WO2019147073A1 - 아데노신 디아미나아제를 이용한 염기 교정 확인 방법 - Google Patents
아데노신 디아미나아제를 이용한 염기 교정 확인 방법 Download PDFInfo
- Publication number
- WO2019147073A1 WO2019147073A1 PCT/KR2019/001104 KR2019001104W WO2019147073A1 WO 2019147073 A1 WO2019147073 A1 WO 2019147073A1 KR 2019001104 W KR2019001104 W KR 2019001104W WO 2019147073 A1 WO2019147073 A1 WO 2019147073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dna
- target
- nucleic acid
- acid molecule
- specific nuclease
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/96—Stabilising an enzyme by forming an adduct or a composition; Forming enzyme conjugates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/34—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving hydrolase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/04—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in cyclic amidines (3.5.4)
- C12Y305/04004—Adenosine deaminase (3.5.4.4)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/30—Phosphoric diester hydrolysing, i.e. nuclease
- C12Q2521/301—Endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/50—Other enzymatic activities
- C12Q2521/531—Glycosylase
Definitions
- the present invention relates to a technique for confirming the base calibration using adenosine deaminase, and includes a composition for confirming the base calibration including adenosine diaminase and a method for confirming the base calibration including a step of treating adenosine diaminase with DNA do.
- Cas9 has DNA insertion and deletion in the target site due to DNA cleavage.
- Cas9 nickase was linked with cystidine deaminase to convert C to T0J (or to a lesser degree, C to G or A) without DNA cleavage, (1993), Nature, 533, 420-424 (2016)).
- cystidine deaminase to convert C to T0J (or to a lesser degree, C to G or A) without DNA cleavage, (1993), Nature, 533, 420-424 (2016)).
- cystidine deaminase is used to convert C to T (or to a lesser degree, C to G or A), or G of the opposite strand corresponding to C to A (or to a lower frequency , Converting G to C or), but there is no known method for converting the purine base, A or, to another base.
- Adenosine deaminase was isolated from the target specific nuclease using digested genomic sequencing (Digenome-seq, a method of distinguishing between cut and spot positions before and after the target specific nuclease treatment) (For example, Cas9 nicase or catalytically deficient Cas9 (dCas9)).
- the present invention provides a method for confirming the base correction of gene scissors, and thereby, the specificity of the base correction by the adenosine base correction gene scissors can be evaluated.
- Adenine Base Editors composed of S. pyogenes-derived Cas9 nicase and E. coli-engineered adenine diaminase enable efficient efficient conversion of adenine / guanine single nucleotides by guided RNA targeting, A mammal such as a human, a rodent such as a mouse, etc.) and / or a plant cell.
- ABE recognizes other f-target genes and other cytotoxic agents such as Cas9, BE (cytosine base editor), and therefore it is necessary to develop an independent method for the efficiency evaluation.
- WGS whole genome sequencing
- the entire dielectric sequencing can be specifically Digenome-seq.
- the Digenome-seq based method provided herein is sensitive enough to capture an ABE non-target location at a substi tut ion frequency of 0.1%. It is also proposed that pre-assembled ABE ribonucleic acid proteins, modified guide RNA, and Sniper-ABE can be used to reduce ABE of f-target activity in animal cells such as humans.
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- a method for producing double strand breaks in DNA using adenine diamidase is a method for producing double strand breaks in DNA using adenine diamidase.
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- a single-strand-specific endonuclease such as endonuclease V or Alkyladenine DNA Glycosyl ase (AAG) and endonuclease VI (combination of endonuclease VI II) to cleave the remaining DNA strands to form a double-strand break;
- a method for detecting a cleavage site or a non-target site of a target-specific nuclease is provided.
- Adenosine deaminase is di-rected evoluti- nated to form adenosine (A) in DNA bases
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- a method for producing double strand breaks in DNA using adenine diamidase is a method for producing double strand breaks in DNA using adenine diamidase.
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- a single-strand specific endonuclease for example, endonuclease V or Alkyl adenine DNA Glycosyl ase (MG) and endonuclease VI II (endonuclease VI 11)
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- a single-strand-specific endonuclease such as endonuclease V or Alkyl adenine DNA Glycosylase (AAG) and endonuclease VI II (a combination of endonuclease VI II) to cleave the remaining DNA strands to form a double-strand break;
- the method of detecting the non-target position (f-target si te) may further comprise, after step (d), if the identified truncated position is not a target position (on-target si te) f-target si te) (step (e)).
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising said nucleic acid molecule
- a nucleic acid molecule encoding a target specific nuclease, a target specific nuclease
- the calibration efficiency verification method of the target specific nuclease is performed after step (d), when the identified truncated position is not an on-target si te, a non-target position of f-target si (step (e)) of determining the degree of severance (non-target position number and / or cutting frequency at the non-target position) at the non-target position and comparing the degree of severity e-1). In this case, it can be judged that the lower the degree of cutting at the non-target position, the higher the correction efficiency and / or the accuracy.
- the comparison subject may be a target specific nuclease for the target sequence of any target DNA and may be, in one example, one of the commonly used or known target specific nuclease (probes, RGEN and guide RNA combinations) It can be any one selected.
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid comprising the nucleic acid molecule; and (2) a target specific nuclease, a target specific nuclease,
- the nucleic acid molecule encoding the cryase, or the plasmid comprising the nucleic acid molecule can be (concurrently) processed into separate cells or isolated DNA, and in one embodiment, the adenosine deaminase and the target specific nuclease fusion (For example, the adenosine deaminase is located at the N-terminal side and the target-specific nucleases can be located at the C terminus), or a nucleic acid sequence encoding the fusion protein or a plasmid containing the nucleic acid sequence .
- a suitable guide RNA according to the target specific nuclease, or
- adenosine deaminase converts the adenosine into inosine in the target site of the strand where the PAM sequence is present (17 to 23 nt site toward the 5 'end of PAM) of the double strand of DNA
- Single strand breaks (ni ck) occur on the opposite strand (complementary strand) of the strand on which the adenosine-inosine conversion occurs (i.e., the strand in which the PAM sequence is present) by the target specific nuclease.
- step (b) when the endonuclease m is used, the adenosine diphosphate of the opposite strand of the strand where the single strand break has occurred (i. E., The strand where the adenosine- The 3 'ends of the nucleotides located within 5 nt, within 4 nt, within 3 nt, within 2 nt, or lnt in the 5' and / or 3 '
- step (b) when the combination of Alkyl adenine DNA Glycosyl ase (endonuclease VI II) is used, the inosine converted in step (a) End of the AP region is generated and endonuclease VIll recognizes the AP region and cleaves the 3 ' and 5 ' ends of the AP region.
- Single strand cleavage also occurs on the strand opposite to that on which single strand cleavage has taken, resulting in double strand
- Another example is
- nucleic acid molecule encoding adenosine deaminase, adenosine deaminase, or a plasmid containing the nucleic acid molecule
- nucleic acid molecule encoding a target-specific nuclease, a target-specific nuclease, or a plasmid comprising said nucleic acid molecule
- the DNA to be subjected to the genetic modification (base correction) and / or cleavage may be a genomic DNA or DNA fragment (PCT product) Lt; / RTI >
- the cell may be a eukaryotic cell such as a mammal such as a human or a mouse.
- the adenosine deaminase and the target specific nuclease may be a fusion protein directly or indirectly linked through a peptide linker or a nucleic acid molecule encoding the fusion protein or a plasmid containing the nucleic acid molecule In the form of It could be used.
- the target specific nuclease may be used or included with the guide RNA.
- adenosine deaminase target specific nuclease
- guide RNA guide RNA
- a conjugate of an adenosine deaminase and a target specific nuclease-linked fusion protein and a guide RNA (ribonucleic acid protein, RNP) or a mixture thereof e.g., introduced into separate cells Or incorporated into the composition or into a separate DNA.
- the plasmid may be any plasmid including an expression system capable of inserting the adenosine deaminase coding gene and / or the inactivated target specific nuclease coding gene and expressing it in a host cell.
- the plasmid includes elements for expression of a target gene and may include a replicate ion origin, a promoter, an operator, a transcription termination terminator, and the like.
- a ribosome binding site e. G., A ribosome binding site
- suitable enzyme site e. G., A restriction enzyme site
- RBS and / or electron regulatory factors.
- the plasmid may be a plasmid, For example, one selected from the group consisting of pcDNA series, pSClOl, pGV1106, pACYC177, ColEl, pKT230, pME290, pBR322, pU phase / 9, pUC6, pBD9, pHC79, pIJ61, pLAFRl, pHV14, pGEX series, pET series, Or more, but is not limited thereto.
- the host cell may be a cell (for example, a eukaryotic cell such as a mammal such as a human cell) to which base correction or double strand breakage is to be introduced by the adenosine deaminase, an adenosine deaminase coding gene and / or an inactivated target (E. G., E. coli, etc.) expressing a specific nuclease coding gene and capable of expressing adenosine deaminase and inactivated target specific nucleases.
- a cell for example, a eukaryotic cell such as a mammal such as a human cell
- an inactivated target E. G., E. coli, etc.
- digested genome sequencing (Digenome-seq) technique refers to sequence analysis of genomes cleaved by nuclease, and the neclease non-target effect f-target ef fect) in in vitro nucl ease-digested who 1 e-genome sequencing. Sequence reads with the same 5 ' ends at the cleavage site of the nuclease, which can be computer-verified by a suitable program (e.g., Di genome program).
- the cleaved dielectric sequencing comprises steps (a), (b), and (c) in the method of genomic DNA sequencing as described above or in the method of detecting the non-target site of nuclease, (A), (b), and (c), or steps (a), (b), (c), and , Or steps (a), (b), (c), and () may be performed by cleaved dielectric sequencing.
- base mutation or base substitution
- a mutation e.g., substitution
- " nucleotide mutation
- Genetic modification and / or genetic modification techniques are techniques that can introduce a target-directed mutation into the genomic sequence of animal and plant cells, including human cells, in which knock-out or knock-in of a particular gene, Or introducing a mutation into a non-coding DNA sequence that does not produce a protein, or the like.
- the method proposed herein is based on the above- And / or to detect non-target positions of the target specific nuclease used in gene correction techniques, which can be usefully useful for developing a target specific nuclease system that specifically operates at the target site.
- adenosine deaminase and a target specific nuclease are treated with a genomic DNA isolated from a separated cell or a living body or cell to cut one strand of DNA, Is a step of cleaving the genomic DNA in vitro (in r / ir) with a nuclease that specifically acts on a specific target.
- the nuclease is produced specifically for the target, it has the possibility of cleaving another site, i.e., a non-target site depending on the specificity.
- the target specific nuclease agent used is cleaved at a target position or a plurality of non-target positions at which the nucleic acid molecule can have an activity with respect to the genomic DNA, lead.
- the isolated genomic DNA may be isolated from non-transformed cells (wild-type cells) and / or cells transformed to have a target-specific nuclease activity or having a nuclease activity, and the ratio of the target specific nuclease ratio And can be used without limitation from the origin depending on the purpose of detecting the target position.
- the cells into which the dielectrics are separated may be selected from among all prokaryotic cells and eukaryotic cells (for example, mammalian cells such as human, eukaryotic plant cells, etc.).
- the adenosine deaminase is an enzyme involved in purine metabolism (EC 3.5.4.4), and adenosine is deaminated (deaminase; All the enzymes that function to convert an amino group into a inosine by substituting a keto group) are collectively referred to as " inosine ".
- the adenosine deaminase may be derived from a prokaryotic or eukaryotic cell, such as an eukaryotic animal (e. G., Fish, amphibian, avian, avian, mammalian, etc.), or from an eukaryotic plant, (E. G., GenBank Access < / RTI > NP_000013.2 (coding gene: NM_000022.3), NP_001308979.1 (coding gene: NM_001322050.1),
- NP_001308980.1 (coding gene: NM_001322051.1), etc.), mouse adenosine deaminase (e.g., GenBank Accession No. (Coding gene: NM_001272052.1), NP_031424.1 (coding gene: NM_007398.4) and the like), E. coli TadA (NP_417054.2), and the like.
- the adenosine deaminase is a protein or DNA encoding it (Which may optionally be included in a suitable recombination vector), or may be used in the form of an mRNA encoding the same.
- Adenosine deaminase may also be used in conjunction with conventional adenosine deaminase, such as, for example, directed evolution ; DE).
- Directed evolution is one of protein engineering methods, which involves mutagenesis (generation of a mutant library), selection process (separation of mutants having the desired function (mutation) among the mutants), and amplification or reproduction (Template generation for the next round), and may be performed in vivo or in vitro.
- the anthropogenic adenosine deaminase is E. coli TadA (NP 417054.2) In E. coli.
- the artificially evolved adenosine deaminase is the 22nd amino acid residue (W) of the N-terminal methionine of NP417054.2 (except for the amino acid sequence, (A), the 107th amino acid residue (H), the 47th amino acid residue (P), the 50th amino acid residue (R), the 83rd amino acid residue (D), 122nd amino acid residue (H), 145th amino acid residue (S), 146th amino acid residue (D)
- NP_417054.2 may be substituted or deleted with other amino acids than the original (wild type) (see Reference Example 2), but the present invention is not limited thereto.
- the target specific nuclease is also referred to as programmable nuclease, and refers to all forms of nuclease that are capable of recognizing and cleaving a specific position on a desired genomic DNA.
- the target specific nuclease used herein can be any nuclease having activity to cleave one strand of DNA double strand.
- the target-specific nuclease is a Cas protein (e.g., Cas9 protein (CRISPR (Clustered regularly interspersed short palindromic repeats) associated protein 9), Cpfl protein (CRISPR from Prevotel la and Francisella 1) Or a nuclease associated with a CRISPR system of the same type H and / or type V (e.g., endonuclease), and the like.
- the target specific nuclease is a target DNA-specific guide RNA for guiding the target portion of the genomic DNA May be further included.
- the guide RNA may be transcribed in vitro, and may be, for example, an oligonucleotide double strand or a plasmid template, but is not limited thereto.
- the target specific nuclease can act as a ribonucleic acid protein (RNP) form by forming a ribonucleic acid-protein complex bound to a guide RNA (RNA-Guided Engineered Nuclease).
- Cas proteins are a major protein component of the CRISPR / Cas system and are capable of forming an activated endonuclease or nickase.
- Cas proteins from Campylobacter such as Campylobacter jejuni, such as Cas9 protein
- Streptococcus species such as Streptococcus thermophilus
- Cas proteins derived from Neisseria meningitidis such as Cas9 protein
- Pasteurella multocida such as Cas9 protein
- Cas proteins derived from the genus Francisella for example, Francisella novicida, such as the Cas9 protein
- the Cpfl protein is an endonuclease of the new CRISPR system distinct from the CRISPR / Cas system, which is relatively small in size and does not require tracrRNA compared to Cas9, and can be acted upon by a single guide show. In addition, it recognizes thymine-rich protospacer-adjacent motif (PAM) sequences and cuts double strands of DNA to produce a cohesive end (cohesive double-strand break).
- PAM thymine-rich protospacer-adjacent motif
- the target specific nuclease may be isolated from microorganisms or artificially or naturally occurring, such as recombinant or synthetic methods.
- the target specific nuclease e.g., Cas9, Cpfl, etc.
- the target specific nuclease may be a recombinant protein made by recombinant DNA.
- Recombinant DNA refers to a DNA molecule artificially created by recombinant methods, such as molecular cloning, to include heterologous or homologous genetic material obtained from various organisms.
- rDNA refers to a DNA molecule artificially created by recombinant methods, such as molecular cloning, to include heterologous or homologous genetic material obtained from various organisms.
- the target-specific nuclease may be a mutated (nonsignified) form of a mutated target-specific nuclease.
- the mutated (inactivated) target-specific nuclease may be meant to be mutated to lose endonuclease activity that cleaves all double strands of DNA, for example, endonuclease activity that cleaves all double strands of DNA
- a mutant target specific nuclease which is mutated so as to lose both the endo-nuclease activity and the niacase activity which are all cleaved.
- the nucleotide conversion for example, conversion to adenosine-inosine
- the diaminase is sequentially or irregularly carried out, (E.g., in the opposite strand of the strand where the PAM is located, in the 5 ' end of the PAM sequence) to the third strand in the opposite strand A nick is introduced at the position between the nucleotide and the fourth nucleotide).
- Such a variation of the target specific nuclease may be that occurring at least in the catalytic domain of the nuclease (e.g., the RuvC catalytic domain in the case of Cas9).
- the mutation is catalytic aspartate residue (catalytic aspartate residue Aspartic acid (D10) of the 986th position, glutamic acid (E762) at the 762nd position, histidine (H840) at the 840th position, asparagine (N854) at the 854th position, asparagine (D986), phenylalanine at position 539 (F539), methionine at position 763 (M763), lysine at position 890 (K890), and the like. have.
- catalytic aspartate residue catalytic aspartate residue Aspartic acid (D10) of the 986th position, glutamic acid (E762) at the 762nd position, histidine (H840) at the 840th position, asparagine (N854) at the 854th position, asparagine (D986), phenylalanine at position 539 (F539), methionine at position 763 (M763), lysine at position 890 (K8
- any other amino acid to be substituted may be alanine, but is not limited thereto.
- the mutated target specific nuclease may be a mutation that results in improved specificity (accuracy) .
- the mutated target specific nuclease is selected from the group consisting of Streptococcus pyoenzens
- One or more amino acid residues selected from the group consisting of F539, M763, and K890 of the Cas9 protein are substituted or deleted with amino acids different from the original amino acid (More specifically, a mutant comprising at least one mutation selected from the group consisting of F539S, M763I, and K890N, for example, a mutant having both F539S, M763I, and K890N mutations (Sniper_Cas9)).
- the mutated (inactivated) target specific nuclease is The DIO of a Cas9 protein (e.g., SwissProt Accession number Q99ZW2 (NP_269215.1)) derived from Streptococcus pyogenes (for example, SwissProt Accession number Q99ZW2 (NP_269215.1)) is substituted with an amino acid different from the original (for example, alanine).
- one or more amino acid residues selected from the group consisting of F539, M763, and K890 may be replaced with amino acids different from the original (for example, F539S, M763I, K890N) to improve the specificity (accuracy).
- the mutation target-specific nuclease may be mutated to recognize a PAM sequence that is different from the wild-type Cas9 protein.
- the mutation target-specific nuclease may include at least one of an aspartic acid (D1135) at position 1135, arginine at position 1335 (R1335), and threonine at position 1337 (T1337) of Cas9 protein from Streptococcus pyoensis
- D1135 aspartic acid
- R1335 arginine at position 1335
- T1337 threonine at position 1337
- all three are replaced with different amino acids
- the NGA language of the PAM sequence (NGG) of wild-type Cas9 is any base selected from A, T, G, and C).
- the mutation target specific nuclease is selected from the amino acid sequence of Cas9 protein from Streptococcus fyiensense,
- D1135, R1335, T1337, or a combination thereof e.g., D1135 + R1335 + T1337
- the 'other amino acids' include, but are not limited to, alanine, isoleucine, leucine, methionine, phenylalanine, proline, trimepophen, valine, aspartic acid, cysteine, glutamine, glycine, serine, threonine, tyrosine, aspartic acid, , Arginine, histidine, lysine, and any of the known variants of the amino acids, amino acids other than the amino acids that the wild-type protein originally has at the mutation position.
- the 'other amino acid' may be alanine, valine, glutamine, or arginine.
- the mutation target specific nuclease is an endonuclease May be one that recognizes a modified Cas9 protein that has lost activity (e. G., Has a niacase activity or lost all endo-nuclease activity and nikase activity), or a PAM sequence that differs from wild-type Cas9.
- the modified Cas9 protein is a Cas9 protein derived from Streptococcus pyogenes,
- a mutation for example, substitution with another amino acid
- a mutation is introduced at one or more positions selected from the group consisting of F539, M763, and K89 ⁇ in addition to the D10 position so that double-strand break endonuclease activity is lost and nica activity Modified Cas9 protein with improved specificity
- the mutation at the D10 position of the CAs9 protein is a D10A mutation (a mutation in which the 10th amino acid of the amino acid of the Cas9 protein is substituted with A; hereinafter, the mutation introduced into Cas9 is represented by the same method)
- the mutations at the H840 position may be the H840A mutation and the mutations at the D1135, R1335, and T1337 positions may be D1135V, R1335Q, and T1337R, respectively, and the mutations at the F539S, M763I, and K890N positions are F539S, M763I, and K890N.
- the nuclease may be an artificial or non-natural occurrence such as isolated from microorganisms or recombinant or synthetic methods.
- the nuclease may be a recombinant protein made by recombinant DNA.
- Recombinant DNA rDNA is artificially engineered by recombinant methods such as molecular cloning to include heterologous or homologous genomic material obtained from various organisms Means a DNA molecule made.
- the recombinant DNA when expressed in an appropriate organism to produce a protein in vivo or in F / iro), the recombinant DNA is selected by selecting a codon optimized for expression in the organism among the codons encoding the protein to be produced And may have a rearranged nucleotide sequence.
- the nuclease may be used in the form of a protein, a nucleic acid molecule (DNA or mRNA) encoding the same, a ribonucleic acid protein bound to a guide RNA, a nucleic acid molecule encoding the ribonucleic acid protein, or a recombinant vector containing the nucleic acid molecule .
- the nuclease or nucleic acid molecule encoding it may be in a form that can be delivered, acted on, and / or expressed in the nucleus.
- the nuclease may be a sibling that is easy to introduce into cells.
- the nuclease may be linked to a cell penetrating peptide and / or protein transduct ion domain.
- the protein transfer domain may be a poly-arginine or a TAT protein derived from HIV, but is not limited thereto.
- Various types of cell penetrating peptide or protein transfer domain other than the above-described examples are well known in the art, so that a person skilled in the art can apply various examples without limitation to the above examples.
- nuclease or encoding nucleic acid molecule may further comprise a nucleotide position signal (NLS) sequence or a sequence encoding the nucleotide signal.
- NLS nucleotide position signal
- an expression cassette comprising a nucleic acid molecule encoding said nuclease may comprise a regulatory sequence such as a promoter sequence for expressing said nuclease, or, in addition, an NLS sequence (e.g., SV40 NLS, etc.) have. Such NLS sequences are well known in the art.
- the nuclease or nucleic acid molecule encoding it may be associated with a tag for isolation and / or purification or a nucleic acid sequence encoding the tag.
- the tag may be appropriately selected from the group consisting of a small peptide tag such as Hi s tag, Fl ag tag, S tag, Glutathione S-transferase tag, MBP (Maltose binding protein) , But is not limited thereto.
- guide RNA refers to a target DNA-specific RNA (for example, a gene capable of hybridizing with a target site of DNA, Proteins, Cpf1, and the like to bind to nucleotides and lead to target DNA.
- the guide RNA may be appropriately selected depending on the kind of nuclease to be complexed and / or the microorganism from which it is derived.
- the guide RNA may be a CRISPR RNA (crRNA) comprising a site capable of hybridizing with a DNA target site;
- iRa-s-act ivat ing crRNA which contains sites that interact with endonucleotides such as Cas protein, Cpf l, and
- a single guide RNA in the form of fusion of the major part of the crRNA and the tracrRNA (for example, the hybridization site of the crRNA and the interaction site of the tracrRNA)
- RNA may be a dual RNA including CRISPR RNA (crRNA) and trans-act ivat ing crRNA (tracrRNA), or a single guide RNA (sgRNA) including a major part of crRNA and tracrRNA.
- crRNA CRISPR RNA
- tracrRNA trans-act ivat ing crRNA
- sgRNA single guide RNA
- the sgRNA comprises a sequence having a sequence complementary to the sequence in the target DNA
- the spacer region (Also referred to as the Spacer region, the target DNA recognition sequence, the base pairing region, etc.) and the hai rpin structure for Cas protein binding. More specifically, it may include a portion having a sequence complementary to the sequence in the target DNA, a hai rpin structure for Cas protein binding, and a terminator sequence.
- the structure described above may be sequentially present in the order of 5 'to 3', but is not limited thereto. Any type of guide RNA can be used in the invention if the guide RNA comprises a major portion of the crRNA and tracrRNA and a complementary portion of the target DNA.
- the Cas9 protein can be used to correct two target genes for target gene correction, namely CRISPR RNA (crRNA) having a nucleotide sequence capable of hybridizing with the target sequence region of the target gene and tranact iat ing crRNA Proteins).
- CRISPR RNA crRNA
- tracrRNAs can be used in the form of double-stranded, double-stranded crRNA: tracrRNA complexes, or in the form of a single guide RNA (sgRNA) linked through a linker.
- sgRNA single guide RNA
- the sgRNA comprises at least a portion which interacts with some or all of the crRNA comprising at least the hybridizable nucleotide sequence of the Cas9 crRNA and the Cas9 protein of the Cas9 tracrRNA Some or all of the containing tracrRNA may be through the nucleotide linker to form a hair-pin structure (a nucleotide linker may correspond to a loop structure).
- the guide RNA specifically the crRNA or sgRNA, comprises a sequence complementary to the sequence in the target DNA, and includes at least one, for example, 1 to 10, at the 5 'end of the crRNA or the upstream region of the sgRNA, , 1-5, or 1-3 additional nucleotides.
- the additional nucleotide may be guanine, but is not limited thereto.
- the guide RNA may contain crRNA, and may be appropriately selected according to the type of Cpf1 protein to be complexed and / or the microorganism derived therefrom.
- the specific sequence of the guide RNA can be appropriately selected according to the kind of the nuclease (Cas9 protein or Cpf 1) (that is, the derived microorganism), and it is easily understood by those skilled in the art There is something.
- the crRNA when the Cas9 protein from Streptococcus pyogenes is used as the target specific nuclease, the crRNA may be represented by the following general formula 1:
- N cas9 is a site that is determined according to the targeting sequence, that is, the sequence of the target sequence of the target gene (that is, a sequence capable of hybridizing with the sequence of the target site), and 1 is included in the targeting sequence
- the site containing the consecutive 12 nucleotides (GUUUUAGAGCUA) located adjacent in the 3 'direction of the target sequence is an essential part of the crRNA,
- X cas9 is a site containing m nucleotides located at the 3 'terminal side of the crRNA (i.e., located adjacent to the 3' direction of the essential part of the crRNA), and m is an integer of 8 to 12, And the m nucleotides may be the same or different, and may be independently selected from the group consisting of A, U, C, and G. 2019/147073 1 »(: 1 ⁇ ⁇ 2019/001104
- 3 < ; nd > 1/3 peach contains the targeting sequence of the egg show and the essential site
- oligonucleotide linker corresponds to the loop structure. More specifically,
- the oligonucleotide linker included in the above requirement may comprise 3 to 5 nucleotides, for example 4 nucleotides,
- the nucleotides may be the same or different and may be independently selected from the group consisting of A, U, C and G.
- the crRNA or sgRNA may additionally contain 1 to 3 guanines (G) at the 5 'end (i.e., at the 5' end of the targeting sequence region of the crRNA).
- G guanines
- the tracrRNA or sgRNA may further comprise a termination site comprising 5 to 7 uracil (U) at the 3 'end of an essential part (60 nt) of the tracrRNA.
- the target sequence of the guide RNA can be obtained by PCR using the PAM (Protospacer Adjacent Mot if) sequence on the target DNA (5.
- PAM Protospacer Adjacent Mot if sequence on the target DNA
- the targeting sequence of the guide RNA capable of hybridizing with the target sequence of the guide show is the DNA strand in which the target sequence is located (i.e., the PAM sequence (5'-NGG-3 'is A, T, G or C)
- a nucleotide sequence having at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or at least 100% complementarity with the nucleotide sequence of the complementary strand of the complementary strand By this means, complementary binding with the nucleotide sequence of the complementary strand is possible.
- the nucleic acid sequence of the target site is represented by the nucleic acid sequence of the strand where the PAM sequence is located in the two DNA strands of the corresponding gene site of the target gene.
- the targeting sequence included in the guide shows the sequence of the target site, Have the same nucleic acid sequence.
- the targeting sequence of the guide RNA and the sequence of the target site (or the sequence of the cleavage site) are represented by the same nucleic acid sequence, except that the U U virtual call is altered.
- the guide RNA may be used (or included in the composition) in the form of a plasmid (or included in the composition) in the form of a plasmid containing DNA encoding it or encoding it.
- on-target si te in the present invention means a position where mutation (cleavage, insertion, and / or deletion) is to be introduced using the target specific nuclease, Can be selected randomly and can be present within the coding sequence of a particular gene, May be present in non-coding DNA sequences that do not produce protein.
- the target-specific nuclease has sequence specificity, so it may act on the target site, but adverse effects on the non-target site (f-target si te) may occur depending on the target sequence.
- a non-target site refers to a site having a sequence that is not the same as the target sequence of the target-specific nuclease but has the activity of the target-specific nuclease. That is, a position that is cleaved by a target specific nuclease other than the target position.
- the non-target location can be used as a concept that includes the actual non-target location for a particular target-specific nuclease, as well as a location that is likely to be a non-target location.
- the non-target position may be any position other than the target position that is cleaved by the target specific nuclease in vitro, including but not limited to.
- gene scissors at locations other than the target site can be caused by various causes.
- gene scissors are likely to work in non-target sequences that are highly homologous to the target site with a target sequence designed for the target site and a mi- ciate mismatch (mi smatch).
- the non-target location may be, but is not limited to, a position with a target sequence and one or more nucleotide mismatches.
- the process of accurately detecting and analyzing non-target positions as well as the activity of the target-specific nuclease at the target site can also be of great importance, which may be important for target-specific, It may be useful for developing cleavage.
- the objective nuclease of the present invention can have nuclease activity in vivo in vivo) and in vitro (Un r / iro), and thus can be used to detect non-target positions of four-dimensional DNA in vitro When applied in vivo, it can be expected to have activity at the same position as the detected non-target position.
- the single-strand-specific endonuclease is treated on the single-stranded show through the step (a), and the remaining DNA strands are cut.
- the single-strand-specific endonuclease may be a generic term for any endonuclease having activity to specifically cleave single-stranded DNA.
- the single-strand-specific endonuclease is a combination of endonuclease V, or endo-nuclease VI with an alkyladenine DNA glycosylase (hMG) ≪ / RTI >
- the endonuclease V may be derived from a prokaryotic or eukaryotic cell such as a fungus, an eukaryotic animal (such as a fish, an amphibian, a reptile, a bird or a mammal), or an eukaryotic plant.
- Examples of the endonuclease V include endonuclease V (NP_418426.2; E. coli ENDOV), and the like.
- the single-stranded specific endonuclease may be one that cleaves a strand con- verted to adenosine-inosine, wherein the adeno-acid is displaced in the 5 'Quot; direction within 5 nt, within 4 nt, within 3 nt, within 2 nt, or within 1 nt of the nucleotide sequence.
- the endonuclease VIII plays a role of removing nucleotides converted by adenine to inosine by adenine diaminase.
- the N-glycosylase activity of removing inosine from the double-stranded DNA and the apurin i c one or more enzymes having all of the AP-lyase activity that cleaves the 3 'and 5' ends of si te (AP si te) can be selected.
- the endonuclease VIII may be selected from the group consisting of E, coli endonuclease VIII (e.g., GenBank Accession No. NP_415242.1, etc.), human endonuclease VIII
- GenBank Accession Nos. BAC06477.1, NP-082623.1, etc. Escherichia coli endonuclease VI1 (For example, GenBank Accession Nos. OBZ49008.1, 0BZ43214.1, 0BZ42025.1, ANJ41661.1, KYL40995.1, KMV55034.1, KMV53379.1, KMV50038.1, KMV40847.1, AQW72152.1 etc.) but it is at least one member selected from the group consisting of, without being limited thereto.
- the alkyl adenine DNA glycosylase is an enzyme that repairs DNA by removing the alkyl at ion-damaged purine base after the hydrolysis of the N-glycosidic bond.
- 3-methyl adenine, 7-methyl adenine, 7- methyl guanine, 1N ethenoadenine and hypoxanthine is an enzyme that repairs DNA by removing the alkyl at ion-damaged purine base after the hydrolysis of the N-glycosidic bond.
- 3-methyl adenine, 7-methyl adenine, 7- methyl guanine, 1N ethenoadenine and hypoxanthine is provided alkyl adenine DNA glycosylase.
- MAG human alkyl adenine DNA glycosylase
- NP_001015052.1 (coding nucleic acid sequence NM_001015052.2), NP_001015054.1 (choline nucleotide sequence NM_001015054.2), NP_002425.2 Nucleic acid sequence < / RTI > J302434.3), and the like, but is not limited thereto.
- the step (c) is a step of performing whole genome sequencing (WGS) using the DNA cleaved through the step (b), wherein a sequence having homology with the sequence of the target position is found, Target specific nuclease at the entire genomic level, as opposed to an indirect method of predicting a non-target location that is predicted to be non-target specific.
- WGS whole genome sequencing
- WGS whole genome sequencing
- next generation sequencing is a chip-based and PCR-based fair-end
- the step (d) comprises the step of determining a position at which the DNA is cleaved in the sequence reading obtained by the total dielectric sequencing, the sequencing data being analyzed to determine the target position of the target specific nuclease and the non- Can be easily detected. Determining the specific position at which the DNA is cleaved from the nucleotide sequence data can be performed by various approaches, and the present invention provides various reasonable methods for determining the position. However, this is merely an example included in the technical idea of the present invention, and the scope of the present invention is not limited by these methods.
- the position where the 5 'end is vertically aligned can indicate the position where the DNA is cleaved.
- vertical aligned is used to refer to a sequence of adjacent Watson strands and Crick strands when analyzing the overall dielectric sequencing results with a program such as BWA / GATK or ISAAC.
- the target specific nuclease has nuclease activity at the target position and the non-target position, and when the position is cleaved, when the nucleotide sequence data is aligned, the common cut regions are respectively located at the 5 'terminus , But they can be arranged in a staggered manner during alignment because there is no 5 'end in the uncut portion.
- the vertically aligned position can be seen as the site cleaved by the target specific nuclease, which may be the target or non-target location of the target specific nuclease.
- alignment means mapping base sequence data to a reference genome and then aligning the bases having the same position in the dielectric to each position.
- any computer program can be used as long as the base sequence data can be arranged in the same manner as described above, and it can be selected from known programs already known in the art or programs tailored to the purpose. In one embodiment, alignment is performed using ISAAC, but the present invention is not limited thereto.
- the position of the DNA cleaved by the target specific nuclease can be determined by a method such as the above-mentioned finding of the position where the 5 'end is vertically aligned as described above, and the cleaved position is determined on- target si te), it can be determined as a non-target position (of f-target si te).
- the sequence identical to the nucleotide sequence designed with the target position of the target specific nuclease is the target position, and the sequence not identical to the nucleotide sequence can be regarded as a non-target position. This is the definition box name of the non-target location described above.
- the non-target position may in particular be composed of a sequence having a homology with the sequence of the target position, and specifically includes a sequence having one or more nucleotide mismatches with the target sequence, more specifically, 1 to 5, 1 to 4, 1 to 3, 1 to 2, or 1 nucleotide mismatch, but is not particularly limited thereto, and the target specific nuclease cleavage It can be included in the scope of the present invention.
- the target position is a guide 15-30 nucleotides sequence, and may additionally include a sequence that is recognized by a target specific nuclease (e.g., a PAM sequence recognized by Cas9 in the case of Cas9).
- the double peak pattern in addition to finding a vertically aligned position at the 5 'end, if the double peak pattern is seen in the 5' end plot, it can be determined as a non-target position if the position is not a target position.
- a graph is drawn by counting the number of nucleotides constituting the 5 'end of the same base at each position in the genomic DNA, a double peak pattern appears at a specific position, and the double peak is cut by the target specific nuclease Because it is represented by each strand of the double strand.
- the genomic DNA is cleaved with a target specific nuclease (e.g., RGEN), followed by total dielectric analysis and aligned with ISAAC, followed by vertical alignment at the cut position and staggered at the non- The alignment pattern was confirmed, and it was confirmed that a unique pattern of the double peak at the cut position is shown when the 5 'end plot is shown.
- a target specific nuclease e.g., RGEN
- Watson strand and Crick strands (Cr i ck strand) corresponding to the nucleotide sequence data (sequence read) respectively where the vertically aligned by more than one non-target locations And 20% or more of the nucleotide sequence data is aligned vertically, and the number of base sequence data having the same 5 'end in each of the Watson's strand and Creek strand is 10 or more, that is, .
- the non-target localization can be performed by treating the target specific nuclease to the genomic DNA in vitro in UnF / fro.
- a non-target effect is also observed in the non-target position (detected substantially in vivo Un FO) detected by the above method.
- this is merely an additional verification process, and thus is not a step that is necessarily essential to the scope of the present invention, and is only a step that can be performed additionally if necessary.
- the term " of f-target ef fect " may be a concept distinct from a non-target position (i.e., f-target si te)
- the concept of a non-target position in the invention refers to a position other than the target position among the positions at which the target-specific nuclease can operate, and the position to be cleaved by the target specific nuclease
- the non-target effect refers to the effect of inducing indentation (or deletion) by target-specific nuclease at non-target sites in the cell.
- Indel " refers to mutations in some bases in the nucleotide sequence of DNA, which are insertions and / or deletions.
- target specific nuclease the non-target location in the present specification can be viewed as a concept involving a non-target indelible position, and is a position that is likely to have a target-specific nuclease activity
- Target position is a candidate of f-target si te
- non-target indel position is a proven non-target position in the present invention. It can also be named as the target position (valued of f-target si te).
- the verification procedure may include, but is not limited to, isolating genomic DNA from cells expressing the target specific nuclease at the non-target location, identifying the indel at a non-target location of the DNA,
- the non-target effect of This can be accomplished by performing an indel check method known in the art, such as T7E1 analysis, mutation detection analysis using Cel-I enzyme, or targeted deep sequencing, to confirm non-target effects.
- the step of verifying the non-target effect may be to directly ascertain whether the indel has occurred at the non-target location. However, even if the indel has not occurred in the in vivo verification process, it should be regarded as an auxiliary means since it has not been confirmed until the indel condition occurs at a frequency below the detectable level.
- the formula for calculating the DNA cleavage score at the position i of each nucleotide is provided based on the alignment pattern of the nucleotide sequence data as follows: 2019/147073 1 »(: 1/10/06 019/001104
- the number of nucleotide sequence data in the above equation means the number of nucleotide leads, and the sequencing depth means the number of sequencing leads at a specific position.
- C may be, but is not limited to, 1 to 1000, 1 to 500, 1 to 100, 1 to 50, 1 to 10, 1 to 5, or 1 to 3.
- the calculated score is 2.5 or more, 0.1 or more, and the C-value is 1, (For example, when it contains PAM (5'-NGN_3 'or 5'-NNG-3') with 0n-target sequence and 10 mismatches or less), the arbitrary position (truncated position) Can be determined as a non-target position.
- the criteria of the score may be appropriately adjusted or changed by a person skilled in the art depending on the purpose.
- the Digenome-seq method provided herein may be performed using a plurality of target specific nuclease agents (e. G., A target specific nuclease comprising a plurality of different guide RNAs with different target sites)
- target specific nuclease agents e. G., A target specific nuclease comprising a plurality of different guide RNAs with different target sites
- this is referred to as "compound digenome-seq "
- the target-specific nuclease may be a mixture of two or more, specifically 2 to 100, target-specific nuclease agents, no.
- RNA-guided engineered nuclease RGEN
- RGEN RNA-guided engineered nuclease
- the method of identifying the f-target may be performed by the method described in Nature Methods 14, 607-614 (2017) and / or SITE-seq (Cameron, P. et al., Mapping the genomic landscape of CRISPR_Cas9 c leavage., Nature methods 14, 600-606 (2017)).
- FIG. 1A schematically illustrates a genomic DNA sequencing process using adenosine deaminase, Cas9 nicase (D10A) (ABE) and endonuclease V.
- FIG. 1A schematically illustrates a genomic DNA sequencing process using adenosine deaminase, Cas9 nicase (D10A) (ABE) and endonuclease V.
- Figure lb shows that adenosine deaminase and Cas9 nicase (D10A) (ABE)
- Alkyladenine DNA Glycosylase (AAG) and endonuclease VIII.
- FIG. 2A shows the results of confirming the generation of DSB (Double Strand Break) through real-time PCR after treating ABE and endonuclease V.
- FIG. 2A shows the results of confirming the generation of DSB (Double Strand Break) through real-time PCR after treating ABE and endonuclease V.
- FIG. 2B shows the results of confirming the generation of DSB through real-time PCR after treating ABE, Alkyladenine DNA Glycosylase (AAG), and endonuclease VIII.
- FIG. 2C shows the result of confirming the base correction after treatment of ABE and endonuclease V or treatment of ABE, Alkyladenine DNA Glycosylase (AAG), and endonuclease VIII.
- FIGS show the results of whole-genome sequencing (WGS) after treatment with ABE and endonuclease V or with ABE, Alkyladenine DNA Glycosylase (AAG), and endonuclease VIll.
- Figure 4 shows the results of identifying the cleavage site after treatment with ABE and endonuclease or with ABE, Alkyladenine DNA Glycosylase (AAG), and Endonuclease VIII.
- FIG. 5 schematically shows a sequence of genome DNA sequence analysis by CIRCLE-seq and SITE_seq.
- FIGS. 6A to 6C are graphs showing the base correction frequency (%) and insertion / deletion frequency (%) obtained using sgRNA having 0 to 4 mismatched bases on the target site, activity ([mismatched sgRNA result value] / [matched sgRNA result value]
- the last three nucleotides at the 3 'end represent the PAM sequence and the small letters represent the mismatched nucleotides, respectively (Means + sem were from three independent experiments).
- FIG. 7A is an overview of the digenome-seq assay using ABE7.10 and Endo V, wherein ABE7.10 catalyzes the conversion of guanine to deoxyinosine and Endo recognizes deoxyinosine,
- the second phosphodiester bond 3 'for inosine is cleaved and the position of the triangle vertex shows the position of phosphodiester bond cleaved by ABE7.10 nickase and Endo V.
- Figure 7b is a schematic diagram of an ABE-mediated digenome-seq workflow
- Figure 7c is a schematic diagram of a Digenome-seq assay using ABE7.10 and hAAG / Endo VIII, wherein ABE7.10 mediates adenine-to-inosine transformation in one strand and nicks in the other strand, AP site (apurinic / apyrimidinic site), Endo VI 11 (DNA glycosylase and AP-lyase) cleaves the AP site, the triangle vertex positions are ABE7.10 nickase and hAAG / Endo VIII Shows site cut by.
- the plots show the results of treatment of ABE7.10 and Endo V with the PCR product containing the ABE7.10 target sequence, showing that the double strand break was expressed in DNA only when both ABE7.10 and Endo were treated.
- Figure 7E shows the results of treatment of ABE7.10, MAG, and Endo VIII with PCR products containing the ABE7.10 target sequence. Only when ABE7.10, MAG, and Endo VIII were all treated, strand breaks are formed.
- Figure shows the results of Sanger sequencing, showing A-to-G conversion by ABE7.10 and DNA cleavage by Endo V.
- Fig. 7g is a graph showing the results of qRT-PCR, showing DNA cleavage by ABE7.10 nickase and Endo V. Fig.
- Figure 7h shows the results of Sanger sequencing, showing the A-to-G conversion when genomic DNA was treated with ABE7.10 and the genomic DNA with ABE7.10,
- a MAG, and Endo VIII shows the conversion of a G-to ⁇ A hangyeongwoo treatment.
- Pottery is a graph showing the results of qRT-PCR, showing the results of digesting the genome show with ABE7.10, hMG, and Endo VIII. 2019/147073 1 »(: 1/10/06 019/001104
- 7J is an IGV image showing the result of straight alignment of sequence reads at the HEK2 target site obtained using WGS data.
- Figure 7k is an IGV image showing the results of the straight alignment of sequencing reads observed after ABE7.10, hAAG, and Endo VIII treatment.
- 8 shows SDS-PAGE analysis of purified ABE7.10 using nickel affinity chromatography and heparin bead chromatography (M: marker, 1 (-): cell lysate before IPTG (isopropyl- Ni-NTA agarose beads after (1), (2), (3), and (4) elution of bound protein, NE: protein fraction eluted from nickel beads, Hp: Heparin Sepharose 6 Fast Flow affinity resins after elution of bound protein, HE: protein fraction after purification using heparin beads. ).
- M marker
- 1 (-) cell lysate before IPTG (isopropyl- Ni-NTA agarose beads after (1), (2), (3), and (4) elution of bound protein
- NE protein fraction eluted from nickel beads
- Hp Heparin Sepharose 6 Fast Flow
- Figure 9 outlines the in vitro DNA cleavage scoring system for Digeneome-seq analysis of ABE.
- Figures 10a and 10b show the ABE7.10 off-target site across the genome identified by Digenome-seq, with 10a being the complete genomic DNA (gray; first region from the center circle) and ABE7.10 + Endo V treated Genome showing DNA cleavage scores obtained through WGS using genomic DNA (blue; second region from the center circle) and ABE7.10 + hAAG + Endo VIII treated genomic DNA (red, third region from the center circle) -wide Circos plots (arrows indicate on-target sites), 10b shows the number of in vitro cleavage sites in the human genome identified using ABE7.10 + Endo V or ABE7.10 + hAAG + Endo VIII Venn diagram.
- Figure 10c is a Sequence logos obtained by comparing DNA sequences in Digenome-captured sites and using WebLogo.
- Figure 11 is an IGV image showing the results of vertical alignment of sequencing reads at the RNF2, TYR03, WEE1, EphB4, HPRT_exon6, and HPRT_exon8 sites.
- Figures 12a and 12b show the complete genomic DNA (gray) (first region at the center) and genomic DNA treated with ABE7.10 / Endo V (blue (12a) and RNF2 (12b) regions using the genomic DNA processed from BE3AUGI / USER (third region from the center circle) or Cas9 (green region from the center circle) The resulting genome-wide Circos plots are shown, and the arrows indicate the on-target sites.
- Figures 12c and 12d are sequence logos obtained by comparing DNA sequences in digenome-captured sites and using WebLogo.
- FIG. 12E is a graph showing the efficiency of base correction at ABE7.10 off-target sites measured using targeted deep sequencing, with three nucleotides at the 3 'end representing the PAM sequence and a lower case representing the mismatched base (Means Sem was from three independent experiments).
- Figure 12f is Scatterplots showing the correlation between the ABE7.10-mediated and Cas9-mediated indel frequencies (above) and between the ABE7.10-mediated and the BE3-mediated substitution frequencies.
- Figure 13a illustrates an exemplary target DNA-complementary RNA sequence of sgRNAs of the commonly used forms (GX 19) , truncated forms (gX 18 or gX 17 ), and extended forms (gX 20 or ggX 20) .
- Figure 13b shows the average of the baseline correction frequencies of ABE7.1o for each strain sgRNA measured by targeted deep sequencing when using modified sgRNA targeting HEK2, RNF2, TK_EphB4, TYR03, WEE1, HPRT-exon6, and HPRT-exon8 Graph.
- Figures 13c and 13d show the frequency (%) of ABE7.10-mediated bases in the on- or off-target sites of HPRT-Exon 6 (13c) and TYR03 and HPRT-exon8 (13d), measured by targeted deep sequencing in HEK293T cells ),
- the heat map shows the relative specificities of the modified sgRNA compared to the case of using the GX 19 sgRNA, and the relative specificity is the specificity of the sgRNA to be measured Target frequency / off-target frequency divided by the on-target frequency / off-target frequency of the GXig sgRNA, and the 3 nucleotides at the 3 'terminal end indicate the PAM sequence,
- the lower case represents a mismatched base.
- Figures 13e and 13f show on- and off-target values at sites on HPRT-Exon 6 (13e) and HPRT-exon8, HEK2, EphB4 and TYR03 (above, 13f) using ABE7.10 and Sniper A law:
- the activity and specificity ratio In the graph and heat map showing, the three nucleotides at the 3 'end represent the PAM sequence, the lower case represents the mismatched base, and the specificity ratio is calculated by the formula: Sniper ABE7.10 specificity (on-target frequency / off-target frequency) / specificity (on-target frequency / off-target frequency) of ABE7.10 (Means ⁇ sem were from three independent experiments).
- Fig. 13g and 13h are in on in part the HPRT-Exon 8 (13g) and HPRT_exon6, HEK2, EphB4 and TYR03 (or more, 13h) of the case that is passed into the furnace and the RNP form when delivered into a cell through a plasmid cell - and off-target activity and specificity ratio, the 3 nucleotides at the 3 'end represent the PAM sequence, the lower case represents the mismatched base, and the specificity ratio is calculated by the following equation (On-target frequency / off-target frequency) / specificity (on-target frequency / off-target frequency) of the RNP transfer (Means + sem were from three independent experiments).
- Figures 13i and 13j show the on- and off-target activity and specificity ratios at the HPRT-exon6 site (13i) and the HPRT-exon8 site (13j) when ABE7.10 or Sniper A alternative 7.10 was used in combination with the modified sgRNA the specificity ratio is calculated by the following equation: Sniper ABE7.10 and variant: (3) the nucleotide at the 3 'end is the PAM sequence, the lower case is the mismatched base, (on-target frequency / off-target frequency) / specificity (on-target frequency / off-target frequency) of ABE7.10.
- Example 1 Genomic DNA sequencing analysis of endonuclease V treated with adenosine deaminase and Cas9 nicase (D10A)
- DNA extracted from cells is mixed with adenosine deaminase-cas9 nicase (D10A, the tenth amino acid of Cas9 is changed from D to A) and the guide RNA complex, one strand of DNA is cleaved and the other strand A I.
- D10A adenosine deaminase-cas9 nicase
- the endonuclease V is treated with the reacted DNA, it recognizes I and induces cleavage.
- DSB double strand break
- the whole double-stranded DNA (DSB) DNA is subjected to an end-repellent process to proceed with full-gene sequencing.
- DNA DNA with two strand breaks
- the sequencing sequence at the truncated portion is a straight al ignment at the on-target position, Target of the adenosine deaminase-Cas9 nicase can be detected (Fig. La).
- Example 2 Analysis of genomic DNA sequence of Alkyl adenine DNA Glycosylase treated with adenosine deaminase and Cas9 nicase treatment Further, adenosine deaminase-Cas9 nicase treated DNA
- FIGS. 2A and 2B After treatment with ABE, endonuclease V or ABE, Alkyl adenine DNA Glycosylase (MG), and endonuclease VIII, D-status was confirmed by real-time PCR and the results are shown in FIGS. . As shown in FIGS. 2A and 2B, when ABE alone was treated, it was found that about 50% of the ABE was cleaved. However, treatment with ABE, endonuclease V or ABE, Alkyl adenine DNA Glycosylase (AAG), endonuclease VIII It can be seen that more than 90% of the DNA target site is cleaved. To verify this phenomenon again, sanger sequencing was performed. After treatment with ABE, we could see that A changed to G.
- AAG Alkyl adenine DNA Glycosylase
- CIRCLE-seq Tsai, SQ et al.
- CIRCLE-seq a highly sensitive in vitro screen for genome-wide CRISPR_Cas9 nuclease off-targets. Nature methods 14, 607-614 (2017)) and SITE-seq (Cameron, P. et al., Mapping the genomic landscape of CRISPR-Cas9 cleavage, Nature methods 14, 600-606 (2017) (See FIG. 5).
- HEK293T cells ATCC CRL-11268, were cultured in DMEM medium supplemented with 10% (v / v) FBS and 1% (v 8) penicillin / streptomycin and HEK293T cells (1.5x10 5 )
- ABE Additional plasmid # 113128; 1.5 g; DNA weight
- BE3 Additional plasmid # 73021; rAPOBECl
- Plasmid Plasmid (0.5 // g) containing 70-80% of the plasmid encoding the plasmid pGFP-XTEN-Cas9n-UGI-NLS; 1.5 // g) or SpCas9 (Addgene plasmid # 43945 NP_269215.1; 0.5 // g) confluency.
- ABE or ABE7.10 is used to refer to the same protein, specifically adenosine deaminase (directed evolution of E. coli TadA (NP417054.2)) and SpCas9 nicase D10A) (a variant Cas9 in which the 1 < th > amino acid residue D of SpCas9 (NP_269215.1) is substituted with A) (refer to Reference Example 2).
- ABE7.10 protein (10 // g) and in vitro transcribed sgRNA (6 // g) or ABE7.10 protein (0.6 / zg) and sgRNA
- TM Neon
- the sgRNA used in the following examples is the target site (in Tables 8-18,
- N cas9 V N cas9
- ABE7.10 was designed as follows (from the N-terminus to the C-terminus):
- MUV VEFS suppression YWMRHALTLMRAWDE rule VPVGAVLVHNNRVIGEGWNRPIGRHDPTAHAEIMALRQ (XiLWQNYI ⁇ IDAUYVTLEPCVMCAGAMIHSRIGRVVFGARDAKTGAAGSLMDVLHHPGMNHRVEITEGIL
- LB Luria-Bertani
- IPTG isopropy1-bD-1-thiogalactopyranoside
- the suspension was then incubated in liquid nitrogen and water bath (37 ° C) alternating 3 times. Cells were lysed by sonication for 9 minutes (5 seconds on, 10 seconds off) and the lysates were cleared by centrifugation at 15,000 g for 20 minutes. The supernatant was incubated with nickel agarose beads (Ni-NTA, QIAGEN) for 60 min at 4 ° C with shaking.
- Ni-NTA nickel agarose beads
- the lysate-resin mixture was loaded on a polypropylene column and washed with three column volumes of wash buffer (50 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich), 35 mM imidazole , 1 mM DTT (GoldBio), 10 yM ZnCl 2 (Sigma-Aldrich), pH 8.0) to wash, and eluting the bound protein half complete heavy liquid (50 mM sodium phosphate (Sigma- Aldrich), 150 mM NaCl (Sigma- Aldrich), 250 mM imidazole (Sigma-Aldrich), 20% glycerol, 1 mM DTT (GoldBio), 10 uM ZnCl and eluted with a 2 (Sigma-Aldrich), pH 8.0).
- wash buffer 50 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich
- the eluted protein fractions were placed in a polypropylene column containing Heparin Sepharose 6 Fast Flow (GE Healthcare) and washed with 3 column volumes of wash buffer (50 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl - Aldrich), 20% glycerol, 1 mM DTT (GoldBio), 10 yM ZnCh (Sigma-Aldrich), pH 8.0).
- wash buffer 50 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl - Aldrich), 20% glycerol, 1 mM DTT (GoldBio), 10 yM ZnCh (Sigma-Aldrich), pH 8.0.
- the bound proteins were eluted with elution buffer (50 mM sodium phosphate (Sigma-Aldrich), 750 mM NaCl (Sigma-Aldrich), 20% glycerol,
- ABE7.10-mediated in vitro digestion of PCR amplicon PCR amplicon containing the target site of the target gene was incubated at 37 for 1 hour with ABE7.10 And incubated with in vitro transcribed HEK2-targeted sgRNA.
- the resulting deaminated product was purified using a PCR purification kit (MGmed).
- the diaminated product 2 was digested with Endo V (NP_418426.2) (40 units) (New England BioLabs) or human Alkyladenine DNA Glycosylase (hAAG; NP_001015052.1, New
- SgRNA was isolated using RNase A (50 // g / ml) The show was purified using DNeasy Blood & Tissue Kit (Qiagen). 3 // g of the purified DNA was incubated with Endo V (40 units) or hAAG (10 units) and Endo VI 11 (20 units) at a reaction volume of 200ra table for 2 hours at 37 ° C and DNeasy Blood & Tissue Kit (Qiagen).
- the purified DNA was incubated with KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems) and analyzed by real-time quantitative PCR (qPCR) to determine whether ABE7.10- and Endo V- or ABE7.10-, hAAG-, and Endo VII I-mediated DNA double-strand breaks (complete genomic DNA fractions were measured by comparative C T analysis).
- KAPA HiFi HotStart PCR polymerase # KK2502; KAPA BIOSYSTEMS was used to amplify the on-target sites and potential off-target sites.
- a deep sequencing library was generated by amplifying the amplicon amplified using a TruSeq HT Dual index-containing primer.
- the generated library was sequenced using Illumina MiniSeq equipped with a paired-end sequencing system.
- Base editing frequencies represent the frequency of modified target regions corrected with one or more edit in the editing window (positions 4-7).
- Computer program source code for calculating substitution and indel frequency can be found at 'https://github.com/ibs_cge/maund'.
- Example 5 Tolerance for mismatched sgRNAs of ABE7.10, BE3, and Cas9
- plasmids coding for sgRNA containing 1 to 4 mismatched bases on the target site and ABE7.10, BE3 (Base Editor plasmid; Addgene plasmid # 73021; rAP0BEC1- (See Reference Example 1), and the target in-depth sequencing of Reference Example 6 was performed to determine the base correction frequency and the insertion rate of the insert (SEQ ID NO: / Indel frequency was measured.
- Target gene HEK2 (on target site: GAACACAAAGCATAGACTGCGGG)
- the ' NGG ' portion of the terminus is a PAM sequence
- the sgRNA sequence is a sequence that is converted to a U in a sequence other than the PAM sequence in the target site sequence;
- Dia Illumination (adenine deaminat ion) - in vitro (/ / z vitro) in order to determine the target site of the genome throughout the ABE7.10 through Digenome seq-dependent
- the resulting double-stranded DNA to the cut (DSB), adenine DSB was generated at the site containing the inosine produced by the inosine.
- Uracil-Specific Excision Reagent (USER), a mixture of E.
- E. coli endo-Nuclease V endo V
- human Alkyladenine DNA Glycosylase MAG
- Endo VIII cleaves phosphodiester linkages at the inosine containing site (See Figs. 7A to 7C).
- Endo V recognizes inosine in DNA and cleaves the second phosphodiester bond 3 to inosine
- hAAG cleaves inosine to produce an apurinic / apyrimidinic site, which is recognized and processed by Endo VIII Leading to a single-strand break.
- the target DNA sequence iHEK2 on-taeger sequence (See Table 8) was treated with recombinant ABE7.10 protein expressed in E. ca // (the result of the purification is shown in Fig. 8) and their sgRNAs, resulting in adenine diamination in one DNA strand (Fig. 7a) and then treated with Endo V (see Fig. 7a) or hMG / Endo VIII (see Fig. 7c) to generate composite D status.
- the results are shown in Figs. 7D and 7E.
- the PCR amplification can be confirmed to be cleaved by ABE7.10 + Endo V ( Figure 7d) or ABE7.10 + hMG / Endo VIII ( Figure 7e).
- the human genome show isolated from HEK293T cells was identified as ABE7.10 + Endo V or ABE7.10 after confirming that D status could be generated at the ABE- + hMG / Endo VIII and analyzed by digenome-seq analysis to determine whether the ABE off-target site across the genome could be identified.
- Human genomic DNA isolated from EK293T cells was digested with ABE7.10 ribonucleoprotein (RNP) (300 nM ABE7.10 and 900 nM sgRNA, targeted to an endogenous chromosomal site
- ⁇ HEK2 on-taeger sequence (See Table 8)) for 8 hours and then incubated for 2 hours with repair enzyme (Endo V or hAAG / Endo VIII).
- repair enzyme Endo V or hAAG / Endo VIII.
- the target site the opposite strand with nick is sequenced Catalyzed DNA cleavage at the ABE-mediated adenine-to-inosine conversion (inosine changed to guanine during PCR amplification) and inosine-containing region (nicked DNA strand not amplifiable) 7F).
- the genomic DNA is treated with ABE7.10 and Endo V, the DNA strand containing inosine is cleaved by endo 5 V.
- DNA strands that are cleaved by Endo V can not be amplified by PCR, but DNA strands that do not have inosine are amplified. As a result, only adenine was observed and no guanine was observed at the correction sites observed by Sanger sequencing (Fig. 7f).
- ABE7.10 RNPs in addition to the HEK2 gene target ABE7.10 RNP used in Example 6, a genomic sequence (Digenome-seq 1.0: site with a DNA cleavage score of 2.5 or higher (Site with a DNA cleavage score of 0.1 or more and less than 10 mismatches with PAM (5'-NGN-3 'or 5'-NNG_3') as non-target location ), And the results are shown in Tables 11 to 18 and FIG.
- Digenome analysis was performed using BE3AUGI or Cas9, respectively, of two of the seven sgRNAs analyzed in this example (HEK2 and RNF2), and the results were compared with the results obtained using ABE7.10 , And Figs. 12A and 12B.
- HEK2 sgRNA was used in combination with ABE7.10, BE3AUGI, and Cas9, respectively, 17, 2, and 24 in vitro cleavage sites were generated (see FIG. 12C).
- RNF2 sgRNA produced 5, 1, and 13 truncation sites, respectively, when used in conjunction with ABE7.10, BE3AUGI, and Cas9 , respectively (see FIG. 12d).
- Such bulge-forming Site is a common feature of BE 3 of f-target si te, but is rarely observed in Cas 9 of f-target si te.
- sgRNA deformation RNP intracellular delivery instead of plasmid
- Sniper-Cas9 E. engineered Cas9 variants obtained from di rected evolutions in coli; Use the E539S, M761I, and K890N mutations for the wild-type SpCas9 amino acid sequence (NP_269215.1), with additional D10A mutations).
- (sgRNA variant) means that the targeting sequence of the sgRNA is a standard GXis sgRNA (Xi 9 : 19 arbitrary nucleotides (each independently selected from A, U (T), C, and G) consisting of 20 nucleotides ;
- the number of X latter meaning any number of nucleotides, hereinafter the same) of the display from the 5 'terminal part of the' X 'in one or two cutting sgRNA (gXis or gX 17 to remove the nucleotides) (g is the target site the corresponding sequence that is inconsistent guanine to, G denotes a guanine matching the corresponding sequence of the target site) or 5 ' referred to as extended sgRNA (gX 2 o or gg3 ⁇ 4o that the terminal further comprises a one or two extra guanine ) Was used to measure the frequency of base correction in HEK293T cells.
- the HPRT exon 6 target sequence as a representative example of the used modified sgRNA is shown in FIG.
- the modified sgRNA for the other gene was prepared with reference to Fig. 13A based on the 0n-target DNA sequence of each gene described in Table 11.
- Figure 13B is a graph showing the average of the baseline correction frequencies of ABE7.10 for each modified sgRNA measured by targeted deep sequencing when using modified sgRNA targeting HEK2, RNF2, TK_EphB4, TYR03, WEE1, HPRT-exon6, and HPRT_exon8.
- the modified sgRNA reduced ABF7.10 of f-target activity in almost all sites while maintaining the targeting activity (in Figure 13d, the y-axis graph shows ggX20, gX20, GX19, gX18, gX17, and untreated).
- the y-axis graph shows ggX20, gX20, GX19, gX18, gX17, and untreated.
- the four verified of f-target si te identified using HPRT Exon 6-targeting GX 19 sgRNA gX 2 o or ggX 2 o sgRNA were compared with existing GX 19 sgRNA- The speci fi c ty test was improved by 2 to 26 times.
- the use of Sniper ABE7.10 in combination with modified sgRNAs can further reduce the off-target effect of ABE7.10 (see Figures 13i and 13j;
- the y-axis graph shows the results of ggX20_Sniper ABE7.10, gX20_Sniper ABE7.10, GX19_Sniper ABE7.10, gX18_Sniper ABE7.10, gX17_Sniper ABE7.10, GX19-ABE7.10, and untreated Lt; / RTI >
- ABE7.10, BE3, and Cas9 can recognize different sets of off-target sites in the human genome, And that it is necessary to use the evaluation tools.
- ABE7.10 diaminase targeting the seven genes tested confirmed that it specifically and specifically catalyzes A-to-G conversion in vitro at a limited number of sites in the human genome. It has also been found that by using modified sgRNAs, ABE7.10 RNPs, and / or Sniper ABE7.10, the off-target activity of ABE7.10 can be reduced or eliminated.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
디아미나아제 및 표적특이적 뉴클레아제를 포함하는 염기 교정용 조성물, 및 상기 염기 교정용 조성물을 이용한 염기 교정 방법 및 유전자 변형 동물 제조 방법이 제공된다.
Description
【발명의 설명】
【발명의 명칭】
아데노신 디아미나아제를이용한염기 교정 확인 방법 【기술분야】
아데노신 디아미나아제 (adenosine deaminase)를 이용한 염기 교정 확인 기술과 관련된 것으로, 아데노신 디아미나아제를 포함하는 염기 교정 확인용 조성물 및 DNA에 아데노신 디아미나아제를 처리하는 단계를 포함하는염기 교정 확인방법이 제공된다.
【발명의 배경이 되는기술】
CRISPR/Cas9 system은 인간세포를 비롯한 여러 동식물의 유전체 교정에 사용되어 왔지만 Cas9의 경우 DNA 절단으로 인해 표적부위에 DNA의 삽입이나결실이 일어나게 된다. 이러한단점을보완하기 위해 최근에 Cas9 니케이즈 (Cas9 nickase)에 시토신 디아미나아제 (cystidine deaminase)를 연결시켜 DNA 절단 없이 C를 T0J)로 (또는 보다 낮은 빈도로, C를 G또는 A로 변환) 변환시키는방법이 개발되었다 (Komor , A.C. , Kim, Y.B., Packer , M.S. , Zuris , J.A. & Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420-424 (2016) 참조) .
하지만시토신 디아미나아제 (cystidine deaminase)를 이용하여 C를 T로 (또는 보다 낮은 빈도로, C를 G 또는 A로 변환) 변환하거나 C에 대응하는 반대쪽 strand의 G를 A로 (또는 보다 낮은 빈도로, G를 C 또는 로 변환)하는것은가능하지만, 퓨린 계열 염기인 A또는 를다른 염기로 바꾸는방법은알려져 있지 않았다.
【발명의 내용】
【해결하고자하는과제】
본 명세서에서는 .절단유전체 시퀀싱 (digested genome sequencing, Digenome-seq; 표적 특이적 뉴클레아제 처리 전과 후를 한 눈에 파악해 잘린 위치를구별하는방식)을 이용하여 아데노신 디아미나아제 (adenosinedeaminase)를 표적특이적 뉴클레아제 (예컨대, Cas9 니케이즈 또는 촉매적으로 결핍된 Cas9 (dCas9) 등)에 결합시켜 만든 아데노신 염기교정
유전자 가위의 염기 교정을 확인하는 방안을 제공하며, 이를 통하여 상기 아데노신 염기교정 유전자 가위에 의한 염기 교정의 특이성을 평가할 수 있다.
S. pyogenes 유래 Cas9 니카아제 및 E. col i 유래 조작된 아데닌디아미나아제로 구성된 Adenine Base Edi tors (ABEs)는 가이드 RNA 표적 방식으로효율적인 아데닌/구아닌 단일 뉴클레오티드 전환을가능하게 하여, 동물 (예컨대, 인간 등의 포유류, 마우스 등의 설치류 등) 및/또는 식물의 세포에서 점돌연변이를 유도하거나 교정한다. ABE는 다른 유전자 교정 수단인 Cas9 , BE (cytosine base edi tor ) 등과 다른 of f-target s i te를 인식하므로, 그 효율성 평가를 위하여 독립적인 방법을 개발하는 것이 필요하다. 본 명세서에서는 전체 유전체 시퀀싱 (whole genome sequencing: WGS)을 통하여, ABE의 유전체 (genome) DNA 전체에 대한표적 특이성을 평가하는 방법을 제시한다. 상기 전체 유전체 시퀀싱은 구체적으로 Digenome-seq일 수 있다. 본 명세서에서 제공되는 Digenome-seq 기반 방법은 0. 1%의 치환 빈도 (subst i tut ion frequency)로 ABE 비표적 위치를 포획하기에 충분한 정도로 민감하다. 또한, 미리 조립된 ABE 리보핵산단백질, 변형된 가이드 RNA, 및 Sniper-ABE를 사용하여 인간등의 동물세포에서 ABE of f-target 활성을감소시킬수 있음을제안한다.
일 예는,
(a) ( 1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나 또는분리된 DNA에 처리하여, DNA이중가닥중하나를절단하는단계; 및
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuclease V) 또는, Alkyl adenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI I I (endonuclease VI I I )의 조합)를처리하여, 나머지 DNA가닥을절단하는단계
를포함하는, 아데닌디아미나아제를사용하여 DNA에 이중가닥절단 (double strand break)를생성하는방법을제공한다.
다른예는,
(a) ( 1) 아데노신 디아미나아제, 이를 암호화하는 핵산분자, 또는 상기 핵산 분자를 포함하는 플라스미드, 및 (2) 표적 특이적 뉴클레아제,
이를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드를 분리된 세포에 도입시키거나또는분리된 DNA에 처리하여, DNA이중가닥중 하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuclease V) 또는,
Alkyladenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI I I (endonuclease VI I I )의 조합)를 처리하여, 나머지 DNA 가닥을 절단하여, 이중 가닥 절단 (double-strand break)를형성하는단계 ; 및
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계
를포함하는 DNA서열분석 방법을제공한다.
다른예는,
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산분자, 또는상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나 또는분리된 DNA에 처리하여, DNA이중가닥중하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 ¥ (endonuclease V) 또는, Alkyladenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI I I (endonuclease VI I I )의 조합)를 처리하여, 나머지 DNA 가닥을 절단하여, 이중 가닥 절단 (double-strand break)를형성하는단계;
(c) 상기 이중가닥 절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계; 및
(d) 상기 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 상기 절단된위치를확인하는단계
를포함하는,
표적 특이적 뉴클레아제의 절단위치 또는 비표적 위치 (of f-target s i te)를검출하는방법을제공한다.
【과제의 해결수단】
아데노신 디아미나아제 (adenosine deaminase)를방향진화 (di rected evolut ion)시킴으로써, DNA의 염기 중 A(adenos ine)를
B
디아미네이션 (deaminat ion)시켜서 K inosine)으로 치환시키는데 성공한 바 있다. 보다 구체적으로, Cas9 니케이즈 (nCas9) 또는 촉매적으로 결핍된 Cas9 (dCas9)에 DNA에 작동할 수 있도록 방향 진화된 아데노신 디아미나아제를 결합시킨 후, 세포 내로 도입하였을 때 DNA의 A가 G(I)로 (또는보다낮은빈도로, A가 C또는 T로변환) 변환하는것을관찰하였다. 이러한 사항에 기초하여, 아데노신 디아미나아제를 DNA 시퀀싱에 적용한기술을제공한다.
일 예는
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산분자, 또는상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나 또는분리된 DNA에 처리하여 , DNA이중가닥중하나를절단하는단계 ; 및
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 세도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuc lease V) 또는,
Alkyl adenine DNA Glycosyl ase (MG)와 엔도뉴클레아제 VI 11 (endonuclease VI 11)의 조합)를처리하여 , 나머지 DNA가닥을절단하는단계
를포함하는, 아데닌디아미나아제를사용하여 DNA에 이중가닥절단 (double strand break)를생성하는방법을제공한다.
다른예는,
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산분자, 또는상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나 또는분리된 DNA에 처리하여 , DNA이중가닥중하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuc lease V) 또는, Alkyl adenine DNA Glycosyl ase (MG)와 엔도뉴클레아제 VI I I (endonuc lease VI 11)의 조합)를 처리하여, 나머지 DNA 가닥을 절단하여 , 이중 가닥 절단 (double-strand break)를형성하는단계; 및
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계
를포함하는 DNA서열분석 방법을제공한다.
다른예는,
(a) (1)아데노신 디아미나아제, 아데노신 디아미나아제를암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산분자, 또는 상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나또는 분리된 DNA에 처리하여 , DNA이중가닥중하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuc lease V) 또는 Alkyl adenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI I I (endonuclease VI I I)의 조합)를 처리하여, 나머지 DNA 가닥을 절단하여, 이중 가닥 절단 (double-strand break)를형성하는단계;
(c) 상기 이중가닥 절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계; 및
(d) 상기 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 상기 절단된위치를확인하는단계
를포함하는,
표적 특이적 뉴클레아제의 절단위치 또는 비표적 위치 (of f-target si te)를검줄하는방법을제공한다.
상기 비표적 위치 (of f-target s i te)를 검출하는 방법은 상기 (d) 단계 이후에, 상기 확인된 절단된 위치가 표적 위치 (on-target si te)가 아닌 경우, 비표적 위치 (of f-target si te)로 판단하는 단계 ((e) 단계)를 추가로포함할수 있다.
다른예는,
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드를분리된 세포에 도입시키거나 또는분리된 DNA에 처리하여, DNA이중가닥중하나를절단하는단계;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuc lease V) 또는
Alkyladenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI I I (endonuc lease VI I I)의 조합)를 처리하여, 나머지 DNA 가닥을 절단하여, 이중 가닥 절단 (double-strand break)를형성하는단계;
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계; 및
(d) 상기 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 상기 절단된위치를확인하는단계
를포함하는,
표적 특이적 뉴클레아제의 교정 효율 및/또는 정확도 확인 방법을 제공한다.
상기 표적 특이적 뉴클레아제의 교정 효율 확인 방법은 상기 (d) 단계 이후에, 상기 확인된 절단된 위치가 표적 위치 (on-target s i te)가 아닌 경우, 비표적 위치 (of f-target si te)로판단하는단계 ((e) 단계) 및 상기 비표적 위치에서의 절단 정도 (비표적 위치 개수 및/또는 비표적 위치에서의 절단 빈도)를 측정하여 비교 대상의 절단 정도와 비교하는 단계 (e-1)를 추가로 포함할 수 있으며, 이 경우, 비표적 위치에서의 절단 정도가 낮을수록 교정 효율 및/또는 정확도가 높다고 판단할 수 있다. 상기 비교 대상은 임의의 표적 DNA의 표적 서열에 대한 표적 특이적 뉴클레아제일 수 있으며, 일 예에서, 통상적으로 사용되거나 이미 알려진 표적 특이적 뉴클레아제 (예칸대, RGEN 및 가이드 RNA 조합)들 중 선택된 어느하나일수있다.
상기 단계 (a)에 있어서, (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드, 및 (2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드는 분리된 세포 또는 분리된 DNA에 동시에 (함께) 처리될 수 있으며, 일 예에서, 아데노신 디아미나아제와 표적 특이적 뉴클레아제가 융합된 융합 단백질 (예컨대, 아데노신 디아미나아제가 N 말단쪽에 위치하고 표적 특이적 뉴클레아제가 C 말단쪼겡 위치할 수 있음), 또느 상기 융합 단백질을 암호화하는 핵산 서열 또는 상기 핵산 서열을 포함하는 플라스미드형태로사용될수 있다. 이 때, (3) 표적 특이적 뉴클레아제에 따라 적절한 가이드 RNA, 또는 이를 암호화하는 핵산 분자 또는 이를 포함하는플라스미드를추가로포함할수있다.
상기 단계 (a)에 있어서, 아데노신 디아미나아제에 의하여, DNA의 이중가닥중 PAM서열이 존재하는 가닥의 표적 부위 (PAM의 5' 말단쪽으로 17 내지 23 nt 부위) 내의 아데노신의 이노신으로의 전환이 일어나고,
표적특이적 뉴클레아제에 의하여 상기 아데노신-이노신 전환이 일어난가닥 (즉, PAM서열이 존재하는가닥)의 반대 가닥 (상보적 가닥)에서 단일 가닥 절단 (ni ck)이 발생한다.
상기 단계 (b)에 있어서, 엔도뉴클레아제 m 사용하는 경우, 상기 단계 (a)에 의하여 단일 가닥 절단이 일어난 가닥의 반대 가닥 (즉, 아데노힌-이노신 전환이 일어난 가닥)의 상기 아데노신이 이노신으로 변환된 변이 위치에서 5’ 방향및/또는 3’ 방향으로 5nt 이내, 4nt 이내, 3 nt 이내, 2nt 이내, 또는 lnt 이내에 위치하는뉴클레오타이드의 3' 말단이 절단된다. 상기 단계 (b)에 있어서, Alkyl adenine DNA Glycosyl ase (쇼쇼이와 엔도뉴클레아제 VI I I (endonuclease VI I I)의 조합을 사용하는 경우, MG에 의하여 상기 단계 (a)에서 전환된 이노신이 제거되어 AP 부위 (apur ini c si te)이 생성되고, 엔도뉴클레아제 VI I I는 상기 AP 부위를 인식하여 AP 부위의 3’ 및 5' 말단을절단한다. 이러한단계 ( 를수행함으로써, 단계 (a)에서 단일 가닥 절단이 일어난 가닥과 반대 가닥에도 단일 가닥 절단이 일어나, 결과적으로이중가닥절단이 생성된다.
다른예는
(1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는 핵산분자, 또는상기 핵산분자를포함하는플라스미드,
(2) 표적 특이적 뉴클레아제, 표적 특이적 뉴클레아제를 암호화하는 핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및
(3) 단일가닥 특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V (endonuclease V) 또는 , Alkyl adenine DNA Glycosylase (AAG)와 엔도뉴클레아제 VI 11 (endonuc lease VI I I)의 조합)
을 포함하는, DNA 이중 가닥 절단 (doubl e strand breaks; DSBs)용 조성물을제공한다.
본 명세서에서 제공된 방법 및 조성물들에 있어서, 유전체 교정 (염기 교정) 및/또는 절단 대상이 되는 DNA는 유전체 (genomic) DNA 또는 DNA 단편 (PCT 산물)일 수 있으며, 세포 내에 존재하거나 세포로부터 분리된 것일 수 있다. 상기 세포는 인간, 마우스 등의 포유류 등과 같은 진핵 세포일 수 있다. 또한, 본 명세서에서 제공된 방법 및 조성물들에 있어서, 아데노신 디아미나아제 및 표적 특이적 뉴클레아제는 직접적 또는 펩타이드 링커를 통하여 간접적으로 연결된 융합 단백질 또는 이를 암호화하는 핵산분자또는 상기 핵산 분자를 포함하는 플라스미드 형태로
사용될수씼다.
또한, 본 명세서에서 제공된 방법 및 조성물들에 있어서, 표적 특이적 뉴클레아제는가이드 RNA와함께사용또는포함될수있다.
이 경우, 아데노신 디아미나아제, 표적 특이적 뉴클레아제, 및 가이드 RNA는
(i ) 아데노신 디아미나아제, 표적 특이적 뉴클레아제, 및 가이드 RNA의 혼합물,
(i i ) 아데노신 디아미나아제 암호화 핵산 서열 또는 이를 포함하는 플라스미드, 표적 특이적 뉴클레아제 암호화핵산서열 또는 이를포함하는 플라스미드, 및 가이드 RNA 또는 이의 암호화 핵산 분자를 포함하는 플라스미드의 혼합물,
(i i i ) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합단백질 및 가이드 RNA의 혼합물,
(iv) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합단백질 암호화핵산분자또는상기 핵산분자를포함하는플라스미드, 및 가이드 RNA 또는 이의 암호화 핵산 분자를 포함하는 플라스미드의 혼합물, 및
(v) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합단백질 및 가이드 RNA의 복합체 (리보핵산단백질, RNP)또는혼합물, 로 이루어진 군에서 선택된 하나 이상의 형태로 사용 (예컨대, 분리된 세포 내로도입 또는 분리된 DNA에 처리 또는조성물에 포함)될 수 있다.
상기 플라스미드는 상기 아데노신 디아미나아제 암호화 유전자 및/또는불활성화된 표적특이적 뉴클레아제 암호화유전자를삽입하고 이를 숙주세포 내에서 발현시킬 수 있는 발현 시스템을 포함하는 모든 플라스미드일 수 있다 . 상기 플라스미드는 목적 유전자 발현을 위한 요소 (elements)를포함하는 것으로, 복제원점 (repl icat ion origin) , 프로모터, 작동 유전자 (operator) , 전사 종결 서열 (terminator) 등을 포함할 수 있고, 숙주 세포의 게놈 내로의 도입을 위한 적절한 효소 부위 (예컨대, 제한 효소 부위) 및/또는 임의로 숙주 세포 내로의 성공적인 도입을 확인하기 위한 선별 마커 및/또는 단백질로의 번역을 위한 리보좀 결합 부위 (ribosome binding si te; RBS) 및/또는 전자 조절 인자 등을 주가로 포함할 수 있다. 상기 플라스미드는 당업계에서 사용되는 플라스미드,
예컨대, pcDNA시리즈, pSClOl, pGV1106, pACYC177, ColEl, pKT230, pME290, pBR322, pU期/ 9, pUC6, pBD9, pHC79, pIJ61, pLAFRl, pHV14, pGEX시리즈, pET시리즈, pUC19 등으로 이루어진 군에서 선택된 1종 이상일 수 있으나, 이에 제한되는 것은 아니다. 상기 숙주세포는 상기 아데노신 디아미나제에 의하여 염기 교정 또는 이중 가닥 절단을 도입하고자 하는 세포 (예컨대, 인간 세포 등의 포유류 등과 같은 진핵 세포) 또는 상기 아데노신 디아미나아제 암호화 유전자 및/또는 불활성화된 표적특이적 뉴클레아제 암호화 유전자를 발현하여 아데노신 디아미나아제 및 불활성화된 표적특이적 뉴클레아제를발현할수 있는모든세포 (예컨대, E. col i 등)들중에서 선택될수있다.
본 명세서에서 절단 유전체 시퀀싱 (digested genome sequencing, Digenome-seq) 기술이라 함은 뉴클레아제에 의해 절단된 유전체의 서열 분석을 의미하는 것으로, 세포에서의 전체 유전체에서의 뉴클레아제 비표적 효과 (of f-target ef fect)를 분석하기 위한 in vitro nucl ease-digested who 1 e-genome sequencing에 적용될 수 있다. 뉴클레아제의 절단 부위에서 동일한 5' 말단을 갖는 sequence reads를 생성하며, 이들은 적절한 프로그램 (예컨대, Di genome program)에 의하여 컴퓨터로 확인 가능하다. 일 예에서, 절단 유전체 시퀀싱은 앞서 기재한 유전체 DNA (genome DNA) 서열 분석 방법 또는 뉴클레아제의 비표적 위치 (of f-target si te)를 검출하는방법에서의 단계 (a) , (b) , 및 (c) , 또는단계 (a) , (b) , (c) , 및 ( 를포함하는것으로정의될 수 있다. 이를다르게 설명하면, 상기 단계 (a) , (b) , 및 (c) , 또는 단계 (a) , (b) , (c) , 및 ( 는 절단 유전체 시퀀싱에 의하여 수행될수 있다.
본 명세서에 사용된 바로서, 용어 "염기 변이 (또는 염기 치환)"은 해당 염기를 포함하는 뉴클레오타이드에 변이 (예컨대, 치환)이 일어난 것을 의미하는 것으로, ’’뉴클레오타이드 변이 (또는 뉴클레오타이드 치환)'’와동일한의미로사용될 수 있으며, 이러한 염기 변이는 대립유전자 중하나또는두개모두에서 일어날수있다.
유전체 교정 및/또는 유전자 교정 기술은, 인간 세포를 비롯한 동식물 세포의 유전체 염기서열에 표적 지향형 변이를 도입할ᅮ수 있는 기술로서, 특정 유전자를 넉아웃 (knock-out) 또는 넉인 (knock-in)하거나, 단백질을 생성하지 않는 비-코딩 DNA 서열에 변이를 도입하는 등 다양한 형태로수행될 수 있다. 본명세서에서 제안되는방법은상기 유전체 교정
및/또는 유전자 교정 기술에 사용되는 표적 특이적 뉴클레아제의 비표적 위치를 검출하는 것으로, 이는 표적 위치에만 특이적으로 작동하는 표적 특이적 뉴클레아제 시스템을개발하는데유용하게 이용될수있다.
상기 (a) 단계는 분리된 세포 또는 생체 또는 세포로부터 분리된 유전체 (genomi c) DNA에 아데노신 디아미나아제와 표적 특이적 뉴클레아제를 처리하여 DNA 이중가닥 중 한 가닥을 절단하는 단계로서, 분리된 유전체 DNA를 시험관 내 { in r/ir)에서 특정 표적에 특이적으로 작용하는 뉴클레아제로 절단하는 단계이다. 상기 뉴클레아제는 표적 특이적으로 제작하였더라도 특이성에 따라 다른 부위, 즉 비표적 위치를 절단할 가능성을 갖는다. 따라서 결과적으로 상기 (a) 단계에 의해, 사용된 표적 특이적 뉴클레아제가 유전체 DNA에 대하여 활성을 가질 수 있는 위치인 표적 위치 또는 다수의 비표적 위치를 절단함으로써 특정 위치가절단된유전체 DNA단편(lead)을얻을수 있다.
상기 분리된 유전체 DNA는 비형질전환 세포 (야생형 세포) 및/또는 표적 특이적 뉴클레아제가 발현되거나 뉴클레아제 활성을 갖도록 형질전환된 세포로부터 분리된 것일 수 있으며, 표적 특이적 뉴클레아제의 비표적 위치를 검출하고자 하는 목적에 따라 그 유래에 제한 없이 사용될 수 있다. 상기 유전체가 분리되는 세포는 모든 원핵 세포 및 진핵세포 (예컨대, 인간 등의 포유동물 세포, 진핵식물 세포 등)들 중에서 선택된 것일수있다.
상기 아데노신 디아미나아제는 퓨린 대사에 관여하는 효소(EC 3.5.4.4)로서, 아데노신을 디아미네이션(deaminat ion; 아미노기를 케토기로 치환)시켜서 이노신 (inos ine)으로 변환시키는 기능하는 모든 효소를 총칭한다. 일 예에서, 아데노신 디아미나아제는원핵세포또는진핵 세포, 예컨대, 진핵 동물 (예컨대, 어류, 양서류, 파중류, 조류, 포유류 등), 또는 진핵 식물 유래의 것일 수 있으며, 예컨대, 인간 아데노신 디아미나아제 (예컨대, GenBank Access i on No. NP_000013.2 (코딩 유전자: NM_000022.3), NP_001308979.1 (코딩 유전자: NM_001322050.1) ,
NP_001308980.1 (코딩 유전자: NM_001322051.1) 등), 마우스 아데노신 디아미나아제 (예컨대, GenBank Access ion No. NP_001258981.1 (코딩 유전자: NM_001272052.1), NP_031424.1 (코딩 유전자: NM_007398.4) 등), E. coli TadA (NP_417054.2) 등으로 이루어진 군에서 선택된 1종 이상일 수 있다. 상기 아데노신 디아미나아제는 단백질 또는 이를 암호화하는 DNA
(임의로 적절한 재조합 벡터에 포함될 수 있음), 또는 이를 암호화하는 mRNA 형태로사용될 수 있다또한 아데노신 디아미나아제는 기존 아데노신 디아미나아제가, 예컨대, E. coli 등의 원핵세포에서, 인위적 진화 (directed evolution; DE) 된 것일 수 있다. 인위적 진화 (directed evolution; 는 단백질 엔지니어링 방법 중 하나로, 돌연변이유발 과정 (변이체 라이브러리 생성), 선별 과정 (상기 변이체를 발현시키고 이 중에서 목적하는 기능 (변이)을 갖는 변이체를 분리), 및 증폭 또는 재생산 과정 (다음 라운드를 위한 템플릿 생성)를포함할수 있고, in vivo (생체 내)또는 in vitro에서 수행될 수 있다. 일 예에서, 상기 인위적 진화된 아데노신 디아미나아제는 E. coli TadA (NP_417054.2)를 E. co//에서 인위적 진화시킨 것일 수 있다. 일 예에서, 상기 인위적 진화된 아데노신 디아미나아제는 NP_417054.2의 N 말단의 메티오닌 ( 을 제외한 아미노산 서열 중, 22번째 아미노산 잔기 (W), 35번째 아미노산 잔기 (H), 47번째 아미노산잔기 (P), 50번째 아미노산잔기 (R), 83번째 아미노산잔기 (L), 105번째 아미노산 잔기 (A), 107번째 아미노산 잔기 (D), 122번재 아미노산 잔기 (H), 145번째 아미노산 잔기 (S), 146번째 아미노산 잔기 (D),
151번째 아미노산잔기 (R), 154번째 아미노산잔기 (E), 155번째 아미노산 잔기 (I), 156번째 아미노산 잔기 (K) (이상, 아미노산 위치는
NP_417054.2의 두 번째 아미노산부터 기산함) 등으로 이루어진 군에서 선택된 하나 이상의 아미노산이 원래 (야생형)와 다른 아미노산으로 치환 또는결실된 것일수있으나 (참고예 2참조), 이에 제한되는것은아니다. 상기 표적 특이적 뉴클레아제는 유전자 가위 (programmable nuclease)라고도 불리며, 목적하는 유전체 DNA상의 특정 위치를 인식하여 절단할수있는모든형태의 뉴클레아제를통칭한다.
본명세서에서 사용된표적 특이적 뉴클레아제는 DNA이중가닥중한 가닥을절단하는활성을갖는모든뉴클레아제일수 있다.
일구체예에서, 상기 표적 특이적 뉴클레아제는 Cas단백질 (예컨대, Cas9단백질 (CRISPR (Clustered regularly interspaced short palindromic repeats) associated protein 9)), Cpfl 단백질 (CRISPR from Prevotel la and Franc i sella 1)등과 같은 타입 H 및/또는 타입 V의 CRISPR시스템에 수반되는 뉴클레아제 (예컨대, 엔도뉴클레아제) 등으로 이루어진 군에서 선택된 1종 이상일 수 있다. 이 경우, 상기 표적 특이적 뉴클레아제는 유전체 DNA의 표적 부위로 안내하기 위한 표적 DNA 특이적 가이드 RNA를
추가로 포함할 수 있다. 상기 가이드 RNA는 생체 외 (in vitro)에서 전사된 (transcribed) 것일 수 있고, 예컨대 올리고뉴클레오티드 이중가닥 또는 플라스미드 주형으로부터 전사된 것일 수 있으나, 이에 제한되지 않는다. 상기 표적 특이적 뉴클레아제는 가이드 RNA에 결합된 리보핵산- 단백질 복합체를 형성 (RNA-Guided Engineered Nuclease)하여 리보핵산 단백질 (RNP) 형태로작용할수 있다.
Cas 단백질은 CRISPR/Cas 시스템의 주요 단백질 구성 요소로, 활성화된 엔도뉴클레아제또는 nickase를형성할수 있는단백질이다.
Cas 단백질 또는 유전자 정보는 N.CBI (National Center for Biotechnology Information)의 GenBank와 같은 공지의 데이터 베이스에서 얻을수있다. 예컨대, 상기 Cas단백질은,
스트렙토코커스 sp. {Streptococcus sp. ) , 예컨대 , 스트렙토코커스 피요게네스 {Streptococcus pyogenes) 유래의 Cas 단백질, 예컨대, Cas9 단백질 (예컨대 , SwissProt Accession number Q99ZW2(NP_269215.1));
캄필로박터 속, 예컨대, 캄필로박터 제주니 {Campylobacter jejuni) 유래의 Cas단백질, 예컨대, Cas9단백질;
스트렙토코커스 속, 예컨대, 스트렙토코커스 써모필러스
{Streptococcus thermophiles) 또는 스트렙토로커스 아우레우스 (Streptocuccus aureus)유래의 Cas단백질, 예컨대, Cas9단백질;
네이세리아 메닝기디티스 {Neisseria meningitidis) 유래의 Cas 단백질, 예컨대, Cas9단백질;
파스테우렐라 [Pasteurella) 속, 예컨대, 파스테우렐라 물토시다 {Pasteurella multocida)유래의 Cas단백질, 예컨대 Cas9단백질;
프란시셀라 (Francisella) 속, 예컨대, 프란시셀라 노비시다 {Francisella novicida)유래의 Cas단백질, 예컨대 Cas9단백질
등으로 이루어진 군에서 선택된 하나 이상일 수 있으나, 이에 제한되는것은아니다.
Cpfl 단백질은상기 CRISPR/Cas 시스템과는 구별되는새로운 CRISPR 시스템의 엔도뉴클레아제로서, Cas9에 비해 상대적으로 크기가 작고 tracrRNA가 필요 없으며, 단일 가이드 쇼에 의해 작용할수 있다. 또한, 티민 (thymine)이 풍부한 PAM (protospacer-adjacent motif) 서열을 인식하고 DNA의 이중 사슬을 잘라 점착종단 (cohesive end; cohesive double-strand break)을생성한다.
예컨대, 상기 Cpfl 단백질은 캔디다투스 iCandidatus) 속, 라치노스피라 (、Lachnospira) 속, 뷰티리비브리오 (、Butyrivibrio) 속, 페레그리니박테리아 (、Peregrinibacteria), 액시도미노코쿠스
(Acidowinococcus) 속, 포르파이로모나스 (Porphyromonas) 속, 프레보텔라 {Prevotella) 속, 프란시셀라 {Francisella) 속, 캔디다투스 메타노플라스마 {Candidatus Methanoplasma) , 또는 유박테리움
{Eubacterium) 속 유래의 것일 수 있고, 예컨대, Parcubacteria bacterium (GWC2011_GWC2_44_17) , Lachnospiraceae bacteri· (MC2017) , Butyrivibrio proteoclasiicus, Peregrinibacteria bacterium (GW2011_GWA_33_10) , Acidawinococcus sp. (BV3L6) , Porphyromonas macacae, Lachnospiraceae bacterium (ND2006) , Porphyromonas crevioricanis, Prevotella disiens, Moraxella bovoculi (237) , Swiihella sp. (SC_K08D17) , Leptospira inadai, Lachnospiraceae bacterium (MA2020) , Francisella novicida (U112) , Candidatus Methanoplasma termitum, Candidatus Paceibacter, Eubacterhmi eligens등의 미생물유래의 것일수있으나, 이에 제한되는것은아니다. 상기 표적 특이적 뉴클레아제는미생물에서 분리된 것 또는재조합적 방법 또는 합성적 방법 등과 같이 인위적 또는 비자연적 생산된 것 (non- natural ly occurring)일 수 있다. 일 예에서 , 상기 표적 특이적 뉴클레아제 (예컨대, Cas9, Cpfl, 등)은 재조합 DNA에 의하여 만들어진 재조합 단백질일 수 있다. 재조합 DAN(Recombinant DNA; rDNA)는 다양한 유기체로부터 얻어진 이종 또는 동종 유전 물질을 포함하가 위하여 분자 클로닝과 같은 유전자 재조합 방법에 의하여 인공적으로 만들어진 DNA 분자를의미한다. 예컨대, 재조합 DNA를 적절한유기체에서 발현시켜 표적 특이적 뉴클레아제를 생산 ( in vivo 또는 in r/iro)하는 경우, 재조합 DNA는제조하고자하는단백질을 암호화하는코돈들중에서 상기 유기체에 발현하기에 최적화된 코돈을 선택하여 재구성된 뉴클레오타이드 서열을 갖는것일수있다.
본 명세서에서 사용된 상기 표적특이적 뉴클레아제는 변이된(불활성화된) 형태의 변이 표적특이적 뉴클레아제일 수 있다. 상기 변이 (불활성화) 표적특이적 뉴클레아제는 DNA 이중 가닥을 모두 절단하는 엔도뉴클레아제 활성을상실하도록 변이된 것을의미할수 있으며, 예컨대, DNA 이중 가닥을 모두 절단하는 엔도뉴클레아제 활성을 상실하고 니카아제 활성을 갖도록 변이된 변이 표적특이적 뉴클레아제 및 DNA 이중 가닥을
모두 절단하는 엔도뉴클레아제 활성과 니카아제 활성을 모두 상실하도록 변이된 변이 표적특이적 뉴클레아제 중에서 선택된 1종 이상일 수 있다. 상기 변이 표적특이적 뉴클레아제가니카아제 활성을갖는 것인 경우, 상기 디아미나제에 의한 염기 변환 (예컨대, 아데노신이 이노신으로 변환)과동시 또는순서와무관하게 순차적으로, 상기 염기 변환이 일어난 가닥또는 그 반대 가닥 (예컨대, 염기 변환이 일어난 가닥의 반대 가닥)에서 단일 가닥 절단 (nick)을 도입시킬 수 있다 (예컨대, PAM이 위치하는 가닥의 반대가닥에서, PAM서열의 5' 말단방향으로 3번째 뉴클레오타이드와 4번째 뉴클레오타이드 사이에 해당하는 위치에 nick이 도입됨) . 이와 같은 표적특이적 뉴클레아제의 변이 (예컨대, 아미노산 치환 등)는 적어도 뉴클레아제의 촉매 활성 도메인 (예컨대, Cas9의 경우 RuvC 촉매 도메인)에서 일어나는 것일 수 있다. 일 예에서, 상기 표적특이적 뉴클레아제가 스트렙토코커스 피요젠스 유래 Cas9 단백질 (SwissProt Accession number Q99ZW2(NP_269215.1))인 경우, 상기 변이는 촉매 활성을 갖는 아스파르트산 잔기 (catalytic aspartate residue; 10번째 위치의 아스파르트산 (D10) 등), 762번째 위치의 글루탐산 (E762) , 840번째 위치의 히스티딘 (H840) , 854번째 위치의 아스파라긴 (N854) , 863번째 위치의 아스파라긴 (N863) , 986번째 위치의 아스파르트산 (D986) , 539번째 위치의 페닐알라닌 (F539) , 763번째 위치의 메티오닌 (M763) , 890번째 위치의 라이신 (K890) 등으로 이루어진 군에서 선택된 하나 이상 임의의 다른 아미노산으로치환된 돌연변이를포함할수 있다. 이 때, 치환되는 임의의 다른아미노산은알라닌 (alanine)일수있지만, 이에 제한되지 않는다. 다른 예에서, 상기 변이된 표적특이적 뉴클레아제는 특이도 (정확도)이 개선되도록 변이가 일어난 것일 수 있다. 일 예에서, 상기 변이된 표적특이적 뉴클레아제는 스트렙토코커스 피요젠스
(Streptococcus pyogenes) 유래의 Cas9 단백질 (예컨대, SwissProt Accession number Q99ZW2(NP_269215.1))의 F539, M763, 및 K890로 이루어진 군에서 선택된 하나 이상의 아미노산 잔기가 원래 아미노산과 상이한 아미노산으로 치환 또는 결실된 변이를 갖는 변이체 (보다 구체적으로, F539S, M763I, 및 K890N로 이루어진 군에서 선택된 하나 이상의 변이를 포함하는 변이체, 에컨대, F539S, M763I, 및 K890N 변이를 모두 갖는 변이체 (Sniper_Cas9))일수있다.
다른 예에서, 상기 변이된 (불활성화된) 표적특이적 뉴클레아제는
스트렙토코커스 피요젠스 {Streptococcus pyogenes) 유래의 Cas9 단백질 (예컨대, SwissProt Accession number Q99ZW2(NP_269215.1))의 DIO이 원래와 다른 아미노산 (예컨대, 알라닌 (A))으로 치환 변이되어 니케이즈 활성을 갖고, 여기에 더하여, F539, M763, 및 K890로 이루어진 군에서 선택된 하나 이상의 아미노산 잔기가 원래와 상이한 아미노산으로 치환 변이 (예컨대 , F539S, M763I , K890N)되어 특이도 (정확도)가개선된 것일 수 있다.
다른 예에서, 상기 변이 표적특이적 뉴클레아제는 야생형 Cas9 단백질과 상이한 PAM서열을 인식하도록 변이된 것일 수 있다. 예컨대, 상기 변이 표적특이적 뉴클레아제는 스트렙토코커스 피요젠스 유래 Cas9 단백질의 1135번째 위치의 아스파르트산 (D1135), 1335번째 위치의 아르기닌 (R1335), 및 1337번째 위치의 트레오닌 (T1337) 중 하나 이상, 예컨대 3개 모두가 다른 아미노산으로 치환되어, 야생형 Cas9의 PAM서열 (NGG)와상이한 NGA어은 A, T, G, 및 C중에서 선택된 임의의 염기임)을 인식하도록변이된 것일수 있다.
일 예에서, 상기 변이 표적특이적 뉴클레아제는 스트렙토코커스 피요젠스유래 Cas9단백질의 아미노산서열중,
(1) DIO, H840, 또는이들의 조합 (D10 + H840);
(2) D10와, F539, M763, 및 K890로 이루어진 군에서 선택된 하나 이상의 조합 (예컨대, D10 + F539 + M763 + K890);
(3) D1135, R1335, T1337, 또는이들의 조합 (예컨대, D1135 + R1335 + T1337) ; 또는
(5) (1)내지 (3)중에서 선택된둘이상의 조합
에서 아미노산치환이 일어난것일수 있다.
본 명세서에 사용된 바로서, 상기 ’다른 아미노산’은, 알라닌, 이소류신, 류신, 메티오닌, 페닐알라닌, 프롤린, 트림토판, 발린, 아스파라긴산, 시스테인, 글루타민, 글리신, 세린, 트레오닌, 티로신, 아스파르트산, 글루탐산, 아르기닌, 히스티딘, 라이신, 상기 아미노산들의 공지된 모든 변형체 중에서, 야생형 단백질이 원래 변이 위치에 갖는 아미노산을 제외한 아미노산들 중에서 선택된 아미노산을 의미한다. 일 예에서, 상기 ’다른아미노산'은 알라닌, 발린, 글루타민, 또는아르기닌일 수있다.
일 예에서, 상기 변이 표적특이적 뉴클레아제는 엔도뉴클레아제
활성을 상실 (예컨대 , 니카아제 활성을 갖거나, 엔도뉴클레아제 활성 및 니카아제 활성을 모두 상실)한 변형 Cas9 단백질, 또는 야생형 Cas9과 상이한 PAM 서열을 인식하는 것일 수 있다. 예컨대, 상기 변형 Cas9 단백질은, 스트렙토코커스 피요제네스 { Streptococcus pyogenes) 유래의 Cas9단백질에 있어서,
(1) D10 또는 H840 위치에 돌연변이 (예컨대, 다른 아미노산으로의 치환)가 도압되어 이중 가닥 절단 엔도뉴클레아제 활성이 상실되고 니카아제 활성을 갖는 변형 Cas9, 또는 스트렙토코커스 피요젠스
( Streptococcus pyogenes) 유래의 Cas9 단백질에 D10 및 H840 위치에 모두 돌연변이 (예컨대, 다른 아미노산으로의 치환)가 도입되어 이중 가닥 절단 엔도뉴클레아제 활성 및 니카아제 활성을모두상실한변형 Cas9단백질;
(2) D10 위치에 더하여 F539, M763, 및 K89◦로 이루어진 군에서 선택된 하나 이상의 위치에 돌연변이 (예컨대, 다른 아미노산으로의 치환)가 도입되어 이중 가닥 절단 엔도뉴클레아제 활성이 상실되고 니카아제 활성을가지면서 특이도가향상된 변형 Cas9단백질;
(3) D1135, R1335 및 T1337 중에서 하나 이상 또는 이들 모두에 돌연변이 (예컨대, 다른 아미노산으로의 치환)가 도입되어 야생형과 상이한 PAM서열을인식하는변형 Cas9단백질 ; 또는
(3) (1)내지 (3)중 2이상이 도입된 변형 Cas9단백질
일수있다.
예컨대, 상기 CAs9 단백질의 D10 위치에서의 돌연변이는 D10A 돌연변이 (Cas9 단백질의 아미노산 중 10번째 아미노산인 D가 A로 치환된 돌연변이를 의미함; 이하, Cas9에 도입된 돌연변이는 동일한 방법으로 표기됨)일 수 있고, 상기 H840위치에서의 돌연변이는 H840A돌연변이일 수 있으며, D1135, R1335, 및 T1337 위치에서의 돌연변이는 각각 D1135V, R1335Q, 및 T1337R일 수 있고, F539S, M763I , 및 K890N 위치에서의 돌연변이는각각 F539S, M763I , 및 K890N일수있다.
상기 뉴클레아제는 미생물에서 분리된 것 또는 재조합적 방법 또는 합성적 방법 등과 같이 인위적 또는 비자연적 생산된 것 (non-natural ly occurr ing)일 수 있다. 일 예에서, 상기 뉴클레아제는 재조합 DNA에 의하여 만들어진 재조합 단백질일 수 있다. 재조합 DAN (Recomb inant DNA; rDNA)는다양한유기체로부터 얻어진 이종또는동종유전 물질을포함하기 위하여 분자 클로닝과 같은 유전자 재조합 방법에 의하여 인공적으로
만들어진 DNA분자를 의미한다. 예컨대, 재조합 DNA를 적절한유기체에서 발현시켜 단백질을 생산 i in vivo또는 in F/iro)하는 경우, 재조합 DNA는 제조하고자 하는 단백질을 암호화 하는 코돈들 중에서 상기 유기체에 발현하기에 최적화된 코돈을 선택하여 재구성된 뉴클레오타이드 서열을 갖는것일수있다.
상기 뉴클레아제는 단백질, 이를 암호화하는 핵산 분자 (DNA 또는 mRNA) , 가이드 RNA와 결합된 리보핵산 단백질, 상기 리보핵산 단백질을 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 재조합 벡터의 형태로사용될수있다.
상기 뉴클레아제 또는 이를 코딩하는 핵산 분자는 핵 내로 전달, 작용, 및/또는발현될수있는형태일수 있다.
상기 뉴클레아제는 세포 내로 도입되기에 용이한 형제일 수 있다. 일 예로, 상기 뉴클레아제는 세포 침투 펩타이드 및/또는 단백질 전달 도메인 (protein transduct ion domain)과 연결될 수 있다. 상기 단백질 전달 도메인은 폴리-아르기닌 또는 HIV 유래의 TAT 단백질일 수 있으나, 이에 제한되지 않는다. 세포 침투 펩타이드 또는 단백질 전달 도메인은 상기 기술된 예 외에도 다양한 종류가 당업계에 공지되어 있으므로, 당업자는상기 예에 제한되지 않고다양한예를적용할수있다.
또한, 상기 뉴클레아제 또는 암호화하는 핵산 분자는 핵 위치 신호 (nuc lear local i zat ion signal , NLS) 서열 또는 이를 암호화하는 서열을 추가로 포함할 수 있다. 따라서, 상기 뉴클레아제를 암호화하는 핵산 분자를 포함하는 발현 카세트는 상기 뉴클레아제를 발현시키기 위한 프로모터 서열 등의 조절 서열, 또는 여기에 더하여, NLS 서열 (예컨대, SV40 NLS 등)을 포함할 수 있다. 상기 NLS 서열은 당업계에 잘 알려져 있다.
상기 뉴클레아제 또는 이를 암호화하는 핵산 분자는 분리 및/또는 정제를 위한 태그 또는 상기 태그를 암호화하는 핵산 서열과 연결될 수 있다. 일 예로, 상기 태그는 Hi s 태그, Fl ag태그, S태그등과같은작은 펩타이드 태그, GST (Glutathione S-transferase) 태그, MBP (Mal tose binding protein) 태그등으로 이루어진 군에서 적절하게 선택될 수 있으나, 이에 제한되지 않는다.
본 발명에서, 용어 "가이드 RNA (guide RNA)"는 표적 DNA 특이적인 RNA (예컨대, DNA의 표적 부위와 혼성화 가능한 시를 의미하며, Cas
단백질, Cpf l 등과 같은 뉴클레오타이드와 결합하여 표적 DNA로 인도하는 역할을한다.
상기 가이드 RNA는 복합체를 형성할 뉴클레아제의 종류 및/또는 그 유래 미생물에 따라서 적절히 선택될수있다. 예컨대, 상기 가이드 RNA는 DNA표적 부위와혼성화가능한부위를포함하는 CRISPR RNA (crRNA);
Cas 단백질, Cpf l 등과 같은 엔도뉴클레오타이드와 상호작용하는 부위를포함하는 ira凡 s-act ivat ing crRNA (tracrRNA) : 및
상기 crRNA및 tracrRNA의 주요부위 (예컨대, crRNA의 혼성화부위 및 tracrRNA의 상호작용 부위)가 융합된 형태의 단일 가이드 RNA (s ingle guide RNA; sgRNA)
로이루어진군에서 선택된 1종이상일수 있으며,
구체적으로 CRISPR RNA (crRNA) 및 trans-act ivat ing crRNA (tracrRNA)를 포함하는 이중 RNA (dual RNA) , 또는 crRNA 및 tracrRNA의 주요부위를포함하는단일 가이드 RNA (sgRNA)일수있다.
상기 sgRNA는 표적 DNA 내 서열과 상보적인 서열을 가지는 부분
(이를 Spacer region, Target DNA recogni t ion sequence , base pai r ing region 등으로도 명명함) 및 Cas 단백질 결합을 위한 hai rpin 구조를 포함할 수 있다. 보다 구체적으로, 표적 DNA 내 서열과 상보적인 서열을 가지는부분, Cas 단백질 결합을 위한 hai rpin구조 및 Terminator 서열을 포함할 수 있다. 상기 기술된 구조는 5’에서 3' 순으로 순차적으로 존재하는것일 수 있으나, 이에 제한되는 것은아니다. 상기 가이드 RNA가 crRNA 및 tracrRNA의 주요 부분 및 표적 DNA의 상보적인 부분을 포함하는 경우라면어떠한형태의 가이드 RNA도본발명에서 사용될수있다.
예컨대, Cas9 단백질을 타겟 유전자 교정을 위하여 두 개의 가이드 RNA, 즉, 표적 유전자의 표적 서열 부위와 혼성화 가능한 뉴클레오타이드 서열을 갖는 CRISPR RNA (crRNA)와 Cas9 단백질와 상호작용하는 tran act ivat ing crRNA (tracrRNA; Cas9 단백질과 상호작용함)를 필요로 하며 , 이들 crRNA와 tracrRNA는 서로 결합된 이중 가닥 crRNA: tracrRNA 복합체 형태, 또는 링커를 통하여 연결되어 단일 가이드 RNA (single guide RNA; sgRNA) 형태로 사용될 수 있다. 일 예에서, Streptococcus pyogenes 유래의 Cas9단백질을사용하는경우, sgRNA는상기 Cas9의 crRNA의 혼성화 가능한 뉴클레오타이드 서열을 적어도 포함하는 crRNA 일부 또는 전부와 상기 Cas9의 tracrRNA의 Cas9 단백질와 상호작용하는 부위를 적어도
포함하는 tracrRNA일부또는전부가뉴클레오타이드 링커를통하여 헤어핀 구조 (stem-loop 구조)를 형성하는 것일 수 있다 (이 때 뉴클레오타이드 링커가루프구조에 해당할수있음).
상기 가이드 RNA, 구체적으로 crRNA 또는 sgRNA는 표적 DNA 내 서열과 상보적인 서열을 포함하며, crRNA 또는 sgRNA의 업스트림 부위, 구체적으로 sgRNA 또는 dualRNA의 crRNA의 5' 말단에 하나 이상, 예컨대, 1-10개, 1-5개, 또는 1-3개의 추가의 뉴클레오티드를포함할수 있다. 상기 추가의 뉴클레오티드는 구아닌 (guanine, «일 수 있으나, 이에 제한되는 것은아니다.
다른 예에서, 상기 뉴클레아제가 Cpf l인 경우, 상기 가이드 RNA는 crRNA을 포함하는 것일 수 있으며, 복합체를 형성할 Cpf l 단백질 종류 및/또는그유래 미생물에 따라서 적절히 선택될수 있다.
상기 가이드 RNA의 구체적 서열은 뉴클레아제 (Cas9 단백질 또는 Cpf l) 의 종류 (즉, 유래 미생물)에 따라서 적절히 선택할수 있으며, 이는 이 발명이 속하는 기술 분야의 통상의 지식을 가진 자가 용이하게 알 수 있는사항이다.
일 예에서, 표적특이적 뉴클레아제로서 Streptococcus pyogenes 유래의 Cas9 단백질을 사용하는 경우, crRNA는 다음의 일반식 1로 표현될 수있다:
5 ' -(Ncas9) ,-(GUUUUAGAGCUA)-(Xcas9)m-3 ' (일반식 1)
상기 일반식 1에서,
Ncas9는 표적화 서열, 즉 표적 유전자(target gene)의 표적 부위(target si te)의 서열에 따라서 결정되는 부위 (즉, 표적 부위의 서열과 혼성화 가능한 서열임)이며, 1은 상기 표적화 서열에 포함된 뉴클레오타이드수를나타내는 것으로 17내지 23, 17내지 22, 18내지 23, 또는 18내지 22의 정수, 예컨대 17, 18, 19, 20, 21, 또는 22일수있고; 상기 표적 서열의 3’ 방향으로 인접하여 위치하는 연속하는 12개의 뉴클레오타이드(GUUUUAGAGCUA)를포함하는 부위는 crRNA의 필수적 부분이고,
Xcas9는 crRNA의 3' 말단쪽에 위치하는 (즉, 상기 crRNA의 필수적 부분의 3' 방향으로 인접하여 위치하는) m개의 뉴클레오타이드를 포함하는 부위로, m은 8 내지 12의 정수, 예컨대 11일 수 있으며, 상기 m개의 뉴클레오타이드들은서로같거나다를수 있으며, 각각독립적으로 A, U, C 및 G로이루어진군에서 선택될수있다.
2019/147073 1»(:1^1{2019/001104
일 예에서, 상기 는 1100](¾1]仰(}를 포함할' 수 있으나 이에 제한되지 않는다.
또한, 상기 別쇼는다음의 일반식 2로표현될수 있다:
5 -(、 요)!)- ( UAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGC) -3 '
(일반식 2)
상기 일반식 2에서,
60개의 뉴클레오타이드
(UAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGC)^

있다.
또한, 3§1¾桃는상기 卵쇼의 표적화서열과필수적 부위를 포함하는

구조 ( 6미-10애 구조)를 형성하는 것일 수 있다 (이 때, 올리고뉴클레오타이드 링커가 루프 구조에 해당함). 보다 구체적으로,

(UAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGC) -3 '
(일반식 3)
상기 일반식 3에서, (比^)!는 표적화 서열로서 앞서 일반식 1에서 설명한바와같다.
상기 크용 요에 포함되는 올리고뉴클레오타이드 링커는 3 내지 5개, 예컨대 4개의 뉴클레오타이드를 포함하는 것일 수 있으며, 상기
뉴클레오타이드들은 서로 같거나 다를 수 있고, A, U, C 및 G로 이루어진 군에서 각각독립적으로선택될수 있다.
상기 crRNA또는 sgRNA는 5 ' 말단 (즉, crRNA의 타겟팅 서열 부위의 5’ 말단)에 1내지 3개의 구아닌(G)을추가로포함할수있다.
상기 tracrRNA 또는 sgRNA는 tracrRNA의 필수적 부분(60nt)의 3’ 말단에 5개 내지 7개의 우라실 (U)을 포함하는 종결부위를 추가로 포함할 수있다.
상기 가이드 RNA의 표적 서열은 표적 DNA 상의 PAM (Protospacer Adj acent Mot i f 서열(5. pyogenes Cas9의 경우, 5 ' -NGG-3 ' 어은 A, T, G, 또는 C임))의 51에 인접하여 위치하는 약 17개 내지 약 23개 또는 약 18개 내지 약 22개, 예컨대 20개의 연속하는핵산서열일수있다.
상기 가이드 쇼의 표적 서열과혼성화가능한가이드 RNA의 표적화 서열은상기 표적 서열이 위치하는 DNA가닥 (즉, PAM서열(5’-NGG-3 ' 어은 A, T, G, 또는 C임)이 위치하는 DNA 가닥)의 상보적인 가닥의 뉴클레오타이드서열과 50%이상, 60%이상, 70%이상, 80%이상, 90%이상, 95% 이상, 99% 이상, 또는 100%의 서열 상보성을 갖는 뉴클레오타이드 서열을 의미하는 것으로, 상기 상보적 가닥의 뉴클레오타이드 서열과 상보적 결합이 가능하다.
본명세서에서, 표적 부위의 핵산서열은표적 유전자의 해당유전자 부위의 두 개의 DNA 가닥 중 PAM 서열이 위치하는 가닥의 핵산 서열로 표시된다. 이 때, 실제로 가이드 RNA가 결합하는 DNA 가닥은 PAM서열이 위치하는 가닥의 상보적 가닥이므로, 상기 가이드 쇼에 포함된 표적화 서열은, RNA특성상 를 U로 변경하는 것을 제외하고, 표적 부위의 서열과 동일한 핵산 서열을 갖게 된다. 따라서, 본 명세서에서, 가이드 RNA의 표적화서열과표적 부위의 서열 (또는 절단부위의 서열)은 와 U가상호 변경되는것을제외하고동일한핵산서열로표시된다.
상기 가이드 RNA는 RNA 형태로 사용 (또는 상기 조성물에 포함)되거나, 이를 암호화하는 DNA를 포함하는 플라스미드 형태로 사용 (또는상기 조성물에 포함)될수 있다.
본 발명에서 용어 "표적 위치 (on-target si te)" 란, 상기 표적 특이적 뉴클레아제를 이용하여 변이(절단, 삽입, 및/또는 결실)를 도입하고자하는 위치를 의미하며, 그목적에 따라 임의로 선택될 수 있는 것으로 특정 유전자의 코딩 서열 내부에 존재할 수 있을 뿐만 아니라,
단백질을생성하지 않는비-코딩 DNA서열에 존재할수도있다.
상기 표적 특이적 뉴클레아제는 서열 특이성 (speci f i ci ty)을 가지므로 표적 위치에 작용하는 것이나, 표적 서열에 따라 비표적 위치 (of f-target si te)에 작용하는부작용이 발생할수도있다.
본 명세서에서, 비표적 위치 (of f-target si te)라함은표적 특이적 뉴클레아제의 표적 서열과 동일하지 않은 서열을 갖지만상기 표적 특이적 뉴클레아제가 활성을 가지는 위치를 말한다. 즉, 표적 위치 이외의, 표적 특이적 뉴클레아제에 의해 절단되는 위치를 말한다. 일 예에서, 상기 비표적 위치는 특정 표적 특이적 뉴클레아제에 대한실제 비표적 위치뿐만 아니라 비표적 위치가 될 가능성이 있는 위치까지 포함하는 개념으로 사용될 수 있다. 상기 비표적 위치는 이에 제한되는 것은 아니나, 시험관 내 ( in iro)에서 표적 특이적 뉴클레아제에 의해 절단되는 표적 위치 이외의 모든위치일수있다.
유전자 가위가 표적 위치 이외의 위치에서도 활성을 가지는 것은 다양한원인에 의해 야기될 수 있다. 예컨대, 표적 위치에 대하여 설계된 표적 서열과 뉴클레오티드 불일치 (mi smatch)를 가지는, 표적 위치와 서열 상동성이 높은 비표적 서열의 경우 유전자 가위가 작동할 가능성이 있다. 상기 비표적 위치는 이에 제한되는 것은 아니나, 표적 서열과 1 이상의 뉴클레오티드불일치 (mi smatch)를가지는위치일수 있다.
이는유전체 내에서 원치 않는유전자의 돌연변이를 야기할수 있어 상기 표적 특이적 뉴클레아제를 사용하는데 심각한 문제가 될 수 있다. 이에, 표적 특이적 뉴클레아제의 표적 위치에서의 활성 못지 않게 비표적 위치를 정확히 검출하여 분석하는 과정 또한 매우 중요할 수 있으며, 이는 비표적 효과 없이 표적 위치에만 특이적으로 작동하는 표적 특이적 뉴클레아제를개발하는데유용하게사용될수있을것이다.
본 발명의 목적상상기 뉴클레아제는 생체 내 Un vivo) 및 시험관 내 Un r/iro)에서 뉴클레아제 활성을 가질 수 있으므로, 시험관 내에사 유전체 DNA의 비표적 위치를 검출하는데 사용될 수 있으며, 이를 생체 내에서 적용하였을 때 상기 검출된 비표적 위치와동일한 위치에도 활성을 가질 것을예상할수있다.
상기 (b) 단계는 상기 (a) 단계를 통하여 한 가닥이 절단된 쇼에 단일가닥 특이적 엔도뉴클레아제를 처리하여, 나머지 DNA 가닥을 절단하는 단계이다.
상기 단일가닥특이적 엔도뉴클레아제는 단일가닥 DNA를 특이적으로 절단하는 활성을 갖는모든 엔도뉴클레아제를총칭하는 것일 수 있다. 일 예에서, 상기 단일가닥 특이적 엔도뉴클레아제는 엔도뉴클레아제 V, 또는 엔도뉴클레아제 VI I I와 알킬아데닌 DNA 글라이코실라아제 (Alkyl adenine DNA Glycosyl ase; hMG)와의 조합 등으로 이루어진 군에서 선택된 1종 이상일 수 있다. 상기 엔도뉴클레아제 V는 원핵 세포 또는 진핵 세포, 예컨대, 균류, 진핵 동물 (예컨대, 어류, 양서류, 파충류, 조류, 포유류 등) , 또는 진핵 식물 유래의 것일 수 있으며, 예컨대, 대장균 유래의 endonuclease V (NP_418426.2; E. col i ENDOV) 등으로 이루어진 군에서 선택된 1종 이상일 수 있다. 상기 단일가닥특이적 엔도뉴클레아제 (예컨대, 엔도뉴클레아제 V)는 아데노신이 이노신으로 변환된 가닥을 절단하는 것일 수 있으며, 상기 아데노산이 이노신으로 변환된 변이 위치에서 5’ 방향 및/또는 3' 방향으로 5nt 이내, 4nt 이내, 3 nt 이내, 2nt 이내, 또는 lnt 이내에 위치하는 뉴클레오타이드의 3' 말단을 절단하는 것이 수 있다. 상기 엔도뉴클레아제 VI I I는 아데닌 디아미나아제에 의하여 아데닌이 이노신으로 변환된 뉴클레오타이드를 제거하는 역할을 하는 것으로, 이노신을 이중가닥 DNA로부터 제거하는 N-glycosylase활성과상기 이노신 제거로 발생한 apur ini c si te (AP si te)의 3’ 및 5' 말단을 절단하는 AP- lyase 활성을 모두 갖는 모든 효소들 중에서 1종 이상 선택될 수 있다. 예컨대, 상기 엔도뉴클레아제 VI I I는 E, coli엔도뉴클레아제 VI I I (예컨대, GenBank Access ion No. NP_415242.1 등) , 인간 엔도뉴클레아제 VI I I
(예컨대, GenBank Accession Nos . BAC06476.1 , NP_001339449.1,
NP_001243481.1, NP_078884.2, NP_001339448.1등) , 마우스 엔도뉴클레아제 VI I I (예컨대, GenBank Accession Nos . BAC06477.1, NP—082623.1 등), Escher ichi a col i 엔도뉴클레아제 VI 1 1 (예컨대, GenBank Accession Nos . OBZ49008.1, 0BZ43214.1, 0BZ42025.1, ANJ41661.1, KYL40995.1, KMV55034.1, KMV53379.1, KMV50038.1, KMV40847.1, AQW72152.1 등) 등으로 이루어진 군에서 선택된 1종 이상일 수 있으나, 이에 제한되는 것은 아니다. 상기 알킬아데닌 DNA 글라이코실라아제는 N-glycosidi c bond의 가수분해를 족매하여 alkyl at ion-damaged퓨린 염기를 제거하여 DNA를 repair하는 효소로, 특히 3-methyl adenine, 7-methyl adenine, 7-methyl guanine, 1N_ ethenoadenine and hypoxanthine 등을 제거한다. 사용 가능한 알킬아데닌
DNA 글라이코실라아제는 인간 알킬아데닌 DNA 글라이코실라아제 (MAG; GenBank Accession no. NP_001015052.1 (코딩 핵산 서열 NM_001015052.2) , NP_001015054.1 (코당 핵산 서열 NM_001015054.2) , NP_002425.2 (코딩 핵산서열 ■J302434.3) 등으로 이루어진 군에서 선택된 1종 이상일 수 있으나이에 제한되는것은아니다.
상기 (c) 단계는 상기 (b) 단계를 통해 절단된 DNA를 이용해 전체 유전체 시퀀싱 (whole genome sequencing, WGS)을수행하는단계로서, 표적 위치의 서열과 상동성을 가지는 서열을 찾아 비표적 위치일 것으로 예측하는 간접적인 방법과 달리 전체 유전체 수준에서 실질적으로 표적 특이적 뉴클레아제에 의해 절단되는비표적 위치를검출하기 위해 수행되는 것이다.
본 발명에서 용어 "전체 유전체 시퀀싱 (whole genome sequencing; WGS)”은 차세대 시퀀싱 (next generation sequencing)에 의한 전장 유전체 시퀀싱을 10 X, 20 X, 40 X 형식으로 여러 배수로 유전체를 읽는 방법을 의미한다. "차세대 시퀀싱 "은 칩 (Chip) 기반 및 PCR 기반 페어드엔드
(paired end) 형식으로 전장 유전체를 조각내고, 상기 조각을 화학적인 반응 (hybridization)에 기초하여 초고속으로 시퀀싱을 수행하는 기술을 의미한다.
상기 (d) 단계는 상기 전체 유전체 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 DNA가 절단된 위치를 결정하는 단계로서 , 시퀀싱 데이터를 분석하여 표적 특이적 뉴클레아제의 표적 위치 및 비표적 위치를 간편하게 검출할 수 있다. 상기 염기서열 데이터로부터 DNA가 절단된특정 위치를 결정하는것은다양한접근방법으로수행될수 있으며, 본 발명에서는 상기 위치를 결정하기 위한 여러 가지의 합리적인 방법들을 제공한다. 그러나 이는 본 발명의 기술적 사상에 포함되는 예시에 불과하며, 본발명의 범위가이들방법에 의해 제한되는것은아니다.
예컨대, 상기 절단된 위치를 결정하기 위한 일례로서, 전체 유전체 시퀀싱을 통해 수득한 염기서열 데이터를 분석 프로그램 (예를 들어, BWA/GATK또는 ISAAC등)을 이용하여 유전체 상의 위치에 따라 정렬하였을 경우, 5' 말단이 수직 정렬된 위치가 DNA가절단된 위치를의미할수 있다. 본 명세서에 사용된 바로서, 상기 용어 "수직 정렬"이란, BWA/GATK 또는 ISAAC 등의 프로그램으로 전체 유전체 시퀀싱 결과를 분석할 때, 인접한 왓슨가닥 (Watson strand)과크릭 가닥 (Crick strand) 각각에 대해, 2개
이상의 염기서열 데이터의 5' 말단이 유전체 상의 동일한위치 (nuc leot ide pos i t ion)에서 시작되는 배열을 말한다. 이로 인하여, 표적 특이적 뉴클레아제에 의해 잘려 동일한 5’ 말단을 갖게 되는 DNA 단편들이 각각 시퀀싱되어 나타나게 된다.
즉, 표적 특이적 뉴클레아제가 표적 위치 및 비표적 위치에 뉴클레아제 활성을 나타내 상기 위치를 절단하는 경우, 염기서열 데이터를 정렬하게 되면 공통적으로 절단된 부위는 각각 그 위치가 5' 말단으로 시작되므로 수직 정렬되나, 절단되지 않은 부위에는 5’ 말단이 존재하지 않으므로정렬 시 스태거드 (staggered) 방식으로배열될수 있다. 따라서, 수직 정렬된 위치를 표적 특이적 뉴클레아제에 의해 절단된 부위로 볼 수 있으며, 이는 곧 표적 특이적 뉴클레아제의 표적 위치 또는 비표적 위치를 의미하는것일수있다.
상기 "정렬"은 표준 염기서열 (reference genome)로 염기서열 데이터를 맵핑한 뒤 , 유전체에서 동일 위치를 가지는 염기들을 각 위치에 맞게 배열하는 것을 의미한다. 따라서, 염기서열 데이터를 상기와 같은 방식으로 정렬할 수 있다면 어떠한 컴퓨터 프로그램도 이용될 수 있으며, 이는 당업계에 이미 알려진 공지의 프로그램이거나 또는 목적에 맞게 제작된 프로그램들 중에서 선택될 수 있다. 일 실시예에서는 ISAAC를 이용하여 정렬을수행하였으나, 이에 제한되는것은아니다.
정렬 결과, 상기 설명한 바와 같은 5’ 말단이 수직 정렬된 위치를 찾는 등의 방법을 통해 표적 특이적 뉴클레아제에 의해 DNA가 절단된 위치를결정할수 있고, 상기 절단된 위치가표적 위치 (on-target s i te)가 아니라면, 비표적 위치 (of f-target si te)로 판단할 수 있다. 다시 말해, 표적 특이적 뉴클레아제의 표적 위치로설계한 염기 서열과동일한서열은 표적 위치이고, 상기 염기 서열과동일하지 않은서열은 비표적 위치로 볼 수 있다. 이는 상기 기술한 비표적 위치의 정의상자명한 것이다. 상기 비표적 위치는 특히, 표적 위치의 서열과 상동성을 가지는 서열로 구성된 것일 수 있고, 구체적으로 표적 서열과 1 개 이상의 뉴클레오티드 불일치 (mi smatch)를가지는서열, 더욱구체적으로표적 위치와 1내지 6개, 1개 내지 5개, 1개 내지 4개, 1개 내지 3개, 1개 내지 2개, 또는 1개의 뉴클레오티드불일치를가지는 것일 수 있으나, 이에 특별히 제한되는 것은 아니고 표적 특이적 뉴클레아제가 절단할 수 있는 위치라면 본 발명의 범위에 포함될 수 있다. 이때, 상기 표적 위치는 가이드■쇼와 상보적인
15 내지 30 뉴클레오티드 서열일 수 있고, 추가적으로 표적 특이적 뉴클레아제가 인식하는 서열 (예컨대, Cas9의 경우 Cas9이 인식하는 PAM 서열)을포함할수있다.
다른예에서, 5' 말단이 수직 정렬된위치를찾는방법 이외에도, 5’ 말단 플롯에서 이중 피크 패턴을 보이는 경우 그 위치가 표적 위치가 아니라면 비표적 위치로 판단할 수 있다. 유전체 DNA 내의 각 위치에 대하여 동일한 염기의 5' 말단을 구성하고 있는 뉴클레오티드 수를 세어 그래프를 그릴 경우, 특정 위치에서 이중 피크 패턴이 나타나게 되는데, 상기 이중 피크는 표적 특이적 뉴클레아제에 의해 절단된 이중 가닥의 각각의 가닥에 의해 나타나는것이기 때문이다.
일 실시예에서, 유전체 DNA를 표적 특이적 뉴클레아제 (예컨대, RGEN)으로 절단한 뒤, 전체 유전체 분석 후 이를 ISAAC로 정렬하여 절단된 위치에서는 수직 정렬, 절단되지 않은 위치에서는 스태거드 방식으로 정렬되는패턴을확인하였으며, 이를 5' 말단플롯으로나타내었을 때 절단 위치에서 이중피크의 독특한패턴이 나타나는것을확인하였다.
나아가 이에 제한되는 것은 아니나, 구체적인 일례로 왓슨 가닥 (Watson strand)과 크릭 가닥 (Cr i ck strand)에 해당하는 염기서열 데이터 (sequence read)가 각각 두 개 이상씩 수직으로 정렬되는 위치를 비표적 위치인 것으로 판단할 수 있고, 또한 20 % 이상의 염기서열 데이터가 수직으로 정렬되고, 각각의 왓슨 가닥 및 크릭 가닥에서 동일한 5' 말단을 가진 염기서열 데이터의 수가 10 이상인 위치가비표적 위치, 즉 절단되는 위치인 것으로판단할수있다.
상기 비표적 위치 확인 (검출)은 시험관 내 Un F/ fro)에서 표적 특이적 뉴클레아제를 유전체 DNA에 처리하여 수행될 수 있다. 이에 상기 방법을 통해 확인 (검출)된 비표적 위치에 대하여 실질적으로 생체 내 Un FO)에서도 비표적 효과가 나타나는지 확인해볼 수 있다. 다만 이는 추가적인 검증 과정에 불과하므로 본 발명의 범위에 필수적으로 수반되는 단계는아니며, 필요에 따라추가적으로수행될수있는단계에 불과하다. 본 명세서에 사용된 바로서, 용어 "비표적 효과 (of f-target ef fect)’’는 비표적 위치 (of f-target si te)와는 구별되는 개념일 수 있다. 즉, 상기 설명한 바와 같이 본 발명에서 비표적 위치라는 개념은 표적 특이적 뉴클레아제가 작동할 수 있는 위치 중 표적 위치가 아닌 위치를 의미하는 것으로, 상기 표적 특이적 뉴클레아제에 의해 절단되는 위치를
말하는 것이나, 비표적 효과는 세포 내 비표적 위치에서 표적 특이적 뉴클레아제에 의해 인델 ( Insert ion and/ or delet ion)이 나타나는 효과를 의미한다.
상기 용어 "인델’’은 DNA의 염기 배열에서 일부 염기가 중간에 삽입되거나 ( insert ion) 및/또는 결실된 (delet ion) 변이를 총칭한다. 또한, 표적 특이적 뉴클레아제에 의해 상기 인델이 일어난 비표적 위치를 비표적 인델 위치라고 한다. 결론적으로, 본 명세서의 비표적 위치는 비표적 인델 위치를 포함하는 개념으로 볼 수 있으며, 표적 특이적 뉴클레아제가 활성을 가질 수 있는 가능성이 있는 위치로 족하며, 반드시 유전자 가위에 의한 인델이 확인되어야 하는 것은 아니다. 한편, 본 발명에서의 비표적 위치는 비표적 후보 위치 (candidate of f-target si te)로, 비표적 인델 위치는 검증된 비표적 위치 (val idated of f-target si te)로도명명될수있다.
구체적으로 상기 검증 과정은, 이에 제한되는 것은 아니나, 상기 비표적 위치에 대한 표적 특이적 뉴클레아제가 발현된 세포로부터 유전체 DNA를 분리하고, 상기 DNA의 비표적 위치에서 인델을 확인하여 비표적 위치에서의 비표적 효과를확인하는것일 수 있다. 이는, T7E1분석, Cel- I 효소를 이용한돌연변이 검출분석 또는표적화딥시퀀싱 (targeted deep sequencing) 등당업계에 공지된 인델 확인 방법을수행하여 비표적 효과를 확인하는 것일 수 있다. 상기 비표적 효과를 확인하는 단계는 비표적 위치에서 인델이 일어났는지를 직접적으로 확인하는 것일 수 있다. 다만, 이러한 생체 내 검증 과정에서 인델이 일어나지 않았다고 하더라도, 이는 검출할 수 있는 수준 이하의 빈도로 인델이 일어날 경우까지 확인한 것은 아니므로어디까지나보조적인수단으로보아야한다.
상기 기술한 것과 같이 수직 정렬된 위치를 확인하거나, 또는 5 ' 말단 플롯에서 이중 피크를 확인하는 것만으로도 비표적 위치를 충분히 검출할 수 있고 이는 고도의 재현성을 가지는 것이나, 불균일 절단 패턴 또는 낮은 시퀀싱 깊이 (depth)를 가지는 일부 위치가 누락될 수 있다는 문제가 있다. 이에 본 명세서에서는 염기서열 데이터의 정렬 패턴을 기반으로 하여, 각 뉴클레오티드의 위치 i (즉 유전체 DNA 상의 뉴클레오타이드 위치)에 DNA 절단 점수를 산출하는 수식을 다음과 같이 제공한다:
2019/147073 1»(:1/10公019/001104
위치에서의점수

상기 수식에서 염기서열 데이터의 수는 뉴클레오타이드 리드 수를 의미하고, 시퀀싱 깊이는특정 위치에서의 시퀀싱 리드수를의미한다.
또한, 상기 수식을통해 기존의 Digenome-seq에서는검출되지 않았던 다수의 추가적인 위치를 검출할 수 있으며, 이를 통해 거짓-양성 위치를 손쉽게 걸러낼 수 있다. 상기 수식에서 C값은 당업자가 임의의 상수를 적용할 수 있는 것으로 본 발명의 실시예에 의해 제한되는 것은 아니다. 일 예에서, 상기 C는 1내지 1000, 1내지 500, 1내지 100, 1내지 50, 1 내지 10, 1 내지 5, 또는 1 내지 3일 수 있으나, 이에 제한되는 것은 아니다. 특히, 이에 제한되는 것은 아니나, 예컨대, 임의의 위치 (절단된 위치)의 염기서열에 있어서, C 값을 1으로 하여 상기 산출된 점수가 2.5점 이상이거나, 0.1 점 이상이며 On-target 서열과 homology를 갖는 경우 (예컨대, 0n-target 서열과 10개 이하의 미스매치를가지고 PAM(5’-NGN_3’ 또는 5'-NNG-3’)을 포함하는 경우) , 상기 임의의 위치(절단된 위치)를 비표적 위치로 판단할 수 있다. 다만, 상기 점수의 기준은 목적에 따라 당업자에 의해 적절히 조정, 변경될수 있다.
일 예에서, 본 명세서에서 제공되는 Digenome-seq 방법은 복수의 표적 특이적 뉴클레아제 (예컨대, 표적 부위가 상이한 가이드 RNA를 다수 포함하는 표적 특이적 뉴클레아제)를 이용하여 수행될 수도 있으며, 본 명세서에서는 이를 "복합 Digenome-seq "로 명명한다. 이 경우, 상기 표적 특이적 뉴클레아제는 2 개 이상, 구체적으로 2 내지 100 개의 표적에 대한 표적 특이적 뉴클레아제를 혼합한 것일 수 있으나, 이에 제한되는 것은
아니다.
상기 복합 Digenome-seq의 경우 각각의 표적 특이적 뉴클레아제에 의해 유전체 DNA가 절단되므로 절단 위치가 어느 유전자 가위에 의해 절단되었는지를 확인하는 것이 중요하다. 이는 표적 위치와의 편집 거리 (edi t di stance)에 따라 비표적 위치를 분류함으로써 달성될 수 있으며, 비표적 위치의 염기 서열이 표적 위치와 상동성을 가진다는 것을 전제로 한다. 이를 통해 각각의 유전자 가위에 대한 표적 및 비표적 위치가 명확하게 구분될수있다.
본 발명의 구체적인 일 실시예에서는. 특정 위치를 표적으로 하는 RGEN (RNA-guided engineered nuc lease)에 대하여, 전체 유전체에서
Digenome-seq를 통해 검출된 비표적 위치 중 표적 위치와의 뉴클레오티드 불일치가 6 개 이하인 상동성 위치가 13,000 개 이하이고, 뉴클레오티드 불일치가 2 개 이하인 상동성 위치를 가지지 않는 경우, 상기 특정 위치를
RGEN의 표적 위치로 선별하는 것이 비표적 효과를 최소화할 수 있음을 확인하였다. 이는 본 발명의 Digenome-seq를 이용하여 표적 위치를 선별하는 바람직한 기준을 확립해가는 과정을 보여주는 일례로서, Digenome-seq를 통해 유전자 가위의 비표적 효과를 최소화 시킬 수 있을 것으로기대된다.
한편, 표적 위치의 서열과 상동성을 가지는 위치의 수는 뉴클레오티드불일치 수준이 증가할수록 Digenome-seq를통해 적은비율로 검출되는 것을 확인하였다. 이는 RGEN의 표적 위치를 선별함에 있어서, 표적 서열과 유전체 내에서 상동성을 가지는 뉴클레오티드 서열이 많을 수록, 특히 고도의 상동성을 가지는 뉴클레오티드 서열이 많을 수록 상대적으로 더욱 특이적이기 때문이다. 이를 통해 선별된 RGEN의 표적 위치는비표적 효과가최소화된 것일수 있다.
다른 예에서, 상기 of f-target 확인 방법은 CIRCLE-seq (ci rcular i zat ion for in vi tro report ing of cl eavage ef fects by sequencing; Nature Methods 14, 607-614 (2017) 참조) 및/또는 SITE-seq (Cameron, P. et al . Mapping the genomi c landscape of CRISPR_Cas9 c leavage. Nature methods 14, 600-606 (2017) 참조)을 통하여도 수행 가능하다 (도 2a및 2b참조) .
【발명의 효과】
본 발명에서 제시되는 유전체 서열 분석 기술에 의하여, 고도의 재현성으로유전체 수준에서 유전자가위의 비표적 위치를검출할수 있어, 표적 특이성이 높은 유전자 가위의 제작 및 이를 위한 연구에 사용될 수 있다.
【도면의 간단한설명】
도 la는 아데노신 디아미나아제와 Cas9 니케이즈 (D10A) (ABE)와 엔도뉴클레아제 V를 사용하는 유전체 DNA 서열 분석 과정을 모식적으로 보여준다.
도 lb는 아데노신 디아미나아제와 Cas9 니케이즈 (D10A) (ABE)와
Alkyladenine DNA Glycosylase (AAG) 및 엔도뉴클레아제 VIII를 사용하는 유전체 DNA서열분석 과정을모식적으로보여준다.
도 2a는 ABE및 엔도뉴클레아제 V를처리한후 real-time PCR을통해 DSB( Double Strand Break)의 생성 여부를확인한결과를보여준다.
도 2b는 ABE, Alkyladenine DNA Glycosylase (AAG) , 및 엔도뉴클레아제 VI II를처리한후 real-time PCR을통해 DSB의 생성 여부를 확인한결과를보여준다.
도 2c는 ABE 및 엔도뉴클레아제 V를 처리하거나 ABE, Alkyladenine DNA Glycosylase (AAG), 및 엔도뉴클레아제 VIII를 처리한 후의 염기 교정을확인한결과이다.
도 3a 및 3b는 ABE 및 엔도뉴클레아제 V를 처리하거나 ABE, Alkyladenine DNA Glycosylase (AAG) , 및 엔도뉴클레아제 VI II를 처리한 후의, 전유전체서열분석 (WGS) 결과를보며준다.
도 4는 ABE 및 엔도뉴클레아제 를 처리하거나 ABE, Alkyladenine DNA Glycosylase (AAG), 및 엔도뉴클레아제 VIII를 처리한 후의, 절단 위치를확인한결과이다.
도 5는 CIRCLE-seq및 SITE_seq에 의한유전체 DNA서열분석 과정을 모식적으로보여준다.
도 6a내지 6c는 on target site에 대하여 0내지 4개의 mismatched 염기를 갖는 sgRNA를 사용하여 얻어진 염기교정 빈도 {%) 및 삽입/결실 빈도 (%)를 보여주는 그래프로, 별표는 하나의 효소에 대한 relative activity ( [mismatched sgRNA 결과값]/ [matched sgRNA 결과값])가 나머지
두 개의 효소보다 3배 이상높은 mismatched sgRNA를 나타내고, 3 ' 말단의 마지막 3개 뉴클레오타이드는 PAM 서열을, 소문자는 mismatched 뉴클레오타이드를 각각 나타낸다 (Means + s.e.m. were from three independent experiments) .
도 7a는 ABE7.10 및 Endo V를 사용하는 Digenome-seq 분석법의 overview로서, ABE7.10가 구아닌의 데옥시이노신(deoxyinosine)으로의 변환을 촉매하고, Endo 이 데옥시이노신을 인식하여 상기 데옥시이노신에 대하여 두 번째 phosphodiester bond 3’를 절단함을 보여주며, 삼각형 꼭지점 위치는 ABE7.10 nickase 및 Endo V에 의하여 절단되는 phosphodiester bond위치를보여준다.
도 7b는 ABE-매개 Digenome-seq workflow의 개략도이고,
도 7c는 ABE7.10 및 hAAG/Endo VIII를 사용하는 Digenome-seq 분석법의 개략도로서, ABE7.10가 하나의 가닥에서 adenine-to-inosine 변환을 매개하고 다른 가닥에서 nick을 생성시키며, MAG이 이노신을 제거하여 AP site (apurinic/apyrimidinic site)을 생성하고, Endo VI 11 (DNA glycosylase and AP-lyase)가 상기 AP site을 절단함을 보여주며, 삼각형 꼭지점 위치는 ABE7.10 nickase 및 hAAG/Endo VIII에 의하여 절단되는 site을보여준다.
도개는 ABE7.10표적 서열을포함하는 PCR산물에 ABE7.10 및 Endo V를 처리한 결과를 보여주는 것으로, ABE7.10와 Endo 가 모두 처리된 경우에만 DNA에 double strand break가형성됨을보여준다.
도 7e는 ABE7.10 표적 서열을 포함하는 PCR산물에 ABE7.10, MAG, 및 Endo VIII를 처리한 결과를 보여주는 것으로, ABE7.10, MAG, 및 Endo VIII가 모두 처리된 경우에만 ■쇼에 double strand break가 형성됨을보여준다.도 가는 Sanger sequencing 결과를 나타낸 것으로, ABE7.10에 의한 A-to-G conversion과 Endo V에 의한 DNA절단을보여준다.
도 7g는 qRT-PCR결과를나타낸그래프로서, ABE7.10 nickase와 Endo V에 의한 DNA절단을보여준다.
도 7h는 Sanger sequencing 결과를 나타낸 것으로, 유전체 DNA를 ABE7.10로 처리한 경우의 A-to-G conversion, 및 유전체 DNA를 ABE7.10,
MAG, 및 Endo VIII로처리한경우의 G-to~A conversion을보여준다.
도기는 qRT-PCR결과를 나타낸 그래프로서, 유전체 쇼를 ABE7.10, hMG, 및 Endo VIII로분해한결과를보여준다.
2019/147073 1»(:1/10公019/001104
도 7j는 WGS data를 사용하여 얻어진 HEK2 target site에서의 sequence reads 의 수직 정렬(straight alignment) 결과를 보여주는 IGV 이미지이다.
도 7k는 ABE7.10, hAAG, 및 Endo VIII 처리 후관찰되는 sequencing reads의 수직 정렬(straight alignment) 결과를보여주는 IGV이미지이다. 도 8은 nickel affinity chromatography 및 heparin bead chromatography를 사용하여 정제된 ABE7.10의 SDS-PAGE 분석 결과를 보여준다 (M: marker , 1(-) : cell lysate before IPTG ( isoproply-l-thio- -D-ga1actopyranoside) induction, 1(+) : cel 1 lysate after IPTG induction, S: soluble lysate fraction, IS: insoluble lysate fraction, FT: flow-through, Wl, W2: waste after washing, Ni : Ni-NTA agarose beads after elution of bound protein, NE: protein fraction eluted from nickel beads, Hp: Heparin Sepharose 6 Fast Flow affinity resins after elution of bound protein, HE: protein fraction after purification using heparin beads. 박스로 표시된 부분은 ABE7.10 단백질 밴드를 나타냄).
도 9는 ABE의 Digenome-seq 분석을 위한 In vitro DNA 절단 점수 시스템을개략적으로보여준다.
도 10a및 10b는 Digenome-seq에 의해 확인된유전체 전역의 ABE7.10 off-target site를 보여주는 것으로, 10a는 온전한 유전체 DNA (회색; 중심원으로부터 첫 번째 영역) 및 ABE7.10 + Endo V 처리된 유전체 DNA (파란색; 중심원으로부터 두 번째 영역), 및 ABE7.10 + hAAG + Endo VIII 처리된 유전체 DNA (붉은색; 중심원으로부터 세 번째 영역)을 사용하여 WGS를 통하여 얻어진 DNA 절단 점수를 보여주는 Genome-wide Circos plots이고 (화살표는 on-target site을 나타냄), 10b는 ABE7.10 + Endo V 또는 ABE7.10 + hAAG + Endo VIII를사용하여 확인된 인간유전체에서의 in vitro절단부위의 개수를보여주는벤다이어그램이다.
도 10c는 Digenome-captured sites에서 DNA 서열을 비교하고 WebLogo를사용하여 얻어진 Sequence logos이다.
도 11은 RNF2, TYR03, WEE1, EphB4, HPRT_exon6, 및 HPRT_exon8 부위에서의 sequencing reads의 수직 정렬 결과를보여주는 IGV이미지이다. 도 12a 및 도 12b는 온전한 유전체 DNA (gray; 중심에서 첫 번째 영역) 및 genomic DNA treated with ABE7.10/Endo V (blue; 중심에서 두
번째 영역), BE3AUGI/USER (red; 중심원으로부터 3번째 영역), 또는 Cas9 (Green; 중심원으로부터 4번째 영역)으로 처리된 유전체 DNA를 사용하여 HEK2 (12a) 및 RNF2 (12b) 부위에 대하여 얻어진 Genome-wide Circos plots을보여주며, 화살표는 on-target부위를나타낸다.
도 12c 및 도 12d는 Digenome-captured sites에서의 DNA 서열을 비교하고 WebLogo를사용하여 얻어진 Sequence logos이다.
도 12e는 targeted deep sequencing을사용하여 측정된 ABE7.10 off- target sites에서의 염기 교정 효율을 나타낸 그래프로서, 3' 말단 끝의 3개 뉴클레오타이드는 PAM 서열을 나타내고, 소문자는 Mismatched base를 나타낸다(Means 士 s.e.m. were from three independent experiments) . 도 12f는 ABE7.10 -매개 치환 빈도와 Cas9 -매개 indel 빈도 간의 상관성 (위쪽) 및 ABE7.10 -매개 치환 빈도와 BE3 -매개 치환 빈도 간의 상관성 (이래쪽)을보여주는 Scatterplots이다.
도 13a는 일반적으로 사용되는 형태 (GX19), 절단된 형태 (gX18 or gX17), 및 연장된 형태 (gX20 또는 ggX20)의 sgRNAs의 표적 DNA-상보적 RNA 서열을예시적으로보여준다.
도 13b는 HEK2, RNF2, TK_EphB4, TYR03, WEE1, HPRT-exon6, 및 HPRT- exon8를 표적으로 하는 변형 sgRNA 사용시 targeted deep sequencing에 의하여 측정된 각 변형 sgRNA별 ABE7.1◦의 염기 교정 빈도평균을 나타낸 그래프이다.
도 13c 및 13d는 HEK293T 세포에서 targeted deep sequencing으로 측정된 HPRT-Exon 6 (13c) 및 TYR03와 HPRT-exon8 (13d)의 on- 또는 off- target sites에서의 ABE7.10 -매개 염기 교정 빈도 (%)를 결과를 보여주는 그래프및 히트맵으로서, 히트맵은 GX19 sgRNA를사용한경우와비교한변형 sgRNA의 상대적 특이도 (relative specificities)를 보여주며, 상대적 특이도 (Specificity ratio)는 측정 대상 sgRNA의 특이도(on-target frequency/off-target frequency)를 GXig sgRNA의 특이도 (on-target frequency/off-target frequency)로 나누어 계산된 값이고, 3' 말단 끝의 3개 뉴클레오타이드는 PAM 서열을 나타내고, 소문자는 Mismatched base를 나타낸다 (Means 士 s.e.m. were from three independent experiments) . 도 13e및 13f는 ABE7.10및 Sniper A則:7.10을사용한경우의 HPRT- Exon 6 (13e) 및 HPRT-exon8, HEK2, EphB4 및 TYR03 (이상, 13f)에 부위에서의 on-및 off-target 활성 및 특이도비율 (specificity ratio)을
보여주는 그래프 및 히트맵으로, 3' 말단 끝의 3개 뉴클레오타이드는 PAM 서열을 나타내고, 소문자는 Mismatched base를 나타내고, specificity ratio는 다음의 수식에 의하여、계산된다: Sniper ABE7.10의 specificity (on-target frequency/off-target frequency)/ABE7.10의 specificity (on- target frequency/off-target frequency) (Means ± s.e.m. were from three independent experiments) .
도 13g및 13h는플라스미드를통하여 세포내로전달되는경우와 RNP 형태로세포내로 전달되는경우의 HPRT-Exon 8 (13g) 및 HPRT_exon6, HEK2, EphB4 및 TYR03 (이상, 13h)에 부위에서의 on- 및 off-target 활성 및 특이도 비율 (specificity ratio)을 보여주는 그래프 및 히트맵으로, 3' 말단 끝의 3개 뉴클레오타이드는 PAM 서열을 나타내고, 소문자는 Mismatched base를 나타내고, specificity ratio는 다음의 수식에 의하여 계산된다: RNP 전달시의 specificity (on-target frequency/off-target frequency)/플라스미드 전달시의 specificity (on-target frequency/off- target frequency) (Means + s.e.m. were from three independent experiments) .
도 13i 및 13j는 ABE7.10 또는 Sniper A別7.10을 변형 sgRNA와 조합하여 사용한 경우의 HPRT-exon6 부위 (13i) 및 HPRT-exon8 부위 (13j)에서의 on- 및 off-target 활성 및 특이도 비율 (specificity ratio)을 보여주는 그래프 및 히트맵으로, 3' 말단 끝의 3개 뉴클레오타이드는 PAM 서열을 나타내고, 소문자는 Mismatched base를 나타내고, specificity ratio는 다음의 수식에 의하여 계산된다: Sniper ABE7.10와 변형 sgRNA의 조합의 specificity (on-target frequency/off- target frequency)/ABE7.10의 specificity (on-target frequency/off- target frequency) (Means 士 s.e.m. were from three independent experiments) .
【발명을실시하기 위한구체적인내용】
이하에서는 실시예를 들어 본 발명을 더욱 구체적으로 설명하고자 하나, 이는 예시적인 것에 불과할 뿐 본 발명의 범위를 제한하고자 함이 아니다. 아래 기재된 실시예들은 발명의 본질적인 요지를 벗어나지 않는 범위에서 변형될수있음은당업자들에게 있어 자명하다.
실시예 1: 아데노신 디아미나아제와 Cas9니케이즈 (D10A) 처리 후, 엔도뉴클레아제 V를처리한경우의 유전체 DNA서열분석
세포에서 추출한 DNA를아데노신 디아미나아제- Cas9니케이즈 (D10A, Cas9의 10번째 아미노산이 D에서 A로 변이된 것)와 guide RNA 복합체와 혼합하게 되면 DNA의 한쪽 가닥이 절단되고 반대쪽 가닥의 A가 I로 바뀌게 된다. 반응시킨 DNA에 엔도뉴클레아제 V (endonuclease 5)를처리하게 되면 I를 인식하여 절단을유도한다. 결국 디아미나아제- Cas9니케이즈 (D10A)와 엔도뉴클레아제 에 의해 DNA의 두 가닥 파손 (DSB; double strand break)이 유도된다 (도 la참조) .
이 후 DNA를 정제한 후, DNA의 두 가닥 파손 (DSB)이 발생한 DNA를 end repai r 과정을 거쳐 전유전체 시퀸싱을 진행한다. DNA의 두 가닥 파손 (DSB)이 발생한 DNA의 경우 잘린 부분에서 시퀀싱에 의한 서열이 on- target 위치에서 직선나열 (straight al ignment)이 됨을 알수 있으며, 전 유전체에서 직선나열 (straight al ignment)이 된 부분을 찾음으로써 아데노신 디아미나아제- Cas9니케이즈의 of f-target을검출할수 있다 (도 la) . 실시예 2: 아데노신 디아미나아제와 Cas9 니케이즈 처리 후, Alkyl adenine DNA Glycosylase (/ 이를처리한경우의 유전체 DNA서열분석 또한, 아데노신 디아미나아제- Cas9 니케이즈가 처리된 DNA에
Alkyl adenine DNA Glycosyl ase (/ 이를 처리하게 되면 I이 제거되면서 AP- si te (apur ini c/apyr imidini c si te)가 만들어 지게 된다. 그후 AP-s i te를 절단할 수 있는 엔도뉴클레아제 VI 11 (endonuc lease 8)을 처리하면 I가 제거되면서 DNA의 두 가닥 파손 (DSB; double strand break)이 유도된다 (도 lb) .
이 후 DNA를 정제한 후, DNA의 두 가닥 파손 (D況)이 발생한 DNA를 end repai r 과정을 거쳐 전유전체 시퀸싱을 진행한다. DNA의 두 가닥 파손 (DSB)이 발생한 DNA의 경우 잘린 부분에서 시퀀싱에 의한 서열이 on- target 위치에서 직선나열 (straight al ignment )이 됨을 알수 있으며, 전 유전체에서 직선나열 (straight al ignment)이 된 부분을 찾음으로써 아데노신 디아미나아제- Cas9니케이즈의 of f-target을검출할수 있다 (도 lb) .
실시예 3: 아데노신 디아미나아제와 Cas9 니케이즈 처리 후, 엔도뉴클레아제 V또는 AAG를처리한경우의 유전체 DNA서열분석
ABE와 엔도뉴클레아제 V 또는 ABE, Alkyl adenine DNA Glycosylase (MG), 엔도뉴클레아제 VII I을 처리한 후 real-time PCR을 통해 D況가 일어나는 지를 확인하여 그 결과를 도 2a 및 2b에 나타내었다. 도 2a 및 2b에 나타난 바와 같이, ABE만 처리하였을 경우에는 50% 정도 절단 되는 것을 알수 있었지만, ABE와 엔도뉴클레아제 V또는 ABE, Alkyl adenine DNA Glycosylase (AAG), 엔도뉴클레아제 VII I을 처리한 후에는 90% 이상의 DNA target site가 절단 된 것을 알 수 있다. 이러한 현상을 다시 한번 검증하기 위해서 sanger sequencing을 진행하였다. ABE를 처리하였을 때는 A가 G로 바뀌는 것을 볼 수 있었으며, ABE와 엔도뉴클레아제 V또는 ABE, Alkyladenine DNA Glycosylase (AAG) , 엔도뉴클레아제 VIII을 처리한 후에는 G로 변했던 부분이 거의 전부 (ABE와 엔도뉴클레아제 V) 또는 일부 (ABE, Alkyladenine DNA Glycosylase (AAG) , 엔도뉴클레아제 VIII)가 A로바뀌는것을확인할수있다. 실시예 4: Whole Genome Sequencing (WGS)수행
상기 실시예 3의 A則:와 엔도뉴클레아제 V 또는 ABE, Alkyladenine DNA Glycosylase (AAG), 엔도뉴클레아제 VII I을 처리한 DNA를 WGS을 진행하여 그 결과를 도 3a, 3b, 및 도 4에 나타내었다. 도 3a에서와 같이, 서열 분석 결과에서 straight alignment를 확인 할수 있다. 도 3b 및 도 4에서와 같이, DNA cleavage scoring system을 이용하여 genome-wide off- target candidates를찾을수 있다.
아데노신 염기교정 유전자 가위의 전유전체에서의 off-target을 프로파일링 하는 방법은 CIRCLE-seql7과 SITE-seql8을 이용하아 응용이 가능하다 (도 5).
아데노신 염기교정 유전자 가위의 전유전체에서의 off-target을 프로파일링 하는 방법은 CIRCLE-seq Tsai , S.Q. et al . CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR_Cas9 nuclease off-targets . Nature methods 14, 607-614 (2017))과 SITE-seq (Cameron, P. et al . Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nature methods 14, 600-606 (2017))을이용하여 응용이 가능하다 (도 5참조).
참고예
1.세포배양및 형질감염
HEK293T 세포 (ATCC CRL-11268)를 10%(v/v) FBS 및 l%(v八) 페니실린/스트렙토 마이신 (Welgene) 보충된 DMEM 배지에서 배양하고, HEK293T 세포(1.5xl05)를 24 -웰 플레이트에 접종하고, Lipofectamine 2000 (Invitrogen; ABE및 BE3의 경우 3m£, SpCas9의 경우 2 사용)을사용하여, ABE (Addgene plasmid #113128; 1.5 g; DNA 중량), BE3 (Addgene plasmid #73021; rAP0BECl-XTEN-Cas9n-UGI-NLS; 1.5//g) 또는 SpCas9 (Addgene plasmid #43945 NP_269215.1; 0.5//g)를 코딩하는 플라스미드를 sgRNA코딩 플라스미드(0.5//g)와함께 70-80% confluency로형질감염시켰다.
하기 실시예에서 상기 ABE 또는 ABE7.10는 서로 동일한 단백질을 지칭하기 위하여 사용되며, 구체적으로 아데노신 디아미나아제 (인위적 진화 (directed evolution) 된 E. coli TadA (NP_417054.2))와 SpCas9 니케이즈 (D10A)(SpCas9 (NP_269215.1)의 1◦번째 아미노산 잔기 D가 A로 치환된 변형 Cas9)가연결된융합단백질을의미한다 (참고예 2참조).
ABE RNP-매개 유전체 교정의 경우, ABE7.10 단백질 (10//g) 및 in vitro전사된 sgRNA (6//g) , 또는 ABE7.10단백질 (0.6/zg) 및 sgRNA ( .2p名) 코딩 플라스미드를 Neon™ Transfection System(Thermo Fisher
Scientific)을 통한 전기천공에 의하여 HEK293T 세포 (1x10s)에 형질감염시켰다. 72 h post-transfection 시점에 DNeasy Blood & Tissue Kit (Qiagen)를사용하여 유전체 DNA를추출하였다.
하기하는 실시예에 사용된 sgRNA는 표적 부위 (표 8 내지 18에서,
' sgRNA표적 부위1 또는 ’DNA seq at a cleavage sites’로표시) 중에서 3’ 말단의 PAM서열 (5’-NGG-3' 어은 A, T, G, 또는 C임))을 제외한서열에서 T를 U로 바꾼 서열을 아래의 일반식 3 중의 표적화 서열 '(Ncas9V로 하여 제작된 것을사용하였다:
5'-(Ncas9)i-(GUUUUAGAGCUA)-(GAAA)-
(UAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGC)-31
(일반식 3; 올리고뉴클레오타이드링커: GAM).
2. ABE7.10단백질의 발현및정제
ABE7.10을다음과같이 설계하였다 (N말단에서 C말단방향):
M況VEFS抑YWMRHALTLMRAWDE則:VPVGAVLVHNNRVIGEGWNRPIGRHDPTAHAEIMALRQ
(XiLWQNYI^IDAUYVTLEPCVMCAGAMIHSRIGRVVFGARDAKTGAAGSLMDVLHHPGMNHRVEITEGIL
ADEX:AALLSDFFRMRRQEIKAQKKAQSSTDSGGSSGGSSGSETPGTSESATPESSGGSSGGS^¾F5¾?F M1RHALTLAKRARDEREVPVGA VL VLNNRVIGEGWNRAIGLHDPTAHAEIMALRQGGL VMQNYRLIDATL YVTFEPCVMCAGAMIHSRIGRVWGVRNAKTGMGSLMDVLHYPGMNHRVEITEGILADECAALLCYFFR MPRQ™@¾4g557gSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGffAVIT DEYKVPSKKFKVLGNTDRHSIKKNLIGALL抑SGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAK VDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMI KFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGE KKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILL SDILRVNTEITKAPLSASMIK及YDEHHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEF YKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEK ILTFRIPYYVGPLARGNSRFAWMTRKSEETITPffNFEEWDKGASAQSFIERMTNFDKNLPNEKVLPKHS LLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTNRKVTVKQLKEDYFKKIECFDSVEISG VEDRFNASLGTYHDLLK11KDKDFLDNEENEDILEDIVLTLTLFEDREMIEERLKTYAHL抑DKVMKQLK RRRYTGffGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLI抑DSLTFKEDIQKAQVSGQGDSLHE HIANLAGSPAIKKGILQTVKWDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERMKRIEEGIKELG SQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDK NRGKSDNVPSEEWKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHVA QILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHA抑AYLNA GTALIKKYP KLESEFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEI VWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDffDPKKYGGFDSPTVAYS VLWAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDL11KLPKYSLFELENGRKR MLASAGELQKGNELALPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRYIL ADANLDKVLSAYNKHRDKPIREQAEN11HLFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSI T6LYETRIDLSQLGGDPKKKRKV
(상기 아미노산서열에서,
(1) 볼드체로 표시된 폴리펩타이드 부분: 야생형 E. coli TadA (NP_417054.2);
(2) 이탤릭체로표시된폴리펩타이드부분: directed evodution된 E. coli TadA (NP_417054.2) (야생형 (NP_417054.2)에서 변이된 아미노산 잔기를이탤릭체 +밑줄로표시함) ;
(3) 밑줄로표시된폴리펩타이드부분 : SpCas9 nickase (D10A) ;
(4) 폴리펩타이드 (1)과 (2)사이 및 폴리펩타이드 (2)와 (3)사이의
올리고펩타이드 (SGGSSGGSSG況TPGTSESATPESSGGSSGGS ): 펩타이드링커; 및
(5)폴리펩타이드 (3)의 C말단에 연결된올리고펩타이드 (PKKKRKV) : SV40 NLS)
상기와 같이 설계된 ABE7.10(N-terminal His purification tag 포함)를 암호화하는 human codon-optimized 유전자가 pET28(Addgene)에 클로닝된 ABE7.10 발현용 플라스미드를 준비하고, 상기 플라스미드를 BL21 Star (DE3)-competent E. coli세포 (NEB)에 형질전환시켰다. 형질전환후, fresh single colony를 37°C에서 진탕하면서 카나마이신 50/zg/mL 함유 Luria-Bertani (LB) 배지 (Thermo Fisher Scientific) 에서 밤새 배양하였다. 1:50으로 희석시킨 pre-cultures 8 를 카나마이신 50 g/mL이 보충된 LB배지 400 가들어있는각각의 플라스크에 접종하한후, 0D60Q=0.650.70이 될 때까지 37°C에서 진탕 배양하고, 얻어진 배양물을 얼음에 보관하였다. IPTG ( isopropy1-b-D-1-thiogalactopyranoside; GoldBio) ImM를사용하여 단백질 발현을유도한후, 배양물을 18 °C에서 16 시간동안진탕배양하였다.
이어지는 단백질 정제 과정은 4°C에서 수행하였다. 상기 얻어진 세포 배양물을 6,000g로 10분 동안 원심분리하여 세포를 수확하고, 용해 완중액 (50 mM sodium phosphate (Sigma-Aldrich), 500 mM NaCl (Sigma- Aldrich), 10 mM imidazole (Sigma-Aldrich) , 1% Triton x-100 (Sigma- Aldrich) , 20% glycerol , 1 mM 1,4-dithiothreitol (DTT; GoldBio) , 1 mM phenylmethylsul fonyl fluoride (PMSF; Sigma-Aldrich) , concentration of 1 mg/mL lysozyme (Sigma-Aldrich) , lOuM Z11CI2 (Sigma-Aldrich) , pH 8.0)에 재현탁시켰다. 그후현탁액을 액체 질소와수조 (37 °C)에서 교대로 3회 반복 배양하였다. 9분 (5초 (on), 10초 (off)) 동안 초음파 처리하여 세포를 용해시키고, 15,000g에서 20분 동안 원심분리하여 용해물을 분리 (cleared)하였다. 상층액을니켈 아가로오스 비드 (Ni-NTA, QIAGEN)와 함께 60분 동안 4°C에서 진탕 배양하였다. 용해물-수지 혼합물을 폴리프로필렌 컬럼에 로딩하고 3 컬럼 부피의 세척 완충액 (50 mM sodium phosphate (Sigma-Aldrich) , 150 mM NaCl (Sigma-Aldrich) , 35 mM imidazole (Sigma-Aldrich) , 20% glycerol , 1 mM DTT (GoldBio) , 10 yM ZnCl2 (Sigma-Aldrich), pH 8.0)으로 세척하고, 결합된 반백질을 용출 완중액 (50 mM sodium phosphate (Sigma-Aldrich) , 150 mM NaCl (Sigma- Aldrich) , 250 mM imidazole (Sigma-Aldrich) , 20% glycerol , 1 mM DTT
(GoldBio), 10 uM ZnCl2 (Sigma-Aldrich) , pH 8.0)로 용출시켰다. 용출된 단백질 분획을 헤파린 아가로오스 비드 (Heparin Sepharose 6 Fast Flow, GE Healthcare)가들어있는폴리프로필렌 컬럼에 넣고 3 컬럼 부피의 세척 완충액 (50 mM sodium phosphate (Sigma-Aldrich) , 150 mM NaCl (Sigma- Aldrich) , 20% glycerol , 1 mM DTT (GoldBio) , 10 yM ZnCh (Sigma-Aldrich) , pH 8.0)으로 세척하였다. 결합된 단백질을 용출 완충액 (50 mM sodium phosphate (Sigma-Aldrich) , 750 mM NaCl (Sigma-Aldrich) , 20% glycerol ,
1 mM DTT (GoldBio), 10 \M ZnCl2 (Sigma-Aldrich), pH 8.0)으로용출시켰다. 생성된 단백질 분획물은 원심분리 컬럼 (Amicon Ultra-4 Contrifugal Filter Devices, Millipore)을 사용하여 6,000g에서 농축시켜, ABE7.10 단백질을정제하였다.
3. PCR amplicon의 ABE7.10 -매개 시험관내 분해 {in vitro digestion) 표적 유전자의 표적 부위를 포함하는 (표 1 내지 11 참조) PCR amplicon을 37 에서 1시간 동안 100m£의 반응 부피로 ABE7.10 단백질 및 시험관내 전사된 ( in vitro transcribed) HEK2 -표적 sgRNA와 함께 배양하였다. 상기 얻어진 디아미네이션(deamination)된 생성물을 PCR 정제 키트 (MGmed)를 사용하여 정제하였다. 디아미네이션된 생성물 2 fig을 엔도뉴클레아제 V (Endo V; NP_418426.2) (40 units) (New England BioLabs) 또는 인간 Alkyladenine DNA Glycosylase (hAAG; NP_001015052.1, New
England BioLabs) (10 units) (New England BioLabs) 및 엔도뉴클레아제 VIII (Endo VIII; NP_415242.1) (20 units) (New England BioLabs)와 함께 200】 의 반응 부피로 37 에서 30분간 배양한후, DNA를 PCR 정제 키트 (MGmed)를 사용하여 다시 정제하고 Bioanalyzer (Agilent)로 분석하여, in vitro digestion결과를측정하였다. 、
4.유전체 DNA (genomic DNA)의 ABE7.10 -매개시험관내분해
제조자 설명서에 따라서_ DNeasy Blood & Tissue Kit (Qiagen)을 사용하여 HEK293T세포로부터 유전체 요를분리하였다. 유전체 DNA 8/zg을 정제된 ABE7.10 단백질 (300nM) 및 sgRNA (900 nM)와 함께 400m요의 반응부피로 37 에서 8시간동안 반응 완충액 (50 mM Tris-HCl (pH 8.0), 25 mM KC1, 2.5 mM MgS04, 0.1 mM EDTA, 10% glycerol, 2 mM DTT, 10 uM ZnCl2)에서 배양하였다. RNase A (50//g/mL)를 이용하여 sgRNA를분리한후,
DNeasy Blood & Tissue Kit (Qiagen)를 사용하여 쇼를 정제하였다. 정제된 DNA 3//g를 Endo V (40 units) 또는 hAAG (10 units) 및 Endo VI 11 (20 units)와 함께 200ra표의 반응부피로 37 °C에서 2시간 동안 배양한 후, DNeasy Blood & Tissue Kit (Qiagen)를사용하여 다시 정제하였다. 정제된 DNA를 KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems)와함께 배양하고 analyzed by real-time quantitative PCR (qPCR)로 분석하여 ABE7.10- 및 Endo V- 또는 ABE7.10-, hAAG-, 및 Endo VII I -매개 DNA 이중가닥절단 ( 을확인하였다. 온전한유전체 DNA분획은 comparative CT 분석법으로측정하였다.
5. 전체 유전체 시퀀싱 (Whole genome sequencing; WGS) 및 digenome sequencing
상기 얻어진 시험관내 분해된 DNA l//g를 Covaris 시스템 (Thermo Fisher Scientific)을 사용하여 400 내지 500 bp 범위로 단편화하고, End Repair Mix (II lumina)^}·함께 배양하여 무딘 말단(blunt end)을 만들었다. 단편화된 DNA를 어댑터 (illumina)로 연결하여 라이브러리를 생성하고, (주)마크로젠에 의뢰하여 HiSeq X Ten Sequencer (Illumina)를사용하여 전체 유전체 시퀀싱 (WGS)을 수행하였다. WGS는 sequencing depth를 30-40x로 하여 수행하였다. 유전체 서열 맵핑에는 Isaac aligner를사용하였다. DNA 절단 부위는 Digenome 1.0 및 Digenome 2.0 프로그램을 사용하여 확인하였다 (Kim, D. , Kim, S. , Kim, S. , Park, J . & Kim, J.S. Genome¬ wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome research 26, 406-415 (2016)). 본 명세서에서 사용된 Digenome 2.0 버전의 소스 코드는 ’https://github.com hizksh八iigenome-toolkit2’에서 확인할수있다.
6. 표적 심층시퀀싱 (Targeted deep sequencing)
KAPA HiFi HotStart PCR polymerase (#KK2502; KAPA BIOSYSTEMS)를 사용하여 표적 부위 (on-target sites) 및 잠재적 비표적 (potential off- target sites) 부위를 증폭시켰다. TruSeq HT Dual index-containing primer를 사용하여 증폭된 앰플리콘을 다시 증폭하여, deep sequencing 라이브러리를생성하였다.
상기 표적 심층 시퀀싱에 사용된 프라이머를 하기의 표 1 내지 7에
2019/147073 1»(:1/10公019/001104 정리하였다:
[표 1]
에 대한프라이머
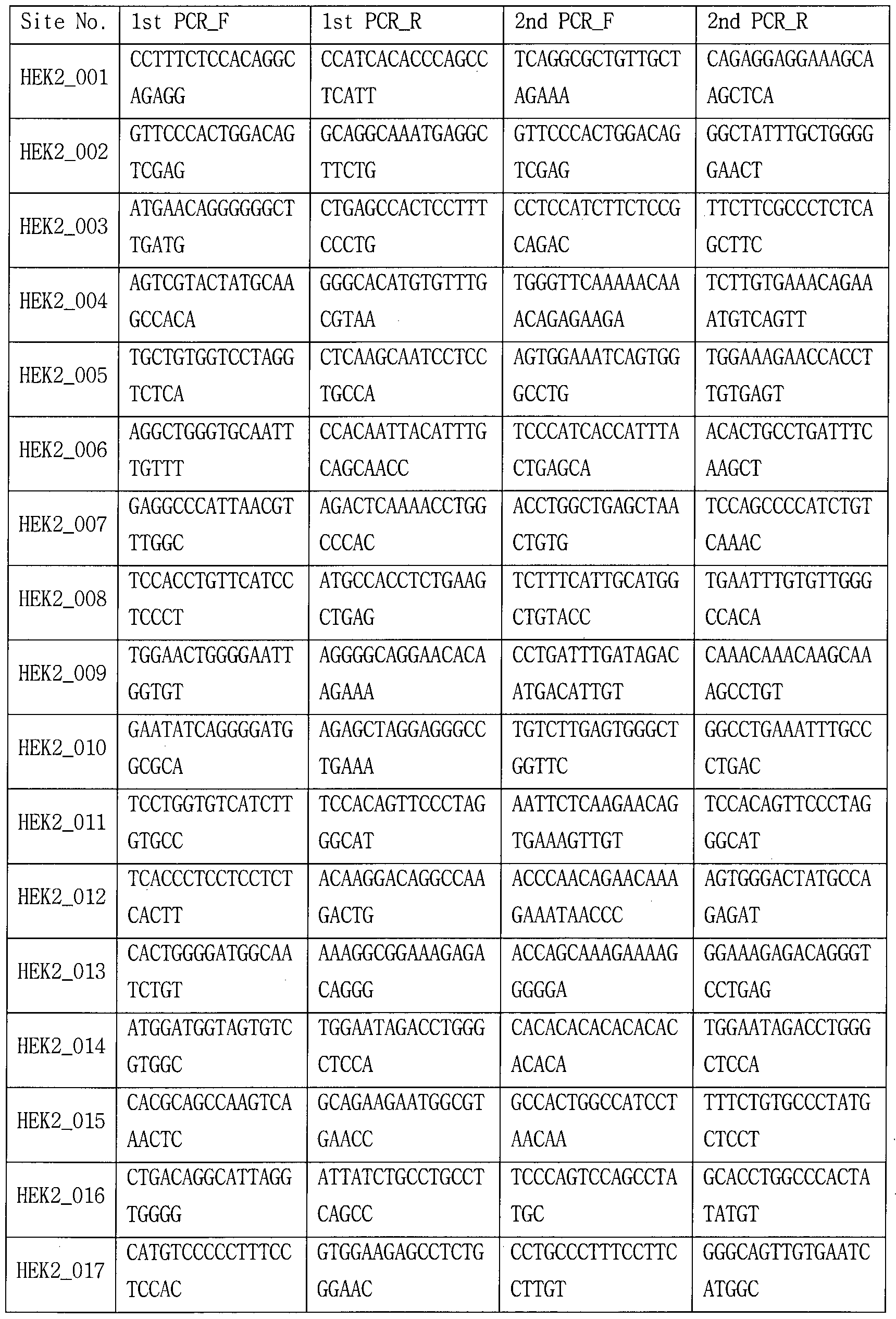


[표 2]
1 2에 대한프라이머


[표 3]
1 1¾)3에 대한프라이머


[표 4]


[표 5]
표1) 4에 대한프라이머


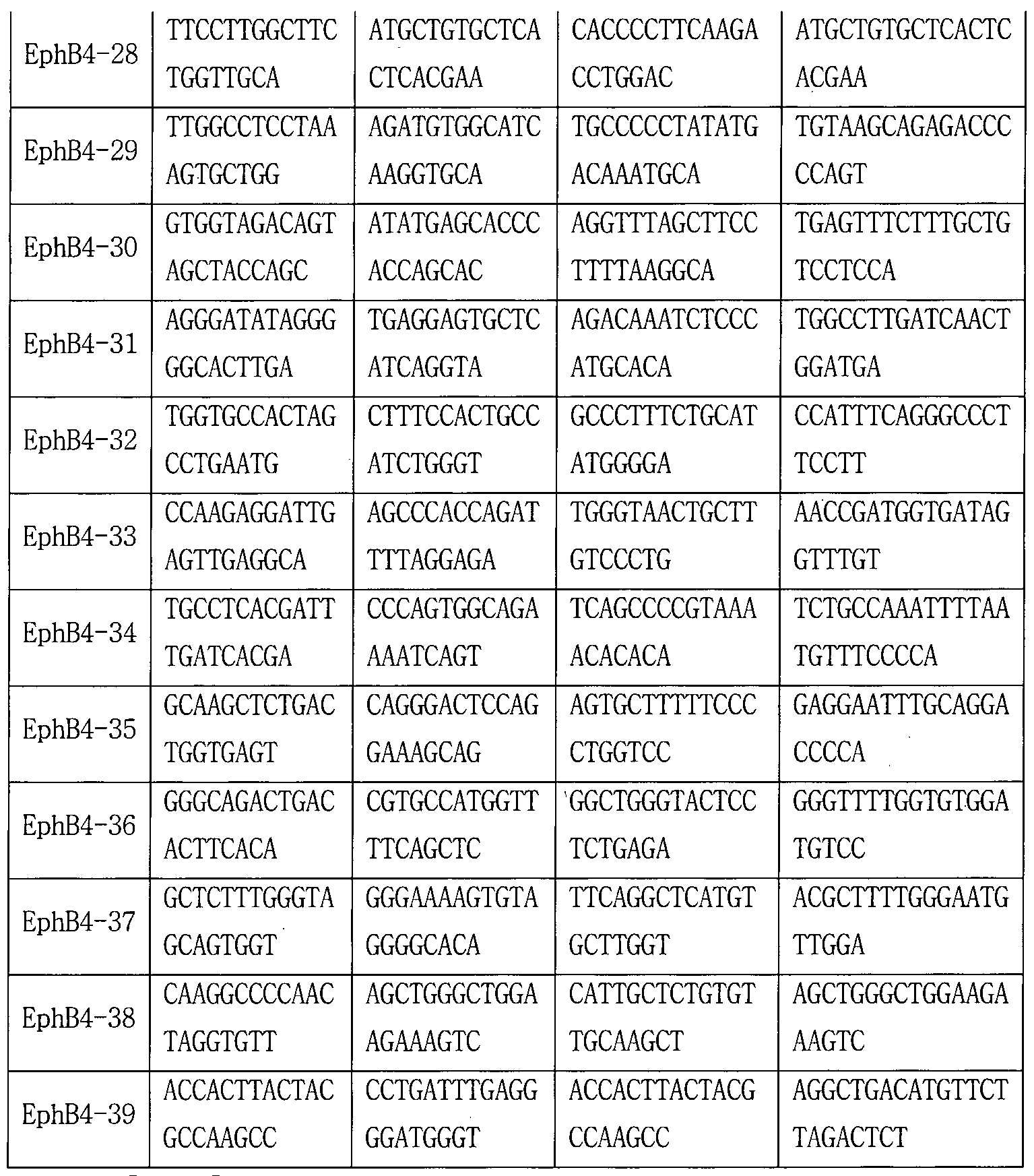
[표 6]
냉)0)116에 대한프라이머


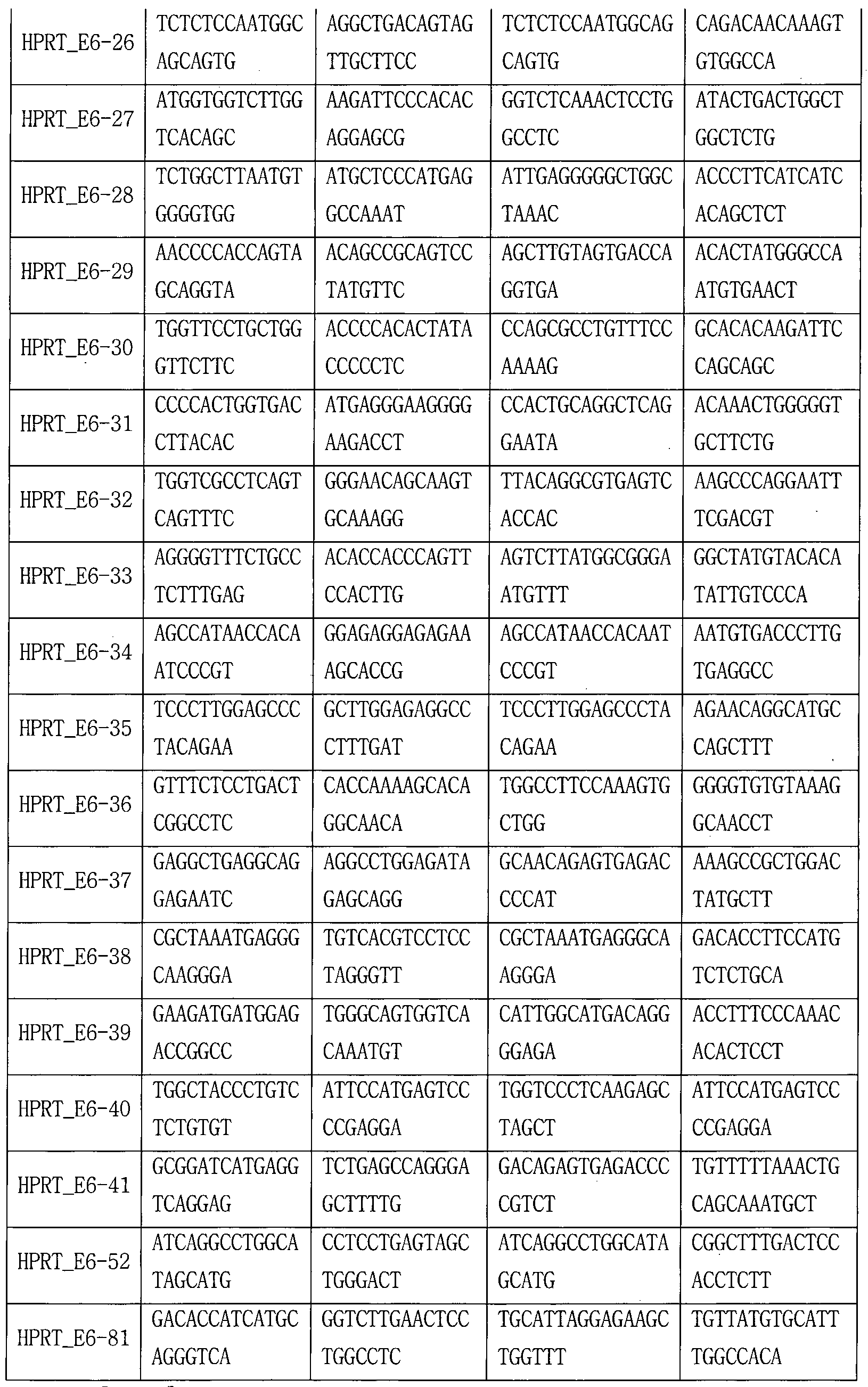
[표 7]


paired-end sequencing system을갖춘 Illumina MiniSeq을사용하여, 상기 생성된 라이브러리를 시퀀싱하였다. 염기교정빈도 (Base editing frequencies)는 editing window (position 4-7) 내의 하나 이상의 edit로 교정된 변형 표적 부위의 빈도를나타낸다. 치환및 indel 빈도를 계산하기 위한 컴퓨터 프로그램 소스 코드는 ’https://github.com/ibs_ cge/maund'에서 확인할수있다. 실시예 5. ABE7.10, BE3, 및 Cas9의 mismatched sgRNAs에 대한허용 오차 (Tolerance)
내재 유전체 부위 (endogenous genomic loci)를 표적으로 하는 일련의 mismatched sgRNAs를 사용하여, ABE7.10, BE3 (cytosine base editor) 및 Cas9의 off-target 활성이 서로 다른지 여부를 조사하였다. 보다 구체적으로, 표적 부위 (on target site)에 대하여 1 내지 4 개의 불일치 염기를 포함하는 sgRNA를 코딩하는 플라스미드와 인간 코돈에 최적화된 ABE7.10, BE3 (Base Editor plasmid; Addgene plasmid #73021; rAP0BEC1-XTEN-Cas9n-UGI-NLS) 또는 SpCas9(NP_269215.1)를 코딩하는 플라스미드를 HEK293T세포에 트랜스펙션하고 (참고예 1참조), 참고예 6의 표적 심층 시퀀싱을 수행하여, 염기 교정 빈도 및 삽입/결실 (Indel) 빈도를측정하였다.
상기 얻어진 염기교정 빈도 (%) 및 삽입/결실 빈도 (%)를도 6a내지
6c, 및 표 8내지 표 10에 나타내었다:
[표 8]
표적 유전자: HEK2(on target site: GAACACAAAGCATAGACTGCGGG)


(상기 표에서,
말단의 'NGG' 부분은 PAM서열이고;
sgRNA서열은 표적 부위 서열 중 PAM 서열을 제외한 서열에서 를 U로변환한서열이고;
on-target sequence에 대한 mismatched base는소문자로표시함; Relative activity = (mismatched sgRNA 결과값/matched sgRNA 결과값);
하기 모든표에서 동일하게 적용됨)
[표 9]
2019/147073 1»(:1/10公019/001104 표적 유전자: 問볘 (011 요


[표 10]
표적 유전자:


3개 또는 4개의 불일치를 갖는 sgRNA와 결합한 경우에는 대부분 활성이 현저하게 감소하였다. 특히, Cas9, ABE7.10 및 BE3의 mismatched sgRNA에 대한관용은서로상이하게 나타났다. 도 6a및 표 8에 나타난바와같이, HEK2 유전자의 표적 부위의 경우, 2 내지 4 개 염기의 mismatch를 갖는 sgRNA사용시 , ABE7.10는비교적 높은관용을갖지만 (활성 유지), Cas9및 BE3는활성이 낮거나불활성이다. 도加및 표 9에 나타난바와같이, HEK3 유전자의 표적 부위의 경우, Cas9는 3-4 위치 (5’에서 3’방향으로의 1에서 23까지의 numbering에 따름)에서 tandem mismatch를 갖는 sgRNA와 함께 (복합체 형성) 사용시 높은 활성을 나타내었다 (fully-matched sgRNA를 사용하는 경우와 비교하여 66%의 활성을 나타냄). 반면, ABE7.10의 경우에는 상기와 동일한 sgRNA와 복합체를 형성하는 경우, fully-matched sgRNA를 사용하는 경우와 비교하여 15%의 비교적 낮은 상대적 활성을 나타내었다. 이러한 결과는 ABE7.10, BE3 및 Cas9가 인간유전체에서 개별 세트의 비표적 부위를 인식함을 제안하고, unbiased 방식으로 유전체 전반의 ABE특이성을결정할수있는방법이 요구됨을시사한다. 실시예 6. genome-wide off-target site확인 (HEK2 gene)
시험관내 (//z vitro) 생성된 DNA 이중가닥절단 (DSB)에 의존하는 Digenome-seq을 통해 ABE7.10의 유전체 전역의 표적 부위를 확인하기 위하여 , 아데닌-디아미네이션(adenine deaminat ion)에 의하여 생성된 이노신(inosine)을 포함하는 부위에서 DSB를 생성시켰다. E. coli Uracil DNA glycosylase (UDG)와 DNA glycosylase-lyase Endonuclease VIII (Endo VIII)의 혼합물인 Uracil-Specific Excision Reagent (USER) (New England Biolabs; https://ww.neb.com/products/m5505-user_enzyme; Uracil DNA glycosylase (UDG) 및 DNA glycosylase-lyase Endonuclease VIII 혼합물과 50 mM KC1 , 5 mM NaCl , 10 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, 1 mM DTT, 175 mg/ml BSA 및 50%(w/v) glycerol 포함) 및 재조합 BE3 delta UGI
단백질 (His6-rAP湖ECl-nCas9 단백질; UGI 도메인을 결여한 BE3 변이형; pET28b-BEl(Addgene plasmid #73018)에서 site directed mutagenesis를 이용하여 dCas9의 A840을 H840로 치환하여 제조)를 사용하여, 사이토신의 디아미네이션 결과물인 우라실을 포함하는 부위에서 DSB를 생성시켜 ( in vitro) , genome-wide BE3 off-target site을 확인하였다 (in an unbiased manner) . 이와 유사하게 , E. coli 엔도뉴클레아제 V (Endo V) 또는 인간 알킬아데닌 DNA 글라이코실라아제 (human Alkyladenine DNA Glycosylase; MAG)와 Endo VIII의 조합이 이노신 포함 부위에서 포스포다이에스테르 결합을 절단하는데 사용될 수 있음을 추론하였다 (도 7a 내지 7c 참조). Endo V는 DNA에서 이노신을 인식하고 이노신에 대하여 두 번째 phosphodiester bond 3를 절단하는 반면, hAAG는 이노신을 소거하여 AP 부위 (apurinic/apyrimidinic site)를 생성하고 이 부위는 Endo VIII에 의해 인식 및 프로세싱되어 단일가닥 절단 (single-strand break)을 유도한다.
상기와 같은 추론을 시험하기 위하여, 표적 DNA 서열 iHEK2 on- taeger sequence; 표 8참조)을포함하는 PCR amplicon을 E. ca//에서 발현 및 정제된 재조합 ABE7.10 단백질 (정제 결과는 도 8 참조) 및 이들의 sgRNA로 처리하여 하나의 DNA 가닥에서 아데닌 디아미네이션을 촉매하고 다른 DNA가닥에서는 nick을생성하고 (도 7a), 그후, Endo V (도 7a참조) 또는 hMG/Endo VIII (도 7c 참조)로 처리하여, composite D況를 생성시켰다. 상기 결과를 도 7d 및 도 7e에 나타내었다. 예상과 같이 , PCR 증폭물은 ABE7.10 + Endo V (도 7d) 또는 ABE7.10 + hMG/Endo VIII (도 7e)에 의해 절단됨을확인할수 있다.
inosine-speci fic DNA repair enzyme을사용하여, 이노신을포함하는 ABE-표적 부위에서 D況가생성될 수 있음을확인한후, HEK293T세포로부터 분리된 인간 유전체 쇼를 ABE7.10 + Endo V 또는 ABE7.10 + hMG/Endo VIII로 in vitro 처리하고 Digenome-seq분석하여 유전체 전역의 ABE off- target site를 확인할 수 있는지 여부를 조사하였다. EK293T 세포로부터 분리된 인간 유전체 DNA를 ABE7.10 ribonucleoprotein (RNP) (300 nM ABE7.10 and 900 nM sgRNA; targeted to an endogenous chromosomal site
{HEK2 on-taeger sequence; 표 8 참조))와 함께 8 시간 동안 배양한 후, repair enzyme (Endo V또는 hAAG/Endo VIII)과함께 2시간더 배양하였다. Sanger 시퀀싱을 이용하여 표적 부위 (nick을 갖는 반대 가닥은 시퀀싱
불가함)에서의 ABE-매개 adenine-to-inosine conversion (PCR 증폭동안 이노신은 구아닌으로 변경됨) 및 이노신-포함 부위 (nicked DNA 가닥은 증폭 불가함)에서의 Endo V-촉매 DNA절단을 확인하였다 (도 7f). 유전체 DNA를 ABE7.10과 Endo V로 처리하면 이노신을 포함하는 DNA 가닥이 엔도 5 V에 의해 절단된다. Endo V가 절단한 DNA가닥은 PCR로 증폭될 수 없지만 이노신을 갖지 않는 DNA가닥은 증폭된다. 그 결과, Sanger sequencing에 의해 관찰된 교정 부위에서는아데닌만관찰되고구아닌은관찰되지 않는다 (도 7f).
또한 ABE7.10 + Endo m사용한 표적 DNA 절단을 검증하기 위해 10 정량적 실시간 PCR을 수행하여, 그: 결과를 도 7g에 나타내었다. Endo V 대신 hAAG및 Endo VIII를사용한경우에도유사한결과가얻어졌다(도 7h 및 도 70. 얻어진 유전체 DNA분해 샘플에 대하여 전체 유전체 시퀀싱 (WGS)을 수행하였다. human reference genome (hgl9)에 대하여 sequencing reads를 맵핑한 후, Integrative Genomics Viewer (IGV)를 사용하여 표적 B 부위 (on-target site)에서의 sequence reads의 정렬을 모니터링하고, sequence reads가무작위 정렬보다는 요의 특성인 일정한(uniform) 정렬을 나타냄을 확인하였다 (도 7j 및 도 7k). 이러한 정렬은 ABE7.10 단독 유전체 DNA또는 미처리 유전체 DNA에서는 관찰되지 않았다 (도 7j 및 도 7k). 이러한 결과는 Digenome-seq이 유전체 범위의 ABE표적 (on-target) 20 부위 및 비표적 (off-target) 부위를 포괄적으로 매핑하는데 사용될 수 있음을보여준다.
인간유전체에서 ABE7.10 off-target sites를 결정하기 위하여, WGS 데이터를 사용하여 유전체 전역에 걸쳐서 각각의 염기쌍 위치에 대한 DNA 절단 점수 (DNA cleavage score)를 할당하고 (도 9 참조), in vitro 25 cleavage sites에 해당하는, 높은 점수를 갖는 부위를 나열하였다 (초기 컷오프점수는 2.5로설정됨(Digenome 1.0에 따름; Kim, D. , Kim, S, , Kim, S. , Park, J . & Kim, J.S. Genome-wide target specificities of CRISPR- Cas9 nucleases revealed by multiplex Digenome-seq. Genome research 26, 406-415 (2016); Digenome 1.0프로그램은서열 상동성을고려하지 않음)).
30 Endo 를 사용하여 17개의 in vitro cleavage sites을, hAAG/Endo VIII를 사용하여 18개의 in vitro cleavage sites을 얻었다. 표적 부위를포함한 16 개의 부위가 서로 다른 두 가지 시험 (Endo V 사용 vs. hAAG/Endo VIII를 사용)에서 공통적으로 확인되었으며(도 10 a 및 도 10b), 이는
Di genome- seq 분석이 높은 재현성과 신뢰성을 가짐을 보여준다. 이들 부위의 DNA 서열을 비교하여 얻은 Sequence logos는 거의 모든 뉴클레오타이드 위치가 ABE7.10디아미나제의 특이성에 기여함을 보여준다 (도 10c참조). 실시예 7. 다양한 유전자 표적시의 genome-wide off-target site 확인
실시예 6에서 사용된 HEK2 유전자 표적 ABE7.10 RNP에 더하여 , 추가의 6개 ABE7.10 RNP를각각사용하여, 유전체 DNA에 대하여 Di genome- seq (Digenome-seq 1.0: DNA cleavage score가 2.5이상인 site를 비표적 위치 후보군으로결정함; (DNA cleavage score가 0.1이상이며 10개 이하의 미스매치를 가지고 PAM (5'-NGN-3' 또는 5’-NNG_3’)을 가지고 있는 site를 비표적 위치로 결정함)을 수행하여, 그 결과를 표 11내지 표 18및 도 11에 나타내었다.
[표 11]
The number of ABE on-target and off-target sites identified by Digenome-seq

[표 12]
[)^1101116 -。찼야아ᄅ선 ( 1(2)




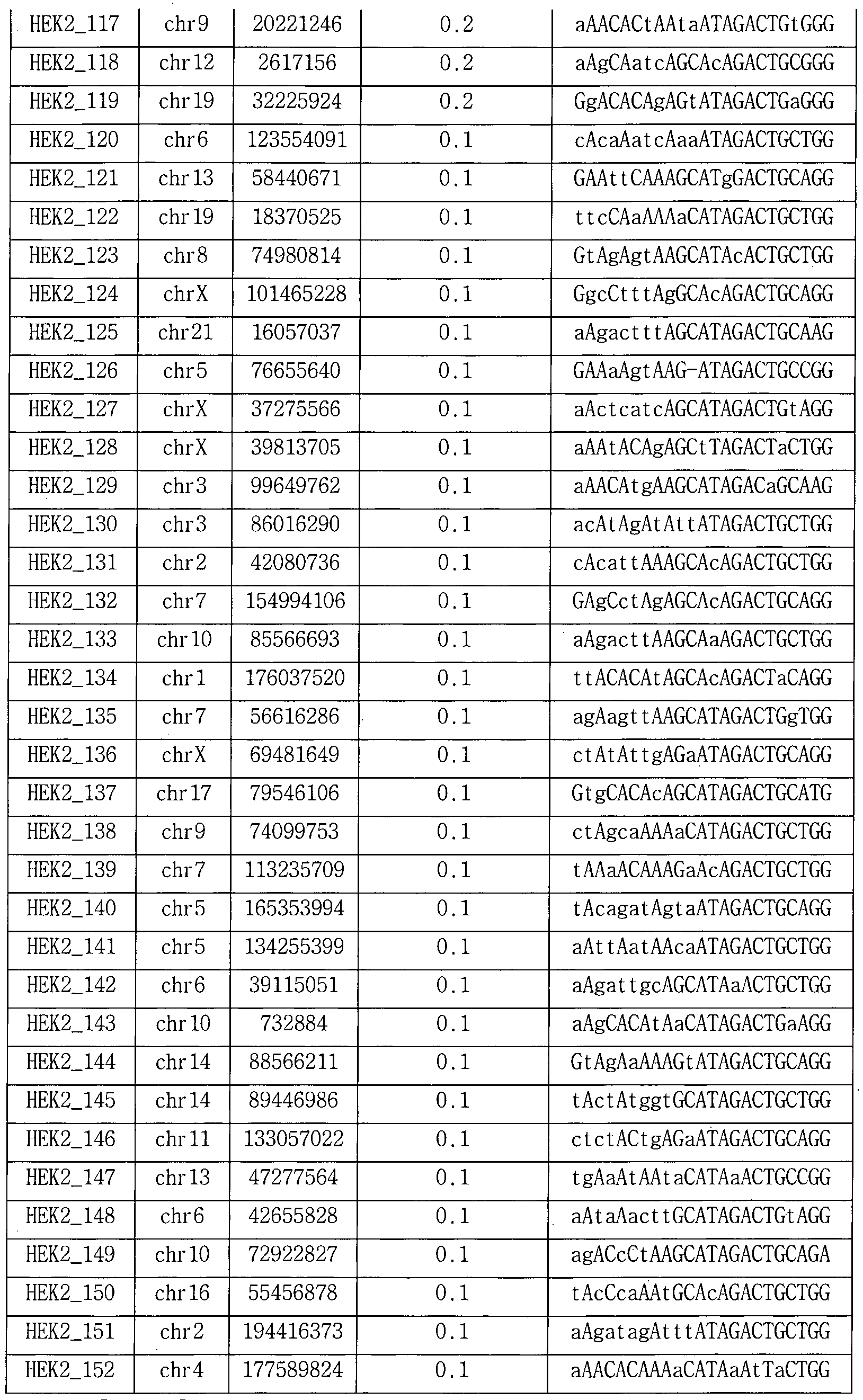
[5.13]
Digenome-captured sites (RNF2)


[5.14]
Digenome-captured sites (TYR03)

[S 15]
Digenome-captured sites (WEE1)


[& 16]
Digenome-captured sites (EphB4)


[3. 17]
Digenome-captured sites (HPRT-Exon6)





[5. 18]
Digenome-captured sites (HPRT-Exon8)


본 실시예에서 분석된 7 개의 sgRNA 중 두 개 (HEK2 및 RNF2)의 sgRNA를 각각 BE3AUGI 또는 Cas9와 함께 사용하여, Digenome 분석을 수행하고, 그결과를 ABE7.10와함께 사용하여 얻어진 결과와비교하여, 도 12a및 도 12b에 나타내었다. HEK2 sgRNA를 ABE7.10, BE3AUGI, 및 Cas9와 각각 결합하여 사용한 경우, 각각 17개, 2개, 및 24개의 in vitro 절단 부위를 생성하였다 (도 12c 참조) . RNF2 sgRNA는 ABE7.10, BE3AUGI, 및 Cas9와각각결합하여 사용된 경우, 각각 5개, 1개, 및 13개의 절단부위를 생성하였다 (도 12d참조) . 2개의 BE3디아미나아제를사용하여 확인 된 3 개의 in vitro 절단 부위는 모두 각각의 ABE7.10 디아미나아제에 의해서도 확인되었다. 대조적으로, ABE7.10와 Cas9를 사용하여 얻은 in vitro절단부위는크게 상이하였다. HEK2 ABE7.1◦으로 얻은 17개 중 10개 부위 (59%)는 Cas9를사용한경우에는동정되지 않았다. RNF2 ABE7.10으로 얻은 5개 중 2개 부위 (40%)는 Cas9를 Cas9를 사용한 경우에는 동정되지 않았다. 이러한 결과는 ABE가 일반적으로 Cas9보다 더 특이적이면서도
Cas9와상이한 off-target site세트를인식할수있음을시사한다.
상기한 7개 sgRNA를 사용하여 targeted amp1 icon sequencing를 통하여 확인된 57개 in vitro cleavage sites에서의 ABE off-target base editing을 검증하였다. 또한, Digenome 2.0 프로그램을 사용하여, PAM과 유사한 서열 (5,-NGN-3 ’또는 V-NNG-S1:)을 포함하고 cutoff score가
>0.1이며, 각각의 on-target sequences와 비교하여 10개 이하의 mismatche를포함하는후보 off-target sites을추가로확인하였다.
ABE7.10과복합체를 형성한 7개의 sgRNA를 이용하여 12내지 152개 사이트 (60±20, on average)를 얻었으며, 이는 Digenome 1.0으로 얻은 7개 on-target 부위와 57개 in vitro 절단 부위 (표 12 내지 18)를 모두 포함하였다. deep sequencing을 통하여 얻어진 절단 점수가 1 이상이거나 서열 상동성이 높은 부위를 포함하여 총 193개 부위에서 7 개의 ABE7.10 다아미나아제의 염기 교정 또는 유전자 교정 빈도를 측정하여, 그 결과를 표 19내지 표 25및도 12e에 나타내었다.
[표 19]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (HEK2; 'DNA sequence at a cleavage sites’은표 12참조)


[표 20]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (RNF2; ’DNA sequence at a cleavage sites’은 표 13참조)

[표 21]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (TYR03; ’DNA sequence at a cleavage sites’은 표 14참조)


[표 22]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (WEE1; ’DNA sequence at a cleavage sites’은표 15참조)

[표 23]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (EphB4; ’DNA sequence at a
2019/147073 1»(:1/10公019/001104 ’은표 16참조)


[표 24]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (HPRT_Exon6; ’DNA sequence at a cleavage sites’은표 17참조)
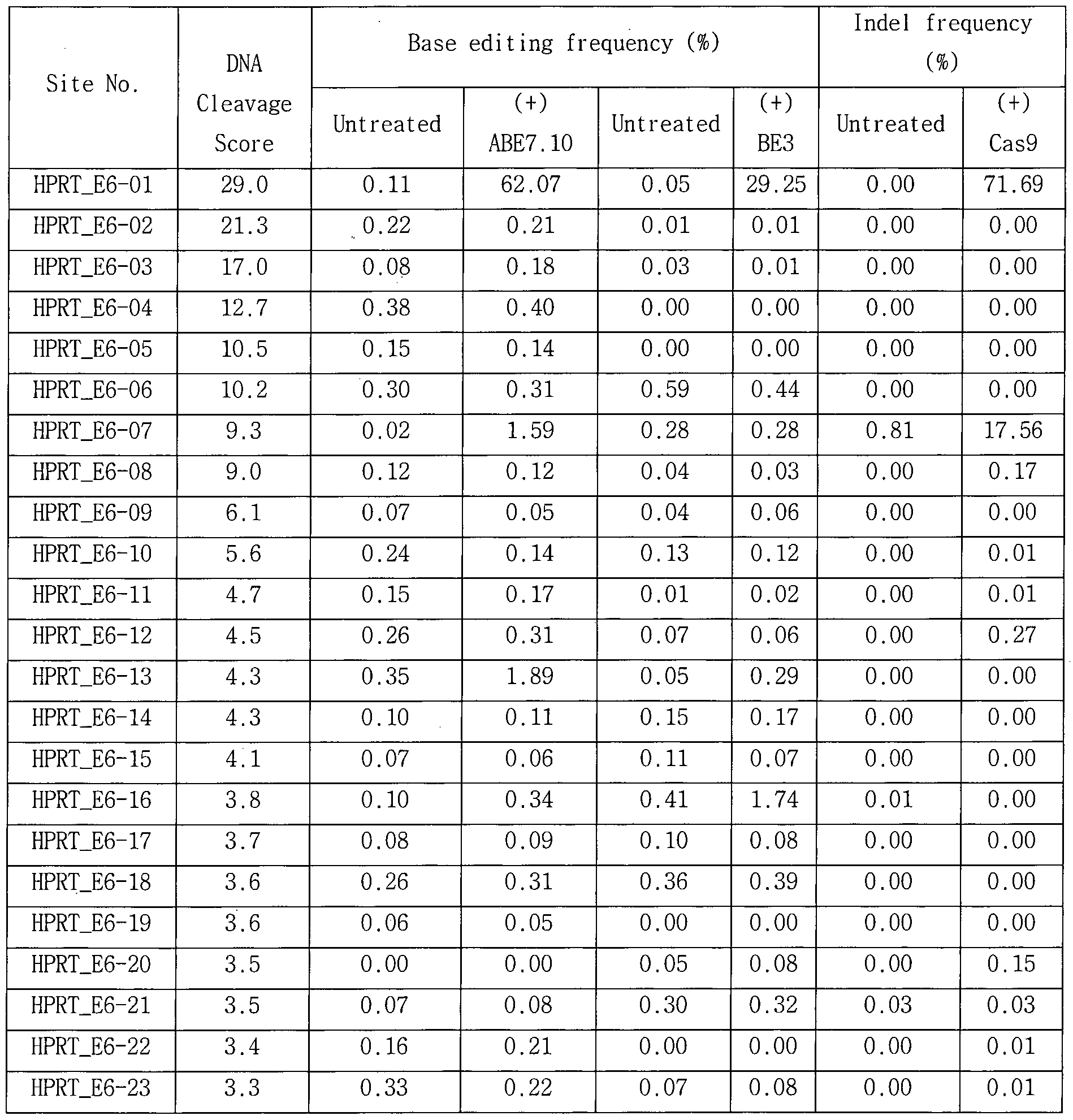

[표 25]
Mutation frequencies of ABE7.10, BE3, and Cas9 at on-target and off-target sites captured by Digenome-seq (HPRT-Exon8; 'DNA sequence at a cleavage sites’은표 18참조)


(일반적으로 0.1~1¾» 범위) 이상의 빈도로 검출되지 않았다. 다른 20개 val idated si tes 중에서 7개 on-target 부위는 29% 내지 72% 사이의 염기 교정 빈도를 나타내었다. 다른 13개의 val idated of f-target si te은 0.1% 내지 7.8% 범위의 염기교정 빈도를 나타내서, 매우 낮은 교정 효율을 보였다. 이러한결과는 Digenome-seq분석법은검출 한계 근처의 매우낮은 교정 빈도를 갖는 ABE of f-target si te까지도 검출 가능할 정도로 충분히 민감한분석법임을보여준다.
상기 시험과 병행하여, 193 부위에서 Cas9 -유도 indel 빈도와 BE3- 유도 치환 빈도를 측정하여 ABE7.10와 비교하여 그 결과를 도 12f에 나타내었다. 도 I2f에 나타난 바와 같이, ABE7.10 활성은, BE3 활성(R2=0.39)보다, Cas9 활성(R2=0.77)과 보다 강하게 연관성을 갖는 것으로 확인되었다. 도 12f의 위쪽 그래프에 있어서, ABE7.10에 의해서는 염기 교정 빈도가비교적 높지만 BE3에 의해서는 염기교정빈도가매우낮은 (0.001% 이하) 6개 부위 (속이 빈 점으로 표시) 중에서, 2 개 부위는 BE3 edi t ing window내에 시토신을포함하지 않는다. 도 12f의 아래쪽그래프에 있어서, 6 개의 부위(속이 빈 점으로 표시)에서 ABE7.10에 의한 염기 교정 빈도가 높지만 Cas9에 의한 indel 빈도는 매우 낮게 (indel 빈도 0.001% 이하) 나타났으며, 이 중 3 개 부위(50%=3/6)는 각각의 on-target 부위와 비교하여 하나의 삽입 또는 결실된 뉴클레오타이드를 가지며, sgRNA와 혼성화 시 DNA 또는 RNA bulge를 형성할 수 있다. 이러한 bulge-형성
부위는 BE3의 of f-target si te의 공통적인 특징이지만 Cas9 of f-target si te에서는 거의 관찰되지 않는다. Digenome-seq을 통해 ABEHO을 사용하여 확인된 193 개의 부위 중 3 개 또는 2 개의 부위는 각각 Cas9 또는 BE3에 의하여 세포 내에서 높은 빈도로 염기 또는 유전자 교정이 일어났지만, ABE7.10에 의해서는 매우 낮은 염기 교정 빈도 (치환 빈도 0.001% 이하)를 나타냈다 (도 12f에서 회색 점으로 표시). 이러한 결과는 ABE7.10, BE3, 및 Cas9가 인간 유전체에서 상이한 세트의 of f-target si te을인식할수있음을재차확인시켜 준다. 실시예 8. ABE off-target효과시험
ABE의 of f-target 효과를 최소화하거나 제거하기 위하여, 다음의 세 가지 방법으로시험하였다: sgRNA변형, 플라스미드 대신 RNP세포내 전달, 및 Sniper-Cas9 (、E. col i 에서 di rected evolut ion를 통하여 얻어진 engineered Cas9 변이체; 야생형 SpCas9 아미노산 서열(NP_269215.1)에 대하여 E539S, M761I , 및 K890N 변이를 가지며, D10A 변이를 추가로 가짐) 사용.
첫째로 (sgRNA변형), sgRNA의 표적화서열이 20개 뉴클레오타이드로 이루어진 표준 GXis sgRNA (Xi9: 19개의 임의의 뉴클레오타이드 (각각 독립적으로 A, U(T), C, 및 G 중에서 선택됨)를 의미함; X 뒤의 숫자는 임의의 뉴클레오타이드 개수를 의미함, 이하 동일함)의 ’X’의 5' 말단 부위로부터 1 또는 2개 뉴클레오타이드를 제거한 절단 sgRNA (gXis 또는 gX17로표시) (g는표적 부위의 대응서열과불일치하는구아닌을, G는표적 부위의 대응 서열과 일치하는 구아닌을 나타냄) 또는 5’ 말단에 1 또는 2 개의 여분의 구아닌을추가로포함하는 연장된 sgRNA(gX2o또는 gg¾o이라고 함)를 사용하여 HEK293T 세포에서의 염기 교정 빈도를 측정하였다. 상기 사용된 변형 sgRNA를 HPRT exon 6 target sequence를 대표로 예시하면 도 13a와같다. 다른유전자에 대한변형 sgRNA는표 11에 기재된 각유전자의 0n-target DNA sequence을기초로도 13a를참조하여 제작하였다.
ABE7.10 및 상기 변형 sgRNA를사용하여 얻어진 HEK293T세포에서의 염기 교정 빈도결과(targeted deep sequencing으로측정됨)를도 13b내지 도 13d및표 26내지 표 32에 나타내었다:
[표 26]
변형 sgRNAs를사용한경우의 ABE7.10 of f-target 효과분석 (HEK2;
2019/147073 1»(:1/10公019/001104
'DNA sequence at a cleavage sites’은표 12참조)


[표 27]
DNA sequence at a cleavage si tes’은표 13참조)

변형 sgRNAs를사용한경우의 ABE7.10 off-target 효과분석 (TYR03;
'DNA sequence at a cleavage sites’은표 14참조)
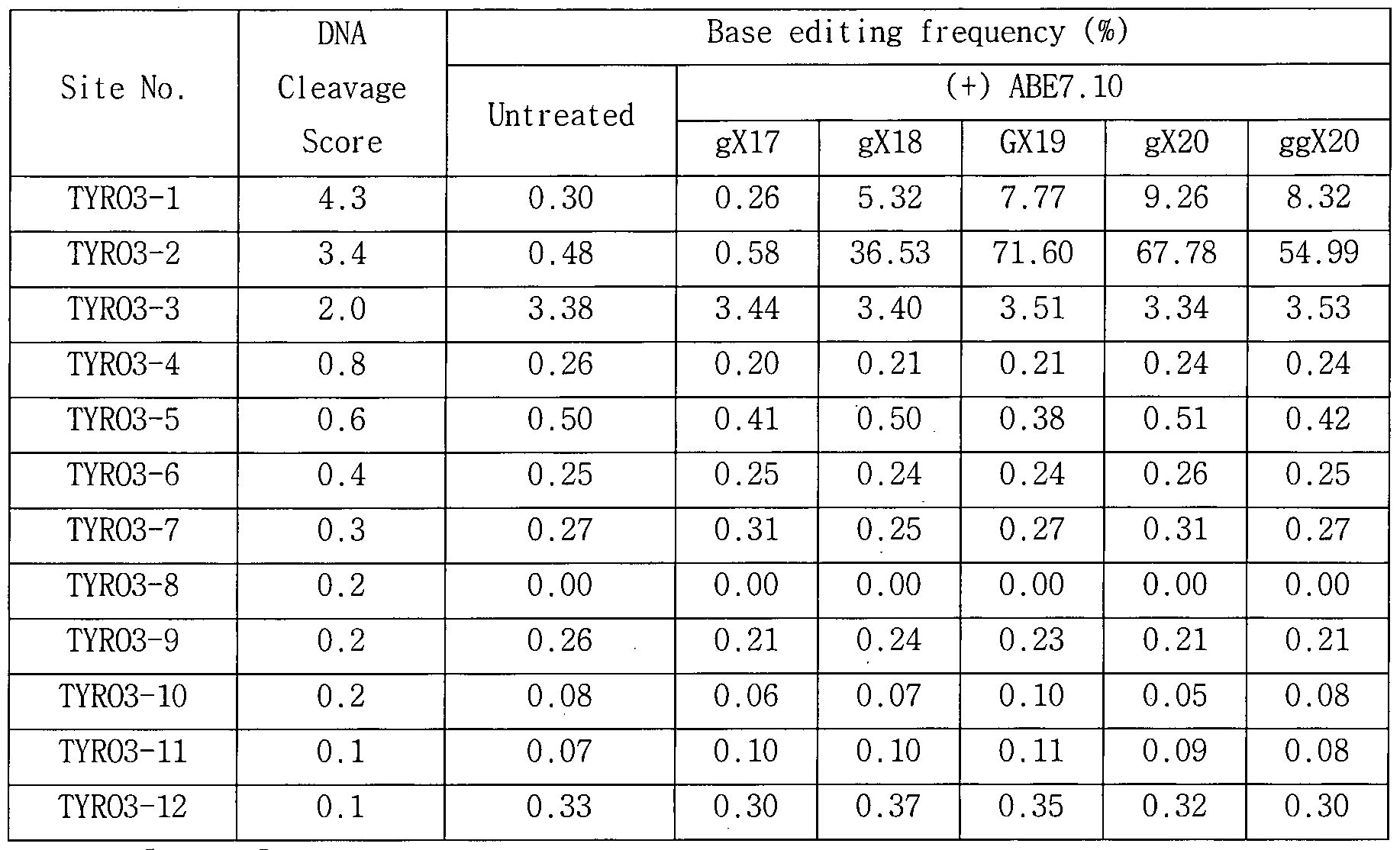
[표 29]
변형 3용別요3를사용한 경우의

DNA sequence at a cleavage sites’은표 15참조)


[표 30]
변형 sgRNAs를사용한경우의 ABE7.10 off-target 효과분석 (EPHB4;
DNA sequence at a cleavage sites'은표 16참조)


[표 31]
변형 3용 쇼3를사용한경우의
 효과분석 0抑奸 -
효과분석 0抑奸 -
Exon6 : 'DNA sequence at a cleavage sites’은표 17참조)
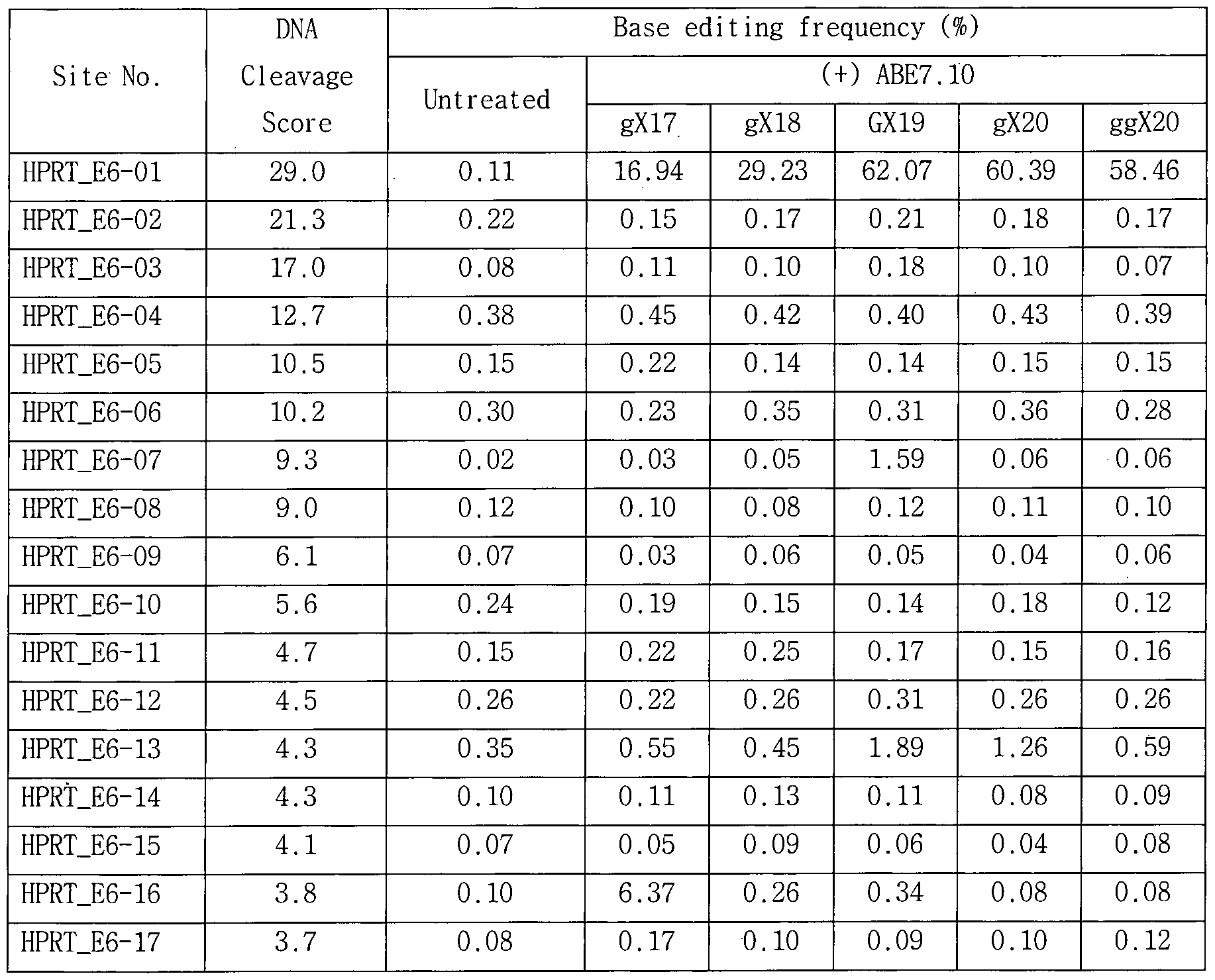

[표 32]
변형 용 쇼를사용한 경우의 새£7.10 卜 융 효과분석 (卵1奸 -
Exon8; 'DNA sequence at a cleavage sites’은표 18참조)


도 13c 및 13d에 나타난 바와 같이, 변형 sgRNA는 표적화 활성은 유지하면서 거의 모든 부위에서 ABE7.10의 of f-target 활성을 감소시켰다 (도 13d에서, y축의 그래프는 위에서부터 순서대로 ggX20, gX20, GX19, gX18, gX17, 및 untreated의 결과를 보여준다) . 예컨대, 도 13c에서와 같이, HPRT Exon 6을 표적으로 하는 GX19 sgRNA를 사용하여 확인된 4 개의 검증된 of f-target si te에 대하여 gX2o 또는 ggX2o sgRNA는 기존 GX19 sgRNA와비교하여 speci f i ci ty rat 가 2내지 26배 향상시켰다. 도 13c 및 13d에 나타난 바와 같이, 절단 sgRNAs는 대부분의 부위에서 ABE7.10의 on target 및 of f-target 활성을 감소시키고, 5’ 말단 근처 또는 그 부근의 불일치가있는부위에서 of f-target 효과를악화시키는것으로나타났다. 또한, 야생형 SpCas9 대신에 Sniper_Cas9가 아데닌 디아미나아제 모이어티와 융합된 Sniper ABE7.10를 사용하여 상기와 동일한 시험을 수행하여, Digenome-seq로 측정된 결과를 도 13e 및 13f (도 13f에서, y축의 그래프는 위에서부터 순서대로 (+) SpCas9 ABE7.10, (+)ABE7.10, 및 untreated의 결과를보여준다) 및 표 33내지 표 39에 나타내었다:
[표 33]
2019/147073 1»(:1/10公019/001104
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome- seq (HEK2; 'DNA sequence at a cleavage sites’은표 12참조)


[표 34]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome-seq (RNF2; 'DNA sequence at a cleavage sites’은표 13참조)


[표 35]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome-seq (TYR03; ’DNA sequence at


[표 36]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome-seq (WEE1; ’DNA sequence at a cleavage sites1은표 15 참조)

[표 37]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome-seq (EphB4; 'DNA sequence at a cleavage sites’은 표 16 참조)


[표 38]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target and off-target sites captured by Digenome-seq (HPRT_Exon6; 'DNA sequence at a cleavage sites1은 표 17 참조)


[ 39]
Mutation frequencies of ABE7.10 and Sniper ABE7.10 at on-target
and off-target sites captured by Digenome-seq (HPRT_Exon8; 'DNA


도 13근 내지 1¾ 및 표 26 내지 32에 나타난 바와 같이, ¾1 라 쇼8£7.10의 사용하는경우 및쇼則:7.10요 를사용하는경우, 각각새묘7.10의 사용하는 경우 및 새묘7.10 플라스미드를 사용하는 경우와 비교하여 염기 교정 특이성을약 7배까지 향상시킬수있다.
또한, Sniper ABE7.10를 변형 sgRNAs와 조합하여 사용함으로써, ABE7.10의 off-target 효과를보다감소시킬 수 있음을 확인하였다(도 13i 및 13j 참조; 도 13i 및 13j에서, y축의 그래프는 위에서부터 순서대로 ggX20_Sniper ABE7.10, gX20_Sniper ABE7.10, GX19_Sniper ABE7.10, gX18_ Sniper ABE7.10, gX17_Sniper ABE7.10, GX19-ABE7.10, 및 untreated의 결과를보여준다).
본실시예를통하여, mismatched sgRNAs , Digenome-seq, 및 targeted amp1 icon sequencing을 사용하여, ABE7.10, BE3, 및 Cas9가 인간 유전체에서 서로 다른 세트의 off-target sites을 인식할 수 있고, 각각 독립적으로 평가 수단을 필요로 함을 확인하였다. 상기 시험된 7 개 유전자를 표적으로 하는 ABE7.10 디아미나아제는 인간 유전체의 제한된 수의 부위에서 매우 특이적으로 in vitro A-to-G conversion을 촉매함을 확인하였다. 또한, 변형 sgRNAs, ABE7.10 RNPs, 및/또는 Sniper ABE7.10를 사용함으로써, ABE7.10의 off-target 활성을 감소시키거나 제거할 수 있음을확인하였다.
Claims
(1) 아데노신 디아미나아제, 이를암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드,
(2)불활성화된표적 특이적 뉴클레아제 , 이를암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드,
(3)가이드 RNA, 및
(4) Alkyl adenine DNA Glycosylase (AAG)와 엔도뉴클레아제 VI I I (endonuclease VI I I )의 조합또는엔도뉴클레아제 V (endonuclease V)
을포함하고,
상기 불활성화된 표적 특이적 뉴클레아제는 DNA 이중 가닥을 모두 절단하는엔도뉴뉴클레아제 활성을상실한것인,
아데닌디아미나제를사용하는 DNA이중가닥절단용조성물.
【청구항 2】
제 1항에 있어서, 상기 불활성화된표적특이적 뉴클레아제는 DNA이중 가닥을 절단하는 엔도뉴클레아제 활성을 상실한 Cas9 단백질 또는 Cpf l 단백질인, 아데닌디아미나아제를사용하는 DNA이중가닥절단용조성물.
【청구항 3]
제 2항에 있어서, 상기 불활성화된 표적특이적 뉴클레아제는 스트렙토코커스 피요젠스 { Streptococcus pyogenes) 유래의 Cas9 단백질에 아미노산 잔기 DIO , H840 , F539 , M763 , 및 K890로 이루어진 군에서 선택된 하나 이상이 원래와 다른 아미노산으로 치환된 돌연변이가 도입된 것인, 아데닌디아미나아제를사용하는 DNA이중가닥절단용조성물.
【청구항 4]
제 1항에 있어서,
상기 아데닌 디아미나아제, 불활성화된 표적특이적 뉴클레아제, 및 가이드 RNA는
( i ) 아데노신 디아미나아제, 표적 특이적 뉴클레아제, 및 가이드 쇼의 혼합물, ( i i ) 아데노신 디아미나아제 암호화 핵산 서열 또는 이를 포함하는플라스미드, 표적 특이적 뉴클레아제 암호화핵산서열 또는 이를 포함하는 플라스미드, 및 가이드 RNA 또는 이의 암호화 핵산 분자를 포함하는 플라스미드의 혼합물, ( i i i ) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 및 가이드 쇼의 혼합물, ( iv)
아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 암호화 핵산분자또는상기 핵산분자를 포함하는 플라스미드, 및 가이드 RNA또는 이의 암호화 핵산분자를 포함하는플라스미드의 혼합물, 및 (v) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 및 가이드 RNA^l 복합체또는혼합물
로 이루어진 군에서 선택된 하나 이상의 형태인, 아데닌 디아미나아제를사용하는 DNA이중가닥절단용조성물.
【청구항 5]
제 1항에 있어서,
상기 불활성화된표적특이적 뉴클레아제는, 스트렙토코커스피요젠스
{ Streptococcus pyogenes)유래의 Cas9단백질에 ,
아미노산 잔기 D10가 원래 아미노산과 다른 아미노산으로 치환된 돌연변이가도입되거나,
아미노산 잔기 D10 및 H840이 모두 원래 아미노산과 다른 아미노산으로치환된돌연변이가도입되거나
아미노산 잔기 F539, M763, 및 K890이 모두 원래 아미노산과 다른 아미노산으로치환된돌연변이가도입된 것인,
아데닌디아미나아제를사용하는 DNA이중가닥절단용조성물.
【청구항 6】
제 1항 내지 제 5항 중 어느 한 항에 있어서, 상기 가이드 RNA는 crRNA와 tracrRNA가 서로 결합된 이중 가닥 crRNA: tracrRNA 복합체, 또는 단일 가닥 가이드 RNA (sgRNA)인, 아데닌 디아미나아제를 사용하는 DNA 이중가닥절단용조성물.
【청구항 7】
( a) ( 1 ) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 불활성화된 표적 특이적 뉴클레아제, 불활성화된 표적 특이적 뉴클레아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드를 분리된 세포에 도입시키거나또는분리된 쇼에 처리하여, DNA이중가닥중 하나를절단하는단계; 및
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 Alkyladenine DNA Glycosyl ase ( 이와 엔도뉴클레아제 VI I I (endonuc lease VI I I)의 조합, 또는 엔도뉴클레아제 V (endonuc lease V)을 처리하여, 나머지 DNA 가닥을
절단하는단계
를포함하는,까데닌디아미나아제를사용하여 DNA에 이중가닥절단 (double strand break)를생성하는방법 .
【청구항 8]
(a) ( 1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 불활성화된 표적 특이적 뉴클레아제, 불활성화된 표적 특이적 뉴클레아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드를 분리된 세포에 도입시키거나또는분리된 DNA에 처리하여, DNA이중가닥중 하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 Alkyl adenine DNA Glycosylase ( 이와 엔도뉴클레아제 VI I I (endonuclease VI I I)의 조합, 또는 엔도뉴클레아제 V (endonuclease V)을 처리하여, 나머지 DNA 가닥을 절단하는단계 ; 및
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계
를 포함하는, 아데닌 디아미나아제에 의하여 염기 교정 (base edi t ing)이 도입된 DNA의 핵산서열분석 방법.
【청구항 9】
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 불활성화된 표적 특이적 뉴클레아제, 불활성화된 표적 특이적 뉴클레아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드를 분리된 세포에 도입시키거나또는분리된 DNA에 처리하여, DNA이중가닥중 하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 Alkyl adenine DNA Glycosylase (MG)와 엔도뉴클레아제 VI I I (endonuc lease VI I I)의 조합, 또는 엔도뉴클레아제 V (endonuclease V)을 처리하여, 나머지 DNA 가닥을 절단하는단계 ;
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계; 및
(d) 상기 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 상기 절단된위치를확인하는단계
를포함하는, 아데닌디아미나아제의 염기 교정 위치 확인방법 .
【청구항 10】
(a) (1) 아데노신 디아미나아제, 아데노신 디아미나아제를 암호화하는핵산분자, 또는상기 핵산분자를포함하는플라스미드, 및 (2) 불활성화된 표적 특이적 뉴클레아제, 불활성화된 표적 특이적 뉴클레아제를 암호화하는 핵산 분자, 또는 상기 핵산 분자를 포함하는 플라스미드를 분리된 세포에 도입시키거나또는분리된 DNA에 처리하여, DNA이중가닥중 하나를절단하는단계 ;
(b) 상기 DNA 이중가닥 중 하나가 절단된 DNA에 Alkyladenine DNA Glycosyl ase (AAG)와 엔도뉴클레아제 VI 11 (endonuc lease VI I I )의 조합, 또는 엔도뉴클레아제 V (endonuclease V)을 처리하여, 나머지 DNA 가닥을 절단하는단계 ;
(c) 상기 이중가닥절단된 DNA에 대하여 전체 유전체 시퀀싱 (whole genome sequencing)을수행하는단계; 및
(d) 상기 시퀀싱으로 수득한 염기서열 데이터 (sequence read)에서 상기 절단된위치를확인하는단계
를포함하는, 아데닌디아미나아제의 비표적 위치 (of f-target si te) 확인 방법.
【청구항 11】
제 7항 내지 제 10항 중 어느 한 항에 있어서, 상기 불활성화된 표적특이적 뉴클레아제는 DNA이중가닥을 절단하는 엔도뉴클레아제 활성을 상실한 Cas9단백질또는 Cpf l단백질인, 방법 .
【청구항 12】
제 7항 내지 제 10항 중 어느 한 항에 있어서, 상기 불활성화된 표적특이적 뉴클레아제는 스트렙토코커스 피요젠스 { Streptococcus pyogenes) 유래의 Cas9단백질에 아미노산잔기 D10 , H840 , F539 , M763 , 및 K890로 이루어진 군에서 선택된 하나 이상이 원래와 다른 아미노산으로 치환된돌연변이가도입된 것인, 방법.
【청구항 13】
제 7항내지 제 10항중어느한항에 있어서,
상기 아데닌 디아미나아제, 불활성화된 표적특이적 뉴클레아제, 및 가이드 RNA는
(0 아데노신 디아미나아제, 표적 특이적 뉴클레아제, 및 가이드
RNA의 혼합물, (ii) 아데노신 디아미나아제 암호화 핵산 서열 또는 이를 포함하는플라스미드, 표적 특이적 뉴클레아제 암호화핵산서열 또는 이를 포함하는 플라스미드, 및 가이드 RNA 또는 이의 암호화 핵산 분자를 포함하는 플라스미드의 혼합물, (iii) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 및 가이드 RNA의 혼합물, (iv) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 암호화 핵산분자또는상기 핵산분자를 포함하는 플라스미드, 및 가이드 RNA또는 이의 암호화 핵산분자를 포함하는 플라스미드의 혼합물, 및 (v) 아데노신 디아미나아제 및 표적 특이적 뉴클레아제가 연결된 융합 단백질 및 가이드 RNA의 복합체또는혼합물
로이루어진군에서 선택된하나이상의 형태로사용되는것인, 방법.
【청구항 14】
제7항내지 제10항중어느한항에 있어서 ,
상기 불활성화된표적특이적 뉴클레아제는, 스트렙토코커스피요젠스 {Streptococcus pyogenes)유래의 Cas9단백질에 ,
아미노산 잔기 D10가 원래 아미노산과 다른 아미노산으로 치환된 돌연변이가도입되거나,
아미노산 잔기 D10 및 H840이 모두 원래 아미노산과 다른 아미노산으로치환된돌연변이가도입되거나
아미노산 잔기 F539, M763, 및 K890이 모두 원래 아미노산과 다른 아미노산으로치환된돌연변이가도입된 것인,
방법 .
【청구항 15】
제7항 내지 제10항 중 어느 한 항에 있어서, 상기 가이드 RNA는 crRNA와 tracrRNA가 서로 결합된 이중 가닥 crRNA:tracrRNA 복합체, 또는 단일 가닥가이드 RNA (sgRNA)인 , 방법 .
【청구항 16】
제7항 내지 제10항 중 어느 한 항에 있어서, 시험관 내 (in vitro)에서 수행되는것인, 방법.
【청구항 17】
제7항내지 제10항중어느한항에 있어서, 상기 단계 (a)의 분리된 DNA는유전체 DNA인, 방법 .
【청구항 18】
제 10항에 있어서, 상기 단계 (d) 이후에,
상기 절단위치가표적 위치 (on-target site)가아닌 경우, 비표적 위치 (off-target site)로판단하는단계를추가로포함하는, 방법.
【청구항 19】
제 10항에 있어서,
상기 단계 ( 에서 확인된 절단 위치는 수득한 염기서열 데이터를 정렬하여 5’ 말단이 수직 정렬된 위치, 또는 5' 말단 플롯에서 이중 피크 패턴을보이는위치인 것인, 방법.
【청구항 20]
제 19항에 있어서, 상기 정렬은 표준 염기서열 (reference genome)로 염기서열 데이터를 맵핑한 뒤, BWA/GATK 또는 ISAAC을 이용하여 수행되는 것인, 방법.
【청구항 21]
제 19항에 있어서, 왓슨 가닥 (Watson strand)과 크릭 가닥 (Crick strand)에 해당하는 염기서열 데이터 (sequence read)가각각두개 이상씩 수직으로 정렬되는 위치를 비표적 위치인 것으로 판단하는 단계를 추가로 포함하는, 방법.
【청구항 22]
제 19항에 있어서, 20 %이상의 염기서열 데이터가수직으로정렬되고, 각각의 왓슨 가닥 및 크릭 가닥에서 동일한 5' 말단을 가진 염기서열 데이터의 수가 10 이상인 위치가 비표적 위치인 것으로 판단하는 단계를 추가로포함하는, 방법.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20180009508 | 2018-01-25 | ||
| KR10-2018-0009508 | 2018-01-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019147073A1 true WO2019147073A1 (ko) | 2019-08-01 |
Family
ID=67396080
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2019/001104 Ceased WO2019147073A1 (ko) | 2018-01-25 | 2019-01-25 | 아데노신 디아미나아제를 이용한 염기 교정 확인 방법 |
Country Status (2)
| Country | Link |
|---|---|
| KR (1) | KR102210700B1 (ko) |
| WO (1) | WO2019147073A1 (ko) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110964742A (zh) * | 2019-12-20 | 2020-04-07 | 北京市农林科学院 | 一种抗除草剂水稻的制备方法 |
| WO2022059928A1 (ko) * | 2020-09-21 | 2022-03-24 | 고려대학교 산학협력단 | 신규의 개량된 염기 편집 또는 교정용 융합단백질 및 이의 용도 |
| KR20220039564A (ko) * | 2020-09-21 | 2022-03-29 | 고려대학교 산학협력단 | 신규의 개량된 염기 편집 또는 교정용 융합단백질 및 이의 용도 |
| CN114317596A (zh) * | 2020-09-30 | 2022-04-12 | 北京市农林科学院 | 一种将植物基因组靶点序列中的a突变为g的方法 |
| WO2023024089A1 (zh) * | 2021-08-26 | 2023-03-02 | 华东师范大学 | 实现a到c和/或a到t碱基突变的碱基编辑系统及其应用 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102258713B1 (ko) * | 2019-07-31 | 2021-05-31 | 한양대학교 산학협력단 | 사이토신 염기교정용 조성물 및 이의 용도 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150166980A1 (en) * | 2013-12-12 | 2015-06-18 | President And Fellows Of Harvard College | Fusions of cas9 domains and nucleic acid-editing domains |
| WO2016022363A2 (en) * | 2014-07-30 | 2016-02-11 | President And Fellows Of Harvard College | Cas9 proteins including ligand-dependent inteins |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017214463A1 (en) * | 2016-06-09 | 2017-12-14 | The Regents Of The University Of California | Compositions and methods for treating cancer and biomarkers to detect cancer stem cell reprogramming and progression |
| KR102547316B1 (ko) * | 2016-08-03 | 2023-06-23 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 아데노신 핵염기 편집제 및 그의 용도 |
-
2019
- 2019-01-25 WO PCT/KR2019/001104 patent/WO2019147073A1/ko not_active Ceased
- 2019-01-25 KR KR1020190009908A patent/KR102210700B1/ko active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150166980A1 (en) * | 2013-12-12 | 2015-06-18 | President And Fellows Of Harvard College | Fusions of cas9 domains and nucleic acid-editing domains |
| WO2016022363A2 (en) * | 2014-07-30 | 2016-02-11 | President And Fellows Of Harvard College | Cas9 proteins including ligand-dependent inteins |
Non-Patent Citations (3)
| Title |
|---|
| GAUDELLI, N. M.: "Programmable base editing of A · T to G · C in genomic DNA without DNA cleavage", NATURE, vol. 551, 25 October 2017 (2017-10-25), pages 464 - 471, XP002785203 * |
| KOMOR, A. C.: "Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage", NATURE, vol. 533, 19 May 2016 (2016-05-19), pages 420 - 424, XP055551781 * |
| ZHENG, Y.: "DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA", NUCLEIC ACIDS RES., vol. 45, no. 6, 28 January 2017 (2017-01-28), pages 3369 - 3377, XP055404026 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110964742A (zh) * | 2019-12-20 | 2020-04-07 | 北京市农林科学院 | 一种抗除草剂水稻的制备方法 |
| WO2022059928A1 (ko) * | 2020-09-21 | 2022-03-24 | 고려대학교 산학협력단 | 신규의 개량된 염기 편집 또는 교정용 융합단백질 및 이의 용도 |
| KR20220039564A (ko) * | 2020-09-21 | 2022-03-29 | 고려대학교 산학협력단 | 신규의 개량된 염기 편집 또는 교정용 융합단백질 및 이의 용도 |
| KR102679001B1 (ko) | 2020-09-21 | 2024-06-28 | 고려대학교 산학협력단 | 신규의 개량된 염기 편집 또는 교정용 융합단백질 및 이의 용도 |
| CN114317596A (zh) * | 2020-09-30 | 2022-04-12 | 北京市农林科学院 | 一种将植物基因组靶点序列中的a突变为g的方法 |
| CN114317596B (zh) * | 2020-09-30 | 2024-01-16 | 北京市农林科学院 | 一种将植物基因组靶点序列中的a突变为g的方法 |
| WO2023024089A1 (zh) * | 2021-08-26 | 2023-03-02 | 华东师范大学 | 实现a到c和/或a到t碱基突变的碱基编辑系统及其应用 |
| CN115725650A (zh) * | 2021-08-26 | 2023-03-03 | 华东师范大学 | 实现a到c和/或a到t碱基突变的碱基编辑系统及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102210700B1 (ko) | 2021-02-02 |
| KR20190090724A (ko) | 2019-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102026421B1 (ko) | 시토신 디아미나제에 의한 dna에서의 염기 교정 확인 방법 | |
| JP6942848B2 (ja) | 遺伝子操作CRISPR−Cas9ヌクレアーゼ | |
| JP7038079B2 (ja) | Crisprハイブリッドdna/rnaポリヌクレオチドおよび使用方法 | |
| US10633642B2 (en) | Engineered CRISPR-Cas9 nucleases | |
| KR102084186B1 (ko) | Dna 단일가닥 절단에 의한 염기 교정 비표적 위치 확인 방법 | |
| WO2019147073A1 (ko) | 아데노신 디아미나아제를 이용한 염기 교정 확인 방법 | |
| AU2015299850B2 (en) | Genome editing using Campylobacter jejuni CRISPR/CAS system-derived RGEN | |
| JP2017533724A (ja) | ゲノムでプログラマブルヌクレアーゼの非標的位置を検出する方法 | |
| Wei et al. | Closely related type II-C Cas9 orthologs recognize diverse PAMs | |
| KR20160036061A (ko) | 뉴클레아제 프로파일링 시스템 | |
| KR102067810B1 (ko) | 크로마틴 dna를 이용한 유전체 서열분석 방법 및 유전체 교정 확인 방법 | |
| KR102567576B1 (ko) | 표적 특이성이 향상된 신규한 Cas9 단백질 변이체 및 이의 용도 | |
| CA2957441C (en) | Genome editing using campylobacter jejuni crispr/cas system-derived rgen | |
| HK40102346A (en) | Crispr hybrid dna/rna polynucleotides and methods of use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19743514 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19743514 Country of ref document: EP Kind code of ref document: A1 |