WO2019107412A1 - 固体分散体 - Google Patents
固体分散体 Download PDFInfo
- Publication number
- WO2019107412A1 WO2019107412A1 PCT/JP2018/043788 JP2018043788W WO2019107412A1 WO 2019107412 A1 WO2019107412 A1 WO 2019107412A1 JP 2018043788 W JP2018043788 W JP 2018043788W WO 2019107412 A1 WO2019107412 A1 WO 2019107412A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- solid dispersion
- hypromellose
- pharmaceutically acceptable
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/146—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to a solid dispersion comprising a compound having high xanthine oxidase inhibitory activity or a pharmaceutically acceptable salt thereof, and a hypromellose derivative.
- Hyperuricemia results in gout or renal failure and is considered as a risk factor for coronary artery disease. A close relationship is also pointed out with the development of lifestyle-related diseases including hypertension. Therefore, treatment of hyperuricemia leads not only to the treatment of gout, but also to the prevention of various lifestyle-related diseases associated with aging. At present, xanthine oxidase inhibitors such as allopurinol or febuxostat are mainly used for the treatment of hyperuricemia. Further, as a compound having a similar mechanism, a compound described in WO 2005/121153 pamphlet (patent document 1) has been reported.
- Patent Document 1 When the present inventors examined a compound having high xanthine oxidase inhibitory activity disclosed in Patent Document 1, there is still room for improvement in in vivo absorbability when the present compound is orally administered to a living body. I found that there is.
- a solid dispersion comprising pyridin-7-yloxy) -3-methylphenyl) -N 6- (4,4-dimethyl-4,5-dihydrooxazol-2-yl) quinazoline-4,6-diamine and a dispersing polymer A method is disclosed.
- R 1 represents an unsubstituted phenyl group or a phenyl group substituted by a substituent, and the substituent is an alkyl group having 1 to 8 carbon atoms, or 1 to 8 carbon atoms substituted by a halogen atom
- R 2 represents a cyano group or a nitro group
- R 3 represents a hydroxyl group
- X represents an oxygen atom or -S (O) n-
- n represents an integer of 0 to 2
- Y represents an oxygen atom or a sulfur atom Or a pharmaceutically acceptable salt thereof and a solid dispersion comprising hyprome
- the solid dispersion of the present invention is useful as a therapeutic agent for hyperuricemia and the like because it exhibits high absorbability and storage stability in vivo.
- R 1 represents an unsubstituted phenyl group or a phenyl group substituted by a substituent.
- Examples of the “C1-C8 alkyl group” as a substituent of the phenyl group represented by R 1 include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, A hexyl group etc. are mentioned, Preferably a methyl group and an ethyl group are mentioned.
- Examples of the “halogen-substituted alkyl group having 1 to 8 carbon atoms” in the phenyl group represented by R 1 include fluoromethyl group, trifluoromethyl group, 1,1-difluoroethyl group, and pentafluoroethyl group. A group etc. are mentioned, Preferably a fluoromethyl group and a trifluoromethyl group are mentioned.
- Examples of the “C1-C8 alkoxy group” having a substituent on the phenyl group represented by R 1 include methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, tert-butoxy and the like. Preferably, a methoxy group is mentioned.
- Examples of the “C 2-8 alkoxycarbonyl group having 2 to 8 carbon atoms” of the substituent in the phenyl group represented by R 1 include methoxycarbonyl group, ethoxycarbonyl group, propoxycarbonyl group, butoxycarbonyl group, tert-butoxycarbonyl group and the like. And preferably a methoxycarbonyl group and an ethoxycarbonyl group.
- Halogen atom in the substituent on the phenyl group represented by R 1 include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom, preferably a fluorine atom, a chlorine atom.
- R 1 an unsubstituted phenyl group is preferable.
- R 2 represents a cyano group and a nitro group, preferably a cyano group.
- X represents an oxygen atom or -S (O) n- , preferably an oxygen atom.
- Y represents an oxygen atom or a sulfur atom, preferably a sulfur atom.
- Examples of the pharmaceutically acceptable salt of the compound represented by the general formula (I) include alkali metal salts such as sodium salt, potassium salt or lithium salt, and potassium salt is preferable.
- the compound of the general formula (I) used for the solid dispersion which is one embodiment of the present invention can be obtained, for example, by the synthesis method described in Patent Document 1.
- preferred compounds include the compounds of Table 1.
- Me represents a methyl group.
- Compound 1 to Compound 14 may form a pharmaceutically acceptable salt, and among them, Compound 3 to Compound 5, Compound 8 to Compound 10, Compound 13 to Compound 14 or a pharmaceutically acceptable compound thereof Acceptable salts are preferred.
- the "hypromellose derivative” represents hypromellose itself (which may be abbreviated as HPMC) and an organic acid ester of hypromellose.
- Hypromellose also referred to as hydroxypropyl methylcellulose, is a mixed ether of methyl and hydroxypropyl groups of cellulose.
- the organic acid which forms an ester with hypromellose include acetic acid, succinic acid and phthalic acid.
- the hypromellose according to the present invention may form an ester with one or more organic acids selected from the organic acids.
- hypromellose derivatives used in the present invention include hypromellose, hypromellose acetate succinate (sometimes abbreviated as HPMCAS), hypromellose phthalate (sometimes abbreviated as HPMCP), etc.
- HPMCAS hypromellose acetate succinate
- HPMCP hypromellose phthalate
- the hypromellose according to the present invention is exemplified by hypromellose having a substitution ratio per monomer unit of 28 to 30% per methoxy group and 7 to 12% per hydroxypropoxy group.
- the hypromellose acetate ester succinate according to the present invention has a substitution ratio per monomer unit of 20 to 26%, preferably 21 to 25% per methoxy group, and preferably 5 to 10% per hydroxypropoxyl group. Is 5 to 9%, 5 to 14%, preferably 7 to 11% per acetyl group, and 4 to 18%, preferably 10 to 14% per succinoyl group, exemplified by hypromellose acetate succinate. Ru.
- the hypromellose phthalate according to the present invention has a substitution ratio per monomer unit of 18 to 24% per methoxy group, 5 to 10% per hydroxypropoxy group, and 21 to 35% per carboxybenzoyl group.
- Hypromellose phthalate ester which is is illustrated. The content of a methoxy group, a hydroxypropoxy group, an acetyl group, a succinoyl group, a carboxybenzoyl group, etc.
- hypromellose derivative is the hypromellose, hypromellose acetate succinate and hypromellose defined in the 17th revision Japanese Pharmacopoeia It can measure by the method based on the measuring method of the substitution degree of phthalic acid ester.
- the viscosity of the hypromellose derivative according to the present invention is not particularly limited as long as the effect of the present invention is obtained, and for example, 2.4 to 204 mPa ⁇ s can be mentioned, and 2.4 to 3.6 mPa ⁇ s is preferable.
- the viscosity of the hypromellose derivative according to the present invention may be measured by the method based on the method of measuring the viscosity of hypromellose, hypromellose acetate ester succinate ester and hypromellose phthalate ester prescribed in the 17th revised Japanese Pharmacopoeia it can.
- the weight ratio of the compound represented by the general formula (I) or the pharmaceutically acceptable salt thereof to the hypromellose derivative can be appropriately adjusted in the range of 1: 0.1 to 1:25.
- One aspect of the weight ratio of the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof to a hypromellose derivative is 1: 0.1 to 1:10, and another aspect is 1: 0.1 to 1: 4, another side is 1: 1 to 1:10, another side is 1: 2 to 1: 5, and still another side is , 1: 3 to 1: 4.
- solid dispersion is a solid wherein the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof forms a system dispersed throughout the hypromellose derivative
- the “solid dispersion” of the present invention is composed of a compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof, a hypromellose derivative, and optionally a pharmaceutically acceptable additive.
- pharmaceutically acceptable additive optionally contained include, for example, additives selected from surfactants, pH adjusters, saccharides, plasticizers and the like. These can be combined appropriately and can be added to the solid dispersion of the present invention in the necessary amount.
- Surfactants which can be used include cationic interface such as sodium bis- (2-ethylhexyl) sulfosuccinate (sodium dokusate), alkyltrimethylammonium bromide (eg cetyltrimethylammonium bromide (cetrimid)) active agents, anionic surface active agents such as sodium lauryl sulfate, polyoxyethylene sorbitan (e.g. Tween (Tween TM 20, 40, 60, 80 or 85)), sorbitan fatty acid esters (e.g., Span TM 20, 40, And nonionic surfactants such as 60, 80 or 85).
- cationic interface such as sodium bis- (2-ethylhexyl) sulfosuccinate (sodium dokusate), alkyltrimethylammonium bromide (eg cetyltrimethylammonium bromide (cetrimid)) active agents, anionic surface active agents such as sodium lauryl sulfate,
- acids such as succinic acid, maleic acid, tartaric acid, citric acid and aspartic acid, and alkalis such as sodium hydroxide and magnesium oxide, silicon dioxide, sodium hydrogencarbonate and L-arginine are used.
- alkalis such as sodium hydroxide and magnesium oxide, silicon dioxide, sodium hydrogencarbonate and L-arginine
- saccharides that can be used include lactose, sucrose, glucose, fructose, sucrose, maltose (malt), reduced maltose, maltitol, mannitol, erythritol, sorbitol, xylitol and the like.
- Plasticizers that can be used include triethyl citrate, polyethylene glycol, triacetin and the like.
- the “solid dispersion” of the present invention may not contain the above-mentioned pharmaceutically acceptable additive, but in the case of including them, for example, a compound represented by the general formula (I) or a compound thereof
- the weight ratio of the pharmaceutically acceptable salt to the surfactant is 1: 0.01 to 1: 2, more preferably 1: 0.02 to 1: 1.5, still more preferably 1: 0.
- the weight ratio of the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof to the pH adjuster is 1: 0.01 to 1: 1.2.
- the pharmaceutically acceptable additive may constitute the dispersed phase of the solid dispersion or may constitute the continuous phase.
- the solid dispersion of the present invention can be produced by a method known per se, and can be produced, for example, using a mixing and pulverizing method (mechanochemical method), a solvent method, a melting method, a heating and kneading melting method and the like.
- the mixed grinding method is a method of mixing a compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof, a hypromellose derivative, and optionally a pharmaceutically acceptable additive, It can carry out by a conventional method using mixers and grinders, such as a ball mill and a hammer mill.
- the solvent method refers to a solvent (organic solvent, water or a mixture thereof) of a compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and a hypromellose derivative, and optionally a pharmaceutically acceptable additive Or the solvent is removed to precipitate the solid dispersion, or the solid dispersion is precipitated in the solvent.
- Solvents can be classified into spray methods (according to embodiments, they can be classified as fluidized bed methods, spray drying methods (also referred to as spray dry methods), rolling bed methods, stirring methods, or supercritical methods), filtration methods, evaporation methods, It can be removed by a method such as a lyophilization method, preferably a spray method, and among these, the spray drying method is particularly preferable.
- the solvent that can be used to produce the solid dispersion of the present invention is preferably a pharmaceutically acceptable solvent, such as ethanol, methanol, 2-propanol, acetone, 2-butanone, methyl isobutyl ketone, tetrahydrofuran (THF ), Tetrahydropyran, 1,4-dioxane, diethyl ether, toluene, acetonitrile, methylene chloride, chloroform, methyl acetate, ethyl acetate, butyl acetate, acetic acid, formic acid, N, N-dimethylformamide, N, N-dimethylacetamide, Dimethyl sulfoxide and the like can be mentioned.
- a pharmaceutically acceptable solvent such as ethanol, methanol, 2-propanol, acetone, 2-butanone, methyl isobutyl ketone, tetrahydrofuran (THF ), Tetrahydropyran, 1,4-
- a solid dispersion can be produced by a method known per se, for example, a compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and hypromellose derivative, and optionally, A pharmaceutically acceptable additive is added to the solvent to form a solution or suspension, and the solution or suspension is finely atomized by centrifugal spraying with a rotating disk or pressure spraying with a pressure nozzle, and this is a drying medium
- the solid dispersion can be obtained by blowing it inside (heated air or nitrogen gas) to give a powdery dry matter.
- the temperature of the drying medium is, for example, 50 to 120 ° C., preferably 50 to 90 ° C.
- the drying medium may be made to flow in a fixed direction, and can be made to flow, for example, as an air flow of 0.2 to 0.6 m 3 / min, preferably 0.3 to 0.5 m 3 / min.
- the precipitation method in the solvent method is preferably a coprecipitation method, and the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and hypromellose derivative, and optionally a pharmaceutically acceptable additive as a solvent Solution or suspension of the compound (I) or pharmaceutically acceptable salt thereof and hypromellose derivative dissolved therein, and optionally added pharmaceutically acceptable additive-insoluble solvent, temperature,
- a solid dispersion can be obtained by precipitating by lowering the dissolution concentration by lowering or the like.
- the melting method is heating the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and hypromellose derivative, and optionally a pharmaceutically acceptable additive to a temperature above the melting point or softening point of the hypromellose derivative
- the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and optionally added pharmaceutically acceptable additives are dissolved or dispersed in the hypromellose derivative by stirring or the like. Then, say the method of quenching. At that time, additives such as plasticizers such as triethyl citrate, polyethylene glycol and triacetin and surfactants can be further added, if desired.
- the production can be carried out using a stirring granulator with a heating device.
- the heat-kneading melting method is a compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof and hypromellose derivative thereof, and optionally a pharmaceutical according to an extruder equipped with a heating device, such as a twin screw extruder, etc.
- a heating device such as a twin screw extruder, etc.
- the solid dispersion of the present invention produced by the above production method can be made into particles of a solid dispersion having any particle diameter by a known method, and the particles of the solid dispersion can be used as powders or granules as they are. It can be used.
- a pharmaceutical composition containing the solid dispersion of the present invention comprises the solid dispersion and a pharmaceutically acceptable additive, and as a pharmaceutically acceptable additive, for example, a binder, a disintegrant, an excipient
- a pharmaceutically acceptable additive for example, a binder, a disintegrant, an excipient
- the pharmaceutical composition of the present invention can be produced by appropriately combining the excipients, lubricants and the like and blending them in the required amount.
- Binders that can be used in the pharmaceutical composition of the present invention include methylcellulose, hydroxyethylcellulose, hydroxypropylcellulose, hypromellose, polyvinylpyrrolidone, gelatin, agar, alginic acid, sodium alginate, partially saponified polyvinyl alcohol, pullulan, partially gelatinized starch , Dextrin, xanthan gum, gum arabic powder and the like.
- Disintegrants which can be used in the pharmaceutical composition of the present invention include, for example, crystalline cellulose, carboxymethylcellulose (carmellose), croscarmellose sodium, carboxymethylcellulose calcium, low substituted hydroxypropylcellulose, crospovidone, hydroxypropyl starch, Examples thereof include starch, partially gelatinized starch, sodium starch glycolate and the like, preferably croscarmellose sodium, sodium starch glycolate and crospovidone, and more preferably crospovidone.
- the amount of the disintegrant to be used is preferably 5 to 30% by weight, more preferably 5 to 15% by weight, based on the total weight of the pharmaceutical composition.
- it is preferably 1 to 10% by weight, more preferably 2 to 6% by weight in the granules for tableting.
- Excipients that can be used in the pharmaceutical composition of the present invention can be incorporated into the kneaded product, the granulated product and the later powder, such as crystalline cellulose, ethyl cellulose, hydroxypropyl cellulose, low substituted hydroxypropyl cellulose, Cellulose such as hydroxypropyl methylcellulose (hypromellose etc.), corn starch, potato starch, wheat starch, rice starch, partially pregelatinized starch, starches such as hydroxypropyl starch, glucose, lactose, sucrose, refined sucrose, powdered sugar, trehalose , Dextrin, sugars such as dextrin, (D-mannitol, xylitol, sorbitol, sugar alcohols such as erythritol, glycerin fatty acid ester, magnesium aluminometasilicate, synthetic hydrotalcite, no Calcium phosphate, precipitated calcium carbonate, calcium silicate, calcium hydrogen phosphate
- Another aspect of the present invention is a solid dispersion of the present invention or a pharmaceutical composition comprising said solid dispersion for use in the treatment of hyperuricemia or gout.
- Yet another aspect of the present invention is the use of the solid dispersion of the present invention for the manufacture of a therapeutic agent for hyperuricemia or gout.
- the dose of the solid dispersion of the present invention or the pharmaceutical composition containing the solid dispersion is the compound represented by the general formula (I) in the solid dispersion of the present invention or the pharmaceutical composition containing the solid dispersion or It can be adjusted according to the content of the pharmaceutically acceptable salt.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Optical Communication System (AREA)
- Glass Compositions (AREA)
- Inorganic Insulating Materials (AREA)
Abstract
Description
本願は、2017年11月28日に、日本に出願された特願2017-227807号に基づき優先権を主張し、その内容をここに援用する。
現在、高尿酸血症の治療には、主にアロプリノール又はフェブキソスタット等のキサンチンオキシダーゼ阻害剤が用いられている。また、同様のメカニズムを有する化合物として、国際公開第2005/121153号パンフレット(特許文献1)記載の化合物が報告されている。
例えば、非特許文献1には、化合物をナノレベルまで微粉化する方法が開示されている。特許文献2には、キサンチン誘導体をメタアクリル酸コポリマーに分散した固体分散体とする方法、特許文献3には、N4-(4-([1,2,4]トリアゾロ[1,5-a]ピリジン-7-イルオキシ)-3-メチルフェニル)-N6-(4,4-ジメチル-4,5-ジヒドロオキサゾール-2-イル)キナゾリン-4,6-ジアミン及び分散ポリマーを含む固体分散体とする方法が開示されている。
しかしながら、固体分散体とすることによって難溶性化合物の生体内における吸収性の改善を図るには、難溶性化合物を非晶質体にする必要があるが、難溶性化合物と高分子の組み合わせによっては、長期保存において、かかる難溶性化合物が非晶質体から結晶体へ転移するため、時間の経過とともに一定の溶解性が得られなくなるなどの問題がある。
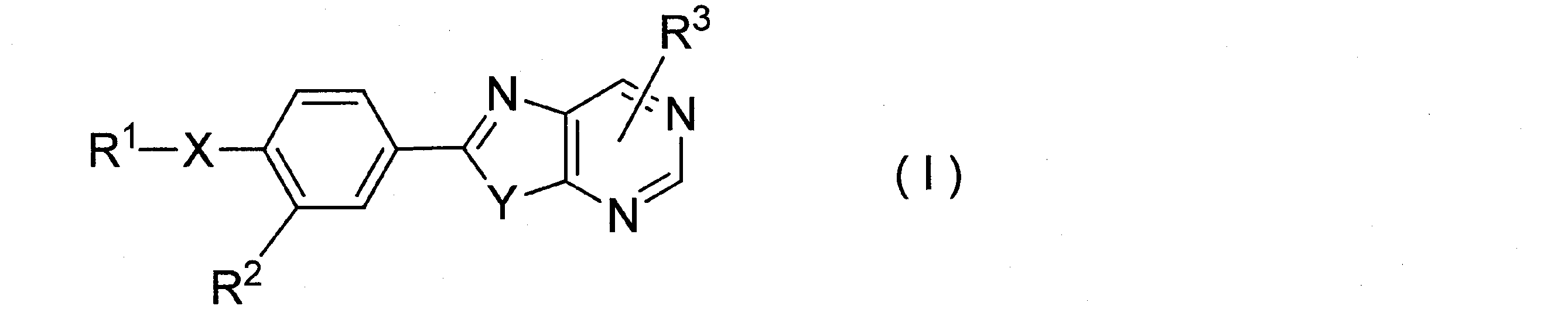
[1]一般式(I)
[2]R1が、無置換のフェニル基、又はハロゲン原子により置換されたフェニル基である[1]記載の固体分散体。
[3]Xが酸素原子である[1]又は[2]記載の固体分散体;
[4]Yが硫黄原子である[1]~[3]のいずれか一項記載の固体分散体;
[5]一般式(I)で表される化合物又はその医薬的に許容され得る塩が非晶質である[1]~[4]のいずれか一項記載の固体分散体;
[6]一般式(I)で表される化合物又はその医薬的に許容され得る塩と、ヒプロメロース誘導体との重量比が、1:0.1~1:25である[1]~[5]のいずれか一項記載の固体分散体;
[7]ヒプロメロース誘導体が、ヒプロメロース酢酸エステルコハク酸エステル又はヒプロメロースフタル酸エステルである[1]~[6]のいずれか一項記載の固体分散体;
[8][1]~[7]のいずれか一項記載の固体分散体を、スプレードライ法で製造することを特徴とする、固体分散体の製造方法;
[9][1]~[7]のいずれか一項記載の固体分散体を含有する医薬組成物;又は
[10]固形製剤である[9]記載の医薬組成物。
一般式(I)で表される化合物において、R1は無置換のフェニル基、又は置換基により置換されたフェニル基を表す。
R1で示されるフェニル基における置換基の「炭素数1~8のアルキル基」としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、ヘキシル基等が挙げられ、好ましくはメチル基、エチル基が挙げられる。

本発明で用いられるヒプロメロース誘導体としては、例えば、ヒプロメロース、ヒプロメロース酢酸エステルコハク酸エステル(HPMCASと略す場合がある)、ヒプロメロースフタル酸エステル(HPMCPと略す場合がある)等が挙げられ、好ましくはヒプロメロース酢酸エステルコハク酸エステル、ヒプロメロースフタル酸エステルである。
本発明に係るヒプロメロース酢酸エステルコハク酸エステルとしては、1モノマー単位当たりの置換割合が、メトキシ基につき20~26%、好ましくは21~25%であり、ヒドロキシプロポキシル基につき5~10%、好ましくは5~9%であり、アセチル基につき5~14%、好ましくは7~11%であり、サクシノイル基につき4~18%、好ましくは10~14%であるヒプロメロース酢酸エステルコハク酸エステルが例示される。
本発明に係るヒプロメロースフタル酸エステルとしては、1モノマー単位当たりの置換割合が、メトキシ基につき18~24%であり、ヒドロキシプロポキシ基につき5~10%、カルボキシベンゾイル基につき、21~35%であるヒプロメロースフタル酸エステルが例示される。
上記ヒプロメロース誘導体における、メトキシ基、ヒドロキシプロポキシ基、アセチル基、サクシノイル基又はカルボキシベンゾイル基等の含有量は、第17改正日本薬局方で規定されているヒプロメロース、ヒプロメロース酢酸エステルコハク酸エステル及びヒプロメロースフタル酸エステルの置換度の測定方法に準拠した方法で測定することができる。
本発明に係るヒプロメロース誘導体の粘度は、第17改正日本薬局方で規定されているヒプロメロース、ヒプロメロース酢酸エステルコハク酸エステル及びヒプロメロースフタル酸エステルの粘度の測定方法に準拠した方法で測定することができる。
本発明の1つの実施態様としては、2-(3-シアノ-4-フェノキシフェニル)-7-ヒドロキシチアゾロ[5,4-d]ピリミジン又はその医薬的に許容され得る塩と、ヒプロメロース誘導体との重量比が、1:0.1~1:25であり、別の実施態様としては、1:0.1~1:10であり、また別の実施態様としては1:0.1~1:4であり、また別の実施態様としては、1:1~1:10であり、更に別の実施態様としては、1:2~1:5であり、更にまた別の実施態様としては、1:3~1:4である固体分散体が挙げられる。
したがって、本発明の「固体分散体」の1つの側面は、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、ヒプロメロース誘導体とを含み、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体が均一に混合された系を形成している固体の組成物である。
本発明の「固体分散体」の別の側面は、一般式(I)で表される化合物又はその医薬的に許容され得る塩が、ヒプロメロース誘導体の全体にわたって分散された系を形成している固体の組成物である。この場合、一般式(I)で表される化合物又はその医薬的に許容され得る塩が分散質として分散相を構成し、ヒプロメロース誘導体が分散媒として連続相を構成している。
使用することができる界面活性剤としては、ビス-(2-エチルヘキシル)スルホコハク酸ナトリウム(ドクセートナトリウム)、臭化アルキルトリメチルアンモニウム(例えば臭化セチルトリメチルアンモニウム(セトリミド))のような陽イオン性界面活性剤、ラウリル硫酸ナトリウムのような陰イオン性界面活性剤、ポリオキシエチレンソルビタン(例えばツウィーン(TweenTM20、40、60、80又は85))、ソルビタン脂肪酸エステル(例えば、SpanTM20、40、60、80又は85)のような非イオン性界面活性剤が挙げられる。
使用することができるpH調整剤としては、コハク酸、マレイン酸、酒石酸、クエン酸、アスパラギン酸等の酸、水酸化ナトリウムや酸化マグネシウム、二酸化ケイ素、炭酸水素ナトリウム、L-アルギニン等のアルカリを用いることができる。
使用することができる糖類としては、乳糖、白糖、ブドウ糖、果糖、ショ糖、マルトース(麦芽糖)、還元麦芽糖、マルチトール、マンニトール、エリスリトール、ソルビトール、キシリトール等が挙げられる。
使用することができる可塑剤としては、クエン酸トリエチル、ポリエチレングリコール、トリアセチン等が挙げられる。
本発明の「固体分散体」は、前記医薬的に許容され得る添加剤を含んでいなくてもよいが、それらを含む場合には、例えば、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、前記界面活性剤との重量比は、1:0.01~1:2、より好ましくは1:0.02~1:1.5、更に好ましくは1:0.03~1:1.2であり、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、前記pH調整剤との重量比は、1:0.01~1:2、より好ましくは1:0.02~1:1.5、更に好ましくは1:0.03~1:1.2であり、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、前記糖類との重量比は、1:0.02~1:20、より好ましくは1:0.15~1:10であり、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、前記可塑剤との重量比は、1:0.02~1:20、より好ましくは1:0.15~1:10である。
本発明の「固体分散体」において、前記医薬的に許容され得る添加剤は、固体分散体の分散相を構成してもよく、連続相を構成してもよい。
本発明において、非晶質は、X線回折において、ハローピークを示すことにより特定することができる。
本発明において、一般式(I)で表される化合物又はその医薬的に許容され得る塩の総重量に対して、50重量%以上が非晶質として存在することが好ましく、80%重量以上が非晶質として存在することがより好ましく、90重量%以上が非晶質として存在することがより好ましく、95重量%以上が非晶質として存在することが更に好ましく、98重量%以上が非晶質として存在することが更に好ましく、100重量%が非晶質であってもよい。上記非晶質の存在率は、X線回折法によって求めることができる。
ここで、混合粉砕法とは、一般式(I)で表される化合物又はその医薬的に許容され得る塩と、ヒプロメロース誘導体と、所望により医薬的に許容され得る添加剤とを混合した後、ボールミル、ハンマーミル等の混合機及び粉砕機を用いて常法で行うことができる。
溶媒法とは、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により医薬的に許容され得る添加剤を溶媒(有機溶媒、水又はその混液)に溶解又は懸濁させ、その後、前記溶媒を除去して固体分散体を析出させるか、又は前記溶媒中、固体分散体を析出させる方法をいう。
溶媒は、噴霧法(実施態様により、流動層法、噴霧乾燥法(スプレードライ法ともいう)、転動層法、攪拌法、又は超臨界法等に分類できる)、ろ過法、エバポレーション法、凍結乾燥法などの方法により除去することができ、好ましくは噴霧法が挙げられ、そのうち噴霧乾燥法が特に好ましい。
本発明の固体分散体を製造するにあたって用いることができる溶媒は、医薬的に許容され得る溶媒が好ましく、例えば、エタノール、メタノール、2-プロパノール、アセトン、2-ブタノン、メチルイソブチルケトン、テトラヒドロフラン(THF)、テトラヒドロピラン、1,4-ジオキサン、ジエチルエーテル、トルエン、アセトニトリル、塩化メチレン、クロロホルム、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸、蟻酸、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルスルホキシド等が挙げられる。
これらの溶媒中、一般式(I)で表される化合物又はその医薬的に許容され得る塩、及び所望により配合される医薬的に許容され得る添加剤は溶解している方が好ましい。
噴霧乾燥法においては、それ自体公知の方法により固体分散体を製造することができ、例えば、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により医薬的に許容され得る添加剤を前記溶媒に加え溶液又は懸濁液とし、前記溶液又は懸濁液を回転円盤による遠心噴霧あるいは圧力ノズルによる加圧噴霧により微細な霧状にし、これを乾燥媒体中(加熱した空気又は窒素ガス)に噴出させ、粉状の乾燥物として、固体分散体を得ることができる。
噴霧乾燥法において、乾燥媒体の温度は、例えば、50~120℃、好ましくは50~90℃である。前記乾燥媒体は、一定方向に流動させていてもよく、例えば、0.2~0.6m3/min、好ましくは0.3~0.5m3/minの風量として流動させることができる。
溶媒法において析出させる方法は共沈法が好ましく、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により医薬的に許容され得る添加剤を溶媒に溶解又は懸濁させ、溶解した前記化合物(I)又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により添加される医薬的に許容され得る添加剤が不溶な溶媒の添加や、温度の低下などで溶解濃度を下げることによって析出させることにより固体分散体を得ることができる。
溶融法とは、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により医薬的に許容され得る添加剤をヒプロメロース誘導体の融点又は軟化点以上に加熱して攪拌等することにより、一般式(I)で表される化合物又はその医薬的に許容され得る塩及び所望により添加される医薬的に許容され得る添加剤を、ヒプロメロース誘導体に溶解又は分散し、ついで急冷する方法をいう。その際、所望によりクエン酸トリエチル、ポリエチレングリコール、トリアセチン等の可塑剤や界面活性剤等の添加剤を更に加えることができる。製造は、加熱装置付き攪拌造粒機を用いて行うことができる。
加熱混練溶融法とは、加熱装置を備えた押し出し機、例えば2軸エクストルーダー等により、一般式(I)で表される化合物又はその医薬的に許容され得る塩及びヒプロメロース誘導体、並びに所望により医薬的に許容され得る添加剤とを加熱・加圧下で混合して、固体分散体を得る方法をいい、得られるプラスチック様の固体分散体を粉砕機を用いて粉砕することにより固体分散体の粉末を得ることができる。
本発明は打錠障害の抑制効果をも有することから、特に固形製剤が錠剤の場合が好ましい。また、これらの固形製剤は、所望によりコーティングを施すことができる。
本発明の1つの側面は、本発明の固体分散体又は前記固体分散体を含む医薬組成物の治療的有効量を、高尿酸血症、又は痛風の治療が必要な対象に投与することを含む高尿酸血症、又は痛風の治療方法である。
本発明の別の側面は、高尿酸血症、又は痛風の治療における使用のための、本発明の固体分散体又は前記固体分散体を含む医薬組成物である。
本発明の更に別の側面は、高尿酸血症、又は痛風の治療薬の製造のための、本発明の固体分散体の使用である。
本発明の固体分散体又は前記固体分散体を含む医薬組成物の投与量は、本発明の固体分散体又は前記固体分散体を含む医薬組成物における、一般式(I)で表される化合物又はその医薬的に許容され得る塩の含有量に応じて調整することができる。また、投与方法、投与対象の年齢、体重、性別、症状、薬剤への感受性等に応じて適宜決定されるが、症状の改善の状況に応じて投与量を調節してよい。
本発明の固体分散体又は前記固体分散体を含む医薬組成物の投与量は、例えば、成人においては、一般式(I)で表される化合物又はその医薬的に許容され得る塩の含有量に換算して、通常、注射剤では1日約0.1mg~100mg,経口投与では1日1mg~2000mgとなるように投与することができるが、年齢、症状等により増減することができる。また、投与回数としては、例えば、1日当たり、1~3回、好ましくは1~2回が挙げられる。
2-(3-シアノ-4-フェノキシフェニル)-7-ヒドロキシチアゾロ[5,4-d]ピリミジン(以下、化合物Aと称する場合がある)250mgをテトラヒドロフランに溶解して100mLとした。表2に示すポリマー各125mgに混合溶液(ジクロロメタン/メタノール=50/15)5mLを添加して溶解させた。化合物A溶液2mLを試験管に入れ、上記ポリマー溶液5mLを添加し、ボルテックスミキサーで混合して均一にした。また、ポリマーを添加しない試料も調製した(比較例9)。これら試料に窒素気流を吹き付けることで溶媒を留去した後、一晩減圧乾燥して化合物Aの固体分散体試料を得た。
実施例1において使用したHPMCASはMF型(すなわち、1モノマー単位当たりの置換割合が、メトキシ基:21.0~25.0%、ヒドロキシプロポキシル基:5.0~9.0%、アセチル基:7.0~11.0%であり、サクシノイル基:10.0~14.0%、粘度:2.4~3.6mPa・s)である。

実施例1及び比較例1~8の固体分散体並びに比較例9の試料に日本薬局方溶出試験第1液(pH1.2)5mL又は第2液(pH6.8)5mLを添加し、ガラス棒又はスパーテルで固体分散体を粉砕した後、37℃で2時間振盪した。0.45μmのフィルターで濾過した試料溶液600μLに直ちに混合溶液(アセトニトリル/水=3/2)400μLを添加し、HPLC((株)島津製作所)により試料溶液中の化合物Aの濃度を測定した。結果を表3に示した。なお、表中の値は繰り返し2回の平均値である。

化合物Aをテトラヒドロフランに溶解し、2.5mg/mLに調製した。表4に示す各ポリマーを混合溶媒(メタノール/ジクロロメタン=3/4)に溶解し、約45mg/mLに調製した。化合物Aとポリマーの重量比が1:25となるように、撹拌しながら上記ポリマー溶液に化合物A溶液を加えた。また、ポリマーを添加しない試料も調製した。直ちにナスフラスコに移し、ロータリーエバポレーター(N-1100、東京理化器械(株))で有機溶剤を留去した。ナスフラスコをデシケーターに移して、真空ポンプで約16時間減圧乾燥し、化合物Aの固体分散体を得た。固体分散体は乾燥後、メノウ乳鉢で粉砕、又はポータブル高速粉砕機(LM-PLUS、大阪ケミカル(株))で粉砕後、篩過(目開き:150μm)した。

絶食下、雄性ラット(8週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例2及び比較例10の固体分散体、比較例11の試料を化合物Aとして10mg/kg、又は30mg/kgの用量で単回経口投与した。対照として、化合物Aの結晶性の原薬を使用した(比較例12)。採取時点は、投与後0.5、1、2、4、8及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=3)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の化合物Aの濃度をHPLC((株)資生堂及び(株)日立ハイテクノロジーズ)により測定した。得られた血漿中濃度推移から最高血漿中濃度到達時間(Tmax)、最高血漿中濃度(Cmax)及び血漿中濃度・時間曲線下面積(AUC)を算出した。結果を表5に示した。なお、表中の値は3例の平均値±標準偏差である。
実施例2及び比較例10の固体分散体、並びに比較例11の試料は原薬に比べ、Cmax、AUCともに高い吸収改善効果を示したことから化合物Aはアモルファス化、さらには固体分散体化することにより吸収性が向上することが示された。また、HPMCASとの固体分散体はEudragitとの固体分散体よりも高い吸収改善効果を示した。

化合物Aをテトラヒドロフランに溶解し、2.5mg/mLに調製した。表6に示す各ポリマーを混合溶媒(メタノール/ジクロロメタン=3/4)に溶解し、約45mg/mLに調製した。化合物Aとポリマーの重量比が1:25となるように混合した後、ロータリーエバポレーター(N-1100、東京理化器械(株))により減圧下、約50℃で溶媒を留去した。得られた1次乾燥物をさらに真空ポンプで2次乾燥(室温/一晩)し、2次乾燥物について適宜ポータブル高速粉砕機(LM-PLUS、大阪ケミカル(株))で粉砕後、篩過(目開き:300μm)した。
実施例3において使用したHPMCASはLG型(すなわち、1モノマー単位当たりの置換割合が、メトキシ基:20.0~24.0%、ヒドロキシプロポキシル基:5.0~9.0%、アセチル基:5.0~9.0%であり、サクシノイル基:14.0~18.0%、粘度:2.4~3.6mPa・s)である。
実施例4において使用したHPMCASはMG型(すなわち、1モノマー単位当たりの置換割合が、メトキシ基:21.0~25.0%、ヒドロキシプロポキシル基:5.0~9.0%、アセチル基:7.0~11.0%であり、サクシノイル基:10.0~14.0%、粘度:2.4~3.6mPa・s)である。
実施例5において使用したHPMCASはHG型(すなわち、1モノマー単位当たりの置換割合が、メトキシ基:22.0~26.0%、ヒドロキシプロポキシル基:6.0~10.0%、アセチル基:10.0~14.0%であり、サクシノイル基:4.0~8.0%、粘度:2.4~3.6mPa・s)である。

絶食下、雄性ラット(7-9週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例3、実施例4及び実施例5の固体分散体を化合物Aとして10mg/kgの用量で単回経口投与した。採取時点は、投与後0.5、1、2、4、8及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=3)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の化合物Aの濃度をHPLC((株)資生堂及び(株)日立ハイテクノロジーズ)により測定した。得られた血漿中濃度推移から最高血漿中濃度到達時間(Tmax)、最高血漿中濃度(Cmax)及び血漿中濃度・時間曲線下面積(AUC)を算出した。結果を表7に示した。なお、表中の値は3例の平均値±標準偏差である。
実施例4の固体分散体において、Cmax、AUCともに最も高い血漿中濃度を示した。

化合物Aをテトラヒドロフランに溶解し、2.5mg/mLに調製した。HPMCAS-MGを混合溶媒(エタノール/水=4/1)に溶解し、約45mg/mLに調製した。化合物Aと表8に示すポリマーの重量比が1:1~1:10となるように混合した後、ペリスタポンプ経由で約5mL/minの速度でスプレードライヤー(GB22、ヤマト科学(株))に汲み上げ、2流体ノズル(直径406又は508μm)から入口温度80℃、出口温度約60℃、乾燥空気風量0.32から0.47m3/min、ノズル噴霧空気圧力1.0から3.1kgf/cm2の条件で噴霧乾燥及び造粒を開始した。得られた1次乾燥物をさらに真空ポンプで2次乾燥(室温/一晩又は室温/一晩・40℃/1日)し、篩過(目開き:300μm)した。

絶食下、雄性ラット(7-9週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例6~8、10~12の固体分散体を化合物Aとして10mg/kgの用量で単回経口投与した。採取時点は、投与後0.5、1、2、4、8及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=3)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の化合物Aの濃度をHPLC((株)資生堂及び(株)日立ハイテクノロジーズ)により測定した。得られた血漿中濃度推移から最高血漿中濃度到達時間(Tmax)、最高血漿中濃度(Cmax)及び血漿中濃度・時間曲線下面積(AUC)を算出した。結果を表9に示した。なお、表中の値は3例の平均値±標準偏差である。
実施例6~8、10~12の固体分散体において、化合物Aに対するHPMCAS-MGの重量比が1、2及び3倍では血漿中濃度は重量比に依存して上昇したが、3、4、5及び10倍の範囲では血漿中濃度に重量比に依存した相違はみられなかった。

固体分散体が経日保管後においても非晶質の特性を保持していることは非常に重要であるため、実施例9~11の固体分散体を40℃/75%RH、開放条件下で保存し、X線回折装置(D2 Phaser、Bruker)を用いて非晶質状態の変化を評価した。結果を図1~3に示した。
実施例9~11の固体分散体を40℃/75%RH、開放条件で保存した場合、検討した7週間までの保存において、経日的な粉末X線回折パターンの変化はみられなかった。
実施例9~11の固体分散体を40℃/75%RH、開放条件下で保存し、非晶質状態の変化をラット吸収性により評価した。
絶食下、雄性ラット(7-9週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例9~11の固体分散体を化合物Aとして10、30mg/kgの用量で単回経口投与した。採取時点は、投与後0.5、1、2、4、8及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=3)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の化合物Aの濃度をHPLC((株)資生堂及び(株)日立ハイテクノロジーズ)により測定した。得られた血漿中濃度推移から最高血漿中濃度到達時間(Tmax)、最高血漿中濃度(Cmax)及び血漿中濃度・時間曲線下面積(AUC)を算出した。結果を表10に示した。なお、表中の値は3例の平均値±標準偏差である。
実施例9~11の固体分散体を40℃/75%RHで開放保存した場合、検討した7週間までの保存において、経日的な血漿中濃度の低下は見られなかった。

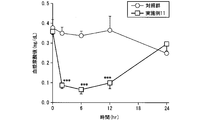
絶食下、雄性ラット(8週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例11の固体分散体を化合物Aとして30mg/kgの用量で単回経口投与した。対照として、1%カルボキシメチルセルロース水溶液を使用した。採取時点は、投与前(0)、投与後2、6、12及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=5)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の尿酸の濃度をHPLC((株)日立ハイテクノロジーズ)により測定した。対照群と実施例11の各時点の血漿尿酸値について、ウェルチのt検定を行った。有意水準はp<0.05(両側)とした。結果を図4に示した。
実施例11の固体分散体を投与後の血漿尿酸値は対照群と比べて有意な低値を示し、その尿酸値低下作用は持続的であった。
化合物Aをテトラヒドロフランに溶解し、2.5mg/mLに調製した。HPMCAS-MGを混合溶媒(エタノール/水=4/1)に溶解し、約45mg/mLに調製した。化合物Aと表11に示すポリマー、界面活性剤(ポリソルベート80:Tween 80、ラウリル硫酸ナトリウム:SLS)の重量比が1:3:0.03となるように混合した後、ペリスタポンプ経由で約5mL/minの速度でスプレードライヤー(GB22、ヤマト科学(株))に汲み上げ、2流体ノズル(直径406又は508μm)から入口温度80℃、出口温度約60℃、乾燥空気風量0.32から0.47m3/min、ノズル噴霧空気圧力1.0から3.1kgf/cm2の条件で噴霧乾燥及び造粒を開始した。得られた1次乾燥物をさらに真空ポンプで2次乾燥(室温/一晩又は室温/一晩・40℃/1日)し、篩過(目開き:300μm)した。

絶食下、雄性ラット(7-9週齢、Crl:CD(SD)、日本チャールス・リバー(株))に、1%カルボキシメチルセルロース水溶液に懸濁した実施例13及び14の固体分散体を化合物Aとして10mg/kgの用量で単回経口投与した。採取時点は、投与後0.5、1、2、4、8及び24時間とし、尾静脈から1時点あたり約300μL採血した(n=3)。得られた血液を1500×g、4℃で15分間遠心分離し、血漿を得た。この血漿中の化合物Aの濃度をHPLC((株)資生堂及び(株)日立ハイテクノロジーズ)により測定した。得られた血漿中濃度推移から最高血漿中濃度到達時間(Tmax)、最高血漿中濃度(Cmax)及び血漿中濃度・時間曲線下面積(AUC)を算出した。実施例8とともに結果を表12に示した。なお、表中の値は3例の平均値±標準偏差である。
実施例13及び14の固体分散体において、界面活性剤を含まない実施例8の血漿中濃度と相違はみられなかった。

実施例5で得られる固体分散体6.0gに、常法によりカルボキシメチルスターチナトリウム2.8g、乳糖3.01g、含水二酸化珪素2.1g、ステアリン酸マグネシウム0.1gを添加し、ビニール袋中にて混合し、ロータリー式打錠機(VELA-5、(株)菊水製作所)にて打錠して錠剤を製造する。
<製剤例2>
実施例5で得られる固体分散体2gに、ステアリン酸マグネシウム0.3g及び乳糖適量を加えて全量23.5gとし、ビニール袋中にて混合し、1号カプセルに手充填してカプセル剤を製造する。
<製剤例3>
実施例5で得られる固体分散体1gに乳糖16.0gを添加し、ビニール袋中にて攪拌混合し、ローラーコンパクター(TF-MINI、フロイント産業(株))にて乾式造粒し、オシレーター(34-C-2、(株)菊水製作所)にて整粒して顆粒剤を製造する。
Claims (10)
- 一般式(I)
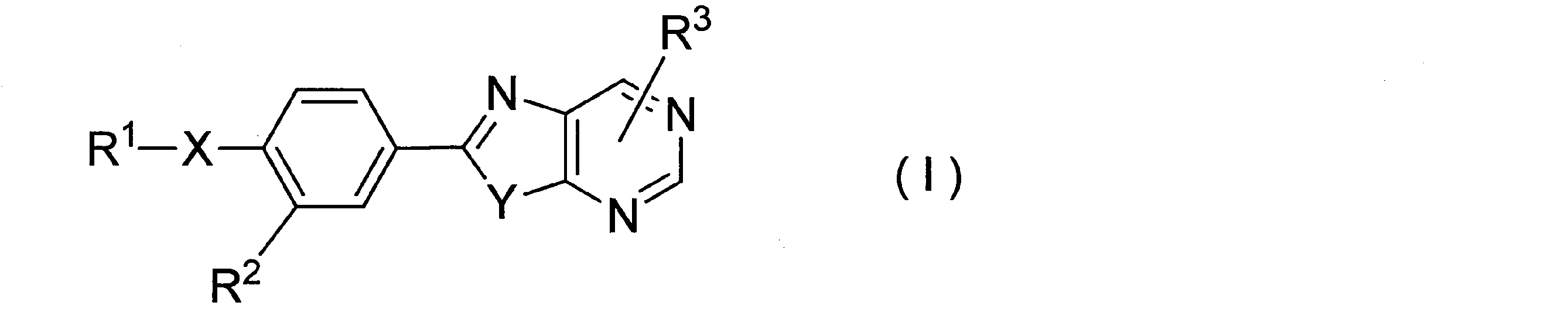
- R1が、無置換のフェニル基、又はハロゲン原子により置換されたフェニル基である請求項1記載の固体分散体。
- Xが酸素原子である請求項1又は2記載の固体分散体。
- Yが硫黄原子である請求項1~3のいずれか一項記載の固体分散体。
- 一般式(I)で表される化合物又はその医薬的に許容され得る塩が非晶質である請求項1~4のいずれか一項記載の固体分散体。
- 一般式(I)で表される化合物又はその医薬的に許容され得る塩と、ヒプロメロース誘導体との重量比が、1:0.1~1:25である請求項1~5のいずれか一項記載の固体分散体。
- ヒプロメロース誘導体が、ヒプロメロース酢酸エステルコハク酸エステル又はヒプロメロースフタル酸エステルである請求項1~6のいずれか一項記載の固体分散体。
- 請求項1~7のいずれか一項記載の固体分散体を、スプレードライ法で製造することを特徴とする、固体分散体の製造方法。
- 請求項1~7のいずれか一項記載の固体分散体を含有する医薬組成物。
- 固形製剤である請求項9記載の医薬組成物。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/765,575 US11234984B2 (en) | 2017-11-28 | 2018-11-28 | Solid dispersion |
| CN202311189665.7A CN117257807A (zh) | 2017-11-28 | 2018-11-28 | 固体分散体 |
| KR1020207014619A KR102728350B1 (ko) | 2017-11-28 | 2018-11-28 | 고체 분산체 |
| ES18882399T ES2929730T3 (es) | 2017-11-28 | 2018-11-28 | Dispersión sólida |
| EP18882399.1A EP3718548B1 (en) | 2017-11-28 | 2018-11-28 | Solid dispersion |
| CN201880076272.6A CN111405900B (zh) | 2017-11-28 | 2018-11-28 | 固体分散体 |
| JP2019557268A JP7217890B2 (ja) | 2017-11-28 | 2018-11-28 | 固体分散体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017227807 | 2017-11-28 | ||
| JP2017-227807 | 2017-11-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019107412A1 true WO2019107412A1 (ja) | 2019-06-06 |
Family
ID=66664040
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/043788 Ceased WO2019107412A1 (ja) | 2017-11-28 | 2018-11-28 | 固体分散体 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US11234984B2 (ja) |
| EP (1) | EP3718548B1 (ja) |
| JP (1) | JP7217890B2 (ja) |
| KR (1) | KR102728350B1 (ja) |
| CN (2) | CN117257807A (ja) |
| ES (1) | ES2929730T3 (ja) |
| TW (1) | TWI780270B (ja) |
| WO (1) | WO2019107412A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020246528A1 (ja) * | 2019-06-04 | 2020-12-10 | 日本ケミファ株式会社 | 痛風又は高尿酸血症の治療薬 |
| WO2020246526A1 (ja) * | 2019-06-04 | 2020-12-10 | 日本ケミファ株式会社 | キサンチンオキシダーゼ阻害剤含有腸溶性製剤 |
| WO2022124325A1 (ja) * | 2020-12-08 | 2022-06-16 | 国立大学法人東京大学 | 細胞内atp増強剤 |
| WO2024177158A1 (ja) | 2023-02-24 | 2024-08-29 | 日本ケミファ株式会社 | P2x4受容体拮抗作用を有する化合物の固体分散体 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5944514B2 (ja) | 1974-09-02 | 1984-10-30 | 北越工業 (株) | 液体処理による液冷式回転圧縮機の運転動力軽減方法 |
| WO2005121153A1 (ja) | 2004-06-14 | 2005-12-22 | Nippon Chemiphar Co., Ltd. | 縮合ピリミジン誘導体、及びキサンチンオキシダーゼ阻害剤 |
| JP3938938B2 (ja) | 1995-07-26 | 2007-06-27 | 協和醗酵工業株式会社 | キサンチン誘導体の固体分散体または固体分散体製剤 |
| WO2016136727A1 (ja) * | 2015-02-24 | 2016-09-01 | 国立大学法人鳥取大学 | 認知症の予防及び/又は治療のための医薬 |
| JP2017227807A (ja) | 2016-06-23 | 2017-12-28 | 株式会社フジクラ | 光レセプタクル |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5944514U (ja) | 1982-09-14 | 1984-03-24 | 日立化成工業株式会社 | 移動床濾過装置 |
| JPH07324086A (ja) * | 1994-05-31 | 1995-12-12 | Sankyo Co Ltd | チアゾリジン誘導体の固体分散体または固体分散体製剤 |
| GB0317615D0 (en) * | 2003-07-28 | 2003-08-27 | Stannah Stairlifts Ltd | Improvements in or relating to stairlifts |
| KR101191071B1 (ko) * | 2004-06-14 | 2012-10-15 | 닛뽕 케미파 가부시키가이샤 | 축합 피리미딘 유도체 및 크산틴 옥시다제 저해제 |
| KR20140037954A (ko) * | 2006-06-22 | 2014-03-27 | 닛뽕 케미파 가부시키가이샤 | 항암제 내성 극복제 |
| US20090264476A1 (en) * | 2008-04-18 | 2009-10-22 | Mckelvey Craig | CB-1 receptor modulator formulations |
| WO2013056108A2 (en) * | 2011-10-14 | 2013-04-18 | Array Biopharma Inc. | Solid dispersion |
| EP2792360A1 (en) * | 2013-04-18 | 2014-10-22 | IP Gesellschaft für Management mbH | (1aR,12bS)-8-cyclohexyl-11-fluoro-N-((1-methylcyclopropyl)sulfonyl)-1a-((3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl)-1,1a,2,2b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide for use in treating HCV |
| US20140363504A1 (en) * | 2013-06-11 | 2014-12-11 | The Beauty Nation Ptd Ltd | Absorbable encapsulated calcium in hypromellose capsule |
| PE20181521A1 (es) * | 2015-12-08 | 2018-09-24 | Ardea Biosciences Inc | Composicion farmaceutica que comprende un potente inhibidor de urat1 |
-
2018
- 2018-11-27 TW TW107142281A patent/TWI780270B/zh active
- 2018-11-28 CN CN202311189665.7A patent/CN117257807A/zh active Pending
- 2018-11-28 ES ES18882399T patent/ES2929730T3/es active Active
- 2018-11-28 CN CN201880076272.6A patent/CN111405900B/zh active Active
- 2018-11-28 US US16/765,575 patent/US11234984B2/en active Active
- 2018-11-28 KR KR1020207014619A patent/KR102728350B1/ko active Active
- 2018-11-28 WO PCT/JP2018/043788 patent/WO2019107412A1/ja not_active Ceased
- 2018-11-28 JP JP2019557268A patent/JP7217890B2/ja active Active
- 2018-11-28 EP EP18882399.1A patent/EP3718548B1/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5944514B2 (ja) | 1974-09-02 | 1984-10-30 | 北越工業 (株) | 液体処理による液冷式回転圧縮機の運転動力軽減方法 |
| JP3938938B2 (ja) | 1995-07-26 | 2007-06-27 | 協和醗酵工業株式会社 | キサンチン誘導体の固体分散体または固体分散体製剤 |
| WO2005121153A1 (ja) | 2004-06-14 | 2005-12-22 | Nippon Chemiphar Co., Ltd. | 縮合ピリミジン誘導体、及びキサンチンオキシダーゼ阻害剤 |
| WO2016136727A1 (ja) * | 2015-02-24 | 2016-09-01 | 国立大学法人鳥取大学 | 認知症の予防及び/又は治療のための医薬 |
| JP2017227807A (ja) | 2016-06-23 | 2017-12-28 | 株式会社フジクラ | 光レセプタクル |
Non-Patent Citations (6)
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020246528A1 (ja) * | 2019-06-04 | 2020-12-10 | 日本ケミファ株式会社 | 痛風又は高尿酸血症の治療薬 |
| JPWO2020246528A1 (ja) * | 2019-06-04 | 2020-12-10 | ||
| WO2020246526A1 (ja) * | 2019-06-04 | 2020-12-10 | 日本ケミファ株式会社 | キサンチンオキシダーゼ阻害剤含有腸溶性製剤 |
| JPWO2020246526A1 (ja) * | 2019-06-04 | 2021-09-13 | 日本ケミファ株式会社 | キサンチンオキシダーゼ阻害剤含有腸溶性製剤 |
| EP3981470A4 (en) * | 2019-06-04 | 2023-07-19 | Nippon Chemiphar Co., Ltd. | ENTERIC COATED FORMULATION COMPRISING A XANTHINE OXIDASE INHIBITOR |
| AU2020287549B2 (en) * | 2019-06-04 | 2026-01-29 | Nippon Chemiphar Co., Ltd. | Therapeutic for gout or hyperuricemia |
| WO2022124325A1 (ja) * | 2020-12-08 | 2022-06-16 | 国立大学法人東京大学 | 細胞内atp増強剤 |
| JPWO2022124325A1 (ja) * | 2020-12-08 | 2022-06-16 | ||
| WO2024177158A1 (ja) | 2023-02-24 | 2024-08-29 | 日本ケミファ株式会社 | P2x4受容体拮抗作用を有する化合物の固体分散体 |
| KR20250167594A (ko) | 2023-02-24 | 2025-12-01 | 닛뽕 케미파 가부시키가이샤 | P2x4 수용체 길항 작용을 갖는 화합물의 고체 분산체 |
| EP4670720A1 (en) | 2023-02-24 | 2025-12-31 | Nippon Chemiphar Co., Ltd. | SOLID DISPERSION OF A COMPOUND HAVING P2X4 RECEPTOR ANTAGONISM |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2019107412A1 (ja) | 2020-11-19 |
| JP7217890B2 (ja) | 2023-02-06 |
| US11234984B2 (en) | 2022-02-01 |
| TW201924688A (zh) | 2019-07-01 |
| EP3718548A1 (en) | 2020-10-07 |
| ES2929730T3 (es) | 2022-12-01 |
| CN111405900B (zh) | 2023-10-10 |
| CN111405900A (zh) | 2020-07-10 |
| EP3718548A4 (en) | 2021-09-01 |
| EP3718548B1 (en) | 2022-10-26 |
| CN117257807A (zh) | 2023-12-22 |
| US20200306251A1 (en) | 2020-10-01 |
| TWI780270B (zh) | 2022-10-11 |
| KR20200092956A (ko) | 2020-08-04 |
| KR102728350B1 (ko) | 2024-11-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5479912B2 (ja) | 複素環式化合物の非晶質体、それを含む固体分散体、薬剤、およびその製造法 | |
| TWI564008B (zh) | 難溶性藥物之溶解性改善製劑 | |
| RU2466717C2 (ru) | Фармацевтический твердый препарат, содержащий бензоазепины, и способ его получения | |
| JP7217890B2 (ja) | 固体分散体 | |
| TWI389915B (zh) | 肝適能之穩定化固態分散及其製備方法 | |
| JP2020518611A (ja) | 水溶解度及びバイオアベイラビリティが改善された組成物 | |
| JP2025061775A (ja) | 痛風又は高尿酸血症の治療薬 | |
| JP6989064B1 (ja) | 固形製剤及びその製造方法 | |
| CN116528840A (zh) | 细胞内atp增强剂 | |
| HK40098529A (zh) | 固体分散体 | |
| JP6903252B2 (ja) | キサンチンオキシダーゼ阻害剤含有腸溶性製剤 | |
| WO2020122244A1 (ja) | 錠剤及びその製造方法 | |
| HK40027115A (en) | Solid dispersion | |
| HK40027115B (zh) | 固体分散体 | |
| HK40073463B (en) | Therapeutic for gout or hyperuricemia | |
| HK40073463A (en) | Therapeutic for gout or hyperuricemia | |
| JP2026067844A (ja) | アパルタミド含有医薬組成物 | |
| HK40000065A (en) | Oral preparation having exceptional elutability |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18882399 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019557268 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20207014619 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018882399 Country of ref document: EP Effective date: 20200629 |