WO2020075663A1 - 樹脂組成物、樹脂硬化物および複合成形体 - Google Patents
樹脂組成物、樹脂硬化物および複合成形体 Download PDFInfo
- Publication number
- WO2020075663A1 WO2020075663A1 PCT/JP2019/039448 JP2019039448W WO2020075663A1 WO 2020075663 A1 WO2020075663 A1 WO 2020075663A1 JP 2019039448 W JP2019039448 W JP 2019039448W WO 2020075663 A1 WO2020075663 A1 WO 2020075663A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- resin composition
- weight
- inorganic filler
- cured product
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3236—Heterocylic compounds
- C08G59/3245—Heterocylic compounds containing only nitrogen as a heteroatom
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/01—Use of inorganic substances as compounding ingredients characterized by their specific function
- C08K3/013—Fillers, pigments or reinforcing additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/38—Boron-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/34—Heterocyclic compounds having nitrogen in the ring
- C08K5/3467—Heterocyclic compounds having nitrogen in the ring having more than two nitrogen atoms in the ring
- C08K5/3477—Six-membered rings
- C08K5/3492—Triazines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/34—Heterocyclic compounds having nitrogen in the ring
- C08K5/3467—Heterocyclic compounds having nitrogen in the ring having more than two nitrogen atoms in the ring
- C08K5/3477—Six-membered rings
- C08K5/3492—Triazines
- C08K5/34926—Triazines also containing heterocyclic groups other than triazine groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10W—GENERIC PACKAGES, INTERCONNECTIONS, CONNECTORS OR OTHER CONSTRUCTIONAL DETAILS OF DEVICES COVERED BY CLASS H10
- H10W40/00—Arrangements for thermal protection or thermal control
- H10W40/20—Arrangements for cooling
- H10W40/25—Arrangements for cooling characterised by their materials
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10W—GENERIC PACKAGES, INTERCONNECTIONS, CONNECTORS OR OTHER CONSTRUCTIONAL DETAILS OF DEVICES COVERED BY CLASS H10
- H10W40/00—Arrangements for thermal protection or thermal control
- H10W40/20—Arrangements for cooling
- H10W40/25—Arrangements for cooling characterised by their materials
- H10W40/255—Arrangements for cooling characterised by their materials having a laminate or multilayered structure, e.g. direct bond copper [DBC] ceramic substrates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/38—Boron-containing compounds
- C08K2003/382—Boron-containing compounds and nitrogen
- C08K2003/385—Binary compounds of nitrogen with boron
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10W—GENERIC PACKAGES, INTERCONNECTIONS, CONNECTORS OR OTHER CONSTRUCTIONAL DETAILS OF DEVICES COVERED BY CLASS H10
- H10W40/00—Arrangements for thermal protection or thermal control
- H10W40/20—Arrangements for cooling
- H10W40/25—Arrangements for cooling characterised by their materials
- H10W40/251—Organics
Definitions
- the present invention relates to a resin composition, a resin cured product, and a composite molded body having a cured product part made of a cured product of the resin composition and a metal part.
- the resin composition, the cured resin product, and the composite molded article of the present invention can be suitably used, for example, as a heat dissipation sheet for power semiconductor devices.
- the power semiconductor device is generally used as a power semiconductor module in which a plurality of semiconductor devices are arranged and packaged on a common heat sink.
- a ceramic substrate with high thermal conductivity such as an alumina substrate or an aluminum nitride substrate is used as a heat dissipation substrate for mounting power semiconductor devices.
- the ceramic substrate has drawbacks that it is easily cracked by impact, and it is difficult to form a thin film and it is difficult to reduce the size.
- Patent Document 1 proposes a heat dissipation resin sheet containing a resin having a Tg of 60 ° C. or lower and a boron nitride filler, in which the content of the boron nitride filler is 30 vol% or more and 60 vol% or less. ing.
- the conventional heat radiation resin sheet made of a resin composition containing an inorganic filler has the following problems when applied to a power semiconductor.
- a solder reflow process is one of the processes for assembling a power semiconductor module.
- the temperature of the members is rapidly increased to melt the solder and bond the metal members together.
- Another problem is that the reliability of the power semiconductor module is reduced due to deterioration of the members used in the module in the reflow process, for example, peeling of the interface between the cured product and the metal, and deterioration of the insulation performance.
- the member absorbs moisture during storage, and the moisture absorption of the member greatly accelerates the deterioration of the member in the solder reflow process, which further deteriorates the performance of the obtained power semiconductor module. ing.
- the present invention provides a resin composition and a resin cured product which have high strength, excellent moisture absorption reflow resistance, and reduced the problem of interfacial peeling due to thermal expansion and contraction when formed into a laminate with a metal plate, and the resin composition.
- An object of the present invention is to provide a composite molded article using a product.
- the present inventor provides a resin composition containing an aggregated inorganic filler, wherein the weight increase rate at 85 ° C. and 85% RH after curing of the resin composition is 0.80% or less, and the inorganic filler is excluded.
- a moisture absorption reflow test in which a resin cured product made of a resin composition having a storage elastic modulus at 200 ° C. of 1.0 ⁇ 10 7 Pa or higher at 200 ° C. is subjected to a reflow test after storage under high temperature and high humidity conditions. It was later found that it has a high insulating property (hereinafter, this characteristic may be referred to as "moisture absorption reflow resistance") and can solve the problem of interfacial peeling.
- the present inventor has also found that the use of a resin cured product having a storage elastic modulus and a weight increase rate in a specific range has excellent moisture absorption reflow resistance and can solve the problem of interfacial peeling.
- the present invention has the following gist.
- a resin composition, wherein the cured product of the resin composition has a storage elastic modulus at 200 ° C. of 1.0 ⁇ 10 7 Pa or more.
- At least the epoxy resin having a biphenyl structure and a weight average molecular weight of 10,000 or more is selected from a structure represented by the following structural formula (1) and a structure represented by the following structural formula (2).
- R 1 and R 2 each represent an organic group, and in the formula (2), R 3 represents a divalent cyclic organic group.
- the content ratio of the epoxy resin having a biphenyl structure and a weight average molecular weight of 10,000 or more is 1% by weight or more and 50% by weight with respect to 100% by weight of solid content in the resin composition excluding the inorganic filler.
- the content ratio of the epoxy resin having three or more epoxy groups per molecule is 10% by weight or more and 50% by weight or less based on 100% by weight of the solid content in the resin composition excluding the inorganic filler.
- the resin composition according to any one of [2] to [5].
- the resin composition of the present invention has high strength, excellent moisture absorption reflow resistance, and has almost no problem of interfacial peeling due to thermal expansion and contraction when formed into a laminate with a metal plate.
- the resin cured product of the present invention has high strength, excellent moisture absorption reflow resistance, and has almost no problem of interfacial peeling due to thermal expansion and contraction when formed into a laminate with a metal plate.
- Such a resin composition and a resin cured product of the present invention, and a composite molded article using this resin composition can be suitably used as a heat dissipation sheet for power semiconductor devices, and provide a highly reliable power semiconductor module. Can be realized.
- the resin composition of the present invention contains a resin and an aggregated inorganic filler, and the weight increase rate at 85 ° C. and 85% RH after curing of the resin composition is 0.80% or less, and the resin composition excluding the inorganic filler.
- the cured product has a storage elastic modulus at 200 ° C. of 1.0 ⁇ 10 7 Pa or more.
- the “resin composition” refers to an uncured composition, for example, a composition in a state before being cured in a molding / pressurizing step or the like. More specifically, examples thereof include a resin composition in the form of a slurry to be used in the coating step described below, a sheet that has been subjected to the coating step, and a sheet that has not been cured after the steps such as coating and drying.
- the “cured resin” means a cured product having an exothermic peak of 10 J / g or less when the temperature is raised from 40 ° C. to 250 ° C. at 10 ° C./min with a differential scanning calorimeter (DSC). It refers to the state.
- DSC differential scanning calorimeter
- the resin composition excluding the inorganic filler refers to a component other than the inorganic filler in the resin composition.
- the inorganic filler will be described later, it includes an aggregated inorganic filler and a non-aggregated inorganic filler (non-aggregated inorganic filler).
- the “solid content” in the resin composition refers to all components other than the solvent in the resin composition.
- the resin composition of the present invention may contain “other components” other than the resin and the aggregated inorganic filler, as long as the effects of the present invention are not impaired.
- Other components include non-aggregated inorganic fillers, curing agents, curing catalysts, solvents, surface treatment agents such as silane coupling agents, insulating carbon components such as reducing agents, viscosity modifiers, dispersants, thixotropic agents, Examples include flame retardants, colorants, organic fillers, organic solvents, and the like.
- a dispersant it becomes possible to form a uniform resin cured product, and it may be possible to improve the thermal conductivity and dielectric breakdown properties of the resulting resin cured product. Specific examples of these "other components” that may be contained in the resin composition of the present invention will be described later.
- the storage elastic modulus at 200 ° C. of the resin composition excluding the inorganic filler of the present invention after curing is 1.0 ⁇ 10 7 Pa or more.
- this storage elastic modulus is less than 1 ⁇ 10 7 Pa, the strength of the resin cured product obtained by curing the resin composition of the present invention (hereinafter simply referred to as “resin cured product”) is low, and therefore the moisture absorption.
- the insulating performance is reduced by the generation of voids inside, or in the case of a composite molded article having a cured part made of a cured product of the resin composition of the present invention and a metal part, the metal part The interface between the hardened material and the cured product may peel off.
- the storage elastic modulus is preferably 1.3 ⁇ 10 7 Pa or higher, more preferably 1.5 ⁇ 10 7 Pa or higher, and 1.7 ⁇ 10 7. More preferably, it is 7 Pa or more.
- the storage elastic modulus is preferably 5 ⁇ 10 9 Pa or less, more preferably 1 ⁇ 10 9 Pa or less, and further preferably 5 ⁇ 10 8 Pa or less. If the storage elastic modulus is less than or equal to the above upper limit, it is possible to suppress the internal stress generated by passing through the moisture absorption reflow test is excessive, cracking of the obtained resin cured product, and between the metal part and the resin cured product part. Interface peeling tends to be suppressed.
- the storage elastic modulus when the storage elastic modulus is in the above range, the cured product of the resin composition easily enters the irregularities of the metal that is the adherend described below, and the cured resin product that enters the irregularities exhibits a strong anchor effect. The adhesiveness between the metal and the resin cured product tends to be improved.
- a rigid structure such as an aromatic ring is introduced into the component constituting the resin-containing component, or a reactive group is added. This can be achieved by introducing a plurality of polyfunctional components to increase the crosslink density of the cured product.
- the curing conditions of the resin composition excluding the inorganic filler of the present invention at the time of measuring the storage elastic modulus are as shown in Examples described later, and the temperature is raised from 25 ° C to 120 ° C at 14 ° C per minute, After holding for 30 minutes at 70 ° C., the temperature is raised to 175 ° C. at 7 ° C. per minute, held at this temperature for 30 minutes, then raised to 200 ° C. at 7 ° C. per minute, and held at this temperature for 10 minutes.
- the method of measuring the storage elastic modulus may be any conventionally known method, but specifically, the method described in the section of Examples below is mentioned.
- the weight increase rate at 85 ° C. and 85% RH is 0.8% or less. If the weight increase rate exceeds 0.8%, it is impossible to solve the problems of the present invention of maintaining high insulation property after moisture absorption reflow test and preventing interfacial peeling. From the viewpoint of maintaining high insulation properties after the moisture absorption reflow test and preventing interfacial peeling, the smaller the weight increase rate, the more preferable it is, 0.75% or less is preferable, and 0.7% or less is more preferable.
- the lower limit of the weight increase rate is not particularly limited, but is, for example, 0.2% or more from the viewpoint of compatibility between strength and insulation performance of the cured resin product and film formability.
- the present invention is based on the finding that the resin composition can solve the problems of the present invention when the weight increase rate at 85 ° C. and 85% RH after curing is within the above specific range.
- a resin composition having a weight increase rate at 85 ° C. and 85% RH in a specific range has, for example, a highly hydrophobic structure such as an aliphatic skeleton or an aromatic ring as a component constituting the resin component in the resin composition. Can be obtained by controlling the rate of increase in weight.
- the rate of weight increase at 85 ° C. and 85% RH after curing of the resin composition of the present invention is measured by the method described in the section of Examples below.
- the resin contained in the resin composition of the present invention is not particularly limited as long as it has a specific storage elastic modulus and a specific weight increase rate after curing.
- a specific storage elastic modulus and a specific weight increase rate after curing For example, those that are cured by heat or light in the presence of a curing agent or a curing catalyst can be mentioned.
- a thermosetting resin is preferable from the viewpoint of easy production.
- the resin examples include epoxy resin, phenol resin, polycarbonate resin, unsaturated polyester resin, urethane resin, melamine resin, urea resin, and maleimide resin.
- epoxy resins are preferable from the viewpoint of viscosity, heat resistance, hygroscopicity, and handleability.
- the epoxy resin for example, an epoxy group-containing silicon compound, an aliphatic epoxy resin, a bisphenol A or F type epoxy resin, a novolac type epoxy resin, an alicyclic epoxy resin, a glycidyl ester type epoxy resin, a polyfunctional epoxy resin, Examples include polymer type epoxy resins.
- the resin composition of the present invention preferably contains the resin in an amount of 5% by weight or more, more preferably 30% by weight or more, and 50% by weight based on 100% by weight of the solid content of the resin composition excluding the inorganic filler. It is more preferable that the content is at least%.
- the resin composition of the present invention more preferably contains 99% by weight or less based on 100% by weight of the solid content of the resin composition excluding the inorganic filler.
- Epoxy resin is a general term for compounds having one or more oxirane rings (epoxy groups) in the molecule.
- the oxirane ring (epoxy group) contained in the epoxy resin may be either an alicyclic epoxy group or a glycidyl group.
- the epoxy resin used in the present invention may be an aromatic oxirane ring (epoxy group) -containing compound. Specific examples thereof include glycidylation of bisphenols such as bisphenol A, bisphenol F, bisphenol AD, bisphenol S, tetramethylbisphenol A, tetramethylbisphenol F, tetramethylbisphenol AD, tetramethylbisphenol S, and tetrafluorobisphenol A.
- bisphenols such as bisphenol A, bisphenol F, bisphenol AD, bisphenol S, tetramethylbisphenol A, tetramethylbisphenol F, tetramethylbisphenol AD, tetramethylbisphenol S, and tetrafluorobisphenol A.
- Bisphenol-type epoxy resin biphenyl-type epoxy resin, dihydroxynaphthalene, epoxy resin obtained by glycidylating divalent phenols such as 9,9-bis (4-hydroxyphenyl) fluorene, 1,1,1-tris (4- Glycidylated epoxy resins such as hydroxyphenyl) methane and tetrakisphenols such as 1,1,2,2-tetrakis (4-hydroxyphenyl) ethane Rishijiru of epoxy resin, phenol novolac, cresol novolac, bisphenol A, novolak, such as novolaks the glycidylated novolak type epoxy resins such as brominated bisphenol A novolak and the like.
- the resin composition of the present invention preferably contains 20% by weight or more, more preferably 45% by weight or more of an epoxy resin in 100% by weight of the resin contained in the resin composition of the present invention.
- an epoxy resin in 100% by weight of the resin contained in the resin composition of the present invention.
- 100% by weight of the epoxy resin may be contained in 100% by weight of all the resin components.
- the resin composition of the present invention preferably contains an epoxy resin having three or more oxirane rings (epoxy groups) in one molecule (hereinafter, sometimes referred to as “multifunctional epoxy resin”).
- multifunctional epoxy resin an epoxy resin having three or more oxirane rings (epoxy groups) in one molecule
- the resin composition of the present invention contains a polyfunctional epoxy resin, it becomes possible to introduce a highly polar oxirane ring (epoxy group) at a high density. Thereby, the effect of physical interaction such as Van der Waals force or hydrogen bond is increased, and the adhesion between the metal and the resin cured product in the composite molded body tends to be improved.
- the inclusion of the polyfunctional epoxy resin tends to make the storage elastic modulus of the resin cured product within the specific range described above, and tends to improve the adhesion between the metal and the resin cured product. Furthermore, by improving the reactivity of the oxirane ring (epoxy group), the amount of hydroxyl groups in the course of the curing reaction tends to be reduced, and the increase in hygroscopicity tends to be suppressed.
- the polyfunctional epoxy resin may be used alone or in combination of two or more.
- the polyfunctional epoxy resin is an epoxy resin having 3 or more oxirane rings (epoxy groups) in one molecule, and preferably an epoxy resin having 4 or more oxirane rings (epoxy groups) in one molecule.
- There is no particular upper limit on the number of oxirane rings (epoxy groups) in one molecule of the polyfunctional epoxy resin but 10 or less is preferable, 8 or less is more preferable, and 6 or less is particularly preferable.
- the number of oxirane rings (epoxy groups) in one molecule is within this range, the adhesion between the metal and the resin cured product is improved, and the increase in hygroscopicity tends to be suppressed.
- the oxirane ring (epoxy group) is more preferably a glycidyl group from the viewpoint of reaction rate and heat resistance.
- the resin composition of the present invention contains a polyfunctional epoxy resin having a plurality of oxirane rings (epoxy groups), particularly a glycidyl group, in one molecule, whereby the crosslink density of the cured product is improved, and the obtained resin cured product is It tends to have higher strength.
- a polyfunctional epoxy resin having a plurality of oxirane rings (epoxy groups), particularly a glycidyl group, in one molecule, whereby the crosslink density of the cured product is improved, and the obtained resin cured product is It tends to have higher strength.
- the resin cured product does not deform or break, and by maintaining its shape, voids such as voids in the resin cured product. Tends to be suppressed.
- the molecular weight of the polyfunctional epoxy resin is not particularly limited, it is preferably 1,000 or less, more preferably 800 or less, and further preferably 600 or less.
- the molecular weight of the polyfunctional epoxy resin is preferably 100 or more, more preferably 150 or more.
- the molecular weight of the polyfunctional epoxy resin is in the above range, it tends to be easy to set the storage elastic modulus at 200 ° C. of the resin cured product after curing the resin composition to 1.0 ⁇ 10 7 Pa or more. .
- polyfunctional epoxy resin specifically, EX321L, DLC301, DLC402 and the like manufactured by Nagase Chemtex can be used.
- the content of the polyfunctional epoxy resin in the resin composition of the present invention is not particularly limited, but it is preferably 5% by weight or more per 100% by weight of the solid content of the resin composition excluding the inorganic filler, and 10% by weight or more. It is more preferable to contain.
- the content is preferably 50% by weight or less, more preferably 40% by weight or less, further preferably 30% by weight or less. Further, it is preferable that the content is 5 wt% or more and 50 wt% or less, further 10 wt% or more and 40 wt% or less, and particularly 10 wt% or more and 30 wt% or less.
- the content of the polyfunctional epoxy resin is equal to or more than the above lower limit value, the above-mentioned effects due to the polyfunctional epoxy resin being contained can be effectively obtained.
- the content of the polyfunctional epoxy resin is not more than the above upper limit value, the hygroscopicity of the resin cured product is suppressed, and the strength performance of the resin cured product is excellent, and these performances can be compatible. It will be possible.
- the resin in the resin composition of the present invention preferably contains an epoxy resin having a biphenyl structure and a weight average molecular weight of 10,000 or more (hereinafter sometimes referred to as “specific epoxy resin”).
- the “organic group” may be any group as long as it contains a carbon atom.
- examples of the organic group include an alkyl group, an alkenyl group, and an aryl group, which may be substituted with a halogen atom, a group having a hetero atom, or another hydrocarbon group.
- the specific epoxy resin having a biphenyl structure and a weight average molecular weight of 10,000 or more is preferably a structure represented by the following structural formula (1) (hereinafter sometimes referred to as “structure (1)”) and the following. It is preferable to further have at least one structure selected from the structures represented by Structural Formula (2) (hereinafter sometimes referred to as “structure (2)”).
- R 1 and R 2 each represent an organic group, and in the formula (2), R 3 represents a divalent cyclic organic group.
- structure (3) an epoxy resin having a structure represented by the following structural formula (3) (hereinafter, may be referred to as “structure (3)”) may be mentioned.
- R 4 , R 5 , R 6 , and R 7 each independently represent an organic group having a molecular weight of 15 or more.
- R 1 and R 2 is preferably an organic group having a molecular weight of 16 or more, particularly 16 to 1000.
- examples thereof include alkyl groups such as ethyl group, propyl group, butyl group, pentyl group, hexyl group and heptyl group, and aryl groups such as phenyl group, tolyl group, xylyl group, naphthyl group and fluorenyl group.
- Both R 1 and R 2 may be an organic group having a molecular weight of 16 or more, one may be an organic group having a molecular weight of 16 or more, and the other may be an organic group having a molecular weight of 15 or less or a hydrogen atom.
- one of R 1 and R 2 is an organic group having a molecular weight of 16 or more and the other is an organic group having a molecular weight of 15 or less, and particularly, one of them is a methyl group and the other is a phenyl group. It is preferable from the viewpoint of facilitating control of handling properties such as, and the strength of the cured product.
- R 3 is a divalent cyclic organic group, and may be an aromatic ring structure such as a benzene ring structure, a naphthalene ring structure or a fluorene ring structure, or cyclobutane, cyclopentane or cyclohexane. It may have an aliphatic ring structure. In addition, they may independently have a substituent such as a hydrocarbon group or a halogen atom.
- the divalent bond of R 3 may be a divalent group at a single carbon atom or a divalent group at a different carbon atom.
- Preferred are divalent aromatic groups having 6 to 100 carbon atoms and divalent groups derived from cycloalkane having 2 to 100 carbon atoms such as cyclopropane and cyclohexane.
- R 3 is a 3,3,5-trimethyl-1,1-cyclohexylene group represented by the following structural formula (4) from the viewpoint of control of handleability such as resin viscosity and strength of the cured product. preferable.
- R 4 , R 5 , R 6 , and R 7 are each independently an organic group having a molecular weight of 15 or more.
- An alkyl group having a molecular weight of 15 to 1000 is preferable, and in particular, all of R 4 , R 5 , R 6 and R 7 are methyl groups from the viewpoint of controllability of handleability such as resin viscosity and strength of the cured product. preferable.
- the specific epoxy resin is preferably an epoxy resin containing one of the structures (1) and (2) and a biphenyl structure, and particularly one of the structures (1) and (2) and the structure ( It is more preferable that the epoxy resin contains 3).
- the specific epoxy resin contains these structures, there is a tendency that the hygroscopicity of the cured product is suppressed and the strength retention performance of the resin composition is compatible.
- Such a specific epoxy resin contains a large amount of hydrophobic hydrocarbon and aromatic structures as compared with a general epoxy resin having a bisphenol A skeleton or a bisphenol F skeleton. Therefore, by incorporating the specific epoxy resin, it is possible to reduce the moisture absorption of the cured product of the resin composition. From the viewpoint of reducing the amount of moisture absorption, the specific epoxy resin preferably contains a large amount of the structures (1), (2) and (3) which are hydrophobic structures.
- the weight average molecular weight of the specific epoxy resin is preferably 10,000 or more, more preferably 20,000 or more, and further preferably 25,000 or more. Further, it is preferably 80,000 or less, more preferably 70,000 or less, and further preferably 60,000 or less.
- the specific epoxy resin is preferably more hydrophobic, and specifically, the epoxy equivalent of the specific epoxy resin is preferably large, preferably 3,000 g / equivalent or more, more preferably 4,000 g / equivalent or more, 000 g / equivalent or more is more preferable.
- the epoxy equivalent of the specific epoxy resin is preferably 20,000 g / equivalent or less, more preferably 5,000 g / equivalent or more and 20,000 g / equivalent or less.
- the weight average molecular weight of the epoxy resin is a polystyrene-equivalent value measured by gel permeation chromatography.
- the epoxy equivalent is defined as "weight of epoxy resin containing one equivalent of epoxy group" and can be measured according to JIS K7236.
- Such specific epoxy resins may be used alone or in combination of two or more.
- the specific epoxy resin may have a plurality of epoxy groups.
- the content of the specific epoxy resin in the resin composition of the present invention is not particularly limited, but is preferably 5% by weight or more and more preferably 10% by weight or more based on 100% by weight of the solid content of the resin composition excluding the inorganic filler. Further, it is preferably 50% by weight or less, more preferably 40% by weight or less.
- the content of the specific epoxy resin is not more than the above upper limit, the storage elastic modulus of the cured product is improved or maintained, and the reflow resistance tends to be improved.
- the content of the specific epoxy resin is not less than the above lower limit, application of the resin composition tends to be easy, and flexibility of the obtained resin cured product tends to be obtained.
- the resin composition of the present invention preferably contains both the specific epoxy resin and the polyfunctional epoxy resin as the epoxy resin from the viewpoint of achieving both high elasticity and low hygroscopicity of the cured product of the resin composition.
- the content ratio of the specific epoxy resin and the polyfunctional epoxy resin is within this range, it becomes easy to control the storage elastic modulus and the weight increase rate within appropriate ranges.
- the resin composition of the present invention may contain an epoxy resin other than the specific epoxy resin and the polyfunctional epoxy resin.
- the epoxy resin other than the specific epoxy resin and the polyfunctional epoxy resin contained in the resin composition of the present invention is not particularly limited, but for example, bisphenols such as bisphenol A type epoxy resin and bisphenol F type epoxy resin are glycidylated. Glycidyl compounds containing aromatic compounds having two hydroxyl groups, such as various bisphenol type epoxy resins, various biphenyl type epoxy resins obtained by glycidylating biphenyls, dihydroxynaphthalene, and 9,9-bis (4-hydroxyphenyl) fluorene.
- Epoxy resin epoxy resin obtained by glycidylating trisphenols such as 1,1,1-tris (4-hydroxyphenyl) methane, tetrakis such as 1,1,2,2-tetrakis (4-hydroxyphenyl) ethane Group phenols
- glycidylated novolak-type epoxy resins such as sidylated epoxy resins, phenol novolacs, cresol novolacs, bisphenol A novolacs, brominated bisphenol A novolacs, and silicone-containing epoxy resins are preferable. .
- the inorganic filler includes an aggregated inorganic filler and a non-aggregated inorganic filler.
- the resin composition of the present invention contains an aggregated inorganic filler.
- the resin composition of the present invention may contain a non-aggregated inorganic filler in addition to the agglomerated inorganic filler.
- Non-aggregated inorganic fillers also include spherical fillers described below.
- the aggregated inorganic filler By containing the aggregated inorganic filler in the resin composition of the present invention, it becomes possible to improve the thermal conductivity and insulation of the resin cured product and control the linear expansion coefficient.
- the aggregated inorganic fillers are deformed by coming into contact with each other, and are brought into contact with each other at the surface, so that more heat conduction paths are formed and a cured resin product with high thermal conductivity can be obtained.
- the aggregated inorganic filler is deformed, voids or voids between the fillers can be effectively removed, and the insulating property is improved.
- the resin composition of the present invention contains a resin and an inorganic filler, and in particular, by using a combination of the epoxy resin and the agglomerated inorganic filler suitable for the aforesaid present invention, even after the agglomerated inorganic filler is deformed in the pressure step described below.
- the deformed state of the filler can be maintained. Furthermore, since the weight increase rate after curing of the resin composition of the present invention is within the above-mentioned specific range, the deformed state of the filler can be maintained even after the moisture absorption reflow step.
- the fillers are in point contact with each other even after the pressurizing step, and a heat conduction path is effectively formed. I can't. Further, in some cases, voids or voids in the gaps between the fillers cannot be removed, and the insulating property is deteriorated.
- Agglomerated morphology of inorganic filler can be confirmed by scanning electron microscope (SEM).
- an electrically insulating one can be used, and examples thereof include those composed of at least one kind of inorganic particles selected from the group consisting of metal carbides, metal oxides and metal nitrides.
- metal carbide examples include silicon carbide, titanium carbide, tungsten carbide and the like.
- metal oxides include magnesium oxide, aluminum oxide, silicon oxide, calcium oxide, zinc oxide, yttrium oxide, zirconium oxide, cerium oxide, ytterbium oxide, sialon (ceramics consisting of silicon, aluminum, oxygen, and nitrogen).
- metal nitrides include boron nitride, aluminum nitride, silicon nitride and the like.
- the agglomerated inorganic filler when used for applications requiring insulation such as power semiconductors, is excellent in insulation with a volume resistivity of 1 ⁇ 10 13 ⁇ ⁇ cm or more, particularly 1 ⁇ 10 14 ⁇ ⁇ cm or more. It is preferably composed of an inorganic compound.
- the inorganic particles constituting the aggregated inorganic filler are preferably made of a metal oxide and / or a metal nitride, because the cured resin has sufficient electric insulation.
- alumina Al 2 O 3, a volume resistivity of 1 ⁇ 10 14 ⁇ ⁇ cm
- aluminum nitride AlN, a volume resistivity of 1 ⁇ 10 14 ⁇ ⁇ cm
- boron nitride BN, volume resistivity 1 ⁇ 10 14 ⁇ ⁇ cm
- silicon nitride Si 3 N 4 , volume resistivity 1 ⁇ 10 14 ⁇ ⁇ cm
- silica SiO 2 , volume resistivity 1 ⁇ ).
- 10 14 ⁇ ⁇ cm) and the like, and among them, alumina, aluminum nitride, boron nitride and silica are preferable, and alumina and boron nitride are particularly preferable.
- the aggregated inorganic filler may be surface-treated with a surface treatment agent.
- a surface treatment agent a known surface treatment agent can be used.
- the aggregating inorganic filler may be used alone or as a mixture of two or more kinds in any combination and ratio.
- the aggregating inorganic filler it is preferable to use the following boron nitride aggregating particles from the viewpoint of effectively exhibiting the above-mentioned effects by using the aggregating inorganic filler.
- the boron nitride agglomerated particles may be used in combination with inorganic fillers having different shapes and types.
- Boron nitride agglomerated particles Boron nitride has high thermal conductivity, but since it is scaly, it has excellent thermal conductivity in the plane direction, but has large thermal resistance in the direction perpendicular to the plane. Aggregated particles obtained by collecting such scaly particles and aggregating them into a spherical shape are preferable because they are excellent in handleability.
- the radial direction of the agglomerated particles is the direction in which the thermal resistance is large.
- the boron nitride agglomerated particles those obtained by aligning the particles of boron nitride in the plane direction so that the radial direction of the agglomerated particles is in the direction of good heat conduction are preferable.
- the boron nitride agglomerated particles also preferably have a card house structure.
- the “card house structure” is, for example, Ceramics 43 No. 2 (published by the Ceramic Society of Japan in 2008), it has a structure in which plate-like particles are not oriented and are laminated in a complicated manner. More specifically, the boron nitride agglomerated particles having a card house structure are aggregates of boron nitride primary particles, and a flat portion and an end face portion of the primary particles are in contact with each other to form, for example, a T-shaped aggregate. It is a boron nitride agglomerated particle having a structure.
- the boron nitride agglomerated particles used in the present invention are particularly preferable.
- the thermal conductivity can be further increased.
- the new Mohs hardness of the boron nitride agglomerated particles is not particularly limited, but 5 or less is preferable. There is no particular lower limit to the new Mohs hardness of the boron nitride agglomerated particles, but it is, for example, 1 or more.
- the particles dispersed in the resin composition are likely to come into surface contact with each other, a heat conduction path between the particles is formed, and the heat conduction of the resin cured product tends to be improved. is there.
- the volume average particle diameter of the boron nitride agglomerated particles is not particularly limited, but is preferably 10 ⁇ m or more, more preferably 15 ⁇ m or more.
- the volume average particle diameter of the boron nitride agglomerated particles is preferably 100 ⁇ m or less, more preferably 90 ⁇ m or less.
- the volume average particle diameter is not less than the above lower limit, the thermal resistance becomes small and the high thermal conductivity tends to be obtained because the interparticle interface is suppressed in the resin composition and the resin cured product.
- the volume average particle diameter is not more than the above upper limit value, the surface smoothness of the resin cured product tends to be obtained.
- the volume average particle diameter of the boron nitride agglomerated particles means the particle diameter when the cumulative volume becomes 50% when a cumulative curve is drawn with the volume of the powder used for measurement as 100%.
- the volume average particle diameter is measured by using a laser diffraction / scattering particle size distribution measuring device or the like for a sample in which agglomerated particles are dispersed in a pure water medium containing sodium hexametaphosphate as a dispersion stabilizer. Examples include a wet measurement method and a dry measurement method in which measurement is performed using "Morphologi" manufactured by Malvern. The same applies to the volume average particle diameter of the spherical inorganic filler described below.
- the content of the aggregated inorganic filler in the resin composition of the present invention is preferably 30% by weight or more, more preferably 40% by weight or more, and 45% by weight in 100% by weight of the solid content of the resin composition. It is more preferable that the above is satisfied.
- the content of the aggregated inorganic filler in the resin composition of the present invention is preferably 99% by weight or less, more preferably 90% by weight or less, and more preferably 80% by weight based on 100% by weight of the solid content of the resin composition. % Or less is more preferable.
- the content of the aggregated inorganic filler is at least the above lower limit, the effect of improving the thermal conductivity and the effect of controlling the linear expansion coefficient by containing the aggregated inorganic filler tend to be sufficiently obtained.
- the content of the aggregated inorganic filler is not more than the above upper limit, the moldability of the resin composition and the resin cured product and the interfacial adhesiveness in the composite molded article tend to be improved.
- the resin composition of the present invention may contain a non-aggregated inorganic filler together with an aggregated inorganic filler as the inorganic filler.
- the non-aggregated inorganic filler preferably has a thermal conductivity of 10 W / m ⁇ K or more, preferably 15 W / m ⁇ K or more, more preferably 20 W / m ⁇ K or more, for example 20 to 30 W / m ⁇ K,
- a spherical inorganic filler having a new Mohs hardness of 3.1 or more, for example, 5 to 10 can be used.
- spherical as used herein may be any that is generally recognized as being spherical, and for example, an average circularity of 0.4 or more may be spherical, and 0.6 or more may be spherical. Usually, the upper limit of the average circularity is 1.
- the circularity can be measured by subjecting the projected image to image processing, for example, FPIA series manufactured by Sysmex Corporation.
- the spherical inorganic filler is preferably at least one selected from the group consisting of alumina, synthetic magnesite, silica, aluminum nitride, silicon nitride, silicon carbide, zinc oxide and magnesium oxide.
- the volume average particle diameter of the spherical inorganic filler is preferably in the range of 0.5 ⁇ m or more and 40 ⁇ m or less. It is considered that when the volume average particle diameter is 0.5 ⁇ m or more, the resin and the inorganic filler can easily flow during heat molding, and the interfacial adhesive force in the composite molded article of the present invention can be increased. Further, when the average particle size is 40 ⁇ m or less, it becomes easy to maintain the dielectric breakdown characteristics of the resin cured product.
- the content ratio of the aggregated inorganic filler and the non-aggregated inorganic filler in the resin composition is not particularly limited, but the weight ratio is 90:10 to 10 : 90 is preferable, and 80: 20 to 20: 80 is more preferable.
- the total content of the aggregated inorganic filler and the non-aggregated inorganic filler in the resin composition of the present invention is preferably 30% by weight or more, and 40% by weight or more in 100% by weight of the solid content of the resin composition. Is more preferable and 50% by weight or more is further preferable.
- the total content of the aggregating inorganic filler and the non-aggregating inorganic filler in the resin composition of the present invention is preferably 99% by weight or less, and 90% by weight or less in 100% by weight of the solid content of the resin composition. It is more preferable that the amount is 80% by weight or less.
- the resin composition of the present invention may contain other components other than the above components as long as the effects of the present invention are not impaired.
- Other components include a compound having a heterocyclic structure containing a nitrogen atom, a curing agent, a curing catalyst, a surface treatment agent such as a silane coupling agent for improving the interfacial adhesion strength between the inorganic filler and the resin, a reducing agent, etc.
- Insulating carbon components, viscosity modifiers, dispersants, thixotropic agents, flame retardants, colorants, organic fillers, organic solvents, thermoplastic resins and the like can be mentioned.
- the presence or absence and the content ratio of other components in the resin composition of the present invention are not particularly limited as long as the effects of the present invention are not significantly impaired.
- the resin composition of the present invention may contain a compound having a heterocyclic structure containing a nitrogen atom.
- nitrogen-containing heterocyclic compound By containing a compound having a heterocyclic structure containing a nitrogen atom (hereinafter, may be referred to as “nitrogen-containing heterocyclic compound”), a resin cured product obtained from the resin composition of the present invention and a metal There is a tendency to exert the effect of improving the adhesion. That is, the nitrogen-containing heterocyclic compound improves the adhesion between the resin composition or the resin cured product and the metal by being located at the interface between the resin composition or the resin cured product when the compound is combined with the metal. . From this viewpoint, the nitrogen-containing heterocyclic compound preferably has a low molecular weight so that the nitrogen-containing heterocyclic compound can easily stay at the interface between the resin composition or the cured resin and the metal.
- the molecular weight of the nitrogen-containing heterocyclic compound is preferably 1,000 or less, more preferably 500 or less.
- the lower limit of the molecular weight of the nitrogen-containing heterocyclic compound is not particularly limited, but is preferably 60 or more, more preferably 70 or more.
- heterocyclic structure of the nitrogen-containing heterocyclic compound examples include structures derived from imidazole, triazine, triazole, pyrimidine, pyrazine, pyridine and azole.
- the nitrogen-containing heterocyclic compound may simultaneously have a plurality of iodine ring structures in one molecule. From the viewpoint of improving the insulating property of the resin composition and the adhesion to a metal, an imidazole compound or a triazine compound is preferable as the nitrogen-containing heterocyclic compound.
- Preferred imidazole compounds and triazine compounds as the nitrogen-containing heterocyclic compound include, for example, 2-ethyl-4-methylimidazole, 2-phenylimidazole, 1-benzyl-2-methylimidazole, 1-benzyl-2-phenylimidazole, 1-cyanoethyl-2-undecylimidazole, 1-cyanoethyl-2-ethyl-4-methylimidazole, 2-phenyl-4-methylimidazole, 1-cyanoethyl-2-phenylimidazolium trimellitate, 2,4-diamino -6- [2'-methylimidazolyl- (1 ')]-ethyl-s-triazine, 2,4-diamino-6- [2'-undecylimidazolyl- (1')]-ethyl-s-triazine, 2,4-diamino-6- [2'-ethyl-4'methylimidazo
- a structure derived from imidazole and a structure derived from triazine are particularly preferable, and a structure derived from triazine is particularly preferable, and the heterocyclic structure of the nitrogen-containing heterocyclic compound is 1,3,5- Structures derived from triazine are particularly preferred. Further, it may be a structure having a plurality of these exemplified structural portions.
- the nitrogen-containing heterocyclic compound tends to have high resin compatibility and a high reaction activation temperature. Therefore, the curing rate and the physical properties after curing can be easily adjusted, which tends to improve the storage stability of the resin composition and further improve the adhesive strength after heat molding.
- the nitrogen-containing heterocyclic compound may contain a curing catalyst described later depending on the structure, and therefore the resin composition of the present invention can contain the nitrogen-containing heterocyclic compound as a curing catalyst.
- the nitrogen-containing heterocyclic compound may be used alone or in combination of two or more.
- the content of the nitrogen-containing heterocyclic compound is preferably 0.001% by weight or more, more preferably 0.1% by weight or more, based on 100% by weight of the solid content of the resin composition excluding the inorganic filler, and 0.5 It is more preferably at least wt%.
- the content of the nitrogen-containing heterocyclic compound is preferably 10% by weight or less, more preferably 7% by weight or less, and even more preferably 5% by weight or less based on 100% by weight of the solid content of the resin composition excluding the inorganic filler. More preferable.
- the content of the nitrogen-containing heterocyclic compound is within the above range, it tends to be easy to control the storage elastic modulus and the weight increase rate within the above-mentioned specific ranges.
- the total amount including the contents thereof be included in the above range.
- the content of the nitrogen-containing heterocyclic compound is at least the above lower limit value, the above-mentioned effects by including this compound can be sufficiently obtained, and when it is at most the above upper limit value, the reaction effectively proceeds, and the crosslinking density Can be improved, the strength can be increased, and the storage stability can be further improved.
- the resin composition of the present invention may contain a curing agent.
- a preferable curing agent is a phenol resin, an acid anhydride having an aromatic skeleton or an alicyclic skeleton, a water addition product of the acid anhydride, or a modified product of the acid anhydride.
- a resin cured product having an excellent balance of heat resistance, moisture resistance and electrical properties.
- the curing agent may be used alone or in combination of two or more.
- the phenol resin is not particularly limited. Specific examples of the phenol resin include phenol novolac, o-cresol novolac, p-cresol novolac, t-butylphenol novolac, dicyclopentadiene cresol, polyparavinylphenol, bisphenol A type novolac, xylylene-modified novolac, decalin-modified novolac and poly. Examples thereof include (di-o-hydroxyphenyl) methane, poly (di-m-hydroxyphenyl) methane, and poly (di-p-hydroxyphenyl) methane.
- a novolac type phenol resin having a rigid main chain skeleton and a phenol resin having a triazine skeleton are used.
- a phenol resin having an allyl group is preferable for improving the flexibility of the uncured resin composition and the toughness of the cured resin.
- phenol resins include MEH-8005, MEH-8000H and NEH-8015 (all manufactured by Meiwa Kasei Co., Ltd.), YLH903 (manufactured by Mitsubishi Chemical Corporation), LA-7052, LA-7054, LA-7751, LA. -1356 and LA-3018-50P (all manufactured by Dainippon Ink and Chemicals, Inc.), PSM6200, PS6313 and PS6492 (manufactured by Gunei Chemical Industry Co., Ltd.) and the like can be mentioned.
- the acid anhydride having an aromatic skeleton, the water addition product of the acid anhydride, or the modified product of the acid anhydride is not particularly limited. Specific examples include SMA resin EF30 and SMA resin EF60 (all manufactured by Sartomer Japan Co., Ltd.), ODPA-M and PEPA (all manufactured by Manac Co., Ltd.), Ricagit MTA-10, Jamaicagit TMTA, Jamaicagit TMEG-.
- RIKAJIT TMEG-500 RIKAJIT TMEG-S
- RIKAJIT TH RIKAJIT MH-700
- RIKAJIT MT-500 RIKAJIT DSDA and RIKAJITT TDA-100
- EPICRON B4400 EPICRON B4400
- EPICLON B570 All of the above are manufactured by Dainippon Ink and Chemicals, Inc. and the like.
- An acid anhydride having an alicyclic skeleton, a water additive of the acid anhydride or a modified product of the acid anhydride is an acid anhydride having a polyalicyclic skeleton, a water additive of the acid anhydride or the acid.
- a modified product of an anhydride, or an acid anhydride having an alicyclic skeleton obtained by an addition reaction of a terpene compound and maleic anhydride, a water addition product of the acid anhydride, or a modified product of the acid anhydride. Is preferred.
- the resin composition of the present invention contains a curing agent is not particularly limited. Moreover, when the resin composition of the present invention contains a curing agent, the content of the curing agent is not particularly limited.
- the solid content of 100% by weight of the resin composition excluding the inorganic filler is preferably 0.5 to 70% by weight, more preferably 0.5 to 55% by weight.
- the content of the curing agent is at least the above lower limit, sufficient curing performance can be obtained, and when it is at most the above upper limit, the reaction effectively proceeds, the crosslink density is improved, and the strength can be increased. It is possible to further improve the film forming property.
- the amount is preferably 0 to 55% by weight based on the epoxy equivalent in the resin composition. Within the above range, the reaction effectively proceeds, the crosslink density can be improved, the strength can be increased, and the film-forming property tends to be further improved.
- the resin composition of the present invention may contain a curing catalyst.
- a curing catalyst In order to adjust the curing rate and the physical properties of the cured product, it is preferable to include a curing catalyst together with the curing agent.
- the curing catalyst is not particularly limited, but is appropriately selected depending on the type of resin and curing agent used.
- Specific examples of the curing catalyst include linear or cyclic tertiary amines, organic phosphorus compounds, quaternary phosphonium salts or diazabicycloalkenes such as organic acid salts.
- organometallic compounds, quaternary ammonium salts, metal halides and the like can also be used.
- the organometallic compounds include zinc octylate, tin octylate, and aluminum acetylacetone complex. These may be used alone or in combination of two or more.
- the curing catalyst is contained in an amount of 0.1 to 10% by weight, particularly 0.1 to 5% by weight based on 100% by weight of the solid content of the resin composition excluding the inorganic filler.
- the content of the curing catalyst is at least the above lower limit value, the progress of the curing reaction can be sufficiently promoted and good curing can be achieved.
- the content of the curing catalyst is at most the above upper limit value, the curing rate will not be too fast, and therefore the storage stability of the resin composition of the present invention can be made good.
- the resin composition of the present invention may contain an organic solvent, for example, in order to improve coatability when a sheet-shaped resin cured product is molded through a coating step.
- organic solvent examples include methyl ethyl ketone, cyclohexanone, propylene glycol monomethyl ether acetate, butyl acetate, isobutyl acetate, propylene glycol monomethyl ether and the like. These organic solvents may be used alone or in combination of two or more.
- the content thereof is appropriately determined according to the handleability of the resin composition at the time of producing a resin cured product, the shape before curing, the drying conditions and the like.
- the organic solvent is used so that the solid content concentration of the resin composition of the present invention is 10 to 90% by weight, particularly 40 to 80% by weight. It is preferable to use.
- the solid content concentration of the resin composition of the present invention is preferably 95% by weight or more, and particularly preferably 98% by weight or more. Is more preferable.
- the resin composition of the present invention may contain a dispersant.
- a dispersant it becomes possible to form a uniform resin cured product, and it is possible to improve the thermal conductivity and dielectric breakdown properties of the resulting resin cured product. There is.
- the dispersant preferably has a functional group containing a hydrogen atom having hydrogen bonding properties.
- the pKa of the functional group containing a hydrogen atom having hydrogen bonding property is preferably in the range of 2 to 10, and more preferably in the range of 3 to 9.
- the pKa is 2 or more, the acidity of the dispersant is in an appropriate range, and the reaction of the epoxy resin in the resin component may be easily suppressed. Therefore, when the uncured molded product is stored, the storage stability tends to be improved.
- the pKa is 10 or less, the function as a dispersant is sufficiently fulfilled, and the cured resin product tends to have sufficiently improved thermal conductivity and dielectric breakdown properties.
- the functional group containing a hydrogen atom having hydrogen bonding property is preferably a carboxyl group or a phosphoric acid group. In this case, the thermal conductivity and dielectric breakdown characteristics of the resin cured product can be further enhanced.
- polyester carboxylic acid, polyether carboxylic acid, polyacrylic carboxylic acid, aliphatic carboxylic acid, polysiloxane carboxylic acid, polyester phosphoric acid, polyether phosphoric acid examples thereof include polyacrylic phosphoric acid, aliphatic phosphoric acid, polysiloxane phosphoric acid, polyester phenol, polyether phenol, polyacrylic phenol, and polysiloxane phenol.
- polyester carboxylic acid, polyether carboxylic acid, polyacrylic carboxylic acid, aliphatic carboxylic acid, polysiloxane carboxylic acid, polyester phosphoric acid, polyether phosphoric acid examples thereof include polyacrylic phosphoric acid, aliphatic phosphoric acid, polysiloxane phosphoric acid, polyester phenol, polyether phenol, polyacrylic phenol, and polysiloxane phenol.
- dispersant only one kind may be used, or two or more kinds may be used in combination.
- the resin composition of the present invention may contain an organic filler and / or a thermoplastic resin.
- the resin composition of the present invention contains an organic filler or a thermoplastic resin, appropriate elongation is imparted to the resin composition, the generated stress is relaxed, and the occurrence of cracks in the temperature cycle test can be suppressed. Sometimes you can.
- thermoplastic resin any generally known thermoplastic resin can be used.
- thermoplastic resin include polyethylene, polypropylene, polystyrene, polyvinyl chloride, (meth) acrylic resin, ethylene-vinyl acetate copolymer, ethylene-vinyl alcohol copolymer and other vinyl-based polymers, polylactic acid resin, polyethylene terephthalate.
- Polyester such as polybutylene terephthalate, nylon, polyamide such as polyamidoamine, polyvinyl acetoacetal, polyvinyl benzal, polyvinyl acetal resin such as polyvinyl butyral resin, ionomer resin, polyphenylene ether, polyphenylene sulfide, polycarbonate, polyether ether ketone, polyacetal , ABS resin, LCP (liquid crystal polymer), fluororesin, urethane resin, silicone resin, various elastomers, It can be mentioned modified products of these resins.
- the thermoplastic resin may be uniform in the resin phase of the cured resin, or may be phase-separated and its shape recognized. In the case of phase separation, the shape of the thermoplastic resin in the cured resin may be particulate or fibrous.
- the thermoplastic resin may be recognized as an organic filler, but in the present invention, the organic filler is a natural product such as wood powder or a modified product. It refers to cellulose, starch, various organic pigments and the like that may be incorporated, and the thermoplastic resin is not included in the organic filler.
- thermoplastic resin When the thermoplastic resin or the organic filler is insoluble in the above-mentioned resin, it is possible to prevent the viscosity of the resin composition from increasing, and improve the smoothness of the sheet surface, for example, when forming it into a sheet shape as described later. it can.
- a thermoplastic resin insoluble in the above-mentioned resin, an organic filler by mixing with a large amount of the inorganic filler at the same time, it is possible to efficiently disperse the component phase that is thermoplastic and well-extended in the resin cured product, Easy to relieve stress. Therefore, it is possible to suppress the occurrence of cracks in the cured resin without lowering the elastic modulus of the cured resin.
- the thermoplastic resin is preferably a polyamide resin such as nylon or a cellulose resin, and particularly preferably a polyamide resin such as nylon.
- the upper limit of the average particle diameter is preferably 100 ⁇ m or less, more preferably 50 ⁇ m or less, and further preferably 30 ⁇ m or less. is there.
- the average particle diameter of the particulate thermoplastic resin is determined by observing the cross section of the cured resin product and averaging the longest diameter of any 20 particles.
- the resin cured product of the present invention is a resin cured product using a resin composition containing an inorganic aggregate filler, and the weight increase rate at 85 ° C. and 85% RH of the resin cured product is 0.80% or less.
- the storage elastic modulus at 200 ° C. after curing of the resin composition excluding the inorganic filler is 1.0 ⁇ 10 7 Pa or more.
- the “resin composition” refers to an uncured resin composition containing an aggregated inorganic filler and a resin. Although not particularly limited, it is preferably a mixture of a thermosetting resin component such as an aggregated inorganic filler and an epoxy resin, and a curing agent that cures it, a curing catalyst that serves as a curing aid, and a complex containing a nitrogen atom. It is more preferable to contain a compound having a ring structure.
- the resin composition described above may be mentioned.
- the resin composition excluding the inorganic filler refers to a component other than the inorganic filler in the resin composition.
- the resin, the curing agent, the curing catalyst, the compound having a nitrogen atom-containing heterocyclic structure, the inorganic filler, the inorganic aggregate filler, and the other components have the same meanings as the resin composition described above, and the preferable ranges and the like are also the same.
- the “resin cured product” of the present invention refers to a product obtained by curing a resin composition. Further, the resin cured product of the present invention is in a state where the exothermic peak obtained when the temperature is raised from 40 ° C. to 250 ° C. at 10 ° C./min by DSC is 10 J / g or less.
- the method for obtaining the cured resin product of the present invention is not particularly limited. For example, a method obtained by curing the resin composition described above can be mentioned.
- the storage elastic modulus at 200 ° C. of the resin composition excluding the inorganic filler after curing is 1.0 ⁇ 10 7 Pa or more.
- the storage elastic modulus is preferably 1.3 ⁇ 10 7 Pa or higher, more preferably 1.5 ⁇ 10 7 Pa or higher, and 1.7 ⁇ 10 7 or higher. More preferably, it is Pa or more.
- the storage elastic modulus is preferably 5 ⁇ 10 9 Pa or less, more preferably 1 ⁇ 10 9 Pa or less, and further preferably 5 ⁇ 10 8 Pa or less.
- the storage elastic modulus When the storage elastic modulus is less than or equal to the above upper limit, it is possible to suppress the internal stress generated by going through the moisture absorption reflow test to be excessive, cracking of the obtained resin cured product, and the interface between the metal and the resin cured product. Peeling tends to be suppressed.
- the storage elastic modulus When the storage elastic modulus is in the above range, the resin cured product easily enters the irregularities of the metal that is the adherend described later, and the resin cured product that enters the irregularities develops a strong anchoring effect, and the metal and the resin cure. Adhesion to objects tends to improve.
- a rigid structure such as an aromatic ring is used as a component constituting the resin composition used for obtaining the resin cured product.
- a polyfunctional component having a plurality of reactive groups to increase the crosslink density of the cured product. It can also be realized by using the above-mentioned resin composition to obtain a resin cured product.
- the method of measuring the storage elastic modulus may be a value measured by any conventionally known method, and specifically, the method described in the section of Examples below can be mentioned.
- the resin cured product (including the inorganic filler) of the present invention has a weight increase rate of 0.8% or less at 85 ° C. and 85% RH. If the weight increase rate exceeds 0.8%, it is impossible to solve the problems of the present invention of maintaining high insulation property and preventing interfacial peeling after the moisture absorption reflow test. From the viewpoints of maintaining high insulation properties and preventing interfacial peeling after the moisture absorption reflow test, the smaller the weight increase rate, the more preferable, and 0.75% or less is preferable, and 0.7% or less is more preferable.
- the lower limit of the weight increase rate is not particularly limited, but is, for example, 0.2% or more from the viewpoint of compatibility between strength and insulation performance of the cured resin product and film formability.
- the present invention has found that the problems of the present invention can be solved by being within the above specific range.
- a method for obtaining a resin cured product having a weight increase rate at 85 ° C. and 85% RH in a specific range for example, introducing a highly hydrophobic structure such as an aliphatic skeleton or an aromatic ring into the constituent components. Can be obtained by controlling the weight increase rate. It can also be realized by using the above-mentioned resin composition to obtain a resin cured product.
- the rate of weight increase of the resin cured product of the present invention at 85 ° C. and 85% RH is measured by the method described in the section of Examples below.
- the sheet-shaped cured resin can be manufactured by a method that is usually used. For example, it can be obtained by preparing the resin composition of the present invention, molding it into a sheet, and curing it.
- the resin composition of the present invention can be obtained by uniformly mixing the inorganic aggregating filler, the resin, and other components added as necessary by stirring or kneading.
- a general kneading device such as a mixer, a kneader, a single-screw kneader or a twin-screw kneader can be used.
- the mixing order of the respective components is arbitrary as long as there is no particular problem such as a reaction or the occurrence of a precipitate, but the following method may be mentioned, for example.
- Resin is mixed and dissolved in an organic solvent (for example, methyl ethyl ketone) to create a resin liquid, and the resulting resin liquid is mixed with a mixture of inorganic coagulation filler and other components, and then the viscosity is adjusted.
- an organic solvent is further added and mixed, and then a curing agent, a curing accelerator, or an additive such as a dispersant is further added and mixed.
- a generally used method can be used.
- the resin composition has plasticity or fluidity
- it can be molded by curing the resin composition in a desired shape, for example, in a state of being housed in a mold.
- injection molding, injection compression molding, extrusion molding, compression molding, or vacuum compression molding can be used.
- the solvent in the resin composition can be removed by a known heating method such as a hot plate, a hot air oven, an IR heating oven, a vacuum dryer or a high frequency heater.
- the sheet-shaped cured resin can also be obtained by shaving the cured resin composition into a desired shape.
- the sheet-shaped cured resin can also be obtained by molding a slurry resin composition into a sheet by a method such as a doctor blade method, a solvent casting method or an extrusion film forming method.
- a slurry resin composition is applied to the surface of a base material to form a coating film (sheet resin composition).
- a coating film is formed on a substrate by a dip method, a spin coating method, a spray coating method, a blade method, or any other method.
- a coating device such as a spin coater, a slit coater, a die coater, or a blade coater can be used for coating the slurry resin composition. With such a coating device, it is possible to uniformly form a coating film having a predetermined film thickness on the base material.
- a copper plate or copper foil described later or a PET film is generally used, but is not limited thereto.
- the coating film formed by applying the resin composition in the form of a slurry is usually at 10 to 150 ° C., preferably 25 to 120 ° C., more preferably 30 to 110 ° C. in order to remove the solvent and low molecular components. Dry at temperature.
- the drying temperature is equal to or lower than the above upper limit, curing of the resin in the resin composition in the slurry state is suppressed, and the resin in the resin composition in the sheet state is fluidized in the subsequent pressing step to remove voids. It tends to be easier.
- the drying temperature is at least the above lower limit, the solvent can be effectively removed and the productivity tends to be improved.
- the drying time is not particularly limited and can be appropriately adjusted depending on the state of the resin composition in a slurry state, the drying environment, and the like.
- the drying time is preferably 1 minute or longer, more preferably 2 minutes or longer, even more preferably 5 minutes or longer, even more preferably 10 minutes or longer, particularly preferably 20 minutes or longer, most preferably 30 minutes or longer.
- the drying time is preferably 24 hours or less, more preferably 10 hours or less, further preferably 4 hours or less, and particularly preferably 2 hours or less.
- the drying time is at least the above lower limit value, the solvent can be sufficiently removed, and the residual solvent tends to be suppressed from becoming a void in the resin cured product.
- productivity tends to be improved and manufacturing cost tends to be suppressed.
- the pressing step is preferably performed by applying a load of 2 MPa or more to the sheet-shaped resin composition on the base material.
- the weight is preferably 5 MPa or more, more preferably 7 MPa or more, and further preferably 9 MPa or more.
- the weight is preferably 1500 MPa or less, more preferably 1000 MPa or less, and further preferably 800 MPa or less.
- the heating temperature of the sheet-shaped resin composition on the substrate in the pressing step is not particularly limited.
- the heating temperature is preferably 10 ° C. or higher, more preferably 20 ° C. or higher, even more preferably 30 ° C. or higher.
- the heating temperature is preferably 300 ° C. or lower, more preferably 250 ° C. or lower, further preferably 200 ° C. or lower, even more preferably 100 ° C. or lower, particularly preferably 90 ° C. or lower.
- the time of the pressurizing step is not particularly limited.
- the time of the pressurizing step is preferably 30 seconds or more, more preferably 1 minute or more, further preferably 3 minutes or more, particularly preferably 5 minutes or more.
- the time of the pressurizing step is preferably 1 hour or less, more preferably 30 minutes or less, further preferably 20 minutes or less.
- the pressurizing time is equal to or less than the above upper limit value, the production time of the resin cured product can be suppressed and the production cost tends to be shortened.
- the pressurizing time is at least the above lower limit value, voids and voids in the resin cured product can be sufficiently removed, and heat transfer performance and withstand voltage characteristics tend to be improved.
- the curing step of completely curing the resin composition of the present invention may be performed under pressure or may be performed without pressure. Moreover, you may perform a pressurization process and a hardening process simultaneously.
- the weight applied when the pressure step and the curing step are performed at the same time is not particularly limited.
- the sheet-shaped resin composition on the substrate is applied with a load of 5 MPa or more, more preferably 7 Pa or more, further preferably 9 MPa or more, particularly preferably 20 MPa or more.
- the weight is preferably 2000 MPa or less, more preferably 1500 MPa or less.
- the pressurizing time when the pressurizing step and the curing step are performed simultaneously is not particularly limited.
- the pressing time is preferably 30 seconds or more, more preferably 1 minute or more, further preferably 3 minutes or more, and particularly preferably 5 minutes or more.
- the pressurizing time is preferably 1 hour or less, more preferably 30 minutes or less, and further preferably 20 minutes or less.
- the pressurizing time is not more than the above upper limit value, the production time of the sheet-shaped resin cured product can be suppressed, and the production cost tends to be shortened.
- the pressurizing time is not less than the above lower limit value, voids and voids in the sheet-shaped resin cured product can be sufficiently removed, and heat transfer performance and withstand voltage characteristics tend to be improved.
- the heating temperature of the sheet-shaped resin composition on the substrate when the pressurizing step and the curing step are performed simultaneously is not particularly limited.
- the heating temperature is preferably 10 ° C. or higher, more preferably 20 ° C. or higher, even more preferably 30 ° C. or higher.
- the heating temperature is preferably 300 ° C or lower, more preferably 250 ° C or lower, further preferably 200 ° C or lower, even more preferably 100 ° C or lower, and particularly preferably 90 ° C or lower.
- the heating temperature of the sheet-shaped resin composition on the substrate when performing only the curing step is not particularly limited.
- the heating temperature is preferably 10 ° C or higher, more preferably 50 ° C or higher, still more preferably 100 ° C or higher.
- the heating temperature is preferably 500 ° C or lower, more preferably 300 ° C or lower, further preferably 200 ° C or lower, still more preferably 180 ° C or lower, and particularly preferably 175 ° C or lower.
- the thickness of the sheet-shaped cured resin thus formed is not particularly limited, but is preferably 50 ⁇ m or more, more preferably 80 ⁇ m or more, and further preferably 100 ⁇ m or more.
- the thickness of the resin cured product is preferably 400 ⁇ m or less, more preferably 300 ⁇ m or less.
- the withstand voltage characteristic is obtained and the breakdown voltage tends to be improved.
- the thickness of the resin cured product is less than or equal to the above upper limit value, the device can be made smaller and thinner, and the thermal resistance of the obtained resin cured product (heat dissipation sheet) tends to be suppressed.
- the composite molded article of the present invention has a cured product part made of a cured product of the resin composition of the present invention and a metal part, which are usually laminated and integrated.
- the metal part may be provided on only one surface of the cured product part of the cured resin product of the present invention, or may be provided on two or more surfaces.
- the resin part may have a metal part only on one surface, or may have the metal part on both surfaces.
- the metal part may be patterned.
- the composite molded article of the present invention can be produced by using the metal part as the base material and forming the resin cured product of the present invention on the base material according to the above method.
- the composite molded article of the present invention is obtained by peeling a sheet-shaped resin composition or a resin cured product formed on a base material different from the metal part from the base material, and then thermocompression bonding on the metal member to be the metal part. It can also be manufactured by
- the sheet-shaped sheet of the present invention is prepared in the same manner as above except that the resin composition of the present invention in slurry form is applied onto a substrate such as PET (polyethylene terephthalate) which may be treated with a release agent.
- a substrate such as PET (polyethylene terephthalate) which may be treated with a release agent.
- the resin composition or the resin cured product is peeled off from the substrate, and the sheet-shaped resin composition or the resin cured product is placed on another metal plate or sandwiched between two metal plates. It may be integrated by applying pressure in the state.
- a metal plate made of copper, aluminum, nickel-plated metal or the like and having a thickness of about 10 ⁇ m to 10 cm can be used.
- the surface of the metal plate may be physically roughened or chemically treated with a surface treatment agent or the like. From the viewpoint of adhesion between the resin composition and the metal plate, it is more preferable that these treatments have been performed.
- the composite molded article of the present invention can be used as a semiconductor device.
- it can be preferably used in a power semiconductor device capable of high output and high density by operating at high temperature.
- Specific epoxy resin having 4 , R 5 , R 6 , R 7 methyl group)
- Inorganic filler 1 Boron nitride agglomerated particles having a card house structure, which is produced according to the method for producing boron nitride agglomerated particles disclosed in Examples of WO 2015/561028.
- New Mohs hardness 2 Volume average particle diameter: 45 ⁇ m
- Inorganic filler 2 spherical alumina particles manufactured by Admatechs Co., Ltd.
- New Mohs hardness 9 Volume average particle diameter: 6.5 ⁇ m Thermal conductivity: 20-30 W / mK
- Curing agent 1 "MEH-8000H” manufactured by Meiwa Kasei Co., Ltd. Phenolic resin hardener
- Curing catalyst 1 "2E4MZ-A” manufactured by Shikoku Chemicals 2,4-Diamino-6- [2'-ethyl-4'-methylimidazolyl- (17 ')]-ethyl-s-triazine (Compound having triazine ring as heterocyclic structure containing nitrogen atom) Molecular weight: 247
- Curing catalyst 2 "C11Z-CN” manufactured by Shikoku Kasei 1-Cyanoethyl-2-undecylimidazole Molecular weight: 275
- Example preparation and measurement / evaluation The method for producing the molded body and the measurement conditions / evaluation methods in Examples and Comparative Examples are as follows.
- Example 1 A mixture was prepared using a rotation and revolution type stirring device so that the inorganic filler in the solid content was 1: 51% by weight, the inorganic filler was 2: 20% by weight, and the components other than the inorganic filler: 29% by weight. At this time, the components other than the inorganic filler in the solid content were adjusted so that the weight ratio was the ratio described in the column of Example 1 in Table 1. Further, when the above mixture was prepared, methyl ethyl ketone and cyclohexanone were used in equal amounts so that the mixture was 63% by weight (solid content concentration) in the coating slurry.
- the obtained slurry resin composition (slurry for a sheet) was applied to a PET base material by a doctor blade method, heated and dried at 60 ° C. for 120 minutes, and then pressed at 42 ° C. and 147 MPa for 10 minutes, A sheet-shaped resin composition having a thickness of 150 ⁇ m was obtained.
- the total content of methyl ethyl ketone and cyclohexanone in the sheet-shaped resin composition was 1% by weight or less.
- the sheet-shaped resin composition was sandwiched between each of the copper plates having a thickness of 500 ⁇ m and a copper plate having a thickness of 2,000 ⁇ m, the surface of which was roughened 100 times each with a # 120 file in advance. Pressing was performed for one minute, then the temperature was raised and pressing was performed at 175 ° C. and 9.8 MPa for 30 minutes.
- the 500 ⁇ m-thick copper plate was patterned by etching the composite molded body containing the copper plate and the resin cured product obtained above by a predetermined method. As for the pattern, two circular patterns with a diameter of 25 mm were left.
- Examples 2 to 3 Comparative Examples 1 to 4> According to the method of Example 1, a mixture was prepared such that the inorganic filler in the solid content was 1: 51% by weight, the inorganic filler was 2: 20% by weight, and the components other than the inorganic filler were 29% by weight. At this time, in the same manner as in Example 1 except that the components other than the inorganic filler in the solid content had the weight ratios shown in Table 1, the sheet-shaped resin composition, the copper plate and the resin, respectively. A composite molded body containing a cured product was obtained.
- the composite molded bodies prepared in the examples and comparative examples were dipped in Fluorinert FC-40 (manufactured by 3M), and then, using an ultra-high voltage withstand tester 7470 (manufactured by Measurement Technology Laboratory Co., Ltd.), copper was patterned on ⁇ 25 mm. The electrode was placed, a voltage of 0.5 kV was applied, the voltage was increased by 0.5 kV every 60 seconds, and measurement was performed until dielectric breakdown.
- the BDV after the hygroscopic reflow test was measured for those having a BDV of 5 kV or more, and the BDV after the hygroscopic reflow test was not performed for the composite molded article (ND) having a BDV of less than 5 kV.
- thermo-hygrostat SH-221 manufactured by ESPEC Corp.
- the temperature was raised from room temperature to 290 ° C in 12 minutes, held at 290 ° C for 10 minutes, and then cooled to room temperature (moisture absorption reflow test). After that, the dielectric breakdown voltage was measured in the same manner as above, and when the BDV was 5 kV or more, “ ⁇ ” and less than 5 kV were expressed as “x”.
- ⁇ Storage elastic modulus of resin composition excluding inorganic filler> Except that no inorganic filler was added, the resin composition was prepared by using a rotation-revolution type stirring device in the same manner as in each Example and Comparative Example, and after heating and drying, the Anton Paar Rheometer "MCR302" was used. Was used to heat cure the uncured resin composition, and the storage elastic modulus at 200 ° C. was measured. An aluminum parallel plate was used for the measurement, and the measurement conditions were a strain of 0.3%, a frequency of 1 Hz, and a gap of 0.5 mm.
- the temperature profile at the time of heat curing starts from 25 ° C, rises to 120 ° C at 14 ° C per minute, holds for 30 minutes after reaching 120 ° C, and then rises to 175 ° C at 7 ° C per minute to 175 ° C. Was reached for 30 minutes, the temperature was further raised to 200 ° C. at 7 ° C. per minute, and the temperature was held for 10 minutes after reaching 200 ° C.
- the storage elastic modulus measured at the time of holding at 200 ° C. for 10 minutes was used for evaluation.
- Table 1 shows the above measurement and evaluation results.
- the cured resin product of the present invention is excellent in withstand voltage performance under high temperature and high humidity conditions and has no problem of interfacial peeling under high temperature and high humidity conditions in the case of forming a composite molding with a metal. .
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Epoxy Resins (AREA)
- Macromonomer-Based Addition Polymer (AREA)
Abstract
Description
本発明の樹脂組成物、樹脂硬化物および複合成形体は、例えばパワー半導体デバイス用の放熱シートとして好適に用いることができる。
パワー半導体デバイスは、一般的には、複数の半導体デバイスを共通のヒートシンク上に配してパッケージングしたパワー半導体モジュールとして利用される。
セラミックス基板の場合は、銅板との焼結により一体化させるため、界面での剥離は起き難い。しかし、放熱樹脂シートでは、硬化膜の加熱圧着により一体化されるため界面剥離を起こし易い。
本発明者はまた、貯蔵弾性率及び重量増加率が特定範囲である樹脂硬化物を用いることにより、吸湿リフロー耐性に優れ、界面剥離の問題を解決することができることを見出した。
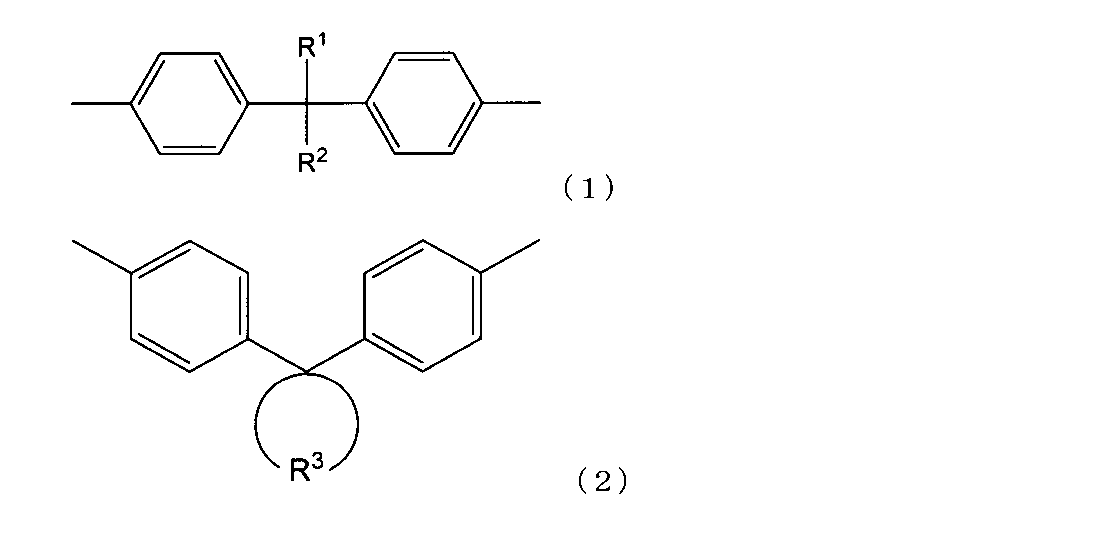
本発明の樹脂組成物は樹脂及び凝集無機フィラーを含み、該樹脂組成物の硬化後の85℃、85%RHでの重量増加率が0.80%以下であり、無機フィラーを除く該樹脂組成物の硬化物の200℃における貯蔵弾性率が1.0×107Pa以上である。
本発明の無機フィラーを除く樹脂組成物の硬化後の200℃における貯蔵弾性率は、1.0×107Pa以上である。この貯蔵弾性率が1×107Pa未満であると、本発明の樹脂組成物を硬化して得られる樹脂硬化物(以下、単に「樹脂硬化物」と称す。)の強度が低く、そのため吸湿リフロー試験において、内部にボイドが発生することで絶縁性能が低下したり、本発明の樹脂組成物の硬化物からなる硬化物部と、金属部とを有する複合成形体にあっては、金属部と硬化物部との界面が剥離したりすることがある。
吸湿リフロー試験後の性能保持の観点から、この貯蔵弾性率は1.3×107Pa以上であることが好ましく、1.5×107Pa以上であることがより好ましく、1.7×107Pa以上であることがさらに好ましい。
一方、この貯蔵弾性率は5×109Pa以下であることが好ましく、1×109Pa以下であることがより好ましく、5×108Pa以下であることがより好ましい。貯蔵弾性率が上記上限値以下であると、吸湿リフロー試験を経ることで発生する内部応力が過剰となることを抑制でき、得られる樹脂硬化物の割れや、金属部と樹脂硬化物部との界面剥離を抑制できる傾向にある。
また、貯蔵弾性率が上記範囲であることで、後述する被着体である金属の凹凸に樹脂組成物の硬化物が入り易くなり、凹凸に入り込んだ樹脂硬化物が強固なアンカー効果を発現し、金属と樹脂硬化物との密着性が向上する傾向にある。
本発明の樹脂組成物(無機フィラーを含む)の硬化後において、85℃、85%RHでの重量増加率が0.8%以下である。この重量増加率が0.8%を超えるものは、吸湿リフロー試験後の高い絶縁性維持、界面剥離の防止という、本発明の課題を解決し得ない。
吸湿リフロー試験後の高い絶縁性維持、界面剥離の防止の観点から、この重量増加率は小さい程好ましく、0.75%以下が好ましく、0.7%以下がより好ましい。重量増加率の下限は特に限定されないが、樹脂硬化物の強度と絶縁性能との両立や製膜性の観点から、例えば0.2%以上である。
本発明は、樹脂組成物は硬化後の85℃、85%RHでの重量増加率が上記特定の範囲であることで、本発明の課題を解決できることを見出したことに基くものである。
本発明の樹脂組成物に含まれる樹脂としては、硬化後に特定の貯蔵弾性率及び重量増加率となるものであれば特に限定されない。例えば、硬化剤や硬化触媒の存在下で熱又は光硬化するものが挙げられる。特に熱硬化性樹脂であることが、製造容易性の点から好ましい。
エポキシ樹脂とは、分子内に1個以上のオキシラン環(エポキシ基)を有する化合物の総称である。エポキシ樹脂に含まれるオキシラン環(エポキシ基)は脂環式エポキシ基、グリシジル基のどちらでも構わない
本発明の樹脂組成物は、一分子内に3個以上のオキシラン環(エポキシ基)を有するエポキシ樹脂(以下、「多官能エポキシ樹脂」と称す場合がある。)を含むことが好ましい。本発明の樹脂組成物が多官能エポキシ樹脂を含むことにより、極性の高いオキシラン環(エポキシ基)を高密度で導入することが可能となる。それにより、ファンデルワールス力や水素結合といった物理的相互作用の効果が増し、複合成形体における金属と樹脂硬化物との密着性を向上させることができる傾向にある。
また、多官能エポキシ樹脂を含むことにより、樹脂硬化物の貯蔵弾性率を前述した特定範囲とし易い傾向にあり、金属と樹脂硬化物との密着性が向上する傾向にある。
さらに、オキシラン環(エポキシ基)の反応性を向上させることで、硬化反応途中の水酸基量を減らし、吸湿性の増加を抑制することができる傾向がある。
本発明の樹脂組成物中の樹脂は、ビフェニル構造を有する重量平均分子量が10,000以上であるエポキシ樹脂(以下、「特定エポキシ樹脂」と称す場合がある。)を含むことが好ましい。
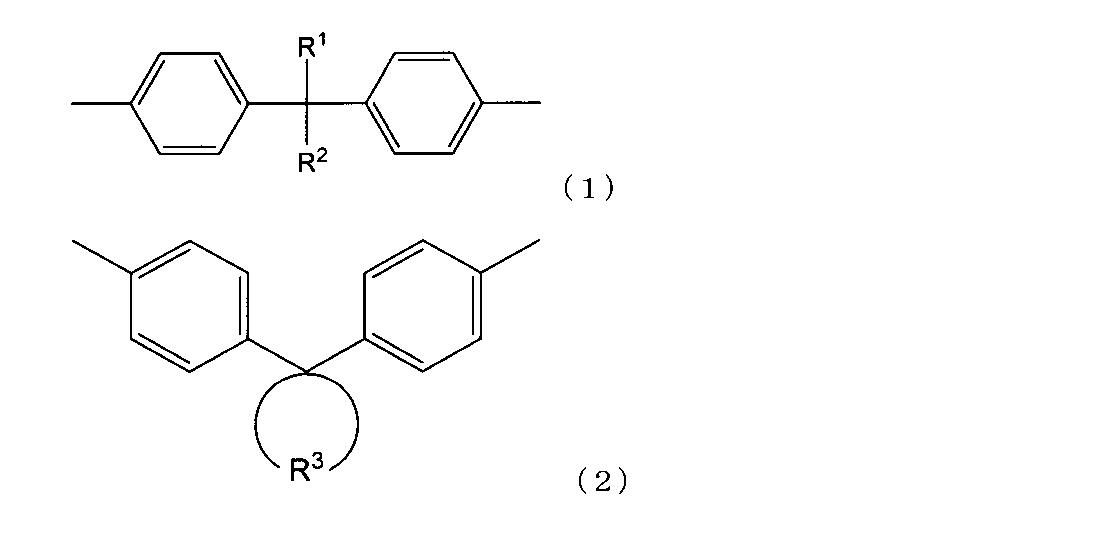
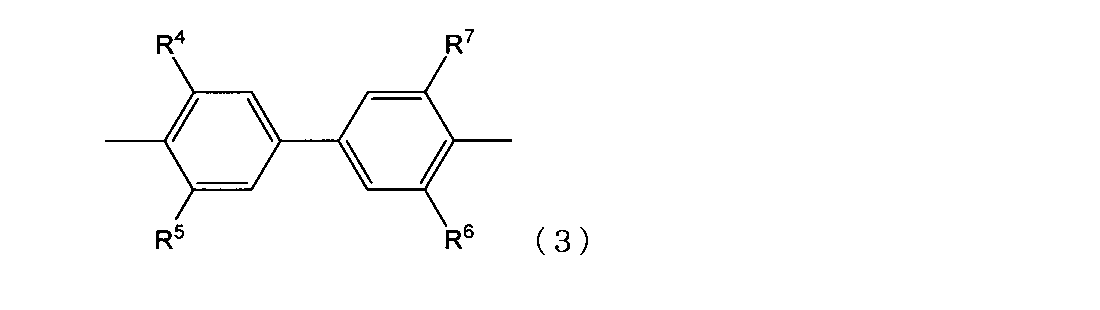
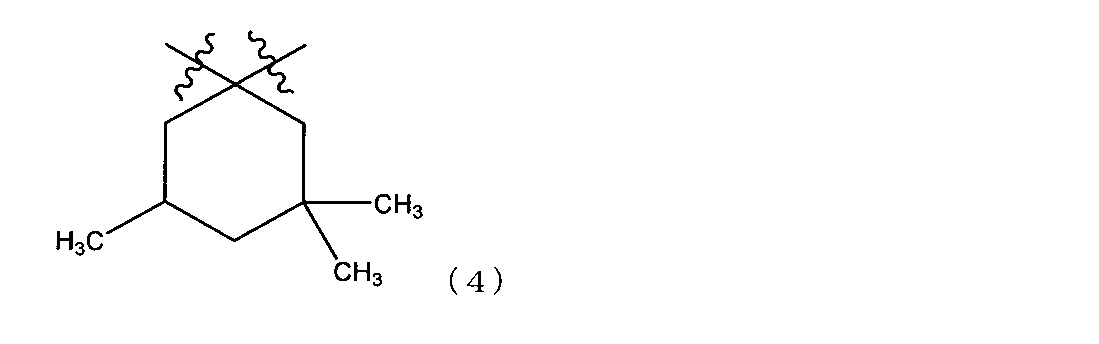
吸湿量を低減するという観点から、特定エポキシ樹脂は疎水性構造である構造(1)、(2)、(3)を多く含むものが好ましい。
エポキシ当量は、「1当量のエポキシ基を含むエポキシ樹脂の重量」と定義され、JIS K7236に準じて測定することができる。
特に本発明の樹脂組成物は、エポキシ樹脂として、特定エポキシ樹脂と多官能エポキシ樹脂とを共に含有することが、樹脂組成物の硬化物の高弾性化と低吸湿性の両立の面で好ましい。
本発明の樹脂組成物は、特定エポキシ樹脂および多官能エポキシ樹脂以外のエポキシ樹脂を含有していてもよい。本発明の樹脂組成物に含まれる特定エポキシ樹脂および多官能エポキシ樹脂以外のエポキシ樹脂としては、特に制限はないが、例えばビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂等のビスフェノール類をグリシジル化した各種ビスフェノール型エポキシ樹脂、ビフェニル類をグリシジル化した各種ビフェニル型のエポキシ樹脂、ジヒドロキシナフタレン、9,9-ビス(4-ヒドロキシフェニル)フルオレンなどの2つの水酸基を有する芳香族性を有する化合物類をグリシジル化したエポキシ樹脂、1,1,1-トリス(4-ヒドロキシフェニル)メタンなどのトリスフェノール類をグリシジル化したエポキシ樹脂、1,1,2,2-テトラキス(4-ヒドロキシフェニル)エタンなどのテトラキスフェノール類をグリシジル化したエポキシ樹脂、フェノールノボラック、クレゾールノボラック、ビスフェノールAノボラック、臭素化ビスフェノールAノボラックなどのノボラック類をグリシジル化したノボラック型エポキシ樹脂、およびシリコーン含有エポキシ樹脂から選ばれる1種又は2種以上が好ましい。
本発明において、無機フィラーは凝集無機フィラー及び非凝集無機フィラーを含む。
本発明の樹脂組成物は、凝集無機フィラーに加え、非凝集無機フィラーを含んでいてもよい。非凝集無機フィラーには、後述する球状フィラーも含まれる。
凝集無機フィラーとしては、電気絶縁性のものが使用でき、金属炭化物、金属酸化物及び金属窒化物からなる群から選ばれる少なくとも1種の無機粒子から構成されるものが挙げられる。
金属酸化物の例としては、酸化マグネシウム、酸化アルミニウム、酸化ケイ素、酸化カルシウム、酸化亜鉛、酸化イットリウム、酸化ジルコニウム、酸化セリウム、酸化イッテルビウム、サイアロン(ケイ素、アルミニウム、酸素、窒素からなるセラミックス)等が挙げられる。
金属窒化物の例としては、窒化ホウ素、窒化アルミニウム、窒化ケイ素等が挙げられる。
凝集無機フィラーは、表面処理剤により表面処理がされていてもよい。表面処理剤は、公知の表面処理剤を用いることができる。
窒化ホウ素は熱伝導性の高いものであるが、鱗片状であるため、面方向には熱伝導性に優れるが、面に垂直な方向には熱抵抗が大きい。このような鱗片状の粒子を集めて球状に凝集させた凝集粒子は取り扱い性にも優れるため好ましい。
窒化ホウ素凝集粒子としては、窒化ホウ素の粒子を面方向に整列させて凝集粒子の径方向が熱伝導のよい方向となるようにしたものが好ましい。
「カードハウス構造」は、例えばセラミックス 43 No.2(2008年 日本セラミックス協会発行)に記載されており、板状粒子が配向せず複雑に積層した構造である。より具体的には、カードハウス構造を有する窒化ホウ素凝集粒子とは、窒化ホウ素一次粒子の集合体であって、一次粒子の平面部と端面部が接触し、例えばT字型の会合体を形成する構造を有する窒化ホウ素凝集粒子である。
体積平均粒子径の測定方法は、分散安定剤としてヘキサメタリン酸ナトリウムを含有する純水媒体中に凝集粒子を分散させた試料に対して、レーザー回折/散乱式粒度分布測定装置などを用いて測定する湿式測定法や、Malvern社製「Morphologi」を用いて測定する乾式測定法が挙げられる。
後述の球状無機フィラーの体積平均粒子径についても同様である。
本発明の樹脂組成物における凝集無機フィラーの含有量は、樹脂組成物の固形分100重量%中に30重量%以上であることが好ましく、40重量%以上であることがより好ましく、45重量%以上であることがさらに好ましい。また、本発明の樹脂組成物における凝集無機フィラーの含有量は、樹脂組成物の固形分100重量%中99重量%以下であることが好ましく、90重量%以下であることがより好ましく、80重量%以下であることがさらに好ましい。凝集無機フィラーの含有量が上記下限値以上であることで、凝集無機フィラーを含有することによる熱伝導性の向上効果や、線膨張係数の制御効果を十分に得ることができる傾向にある。凝集無機フィラーの含有量が上記上限値以下であることで、樹脂組成物及び樹脂硬化物の成形性や複合成形体における界面接着性が向上する傾向にある。
本発明の樹脂組成物は、無機フィラーとして凝集無機フィラーと共に非凝集無機フィラーを含んでいてもよい。
本発明の樹脂組成物には、本発明の効果を損なうことのない範囲において、上記以外のその他の成分が含まれていてもよい。その他の成分としては、窒素原子を含有する複素環構造を有する化合物、硬化剤、硬化触媒、無機フィラーと樹脂との界面接着強度を改善するシランカップリング剤などの表面処理剤、還元剤等の絶縁性炭素成分、粘度調整剤、分散剤、チキソ性付与剤、難燃剤、着色剤、有機フィラー、有機溶剤、熱可塑性樹脂等が挙げられる。
本発明の樹脂組成物のその他の成分の含有の有無及び含有割合は、本発明の効果を著しく損なわない範囲であれば特に限定されない。
本発明の樹脂組成物は、窒素原子を含有する複素環構造を有する化合物を含有していてもよい。
窒素含有複素環化合物の分子量は1,000以下であることが好ましく、500以下であることがより好ましい。窒素含有複素環化合物の分子量の下限は特に限定されないが、60以上が好ましく、70以上がより好ましい。
窒素含有複素環化合物は、1分子中に複数の復素環構造を同時に有していても構わない。
樹脂組成物の絶縁性、金属との密着性の向上の観点から、窒素含有複素環化合物としてはイミダゾール系化合物やトリアジン系化合物が好ましい。
本発明の樹脂組成物は硬化剤を含んでいてもよい。
樹脂組成物の柔軟性および難燃性のより一層の向上、樹脂硬化物の力学物性および耐熱性向上のためには、剛直な主鎖骨格を持つノボラック型フェノール樹脂やトリアジン骨格を有するフェノール樹脂が好ましい。
未硬化の樹脂組成物の柔軟性および樹脂硬化物の靭性向上のためにはアリル基を有するフェノール樹脂が好ましい。
また、樹脂組成物に硬化剤を含む場合、樹脂組成物中のエポキシ当量に対して0~55重量%当量であることが好ましい。上記範囲であることで、反応が効果的に進行し、架橋密度を向上させ、強度を増すことができ、さらに製膜性が向上する傾向にある。
本発明の樹脂組成物は、硬化触媒を含んでいてもよい。硬化速度や硬化物の物性などを調整するために、上記硬化剤と共に硬化触媒を含有することは好ましい。
有機金属化合物類としては、オクチル酸亜鉛、オクチル酸錫又はアルミニウムアセチルアセトン錯体等が挙げられる。
これらは1種を単独で用いてもよく、2種以上を混合して用いてもよい。
本発明の樹脂組成物は、例えば、塗布工程を経てシート状の樹脂硬化物を成形する際の塗布性の向上のために、有機溶剤を含有していてもよい。
これらの有機溶剤は、1種のみを用いてもよく、2種以上を併用してもよい。
本発明の樹脂組成物が、後述する塗布工程に供するスラリー状である場合、有機溶剤は本発明の樹脂組成物の固形分濃度が10~90重量%、特に40~80重量%となるように用いることが好ましい。
本発明の樹脂組成物が、塗布及び乾燥等の工程を経たシート状の場合、本発明の樹脂組成物の固形分濃度は95重量%以上であることが好ましく、特に98重量%以上であることがより好ましい。
本発明の樹脂組成物は、分散剤を含んでいてもよい。本発明の樹脂組成物に分散剤が含まれていることで、均一な樹脂硬化物を形成することが可能となり、得られる樹脂硬化物の熱伝導性および絶縁破壊特性を向上させることができる場合がある。
分散剤は、1種のみが用いられてもよく、2種以上が併用されてもよい。
本発明の樹脂組成物は、有機フィラー及び/又は熱可塑性樹脂を含んでいてもよい。本発明の樹脂組成物が有機フィラーや熱可塑性樹脂を含むことで、樹脂組成物に適度な伸び性が付与され、発生する応力が緩和され、温度サイクル試験でのクラックの発生を抑制することができる場合がある。
これらの観点から、熱可塑性樹脂としては、ナイロンなどのポリアミド樹脂やセルロース樹脂などが好ましく、特にナイロンなどのポリアミド樹脂が好ましい
粒子状の熱可塑性樹脂の平均粒子径は、樹脂硬化物の断面を観察し、任意の20個の粒子の最長径の平均値により定める。
本発明の樹脂硬化物は、無機凝集フィラーを含む樹脂組成物を用いた樹脂硬化物であって、該樹脂硬化物の85℃、85%RHでの重量増加率が0.80%以下であり、無機フィラーを除く該樹脂組成物の硬化後の200℃における貯蔵弾性率が1.0×107Pa以上のものである。
また、無機フィラーを除く樹脂組成物とは、樹脂組成物中の無機フィラー以外の成分を指す。樹脂、硬化剤、硬化触媒、窒素原子を含有する複素環構造を有する化合物、無機フィラー、無機凝集フィラー、その他成分については前述の樹脂組成物と同義であり、好ましい範囲なども同義である。
本発明の樹脂硬化物を得る方法は特に限定されない。例えば、上述した樹脂組成物を硬化させて得る方法が挙げられる。
無機フィラーを除く該樹脂組成物の硬化後の200℃における貯蔵弾性率が1.0×107Pa以上である。吸湿リフロー試験後の性能保持の観点から、貯蔵弾性率は1.3×107Pa以上であることが好ましく、1.5×107Pa以上であることがより好ましく、1.7×107Pa以上であることがさらに好ましい。一方、貯蔵弾性率は5×109Pa以下であることが好ましく、1×109Pa以下であることがより好ましく、5×108Pa以下であることがより好ましい。
貯蔵弾性率が上記上限値以下であることで、吸湿リフロー試験を経ることで発生する内部応力が過剰となることを抑制でき、得られる樹脂硬化物の割れや、金属と樹脂硬化物との界面剥離を抑制できる傾向にある。
貯蔵弾性率が上記範囲であることで、後述する被着体である金属の凹凸に樹脂硬化物が入り易くなり、凹凸に入り込んだ樹脂硬化物が強固なアンカー効果を発現し、金属と樹脂硬化物との密着性が向上する傾向にある。
本発明の樹脂硬化物(無機フィラーを含む)は、85℃、85%RHでの重量増加率が0.8%以下である。この重量増加率が0.8%を超えるものは、吸湿リフロー試験後に高い絶縁性維持、界面剥離の防止という、本発明の課題を解決し得ない。
吸湿リフロー試験後に高い絶縁性維持、界面剥離の防止の観点から、この重量増加率は小さい程好ましく、0.75%以下が好ましく、0.7%以下がより好ましい。
重量増加率の下限は特に限定されないが、樹脂硬化物の強度と絶縁性能との両立や製膜性の観点から、例えば0.2%以上である。
樹脂硬化物の重量増加の要因は様々考えられるが、吸湿による重量増加をコントロールすることが重要である。本発明は上記特定の範囲であることで、本発明の課題を解決できることを見出したものである。
本発明の樹脂組成物及び本発明の樹脂硬化物を製造する方法を、本発明の樹脂組成物よりなるシート状の樹脂硬化物の製造方法を例示して説明する。
樹脂を有機溶剤(例えば、メチルエチルケトン)に混合、溶解させて樹脂液を作成し、得られた樹脂液に、無機凝集フィラー、その他の成分を十分混合したものを加えて混合し、その後、粘度調整用として更に有機溶剤を加えて混合した後に、更に、硬化剤や硬化促進剤、或いは、分散剤等の添加剤を加えて混合する。
例えば、樹脂組成物が可塑性や流動性を有する場合、該樹脂組成物を所望の形状で、例えば型へ収容した状態で硬化させることによって成形することができる。この場合、射出成形、射出圧縮成形、押出成形、圧縮成形、真空圧縮成形を利用することができる。
まず基材の表面に、スラリー状の樹脂組成物を塗布して塗膜(シート状の樹脂組成物)を形成する。
スラリー状の樹脂組成物を塗布することにより形成された塗膜を、溶剤や低分子成分の除去のために、通常10~150℃、好ましくは25~120℃、より好ましくは30~110℃の温度で乾燥する。
乾燥温度が上記上限値以下であることで、スラリー状の樹脂組成物中の樹脂の硬化が抑制され、その後の加圧工程でシート状の樹脂組成物中の樹脂が流動ししボイドを除去しやすくなる傾向にある。乾燥温度が上記下限値以上であることで、効果的に溶剤を取り除くことができ生産性が向上する傾向にある。
乾燥時間が上記下限値以上であることで、十分に溶剤が除去でき、残留溶剤が樹脂硬化物内のボイドとなることを抑制できる傾向にある。乾燥時間が上記上限値以下であることで、生産性が向上し、製造コストを抑制できる傾向にある。
乾燥工程の後には、凝集無機フィラー同士を接合させ熱伝導パスを形成する目的、シート内のボイドや空隙をなくす目的、基材との密着性を向上させる目的等から、得られたシート状の樹脂組成物に加圧工程を行うことが望ましい。
加圧時の加重を上記上限値以下とすることにより、凝集無機フィラーの二次粒子が破壊することなく、シート状の樹脂硬化物中に空隙などがない高い熱伝導性を有するシートを得ることができる。加重を上記下限値以上とすることにより、凝集無機フィラー間の接触が良好となり、熱伝導パスを形成しやすくなるため、高い熱伝導性を有する樹脂硬化物を得ることができる。
この温度範囲で加圧工程を行うことにより、シート状の樹脂組成物中の樹脂の溶融粘度を低下させることができ、樹脂硬化物内のボイドや空隙をより低減することができる。また、上記上限値以下で加熱することで、シート状の樹脂組成物及び樹脂硬化物中の有機成分の分解、残留溶剤により発生するボイドを抑制できる傾向にある。
加圧時間が上記上限値以下であることで、樹脂硬化物の製造時間が抑制でき、生産コストを短縮できる傾向にある。加圧時間が上記下限値以上であることで、樹脂硬化物内の空隙やボイドを十分に取り除くことができ、熱伝達性能や耐電圧特性を向上できる傾向にある。
本発明の樹脂組成物を完全に硬化反応させる硬化工程は、加圧下で行ってもよく、無加圧で行ってもよい。また、加圧工程と硬化工程を同時に行ってもよい。
加圧工程と硬化工程を同時に行う場合の加重を上記上限値以下とすることにより、凝集無機フィラーの二次粒子が破壊することなく、シート状の樹脂硬化物中に空隙などがない高い熱伝導性を有するシート状硬化物を得ることができる。また、加重を上記下限値以上とすることにより、凝集無機フィラー間の接触が良好となり、熱伝導パスを形成しやすくなるため、高い熱伝導性を有する樹脂硬化物を得ることができる。
加圧時間が上記上限値以下であることで、シート状の樹脂硬化物の製造時間が抑制でき、生産コストを短縮できる傾向にある。加圧時間が上記下限値以上であることで、シート状の樹脂硬化物内の空隙やボイドを十分に取り除くことができ、熱伝達性能や耐電圧特性を向上できる傾向にある。
加熱温度を上記下限値以上とすることにより、シート状の樹脂組成物中の樹脂の溶融粘度を低下させることができ、樹脂硬化物内のボイドや空隙をなくすことができる。加熱温度を上記上限値以下とすることで、シート状の樹脂組成物及びシート状の樹脂硬化物中の有機成分の分解、残留溶剤により発生するボイドを抑制できる傾向にある。
加熱温度をこの温度範囲とすることにより、樹脂の硬化反応を効果的に進行させる。加熱温度が上記上限値以下であることで樹脂の熱劣化を防止する。加熱温度が上記下限値以上であることで、樹脂の硬化反応をより効果的に進行させる。
樹脂硬化物の厚さが上記下限値以上であることで、耐電圧特性が得られ、絶縁破壊電圧が向上する傾向にある。樹脂硬化物の厚さが上記上限値以下であることで、デバイスの小型化や薄型化が達成でき、得られる樹脂硬化物(放熱シート)の熱抵抗を抑制できる傾向にある。
本発明の複合成形体は、本発明の樹脂組成物の硬化物からなる硬化物部と金属部とを有し、通常これらが積層一体化されてなるものである。
金属板の表面は物理的に粗化処理がなされていてもよいし、化学的に表面処理剤等で処理されていてもよい。樹脂組成物と金属板の密着の観点から、これらの処理がなされていることがより好ましい
本発明の複合成形体は半導体デバイスとして用いることができる。特に、高温で作動させることにより高出力かつ高密度化が可能なパワー半導体デバイスにおいて好適に用いることができる。
下記の実施例における各種の条件や評価結果の値は、本発明の実施態様における好ましい範囲と同様に、本発明の好ましい範囲を示すものであり、本発明の好ましい範囲は前記した実施態様における好ましい範囲と下記実施例の値または実施例同士の値の組合せにより示される範囲を勘案して決めることができる。
実施例および比較例で用いた原材料は以下の通りである。
樹脂成分1:特開2006-176658号公報の実施例に開示されるエポキシ樹脂の製造方法に準拠して製造した、構造(2)(R3=構造(4))および構造(3)(R4,R5,R6,R7=メチル基)を有する特定エポキシ樹脂
ポリスチレン換算の重量平均分子量:30,000
エポキシ当量:9,000g/当量
樹脂成分2:特開2003-342350号公報の実施例に開示されるエポキシ樹脂の製造方法に準拠して製造した、構造(1)(R1=メチル基、R2=フェニル基)および構造(3)(R4,R5,R6,R7=メチル基)を有する特定エポキシ樹脂
ポリスチレン換算の重量平均分子量:39,000
エポキシ当量:13,000g/当量
樹脂成分3:三菱ケミカル社製 一分子当たりエポキシ基を2個有する構造を含むビスフェノールF型固形エポキシ樹脂
ポリスチレン換算の重量平均分子量:60,000
樹脂成分4:三菱ケミカル社製 一分子当たりエポキシ基を2個有する構造を含むビスフェノールA型液状エポキシ樹脂
分子量:約370
樹脂成分5:ナガセケムテックス社製 一分子当たりグリシジル基を4個以上有する構造を含む多官能エポキシ樹脂
分子量:約400
樹脂成分6:三菱ケミカル社製 一分子当たりエポキシ基を2個有する構造を含む水添ビスフェノールA型液状エポキシ樹脂
分子量 約410
樹脂成分7:三菱ケミカル社製 一分子当たりエポキシ基を3個以上有する構造を含むp-アミノフェノール型液状多官能エポキシ樹脂
分子量:約290
無機フィラー1:国際公開第2015/561028号の実施例に開示される窒化ホウ素凝集粒子の製造方法に準拠して製造した、カードハウス構造を有する窒化ホウ素凝集粒子
新モース硬度:2
体積平均粒子径:45μm
無機フィラー2:アドマテックス社製、球状アルミナ粒子
新モース硬度:9
体積平均粒子径:6.5μm
熱伝導率:20~30W/m・K
硬化剤1:明和化成社製「MEH-8000H」
フェノール樹脂系硬化剤
硬化触媒1:四国化成社製「2E4MZ-A」
2,4-ジアミノ-6-[2’-エチル-4’-メチルイミダゾリル-(17’)]-エチル-s-トリアジン
(窒素原子を含有する複素環構造としてトリアジン環を有する化合物)
分子量:247
硬化触媒2:四国化成社製「C11Z-CN」
1-シアノエチル-2-ウンデシルイミダゾール
分子量:275
実施例と比較例における成形体の作成方法、および測定条件・評価方法は以下の通りである。
自転公転式撹拌装置を用いて、固形分中の無機フィラー1:51重量%、無機フィラー2:20重量%、無機フィラー以外の成分:29重量%になるように混合物を調製した。このとき、固形分中の無機フィラー以外の成分の内訳は重量比で表1の実施例1の欄に記載の比率となるように調整した。また上記混合物を調製する際、上記混合物が塗布スラリーのうち63重量%(固形分濃度)となるように、メチルエチルケトンとシクロヘキサノンを等量ずつ用いた。
実施例1の方法に準拠し、固形分中の無機フィラー1:51重量%、無機フィラー2:20重量%、無機フィラー以外の成分:29重量%になるように混合物を調製した。このとき、固形分中の無機フィラー以外の成分の内訳が表1に示す重量比となるようにしたこと以外は、実施例1と同様にして、それぞれシート状の樹脂組成物と、銅板及び樹脂硬化物を含む複合成形体を得た。
<吸湿リフロー試験前のBDV>
実施例および比較例で作成した複合成形体をフロリナートFC-40(3M社製)に浸し、超高電圧耐圧試験器7470(計測技術研究所社製)を用いて、パターニングしたφ25mmの銅上に電極を置いて、0.5kV電圧を印加し、60秒おきに0.5kVずつ昇圧していき、絶縁破壊に到るまで測定を実施した。BDVが5kV以上のものについて、吸湿リフロー試験後のBDVの測定を実施し、BDVが5kV未満の複合成形体(N.D.)については吸湿リフロー試験後のBDVの測定は行わなかった。
実施例および比較例で作成した複合成形体を恒温恒湿機SH-221(エスペック社製)を用いて85℃、85%RHの環境に3日保管した後、30分以内に窒素雰囲気下において室温から290℃まで12分で昇温し、290℃で10分保持した後、室温まで冷却した(吸湿リフロー試験)。その後、上記と同様に絶縁破壊電圧を測定し、BDVが5kV以上である場合に「〇」、5kV未満を「×」と表記した。
実施例および比較例で作成した複合成形体を上記と同様に吸湿リフロー試験した後、超音波映像装置FinSAT(FS300III)(日立パワーソリューションズ製)により、銅板とシート状の樹脂硬化物との界面を観察した。測定には周波数50MHzのプローブを用い、ゲイン30dB、ピッチ0.2mmとし、試料を水中に置いて実施した。界面に剥離が認められないものを「○」、界面剥離が認められたものを「×」と表記した。
実施例および比較例で作成したシート状の樹脂硬化物を6cm×7cmの試験片に切り出し、150℃で1時間で乾燥し、重量aを測定した。さらにそれらのシート状樹脂硬化物を恒温恒湿機SH-221(エスペック社製)を用いて85℃、85%RHの環境に一定時間保管し、経時で重量を測定し、一定の重量(恒量)bになるまで保管した。下記式で重量増加率を算出した。
重量増加率(%)=(b-a)/a ×100
無機フィラーを配合しないこと以外は、それぞれ各実施例および比較例と同様に、自転公転式撹拌装置を用いて樹脂組成物を調製し、加熱乾燥を行った後にアントンパール社製のレオメーター「MCR302」を用いて、未硬化の樹脂組成物を加熱硬化させ、200℃での貯蔵弾性率を測定した。
測定にはアルミニウム製のパラレルプレートを使用し、測定条件は歪を0.3%、周波数を1Hz、ギャップを0.5mmとした。
加熱硬化時の温度プロファイルは25℃から開始し、毎分14℃で120℃まで昇温し、120℃に到達後30分間保持、続けて毎分7℃で175℃まで昇温し、175℃に到達後30分間保持、さらに毎分7℃で200℃まで昇温し、200℃に到達後10分間保持した。この200℃で10分間保持時に測定した貯蔵弾性率を評価に用いた。

本出願は、2018年10月11日付で出願された日本特許出願2018-192691に基づいており、その全体が引用により援用される。
Claims (13)
- 樹脂及び凝集無機フィラーを含む樹脂組成物であって、
該樹脂組成物の硬化後の85℃、85%RHでの重量増加率が0.80%以下であり、
無機フィラーを除く該樹脂組成物の硬化物の200℃における貯蔵弾性率が1.0×107Pa以上である、樹脂組成物。 - 前記樹脂組成物が、一分子当たりエポキシ基を3つ以上有するエポキシ樹脂を含むものである、請求項1に記載の樹脂組成物。
- 前記樹脂組成物が、ビフェニル構造を有する重量平均分子量が10,000以上であるエポキシ樹脂を含むものである、請求項1又は2に記載の樹脂組成物。
- 前記ビフェニル構造を有する重量平均分子量が10,000以上であるエポキシ樹脂が、更に下記構造式(1)で表される構造および下記構造式(2)で表される構造から選ばれる少なくとも一つの構造を有するものである、請求項3に記載の樹脂組成物。

- 前記ビフェニル構造を有する重量平均分子量が10,000以上であるエポキシ樹脂の含有割合が、無機フィラーを除く該樹脂組成物中の固形分100重量%に対して1重量%以上50重量%以下である、請求項3又は4に記載の樹脂組成物。
- 前記一分子当たりエポキシ基を3つ以上有するエポキシ樹脂の含有割合が、無機フィラーを除く該樹脂組成物中の固形分100重量%に対して10重量%以上50重量%以下である、請求項2~5のいずれか1項に記載の樹脂組成物。
- 前記一分子当たりエポキシ基を3つ以上有するエポキシ樹脂の分子量が800以下である、請求項2~6のいずれか1項に記載の樹脂組成物。
- 更に、窒素原子を含有する複素環構造を有する化合物を含む、請求項1~7のいずれか1項に記載の樹脂組成物。
- 前記凝集無機フィラーが窒化ホウ素凝集粒子である、請求項1~8のいずれか1項に記載の樹脂組成物。
- 前記窒化ホウ素凝集粒子がカードハウス構造を有するものである、請求項9に記載の樹脂組成物。
- 請求項1~10のいずれか1項に記載の樹脂組成物の硬化物からなる硬化物部と、金属部とを有する、複合成形体。
- 請求項11に記載の複合成形体を有する、半導体デバイス。
- 樹脂及び凝集無機フィラーを含む樹脂組成物を用いた樹脂硬化物であって、
85℃、85%RHでの重量増加率が0.80%以下であり、
無機フィラーを除く該樹脂組成物の硬化後の200℃における貯蔵弾性率が1.0×107Pa以上である、樹脂硬化物。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980063537.3A CN112771123B (zh) | 2018-10-11 | 2019-10-07 | 树脂组合物、树脂固化物及复合成形体 |
| EP19871054.3A EP3865543B1 (en) | 2018-10-11 | 2019-10-07 | Resin composition, resin cured product, and composite molded body |
| KR1020217008319A KR102799378B1 (ko) | 2018-10-11 | 2019-10-07 | 수지 조성물, 수지 경화물 및 복합 성형체 |
| JP2020551138A JP7543909B2 (ja) | 2018-10-11 | 2019-10-07 | 樹脂組成物、樹脂硬化物および複合成形体 |
| US17/206,389 US12264229B2 (en) | 2018-10-11 | 2021-03-19 | Resin composition, resin cured product, and composite molded body |
| JP2024125962A JP7772151B2 (ja) | 2018-10-11 | 2024-08-01 | 樹脂組成物、樹脂硬化物および複合成形体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018192691 | 2018-10-11 | ||
| JP2018-192691 | 2018-10-11 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/206,389 Continuation US12264229B2 (en) | 2018-10-11 | 2021-03-19 | Resin composition, resin cured product, and composite molded body |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020075663A1 true WO2020075663A1 (ja) | 2020-04-16 |
Family
ID=70164932
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/039448 Ceased WO2020075663A1 (ja) | 2018-10-11 | 2019-10-07 | 樹脂組成物、樹脂硬化物および複合成形体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US12264229B2 (ja) |
| EP (1) | EP3865543B1 (ja) |
| JP (2) | JP7543909B2 (ja) |
| KR (1) | KR102799378B1 (ja) |
| CN (1) | CN112771123B (ja) |
| TW (1) | TWI824032B (ja) |
| WO (1) | WO2020075663A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022210686A1 (ja) * | 2021-03-29 | 2022-10-06 | ||
| JP2022151867A (ja) * | 2021-03-26 | 2022-10-07 | 三菱ケミカル株式会社 | シート硬化物の製造方法及び複合成形体の製造方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20230122014A (ko) * | 2020-12-22 | 2023-08-22 | 디아이씨 가부시끼가이샤 | 경화성 수지, 경화성 수지 조성물, 및, 경화물 |
| CN118382656A (zh) * | 2022-03-28 | 2024-07-23 | 三菱化学株式会社 | 热固性树脂组合物、树脂固化物和复合成型体 |
| CN115572457A (zh) * | 2022-07-06 | 2023-01-06 | 江苏澳盛复合材料科技有限公司 | 一种耐环境抗老化的环氧树脂体系 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003342350A (ja) | 2002-05-30 | 2003-12-03 | Japan Epoxy Resin Kk | 高分子量エポキシ樹脂、電気積層板用樹脂組成物及び電気積層板 |
| JP2006176658A (ja) | 2004-12-22 | 2006-07-06 | Japan Epoxy Resin Kk | ポリエーテルポリオール樹脂、硬化性樹脂組成物及びその硬化物 |
| JP2006257400A (ja) * | 2005-02-18 | 2006-09-28 | Hitachi Chem Co Ltd | 新規化合物とその製造方法、及び新規化合物を含む硬化性樹脂、エポキシ樹脂組成物、電子部品装置 |
| JP2012136689A (ja) * | 2010-10-18 | 2012-07-19 | Mitsubishi Chemicals Corp | 三次元集積回路用の層間充填材組成物、塗布液及び三次元集積回路の製造方法 |
| JP2013145840A (ja) * | 2012-01-16 | 2013-07-25 | Mitsubishi Chemicals Corp | 三次元集積回路の層間充填層形成用塗布液、及び三次元集積回路の製造方法 |
| JP2013151672A (ja) * | 2011-12-26 | 2013-08-08 | Mitsubishi Chemicals Corp | エポキシ樹脂、エポキシ樹脂組成物及び硬化物 |
| JP2013221120A (ja) * | 2012-04-18 | 2013-10-28 | Mitsubishi Chemicals Corp | エポキシ樹脂組成物および該エポキシ樹脂組成物を硬化させてなる硬化物 |
| JP2015006980A (ja) * | 2013-05-27 | 2015-01-15 | 三菱化学株式会社 | 窒化ホウ素凝集粒子、凝集bn粒子含有樹脂組成物及び放熱シート |
| JP2015067816A (ja) * | 2013-09-30 | 2015-04-13 | 明和化成株式会社 | エポキシ樹脂組成物、封止材、その硬化物、及びフェノール樹脂 |
| WO2015061028A1 (en) | 2013-10-22 | 2015-04-30 | Pepsico, Inc. | D-psicose in zero or low calorie frozen beverages |
| JP2016536403A (ja) * | 2014-04-02 | 2016-11-24 | 廣東生益科技股▲ふん▼有限公司Shengyi Technology Co.,Ltd. | ノンハロゲン樹脂組成物及びその用途 |
| JP2016219619A (ja) * | 2015-05-21 | 2016-12-22 | 日東電工株式会社 | 接着シート、ダイシングテープ一体型接着シート、フィルム、半導体装置の製造方法および半導体装置 |
| JP2017036415A (ja) | 2015-08-12 | 2017-02-16 | 三菱化学株式会社 | 放熱樹脂シート及び該放熱樹脂シートを含むデバイス |
| JP2018192691A (ja) | 2017-05-17 | 2018-12-06 | オリンパス株式会社 | 複合成形体 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0817060B2 (ja) * | 1989-08-18 | 1996-02-21 | 株式会社日立製作所 | 電気絶縁線輪、回転電機及びその製造方法 |
| CN101962465A (zh) * | 2009-07-21 | 2011-02-02 | 聚鼎科技股份有限公司 | 导热电绝缘高分子材料及包含该导热电绝缘高分子材料的散热基板 |
| JP2011070930A (ja) * | 2009-09-25 | 2011-04-07 | Sekisui Chem Co Ltd | 多層絶縁シート及び積層構造体 |
| JP2011241245A (ja) * | 2010-05-14 | 2011-12-01 | Mitsubishi Chemicals Corp | エポキシ樹脂組成物および硬化物 |
| TWI616398B (zh) * | 2011-11-29 | 2018-03-01 | Mitsubishi Chemical Corporation | 含有氮化硼之組成物、以及具有由該組成物所構成的層之三維積體電路 |
| CN104220533B (zh) * | 2012-03-30 | 2016-09-21 | 昭和电工株式会社 | 固化性散热组合物 |
| JP6595336B2 (ja) * | 2013-06-25 | 2019-10-23 | 味の素株式会社 | 樹脂組成物 |
| TWI629306B (zh) * | 2013-07-19 | 2018-07-11 | Ajinomoto Co., Inc. | Resin composition |
| JP2015189884A (ja) * | 2014-03-28 | 2015-11-02 | 日立化成株式会社 | 熱硬化性樹脂組成物、樹脂シート、プリプレグ及び積層板 |
| DE112016003257T5 (de) * | 2015-07-21 | 2018-04-05 | Sumitomo Bakelite Co., Ltd. | Wärmeleitende harzzusammensetzung, wärmeleitende folie und halbleiterbauelement |
-
2019
- 2019-10-07 JP JP2020551138A patent/JP7543909B2/ja active Active
- 2019-10-07 CN CN201980063537.3A patent/CN112771123B/zh active Active
- 2019-10-07 WO PCT/JP2019/039448 patent/WO2020075663A1/ja not_active Ceased
- 2019-10-07 EP EP19871054.3A patent/EP3865543B1/en active Active
- 2019-10-07 KR KR1020217008319A patent/KR102799378B1/ko active Active
- 2019-10-09 TW TW108136587A patent/TWI824032B/zh active
-
2021
- 2021-03-19 US US17/206,389 patent/US12264229B2/en active Active
-
2024
- 2024-08-01 JP JP2024125962A patent/JP7772151B2/ja active Active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003342350A (ja) | 2002-05-30 | 2003-12-03 | Japan Epoxy Resin Kk | 高分子量エポキシ樹脂、電気積層板用樹脂組成物及び電気積層板 |
| JP2006176658A (ja) | 2004-12-22 | 2006-07-06 | Japan Epoxy Resin Kk | ポリエーテルポリオール樹脂、硬化性樹脂組成物及びその硬化物 |
| JP2006257400A (ja) * | 2005-02-18 | 2006-09-28 | Hitachi Chem Co Ltd | 新規化合物とその製造方法、及び新規化合物を含む硬化性樹脂、エポキシ樹脂組成物、電子部品装置 |
| JP2012136689A (ja) * | 2010-10-18 | 2012-07-19 | Mitsubishi Chemicals Corp | 三次元集積回路用の層間充填材組成物、塗布液及び三次元集積回路の製造方法 |
| JP2013151672A (ja) * | 2011-12-26 | 2013-08-08 | Mitsubishi Chemicals Corp | エポキシ樹脂、エポキシ樹脂組成物及び硬化物 |
| JP2013145840A (ja) * | 2012-01-16 | 2013-07-25 | Mitsubishi Chemicals Corp | 三次元集積回路の層間充填層形成用塗布液、及び三次元集積回路の製造方法 |
| JP2013221120A (ja) * | 2012-04-18 | 2013-10-28 | Mitsubishi Chemicals Corp | エポキシ樹脂組成物および該エポキシ樹脂組成物を硬化させてなる硬化物 |
| JP2015006980A (ja) * | 2013-05-27 | 2015-01-15 | 三菱化学株式会社 | 窒化ホウ素凝集粒子、凝集bn粒子含有樹脂組成物及び放熱シート |
| JP2015067816A (ja) * | 2013-09-30 | 2015-04-13 | 明和化成株式会社 | エポキシ樹脂組成物、封止材、その硬化物、及びフェノール樹脂 |
| WO2015061028A1 (en) | 2013-10-22 | 2015-04-30 | Pepsico, Inc. | D-psicose in zero or low calorie frozen beverages |
| JP2016536403A (ja) * | 2014-04-02 | 2016-11-24 | 廣東生益科技股▲ふん▼有限公司Shengyi Technology Co.,Ltd. | ノンハロゲン樹脂組成物及びその用途 |
| JP2016219619A (ja) * | 2015-05-21 | 2016-12-22 | 日東電工株式会社 | 接着シート、ダイシングテープ一体型接着シート、フィルム、半導体装置の製造方法および半導体装置 |
| JP2017036415A (ja) | 2015-08-12 | 2017-02-16 | 三菱化学株式会社 | 放熱樹脂シート及び該放熱樹脂シートを含むデバイス |
| JP2018192691A (ja) | 2017-05-17 | 2018-12-06 | オリンパス株式会社 | 複合成形体 |
Non-Patent Citations (2)
| Title |
|---|
| "Ceramic", vol. 43, 2008, THE CERAMIC SOCIETY OF JAPAN. |
| See also references of EP3865543A4 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022151867A (ja) * | 2021-03-26 | 2022-10-07 | 三菱ケミカル株式会社 | シート硬化物の製造方法及び複合成形体の製造方法 |
| JP7800253B2 (ja) | 2021-03-26 | 2026-01-16 | 三菱ケミカル株式会社 | シート硬化物の製造方法及び複合成形体の製造方法 |
| JPWO2022210686A1 (ja) * | 2021-03-29 | 2022-10-06 | ||
| JP7806785B2 (ja) | 2021-03-29 | 2026-01-27 | 三菱ケミカル株式会社 | 樹脂組成物、シート硬化物、複合成形体及び半導体デバイス |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202033668A (zh) | 2020-09-16 |
| JP7543909B2 (ja) | 2024-09-03 |
| EP3865543A1 (en) | 2021-08-18 |
| KR20210075979A (ko) | 2021-06-23 |
| EP3865543A4 (en) | 2021-11-17 |
| US20210206943A1 (en) | 2021-07-08 |
| TWI824032B (zh) | 2023-12-01 |
| JP7772151B2 (ja) | 2025-11-18 |
| JPWO2020075663A1 (ja) | 2021-09-02 |
| EP3865543B1 (en) | 2025-12-03 |
| KR102799378B1 (ko) | 2025-04-22 |
| US12264229B2 (en) | 2025-04-01 |
| JP2024149637A (ja) | 2024-10-18 |
| CN112771123A (zh) | 2021-05-07 |
| CN112771123B (zh) | 2023-08-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7772151B2 (ja) | 樹脂組成物、樹脂硬化物および複合成形体 | |
| EP4053213B1 (en) | Resin composition, cured product, composite molded body and semiconductor device | |
| US20240010814A1 (en) | Resin composition, cured product sheet, composite molded body, and semiconductor device | |
| WO2023189030A1 (ja) | 熱硬化性樹脂組成物、樹脂硬化物および複合成形体 | |
| JP7383971B2 (ja) | 樹脂組成物、樹脂硬化物および複合成形体 | |
| US20250019489A1 (en) | Thermosetting resin composition, thermally conductive resin sheet, heat-dissipating layered product, heat-dissipating circuit board, semiconductor device and power module | |
| JP2025103021A (ja) | 樹脂組成物層の製造方法、該製造方法で得られた樹脂組成物層及び該樹脂組成物層を含む複合成形体 | |
| JP2023152859A (ja) | 放熱シート積層体、及び放熱シート積層体の製造方法 | |
| JP7800253B2 (ja) | シート硬化物の製造方法及び複合成形体の製造方法 | |
| TWI915323B (zh) | 樹脂組合物、硬化物、複合成形體、半導體裝置 | |
| JP2023145370A (ja) | 熱硬化性樹脂組成物、熱硬化性樹脂シート、絶縁シート及び半導体装置 | |
| JP2023145355A (ja) | 熱硬化性樹脂組成物、熱硬化性樹脂シート、絶縁シート及び半導体装置 | |
| JP2024134539A (ja) | 熱硬化性組成物、硬化物、熱伝導性シート及び複合成形体 | |
| JP2024137893A (ja) | 熱硬化性組成物、シート状硬化物、複合成形体及び半導体デバイス | |
| JP2023147228A (ja) | 熱硬化性樹脂組成物、硬化物、複合成形体および半導体デバイス | |
| WO2025197972A1 (ja) | 熱硬化性組成物、熱硬化性シートの製造方法、熱伝導性シート、放熱積層体、放熱性回路基板及びパワー半導体装置 | |
| JP2022151865A (ja) | 積層構造体およびその製造方法 | |
| JP2023102284A (ja) | 熱伝導性樹脂組成物、熱伝導性樹脂シート、放熱積層体、放熱性回路基板、半導体装置およびパワーモジュール | |
| CN118355074A (zh) | 热固性树脂组合物、导热性树脂片、散热层叠体、散热性电路基板、半导体装置及功率模块 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19871054 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020551138 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019871054 Country of ref document: EP Effective date: 20210511 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2019871054 Country of ref document: EP |