WO2019022018A1 - 多型検出法 - Google Patents
多型検出法 Download PDFInfo
- Publication number
- WO2019022018A1 WO2019022018A1 PCT/JP2018/027535 JP2018027535W WO2019022018A1 WO 2019022018 A1 WO2019022018 A1 WO 2019022018A1 JP 2018027535 W JP2018027535 W JP 2018027535W WO 2019022018 A1 WO2019022018 A1 WO 2019022018A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sequence
- sequence data
- length
- data
- target
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

















Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/10—Sequence alignment; Homology search
Definitions
- the present invention relates to the field of information processing of sequence information, especially sequence information of biomolecules such as genomes.
- next-generation sequencers whole genome sequence information of organisms has come to be obtained.
- polymorphism information from the sequence information of the next-generation sequencer and examining the association with the phenotype, it leads to the identification of the gene responsible for the phenotype.
- Acquisition of accurate polymorphism information is a basic technology that is required in a wide range of fields, such as diagnosis of human genetic diseases, identification of species and varieties, etc., as well as crop breeding. If type information can be obtained, the impact is large.
- Polymorphism detection using nucleotide sequence data from the next-generation sequencer is performed by first obtaining the positional information and mismatch information on the reference sequence using a mapping program such as bwa or bowtie for the sequence data.
- a mapping program such as bwa or bowtie for the sequence data.
- polymorphism information such as SNP and indel is extracted by polymorphism extraction programs such as Samtools and GATK.
- methods are provided for detecting polymorphisms between two or more sequences.
- the method of the present invention enables the detection of polymorphisms between multiple sequence data without considering the position in the full-length sequence of individual sequences in the sequence data.
- the methods of the invention detect polymorphisms without the need for linking individual sequences (e.g., short reads from next-generation sequencers) in the sequence data into longer sequences (e.g., assembly).
- One feature is that it can be done.
- the present invention provides the following items.
- (Item 1) A method for detecting polymorphism in control sequence data in target sequence data, comprising: a) providing the frequency of occurrence of each subsequence of the subset of subsequences of length k of the target sequence data; b) providing the frequency of occurrence of each subsequence of the subset of subsequences of length k of the control sequence data; c) comparing the target sequence with a control sequence and detecting polymorphism based on the comparison of the distribution of the occurrence frequency, where k is any one of the target sequence and the control sequence, which is shorter A method that is an integer less than or equal to the total length of the other.
- the method further includes the step of calculating the distribution of the appearance frequency for the portion of length x for each sequence common to the sequence portion of length kx in the partial array, wherein x is less than k
- the method according to the above item which is a positive integer.
- the comparison includes a difference in the distribution of the frequency of occurrence of the portion of length x in the sequences common to the sequence portion of length k-x in the partial array Method described.
- (Item 4) In any of the items described above, including the step of grouping sequence portions of length kx in the partial sequence into unique sequences, wherein x is a positive integer less than k. Method described.
- the target sequence data and the control sequence data are base sequence data derived from the genome of an organism, and the k is a length that excludes accidental identity at different places in the genome of the organism. , A method according to any of the preceding items.
- the target sequence data and the control sequence data are base sequence data, and a sequence of the portion of length x in the subset of the target sequence data, which has the highest frequency in the subset of the control sequence data
- the array of the length x part is detected as a homopolymorphism in the target sequence data , A method according to any of the preceding items.
- the target sequence data and the control sequence data are base sequence data, and the sequence of the portion of length x in the target subset has the same length x as the most frequent one in the subset of control sequence data If there is an array of a portion of x and there is an array portion of length k x in which an array of a portion of length x different from the most frequent one in the subset of control sequence data is present, the length x
- the method according to any of the above items, wherein the sequence of part of is detected as heteropolymorphism in the subject sequence data.
- the partial array whose frequency of occurrence is lower is noise as compared with the frequency of occurrence predicted from the amount of target array data.
- the target sequence data and the control sequence data are base sequence data, and the partial sequence having an appearance frequency of less than [(target sequence data amount) ⁇ (1-accuracy)] / (target genome size) +1 is noise The method according to any one of the above items.
- the target sequence data is sequence data obtained from an individual, and the control sequence data is another individual of the same type as the individual, or sequence data obtained from a database. How to describe.
- the item of the item, wherein the target sequence data is sequence data obtained from a tissue sample of an individual, and the control sequence data is sequence data obtained from another tissue of the individual or a database.
- the method described in either. (Item 22) The target sequence data is sequence data obtained from a cell sample, and the control sequence data is sequence data obtained from another cell or a database.
- Method. (Item 23) In any of the items, the polymorphism is substitution, insertion, deletion, copy number variation (CNV), short tandem repeat polymorphism (STRP), inversion or translocation. Method described.
- the polymorphism is a substitution.
- the method according to any of the items above, further comprising the step of identifying the position of the polymorphism in the reference sequence relative to the target sequence.
- the target sequence data and the control sequence data are nucleotide sequence data derived from the genome of an organism, and further comprising the step of specifying the position of the polymorphism on the genome. the method of.
- the method further includes the step of comparing and checking the detected polymorphic site with target sequence data and / or control sequence data using a query sequence set generated from the reference sequence or control sequence, The method described in any of the items.
- the target sequence data and the control sequence data are nucleotide sequence data, and for the detected polymorphic site, a reference sequence relative to the sequence data of the complementary strand of the target sequence data and / or the control sequence data Or the method described in any of the above items, further comprising the step of comparing and confirming with a set of query sequences generated from a control sequence.
- the target sequence data and the control sequence data are base sequence data, and for a sequence data having a mutated base of the target sequence data and / or the control sequence data for the detected polymorphic site.
- the method according to any of the above items further comprising the step of comparing and confirming with a query sequence set generated from a reference sequence or a control sequence.
- the method according to any of the items, wherein the target sequence data and the control sequence data are nucleotide sequence data derived from the genome of an organism, and the sequence of the genome is unknown.
- (Item X1) Any of the items described above, further including assigning, as an identifier of the polymorphism, a sequence containing at least a part of a non-polymorphic part in a partial sequence containing polymorphism to control sequence data in target sequence data Or the method described in paragraph 1.
- (Item X2) The method according to any of the items above, including mapping the polymorphism identifier to a reference sequence and locating the polymorphism on a reference.
- a program for causing a computer to execute a method of detecting polymorphism in control sequence data in target sequence data a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; d) comparing the target sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- a recording medium storing a program for causing a computer to execute a method of detecting polymorphism of control sequence data in target sequence data, wherein the method comprises a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; d) comparing the target sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- (Item 35A) A recording medium according to the item, having the feature described in any one or more of the items.
- (Item 36) A step in which the method displays a sequence (which may be the entire partial sequence) including at least a portion of a non-polymorphic portion in the partial sequence as a name of the detected polymorphism.
- a system for detecting polymorphism in control sequence data in target sequence data comprises: A sequence data processing unit configured to provide an appearance frequency of each partial sequence of the target sequence data and a subset of the length k of the control sequence data, wherein k is the target sequence and An array data processing unit having a length equal to or less than the total length of the control array; A sequence data calculator configured to compare the target sequence and the control sequence, and to detect polymorphism based on the difference in the distribution of the frequency of occurrence.
- a sequence data calculator configured to compare the target sequence and the control sequence, and to detect polymorphism based on the difference in the distribution of the frequency of occurrence.
- Insertion, deletion, inversion in the target sequence data by the step of comparing the characters between the partial sequence sites with the characters on the corresponding control sequence and sequentially comparing them starting from the partial sequence site and detecting the mismatched sites.
- a process of detecting translocations or substitutions. 39A. A method according to any of the preceding items having the features described in any one or more of the items.
- (Item 40) A method for detecting polymorphism in reference sequence data in target sequence data, comprising the step of creating, from the reference sequence data, a k-length partial array set of reference sequences associated with each position information.
- the method further includes the step of detecting a non-matching portion by comparing characters on the partial sequence site in the target sequence data with characters on the corresponding control sequence, when the positional relationship is not different, A process further comprising the step of determining that a substitution exists if there is a non-coincidence site; Method simultaneously, in parallel, or sequentially. 40A. The method of claim 40 having the features described in any one or more of the items.
- (Item A1) A comparison method of target array data and control array data, Assigning a sequence including at least a portion of a non-polymorphic portion in a partial sequence including polymorphism to control sequence data in target sequence data as an identifier of the polymorphism.
- (Item A1A) A method according to any of the preceding items having the features described in any one or more of the items. A2. The method of any of the above items, including mapping the polymorphic identifier to a reference sequence and locating the polymorphic location on a reference.
- the invention makes it possible to detect polymorphisms, in particular substitutions, precisely between two or more sequences without having to consider the position in the full-length sequence.
- substitution mutations using k-length sequences
- one major feature is that polymorphism detection can be performed before genome mapping. And, even in organisms where the reference sequence does not exist, polymorphism detection is possible, and k-mer itself can be used as the name of polymorphism. Therefore, it is possible to bring about a major change in genetic analysis such as linkage analysis. There is.
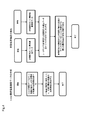
- FIG. 1 is a flow diagram illustrating one embodiment of the method of the present invention.
- FIG. 2 is a flow diagram illustrating one embodiment of the method of the present invention.
- FIG. 3 is a flow diagram illustrating one embodiment of the method of the present invention. In FIG.
- FIG. 4 is a flow diagram illustrating one embodiment of the method of the present invention.
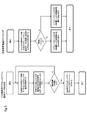
- FIG. 4 shows an example of a process of comparing a target sequence and a control sequence and detecting polymorphism based on the comparison of the distribution of the frequency of appearance.
- FIG. 5 is a flow diagram illustrating one embodiment of the method of the present invention. In FIG. 5, an example of the process of specifying the position of polymorphism in the reference sequence is shown.
- FIG. 6 is a flow diagram illustrating one embodiment of the method of the present invention. In FIG.
- FIG. 7 shows a part of comparison results of k-mer sequences between the control sequence (N1) and the target sequence (N1S7).
- the subsequences of the control sequence and the k-length subsequence subset of the subject sequence, and the frequency of occurrence of each of the subsequences in the control sequence and the k length subsequence subset of the subject sequence are shown.
- the comparison indicates that the sequence derived from the control sequence is not detected in the target sequence, and conversely, the sequence derived from the target sequence is not detected in the reference sequence, thus indicating that the polymorphism can be detected.
- FIG. 7 is an example of the results observed when the mutation is homozygous.
- the k-mer partial sequence of the reference sequence in FIG. 7 corresponds to SEQ ID NO: 1 to 60 respectively from the top, and the k-mer portion of the target sequence corresponds to SEQ ID NO: 1 to 20, 61 to 80, 40 to 60 from the top Do.
- FIG. 8 shows a part of comparison results of k-mer sequences between the control sequence (N1) and the target sequence (N1S7).
- the subsequences of the control sequence and the k-length subsequence subset of the subject sequence, and the frequency of occurrence of each of the subsequences in the control sequence and the k length subsequence subset of the subject sequence are shown.
- the comparison indicates that the sequence derived from the control sequence is not detected in the target sequence, and conversely, the sequence derived from the target sequence is not detected in the reference sequence, thus indicating that the polymorphism can be detected.
- the underlined bases differ between the control and the subject, indicating polymorphism.
- FIG. 8 is an example of the result observed when the mutation is heterozygous.
- FIG. 9 shows a sequence corresponding to the sequence starting from the corresponding position of the rice reference sequence by aligning a partial sequence subset of the k-mer sequence of the control sequence (N1) and the subject sequence (N1S5, N1S6, N1S7, N1S10) It is a figure which shows the result of having calculated
- FIG. 10 shows a part of the results of detection and confirmation of polymorphisms by polymorphic edge detection between the control sequence (N1) and the subject sequence (N1S5, N1S6, N1S7, N1S10).
- Chr indicates a chromosome number
- Pos indicates a position on the chromosome
- Ref indicates a base of the position in the reference sequence
- Alt indicates a base of the position where a mutation is present.
- the column of Polymorphic Edge Detection shows the frequency of appearance of the final base in the 20-mer partial sequence of Control sequence (N1) and the frequency of appearance of the final base in the 20-mer partial sequence of each target (Target) sequence. The portion where the most frequent final base differs is detected as a polymorphism.
- the column of Verify shows the frequency of occurrence of Control sequence data and Target sequence data for a query sequence set (Ref: wild type query set, Alt: variant query set) created from the reference sequence.
- the genotype of each sample at each site finally determined is shown in the Genotype column (M: homo mutation, H: hetero mutation, W: wild type).
- FIG. 11 shows a part of the result of detection and confirmation of polymorphism by Polymorphic Edge Detection between the control sequence (N1) and the subject sequence (N1S5, N1S6, N1S7, N1S10).
- Chr indicates a chromosome number
- Pos indicates a position on the chromosome
- Ref indicates a base of the position in the reference sequence
- Alt indicates a base of the position where a mutation is present.
- the column of Polymorphic Edge Detection shows the frequency of appearance of the final base in the 20-mer partial sequence of Control sequence (N1) and the frequency of appearance of the final base in the 20-mer partial sequence of each target (Target) sequence. The portion where the most frequent final base differs is detected as a polymorphism.
- the column of Verify indicates the frequency of occurrence of Control sequence data and Target sequence data for the query sequence set generated from the reference sequence. The genotype of each sample at each site finally determined is shown in the Genotype column (M: homo mutation, H: hetero mutation, W: wild type).
- FIG. 12 is a figure which shows the result of having amplified the target polymorphic part by PCR method about the rice sample of each generation, and having confirmed the base sequence by the Sanger method.
- the genotype of each sample is shown as M: homomutation, H: heteromutation, W: wild type.
- Chr indicates a chromosome number
- Pos indicates a position on the chromosome
- Ref indicates a base of the position in the reference sequence
- Alt indicates a base of the position where a mutation is present. It can be seen that a hetero mutation occurs while superposing generations with N1, N1 S1, N1 S2, N1 S3, N1 S4, N1 S5, N1 S6, N1 S7, N1 S8, N1 S9, N1 S10, and then a mode of fixation as a homo mutation is observed.
- FIG. 13 shows a part of the analysis result of detecting polymorphisms to reference (control) sequences generated from reference genome sequence data, using sequence data of Yoruba male (NA18507) in Nigeria.
- the target sequence data was downloaded and used data already analyzed by Illumina's next-generation sequencer and registered and released to NCBI.
- FIG. 14 is a schematic view of the creation of a query sequence set in the confirmation step that can be performed in the present invention.
- the upper sequence is a reference sequence, and a T indicated by underlined bold indicates a polymorphic site detected.
- a set of partial sequences containing the polymorphic site can be generated to be a query sequence set.
- FIG. 15A schematically illustrates an embodiment of the system of the present invention.
- FIG. 15B schematically illustrates a further embodiment of the system of the present invention.
- FIG. 16 schematically shows an embodiment of the method of the present invention.
- FIG. 17 shows a part of the results of detection of copy number variation (CNV) according to the method of the present invention.
- the copy number variation site has been started from the 26694795 position of rice chromosome 7 (indicated by an arrow in the figure).
- the copy number variation site detected corresponds to the retrotransposon Tos17. Since this transposon is 4.1 kb, only the part of the first junction is shown in FIG.
- FIG. 18 is a flow diagram showing an embodiment in the case of combining the polymorphism detection flow using the frequency of the k-mer sequence and the polymorphism detection flow using the positional relationship of partial arrays.
- array refers to a plurality of variables, each of which takes some value, which further includes information of the order of the plurality of variables. Typically, it is displayed as a character string.
- target sequence refers to any sequence for which polymorphism is to be detected, and may also be referred to as “target”, “target sequence”, or “target” in the present specification.
- control sequence refers to any sequence used as a reference for detecting differences from the sequence as polymorphisms, and as used herein, “control”, “reference sequence”, It may be written as “comparison sequence” or "control”.
- polymorphism refers to any portion of the subject sequence that differs from the control sequence.
- mutation can also be used in the same meaning.
- reference sequence refers to a sequence that can be treated as a full length sequence of the subject sequence and / or control sequence. Which sequence is to be used as a full-length sequence is appropriately determined according to the sequence used as the target sequence and / or control sequence, and is not limited to the exemplified ones, but is present in, for example, a database on the web A whole genome sequence, a chromosome full length sequence, a gene full length sequence, a plasmid full length sequence, an exon full length sequence, a protein full length sequence or the like can be used as a reference sequence.
- sequence data refers to data that gives information about a certain sequence. Typically, the sequence itself can also be referred to as sequence data, and data giving information on a part of the sequence (eg, analysis data obtained by sequencing of genomic sequences) is also included as sequence data.
- partial sequence of a sequence refers to any sequence contained in the sequence.
- subset refers to any subset of a set of sequences and a set of subsequences of those sequences.
- next-generation sequencing is a sequencing technique that parallelizes the sequencing process to generate tens to hundreds of millions of sequencing data in a single run.
- Next-generation sequencer refers to an apparatus for performing next-generation sequencing.
- coverage refers to how many times the amount of sequence data corresponds to the total sequence length. It may be called “coverage”, “-double reading”, etc.
- sequence structure refers to a series of physically separated sequences in a sequence.
- sequence construct each of the chromosomes can be referred to as a sequence construct.
- translocation refers to a polymorphism in which a partial sequence on one sequence structure is moved on another sequence structure in a sequence having a plurality of sequence structures.
- junction refers to the boundaries between identical and non-identical parts of two sequences which are partially identical.
- identifier refers to a name given to distinguish one polymorphism from another polymorphism. Generally, although it is often described by the start position and type of polymorphism, the identifier described herein can be used.
- edge refers to the end of the portion of the sequence that contains the polymorphism.
- the present invention provides a method for detecting polymorphism in control sequence data in target sequence data.
- the method comprises the steps of: a) providing the frequency of occurrence of each subsequence of the subset of length k subsequences of the subject sequence data; b) each of the subsets of length k subsequences of the control sequence data And c) comparing the target sequence with a control sequence and detecting polymorphism based on the comparison of the distribution of the occurrence frequency, wherein k Provides a method, which is an integer less than or equal to the total length of the subject sequence and the control sequence, whichever is shorter.
- An exemplary flow of the invention is illustrated in FIG.
- the method of the present invention can detect polymorphisms by direct comparison from two sequence data (for example, next-generation sequencer analysis results) of target sequence data and control sequence data. Fundamentally different.
- polymorphism detection in a base sequence a method of direct comparison without consideration of the position on the genome is considered to be novel.
- the method of the present invention is characterized by obtaining a set of partial sequences of constant length (k length) from sequence data. In one embodiment, the method of the present invention is characterized by obtaining, from the sequence data, the frequency distribution of each partial sequence in the set of k-long partial sequences. In some embodiments, from the array data, a set of k-length subarrays is created while shifting the arrays one by one.
- data having the same array of kx (x is 1, etc.) is sorted to detect the frequency of different portions (corresponding to the x-length portions).
- the X-length portion is not limited in position in the partial sequence, and may be the central portion in the sequence. However, making the X-length part the end of the partial sequence (for example, the 3 'end or 5' end in the base sequence) is preferable in order to remarkably simplify and speed up the processing such as sorting.
- PED polymorphic edge detection
- k it is possible to cite an arbitrary value up to the length of each sequence of sequence data (for example, each short read of next-generation sequencer), for example, about 500, about 400, About 300, about 200, about 100, about 50, about 40, about 30, about 25, about 20, about 15, and the like can be mentioned.
- the data of k-mer sequence increases exponentially (for example, in the case of a base sequence, the combination of bases is quadrupled for each increase of k by one base).
- the letter of part x of the data in which the k-x length sequence is the same is different among comparison objects, it is considered that the letter includes polymorphism (mutation). Insertion and deletion mutations can also detect the terminal letters of mutations. For example, it is considered that polymorphism (mutation) is included in the bases when the bases of the part of x of the data in which the base sequences of the kx length are the same differ among comparison objects.
- data is calculated that is ordered by the number of occurrences for the same sequence.
- This process can be conveniently performed using a computer, for example on Unix: It can be implemented as follows, and can generate sorted data (in strings) and numerical data indicating frequency. Furthermore, when combining target and control frequency data into the same k-mer, it is possible to use, for example, join command etc. in Unix.
- sequence data it is also possible to assess differences in the sequence data by comparing the number of occurrences of the sequence with the coverage of the sequence data. For example, for sequence data from sequence analysis with a data amount of 40 times that of a genome sequence, one having an appearance frequency of 1 can be considered as noise.
- the present invention as a method for directly detecting polymorphisms from nucleotide sequence data obtained from a next-generation sequencer, it becomes possible to detect polymorphisms between two types of samples and between reference sequences and samples. . Further, since the partial sequence of length k and the partial sequence of k ⁇ x are unique sequences without duplication, the sequence itself can be used as a polymorphic identifier (name). Therefore, even when the positional relationship of polymorphisms on the genome can not be determined because the reference genome sequence is not determined, it can be used as a universally unique unique polymorphism identifier.
- a method of comparing target sequence data with control sequence data wherein at least a portion of a non-polymorphic portion in a partial sequence containing polymorphism to control sequence data in target sequence data
- a method is provided, including assigning the containing sequence as a polymorphic identifier. Also, it is possible to map polymorphism identifiers to a reference sequence to specify the position of polymorphisms on the reference.
- the identifier may include, in addition to at least part of the non-polymorphic part, the polymorphism itself. Identifiers including polymorphic bases can be used for linkage analysis although mapping to a reference sequence is difficult. For example, AAACCACTTCACGTTTCCA A AAACCACTTCACGTTTCCA G In the polymorphic example, The expression “A / G heterotype” of type A AAACC ACT TC ACGTTTTCCA of AAACC ACT TC ACGT TTCCA is an example of the description.
- AAACCACTTCACGTTTCCAA type As an example of the notation method including polymorphism, AAACCACTTCACGTTTCCAA type, AAACCACTTCACGTTTCCAG type, And heterotype is AAACCACTTCACGTTTCCAA / AAACCACTTCACGTTTCCAG It is possible to write two types at the same time.
- One embodiment of the present invention is a method of detecting polymorphism in control sequence data in target sequence data.
- the method comprises the step of providing the frequency of occurrence of each subsequence of the subset of the subsequence of length k of the target sequence data. k is an integer equal to or less than the total length of the target sequence and the control sequence, whichever is shorter.
- the method comprises the step of providing the frequency of occurrence of each subsequence of the subset of the subsequence of length k of the control sequence data.
- the method comprises the steps of comparing a subject sequence with a control sequence and detecting polymorphism based on the comparison of the distribution of the frequency of occurrence. By such a step, polymorphisms can be detected by comparing sequence data without considering positions in the full-length sequence and without linking the sequences.
- the method of the present invention further comprises the step of calculating the distribution of the frequency of occurrence for the portion of length x for each sequence common to the portion of length kx in the partial sequence.
- x is a positive integer less than k.
- the comparison of the distribution of the frequency of occurrence may include the comparison of the distribution of the frequency of occurrence of the portion of length x in the sequences common to the sequence portion of length k-x in the partial sequences. This makes it possible to speed up the process of polymorphism detection.
- the method of the present invention comprises the step of grouping sequence portions of length kx in the partial sequence into unique sequences. This may include, for example, sorting the length kx array portion (eg, sorting the length kx array portion as a character string).
- the value of k is a length that excludes accidental identity in the subject sequence data or the like.
- the k may be a length that excludes accidental identity at different places in the genome of the organism. . This makes it possible to make the detection of polymorphism more accurate.
- the length x is not limited, but is preferably 1 to 3, more preferably 1 to 2, and more preferably 1.
- the portion of length x is present at the end of the partial sequence.
- the portion of the length x may be at the 3 'end or 5' end of the partial sequence. Taking a portion of length x at the end of the partial sequence is desirable for speeding-up and simplification of the comparison process.
- the following polymorphisms can be detected by comparing the difference in the distribution of the appearance frequency.
- the sequence of the portion of length x when the appearance frequency of the sequence of the portion of length x differs between the subset of control sequence data and the subset of the target sequence data, the sequence of the portion of length x is targeted It detects as polymorphism with respect to control sequence data in sequence data.
- a sequence portion of length kx in which the most frequent ones of the sequences of the portion of length x differ between the subset of control sequence data and the subset of target sequence data If there is, the sequence of the portion of length x is detected as a polymorphism in the subject sequence data.
- the target sequence data and the control sequence data are nucleotide sequence data, and a sequence of the portion of length x in the subset of the target sequence data, which is the most frequent in the subset of the control sequence data If there is an array portion of length k-x in which the sequence of the portion of length x identical to that of the object is only a count below the noise, the array of the portion of length x is regarded as a homopolymorphism in the target sequence data. To detect. Noise can be determined using criteria as described later herein.
- the target sequence data and the control sequence data are nucleotide sequence data, and a sequence of the portion of length x in the target subset that is identical in length to the most frequent one in the subset of control sequence data If there is a sequence of a portion of size x and there is a portion of sequence of length k-x where a sequence of a portion of length x different from the most frequent one in the subset of control sequence data is present The sequence of the portion of x is detected as a heteropolymorphism in the sequence data of interest.
- the target sequence data and the control sequence data are base sequence data
- the target sequence data amount, the expected error rate in the sequence data for example, the nominal value of the manual / apparatus
- the target sequence total length size For example, in consideration of genome size, etc., it is possible to use the expected noise count degree or the partial sequence of the frequency of occurrence less than the expected noise count as the noise.
- the expected value of the frequency of the generated noise is int (how many times the genome has been read x (1-precision) + 1).
- the precision is one.
- the target sequence data and the control sequence data are base sequence data
- the array can be noise.
- the integer value carried forward can be used as the noise prediction value, and can be a integer value carried forward by adding 1 in the int function.
- a partial sequence having a high frequency of occurrence can be excluded as a repeat sequence site as compared to the expected frequency of occurrence. For example, a partial sequence that exceeds twice the coverage (cover rate) of the entire target sequence of the target sequence data can be excluded from analysis.
- the target sequence data and / or control sequence data is nucleotide sequence data obtained by next-generation sequencing.
- mapping to a reference and assembly of sequences have conventionally been required, and the uncertainty caused by such steps greatly hinders the detection of polymorphism. Therefore, it can be said that it is particularly advantageous to use the method of the present invention which does not require such steps on sequence data obtained from next-generation sequencing.
- the target sequence data and the control sequence data are not limited, but in order to detect polymorphism, it is desirable that they be sequence data for sequences having a certain commonality.
- the method of obtaining the sequences may be identical or different, and it is possible to compare the data obtained by sequencing or to compare the data obtained from a database or the like. It is also possible to make comparisons between data obtained by Sing and data obtained from a database or the like.
- the subject sequence data is sequence data obtained from an individual, and the control sequence data is sequence data obtained from another individual homologous to the individual or from a database.
- the subject sequence data is sequence data obtained from a tissue sample of an individual and the control sequence data is sequence data obtained from another tissue or database of the individual.
- the subject sequence data is sequence data obtained from a cell sample and the control sequence data is sequence data obtained from another cell or database.
- the method of the present invention does not require the information of full-length sequence, it can be used also in a database etc. when full-length sequence is not known, for example, target sequence data and control sequence data are derived from genome of organism In the case of base sequence data, the sequence of the genome may be unknown.
- Polymorphs that can be detected by the methods of the present invention include substitutions, insertions, deletions, Copy Number Variation (CNV), short tandem repeat polymorphism (STRP), inversions and translocations.
- the method of the present invention is also capable of simultaneously detecting any combination of the plurality of polymorphisms.
- the method of the present invention is also capable of detecting all of the plurality of polymorphisms simultaneously.
- the method of the present invention can exert very high detection power.
- the method of the present invention can further include the step of specifying the position of the polymorphism in the reference sequence to the subject sequence.
- the method may further comprise the step of identifying the position of the polymorphism on the genome.
- This location specification allows the method of the present invention to detect polymorphisms in association with surrounding sequences (eg, polymorphisms in the x-long portion are associated with sequences in the kx). It is possible to simply carry out by searching the reference sequence.
- the method of the invention may further comprise the step of identifying the detected polymorphism.
- the confirmation can be performed, for example, by comparing the target sequence data and / or the control sequence data, using the query sequence set generated from the reference sequence or the control sequence, for the detected polymorphic site.
- the query sequence set is a variant query sequence set in which the character of the site corresponding to the polymorphism is replaced with a different character in the reference sequence or control sequence, and / or the character of the site corresponding to the polymorphism in the reference sequence or control sequence May comprise a wild-type query sequence set without replacement of
- the method of the present invention refers to the sequence data of the complementary strand of the target sequence data and / or control sequence data for the detected polymorphic site. It may further include the step of comparing and confirming with the set of query sequences generated from the sequence or control sequence.
- the method of the present invention refers to the sequence data of the alleles of the target sequence data and / or the control sequence data with respect to the detected polymorphism site. It may further include the step of comparing and confirming with the set of query sequences generated from the sequence or control sequence.
- allele sequence data sequence data having a mutant-type base to wild-type can be used regardless of the presence or absence of the actual gene.
- the method of the present invention may comprise the step of obtaining target sequence data or control sequence data from experimental results or a database.
- the sequence data itself need not necessarily be acquired, but it is also possible to acquire and execute a subset of the array data and / or frequency distribution data in the array data or a subset of the array data is there.
- the present invention provides a program for implementing a method for causing a computer to execute the method of detecting polymorphism of the present invention, a recording medium recording the program, and a system for realizing the same.
- Optional features that can be employed herein can employ any of the features described in the description of the method of detecting polymorphism or a combination thereof.
- a program for causing a computer to execute a method of detecting polymorphism in control sequence data in target sequence data comprising a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; and d) comparing the subject sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- the step of displaying the sequence including at least a part of the non-polymorphic part in the partial sequence (which may be the entire partial sequence) as the name of the detected polymorphism
- a program is
- a recording medium storing a program for causing a computer to execute a method of detecting polymorphism in control sequence data in target sequence data, the method comprising: a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; and d) comparing the subject sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- the step of displaying a sequence comprising at least a portion of the non-polymorphic portion in the partial sequence (which may be the entire partial sequence) as the name of the detected polymorphism is further described.
- a recording medium is provided, including.
- a system for detecting polymorphism in control sequence data in target sequence data comprising: A sequence data processing unit configured to provide an appearance frequency of each partial sequence of the target sequence data and a subset of the length k of the control sequence data, wherein k is the target sequence and An array data processing unit having a length equal to or less than the total length of the control array;
- a system comprising: a sequence data calculator configured to compare an object sequence with a control sequence and to detect polymorphism based on the difference in distribution of the frequency of occurrence.
- the system further comprising display means for displaying the sequence including the at least a part of the non-polymorphic part in the partial sequence as the name of the detected polymorphism).
- An exemplary embodiment of the present invention is a method according to the following steps.
- sequence data obtain a subset of subsequences of length k. For example, a set of k-length sequences is obtained while shifting the base sequence data one base at a time from the end.
- sequence data of the rice individual obtained by the next-generation sequencer data volume is 40 times that of the rice genome, and the read length is 100 bases
- sequence set obtained with k lengths of 20 bases explain.
- the obtained sequence set if bases at the 3 'end of the data whose 5' end to the k-1 length sequence are the same among the samples to be compared differ between the targets to be compared, the bases are polymorphic. . (In the above example, SEQ ID NOS: 191 to 201 are from the first line, respectively.)
- the first line is the base sequence obtained from the next generation sequencer, and the following line shows the partial sequence of k length.
- a sequence set is obtained with k bases of 20 bases.
- Occurrence frequency data of the obtained partial sequence set is obtained. That is, data is obtained that is ordered by the number of occurrences for the same sequence in the partial sequence set. (In the above example, from the first line, they are SEQ ID NOS: 202 to 211, respectively.)
- the array set is arranged in descending order, and the number of occurrences is displayed on the right of the array.
- the analysis is 40 times that of the genome, it is possible to judge that a sequence having about one appearance frequency is noise.
- the sequence of about 40 to 50 times is derived from a unique sequence on the genome, and the 89-occurring sequences are considered to be present at two places on the genome.
- the distribution of the appearance frequency is calculated for the portion of length x for each sequence common to the portion of the array of length kx in the partial array.
- they are SEQ ID NOS: 212 to 220, respectively.
- the polymorphic position can be determined from the genomic position corresponding to the first 19 bases.
- the genomic reference sequence is known
- the polymorphic position can be determined from the genomic position corresponding to the first 19 bases.
- both the normal strand and the complementary strand are detected at the same position, it is a single nucleotide polymorphism.
- Detection of insertion / deletion polymorphisms is also possible in order to detect the terminal part of the sequence to be polymorphic. Since the first 19 bases are designated as the name of polymorphism and the last base as a genotype, they can be used as the only notation indicating the polymorphism.
- the entire k-mer (eg, 20 bases), including polymorphisms, can be used as one name.
- k 5
- notation such as ACGTA type and ACGTT type is possible.
- the method of the present invention can be carried out by appropriately adopting the steps shown in FIG. 1 according to the flow as shown in FIG.
- one feature of the present invention is that in the detection of polymorphism relative to control sequence data in the subject sequence data, each of the subsets of the partial sequence of length k of the subject sequence data, or each of the subsets of the partial sequences It is to use the appearance frequency of the partial sequence of
- k is an integer equal to or less than the total length of either the target sequence or the control sequence, whichever is shorter.
- a partial array of length k can be generated by cutting out from target array data, control array data, reference array data, and the like. For example, it can be generated by cutting out a k-length array at regular intervals, and can be segmented at intervals of one character, every two characters, every three characters, or more to generate a partial array set. Alternatively, it is also possible to randomly select and cut out positions from target sequence data, control sequence data, reference sequence data and the like. If it is desirable to generate all k-length subsequences, subsets of k-length subsequences can be generated while shifting the cutout position for each character.
- the length k is desirably a length that excludes accidental identity in the subject sequence, control sequence and / or reference sequence. By excluding the same by chance, it is possible to analyze more accurately, reducing the possibility of detecting, as a difference from the control sequence, one in which a different sequence was accidentally included in another part of the target sequence. It is desirable that the expected value for which a sequence of k length and a sequence identical to the same sequence happen to appear in the target sequence, control sequence and / or reference sequence is less than 1.
- v type of value that each variable can take in the array
- L total length (number of variables included) of the sequence to be analyzed (target sequence, control sequence and / or reference sequence) It is considered that k is a length that excludes the same by chance when> L.
- the total length of the sequence is expressed in an order such as 10 ⁇ L, take the logarithm of both sides, and if k> L / log (v), consider that k is the length by chance to exclude the same Be
- v 4 because four characters are considered as the value of the variable.
- the size of the human genome is 3.1 ⁇ 10 9, and if there is an 10 9 attrib-long atrandom base sequence, 9 / log (4) ⁇ 15 bases is the smallest chance to eliminate coincidence. It is considered to be k length.
- the method of the present invention comprises the step of providing the frequency of occurrence of the partial sequence of each of the subsets of length k of the target sequence data and / or control sequence data.
- the frequency of appearance of each partial sequence of the subset of the partial sequence of length k is a pair of frequency data for the unique k-length partial sequence data (for example, partial array in column 1, column 2 in Frequencies may be provided as matrix data provided.
- the appearance frequency of the partial array is not limited as long as the array and its frequency are associated, the format is not limited, and the array data having the corresponding identifier and the frequency data may be separately output. It is possible. Also, the appearance frequency of the partial sequence can be provided by acquiring already existing data.
- the step of obtaining the frequency of appearance of each partial sequence of the subset of the partial sequence of length k can be conveniently performed using a computer, for example, in Unix: It can be implemented as
- the site where polymorphism is present is a unique sequence site on genome, for example, there is a single base substitution
- k-mer partial sequence including the substitution is present in a subset of the subject sequence data.
- the target sequence data and the control sequence data are base sequence data
- the target sequence data amount, the expected error rate in the sequence data for example, the nominal value of the manual / apparatus
- the target sequence total length size For example, in consideration of genome size, etc., it is possible to use the expected noise count degree or the partial sequence of the frequency of occurrence less than the expected noise count as the noise.
- the expected value of the frequency of the generated noise is int (how many times the genome has been read x (1-precision) + 1).
- noise of one base is introduced. Since the normal analysis is about 40 times the reading of the genome, the noise is considered to be 1 base or less, but since the count is an integer value, the read error is counted as 1. That is, it is considered that counting of two or more bases is likely to have some cause other than noise. However, although there is a low probability, there is a possibility that noise may be introduced twice or more to the same base.
- the expected noise count is, for example, int (how many times the genome has been read x (1-precision) + 1). The accuracy in this case can be calculated as 1 for 100% and 0.99 for 99%. Note that int (X) is a function that returns the largest integer less than X.
- a partial array with a count of 1 is determined as noise. For example, when the target sequence data and the control sequence data are base sequence data, partial sequences with an appearance frequency of less than [(target sequence data amount) ⁇ (1-accuracy)] / (target genome size) +1 are considered as noise. By doing this, it is possible to eliminate a partial arrangement of the frequency of occurrence below the expected noise count as noise.
- CNV copy number mutations
- a partial sequence having a high frequency of occurrence can be excluded as a repeat sequence site as compared to the expected frequency of occurrence. For example, a partial sequence that exceeds twice the coverage (cover rate) of the entire target sequence of the target sequence data can be excluded from analysis.
- a partial sequence with the same frequency of occurrence is considered to be a sequence derived from a unique sequence in the entire length of the target sequence.
- the partial sequence with an appearance frequency of about twice is considered to be a sequence derived from a sequence existing at two places in the entire length of the target sequence.
- the partial sequence is derived from the repeat sequence site.
- the sequence having an occurrence frequency of about 40 to 50 times is derived from a unique sequence on the genome, and the 89 occurrence sequences are present at two places on the genome It is thought that The generation of the appearance frequency data of the subsets of the k-length array and / or each partial array can be performed, for example, by employing the process illustrated in FIG.
- the method of the present invention is characterized by calculating the distribution of the frequency of occurrence for the portion of length x for each sequence in which the portion of length kx in the partial array of k lengths is common.
- x is a positive integer less than k. It is possible to significantly reduce the computational complexity by looking at the difference in letters of part of the k-mer (part of the x length) rather than the full length of length k.
- the length x is not limited, but is preferably 1 to 2, more preferably 1.
- the method of the present invention comprises the step of grouping sequence portions of length kx in the partial sequence into unique sequences. This may include, for example, sorting the length kx array portion (eg, sorting the length kx array portion as a character string).
- the distribution of the frequency of occurrence for the portion of length x for each sequence common to the sequence of length kx in the partial sequences can be calculated from the frequency of occurrence of the partial sequence of length k.
- the array common to the array portion of kx is the array portion of kx
- the appearance frequency for the portion of length x for each sequence common to the sequence portion of length kx corresponds to the frequency data of the corresponding partial sequence of length k.
- the portion of length x is present at the end of the partial sequence.
- the portion of the length x may be at the 3 'end or 5' end of the partial sequence. Taking a portion of length x at the end of the partial sequence is desirable for speeding-up and simplification of the comparison process.
- the site where the polymorphism is present is a unique sequence site on the genome, for example, if a substitution is present, the k-mer partial sequence containing the substitution is present in a subset of the subject sequence data, but the control It is considered not to exist in a subset of sequence data. It is considered that this result is obtained for all k-mers containing the substitution mutation, and as a result, a difference in appearance frequency is observed in 2 ⁇ k-1 k-mer partial sequences containing the substitution mutation site, Polymorphism can be detected. Such analysis can be performed, for example, by employing a process as shown in FIG.
- the sequence of the portion of length x when the appearance frequency of the sequence of the portion of length x differs between the subset of control sequence data and the subset of the target sequence data, the sequence of the portion of length x is targeted It detects as polymorphism with respect to control sequence data in sequence data. In one embodiment, a sequence portion of length kx in which the most frequent ones of the sequences of the portion of length x differ between the subset of control sequence data and the subset of target sequence data If there is, the sequence of the portion of length x is detected as a polymorphism in the subject sequence data.
- the target sequence data and the control sequence data are nucleotide sequence data, and a sequence of the portion of length x in the subset of the target sequence data, which is the most frequent in the subset of the control sequence data If there is an array portion of length kx in which the sequence of the portion of length x identical to that of the object is only a count below the noise, the array of the portion of length x is regarded as a homopolymorphism in the target sequence data To detect.
- the target sequence data and the control sequence data are nucleotide sequence data, and a sequence of the portion of length x in the target subset that is identical in length to the most frequent one in the subset of control sequence data If there is a sequence of a portion of size x and there is a portion of sequence of length k-x where a sequence of a portion of length x different from the most frequent one in the subset of control sequence data is present The sequence of the portion of x is detected as a heteropolymorphism in the sequence data of interest.
- the comparison of the frequency data of the set of k-length arrays can be performed, for example, by the process as shown in FIG.
- control and target When processing a file of the k-1 long sequence and the frequency of the last base, control and target This can be done by outputting the frequency of putting k-1 array, control and target in one line by command of.
- Conditions for examining each line of this output result include, but are not limited to, there are two or more bases each having one or less count in both control and target, and 10 or more counts were shown in control or target. When there is one or two cases where the count of the target or control base corresponding to the base is 1 or less, it can be assumed that the boundary of polymorphism is detected.
- the first count in the control sequence and / or the subset of the partial sequence of the subject sequence exceeds the first threshold, and the corresponding second count in the subset of the control sequence and / or the partial sequence of the subject sequence (Ie, if the first count is of the control array, the second count is of the target array, and if the first count is of the target array, the second count is of the control array ) Is below the second threshold, it may be assumed that a boundary of polymorphism is detected.
- the first threshold varies depending on the coverage of array data, but can be set in the range of 10 to 50, for example.
- the first threshold can be set, for example, in the range of 10 to 40, 10 to 30, 10 to 20, or 10 to 15. If the coverage of the sequence data becomes large, the first threshold can also be set large. For example, in human genome analysis, there is also data about 200 times read, but in this case, 200 is set as the first threshold. It can be used.
- the first threshold may be the count at which the array is actually present in consideration of coverage, for example, about 100%, about 90%, about 80%, about 70%, about 60%, about Values such as 40%, about 30%, or about 20% can be used.
- the second threshold also varies depending on the coverage of array data, but can be set in the range of 1 to 7.
- the second threshold may be, for example, 1, 2, 3, 4, 5, 6 or 7.
- Array As the target sequence, control sequence and / or reference sequence of the present invention, any sequence that can generate polymorphism can be used. Note that a reference sequence can be used as a control sequence.
- the subject sequence, control sequence and / or reference sequence is a biological sequence, eg, a base sequence (including sequences such as DNA, RNA and their analogs), an amino acid sequence Or a sugar chain sequence or the like. Examples of biological sequences include, for example, genomic sequences, chromosomal sequences, gene sequences, plasmid sequences, exon sequences, protein sequences and the like.
- the target sequence data and the control sequence data are not limited, but in order to detect polymorphism, it is desirable that they be sequence data for sequences having a certain commonality.
- the method of obtaining the sequences may be identical or different, and it is possible to compare the data obtained by sequencing or to compare the data obtained from a database or the like. It is also possible to make comparisons between data obtained by Sing and data obtained from a database or the like.
- the subject sequence data is sequence data obtained from an individual, and the control sequence data is sequence data obtained from another individual homologous to the individual or from a database.
- the subject sequence data is sequence data obtained from a tissue sample of an individual and the control sequence data is sequence data obtained from another tissue or database of the individual.
- the subject sequence data is sequence data obtained from a cell sample and the control sequence data is sequence data obtained from another cell or database.
- the method of the present invention does not require the information of full-length sequence, it can be used also in a database etc. when full-length sequence is not known, for example, target sequence data and control sequence data are derived from the genome of an organism In the case of base sequence data, the sequence of the genome may be unknown.
- polymorphism detection by direct comparison of sequences between a target (target) and a control in next-generation sequencer read data has not been possible with existing technologies.
- a reference sequence polymorphisms can be mapped onto the genome, but in the absence of a reference (eg, an organism for which a reference genomic sequence has not been created), between the subject and the control without using the reference information.
- the ability to detect polymorphisms in If polymorphisms linked to phenotypic segregation are detected in F2 segregating populations in organisms without a reference, DNA markers corresponding to phenotypes can be obtained even if the position on the genome is unknown, The range is considered to be very broad. In fact, although positional information on the genome is important for breeding, it is possible to use it for selective breeding if selection with a DNA marker linked to excellent traits is possible even if the positional information is unknown.
- the target sequence data and / or control sequence data used in the method of the present invention is nucleotide sequence data obtained by sequencing.
- Sequencing methods include the Sanger method, Maxam-Gilbard method, single molecule real time sequencing (eg, Pacific Biosciences, Menlo Park, California), ion semiconductor sequencing (eg, Ion Torrent, South San Francisco, California), Pyrosequencing (eg, 454, Branford, Connecticut), sequencing by ligation (eg, SOLiD sequencing of Life Technologies, Carlsbad, California), sequencing by synthetic and reversible terminators (eg, Illumina, San Diego, Californi) ), Nucleic imaging techniques such as transmission electron microscopy, and the like nanopore sequencing.
- the subject sequence data and / or control sequence data used in the methods of the invention may be sequence data obtained by next-generation sequencing.
- Next generation sequencing includes sequencing bi synthesis, pyro sequencing, sequencing by ligation, ion semiconductor sequencing, nanopore sequencing and the like.
- the accuracy is limited by mapping to a reference or assembly, and it is considered that great benefits can be obtained when using the method of the present invention.
- the subject sequence data and / or control sequence data used in the method of the present invention is a dinitrophenylation method, a hydrazinolysis method, a carboxypeptidase method, an Edman method or an apparatus for automating those methods (peptide sequencer or Amino acid sequence data obtained from a method using a protein sequencer, a method using mass spectrometry (for example, tandem mass spectrometry (MS / MS)) (for example, sequence tag method), and the like.
- mass spectrometry for example, tandem mass spectrometry (MS / MS)
- sequence tag method for example, sequence tag method
- target sequence data and / or control sequence data of the present invention are derived is not limited as long as it has biological sequences.
- animals include humans or non-human mammals (eg, mice, rats, rabbits, sheep, pigs, cattle, horses, cats, dogs, monkeys, chimpanzees), birds, reptiles, amphibians, fish, etc. Vertebrates and invertebrates, for example, insects, linear animals and the like.
- plants include rice, wheat, corn, potato, barley, sweet potato, buckwheat, Arabidopsis thaliana, Lotus pea, tomato, cucumber, cabbage, Chinese cabbage, eggplant, sugar cane, sorghum, apple, orange, banana, peach, poplar, pine, cedar, Angiosperms, gymnosperms, ferns, mosses, algae and the like can be mentioned.
- fungi, bacteria, viruses and the like may be used.
- target sequence data and / or control sequence data derived from parts of these organisms such as tissues, cells, etc. and detect polymorphisms.
- the method of the present invention can be used, for example, for detection of polymorphisms such as substitution, insertion, deletion, copy number mutation, short tandem repeat polymorphism (STRP), inversion or translocation. Since the edge portion of the mutation is detected, if there is a difference in the sequence of length x as a result of insertion / deletion, the edge portion can be detected. It is also possible to detect short tandem repeat polymorphism (STRP) if it fits within the k-mer. STR (short tandem repeat) is also called a microsatellite, and a sequence consisting of 2 to 7 bases repeats 2 to several times, and polymorphism is observed in this frequency. Copy number variation (CNV) can also be detected by the frequency of occurrence of partial sequences. From the viewpoint of edge detection, it is also possible to detect inverted or transposed edges.
- polymorphisms such as substitution, insertion, deletion, copy number mutation, short tandem repeat polymorphism (STRP), inversion or translocation. Since the edge portion of the
- the method of the present invention can exert very high detection power.
- the method of the present invention may further include the step of specifying the position of the polymorphism in the reference sequence to the subject sequence.
- the method may further comprise the step of identifying the position of the polymorphism on the genome.
- the search for the reference sequence can be performed, for example, by preparing reference genome sequence data for binary search by the process as shown in FIG. 5, and then searching by mapping polymorphic boundary bases by binary search.
- a method can be used in which mapping is performed by outputting the position and orientation in the reference array of the partial array in the target array data by the unix join command. More specifically, a method of determining the position of a target sequence on a control sequence, comprising the steps of: a) outputting the position and orientation in the sequence and control sequence for a plurality of k-length subsequences in the control sequence; B) comparing the sequences obtained in c) a) and b) with the step of outputting the sequence and the position in the subject sequence for a plurality of subsequences of k length in the subject sequence, and comparing them with identical subsequences
- the method may be used, including the step of correlating the position in the corresponding control sequence with the position in the subject sequence, where k is a length not exceeding the length of the subject sequence.
- the method of the invention may further comprise the step of identifying the detected polymorphism.
- the confirmation can be performed, for example, by comparing the target sequence data and / or the control sequence data, using the query sequence set generated from the reference sequence or the control sequence, for the detected polymorphic site.
- the query sequence set is a variant query sequence set in which the character of the site corresponding to the polymorphism is replaced with a different character in the reference sequence or control sequence, and / or the character of the site corresponding to the polymorphism in the reference sequence or control sequence May comprise a wild-type query sequence set without replacement of
- the method of the present invention refers to the sequence data of the complementary strand of the target sequence data and / or control sequence data for the detected polymorphic site. It may further include the step of comparing and confirming with the set of query sequences generated from the sequence or control sequence.
- the target sequence data and the control sequence data are nucleotide sequence data
- the method of the present invention refers to the sequence data of the alleles of the target sequence data and / or the control sequence data with respect to the detected polymorphism site. It may further include the step of comparing and confirming with the set of query sequences generated from the sequence or control sequence. As an example, the step of confirming can be performed by appropriately adopting the step shown in FIG. 6 in accordance with the flow shown in FIG.
- allele sequence data sequence data having a mutant-type base to wild-type can be used regardless of the presence or absence of the actual gene.
- control sequence is obtained from a wild type (that is, almost identical to the reference genome sequence) or the case where the control sequence is a sequence having the same length as the target sequence from the reference genome sequence is exemplified.
- the base sequence length of the short read read by the next-generation sequencer is L
- the position from L-1 base position to L-1 base position A sequence of 2L-1 bases in length is obtained, and a substituted sequence and a control non-substituted sequence in which the polymorphic base position is substituted with the presumed polymorphic base are created.
- An L-length query sequence set is created while shifting each of the substituted and non-replaced sequences by one base each (for example, as illustrated in FIG. 14). Using the individual query sequences, obtain counts that exactly match the subject and control sequences.
- the count of the target sequence is most, and in the case of a heterozygous mutation, the coincidence count to the target sequence and the control sequence is considered to be approximately half.
- a non-replacement array basically the control array is mostly hit. If a non-replacement sequence hits the target sequence, it is judged that it is not polymorphic and can be excluded.
- Sequences of control sequences and target sequences and complementary chains are also combined to search for control sequences and target sequences, and then sorted in the lexicographic order, using the data set in which identical sequences are combined into one, query sequence by binary search method Search for
- the complementary strand of each read can also be sorted and uniquely processed.
- Processing of sort and uniq before excising k-mer may cause a step of PCR amplification in the sequence reaction (some kits may not be included), and the same sequence may appear in the read data multiple times. It may be preferable in dealing with what may come. If processed as it is, the k-mer distribution may be distorted. Since a read that contains the same sequence but contains N may be recognized as another and may cause distortion, the sequence that does not contain N, and that sequence and its complementary strand sequence are sorted and uniq-processed It is possible to obtain mer.
- k-mer is used to detect polymorphism and map to k-mer It is possible.
- the lengths of the sequence data be uniform. According to the findings of the present inventors, if polymorphisms were detected in both the normal strand and the complementary strand at the stage of mapping the polymorphism with the k-1 sequence prior to the confirmation step, it is almost certainly in SNPs I know that there is. Although there may be cases in which either the forward strand or the complementary strand is in the repeat region and only one side can be detected, it may actually be a SNP, but in such a case, the actual SNP can be obtained by binary search of the sort_uniq sequence. It may be possible to determine whether or not.
- the lengths of the short read at start (base sequence data obtained by the next-generation sequencer) be uniform.
- the sequence lengths of the subject and the control do not have to be the same, and if the length is constant in each of the subject sequence data and the control sequence data, confirmation can preferably be performed by a binary search.
- confirmation can be made as follows.
- a partial sequence is cut out so as to include a mutation site at the L length of the target sequence from the reference sequence, and a set substituted with mutations and a set not substituted are created, sorted and output together with descriptions of positional relationship, presence or absence of mutation, etc.
- This data and the target sequence sorted by this process are processed (or appropriate equivalent processing) with the unix command to select wild-type and mutant-type sequences contained in the target sequence, and the number of sequences per mutation site Examine. After sorting the selected array, you can count the number of arrays with the uniq-c command. The same operation is performed on the control sequence (L 'length).
- the lead sequence obtained from this individual can be used as a control sequence. If there is no control individual, it is possible to use a sequence created by cutting L length from the reference sequence as a control sequence. If the target and control sequences differ in length, one can generate data sets of mutant and wild-type corresponding to the respective lengths and find out the corresponding numbers.
- Illumina's next-generation sequencer for example, HiSeq
- HiSeq next-generation sequencer
- the present invention can be applied to sequence data based on short reads having variation in length or sequence data that is not a set of short reads (for example, sequences obtained by the Sanger method).
- a binary search is performed using data that has been processed to make the lengths uniform (for example, select and cut out an L-length sequence that maximizes the quality score during reading, cut out an L-length sequence from one end, etc.)
- sequence data read
- BLAST target sequence database
- Speeding up is desirable. As a method for speeding up, there are hardware methods such as performing calculations all on memory, and placing files on a high speed disk such as an SSD. In terms of software, instead of simply performing a binary search on a sorted file, the file to be searched can be subjected to Burrows-Wheeler conversion to achieve higher speed.
- the present invention provides a program for implementing a method for causing a computer to execute the method of detecting polymorphism of the present invention, a recording medium recording the program, and a system for realizing the same.
- Optional features that can be employed herein can employ any of the features described in the description of the method of detecting polymorphism or a combination thereof.
- a program for causing a computer to execute a method of detecting polymorphism in control sequence data in target sequence data comprising a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; and d) comparing the subject sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- the step of displaying a sequence comprising at least a portion of the non-polymorphic portion in the partial sequence (which may be the entire partial sequence) as the name of the detected polymorphism is further described.
- the program is provided. The program may be written in any language.
- a recording medium storing a program for causing a computer to execute a method of detecting polymorphism in control sequence data in target sequence data, the method comprising: a) storing, on a computer, a subset of the partial sequence of length k of the target sequence data, wherein k is a length equal to or less than the total length of the target sequence and the control sequence; b) calculating the frequency of appearance of each partial sequence of the subset of length k of the target sequence data; c) storing the frequency of occurrence of each partial sequence in a subset of the length k subsequences of the control sequence data in a computer; and d) comparing the subject sequence with a control sequence, and detecting polymorphism based on the difference in distribution of the frequency of occurrence.
- the step of displaying a sequence comprising at least a portion of the non-polymorphic portion in the partial sequence (which may be the entire partial sequence) as the name of the detected polymorphism is further described.
- a recording medium is provided, including.
- the program may be written in any language.
- the recording medium may be an external storage device such as a ROM, HDD, magnetic disk, flash memory such as USB memory, etc., which may be stored internally.
- a system for detecting polymorphism in control sequence data in target sequence data comprising: each of the target sequence data and a subset of a length k subsequence of the control sequence data An array data processing unit configured to provide an appearance frequency of the partial array, wherein k is a length equal to or less than a total length of the target sequence and the control sequence, And a sequence data calculator configured to compare with a control sequence and to detect polymorphism based on the difference in distribution of the frequency of occurrence.
- the method further comprises display means for displaying a sequence (which may be the entire partial sequence) including at least a part of the non-polymorphic portion in the partial sequence as the name of the detected polymorphism.
- FIG. 15A shows the case where it is realized by a single system, it is understood that the case where it is realized by a plurality of systems is also included in the scope of the present invention.
- a system 1000 includes a CPU 1001 incorporated in a computer system and a RAM 1003 via a system bus 1020, an external storage device 1005 such as a ROM, a HDD, a flash disk such as a magnetic disk or USB memory, and an input / output interface (I / F ) 1025 are connected.
- An input device 1009 such as a keyboard and a mouse, an output device 1007 such as a display, and a communication device 1011 such as a modem are connected to the input / output I / F 1025, respectively.
- the external storage device 1005 includes an information database storage unit 1030 and a program storage unit 1040. Each is a fixed storage area secured in the external storage device 1005.
- this storage device is input by inputting various commands (commands) via the input device 1009 or by receiving a command via the communication I / F, the communication device 1011 or the like.
- a software program installed in 1005 is called, expanded, and executed on the RAM 1003 by the CPU 1001 to detect polymorphisms in the target array data of the present invention in cooperation with the OS (operation system). It is supposed to play the function of the method. Of course, it is possible to implement the present invention by a mechanism other than such cooperation.
- a step of storing a subset of a partial sequence of length k of target sequence data in a computer wherein k is a length equal to or less than the total length of the target sequence and control sequence
- the target array data and / or the partial array data of length k of the target array data is input via the input device 1009, or is input via the communication I / F, the communication device 1011, etc.
- it may be stored in the database storage unit 1030.
- the program stored in the program storage unit 1040 or various instructions (commands) via the input device 1009 Can be executed by the software program installed in the external storage device 1005 by inputting a command or by receiving a command via the communication I / F or the communication device 1011 or the like.
- the appearance frequency calculated in advance can be input through the input device 1009.
- the appearance frequency data may be output through the output device 1007 or may be stored in the external storage device 1005 such as the information database storage unit 1030.
- the step of comparing the target sequence and the control sequence and detecting the polymorphism based on the difference in the distribution of the appearance frequency can be performed by the program stored in the program storage unit 1040 or various commands via the input device 1009.
- the program stored in the program storage unit 1040 or various commands via the input device 1009.
- the software program installed in the external storage device 1005 can execute the command.
- the above calculation results are stored in the database storage unit 1030 in association with information on the sequence, for example, biological information, biochemical information, medical information, for example, known information such as a disease, a disorder, or biological information. It is also good.
- association may be made as data available through the network (Internet, intranet, etc.) as a link of the network as it is.
- the computer program stored in the program storage unit 1040 may be a computer having the processing system described above, for example, provision of array data, provision of partial array subsets, calculation of appearance frequency data, comparison of appearance frequency data, polymorphism
- the system is configured as a system for performing processing such as detection and confirmation of polymorphism.
- Each of these functions is an independent computer program, its module, routine, or the like, and is executed by the CPU 1001 to configure the computer as each system or apparatus.
- each function in each system cooperates to constitute each system, but the program for this processing is also an external storage device or a communication device, respectively. It may be provided via an input device.
- the method of the present invention may be implemented by a computing system having a cluster structure.
- the system is in a cluster configuration, consisting of heads and nodes.
- the node can use an SSD for the main storage to speed up the search.
- one head can be operated by a plurality of nodes (for example, 12).
- the computing system has a cluster structure and mounts a mass storage device (HDD) on a main computer (cluster head) to store analysis data and results. From the cluster head, the divided data is sent to each node and calculation is performed, and the result is aggregated into the cluster head.
- HDD mass storage device
- Both the cluster head and the nodes can be equipped with a central control element (CPU) and a memory (RAM), and data can be communicated via a communication interface (NIC).
- a solid state drive (SSD) can be used as a main storage device for high speed search processing of nodes.
- the CPU, RAM, SSD and the like mounted on each node may be shared with other nodes or physically separated.
- the output files (reads) are sorted in lexicographic order, and identical sequences are combined into one.
- the unix command is as follows.
- the k-mer (here 20 bases) sequence is repeatedly output (k-mer_file) until it reaches the 3' end of the target base sequence.
- the output k-mer sequences are sorted in lexical order, and the same sequences are put together into one, and a file in which the number of occurrences is written together with the sequences is created.
- the unix command is as follows.
- a filter program may be output in the order of array ⁇ frequency after a command such as uniq-c.
- k-mer_count_file For each row of data in k-mer_count_file, a sequence of k-1 bases is obtained from the 5 'end of the sequence, and the base at the 3' end, that is, the kth base is represented as the number of occurrences of A, C, G, T Converted to k-1 mer array A number of times C number of times G number of times T number of times is output.
- Such data are respectively obtained from next-generation sequencer sequence data obtained from target and control samples.
- the data created by the control and target 5 methods are organized into k-1 mer sequences to create data.
- the unix command is as follows
- the base following ACTTTCTTCAAGGTCTGTT (SEQ ID NO: 225) is G, but in target, it is C. That is, the polymorphism following the unique identifier (name) of k-1 mer can be represented by the notation of G type or C type. The number corresponding to each base is the number of independent reads for which this polymorphism was detected.
- the name of the polymorphism can be represented by the k-1 mer sequence itself, and the genotype will be the subsequent polymorphic base. This data set can be used to perform association analysis on phenotypes.
- the binary position search method can determine the genomic position of the k-1 mer sequence.
- the positions of the same sequences are also known in a plurality of lines, so that even if the target sequence is in the repeat area, corresponding candidate areas are listed and output in the search.
- the reference sequence is a sequence that has the same length as the sequence created in 2 for every 2 bases from the 5 'end, and the data that has been subjected to sort and uniq treatment with the complementary strand Create This reference and target sort_uniq data will be searched.
- a sequence set (query set to be searched) of the same length as the sequence created in 2 including the polymorphic position is cut out.
- This sequence set is a sequence set including bases at polymorphic positions at all positions from the 5 'end to the 3' end of the sequence.
- the number of sequence sets is identical to the sequence length.
- a sequence set (target set) is created in which bases at polymorphic positions are replaced with predicted polymorphic bases. The creation of the query array set is illustrated in FIG.
- the reference set and the target set are used as queries, and the reference genome and the sort_uniq sequence created in 2 are respectively searched (for example, a method using a binary search method or a join command), and the sort_uniq sequence is generated for each set. Find out how many matches.
- the reference set only hits the sort_uniq array of references.
- homozygous mutations should be detected only from the target sort_uniq data, and heterozygous mutations should be detected from both the reference and the target sort_uniq data . In this way, when a polymorphism showing a search value that matches the expectation is extracted, it is possible to output a result as follows.
- a process useful for detecting substitutions, insertions, deletions, inversions or translocations includes the step of locating at least two partial sequences in the sequence of the subject sequence data on a control sequence.
- a partial sequence of k length can be used as the partial sequence.
- the control sequence is a sequence that can specify positional information on the sequence, and more preferably, the control sequence is a reference sequence.
- the process may include the step of comparing the positional relationship between partial sequences in the target sequence data with the positional relationship between partial sequences on the control sequence.
- the positional relationship between partial sequences in the target sequence data and the positional relationship between partial sequences on the control sequence are different, it can be determined that there is a target polymorphism.
- a partial sequence is present on a different sequence structure of a control sequence, it is determined that a translocation has occurred, the partial sequence is present on the same sequence structure of a control sequence, and If is different from that on the target sequence data, it is determined that an inversion exists, a partial sequence is present on the same sequence structure of the control sequence, and the direction is the same as that on the target sequence data If the partial sequence distance is shorter than the target sequence data distance on the control sequence, it is determined that a deletion is present, and / or the partial sequence is present on the same sequence structure of the control sequence.
- the orientation is the same as that on the target sequence data and the distance between the partial sequences is longer than the distance on the target sequence data on the control sequence, then it is judged that an insertion is present. It may include. If the positional relationship is not different, processing may be terminated, or it may be determined that there is no target polymorphism, and characters between partial array sites in the target array data are compared with characters on the corresponding control array. Then, the step of detecting the non-coincidence site may be further performed, and if the non-coincidence site exists, it may be determined that the substitution is present.
- the process determines that there is a target polymorphism, and characters between partial array sites in the target array data are sequentially identified starting from the character on the corresponding control array and the partial array site. It may include a step of comparing and detecting a mismatched site. By this step, it is possible to detect the borderline base of the detected polymorphism.
- a method for detecting polymorphism in control sequence data in target sequence data comprising: (1) a) providing the frequency of appearance of each partial sequence of a subset of the length k subsequences of the target sequence data; b) providing the frequency of occurrence of each subsequence of the subset of subsequences of length k of the control sequence data; c) comparing the target sequence with the control sequence and detecting polymorphism based on the comparison of the frequency of occurrence, substitution, copy number polymorphism, STRP, insertion, deletion in the target sequence data; A process of detecting inversion or translocation; (2) a) specifying the position on the control sequence of at least two partial sequences in the sequence of the target sequence data; b) comparing the positional relationship between the partial sequences in the target sequence data with the positional relationship between the partial sequences on the control sequence; c) When the positional relationship between the partial sequences in the target sequence data and
- Insertion, deletion, inversion in the target sequence data by the step of comparing the characters between the partial sequence sites with the characters on the corresponding control sequence and sequentially comparing them starting from the partial sequence site and detecting the mismatched sites. , A process of detecting a translocation or substitution is provided.
- a method for detecting polymorphisms in reference sequence data in target sequence data comprising the step of creating, from the reference sequence data, a k-length partial array set of reference sequences associated with each position information, (A1) generating a subset of a subsequence of length k of the target sequence data, and providing a frequency of appearance of a unique subsequence of length k; (A2) providing the frequency of appearance of unique length k subsequences of the k-length subsequence set of the reference sequence; (A3) comparing the target sequence with the reference sequence, and detecting insertion, deletion, substitution, copy number variation, STRP, inversion or translocation based on the comparison of the distribution of the frequency of occurrence; (B1) a binary search is performed on the k-length subsequence set of the reference sequence by using at least two k-length subsequences in the sequence of the target sequence data as a query; Identifying the position on
- the method further includes the step of detecting a non-matching portion by comparing characters on the partial sequence site in the target sequence data with characters on the corresponding control sequence, when the positional relationship is not different,
- a process further comprising the step of determining that a substitution exists if there is a non-coincidence site; Are provided simultaneously, in parallel, or sequentially.
- Example 1 Rice SNP detection and verification
- Example 2 Rice SNP detection and verification
- Example One seed of the rice cultivar Nipponbare was N1 individual, germinated and grown, and the leaves were sampled. The seeds grown on N1 individuals were designated as N1S1, and one seed was germinated and grown to sample leaves. The seeds grown on N1S1 individuals were designated as N1S2, and leaves and seeds were sampled similarly until the N1S10 generation.
- next-generation sequencing Whole-genome analysis was performed on next-generation sequencers on leaf samples of N1, N1S5, N1S6, N1S7, and N1S10. The sequencer was analyzed at the pair end using HiSeq 2000 manufactured by Illumina company. The read length was 100 bases for N1, N1S5, N1S6, and N1S7, and 101 bases for only N1S10.
- a sequence library was used to form a cluster serving as a template for sequences, and the base sequence of the template DNA was obtained. Analysis of the sequence data was performed using a software provided to make a base call and output as a fastq format file.
- N1 and N1S6 were performed under the conditions according to the manufacturer's manual as follows. Table 5: Manual name and version number used for each task Table 6: Equipment used for cluster formation, sequence and sequence analysis, reagents, software
- N1S5, N1S7, N1S10 was performed under the conditions according to the manufacturer's manual as follows.
- Table 7 Manual name and version number used for each task
- Table 8 Equipment used for cluster formation, sequence and sequence analysis, reagents, software
- the output files (reads) are sorted lexicographically and the same sequence is put together into one.
- the unix command was as follows.
- the outputted k-mer sequences were sorted in lexical order, and the same sequences were put together to create a file in which the number of occurrences was written together with the sequences.
- the unix command was as follows.
- k-mer sequences were aligned for control sequence: N1 and target sequence: N1S7 to detect polymorphism.
- site where polymorphism is present is a unique sequence site on genome, for example, single base substitution exists there, k-mer sequence including the substitution is present in the target sequence, but the control It is considered to be absent in the array. Therefore, in the k-mer sequence containing the substitution mutation, the occurrence is different in the k k-mer sequences including the substitution mutation site because the k-mer sequence is present in the target sequence and is not present in the control sequence. it is conceivable that.
- the alignment of the k-mer sequence was aligned along the reference sequence.
- a sequence of k-1 bases is obtained from the 5 'end of the sequence, and the base at the 3' end, that is, the kth base is represented as the number of occurrences of A, C, G, T Converted to It is output in the form of "number of times of array A of k-1 mer, number of times of C, number of times of G, number of times of T".
- next generation sequencing data 40 to 50 times the size of the rice genome was targeted, and therefore, when the frequency of the base exceeded 100, it was excluded as a repeat sequence site.
- next generation sequencing data 40 to 50 times the size of the rice genome was targeted, and therefore, when the frequency of the base exceeded 100, it was excluded as a repeat sequence site.
- the reference genome (rice reference genome (IRGSP1.0)) is excised with a k-mer while shifting each chromosome by 1 base from the 5 'side in the same manner as in the above step.
- the data in which the chromosome number, the position, and the direction were described on the same line was created, and arranged in lexicographic order with the k-mer sequence.
- the genomic position of the sequence of polymorphic k-1 mer was determined by binary search.
- sequence set (a query set to be searched) having the same length as the read length including the polymorphic position was cut out.
- This sequence set is a sequence set including bases at polymorphic positions at all positions from the 5 'end to the 3' end of the sequence.
- the number of sequence sets is identical to the sequence length.
- a sequence set (target set) was created in which bases at polymorphic positions were replaced with predicted polymorphic bases.
- a reference set, a target set as a query, a reference genome, and a file (reads) output from a Fastq format file obtained from the next-generation sequencer are sorted in lexical order, and a sort_uniq sequence that combines the same sequence into one Were each searched by binary search, and it was checked how many sort_uniq sequences matched each set.
- the reference set only hits the sort_uniq array of references.
- homozygous mutations should be detected only from the target sort_uniq data, and heterozygous mutations should be detected from both the reference and the target sort_uniq data . In this way, polymorphisms that showed search values that matched the expectation were extracted.
- Genotype confirmation was performed by amplifying the region by PCR and determining by Sanger method.
- the primer sequences used for amplification of each region were as follows.
- the reaction conditions for the PCR reaction were as follows.
- the reaction cycle was carried out with 30 cycles of 94 ° C. for 0.5 minutes, 60 ° C. for 0.5 minutes and 72 ° C. for 1 minute.
- the amplified DNA fragments are separated by 1% agarose gel electrophoresis and stained with ethidium bromide at a concentration of 0.5 ⁇ g / ml to cut out a band that emits fluorescence with a long wavelength ultraviolet lamp (365 nm), and use Wizard (Promega) Purification of the fragment was performed with SV Gel and PCR Clean-Up System (Cat. # A9282).
- the number of reads (total data) for each sample is as follows: It was.
- sort_uniq is the result of sorting the data of N-free read and its complementary strand and then unifying the same sequence in uniq.
- This data unlike k-mer data, is array-only data and does not include frequency data.
- the position of the chromosome the frequency of 20-mer of N1, N1S5, N1S6, N1S7, N1S10 is shown.
- the heterozygote at N1S7 and the mutant homozygote at N1S10 show that the wild-type 20-mer is zero. That is, from the change in the appearance frequency of the k-mer sequence, it was possible to detect that a hetero mutation occurred in N1S7 and a homo mutation occurred in N1S10.
- the method of the present invention can detect polymorphism between generations in detail.
- polymorphisms detected by Polymorphic Edge Detection are also confirmed by the Sanger method, polymorphisms can be detected between sequence data without the need for reference sequences (genomic reference sequences). It has been demonstrated that it can.
- Example 2 Sequence analysis of Yoruba male (NA18507) in Nigeria)
- Human genome reference hg38 was used as control sequence data.
- sequences the chromosomal data of chr1 to chr22 and chrX, chrY and chrM were downloaded and used from ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/. Data with comments on file names such as alt and v1 is excluded.
- next-generation sequence sequence data NA18507 of human genome was downloaded and used.
- the sequence data was analyzed by a next-generation sequencer manufactured by Illumina, and registered and published in NCBI. The data was downloaded and used.
- the URL of the experiment ID of the base sequence set was https://www.ncbi.nlm.nih.gov/sra/SRX016231 and the accession number of the sequence was in the range of SRR034939 to SRR034975.
- FIG. 1 A part of the results is shown in FIG.
- the wild type / mutant bases detected in the complementary strand (r) are shown converted to a normal strand.
- the respective bases are shown side by side.
- the numbers of A, C, G, T of the reference sequence x of interest indicate the frequency of each base of the sequence x following the k-1 sequence.
- sequence data origin There were two types of samples of sequence data origin, and the sequence data names and sample contents were as follows.
- SRR2096532 Control Blood (Normal) SRR 2096535 Follicular lymphoma (9690/3: Follicular lymphoma) Number of reads (sequence length 101 bases)
- SRR 2096532 1300353764 SRR 2096535 1339310760 Number of arrays of sort_uniq SRR 2096532 2056683322
- SRR 2096532 normal tissue
- SRR 2096535 tumor tissue
- the target has 5 or more leads, 1 or less wild type, and 1 or less mutations in the control. If the wild type has 5 or more reads, mark it as a homozygous mutation (M), and the ratio of the variant to the total number of reads of the target is greater than 0.3 or less than 0.7, and the variant is the control.
- M homozygous mutation
- H heterozygote
- Human genome reference hg38 was used as reference sequence data.
- sequences the chromosomal data of chr1 to chr22 and chrX, chrY and chrM were downloaded and used from ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/. Data with comments on file names such as alt and v1 is excluded.
- the number of loci subjected to Verify (confirmation step) in PED was 22601, of which substitution mutations were detected in both normal strand and complementary strand at locus 514. Although the result of Verify was detected in only one strand, it was predicted that there was a substitution mutation at the 1042 locus by combining the loci judged to be positive. All 1042 loci detected by PED were heterozygous. This is consistent with the fact that the possibility of homozygous mutation is considered to be extremely low in principle because it is a comparison of sequences from blood and cancer tissue of the same person. As PED compares the target and control directly before mapping, the number of target / control SNPs is not output.
- the method of the present invention is extremely high in both the capture rate and capture accuracy of polymorphism as compared to the method of mapping to the reference genome and then examining the difference.
- Example 4 Detection of copy number variation
- Cell culture is carried out from one seed of the same generation as N1S6 in Example 1, and DNA is extracted from the leaf which is redifferentiated one month, three months, and five months later and made into an individual of rice, 1M1, 3M1, 5M1 respectively Used as a sample.
- Individuals cultured for 5 months with seeds of the same generation as the N1 seed in Example 1 were redifferentiated, and DNA was extracted from individuals generated by self-fermentation for 4 generations and used as samples of TTM2 and TTM5.
- Sequence data were acquired from the extracted DNA by a next-generation sequencer. The sequencing protocol was similar to that in Example 1.
- N1S5, N1S6, N1S7, and N1S10 of Example 1 were used as target arrangement data, and N1 as reference arrangement data.
- accession number, the number of reads, and the number of sequences of sort_uniq for these sequence data are shown in the following table.
- the TTM5 data is divided into two accession numbers, SRR556174 and SRR556175.
- TTM5's sort_uniq combines two reads into one file.
- the sequence of the retrotransposon Tos17 starts from this position. Two copies of this transposon exist in the genome, and the terminal part of each transposon has the same sequence (Long Terminal Repeat, LTR). Therefore, it is considered that a value of about four times that of the previous one from 26694795 of chromosome 7 appeared at N1 to N1S10. Since the total length of this transposon is 4.1 kb, only the portion of the first junction is shown in FIG.
- TTM2 and TTM5 are DNA of individuals generated by self-fertilizing four generations of individuals differentiated by culturing seeds of the same generation as N1 seeds for 5 months, and in this case also, an increase in copy number is observed.
- Tos17 is known as a rice transposon that is activated and transferred only during culture. Since Tos17 is a retrotransposon, transfer of a copy of Tos17 increases the copy number of Tos17 on the genome without the original being excised and transferred. Therefore, Tos17 has been known to metastasize in culture and to increase copy number.
- the method of the present invention demonstrates that it is possible to detect copy number variation.
- copy number variation is considered to be able to be used for quality control in cultured cells (eg, iPS cells etc.).
- cultured cells eg, iPS cells etc.
- iPS human cultured cells
- SEQ ID NO: 1 to 60 k-mer reference sequence of FIG. 7
- SEQ ID NO: 61 to 80 k-mer target sequence of FIG. 7 (portion in which a mutation exists)
- SEQ ID NO: 81 to 140 k-mer reference sequence of FIG. 8
- SEQ ID NO: 141 to 160 k-mer target sequence of FIG. 8 (portion in which a mutation is present)
- SEQ ID NO: 161 to 190: k-1 (k 20) in FIG.
- SEQ ID NO: 191 to 221 Sequence used by (specific example) SEQ ID NO: 222 to 232: (exemplified calculation flow) SEQ ID NO: 233 to 266: SEQ ID NO: 267 to 275 of the primer used in Example 1: Sequence of FIG.
Landscapes
- Bioinformatics & Cheminformatics (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Physics & Mathematics (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Computational Biology (AREA)
- Analytical Chemistry (AREA)
- Evolutionary Biology (AREA)
- General Health & Medical Sciences (AREA)
- Medical Informatics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Theoretical Computer Science (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Information Retrieval, Db Structures And Fs Structures Therefor (AREA)
Abstract
本発明において、2つ以上の配列の間における多型を検出する方法が提供される。本発明の方法は、配列データにおける個々の配列の全長配列における位置を考慮することなく、複数の配列データ間での多型の検出を可能にする。本発明の方法は、配列データ中の個々の配列(例えば、次世代シーケンサーからのショートリード)を連結してより長い配列とすること(例えば、アセンブリ)を必要とせずに、多型を検出することができることを1つの特徴とする。
Description
本発明は、配列情報、とりわけ、ゲノム等の生体分子の配列情報の情報処理の分野に関する。
次世代シーケンサーの出現により、生物の全ゲノム配列情報が得られるようになった。次世代シーケンサーの配列情報から多型情報を得て、表現型との関連を調べることにより、その表現型の原因となる遺伝子の特定につながる。正確な多型情報の取得は、作物育種のみならず、ヒトの遺伝病の診断、生物種・品種等の特定等、幅広い分野で必要とされる基盤技術であり、これまでにない精度で多型情報が得られれば、そのインパクトは大きい。
次世代シーケンサーからの塩基配列データを用いた多型の検出は、まず最初に配列データをbwa、またはbowtieのようなマッピングプログラムを用いてリファレンス配列上の位置情報とミスマッチの情報を得て、次に、SamtoolsやGATK等の多型抽出プログラムでSNPやindel等の多型情報を抽出するのが一般的である。
これらの方法では、多型の可能性のある部分は可能な限り出力するため、多くのノイズを含みこれらの技術のみでは、正確な多型解析が困難である。マイクロアレイやDNAチップ等の別の技術を併用して用いられているというのが現状である。
本発明において、2つ以上の配列の間における多型を検出する方法が提供される。本発明の方法は、配列データにおける個々の配列の全長配列における位置を考慮することなく、複数の配列データ間での多型の検出を可能にする。本発明の方法は、配列データ中の個々の配列(例えば、次世代シーケンサーからのショートリード)を連結してより長い配列とすること(例えば、アセンブリ)を必要とせずに、多型を検出することができることを1つの特徴とする。
例えば、本発明は以下の項目を提供する。
(項目1) 対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
を包含し、ここで、kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である、方法。
(項目2) 前記部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出する工程をさらに含み、ここで、xはk未満の正の整数である、前記項目に記載の方法。
(項目3) 前記比較が、前記部分配列中の長さk-xの配列部分が共通する配列における、長さxの部分の出現頻度の分布の差異の比較を含む、前記項目のいずれかに記載の方法。
(項目4) 前記部分配列中の長さk-xの配列部分を、ユニークな配列ごとにグルーピングする工程を含み、ここで、xはk未満の正の整数である、前記項目のいずれかに記載の方法。
(項目5) 前記長さk-xの配列部分をソートする工程を含む、前記項目のいずれかに記載の方法。
(項目6) 前記長さk-xの配列部分を文字列としてソートする工程を含む、前記項目のいずれかに記載の方法。
(項目7) 前記kが、前記対象配列における偶然同一を排除する長さである、前記項目のいずれかに記載の方法。
(項目8) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記kが、前記生物のゲノムにおいて、異なる箇所での偶然同一を排除する長さである、前記項目のいずれかに記載の方法。
(項目9) 長さxが1~2である、前記項目のいずれかに記載の方法。
(項目10) 長さxが1である、前記項目のいずれかに記載の方法。
(項目11) 前記長さxの部分が、前記部分配列の末端に存在する、前記項目のいずれかに記載の方法。
(項目12) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記長さxの部分が、前記部分配列の3’末端である、前記項目のいずれかに記載の方法。
(項目13) 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列の出現頻度が異なる場合、該長さxの部分の配列を、対象配列データにおけるコントロール配列データに対する多型として検出する、前記項目のいずれかに記載の方法。
(項目14) 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列で最も高頻度のものが異なっている長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおける多型として検出する、前記項目のいずれかに記載の方法。
(項目15) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記対象配列データのサブセットにおける前記長さxの部分の配列で、前記コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列がノイズ以下のカウントしか存在しない長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるホモ多型として検出する、前記項目のいずれかに記載の方法。
(項目16) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、対象サブセットにおける前記長さxの部分の配列で、コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列が存在し、かつ、コントロール配列データのサブセットにおける最も高頻度のものと異なる長さxの部分の配列が存在する長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるヘテロ多型として検出する、前記項目のいずれかに記載の方法。
(項目17) 対象配列データ量から予測される出現頻度と比較して、前記出現頻度が少ない部分配列をノイズとする、前記項目のいずれかに記載の方法。
(項目18) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、[(対象配列データ量)×(1-精度)]/(対象ゲノムサイズ)+1未満の出現頻度の部分配列をノイズとする、前記項目のいずれかに記載の方法。
(項目19) 前記対象配列データが、次世代シーケンシングによって得られた塩基配列データである、前記項目のいずれかに記載の方法。
(項目20) 前記対象配列データが、個体から得られた配列データであり、前記コントロール配列データが、該個体と同種の別の個体、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目21) 前記対象配列データが、個体の組織試料から得られた配列データであり、前記コントロール配列データが、該個体の別の組織、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目22) 前記対象配列データが、細胞試料から得られた配列データであり、前記コントロール配列データが、別の細胞、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目23) 前記多型が、置換、挿入、欠失、コピー数多型(Copy Number Variation, CNV)、STRP(short tandem repeat polymorphism)、逆位または転座である、前記項目のいずれかに記載の方法。
(項目24) 前記多型が、置換である、前記項目のいずれかに記載の方法。
(項目25) 前記対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目26) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記多型のゲノム上の位置を特定する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目27) 検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目28) 前記クエリ配列セットが、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セットを含む、前記項目のいずれかに記載の方法。
(項目29) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの相補鎖の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目30) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの変異型の塩基を有する配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目31) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記ゲノムの配列が不明である、前記項目のいずれかに記載の方法。
(項目32) 実験結果またはデータベースから対象配列データまたはコントロール配列データを取得する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目X1) 対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることをさらに含む、前記項目のいずれか1項に記載の方法。
(項目X2) 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、前記項目のいずれかに記載の方法。
(項目33) 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムであって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラム。
(項目33A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載のプログラム。
(項目34) 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する工程をさらに含む、前記項目のいずれかに記載のプログラム。
(項目35) 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムを格納する記録媒体であって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体。
(項目35A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の記録媒体。
(項目36) 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する工程をさらに含む、前記項目のいずれかに記載の記録媒体。
(項目37) 対象配列データにおいてコントロール配列データに対する多型を検出するためのシステムであって、該システムは、
該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部と
を備える、システム。
(項目37A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載のシステム。
(項目38) 前記システムが、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する表示手段をさらに含む、前記項目のいずれかに記載のシステム。
(項目39) 対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
(1)a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
によって、対象配列データにおける置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するプロセスと、
(2)a)該対象配列データの配列中の少なくとも2ヶ所の部分配列の、該コントロール配列上の位置を特定する工程と、
b)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とを比較する工程と、
c)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とが異なっている場合、目的とする多型があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程と
によって、対象配列データにおける挿入、欠失、逆位、転座または置換を検出するプロセスと
を包含する、方法。
(項目39A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目40) 対象配列データにおいてリファレンス配列データに対する多型を検出する方法であって、リファレンス配列データから、各々の位置情報と関連付けられたリファレンス配列のk長の部分配列セットを作成する工程を含み、さらに、
(A1)該対象配列データの長さkの部分配列のサブセットを生成し、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A2)該リファレンス配列のk長の部分配列セットの、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A3)該対象配列と該リファレンス配列とを比較し、該出現頻度の分布の比較に基づいて、挿入、欠失、置換、コピー数多型、STRP、逆位または転座を検出する工程とを包含するプロセスと
(B1)該対象配列データの配列中の少なくとも2ヶ所のk長の部分配列をクエリとして、該リファレンス配列のk長の部分配列セットに対して検索を行い、該少なくとも2ヶ所の部分配列の、リファレンス配列上の位置を特定する工程と、
(B2)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とを比較する工程と、
(B3)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とが異なっている場合、挿入、欠失、逆位または転座があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程を包含し、必要に応じて、
(B4)該位置関係が異ならない場合に、該対象配列データにおける該部分配列部位間の文字を、対応する前記コントロール配列上の文字と比較して不一致となる部位を検出する工程をさらに含み、不一致となる部位が存在する場合、置換が存在すると判定する工程をさらに含む、プロセスと、
を、同時に、並行して、または逐次的に行うことを特徴とする、方法。
(項目40A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目A1) 対象配列データとコントロール配列データとの比較方法であって、
対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることを含む、方法。
(項目A1A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目A2) 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、前記項目のいずれかに記載の方法。
(項目1) 対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
を包含し、ここで、kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である、方法。
(項目2) 前記部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出する工程をさらに含み、ここで、xはk未満の正の整数である、前記項目に記載の方法。
(項目3) 前記比較が、前記部分配列中の長さk-xの配列部分が共通する配列における、長さxの部分の出現頻度の分布の差異の比較を含む、前記項目のいずれかに記載の方法。
(項目4) 前記部分配列中の長さk-xの配列部分を、ユニークな配列ごとにグルーピングする工程を含み、ここで、xはk未満の正の整数である、前記項目のいずれかに記載の方法。
(項目5) 前記長さk-xの配列部分をソートする工程を含む、前記項目のいずれかに記載の方法。
(項目6) 前記長さk-xの配列部分を文字列としてソートする工程を含む、前記項目のいずれかに記載の方法。
(項目7) 前記kが、前記対象配列における偶然同一を排除する長さである、前記項目のいずれかに記載の方法。
(項目8) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記kが、前記生物のゲノムにおいて、異なる箇所での偶然同一を排除する長さである、前記項目のいずれかに記載の方法。
(項目9) 長さxが1~2である、前記項目のいずれかに記載の方法。
(項目10) 長さxが1である、前記項目のいずれかに記載の方法。
(項目11) 前記長さxの部分が、前記部分配列の末端に存在する、前記項目のいずれかに記載の方法。
(項目12) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記長さxの部分が、前記部分配列の3’末端である、前記項目のいずれかに記載の方法。
(項目13) 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列の出現頻度が異なる場合、該長さxの部分の配列を、対象配列データにおけるコントロール配列データに対する多型として検出する、前記項目のいずれかに記載の方法。
(項目14) 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列で最も高頻度のものが異なっている長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおける多型として検出する、前記項目のいずれかに記載の方法。
(項目15) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記対象配列データのサブセットにおける前記長さxの部分の配列で、前記コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列がノイズ以下のカウントしか存在しない長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるホモ多型として検出する、前記項目のいずれかに記載の方法。
(項目16) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、対象サブセットにおける前記長さxの部分の配列で、コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列が存在し、かつ、コントロール配列データのサブセットにおける最も高頻度のものと異なる長さxの部分の配列が存在する長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるヘテロ多型として検出する、前記項目のいずれかに記載の方法。
(項目17) 対象配列データ量から予測される出現頻度と比較して、前記出現頻度が少ない部分配列をノイズとする、前記項目のいずれかに記載の方法。
(項目18) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、[(対象配列データ量)×(1-精度)]/(対象ゲノムサイズ)+1未満の出現頻度の部分配列をノイズとする、前記項目のいずれかに記載の方法。
(項目19) 前記対象配列データが、次世代シーケンシングによって得られた塩基配列データである、前記項目のいずれかに記載の方法。
(項目20) 前記対象配列データが、個体から得られた配列データであり、前記コントロール配列データが、該個体と同種の別の個体、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目21) 前記対象配列データが、個体の組織試料から得られた配列データであり、前記コントロール配列データが、該個体の別の組織、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目22) 前記対象配列データが、細胞試料から得られた配列データであり、前記コントロール配列データが、別の細胞、またはデータベースから得られた配列データである、前記項目のいずれかに記載の方法。
(項目23) 前記多型が、置換、挿入、欠失、コピー数多型(Copy Number Variation, CNV)、STRP(short tandem repeat polymorphism)、逆位または転座である、前記項目のいずれかに記載の方法。
(項目24) 前記多型が、置換である、前記項目のいずれかに記載の方法。
(項目25) 前記対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目26) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記多型のゲノム上の位置を特定する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目27) 検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目28) 前記クエリ配列セットが、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セットを含む、前記項目のいずれかに記載の方法。
(項目29) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの相補鎖の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目30) 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの変異型の塩基を有する配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目31) 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記ゲノムの配列が不明である、前記項目のいずれかに記載の方法。
(項目32) 実験結果またはデータベースから対象配列データまたはコントロール配列データを取得する工程をさらに含む、前記項目のいずれかに記載の方法。
(項目X1) 対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることをさらに含む、前記項目のいずれか1項に記載の方法。
(項目X2) 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、前記項目のいずれかに記載の方法。
(項目33) 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムであって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラム。
(項目33A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載のプログラム。
(項目34) 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する工程をさらに含む、前記項目のいずれかに記載のプログラム。
(項目35) 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムを格納する記録媒体であって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体。
(項目35A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の記録媒体。
(項目36) 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する工程をさらに含む、前記項目のいずれかに記載の記録媒体。
(項目37) 対象配列データにおいてコントロール配列データに対する多型を検出するためのシステムであって、該システムは、
該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部と
を備える、システム。
(項目37A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載のシステム。
(項目38) 前記システムが、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する表示手段をさらに含む、前記項目のいずれかに記載のシステム。
(項目39) 対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
(1)a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
によって、対象配列データにおける置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するプロセスと、
(2)a)該対象配列データの配列中の少なくとも2ヶ所の部分配列の、該コントロール配列上の位置を特定する工程と、
b)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とを比較する工程と、
c)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とが異なっている場合、目的とする多型があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程と
によって、対象配列データにおける挿入、欠失、逆位、転座または置換を検出するプロセスと
を包含する、方法。
(項目39A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目40) 対象配列データにおいてリファレンス配列データに対する多型を検出する方法であって、リファレンス配列データから、各々の位置情報と関連付けられたリファレンス配列のk長の部分配列セットを作成する工程を含み、さらに、
(A1)該対象配列データの長さkの部分配列のサブセットを生成し、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A2)該リファレンス配列のk長の部分配列セットの、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A3)該対象配列と該リファレンス配列とを比較し、該出現頻度の分布の比較に基づいて、挿入、欠失、置換、コピー数多型、STRP、逆位または転座を検出する工程とを包含するプロセスと
(B1)該対象配列データの配列中の少なくとも2ヶ所のk長の部分配列をクエリとして、該リファレンス配列のk長の部分配列セットに対して検索を行い、該少なくとも2ヶ所の部分配列の、リファレンス配列上の位置を特定する工程と、
(B2)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とを比較する工程と、
(B3)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とが異なっている場合、挿入、欠失、逆位または転座があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程を包含し、必要に応じて、
(B4)該位置関係が異ならない場合に、該対象配列データにおける該部分配列部位間の文字を、対応する前記コントロール配列上の文字と比較して不一致となる部位を検出する工程をさらに含み、不一致となる部位が存在する場合、置換が存在すると判定する工程をさらに含む、プロセスと、
を、同時に、並行して、または逐次的に行うことを特徴とする、方法。
(項目40A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目A1) 対象配列データとコントロール配列データとの比較方法であって、
対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることを含む、方法。
(項目A1A) 前記項目のいずれか1つまたは複数に記載される特徴を有する、前記項目に記載の方法。
(項目A2) 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、前記項目のいずれかに記載の方法。
本発明において、上記1または複数の特徴は、明示された組み合わせに加え、さらに組み合わせて提供され得ることが意図される。本発明のなおさらなる実施形態および利点は、必要に応じて以下の詳細な説明を読んで理解すれば、当業者に認識される。
本発明により、2つ以上の配列の間で、全長配列における位置を考慮する必要なく、正確に多型、特に置換を検出することができる。k長配列を用いた置換変異の検出に関しては、ゲノムマッピングを行う前に多型検出ができることが1つの大きな特徴となる。そして、リファレンス配列が存在しない生物でも多型検出が可能であり、k-mer自体を多型の名称として用いることが可能であるため、それゆえ連鎖解析等の遺伝解析で大きな変革をもたらす可能性がある。
以下、本発明を最良の形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の冠詞(例えば、英語の場合は「a」、「an」、「the」など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当該分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
(定義)
以下に本明細書において特に使用される用語の定義および/または基本的技術内容を適宜説明する。
以下に本明細書において特に使用される用語の定義および/または基本的技術内容を適宜説明する。
本明細書において、「配列」とは、各々が何らかの値を取る複数の変数であって、それら複数の変数の順序の情報をさらに含むものをいう。代表的には文字列で表示される。
本明細書において、「対象配列」とは、多型を検出しようとする任意の配列をいい、本明細書においては、「ターゲット」、「ターゲット配列」、「target」とも表記する場合がある。
本明細書において、「コントロール配列」とは、その配列との差異を多型として検出するための基準として用いられる任意の配列をいい、本明細書においては、「コントロール」、「参照配列」、「比較配列」、「control」とも表記する場合がある。
本明細書において、「多型(polymorphism)」とは、対象配列中においてコントロール配列と異なっている任意の部分を指す。本明細書において、「変異」も同様の意味で使用することができる。
本明細書において、「リファレンス(reference)配列」とは、対象配列および/またはコントロール配列の全長の配列として扱うことができる配列を指す。いかなる配列を全長配列とするかは、対象配列および/またはコントロール配列として用いる配列に応じて適宜決定されるものであり、例示されるものに限定されないが、例えば、ウェブ上のデータベース等に存在する、全ゲノム配列、染色体全長配列、遺伝子全長配列、プラスミド全長配列、エクソン全長配列、タンパク質全長配列などをリファレンス配列として用いることができる。
本明細書において、「配列データ」とは、ある配列についての情報を与えるデータをいう。代表的には、配列そのものも配列データということができ、また、配列の一部について情報を与えるデータ(例えば、ゲノム配列に対するシーケンシングによる解析データ)も配列データとして包含される。
本明細書において、ある配列の「部分配列」とは、その配列に含まれる任意の配列をいう。
本明細書において、「サブセット」とは、配列の集合と、それらの配列の部分配列の集合とを合わせた集合の任意の部分集合をいう。
本明細書において、「次世代シーケンシング」とは、配列決定プロセスを並列化し、一度のランで数千万から数億の配列データを生成するシーケンシング技法である。「次世代シーケンサー」とは、次世代シーケンシングを行うための機器を指す。
「偶然同一を排除する」とは、ある配列と、偶然に同一の配列が出現する期待値を1未満にすることをいう。
本明細書において、「カバレッジ」とは、配列データの量が、配列全長の何倍に相当しているかを指す。「カバー率」、「~倍の読み」などと称される場合もある。
本明細書において、「配列構造体」とは、配列中における、物理的に分離された一連の配列をいう。例えば、ゲノム配列の文脈では、染色体のそれぞれは配列構造体ということができる。
本明細書において、「転座」とは、複数の配列構造体を有する配列中で、ある配列構造体上の部分配列が、他の配列構造体上に移動している多型をいう。
本明細書において、「ジャンクション」とは、一部が同一である2つの配列について、同一である部分と同一でない部分の境界を指す。
本明細書において、「識別子」とは、ある多型を他の多型と区別するために付される名称を指す。一般的には、多型の開始位置と型で記載されることが多いが、本明細書において記載される識別子を用いることができる。
本明細書において、「エッジ」とは、配列において多型を含む部分の末端をさす。
(好ましい実施形態)
以下に本発明の好ましい実施形態を説明する。以下に提供される実施形態は、本発明のよりよい理解のために提供されるものであり、本発明の範囲は以下の記載に限定されるべきでないことが理解される。従って、当業者は、本明細書中の記載を参酌して、本発明の範囲内で適宜改変を行うことができることは明らかである。また、本発明の以下の実施形態は単独でも使用されあるいはそれらを組み合わせて使用することができることが理解される。
以下に本発明の好ましい実施形態を説明する。以下に提供される実施形態は、本発明のよりよい理解のために提供されるものであり、本発明の範囲は以下の記載に限定されるべきでないことが理解される。従って、当業者は、本明細書中の記載を参酌して、本発明の範囲内で適宜改変を行うことができることは明らかである。また、本発明の以下の実施形態は単独でも使用されあるいはそれらを組み合わせて使用することができることが理解される。
なお、以下で説明する実施の形態は、いずれも包括的または具体的な例を示すものである。以下の実施の形態で示される数値、形状、材料、構成要素、構成要素の配置位置及び接続形態、ステップ、ステップの順序などは、一例であり、請求の範囲を限定する主旨ではない。また、以下の実施の形態における構成要素のうち、最上位概念を示す独立請求項に記載されていない構成要素については、任意の構成要素として説明される。
(本発明の多型検出の概要)
本発明は、対象配列データにおいてコントロール配列データに対する多型を検出する方法を提供する。この方法は、a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程とを包含し、ここで、kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である、方法を提供する。本発明の例示的なフローは図16に説明されている。
本発明は、対象配列データにおいてコントロール配列データに対する多型を検出する方法を提供する。この方法は、a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程とを包含し、ここで、kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である、方法を提供する。本発明の例示的なフローは図16に説明されている。
本発明の方法は、対象配列データと、コントロール配列データとの2つの配列データ(例えば、次世代シーケンサー解析結果)からの直接比較により多型を検出することが可能である点で、従来法と根本的に異なる。とりわけ、塩基配列における多型検出において、ゲノム上の位置の考慮なしに直接比較する方法は、新規なものであると考えられる。
1つの実施形態において、本発明の方法は、配列データから、一定長(k長)の部分配列のセットを得ることを1つの特徴とする。1つの実施形態において、本発明の方法は、配列データから、k長の部分配列のセットにおける各部分配列の頻度分布を得ることを1つの特徴とする。一部の実施形態では、配列データから、配列を1つずつずらしながらk長の部分配列のセットを作成する。
1つの実施形態では、k長の配列のうち、k-x(xは1など)の配列が同じであるデータをソートし、異なる部分(x長の部分に該当)の頻度を検出する。X長の部分は、部分配列中での位置は限定されず、配列中の中央部でもよい。しかしながら、X長の部分を、部分配列の末端(例えば、塩基配列においては3’末端または5’末端)にすることは、ソートなどの処理を顕著に簡便化・高速化するため、好ましい。このように、k長の配列のうち末端部で異なる部分(x長の部分に該当)の頻度を検出する場合、本明細書において、「Polymorphic Edge Detection(PED)」または「edge detection」と呼称される場合がある。
ここで、kの値としては、配列データの各配列(例えば、次世代シーケンサーの各々のショートリード)の長さを上限とした任意の値を挙げることができ、例えば、約500、約400、約300、約200、約100、約50、約40、約30、約25、約20、約15等を挙げることができる。kが増加することにより、k-mer配列のデータは指数的に増加する(例えば、塩基配列の場合、kが1塩基増えるごとに塩基の組み合わせは4倍になる)ため、例えば、塩基配列の場合、k=20~25程度が好ましいが、理論上は、例えば、k=500等でも用いることが可能であり、制限されるものではない。ヒトの場合であると、k=17以下だと偶然一致が生じる確率が高くなるが、ゲノムサイズが小さな生物であれば、例えば、k=15などより小さなk値を用いることも可能である。1つの実施形態ではk=20を用いる。
k-x長の配列が同じであるデータのxの部分の文字が比較対象間で異なる場合、その文字に多型(変異)が含まれると考えられる。挿入・欠失変異も変異の末端文字を検出できる。例えば、k-x長の塩基配列が同じであるデータのxの部分の塩基が比較対象間で異なる場合、その塩基に多型(変異)が含まれると考えられる。
一部の実施形態において、得られた配列セットのうち、同じ配列に関して出現回数で整理したデータを算出する。この工程は、計算機を使用して簡便に行うことができ、例えば、Unixでは以下:
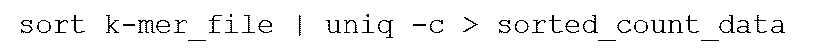
のように実装することができ、(文字列で)ソートされた配列と頻度を示す数値のデータを生成することができる。さらに、対象とコントロールの頻度データを同一k-merでまとめる際に、例えば、Unixではjoinコマンド等を使用して行うことが可能である。
のように実装することができ、(文字列で)ソートされた配列と頻度を示す数値のデータを生成することができる。さらに、対象とコントロールの頻度データを同一k-merでまとめる際に、例えば、Unixではjoinコマンド等を使用して行うことが可能である。
配列出現回数を、配列データのカバレッジ(何倍読みか)と比較することによって、配列データにおける差異を評価することも可能である。例えば、ゲノム配列に対して40倍のデータ量の配列解析からの配列データに対して、出現頻度が1となるようなものはノイズと考えることができる。
本発明は、特に、「置換」多型(長さが変わらない=欠失挿入ではない)の検出には、極めて高い効果を発揮する。多型部位が多コピーの場合は位置の特定ができない場合があり得るが、それでもなお、多型自体の検出は可能であり、多型に名称を付して特定できる。そのため、例えば、検出された各多型を、形質との関係を調べる多型マーカーとして使用することが可能であり、診断・育種・鑑定・品質管理(例えば、iPS細胞の品質管理)・分類・検査にも応用可能である。
本発明を、次世代シーケンサーから得られた塩基配列データから直接多型を検出する方法として用いることで、2種類のサンプル間、およびリファレンス配列とサンプルとの間の多型の検出が可能となる。また、長さkの部分配列、k-xの部分配列は重複のないユニークな配列なので、配列自体を多型の識別子(名称)として利用可能である。このため、リファレンスゲノム配列が決定されていないため多型のゲノム上の位置関係が判別できない場合においても、世界共通の一意の多型の識別子として利用できる。本発明の1つの実施形態において、対象配列データとコントロール配列データとの比較方法であって、対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、多型の識別子として割り当てることを含む、方法が提供される。また、多型の識別子をリファレンス配列にマッピングし、リファレンス上の多型の位置を特定することが可能である。
識別子は、多型ではない部分の少なくとも一部に加えて、多型自体も含み得る。多型塩基を含めた識別子は、リファレンス配列へのマッピングは難しいものの、連鎖解析に用いることが可能である。
例えば、
AAACCACTTCACGTTTCCA A
AAACCACTTCACGTTTCCA G
という多型の例では、
AAACCACTTCACGTTTCCAのA型
AAACCACTTCACGTTTCCAのG型
AAACCACTTCACGTTTCCAのA/Gのヘテロ型
という表現が記載の一例である。
多型を含めた表記の仕方の例としては、
AAACCACTTCACGTTTCCAA型、
AAACCACTTCACGTTTCCAG型、
そして、ヘテロ型は、
AAACCACTTCACGTTTCCAA/AAACCACTTCACGTTTCCAG
のように、2つの型を併記することが可能である。
例えば、
AAACCACTTCACGTTTCCA A
AAACCACTTCACGTTTCCA G
という多型の例では、
AAACCACTTCACGTTTCCAのA型
AAACCACTTCACGTTTCCAのG型
AAACCACTTCACGTTTCCAのA/Gのヘテロ型
という表現が記載の一例である。
多型を含めた表記の仕方の例としては、
AAACCACTTCACGTTTCCAA型、
AAACCACTTCACGTTTCCAG型、
そして、ヘテロ型は、
AAACCACTTCACGTTTCCAA/AAACCACTTCACGTTTCCAG
のように、2つの型を併記することが可能である。
本発明の1つの実施形態は、対象配列データにおいてコントロール配列データに対する多型を検出する方法である。1つの実施形態において、当該方法は、該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程を含む。kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である。1つの実施形態において、当該方法は、該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程を含む。1つの実施形態において、当該方法は、対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程とを含む。このような工程によって、全長配列における位置を考慮せず、また、配列を連結することなく配列データを比較し、多型を検出することができる。
さらなる実施形態において、本発明の方法は、部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出する工程をさらに含む。xはk未満の正の整数である。この場合において、出現頻度の分布の比較として、前記部分配列中の長さk-xの配列部分が共通する配列における、長さxの部分の出現頻度の分布の差異の比較が含まれ得る。これにより、多型検出の処理を高速化することが可能である。
一部の実施形態において、本発明の方法は、前記部分配列中の長さk-xの配列部分を、ユニークな配列ごとにグルーピングする工程を含む。これには、例えば、前記長さk-xの配列部分をソートする工程(例えば、前記長さk-xの配列部分を文字列としてソートする工程)が含まれ得る。
一部の実施形態において、kの値は、前記対象配列データ等における偶然同一を排除する長さである。例えば、前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データである場合、前記kは、前記生物のゲノムにおいて、異なる箇所での偶然同一を排除する長さであり得る。これにより、多型の検出をより正確なものとすることが可能である。
長さxは、限定されるものではないが、好ましくは1~3であり、さらに好ましくは1~2であり、より好ましくは1である。
1つの実施形態では、前記長さxの部分が、前記部分配列の末端に存在する。例えば、前記対象配列データおよび前記コントロール配列データが塩基配列データである場合、前記長さxの部分は、前記部分配列の3’末端または5’末端であり得る。長さxの部分を部分配列の末端にとることは、比較処理の高速化・簡便化にとって望ましい。
出現頻度の分布の差異の比較により、例えば、以下のような多型の検出が可能である。1つの実施形態では、前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列の出現頻度が異なる場合、該長さxの部分の配列を、対象配列データにおけるコントロール配列データに対する多型として検出する。1つの実施形態では、前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列で最も高頻度のものが異なっている長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおける多型として検出する。
1つの実施形態では、前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記対象配列データのサブセットにおける前記長さxの部分の配列で、前記コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列がノイズ以下のカウントしか存在しない長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるホモ多型として検出する。ノイズは、本明細書において後述されるような基準を用いて判定することができる。1つの実施形態では、前記対象配列データおよび前記コントロール配列データが塩基配列データであり、対象サブセットにおける前記長さxの部分の配列で、コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列が存在し、かつ、コントロール配列データのサブセットにおける最も高頻度のものと異なる長さxの部分の配列が存在する長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるヘテロ多型として検出する。
一部の実施形態において、本発明の方法において、予測される出現頻度と比較して、出現頻度が少ない部分配列をノイズとすることが可能である。これにより、機械的に生じた差異と、実際に生じている多型とを識別して検出することが可能である。例えば、前記対象配列データおよび前記コントロール配列データが塩基配列データである場合には、対象配列データ量、配列データにおける予想されるエラー率(例えば、マニュアル・機器の公称値)、対象配列全長サイズ(例えば、ゲノムサイズ)等を考慮して、予想されるノイズのカウント程度、または予想されるノイズのカウント以下の出現頻度の部分配列をノイズとすることができる。1つの予測としては、生じるノイズの頻度の期待値は、int(ゲノムを何倍読んだか×(1-精度)+1)」となる。エラーが存在しない場合、精度は1になる。1つの実施形態では、対象配列データおよび前記コントロール配列データが塩基配列データである場合に、[(対象配列データ量)×(1-精度)]/(対象ゲノムサイズ)+1未満の出現頻度の部分配列をノイズとすることが可能である。
例えば、エラー率が0.001の場合は着目した塩基に関して1カウントでも出現する確率は0.001になるためほぼ0と考えられるが、1000塩基の範囲で見れば、どれか1塩基にエラーが含まれる計算となる。そのため、繰り上げた整数値をノイズの予測値とすることができると考えられ、int関数中で1を加えることによって繰り上げた整数値とすることができる。
あるいは、本発明の方法において、予測される出現頻度と比較して、出現頻度が多い部分配列を、リピート配列部位として除外することができる。例えば、対象配列データの対象配列全長のカバレッジ(カバー率)の2倍を超えるような部分配列を解析から除外することができる。
1つの実施形態では、前記対象配列データおよび/またはコントロール配列データが、次世代シーケンシングによって得られた塩基配列データである。次世代シーケンシングにおける多型の検出においては、従来、リファレンスへのマッピングや、配列のアセンブリが必要とされており、このような工程で生じる不確実性によって、多型の検出が大きく阻害されていたため、このような工程を必要としない本発明の方法を次世代シーケンシングから得られた配列データに対して用いることは特に有利なものであるということができる。
対象配列データおよびコントロール配列データは、限定されるものではないが、多型を検出する上では、一定の共通性を持つ配列についての配列データであることが望ましい。しかしながら、配列の取得方法については各々同一でも異なっていてもよく、シーケンシングによって得られたデータ間での比較を行うことも、データベース等から得られたデータ間での比較を行うことも、シーケンシングによって得られたデータとデータベース等から得られたデータとの間での比較を行うことも可能である。
1つの実施形態では、対象配列データが、個体から得られた配列データであり、コントロール配列データが、該個体と同種の別の個体、またはデータベースから得られた配列データである。1つの実施形態では、対象配列データが、個体の組織試料から得られた配列データであり、コントロール配列データが、該個体の別の組織、またはデータベースから得られた配列データである。1つの実施形態では、対象配列データが、細胞試料から得られた配列データであり、コントロール配列データが、別の細胞、またはデータベースから得られた配列データである。
本発明の方法は、全長配列の情報を必要としないため、データベース等において例えば全長配列が公知でない場合にも用いることができ、例えば、対象配列データおよびコントロール配列データは、生物のゲノムに由来する塩基配列データである場合、前記ゲノムの配列が不明であってもよい。
本発明の方法によって検出できる多型としては、置換、挿入、欠失、コピー数変異(Copy Number Variation, CNV)、STRP(short tandem repeat polymorphism)、逆位および転座が挙げられる。1つの実施形態において、本発明の方法は、上記複数の多型の任意の組み合わせを同時に検出することも可能である。さらなる実施形態において、本発明の方法は、上記複数の多型の全てを同時に検出することも可能である。特に、多型が置換である場合には、本発明の方法は、非常に高い検出力を発揮することが可能である。
対象配列に対するリファレンス配列が存在する場合、本発明の方法は、前記対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含むことができる。例えば、対象配列データおよびコントロール配列データが、生物のゲノムに由来する塩基配列データである場合、多型のゲノム上の位置を特定する工程をさらに含むことができる。この位置の特定は、本発明の方法が、多型を周囲の配列と関連づけて検出する(例えば、x長部分の多型がk-x長の配列と関連付けられる)ことを可能にしているため、リファレンス配列に対して検索を行うことにより、簡便に行うことが可能である。
本発明の方法は、検出した多型について確認する工程をさらに含むことができる。確認は、例えば、検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行うことによって行うことができる。クエリ配列セットは、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セット、および/またはリファレンス配列またはコントロール配列において前記多型に該当する部位の文字を置換していない野生型クエリ配列セットを含み得る。
本発明の方法は、対象配列データおよびコントロール配列データが塩基配列データである場合、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの相補鎖の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含むことができる。本発明の方法は、対象配列データおよびコントロール配列データが塩基配列データである場合、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの対立遺伝子の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含むことができる。ここで、対立遺伝子の配列データとして、実際の遺伝子の存在の有無とは関係なく、野生型に対する変異型の塩基を有する配列データを用いることができる。
本発明の方法は、実験結果またはデータベースから対象配列データまたはコントロール配列データを取得する工程を含んでもよい。また、本発明の方法においては、必ずしも配列データそのものを取得する必要はなく、配列データのサブセット、および/または配列データもしくは配列データのサブセットにおける頻度分布のデータを取得して実行することも可能である。
1つの局面において、本発明は、本発明の多型を検出する方法をコンピュータに実施させるための方法を実装するプログラム、該プログラムを記録した記録媒体、およびこれを実現するためのシステムを提供する。ここで採用され得る任意の特徴は本明細書の多型を検出する方法の説明に記載される任意の特徴またはその組み合わせを採用することができる。
したがって、1つの実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムであって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラムが提供される。さらなる実施形態において、上記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、プログラムが提供される。
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラムが提供される。さらなる実施形態において、上記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、プログラムが提供される。
別の実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムを格納する記録媒体であって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体が提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、記録媒体が提供される。
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体が提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、記録媒体が提供される。
別の実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出するためのシステムであって、該システムは、
該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部と
を備える、システムが提供される。さらなる実施形態において、前記部分配列中の多型ではない部分の少なくとも一部を含む配列前記部分配列全体であり得る)を、検出された前記多型の名称として表示する表示手段をさらに含む、システムが提供される。
該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部と
を備える、システムが提供される。さらなる実施形態において、前記部分配列中の多型ではない部分の少なくとも一部を含む配列前記部分配列全体であり得る)を、検出された前記多型の名称として表示する表示手段をさらに含む、システムが提供される。
(具体的な例)
本発明の例示的な実施形態は、以下のような工程による方法である。
本発明の例示的な実施形態は、以下のような工程による方法である。
1.配列データについて、長さkの部分配列のサブセットを得る。例えば、塩基配列データを端から1塩基ずつずらしながら、k長の配列のセットを得る。以下の例では、次世代シーケンサーで得られたイネ個体の配列データ(データ量はイネゲノムの40倍、リード長は100塩基)を用いて、k長を20塩基として得られた配列セットを用いて説明する。得られた配列セットに関して、比較対象のサンプル間で5’末端からk-1長の配列が同じであるデータの3’末端の塩基が比較対象の間で異なる場合、その塩基は多型である。

(上記例において、1行目から各々配列番号191~201である。)
(上記例において、1行目から各々配列番号191~201である。)
最初の行が次世代シーケンサーから得られた塩基配列であり、以下の行がk長の部分配列を示す。この場合k長を20塩基で配列セットを得ている。
2.得られた部分配列セットの出現頻度データを得る。すなわち、部分配列セットのうちの同じ配列に関して、出現回数で整理したデータを得る。

(上記例において、1行目から各々配列番号202~211である。)
(上記例において、1行目から各々配列番号202~211である。)
配列セットを降順に整列して出現回数を配列の右に表示している。この例では、ゲノムの40倍の解析なので、出現回数が1回程度の配列はノイズであると判断することができる。40~50回程度の配列は、ゲノム上のユニークな配列由来であり、89回出現した配列は、ゲノム上2ヶ所存在していると考えられる。
3.部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出する。この例では、得られたk塩基の部分配列(k=20)の頻度データをもとに、最初の19塩基(k-x、x=1)に対する最後の1塩基のACGTそれぞれの塩基の出現頻度データに変換する。

(上記例において、1行目から各々配列番号212~220である。)
(20塩基の頻度データを最初の19塩基と最後のACGTの塩基の頻度一覧に変換する。)
(上記例において、1行目から各々配列番号212~220である。)
(20塩基の頻度データを最初の19塩基と最後のACGTの塩基の頻度一覧に変換する。)
4.部分配列中の長さk-xの配列部分が共通する配列における、長さxの部分の出現頻度の分布の差異を比較する。例えば、コントロールと調べたい対象の配列データ(ターゲット)からの頻度データを19merの配列でまとめた一覧を作成する。多型がない場合、最後の塩基の頻度は同じ塩基で最多となる。最後の塩基の頻度でコントロールと対象で塩基が異なる場合は、その塩基が多型である。
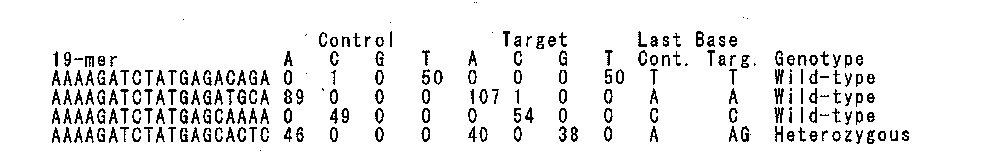
(上記例において、1行目から各々配列番号213、215、217、および218である。)
(上記例において、1行目から各々配列番号213、215、217、および218である。)
コントロールと調べたい対象の最後の塩基の出現頻度の一覧。この場合、AAAAGATCTATGAGCACTC(配列番号218)の次にはコントロールではAのみであるが、対象ではAとGのヘテロザイガスであることがわかる。また、ホモザイガスの多型が生じる場合には、以下のように、最後の塩基として出現するものが異なり、検出することができる。

このように、ゲノム上の位置が不明でも、最初の19塩基で表記される配列位置に続く塩基の多型を網羅することができる。ゲノムリファレンス配列が既知の場合、最初の19塩基に対応するゲノム位置から多型の位置を決めることができる。順鎖、相補鎖ともに同じ位置に検出された場合は一塩基多型である。多型となる配列の末端部分を検出するため、挿入・欠失多型の検出も可能である。最初の19塩基を多型の名称、最後の塩基を遺伝子型として表記するため、その多型を示す唯一の表記法として利用できる。あるいは、多型も含めてk-mer(例えば、20塩基)全体で一つの名称とすることも可能である。例えばk=5の場合には、ACGTA型とACGTT型といった表記が可能である。本発明の方法は、図1に示されるようなフローにしたがって、図1に示される工程を適宜採用することによって実行することが可能である。
(k-mer)
1つの実施形態において、本発明の1つの特徴は、対象配列データにおけるコントロール配列データに対する多型の検出において、該対象配列データの長さkの部分配列のサブセット、または該部分配列のサブセットの各々の部分配列の出現頻度を用いることである。ここで、kは対象配列およびコントロール配列のいずれか短いほうの全長以下の整数である。
1つの実施形態において、本発明の1つの特徴は、対象配列データにおけるコントロール配列データに対する多型の検出において、該対象配列データの長さkの部分配列のサブセット、または該部分配列のサブセットの各々の部分配列の出現頻度を用いることである。ここで、kは対象配列およびコントロール配列のいずれか短いほうの全長以下の整数である。
長さkの部分配列は、対象配列データ、コントロール配列データ、リファレンス配列データ等から切り出すことによって生成することができる。例えば、一定間隔でk長の配列を切り出すことによって生成することができ、1文字ごと、2文字ごと、3文字ごと、またはそれ以上の間隔で切り出して部分配列セットを生成することができる。あるいは、対象配列データ、コントロール配列データ、リファレンス配列データ等から、ランダムに位置を選択して切り出すことも可能である。全てのk長部分配列を生成することが望ましい場合には、1文字ごとに切り出し位置をずらしながらk長部分配列のサブセットを生成することができる。
長さkは、対象配列、コントロール配列および/またはリファレンス配列における偶然同一を排除する長さであることが望ましい。偶然同一を排除することによって、異なる配列が対象配列の別の箇所に偶然含まれていたものをコントロール配列との差異として検出する可能性を低減し、より正確に解析することができる。k長のある配列と、偶然に同一の配列が、対象配列、コントロール配列および/またはリファレンス配列に出現する期待値を1未満にすることが望ましい。
一般的に、v:配列において各変数が取り得る値の種類、L:解析対象とする配列(対象配列、コントロール配列および/またはリファレンス配列)の全長(含まれる変数の数)として、v^k>Lとなる場合に、kが偶然同一を排除する長さであると考えられる。あるいは、配列全長が10^Lのようなオーダーで表される場合、両辺の対数をとり、k>L/log(v)を満たす場合に、kが偶然同一を排除する長さであると考えられる。
例えば、配列データが塩基配列データである場合、4つの文字が変数の値として考えられるため、v=4である。例えば、ヒトゲノムは3.1×10の9乗のサイズであり、仮に10の9乗長のアトランダムな塩基配列があった場合、9/log(4)≒15塩基が偶然一致を排除できる最小k長と考えられる。ゲノムサイズが異なる生物に対しても、例えば、10/log(4)の場合はk=17で偶然一致を排除できると考えられる。長いほど偶然一致の配列が生じる可能性を減少させることができるが、データサイズがその分大きくなる。
(頻度)
1つの実施形態において、本発明の方法は、対象配列データおよび/またはコントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程を含む。
1つの実施形態において、本発明の方法は、対象配列データおよび/またはコントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程を含む。
長さkの部分配列のサブセットの各々の部分配列の出現頻度は、ユニークなk長の部分配列データに対する頻度がペアになったデータとして(例えば、1列目に部分配列、2列目にその頻度が提供される行列データとして)提供されてもよい。部分配列の出現頻度は、配列とその頻度とが関連付けられているものであれば、その形式は限定されるものではなく、対応する識別子を有する配列データと頻度データとを別個に出力することも可能である。また、部分配列の出現頻度は、既に存在するデータを取得することによって提供することも可能である。
長さkの部分配列のサブセットの各々の部分配列の出現頻度を得る工程は、計算機を使用して簡便に行うことができ、例えば、Unixでは以下:

のように実装することができる。
のように実装することができる。
多型が存在している部位がゲノム上でユニークな配列部位であり、例えば1塩基置換が存在している場合、その置換を含むk-mer部分配列は対象配列データのサブセットには存在するが、コントロール配列データのサブセットには存在しないと考えられる。当該置換変異を含むk-merではすべてこの結果が得られると考えられ、結果的に当該置換変異部位を含む2×k-1個のk-mer部分配列で出現頻度の差異が観察され、当該多型を検出することができる。
一部の実施形態において、本発明の方法において、予測される出現頻度と比較して、出現頻度が少ない部分配列をノイズとすることが可能である。これにより、機械的に生じた差異と、実際に生じている多型とを識別して検出することが可能である。例えば、前記対象配列データおよび前記コントロール配列データが塩基配列データである場合には、対象配列データ量、配列データにおける予想されるエラー率(例えば、マニュアル・機器の公称値)、対象配列全長サイズ(例えば、ゲノムサイズ)等を考慮して、予想されるノイズのカウント程度、または予想されるノイズのカウント以下の出現頻度の部分配列をノイズとすることができる。1つの予測としては、生じるノイズの頻度の期待値は、int(ゲノムを何倍読んだか×(1-精度)+1)」となる。
理論的には、シーケンサーの精度が99%でゲノムの100倍読みした場合は、1塩基のノイズが入ることになる。通常の解析はゲノムの40倍読み程度であるため、ノイズは1塩基以下と考えられるが、カウントは整数値となるため、リードエラーは1とカウントされる。すなわち、2塩基以上のカウントはノイズ以外の何らかの要因がある可能性が高いと考えられる。ただし、低い確率だが、同一塩基に2回以上ノイズが入る可能性はある。予想されるノイズのカウントは、例えば、int(ゲノムを何倍読んだか×(1-精度)+1)である。この場合の精度は100%の場合1、99%の場合0.99として計算できる。なお、int(X)は、X未満の最大の整数を返す関数である。
エラー率(精度)が完全に0でない場合には、例えば0.00001でも10万か所のうち1ヶ所は1のカウントが生じると考えられる。そのため、この場合、結局1はノイズである可能性が高いと考えられるが、2はノイズにしては高すぎると言える。そのため、int(ゲノムを何倍読んだか×(1-精度)+1)で繰り上げて整数にすることにより、予測値を算出することが可能である。1つの実施形態では、カウントが1となる部分配列をノイズと判定する。例えば、対象配列データおよび前記コントロール配列データが塩基配列データである場合に、[(対象配列データ量)×(1-精度)]/(対象ゲノムサイズ)+1未満の出現頻度の部分配列をノイズとすることにより、予想されるノイズのカウント以下の出現頻度の部分配列をノイズとして排除することができる。
頻度の算出から、コピー数変異(CNV)の検出も可能である。例えば、コントロール配列と比較して約2倍以上の頻度が連続して検出された場合にCNVと判定することができる。頻度の基準は、約2.5倍以上などと厳しくすることによって、誤検出を減少させることもできる。
あるいは、本発明の方法において、予測される出現頻度と比較して、出現頻度が多い部分配列を、リピート配列部位として除外することができる。例えば、対象配列データの対象配列全長のカバレッジ(カバー率)の2倍を超えるような部分配列を解析から除外することができる。
例えば、配列データ量の配列全長に対するカバレッジと比較し、同程度の出現頻度の部分配列は、対象配列の全長におけるユニークな配列に由来している配列と考えられる。さらに、配列データ量の配列全長に対するカバレッジと比較し、2倍程度の出現頻度の部分配列は、対象配列の全長において2箇所存在する配列に由来している配列と考えられる。
kが偶然同一を排除している場合、それを上回る場合には、部分配列が、リピート配列部位に由来しているものと判断することができる。例えば、ゲノムの40倍のカバレッジのシーケンシングデータにおいて、出現頻度が40~50回程度の配列は、ゲノム上のユニークな配列由来であり、89回出現した配列は、ゲノム上2ヶ所存在していると考えられる。k長配列のサブセットおよび/または各部分配列の出現頻度データの作成は、一例としては、図3に例示されるような工程を採用して行うことができる。
(k-x)
1つの実施形態では、本発明の方法は、k長の部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出することを特徴とする。xはk未満の正の整数である。長さkの全長ではなく、k-merの一部(x長の部分)の文字の差異を見ることによって、計算量を顕著に減少させることが可能である。長さxは、限定されるものではないが、好ましくは1~2であり、より好ましくは1である。
1つの実施形態では、本発明の方法は、k長の部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出することを特徴とする。xはk未満の正の整数である。長さkの全長ではなく、k-merの一部(x長の部分)の文字の差異を見ることによって、計算量を顕著に減少させることが可能である。長さxは、限定されるものではないが、好ましくは1~2であり、より好ましくは1である。
一部の実施形態において、本発明の方法は、前記部分配列中の長さk-xの配列部分を、ユニークな配列ごとにグルーピングする工程を含む。これには、例えば、前記長さk-xの配列部分をソートする工程(例えば、前記長さk-xの配列部分を文字列としてソートする工程)が含まれ得る。
部分配列中の長さk-xの配列部分が共通する配列ごとの、長さxの部分についての出現頻度の分布は、長さkの部分配列の出現頻度から算出できる。配列の文字の種数をv(配列において各変数が取り得る値の種類)とした場合、長さkの部分配列において、k-xの配列部分が共通する配列が、k-xの配列部分が共通する配列ごとにv^x種生じる。例えば、配列が塩基配列であり、x=1とした場合、長さkの配列のセットの中には、k-1の配列部分が共通する配列ごとに、xに対応する部分がA、C、G、Tである4種の配列が存在している。長さk-xの配列部分が共通する配列ごとの、長さxの部分についての出現頻度は、それぞれに対応する長さkの部分配列の頻度データに対応する。
1つの実施形態では、前記長さxの部分が、前記部分配列の末端に存在する。例えば、前記対象配列データおよび前記コントロール配列データが塩基配列データである場合、前記長さxの部分は、前記部分配列の3’末端または5’末端であり得る。長さxの部分を部分配列の末端にとることは、比較処理の高速化・簡便化にとって望ましい。このように、k長の配列のうち末端部で異なる部分(x長の部分に該当)の頻度を検出する場合、対象となる配列の多型部位の「エッジ」(置換の場合はその位置そのものに該当するが、挿入・欠失変異の場合はその縁(エッジ)に該当する)を検出しているというように理解することができるため、本明細書において、「Polymorphic Edge Detection(PED)」または「edge detection」と呼称される場合がある。
(比較・多型の検出)
出現頻度の分布の差異の比較により、例えば、以下のような多型の検出が可能である。
出現頻度の分布の差異の比較により、例えば、以下のような多型の検出が可能である。
多型が存在している部位がゲノム上でユニークな配列部位であり、例えば置換が存在している場合、その置換を含むk-mer部分配列は対象配列データのサブセットには存在するが、コントロール配列データのサブセットには存在しないと考えられる。当該置換変異を含むk-merではすべてこの結果が得られると考えられ、結果的に当該置換変異部位を含む2×k-1個のk-mer部分配列で出現頻度の差異が観察され、当該多型を検出することができる。そのような解析は、一例としては、図6に示されるような工程を採用して行うことが可能である。
1つの実施形態では、前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列の出現頻度が異なる場合、該長さxの部分の配列を、対象配列データにおけるコントロール配列データに対する多型として検出する。1つの実施形態では、前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列で最も高頻度のものが異なっている長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおける多型として検出する。
1つの実施形態では、前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記対象配列データのサブセットにおける前記長さxの部分の配列で、前記コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列がノイズ以下のカウントしか存在しない長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるホモ多型として検出する。1つの実施形態では、前記対象配列データおよび前記コントロール配列データが塩基配列データであり、対象サブセットにおける前記長さxの部分の配列で、コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列が存在し、かつ、コントロール配列データのサブセットにおける最も高頻度のものと異なる長さxの部分の配列が存在する長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるヘテロ多型として検出する。k長配列のセットの頻度データの比較は、一例としては、図4に示されるような工程によって行うことが可能である。
このような比較は、1つの例としては、k-1長配列と最後の塩基の頻度のファイル、controlとtargetを処理する場合、
のコマンドでk-1配列とcontrolとtargetを1行にまとめた頻度を出力することによって行うことができる。この出力結果の各行を調べる条件としては、限定されるものではないが、control、targetの両方でカウントが1塩基以下の塩基が2個以上存在し、controlあるいはtargetで10以上のカウントを示した塩基に対応するtargetあるいはcontrolの塩基のカウントが1以下である事例が1ないし2回ある場合、多型の境界を検出したとすることができる。
のコマンドでk-1配列とcontrolとtargetを1行にまとめた頻度を出力することによって行うことができる。この出力結果の各行を調べる条件としては、限定されるものではないが、control、targetの両方でカウントが1塩基以下の塩基が2個以上存在し、controlあるいはtargetで10以上のカウントを示した塩基に対応するtargetあるいはcontrolの塩基のカウントが1以下である事例が1ないし2回ある場合、多型の境界を検出したとすることができる。
すなわち、コントロール配列および/または対象配列の部分配列のサブセットにおける第1のカウントが第1の閾値を超えており、かつ、コントロール配列および/または対象配列の部分配列のサブセットにおける対応する第2のカウント(すなわち、第1のカウントがコントロール配列のものである場合、第2のカウントは対象配列のものであり、第1のカウントが対象配列のものである場合、第2のカウントはコントロール配列のものである)が第2の閾値を下回る場合、多型の境界を検出したとすることができる。
第1の閾値は、配列データのカバレッジによって変動するが、例えば、10~50の範囲で設定することができる。第1の閾値は、例えば、10~40、10~30、10~20、または10~15の範囲で設定することができる。配列データのカバレッジが大きくなる場合には、第1の閾値も大きく設定することができ、例えば、ヒトゲノム解析では200倍読み程度のデータも存在するが、この場合は、第1の閾値として200を使用することができる。カバレッジを考慮して実際にその配列が存在しているといえるカウントを第1の閾値としてよく、例えば、カバレッジの約100%、約90%、約80%、約70%、約60%、約40%、約30%、または約20%等の値を用いることができる。
第2の閾値も同様に配列データのカバレッジによって変動するが、1~7の範囲で設定することができる。第2の閾値は、例えば、1、2、3、4、5、6または7であり得る。
(配列)
本発明の対象配列、コントロール配列および/またはリファレンス配列としては、多型が生じ得る任意の配列を用いることができる。なお、コントロール配列として、リファレンス配列を用いることが可能である。代表的な実施形態では、対象配列、コントロール配列および/またはリファレンス配列は、生物学的配列であり、例えば、塩基配列(DNA、RNA、およびそれらのアナログ等の配列が包含される)、アミノ酸配列、または糖鎖配列等である。生物学的配列の例としては、例えば、ゲノム配列、染色体配列、遺伝子配列、プラスミド配列、エクソン配列、タンパク質配列等が挙げられる。
本発明の対象配列、コントロール配列および/またはリファレンス配列としては、多型が生じ得る任意の配列を用いることができる。なお、コントロール配列として、リファレンス配列を用いることが可能である。代表的な実施形態では、対象配列、コントロール配列および/またはリファレンス配列は、生物学的配列であり、例えば、塩基配列(DNA、RNA、およびそれらのアナログ等の配列が包含される)、アミノ酸配列、または糖鎖配列等である。生物学的配列の例としては、例えば、ゲノム配列、染色体配列、遺伝子配列、プラスミド配列、エクソン配列、タンパク質配列等が挙げられる。
対象配列データおよびコントロール配列データは、限定されるものではないが、多型を検出する上では、一定の共通性を持つ配列についての配列データであることが望ましい。しかしながら、配列の取得方法については各々同一でも異なっていてもよく、シーケンシングによって得られたデータ間での比較を行うことも、データベース等から得られたデータ間での比較を行うことも、シーケンシングによって得られたデータとデータベース等から得られたデータとの間での比較を行うことも可能である。
1つの実施形態では、対象配列データが、個体から得られた配列データであり、コントロール配列データが、該個体と同種の別の個体、またはデータベースから得られた配列データである。1つの実施形態では、対象配列データが、個体の組織試料から得られた配列データであり、コントロール配列データが、該個体の別の組織、またはデータベースから得られた配列データである。1つの実施形態では、対象配列データが、細胞試料から得られた配列データであり、コントロール配列データが、別の細胞、またはデータベースから得られた配列データである。
本発明の方法は、全長配列の情報を必要としないため、データベース等において例えば全長配列が公知でない場合にも用いることができ、例えば、対象配列データおよびコントロール配列データは、生物のゲノムに由来する塩基配列データである場合、前記ゲノムの配列が不明であってもよい。
例えば、次世代シーケンサーのリードデータで対象(ターゲット)とコントロール間の配列の直接比較による多型検出は既存の技術では不可能であった。リファレンス配列がある場合は、多型をゲノム上にマップすることができるが、リファレンスが存在しない場合(例えば、リファレンスゲノム配列が作成されていない生物)で、リファレンス情報を用いることなく対象とコントロール間での多型を検出できるというのは画期的である。リファレンスのない生物でのF2分離集団で表現型の分離と連鎖する多型を検出すれば、ゲノム上の位置が不明であったとしても、表現型に対応するDNAマーカーを得ることができ、応用範囲は非常に広いと考えられる。実際、育種にはゲノム上の位置情報も重要ではあるが、位置情報が不明でも優良形質にリンクしているDNAマーカーでの選抜ができれば、選抜育種への利用が十分に可能である。
同一個体(例えば、ヒト)の正常組織と変異を含み得る組織(例えば、癌組織)の配列を直接比較して多型を検出することも可能で、いったんリファレンスゲノムにマップしてから差を調べる方法に比べて、多型の捕捉率、捕捉精度ともに非常に高くなる。
1つの実施形態では、本発明の方法で用いる対象配列データおよび/またはコントロール配列データは、シーケンシングによって得られた塩基配列データである。シーケンシングの手法としては、サンガー法、マクサム・ギルバード法、単一分子リアルタイムシーケンシング(例えば、Pacific Biosciences、Menlo Park、California)、イオン半導体シーケンシング(例えば、Ion Torrent、South San Francisco、California)、パイロシーケンシング(例えば、454、Branford、Connecticut)、ライゲーションによるシーケンシング(例えば、Life Technologies、Carlsbad、CaliforniaのSOLiDシーケンシング)、合成および可逆性ターミネーターによるシーケンシング(例えば、Illumina、San Diego、California)、透過型電子顕微鏡法などの核酸イメージング技術、ナノポアシーケンシングなどがある。
1つの実施形態では、本発明の方法で用いる対象配列データおよび/またはコントロール配列データは、次世代シーケンシングによって得られた配列データであり得る。次世代シーケンシングとしては、シーケンシングバイシンセシス、パイロシーケンシング、ライゲーションによるシーケンシング、イオン半導体シーケンシング、ナノポアシーケンシング等が挙げられる。次世代シーケンシングデータからの多型の検出においては、リファレンスへのマッピングやアセンブリによって精度が制限されていたため、本発明の方法を用いた場合に大きな利益が得られると考えられる。
1つの実施形態では、本発明の方法で用いる対象配列データおよび/またはコントロール配列データは、ジニトロフェニル化法、ヒドラジン分解法、カルボキシペプチダーゼ法、エドマン法もしくはそれらの方法を自動化する装置(ペプチドシーケンサーあるいはプロテインシーケンサー)を用いる方法、質量分析(例えば、タンデム質量分析計(MS/MS))を用いた方法(例えば、シーケンスタグ法)等から得られたアミノ酸配列データである。
本発明の対象配列データおよび/またはコントロール配列データの由来となる生物種としては、生物学的配列を有するものである以上は何ら制限されない。一部を例示すると、動物としては、ヒトもしくは非ヒト哺乳動物(例えば、マウス、ラット、ウサギ、ヒツジ、ブタ、ウシ、ウマ、ネコ、イヌ、サル、チンパンジー)、鳥類、爬虫類、両生類、魚類等の脊椎動物、無脊椎動物、例えば、昆虫、線形動物などを挙げることができる。植物としては、イネ、コムギ、トウモロコシ、ジャガイモ、オオムギ、サツマイモ、ソバ、シロイヌナズナ、ミヤコグサ、トマト、キュウリ、キャベツ、白菜、ナス、サトウキビ、ソルガム、リンゴ、ミカン、バナナ、桃、ポプラ、松、杉、被子植物、裸子植物、シダ、コケ、藻類などを挙げることができる。その他、真菌、細菌、ウイルス等でもよい。
さらに、これらの生物の一部分、例えば、組織、細胞等に由来する対象配列データおよび/またはコントロール配列データを解析し、多型を検出することも可能である。
(変異)
本発明の方法は、例えば、置換、挿入、欠失、コピー数変異、STRP(short tandem repeat polymorphism)、逆位または転座等の多型の検出に用いることができる。変異のエッジの部分が検出されるため、挿入・欠失の結果、長さxの配列に違いがあればそのエッジ部分を検出することができる。k-mer内に収まりきる場合であれば、STRP(short tandem repeat polymorphism)を検出することも可能である。STR(short tandem repeat)は、マイクロサテライトとも称され、2~7塩基からなる配列が2~数十回反復するもので、この回数に多型が見られる。部分配列の出現頻度によって、コピー数多型(CNV)を検出することもできる。エッジ検出という観点からは、逆位、転座のエッジも検出することが可能である。
本発明の方法は、例えば、置換、挿入、欠失、コピー数変異、STRP(short tandem repeat polymorphism)、逆位または転座等の多型の検出に用いることができる。変異のエッジの部分が検出されるため、挿入・欠失の結果、長さxの配列に違いがあればそのエッジ部分を検出することができる。k-mer内に収まりきる場合であれば、STRP(short tandem repeat polymorphism)を検出することも可能である。STR(short tandem repeat)は、マイクロサテライトとも称され、2~7塩基からなる配列が2~数十回反復するもので、この回数に多型が見られる。部分配列の出現頻度によって、コピー数多型(CNV)を検出することもできる。エッジ検出という観点からは、逆位、転座のエッジも検出することが可能である。
特に、多型が置換である場合には、本発明の方法は、非常に高い検出力を発揮することが可能である。
(位置の特定)
対象配列に対するリファレンス配列が存在する場合、本発明の方法は、対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含むことができる。例えば、対象配列データおよびコントロール配列データが、生物のゲノムに由来する塩基配列データである場合、多型のゲノム上の位置を特定する工程をさらに含むことができる。この位置の特定は、本発明の方法が、多型を周囲の配列と関連づけて検出する(例えば、x長部分の多型がk-x長の配列と関連付けられる)ことを可能にしているため、リファレンス配列に対して検索を行うことにより、簡便に行うことが可能である。
対象配列に対するリファレンス配列が存在する場合、本発明の方法は、対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含むことができる。例えば、対象配列データおよびコントロール配列データが、生物のゲノムに由来する塩基配列データである場合、多型のゲノム上の位置を特定する工程をさらに含むことができる。この位置の特定は、本発明の方法が、多型を周囲の配列と関連づけて検出する(例えば、x長部分の多型がk-x長の配列と関連付けられる)ことを可能にしているため、リファレンス配列に対して検索を行うことにより、簡便に行うことが可能である。
リファレンス配列に対する検索は、一例としては、図5に示されるような工程によって二分検索用リファレンスゲノム配列データを作成し、その後二分検索によって多型境界塩基のマッピングによって検索を行うことができる。
他の方法として、unixのjoinコマンドによって対象配列データ中の部分配列の、リファレンス配列における位置および向きを出力することによってマッピングを行う方法を用いることができる。より詳細には、対象配列のコントロール配列上の位置を決定する方法であって、a)コントロール配列中の複数のk長の部分配列について、配列およびコントロール配列中の位置と向きを出力する工程と、b)対象配列中の複数のk長の部分配列について、配列および対象配列中の位置を出力する工程と、c)a)およびb)で得られた配列を比較し、同一の部分配列に対応するコントロール配列中の位置と対象配列中の位置とを対応付ける工程とを含み、ここで、kは、対象配列の長さを超えない長さである、方法を使用することができる。当該方法については、本出願人により本出願と同日に出願された「挿入・欠失・逆位・転座・置換検出法」との名称の出願(整理番号NG012PCT/F5-18PCT075)を参照することができる。
(確認(verify))
本発明の方法は、検出した多型について確認する工程をさらに含むことができる。確認は、例えば、検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行うことによって行うことができる。クエリ配列セットは、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セット、および/またはリファレンス配列またはコントロール配列において前記多型に該当する部位の文字を置換していない野生型クエリ配列セットを含み得る。
本発明の方法は、検出した多型について確認する工程をさらに含むことができる。確認は、例えば、検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行うことによって行うことができる。クエリ配列セットは、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セット、および/またはリファレンス配列またはコントロール配列において前記多型に該当する部位の文字を置換していない野生型クエリ配列セットを含み得る。
本発明の方法は、対象配列データおよびコントロール配列データが塩基配列データである場合、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの相補鎖の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含むことができる。本発明の方法は、対象配列データおよびコントロール配列データが塩基配列データである場合、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの対立遺伝子の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含むことができる。確認する工程は、一例としては、図6に示されるフローに従って、図6に示される工程を適宜採用して行うことができる。ここで、対立遺伝子の配列データとして、実際の遺伝子の存在の有無とは関係なく、野生型に対する変異型の塩基を有する配列データを用いることができる。
以下、コントロール配列が野生型(つまりリファレンスゲノム配列とほぼ同一である)から得られた場合、または、コントロール配列が、リファレンスゲノム配列から対象配列と同一長で作成された配列である場合について例示する。
次世代シーケンサーで読まれたショートリードの塩基配列長がLの場合、リファレンスゲノム配列の対象となる多型塩基位置を起点として、L-1塩基前の位置からL-1塩基後の位置までの2L-1塩基長の配列を得て、多型塩基位置を推定された多型塩基に置換した置換配列とコントロールの非置換配列を作成する。置換配列、および、非置換配列それぞれを1塩基ずつずらしながらL長のクエリ配列セットを作成する(例えば、図14に例示される)。個々のクエリ配列を用いて対象配列とコントロール配列に対して完全一致するカウントを取得する。ホモ型変異の場合は変異を導入したクエリ配列では対象配列のカウントが大部分になり、ヘテロ型変異の場合は、対象配列とコントロール配列への一致カウントが概ね半分ずつとなると考えられる。非置換配列をクエリにした場合は、基本的には大部分コントロール配列にヒットする。非置換配列が対象配列にヒットする場合は、多型ではないと判断し、除外することが可能である。
コントロール配列、対象配列の検索のため、コントロール配列、対象配列、それぞれ相補鎖の配列も合わせたのち辞書順にソートして同一配列は一つにまとめたデータセットを用いて、二分探索法によりクエリ配列を検索する。実施形態において、Fastqファイルからの塩基配列データをソートしてユニークなデータセットにする時点で、つまり最初の段階で、各リードの相補鎖も一緒にソートしてユニーク処理することも可能である。
k-merを切り出す前にsortとuniqの処理をすることは、シーケンス反応時にPCR増幅のステップが入る場合があり(入らないキットも存在する)、同一の配列が複数回リードデータの中に出てくる場合があることに対処する上で好ましい場合がある。そのまま処理するとk-merの分布が歪む可能性がある。同一配列ではあるがNが含まれるようなリードも別物と認識されて歪む原因になり得るため、Nを含まない配列で、その配列とその相補鎖配列をsortしてuniq処理した配列からk-merを得ることが可能である。
歪むことを許容する場合、もとの配列の長さは揃っている必要はなく、サンガー法で得られたような長さが一定でない配列データでもk-merにして多型検出、マッピングまで行うことが可能である。
確認工程における、sort_uniq配列を二分検索して、変異型と野生型のリード数を調べる工程では、配列データの長さが揃っていることが好ましい。本発明者らの知見によれば、確認工程の前にk-1配列で多型をマップした段階で、順鎖と相補鎖の両方で多型が検出された場合は、ほぼ間違いなくSNPであることがわかっている。順鎖、相補鎖のどちらかがリピート領域にあって片側しか検出できないものでも実際にSNPである場合があるが、このような場合、sort_uniq配列を二分検索して確認することで、実際のSNPかどうかの判断をすることができる場合があり得る。二分検索による確認を行って捕捉率を上げるためには、スタートのショートリード(次世代シーケンサーで得られた塩基配列データ)の長さが揃っていることが好ましい場合がある。対象とコントロールの配列長が同一である必要はなく、対象配列データとコントロール配列データそれぞれの中で長さが一定であれば、好適に二分検索によって確認を行うことができる。
あるいは、以下のように確認を行うことができる。リファレンス配列から対象配列のL長で変異部位を含むようにして部分配列を切り出し、変異に置換したセットと、置換しないセットを作り、位置関係、変異の有無等の記載と一緒にソートして出力する。このデータとソートした対象配列を、unixコマンドのjoinで処理(または適切な等価な処理)して、対象配列中に含まれる、野生型と変異型の配列を選び、配列数を変異部位ごとに調べる。選んだ配列を、sortした後、uniq-cのコマンドで配列数を数えることができる。同様の操作を、コントロール配列(L’長)に対しても行う。対象個体に対するコントロール個体がある場合は、この個体から得られたリード配列をコントロール配列として用いることができる。コントロール個体がない場合は、リファレンス配列から、L長で切り出して作った配列をコントロール配列として使うことが可能である。対象配列およびコントロール配列で長さが違う場合は、それぞれの長さに対応する変異型と野生型のデータセットを作り、対応する個数を調べることができる。当該方法については、本出願人により本出願と同日に出願された「挿入・欠失・逆位・転座・置換検出法」との名称の出願(整理番号NG012PCT/F5-18PCT075)を参照することができる。
例えば、イルミナ社の次世代シーケンサー(例えば、HiSeq)は同じ長さの配列を出力するため、対象配列データにおいて、特に長さをそろえる処理をする必要はない。この場合、対象配列データと同じ長さを有するクエリ配列のセットを作成することが可能であるため、直接二分検索を行うことができ、確実な結果を得る上で有利であり得る。
長さにばらつきがあるショートリードによる配列データあるいはショートリードの集合ではない配列データ(例えば、サンガー法で得られた配列)でも、本発明において適用可能である。確認工程においては、長さを揃える処理(例えば、リード中のクオリティスコアが最大となるL長配列を選択して切り出す、一端からL長配列を切り出す等)の処理をしたデータを用いて二分検索を行うことが可能である。あるいは、配列データ(リード)をBLASTのターゲット配列(データベース)にして、配列データとは長さの異なるクエリ配列を検索して数を数えることも可能である。
コンピュータで計算させる場合、sort_uniq配列や二分探索用のリファレンス配列に対して二分探索で完全一致の配列を探索する場合に、ファイルサイズが大きくなる場合が多いため、高速のシステムを利用するか、適切に高速化を図ることが好ましい。高速化の手法としては、すべてオンメモリで計算を行う、SSD等高速ディスクにファイルを置く等、ハード的な手法が存在する。ソフト的には、単純にソートされたファイルに対して二分検索を行うのではなく、検索対象のファイルをBurrows-Wheeler変換してより高速化を行うこともできる。
(プログラム、記録媒体およびシステム)
1つの局面において、本発明は、本発明の多型を検出する方法をコンピュータに実施させるための方法を実装するプログラム、該プログラムを記録した記録媒体、およびこれを実現するためのシステムを提供する。ここで採用され得る任意の特徴は本明細書の多型を検出する方法の説明に記載される任意の特徴またはその組み合わせを採用することができる。
1つの局面において、本発明は、本発明の多型を検出する方法をコンピュータに実施させるための方法を実装するプログラム、該プログラムを記録した記録媒体、およびこれを実現するためのシステムを提供する。ここで採用され得る任意の特徴は本明細書の多型を検出する方法の説明に記載される任意の特徴またはその組み合わせを採用することができる。
したがって、1つの実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムであって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラムが提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、プログラムが提供される。プログラムはどのような言語で記述されてもよい。
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラムが提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、プログラムが提供される。プログラムはどのような言語で記述されてもよい。
別の実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムを格納する記録媒体であって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体が提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、記録媒体が提供される。プログラムはどのような言語で記述されてもよい。1つの実施形態では、記録媒体は、内部に格納され得るROMやHDD、磁気ディスク、USBメモリ等のフラッシュメモリなどの外部記憶装置でありうる。
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体が提供される。さらなる実施形態において、方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る)を、検出された前記多型の名称として表示する工程をさらに含む、記録媒体が提供される。プログラムはどのような言語で記述されてもよい。1つの実施形態では、記録媒体は、内部に格納され得るROMやHDD、磁気ディスク、USBメモリ等のフラッシュメモリなどの外部記憶装置でありうる。
別の実施形態において、対象配列データにおいてコントロール配列データに対する多型を検出するためのシステムであって、該システムは、該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部とを備える、システムが提供される。さらなる実施形態において、前記部分配列中の多型ではない部分の少なくとも一部を含む配列(前記部分配列全体であり得る。)を、検出された前記多型の名称として表示する表示手段をさらに含む、システムが提供される。
次に、図15Aの機能ブロック図を参照して、本発明のシステム1の構成を説明する。なお、本図においては、単一のシステムで実現した場合を示しているが、複数のシステムで実現される場合も本発明の範囲に包含されることが理解される。
本発明のシステム1000は、コンピュータシステムに内蔵されたCPU1001にシステムバス1020を介してRAM1003、ROMやHDD、磁気ディスク、USBメモリ等のフラッシュメモリなどの外部記憶装置1005及び入出力インターフェース(I/F)1025が接続されて構成される。入出力I/F1025には、キーボードやマウスなどの入力装置1009、ディスプレイなどの出力装置1007、及びモデムなどの通信デバイス1011がそれぞれ接続されている。外部記憶装置1005は、情報データベース格納部1030とプログラム格納部1040とを備えている。何れも、外部記憶装置1005内に確保された一定の記憶領域である。
このようなハードウェア構成において、入力装置1009を介して各種の指令(コマンド)が入力されることで、又は通信I/Fや通信デバイス1011等を介してコマンドを受信することで、この記憶装置1005にインストールされたソフトウェアプログラムがCPU1001によってRAM1003上に呼び出されて展開され実行されることで、OS(オペレーションシステム)と協働して本発明の対象配列データにおいてコントロール配列データに対する多型を検出する方法の機能を奏するようになっている。もちろん、このような協働する場合以外の仕組みでも本発明を実装することは可能である。
本発明の実装において、対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程を行う際に、対象配列データおよび/または対象配列データの長さkの部分配列のデータは、入力装置1009を介して入力され、あるいは、通信I/Fや通信デバイス1011等を介して入力されるか、あるいは、データベース格納部1030に格納されたものであってもよい。次に、対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程は、プログラム格納部1040に格納されたプログラム、または、入力装置1009を介して各種の指令(コマンド)が入力されることで、又は通信I/Fや通信デバイス1011等を介してコマンドを受信することで、この外部記憶装置1005にインストールされたソフトウェアプログラムによって実行することができる。あるいは、あらかじめ算出されている出現頻度を、入力装置1009を介して入力することができる。出現頻度データは、出力装置1007を通じて出力されるかまたは情報データベース格納部1030等の外部記憶装置1005に格納されてもよい。
次に、コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程を行う差異、コントロール配列データ、コントロール配列データの長さkの部分配列のサブセットのデータ、または部分配列の出現頻度のデータは、入力装置1009を介して入力され、あるいは、通信I/Fや通信デバイス1011等を介して入力されるか、あるいは、データベース格納部1030に格納されたものであってもよく、プログラム格納部1040に格納されたプログラム、または、入力装置1009を介して各種の指令(コマンド)が入力されることで、又は通信I/Fや通信デバイス1011等を介してコマンドを受信することで、この外部記憶装置1005にインストールされたソフトウェアプログラムによってこれらのデータを処理してコントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度を提供してもよい。
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程は、プログラム格納部1040に格納されたプログラム、または、入力装置1009を介して各種の指令(コマンド)が入力されることで、又は通信I/Fや通信デバイス1011等を介してコマンドを受信することで、この外部記憶装置1005にインストールされたソフトウェアプログラムによって実行することができる。
データベース格納部1030には、これらのデータや計算結果、もしくは通信デバイス1011等を介して取得した情報が随時書き込まれ、更新される。各入力配列セット中の各々の配列、参照データベースの各遺伝子情報ID等の情報を各マスタテーブルで管理することにより、蓄積対象となるサンプルに帰属する情報を、各マスタテーブルにおいて定義されたIDにより管理することが可能となる。
データベース格納部1030には、上記計算結果は、配列に関する情報、例えば、生物学的情報、生化学的情報、医学的情報、例えば疾患、障害、生体情報等の既知の情報と関連付けて格納されてもよい。このような関連付けは、ネットワーク(インターネット、イントラネット等)を通じて入手可能なデータをそのまままたはネットワークのリンクとしてなされてもよい。
また、プログラム格納部1040に格納されるコンピュータプログラムは、コンピュータを、上記した処理システム、例えば、配列データの提供、部分配列サブセットの提供、出現頻度データの算出、出現頻度データの比較、多型の検出、多型の確認などの処理を実施するシステムとして構成するものである。これらの各機能は、それぞれが独立したコンピュータプログラムやそのモジュール、ルーチンなどであり、上記CPU1001によって実行されることでコンピュータを各システムや装置として構成させるものである。なお、本発明の例示においては、それぞれのシステムにおける各機能が協働してそれぞれのシステムを構成しているものとするが、この処理のためのプログラムもまた、それぞれ外部記憶装置または通信デバイスまたは入力装置を介して提供されうる。
本発明がシステムとして構成される場合は、対象配列データおよび/またはコントロール配列データ、それらの長さkのサブセットのデータ、ならびに/あるいはそれらの出現頻度データの提供は、まとめて配列データ処理部としてもよい。また、出現頻度の分布の比較および多型の検出は、配列データ計算部としてまとめてもよい。
また、図15Bに示されるように、クラスター構造を有する計算システムによって本発明の方法を実装してもよい。1つの実施形態では、システムはクラスター構成であり、ヘッドとノードからなる。ノードは検索の高速化を図るため、主記憶装置にSSDを用いることができる。1つの実施形態では、ヘッド1台に対して複数のノード(例えば12台)で運用することができる。1つの実施形態では、計算システムはクラスター構造を持ち、主コンピュータ(クラスターヘッド)に大容量記憶装置(HDD)を搭載して解析データおよび結果を保存する。クラスターヘッドより、分割したデータを各ノードに送り計算を実行し、結果をクラスターヘッドに集約する。クラスターヘッド、ノード共に、中央制御素子(CPU)、メモリ(RAM)を搭載し、通信インターフェース(NIC)を介してデータの通信を行い得る。ノードには高速での検索処理をするため、ソリッドステートドライブ(SSD)を主記憶装置とすることができる。各ノードに搭載されるCPU、RAM、SSD等は、他のノードと共有されてもよく、物理的に分離していてもよい。
(例示的計算フロー)
本発明は、例えば、以下のフローにおいて実施することができる。
本発明は、例えば、以下のフローにおいて実施することができる。
1.次世代シーケンサーから得られたFastq形式のファイルより、配列内にNを含まない塩基配列を選んで、相補鎖の配列と合わせて、それぞれ出力する。
2.出力されたファイル(reads)を辞書順にソートし同一の配列は一つにまとめる。
unixコマンドは以下の通り。

unixコマンドは以下の通り。
相同な配列がゲノム上に複数存在する部分に対しての解析を行う場合は、readsに対してsortのみを行い、uniq処理は行わない場合もあり得る。例えば、マルチコピー領域に対してのCNVを検出する場合にはsortのみを行うことが有利であり得る。
3.ソートされた各塩基配列をそれぞれ5’末端から1塩基ずつずらしながら、k-mer(ここでは20塩基)の配列を対象の塩基配列の3’末端に到達するまで繰り返し出力(k-mer_file)する。
4.出力されたk-merの配列を辞書順にソートし、同一配列は一つにまとめて、出現回数を配列と共に表記したファイルを作成する。
unixコマンドは以下の通り。

unixコマンドは以下の通り。
uniq -cコマンドなどのように、出力結果が頻度→配列の順になる場合、uniq-c等のコマンドの後に配列→頻度の順に出力されるフィルタープログラムを通してもよい。
5.k-mer_count_fileの各行のデータに対して、配列の5’末端よりk-1塩基の配列を得て、3’末端の塩基すなわちk番目の塩基をA、C、G、Tの出現回数として表記したデータに変換する。
k-1merの配列 Aの回数 Cの回数 Gの回数 Tの回数
という形式で出力される。

k-1merの配列 Aの回数 Cの回数 Gの回数 Tの回数
という形式で出力される。
6.このようなデータを対象(target)と比較(control)のサンプルから得られた次世代シーケンサーの配列データからそれぞれ得る。
7.controlとtargetの5の方法で作成したデータをk-1merの配列でまとめたデータを作成する。
unixコマンドは以下の通り

unixコマンドは以下の通り
8.joinコマンドで得られたデータでcontrolとtargetで異なる塩基でカウントが示されるデータを選び出す。

この例では、controlでは、ACTTTCTTCAAGGTCTGTT(配列番号225)に続く塩基はGであるが、targetではCである。つまり、k-1merのユニークな識別子(名称)に続く塩基がG型、あるいはC型という表記で多型を表現することができる。それぞれの塩基に対応する数は、この多型が検出された独立のreadの数である。
9.このk-1merの識別子をリファレンスゲノム上にマッピングすれば、容易に多型位置を決めることができる。
10.本法は配列情報をリファレンスゲノムにマップする前にcontrolとtarget間の多型を検出するので、リファレンスゲノムが未知の生物種でも多型を検出できる。
11.ゲノム上の場所が決められない場合は、多型の名称はk-1merの配列自身で表すことができ、遺伝子型はそれに続く多型塩基となる。このデータセットを用いて、表現型に対するアソシエーション解析が可能である。
12.リファレンスゲノムへのマッピングには、リファレンスゲノムを3の方法と同様に各染色体を5’側から1塩基ずつずらしながらk-merで切り出し、k-mer配列、染色体番号、位置、向きを同一行に表記したデータをk-merの配列で辞書順に整列したデータに対して、二分検索法によりk-1merの配列のゲノム上の位置を決めることができる。

1,2行目のように同一配列も複数行でそれぞれの位置がわかるので、対象配列がリピート領域に存在しても、検索では対応する候補領域が列挙されて出力される。
今回は、複数の位置情報が出力された場合は、位置不定として多型情報の出力から除外している。複数の位置のどれかという出力の仕方も可能である。
13.リファレンスゲノムにマップされた多型の確認を行う。2で作成されたsort_uniq配列(ターゲット)と同様に、リファレンス配列も5’末端から2塩基ごとに2で作成された配列と同じ長さの配列を切り出し、相補鎖とともにsort,uniq処理をしたデータを作成する。このリファレンスとターゲットのsort_uniqデータが検索の対象となる。
次に、リファレンスゲノム配列から、多型位置を含む2で作成された配列と同じ長さの配列セット(検索を行うクエリセット)を切り出す。この配列セットは配列の5’末端から3’末端までのすべての位置で多型位置の塩基を含む配列セットである。配列セット数は配列長と同一となる。リファレンスの配列セット(リファレンスセット)に対して、多型位置の塩基を予想された多型塩基に置換した配列セット(ターゲットセット)を作成する。クエリ用配列セットの作成は、図14に例示される。
14.リファレンスセット、ターゲットセットをクエリにして、リファレンスゲノム、及び、2で作成されたsort_uniq配列をそれぞれ、検索(例えば、二分検索法またはjoinコマンドを用いた方法)し、それぞれのセットに対してsort_uniq配列が何個マッチするかを調べる。リファレンスセットではリファレンスのsort_uniq配列のみにヒットする。これに対して、ターゲットセットでは、ホモ型の変異の場合は、ターゲットのsort_uniqデータのみから検出され、ヘテロ型の変異の場合は、リファレンスと、ターゲットのsort_uniqデータの両方から検出されるはずである。このようにして、予想と一致する検索値を示した多型を抽出すると、以下のように結果を出力することが可能である。
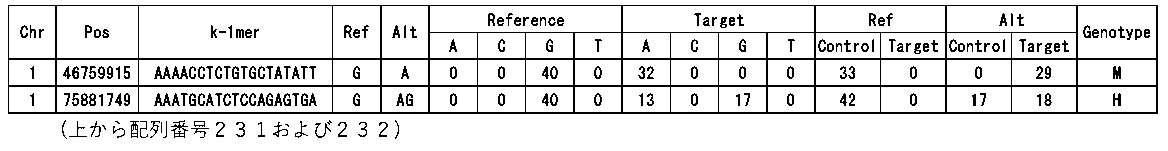
(組み合わせ)
本明細書において、上述のとおり置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するのに有用な方法を記載しているが、かかるプロセスは、置換、挿入、欠失、逆位または転座を検出するのに有用な以下に記載するプロセスと組み合わせて行うことができる。例えば、組み合わせた方法は、図18に示されるようなフローに従って実行することが可能である。
本明細書において、上述のとおり置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するのに有用な方法を記載しているが、かかるプロセスは、置換、挿入、欠失、逆位または転座を検出するのに有用な以下に記載するプロセスと組み合わせて行うことができる。例えば、組み合わせた方法は、図18に示されるようなフローに従って実行することが可能である。
このようにプロセスを組み合わせることで、配列に存在し得る多くの種類の多型を網羅的に高い検出力で検出することが可能である。このようなプロセスの組み合わせは、例えば、複数のプロセスを同時に、並行して、または逐次的に行うことによって達成することが可能である。例えば、図15Bに示されるようなクラスター構造を有する計算システムによって、異なるノードを用いてそれぞれのプロセスを行うことにより、組み合わせの方法を実装することができる。
置換、挿入、欠失、逆位または転座を検出するのに有用なプロセスとしては、対象配列データの配列中の少なくとも2ヶ所の部分配列の、コントロール配列上の位置を特定する工程を含むプロセスがある。ここで、部分配列は、k長の部分配列を用いることができる。好ましくは、コントロール配列は、配列上の位置情報が特定できる配列であり、より好ましくは、コントロール配列はリファレンス配列である。
プロセスは、対象配列データにおける部分配列間の位置関係と、コントロール配列上の部分配列間の位置関係とを比較する工程を含み得る。ここで、対象配列データにおける部分配列間の位置関係と、コントロール配列上の部分配列間の位置関係とが異なっている場合、目的とする多型があると判定することができる。例えば、部分配列が、コントロール配列の異なる配列構造体上に存在する場合、転座が生じていると判定すること、部分配列が、コントロール配列の同一の配列構造体上に存在し、かつ、向きが対象配列データ上のものと異なっている場合、逆位が存在すると判定すること、部分配列が、コントロール配列の同一の配列構造体上に存在し、向きが対象配列データ上のものと同一であり、部分配列の距離が、コントロール配列上で対象配列データ上の距離より短い場合、欠失が存在すると判定すること、および/または部分配列が、コントロール配列の同一の配列構造体上に存在し、向きが対象配列データ上のものと同一であり、部分配列の距離が、コントロール配列上で対象配列データ上の距離より長い場合、挿入が存在すると判定することを含み得る。位置関係が異ならない場合、処理を終了してもよく、目的とする多型はないと判定してもよく、対象配列データにおける部分配列部位間の文字を、対応するコントロール配列上の文字と比較して不一致となる部位を検出する工程をさらに行い、不一致となる部位が存在する場合、置換が存在すると判定してもよい。
プロセスは、位置関係が異なっている場合、目的とする多型があると判定し、対象配列データにおける部分配列部位間の文字を、対応するコントロール配列上の文字と、部分配列部位を始点として順次比較して不一致となる部位を検出する工程を含み得る。かかる工程により、検出した多型の境界塩基を検出することができる。
例えば、このようなプロセスを組み合わせた場合、本発明の1つの実施形態では、
対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
(1)a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
によって、対象配列データにおける置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するプロセスと、
(2)a)該対象配列データの配列中の少なくとも2ヶ所の部分配列の、該コントロール配列上の位置を特定する工程と、
b)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とを比較する工程と、
c)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とが異なっている場合、目的とする多型があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程と
によって、対象配列データにおける挿入、欠失、逆位、転座または置換を検出するプロセスと
を包含する、方法が提供される。
対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
(1)a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
によって、対象配列データにおける置換、コピー数多型、STRP、挿入、欠失、逆位または転座を検出するプロセスと、
(2)a)該対象配列データの配列中の少なくとも2ヶ所の部分配列の、該コントロール配列上の位置を特定する工程と、
b)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とを比較する工程と、
c)対象配列データにおける該部分配列間の位置関係と、コントロール配列上の該部分配列間の位置関係とが異なっている場合、目的とする多型があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程と
によって、対象配列データにおける挿入、欠失、逆位、転座または置換を検出するプロセスと
を包含する、方法が提供される。
本発明のさらなる実施形態では、例えば、
対象配列データにおいてリファレンス配列データに対する多型を検出する方法であって、リファレンス配列データから、各々の位置情報と関連付けられたリファレンス配列のk長の部分配列セットを作成する工程を含み、さらに、
(A1)該対象配列データの長さkの部分配列のサブセットを生成し、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A2)該リファレンス配列のk長の部分配列セットの、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A3)該対象配列と該リファレンス配列とを比較し、該出現頻度の分布の比較に基づいて、挿入、欠失、置換、コピー数多型、STRP、逆位または転座を検出する工程とを包含するプロセスと
(B1)該対象配列データの配列中の少なくとも2ヶ所のk長の部分配列をクエリとして、該リファレンス配列のk長の部分配列セットに対して二分検索を行い、該少なくとも2ヶ所の部分配列の、リファレンス配列上の位置を特定する工程と、
(B2)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とを比較する工程と、
(B3)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とが異なっている場合、挿入、欠失、逆位または転座があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程を包含し、必要に応じて、
(B4)該位置関係が異ならない場合に、該対象配列データにおける該部分配列部位間の文字を、対応する前記コントロール配列上の文字と比較して不一致となる部位を検出する工程をさらに含み、不一致となる部位が存在する場合、置換が存在すると判定する工程をさらに含む、プロセスと、
を、同時に、並行して、または逐次的に行うことを特徴とする、方法が提供される。
対象配列データにおいてリファレンス配列データに対する多型を検出する方法であって、リファレンス配列データから、各々の位置情報と関連付けられたリファレンス配列のk長の部分配列セットを作成する工程を含み、さらに、
(A1)該対象配列データの長さkの部分配列のサブセットを生成し、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A2)該リファレンス配列のk長の部分配列セットの、ユニークな長さkの部分配列の出現頻度を提供する工程と、
(A3)該対象配列と該リファレンス配列とを比較し、該出現頻度の分布の比較に基づいて、挿入、欠失、置換、コピー数多型、STRP、逆位または転座を検出する工程とを包含するプロセスと
(B1)該対象配列データの配列中の少なくとも2ヶ所のk長の部分配列をクエリとして、該リファレンス配列のk長の部分配列セットに対して二分検索を行い、該少なくとも2ヶ所の部分配列の、リファレンス配列上の位置を特定する工程と、
(B2)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とを比較する工程と、
(B3)該対象配列データにおける該部分配列間の位置関係と、該リファレンス配列上の該部分配列間の位置関係とが異なっている場合、挿入、欠失、逆位または転座があると判定し、該対象配列データにおける該部分配列部位間の文字を、対応するコントロール配列上の文字と、該部分配列部位を始点として順次比較して不一致となる部位を検出する工程を包含し、必要に応じて、
(B4)該位置関係が異ならない場合に、該対象配列データにおける該部分配列部位間の文字を、対応する前記コントロール配列上の文字と比較して不一致となる部位を検出する工程をさらに含み、不一致となる部位が存在する場合、置換が存在すると判定する工程をさらに含む、プロセスと、
を、同時に、並行して、または逐次的に行うことを特徴とする、方法が提供される。
(一般技術)
本明細書において用いられる分子生物学的手法、生化学的手法、微生物学的手法、バイオインフォマティクスは、当該分野において公知であり、周知でありまたは慣用される任意のものが使用され得る。
本明細書において用いられる分子生物学的手法、生化学的手法、微生物学的手法、バイオインフォマティクスは、当該分野において公知であり、周知でありまたは慣用される任意のものが使用され得る。
本明細書において「または」は、文章中に列挙されている事項の「少なくとも1つ以上」を採用できるときに使用される。「もしくは」も同様である。本明細書において「2つの値」の「範囲内」と明記した場合、その範囲には2つの値自体も含む。
本明細書において引用された、科学文献、特許、特許出願などの参考文献は、その全体が、各々具体的に記載されたのと同じ程度に本明細書において参考として援用される。
以上、本発明の理解を容易にするために好ましい実施形態を示して説明してきた。以下に、実施例に基づいて本発明を説明するが、上述の説明および以下の実施例は、例示の目的のみに提供され、本発明を限定する目的で提供したのではない。従って、本発明の範囲は、本明細書に具体的に記載された実施形態にも実施例にも限定されず、特許請求の範囲によってのみ限定される。
(実施例1:イネSNP検出および検証)
(概要)
本発明の方法を用いて、以下のイネ配列データを用いてイネのSNPの検出および検証を行った。
対象:N1S5、N1S6、N1S7、N1S10
コントロール:N1
リファレンス:イネリファレンスゲノム(IRGSP1.0)
なお、本明細書において、サンプルの表記は、大文字で表記しても小文字で表記しても同じものを指すことに留意されたい。
(概要)
本発明の方法を用いて、以下のイネ配列データを用いてイネのSNPの検出および検証を行った。
対象:N1S5、N1S6、N1S7、N1S10
コントロール:N1
リファレンス:イネリファレンスゲノム(IRGSP1.0)
なお、本明細書において、サンプルの表記は、大文字で表記しても小文字で表記しても同じものを指すことに留意されたい。
(材料および方法)
(サンプル)
イネ品種日本晴の種子1粒をN1個体とし、発芽させて育てて葉をサンプリングした。N1個体に実った種子をN1S1とし、その種子1粒を発芽させて育てて葉をサンプリングした。N1S1個体に実った種子をN1S2とし、同様にして、N1S10世代まで、葉と種子をサンプリングした。
(サンプル)
イネ品種日本晴の種子1粒をN1個体とし、発芽させて育てて葉をサンプリングした。N1個体に実った種子をN1S1とし、その種子1粒を発芽させて育てて葉をサンプリングした。N1S1個体に実った種子をN1S2とし、同様にして、N1S10世代まで、葉と種子をサンプリングした。
(次世代シーケンシング)
N1、N1S5、N1S6、N1S7、N1S10の葉のサンプルについて、次世代シーケンサーでの全ゲノム解析を行った。シーケンサーは、Illumina社のHiSeq2000を用い、ペアエンドで解析した。リード長はN1、N1S5、N1S6、N1S7は100塩基、N1S10のみ101塩基であった。
N1、N1S5、N1S6、N1S7、N1S10の葉のサンプルについて、次世代シーケンサーでの全ゲノム解析を行った。シーケンサーは、Illumina社のHiSeq2000を用い、ペアエンドで解析した。リード長はN1、N1S5、N1S6、N1S7は100塩基、N1S10のみ101塩基であった。
シーケンスライブラリーを用いて、シーケンスの鋳型となるクラスターを形成し、鋳型DNAの塩基配列を取得した。シーケンスデータの解析は付属のソフトウェアを使用しベースコールを行い、fastq形式ファイルとして出力した。
N1、N1S6の解析は以下のとおり製造業者のマニュアルに従った条件で行った。
表5:各作業に使用したマニュアル名、バージョン番号

表6:クラスター形成、シーケンス及びシーケンス解析に使用した装置、試薬、ソフトウェア

表5:各作業に使用したマニュアル名、バージョン番号
表6:クラスター形成、シーケンス及びシーケンス解析に使用した装置、試薬、ソフトウェア
N1S5、N1S7、N1S10の解析は以下のとおり製造業者のマニュアルに従った条件で行った。
表7:各作業に使用したマニュアル名、バージョン番号

表8:クラスター形成、シーケンス及びシーケンス解析に使用した装置、試薬、ソフトウェア

表7:各作業に使用したマニュアル名、バージョン番号
表8:クラスター形成、シーケンス及びシーケンス解析に使用した装置、試薬、ソフトウェア
(多型の検出)
各サンプルについて次世代シーケンサーから得られたFastq形式のファイルより、配列内にNを含まない塩基配列を選んで、相補鎖の配列と合わせて、それぞれ出力した。
各サンプルについて次世代シーケンサーから得られたFastq形式のファイルより、配列内にNを含まない塩基配列を選んで、相補鎖の配列と合わせて、それぞれ出力した。
出力されたファイル(reads)を辞書順にソートし同一の配列は一つにまとめた。unixコマンドは以下の通りであった。

ソートされた各塩基配列をそれぞれ5’末端から1塩基ずつずらしながら、k-mer(本実施例では20塩基)の配列を対象の塩基配列の3’末端に到達するまで繰り返し出力(k-mer_file)した。
出力されたk-merの配列を辞書順にソートし、同一配列は一つにまとめて、出現回数を配列と共に表記したファイルを作成した。unixコマンドは以下の通りであった。

ここで、コントロール配列:N1、対象配列:N1S7について、k-mer配列を整列させ、多型の検出を行った。多型が存在している部位が、ゲノム上でユニークな配列部位であり、そこに例えば一塩基置換が存在している場合、その置換を含むk-mer配列は対象配列では存在するが、コントロール配列では存在しないと考えられる。したがって、当該置換変異を含むk-mer配列では、対象配列では存在し、コントロール配列で存在しないため、結果的に当該置換変異部位を含むk個のk-mer配列で出現頻度の差異が見られると考えられる。k-mer配列の整列は、リファレンス配列に沿う形で整列させた。
k-mer_count_fileの各行のデータに対して、配列の5’末端よりk-1塩基の配列を得て、3’末端の塩基すなわちk番目の塩基をA、C、G、Tの出現回数として表記したデータに変換した。「k-1merの配列 Aの回数 Cの回数 Gの回数 Tの回数」という形式で出力した。
コントロール配列と対象配列について、上記工程で出力したデータをk-1merの配列でまとめたデータを作成した。unixコマンドは以下の通りであった。

さらに、joinコマンドで得られたデータでcontrolとtargetで異なる塩基でカウントが示されるデータを検出した。本実施例においては、イネゲノムの40~50倍読みの次世代シーケンシングデータを対象にしたため、塩基の頻度が100を超える場合はリピート配列部位として除外した。次にcontrol、targetの両方でカウントが1塩基以下の塩基が2個以上存在し、controlあるいはtargetで10以上のカウントを示した塩基に対応するtargetあるいはcontrolの塩基のカウントが1以下である事例が1ないし2回ある場合、多型の境界を検出したものとし、当該事例が生じているk-1merの配列を、多型部位を含むものとして検出した。
リファレンスゲノムへのマッピングのために、リファレンスゲノム(イネリファレンスゲノム(IRGSP1.0))を上記工程と同様に各染色体を5’側から1塩基ずつずらしながらk-merで切り出し、k-mer配列、染色体番号、位置、向きを同一行に表記したデータを作成し、k-merの配列で辞書順に整列させた。当該データに対して、二分検索法により多型k-1merの配列のゲノム上の位置を決定した。
リファレンスゲノムにマップされた多型の確認を行った。上記工程で作成されたsort_uniq配列(ターゲット)と同様に、リファレンス配列も5’末端から2塩基ごとにリード長と同じ長さの配列を切り出し、相補鎖とともにsort、uniq処理をしたデータを作成した。このリファレンスとターゲットのsort_uniqデータを検索の対象とした。
次に、リファレンスゲノム配列から、多型位置を含むリード長と同じ長さの配列セット(検索を行うクエリセット)を切り出した。この配列セットは配列の5’末端から3’末端までのすべての位置で多型位置の塩基を含む配列セットである。配列セット数は配列長と同一となる。リファレンスの配列セット(リファレンスセット)に対して、多型位置の塩基を予想された多型塩基に置換した配列セット(ターゲットセット)を作成した。
リファレンスセット、ターゲットセットをクエリにして、リファレンスゲノム、及び、次世代シーケンサーから得られたFastq形式のファイルより出力されたファイル(reads)を辞書順にソートし同一の配列を一つにまとめたsort_uniq配列をそれぞれ、二分検索法で検索し、それぞれのセットに対してsort_uniq配列が何個マッチするかを調べた。リファレンスセットではリファレンスのsort_uniq配列のみにヒットする。これに対して、ターゲットセットでは、ホモ型の変異の場合は、ターゲットのsort_uniqデータのみから検出され、ヘテロ型の変異の場合は、リファレンスと、ターゲットのsort_uniqデータの両方から検出されるはずである。このようにして、予想と一致する検索値を示した多型を抽出した。
(ジェノタイプの確認)
ジェノタイプの確認は、PCRで当該領域を増幅し、サンガー法で決定した。
ジェノタイプの確認は、PCRで当該領域を増幅し、サンガー法で決定した。
各領域の増幅に用いたプライマー配列は以下のとおりであった。

PCR反応の反応条件は以下のとおりであった。

反応サイクルは、94℃ 0.5分、60℃ 0.5分、72℃ 1分を30サイクルとして行った。
増幅したDNA断片を1%アガロースゲル電気泳動で分離し、0.5μg/mlの濃度のエチジウムブロマイドで染色して、長波長紫外線ランプ(365nm)で蛍光を発するバンドを切り出し、Promega社のWizard(登録商標) SV Gel and PCR Clean-Up System(Cat.# A9282)で断片の精製を行った。
サンガー法でのSNPを含む塩基配列の確認
精製された断片をBigDye(登録商標) Terminator v3.1 Cycle Sequencing Kit(Thermo Fisher Scientific Cat.# 4337455)で反応を行い、DNAシーケンサー ABI PRISM 3130xlで塩基配列の確認を行った。
精製された断片をBigDye(登録商標) Terminator v3.1 Cycle Sequencing Kit(Thermo Fisher Scientific Cat.# 4337455)で反応を行い、DNAシーケンサー ABI PRISM 3130xlで塩基配列の確認を行った。
(結果)
(シーケンシング)
イネ個体(N1、N1S5、N1S6、N1S7、N1S10)を次世代シーケンサーで解析した結果の塩基配列データは、DDBJに送信されており、以下のアクセッション番号で登録されている。

(シーケンシング)
イネ個体(N1、N1S5、N1S6、N1S7、N1S10)を次世代シーケンサーで解析した結果の塩基配列データは、DDBJに送信されており、以下のアクセッション番号で登録されている。
各サンプルについてのリード数(総データ数)は、以下:

のとおりであった。
のとおりであった。
出力されたFastqファイルを処理したsort_uniqのデータ数は以下:

のとおりであった。sort_uniqは、Nを含まないリードとその相補鎖のデータをsortしたのちuniqで同一配列を一つにまとめたものである。このデータは、k-merのデータと異なり配列のみのデータであり、頻度の数値データは含まない。
のとおりであった。sort_uniqは、Nを含まないリードとその相補鎖のデータをsortしたのちuniqで同一配列を一つにまとめたものである。このデータは、k-merのデータと異なり配列のみのデータであり、頻度の数値データは含まない。
(多型の検出)
コントロール配列:N1、対象配列:N1S7について、k-mer配列をリファレンス配列に沿って整列させ、多型の検出を行った結果は、図7および8に示される。下線を付された塩基がコントロールと対象で異なっており、多型が検出されたことが示されている。図9においては、コントロール配列(N1)と対象配列(N1S5、N1S6、N1S7、N1S10)のk-mer配列の部分配列サブセットを整列させることによって、イネリファレンス配列の対応する位置から始まる配列と対応する配列の出現頻度を求めた結果が示される。染色体番号、染色体の位置に続いて、N1、N1S5、N1S6、N1S7、N1S10の20-merの頻度が示される。N1S7でヘテロ、N1S10でミュータントホモになり、野生型の20-merがゼロになっていることがわかる。すなわち、k-mer配列の出現頻度の変化から、N1S7においてヘテロ変異が生じ、N1S10においてホモ変異となったことを検出することができた。
コントロール配列:N1、対象配列:N1S7について、k-mer配列をリファレンス配列に沿って整列させ、多型の検出を行った結果は、図7および8に示される。下線を付された塩基がコントロールと対象で異なっており、多型が検出されたことが示されている。図9においては、コントロール配列(N1)と対象配列(N1S5、N1S6、N1S7、N1S10)のk-mer配列の部分配列サブセットを整列させることによって、イネリファレンス配列の対応する位置から始まる配列と対応する配列の出現頻度を求めた結果が示される。染色体番号、染色体の位置に続いて、N1、N1S5、N1S6、N1S7、N1S10の20-merの頻度が示される。N1S7でヘテロ、N1S10でミュータントホモになり、野生型の20-merがゼロになっていることがわかる。すなわち、k-mer配列の出現頻度の変化から、N1S7においてヘテロ変異が生じ、N1S10においてホモ変異となったことを検出することができた。
さらに、最終的に上記手順によって、コントロール配列(N1)と対象配列(N1S5、N1S6、N1S7、N1S10)との間で検出された多型の一部を、図10および11に示した。これらの結果は、リファレンス配列を用いた確認と一致していた。Wが野生型、Hがヘテロ型、Mがミュータントホモ型を示す。
サンガー法により確認した各サンプルにおける多型を、図12に示す。N1、N1S1、N1S2、N1S3、N1S4、N1S5、N1S6、N1S7、N1S8、N1S9、N1S10と世代を重ねながら、ヘテロ変異が生じ、その後ホモ変異として定着する様子が観察でき、この結果は、本発明の方法によって検出された多型とよく一致していた(図12)。
したがって、本発明の方法によって、世代間に生じた多型を詳細に検出できることが示された。また、Polymorphic Edge Detectionによって検出された多型が、サンガー法によっても確認されていることから、リファレンス配列(ゲノムリファレンス配列)を必要とせずに、配列データ間での多型の検出を行うことができることが実証された。
(実施例2:ナイジェリアのヨルバ族男性(NA18507)の配列解析)
(材料および方法)
コントロール配列データとして、ヒトゲノムリファレンスhg38を用いた。配列は、ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/よりchr1~chr22とchrX、chrY、chrMの染色体データをダウンロードして用いた。alt、v1等のファイル名にコメントが付いているデータは除外した。
(材料および方法)
コントロール配列データとして、ヒトゲノムリファレンスhg38を用いた。配列は、ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/よりchr1~chr22とchrX、chrY、chrMの染色体データをダウンロードして用いた。alt、v1等のファイル名にコメントが付いているデータは除外した。
対象配列データとしては、ヒトゲノムの次世代シーケンス配列データNA18507をダウンロードして用いた。この配列データは、Illumina社の次世代シーケンサーで解析が行われたものであり、NCBIに登録・公開されており、当該データをダウンロードして用いた。塩基配列セットの実験IDのURLは、https://www.ncbi.nlm.nih.gov/sra/SRX016231であり、配列のアクセッション番号は、SRR034939~SRR034975の範囲であった。
情報処理は、実施例1の(多型の検出)と同様に行った。
(結果)
結果の一部を、図13に示す。相補鎖(r)で検出された野生型・変異型の塩基は順鎖に変換して表示している。ヘテロ型の場合はそれぞれの塩基を並べて示している。参照、対象の配列xのA、C、G、Tの数はk-1配列に続く配列xの各塩基の頻度を示している。P~Q列に関しては、野生型あるいは変異型の塩基をもつ参照あるいは対象配列の数を示している。
結果の一部を、図13に示す。相補鎖(r)で検出された野生型・変異型の塩基は順鎖に変換して表示している。ヘテロ型の場合はそれぞれの塩基を並べて示している。参照、対象の配列xのA、C、G、Tの数はk-1配列に続く配列xの各塩基の頻度を示している。P~Q列に関しては、野生型あるいは変異型の塩基をもつ参照あるいは対象配列の数を示している。
順鎖、逆鎖の双方で同じ変異が検出された場合は、ほぼ間違いなくSNPである。片側の鎖のみで検出される原因は、逆の鎖の配列がゲノム上でユニークでなく、一意で検出できなかったため、あるいは、挿入、欠失、転座、逆位の境界塩基を検出したためであると考えられる。
本発明の方法は、ヒトゲノムについても、正確に多型を検出できることが実証された。また、コントロール配列として、データベースから取得したリファレンス配列を用いることができることも示される。
(実施例3:同一個体の組織間での多型の検出)
(概要)
本発明の方法により、同一個体の組織間での多型の検出が可能であることを実証する。
(概要)
本発明の方法により、同一個体の組織間での多型の検出が可能であることを実証する。
(材料および方法)
NCBIのSRAよりfastq-dumpを用いて配列データを取得した。本データは、Texas Cancer Research Biobank Open Access Data Sharing: Genome Projectが登録したデータであり、詳細データについて、以下のURL:https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP060654において提供されている(Becnel, L. et al. NCBI Sequence Read Archive PRJNA284598 (2015).)。本発明の方法により、前記配列データにおいて多型を検出し、同一個体の正常組織と腫瘍組織との間の多型を本発明の方法によって検出できるかを検証した。
NCBIのSRAよりfastq-dumpを用いて配列データを取得した。本データは、Texas Cancer Research Biobank Open Access Data Sharing: Genome Projectが登録したデータであり、詳細データについて、以下のURL:https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP060654において提供されている(Becnel, L. et al. NCBI Sequence Read Archive PRJNA284598 (2015).)。本発明の方法により、前記配列データにおいて多型を検出し、同一個体の正常組織と腫瘍組織との間の多型を本発明の方法によって検出できるかを検証した。
配列データの起源のサンプルは2種類で、配列データ名とサンプルの内容は以下の通りであった。
SRR2096532 コントロール血液 (Normal)
SRR2096535 濾胞性リンパ腫 (9690/3: Follicular lymphoma)
リード数 (配列長101塩基)
SRR2096532 1300353764
SRR2096535 1339310760
sort_uniqの配列数
SRR2096532 2056683322
SRR2096535 2181081390
SRR2096532 コントロール血液 (Normal)
SRR2096535 濾胞性リンパ腫 (9690/3: Follicular lymphoma)
リード数 (配列長101塩基)
SRR2096532 1300353764
SRR2096535 1339310760
sort_uniqの配列数
SRR2096532 2056683322
SRR2096535 2181081390
SRR2096532(正常組織)をコントロール配列データとして用い、SRR2096535(腫瘍組織)を対象配列データとして用いて解析を行った。
情報処理は、実施例2の(多型の検出)と同様に行った。
なお、本実施例においては、確認(verify)工程で、検出されたそれぞれの変異に対して、ターゲットでは、変異型が5リード以上、野生型が1リード以下、コントロールでは変異型が1リード以下、野生型が5リード以上の場合、ホモ型変異(M)とマークし、ターゲットのリード総数に対する変異型の割合が0.3より大きいか、0.7より小さく、かつ、コントロールで変異型が1リード以下、かつ、ターゲットで野生型リードが5以上の場合、ヘテロ型変異(H)とした。
[従来法]
この分野で一般的に広く用いられている、Samtoolsを用いて同じデータを処理した。
従来法による解析は、以下の工程によって行った。工程0は準備で1回のみ実施し、工程1から5はサンプル毎に実施した。
0.準備:リファレンス配列にインデックス付加
1.ショートリードデータのマッピング
2.SAM形式をBAMに変換(マッピング位置でソートも)
3.Samtoolsで多型部位の検出
この分野で一般的に広く用いられている、Samtoolsを用いて同じデータを処理した。
従来法による解析は、以下の工程によって行った。工程0は準備で1回のみ実施し、工程1から5はサンプル毎に実施した。
0.準備:リファレンス配列にインデックス付加
1.ショートリードデータのマッピング
2.SAM形式をBAMに変換(マッピング位置でソートも)
3.Samtoolsで多型部位の検出
リファレンス配列データとして、ヒトゲノムリファレンスhg38を用いた。配列は、ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/よりchr1~chr22とchrX、chrY、chrMの染色体データをダウンロードして用いた。alt、v1等のファイル名にコメントが付いているデータは除外した。
(結果)
本発明の方法(PED)および従来法(bwa+Samtools)で検出された多型の数は以下のとおりであった。
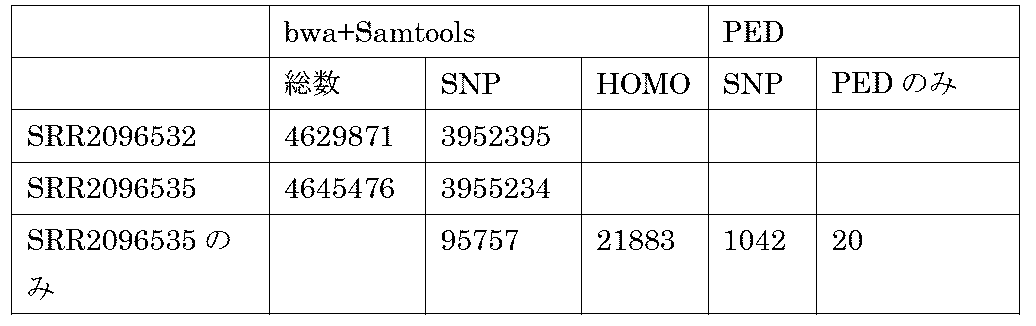
本発明の方法(PED)および従来法(bwa+Samtools)で検出された多型の数は以下のとおりであった。
PEDでVerify(確認工程)にかけた座位数は22601で、そのうち順鎖、相補鎖共に置換変異が検出されたのは、514座位であった。Verifyの結果片側鎖のみで検出されたものの、ポジティブと判断された座位を合わせて1042座位に置換変異があると予想された。PEDで検出された1042座位はすべてヘテロ型であった。このことは、同一人物の血液と癌組織からの配列の比較であるため、原理的にホモ型変異が起こる可能性は非常に低いと考えられることと整合する。PEDではマッピングする前に直接対象とコントロールの比較を行っているため、対象・コントロールのSNP数は出力されない。
PEDで検出されたが、bwa+Samtoolsで検出できなかったSNPは20であった。bwa+Samtoolsではホモ型変異も多く検出されており、結果に非常に多くのノイズを含んでいると考えられる。
(考察)
同一人物からの組織間の比較なので、ミュータントホモはあり得ないと予想されていたところ、予想通りにヘテロ型のみが検出された。この精度で多型を検出できる系は、他にはなく、本発明の方法は従来技術に対して顕著に有利なものであると考えられる。
同一人物からの組織間の比較なので、ミュータントホモはあり得ないと予想されていたところ、予想通りにヘテロ型のみが検出された。この精度で多型を検出できる系は、他にはなく、本発明の方法は従来技術に対して顕著に有利なものであると考えられる。
上記の結果から、同一個体の正常組織と癌組織の配列を直接比較して多型を検出することも可能であることが示された。本発明の方法は、いったんリファレンスゲノムにマップしてから差を調べる方法に比べて、多型の捕捉率、捕捉精度ともに非常に高くなる。
PEDではマッピングの前に、リード同士の比較からSNPを検出するので確度の高いSNPが得られる。本実施例で得られた結果を、挿入・欠失を検出するプログラムの結果と併せて考慮すると、癌細胞では、置換変異よりむしろ、二重鎖切断の後の除去修復に起因すると考えられる挿入・欠失変異の頻度が高いと考えられる。
(実施例4:コピー数多型の検出)
(材料および方法)
実施例1におけるN1S6と同じ世代の種子1粒から細胞培養を行い、1ヶ月、3ヶ月、5ヶ月後に再分化してイネの個体にした葉よりDNAを抽出し、それぞれ1M1、3M1、5M1のサンプルとして用いた。実施例1におけるN1種子と同じ世代の種子を5ヶ月培養して再分化した個体を4世代自殖した個体からDNAを抽出し、TTM2とTTM5のサンプルとして用いた。抽出したDNAから次世代シーケンサーによって配列データを取得した。シーケンシングのプロトコルは実施例1におけるものと同様であった。これらの配列データおよび実施例1のN1S5、N1S6、N1S7、N1S10の配列データを対象配列データとして、N1を参照配列データとして用いた。これらの配列データについてのアクセッション番号、リード数、sort_uniqの配列数は以下の表に示される。なお、TTM5のデータは、SRR556174とSRR556175の2つのアクセッション番号に分割されている。TTM5のsort_uniqは2つのリードを合わせて一つのファイルとして作成した。
(材料および方法)
実施例1におけるN1S6と同じ世代の種子1粒から細胞培養を行い、1ヶ月、3ヶ月、5ヶ月後に再分化してイネの個体にした葉よりDNAを抽出し、それぞれ1M1、3M1、5M1のサンプルとして用いた。実施例1におけるN1種子と同じ世代の種子を5ヶ月培養して再分化した個体を4世代自殖した個体からDNAを抽出し、TTM2とTTM5のサンプルとして用いた。抽出したDNAから次世代シーケンサーによって配列データを取得した。シーケンシングのプロトコルは実施例1におけるものと同様であった。これらの配列データおよび実施例1のN1S5、N1S6、N1S7、N1S10の配列データを対象配列データとして、N1を参照配列データとして用いた。これらの配列データについてのアクセッション番号、リード数、sort_uniqの配列数は以下の表に示される。なお、TTM5のデータは、SRR556174とSRR556175の2つのアクセッション番号に分割されている。TTM5のsort_uniqは2つのリードを合わせて一つのファイルとして作成した。
情報処理は、実施例1の(多型の検出)と同様に行い、参照配列データと、対象配列データの間で出現頻度が大きく異なったk-merを検出した。コントロールのN1より2倍程度以上の頻度が連続して検出された場合にCNVと判定した。誤検出も出てくるので、2.5倍以上と厳しくすることもできる。
(結果)
結果の一部を図17に示した。第7染色体の26694795位置(図7中矢印で示される)に対応するk-mer配列から、それまでの位置に対応するk-mer配列の出現頻度の4倍程度の値が、N1~N1S10で現れていることが分かる。
結果の一部を図17に示した。第7染色体の26694795位置(図7中矢印で示される)に対応するk-mer配列から、それまでの位置に対応するk-mer配列の出現頻度の4倍程度の値が、N1~N1S10で現れていることが分かる。
この位置からレトロトランスポゾンTos17の配列が始まる。このトランスポゾンはゲノムに2コピー存在しており、それぞれのトランスポゾンの末端部分は同じ配列(Long Terminal Repeat、LTR)を有する。そのため、第7染色体の26694795より、それまでの4倍程度の値が、N1~N1S10で現れたと考えられる。このトランスポゾンの全長は4.1kbあるため、図17には最初のジャンクションの部分のみ示している。
1M1、3M1、5M1はN1S6と同じ世代の種子1粒から細胞培養を行い、それぞれ1ヶ月、3ヶ月、5ヶ月後に再分化してイネの個体にした葉よりDNAを抽出して解析したものであり、図17に示される結果から、培養時間に応じてカウントが増えていることがわかる。これは、培養時間に比例してトランスポゾンが転移してコピー数が増加し、コピー数多型(CNV)が生じたためと考えられる。TTM2とTTM5はN1種子と同じ世代の種子を5ヶ月培養して再分化した個体を4世代自殖した個体のDNAであり、こちらも同様にコピー数の増加が認められる。
(考察)
Tos17は培養時のみ活性化されて転移するイネのトランスポゾンとして知られている。Tos17はレトロトランスポゾンなので、オリジナルは切り出されて転移することなく、Tos17のコピーの転移によってゲノム上のTos17のコピー数が増加する。そのため、Tos17は培養で転移してコピー数が増えることが以前から知られている。
Tos17は培養時のみ活性化されて転移するイネのトランスポゾンとして知られている。Tos17はレトロトランスポゾンなので、オリジナルは切り出されて転移することなく、Tos17のコピーの転移によってゲノム上のTos17のコピー数が増加する。そのため、Tos17は培養で転移してコピー数が増えることが以前から知られている。
本発明の方法によって、コピー数の変異を検出した結果、理論とよく一致して、培養時間が長くなるにつれて、Tos17のコピー数が増加していることが図17に示されるカウントから理解される。
したがって、本発明の方法によって、コピー数多型を検出することが可能であることが実証される。
例えば、このようなコピー数多型の検出は、培養細胞(例えば、iPS細胞等)において、品質の管理に用いることが可能であると考えられる。上記イネの培養細胞と同様にトランスポゾン等によるコピー数変異が、例えば、ヒトの培養細胞(iPS等)で観察されている場合には、例えば、治療に用いるのは危険である可能性が高いという判断に用いることが可能である。
(関連出願)
本出願は、2017年7月24日に出願された特願2017-142781号の優先権の利益を主張し、当該出願は、全ての目的において、その開示全体が本明細書において参考として援用される。さらに、本明細書において、本出願人により本出願と同日に出願された「挿入・欠失・逆位・転座・置換検出法」との名称の出願(整理番号NG012PCT/F5-18PCT075)およびその基礎出願である2017年7月24日に出願された特願2017-142782号(整理番号J1-17369162)は、全ての目的において、その開示全体が本明細書において参考として援用される。
本出願は、2017年7月24日に出願された特願2017-142781号の優先権の利益を主張し、当該出願は、全ての目的において、その開示全体が本明細書において参考として援用される。さらに、本明細書において、本出願人により本出願と同日に出願された「挿入・欠失・逆位・転座・置換検出法」との名称の出願(整理番号NG012PCT/F5-18PCT075)およびその基礎出願である2017年7月24日に出願された特願2017-142782号(整理番号J1-17369162)は、全ての目的において、その開示全体が本明細書において参考として援用される。
塩基配列解析で多型を検出するすべての分野で利用が可能で、DNA育種利用の他、臨床検査、iPS細胞の検査、メタゲノム解析、発現解析等、幅広い分野で利用することができる。
配列番号1~60:図7のk-mer参照配列
配列番号61~80:図7のk-mer対象配列(変異が存在する部分)
配列番号81~140:図8のk-mer参照配列
配列番号141~160:図8のk-mer対象配列(変異が存在する部分)
配列番号161~190:図13のk-1(k=20)配列
配列番号191~221:(具体的な例)で用いられた配列
配列番号222~232:(例示的計算フロー)で用いられた配列
配列番号233~266:実施例1で用いられたプライマーの配列
配列番号267~275:図11の配列
配列番号61~80:図7のk-mer対象配列(変異が存在する部分)
配列番号81~140:図8のk-mer参照配列
配列番号141~160:図8のk-mer対象配列(変異が存在する部分)
配列番号161~190:図13のk-1(k=20)配列
配列番号191~221:(具体的な例)で用いられた配列
配列番号222~232:(例示的計算フロー)で用いられた配列
配列番号233~266:実施例1で用いられたプライマーの配列
配列番号267~275:図11の配列
Claims (42)
- 対象配列データにおいてコントロール配列データに対する多型を検出する方法であって、
a)該対象配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
b)該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供する工程と、
c)対象配列とコントロール配列とを比較し、該出現頻度の分布の比較に基づいて、多型を検出する工程と
を包含し、ここで、kは該対象配列および該コントロール配列のいずれか短いほうの全長以下の整数である、方法。 - 前記部分配列中の長さk-xの配列部分が共通する配列ごとに、長さxの部分について出現頻度の分布を算出する工程をさらに含み、ここで、xはk未満の正の整数である、請求項1に記載の方法。
- 前記比較が、前記部分配列中の長さk-xの配列部分が共通する配列における、長さxの部分の出現頻度の分布の差異の比較を含む、請求項2に記載の方法。
- 前記部分配列中の長さk-xの配列部分を、ユニークな配列ごとにグルーピングする工程を含み、ここで、xはk未満の正の整数である、請求項1~3のいずれか1項に記載の方法。
- 前記長さk-xの配列部分をソートする工程を含む、請求項4に記載の方法。
- 前記長さk-xの配列部分を文字列としてソートする工程を含む、請求項5に記載の方法。
- 前記kが、前記対象配列における偶然同一を排除する長さである、請求項1~6のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記kが、前記生物のゲノムにおいて、異なる箇所での偶然同一を排除する長さである、請求項1~7のいずれか1項に記載の方法。
- 長さxが1~2である、請求項2~8のいずれか1項に記載の方法。
- 長さxが1である、請求項9に記載の方法。
- 前記長さxの部分が、前記部分配列の末端に存在する、請求項2~10のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記長さxの部分が、前記部分配列の3’末端である、請求項9に記載の方法。
- 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列の出現頻度が異なる場合、該長さxの部分の配列を、対象配列データにおけるコントロール配列データに対する多型として検出する、請求項2~12のいずれか1項に記載の方法。
- 前記コントロール配列データのサブセットと前記対象配列データのサブセットとの間で、前記長さxの部分の配列で最も高頻度のものが異なっている長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおける多型として検出する、請求項2~13のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、前記対象配列データのサブセットにおける前記長さxの部分の配列で、前記コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列がノイズ以下のカウントしか存在しない長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるホモ多型として検出する、請求項2~14のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、対象サブセットにおける前記長さxの部分の配列で、コントロール配列データのサブセットにおける最も高頻度のものと同一の長さxの部分の配列が存在し、かつ、コントロール配列データのサブセットにおける最も高頻度のものと異なる長さxの部分の配列が存在する長さk-xの配列部分が存在する場合、該長さxの部分の配列を、対象配列データにおけるヘテロ多型として検出する、請求項2~15のいずれか1項に記載の方法。
- 対象配列データ量から予測される出現頻度と比較して、前記出現頻度が少ない部分配列をノイズとする、請求項1~16のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、[(対象配列データ量)×(1-精度)]/(対象ゲノムサイズ)+1未満の出現頻度の部分配列をノイズとする、請求項17に記載の方法。
- 前記対象配列データが、次世代シーケンシングによって得られた塩基配列データである、請求項1~18のいずれか1項に記載の方法。
- 前記対象配列データが、個体から得られた配列データであり、前記コントロール配列データが、該個体と同種の別の個体、またはデータベースから得られた配列データである、請求項1~19のいずれか1項に記載の方法。
- 前記対象配列データが、個体の組織試料から得られた配列データであり、前記コントロール配列データが、該個体の別の組織、またはデータベースから得られた配列データである、請求項1~20のいずれか1項に記載の方法。
- 前記対象配列データが、細胞試料から得られた配列データであり、前記コントロール配列データが、別の細胞、またはデータベースから得られた配列データである、請求項1~21のいずれか1項に記載の方法。
- 前記多型が、置換、挿入、欠失、コピー数多型(Copy Number Variation, CNV)、STRP(short tandem repeat polymorphism)、逆位または転座である、請求項1~22のいずれか1項に記載の方法。
- 前記多型が、置換である、請求項23に記載の方法。
- 前記対象配列に対するリファレンス配列における前記多型の位置を特定する工程をさらに含む、請求項1~24のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記多型のゲノム上の位置を特定する工程をさらに含む、請求項1~25のいずれか1項に記載の方法。
- 検出された多型の部位について、リファレンス配列またはコントロール配列から作成したクエリ配列セットを用いて、対象配列データおよび/またはコントロール配列データとの比較を行い確認する工程をさらに含む、請求項25または26に記載の方法。
- 前記クエリ配列セットが、リファレンス配列またはコントロール配列において前記多型に該当する部位の文字を異なる文字に置換した変異型クエリ配列セットを含む、請求項27に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの相補鎖の配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、請求項27または28に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが塩基配列データであり、検出された多型の部位について、対象配列データおよび/またはコントロール配列データの変異型の塩基を有する配列データに対して、リファレンス配列またはコントロール配列から作成したクエリ配列セットとの比較を行い確認する工程をさらに含む、請求項27~29のいずれか1項に記載の方法。
- 前記対象配列データおよび前記コントロール配列データが、生物のゲノムに由来する塩基配列データであり、前記ゲノムの配列が不明である、請求項1~30のいずれか1項に記載の方法。
- 実験結果またはデータベースから対象配列データまたはコントロール配列データを取得する工程をさらに含む、請求項1~31のいずれか1項に記載の方法。
- 対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることをさらに含む、請求項1~32のいずれか1項に記載の方法。
- 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、請求項33に記載の方法。
- 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムであって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、プログラム。 - 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列を、検出された前記多型の名称として表示する工程をさらに含む、請求項35に記載のプログラム。
- 対象配列データにおいてコントロール配列データに対する多型を検出する方法をコンピュータに実行させるためのプログラムを格納する記録媒体であって、該方法は、
a)該対象配列データの長さkの部分配列のサブセットをコンピュータに保存する工程であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、工程と、
b)該対象配列データの長さkのサブセットの各々の部分配列の出現頻度を算出する工程と、
c)該コントロール配列データの長さkの部分配列のサブセットにおける各々の部分配列の出現頻度をコンピュータに保存する工程と、
d)対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程と
を包含する、記録媒体。 - 前記方法が、前記部分配列中の多型ではない部分の少なくとも一部を含む配列を、検出された前記多型の名称として表示する工程をさらに含む、請求項37に記載の記録媒体。
- 対象配列データにおいてコントロール配列データに対する多型を検出するためのシステムであって、該システムは、
該対象配列データおよび該コントロール配列データの長さkの部分配列のサブセットの各々の部分配列の出現頻度を提供するように構成された配列データ処理部であって、ここで、kは対象配列およびコントロール配列の全長以下の長さである、配列データ処理部と、
対象配列とコントロール配列とを比較し、該出現頻度の分布の差異に基づいて、多型を検出する工程とを行うように構成された、配列データ計算部と
を備える、システム。 - 前記システムが、前記部分配列中の多型ではない部分の少なくとも一部を含む配列を、検出された前記多型の名称として表示する表示手段をさらに含む、請求項39に記載のシステム。
- 対象配列データとコントロール配列データとの比較方法であって、
対象配列データにおけるコントロール配列データに対する多型を含む部分配列中の多型ではない部分の少なくとも一部を含む配列を、該多型の識別子として割り当てることを含む、方法。 - 前記多型の識別子をリファレンス配列にマッピングし、リファレンス上の該多型の位置を特定することを含む、請求項41に記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019532603A JP7166638B2 (ja) | 2017-07-24 | 2018-07-23 | 多型検出法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017142781 | 2017-07-24 | ||
| JP2017-142781 | 2017-07-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019022018A1 true WO2019022018A1 (ja) | 2019-01-31 |
Family
ID=65039682
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/027535 Ceased WO2019022018A1 (ja) | 2017-07-24 | 2018-07-23 | 多型検出法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP7166638B2 (ja) |
| TW (1) | TW201920682A (ja) |
| WO (1) | WO2019022018A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20220070452A (ko) * | 2019-09-30 | 2022-05-31 | 롱가스 테크놀로지즈 피티와이 엘티디 | 돌연변이들을 포함하는 동일한 서열로부터 2개의 돌연변이된 서열 판독물이 유래할 확률과 상관된 측정치를 결정하기 위한 방법 |
| CN115910197A (zh) * | 2021-12-29 | 2023-04-04 | 上海智峪生物科技有限公司 | 基因序列处理方法、装置、存储介质及电子设备 |
| CN118658524A (zh) * | 2024-06-07 | 2024-09-17 | 天津必佳生物技术有限公司 | 一种用于动物饲料的嵌合溶菌酶基因序列数据分析方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001112486A (ja) * | 1999-08-05 | 2001-04-24 | Takeda Chem Ind Ltd | 遺伝子解析結果の記録方法 |
| JP2001167123A (ja) * | 1999-12-13 | 2001-06-22 | Iyaku Bunshi Sekkei Kenkyusho:Kk | 遺伝子配列多型クラスの管理方法 |
| JP2008165375A (ja) * | 2006-12-27 | 2008-07-17 | Canon Inc | 塩基配列を識別する変異セットの選別法 |
| JP2016504667A (ja) * | 2012-11-26 | 2016-02-12 | コーニンクレッカ フィリップス エヌ ヴェKoninklijke Philips N.V. | 患患者固有の関連性評価を用いた変異と疾患の関連付けを使用する診断的遺伝子分析 |
| JP2017045451A (ja) * | 2012-03-29 | 2017-03-02 | 三菱レイヨン株式会社 | βグロビン遺伝子の変異を検出するためのマイクロアレイ及びその検出方法 |
-
2018
- 2018-07-23 WO PCT/JP2018/027535 patent/WO2019022018A1/ja not_active Ceased
- 2018-07-23 JP JP2019532603A patent/JP7166638B2/ja active Active
- 2018-07-23 TW TW107125394A patent/TW201920682A/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001112486A (ja) * | 1999-08-05 | 2001-04-24 | Takeda Chem Ind Ltd | 遺伝子解析結果の記録方法 |
| JP2001167123A (ja) * | 1999-12-13 | 2001-06-22 | Iyaku Bunshi Sekkei Kenkyusho:Kk | 遺伝子配列多型クラスの管理方法 |
| JP2008165375A (ja) * | 2006-12-27 | 2008-07-17 | Canon Inc | 塩基配列を識別する変異セットの選別法 |
| JP2017045451A (ja) * | 2012-03-29 | 2017-03-02 | 三菱レイヨン株式会社 | βグロビン遺伝子の変異を検出するためのマイクロアレイ及びその検出方法 |
| JP2016504667A (ja) * | 2012-11-26 | 2016-02-12 | コーニンクレッカ フィリップス エヌ ヴェKoninklijke Philips N.V. | 患患者固有の関連性評価を用いた変異と疾患の関連付けを使用する診断的遺伝子分析 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20220070452A (ko) * | 2019-09-30 | 2022-05-31 | 롱가스 테크놀로지즈 피티와이 엘티디 | 돌연변이들을 포함하는 동일한 서열로부터 2개의 돌연변이된 서열 판독물이 유래할 확률과 상관된 측정치를 결정하기 위한 방법 |
| JP2022550013A (ja) * | 2019-09-30 | 2022-11-30 | ロンガス テクノロジーズ ピーティーワイ リミテッド | 2つの変異シーケンスリードが、同一の変異を含むシーケンスに由来する確率と相関する尺度を決定するための方法 |
| JP7636400B2 (ja) | 2019-09-30 | 2025-02-26 | イルミナ シンガポール ピーティーイー リミテッド | 2つの変異シーケンスリードが、同一の変異を含むシーケンスに由来する確率と相関する尺度を決定するための方法 |
| KR102903933B1 (ko) | 2019-09-30 | 2025-12-23 | 일루미나 싱가포르 피티이 엘티디 | 돌연변이들을 포함하는 동일한 서열로부터 2개의 돌연변이된 서열 판독물이 유래할 확률과 상관된 측정치를 결정하기 위한 방법 |
| CN115910197A (zh) * | 2021-12-29 | 2023-04-04 | 上海智峪生物科技有限公司 | 基因序列处理方法、装置、存储介质及电子设备 |
| CN115910197B (zh) * | 2021-12-29 | 2024-03-22 | 上海智峪生物科技有限公司 | 基因序列处理方法、装置、存储介质及电子设备 |
| CN118658524A (zh) * | 2024-06-07 | 2024-09-17 | 天津必佳生物技术有限公司 | 一种用于动物饲料的嵌合溶菌酶基因序列数据分析方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201920682A (zh) | 2019-06-01 |
| JPWO2019022018A1 (ja) | 2020-05-28 |
| JP7166638B2 (ja) | 2022-11-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Larson et al. | A clinician’s guide to bioinformatics for next-generation sequencing | |
| Kockum et al. | Overview of genotyping technologies and methods | |
| Springer et al. | The maize W22 genome provides a foundation for functional genomics and transposon biology | |
| Kumar et al. | SNP discovery through next-generation sequencing and its applications | |
| US20060286566A1 (en) | Detecting apparent mutations in nucleic acid sequences | |
| Lange et al. | Analysis pipelines for cancer genome sequencing in mice | |
| Vezzulli et al. | Molecular mapping of grapevine genes | |
| US20260018252A1 (en) | Machine learning techniques for analysis of structural variants | |
| JP7166638B2 (ja) | 多型検出法 | |
| CN112289384A (zh) | 一种柑橘全基因组kasp标记库的构建方法及应用 | |
| CN114842907B (zh) | 基于高通量全基因组测序的多亲本作物基因型鉴定 | |
| Roy et al. | NGS-μsat: bioinformatics framework supporting high throughput microsatellite genotyping from next generation sequencing platforms | |
| JP2022549823A (ja) | キットおよびキットの使用方法 | |
| CN118957105A (zh) | 一种用于肉牛血统鉴定的液相芯片及设计方法、应用 | |
| CN114566214B (zh) | 检测基因组缺失插入变异的方法及检测装置和计算机可读存储介质与应用 | |
| JP7122006B2 (ja) | 挿入・欠失・逆位・転座・置換検出法 | |
| Rachappanavar et al. | Analytical pipelines for the GBS analysis | |
| JP7444488B2 (ja) | 混入検出法 | |
| JP7819961B2 (ja) | 転移因子検出法 | |
| Fletcher et al. | AFLAP: Assembly-Free Linkage Analysis Pipeline using k-mers from whole genome sequencing data | |
| Lübberstedt et al. | Markers and Sequencing | |
| Clarke | Bioinformatics challenges of high-throughput SNP discovery and utilization in non-model organisms | |
| Ko et al. | Variant detection and annotation workflows using the GATK pipeline | |
| CN120945074A (zh) | 一种鸡bf2基因快速分型检测方法及其应用 | |
| CN115956129A (zh) | 遗传信息分析系统和遗传信息分析方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2019532603 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18838697 Country of ref document: EP Kind code of ref document: A1 |