WO2020050102A1 - 癒着防止材 - Google Patents
癒着防止材 Download PDFInfo
- Publication number
- WO2020050102A1 WO2020050102A1 PCT/JP2019/033549 JP2019033549W WO2020050102A1 WO 2020050102 A1 WO2020050102 A1 WO 2020050102A1 JP 2019033549 W JP2019033549 W JP 2019033549W WO 2020050102 A1 WO2020050102 A1 WO 2020050102A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- agent
- adhesion
- gelatin
- gelatin derivative
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images











Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/04—Macromolecular materials
- A61L31/043—Proteins; Polypeptides; Degradation products thereof
- A61L31/045—Gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/14—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L31/148—Materials at least partially resorbable by the body
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/20—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices containing or releasing organic materials
- A61L2300/252—Polypeptides, proteins, e.g. glycoproteins, lipoproteins, cytokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/424—Anti-adhesion agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2430/00—Materials or treatment for tissue regeneration
- A61L2430/40—Preparation and treatment of biological tissue for implantation, e.g. decellularisation, cross-linking
Definitions
- the present invention relates to an anti-adhesion material for living tissue, and more particularly, to a bio-absorbable anti-adhesion material mainly comprising a gelatin derivative having a predetermined hydrophobic group.
- the anti-adhesion material is defined as a bioabsorbable synthetic material used directly at the application site during surgery for the purpose of reducing post-operative adhesion (see “Specific Medical Materials and Their Material Prices (Material Price Standard)” 2016) (Ministry of Health, Labor and Welfare Notification No. 402), which is generally referred to as an “adhesion-preventive absorbent barrier”.
- the adhesion preventing material includes a sheet type which is applied by being adhered to a surgical site, and a spray type which is applied in a liquid state or prepared in a liquid state at the time of use by spraying.
- Examples of the former include those mainly containing sodium hyaluronate and carboxymethylcellulose (Seprafilm (trademark), Kaken Pharmaceutical Co., Ltd.), those mainly containing gelatin (Zelfilm (trademark), Pfizer Corporation, and Patent Literature 1), and an example of the latter is one comprising two agents, N-hydroxysuccinimide dextrin and sodium carbonate / sodium bicarbonate (Adspray TM, Terumo Corporation).
- the adhesion preventing material needs to cover and protect the tissue surface, but it is difficult to apply a film-like material along the complicated shape of the surgical site.
- the operability of a sepra film, once wetted with water, is significantly reduced, so that it is practically impossible to correct the position.
- the spray type can be applied along the surgical site and more widely.
- a surgical sealant in which a gelatin derivative having a hydrophobic group introduced therein (hereinafter, sometimes referred to as “hydrophobized gelatin”) and a hardening agent are applied to a tissue by spraying.
- hydrophobized gelatin a gelatin derivative having a hydrophobic group introduced therein
- Patent Documents 2 and 3 a hardening agent
- the sealant has a high affinity for tissue by having a hydrophobic group, and has a significantly higher sealing strength than gelatin or fibrin sealant not having the hydrophobic group.
- a first agent containing a gelatin derivative wherein the gelatin derivative comprises: (A) including a structure represented by the following formula, GltnNH-CHR 1 R 2
- Gltn is a gelatin residue
- R 1 is an alkyl group having 5 to 17 carbon atoms
- R 2 is a hydrogen atom or an alkyl group having 5 to 17 carbon atoms
- (B) imino group / amino group (molar ratio) is from 1/99 to 30/70
- C having a weight average molecular weight of 10,000 to 50,000
- An adhesion preventing material comprising a first agent and (2) a second agent containing a crosslinking agent for the gelatin derivative.
- the anti-adhesion material can be applied to the tissue by spraying, it can be easily applied to a surgical site having a complicated shape.
- the cured film of the anti-adhesion material exhibits excellent anti-adhesion properties. Since it has high adhesiveness to a surgical site, it can be used as an adhesive anti-adhesion material or as a surgical sealant having anti-adhesion properties.
- the gelatin derivative as a main ingredient can be produced in an aqueous solvent and is not only safe in the production environment and in the body, but also can be synthesized in a single step with high yield.
- FIG. 1 is a graph showing the number of cell adhesion.
- FIG. 2 is a schematic diagram of the device used for the cell permeability test.
- FIG. 3 is a graph showing cell permeability.
- FIG. 4a is a cross-sectional photograph of the tissue at the surgical site one week after the operation.
- FIG. 4b is a cross-sectional photograph of the tissue at the surgical site two weeks after the operation. It is a graph which shows the result of burst strength. It is a cross-sectional photograph of a test piece after burst strength measurement.
- It is a schematic diagram of the apparatus used for the protein permeability test. It is a graph which shows a protein permeation ratio. It is a graph which shows a time-dependent change of a swelling degree. It is a graph which shows the weight reduction by an enzyme literature.
- the anti-adhesion material of the present invention is a two-part type consisting of a first part containing a gelatin derivative as a main agent for forming a substantially cured film and a second part containing a crosslinking agent for the gelatin derivative.
- the first agent and the second agent are separately packaged and provided, and are mixed at the time of use.
- the first agent contains a gelatin derivative.
- the gelatin derivative has a hydrophobic group linked via an imino group, ie, -NH-, and includes a structure represented by the following formula.
- GltnNH-CHR 1 R 2 In the above formula, Gltn is a gelatin residue, R 1 is an alkyl group having 5 to 17 carbon atoms, and R 2 is a hydrogen atom or an alkyl group having 5 to 17 carbon atoms. N is mainly derived from the ⁇ -amino group of lysine (Lys) in gelatin. Preferably, R 2 is a hydrogen atom.
- the NH structure of the formula (1) can be detected by, for example, a band near 3300 cm ⁇ 1 in the FT-IR spectrum.
- R 2 is an alkyl group having 5 to 17 carbon atoms, it may be the same as or different from R 1 .
- the alkyl group may contain a branch.
- Examples of the alkyl group include a hexyl group, an octyl group (or a capryl group), a nonyl group (or a pelargonyl group), a dodecyl group (or a lauryl group), a tetradecyl group (or a myristyl group), and the like.
- R 1 is a linear alkyl group having 5 to 11 carbon atoms
- R 2 is a hydrogen atom.
- the derivatization ratio in the gelatin derivative is 1 to 20 mol%, preferably 5 to 10 mol%, in terms of mol% of the imino group to which the alkyl group is bonded, based on the amount of amino groups in the raw material gelatin.
- the imino group / amino group (molar ratio) in the obtained gelatin derivative is from 1/99 to 20/80, preferably from 5/95 to 10/90.
- the derivatization ratio can be determined by quantifying the amount of amino group in the raw gelatin after bonding the amino group and the alkyl group by a 2,4,6-trinitrobenzenesulfonic acid method (TNBS method), or by NMR. It can be determined by identifying and quantifying an alkyl group by the method described above.
- TNBS method 2,4,6-trinitrobenzenesulfonic acid method
- the raw material gelatin may be any of natural, synthetic, fermented or obtained by genetic recombination, and preferably gelatin derived from animals such as pigs and cows and fish derived from pollock and the like.
- any of acid-treated gelatin, alkali-treated gelatin and genetically modified gelatin may be used, but alkali-treated gelatin is preferred, and low endotoxin-modified gelatin is more preferred.
- the molecular weight of the gelatin is preferably in the range where the weight average molecular weight (Mw) of the gelatin derivative is 10,000 to 100,000, and more preferably in the range where it is 10,000 to 50,000. More preferred.
- the molecular weight can be measured by gel permeation chromatography (GPC) according to a standard method.
- the first agent may contain non-derivatized gelatin in addition to the above gelatin derivative.
- the gelatin the above-mentioned various gelatins can be used.
- the amount of underivatized gelatin is 0-99 wt%, preferably 0-50 wt% of the total weight with the gelatin derivative.
- the first agent may further contain an aqueous solvent for dissolving or dispersing the gelatin derivative.
- an aqueous solvent for dissolving or dispersing the gelatin derivative.
- the gelatin derivative may be dissolved or dispersed in the aqueous solvent and provided as an aqueous liquid (hereinafter sometimes simply referred to as “aqueous solution”).
- aqueous solution a buffer containing various acids such as ultrapure water, physiological saline, boric acid, phosphoric acid, and carbonic acid and a salt thereof, or a mixture thereof can be used.
- a borate buffer of pH 8-11, more preferably pH 9-10 is used.
- the aqueous solvent is used in such an amount that the gelatin derivative is 10 to 80 wt / v%, preferably 15 to 30 wt / v%.
- the total weight with the gelatin derivative is such that the above concentration is obtained.
- the second agent is a crosslinking agent for a gelatin derivative, and forms a structure, for example, a film, which is insoluble in body fluids such as water and blood by crosslinking.
- a crosslinking agent at least one of those having at least two functional groups in the molecule that are reactive with amino groups in gelatin, mainly primary amino groups in side chains, is used.
- cross-linking agents include genipin, polybasic acids activated with N-hydroxysuccinimide or N-hydroxysulfosuccinimide, aldehyde compounds, acid anhydrides, and diisothiocyanate.
- Polybasic acids include tartaric acid, citric acid, malic acid, glutaric acid, glutamic acid, aspartic acid, oxaloacetic acid, cis-aconitic acid, 2-ketoglutaric acid, polytartaric acid, polycitric acid, polymalic acid, polyglutamic acid, polyaspartic acid Carboxymethylated dextrin, carboxymethylated dextran, carboxymethylated starch, carboxymethylated cellulose, carboxymethylated chitosan, carboxymethylated pullulan and the like.
- Imidyl glutarate (DSG), disuccinimidyl suberate (DSS), disuccinimidyl tartrate (DST) and the like can be used.
- a polybasic acid ester of polyethylene glycol or polyethylene glycol ether, wherein at least one of the carboxyl groups of the polybasic acid which has not been reacted with polyethylene glycol is active esterified for example, 4, 7, 10, 13 , 16-Pentaoxanonadecanedioic acid di (N-succinimidyl), and polyethylene glycol di (succinimidyl succinate) represented by the following formula (SS-PEG-SS): (N is a number such that Mw is about 10,000 to 20,000); Further, pentaerythritol-polyethylene glycol ether tetrasuccinimidyl glutarate (4S-PEG) represented by the following formula: (N is a number such that Mw is about 3,000 to 30,000, preferably about 5,000 to 27,000, more preferably about 10,000 to 20,000); Is mentioned.
- aldehyde compound examples include aldehyde group-introduced polysaccharides in which two or more aldehyde groups are introduced in one molecule, such as aldehyde group-introduced starch, aldehyde group-introduced dextran, aldehyde group-introduced dextrin, and aldehyde group-introduced hyaluronic acid.
- aldehyde group-introduced starch aldehyde group-introduced dextran, aldehyde group-introduced dextrin, and aldehyde group-introduced hyaluronic acid.
- the anhydride examples include glutaric anhydride, maleic anhydride, and succinic anhydride
- diisothiocyanate examples include hexamethylene diisothiocyanate.
- crosslinking agents have a functional group in the crosslinking agent, for example, an ester group activated with N-hydroxysuccinimide in an amount of 0.2 to 3 equivalents, preferably 0.1 to 1 equivalent of the amino group of the gelatin derivative. It is used in an amount of 2 to 2 equivalents, more preferably 0.2 to 1.0 equivalent, most preferably 0.2 to 0.6.
- a mixture of two or more cross-linking agents may be used, and in such a case, the total equivalent weight of the cross-linking agents falls within the above range.
- the second agent may further include an aqueous solvent for dissolving the crosslinking agent.
- the crosslinking agent and the aqueous solvent are provided in separate containers, and at the time of use, about 2 hours before or after use, an appropriate amount of the two are mixed to form an aqueous solution (hereinafter sometimes simply referred to as “aqueous solution”). It is preferred to use.
- aqueous solution those described above for the first agent can be used.
- a phosphate buffer of pH 3-8, more preferably pH 4-6 is used.
- the ionic strength of both aqueous solvents is adjusted so that when the aqueous solution of the first agent and the aqueous solution of the second agent are mixed in the same volume, the pH becomes 8 to 10.
- the first aqueous solution is a borate buffer having a pH of 9 and an ionic strength of 0.05 to 0.1
- the second aqueous solution is a phosphate buffer having a pH of 4 and an ionic strength of 0.01 to 0.03.
- the pH can be adjusted to 8 to 10.
- the first aqueous solution may be a borate buffer having a pH of 10 and an ionic strength of 0.05 to 0.1
- the second aqueous solution may be a phosphate buffer having a pH of 4 and an ionic strength of 0.01 to 0.07.
- the concentration of the crosslinking agent in the second agent is the equivalent of the functional group in the second agent to the equivalent of the amino group in the first agent, that is, (functional group equivalent in the second agent / amino group equivalent in the first agent). Is adjusted to be within the above range.
- a mixture of two or more cross-linking agents may be used, and in such a case, the total amount thereof is in the above range.
- the first agent and / or the second agent may further contain various additives in amounts not to impair the object of the present invention.
- the additive include a colorant, a pH adjuster, a viscosity adjuster, and a preservative.
- a coloring agent for example, brilliant blue, is added to the aqueous solution of the first or second agent so that the application site of the adhesion preventing material can be easily recognized.
- the amount added may be, for example, 10 to 100 ⁇ g / mL.
- the anti-adhesion material of the present invention can be obtained by separately preparing the first agent and the second agent, packaging them, and sterilizing them as desired.
- Method for Preparing First Agent (1) Preparation of aqueous solution of raw material gelatin The starting material gelatin is dissolved in an aqueous solvent by heating at 40 to 90 ° C. in an amount of 5 to 50 wt / v%.
- aqueous solvent a mixture of water and a water-soluble organic solvent is used.
- the water-soluble organic solvent an alcohol or ester having 1 to 3 carbon atoms can be used, and ethanol is preferably used.
- a derivatizing agent having an alkyl group to be introduced is added to the aqueous gelatin solution obtained in the step (1), and the mixture is stirred for a predetermined time to react.
- the derivatizing agent the above-mentioned aldehyde or ketone having an alkyl group, for example, dodecanal, tetradecanal, decylethylketone is used.
- the reaction temperature is 30 to 80 ° C., and the reaction time is 0.5 to 12 hours.
- Gelatin can be obtained.
- the amount of aldehyde used is 1 to 4 times the stoichiometric amount corresponding to the desired derivatization ratio. More preferably, it is 1 to 2 times.
- the Schiff base is reduced.
- a known reducing agent such as sodium cyanoborohydride (NaBH 3 CN), sodium triacetoxyborohydride (NaBH (OAc) 3 ), 2-picoline borane, pyridine borane and the like can be used.
- 2-picoline borane is preferred.
- Picoline borane is stable, and can perform a reductive amination reaction of an aldehyde or a ketone in an aqueous solvent in one step (one pot). Also, a yield of 80-90% can be achieved, which is significantly higher than that of sodium cyanoborohydride of 70-75%.
- the amount of 2-picoline borane used is preferably 1 to 3 equivalents relative to the equivalent of the derivatized drug.
- the gelatin derivative obtained in the step (3) is provided in a form of a powder or in the form of an aqueous solution in an aqueous solvent such as a borate buffer and the like.
- an aqueous solvent such as a borate buffer and the like.
- a glass or plastic vial, bottle, dispenser, syringe, or the like can be used. If desired, underivatized gelatin and other additives may be added.
- the gelatin derivative is provided in powder form, the container is separately filled with an aqueous solvent such as a borate buffer and provided.
- the gelatin derivative When the gelatin derivative is provided in the form of an aqueous solution, it may be filled in one of a double syringe type dispenser or the like which can be used for applying the anti-adhesion material to the affected part and which can mix both agents at the tip.
- the adhesion preventing material of the present invention may include a double syringe type dispenser, a mixing vial, a spare aqueous solvent, and the like as accessories.
- Each of the above-mentioned crosslinking agents exemplified as the second agent may be synthesized by a known method, or a commercially available one may be used.
- the crosslinking agent and an aqueous solvent for dissolving the crosslinking agent, such as a phosphate buffer, are provided in separate containers, for example, the crosslinking agent is placed in a glass vial, and the aqueous solvent is placed in a plastic bottle. If desired, additives may be added.
- the amount of the first agent and the amount of the second agent are provided in such an amount that (equivalent of the functional group of the crosslinking agent / equivalent of the amino group of the gelatin derivative) becomes 0.2 to 2,
- the agent and the second agent may be provided in a form that can be independently replenished.
- Each of the second agents in the form of a combination of the powdered crosslinking agent and an aqueous solvent for dissolving the crosslinking agent filled in a bottle or the like is sterilized.
- the sterilization method of the gelatin derivative powder and the cross-linking agent in powder form is preferably radiation sterilization.
- the radiation examples include an electron beam, a gamma ray, and bremsstrahlung, and electron beam sterilization is preferable.
- the total absorbed dose may be 20 kGy or more, and preferably 25 kGy or more, which has been widely used in the past (14th revised Japanese Pharmacopoeia, Part 2, Reference Information, page 1235, right column, 2.2 Radiation Method). 45 kGy.
- the total absorbed dose should be 20 kGy or more. Even if the electron beam is divided into a plurality of irradiations in order to prevent damage to the gelatin derivative and the crosslinking agent, the sterilization effect remains unchanged.
- irradiation with 10 kGy may be performed three times.
- Autoclave or filter sterilization is preferable as a method for sterilizing the aqueous solution of gelatin derivative.
- the present invention includes a method for producing an anti-adhesion material film including a step of applying a first agent and a second agent to a diseased part of a human or non-human animal subject, and a method for producing the anti-adhesion material film Also provided is a method for preventing adhesion of the subject.
- the anti-adhesion material of the present invention can be applied to ruptured parts of tubular tissues such as skin, blood vessels, tendons, nerves, intestines, and lymph vessels, liver, pancreas, and heart. Among them, it is suitably applied to moist tissues such as blood vessels and lungs.
- the second agent is preferably used as an aqueous solution immediately before use as described above.
- the concentration of the crosslinking agent at that time is as described above.
- the obtained second solution is applied to the empty syringe of the double syringe type dispenser already filled with the first solution and applied, or sprayed with an air-assisted spray having a double syringe. Apply to affected area. After application, a film is formed within several minutes to 10 minutes, and the affected part can be sealed.
- a gelatin derivative was prepared by the method described in Patent Document 3 (Japanese Patent No. 5995128). As an example, a method for preparing gelatin derivative 1 using octanal will be described. 30 g of Alaska pollack-derived gelatin (Mw 33,000, manufactured by Nitta Gelatin Co., Ltd.) was dissolved in 105 mL of water at 50 ° C., and a stoichiometric amount of n-octanal corresponding to 10 mol% of amino groups of the gelatin was added.
- Infrared spectrum (FTIR-8400, Shimadzu Corporation) and 13 C-NMR spectrum (AL300, JEOL Ltd.) of gelatin derivative 1 were measured to confirm derivatization.
- FTIR-8400 Shimadzu Corporation
- 13 C-NMR spectrum A300, JEOL Ltd.
- secondary amines in 3280 cm -1 the peak increase methylene group to 2936cm -1 and 2879cm -1 are peak derived from primary carbon in 17-17.5ppm the 13 C-NMR spectra, respectively And it was confirmed that the alkyl group was introduced.
- the derivatization ratio was measured by the following method. Gelatin derivative 1 was dissolved in a dimethylformamide / water mixed solvent at a volume ratio of 1: 1 to obtain a 0.1 w / v% solution. An aqueous solution of 0.1 v / v% triethylamine and an aqueous solution of 0.1 w / v% trinitrobenzenesulfonic acid are added to the solution, and the mixture is stirred for about 1 minute to react trinitrobenzenesulfonic acid with the amino group in the gelatin derivative. The absorbance at 340 nm was measured using a microplate reader (SPARC @ 10M, Tecan Japan KK). The derivatization ratio of gelatin derivative 1 was 7.2 mol%.
- the gelatin derivatives shown in Table 1 were prepared in the same manner as the above-mentioned method of preparing the gelatin derivative, except that n-octanal was changed to n-decanal or n-dodecanal, and gelatin was changed to Mw 38,000. .
- “C8” represents gelatin derivatized with a C8 aldehyde (in the formula (1), R 1 is a heptyl group and R 2 is a hydrogen atom).
- Each of the above gelatin derivatives was dissolved in a 0.1 M borate buffer at pH 9.5 at a concentration of 15 w / v% to prepare a first agent.
- second agent pentaerythritol-polyethylene glycol ether tetrasuccinimidyl glutarate (4S-PEG, manufactured by NOF CORPORATION) was used. Immediately before each of the following evaluations, when mixed in a 0.01 M phosphate buffer at pH 4.0 at the same volume as the first agent, 0.4 equivalent to 1 equivalent of the remaining amino groups of the first agent (Example) 4S-PEG was dissolved so as to obtain 0.5 equivalent.
- Cell permeability was evaluated using a cell culture insert (Transwell insert, PTFE membrane caliber 8.0 ⁇ m, Corning International Co., Ltd.) as schematically shown in FIG.
- a cell culture insert Transwell insert, PTFE membrane caliber 8.0 ⁇ m, Corning International Co., Ltd.
- 100 ⁇ L each of the first agent and the second agent of Example 1 and Comparative Example 1 and Comparative Examples 2 and 3 having the same area were applied to form a film. Irradiated for 1 hour and sterilized.
- L929 fibroblasts were seeded on the membrane (1.0 ⁇ 10 6 cells / well) and cultured at 37 ° C. in RPMI-1640 medium containing 10% fetal bovine serum and 1% penicillin / streptomycin solution under carbon dioxide gas. did. Twenty-four hours later, the number of cells permeated to the lower part of the well was counted, and the ratio (%) to the untreated case where no membrane was formed was determined. The results are shown in FIG.
- the Example has significantly lower cell permeability than the Sepra film, which is considered to contribute to the anti-adhesion property.
- Rats (Wistar, 7 weeks female, Charles River Japan Co., Ltd.) were laparotomized to prepare a cecal peritoneal wall defect model.
- the cecal wall (Cecum) was rubbed with medical gauze to bleed and damage the cecal wall.
- the peritoneal wall (Abdominal wall) facing the injured part was excised to a predetermined size (1.0 ⁇ 2.0 cm) with a medical knife to prepare a peritoneal wall defect.
- the anti-adhesion material of Example 1 and Comparative Examples 1 to 3 was applied to both the peritoneal wall defect and the cecal injury so as to cover the entire area of the wound, and the laparotomy was sutured.
- sealing strength pressure resistance
- porcine large intestine ⁇ 30 mm
- an adhesion preventing material film having a thickness of 1.0 mm and a diameter of 15 mm was prepared. After the application, pressure bonding was performed for 10 minutes under a load of 5.0 g / mm 2 , and physiological saline at 37 ° C.
- tissue of the test piece after rupture was fixed with a neutral buffered formalin solution and then stained with hematoxylin and eosin to observe the interface between the tissue and the sample (FIG. 6).
- the adhesion preventing material of the present invention exhibited a remarkably high sealing strength as compared with other samples, and the results shown in Patent Document 3 were confirmed again.
- the fibrin membrane and the Sepra film were separated at the interface between the membrane and the porcine large intestine.
- the adhesion preventive film itself was cohesively broken. From this, it can be seen that the adhesion preventing material of the present invention is also excellent in adhesive strength.
- Example 1 showed significantly lower protein permeability (about 20%) as compared to the fibrin membrane and Sepra film, and most of the protein remained on the surface. It is considered that this protein permeation inhibitory action contributes to the anti-adhesion property.
- the non-derivatized gelatin membrane also had low transmission, but the protein was spread throughout its thickness.
- the membrane was immersed in a physiological saline solution at 37 ° C., and the degree of swelling was calculated from the change in the weight (W s ) of the anti-adhesion material membrane according to the elapsed time according to the following equation, and was examined.
- Swelling degree (%) ⁇ (W s ⁇ W d ) / W d ⁇ ⁇ 100
- FIG. 9 shows the change in the degree of swelling with time. No significant difference was found between the two samples within the time of the test.
- Example 1 and Comparative Example 1 1000 ⁇ L of each of the first agent and the second agent was poured between two glass plates each having a 1.0 mm thick silicone rubber as a spacer, and a plate-shaped cured product of the adhesion preventing material was poured. It was created. The obtained cured product of the adhesion preventing material was hollowed out with a punch having a diameter of 10 mm to prepare a circular film having a thickness of 1.0 mm and a diameter of 10 mm, and the initial weight (W t0 ) was measured.
- Specimens were immersed in Tris-HCl buffer (2.5 mM CaCl 2 , pH 7.4) overnight to prevent calcium salt formation with phosphate buffered saline (PBS).
- Collagenase was dissolved in Tris-HCl buffer at a concentration of 100 ⁇ g / mL, and the test piece was immersed in 2 mL of collagenase solution. From the change in the weight (W t ) of the adhesion preventing material film with the lapse of time, the weight (%) remaining without being decomposed was calculated and examined by the following equation.
- Weight (%) (W t / W t0 ) ⁇ 100
- W t0 is the initial weight
- W t is the weight at time t after immersion.
- FIG. 10 it was confirmed that the decomposition time of the membrane of the example was longer than that of the comparative example, and it was possible to protect the operation part for a longer time.
- the anti-adhesion material of the present invention can be applied to a surgical site having a complicated shape, and is very suitable for clinical use as an anti-adhesion material having an adhesive property or as a surgical sealant having an anti-adhesion property. Further, the adhesion preventing method of the present invention can be easily applied to a site which is difficult with the conventional method.
Landscapes
- Health & Medical Sciences (AREA)
- Heart & Thoracic Surgery (AREA)
- Surgery (AREA)
- Vascular Medicine (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Materials For Medical Uses (AREA)
Abstract
癒着防止性に優れ、手術部位の複雑な形状に沿って貼付することが容易な癒着防止材及び該癒着防止材を施与する工程を含む癒着防止法を提供する。 (1)ゼラチン誘導体を含む第1剤であって、該ゼラチン誘導体は、 (a)下記式で表される構造を含み、 GltnNH-CHR1R2 上式においてGltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子または炭素数5~17のアルキル基である; (b)イミノ基/アミノ基(モル比)が1/99~30/70であり; (c)重量平均分子量が10,000~50,000である、 第1剤、及び (2)該ゼラチン誘導体の架橋剤を含む第2剤 からなる癒着防止材。
Description
本発明は、生体組織の癒着防止材に関し、詳細には所定の疎水性基を有するゼラチン誘導体を主剤とする生体吸収性の癒着防止材にする。
癒着防止材は、手術後の癒着の軽減を目的に、手術時に適用部位に直接使用する生体吸収性の合成材料として定義され(「特定医療材料及びその材料価格(材料価格基準)」平成28年厚生労働省告示第402号)、一般的には「癒着防止吸収性バリア」と呼ばれる。癒着防止材には、手術部位に貼付して施与されるシート型と、液状もしくは使用時に液状に調製されて、噴霧により施与されるスプレー型がある。前者の例として、ヒアルロン酸ナトリウムとカルボキシメチルセルロースを主成分とするもの(セプラフィルム(商標)、科研製薬株式会社)、ゼラチンを主成分とするもの(ゼルフィルム(商標)、ファイザー株式会社、及び特許文献1)、後者の例としてN-ヒドロキシスクシンイミド化デキストリンと炭酸ナトリウム/炭酸水素ナトリウムの2剤から成るもの(アドスプレー(商標)、テルモ株式会社)がある。
癒着防止材には、組織表面を覆って保護することが必要とされるが、フィルム状のものは手術部位の複雑な形状に沿って貼付することが困難である。またセプラフィルムは一旦水に濡れると著しく操作性が低下するため、位置を修正することは実際上不可能である。その点、スプレー型のものは手術部位に沿って、且つより広く施与することができる。
ところで、本発明者らは、疎水性基を導入したゼラチン誘導体(以下「疎水化ゼラチン」という場合がある)と硬化剤とをスプレーにより組織に施与する外科用シーラントの開発を進めてきた(例えば特許文献2、特許文献3)。同シーラントは、疎水性基を有することによって組織への親和性が高く、該疎水基を有しないゼラチンやフィブリンシーラントに比べて顕著に高いシーリング強度を有する。
上記シーラントは組織への高い接着性から、手術部位以外の組織への接着が危惧された。しかし、驚くことに、該シーラントは硬化されて膜となった後には、優れた癒着防止効果を奏することを見出し、本発明を完成した。
即ち、本発明は、下記のものである:
(1)ゼラチン誘導体を含む第1剤であって、該ゼラチン誘導体は、
(a)下記式で表される構造を含み、
GltnNH-CHR1R2
上式においてGltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子または炭素数5~17のアルキル基である;
(b)イミノ基/アミノ基(モル比)が1/99~30/70であり;
(c)重量平均分子量が10,000~50,000である、
第1剤、及び
(2)該ゼラチン誘導体の架橋剤を含む第2剤
からなる癒着防止材。
(1)ゼラチン誘導体を含む第1剤であって、該ゼラチン誘導体は、
(a)下記式で表される構造を含み、
GltnNH-CHR1R2
上式においてGltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子または炭素数5~17のアルキル基である;
(b)イミノ基/アミノ基(モル比)が1/99~30/70であり;
(c)重量平均分子量が10,000~50,000である、
第1剤、及び
(2)該ゼラチン誘導体の架橋剤を含む第2剤
からなる癒着防止材。
癒着防止材は噴霧により組織に施与することができるので、複雑な形状の手術部位でも容易に施与することができる。該癒着防止材の硬化膜は、優れた癒着防止性を示す。手術部位への接着性も高いので、接着性癒着防止材として、又は癒着防止性を有する外科用シーラントとしても使用できる。また、主剤であるゼラチン誘導体は水性溶媒中で作ることができ、製造環境及び体内において安全であるだけでなく、一段工程で簡易に且つ高い収率で合成することができる。
本発明の癒着防止材は、実質的に硬化膜を形成する主剤としてゼラチン誘導体を含む第1剤と該ゼラチン誘導体の架橋剤を含む第2剤からなる、2剤型である。第1剤と第2剤は別々に包装されて供され、使用に際して混合される。以下、それらの詳細について説明する。
<第1剤>
本発明の癒着防止材において、第1剤はゼラチン誘導体を含む。該ゼラチン誘導体はイミノ基、即ち、-NH-、を介して結合された疎水性基を有し、下記式示される構造を含む。
GltnNH-CHR1R2
上式において、Gltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子又は炭素数5~17のアルキル基である。Nは、ゼラチン中の主としてリジン(Lys)のε-アミノ基由来である。好ましくは、R2が水素原子である。該式(1)のNH構造は、例えばFT‐IRスペクトルにおいて3300cm-1付近のバンドにより検出することができる。
<第1剤>
本発明の癒着防止材において、第1剤はゼラチン誘導体を含む。該ゼラチン誘導体はイミノ基、即ち、-NH-、を介して結合された疎水性基を有し、下記式示される構造を含む。
GltnNH-CHR1R2
上式において、Gltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子又は炭素数5~17のアルキル基である。Nは、ゼラチン中の主としてリジン(Lys)のε-アミノ基由来である。好ましくは、R2が水素原子である。該式(1)のNH構造は、例えばFT‐IRスペクトルにおいて3300cm-1付近のバンドにより検出することができる。
R2が炭素数5~17のアルキル基である場合、R1と同じでも互いに異なっていてもよい。該アルキル基は分岐を含んでいてもよい。該アルキル基の例としては、ヘキシル基、オクチル基(又はカプリル基)、ノニル基(又はペラルゴルニル基)、ドデジシル基(又はラウリル基)、テトラデシル基(又はミリスチル基)等が挙げられる。好ましくは、R1が炭素数5~11の直鎖アルキル基であり、R2が水素原子である。
該ゼラチン誘導体中の誘導化率は、アルキル基が結合されたイミノ基の、原料ゼラチン中のアミノ基量に対するモル%で、1~20モル%、好ましくは5~10モル%である。言い換えれば、得られたゼラチン誘導体におけるイミノ基/アミノ基(モル比)は、1/99~20/80であり、好ましくは5/95~10/90である。該誘導化率は、原料ゼラチン中のアミノ基と、アルキル基を結合した後のアミノ基量を、2,4,6-トリニトロベンゼンスルホン酸法(TNBS法)によって定量することで、或いは、NMR等によりアルキル基の同定及び定量を行うことによって求めることができる。
原料ゼラチンは、天然由来、合成、発酵又は遺伝子組換えにより得られるゼラチンのいずれであってもよく、好ましくはブタ、ウシ等の動物由来、スケトウダラ等の魚由来のゼラチンが使用される。また、酸処理ゼラチン、アルカリ処理ゼラチン、遺伝子組み換えゼラチンのいずれであってもよいが、好ましくはアルカリ処理ゼラチンであり、より好ましくは低エンドトキシン化ゼラチンである。また、該ゼラチンの分子量の範囲は、ゼラチン誘導体の重量平均分子量(Mw)が10,000~100,000となる範囲であることが好ましく、10,000~50,000となる範囲であることがより好ましい。該分子量は、ゲル浸透クロマトグラフィー(GPC)により定法に従い測定することができる。
第1剤は、上記ゼラチン誘導体に加えて、誘導体化されていないゼラチンを含んでもよい。該ゼラチンとしては、上述の各種ゼラチンを用いることができる。誘導体化されていないゼラチンの量は、ゼラチン誘導体との合計重量の0~99wt%であり、好ましくは、0~50wt%である。
第1剤は、該ゼラチン誘導体を溶解又は分散するための水性溶媒をさらに含んでよい。利便性の点から、該ゼラチン誘導体を該水性溶媒に溶解又は分散して水性液(以下、単に「水溶液」という場合がある)として供してもよい。該水性溶媒としては、超純水、生理食塩水、ホウ酸、リン酸、炭酸等各種酸とその塩を含む緩衝液又はこれらの混合物を用いることができる。好ましくはpH8~11、より好ましくはpH9~10のホウ酸緩衝液が使用される。該水性溶媒は、ゼラチン誘導体が10~80wt/v%、好ましくは15~30wt/v%となるような量で使用される。誘導体化されていないゼラチンを含む場合には、ゼラチン誘導体との合計重量が上記濃度となる量である。
<第2剤>
本発明において、第2剤はゼラチン誘導体の架橋剤であり、架橋により水、血液等の体液に不溶性の構造体、例えば膜を形成する。該架橋剤としては、ゼラチン中のアミノ基、主として側鎖の第一級アミノ基、と反応性の官能基を分子中に少なくとも2つ以上有するものの少なくとも一種が使用される。架橋剤の例としては、ゲニピン、N-ヒドロキシスクシンイミドもしくはN-ヒドロキシスルホスクシンイミドで活性化された多塩基酸、アルデヒド化合物、酸無水物、及びジイソチオシアンネートが挙げられる。
本発明において、第2剤はゼラチン誘導体の架橋剤であり、架橋により水、血液等の体液に不溶性の構造体、例えば膜を形成する。該架橋剤としては、ゼラチン中のアミノ基、主として側鎖の第一級アミノ基、と反応性の官能基を分子中に少なくとも2つ以上有するものの少なくとも一種が使用される。架橋剤の例としては、ゲニピン、N-ヒドロキシスクシンイミドもしくはN-ヒドロキシスルホスクシンイミドで活性化された多塩基酸、アルデヒド化合物、酸無水物、及びジイソチオシアンネートが挙げられる。
多塩基酸としては、酒石酸、クエン酸、リンゴ酸、グルタル酸、グルタミン酸、アスパラギン酸、オキサロ酢酸、cis-アコニット酸、2-ケトグルタル酸、ポリ酒石酸、ポリクエン酸、ポリリンゴ酸、ポリグルタミン酸、ポリアスパラギン酸、カルボキシメチル化デキストリン、カルボキシメチル化デキストラン、カルボキシメチル化デンプン、カルボキシメチル化セルロース、カルボキシメチル化キトサン、カルボキシメチル化プルラン等が例示され、これらのカルボキシル基が活性エステル化されたもの、例えばジスクシンイミジルグルタレート(DSG)、ジスクシンイミジルスベレート(DSS)、ジスクシンイミジルタートレート(DST)等を使用することができる。
また、ポリエチレングリコールもしくはポリエチレングリコールエーテルの、多塩基酸エステルで、該多塩基酸の、ポリエチレングリコールと反応していないカルボキシル基の少なくとも1つが活性エステル化されたもの、例えば4,7,10,13,16-ペンタオキサノナデカン二酸ジ(N-スクシンイミジル)、及び下記式で表されるポリエチレングリコール ジ(スクシンイミジル スクシネート)(SS-PEG-SS):

(nはMwが約10,000~20,000となる数);
さらに、下記式で表されるペンタエリスリトール‐ポリエチレングリコールエーテル テトラスクシンイミジル グルタレート(4S-PEG):

(nはMwが約3,000~30,000、好ましくは約5,000~27,000、より好ましくは約10,000~20,000となる数);
が挙げられる。
さらに、下記式で表されるペンタエリスリトール‐ポリエチレングリコールエーテル テトラスクシンイミジル グルタレート(4S-PEG):
が挙げられる。
アルデヒド化合物としては、1分子中に2つ以上のアルデヒド基が導入された、アルデヒド基導入多糖類、例えばアルデヒド基導入デンプン、アルデヒド基導入デキストラン、アルデヒド基導入デキストリン及びアルデヒド基導入ヒアルロン酸が、酸無水物としては、無水グルタル酸、無水マレイン酸、及び無水コハク酸が、ジイソチオシアンネートとしてはヘキサメチレンジイソチオシアネート等が例示される。これらのうち、上記活性化ポリエチレングリコール多塩基酸エステル、及びアルデヒド基導入多糖類が好ましく使用される。
これらの架橋剤は、ゼラチン誘導体のアミノ基1当量に対して、該架橋剤中の官能基、例えばN-ヒドロキシスクシンイミドで活性化されたエステル基、が0.2~3当量、好ましくは0.2~2当量、より好ましくは0.2~1.0当量、最も好ましくは0.2~0.6となる量で使用される。2種以上の架橋剤の混合物を用いてもよく、その場合はそれらの合計当量が上記範囲となる量とする。
第2剤も、該架橋剤を溶解するための水性溶媒をさらに含んでよい。但し、該架橋剤と該水性溶媒とを別々の容器で供し、使用に際して、使用の約2時間前以後に、両者を適量混合して水性溶液(以下、単に「水溶液」という場合がある)として使用することが好ましい。該水性溶媒については、第1剤について上記したものを使用することができる。好ましくは、pH3~8、より好ましくはpH4~6のリン酸緩衝液が使用される。最も好ましくは、第1剤の水溶液と第2剤の水溶液を同体積で混合した際に、pHが8~10となるように双方の水性溶媒のイオン強度が調整される。例えば、第1剤水溶液をpH9、イオン強度0.05~0.1のホウ酸緩衝液とし、第2剤水溶液をpH4、イオン強度0.01~0.03のリン酸緩衝液とすることで、同体積で混合した際に8~10のpHとすることができる。又は、第1剤水溶液をpH10、イオン強度0.05~0.1のホウ酸緩衝液として、第2剤水溶液をpH4、イオン強度0.01~0.07のリン酸緩衝液としてもよい。
第2剤中の架橋剤濃度は、第1剤中のアミノ基の当量に対する第2剤中の官能基の当量、即ち(第2剤中の官能基当量/第1剤中のアミノ基当量)が、上記範囲になるように調整される。2種以上の架橋剤の混合物を用いてもよく、その場合はそれらの合計が上記範囲となる量とする。
<添加剤>
上記第1剤及び/又は第2剤は、各種添加剤を本発明の目的を阻害しない量でさらに含んでよい。該添加剤としては、着色料、pH調整剤、粘度調整剤、保存剤等が挙げられる。好ましくは、癒着防止材の適用箇所が分かり易いように、第1剤あるいは2剤水溶液中に着色料、例えばブリリアントブルーを添加する。添加量は、例えば10~100μg/mLであってよい。
上記第1剤及び/又は第2剤は、各種添加剤を本発明の目的を阻害しない量でさらに含んでよい。該添加剤としては、着色料、pH調整剤、粘度調整剤、保存剤等が挙げられる。好ましくは、癒着防止材の適用箇所が分かり易いように、第1剤あるいは2剤水溶液中に着色料、例えばブリリアントブルーを添加する。添加量は、例えば10~100μg/mLであってよい。
<製造方法>
本発明の癒着防止材は、第1剤と第2剤を個別に調製して包装し、所望により滅菌することによって得ることができる。
[第1剤の調製法]
(1)原料ゼラチン水性溶液の調製
出発材料のゼラチンを5~50wt/v%となる量で、40~90℃で加熱して水性溶媒に溶解する。該水性溶媒としては、水と水溶性有機溶媒との混合物を用いる。該水溶性有機溶媒としては、炭素数1~3のアルコール、エステル等を用いることができ、好ましくはエタノールが使用される。
本発明の癒着防止材は、第1剤と第2剤を個別に調製して包装し、所望により滅菌することによって得ることができる。
[第1剤の調製法]
(1)原料ゼラチン水性溶液の調製
出発材料のゼラチンを5~50wt/v%となる量で、40~90℃で加熱して水性溶媒に溶解する。該水性溶媒としては、水と水溶性有機溶媒との混合物を用いる。該水溶性有機溶媒としては、炭素数1~3のアルコール、エステル等を用いることができ、好ましくはエタノールが使用される。
(2)誘導体化
工程(1)で得られたゼラチン水溶液に、導入するアルキル基を有する誘導体化薬剤を添加し、所定時間撹拌して反応させる。該誘導体化薬剤としては、上記アルキル基を有するアルデヒドもしくはケトン、例えばドデカナール、テトラデカナール、デシルエチルケトンが使用される。反応温度は30~80℃、反応時間は0.5~12時間であり、通常、撹拌するだけでゼラチンのアミノ基にシッフ塩基(GltnN=CR1R2)を介してアルキル基が結合されたゼラチンを得ることができる。アルデヒドの使用量は、所望の誘導化率に相当する化学量論量に対して1~4倍とする。より好ましくは、1~2倍とする。
工程(1)で得られたゼラチン水溶液に、導入するアルキル基を有する誘導体化薬剤を添加し、所定時間撹拌して反応させる。該誘導体化薬剤としては、上記アルキル基を有するアルデヒドもしくはケトン、例えばドデカナール、テトラデカナール、デシルエチルケトンが使用される。反応温度は30~80℃、反応時間は0.5~12時間であり、通常、撹拌するだけでゼラチンのアミノ基にシッフ塩基(GltnN=CR1R2)を介してアルキル基が結合されたゼラチンを得ることができる。アルデヒドの使用量は、所望の誘導化率に相当する化学量論量に対して1~4倍とする。より好ましくは、1~2倍とする。
次いで、該シッフ塩基を還元する。還元剤としてはシアノ水素化ホウ素ナトリウム(NaBH3CN)、水素化トリアセトキシホウ素ナトリウム(NaBH(OAc)3)、2-ピコリンボラン、ピリジンボラン等の、公知の還元剤を使用することができる。これらのうち、2-ピコリンボランが好ましい。ピコリンボランは安定性であり、水性溶媒中でアルデヒドもしくはケトンの還元アミノ化反応を一段(ワンポット)で行うことが可能である。また、80~90%の収率を達成することができ、これはシアノ水素化ホウ素ナトリウムが70~75%であるのに比べて顕著に高い。2-ピコリンボランの使用量は、誘導体化薬剤の当量に対して1~3当量であることが好ましい。
(3)精製
工程(2)で得られた反応溶液に、大過剰の貧溶媒、例えば冷エタノールを加えて、ゼラチン誘導体を沈殿させる。該沈殿を濾別した後、エタノール等で洗浄して、最終生成物を得る。
工程(2)で得られた反応溶液に、大過剰の貧溶媒、例えば冷エタノールを加えて、ゼラチン誘導体を沈殿させる。該沈殿を濾別した後、エタノール等で洗浄して、最終生成物を得る。
(4)第1剤の調製
工程(3)で得られたゼラチン誘導体は粉末状の形態で、又はホウ酸緩衝液等の水性溶媒中の水溶液の形態で、容器に充填して供する。容器としては、ガラス製又はプラスチック製の、バイアル、ボトル、ディスペンサ、シリンジ等を用いることができる。所望により、誘導体化されていないゼラチン、その他添加剤を添加してよい。ゼラチン誘導体を粉末状で供する場合には、別途、ホウ酸緩衝液等の水性溶媒を容器に充填して供する。ゼラチン誘導体を水溶液で供する場合には、癒着防止材を患部に適用する際に使用する、先端部で両剤を混合することができるダブルシリンジ型ディスペンサ等の一方に充填してもよい。なお、本発明の癒着防止材が、ダブルシリンジ型ディスペンサや、混合用のバイアル、予備の水性溶媒等を付属品として含んでもよいことは言うまでもない。
工程(3)で得られたゼラチン誘導体は粉末状の形態で、又はホウ酸緩衝液等の水性溶媒中の水溶液の形態で、容器に充填して供する。容器としては、ガラス製又はプラスチック製の、バイアル、ボトル、ディスペンサ、シリンジ等を用いることができる。所望により、誘導体化されていないゼラチン、その他添加剤を添加してよい。ゼラチン誘導体を粉末状で供する場合には、別途、ホウ酸緩衝液等の水性溶媒を容器に充填して供する。ゼラチン誘導体を水溶液で供する場合には、癒着防止材を患部に適用する際に使用する、先端部で両剤を混合することができるダブルシリンジ型ディスペンサ等の一方に充填してもよい。なお、本発明の癒着防止材が、ダブルシリンジ型ディスペンサや、混合用のバイアル、予備の水性溶媒等を付属品として含んでもよいことは言うまでもない。
[第2剤の調製法]
第2剤として例示した上記各架橋剤は、公知の方法で合成してもよいし、市販されているものを使用してもよい。該架橋剤と、それを溶解するための、例えばリン酸緩衝液等の水性溶媒を別々の容器、例えば架橋剤をガラス製のバイアルに、水性溶媒をプラスチックボトルに入れて供する。所望により、添加剤を添加してよい。第1剤の量と該第2剤の量は、(該架橋剤の官能基の当量/該ゼラチン誘導体のアミノ基の当量)が0.2~2となる量で供されるが、第1剤と第2剤を夫々独立に補充できる形態で供されてもよい。
第2剤として例示した上記各架橋剤は、公知の方法で合成してもよいし、市販されているものを使用してもよい。該架橋剤と、それを溶解するための、例えばリン酸緩衝液等の水性溶媒を別々の容器、例えば架橋剤をガラス製のバイアルに、水性溶媒をプラスチックボトルに入れて供する。所望により、添加剤を添加してよい。第1剤の量と該第2剤の量は、(該架橋剤の官能基の当量/該ゼラチン誘導体のアミノ基の当量)が0.2~2となる量で供されるが、第1剤と第2剤を夫々独立に補充できる形態で供されてもよい。
<滅菌>
次いで、ディスペンサ等に充填された水溶液の形態の第1剤又はバイアル等に充填されたゼラチン誘導体粉末とボトル等に充填された水性溶媒との組み合わせの形態の第1剤、及び、バイアル等に充填された粉末形態の架橋剤とボトル等に充填された該架橋剤を溶解するための水性溶媒との組み合わせの形態の第2剤を夫々滅菌する。ゼラチン誘導体粉末および粉末形態の架橋剤の滅菌法は、放射線滅菌が好ましい。該放射線としては、電子線、ガンマ線、制動放射線が挙げられ、電子線滅菌が好ましい。総吸収線量としては、従来広く用いられている(第十四改正日本薬局方、第二部、参考情報、第1235頁、右欄、2.2 放射線法)20kGy以上であればよく、好ましくは25kGy~45kGyである。総吸収線量が20kGy以上となればよく、ゼラチン誘導体や架橋剤への損傷を防ぐために、電子線を複数回に分けて照射しても、滅菌効果には変わりはない。例えば総吸収線量30kGyで滅菌する場合には、10kGyの照射を3回照射してもよい。ゼラチン誘導体水溶液の滅菌法は、オートクレーブあるいはフィルター滅菌が好ましい。
次いで、ディスペンサ等に充填された水溶液の形態の第1剤又はバイアル等に充填されたゼラチン誘導体粉末とボトル等に充填された水性溶媒との組み合わせの形態の第1剤、及び、バイアル等に充填された粉末形態の架橋剤とボトル等に充填された該架橋剤を溶解するための水性溶媒との組み合わせの形態の第2剤を夫々滅菌する。ゼラチン誘導体粉末および粉末形態の架橋剤の滅菌法は、放射線滅菌が好ましい。該放射線としては、電子線、ガンマ線、制動放射線が挙げられ、電子線滅菌が好ましい。総吸収線量としては、従来広く用いられている(第十四改正日本薬局方、第二部、参考情報、第1235頁、右欄、2.2 放射線法)20kGy以上であればよく、好ましくは25kGy~45kGyである。総吸収線量が20kGy以上となればよく、ゼラチン誘導体や架橋剤への損傷を防ぐために、電子線を複数回に分けて照射しても、滅菌効果には変わりはない。例えば総吸収線量30kGyで滅菌する場合には、10kGyの照射を3回照射してもよい。ゼラチン誘導体水溶液の滅菌法は、オートクレーブあるいはフィルター滅菌が好ましい。
<組織への適用方法等>
本発明は、ヒト又はヒト以外の動物である対象の患部に、第1剤及び第2剤を施与する工程を含む癒着防止材膜の製造方法、及びその癒着防止材膜の製造方法を含む、対象の癒着防止方法も提供する。本発明の癒着防止材は、皮膚、血管、腱、神経、腸、及びリンパ管等の管状組織、肝臓、膵臓、及び心臓などの臓器の断裂部分に適用することができる。なかでも、湿潤組織、例えば、血管、肺等に好適に適用される。癒着防止材の適用方法としては、上述のとおり第2剤を、好ましくは使用直前に水溶液とする。その際の架橋剤の濃度は、既に述べたとおりである。得られた第2剤水溶液を、既に第1剤水溶液が充填されているダブルシリンジ型ディスペンサの空いている方のシリンジに充填して適用し、又は、ダブルシリンジを備えるエアアシストスプレーで噴霧して患部に施与する。施与後、数分~10分程度で膜が形成され、患部を封じることができる。
本発明は、ヒト又はヒト以外の動物である対象の患部に、第1剤及び第2剤を施与する工程を含む癒着防止材膜の製造方法、及びその癒着防止材膜の製造方法を含む、対象の癒着防止方法も提供する。本発明の癒着防止材は、皮膚、血管、腱、神経、腸、及びリンパ管等の管状組織、肝臓、膵臓、及び心臓などの臓器の断裂部分に適用することができる。なかでも、湿潤組織、例えば、血管、肺等に好適に適用される。癒着防止材の適用方法としては、上述のとおり第2剤を、好ましくは使用直前に水溶液とする。その際の架橋剤の濃度は、既に述べたとおりである。得られた第2剤水溶液を、既に第1剤水溶液が充填されているダブルシリンジ型ディスペンサの空いている方のシリンジに充填して適用し、又は、ダブルシリンジを備えるエアアシストスプレーで噴霧して患部に施与する。施与後、数分~10分程度で膜が形成され、患部を封じることができる。
以下、本発明を実施例により説明するが、本発明はこれらに限定されるものではない。
<ゼラチン誘導体の調製>
特許文献3(特許第5995128号)に記載の方法でゼラチン誘導体を調製した。例として、オクタナールを用いてゼラチン誘導体1を調製した方法を示す。
スケトウダラ由来のゼラチン(Mw33,000、新田ゼラチン(株)製)30gを50℃で水105mLに溶解したところへ、該ゼラチンのアミノ基の10モル%に相当する化学量論量のn-オクタナール139μLを40mLのエタノールに溶解した溶液を加え、50℃で1時間攪拌した。そこへ、2-ピコリンボラン143μLを5mLのエタノールに溶解した溶液を加えた。得られた反応混合物を50℃で17時間攪拌した後、反応混合物の10倍体積量の冷エタノール中に該反応混合物を滴下して、ゼラチン誘導体を沈殿させ、吸引濾過を行った。得られた沈殿物を冷エタノールで3回洗浄し後、濾別した。得られた濾過物を減圧乾燥して、ゼラチン誘導体1を得た。
<ゼラチン誘導体の調製>
特許文献3(特許第5995128号)に記載の方法でゼラチン誘導体を調製した。例として、オクタナールを用いてゼラチン誘導体1を調製した方法を示す。
スケトウダラ由来のゼラチン(Mw33,000、新田ゼラチン(株)製)30gを50℃で水105mLに溶解したところへ、該ゼラチンのアミノ基の10モル%に相当する化学量論量のn-オクタナール139μLを40mLのエタノールに溶解した溶液を加え、50℃で1時間攪拌した。そこへ、2-ピコリンボラン143μLを5mLのエタノールに溶解した溶液を加えた。得られた反応混合物を50℃で17時間攪拌した後、反応混合物の10倍体積量の冷エタノール中に該反応混合物を滴下して、ゼラチン誘導体を沈殿させ、吸引濾過を行った。得られた沈殿物を冷エタノールで3回洗浄し後、濾別した。得られた濾過物を減圧乾燥して、ゼラチン誘導体1を得た。
ゼラチン誘導体1の赤外線スペクトル(FTIR-8400、島津製作所株式会社)及び13C-NMRスペクトル(AL300、日本電子株式会社)を測定し、誘導体化を確認した。赤外線スペクトルでは、3280cm-1において第二級アミン、2936cm-1及び2879cm-1にメチレン基のピーク増加が、13C-NMRスペクトルでは17-17.5ppmに第一級炭素由来のピークが、夫々、認められ、アルキル基が導入されたことが確認された。
誘導化率を以下の方法で測定した。ゼラチン誘導体1を体積比1:1のジメチルホルムアミド/水混合溶媒中に溶解して0.1w/v%溶液を得た。該溶液に0.1v/v%トリエチルアミン水溶液及び0.1w/v%トリニトロベンゼンスルホン酸水溶液を加えて、約1分間攪拌してトリニトロベンゼンスルホン酸を該ゼラチン誘導体中のアミノ基と反応させた後、マイクロプレートリーダー(SPARK 10M、テカンジャパン株式会社)で340nmの吸光度を測定した。ゼラチン誘導体1の誘導化率は、7.2モル%であった。
n-オクタナールをn-デカナール又はn-ドデカナールに変えたこと、及びMw38,000のゼラチンに変えたことを除き、上記ゼラチン誘導体の調製方法と同様の方法で、表1に示すゼラチン誘導体を調製した。なお、表1等において、例えば「C8」はC8アルデヒドによって誘導化されたゼラチン(式(1)においてR1がヘプチル基であり、R2が水素原子)を表す。

上記各ゼラチン誘導体を、pH9.5の0.1Mホウ酸緩衝液に15w/v%の濃度で溶解し、第1剤とした。
<第2剤の調製>
第2剤として、ペンタエリスリトール‐ポリエチレングリコールエーテル テトラスクシンイミジル グルタレート(4S-PEG、日油(株)社製)を用いた。下記各評価の直前に、pH4.0の0.01Mリン酸緩衝液に、第1剤と同体積で混合した際に第1剤の残存アミノ基1当量に対して0.4当量(実施例4では0.5当量)となるように4S-PEGを溶解した。
第2剤として、ペンタエリスリトール‐ポリエチレングリコールエーテル テトラスクシンイミジル グルタレート(4S-PEG、日油(株)社製)を用いた。下記各評価の直前に、pH4.0の0.01Mリン酸緩衝液に、第1剤と同体積で混合した際に第1剤の残存アミノ基1当量に対して0.4当量(実施例4では0.5当量)となるように4S-PEGを溶解した。
<膜の調製法>
ダブルシリンジ型ディスペンサで同体積の第1剤と第2剤を各試験における基材上に押し出して、実施例の膜を組織上に塗布した。また、表2に示すように、比較例としては誘導化前のゼラチン(比較例1:Org)と、フィブリン(比較例2:fibrin;商品名ボルヒール、化学及び血清療法研究所製)、セプラフィルム(比較例3:HA/CMC Film;商標、科研製薬株式会社)を、夫々の添付文書に従って用いた。比較例2及び3は実施例と同面積になる量で用いた。

なお、図3等において、比較例2は”Fibrin”, 比較例3は“HA/CMC”と表記する。
ダブルシリンジ型ディスペンサで同体積の第1剤と第2剤を各試験における基材上に押し出して、実施例の膜を組織上に塗布した。また、表2に示すように、比較例としては誘導化前のゼラチン(比較例1:Org)と、フィブリン(比較例2:fibrin;商品名ボルヒール、化学及び血清療法研究所製)、セプラフィルム(比較例3:HA/CMC Film;商標、科研製薬株式会社)を、夫々の添付文書に従って用いた。比較例2及び3は実施例と同面積になる量で用いた。
癒着防止性を評価するために、インビトロでの細胞接着試験、細胞透過試験及びインビボ癒着防止試験を行った。
<細胞接着性>
48ウェルプレートの底面上に、実施例2~5、及び比較例1の第1剤と第2剤を夫々150μLずつ施与して膜を形成し、紫外線を1時間照射して滅菌した。膜上にL929線維芽細胞を播種し(1.0×105細胞/ウェル)、炭酸ガス下、10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシン溶液を含むRPMI-1640培地中、37℃で培養した。24時間後、細胞をD-PBSで洗浄し、WST-8アッセイキットを用いて膜上に付着した細胞数を計測した。コントロールとして、膜の無いものを用いた。
<細胞接着性>
48ウェルプレートの底面上に、実施例2~5、及び比較例1の第1剤と第2剤を夫々150μLずつ施与して膜を形成し、紫外線を1時間照射して滅菌した。膜上にL929線維芽細胞を播種し(1.0×105細胞/ウェル)、炭酸ガス下、10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシン溶液を含むRPMI-1640培地中、37℃で培養した。24時間後、細胞をD-PBSで洗浄し、WST-8アッセイキットを用いて膜上に付着した細胞数を計測した。コントロールとして、膜の無いものを用いた。
結果を図1に示す。同図から分かるように、コントロールに比べて、いずれの実施例も細胞の接着が少なく、インビトロでの優れた癒着防止性を示唆した。
<細胞透過性>
図2に模式的に示すような細胞培養インサート(トランズウェルインサート、PTFEメンブレン口径8.0μm、コーニングインターナショナル株式会社)を用いて、細胞透過性を評価した。インサートの底面上に、実施例1及び比較例1の第1剤と第2剤を夫々100μLずつ、及び同面積となる量の比較例2、3を施与して膜を形成し、紫外線を1時間照射して滅菌した。膜上にL929線維芽細胞を播種し(1.0×106細胞/ウェル)、炭酸ガス下、10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシン溶液を含むRPMI-1640培地中、37℃で培養した。24時間後、ウェル下部に透過した細胞数を計測し、膜を形成しない未処理の場合に対する割合(%)を求めた。結果を図3に示す。
図2に模式的に示すような細胞培養インサート(トランズウェルインサート、PTFEメンブレン口径8.0μm、コーニングインターナショナル株式会社)を用いて、細胞透過性を評価した。インサートの底面上に、実施例1及び比較例1の第1剤と第2剤を夫々100μLずつ、及び同面積となる量の比較例2、3を施与して膜を形成し、紫外線を1時間照射して滅菌した。膜上にL929線維芽細胞を播種し(1.0×106細胞/ウェル)、炭酸ガス下、10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシン溶液を含むRPMI-1640培地中、37℃で培養した。24時間後、ウェル下部に透過した細胞数を計測し、膜を形成しない未処理の場合に対する割合(%)を求めた。結果を図3に示す。
図3から分かるように、実施例はセプラフィルムに比べて細胞の透過性が有意に低く、この事が癒着防止性に寄与しているものと考えられる。
<インビボ癒着防止性評価>
ラット(Wistar、7週雌、日本チャールス・リバー株式会社)を開腹して盲腸腹膜壁欠損モデルを用意した。盲腸壁(Cecum)を医療用ガーゼで擦って出血させ、盲腸壁を損傷させた。該損傷個所に対向する腹膜壁(Abdominal wall)を医療用ナイフで所定の大きさ(1.0×2.0cm)切除することで腹膜壁欠損を用意した。腹膜壁欠損と盲腸損傷の双方に実施例1及び比較例1~3の癒着防止材を、傷の全範囲を覆うように塗布した後、開腹部を縫合した。
ラット(Wistar、7週雌、日本チャールス・リバー株式会社)を開腹して盲腸腹膜壁欠損モデルを用意した。盲腸壁(Cecum)を医療用ガーゼで擦って出血させ、盲腸壁を損傷させた。該損傷個所に対向する腹膜壁(Abdominal wall)を医療用ナイフで所定の大きさ(1.0×2.0cm)切除することで腹膜壁欠損を用意した。腹膜壁欠損と盲腸損傷の双方に実施例1及び比較例1~3の癒着防止材を、傷の全範囲を覆うように塗布した後、開腹部を縫合した。
手術1週間後及び2週間後に、ラットを犠死させ手術部位の組織3カ所の断面の試験片を採取し、10%ホルマリン中性緩衝液中で固定して、ヘマトキシリン・エオジン染色し、光学顕微鏡により観察した。1週間後の写真を図4aに、2週間後の写真を図4bに示す。図中、“Untreated”は、欠損部に何も適用しなかったことを示す。各試験片の癒着(Adhesion)を表3の基準で評価した結果を表4、5に示す。スコアが低い試験片数が多いほど癒着が少ないことを示す。


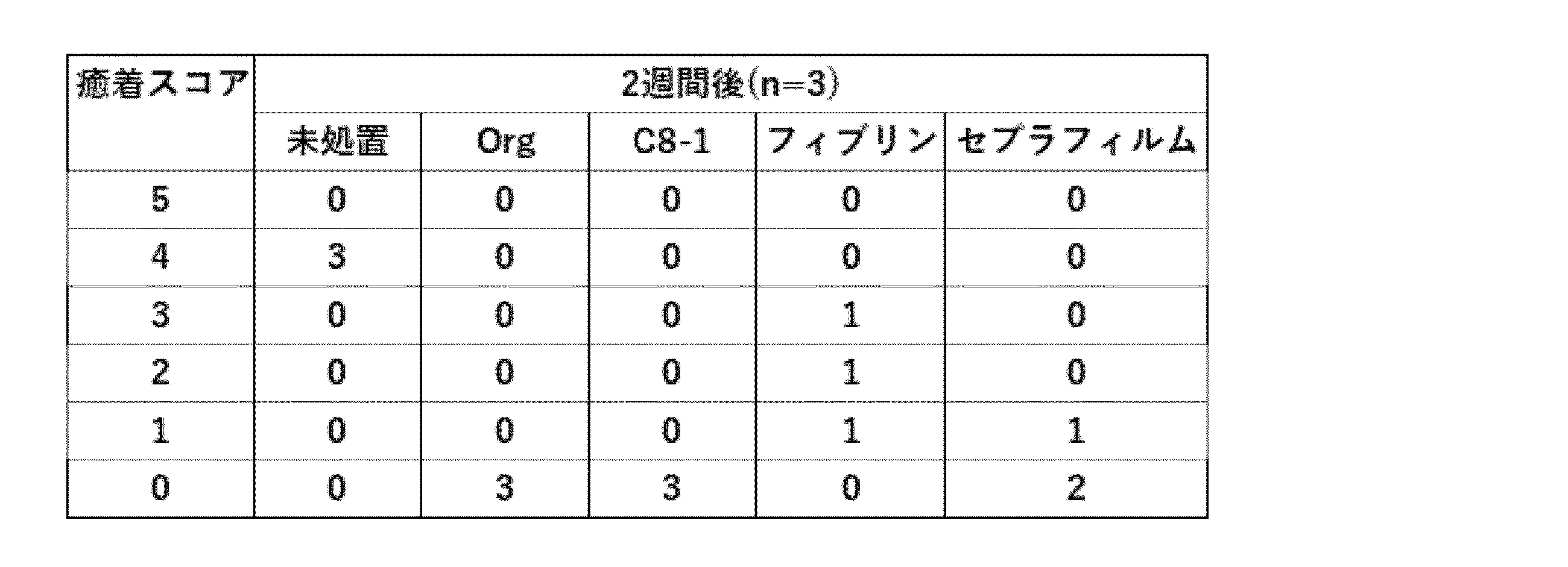
表4、5に示すように、未処置試料及びフィブリンを適用した組織には顕著に癒着が認められたが、実施例1は2週間後においても全く癒着が無く、優れた癒着防止性が確認された。
<シーリング強度>
ASTM(F2392-04)に従い、ブタ大腸(φ30mm)を基材として用い、シーリング強度(耐圧強度)を測定した。実施例1、比較例1~3の第1剤と第2剤をそれぞれ100μLずつ混合し(架橋剤中のスクシンイミドエステル基の当量/ゼラチン誘導体中のアミノ基の当量=0.4)、前記ブタ大腸に塗布することで、厚さ1.0mm、直径15mmの癒着防止材膜を調製した。塗布後、5.0g/mm2の荷重により10分間の圧着を行った後、37℃の生理食塩水を2ml/分で流し、破裂したときの圧力の測定を行った。結果を図5に示す。また、破裂した後の試験片組織を中性緩衝ホルマリン液で固定後ヘマトキシリン・エオジン染色を行い、組織と試料との界面の観察を行った(図6)。
ASTM(F2392-04)に従い、ブタ大腸(φ30mm)を基材として用い、シーリング強度(耐圧強度)を測定した。実施例1、比較例1~3の第1剤と第2剤をそれぞれ100μLずつ混合し(架橋剤中のスクシンイミドエステル基の当量/ゼラチン誘導体中のアミノ基の当量=0.4)、前記ブタ大腸に塗布することで、厚さ1.0mm、直径15mmの癒着防止材膜を調製した。塗布後、5.0g/mm2の荷重により10分間の圧着を行った後、37℃の生理食塩水を2ml/分で流し、破裂したときの圧力の測定を行った。結果を図5に示す。また、破裂した後の試験片組織を中性緩衝ホルマリン液で固定後ヘマトキシリン・エオジン染色を行い、組織と試料との界面の観察を行った(図6)。
図5に示すとおり、本発明の癒着防止材は、他の試料に比べて顕著に高いシーリング強度を示し、特許文献3に示した結果が再確認された。また、図6に示すように、フィブリン膜とセプラフィルムは、膜とブタ大腸との界面で剥離したが、本発明の癒着防止材は、癒着防止材膜自体が凝集破壊された。ここから、本発明の癒着防止材は接着強度にも優れることが分かる。
<タンパク質透過性>
図7に示すような細胞培養インサート(トランズウェルインサート、PTFEメンブレン口径8.0μm、コーニングインターナショナル株式会社)を用い、D.Nwa et al.,J.Biomed.Mater.Res.B, 2013,101B,1251-1258記載の方法に従い、タンパク質透過性を調べた。インサートの底面上に、第1剤と第2剤を夫々100μLずつ施与して膜を形成し、紫外線を1時間照射して滅菌した。インサートを0.9mLのD-PBS中に浸漬した。フルオロセインイソシアネート標識アルブミンの0.1w/v%水溶液の0.1mLを該膜上に滴下した。24時間後の透過率を、該インサートの裏面の蛍光測定と画像解析システム(IVIS Lumina II、パーキンエルマ-社)を用いて求めた。結果を図8に示す。
図7に示すような細胞培養インサート(トランズウェルインサート、PTFEメンブレン口径8.0μm、コーニングインターナショナル株式会社)を用い、D.Nwa et al.,J.Biomed.Mater.Res.B, 2013,101B,1251-1258記載の方法に従い、タンパク質透過性を調べた。インサートの底面上に、第1剤と第2剤を夫々100μLずつ施与して膜を形成し、紫外線を1時間照射して滅菌した。インサートを0.9mLのD-PBS中に浸漬した。フルオロセインイソシアネート標識アルブミンの0.1w/v%水溶液の0.1mLを該膜上に滴下した。24時間後の透過率を、該インサートの裏面の蛍光測定と画像解析システム(IVIS Lumina II、パーキンエルマ-社)を用いて求めた。結果を図8に示す。
図8に示すように、実施例1はフィブリン膜及びセプラフィルムに比べて顕著に低いタンパク質透過率(約20%)を示し、大半のタンパク質は表面上に留まっていた。このタンパク質透過抑制作用が、癒着防止性に寄与しているものと考えられる。誘導化されていないゼラチン膜も透過率は低かったが、タンパク質が厚み全体に広がっていた。
<膨潤度>
実施例1、比較例1の第1剤と第2剤を夫々1000μLずつ、厚さ0.5mmのシリコーンゴムをスペーサーとする2枚のガラス板間に流し込み、板状の癒着防止材硬化物を作成した。得られた癒着防止材硬化物を直径4mmのポンチでくりぬくことにより、厚さ0.5mm、直径4mmの円形膜を調製した。凍結乾燥した後の重さ(Wd)を測定した。膜を37℃の生理食塩水中に浸漬し、経過時間による癒着防止材膜の重量(Ws)の変化から下記式に従い膨潤度を算出して調べた。
膨潤度(%)={(Ws-Wd)/Wd}×100
図9に時間による膨潤度の変化を示す。試験を行った時間内では両者の試料に有意差は見られなかった。
実施例1、比較例1の第1剤と第2剤を夫々1000μLずつ、厚さ0.5mmのシリコーンゴムをスペーサーとする2枚のガラス板間に流し込み、板状の癒着防止材硬化物を作成した。得られた癒着防止材硬化物を直径4mmのポンチでくりぬくことにより、厚さ0.5mm、直径4mmの円形膜を調製した。凍結乾燥した後の重さ(Wd)を測定した。膜を37℃の生理食塩水中に浸漬し、経過時間による癒着防止材膜の重量(Ws)の変化から下記式に従い膨潤度を算出して調べた。
膨潤度(%)={(Ws-Wd)/Wd}×100
図9に時間による膨潤度の変化を示す。試験を行った時間内では両者の試料に有意差は見られなかった。
<酵素分解性>
実施例1及び比較例1について、第1剤と第2剤を夫々1000μLずつ、厚さ1.0mmのシリコーンゴムをスペーサーとする2枚のガラス板間に流し込み、板状の癒着防止材硬化物を作成した。得られた癒着防止材硬化物を直径10mmのポンチでくりぬくことにより、厚さ1.0mm、直径10mmの円形膜を調製し、初期重量(Wt0)を測定した。リン酸緩衝生理食塩水(PBS)とのカルシウム塩形成を防ぐだめに、試験片をトリス塩酸緩衝液(2.5mM CaCl2、pH7.4)に一晩浸漬した。トリス塩酸緩衝液に100μg/mLの濃度でコラゲナーゼを溶解し、試験片を2mLのコラゲナーゼ溶液中に浸漬した。経過時間による癒着防止材膜の重量(Wt)の変化から下記式により分解されずに残った重量(%)を算出して調べた。
重量(%)=(Wt/Wt0)×100
上式においてWt0は初期重量、Wtは浸漬後t時間における重量である。結果を図10に示す。
図10に示すように、実施例の膜は比較例に比べて分解時間が長く、手術部をより長時間保護することができることが確認された。
実施例1及び比較例1について、第1剤と第2剤を夫々1000μLずつ、厚さ1.0mmのシリコーンゴムをスペーサーとする2枚のガラス板間に流し込み、板状の癒着防止材硬化物を作成した。得られた癒着防止材硬化物を直径10mmのポンチでくりぬくことにより、厚さ1.0mm、直径10mmの円形膜を調製し、初期重量(Wt0)を測定した。リン酸緩衝生理食塩水(PBS)とのカルシウム塩形成を防ぐだめに、試験片をトリス塩酸緩衝液(2.5mM CaCl2、pH7.4)に一晩浸漬した。トリス塩酸緩衝液に100μg/mLの濃度でコラゲナーゼを溶解し、試験片を2mLのコラゲナーゼ溶液中に浸漬した。経過時間による癒着防止材膜の重量(Wt)の変化から下記式により分解されずに残った重量(%)を算出して調べた。
重量(%)=(Wt/Wt0)×100
上式においてWt0は初期重量、Wtは浸漬後t時間における重量である。結果を図10に示す。
図10に示すように、実施例の膜は比較例に比べて分解時間が長く、手術部をより長時間保護することができることが確認された。
本発明の癒着防止材は複雑な形状の手術部位に適用することができ、接着性を有する癒着防止材として、又は癒着防止性を有する外科用シーラントとして臨床での使用に大変適する。また、本発明の癒着防止法は、従来の方法では難しい部位に簡易に施すことができる。
Claims (7)
- (1)ゼラチン誘導体を含む第1剤であって、該ゼラチン誘導体は、
(a)下記式で表される構造を含み、
GltnNH-CHR1R2
上式においてGltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子または炭素数5~17のアルキル基である;
(b)イミノ基/アミノ基(モル比)が1/99~30/70であり;
(c)重量平均分子量が10,000~50,000である、
第1剤、及び
(2)該ゼラチン誘導体の架橋剤を含む第2剤
からなる癒着防止材。 - 該ゼラチンが、スケトウダラ由来のゼラチンである、請求項1記載の癒着防止材。
- R1が炭素数5~11のアルキル基であり、R2は水素原子である、請求項1又は2記載の癒着防止材。
- 該架橋剤が、少なくとも2つの活性化されたエステル基を有する、ポリエチレングリコールエーテル多塩基酸エステルである、請求項1~3のいずれか1項記載の癒着防止材。
- 組織接着性癒着防止材である、請求項1~4のいずれか1項記載の癒着防止材。
- ヒト以外の動物である対象の患部の癒着を防止する方法であって、
該患部に癒着防止材を施与する工程を含み、該癒着防止材が、
(1)ゼラチン誘導体を含む第1剤であって、該ゼラチン誘導体は、
(a)下記式で表される構造を含み、
GltnNH-CHR1R2
上式においてGltnはゼラチン残基であり、R1は炭素数5~17のアルキル基であり、R2は水素原子または炭素数5~17のアルキル基である;
(b)イミノ基/アミノ基(モル比)が1/99~30/70であり;
(c)重量平均分子量が10,000~50,000である、
第1剤、及び
(2)該ゼラチン誘導体の架橋剤を含む第2剤
からなる、癒着を防止する方法。 - 該患部に癒着防止材を施与する工程が、ダブルシリンジ型ディスペンサ又はダブルシリンジを備えるエアアシストスプレーにより、第1剤及び第2剤を噴霧する工程を含む、請求項6記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19858375.9A EP3848066B1 (en) | 2018-09-05 | 2019-08-27 | Adhesion prevention material |
| US17/268,078 US20210316046A1 (en) | 2018-09-05 | 2019-08-27 | Adhesion prevention material |
| JP2020541148A JP7127888B2 (ja) | 2018-09-05 | 2019-08-27 | 癒着防止材 |
| CN201980049853.5A CN112512602A (zh) | 2018-09-05 | 2019-08-27 | 防粘连材料 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-165810 | 2018-09-05 | ||
| JP2018165810 | 2018-09-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020050102A1 true WO2020050102A1 (ja) | 2020-03-12 |
Family
ID=69722677
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/033549 Ceased WO2020050102A1 (ja) | 2018-09-05 | 2019-08-27 | 癒着防止材 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20210316046A1 (ja) |
| EP (1) | EP3848066B1 (ja) |
| JP (1) | JP7127888B2 (ja) |
| CN (1) | CN112512602A (ja) |
| WO (1) | WO2020050102A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2023181563A1 (ja) * | 2022-03-23 | 2023-09-28 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115873529B (zh) * | 2022-12-30 | 2024-08-02 | 江苏斯迪克新材料科技股份有限公司 | 非硅离型力、熔点可调控的离型剂及其制备方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011025013A (ja) * | 2009-06-25 | 2011-02-10 | Doshisha | 癒着防止材及びその製造方法 |
| JP2013226166A (ja) | 2012-04-24 | 2013-11-07 | Gunze Ltd | 生体吸収性癒着防止材料 |
| WO2014112208A1 (ja) | 2013-01-18 | 2014-07-24 | 独立行政法人物質・材料研究機構 | 組織接着剤及びその製造方法 |
| JP5995128B1 (ja) | 2016-01-20 | 2016-09-21 | 国立研究開発法人物質・材料研究機構 | 外科用シーラント |
| WO2019045081A1 (ja) * | 2017-09-04 | 2019-03-07 | 国立研究開発法人物質・材料研究機構 | 架橋ゼラチン誘導体粒子を含む創傷被覆材 |
| JP2019051189A (ja) * | 2017-09-19 | 2019-04-04 | 国立研究開発法人物質・材料研究機構 | ゼラチン誘導体創傷被覆材 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3502272B2 (ja) * | 1997-09-01 | 2004-03-02 | 株式会社ジェイ・エム・エス | 生体組織接着性医用材料及びその製造法 |
| JP4226299B2 (ja) * | 2002-10-22 | 2009-02-18 | 日本水産株式会社 | 魚由来ゼラチンペプチドの製造方法 |
| US7273896B2 (en) * | 2003-04-10 | 2007-09-25 | Angiotech Pharmaceuticals (Us), Inc. | Compositions and methods of using a transient colorant |
| DK2136850T3 (da) * | 2007-04-13 | 2012-04-10 | Kuros Biosurgery Ag | Polymervævforsegling |
| JP2012095769A (ja) * | 2010-10-29 | 2012-05-24 | Gunze Ltd | 医療用接着剤及び医療用材料 |
| CN102068719A (zh) * | 2011-01-18 | 2011-05-25 | 复旦大学 | 由物理交联水凝胶组合物构成的防粘连材料及其制备方法与应用 |
| WO2015049800A1 (ja) * | 2013-10-04 | 2015-04-09 | ニチバン株式会社 | 生体内付着性の医療用フィルム |
| WO2015076252A1 (ja) * | 2013-11-22 | 2015-05-28 | 独立行政法人物質・材料研究機構 | 組織接着性多孔質膜、その製造方法及び組織接着性多孔質膜テープ |
| CN104307052B (zh) * | 2014-10-27 | 2017-02-22 | 爱美客技术发展股份有限公司 | 医用可注射防粘连凝胶及其制备方法 |
-
2019
- 2019-08-27 WO PCT/JP2019/033549 patent/WO2020050102A1/ja not_active Ceased
- 2019-08-27 CN CN201980049853.5A patent/CN112512602A/zh active Pending
- 2019-08-27 US US17/268,078 patent/US20210316046A1/en not_active Abandoned
- 2019-08-27 JP JP2020541148A patent/JP7127888B2/ja active Active
- 2019-08-27 EP EP19858375.9A patent/EP3848066B1/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011025013A (ja) * | 2009-06-25 | 2011-02-10 | Doshisha | 癒着防止材及びその製造方法 |
| JP2013226166A (ja) | 2012-04-24 | 2013-11-07 | Gunze Ltd | 生体吸収性癒着防止材料 |
| WO2014112208A1 (ja) | 2013-01-18 | 2014-07-24 | 独立行政法人物質・材料研究機構 | 組織接着剤及びその製造方法 |
| JP5995128B1 (ja) | 2016-01-20 | 2016-09-21 | 国立研究開発法人物質・材料研究機構 | 外科用シーラント |
| WO2017126390A1 (ja) * | 2016-01-20 | 2017-07-27 | 国立研究開発法人物質・材料研究機構 | 外科用シーラント |
| WO2019045081A1 (ja) * | 2017-09-04 | 2019-03-07 | 国立研究開発法人物質・材料研究機構 | 架橋ゼラチン誘導体粒子を含む創傷被覆材 |
| JP2019051189A (ja) * | 2017-09-19 | 2019-04-04 | 国立研究開発法人物質・材料研究機構 | ゼラチン誘導体創傷被覆材 |
Non-Patent Citations (1)
| Title |
|---|
| ITO, TAICHI ET AL.: "Formation of a Biocompatible Film in vivo -from Peritoneal Adhesion to Drug Delivery System", MEMBRANE, vol. 36, no. 2, 2011, pages 63 - 70, XP009526848, ISSN: 0385-1036 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2023181563A1 (ja) * | 2022-03-23 | 2023-09-28 | ||
| WO2023181563A1 (ja) * | 2022-03-23 | 2023-09-28 | 国立研究開発法人物質・材料研究機構 | 癒着防止材、及び癒着防止材の製造方法 |
| JP7802409B2 (ja) | 2022-03-23 | 2026-01-20 | 国立研究開発法人物質・材料研究機構 | 癒着防止材、及び癒着防止材の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210316046A1 (en) | 2021-10-14 |
| CN112512602A (zh) | 2021-03-16 |
| EP3848066B1 (en) | 2024-03-13 |
| EP3848066A1 (en) | 2021-07-14 |
| EP3848066A4 (en) | 2022-06-08 |
| JP7127888B2 (ja) | 2022-08-30 |
| JPWO2020050102A1 (ja) | 2021-08-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5995128B1 (ja) | 外科用シーラント | |
| EP2214730B1 (en) | Dextran-based polymer tissue adhesive for medical use | |
| KR101250543B1 (ko) | 수화겔, 이의 제조 방법 및 용도 | |
| Chichiricco et al. | In situ photochemical crosslinking of hydrogel membrane for guided tissue regeneration | |
| CN106075549B (zh) | 组织封闭胶组合物,组织封闭胶及其制备方法和应用 | |
| Mizuta et al. | Enhanced sealing by hydrophobic modification of Alaska Pollock‐derived gelatin‐based surgical sealants for the treatment of pulmonary air leaks | |
| EP2210917A1 (en) | -1,3-glucan-derived polyaldehyde/polyamine hydrogel | |
| US12337075B2 (en) | Extracellular matrix-based bioadhesive | |
| KR20010013105A (ko) | 콜라겐 겔 | |
| JP2019216755A (ja) | 止血材 | |
| EP4480507A1 (en) | Use of polysaccharide derivative | |
| Rekulapally et al. | Tissue engineering of collagen scaffolds crosslinked with plant based polysaccharides | |
| JP7127888B2 (ja) | 癒着防止材 | |
| JP6367979B2 (ja) | 外科用シーラント | |
| US12403646B2 (en) | Method for 3D printing with a bio-ink | |
| Willems et al. | Hydrogels based on oxidized cellulose sulfates and carboxymethyl chitosan: studies on intrinsic gel properties, stability, and biocompatibility | |
| CN106075550B (zh) | 医用组织封闭胶组合物,医用组织封闭胶及其制备方法和应用 | |
| JP2023161851A (ja) | 接着剤、創傷被覆材、癒着防止材、止血材、シーラント、及び噴霧キット | |
| Sternberg et al. | Development of a biodegradable tissue adhesive based on functionalized 1, 2‐ethylene glycol bis (dilactic acid). I. | |
| KR102630678B1 (ko) | 아민-말단화 된 페놀 유도체가 수식된 알데히드 치환 히알루론산 유도체 및 그의 용도 | |
| JP4397015B2 (ja) | ゲル化組成物 | |
| JPWO2006016600A1 (ja) | 酒石酸誘導体及び該誘導体により合成された高分子架橋体 | |
| JP2004503483A (ja) | 低分子量ポリマー組成物 | |
| JP2005290147A (ja) | 反応性多分岐多糖誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19858375 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020541148 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019858375 Country of ref document: EP Effective date: 20210406 |