WO2019131270A1 - 活性炭およびそれを用いた金属担持活性炭、並びに水素化反応触媒 - Google Patents
活性炭およびそれを用いた金属担持活性炭、並びに水素化反応触媒 Download PDFInfo
- Publication number
- WO2019131270A1 WO2019131270A1 PCT/JP2018/046301 JP2018046301W WO2019131270A1 WO 2019131270 A1 WO2019131270 A1 WO 2019131270A1 JP 2018046301 W JP2018046301 W JP 2018046301W WO 2019131270 A1 WO2019131270 A1 WO 2019131270A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- activated carbon
- metal
- supported
- present
- palladium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
- B01J37/0207—Pretreatment of the support
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/18—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/33—Electric or magnetic properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/318—Preparation characterised by the starting materials
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/336—Preparation characterised by gaseous activating agents
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/342—Preparation characterised by non-gaseous activating agents
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/354—After-treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/16—Pore diameter
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/90—Other properties not specified above
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/30—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds
- C07C209/32—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups
- C07C209/36—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups by reduction of nitro groups bound to carbon atoms of six-membered aromatic rings in presence of hydrogen-containing gases and a catalyst
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/43—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C211/44—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to only one six-membered aromatic ring
- C07C211/45—Monoamines
- C07C211/46—Aniline
Definitions
- the present invention relates to activated carbon for supporting a catalyst and metal-supported activated carbon using the same.
- the catalytic hydrogenation reaction using a heterogeneous catalyst is one of the important processes of the chemical industry and is widely used in industry.
- support carbon materials, such as activated carbon is widely utilized by industrial processes, such as a hydrogenation reaction and a dehydrogenation reaction.
- the carbon material used for the catalyst support is often subjected to an oxidation treatment in order to enhance the reaction efficiency such as the hydrogenation reaction by atomization of the supported metal catalyst.
- an oxidation treatment in order to enhance the reaction efficiency such as the hydrogenation reaction by atomization of the supported metal catalyst.
- the surface functional group is formed by performing heat treatment (oxidation treatment) of activated carbon in air at 300 to 500 ° C. in advance and then performing ion exchange, and the supported catalyst (metal) is micronized. It is stated that it carries out and raises the catalytic reaction efficiency.
- the activated carbon according to one aspect of the present invention is characterized in that the conductivity obtained by powder resistance measurement under a load of 12 kN is 3.5 S / cm or more, and the oxygen content is 3.0 mass% or more. Do.
- the present invention has been made in view of the above-mentioned problems, and it is possible to improve the catalyst performance without increasing the specific surface area while the fineness of the metal is equivalent to the conventional one, and the activated carbon for catalyst support And it aims at providing a metal support activated carbon using it.
- the activated carbon of the present embodiment is characterized in that the conductivity obtained by powder resistance measurement under a load of 12 kN is 3.5 S / cm or more, and the oxygen content is 3.0 mass% or more.
- the activated carbon of the present embodiment can exhibit very excellent catalytic performance by the above configuration.
- the catalyst performance can be improved without increasing the specific surface area, while the fineness of the metal is comparable to the conventional one. That is, according to the present invention, it is possible to provide a catalyst-supporting activated carbon having excellent catalytic performance while suppressing the cost.
- the conductivity of the activated carbon of the present embodiment is 3.5 S / cm or more as described above. By setting the conductivity within the above range, it is presumed that the interaction between the metal catalyst described later and the activated carbon changes, and the catalyst performance is improved.
- the more preferable conductivity is 5.0 S / cm or more, and more preferably 6.0 S / cm or more.
- the upper limit of the conductivity is not particularly limited, but if it is too large, it takes time for activation treatment due to excessive development of the carbon structure, which is economically unpreferable. In addition, if the conductivity is too high, the oxidation treatment may be difficult. In that case, the degree of dispersion of the metal catalyst is lowered, which is not preferable. Therefore, the conductivity of the activated carbon of the present embodiment is preferably 15 S / cm or less, more preferably 13 S / cm or less, and still more preferably 10 S / cm or less.
- conductivity means conductivity obtained by powder resistance measurement under a load of 12 kN in activated carbon ground to have a predetermined particle size (particle size distribution). Since the particle size of the measurement sample greatly affects the measurement of the conductivity, it can be specifically measured by the measurement method described in the examples described later.
- the oxygen content of the activated carbon of the present embodiment is 3.0% by mass or more.
- the oxygen content is 3.0% by mass or more, the degree of dispersion of the metal catalyst is sufficiently obtained (fineization of the metal catalyst becomes sufficient), and the reaction efficiency of the hydrogenation reaction is improved.
- a more preferable oxygen content is 4.0 mass% or more, and more preferably 5.0 mass% or more.
- the upper limit of the oxygen content in the activated carbon of the present embodiment is not particularly limited, but if it is too large, carbon consumption increases and the yield of activated carbon decreases, which is not preferable from an economic viewpoint. Further, when the oxygen content is too large, the hardness of the activated carbon is lowered, and the metal supported is separated by pulverization, which is also not preferable in that respect. Therefore, the oxygen content of the activated carbon of the present invention is preferably 20.0% by mass or less, more preferably 15.0% by mass or less, and still more preferably 10.0% by mass or less.
- the "oxygen content” means the amount of oxygen in the pulverized and dried activated carbon, and specifically, it can be measured by the measurement method described in the examples described later.
- the BET specific surface area of the activated carbon of the present embodiment is not particularly limited, but is preferably 1000 m 2 / g or more, more preferably 1300 m 2 / g or more, and further preferably 1600 m 2 / g or more. preferable.
- the BET specific surface area is 1000 m 2 / g or more, it is considered that the adsorption of the reaction compound is sufficiently obtained, and the reaction efficiency of the hydrogenation reaction is improved.
- the upper limit of the BET specific surface area is not particularly limited, but if the amount is too large, the yield of the activated carbon obtained is lowered, which is not preferable from the economic viewpoint. Therefore, the BET specific surface area of the activated carbon of the present invention is preferably 2200 m 2 / g or less, more preferably 2100 m 2 / g or less, and still more preferably 2000 m 2 / g or less.
- the specific surface area refers to the BET specific surface area calculated by the nitrogen adsorption method.
- the measurement method of this specific surface area can be measured by a known method, and for example, a method of measuring from a nitrogen adsorption isotherm and calculating from the obtained adsorption isotherm can be mentioned. More specifically, it can be measured by the method described in the examples.
- the micropore average pore diameter of the activated carbon of the present embodiment is not particularly limited, but is preferably 1.55 nm or more, more preferably 1.60 nm or more, and still more preferably 1.65 nm or more. If the micropore average pore diameter is 1.55 nm or more, the diffusion of the reaction compound in the catalyst particles is improved, and it is considered that the hydrogenation reaction efficiency is improved.
- the upper limit of the micropore average pore diameter is not particularly limited, but if it becomes too large, the yield of the activated carbon obtained decreases, which is not preferable from an economic viewpoint. Therefore, the micropore average pore diameter of the activated carbon of the present invention is preferably 1.90 nm or less, more preferably 1.85 nm or less, and still more preferably 1.80 nm or less.
- the micropore average pore size is calculated by a nitrogen adsorption method and can be measured by a known method.
- a specific measurement method for example, a method of measuring a nitrogen adsorption isotherm and calculating from the obtained adsorption isotherm may be mentioned. More specifically, it can be measured by the method described in the examples.
- the shape of the activated carbon is not particularly limited, but is preferably any of granular, powdery, fibrous, pelleted and spherical.
- the shape of the activated carbon can be appropriately selected depending on the application, but is usually in the form of granular or powder, and in particular, it is preferable to be in the form of powder having high loading capacity per volume.
- a jaw crusher for example, a jaw crusher, a hammer mill, a pin mill, a roller mill, a rod mill, a ball mill, a jet mill and the like can be used.
- the size of the activated carbon is preferably about 150 ⁇ m to 5 mm in average particle diameter (D50) in the case of granular or the like, and in the case of powder or the like, the average particle diameter (D50) is in the range of 1 to It is preferably about 100 ⁇ m.
- the numerical value of D50 is a value measured by the laser diffraction measurement method in the same manner as in the examples described later, and for example, the line is measured by a wet particle size distribution measuring apparatus It will be.
- the activated carbon of the present embodiment as described above dry-distills a carbonaceous material, heat-treats the obtained dry-distilled product at a temperature of 1100 ° C. or higher, and then is activated in a mixed gas atmosphere containing water vapor, nitrogen and carbon dioxide Then, it can be obtained by oxidation treatment in an oxidative atmosphere. That is, the present invention also encompasses a method for producing activated carbon including at least a step of heat-treating a dry-cut product of a carbonaceous material at a temperature of 1100 ° C. or higher, an activation treatment step, and an oxidation treatment step.
- the carbonaceous material can be selected from all known materials, for example, plants (eg coconut shell, rattan, sweet potato, wood, etc.), natural polymers (starch, cellulose, lignin etc.), semi-synthetic polymers (cellulose) Examples thereof include esters, cellulose ethers, lignin resins, etc., synthetic polymers (phenol resins, furan resins, epoxy resins, etc.), natural minerals, etc. These raw materials can be used alone or in combination of two or more. Preferred raw materials are plant raw materials such as wood, and more preferred are coconut shells low in impurities.
- coconut husks obtained from these palms may be used alone or in combination of two or more.
- coconut husks derived from coconut or palm palm which are biomass wastes which are used as food, detergent raw materials, biodiesel raw materials, etc. and are produced in large quantities are particularly preferable because they are easily available and inexpensive.
- char generally refers to a powdery solid rich in carbon content that is generated without melting and softening when heating coal, but here, carbon content that is generated without heating and melting the organic matter. It also refers to a powdery solid rich in
- the method for producing char from coconut shell is not particularly limited, and can be produced using methods known in the art.
- coconut husks serving as a raw material are, for example, inert gases such as nitrogen, carbon dioxide, helium, argon, carbon monoxide or fuel exhaust gas, mixed gases of these inert gases, or others mainly comprising these inert gases
- inert gases such as nitrogen, carbon dioxide, helium, argon, carbon monoxide or fuel exhaust gas
- mixed gases of these inert gases or others mainly comprising these inert gases
- the heat treatment of the dry distillate can be carried out by heating the dry distillate at a temperature of 1100 ° C. or higher, preferably at a temperature of 1200 ° C. or higher, while blocking oxygen or air.
- the heat treatment temperature is too low, the conductivity of the activated carbon is lowered, the interaction between the metal catalyst and the activated carbon becomes insufficient, and the catalyst performance is lowered.
- the higher the heat treatment temperature the higher the conductivity of the activated carbon, but the longer the activation time for obtaining a sufficient specific surface area is not preferable because the production cost increases. Therefore, the upper limit of the heat treatment temperature is preferably 1500 ° C. or less.
- the means for heating is not particularly limited, but can be performed using, for example, an electric furnace or the like.
- the activation treatment can be carried out by a general method in the technical field to which the present invention belongs, and mainly includes two kinds of treatment methods, gas activation treatment and drug activation treatment.
- a method of heating an activated carbon precursor in the presence of water vapor, carbon dioxide, air, oxygen, combustion gas, or a mixed gas thereof is known.
- an activator such as zinc chloride, calcium chloride, phosphoric acid, sulfuric acid, sodium hydroxide, potassium hydroxide, magnesium hydroxide, calcium hydroxide and the like are mixed with the activated carbon precursor and inactivated. It is known to heat in a gas atmosphere. In the present embodiment, it is preferable to use a gas activation treatment, since the drug activation requires a step of removing the remaining drug, and the manufacturing method becomes complicated.
- the gas activation treatment is carried out in a mixed atmosphere of water vapor, nitrogen and carbon dioxide at a temperature of 850 ° C. or higher, preferably 850 to 1000 ° C. (eg, 850 to 950 ° C.) using a fluidized bed, multistage furnace, rotary furnace or the like. Can be done with By activating the mixture under the atmosphere, the dry distillate is partially gasified to obtain activated carbon.
- the gas for gasifying a part of the dry matter of the carbonaceous material (a mixed gas consisting of steam, nitrogen and carbon dioxide) burns natural gas, petroleum or other combustibles including hydrocarbon. It can also be obtained by
- the activation temperature usually fluctuates in the range of about ⁇ 25 ° C. in many cases.
- the activation time is not particularly limited, but may be about 0.5 to 48 hours, preferably 1 to 24 hours, and more preferably 2 to 20 hours (eg, 6 to 12 hours). If the activation time is too short, a sufficient specific surface area can not be obtained, the catalyst performance after supporting the metal decreases, and if too long, the productivity may decrease.
- the partial pressure of gas is not particularly limited either, but it is 7.5 to 40%, preferably 10 to 30% (eg, 10 to 20%) partial pressure of steam, 10 to 50% of carbon dioxide partial pressure, preferably 15 to 45% (For example, 20 to 40%), nitrogen partial pressure 30 to 80%, preferably 40 to 70% (for example, 45 to 65%), gas partial pressure is 10 to 40% water vapor partial pressure, carbon dioxide
- the partial pressure may be about 10 to 40% and the nitrogen partial pressure about 40 to 80%.
- the total pressure of the gas is usually 1 atm (about 0.1 MPa).
- the total gas supply amount (flow rate) is not particularly limited, but it is about 1 to 50 L / min, preferably about 1 to 25 L / min, with respect to 100 g of the dry distillate raw material.
- the manufacturing process of the activated carbon of the present embodiment may include an acid washing process.
- the acid washing step is a step for removing impurities such as metal components contained in the activated carbon by washing the activated carbon after the activation treatment with a washing solution containing an acid.
- the acid cleaning can be performed, for example, by immersing the raw material activated carbon in a cleaning solution containing an acid.
- the raw activated carbon may be washed with hydrochloric acid and then washed with water, or the water washing and the acid washing may be appropriately combined, such as repeating acid washing and water washing.
- the acid cleaning solution examples include inorganic acids such as hydrochloric acid, sulfuric acid and nitric acid, and organic acids such as formic acid, acetic acid, propionic acid, oxalic acid and saturated carboxylic acids such as tartaric acid and citric acid, and aromatic carboxylic acids such as benzoic acid and terephthalic acid. It is preferred to use an acid, among which washing with hydrochloric acid is more preferred.
- the concentration of hydrochloric acid is preferably 0.1 to 3.0% by mass, and more preferably 0.3 to 1.0% by mass. If the concentration of hydrochloric acid is too low, it is necessary to increase the number of times of pickling to remove impurities. Conversely, if it is too high, the amount of residual hydrochloric acid will increase. The process can be performed, which is preferable from the viewpoint of productivity.
- the liquid temperature at the time of pickling and washing with water is not particularly limited, but it is preferably 0 to 98 ° C., more preferably 10 to 95 ° C., and still more preferably 15 to 90 ° C. If the temperature of the cleaning solution at the time of immersing the raw material activated carbon is within the above-mentioned range, it is desirable because it is possible to perform the cleaning with the load on the apparatus suppressed for a practical time.
- the method for producing activated carbon of the present embodiment includes an oxidation treatment step.
- the oxidation treatment step is a step of increasing the oxygen content of the activated carbon by oxidizing the activated carbon in an oxidizing atmosphere.
- oxidizing agents such as a hydrogen-peroxide solution, nitric acid, potassium permanganate
- Oxidation treatment using oxygen can be performed using a fluidized bed, multistage furnace, rotary furnace or the like similar to activation treatment, and can be performed at a temperature of 400 ° C. or higher, preferably 400 to 600 ° C. . If the oxidation treatment temperature is less than 400 ° C., the oxidation of the activated carbon does not proceed sufficiently, and the oxidation of the activated carbon proceeds rapidly at 600 ° C. or more, carbon consumption becomes intense and the yield decreases, which is not preferable.
- the oxidation treatment time is not particularly limited, but may be about 0.1 to 3 hours, preferably 0.2 to 2 hours, and more preferably 0.3 to 1 hour.
- the oxidation treatment time is too short, the oxidation of the activated carbon does not proceed sufficiently, and when it is too long, productivity decreases.
- the gas partial pressure is not particularly limited, but may be about 1 to 15% oxygen partial pressure, 5 to 15% steam partial pressure, 5 to 15% carbon dioxide partial pressure, and about 50 to 80% nitrogen partial pressure.
- the total pressure of the gas is usually 1 atm (about 0.1 MPa).
- the total gas supply amount (flow rate) is not particularly limited, but it is about 1 to 100 L / min, preferably about 1 to 50 L / min, with respect to 50 g of the activated material raw material.
- Metal-supported activated carbon The metal-supported activated carbon of the present embodiment is characterized in that the metal serving as a catalyst is supported on the activated carbon as described above.
- the metal to be supported is not particularly limited, but includes metals used as catalysts for hydrogenation reaction and dehydrogenation reaction. Specific examples thereof include palladium, platinum, ruthenium, rhodium, osmium, iridium, nickel, cobalt, rhenium, vanadium, tungsten, molybdenum, iron, titanium and the like, and platinum group elements (palladium, platinum, ruthenium, rhodium) (Osmium, iridium), nickel and iron are more preferable, and palladium or platinum is preferable among them. These can be used alone or in combination of two or more.
- the amount of metal supported in the metal-supported activated carbon of the present embodiment is not particularly limited, but is preferably 0.1 to 50% by mass, and particularly preferably 0.5 to 10% by mass.
- the metal-supported activated carbon of the present embodiment can be prepared by a known method.
- a precursor of a metal serving as a catalyst can be manufactured by a method of adsorption treatment on activated carbon as described above and reduction treatment.
- metal chlorides bromides, fluorides, hydroxides, nitrates, acetates, carbonates, sulfates, and ammonium salts.
- a precursor of a palladium catalyst palladium chloride, palladium nitrate, palladium acetate can be mentioned.
- adsorbing a metal precursor to activated carbon for example, (i) an impregnation method in which activated carbon is suspended in a precursor solution of a metal component and then the solvent is distilled off, (ii) the precursor solution A precipitation method in which activated carbon is suspended and brought into contact with a precipitant to form precipitates such as metal hydroxide on the activated carbon surface, (iii) ion exchange method in which metal ions are ion-exchanged with acid points and base points of activated carbon, iv) Spray method in which the precursor solution is spray-impregnated under reduced pressure, (v) activated carbon is exhausted, and then the precursor solution is added little by little to impregnate the same volume as the pore volume of the activated carbon Can be used.
- the impregnation method, the precipitation method and the ion exchange method are preferable, and the impregnation method and the precipitation method are more preferable.
- the order of adsorption in the case of adsorbing the precursors of plural types of metal components on activated carbon, and the precursors of the metal components may be adsorbed simultaneously or the precursors of the respective components may be adsorbed individually.
- the metal-supported activated carbon can be obtained by reduction treatment.
- the reduction treatment method is not limited to either the liquid phase method or the gas phase method, but the liquid phase method is preferable.
- a reducing agent to be used hydrogen, formaldehyde, methanol, sodium borohydride, hydrazine and the like can be mentioned.
- a solvent water is preferred, and other water-miscible solvents may be used in combination.
- the reduction temperature is preferably room temperature to 100 ° C.
- the metal-supported activated carbon of the present embodiment can be suitably used for a hydrogenation reaction catalyst.
- the method for carrying out the hydrogenation reaction using the metal-supported activated carbon of the present embodiment is not particularly limited, and the hydrogen source and the metal-supported activated carbon are brought into contact with the object (reaction substrate) to carry out the hydrogenation reaction in the object Or it should just be able to cause a dehydrogenation reaction.
- the hydrogen source examples include reducing gases such as hydrogen; alcohols such as methanol, ethanol and propanol; hydrazines such as hydrazine, methylhydrazine, allylhydrazine and phenylhydrazine and their derivatives and salts thereof.
- reducing gases such as hydrogen
- alcohols such as methanol, ethanol and propanol
- hydrazines such as hydrazine, methylhydrazine, allylhydrazine and phenylhydrazine and their derivatives and salts thereof.
- hydrogen is preferably used.
- the amount of the hydrogen source used is preferably 10 to 2000 moles relative to 1 mole of the target (reaction substrate).
- the use amount of the metal-supported activated carbon of the present embodiment in the hydrogenation reaction is preferably, for example, an amount such that the amount of supported metal is 0.0001 to 1 mole with respect to 1 mole of the target (reaction substrate).
- the metal-supported activated carbon of the present embodiment is very useful as a catalyst for the hydrogenation reaction, and therefore exhibits excellent effects in various industrial processes.
- the conductivity obtained by powder resistance measurement under a load of 12 kN is 3.5 S / cm or more, and the oxygen content is 3.0 mass% or more It features.
- Such a configuration can provide activated carbon that is very excellent in catalyst performance.
- the said activated carbon is coconut shell origin. Thereby, it is thought that the above-mentioned effect can be acquired more certainly.
- the metal-supported activated carbon according to another aspect of the present invention is characterized in that a metal is supported on the above-mentioned activated carbon.
- a metal is supported on the above-mentioned activated carbon.
- the metal is palladium. Thereby, it is thought that the above-mentioned effect can be acquired more certainly.
- the metal-supported activated carbon is considered to exert more effects by being used as a hydrogenation reaction catalyst.
- the conductivity of the activated carbon was measured using a powder resistivity measuring unit “MCP-PD51” manufactured by Mitsubishi Chemical Analytech Co., Ltd. Since the particle size of the measurement sample largely affects the conductivity measurement, the 10% particle size (D10) of the volume-based cumulative distribution of activated carbon is about 1 to 3 ⁇ m, and the 50% particle size of the volume-based cumulative distribution (D10) Pulverization was performed so that the D50) was about 5 to 8 ⁇ m, and the 90% particle diameter (D90) of the volume-based cumulative distribution was about 10 to 20 ⁇ m, and the conductivity of the activated carbon pellet was measured when a load of 12 kN was applied.
- MCP-PD51 powder resistivity measuring unit manufactured by Mitsubishi Chemical Analytech Co., Ltd. Since the particle size of the measurement sample largely affects the conductivity measurement, the 10% particle size (D10) of the volume-based cumulative distribution of activated carbon is about 1 to 3 ⁇ m, and the 50% particle size of the volume-based cumulative distribution (D10) Pulverization was performed so
- the particle size of the pulverized activated carbon was measured by a laser diffraction measurement method. That is, the activated carbon to be measured was put in ion exchange water together with a surfactant, ultrasonic vibration was applied to prepare a uniform dispersion, and the measurement was carried out using Microtrac MT3300EX-II manufactured by Microtrac Bell.
- a surfactant "polyoxyethylene (10) octyl phenyl ether” manufactured by Wako Pure Chemical Industries, Ltd. was used. The analysis conditions are shown below.
- Example 1 100 g of coconut rattle was introduced into the furnace, and the air was shut off to conduct heat treatment at 1300 ° C. Thereafter, 80 g of the heat-treated product is put into a fluidized-bed furnace, and a mixed gas of 15% water vapor partial pressure, 11% carbon dioxide partial pressure and 74% nitrogen partial pressure is used at a total pressure of 1 atmosphere and a flow rate of 20 L / min.
- the catalyst was supplied to the inside, and the activation time was adjusted by adjusting the activation time so that the benzene adsorption performance was 21.8% under the condition of the activation temperature of 950 ° C.
- the activated product is pickled using hydrochloric acid (concentration: 1 N, diluted solution: ion-exchanged water) for 30 minutes at a temperature of 70 ° C., and then the remaining acid is removed, so sufficient with ion-exchanged water
- the reaction product was washed with water and dried to obtain acid-washed activated carbon.
- the acid-washed activated carbon is introduced into a fluidized furnace with an in-furnace temperature of 500 ° C., and a mixed gas of 7% oxygen partial pressure, 11% steam partial pressure, 8% carbon dioxide partial pressure and 74% nitrogen partial pressure
- the gas was supplied into the furnace at a total pressure of 1 atmosphere and a flow rate of 30 L / min, and oxidation treatment was performed until the yield became 90% while raising the heat treatment temperature to 550 ° C. to obtain activated carbon.
- the specific surface area, conductivity and oxygen content (O content) of the obtained activated carbon were as shown in Table 1.
- Example 2 Activated carbon was produced in the same manner as in Example 1 except that the activation time was adjusted so that the benzene adsorption performance after activation was 30.9%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 3 Activated carbon was produced in the same manner as in Example 1 except that the activation time was adjusted so that the benzene adsorption performance after activation was 45.0%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 4 Activated carbon was produced in the same manner as in Example 1 except that the activation time was adjusted so that the benzene adsorption performance after activation was 59.6%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 5 Activated carbon was produced in the same manner as in Example 1 except that the heat treatment temperature of the coconut shell was adjusted to 1200 ° C., and the activation time was adjusted so that the benzene adsorption performance after activation was 31.1%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 6 Activated carbon was produced in the same manner as in Example 1 except that the heat treatment temperature of the coconut shell was adjusted to 1100 ° C. and the activation time was adjusted so that the benzene adsorption performance after activation was 29.9%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 7 Activated carbon was prepared in the same manner as in Example 1, except that the activation time was adjusted so that the benzene adsorption performance after activation was 30.9%, and oxidation treatment was performed until the yield was 95.4%. .
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Activated carbon was produced in the same manner as in Example 1 except that the coconut glass was put into a furnace without heat treatment and the activation time was adjusted so that the benzene adsorption performance after activation was 21.2%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 2 Activated carbon was produced in the same manner as in Example 1 except that the coconut glass was put into a furnace without heat treatment and the activation time was adjusted so that the benzene adsorption performance after activation was 31.6%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 3 Activated carbon was produced in the same manner as in Example 1 except that the coconut glass was put into a furnace without heat treatment, and the activation time was adjusted so that the benzene adsorption performance after activation was 46.0%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Example 4 Activated carbon was produced in the same manner as in Example 1 except that the coconut glass was put into a furnace without heat treatment and the activation time was adjusted so that the benzene adsorption performance after activation was 60.9%.
- the specific surface area, conductivity and oxygen content of the obtained activated carbon were as shown in Table 1.
- Activated carbon of each example and each comparative example was pulverized to obtain powdered activated carbon.
- 1 g of powdered activated carbon was added to 20 ml of ion exchanged water to prepare a slurry.
- 0.0168 g of palladium chloride was dissolved in 20 ml of 0.1 N hydrochloric acid, and 1N sodium hydroxide solution was added to adjust the pH to about 3.8 to 4.2. While stirring the activated carbon slurry solution, the pH-adjusted palladium chloride solution was added, and the mixture was stirred for 15 minutes.
- the palladium-supported activated carbons of the respective Examples and Comparative Examples obtained above are vacuum dried at 120 ° C. for 2 hours, 0.1 g of the activated carbon is put into a decomposition vessel, and 10 ml of 60% nitric acid is added and mixed, and then microwave sample pretreatment The sample was dissolved using an apparatus (Discover SP-D80 manufactured by CEM). The solution was taken out and the solution was brought up to 25 ml to prepare a measurement solution, which was then analyzed by an ICP emission spectrophotometer (ICPE-9800, manufactured by Shimadzu Corporation). The amount of supported palladium was determined from the obtained value and a calibration curve prepared from a palladium standard solution of known concentration.
- ICPE-9800 ICP emission spectrophotometer
- the palladium dispersion was measured by CO pulse method using Micro-track Bell Co. Bel-CAT II.
- the palladium-supported activated carbon of each Example and each Comparative Example was filled in a measurement container made of quartz and set in the apparatus.
- the pretreatment was performed according to the following procedure. Helium gas was flowed at 50 mL / min, the temperature was raised to 100 ° C. at a heating rate of 5 ° C./min, and held for 15 minutes. Thereafter, reduction treatment was performed by circulating hydrogen gas at 50 mL / min for 20 minutes.
- helium gas was again circulated at 50 mL / min, and the inside of the measurement container was cooled to 50 ° C.
- CO pulse measurement was performed.
- the amount of adsorbed CO was measured at a measurement temperature of 50 ° C. using 10% CO / He as the adsorption gas.
- the degree of dispersion of palladium on palladium-on-activated carbon was measured from the obtained amount of CO adsorption and the amount of palladium supported calculated by ICP measurement.
- the present invention has wide industrial applicability in the technical field related to activated carbon and catalyst-supported activated carbon using the same.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
- Carbon And Carbon Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
本実施形態の活性炭は、荷重12kNにおける粉体抵抗測定によって得られる導電率が3.5S/cm以上であり、かつ、酸素含有量が3.0質量%以上であることを特徴とする。
上述したような本実施形態の活性炭は、炭素質材料を乾留し、得られた乾留品を1100℃以上の温度で熱処理し、その後、水蒸気、窒素及び二酸化炭素を含む混合ガス雰囲気下で賦活処理した後に、酸化性雰囲気中で酸化処理をすることにより得ることができる。すなわち、本発明には、炭素質材料の乾留品を1100℃以上の温度で熱処理する工程、賦活処理工程、及び、酸化処理工程を少なくとも含む、活性炭の製造方法も包含される。
炭素質材料としては、公知の材料全てから選択でき、例えば、植物(ヤシガラ、籾ガラ、珈琲豆滓、木材など)、天然高分子(デンプン、セルロース、リグニン類など)、半合成高分子(セルロースエステル類、セルロースエーテル類、リグニン樹脂など)、合成高分子(フェノール系樹脂、フラン系樹脂、エポキシ樹脂など)、天然鉱物などで例示できる。これらの原料は単独で又は二種以上組み合わせて使用できる。好ましい原料は木材などの植物原料であり、不純物の少ないヤシガラがより好ましい。
乾留品の熱処理は、乾留物を、酸素又は空気を遮断して、1100℃以上の温度、好ましくは1200℃以上の温度で加熱することにより行うことができる。この熱処理温度が低すぎると、活性炭の導電率が低くなり、金属触媒と活性炭との相互作用が不十分となって、触媒性能が低下する。また、熱処理温度が高くなればなるほど活性炭の導電率は高まるが、十分な比表面積を得るための賦活時間が長くなることによって製造コストが増大するため好ましくない。よって、熱処理温度の上限は、1500℃以下であることが好ましい。
賦活処理は、本発明が属する技術分野において一般的な方法により行うことができ、主に、ガス賦活処理と薬剤賦活処理の2種類の処理方法を挙げることができる。
賦活時間は、特に限定はされないが、0.5~48時間、好ましくは1~24時間、さらに好ましくは2~20時間(例えば、6~12時間)程度であってもよい。賦活時間が短すぎると、十分な比表面積が得られず、金属担持後の触媒性能が低下し、長すぎると生産性が低下するおそれがある。
ガス分圧も特に限定はされないが、水蒸気分圧7.5~40%、好ましくは10~30%(例えば、10~20%)、二酸化炭素分圧10~50%、好ましくは15~45%(例えば、20~40%)、窒素分圧30~80%、好ましくは40~70%(例えば、45~65%)程度であり、ガス分圧は、水蒸気分圧10~40%、二酸化炭素分圧10~40%及び窒素分圧40~80%程度であってもよい。なお、ガスの全圧は、通常、1気圧(約0.1MPa)である。
また、総ガス供給量(流量)は、特に限定されないが、乾留品原料100gに対して、1~50L/分、好ましくは1~25L/分程度である。
(酸洗浄工程)
本実施形態の活性炭の製造工程は、酸洗浄工程を含んでも良い。酸洗浄工程は、賦活処理後の活性炭を、酸を含む洗浄液により洗浄することにより、活性炭中に含まれる金属成分等の不純物を除去するための工程である。酸洗浄は、例えば、酸を含む洗浄液に、原料活性炭を浸漬することにより行うことができる。酸洗浄工程では、原料活性炭を塩酸で洗浄後、水洗してもよく、酸洗と水洗を繰り返すなど、水洗と酸洗を適宜組合せてもよい。
酸洗浄液には、塩酸、硫酸、硝酸等の無機酸や、ギ酸、酢酸、プロピオン酸、シュウ酸及び酒石酸、クエン酸等の飽和カルボン酸、安息香酸及びテレフタル酸等の芳香族カルボン酸等の有機酸を用いることが好ましく、中でも、塩酸による洗浄がより好ましい。酸洗浄液として塩酸を用いる場合、塩酸の濃度は0.1~3.0質量%であることが好ましく、0.3~1.0質量%であることがより好ましい。塩酸濃度が低過ぎると、不純物を除去するために酸洗回数を増やす必要があり、逆に高過ぎると、残留する塩酸が多くなることから、上記範囲の濃度とすることにより、効率よく酸洗浄工程を行うことができ、生産性の面から好ましい。
酸洗や水洗をする際の液温度は特に限定されるものではないが、好ましくは0~98℃であり、より好ましくは10~95℃であり、さらに好ましくは15~90℃である。原料活性炭を浸漬する際の洗浄液の温度が上記範囲内であれば、実用的な時間かつ装置への負荷を抑制した洗浄の実施が可能となるため望ましい。
(酸化処理工程)
本実施形態の活性炭の製造方法は、酸化処理工程を含む。酸化処理工程は、活性炭を、酸化性雰囲気中で酸化処理することにより、活性炭の酸素含有量を高める工程である。具体的には、酸素を含む混合ガス雰囲気で熱処理する方法や、過酸化水素水、硝酸、過マンガン酸カリウム等の酸化剤で処理する方法が挙げられる。
酸素を用いた酸化処理は、賦活処理と同じような流動層、多段炉、回転炉などを用いて行うことができ、400℃以上の温度、好ましくは400~600℃の温度で行うことができる。酸化処理温度が400℃未満であると、活性炭の酸化が十分に進行せず、また600℃以上では活性炭の酸化が急速に進行し、炭素消費が激しくなって収率が低下するため好ましくない。
酸化処理時間は、特に限定はされないが、0.1~3時間、好ましくは0.2~2時間、より好ましくは0.3~1時間程度であってもよい。酸化処理時間が短すぎると、活性炭の酸化が十分に進行せず、長すぎると生産性の低下が生じる。
ガス分圧は、特に限定はされないが、酸素分圧1~15%、水蒸気分圧5~15%、二酸化炭素分圧5~15%及び窒素分圧50~80%程度であってもよい。なお、ガスの全圧は、通常、1気圧(約0.1MPa)である。
また、総ガス供給量(流量)は、特に限定されないが、賦活品原料50gに対して、1~100L/分、好ましくは1~50L/分程度である。
[金属担持活性炭]
本実施形態の金属担持活性炭は、上述したような活性炭に触媒となる金属が担持されていることを特徴とする。
担持する金属は、特には限定されないが、水素化反応や脱水素反応の触媒として用いられる金属が挙げられる。具体的には、例えば、パラジウム、白金、ルテニウム、ロジウム、オスミウム、イリジウム、ニッケル、コバルト、レニウム、バナジウム、タングステン、モリブデン、鉄、チタンなどが挙げられ、白金族元素(パラジウム、白金、ルテニウム、ロジウム、オスミウム、イリジウム)やニッケル、鉄がより好ましく、中でもパラジウムまたは白金が好適である。これらは単独で使用することも、複数を組み合わせて使用することもできる。
本実施形態の金属担持活性炭における、金属の担持量は、特には限定されないが、0.1~50質量%、特に0.5~10質量%とするのが好ましい。
本実施形態の金属担持活性炭は、公知の方法により調製することができる。例えば、触媒となる金属の前駆体を、上述したような活性炭に吸着後、還元処理を行う方法で製造できる。
本実施形態の金属担持活性炭に使用できる金属(金属成分)の前駆体としては、例えば、金属の塩化物、臭化物、フッ化物、水酸化物、硝酸塩、酢酸塩、炭酸塩、硫酸塩、アンモニウム塩等が挙げられ、これらを単独で又は2種以上を任意の割合で混合して用いることができる。例えば、パラジウム触媒の前駆体としては、塩化パラジウム、硝酸パラジウム、酢酸パラジウムが挙げられる。
金属の前駆体を活性炭に吸着させる方法としては、例えば、(i)金属成分の前駆体溶液に、活性炭を懸濁させた後、溶媒を留去する含浸法、(ii)前記前駆体溶液に活性炭を懸濁させ、沈殿剤と接触させて金属水酸化物等の沈殿を活性炭表面に生成させる沈殿法、(iii)活性炭の酸点や塩基点に金属イオンをイオン交換するイオン交換法、(iv)減圧状態で前記前駆体溶液を噴霧含浸するスプレー法、(v)活性炭を排気した後、前記前駆体溶液を少量ずつ加え、活性炭の細孔容積と同容積分を含浸するIncipient Wetness法等を用いることができる。これらの中でも、金属成分の分散性及び作業性の観点から、含浸法、沈殿法、イオン交換法が好ましく、含浸法、沈殿法がより好ましい。活性炭に複数種の金属成分の前駆体を吸着させる場合の吸着順序については特に制限がなく、金属成分の前駆体を同時に吸着させても、各成分の前駆体を個別に吸着させてもよい。
活性炭に金属成分の前駆体を吸着させた後、還元処理を行うことで、金属担持活性炭を得ることができる。還元処理方法としては液相法、気相法のいずれでも限定されないが、液相法が好ましい。使用する還元剤としては、水素、ホルムアルデヒド、メタノール、水素化ホウ素ナトリウム、ヒドラジン等が挙げられる。溶媒としては水が好ましく、その他の水と混和する溶媒を併用してもよい。還元温度としては室温~100℃が好ましい。
本実施形態の金属担持活性炭は、水素化反応触媒に好適に使用することができる。本実施形態の金属担持活性炭を使用して水素化反応を行う方法としては、特に限定はされず、対象物(反応基質)に水素源と金属担持活性炭を接触させて該対象物において水素化反応または脱水素反応を起こすことができればよい。
水素源としては、水素等の還元性ガス;メタノール、エタノール、プロパノール等のアルコール;ヒドラジン、メチルヒドラジン、アリルヒドラジン、フェニルヒドラジン等のヒドラジン及びその誘導体とそれらの塩等が挙げられる。これら水素源のうち、水素が好ましく用いられる。
前記水素源の使用量は、対象物(反応基質)1モルに対して、10~2000モルであることが好ましい。
水素化反応における本実施形態の金属担持活性炭の使用量は、例えば、対象物(反応基質)1モルに対して、担持金属量が0.0001~1モルとなる量であることが好ましい。
本実施形態の金属担持活性炭は、水素化反応の触媒として非常に有用であるため、様々な工業プロセスにおいて優れた効果を発揮する。
三菱化学アナリテック社製、粉体抵抗率測定ユニット「MCP-PD51」を使用し、活性炭の導電率を測定した。導電率の測定には、測定試料の粒径が大きく影響するため、活性炭の体積基準の累積分布の10%粒子径(D10)が1~3μm程度、体積基準の累積分布の50%粒子径(D50)が5~8μm程度、体積基準の累積分布の90%粒子径(D90)が10~20μm程度になるよう粉砕し、荷重を12kNかけた際の活性炭ペレットの導電率を測定した。なお、粉砕した活性炭の粒径はレーザー回折測定法により測定した。すなわち、測定対象である活性炭を界面活性剤と共にイオン交換水中に入れ、超音波振動を与え均一分散液を作製し、マイクロトラック・ベル社製のMicrotrac MT3300EX-IIを用いて測定した。界面活性剤には、和光純薬工業株式会社製の「ポリオキシエチレン(10)オクチルフェニルエーテル」を用いた。分析条件を以下に示す。
測定回数:3回
測定時間:30秒
分布表示:体積
粒径区分;標準
計算モード:MT3000II
溶媒名:WATER
測定上限:2000μm、測定下限:0.021μm
残分比:0.00
通過分比:0.00
残分比設定:無効
粒子透過性:吸収
粒子屈折率:N/A
粒子形状:N/A
溶媒屈折率:1.333
DV値:0.0100~0.0500
透過率(TR):0.750~0.920
粉砕した活性炭を120℃で2時間真空乾燥した後、ELEMENTAR社製Vario EL IIIを使用し、基準物質に安息香酸を用いて、活性炭の酸素含有量を測定した。
マイクロトラック・ベル(株)製のBELSORP-maxを使用し、試料となる活性炭を窒素気流下(窒素流量:50mL/分)にて300℃で3時間加熱した後、77Kにおける活性炭の窒素吸脱着等温線を測定した。得られた吸脱着等温線からBET式により多点法による解析を行い、得られた曲線の相対圧P/P0=0.01~0.1の領域での直線から比表面積を算出した。
上記活性炭の比表面積の測定方法に従って得られた窒素吸脱着等温線を、MP法により解析し、得られたミクロ孔細孔容積およびミクロ孔比表面積から、下式に従ってミクロ孔平均細孔径を算出した。
D=4000×V/S
(式中、D:ミクロ孔平均細孔径(nm),V:ミクロ孔細孔容積(mL/g),S:ミクロ孔比表面積(m2/g)を表す)
日本工業規格における活性炭試験方法JISK1474(1991年)に準拠して、賦活処理後の活性炭のベンゼン吸着性能を測定した。25℃において、粒状試料に溶剤飽和濃度の1/10となる溶剤蒸気を含む空気を通し、質量が一定となったときの試料の増量から平衡吸着性能を求めた。
ヤシガラチャー100gを炉内に投入し、空気を遮断して1300℃の条件下で熱処理した。その後に、熱処理品80gを流動炉に投入し、水蒸気分圧15%、二酸化炭素分圧11%、窒素分圧74%の混合ガスを、ガスの全圧1気圧でかつ流量20L/minで炉内に供給し、賦活温度950℃の条件下で、ベンゼン吸着性能が21.8%となるように賦活時間を調整して賦活処理を行った。次に、賦活処理品を、塩酸(濃度:1規定、希釈液:イオン交換水)を用いて、温度70℃で30分間酸洗した後、残留した酸を除去するため、イオン交換水で十分に水洗、乾燥して、酸洗浄活性炭を得た。次に、酸洗浄活性炭を、炉内温度を500℃とした流動炉に投入し、酸素分圧7%、水蒸気分圧11%、二酸化炭素分圧8%、窒素分圧74%の混合ガスを、ガスの全圧1気圧でかつ流量30L/minで炉内に供給し、熱処理温度を550℃まで昇温させながら、収率が90%となるまで酸化処理を行い、活性炭を得た。得られた活性炭の比表面積、導電率および酸素含有量(O含有量)は表1に示す通りであった。
賦活後のベンゼン吸着性能が30.9%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
賦活後のベンゼン吸着性能が45.0%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
賦活後のベンゼン吸着性能が59.6%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーの熱処理温度を1200℃とし、賦活後のベンゼン吸着性能が31.1%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーの熱処理温度を1100℃とし、賦活後のベンゼン吸着性能が29.9%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
賦活後のベンゼン吸着性能が30.9%となるように賦活時間を調整し、収率が95.4%となるまで酸化処理をした以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーを熱処理することなく炉に投入し、賦活後のベンゼン吸着性能が21.2%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーを熱処理することなく炉に投入し、賦活後のベンゼン吸着性能が31.6%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーを熱処理することなく炉に投入し、賦活後のベンゼン吸着性能が46.0%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
ヤシガラチャーを熱処理することなく炉に投入し、賦活後のベンゼン吸着性能が60.9%となるように賦活時間を調整した以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。
賦活後のベンゼン吸着性能が31.0%となるように賦活時間を調整し、空気酸化処理を行わなかった以外は、実施例1と同様の方法で活性炭を作製した。得られた活性炭の比表面積、導電率および酸素含有量は表1に示す通りであった。

実施例1~7及び比較例1~5で得られた活性炭を用い、以下のパラジウム担持方法に従ってパラジウム担持活性炭を作製した。そして、作製したパラジウム担持活性炭のパラジウム担持量、パラジウム分散度、及びニトロベンゼンの水素化性能を、下記方法に従って測定した。得られた結果を表2に示す。
各実施例および各比較例の活性炭を粉砕して、粉末状活性炭とした。粉末状活性炭1gをイオン交換水20mlに加えて、スラリーを調製した。一方、塩化パラジウム0.0168gを0.1N塩酸20mlに溶解させた後、1N水酸化ナトリウム溶液を添加して、pHを3.8~4.2程度に調整した。活性炭スラリー溶液を攪拌しながら、pH調整後の塩化パラジウム溶液を加え、15分間攪拌した。その後、10%炭酸水素ナトリウム飽和溶液を加えて、溶液のpHを7とした後、更に1時間攪拌を行った。その後、37%ホルムアルデヒド溶液を0.8g添加し、オイルバス中100℃で5時間還流してパラジウムの還元を行った。還元後、触媒を吸引濾過機で濾過し、イオン交換水で十分に洗浄した。洗浄後、120℃で真空乾燥を行い、パラジウム担持活性炭を得た。
上記で得られた各実施例および各比較例のパラジウム担持活性炭を120℃で2時間真空乾燥した後、分解容器に0.1g入れ、60%硝酸10mlを加え混ぜた後、マイクロウェーブ試料前処理装置(CEM社製、DiscoverSP-D80)を用いて試料を溶解した。その溶解液を取り出し、25mlにメスアップして測定溶液を調製した後、ICP発光分光分析装置((株)島津製作所製、ICPE-9800)にて分析した。得られた値と既知濃度のパラジウム標準液から作成した検量線より、パラジウム担持量を求めた。
マイクロトラック・ベル株式会社製Bel-CATIIを用いて、COパルス法によりパラジウム分散度を測定した。各実施例および各比較例のパラジウム担持活性炭を石英製の測定容器に充填し、装置内にセットした。前処理を以下の手順で行った。ヘリウムガスを50mL/分で流通し、5℃/分の昇温速度で100℃に昇温して15分保持した。その後、水素ガスを50mL/分で20分流通する事で、還元処理を行った。還元処理後、再度ヘリウムガスを50mL/分で流通し、測定容器内が50℃になるまで冷却した。前処理後、COパルス測定を行った。吸着ガスには10%CO/Heを用い、測定温度50℃の条件で、CO吸着量を測定した。得られたCO吸着量、及びICP測定によって算出したパラジウム担持量から、パラジウム担持活性炭のパラジウム分散度を測定した。
株式会社石井理化機器製作所製の中圧還元装置「CH-200」を用いて評価した。反応容器内にニトロベンゼン4.2ml、2-プロパノール25mlを加えて混合後、上記で得られた各実施例および各比較例のパラジウム担持活性炭をそれぞれ50mg添加し、溶液中に分散させた。反応容器を装置内にセットし、容器内を水素ガスで十分置換後、40℃に昇温した。15分間攪拌して安定化後、初期圧0.35MPaとした水素ガスを導入し、反応容器内水素圧の経時変化を測定することで、水素化性能を評価した。反応20分後での初期水素圧からの水素圧変化量を読み取り、活性炭の比表面積で割った値を比較した。本実施例では、この値が1.7E-05以上を合格とする。
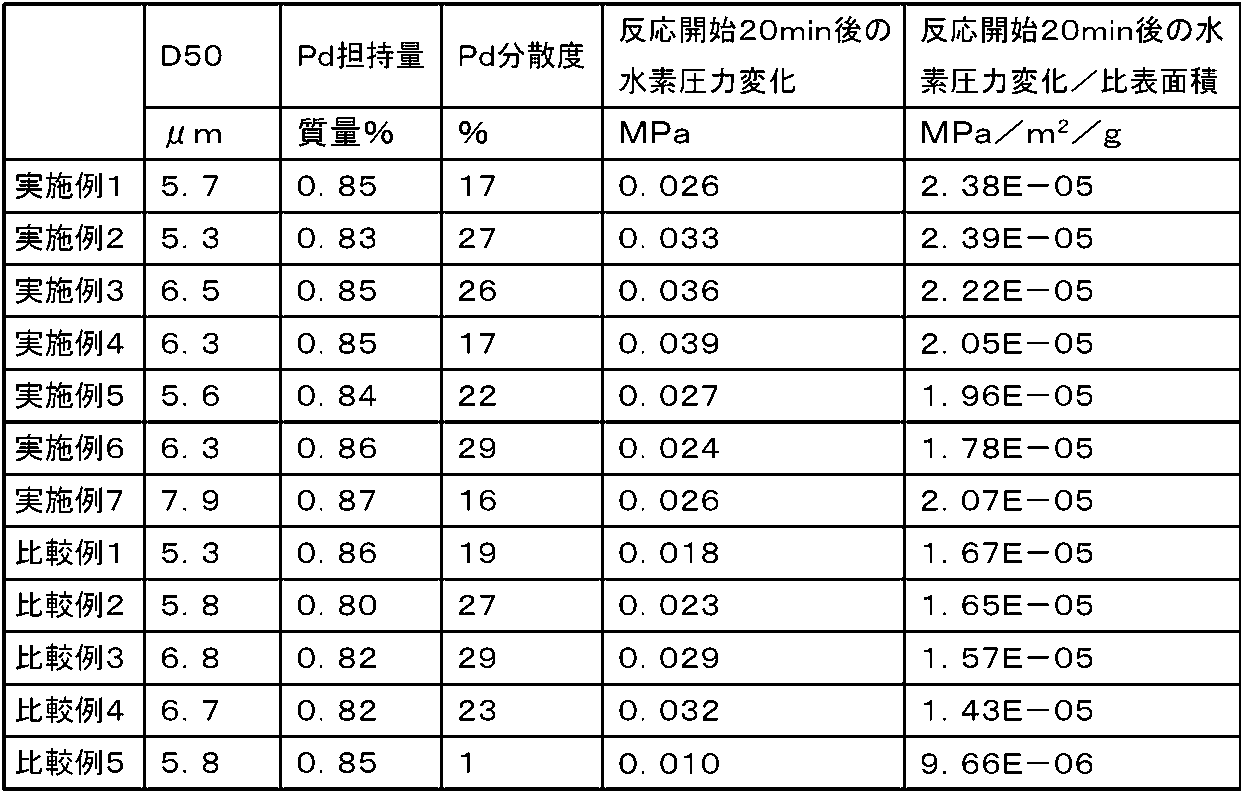
表2の結果から明らかなように、比較例1~5で得られた活性炭をパラジウム担体として使用した場合は、比表面積に対する反応開始20分における水素圧変化量は小さい。
Claims (5)
- 荷重12kNにおける粉体抵抗測定によって得られる導電率が3.5S/cm以上であり、かつ、酸素含有量が3.0質量%以上である、活性炭。
- 前記活性炭がヤシガラ由来である、請求項1に記載の活性炭。
- 請求項1または2に記載の活性炭に金属が担持されている金属担持活性炭。
- 前記金属がパラジウムである、請求項3に記載の金属担持活性炭。
- 請求項3または4に記載の金属担持活性炭を用いた水素化反応触媒。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019563011A JP7186182B2 (ja) | 2017-12-25 | 2018-12-17 | 活性炭およびそれを用いた金属担持活性炭、並びに水素化反応触媒 |
| KR1020207020112A KR102679769B1 (ko) | 2017-12-25 | 2018-12-17 | 활성탄 및 그것을 사용한 금속 담지 활성탄, 그리고 수소화 반응 촉매 |
| US15/733,219 US10974224B2 (en) | 2017-12-25 | 2018-12-17 | Activated carbon, metal-carrying activated carbon using same and hydrogenation reaction catalyst |
| CN201880083117.7A CN111511682B (zh) | 2017-12-25 | 2018-12-17 | 活性炭、使用其的担载金属的活性炭、以及氢化反应催化剂 |
| EP18894668.5A EP3712108B1 (en) | 2017-12-25 | 2018-12-17 | Activated carbon, metal-carrying activated carbon using same and hydrogenation reaction catalyst |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017-247439 | 2017-12-25 | ||
| JP2017247439 | 2017-12-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019131270A1 true WO2019131270A1 (ja) | 2019-07-04 |
Family
ID=67063554
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/046301 Ceased WO2019131270A1 (ja) | 2017-12-25 | 2018-12-17 | 活性炭およびそれを用いた金属担持活性炭、並びに水素化反応触媒 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10974224B2 (ja) |
| EP (1) | EP3712108B1 (ja) |
| JP (1) | JP7186182B2 (ja) |
| KR (1) | KR102679769B1 (ja) |
| CN (1) | CN111511682B (ja) |
| TW (1) | TWI770335B (ja) |
| WO (1) | WO2019131270A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023533579A (ja) * | 2020-07-13 | 2023-08-03 | 万華化学集団股▲フン▼有限公司 | 銅系水素化触媒の調製方法、それで調製された触媒及び使用 |
| JP7597901B1 (ja) | 2023-11-29 | 2024-12-10 | 大阪ガスケミカル株式会社 | 炭素質材料及びその製造方法、吸着フィルター、浄水器カートリッジ、浄水器、並びに水浄化設備 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI753313B (zh) * | 2019-10-18 | 2022-01-21 | 國立成功大學 | 鎳鐵合金氫化觸媒及其製作方法 |
| CN113735122B (zh) * | 2021-08-17 | 2023-07-07 | 山东利特纳米技术有限公司 | 一种疏水活性炭的制备方法 |
| CN116020411A (zh) * | 2021-10-27 | 2023-04-28 | 中国石油化工股份有限公司 | 纳米金属复合吸附材料及其制备方法和应用以及co的吸附和脱附方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06269667A (ja) | 1993-03-19 | 1994-09-27 | Tanaka Kikinzoku Kogyo Kk | 貴金属担持活性炭触媒の製法 |
| JP2001035522A (ja) * | 1999-07-19 | 2001-02-09 | Honda Motor Co Ltd | 燃料電池用改質器 |
| WO2008053919A1 (en) * | 2006-11-02 | 2008-05-08 | Kuraray Chemical Co., Ltd | Activated carbon and process for production thereof, nonaqueous type polarizable electrodes and electric double-layer capacitors |
| JP2011136937A (ja) * | 2009-12-28 | 2011-07-14 | Showa Denko Kk | 酢酸n−プロピルの製造方法 |
| WO2011125504A1 (ja) * | 2010-03-31 | 2011-10-13 | クラレケミカル株式会社 | 活性炭及びその用途 |
| JP2013163629A (ja) * | 2012-02-13 | 2013-08-22 | Kuraray Chemical Co Ltd | 活性炭及びその用途 |
| JP2016159256A (ja) * | 2015-03-04 | 2016-09-05 | 株式会社クレハ | 触媒複合体 |
| WO2018116842A1 (ja) * | 2016-12-20 | 2018-06-28 | 株式会社クラレ | 多孔質炭素材料並びにその製造方法と用途 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04342408A (ja) * | 1991-05-17 | 1992-11-27 | Koa Oil Co Ltd | 活性炭およびその製造方法 |
| WO1996030318A1 (en) * | 1995-03-30 | 1996-10-03 | Nippon Sanso Corporation | Porous carbonaceous material, process for producing the same, and use thereof |
| KR100451646B1 (ko) * | 2000-01-05 | 2004-10-08 | 니폰 쇼쿠바이 컴파니 리미티드 | 배수처리용 촉매, 그의 제조방법 및 배수의 처리방법 |
| DE10003317A1 (de) * | 2000-01-27 | 2001-08-02 | Degussa | Verfahren zur Durchführung homogen katalysierter Umsetzungen |
| JP2004025023A (ja) * | 2002-06-25 | 2004-01-29 | Toyobo Co Ltd | 活性炭担体、触媒担持活性炭およびそれらの製造方法 |
| JP4239488B2 (ja) * | 2002-06-25 | 2009-03-18 | 東洋紡績株式会社 | 活性炭担体、触媒担持活性炭およびそれらの製造方法 |
| KR100928871B1 (ko) * | 2008-03-24 | 2009-11-30 | 방영석 | 활성탄에 담지된 팔라듐 촉매 및 그의 제조방법 |
| AU2011343489A1 (en) * | 2010-12-16 | 2013-08-01 | Energia Technologies, Inc. | Catalysts, methods of preparation of catalyst, methods of deoxygenation, and systems for fuel production |
| CN102626620A (zh) * | 2012-03-24 | 2012-08-08 | 中国石油化工股份有限公司 | 负载深度可控钯碳催化剂的制备方法 |
| CN103811752A (zh) * | 2012-11-13 | 2014-05-21 | 海洋王照明科技股份有限公司 | 铅碳电池负极铅膏及其制备方法、铅碳电池负极板与铅碳电池 |
| CN106794989B (zh) * | 2014-09-02 | 2020-03-31 | 株式会社可乐丽 | 源自植物的碳前体的纯化方法 |
-
2018
- 2018-12-17 CN CN201880083117.7A patent/CN111511682B/zh active Active
- 2018-12-17 JP JP2019563011A patent/JP7186182B2/ja active Active
- 2018-12-17 KR KR1020207020112A patent/KR102679769B1/ko active Active
- 2018-12-17 US US15/733,219 patent/US10974224B2/en active Active
- 2018-12-17 WO PCT/JP2018/046301 patent/WO2019131270A1/ja not_active Ceased
- 2018-12-17 EP EP18894668.5A patent/EP3712108B1/en active Active
- 2018-12-19 TW TW107145830A patent/TWI770335B/zh active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06269667A (ja) | 1993-03-19 | 1994-09-27 | Tanaka Kikinzoku Kogyo Kk | 貴金属担持活性炭触媒の製法 |
| JP2001035522A (ja) * | 1999-07-19 | 2001-02-09 | Honda Motor Co Ltd | 燃料電池用改質器 |
| WO2008053919A1 (en) * | 2006-11-02 | 2008-05-08 | Kuraray Chemical Co., Ltd | Activated carbon and process for production thereof, nonaqueous type polarizable electrodes and electric double-layer capacitors |
| JP2011136937A (ja) * | 2009-12-28 | 2011-07-14 | Showa Denko Kk | 酢酸n−プロピルの製造方法 |
| WO2011125504A1 (ja) * | 2010-03-31 | 2011-10-13 | クラレケミカル株式会社 | 活性炭及びその用途 |
| JP2013163629A (ja) * | 2012-02-13 | 2013-08-22 | Kuraray Chemical Co Ltd | 活性炭及びその用途 |
| JP2016159256A (ja) * | 2015-03-04 | 2016-09-05 | 株式会社クレハ | 触媒複合体 |
| WO2018116842A1 (ja) * | 2016-12-20 | 2018-06-28 | 株式会社クラレ | 多孔質炭素材料並びにその製造方法と用途 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3712108A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023533579A (ja) * | 2020-07-13 | 2023-08-03 | 万華化学集団股▲フン▼有限公司 | 銅系水素化触媒の調製方法、それで調製された触媒及び使用 |
| JP7594082B2 (ja) | 2020-07-13 | 2024-12-03 | 万華化学集団股▲フン▼有限公司 | 銅系水素化触媒の調製方法、それで調製された触媒及び使用 |
| JP7597901B1 (ja) | 2023-11-29 | 2024-12-10 | 大阪ガスケミカル株式会社 | 炭素質材料及びその製造方法、吸着フィルター、浄水器カートリッジ、浄水器、並びに水浄化設備 |
| WO2025115889A1 (ja) * | 2023-11-29 | 2025-06-05 | 大阪ガスケミカル株式会社 | 炭素質材料及びその製造方法、吸着フィルター、浄水器カートリッジ、浄水器、並びに水浄化設備 |
| JP2025087184A (ja) * | 2023-11-29 | 2025-06-10 | 大阪ガスケミカル株式会社 | 炭素質材料及びその製造方法、吸着フィルター、浄水器カートリッジ、浄水器、並びに水浄化設備 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111511682A (zh) | 2020-08-07 |
| JP7186182B2 (ja) | 2022-12-08 |
| EP3712108B1 (en) | 2025-11-05 |
| EP3712108A4 (en) | 2021-07-28 |
| CN111511682B (zh) | 2023-12-29 |
| US10974224B2 (en) | 2021-04-13 |
| TW201930190A (zh) | 2019-08-01 |
| EP3712108A1 (en) | 2020-09-23 |
| JPWO2019131270A1 (ja) | 2020-12-24 |
| US20200384444A1 (en) | 2020-12-10 |
| KR102679769B1 (ko) | 2024-06-28 |
| TWI770335B (zh) | 2022-07-11 |
| KR20200100692A (ko) | 2020-08-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019131270A1 (ja) | 活性炭およびそれを用いた金属担持活性炭、並びに水素化反応触媒 | |
| CN107199047B (zh) | 一种分散于sba-15孔道内的镍基甲烷化催化剂及其制备和应用 | |
| CN102527377B (zh) | 一种浸渍-可控还原法制备的CO羰化制草酸酯用高效纳米Pd催化剂 | |
| JP6450352B2 (ja) | 多孔質炭素材料、及びその製造方法、並びに合成反応用触媒 | |
| CN106622327A (zh) | 一种氮掺杂多孔碳负载金属的催化剂及其制备方法和用途 | |
| CN109331860B (zh) | 一种用于空气净化的低铂合金复合纳米光催化剂及其制备方法和应用 | |
| CN110302769A (zh) | 一种催化剂载体、负载型催化剂及其制备方法和用途 | |
| KR20090037949A (ko) | 지지된 팔라듐/탄소 촉매 및 그의 제조방법 | |
| JP2014208328A (ja) | カーボンナノチューブ合成用触媒、カーボンナノチューブ集合体、及びその製造方法 | |
| CN104368358A (zh) | 一种适用于琥珀酸加氢反应的催化剂及其制备和加氢反应方法 | |
| JP4787968B2 (ja) | ナノ金属または金属酸化物担持活性炭の高効率製造方法 | |
| CN109876832B (zh) | 一种合成呋喃甲胺的催化剂及其制备方法 | |
| CN109876866A (zh) | 一种用于芳香醛合成芳香胺的催化剂及其制备方法 | |
| CN105579131A (zh) | 用于直接合成过氧化氢的催化剂 | |
| CN103816895A (zh) | 一种负载型贵金属甲烷化催化剂及制法和应用 | |
| CN105903466A (zh) | 一种合成草酸二甲酯用催化剂及其制备方法 | |
| Li et al. | Effect of metal dispersion on the hydrogenation of 2-amyl anthraquinone over Pd/Al 2 O 3 catalyst | |
| Liu et al. | A novel carbothermal method for the preparation of nano-sized WC on high surface area carbon | |
| Pampararo et al. | Nanostructured Bimetallic (Cu, Ni)/SiO2 Catalysts for the Dehydrogenation of Ethanol to Acetaldehyde | |
| CN109876791B (zh) | 一种臭氧氧化催化剂及其制备方法 | |
| JP6646088B2 (ja) | 多孔質炭素材料、及びその製造方法、並びに合成反応用触媒 | |
| CN115672317A (zh) | 一种提高Pd(OH)2/C催化剂氢解脱苄活性的方法 | |
| CN117816152B (zh) | 一种选择性脱苄催化剂的制备方法 | |
| CN119951507B (zh) | 铁/碳复合材料及制法和在5-hmf氢解反应中的应用 | |
| CN111036207A (zh) | 加氢催化剂及其制备方法以及甘油加氢方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18894668 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019563011 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018894668 Country of ref document: EP Effective date: 20200618 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20207020112 Country of ref document: KR Kind code of ref document: A |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2018894668 Country of ref document: EP |