, more preferably 70 to 90 ° C, and still more preferably 80 to 90 ° C.
- Tg glass transition temperature
- liquidity of polyester falls at the time of heat sealing, and there exists a possibility that the heat sealing property in low temperature may be impaired.
- the glass transition temperature is less than 60 ° C., there is a possibility that sufficient sealing strength cannot be obtained or the flavor barrier property is lowered.
- Examples of a method for forming the polyester film include a method of extruding the polyester by a T-die method, an inflation method, or the like.
- the intrinsic viscosity of the polyester used for extrusion molding is preferably in the range of 0.6 to 1.3 dl / g in the T-die method, and in the range of 0.8 to 1.3 dl / g in the inflation method. It is preferable.
- Examples of the extruder used for extruding the polyester include a single screw extruder, a twin screw extruder, a vent extruder, and a tandem extruder.
- the temperature of the polyester at the time of extrusion molding is preferably set to a temperature within the range of (melting point of the polyester + 10 ° C.) to (melting point of the polyester resin + 80 ° C.). It is preferable to obtain an unstretched film by extruding the melted polyester into a film and then rapidly cooling and solidifying it using a cooling drum or the like. Thereby, crystallization of polyester in the obtained film is suppressed.
- the temperature of the cooling drum is preferably set to a temperature in the range of (glass transition point of the polyester—30 ° C.) to (glass transition point of the polyester + 10 ° C.). Scraps such as trim generated during film production can be collected and reused.
- Examples of methods for stretching the unstretched film thus obtained include a tenter stretching method, a tubular stretching method, and a roll stretching method.
- the stretching temperature is preferably set to a temperature within the range of (glass transition point of the polyester + 5 ° C.) to (glass transition point of the polyester + 40 ° C.).
- the sealant film of the present invention is preferably a stretched film having a uniaxial stretch ratio of 1.1 to 4.5 times.
- a stretched film having a uniaxial stretching ratio of 1.1 to 4.5 times it is possible to improve mechanical properties, particularly tensile elongation, while maintaining excellent heat sealability and gas barrier properties.
- the uniaxial stretching ratio is less than 1.1, the tensile elongation may be insufficient.
- the uniaxial stretching ratio is more preferably 1.5 times or more.
- the uniaxial stretching ratio exceeds 4.5 times, the tensile elongation and heat sealability may be insufficient.
- the uniaxial stretching ratio is more preferably 3.5 times or less, and further preferably 2.5 times or less.
- Biaxial stretching may be simultaneous biaxial stretching or sequential biaxial stretching.
- the plane stretching ratio during biaxial stretching is preferably 1.1 to 9.0 times. If the surface draw ratio is less than 1.1 times, the tensile elongation may be insufficient.
- the area magnification is more preferably 1.5 times or more. On the other hand, when the surface draw ratio exceeds 9.0 times, the tensile elongation and heat sealability may be insufficient.
- the area magnification is more preferably 7 times or less, and further preferably 5 times or less.
- the obtained stretched film may be further heat-treated (heat-set). If the heat shrinkage of the stretched film is too high, it may be difficult to handle depending on the application. In such a case, the heat shrinkage rate can be reduced by heat-treating the stretched film at a temperature exceeding the stretching temperature.
- the heat treatment temperature at this time is preferably 120 ° C. or lower.
- the sealant film of the present invention needs to be a stretched film having a shrinkage ratio in the maximum shrinkage direction of 6% or more when left at 125 ° C. for 20 seconds.
- the shrinkage rate increases when the film is stretched.
- the shrinkage rate is preferably 10% or more, and more preferably 15% or more.
- the shrinkage ratio is more preferably 30% or more.
- the shrinkage rate is preferably 70% or less, and more preferably 65% or less.
- the heat shrinkage rate is preferably 50% or less, and more preferably 48% or less.
- the shrinkage rate is determined by the following method. The sealant film is cut into 80 mm squares, and the flow direction and the stretching direction during film formation are marked on the film. The heat treatment is performed by allowing the sealant film to stand at 125 ° C. for 20 seconds. At this time, the shrinkage rate in the direction in which the shrinkage rate of the film is high is defined as the shrinkage rate in the maximum shrinkage direction.
- MD is the maximum shrinkage direction
- TD may be the maximum contraction direction
- the shrinkage rate can be determined by the above method after heat treating the sealant film cut in a circular shape in advance and confirming the maximum shrinkage direction.
- the shrinkage rate in the direction orthogonal to the maximum shrinkage direction when the sealant film of the present invention is allowed to stand at 125 ° C. for 20 seconds is less than 10%, 5% or less is more preferable.
- the crystallinity of the sealant film of the present invention needs to be less than 14%.
- the crystallinity of the sealant film is the crystallinity of the polyester in the stretched film.
- the crystallinity is preferably 10% or less, and more preferably 8% or less.
- the crystallinity may be 0% or more. It is also preferable that the intrinsic viscosity of the polyester in the sealant film of the present invention is in the range of 0.5 to 1.3 dl / g.
- the tensile elongation of the sealant film measured by conducting a tensile test under the conditions of a temperature of 23 ° C., a relative humidity of 65%, and a tensile speed of 50 mm / min is preferably 130% or more.
- the tensile elongation may be 130% or more in at least one direction of the sealant film.
- the sealant film having such a tensile elongation is suitably used as the innermost layer of various packaging materials.
- the tensile elongation is more preferably 200% or more, and further preferably 250% or more.
- the sealant film of the present invention is excellent in mechanical properties, particularly tensile elongation, and also excellent in heat sealability, non-adsorption property and gas barrier property. Therefore, the sealant film is suitably used as the innermost layer of packaging materials for foods, beverages, medicines, cosmetics, medical instruments and the like.
- the sealant film of the present invention has a layer made of the above polyester on at least the outermost surface, but when used as a single layer packaging material, the thickness is usually 20 to 80 ⁇ m. When the sealant film of the present invention is used as the innermost layer of a multilayer packaging material, the thickness of the sealant film is usually 3 to 20 ⁇ m.
- examples of other layers to be laminated with the polyester layer used in the present invention include paper, resin films other than the sealant film, and metal plates.
- other polyesters other than the polyester used in the present invention are used as other layers, and the polyester used in the present invention and the other layers are used. It is a preferred embodiment of the present invention that it is a co-extruded and co-stretched multilayer film.
- polyethylene terephthalate, polyethylene terephthalate isophthalate copolymer, polyethylene-cyclohexanedimethylene-terephthalate copolymer, polyethylene terephthalate or a dry blend of polyethylene terephthalate isophthalate copolymer and polyethylene furanoate, etc. are suitable. Used for.
- a method for coextrusion molding the same method as that described in the above extrusion molding can be used.
- a method of co-stretching the method similar to the method demonstrated in the method of extending
- Intrinsic viscosity The intrinsic viscosity of the polyester was measured at a temperature of 30 ° C. using an equal mass mixture of phenol and 1,1,2,2-tetrachloroethane as a solvent.
- Tg Glass transition temperature
- the glass transition temperature (Tg) of the polyester was measured using a differential scanning calorimeter (TA Q2000 manufactured by TA Instruments). Glass transition from data when polyester was heated to 280 ° C at a heating rate of 10 ° C / min, then rapidly cooled to 30 ° C at -50 ° C / min, and then heated again at a heating rate of 10 ° C / min. The temperature (Tg) was calculated.
- Tensile test A sealant film having a thickness of 40 to 60 ⁇ m was cut into strips having a length of 100 mm and a width of 15 mm so that the flow direction (MD) during film formation was the longitudinal direction. Using an autograph (manufactured by Shimadzu Corporation), a tensile test of the test piece at a temperature of 23 ° C., a relative humidity of 65%, and a tensile speed of 50 mm / min. Young's modulus and tensile elongation (breaking elongation) were determined.
- the sealing temperature when the peel strength was 5.0 N / 15 mm or more was defined as the heat-sealable temperature. Even when heat sealing was performed at 150 ° C., if the peel strength was not 5.0 N / 15 mm or more, heat sealing was not possible.
- Oxygen permeability was measured using an oxygen permeability measuring device ("MOCON OX-TRAN 2/20" manufactured by Modern Control). Specifically, the film was set in a measuring apparatus and measured at a temperature of 20 ° C. and a humidity of 65% RH.
- Example 1 Melt polycondensation 2,5-furandicarboxylic acid (FDCA) 100 parts by mass, ethylene glycol (EG) 47.7 parts by mass, germanium dioxide (GeO 2 ) 0.012 parts by mass and phosphorous acid 0.012 parts by mass A slurry consisting of parts was prepared, and an oligomer was produced by heating to 190 ° C. under pressure (gauge pressure of 0.25 MPa) while feeding the slurry to an esterification tank over 2 hours. The obtained oligomer was transferred to a polycondensation tank and subjected to melt polycondensation under 0.1 kPa at 270 ° C.
- FDCA Melt polycondensation 2,5-furandicarboxylic acid
- copolyester having an intrinsic viscosity of 0.7 dL / g.
- the obtained copolymer polyester was extruded in a strand form from a nozzle, cooled in warm water at 30 ° C., then cut into a cylindrical shape (diameter: about 2.5 mm, length: about 2.5 mm), and copolymer polyester pellets ( Melt polymerization pellets) were obtained.
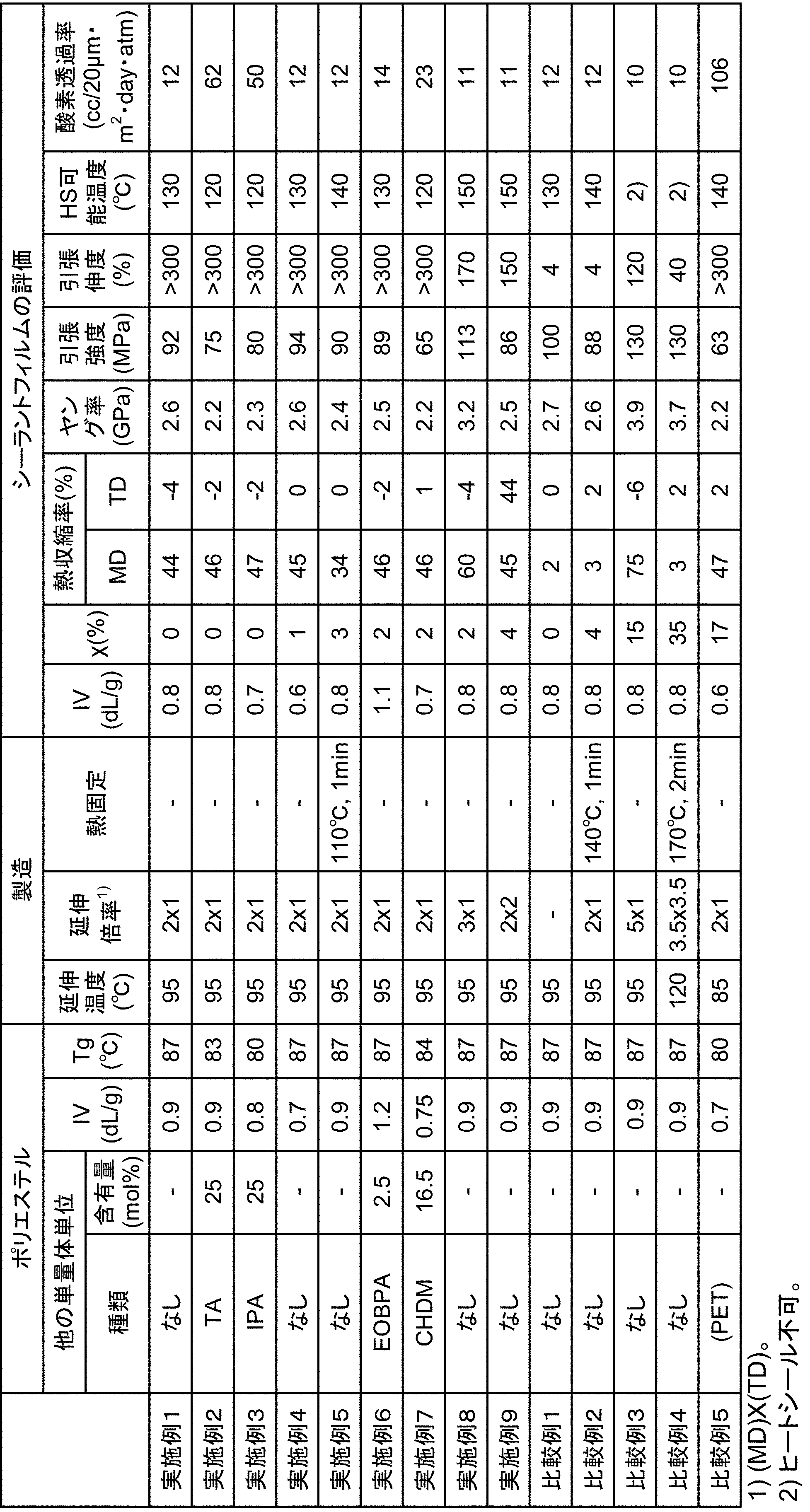
- the obtained stretched film was measured for intrinsic viscosity, crystallinity, thermal shrinkage, tensile test, heat seal test, and oxygen permeability. The results are shown in Table 1.
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- the heat shrinkage rate in the transverse direction (TD) is also shown in Table 1.
- Example 2 From 48.5 parts by mass of 2,5-furandicarboxylic acid, 51.5 parts by mass of terephthalic acid, 46.25 parts by mass of ethylene glycol, 0.012 parts by mass of germanium dioxide (GeO 2 ), and 0.012 parts by mass of phosphorous acid The resulting slurry was heated to 220 ° C. under pressure (gauge pressure of 0.25 MPa) while feeding into the esterification tank over 2 hours to carry out the esterification reaction to produce an oligomer. The obtained oligomer was transferred to a polycondensation tank and subjected to melt polycondensation at 0.1 kPa and 270 ° C.
- copolyester having an intrinsic viscosity of 0.9 dL / g.
- the obtained copolymer polyester was extruded in a strand form from a nozzle, cooled in warm water at 30 ° C., then cut into a cylindrical shape (diameter: about 2.5 mm, length: about 2.5 mm), and copolymer polyester pellets ( Melt polymerization pellets) were obtained.
- the ratio of monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- 2,5-furandicarboxylic acid unit: terephthalic acid unit: ethylene glycol unit: diethylene glycol unit 25: 25: 48 .75: 1.25 (molar ratio).
- the glass transition temperature (Tg) of the obtained copolymer polyester was 83 degreeC.
- Tg glass transition temperature
- the results are shown in Table 1.
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 3 From 48.5 parts by mass of 2,5-furandicarboxylic acid, 51.5 parts by mass of isophthalic acid, 46.25 parts by mass of ethylene glycol, 0.012 parts by mass of germanium dioxide (GeO 2 ), and 0.012 parts by mass of phosphorous acid A melt-polymerized pellet was obtained in the same manner as in Example 2 except that the resulting slurry was used. The intrinsic viscosity of the copolyester thus obtained was 0.8 dL / g. The ratio of monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- Tg glass transition temperature
- a stretched film was prepared and evaluated in the same manner as in Example 2 except that the obtained copolyester was used. The results are shown in Table 1. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 4 In the same manner as in Example 1, melt-polymerized pellets were obtained.
- the intrinsic viscosity of the copolyester thus obtained was 0.7 dL / g.
- the ratio of the monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- Tg glass transition temperature
- Preparation and evaluation of stretched film in the same manner as in Example 2 except that the obtained copolyester was used, and pellets dried under vacuum at 0.1 kPa for 2 days at 75 ° C. were used during film formation. Went.
- the results are shown in Table 1.
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 5 After the film was stretched, a stretched film was prepared and evaluated in the same manner as in Example 1 except that it was subsequently heat treated at 110 ° C. for 1 minute. The results are shown in Table 1.
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 6 2,5-furandicarboxylic acid 100 parts by mass, ethylene glycol 45.3 parts by mass, bisphenol A ethylene oxide adduct (EOBPA) 10.1 parts by mass, germanium dioxide (GeO 2 ) 0.012 parts by mass and phosphorous acid 0
- a solid phase polymerization pellet was obtained in the same manner as in Example 1 except that a slurry consisting of 0.012 parts by mass was used and the solid phase polymerization time was 200 hours.
- the intrinsic viscosity of the copolyester thus obtained was 1.2 dL / g.
- the ratio of the monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- Tg glass transition temperature
- a stretched film was prepared and evaluated in the same manner as in Example 1 except that the obtained copolyester was used. The results are shown in Table 1. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 7 2,5-furandicarboxylic acid 100 parts by mass, 1,4-cyclohexanedimethanol (CHDM) 37.1 parts by mass, ethylene glycol 31.9 parts by mass, germanium dioxide (GeO 2 ) 0.012 parts by mass and phosphorous acid
- CHDM 1,4-cyclohexanedimethanol
- ethylene glycol 31.9 parts by mass
- germanium dioxide GeO 2
- phosphorous acid phosphorous acid
- a melt-polymerized pellet was obtained in the same manner as in Example 2 except that 0.012 parts by mass of slurry was used and solid phase polymerization was not performed.
- the intrinsic viscosity of the copolyester thus obtained was 0.75 dL / g.
- the ratio of the monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- Example 8 A stretched film was prepared and evaluated in the same manner as in Example 1 except that the film thickness during film formation was changed to 150 ⁇ m and the stretch ratio was changed to 3 times. The results are shown in Table 1. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 9 The film thickness at the time of film formation was changed to 200 ⁇ m, and the stretching operation was changed to pantograph biaxial stretching [stretching temperature: 95 ° C., stretching ratio: flow direction (MD) 2 times, transverse direction (TD) 2 times].
- a stretched film was prepared and evaluated in the same manner as in Example 1 except for the above. The results are shown in Table 1.
- the maximum shrinkage direction in the heat shrinkage measurement was the flow direction during film formation.
- Example 10 Except for changing the production method of the stretched film from uniaxial stretching by pantograph to roll stretching method (preheating roll temperature 90 ° C., stretching roll temperature 95 ° C., heat setting roll temperature 95 ° C., stretching ratio 2 times), the same as in Example 1.
- a stretched film was prepared and evaluated. The results are shown in Table 2.
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 11 A stretched film was prepared and evaluated in the same manner as in Example 10 except that the heat setting roll temperature was changed to 110 ° C. The results are shown in Table 2. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Example 12 2,5-furandicarboxylic acid 99.9 parts by mass, trimellitic anhydride (TMA) 0.1 part by mass, ethylene glycol 44.8 parts by mass, germanium dioxide (GeO 2 ) 0.012 parts by mass and phosphorous acid 0
- TMA trimellitic anhydride
- ethylene glycol 44.8 parts by mass ethylene glycol 44.8 parts by mass
- germanium dioxide (GeO 2 ) 0.012 parts by mass
- phosphorous acid 0 A solid phase polymerization pellet was obtained in the same manner as in Example 1 except that a slurry consisting of 0.012 parts by mass was used.
- the intrinsic viscosity of the copolyester thus obtained was 0.9 dL / g.
- the ratio of the monomer components constituting the copolymerized polyester was confirmed in the same manner as in Example 1.
- Comparative Example 1 An unstretched film was obtained in the same manner as in Example 1 except that the film thickness at the time of film formation was changed to 50 ⁇ m. The obtained unstretched film was evaluated in the same manner as in Example 1. The results are shown in Table 1.
- Comparative Example 2 A stretched film was prepared and evaluated in the same manner as in Example 5 except that the heat treatment temperature was changed to 140 ° C. The results are shown in Table 1. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Comparative Example 3 A stretched film was prepared and evaluated in the same manner as in Example 1 except that the film thickness during film formation was changed to 250 ⁇ m and the stretch ratio was changed to 5 times. The results are shown in Table 1. The maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- Comparative Example 4 An unstretched film was produced in the same manner as in Example 1 except that the sheet thickness at the time of film formation was changed to 500 ⁇ m. The unstretched film was used, and the stretching operation was changed to pantograph biaxial stretching [stretching temperature: 120 ° C., stretching ratio: flow direction (MD) 3.5 times, transverse direction (TD) 3.5 times]. A stretched film was prepared and evaluated in the same manner as in Example 1 except that the film was further heat treated at 170 ° C. for 2 minutes after being stretched. The results are shown in Table 1. The maximum shrinkage direction in the heat shrinkage measurement was the flow direction during film formation.
- Example 13 (1) Melt polycondensation A slurry consisting of 85.0 parts by mass of terephthalic acid (TA), 15.0 parts by mass of isophthalic acid (IPA) and 44.8 parts by mass of ethylene glycol (EG) is prepared under pressure (gauge pressure 0). The oligomer was produced by carrying out esterification reaction by heating to 250 ° C. at .25 MPa). The obtained oligomer was transferred to a polycondensation tank, and 0.012 parts by mass of germanium dioxide (GeO 2 ) and 0.012 parts by mass of phosphorous acid were added thereto. Under 0.1 kPa, melt polycondensation was performed at 280 ° C.
- TA terephthalic acid
- IPA isophthalic acid
- EG ethylene glycol
- copolyester having an intrinsic viscosity of 0.7 dL / g.
- the obtained copolyester was extruded into a strand form from a nozzle, cooled with water, and then cut into a cylindrical shape (diameter: about 2.5 mm, length: about 2.5 mm) to obtain a copolyester pellet (melt polymerized pellet). It was.
- melt polymerization pellets obtained as described above were put into a rolling vacuum solid phase polymerization apparatus, and precrystallization was performed at 120 ° C. for 10 hours under 0.1 kPa. .
- the first layer thickness is 80 ⁇ m
- the second layer thickness is 20 ⁇ m
- the width is A 300 mm unstretched coextruded film.
- the obtained unstretched coextruded film was cut into 100 mm squares, fixed on all sides by chucking, and subjected to pantograph uniaxial stretching [stretching ratio: 2 times, stretching direction: flow direction during film formation (MD)] at 95 ° C.
- the obtained stretched film was measured for intrinsic viscosity, crystallinity, thermal shrinkage, tensile test, heat seal test, and oxygen permeability.
- the second layer side was the heat seal surface.
- the obtained stretched film has an intrinsic viscosity of 0.8 dL / g, a crystallinity of 0%, and a heat shrinkage ratio of 26% in the stretching direction (flow direction during film formation) and 2% in the transverse direction (TD).
- the maximum shrinkage direction in the measurement of the heat shrinkage rate was the stretching direction (flow direction during film formation).
- the tensile strength in the flow direction (MD) was 72 MPa
- the Young's modulus was 2.2 GPa
- the tensile elongation (breaking elongation) was greater than 300%.
- the heat sealable temperature was 130 ° C.
- the obtained stretched film had an oxygen permeability of 40 cc / 20 ⁇ m ⁇ m 2 ⁇ day ⁇ atm. The results are shown in Table 3.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Mechanical Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Polyesters Or Polycarbonates (AREA)
Abstract
Description

ポリエステルの固有粘度は、フェノールと1,1,2,2-テトラクロロエタンとの等質量混合物を溶媒として用いて、温度30℃にて測定した。
示差走査熱量計(TA インスツルメント製TA Q2000型)を用いてポリエステルのガラス転移温度(Tg)を測定した。ポリエステルを昇温速度10℃/分で280℃まで昇温した後、-50℃/分で30℃まで急冷してから、再び昇温速度10℃/分で昇温したときのデータからガラス転移温度(Tg)を算出した。
前記示差走査熱量計を用いてフィルム中のポリエステルの結晶化度(χ)を求めた。ポリエステルフィルムを昇温速度10℃/分で昇温させることにより、融解エンタルピー(ΔHm(J/g)及び結晶化エンタルピーΔHc(J/g)を測定した。このとき測定されたΔHm及びΔHc、並びにポリエステルの完全結晶体融解熱量[140.1(J/g)]を用いて、下記式から結晶化度χ(%)を求めた。
χ(%)=[(ΔHm-ΔHc)/140.1]×100
厚み40~60μmのシーラントフィルムを80mm角の正方形に切り出し、製膜時の流れ方向(MD)および延伸方向をフィルムに印した。フィルムを125℃に保持された恒温装置中に20秒間静置した後、最大収縮方向における収縮率を求めた。
厚み40~60μmのシーラントフィルムを、製膜時の流れ方向(MD)が長手方向となるように、長さ100mm、幅15mmの短冊状に切り出し試験片を得た。オートグラフ(島津製作所製)を用いて、温度23℃、相対湿度65%、引張速度50mm/分にて前記試験片の引張試験を行うことにより、シーラントフィルムの流れ方向(MD)における引張強度、ヤング率及び引張伸度(破断伸度)を求めた。
幅15mmの短冊状に切り出した厚み40~60μmのシーラントフィルム2枚を重ね合わせ、YSS式ヒートシーラー(安田精機製作所製)を用いて、70~150℃の温度条件(10℃間隔)で、0.2MPaにて2秒間ヒートシールを行うことにより、異なる温度でヒートシールされたサンプルを作製した。オートグラフ(島津製作所製)を用いて、引張速度50mm/分にて各サンプルの剥離試験を行うことにより、2枚のフィルムを引き剥がすのに必要なピール強度(180度ピール)を測定した。ピール強度が5.0N/15mm以上となるときのシール温度をヒートシール可能温度とした。150℃でヒートシールした場合でもピール強度が5.0N/15mm以上とならなかった場合、ヒートシール不可とした。
酸素透過度は、酸素透過量測定装置(モダンコントロール社製「MOCON OX-TRAN2/20」)を用いて測定した。具体的には、測定装置にフィルムをセットし、温度20℃、湿度65%RHにて測定した。
(1)溶融重縮合
2,5-フランジカルボン酸(FDCA)100質量部、エチレングリコール(EG)47.7質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーをつくり、スラリーをエステル化槽に2時間かけてフィードしながら加圧下(ゲージ圧0.25MPa)で190℃に加熱してエステル化反応を行ってオリゴマーを製造した。得られたオリゴマーを重縮合槽に移し、これに0.1kPa下、270℃で120分間溶融重縮合させて、固有粘度0.7dL/gの共重合ポリエステルを得た。得られた共重合ポリエステルをノズルからストランド状に押出し30℃の温水中で冷却した後、円柱状(直径約2.5mm、長さ約2.5mm)に切断して、共重合ポリエステルのペレット(溶融重合ペレット)を得た。
以上のようにして得られた溶融重合ペレットを転動式真空固相重合装置に投入し、0.1kPa下、130~140℃で3時間予備結晶化を行った。
前記予備結晶化の後に、温度を上昇させて、0.1kPa下、190~200℃で100時間固相重合させて、固相重合ペレットを得た。得られた共重合ポリエステルの固有粘度は0.9dL/gであった。得られたポリエステルを構成する単量体成分の比率を1H-NMRスペクトル(装置:日本電子社製「JNM-GX-500型」、溶媒:重水素化トリフルオロ酢酸)により確認したところ、2,5-フランジカルボン酸単位:エチレングリコール単位:ジエチレングリコール単位=50:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は87℃であった。ポリエステルを構成する単量体成分の比率および、ガラス転移温度は固相重合の前後で変化しなかった。
得られた固相重合ペレットを120℃の乾燥機で終夜乾燥させたのち、一軸混練(シリンダー温度:260~280℃)させてから、T-ダイから85℃の冷却ロール上に押し出し、厚さ100μm、幅300mmの未延伸フィルムを作製した。得られた未延伸フィルムを100mm角に切り出し、四方をチャック固定し、95℃でパンタグラフ一軸延伸[延伸倍率:2倍、延伸方向:製膜時の流れ方向(MD)]を行った。得られた延伸フィルムの固有粘度の測定、結晶化度測定、熱収縮率測定、引張試験、ヒートシール試験及び酸素透過率測定を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。横方向(TD)の熱収縮率も合わせて表1に示す。
2,5-フランジカルボン酸48.5質量部、テレフタル酸51.5質量部、エチレングリコール46.25質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーをエステル化槽に2時間かけてフィードしながら加圧下(ゲージ圧0.25MPa)で220℃に加熱してエステル化反応を行ってオリゴマーを製造した。得られたオリゴマーを重縮合槽に移し、これに0.1kPa下、270℃で200分間溶融重縮合させて、固有粘度0.9dL/gの共重合ポリエステルを得た。得られた共重合ポリエステルをノズルからストランド状に押出し30℃の温水中で冷却した後、円柱状(直径約2.5mm、長さ約2.5mm)に切断して、共重合ポリエステルのペレット(溶融重合ペレット)を得た。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:テレフタル酸単位:エチレングリコール単位:ジエチレングリコール単位=25:25:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は83℃であった。得られた共重合ポリエステルを用いたこと、フィルム製膜時に75℃で2日間0.1kPa下で真空乾燥させたペレットを用いたこと以外は、実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
2,5-フランジカルボン酸48.5質量部、イソフタル酸51.5質量部、エチレングリコール46.25質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーを用いたこと以外は実施例2と同様にして溶融重合ペレットを得た。こうして得られた共重合ポリエステルの固有粘度は0.8dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:イソフタル酸単位:エチレングリコール単位:ジエチレングリコール単位=25:25:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は80℃であった。得られた共重合ポリエステルを用いたこと、以外は、実施例2と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
実施例1と同様にして溶融重合ペレットを得た。こうして得られた共重合ポリエステルの固有粘度は0.7dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:エチレングリコール単位:ジエチレングリコール単位=50:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は87℃であった。得られた共重合ポリエステルを用いたこと、フィルム製膜時に75℃で2日間0.1kPa下で真空乾燥させたペレットを用いたこと以外は、実施例2と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
フィルムを延伸した後、引き続き110℃で1分間熱処理したこと以外は実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
2,5-フランジカルボン酸100質量部、エチレングリコール45.3質量部、ビスフェノールAエチレンオキサイド付加物(EOBPA)10.1質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーを用いたこと、固相重合時間を200時間としたこと以外は、実施例1と同様にして固相重合ペレットを得た。こうして得られた共重合ポリエステルの固有粘度は1.2dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:エチレングリコール単位:ビスフェノールAエチレンオキサイド付加物由来の単位:ジエチレングリコール単位=50:46.25:2.5:1.25(モル比)であった。また、得られたポリエステルのガラス転移温度(Tg)は87℃であった。得られた共重合ポリエステルを用いたこと以外は、実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
2,5-フランジカルボン酸100質量部、1,4-シクロヘキサンジメタノール(CHDM)37.1質量部、エチレングリコール31.9質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーを用いたこと、固相重合を行わなかったこと以外は、実施例2と同様にして、溶融重合ペレットを得た。こうして得られた共重合ポリエステルの固有粘度は0.75dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:CHDM単位:エチレングリコール単位:ジエチレングリコール単位=50:16.5:32.0:1.5(モル比)であった。また、得られたポリエステルのガラス転移温度(Tg)は84℃であった。得られた共重合ポリエステルを用いたこと及び押し出し時のシリンダー温度を240~260℃に変更したこと以外は、実施例2と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
製膜時の膜厚を150μmに変更したこと、延伸倍率を3倍に変更したこと以外は実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
製膜時の膜厚を200μmに変更したこと、延伸操作をパンタグラフ二軸延伸[延伸温度:95℃、延伸倍率:流れ方向(MD)2倍、横方向(TD)2倍]に変更したこと以外は実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、製膜時の流れ方向であった。
延伸フィルムの作製方法をパンタグラフによる一軸延伸からロール延伸法(予熱ロール温度90℃、延伸ロール温度95℃、熱固定ロール温度95℃、延伸倍率2倍)に変更した以外は、実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表2に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
熱固定ロール温度を110℃に変更したこと以外は実施例10と同様にして延伸フィルムの作製及び評価を行った。結果を表2に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
2,5-フランジカルボン酸99.9質量部、無水トリメリット酸(TMA)0.1質量部、エチレングリコール44.8質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーを用いたこと以外は、実施例1と同様にして固相重合ペレットを得た。こうして得られた共重合ポリエステルの固有粘度は0.9dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、2,5-フランジカルボン酸単位:トリメリット酸単位:エチレングリコール単位:ジエチレングリコール単位=49.9:0.05:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は87℃であった。実施例10と同様にして延伸フィルムの作製及び評価を行った。結果を表2に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
製膜時の膜厚を50μmに変更したこと以外は実施例1と同様にして未延伸フィルムを得た。得られた未延伸フィルムの評価を実施例1と同様にして行った。結果を表1に示す。
熱処理温度を140℃に変更したこと以外は実施例5と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
製膜時の膜厚を250μmに変更したこと、延伸倍率を5倍に変更したこと以外は実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。
製膜時のシート厚みを500μmに変更したこと以外は実施例1と同様にして未延伸フィルムを作製した。当該未延伸フィルムを用いたこと、延伸操作をパンタグラフ二軸延伸[延伸温度:120℃、延伸倍率:流れ方向(MD)3.5倍、横方向(TD)3.5倍]に変更したこと、及び延伸した後さらに170℃で2分間熱処理したこと以外は実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、製膜時の流れ方向であった。
テレフタル酸100質量部及びエチレングリコール44.8質量部、二酸化ゲルマニウム(GeO2)0.012質量部及び亜リン酸0.012質量部からなるスラリーを用いたこと、エステル化温度を250℃にしたこと、重縮合を280℃で90分間行ったこと以外は、実施例1と同様にして溶融重合ペレットを得た。こうして得られたポリエステルの固有粘度は0.7dL/gであった。当該ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、テレフタル酸単位:エチレングリコール単位:ジエチレングリコール単位=50:48.75:1.25(モル比)であった。また、得られたポリエステルのガラス転移温度(Tg)は80℃であった。得られたポリエステルを用いたこと以外は、実施例2と同様にして未延伸フィルムを作製した。当該未延伸フィルムを用いたこと及び延伸温度を85℃に変更したこと以外は、実施例1と同様にして延伸フィルムの作製及び評価を行った。結果を表1に示す。熱収縮率測定における最大収縮方向は、製膜時の流れ方向であった。
(1)溶融重縮合
テレフタル酸(TA)85.0質量部、イソフタル酸(IPA)15.0質量部及びエチレングリコール(EG)44.8質量部からなるスラリーをつくり、加圧下(ゲージ圧0.25MPa)で250℃に加熱してエステル化反応を行ってオリゴマーを製造した。得られたオリゴマーを重縮合槽に移し、これに二酸化ゲルマニウム(GeO2)を0.012質量部、亜リン酸を0.012質量部添加した。0.1kPa下、280℃で90分間溶融重縮合させて、固有粘度0.7dL/gの共重合ポリエステルを得た。得られた共重合ポリエステルをノズルからストランド状に押出し水冷した後、円柱状(直径約2.5mm、長さ約2.5mm)に切断して、共重合ポリエステルのペレット(溶融重合ペレット)を得た。
以上のようにして得られた溶融重合ペレットを転動式真空固相重合装置に投入し、0.1kPa下、120℃で10時間予備結晶化を行った。
前記予備結晶化の後に、温度を上昇させて、0.1kPa下、185℃で20時間固相重合させて、固相重合ペレット(ペレットA)を得た。得られた共重合ポリエステルの固有粘度は0.9dL/gであった。当該共重合ポリエステルを構成する単量体成分の比率を実施例1と同様にして確認したところ、テレフタル酸単位:イソフタル酸単位:エチレングリコール単位:ジエチレングリコール単位=42.5:7.5:48.75:1.25(モル比)であった。また、得られた共重合ポリエステルのガラス転移温度(Tg)は75℃であった。
ペレットAと実施例1で得られた固相重合ペレット(ペレットB)を120℃の乾燥機で終夜乾燥させたのち、ペレットAを80重量分率に対しペレットBを20質量分率混合したものを第一層側の押出機で一軸混練(シリンダー温度:260~280℃)させ、ペレットBを第二層側の押出機で一軸混練(シリンダー温度:260~280℃)させてから、フィードブロックでそれらの溶融樹脂を合流させたのち、T-ダイから85℃の冷却ロール上に第二層が冷却ロールに接触するように押し出し、第一層厚さ80μm、第二層厚さ20μm、幅300mmの未延伸共押出フィルムを作製した。得られた未延伸共押出フィルムを100mm角に切り出し、四方をチャック固定し、95℃でパンタグラフ一軸延伸[延伸倍率:2倍、延伸方向:製膜時の流れ方向(MD)]を行った。得られた延伸フィルムの固有粘度の測定、結晶化度測定、熱収縮率測定、引張試験、ヒートシール試験及び酸素透過率測定を行った。ヒートシール試験は第二層側をヒートシール面とした。得られた延伸フィルムの固有粘度は0.8dL/g、結晶化度は0%、熱収縮率は延伸方向(製膜時の流れ方向)が26%、横方向(TD)が2%であり熱収縮率測定における最大収縮方向は、延伸方向(製膜時の流れ方向)であった。引張試験の結果、流れ方向(MD)における引張強度は72MPa、ヤング率は2.2GPa、引張伸度(破断伸度)は300%より大きかった。ヒートシール試験の結果、ヒートシール可能温度は130℃であった。また、得られた延伸フィルムの酸素透過率は40cc/20μm・m2・day・atmであった。結果を表3に示す。


Claims (7)
- ポリエステルからなる層を少なくとも最表層に有するシーラントフィルムであって、
前記ポリエステルが、2,5-フランジカルボン酸単位を20~50モル%、エチレングリコール単位、1,3-プロパンジオール単位及び1,4-ブタンジオール単位からなる群から選択される少なくとも1種のジオール単位を18~49.5モル%、及びジエチレングリコール単位を0.5~2.5モル%含有し、
前記シーラントフィルムの結晶化度が14%未満であり、かつ
前記シーラントフィルムが、125℃で20秒間静置した場合における最大収縮方向の収縮率が6%以上である延伸フィルムであることを特徴とするシーラントフィルム。 - 前記ポリエステルが、更に炭素数5以上の他のコモノマー単位を0.01~30モル%含有する請求項1記載のシーラントフィルム。
- 一軸延伸倍率が1.1~4.5倍である請求項1又は2記載のシーラントフィルム。
- 面延伸倍率が1.1~9.0倍である請求項1~3のいずれか記載のシーラントフィルム。
- 温度23℃、相対湿度65%、引張速度50mm/分の条件下で引張試験を行うことにより測定される引張伸度が130%以上である請求項1~4のいずれか記載のシーラントフィルム。
- 前記ポリエステルを押出成形することによってフィルムを得た後、該フィルムを一軸延伸する請求項1~5のいずれか記載のシーラントフィルムの製造方法。
- 前記ポリエステルを押出成形することによってフィルムを得た後、該フィルムを二軸延伸する請求項1~5のいずれか記載のシーラントフィルムの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780043353.1A CN109476857B (zh) | 2016-07-15 | 2017-07-13 | 密封剂膜和其制备方法 |
| US16/314,119 US11332574B2 (en) | 2016-07-15 | 2017-07-13 | Sealant film and method for producing same |
| JP2018527654A JP6890587B2 (ja) | 2016-07-15 | 2017-07-13 | シーラントフィルム及びその製造方法 |
| EP17827695.2A EP3486275A4 (en) | 2016-07-15 | 2017-07-13 | SEALING FILM AND METHOD FOR THE PRODUCTION THEREOF |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016140832 | 2016-07-15 | ||
| JP2016-140832 | 2016-07-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018012572A1 true WO2018012572A1 (ja) | 2018-01-18 |
Family
ID=60952116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/025497 Ceased WO2018012572A1 (ja) | 2016-07-15 | 2017-07-13 | シーラントフィルム及びその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11332574B2 (ja) |
| EP (1) | EP3486275A4 (ja) |
| JP (1) | JP6890587B2 (ja) |
| CN (1) | CN109476857B (ja) |
| WO (1) | WO2018012572A1 (ja) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019244797A1 (ja) * | 2018-06-22 | 2019-12-26 | 三菱ケミカル株式会社 | 非晶質ポリエステル樹脂、接着剤、塗料組成物、積層体 |
| JP2020082615A (ja) * | 2018-11-29 | 2020-06-04 | 三菱ケミカル株式会社 | シュリンクフィルム |
| JPWO2022004811A1 (ja) * | 2020-07-01 | 2022-01-06 | ||
| US11312830B2 (en) | 2016-03-30 | 2022-04-26 | Toyobo Co., Ltd. | Polyester film |
| US11318662B2 (en) | 2015-12-28 | 2022-05-03 | Toyobo Co., Ltd. | Layered polyester film |
| US11325362B2 (en) | 2015-12-28 | 2022-05-10 | Toyobo Co., Ltd. | Layered polyester film |
| US11325363B2 (en) | 2017-03-01 | 2022-05-10 | Toyobo Co., Ltd. | Laminate including polyester film having furandicarboxylate unit and heat-sealable resin layer, and packaging bag |
| US11511473B2 (en) * | 2017-03-01 | 2022-11-29 | Toyobo Co., Ltd. | Method for producing polyester film having furandicarboxylate unit |
| JP2023540343A (ja) * | 2020-09-17 | 2023-09-22 | コーロン インダストリーズ インク | ポリエステル重合体 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110128797A (zh) * | 2019-05-13 | 2019-08-16 | 无锡风鹏新材料科技有限公司 | 一种高温尺寸稳定性优异的双向拉伸聚酯薄膜及其生产方法 |
| JP7532807B2 (ja) * | 2020-02-28 | 2024-08-14 | Toppanホールディングス株式会社 | 積層体 |
| CN113501945B (zh) * | 2021-07-28 | 2022-07-12 | 浙江大学 | 一种高强度高韧性高阻隔性的无规共聚酯及其制备方法 |
| CN116100900B (zh) * | 2022-10-09 | 2025-05-16 | 安徽国风新材料股份有限公司 | 一种高热封强度直线易撕聚酯薄膜及其制备方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007146153A (ja) | 2005-11-07 | 2007-06-14 | Canon Inc | 高分子化合物およびその合成方法 |
| JP2008291244A (ja) * | 2007-04-24 | 2008-12-04 | Mitsubishi Chemicals Corp | フラン構造を含むポリエステル樹脂の製造方法 |
| JP2011520005A (ja) * | 2008-05-08 | 2011-07-14 | ノバモント・ソシエタ・ペル・アチオニ | 脂肪−芳香族生分解性ポリエステル |
| JP2012229395A (ja) * | 2011-04-11 | 2012-11-22 | Canon Inc | プラスチックフィルム |
| JP2014118457A (ja) * | 2012-12-14 | 2014-06-30 | Nippon Ester Co Ltd | ポリエステル樹脂組成物及びそれからなるダイレクトブロー成形品 |
| JP2014530948A (ja) * | 2011-10-24 | 2014-11-20 | フラニクス テクノロジーズビー. ブイ. | ボトル、フィルムまたは繊維用途に使用されるポリマー骨格内に2,5−フランジカルボキシレート部分を有するポリマー生成物を調製するためのプロセス |
| JP2015506389A (ja) * | 2011-12-29 | 2015-03-02 | ナチュラ コスメティコス ソシエダッド アノニマ | 2,5−フランジカルボン酸からのポリ(エチレン2,5−フランジカルボキシレート)の製造方法、その使用、そのポリエステル化合物及び配合物 |
| JP2015507684A (ja) * | 2012-01-04 | 2015-03-12 | ペプシコ, インコーポレイテッドPepsiCo Inc. | バイオマスから調製された2,5−フランジカルボン酸系ポリエステル |
| JP2015511662A (ja) * | 2012-03-30 | 2015-04-20 | ロケット フレールRoquette Freres | ポリマー、それを合成するためのプロセスおよびそれを含む組成物 |
| JP2015514151A (ja) * | 2012-03-30 | 2015-05-18 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニーE.I.Du Pont De Nemours And Company | ポリエステルおよびそれから製造される物品 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10137625B2 (en) * | 2011-07-08 | 2018-11-27 | Toray Plastics (America), Inc. | Biaxially oriented bio-based polyester films and laminates |
| US20150353692A1 (en) * | 2012-12-20 | 2015-12-10 | Dow Global Technologies Llc | Barrier films of fdca-based polyesters |
| CN104955646B (zh) * | 2012-12-20 | 2017-05-24 | 陶氏环球技术有限责任公司 | 基于fdca的聚酯的多层膜 |
| WO2014175313A1 (ja) * | 2013-04-26 | 2014-10-30 | 東洋紡株式会社 | シーラント用途のポリエステル系フィルム、積層体及び包装袋 |
| EP3186064B1 (en) * | 2014-08-25 | 2021-12-29 | Furanix Technologies B.V. | Process for producing an oriented film comprising poly(ethylene-2,5-furandicarboxylate) |
-
2017
- 2017-07-13 WO PCT/JP2017/025497 patent/WO2018012572A1/ja not_active Ceased
- 2017-07-13 CN CN201780043353.1A patent/CN109476857B/zh active Active
- 2017-07-13 JP JP2018527654A patent/JP6890587B2/ja active Active
- 2017-07-13 US US16/314,119 patent/US11332574B2/en active Active
- 2017-07-13 EP EP17827695.2A patent/EP3486275A4/en not_active Withdrawn
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007146153A (ja) | 2005-11-07 | 2007-06-14 | Canon Inc | 高分子化合物およびその合成方法 |
| JP2008291244A (ja) * | 2007-04-24 | 2008-12-04 | Mitsubishi Chemicals Corp | フラン構造を含むポリエステル樹脂の製造方法 |
| JP2011520005A (ja) * | 2008-05-08 | 2011-07-14 | ノバモント・ソシエタ・ペル・アチオニ | 脂肪−芳香族生分解性ポリエステル |
| JP2012229395A (ja) * | 2011-04-11 | 2012-11-22 | Canon Inc | プラスチックフィルム |
| JP2014530948A (ja) * | 2011-10-24 | 2014-11-20 | フラニクス テクノロジーズビー. ブイ. | ボトル、フィルムまたは繊維用途に使用されるポリマー骨格内に2,5−フランジカルボキシレート部分を有するポリマー生成物を調製するためのプロセス |
| JP2015506389A (ja) * | 2011-12-29 | 2015-03-02 | ナチュラ コスメティコス ソシエダッド アノニマ | 2,5−フランジカルボン酸からのポリ(エチレン2,5−フランジカルボキシレート)の製造方法、その使用、そのポリエステル化合物及び配合物 |
| JP2015507684A (ja) * | 2012-01-04 | 2015-03-12 | ペプシコ, インコーポレイテッドPepsiCo Inc. | バイオマスから調製された2,5−フランジカルボン酸系ポリエステル |
| JP2015511662A (ja) * | 2012-03-30 | 2015-04-20 | ロケット フレールRoquette Freres | ポリマー、それを合成するためのプロセスおよびそれを含む組成物 |
| JP2015514151A (ja) * | 2012-03-30 | 2015-05-18 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニーE.I.Du Pont De Nemours And Company | ポリエステルおよびそれから製造される物品 |
| JP2014118457A (ja) * | 2012-12-14 | 2014-06-30 | Nippon Ester Co Ltd | ポリエステル樹脂組成物及びそれからなるダイレクトブロー成形品 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3486275A4 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11325362B2 (en) | 2015-12-28 | 2022-05-10 | Toyobo Co., Ltd. | Layered polyester film |
| US11318662B2 (en) | 2015-12-28 | 2022-05-03 | Toyobo Co., Ltd. | Layered polyester film |
| US11312830B2 (en) | 2016-03-30 | 2022-04-26 | Toyobo Co., Ltd. | Polyester film |
| US11511473B2 (en) * | 2017-03-01 | 2022-11-29 | Toyobo Co., Ltd. | Method for producing polyester film having furandicarboxylate unit |
| US11325363B2 (en) | 2017-03-01 | 2022-05-10 | Toyobo Co., Ltd. | Laminate including polyester film having furandicarboxylate unit and heat-sealable resin layer, and packaging bag |
| EP3812413A4 (en) * | 2018-06-22 | 2021-08-18 | Mitsubishi Chemical Corporation | AMORPHIC POLYESTER RESIN, ADHESIVE, COATING COMPOSITION AND MULTI-LAYER BODY |
| WO2019244797A1 (ja) * | 2018-06-22 | 2019-12-26 | 三菱ケミカル株式会社 | 非晶質ポリエステル樹脂、接着剤、塗料組成物、積層体 |
| JPWO2019244797A1 (ja) * | 2018-06-22 | 2021-05-06 | 三菱ケミカル株式会社 | 非晶質ポリエステル樹脂、接着剤、塗料組成物、積層体 |
| CN112272682A (zh) * | 2018-06-22 | 2021-01-26 | 三菱化学株式会社 | 非晶质聚酯树脂、粘接剂、涂料组合物、层叠体 |
| JP7294133B2 (ja) | 2018-06-22 | 2023-06-20 | 三菱ケミカル株式会社 | 非晶質ポリエステル樹脂、接着剤、塗料組成物、積層体 |
| JP2020082615A (ja) * | 2018-11-29 | 2020-06-04 | 三菱ケミカル株式会社 | シュリンクフィルム |
| JP2023095930A (ja) * | 2018-11-29 | 2023-07-06 | 三菱ケミカル株式会社 | シュリンクフィルム |
| JP7533681B2 (ja) | 2018-11-29 | 2024-08-14 | 三菱ケミカル株式会社 | シュリンクフィルム |
| WO2022004811A1 (ja) * | 2020-07-01 | 2022-01-06 | キリンホールディングス株式会社 | ポリエチレンフラノエート、高粘度ポリエチレンフラノエートの製造方法、ポリエステル組成物、ポリエステル製ボトル、ポリエステル製ボトルの製造方法及び飲料製品 |
| JPWO2022004811A1 (ja) * | 2020-07-01 | 2022-01-06 | ||
| US12570792B2 (en) | 2020-07-01 | 2026-03-10 | Kirin Holdings Kabushiki Kaisha | Polyethylene furanoate, highly-viscous polyethylene furanoate manufacturing method, polyester composition, polyester bottle, polyester bottle manufacturing method, and beverage product |
| JP2023540343A (ja) * | 2020-09-17 | 2023-09-22 | コーロン インダストリーズ インク | ポリエステル重合体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3486275A4 (en) | 2020-03-04 |
| US11332574B2 (en) | 2022-05-17 |
| CN109476857B (zh) | 2022-07-29 |
| JPWO2018012572A1 (ja) | 2019-04-25 |
| JP6890587B2 (ja) | 2021-06-18 |
| CN109476857A (zh) | 2019-03-15 |
| US20190225745A1 (en) | 2019-07-25 |
| EP3486275A1 (en) | 2019-05-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6890587B2 (ja) | シーラントフィルム及びその製造方法 | |
| CN108883569B (zh) | 密封膜及其制造方法 | |
| CN107868265B (zh) | 用于收缩膜应用的用聚合增塑剂塑化的共聚酯 | |
| JP6440336B2 (ja) | フランジカルボン酸ユニットを有するポリエステルフィルム | |
| KR20160002852A (ko) | 실란트 용도의 폴리에스테르계 필름, 적층체 및 포장 주머니 | |
| CN109996829A (zh) | 聚酯、其制造方法和由其形成的成型品 | |
| WO2003037956A9 (en) | Crystalline polyglycolic acid, polyglycolic acid composition and processes for production of both | |
| JP7251663B2 (ja) | 二軸配向ポリエステルフィルム、及びその製造方法 | |
| JP2002518536A (ja) | ガスバリヤー性の改良されたポリエステル/フェニレンジ(オキシ酢酸)コポリエステルブレンド | |
| KR970006674B1 (ko) | 공중합 폴리에스테르 | |
| TW202225253A (zh) | 可收縮聚酯膜 | |
| JP4563090B2 (ja) | ポリエステル系樹脂組成物、該樹脂組成物からなる熱収縮性ポリエステル系フィルム、成形品および容器 | |
| JP2002020471A (ja) | 共重合ポリエステル樹脂 | |
| JP2001288283A (ja) | 熱収縮性ポリエステル系フィルム | |
| JP7466846B2 (ja) | ポリエステル樹脂、並びに、当該ポリエステル樹脂を含む成形体、延伸フィルム及びボトル | |
| JP2004034451A (ja) | 熱収縮性ポリエステル系フィルムの製造方法 | |
| KR101705243B1 (ko) | 백색 열수축성 적층 필름 및 이를 포함하는 라벨 | |
| JP2002020470A (ja) | 共重合ポリエステル樹脂 | |
| JP4502091B2 (ja) | 熱収縮性ポリエステル系フィルム | |
| KR101750925B1 (ko) | 백색 열수축성 적층 필름 및 이를 포함하는 라벨 | |
| KR20170028620A (ko) | 백색 열수축성 적층 필름 및 이를 이용한 열수축성 라벨 | |
| JP2011094159A (ja) | 熱収縮性ポリエステル系フィルムおよびその製造方法 | |
| JP2652878B2 (ja) | ポリエステル中空成形体 | |
| JPH02150419A (ja) | ガスバリヤー性に優れたポリエステル | |
| JP2002201346A (ja) | ポリエステル樹脂組成物、その製法および用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018527654 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17827695 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017827695 Country of ref document: EP Effective date: 20190215 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2017827695 Country of ref document: EP |