WO2020179659A1 - 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 - Google Patents
置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 Download PDFInfo
- Publication number
- WO2020179659A1 WO2020179659A1 PCT/JP2020/008278 JP2020008278W WO2020179659A1 WO 2020179659 A1 WO2020179659 A1 WO 2020179659A1 JP 2020008278 W JP2020008278 W JP 2020008278W WO 2020179659 A1 WO2020179659 A1 WO 2020179659A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- iron
- substituted
- magnetic particle
- iron oxide
- particle powder
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G51/00—Compounds of cobalt
- C01G51/40—Complex oxides containing cobalt and at least one other metal element
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G49/00—Compounds of iron
- C01G49/02—Oxides; Hydroxides
- C01G49/06—Ferric oxide [Fe2O3]
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/10—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials non-metallic substances, e.g. ferrites, e.g. [(Ba,Sr)O(Fe2O3)6] ferrites with hexagonal structure
- H01F1/11—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials non-metallic substances, e.g. ferrites, e.g. [(Ba,Sr)O(Fe2O3)6] ferrites with hexagonal structure in the form of particles
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/12—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials
- H01F1/34—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites
- H01F1/342—Oxides
- H01F1/344—Ferrites, e.g. having a cubic spinel structure (X2+O)(Y23+O3), e.g. magnetite Fe3O4
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/12—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials
- H01F1/34—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites
- H01F1/36—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites in the form of particles
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/12—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials
- H01F1/34—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites
- H01F1/36—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites in the form of particles
- H01F1/37—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of soft-magnetic materials non-metallic substances, e.g. ferrites in the form of particles in a bonding agent
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K9/00—Screening of apparatus or components against electric or magnetic fields
- H05K9/0073—Shielding materials
- H05K9/0075—Magnetic shielding materials
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K9/00—Screening of apparatus or components against electric or magnetic fields
- H05K9/0073—Shielding materials
- H05K9/0081—Electromagnetic shielding materials, e.g. EMI, RFI shielding
- H05K9/0083—Electromagnetic shielding materials, e.g. EMI, RFI shielding comprising electro-conductive non-fibrous particles embedded in an electrically insulating supporting structure, e.g. powder, flakes, whiskers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y25/00—Nanomagnetism, e.g. magnetoimpedance, anisotropic magnetoresistance, giant magnetoresistance or tunneling magnetoresistance
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/74—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by peak-intensities or a ratio thereof only
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/42—Magnetic properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2265—Oxides; Hydroxides of metals of iron
- C08K2003/2272—Ferric oxide (Fe2O3)
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/01—Magnetic additives
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/10—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials non-metallic substances, e.g. ferrites, e.g. [(Ba,Sr)O(Fe2O3)6] ferrites with hexagonal structure
- H01F1/11—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials non-metallic substances, e.g. ferrites, e.g. [(Ba,Sr)O(Fe2O3)6] ferrites with hexagonal structure in the form of particles
- H01F1/113—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials non-metallic substances, e.g. ferrites, e.g. [(Ba,Sr)O(Fe2O3)6] ferrites with hexagonal structure in the form of particles in a bonding agent
Definitions
- the present invention relates to a substitution type ⁇ iron oxide magnetic particle powder suitable for high density magnetic recording media, radio wave absorbers, etc., and in particular, contains a non-magnetic ⁇ type iron-based oxide which is a different phase for substitution type ⁇ iron oxide.
- the present invention relates to a reduced amount of substituted ⁇ iron oxide magnetic particle powder and a method for producing the same.
- an oxide obtained by substituting a part of the Fe site of ⁇ -Fe 2 O 3 with another metal element is the same as an ⁇ type iron-based oxide and has the same crystal system as that of ⁇ -Fe 2 O 3 .
- Substituted ⁇ -iron oxide particles may be referred to as ⁇ -type iron oxides.
- ⁇ -Fe 2 O 3 is an extremely rare phase among iron oxides, but at room temperature, particles of nanometer order have a huge coercive force (Hc) of about 20 kOe (1.59 ⁇ 10 6 A/m). ), a study on a manufacturing method for synthesizing ⁇ -Fe 2 O 3 in a single phase has been made conventionally (Patent Document 1). However, when ⁇ -Fe 2 O 3 is used for a magnetic recording medium, there is currently no corresponding material for a magnetic head having a high level of saturation magnetic flux density, and therefore ⁇ -Fe 2 O is practically used. It is necessary to replace a part of the Fe site of No.
- Patent Document 2 since magnetic particles of ⁇ -type iron-based oxide are extremely fine, some of the Fe sites of ⁇ -Fe 2 O 3 are made heat-resistant in order to improve environmental stability and thermal stability.
- A is a divalent metal element such as Co, Ni, Mn, and Zn
- B is a tetravalent metal element such as Ti
- C is a trivalent metal element such as In, Ga, and Al.
- Patent Document 3 Since ⁇ -Fe 2 O 3 and ⁇ -type iron-based oxides are not thermodynamically stable phases, a special method is required for their production.
- Patent Documents 1 to 3 fine crystals of iron oxyhydroxide produced by the liquid phase method or iron oxyhydroxide containing a substituent are used as a precursor, and silicon oxide is used as the precursor by the sol-gel method.
- a method for producing an ⁇ -Fe 2 O 3 or ⁇ -type iron oxide that is heat-treated after coating is disclosed.
- As a liquid phase method a reverse micelle method using an organic solvent as a reaction medium and an aqueous solution as a reaction medium are disclosed. Each method is disclosed using only one.
- Patent Documents 1 to 3 are non-magnetic ⁇ -type iron-based oxides as impurities. It contains a considerable amount of.
- Patent Document 6 discloses a technique for reducing the amount of ⁇ -type iron-based oxide as an impurity contained in the substituted ⁇ -iron oxide magnetic particle powder.
- Patent Document 7 discloses a method for producing an ⁇ -type iron-based oxide, which coats a silicon oxide with a sol-gel method in a wide pH range.
- JP 2008-174405 A International Publication No. 2008/029861 International Publication No. 2008/149785 JP, 2008-277726, A JP, 2009-224414, A JP, 2016-130208, A Japanese Patent Laid-Open No. 2018-092691
- the ⁇ -type iron-based oxide produced by the production method disclosed in Patent Document 4 in which a part of the Fe sites is replaced is an impurity compared to the ⁇ -type iron-based oxide produced by the conventional method.
- the content of ⁇ -type iron oxide is reduced.
- the ⁇ -type iron-based oxide is a metastable phase, and when the substitution amount of Fe by another metal element is small, ⁇ -Fe 2 O is used even if the production method disclosed in Patent Document 4 is used. It became difficult to take the same space group as in No. 3, and the reduction of the content of ⁇ -type iron-based oxides was sometimes insufficient.
- the technical problem to be solved in the present invention is a method for producing substituted ⁇ -iron oxide magnetic particle powder and substituted ⁇ -iron oxide magnetic particle powder in which the content of non-magnetic ⁇ -type iron oxide is reduced. Is to provide.
- the present inventors have conducted diligent research focusing on the fact that in order to obtain a substituted ⁇ -iron oxide magnetic particle powder, it is necessary to heat the precursor of the magnetic particle powder in a state of being coated with a silicon oxide.
- a hydrolyzable silicon compound used for coating By adding a hydrolyzable silicon compound used for coating to an aqueous solution containing the precursor at a pH of 2.0 or more and 7.0 or less, the content of ⁇ -type iron-based oxide was reduced. It turned out that it could be done.
- the present inventors have completed the present invention described below.
- the substitution amount of Fe by another metal element defined by Me/(Fe+Me) is 0.08.
- a substitutional type ⁇ iron oxide magnetic particle powder which is 0.17 or less and the content of ⁇ -type iron-based oxide measured by X-ray diffraction is 3% or less.
- the other metal element that partially replaces the Fe site is preferably one or more selected from Co, Ti, and Ga and Al.
- an ⁇ -type iron-based oxide containing Co and Ti and at least one selected from Ga and Al as another metal element that partially replaces the Fe site is a suitable target.
- the present invention also provides a green compact made of the above-mentioned substituted ⁇ -iron oxide magnetic particle powder.
- the present invention also provides a radio wave absorber in which the above-mentioned substituted ⁇ -iron oxide magnetic particle powder is dispersed in a resin or rubber.
- an acidic aqueous solution containing trivalent iron ions and metal ions partially replacing the Fe sites is used as a raw material solution, and an alkali is added to the raw material solution to adjust the pH to 8.0 or higher.
- a dispersion liquid containing iron oxyhydroxide or a mixture of iron oxyhydroxide and a hydroxide of a substituted metal element and the silicon compound described above is maintained at a pH of 8.0 or more and 10.0 or less, and oxywater containing
- a part of the Fe site of ⁇ -Fe 2 O 3 including a aging step of coating the chemical reaction product of the silicon compound with iron oxide or a mixture of oxyhydroxide and a hydroxide of a substituted metal element, etc.
- both the addition of the alkali and the addition of the silicon compound may be carried out intermittently, or the addition of the alkali may be carried out intermittently and the addition of the silicon compound may be carried out continuously.
- the other metal element partially substituting the Fe site is at least one selected from Co, Ti, and Ga and Al.
- Co and Ti are contained as other metal elements that partially replace Fe sites, and one or more selected from Ga and Al are contained.
- the present invention also provides a method for producing a green compact, which obtains a green compact by compression molding the above-mentioned substituted iron oxide magnetic particle powder.
- the production method of the present invention is for producing a substitutional type ⁇ iron oxide magnetic particle powder mainly containing an ⁇ type iron-based oxide in which a part of the Fe sites of ⁇ -Fe 2 O 3 is substituted with another metal element.
- the magnetic particle powder contains different phases, which are unavoidable impurities in its production.
- the heterogeneous phase is mainly ⁇ -type iron-based oxide
- the iron oxide magnetic particle powder obtained by the present invention is substantially composed of ⁇ -type iron-based oxide magnetic particles and ⁇ -type iron-based oxide.
- An object of the present invention is to reduce the content of ⁇ -type iron-based oxide that is a different phase.
- a partially substituted product in which a part of the Fe site of ⁇ -Fe 2 O 3 is replaced with another metal element has an ⁇ structure is determined by X-ray diffraction (XRD), high-speed electron diffraction (HEED), etc. It is possible to confirm using.
- XRD X-ray diffraction
- HEED high-speed electron diffraction
- the ⁇ type and ⁇ type iron-based oxides are identified by XRD.
- the following are examples of partially substituted products that can be produced by the production method of the present invention. It is represented by the general formula ⁇ -C z Fe 2-z O 3 (where C is one or more trivalent metal elements selected from In, Ga, and Al).
- Formula ⁇ -A x B y Fe 2 -x-y O 3 (where A is Co, Ni, Mn, 1 or more divalent metal element selected from Zn, B is selected Ti, Sn, One or more kinds of tetravalent metal elements).
- General formula ⁇ -A x C z Fe 2-x-z O 3 (where A is one or more divalent metal elements selected from Co, Ni, Mn, Zn, and C is from In, Ga, Al. One or more selected trivalent metal elements).
- General formula ⁇ -B y C z Fe 2-yz O 3 (where B is one or more tetravalent metal elements selected from Ti and Sn, C is selected from In, Ga and Al 1 Those represented by (trivalent metal elements above the species).
- Formula ⁇ -A x B y C z Fe 2-x-y-z O 3 (where A is Co, Ni, Mn, 1 or more divalent metal element selected from Zn, B is Ti, One or more tetravalent metal elements selected from Sn, and C is one or more trivalent metal elements selected from In, Ga, and Al).
- the type in which only the C element is substituted has the advantage that the coercive force of the magnetic particles can be arbitrarily controlled and that the same space group as ⁇ -Fe 2 O 3 can be easily obtained, but when the thermal stability is poor.
- the thermal stability of the obtained substituted ⁇ -iron oxide magnetic particle powder is slightly inferior, so that it is preferable to replace them with A and / or B elements at the same time.
- the three-element substitution type of A, B, and C has the best balance of the above-mentioned characteristics, is excellent in heat resistance, is easy to obtain a single phase, and has controllability of coercive force.
- the production method of the present invention can be applied to any of the above-mentioned substitution type iron oxide magnetic particles.
- the production method of the present invention which will be described later, can be applied to any value of the substitution amount of the metal element that replaces the Fe site, but is applied at a substitution amount that easily forms an ⁇ -type iron oxide. Is effective. Specifically, when the number of moles of Fe contained in the substituted ⁇ iron oxide magnetic particle powder is Fe and the number of moles of all metal elements substituted with Fe sites is Me, it is defined as Me / (Fe + Me).
- the substituted ⁇ iron oxide magnetic particle powder obtained by the present invention functions as a radio wave absorber having an excellent radio wave absorbing ability by forming a packed structure of the powder particles.
- the packed structure referred to here means that each particle constitutes a three-dimensional structure in a state where the particles are in contact with each other or in close proximity to each other. It is necessary to maintain the packed structure in order to put the radio wave absorber into practical use.
- a method of compression-molding the substitution type ⁇ iron oxide magnetic particle powder into a green compact, or a filling structure by fixing the substitution type ⁇ iron oxide magnetic particle powder using a non-magnetic polymer compound as a binder The method of forming is mentioned.
- the substituted ⁇ -iron oxide magnetic particle powder is mixed with a non-magnetic polymer base material to obtain a kneaded product.
- the blending amount of the radio wave absorbing material powder in the kneaded material is preferably 60% by mass or more. The larger the amount of the radio wave absorbing material powder, the more advantageous it is in improving the radio wave absorbing characteristics, but if it is too large, it becomes difficult to knead with the polymer base material, so care must be taken.
- the blending amount of the radio wave absorbing material powder can be 80 to 95% by mass or 85 to 95% by mass.
- polymer base material various materials satisfying heat resistance, flame retardancy, durability, mechanical strength, and electrical characteristics can be used depending on the usage environment.
- an appropriate resin nylon or the like
- gel silicone gel or the like
- thermoplastic elastomer rubber or the like
- two or more kinds of polymer compounds may be blended and used as a base material.
- the average particle size of the iron oxide magnetic particle powder obtained by the production method of the present invention is not particularly specified, but it is preferable that each particle is fine enough to have a single magnetic domain structure. Usually, particles having an average particle size measured by a transmission electron microscope of 10 nm or more and 40 nm or less are obtained.
- an acidic aqueous solution (hereinafter referred to as a raw material solution) containing trivalent iron ions as a starting material of iron-based oxide magnetic particle powder and metal ions of a metal element that finally replaces Fe sites. .) Is used.
- a divalent Fe ion is used instead of the trivalent Fe ion as a starting material
- a divalent iron hydrate oxide or a divalent iron hydrate oxide is used as a precipitate in addition to the trivalent iron hydrate oxide. Since a mixture containing magnetite and the like is produced and the shape of the iron-based oxide particles finally obtained varies, the content of the ⁇ -type iron-based oxide as in the present invention is reduced.
- Substituted ⁇ iron oxide magnetic particle powder cannot be obtained.
- acidity means that the pH of the liquid is less than 7.0.
- a water-soluble inorganic acid salt such as nitrate, sulfate, or chloride from the viewpoint of availability and price.
- these metal salts When these metal salts are dissolved in water, the metal ions dissociate and the aqueous solution becomes acidic.
- alkali is added to the acidic aqueous solution containing metal ions to neutralize it, a mixture of iron oxyhydroxide and hydroxide of a substituent or oxyhydroxide in which a part of Fe site is substituted with another metal element.
- a precipitate of iron (hereinafter, these are collectively referred to as iron oxyhydroxide containing a substituent) is obtained.
- iron oxyhydroxide containing these substituents is used as a precursor of the substituted ⁇ iron oxide magnetic particle powder.
- the total metal ion concentration in the raw material solution is not particularly specified in the present invention, but is preferably 0.01 mol / L or more and 0.5 mol / L or less. If it is less than 0.01 mol / L, the amount of the substituted ⁇ -iron oxide magnetic particle powder obtained in one reaction is small, which is economically unfavorable. If the total metal ion concentration exceeds 0.5 mol/L, rapid precipitation of hydroxide causes the reaction solution to gel easily, which is not preferable.
- a dispersion liquid containing a precipitate of iron oxyhydroxide containing a substitution element is prepared.
- the hydroxide of trivalent iron ions is mainly composed of oxyhydroxide.
- the pH of the dispersion liquid is set to 8.0 or higher in order to complete the precipitation formation of the hydroxide of the substitution metal element, for example, Co, and to accelerate the condensation reaction of the silanol derivative which is a hydrolysis product. This is because.
- the upper limit of the pH reached in the neutralization step is not particularly specified, but the effect of neutralization is saturated and the effect of promoting the condensation reaction of the silanol derivative described later is reduced. It is preferably 0.
- the alkali used for the neutralization may be any of alkali metal or alkaline earth hydroxides, aqueous ammonia, and ammonium salts such as ammonium hydrogencarbonate, but the final heat treatment is ⁇ -type iron-based oxidation. It is preferable to use aqueous ammonia or ammonium hydrogen carbonate, which does not leave impurities when made into a product.
- alkalis may be added as solids to the aqueous solution of the starting material, but from the viewpoint of ensuring the uniformity of the reaction, it is preferable to add them in the state of aqueous solution.
- a precipitate of iron oxyhydroxide containing a substitution element is deposited. Stir by known mechanical means.
- the addition of alkali to the raw material solution may be carried out continuously from the start to the end of the addition. Further, the addition of the alkali may be interrupted and a predetermined pH holding time may be provided before the pH of the dispersion liquid reaches 8.0.
- the pH holding time can be set a plurality of times, and the addition of alkali can be performed intermittently.
- the number of times the pH holding time is provided, that is, the number of times the alkali addition is interrupted is preferably 3 times or less in order to avoid complication of the manufacturing process.
- the reaction temperature during the neutralization treatment is 5 ° C. or higher and 60 ° C. or lower. If the reaction temperature is less than 5 ° C., the cost of cooling increases, which is not preferable. If the temperature exceeds 60 ° C., ⁇ -type oxides having a different phase are likely to be formed in the end, which is not preferable. More preferably, it is 10 ° C. or higher and 40 ° C.
- the iron oxyhydroxide containing a substituent which is a precursor of the substituted ⁇ -iron oxide magnetic particle powder produced in the above step is ⁇ -type iron even if it is heat-treated as it is. Since it is difficult to change the phase to a system oxide, it is necessary to coat iron oxyhydroxide containing a substituent with a chemical reaction product obtained by a hydrolysis reaction and a condensation reaction of a silicon compound prior to the heat treatment.
- the chemical reaction product of the silicon compound means not only a silicon oxide having a stoichiometric composition but also a non-stoichiometric composition such as a silanol derivative or a polysiloxane structure which will be described later, and a silicon oxide obtained by heat treatment. It is used as a general term for things that have changed into things.
- the sol-gel method is used as a method for coating the chemical reaction product of the silicon compound, the neutralization treatment of the raw material solution is completed, and the pH of the reaction solution is alkaline. After that, a silicon compound having a hydrolyzing group is added to the reaction solution.
- the sol-gel method is also used as the coating method for the silicon oxide, but the pH of the reaction solution is 2.0 or more on the acidic side before the neutralization of the raw material solution is completed. It is characterized in that the addition of the silicon compound having a hydrolyzing group is started at a time in the range of .0 or less. The end time of the addition of the silicon compound will be described later.
- a silicon compound having a hydrolyzing group such as tetraethoxysilane (TEOS), tetramethoxysilane (TMS) and other alcoholicylans in a dispersion containing iron oxyhydroxide containing a substituent, and A silane compound such as various silane coupling agents is added to cause a hydrolysis reaction under stirring, and the generated silanol derivative is condensed to form a polysiloxane bond. Cover the surface.
- the present inventors have started the addition of the above-mentioned silicon compound having a hydrolyzing group at pH 2.0 or more and 7.0 or less, and the ⁇ -type iron contained in the finally obtained substituted ⁇ -iron oxide magnetic particle powder.
- the rate of the hydrolysis reaction of the silicon compound having a hydrolysis group and the condensation reaction of the silanol derivative which is a hydrolysis product changes depending on the pH of the reaction system.
- the hydrolysis reaction rate is generally large in the low pH region on the acidic side, decreases with increasing pH, and increases again in the high pH region on the alkaline side.
- the rate of the condensation reaction is small in the low pH range on the acidic side, increases with increasing pH, and increases in the pH range on the neutral to alkaline side.
- a silicon compound having a hydrolyzing group is added to a dispersion containing a precipitate of iron oxyhydroxide containing a substituent in a low pH region on the acidic side, the hydrolysis of the silicon compound proceeds rapidly, and the organic component becomes a component. While a small amount of silanol derivative is produced, the condensation reaction of the produced silanol derivative does not proceed.
- the silanol derivative since the silanol derivative has an OH group which is a hydrophilic group and is uniformly distributed in an aqueous solution, the precipitation of iron oxyhydroxide containing a substitution element and the silanol derivative coexist in the dispersion liquid. It is considered to be in a state. After that, when the pH of the dispersion is further increased, the condensation reaction of the silanol derivative becomes dominant, so that the precipitate of iron oxyhydroxide containing the substitution element is uniformly coated with the silanol derivative or its condensation reaction product. Become. Therefore, it is considered that the content of ⁇ -type iron oxide contained in the substituted ⁇ -iron oxide magnetic particle powder finally obtained by heat treatment is reduced.
- Patent Document 5 discloses that the silicon oxide is coated by the sol-gel method in a wide pH range, but in that case, the addition of the silicon compound is constant after the completion of the neutralization treatment. As in the present invention, the technical idea of considering both the hydrolysis reaction rate and the condensation reaction rate of the silicon compound is not disclosed.
- the pH at the start of addition of the silicon compound having a hydrolyzable group is preferably 2.0 or higher. If the pH is less than 2.0, the precipitation formation of the hydroxide of trivalent iron ions contained in the raw material solution, which is the main component of the substituted iron oxide magnetic particle powder, may be insufficient.
- the pH at the time of starting the addition is more preferably 3.0 or higher. Further, the pH at the time of starting the addition of the silicon compound having a hydrolyzing group is preferably 7.0 or less. If the pH exceeds 7.0, the hydrolysis reaction will be delayed and the silanol derivative will be insufficiently produced.
- the pH at the time of starting the addition is preferably 6.0 or less, more preferably 4.0 or less.
- Addition of the silicon compound having a hydrolyzing group is started when the pH of the raw material solution reaches a desired value in the neutralization step.
- the addition of the silicon compound may be carried out continuously from the start to the end of the addition.
- continuous includes adding the entire amount of the silicon compound added to the dispersion liquid to the dispersion liquid at one time. Further, the addition of the silicon compound may be divided into a plurality of times and may be performed intermittently.
- the amount of the silicon compound added to the dispersion must satisfy the following two conditions at the same time.
- the first condition is the amount of silicon compound added to the dispersion having a pH of 2.0 or more and 7.0 or less.
- the number of moles of the silicon compound added to the dispersion having a pH of 2.0 or more and 7.0 or less was S1
- the number of moles of Fe ions contained in the raw material solution was F
- the total number of moles of substituted metal element ions was M.
- S1 / (F + M) is set to 0.01 or more and 10.0 or less.
- S1/(F+M) is less than 0.01
- the amount of the silanol derivative coexisting with the precipitation of the iron oxyhydroxide containing the substitution element is small, and the precipitation of the iron oxyhydroxide containing the substitution element causes the silanol derivative or its condensation reaction. It is not preferable because the product reduces the effect of uniformly coating.
- S1 / (F + M) is preferably 10.0 or less.
- the second condition is the amount of silicon compound added throughout the manufacturing process.
- the total number of moles of the silicon compound to be added is S2
- the number of moles of Fe ions contained in the raw material solution is F
- the total number of moles of substituted metal element ions is M
- S2/(F+M) is 0.50 or more 10 It shall be .0 or less.
- S2 is less than 0.50, the amount of the chemical reaction product of the silicon compound coated on the surface of the precipitate of iron oxyhydroxide containing a substituent is reduced, and as a result, the ⁇ -type iron oxide is produced. There is a demerit that it becomes easy to generate, which is not preferable.
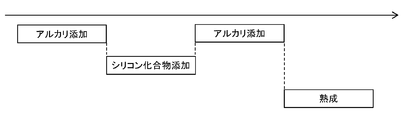
- FIG. 1 schematically illustrates the time course of an embodiment in which both the addition of alkali and the addition of a silicon compound are continuously performed.
- the addition of the silicon compound is started when the pH of the raw material solution reaches a predetermined pH in the range of 2.0 or more and 7.0 or less.
- the neutralization step and the silicon compound addition step are not continuous steps in time but parallel steps.
- the addition of the silicon compound may be continued after the addition of the alkali is finished, or even when the addition of the alkali is finished, it is finished when the pH is 7.0 or less. It doesn't matter.
- the silicon compound is added even after the completion of the alkali addition, it is preferable to finish the addition of the silicon compound within 120 minutes after the completion of the alkali addition in consideration of the time of the entire production process.
- the aging step described later is provided.
- the addition of the silicon compound may be divided into several times and may be performed intermittently.
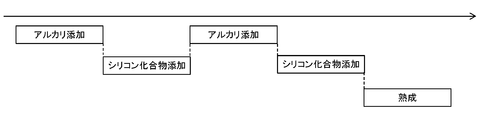
- FIG. 2 schematically illustrates the time course of an example of an embodiment in which the addition of alkali is interrupted in the middle of the neutralization step.
- FIG. 3 schematically illustrates a time course of another example of the embodiment in which the alkali addition is once interrupted.
- the addition of the silicon compound is started when the pH of the raw material solution reaches a predetermined pH in the range of 2.0 or more and 7.0 or less, but the end time of the addition is after the end of the neutralization step.
- the form of addition of the silicone compound is continuous.
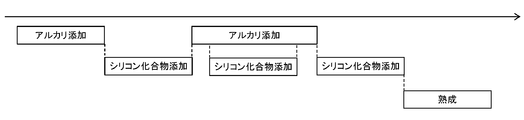
- FIGS. 4 and 5 schematically illustrate the passage of time in an example of an embodiment in which the addition of alkali is interrupted once and the silicon compound is added intermittently.
- the addition of the silicon compound is carried out in two steps in FIG. 4 and in three steps in FIG.
- FIG. 6 schematically illustrates the passage of time in an example of an embodiment in which the addition of alkali is interrupted twice and the addition of the silicon compound is intermittently performed three times.
- the production method of the present invention is not limited to the embodiments illustrated in FIGS. 1 to 6, and the addition of alkali in the neutralization step and the addition of the silicon compound in the silicon compound addition step. It is possible to arbitrarily combine the forms of.
- this coating layer covers almost the entire surface of the precipitate of iron oxyhydroxide containing the substituting element, but within the range where the effect of the present invention can be achieved, the surface of the precipitate of iron oxyhydroxide containing the substituting element.
- the presence of uncoated parts in is acceptable.
- the aging time is preferably 1 h or more and 24 hours or less. If the holding time is less than 1 h, the coating of the precipitation of iron oxyhydroxide containing a substitution element by condensation of the silanol derivative is not completed, and ⁇ -type iron-based oxides are easily generated. Is saturated, which is not preferable.
- the aging time is set after the addition of the silicon compound is completed when the addition of the silicon compound is continued even after the completion of the neutralization step, and when the addition of the silicon compound is completed before the completion of the neutralization step, the addition of alkali is performed. It is the time after the end of.
- the steps after the aging step can use the same steps as the conventional production methods described in Patent Documents 1 to 4, for example. .. Specifically, the following steps can be mentioned.
- Heating process In the production method of the present invention, iron oxyhydroxide containing a substituent coated with the condensation reaction product of the silanol derivative is recovered by a known solid-liquid separation method and then heat-treated to form an ⁇ type. To obtain the iron-based oxide of. A washing and drying steps may be provided before the heat treatment. The heat treatment is performed in an oxidizing atmosphere, but the oxidizing atmosphere may be an atmospheric atmosphere. Heating can be carried out in a range of approximately 700 ° C.
- the heat treatment is preferably performed at 900 ° C. or higher and 1200 ° C. or lower, more preferably 950 ° C. or higher and 1150 ° C. or lower.
- the heat treatment time can be adjusted in the range of 0.5H or more and 10H or less, but good results are likely to be obtained in the range of 2H or more and 5H or less.
- the presence of the silicon-containing substance covering the particles has an advantageous effect on causing the phase change to the ⁇ -type iron-based oxide instead of the phase change to the ⁇ -type iron-based oxide.
- the silicon oxide coating has an effect of preventing sintering of iron oxyhydroxide crystals containing a substituent during heat treatment.
- the silicon oxide coating may be removed after the heat treatment.
- composition analysis by radio frequency inductively coupled plasma emission spectroscopy (ICP)
- the composition analysis of the obtained substitution type ⁇ iron oxide magnetic particle powder was performed by a dissolution method.
- ICP-720ES manufactured by Agilent Technologies was used for composition analysis, and the measurement wavelength (nm) was Fe; 259.940 nm, Ga; 294.363 nm, Co; 230.786 nm, Ti; 336.122 nm, Si; 288. At 158 nm.
- the BET specific surface area was determined by the BET one-point method using a Macsorb model-1210 manufactured by Mountech Co., Ltd.
- the TEM observation of the substituted ⁇ -iron oxide magnetic particle powder obtained by the production method of the present invention was carried out under the following conditions. JEM-1011 manufactured by JEOL Ltd. was used for TEM observation. For particle observation, TEM photographs taken at a magnification of 10,000 times and a magnification of 100,000 times were used. (Use the one after removing the silicon oxide coating). -Measurement of average particle size and particle size distribution evaluation (coefficient of variation (%))- Digitization was used for TEM average particle size and particle size distribution evaluation (coefficient of variation (%)). As image processing software, Mac-View Ver.4.0 was used.
- the particle size of a particle is calculated as the length of the long side of the rectangle having the smallest area among the rectangles circumscribing the particle.
- the number 200 or more were measured.
- the selection criteria for the particles to be measured were as follows. [1] Some particles that are outside the visual field of the photograph are not measured. [2] Particles with a well-defined contour and existing independently are measured. [3] Even if the particle shape deviates from the average particle shape, particles that are independent and can be measured as a single particle are measured. [4] Particles that overlap each other, but the boundaries between the particles are clear, and the shape of the entire particle can be determined are measured as individual particles.
- Example 1 In a 5 L reaction tank, 2833.21 g of pure water, 283.26 g of 99% ferric nitrate nonahydrate (III), 56.36 g of Ga(III) nitrate solution having a Ga concentration of 11.55 mass%, and a purity of 97 6.25 g of% cobalt nitrate (II) hexahydrate and 6.61 g of titanium (IV) sulfate having a Ti concentration of 15.1 mass% were dissolved in an air atmosphere while mechanically stirring with a stirring blade to prepare a raw material solution. (Procedure 1). The pH of this raw material solution was about 1.
- the Roman numeral in parentheses after the reagent name indicates the valence of the metal element.
- 29.85 g of 22.30 mass% aqueous ammonia solution was added over 90 minutes while mechanically stirring the raw material solution at 20° C. with a stirring blade (procedure 2).
- 519.22 g of tetraethoxysilane (TEOS) having a purity of 95.0 mass% was concurrently used as a silicon compound having a hydrolyzing group.
- TEOS tetraethoxysilane
- the amount of Si element contained in the tetraethoxysilane added to the dispersion having a pH of 2.0 or more and 7.0 or less, and the amount of iron, gallium, cobalt, or titanium ions contained in the raw material solution The molar ratio of S1 / (F + M) is 0.34, and the total amount of Si elements contained in tetraethoxysilane dropped in the dispersion and the amount of iron, gallium, cobalt, and titanium ions contained in the raw material solution.
- the molar ratio S2 / (F + M) of is 2.84.
- step 3 The slurry obtained in step 3 is filtered, and the water of the iron oxyhydroxide precipitate containing the substitution element coated with the obtained chemical reaction product of the silicon compound is drained as much as possible and then dispersed again in pure water. Repulp washed. The washed slurry was filtered again and the resulting cake was dried in the air at 110 ° C. (step 4).
- the dried product obtained in Procedure 4 was heat-treated at 1090° C. for 4 hours in the atmosphere using a box-type firing furnace to obtain iron oxide magnetic powder coated with silicon oxide (Procedure 5).
- the chemical reaction product of the silicon compound is dehydrated and converted into an oxide when heat-treated in an air atmosphere.
- Table 1 shows the production conditions such as the preparation conditions of the raw material solution of this example. Table 1 also shows the production conditions of other examples and comparative examples.
- the heat-treated powder obtained in step 5 obtained by heat-treating the precipitate of iron oxyhydroxide containing a substituent coated with the chemical reaction product of the silicon compound was subjected to heat treatment in a 17.58 mass% NaOH aqueous solution at about 60 ° C., 24. After stirring for a time, the silicon oxide coating on the particle surface was removed (procedure 6). Then, the slurry was washed using a centrifuge until the conductivity of the slurry became 500 mS / m or less, filtered through a membrane filter, and then dried. Chemical analysis, XRD measurement and XRD measurement of the composition of the obtained iron-based oxide magnetic powder were performed. It was used for measurement of magnetic properties. Table 2 shows the measurement results.
- Table 2 also shows the physical property values of the substituted ⁇ -iron oxide magnetic particle powder obtained in the other examples and comparative examples.
- the substituted ⁇ iron oxide magnetic particle powder according to Example 1 was subjected to XRD measurement, and the ⁇ phase content was determined to be 1.3%. This value is for the substituted ⁇ -iron oxide powder obtained in Comparative Example 1 in which an intermediate aging step was provided after the pH was 8.9 after the addition of the ammonia solution, which will be described later, and then TEOS was added in the state of pH 8.9. It's better than that.
- chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Example 2 As Example 2, the temperature at the time of adding the aqueous ammonia solution was 30° C., the adding time of the aqueous ammonia solution was 30 min, the starting pH of TEOS addition was 6.0, the addition rate of TEOS was 5.88 g/min during the addition of the aqueous ammonia solution, After the addition of the aqueous ammonia solution was completed, a substitutional type ⁇ iron oxide magnetic particle powder was obtained by the same procedure as in Example 1 except that the amount was 22.47 g/min.
- the pH at the end of the addition of the aqueous ammonia solution was 8.9, and the pH of the reaction solution at the end of the addition of TEOS and the pH of the reaction solution during the stirring for 20 hours were both 8.8. there were.
- the substituted ⁇ iron oxide magnetic particle powder according to Example 2 was subjected to XRD measurement, and the ⁇ phase content was determined to be 3.0%. This value is determined for the substituted ⁇ -iron oxide magnetic particle powder obtained in Comparative Example 3 in which a aging step was performed for 30 minutes after the pH became 8.9 after the addition of the ammonia solution, which will be described later, and then TEOS was added in the state of pH 8.9. It is better than that of.
- chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Comparative Example 1 As Comparative Example 1, substituted ⁇ iron oxide magnetic particles were added in the same procedure as in Example 1 except that TEOS was added when the pH reached 8.9 by adding an aqueous ammonia solution, and then an aging step was provided. I got the powder. The pH at the end of the ammonia addition was 8.9, and the pH of the reaction liquid at the end of the TEOS addition and the pH of the reaction liquid during the stirring for 20 h were both 8.8.
- the substitution type ⁇ iron oxide magnetic particle powder according to Comparative Example 1 was subjected to XRD measurement, and the content of ⁇ phase was determined to be 3.6%, which was a high value as compared with Examples 1 to 6. In addition, chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Comparative Example 3 As Comparative Example 3, a aging step was provided after adding an aqueous ammonia solution to a pH of 8.9, and then TEOS was added in a state of pH 8.9. Substitution-type ⁇ -oxidation was carried out in the same procedure as in Example 2. Iron magnetic particle powder was obtained. The pH of the reaction solution at the end of addition of TEOS and the pH of the reaction solution during the stirring for 20 hours were both 8.8. The substitution type ⁇ iron oxide magnetic particle powder according to Comparative Example 3 was subjected to XRD measurement, and the content of ⁇ phase was determined to be 5.4%, which was a high value as compared with Examples 1 to 6. In addition, chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Example 3 As Example 3, after adding the aqueous ammonia solution to pH 4.0 (first neutralization step), the addition of the aqueous ammonia solution was stopped, the addition of TEOS was immediately started without providing an intermediate aging step, and the addition of TEOS was performed. Substituted ⁇ -iron oxide magnetic particle powder was obtained in the same procedure as in Example 1 except that the remaining aqueous ammonia solution was added (second neutralization step) after completion. The pH of the reaction solution at the end of the second neutralization step and the pH of the reaction solution during the stirring for 20 hours were both 8.8. The substitution type ⁇ iron oxide magnetic particle powder according to Example 3 was subjected to XRD measurement, and the content rate of the ⁇ phase was 0%.
- Example 4 In a 1 L reaction tank, in pure water 737.71 g, ferric sulfate (III) sulfate solution 104.81 g with Fe concentration 11.65 mass%, nitric acid Ga (III) solution 14.32 g with Ga concentration 11.55 mass%, purity 1.91 g of 97% cobalt nitrate (II) hexahydrate and 2.02 g of titanium (IV) sulfate having a Ti concentration of 15.1 mass% were dissolved in an air atmosphere while mechanically stirring with a stirring blade (procedure 1). .. The pH of this solution was about 1.
- 15.00 g of a 22.30 mass% aqueous ammonia solution was added in 1.9 min while mechanically stirring the prepared solution under the condition of 30 ° C. with a stirring blade (first neutralization step). Then, after the completion of the dropping, stirring was continued for 30 minutes to mature the produced precipitate (intermediate aging step). At that time, the pH of the slurry containing the precipitate was 2.0 (procedure 2).
- TEOS tetraethoxysilane
- Procedure 2 While stirring the slurry obtained in Procedure 2, 158.88 g of tetraethoxysilane (TEOS) having a purity of 95.0 mass% was added dropwise at 30° C. in the air over 10 minutes. After the addition of TEOS was completed, 62.77 g of a 22.30 mass% ammonia solution was added in 8.1 min (second neutralization step). The pH after the second neutralization step was 8.8. After that, stirring was continued for 20 hours, and the precipitate was coated with the chemical reaction product of the silicon compound (procedure 3). The pH of the reaction solution during the stirring for 20 hours was 8.8.
- TEOS tetraethoxysilane
- the molar ratio of S1 / (F + M) is 2.84, and the total amount of Si elements contained in tetraethoxysilane dropped in the dispersion and the amount of iron, gallium, cobalt, and titanium ions contained in the raw material solution.
- the substituted ⁇ iron oxide powder according to Example 4 was subjected to XRD measurement, and the ⁇ phase content was determined to be 0%. This value is superior to those for the substituted ⁇ -oxidized magnetic particle powder obtained in Comparative Examples 4 and 5 described later.
- chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Example 5 As Example 5, the addition amount and the addition time of the aqueous ammonia solution used in the first neutralization step are 51.00 g and 6.5 min, the pH after the first neutralization step is 3.0, and the second neutralization step is performed. Substituted ⁇ iron oxide magnetic particle powder was obtained in the same procedure as in Example 4 except that the addition amount and addition time of the ammonia solution used were 27.24 g and 3.5 min. The pH of the reaction solution at the end of the second neutralization step and the pH of the reaction solution during the stirring for 20 hours were both 8.8. The substitution type ⁇ iron oxide magnetic particle powder according to Example 5 was subjected to XRD measurement, and the content of the ⁇ phase was determined to be 0%. This value is superior to those for the substituted ⁇ -iron oxide magnetic particles obtained in Comparative Examples 4 and 5 described later. In addition, chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Example 6 As Example 6, the addition amount and the addition time of the aqueous ammonia solution used in the first neutralization step were 53.00 g and 6.8 min, the pH after the first neutralization step was 4.0, and the second neutralization step was performed. Substituted ⁇ iron oxide magnetic particle powder was obtained in the same procedure as in Example 4 except that the addition amount and addition time of the ammonia solution used were 24.78 g and 3.2 min. The pH of the reaction solution after the second neutralization step and the pH of the reaction solution during the stirring for 20 hours were 8.8. The substituted ⁇ iron oxide powder according to Example 5 was subjected to XRD measurement, and the ⁇ phase content was determined to be 0%. This value is superior to those for the substituted ⁇ -iron oxide magnetic particle powder obtained in Comparative Examples 4 and 5 described later. In addition, chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 1.
- Example 7 In a 1-liter reaction tank, in 746.26 g of pure water, 102.73 g of a ferric sulfate (III) solution having a Fe concentration of 11.65 mass%, purity of 98% aluminum nitrate (III) nonahydrate 10.74 g, purity of 97 % Cobalt(II) nitrate hexahydrate (1.91 g) and titanium sulfate (IV) having a Ti concentration of 15.1 mass% (2.02 g) were dissolved in an air atmosphere with mechanical stirring using a stirring blade (procedure 1). The pH of this solution was about 1.
- 51.59 g of a 22.30 mass% aqueous ammonia solution was added in 60 minutes (first neutralization step) while mechanically stirring the prepared solution under the condition of 30 ° C. with a stirring blade. After the completion of the dropping, stirring was continued for 10 minutes to ripen the produced precipitate (intermediate aging step). At that time, the pH of the slurry containing the precipitate was 3.0 (procedure 2).
- TEOS tetraethoxysilane
- 27.09 g of a 22.30 mass% ammonia solution was added in 14 minutes (second neutralization step).
- the pH after the second neutralization step was 8.8.
- stirring was continued for 20 hours, and the precipitate was coated with the chemical reaction product of the silicon compound (procedure 3).
- the pH of the reaction solution during the stirring for 20 hours was 8.8.
- the molar ratio S1/(F+M) is 2.84, and the total amount of Si element contained in the tetraethoxysilane dropped into the dispersion and the amount of iron, aluminum, cobalt, and titanium ions contained in the raw material solution.
- the molar ratio S2/(F+M) is 2.84.
- Comparative Example 4 As Comparative Example 4, a substituted ⁇ -iron oxide powder was obtained in the same procedure as in Example 6 except that the second neutralization step was not carried out.
- the pH of the reaction solution at the end of the addition of TEOS and the pH of the reaction solution during the stirring for 20 hours were 4.0.
- the substitution type ⁇ iron oxide magnetic particle powder according to Comparative Example 4 was subjected to XRD measurement, and the content rate of the ⁇ phase was determined to be 87.5%, which was a high value as compared with Examples 1 to 7.
- chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
- Comparative Example 5 As Comparative Example 5, Comparative Example except that the addition amount and addition time of the aqueous ammonia solution used in the first neutralization step were 57.00 g and 7.3 min, and the pH at the end of the first neutralization step was 6.0. Substituted ⁇ iron oxide magnetic particle powder was obtained in the same procedure as in 4. The pH of the reaction solution at the end of the addition of TEOS and the pH of the reaction solution during the stirring for 20 hours were 6.0. The substitution type ⁇ iron oxide magnetic particle powder according to Comparative Example 5 was subjected to XRD measurement, and the content of ⁇ phase was determined to be 36.9%, which was a high value as compared with Examples 1 to 7. In addition, chemical analysis of the composition and evaluation of magnetic properties were performed.
- Example 8 Substitution type ⁇ iron oxide magnetic particle powder was obtained by the same procedure as in Example 5 except that the stirring time in the intermediate aging step was 10 min, and tetraethoxysilane (TEOS) was added over 5 min.
- the pH of the reaction solution at the end of the addition of TEOS was 3.0, and the pH of the reaction solution during the stirring for 20 hours was 8.6.
- the ⁇ phase content of the obtained substituted ⁇ iron oxide magnetic particle powder was 0%, and the radio wave absorption characteristics were measured by the method described above.
- the maximum absorption frequency of the green compact in the frequency range of 50 GHz to 100 GHz was 67.2 GHz
- the transmission attenuation per unit thickness was 4.6 dB / mm.
- the charged solution was mechanically stirred by a stirring blade, and 388.91 g of 22.30 mass% aqueous ammonia solution was added in 10 minutes (first neutralization step), After the completion of the dropping, stirring was continued for 30 minutes to ripen the produced precipitate (intermediate aging step). At that time, the pH of the slurry containing the precipitate was 8.6 (procedure 2).
- Example 9 Substitution type ⁇ iron oxide powder was obtained by the same procedure as in Example 7 except that 101.50 g of ferric (III) sulfate solution and 11.7 g of aluminum (III) nitrate nonahydrate were used.
- the pH of the reaction solution at the end of the addition of TEOS was 3.0, and the pH of the reaction solution during the stirring for 20 hours was 8.6.
- Chemical analysis of the composition, magnetic properties and radio wave absorption properties were evaluated. The measurement results are also shown in Table 2.
- the molar ratio S2 / (F + M) is 2.84.
- a substituted ⁇ iron oxide magnetic particle powder was obtained by the same procedure as in Example 1.
- the substitution type ⁇ iron oxide powder according to Comparative Example 7 was subjected to XRD measurement, and the content rate of the ⁇ phase was determined to be 8.7%, which was a high value as compared with Additional Example 2.
- chemical analysis, magnetic characteristics and radio wave absorption characteristics were evaluated. The measurement results are also shown in Table 2.
- Comparative Example 8 This comparative example is an experimental example corresponding to the conventional manufacturing method of the composition ratio of Example 7. Additional Comparative Example 2 except that the amount of the metal salt to be added was 102.73 g of a ferric sulfate (III) solution having an Fe concentration of 11.65 mass% and 10.74 g of a 98% pure Al (III) nitrate nine hydrate. Substituted ⁇ iron oxide magnetic particle powder was obtained by the same procedure. The pH of the reaction solution at the end of the addition of TEOS and the pH of the reaction solution during the stirring for 20 hours were both 8.6.
- III ferric sulfate
- the substitution type ⁇ iron oxide magnetic particle powder according to the comparative example was subjected to XRD measurement, and the content rate of the ⁇ phase was determined to be 8.1%, which was a high value as compared with Example 7.
- chemical analysis of the composition and evaluation of magnetic properties were performed. The measurement results are also shown in Table 2.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Power Engineering (AREA)
- Dispersion Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Physics & Mathematics (AREA)
- Electromagnetism (AREA)
- Compounds Of Iron (AREA)
- Hard Magnetic Materials (AREA)
Abstract
【課題】非磁性のαタイプの鉄系酸化物の含有量を低減した、ε-Fe2O3のFeサイトの一部を他の金属元素で置換した置換型ε酸化鉄磁性粒子粉および置換型ε酸化鉄磁性粒子粉の製造方法を提供する。 【解決手段】3価の鉄イオンとFeサイトを一部置換する金属のイオンを含む酸性の水溶液をpH2.0以上7.0以下まで中和した後、生成した置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む分散液に加水分解基を持つシリコン化合物を添加し、引き続いて分散液をpH8.0以上まで中和した後保持して熟成を行い、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物にシリコン化合物の化学反応生成物を被覆して加熱することにより、αタイプの鉄系酸化物の含有量を低減した置換型ε酸化鉄磁性粒子粉が得られる。
Description
本発明は、高密度磁気記録媒体、電波吸収体等に好適な置換型ε酸化鉄磁性粒子粉、特に、置換型ε酸化鉄にとっては異相である非磁性のαタイプの鉄系酸化物の含有量が低減された置換型ε酸化鉄磁性粒子粉およびその製造方法に関する。なお、本明細書では、ε-Fe2O3のFeサイトの一部を他の金属元素で置換した酸化物をεタイプの鉄系酸化物、結晶系がα-Fe2O3のそれと同一の置換型α酸化鉄粒子をαタイプの鉄系酸化物とそれぞれ呼ぶことがある。
ε-Fe2O3は酸化鉄の中でも極めて稀な相であるが、室温において、ナノメートルオーダーのサイズの粒子が20kOe(1.59×106A/m)程度の巨大な保磁力(Hc)を示すため、ε-Fe2O3を単相で合成する製造方法の検討が従来よりなされてきている(特許文献1)。しかし、ε-Fe2O3を磁気記録媒体に用いた場合、現時点ではそれに対応する、高レベルの飽和磁束密度を有する磁気ヘッド用の材料が存在しないため、実用的にはε-Fe2O3のFeサイトの一部をAl、Ga、In等の3価の金属で置換し、保磁力を調整する必要があり、電波吸収材料として使用する場合にも、要求される吸収波長に応じてFeサイトの置換量を変化させる必要がある(特許文献2)。
一方、εタイプの鉄系酸化物の磁性粒子は極めて微細であるため、耐環境安定性、熱安定性の向上のために、ε-Fe2O3のFeサイトの一部を、耐熱性に優れた他の金属で置換することも検討されており、一般式ε-AxByFe2-x-yO3またはε-AxByCzFe2-x-y-zO3(ここでAはCo、Ni、Mn、Zn等の2価の金属元素、BはTi等の4価の金属元素、CはIn、Ga、Al等の3価の金属元素)で表される、耐環境安定性、熱安定性にも優れた各種のε-Fe2O3の一部置換体が提案されている(特許文献3)。
ε-Fe2O3およびεタイプの鉄系酸化物は熱力学的な安定相ではないため、その製造には特殊な方法を必要とする。上述の特許文献1~3には、液相法で生成したオキシ水酸化鉄もしくは置換元素を含むオキシ水酸化鉄の微細結晶を前駆体として用い、その前駆体にゾル-ゲル法によりシリコン酸化物を被覆した後に熱処理するε-Fe2O3またはεタイプの鉄系酸化物の製造方法が開示されており、液相法としては反応媒体として有機溶媒を用いる逆ミセル法と、反応媒体として水溶液のみを用いる方法がそれぞれ開示されている。
また、前記のε-Fe2O3およびεタイプの鉄系酸化物は、例えば特許文献4~5において、100GHzを超える高周波域で電波吸収のピークを有することが示されており、電波吸収体としての用途も期待されている。
しかし、特許文献1~3に開示された製造方法により得られる磁性粒子粉は、ε-Fe2O3およびεタイプの鉄系酸化物以外に、不純物として非磁性のαタイプの鉄系酸化物を相当量含むものである。
特許文献6には、置換型ε酸化鉄磁性粒子粉に含まれる、不純物としてのαタイプの鉄系酸化物の量を低減させる技術が開示されている。
特許文献7には、広い範囲のpH領域でゾル-ゲル法によりシリコン酸化物を被覆する、εタイプの鉄系酸化物の製造方法が開示されている。
一方、εタイプの鉄系酸化物の磁性粒子は極めて微細であるため、耐環境安定性、熱安定性の向上のために、ε-Fe2O3のFeサイトの一部を、耐熱性に優れた他の金属で置換することも検討されており、一般式ε-AxByFe2-x-yO3またはε-AxByCzFe2-x-y-zO3(ここでAはCo、Ni、Mn、Zn等の2価の金属元素、BはTi等の4価の金属元素、CはIn、Ga、Al等の3価の金属元素)で表される、耐環境安定性、熱安定性にも優れた各種のε-Fe2O3の一部置換体が提案されている(特許文献3)。
ε-Fe2O3およびεタイプの鉄系酸化物は熱力学的な安定相ではないため、その製造には特殊な方法を必要とする。上述の特許文献1~3には、液相法で生成したオキシ水酸化鉄もしくは置換元素を含むオキシ水酸化鉄の微細結晶を前駆体として用い、その前駆体にゾル-ゲル法によりシリコン酸化物を被覆した後に熱処理するε-Fe2O3またはεタイプの鉄系酸化物の製造方法が開示されており、液相法としては反応媒体として有機溶媒を用いる逆ミセル法と、反応媒体として水溶液のみを用いる方法がそれぞれ開示されている。
また、前記のε-Fe2O3およびεタイプの鉄系酸化物は、例えば特許文献4~5において、100GHzを超える高周波域で電波吸収のピークを有することが示されており、電波吸収体としての用途も期待されている。
しかし、特許文献1~3に開示された製造方法により得られる磁性粒子粉は、ε-Fe2O3およびεタイプの鉄系酸化物以外に、不純物として非磁性のαタイプの鉄系酸化物を相当量含むものである。
特許文献6には、置換型ε酸化鉄磁性粒子粉に含まれる、不純物としてのαタイプの鉄系酸化物の量を低減させる技術が開示されている。
特許文献7には、広い範囲のpH領域でゾル-ゲル法によりシリコン酸化物を被覆する、εタイプの鉄系酸化物の製造方法が開示されている。
上述の特許文献4に開示された製造方法により製造されたFeサイトの一部を置換したεタイプの鉄系酸化物は、従来法により製造されたεタイプの鉄系酸化物と比較して不純物であるαタイプの鉄系酸化物の含有量が低減されたものである。しかし、εタイプの鉄系酸化物は準安定相であり、他の金属元素によるFeの置換量が少ない場合には、特許文献4に開示された製造方法を用いても、ε-Fe2O3と同じ空間群を取ることが困難になり、αタイプの鉄系酸化物の含有量の低減が不十分になることがあった。
αタイプの鉄系酸化物は非磁性であるため、置換型ε酸化鉄磁性粒子粉を電波吸収材料として使用した場合、電波吸収特性に寄与せず、磁気記録媒体に使用した場合にも、記録密度を高めることに寄与しないので、その含有量を低減する必要がある。
すなわち、本発明において解決すべき技術課題とは、非磁性のαタイプの鉄系酸化物の含有量を低減した置換型ε酸化鉄磁性粒子粉および置換型ε酸化鉄磁性粒子粉の製造方法を提供することである。
αタイプの鉄系酸化物は非磁性であるため、置換型ε酸化鉄磁性粒子粉を電波吸収材料として使用した場合、電波吸収特性に寄与せず、磁気記録媒体に使用した場合にも、記録密度を高めることに寄与しないので、その含有量を低減する必要がある。
すなわち、本発明において解決すべき技術課題とは、非磁性のαタイプの鉄系酸化物の含有量を低減した置換型ε酸化鉄磁性粒子粉および置換型ε酸化鉄磁性粒子粉の製造方法を提供することである。
本発明者等は、置換型ε酸化鉄磁性粒子粉を得るためには、シリコン酸化物を被覆した状態で当該磁性粒子粉の前駆体を加熱する必要があることに着目して鋭意研究を行ったところ、被覆に用いる加水分解基を持つシリコン化合物を、pH2.0以上7.0以下の時点で当該前駆体を含む水溶液に添加することにより、αタイプの鉄系酸化物の含有量を低減できることが判明した。
以上の知見を基に、本発明者等は、以下に述べる本発明を完成させた。
以上の知見を基に、本発明者等は、以下に述べる本発明を完成させた。
上記の課題を解決するために、本発明においては、
ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉であって、前記置換型ε酸化鉄磁性粒子粉に含まれるFeのモル数をFe、Feサイトを置換した全金属元素のモル数をMeとしたとき、Me/(Fe+Me)で定義される他の金属元素によるFeの置換量が0.08以上0.17以下であり、かつ、X線回折法により測定されるαタイプの鉄系酸化物の含有率が3%以下である、置換型ε酸化鉄磁性粒子粉が提供される。
前記のFeサイトを一部置換する他の金属元素は、Co、Tiならびに、GaおよびAlから選ばれる1種以上であることが好ましい。例えば、Feサイトを一部置換する他の金属元素として、CoおよびTiを含み、かつGaおよびAlから選ばれる1種以上を含むεタイプの鉄系酸化物が好適な対象となる。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉からなる圧粉体が提供される。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉が樹脂またはゴムに分散されてなる電波吸収体が提供される。
本発明においてはまた、原料溶液として3価の鉄イオンと前記Feサイトを一部置換する金属のイオンを含む酸性の水溶液を用い、前記の原料溶液にアルカリを添加してpH8.0以上10.0以下の範囲まで中和し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む分散液を得る中和工程と、前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む前記の分散液に、加水分解基を持つシリコン化合物を添加するシリコン化合物添加工程と、前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物と前記のシリコン化合物を含む分散液を、pH8.0以上10.0以下で保持し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物に前記シリコン化合物の化学反応生成物を被覆する熟成工程と、を含む、ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉の製造方法であって、前記の加水分解基を持つシリコン化合物の添加を、前記の中和工程において分散液のpHが2.0以上7.0以下の範囲内にある時点で開始し、pH2.0以上7.0以下の分散液に添加する前記のシリコン化合物のモル数をS1、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S1/(F+M)が0.01以上10.0以下であり、かつ、前記のシリコン化合物の全添加モル数をS2としたとき、S2/(F+M)が0.50以上10.0以下である、置換型ε酸化鉄磁性粒子粉の製造方法が提供される。
前記の製造方法において、前記の中和工程におけるアルカリ添加と、前記のシリコン化合物添加工程におけるシリコン化合物の添加を、いずれも連続的に行っても良く、アルカリ添加を連続的に、シリコン化合物の添加を間歇的に行っても良く、アルカリ添加とシリコン化合物の添加をいずれも間歇的に行っても良く、アルカリ添加を間歇的に、シリコン化合物の添加を連続的に行っても構わない。
また、前記の製造方法においては、前記のFeサイトを一部置換する他の金属元素がCo、Tiならびに、GaおよびAlから選ばれる1種以上であることが好ましい。例えば、Feサイトを一部置換する他の金属元素として、CoおよびTiを含み、かつGaおよびAlから選ばれる1種以上を含むことが好ましい。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉を圧縮成形して圧粉体を得る、圧粉体の製造方法が提供される。
ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉であって、前記置換型ε酸化鉄磁性粒子粉に含まれるFeのモル数をFe、Feサイトを置換した全金属元素のモル数をMeとしたとき、Me/(Fe+Me)で定義される他の金属元素によるFeの置換量が0.08以上0.17以下であり、かつ、X線回折法により測定されるαタイプの鉄系酸化物の含有率が3%以下である、置換型ε酸化鉄磁性粒子粉が提供される。
前記のFeサイトを一部置換する他の金属元素は、Co、Tiならびに、GaおよびAlから選ばれる1種以上であることが好ましい。例えば、Feサイトを一部置換する他の金属元素として、CoおよびTiを含み、かつGaおよびAlから選ばれる1種以上を含むεタイプの鉄系酸化物が好適な対象となる。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉からなる圧粉体が提供される。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉が樹脂またはゴムに分散されてなる電波吸収体が提供される。
本発明においてはまた、原料溶液として3価の鉄イオンと前記Feサイトを一部置換する金属のイオンを含む酸性の水溶液を用い、前記の原料溶液にアルカリを添加してpH8.0以上10.0以下の範囲まで中和し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む分散液を得る中和工程と、前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む前記の分散液に、加水分解基を持つシリコン化合物を添加するシリコン化合物添加工程と、前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物と前記のシリコン化合物を含む分散液を、pH8.0以上10.0以下で保持し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物に前記シリコン化合物の化学反応生成物を被覆する熟成工程と、を含む、ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉の製造方法であって、前記の加水分解基を持つシリコン化合物の添加を、前記の中和工程において分散液のpHが2.0以上7.0以下の範囲内にある時点で開始し、pH2.0以上7.0以下の分散液に添加する前記のシリコン化合物のモル数をS1、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S1/(F+M)が0.01以上10.0以下であり、かつ、前記のシリコン化合物の全添加モル数をS2としたとき、S2/(F+M)が0.50以上10.0以下である、置換型ε酸化鉄磁性粒子粉の製造方法が提供される。
前記の製造方法において、前記の中和工程におけるアルカリ添加と、前記のシリコン化合物添加工程におけるシリコン化合物の添加を、いずれも連続的に行っても良く、アルカリ添加を連続的に、シリコン化合物の添加を間歇的に行っても良く、アルカリ添加とシリコン化合物の添加をいずれも間歇的に行っても良く、アルカリ添加を間歇的に、シリコン化合物の添加を連続的に行っても構わない。
また、前記の製造方法においては、前記のFeサイトを一部置換する他の金属元素がCo、Tiならびに、GaおよびAlから選ばれる1種以上であることが好ましい。例えば、Feサイトを一部置換する他の金属元素として、CoおよびTiを含み、かつGaおよびAlから選ばれる1種以上を含むことが好ましい。
本発明においてはまた、前記の置換型ε酸化鉄磁性粒子粉を圧縮成形して圧粉体を得る、圧粉体の製造方法が提供される。
以上、本発明の製造方法を用いることにより、αタイプの鉄系酸化物の含有量を低減した置換型ε酸化鉄磁性粒子粉、およびそれを用いた圧粉体、電波吸収体を得ることができる。
[酸化鉄磁性粒子粉]
本発明の製造方法は、ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉を製造するためのものであり、当該磁性粒子粉には、その製造上不可避的な不純物である異相が混在する。異相は主としてαタイプの鉄系酸化物であり、本発明により得られる酸化鉄磁性粒子粉は実質的にεタイプの鉄系酸化物磁性粒子とαタイプの鉄系酸化物からなる。本発明の目的は、異相であるαタイプの鉄系酸化物の含有量の低減である。
ε-Fe2O3のFeサイトの一部を他の金属元素で置換した一部置換体がε構造を有するかどうかについては、X線回折法(XRD)、高速電子回折法(HEED)等を用いて確認することが可能である。本発明においては、εタイプおよびαタイプの鉄系酸化物の同定は、XRDによって行っている。
本発明の製造方法により製造が可能な一部置換体については、以下が挙げられる。
一般式ε-CzFe2-zO3(ここでCはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-AxByFe2-x-yO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、BはTi、Snから選択される1種以上の4価の金属元素)で表されるもの。
一般式ε-AxCzFe2-x-zO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-ByCzFe2-y-zO3(ここでBはTi、Snから選択される1種以上の4価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-AxByCzFe2-x-y-zO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、BはTi、Snから選択される1種以上の4価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
本発明の製造方法は、ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉を製造するためのものであり、当該磁性粒子粉には、その製造上不可避的な不純物である異相が混在する。異相は主としてαタイプの鉄系酸化物であり、本発明により得られる酸化鉄磁性粒子粉は実質的にεタイプの鉄系酸化物磁性粒子とαタイプの鉄系酸化物からなる。本発明の目的は、異相であるαタイプの鉄系酸化物の含有量の低減である。
ε-Fe2O3のFeサイトの一部を他の金属元素で置換した一部置換体がε構造を有するかどうかについては、X線回折法(XRD)、高速電子回折法(HEED)等を用いて確認することが可能である。本発明においては、εタイプおよびαタイプの鉄系酸化物の同定は、XRDによって行っている。
本発明の製造方法により製造が可能な一部置換体については、以下が挙げられる。
一般式ε-CzFe2-zO3(ここでCはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-AxByFe2-x-yO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、BはTi、Snから選択される1種以上の4価の金属元素)で表されるもの。
一般式ε-AxCzFe2-x-zO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-ByCzFe2-y-zO3(ここでBはTi、Snから選択される1種以上の4価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
一般式ε-AxByCzFe2-x-y-zO3(ここでAはCo、Ni、Mn、Znから選択される1種以上の2価の金属元素、BはTi、Snから選択される1種以上の4価の金属元素、CはIn、Ga、Alから選択される1種以上の3価の金属元素)で表されるもの。
ここでC元素のみで置換したタイプは、磁性粒子の保磁力を任意に制御できることに加え、ε-Fe2O3と同じ空間群を得易いという利点を有するが、熱的安定性に劣る場合がある。特にCとしてGaおよびAlを用いた場合には、得られた置換型ε酸化鉄磁性粒子粉の熱的安定性がやや劣るので、さらにAおよび/またはB元素で同時に置換することが好ましい。A、BおよびCの三元素置換タイプは、上述の特性のバランスが最も良く取れたもので、耐熱性、単一相の得易さ、保磁力の制御性に優れるものであり、CとしてGaおよびAlを用いる場合には、CoおよびTiも同時に置換することが好ましい。
なお、本発明の製造方法は、上述したいずれの置換タイプの酸化鉄磁性粒子についても適用可能である。
後述する本発明の製造方法は、前記のFeサイトを置換する金属元素の置換量がいかなる値であっても適用可能であるが、αタイプの鉄系酸化物が生成し易い置換量で適用するのが効果的である。具体的には、前記置換型ε酸化鉄磁性粒子粉に含まれるFeのモル数をFe、Feサイトを置換した全金属元素のモル数をMeとしたとき、Me/(Fe+Me)で定義される他の金属元素によるFeの置換量が0.08以上0.17以下で適用した場合、従来法では得られなかった、XRDにより測定されるαタイプの鉄系酸化物の含有率が3%以下の置換型ε酸化鉄磁性粒子粉を得ることができる。
なお、本発明の製造方法は、上述したいずれの置換タイプの酸化鉄磁性粒子についても適用可能である。
後述する本発明の製造方法は、前記のFeサイトを置換する金属元素の置換量がいかなる値であっても適用可能であるが、αタイプの鉄系酸化物が生成し易い置換量で適用するのが効果的である。具体的には、前記置換型ε酸化鉄磁性粒子粉に含まれるFeのモル数をFe、Feサイトを置換した全金属元素のモル数をMeとしたとき、Me/(Fe+Me)で定義される他の金属元素によるFeの置換量が0.08以上0.17以下で適用した場合、従来法では得られなかった、XRDにより測定されるαタイプの鉄系酸化物の含有率が3%以下の置換型ε酸化鉄磁性粒子粉を得ることができる。
[圧粉体および電波吸収体]
本発明により得られる置換型ε酸化鉄磁性粒子粉は、その粉体粒子の充填構造を形成させることによって、優れた電波吸収能を有する電波吸収体として機能する。ここでいう充填構造は、粒子同士が接しているかまたは近接している状態で各粒子が立体構造を構成しているものを意味する。電波吸収体の実用に供するためには充填構造を維持させる必要がある。その手法として、例えば置換型ε酸化鉄磁性粒子粉を圧縮成形して圧粉体にする方法や、非磁性高分子化合物をバインダーとして、置換型ε酸化鉄磁性粒子粉を固着させることによって充填構造を形成させる方法が挙げられる。
バインダーを用いる方法の場合、置換型ε酸化鉄磁性粒子粉を非磁性の高分子基材と混合して混練物を得る。混練物中における電波吸収材料粉体の配合量は60質量%以上とすることが好ましい。電波吸収材料粉体の配合量が多いほど電波吸収特性を向上させる上で有利となるが、あまり多いと高分子基材との混練が難しくなるので注意を要する。例えば電波吸収材料粉体の配合量は80~95質量%あるいは85~95質量%とすることができる。
高分子基材としては、使用環境に応じて、耐熱性、難燃性、耐久性、機械的強度、電気的特性を満足する各種のものが使用できる。例えば、樹脂(ナイロン等)、ゲル(シリコーンゲル等)、熱可塑性エラストマー、ゴムなどから適切なものを選択すれば良い。また2種以上の高分子化合物をブレンドして基材としてもよい。
本発明により得られる置換型ε酸化鉄磁性粒子粉は、その粉体粒子の充填構造を形成させることによって、優れた電波吸収能を有する電波吸収体として機能する。ここでいう充填構造は、粒子同士が接しているかまたは近接している状態で各粒子が立体構造を構成しているものを意味する。電波吸収体の実用に供するためには充填構造を維持させる必要がある。その手法として、例えば置換型ε酸化鉄磁性粒子粉を圧縮成形して圧粉体にする方法や、非磁性高分子化合物をバインダーとして、置換型ε酸化鉄磁性粒子粉を固着させることによって充填構造を形成させる方法が挙げられる。
バインダーを用いる方法の場合、置換型ε酸化鉄磁性粒子粉を非磁性の高分子基材と混合して混練物を得る。混練物中における電波吸収材料粉体の配合量は60質量%以上とすることが好ましい。電波吸収材料粉体の配合量が多いほど電波吸収特性を向上させる上で有利となるが、あまり多いと高分子基材との混練が難しくなるので注意を要する。例えば電波吸収材料粉体の配合量は80~95質量%あるいは85~95質量%とすることができる。
高分子基材としては、使用環境に応じて、耐熱性、難燃性、耐久性、機械的強度、電気的特性を満足する各種のものが使用できる。例えば、樹脂(ナイロン等)、ゲル(シリコーンゲル等)、熱可塑性エラストマー、ゴムなどから適切なものを選択すれば良い。また2種以上の高分子化合物をブレンドして基材としてもよい。
[平均粒子径]
本発明においては、本発明の製造法により得られる酸化鉄磁性粒子粉の平均粒子径は特に規定するものではないが、各粒子が単磁区構造となる程度に微細であることが好ましい。通常は、透過電子顕微鏡で測定した平均粒子径が10nm以上40nm以下のものが得られる。
本発明においては、本発明の製造法により得られる酸化鉄磁性粒子粉の平均粒子径は特に規定するものではないが、各粒子が単磁区構造となる程度に微細であることが好ましい。通常は、透過電子顕微鏡で測定した平均粒子径が10nm以上40nm以下のものが得られる。
[出発物質および前駆体]
本発明の製造方法においては、鉄系酸化物磁性粒子粉の出発物質として3価の鉄イオンと最終的にFeサイトを置換する金属元素の金属イオンを含む酸性の水溶液(以下、原料溶液と言う。)を用いる。もし、出発物質として3価のFeイオンに替えて2価のFeイオンを用いた場合には、沈殿物として3価の鉄の水和酸化物のほかに2価の鉄の水和酸化物やマグネタイト等をも含む混合物が生成し、最終的に得られる鉄系酸化物粒子の形状にバラつきが生じてしまうため、本発明のようなαタイプの鉄系酸化物の含有量が低減された、置換型ε酸化鉄磁性粒子粉を得ることができない。ここで、酸性とは液のpHが7.0未満であることを指す。これらの鉄イオンもしくは置換元素の金属イオンの供給源としては、入手の容易さおよび価格の面から、硝酸塩、硫酸塩、塩化物のような水溶性の無機酸塩を用いることが好ましい。これらの金属塩を水に溶解すると、金属イオンが解離し、水溶液は酸性を呈する。この金属イオンを含む酸性水溶液にアルカリを添加して中和すると、オキシ水酸化鉄と置換元素の水酸化物の混合物、もしくは、Feサイトの一部を他の金属元素で置換されたオキシ水酸化鉄(本明細書では、以下これらを、置換元素を含むオキシ水酸化鉄と総称する。)の沈殿が得られる。本発明の製造方法においては、これらの置換元素を含むオキシ水酸化鉄を置換型ε酸化鉄磁性粒子粉の前駆体として用いる。
原料溶液中の全金属イオン濃度は、本発明では特に規定するものではないが、0.01mol/L以上0.5mol/L以下が好ましい。0.01mol/L未満では1回の反応で得られる置換型ε酸化鉄磁性粒子粉の量が少なく、経済的に好ましくない。全金属イオン濃度が0.5mol/Lを超えると、急速な水酸化物の沈澱発生により、反応溶液がゲル化しやすくなるので好ましくない。
本発明の製造方法においては、鉄系酸化物磁性粒子粉の出発物質として3価の鉄イオンと最終的にFeサイトを置換する金属元素の金属イオンを含む酸性の水溶液(以下、原料溶液と言う。)を用いる。もし、出発物質として3価のFeイオンに替えて2価のFeイオンを用いた場合には、沈殿物として3価の鉄の水和酸化物のほかに2価の鉄の水和酸化物やマグネタイト等をも含む混合物が生成し、最終的に得られる鉄系酸化物粒子の形状にバラつきが生じてしまうため、本発明のようなαタイプの鉄系酸化物の含有量が低減された、置換型ε酸化鉄磁性粒子粉を得ることができない。ここで、酸性とは液のpHが7.0未満であることを指す。これらの鉄イオンもしくは置換元素の金属イオンの供給源としては、入手の容易さおよび価格の面から、硝酸塩、硫酸塩、塩化物のような水溶性の無機酸塩を用いることが好ましい。これらの金属塩を水に溶解すると、金属イオンが解離し、水溶液は酸性を呈する。この金属イオンを含む酸性水溶液にアルカリを添加して中和すると、オキシ水酸化鉄と置換元素の水酸化物の混合物、もしくは、Feサイトの一部を他の金属元素で置換されたオキシ水酸化鉄(本明細書では、以下これらを、置換元素を含むオキシ水酸化鉄と総称する。)の沈殿が得られる。本発明の製造方法においては、これらの置換元素を含むオキシ水酸化鉄を置換型ε酸化鉄磁性粒子粉の前駆体として用いる。
原料溶液中の全金属イオン濃度は、本発明では特に規定するものではないが、0.01mol/L以上0.5mol/L以下が好ましい。0.01mol/L未満では1回の反応で得られる置換型ε酸化鉄磁性粒子粉の量が少なく、経済的に好ましくない。全金属イオン濃度が0.5mol/Lを超えると、急速な水酸化物の沈澱発生により、反応溶液がゲル化しやすくなるので好ましくない。
[中和工程]
本発明の製造方法においては、原料溶液にアルカリを添加し、そのpHが8.0以上10.0以下になるまで中和し、置換元素を含むオキシ水酸化鉄の沈殿物を含む分散液を得る。なお、3価の鉄イオンの水酸化物は主としてオキシ水酸化物からなる。ここで分散液のpHを8.0以上にするのは、置換金属元素、例えばCo、の水酸化物の沈殿生成を完了させるためと、加水分解生成物であるシラノール誘導体の縮合反応を促進させるためである。本発明の製造方法において、中和工程の到達pHの上限は特に規定するものではないが、中和の効果が飽和し、後記するシラノール誘導体の縮合反応の促進の効果が低下するので、10.0とすることが好ましい。
中和に用いるアルカリとしては、アルカリ金属またはアルカリ土類の水酸化物、アンモニア水、炭酸水素アンモニウムなどのアンモニウム塩のいずれであっても良いが、最終的に熱処理してεタイプの鉄系酸化物としたときに不純物が残りにくいアンモニア水や炭酸水素アンモニウムを用いることが好ましい。これらのアルカリは、出発物質の水溶液に固体で添加しても構わないが、反応の均一性を確保する観点からは、水溶液の状態で添加することが好ましい。
前記のように、原料溶液にアルカリを添加して中和処理を行うと、置換元素を含むオキシ水酸化鉄の沈澱物が析出するので、中和処理中は前記の沈殿物を含む分散液を公知の機械的手段により撹拌する。
原料溶液へのアルカリの添加は、添加を開始してから終了するまで、連続的に行っても良い。また、分散液のpHが8.0に到達する以前に、アルカリの添加を中断し、所定のpH保持時間を設けても良い。その場合、pH保持時間を複数回設け、アルカリの添加を間歇的に行うことができる。なお、pH保持時間を設ける回数、すなわち、アルカリの添加を中断する回数は、製造工程の煩雑化を避けるために3回以下とすることが好ましい。
本発明の製造方法においては、中和処理時の反応温度は5℃以上60℃以下とする。反応温度が5℃未満では冷却によるコストが増大するため好ましくない。60℃を超えると最終的に異相であるαタイプの酸化物が生成し易くなるので好ましくない。より好ましくは10℃以上40℃以下である。上述した特許文献4に記載された製造方法の場合、中和処理は5℃以上25℃以下で行う必要があり、反応時に冷凍機を用いる必要があったが、本発明の製造方法においては、常温以上の反応温度で中和処理を行うことも可能である。
なお、本明細書に記載のpHの値は、JIS Z8802に基づき、ガラス電極を用いて測定した。pH標準液として、測定するpH領域に応じた適切な緩衝液を用いて校正したpH計により測定した値をいう。また、本明細書に記載のpHは、温度補償電極により補償されたpH計の示す測定値を、反応温度条件下で直接読み取った値である。
本発明の製造方法においては、原料溶液にアルカリを添加し、そのpHが8.0以上10.0以下になるまで中和し、置換元素を含むオキシ水酸化鉄の沈殿物を含む分散液を得る。なお、3価の鉄イオンの水酸化物は主としてオキシ水酸化物からなる。ここで分散液のpHを8.0以上にするのは、置換金属元素、例えばCo、の水酸化物の沈殿生成を完了させるためと、加水分解生成物であるシラノール誘導体の縮合反応を促進させるためである。本発明の製造方法において、中和工程の到達pHの上限は特に規定するものではないが、中和の効果が飽和し、後記するシラノール誘導体の縮合反応の促進の効果が低下するので、10.0とすることが好ましい。
中和に用いるアルカリとしては、アルカリ金属またはアルカリ土類の水酸化物、アンモニア水、炭酸水素アンモニウムなどのアンモニウム塩のいずれであっても良いが、最終的に熱処理してεタイプの鉄系酸化物としたときに不純物が残りにくいアンモニア水や炭酸水素アンモニウムを用いることが好ましい。これらのアルカリは、出発物質の水溶液に固体で添加しても構わないが、反応の均一性を確保する観点からは、水溶液の状態で添加することが好ましい。
前記のように、原料溶液にアルカリを添加して中和処理を行うと、置換元素を含むオキシ水酸化鉄の沈澱物が析出するので、中和処理中は前記の沈殿物を含む分散液を公知の機械的手段により撹拌する。
原料溶液へのアルカリの添加は、添加を開始してから終了するまで、連続的に行っても良い。また、分散液のpHが8.0に到達する以前に、アルカリの添加を中断し、所定のpH保持時間を設けても良い。その場合、pH保持時間を複数回設け、アルカリの添加を間歇的に行うことができる。なお、pH保持時間を設ける回数、すなわち、アルカリの添加を中断する回数は、製造工程の煩雑化を避けるために3回以下とすることが好ましい。
本発明の製造方法においては、中和処理時の反応温度は5℃以上60℃以下とする。反応温度が5℃未満では冷却によるコストが増大するため好ましくない。60℃を超えると最終的に異相であるαタイプの酸化物が生成し易くなるので好ましくない。より好ましくは10℃以上40℃以下である。上述した特許文献4に記載された製造方法の場合、中和処理は5℃以上25℃以下で行う必要があり、反応時に冷凍機を用いる必要があったが、本発明の製造方法においては、常温以上の反応温度で中和処理を行うことも可能である。
なお、本明細書に記載のpHの値は、JIS Z8802に基づき、ガラス電極を用いて測定した。pH標準液として、測定するpH領域に応じた適切な緩衝液を用いて校正したpH計により測定した値をいう。また、本明細書に記載のpHは、温度補償電極により補償されたpH計の示す測定値を、反応温度条件下で直接読み取った値である。
[シリコン化合物の添加工程]
本発明の製造方法においては、前記の工程で生成した置換型ε酸化鉄磁性粒子粉の前駆体である置換元素を含むオキシ水酸化鉄は、そのままの状態で熱処理を施してもεタイプの鉄系酸化物に相変化しにくいので、熱処理に先立って置換元素を含むオキシ水酸化鉄にシリコン化合物の加水分解反応および縮合反応により得られた化学反応生成物による被覆を施す必要がある。なおここでシリコン化合物の化学反応生成物とは、化学量論組成のシリコン酸化物だけではなく、後述するシラノール誘導体やポリシロキサン構造等の非量論組成のもの、また加熱処理を施してシリコン酸化物に変化したもの等の総称として使用する。
特許文献1~4に記載の製造方法においては、シリコン化合物の化学反応生成物の被覆法としてゾル-ゲル法を用いており、原料溶液の中和処理が完了し、反応溶液のpHがアルカリ側になった後に加水分解基を持つシリコン化合物を反応溶液に添加している。一方、本発明の製造方法においては、シリコン酸化物の被覆法として同じくゾル-ゲル法を用いるが、原料溶液の中和が完了する以前の、反応溶液のpHが酸性側のpH2.0以上7.0以下の範囲にある時点において加水分解基を持つシリコン化合物の添加を開始することを特徴とする。なお、シリコン化合物添加の終了時期については後述する。
ゾル-ゲル法の場合、置換元素を含むオキシ水酸化鉄含む分散液に、加水分解基を持つシリコン化合物、例えばテトラエトキシシラン(TEOS)、テトラメトキシシラン(TMOS)等のアルコシキシラン類や、各種のシランカップリング剤等のシラン化合物を添加して撹拌下で加水分解反応を生起させ、生成したシラノール誘導体を縮合してポリシロキサン結合を形成させることにより、置換元素を含むオキシ水酸化鉄の表面を被覆する。
本発明の製造方法においては、前記の工程で生成した置換型ε酸化鉄磁性粒子粉の前駆体である置換元素を含むオキシ水酸化鉄は、そのままの状態で熱処理を施してもεタイプの鉄系酸化物に相変化しにくいので、熱処理に先立って置換元素を含むオキシ水酸化鉄にシリコン化合物の加水分解反応および縮合反応により得られた化学反応生成物による被覆を施す必要がある。なおここでシリコン化合物の化学反応生成物とは、化学量論組成のシリコン酸化物だけではなく、後述するシラノール誘導体やポリシロキサン構造等の非量論組成のもの、また加熱処理を施してシリコン酸化物に変化したもの等の総称として使用する。
特許文献1~4に記載の製造方法においては、シリコン化合物の化学反応生成物の被覆法としてゾル-ゲル法を用いており、原料溶液の中和処理が完了し、反応溶液のpHがアルカリ側になった後に加水分解基を持つシリコン化合物を反応溶液に添加している。一方、本発明の製造方法においては、シリコン酸化物の被覆法として同じくゾル-ゲル法を用いるが、原料溶液の中和が完了する以前の、反応溶液のpHが酸性側のpH2.0以上7.0以下の範囲にある時点において加水分解基を持つシリコン化合物の添加を開始することを特徴とする。なお、シリコン化合物添加の終了時期については後述する。
ゾル-ゲル法の場合、置換元素を含むオキシ水酸化鉄含む分散液に、加水分解基を持つシリコン化合物、例えばテトラエトキシシラン(TEOS)、テトラメトキシシラン(TMOS)等のアルコシキシラン類や、各種のシランカップリング剤等のシラン化合物を添加して撹拌下で加水分解反応を生起させ、生成したシラノール誘導体を縮合してポリシロキサン結合を形成させることにより、置換元素を含むオキシ水酸化鉄の表面を被覆する。
本発明者等は、前記の加水分解基を持つシリコン化合物の添加をpH2.0以上7.0以下で開始すると、最終的に得られる置換型ε酸化鉄磁性粒子粉に含まれるαタイプの鉄系酸化物の含有率を低減することが可能であることを見出したが、その理由は以下のように考えられる。
前記の加水分解基を持つシリコン化合物の加水分解反応と、加水分解生成物であるシラノール誘導体の縮合反応の速度は、反応系のpHに依存して変化する。加水分解反応速度は一般に、酸性側の低pH領域で大きく、pHの上昇とともに低下し、アルカリ性側の高pH域で再び増大する。これに対して縮合反応の速度は、酸性側の低pH領域で小さく、pHの上昇とともに増加し、中性からアルカリ性側のpH域で大きくなる。
置換元素を含むオキシ水酸化鉄の沈殿を含む分散液に、酸性側の低pH領域で加水分解基を持つシリコン化合物を添加すると、前記のシリコン化合物の加水分解が急速に進行し、有機成分の少ないシラノール誘導体が生成する一方、生成したシラノール誘導体の縮合反応は進行しない。ここで、シラノール誘導体は親水基であるOH基を有し、水溶液中で均一に分布するため、置換元素を含むオキシ水酸化鉄の沈殿とシラノール誘導体が分散液中で均一に分散して共存する状態になると考えられる。
その後、分散液のpHを更に上昇させると、シラノール誘導体の縮合反応が優勢になるため、置換元素を含むオキシ水酸化鉄の沈殿がシラノール誘導体またはその縮合反応生成物により均一に被覆されることになる。そのため、最終的に熱処理を施して得られる置換型ε酸化鉄磁性粒子粉に含まれるαタイプの鉄系酸化物の含有率が低減されるものと考えられる。
なお、前記の特許文献5には、広い範囲のpH領域でゾル-ゲル法によりシリコン酸化物を被覆することが開示されているが、その場合、シリコン化合物の添加は中和処理の終了後に一定のpHで行われており、本発明のように、シリコン化合物の加水分解反応速度と縮合反応速度の両方を考慮するとの技術思想は開示されていない。
前記の加水分解基を持つシリコン化合物の加水分解反応と、加水分解生成物であるシラノール誘導体の縮合反応の速度は、反応系のpHに依存して変化する。加水分解反応速度は一般に、酸性側の低pH領域で大きく、pHの上昇とともに低下し、アルカリ性側の高pH域で再び増大する。これに対して縮合反応の速度は、酸性側の低pH領域で小さく、pHの上昇とともに増加し、中性からアルカリ性側のpH域で大きくなる。
置換元素を含むオキシ水酸化鉄の沈殿を含む分散液に、酸性側の低pH領域で加水分解基を持つシリコン化合物を添加すると、前記のシリコン化合物の加水分解が急速に進行し、有機成分の少ないシラノール誘導体が生成する一方、生成したシラノール誘導体の縮合反応は進行しない。ここで、シラノール誘導体は親水基であるOH基を有し、水溶液中で均一に分布するため、置換元素を含むオキシ水酸化鉄の沈殿とシラノール誘導体が分散液中で均一に分散して共存する状態になると考えられる。
その後、分散液のpHを更に上昇させると、シラノール誘導体の縮合反応が優勢になるため、置換元素を含むオキシ水酸化鉄の沈殿がシラノール誘導体またはその縮合反応生成物により均一に被覆されることになる。そのため、最終的に熱処理を施して得られる置換型ε酸化鉄磁性粒子粉に含まれるαタイプの鉄系酸化物の含有率が低減されるものと考えられる。
なお、前記の特許文献5には、広い範囲のpH領域でゾル-ゲル法によりシリコン酸化物を被覆することが開示されているが、その場合、シリコン化合物の添加は中和処理の終了後に一定のpHで行われており、本発明のように、シリコン化合物の加水分解反応速度と縮合反応速度の両方を考慮するとの技術思想は開示されていない。
加水分解基を持つシリコン化合物の添加を開始する時点のpHは、2.0以上であることが好ましい。pHが2.0未満では、置換型ε酸化鉄磁性粒子粉の主成分である、原料溶液中に含まれる3価の鉄イオンの水酸化物の沈殿形成が不十分になる可能性がある。添加を開始する時点のpHは、3.0以上であることがより好ましい。また、加水分解基を持つシリコン化合物の添加を開始する時点のpHは、7.0以下であることが好ましい。pHが7.0を超すと加水分解反応が遅くなり、シラノール誘導体の生成が不十分になるため、置換元素を含むオキシ水酸化鉄の沈殿とシラノール誘導体が分散液中で均一に分散して共存する状態が得られなくなり、置換元素を含むオキシ水酸化鉄の沈殿がシラノール誘導体またはその縮合反応生成物により均一に被覆され難くなる。添加を開始する時点のpHは6.0以下が好ましく、4.0以下がさらに好ましい。
加水分解基を持つシリコン化合物の添加は、中和工程において原料溶液のpHが所望の値になった時点で開始する。シリコン化合物の添加は、添加を開始してから終了するまで連続的に行っても良い。ここで連続的とは、分散液に添加するシリコン化合物の全量を、分散液に一度に添加することを含む。また、シリコン化合物の添加を複数回に分け、間歇的に行っても構わない。
加水分解基を持つシリコン化合物の添加は、中和工程において原料溶液のpHが所望の値になった時点で開始する。シリコン化合物の添加は、添加を開始してから終了するまで連続的に行っても良い。ここで連続的とは、分散液に添加するシリコン化合物の全量を、分散液に一度に添加することを含む。また、シリコン化合物の添加を複数回に分け、間歇的に行っても構わない。
本発明の製造方法においては、分散液に添加するシリコン化合物の量は、以下の二つの条件を同時に満足する必要がある。
第一の条件は、pH2.0以上7.0以下の分散液に添加するシリコン化合物の量である。pH2.0以上7.0以下の分散液に添加する前記のシリコン化合物のモル数をS1、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S1/(F+M)を0.01以上10.0以下とする。S1/(F+M)が0.01未満であると置換元素を含むオキシ水酸化鉄の沈殿と共存するシラノール誘導体の量が少なく、置換元素を含むオキシ水酸化鉄の沈殿をシラノール誘導体またはその縮合反応生成物により均一に被覆する効果が低下するので、好ましくない。シリコン化合物の添加量を多くすると、後述の加熱工程とシリコン酸化物の除去工程の処理量が増大し、製造コストが増大するので、S1/(F+M)は10.0以下であることが好ましい。
第二の条件は、製造工程全体で添加するシリコン化合物の量である。添加するシリコン化合物の全モル数をS2、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S2/(F+M)を0.50以上10.0以下とする。S2が0.50未満であると、置換元素を含むオキシ水酸化鉄の沈殿の表面に被覆されるシリコン化合物の化学反応生成物の被覆量が少なくなり、その結果αタイプの鉄系酸化物が生成しやすくなるデメリットがあり、好ましくない。またS2/(F+M)が10.0を超えると後述の加熱工程とシリコン酸化物の除去工程の処理量が増大し、製造コストが増大するので、好ましくない。
なお、シリコン化合物の全量をpH2.0以上7.0以下の範囲で添加すると、S1=S2となる。
第一の条件は、pH2.0以上7.0以下の分散液に添加するシリコン化合物の量である。pH2.0以上7.0以下の分散液に添加する前記のシリコン化合物のモル数をS1、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S1/(F+M)を0.01以上10.0以下とする。S1/(F+M)が0.01未満であると置換元素を含むオキシ水酸化鉄の沈殿と共存するシラノール誘導体の量が少なく、置換元素を含むオキシ水酸化鉄の沈殿をシラノール誘導体またはその縮合反応生成物により均一に被覆する効果が低下するので、好ましくない。シリコン化合物の添加量を多くすると、後述の加熱工程とシリコン酸化物の除去工程の処理量が増大し、製造コストが増大するので、S1/(F+M)は10.0以下であることが好ましい。
第二の条件は、製造工程全体で添加するシリコン化合物の量である。添加するシリコン化合物の全モル数をS2、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S2/(F+M)を0.50以上10.0以下とする。S2が0.50未満であると、置換元素を含むオキシ水酸化鉄の沈殿の表面に被覆されるシリコン化合物の化学反応生成物の被覆量が少なくなり、その結果αタイプの鉄系酸化物が生成しやすくなるデメリットがあり、好ましくない。またS2/(F+M)が10.0を超えると後述の加熱工程とシリコン酸化物の除去工程の処理量が増大し、製造コストが増大するので、好ましくない。
なお、シリコン化合物の全量をpH2.0以上7.0以下の範囲で添加すると、S1=S2となる。
[本発明の実施態様]
上述のように、本発明の製造方法においては、中和工程におけるアルカリの添加と、シリコン化合物添加工程におけるシリコン化合物の添加を、それぞれ連続的または間歇的に行うことができる、したがって、本発明の製造方法においては、アルカリの添加とシリコン化合物の添加方法の組み合わせにより、様々な実施態様を取ることが可能である。以下に本発明の実施態様の幾つかを例示するが、本発明の製造方法は、以下に記述する実施態様に限定されるものではない。
図1に、アルカリの添加とシリコン化合物の添加の両方を連続的に行う実施態様の時間経過を模式的に例示する。この場合、アルカリの添加を開始した後、原料溶液のpHが2.0以上7.0以下の範囲にある所定のpHに到達した時点でシリコン化合物の添加を開始する。本実施態様の場合、中和工程とシリコン化合物添加工程は、時間的に連続する工程ではなく、並行する工程である。本図に示すように、シリコン化合物の添加は、アルカリの添加が終了した後に継続しても良く、アルカリの添加が終了した時点で終了しても、pHが7.0以下の時点で終了しても構わない。アルカリ添加の終了後もシリコン化合物の添加を行う場合には、製造プロセス全体の時間を考慮し、アルカリ添加の終了後120min以内にシリコン化合物の添加を終了することが好ましい。シリコン化合物の添加が終了した後、後記の熟成工程を設ける。
なお、特に図示しないが、図1の実施態様において、シリコン化合物の添加を数回に分けて、間歇的に行っても構わない。
上述のように、本発明の製造方法においては、中和工程におけるアルカリの添加と、シリコン化合物添加工程におけるシリコン化合物の添加を、それぞれ連続的または間歇的に行うことができる、したがって、本発明の製造方法においては、アルカリの添加とシリコン化合物の添加方法の組み合わせにより、様々な実施態様を取ることが可能である。以下に本発明の実施態様の幾つかを例示するが、本発明の製造方法は、以下に記述する実施態様に限定されるものではない。
図1に、アルカリの添加とシリコン化合物の添加の両方を連続的に行う実施態様の時間経過を模式的に例示する。この場合、アルカリの添加を開始した後、原料溶液のpHが2.0以上7.0以下の範囲にある所定のpHに到達した時点でシリコン化合物の添加を開始する。本実施態様の場合、中和工程とシリコン化合物添加工程は、時間的に連続する工程ではなく、並行する工程である。本図に示すように、シリコン化合物の添加は、アルカリの添加が終了した後に継続しても良く、アルカリの添加が終了した時点で終了しても、pHが7.0以下の時点で終了しても構わない。アルカリ添加の終了後もシリコン化合物の添加を行う場合には、製造プロセス全体の時間を考慮し、アルカリ添加の終了後120min以内にシリコン化合物の添加を終了することが好ましい。シリコン化合物の添加が終了した後、後記の熟成工程を設ける。
なお、特に図示しないが、図1の実施態様において、シリコン化合物の添加を数回に分けて、間歇的に行っても構わない。
図2に、中和工程の途中でアルカリの添加を中断する実施態様の一例の時間経過を模式的に例示する。この場合、アルカリ添加の中断は一度であり、アルカリ添加を中断したpH保持時間内にシリコン化合物の全量を連続的に添加しており、S1=S2となる。
図3に、アルカリ添加の中断を一度行う実施態様の他の一例の時間経過を模式的に例示する。この場合、シリコン化合物の添加は原料溶液のpHが2.0以上7.0以下の範囲にある所定のpHに到達した時点で開始するが、添加の終了時点は中和工程の終了後であり、シリコン化合物の添加の形態は連続的である。
図4および図5に、アルカリ添加の中断を一度行い、シリコン化合物の添加を間歇的に行う実施態様の例の時間経過を模式的に例示する。シリコン化合物の添加を、図4では二回に分けて、図5では三回に分けて行っている。
図6に、アルカリ添加の中断を二度行い、シリコン化合物の添加を間歇的に三度行う実施態様の例の時間経過を模式的に例示する。
なお、上述のように、本発明の製造方法は、図1~図6に例示した実施態様に限定されるものではなく、中和工程におけるアルカリの添加と、シリコン化合物添加工程におけるシリコン化合物の添加の形態を任意に組み合わせることが可能である。
図3に、アルカリ添加の中断を一度行う実施態様の他の一例の時間経過を模式的に例示する。この場合、シリコン化合物の添加は原料溶液のpHが2.0以上7.0以下の範囲にある所定のpHに到達した時点で開始するが、添加の終了時点は中和工程の終了後であり、シリコン化合物の添加の形態は連続的である。
図4および図5に、アルカリ添加の中断を一度行い、シリコン化合物の添加を間歇的に行う実施態様の例の時間経過を模式的に例示する。シリコン化合物の添加を、図4では二回に分けて、図5では三回に分けて行っている。
図6に、アルカリ添加の中断を二度行い、シリコン化合物の添加を間歇的に三度行う実施態様の例の時間経過を模式的に例示する。
なお、上述のように、本発明の製造方法は、図1~図6に例示した実施態様に限定されるものではなく、中和工程におけるアルカリの添加と、シリコン化合物添加工程におけるシリコン化合物の添加の形態を任意に組み合わせることが可能である。
[熟成工程]
pHを8.0以上にしても、シラノール誘導体の縮合反応は緩やかに進行するので、前記の中和工程およびシリコン化合物添加工程を経て得られた、置換元素を含むオキシ水酸化鉄とシリコン化合物の化学反応生成物を含む分散液をpH8.0以上で保持して熟成し、シラノール誘導体の縮合反応を進行させる。その結果として、置換元素を含むオキシ水酸化鉄の沈殿の表面に、シラノール誘導体の縮合反応生成物の均一な被覆層が形成される。この被覆層は、置換元素を含むオキシ水酸化鉄の沈殿物表面のほぼ全面を覆っていると考えられるが、本発明の効果を達成できる範囲において、置換元素を含むオキシ水酸化鉄の沈殿表面における未被覆の部分の存在は許容される。前記の熟成時間は1h以上24h以下であることが好ましい。保持時間が1h未満では、置換元素を含むオキシ水酸化鉄の沈殿のシラノール誘導体の縮合による被覆が完了しておらず、αタイプの鉄系酸化物が生成し易く、24hを超えると熟成の効果が飽和するので好ましくない。なお、前記の熟成時間は、シリコン化合物の添加が中和工程終了後も継続される場合はシリコン化合物添加の終了後、シリコン化合物の添加が中和工程終了前に完了する場合には、アルカリ添加の終了後の時間である。
pHを8.0以上にしても、シラノール誘導体の縮合反応は緩やかに進行するので、前記の中和工程およびシリコン化合物添加工程を経て得られた、置換元素を含むオキシ水酸化鉄とシリコン化合物の化学反応生成物を含む分散液をpH8.0以上で保持して熟成し、シラノール誘導体の縮合反応を進行させる。その結果として、置換元素を含むオキシ水酸化鉄の沈殿の表面に、シラノール誘導体の縮合反応生成物の均一な被覆層が形成される。この被覆層は、置換元素を含むオキシ水酸化鉄の沈殿物表面のほぼ全面を覆っていると考えられるが、本発明の効果を達成できる範囲において、置換元素を含むオキシ水酸化鉄の沈殿表面における未被覆の部分の存在は許容される。前記の熟成時間は1h以上24h以下であることが好ましい。保持時間が1h未満では、置換元素を含むオキシ水酸化鉄の沈殿のシラノール誘導体の縮合による被覆が完了しておらず、αタイプの鉄系酸化物が生成し易く、24hを超えると熟成の効果が飽和するので好ましくない。なお、前記の熟成時間は、シリコン化合物の添加が中和工程終了後も継続される場合はシリコン化合物添加の終了後、シリコン化合物の添加が中和工程終了前に完了する場合には、アルカリ添加の終了後の時間である。
本発明の置換型ε酸化鉄磁性粒子粉の製造方法においては、前記の熟成工程以降の工程は、例えば特許文献1~4に記載されている、従来の製造方法と同じ工程を用いることができる。具体的には、以下のような工程が挙げられる。
[加熱工程]
本発明の製造方法においては、前記のシラノール誘導体の縮合反応生成物で被覆した置換元素を含むオキシ水酸化鉄を、公知の固液分離法を用いて回収した後、加熱処理を施してεタイプの鉄系酸化物を得る。加熱処理前に、洗浄、乾燥の工程を設けても良い。加熱処理は酸化雰囲気中で行われるが、酸化雰囲気としては大気雰囲気で構わない。加熱は概ね700℃以上1300℃以下の範囲で行うことができるが、加熱温度が高いと熱力学安定相であるα-Fe2O3(ε-Fe2O3に対して不純物である)が生成し易くなるので、好ましくは900℃以上1200℃以下、より好ましくは950℃以上1150℃以下で加熱処理を行う。
熱処理時間は0.5H以上10H以下程度の範囲で調整可能であるが、2H以上5H以下の範囲で良好な結果が得られやすい。なお、粒子を覆うシリコン含有物質の存在がαタイプの鉄系酸化物への相変化ではなくεタイプの鉄系酸化物への相変化を引き起こす上で有利に作用するものと考えられる。またシリコン酸化物被覆は、置換元素を含むオキシ水酸化鉄結晶同士の加熱処理時の焼結を防止する作用を有する。
εタイプの鉄系酸化物磁性粒子粉がシリコン酸化物による被覆を必要としない場合には、加熱処理後に前記のシリコン酸化物被覆を除去すれば良い。
[加熱工程]
本発明の製造方法においては、前記のシラノール誘導体の縮合反応生成物で被覆した置換元素を含むオキシ水酸化鉄を、公知の固液分離法を用いて回収した後、加熱処理を施してεタイプの鉄系酸化物を得る。加熱処理前に、洗浄、乾燥の工程を設けても良い。加熱処理は酸化雰囲気中で行われるが、酸化雰囲気としては大気雰囲気で構わない。加熱は概ね700℃以上1300℃以下の範囲で行うことができるが、加熱温度が高いと熱力学安定相であるα-Fe2O3(ε-Fe2O3に対して不純物である)が生成し易くなるので、好ましくは900℃以上1200℃以下、より好ましくは950℃以上1150℃以下で加熱処理を行う。
熱処理時間は0.5H以上10H以下程度の範囲で調整可能であるが、2H以上5H以下の範囲で良好な結果が得られやすい。なお、粒子を覆うシリコン含有物質の存在がαタイプの鉄系酸化物への相変化ではなくεタイプの鉄系酸化物への相変化を引き起こす上で有利に作用するものと考えられる。またシリコン酸化物被覆は、置換元素を含むオキシ水酸化鉄結晶同士の加熱処理時の焼結を防止する作用を有する。
εタイプの鉄系酸化物磁性粒子粉がシリコン酸化物による被覆を必要としない場合には、加熱処理後に前記のシリコン酸化物被覆を除去すれば良い。
[高周波誘導結合プラズマ発光分光分析法(ICP)による組成分析]
溶解法により、得られた置換型ε酸化鉄磁性粒子粉の組成分析を行った。組成分析にあたっては、アジレントテクノロジー製ICP-720ESを使用し、測定波長(nm)についてはFe;259.940nm、Ga;294.363nm、Co;230.786nm、Ti;336.122nm、Si;288.158nm、にて行った。
溶解法により、得られた置換型ε酸化鉄磁性粒子粉の組成分析を行った。組成分析にあたっては、アジレントテクノロジー製ICP-720ESを使用し、測定波長(nm)についてはFe;259.940nm、Ga;294.363nm、Co;230.786nm、Ti;336.122nm、Si;288.158nm、にて行った。
[磁気ヒステリシス曲線(バルクB-H曲線)の測定]
振動試料型磁力計VSM(東英工業社製VSM-P7)を用い、印加磁場1193kA/m(15kOe)、M測定レンジ0.005A・m2(5emu)、ステップビット140bit、時定数0.03sec、ウエイトタイム0.1secで磁気特性を測定した。B-H曲線により、保磁力Hc、飽和磁化σsについて評価を行った。
振動試料型磁力計VSM(東英工業社製VSM-P7)を用い、印加磁場1193kA/m(15kOe)、M測定レンジ0.005A・m2(5emu)、ステップビット140bit、時定数0.03sec、ウエイトタイム0.1secで磁気特性を測定した。B-H曲線により、保磁力Hc、飽和磁化σsについて評価を行った。
[X線回折法(XRD)による結晶性の評価]
得られた試料を粉末X線回折(XRD:リガク社製試料水平型多目的X線回折装置 Ultima IV、線源CuKα線、電圧40kV、電流40mA、2θ=10°以上70°以下)に供した。得られた回折パターンについて、統合粉末X線解析ソフトウェア(PDXL2:リガク社製)を用いICSD(無機結晶構造データベース)のNo.173025:Iron(III)Oxide-Epsilon、No.82134:Hematiteをもとにしてリートベルト解析による評価を行い、結晶構造確認とα相の含有率を確認した。
得られた試料を粉末X線回折(XRD:リガク社製試料水平型多目的X線回折装置 Ultima IV、線源CuKα線、電圧40kV、電流40mA、2θ=10°以上70°以下)に供した。得られた回折パターンについて、統合粉末X線解析ソフトウェア(PDXL2:リガク社製)を用いICSD(無機結晶構造データベース)のNo.173025:Iron(III)Oxide-Epsilon、No.82134:Hematiteをもとにしてリートベルト解析による評価を行い、結晶構造確認とα相の含有率を確認した。
[BET比表面積]
BET比表面積は、株式会社マウンテック製のMacsorb model-1210を用いて、BET一点法により求めた。
BET比表面積は、株式会社マウンテック製のMacsorb model-1210を用いて、BET一点法により求めた。
本発明の製造法により得られた置換型ε酸化鉄磁性粒子粉のTEM観察は、以下の条件で行った。TEM観察には日本電子株式会社製JEM-1011を使用した。粒子観察については、倍率10,000倍、倍率100,000倍で撮影したTEM写真を用いた。(シリコン酸化物被覆を除去後のものを使用)。
-平均粒子径、粒度分布評価(変動係数(%))の測定-
TEM平均粒子径、粒度分布評価(変動係数(%))にはデジタイズを使用した。画像処理ソフトとして、Mac-View Ver.4.0を使用した。この画像ソフトを使用した場合、ある粒子の粒子径は、その粒子に外接する長方形のうち、面積が最小となる長方形の長辺の長さとして算出される。個数については200個以上を測定した。
透過型電子顕微鏡写真上に映っている粒子のうち、測定する粒子の選定基準は次のとおりとした。
[1] 粒子の一部が写真の視野の外にはみだしている粒子は測定しない。
[2] 輪郭がはっきりしており、孤立して存在している粒子は測定する。
[3] 平均的な粒子形状から外れている場合でも、独立しており単独粒子として測定が可能な粒子は測定する。
[4] 粒子同士に重なりがあるが、両者の境界が明瞭で、粒子全体の形状も判断可能な粒子は、それぞれの粒子を単独粒子として測定する。
[5] 重なり合っている粒子で、境界がはっきりせず、粒子の全形も判らない粒子は、粒子の形状が判断できないものとして測定しない。
以上の基準で選定された粒子の粒子径の個数平均値を算出し、置換型ε酸化鉄磁性粒子粉のTEM観察による平均粒子径とした。
-平均粒子径、粒度分布評価(変動係数(%))の測定-
TEM平均粒子径、粒度分布評価(変動係数(%))にはデジタイズを使用した。画像処理ソフトとして、Mac-View Ver.4.0を使用した。この画像ソフトを使用した場合、ある粒子の粒子径は、その粒子に外接する長方形のうち、面積が最小となる長方形の長辺の長さとして算出される。個数については200個以上を測定した。
透過型電子顕微鏡写真上に映っている粒子のうち、測定する粒子の選定基準は次のとおりとした。
[1] 粒子の一部が写真の視野の外にはみだしている粒子は測定しない。
[2] 輪郭がはっきりしており、孤立して存在している粒子は測定する。
[3] 平均的な粒子形状から外れている場合でも、独立しており単独粒子として測定が可能な粒子は測定する。
[4] 粒子同士に重なりがあるが、両者の境界が明瞭で、粒子全体の形状も判断可能な粒子は、それぞれの粒子を単独粒子として測定する。
[5] 重なり合っている粒子で、境界がはっきりせず、粒子の全形も判らない粒子は、粒子の形状が判断できないものとして測定しない。
以上の基準で選定された粒子の粒子径の個数平均値を算出し、置換型ε酸化鉄磁性粒子粉のTEM観察による平均粒子径とした。
[電波吸収特性測定]
置換型ε酸化鉄粉体1.2gを28MPa(20kN)で加圧成形して直径13mm、厚さ3mmの圧粉体を得た。得られた圧粉体に対して、テラヘルツ波時間領域分光法にて透過減衰量測定を行った。具体的には、アドバンテスト社製のテラヘルツ分光システムTAS7400SLを用い、圧粉体をサンプルホルダーに置いた場合とブランクの場合との測定をおこなった。測定条件は以下の通りとした。
・サンプルホルダー径:φ10mm
・Measurement Mode:Transmission
・Frequency Resolution:1.9GHz
・Vertical Axis:Absorbance
・Horizontal Axis:Frequency[THz]
・Cumulated Number(Sample):2048
・Cumulated Number(Background):2048
観測されたサンプルの信号波形およびブランクの参照波形を2112psまで拡張してフーリエ変換し、得られたフーリエスペクトル(各々、Sref、Ssigとする。)の比(Ssig/Sref)を求め、サンプルホルダーに置かれた圧粉体の透過減衰量を算定した。
置換型ε酸化鉄粉体1.2gを28MPa(20kN)で加圧成形して直径13mm、厚さ3mmの圧粉体を得た。得られた圧粉体に対して、テラヘルツ波時間領域分光法にて透過減衰量測定を行った。具体的には、アドバンテスト社製のテラヘルツ分光システムTAS7400SLを用い、圧粉体をサンプルホルダーに置いた場合とブランクの場合との測定をおこなった。測定条件は以下の通りとした。
・サンプルホルダー径:φ10mm
・Measurement Mode:Transmission
・Frequency Resolution:1.9GHz
・Vertical Axis:Absorbance
・Horizontal Axis:Frequency[THz]
・Cumulated Number(Sample):2048
・Cumulated Number(Background):2048
観測されたサンプルの信号波形およびブランクの参照波形を2112psまで拡張してフーリエ変換し、得られたフーリエスペクトル(各々、Sref、Ssigとする。)の比(Ssig/Sref)を求め、サンプルホルダーに置かれた圧粉体の透過減衰量を算定した。
[実施例1]
5L反応槽にて、純水3813.21gに、純度99%硝酸第二鉄9水和物(III)283.26g、Ga濃度11.55mass%の硝酸Ga(III)溶液56.36g、純度97%硝酸コバルト(II)6水和物6.25g、Ti濃度15.1mass%の硫酸チタン(IV)6.61gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解し、原料溶液とした(手順1)。この原料溶液のpHは約1であった。この原料溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.677:0.223:0.050:0.050である。なお、試薬名の後の括弧内のローマ数字は、金属元素の価数を表している。
大気雰囲気中、この原料溶液を20℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液294.85gを90minかけて添加した(手順2)。
60min後に、アンモニア水溶液を添加中の反応液のpHが4.0になった段階で、加水分解基をもつシリコン化合物として純度95.0mass%のテトラエトキシシラン(TEOS)519.22gを並行して添加開始し、30minかけて滴下した。アンモニア水溶液を全て添加した後、20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で置換元素を含むオキシ水酸化鉄の沈殿物を被覆した(手順3)。アンモニア水溶液の添加終了時点の反応液のpH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。なお、この条件下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0.34であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
手順3で得られたスラリーを濾過し、得られたシリコン化合物の化学反応生成物で被覆した置換元素を含むオキシ水酸化鉄の沈殿物の水分をできるだけ切ってから純水中に再度分散させ、リパルプ洗浄した。洗浄後のスラリーを再度濾過し、得られたケーキを大気中110℃で乾燥した(手順4)。
手順4で得られた乾燥品を、箱型焼成炉を用い、大気中1090℃で4h加熱処理し、シリコン酸化物で被覆された鉄系酸化物磁性粉を得た(手順5)。なお、前記のシリコン化合物の化学反応生成物は、大気雰囲気で熱処理した際に、脱水して酸化物に変化する。
本実施例の原料溶液の仕込み条件等の製造条件を、表1に示す。表1には他の実施例および比較例の製造条件も併せて示してある。
5L反応槽にて、純水3813.21gに、純度99%硝酸第二鉄9水和物(III)283.26g、Ga濃度11.55mass%の硝酸Ga(III)溶液56.36g、純度97%硝酸コバルト(II)6水和物6.25g、Ti濃度15.1mass%の硫酸チタン(IV)6.61gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解し、原料溶液とした(手順1)。この原料溶液のpHは約1であった。この原料溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.677:0.223:0.050:0.050である。なお、試薬名の後の括弧内のローマ数字は、金属元素の価数を表している。
大気雰囲気中、この原料溶液を20℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液294.85gを90minかけて添加した(手順2)。
60min後に、アンモニア水溶液を添加中の反応液のpHが4.0になった段階で、加水分解基をもつシリコン化合物として純度95.0mass%のテトラエトキシシラン(TEOS)519.22gを並行して添加開始し、30minかけて滴下した。アンモニア水溶液を全て添加した後、20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で置換元素を含むオキシ水酸化鉄の沈殿物を被覆した(手順3)。アンモニア水溶液の添加終了時点の反応液のpH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。なお、この条件下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0.34であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
手順3で得られたスラリーを濾過し、得られたシリコン化合物の化学反応生成物で被覆した置換元素を含むオキシ水酸化鉄の沈殿物の水分をできるだけ切ってから純水中に再度分散させ、リパルプ洗浄した。洗浄後のスラリーを再度濾過し、得られたケーキを大気中110℃で乾燥した(手順4)。
手順4で得られた乾燥品を、箱型焼成炉を用い、大気中1090℃で4h加熱処理し、シリコン酸化物で被覆された鉄系酸化物磁性粉を得た(手順5)。なお、前記のシリコン化合物の化学反応生成物は、大気雰囲気で熱処理した際に、脱水して酸化物に変化する。
本実施例の原料溶液の仕込み条件等の製造条件を、表1に示す。表1には他の実施例および比較例の製造条件も併せて示してある。
手順5で得られた、シリコン化合物の化学反応生成物で被覆した置換元素を含むオキシ水酸化鉄の沈殿物に熱処理を施した熱処理粉を、17.58mass%NaOH水溶液中で約60℃、24時間撹拌し、粒子表面のシリコン酸化物被覆の除去処理を行った(手順6)。次いで、遠心分離器を用いてスラリーの導電率が500mS/m以下になるまで洗浄し、メンブレンフィルターでろ過した後に乾燥し、得られた鉄系酸化物磁性粉の組成の化学分析、XRD測定および磁気特性の測定等に供した。それらの測定結果を表2に示す。表2には他の実施例および比較例で得られた置換型ε酸化鉄磁性粒子粉の物性値も併せて示してある。
当該実施例1に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ1.3%であった。この値は後述するアンモニア溶液添加後pH8.9になった後に中間の熟成工程を設け、その後pH8.9の状態でTEOSを添加した比較例1により得られた置換型ε酸化鉄粉体についてのそれよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
当該実施例1に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ1.3%であった。この値は後述するアンモニア溶液添加後pH8.9になった後に中間の熟成工程を設け、その後pH8.9の状態でTEOSを添加した比較例1により得られた置換型ε酸化鉄粉体についてのそれよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[実施例2]
実施例2として、アンモニア水溶液の添加時の温度を30℃、アンモニア水溶液の添加時間を30min、TEOSの添加開始pHを6.0、TEOSの添加速度をアンモニア水溶液添加中は5.88g/min、 アンモニア水溶液添加終了後は22.47g/minとした以外は実施例1と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、アンモニア水溶液の添加終了時点のpHは8.9であり、TEOSの添加終了時点の反応液pH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。当該実施例2に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ3.0%であった。この値は後述するアンモニア溶液添加後pH8.9になった後に熟成工程を30min間設け、その後pH8.9の状態でTEOSを添加した比較例3により得られた置換型ε酸化鉄磁性粒子粉についてのそれよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
実施例2として、アンモニア水溶液の添加時の温度を30℃、アンモニア水溶液の添加時間を30min、TEOSの添加開始pHを6.0、TEOSの添加速度をアンモニア水溶液添加中は5.88g/min、 アンモニア水溶液添加終了後は22.47g/minとした以外は実施例1と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、アンモニア水溶液の添加終了時点のpHは8.9であり、TEOSの添加終了時点の反応液pH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。当該実施例2に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ3.0%であった。この値は後述するアンモニア溶液添加後pH8.9になった後に熟成工程を30min間設け、その後pH8.9の状態でTEOSを添加した比較例3により得られた置換型ε酸化鉄磁性粒子粉についてのそれよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[比較例1]
比較例1として、アンモニア水溶液を添加してpHが8.9になった時点でTEOSを添加し、その後に熟成工程を設けた以外は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、アンモニア添加終了時点のpHは8.9であり、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。当該比較例1に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ3.6%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
比較例1として、アンモニア水溶液を添加してpHが8.9になった時点でTEOSを添加し、その後に熟成工程を設けた以外は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、アンモニア添加終了時点のpHは8.9であり、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間を通じての反応液のpHはいずれも8.8であった。当該比較例1に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ3.6%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[比較例2]
比較例2として、アンモニア水溶液の添加時の温度を25℃、アンモニア水溶液の添加時間を60min、アンモニア水溶液添加後、分散液のpHが8.9になった後に中間の熟成工程を30min間設け、その後pH8.9の状態でTEOSを添加した以外は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった当該比較例2に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ7.4%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
比較例2として、アンモニア水溶液の添加時の温度を25℃、アンモニア水溶液の添加時間を60min、アンモニア水溶液添加後、分散液のpHが8.9になった後に中間の熟成工程を30min間設け、その後pH8.9の状態でTEOSを添加した以外は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった当該比較例2に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ7.4%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[比較例3]
比較例3として、アンモニア水溶液を添加してpHが8.9になった後に熟成工程を設け、その後pH8.9の状態でTEOSを添加した以外は実施例2と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該比較例3に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ5.4%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
以上の結果から、中和処理を連続的に行った場合、TEOSの添加開始時期をpH2.0以上7.0以下とする効果は明らかである。また、実施例2の結果が示すように、本発明の製造方法の場合、中和処理を常温より高い30℃で行った場合でも、α相の含有率の低い置換型ε酸化鉄磁性粒子粉が得られる。
比較例3として、アンモニア水溶液を添加してpHが8.9になった後に熟成工程を設け、その後pH8.9の状態でTEOSを添加した以外は実施例2と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該比較例3に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ5.4%と実施例1~6と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
以上の結果から、中和処理を連続的に行った場合、TEOSの添加開始時期をpH2.0以上7.0以下とする効果は明らかである。また、実施例2の結果が示すように、本発明の製造方法の場合、中和処理を常温より高い30℃で行った場合でも、α相の含有率の低い置換型ε酸化鉄磁性粒子粉が得られる。
[実施例3]
実施例3として、アンモニア水溶液をpH4.0になるまで添加(第一の中和工程)した後にアンモニア水溶液の添加を止め、中間の熟成工程を設けず、直ちにTEOSの添加を開始し、TEOS添加終了後に残りのアンモニア水溶液を添加(第二の中和工程)した以外は実施例1と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該実施例3に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する、アンモニア水溶液をpH4.0または6.0になるまで添加(第一の中和工程)した後にアンモニア溶液の添加を止め、中間の熟成工程30minを設け、その後TEOS添加開始するが、TEOS添加終了後に第二中和工程を設けなかった比較例4および5により得られた置換型ε酸化鉄磁性粒子粉についてのそれらの値よりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
実施例3として、アンモニア水溶液をpH4.0になるまで添加(第一の中和工程)した後にアンモニア水溶液の添加を止め、中間の熟成工程を設けず、直ちにTEOSの添加を開始し、TEOS添加終了後に残りのアンモニア水溶液を添加(第二の中和工程)した以外は実施例1と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該実施例3に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する、アンモニア水溶液をpH4.0または6.0になるまで添加(第一の中和工程)した後にアンモニア溶液の添加を止め、中間の熟成工程30minを設け、その後TEOS添加開始するが、TEOS添加終了後に第二中和工程を設けなかった比較例4および5により得られた置換型ε酸化鉄磁性粒子粉についてのそれらの値よりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[実施例4]
1L反応槽にて、純水737.71gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液104.81g、Ga濃度11.55mass%の硝酸Ga(III)溶液14.32g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.02gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.714:0.186:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液15.00gを1.9minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは2.0であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを10minかけて滴下した。TEOS添加終了後に22.30mass%のアンモニア溶液62.77gを8.1minで添加(第二の中和工程)した。第二中和工程後のpHは8.8であった。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.8であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は2.84であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)も2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該実施例4に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
1L反応槽にて、純水737.71gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液104.81g、Ga濃度11.55mass%の硝酸Ga(III)溶液14.32g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.02gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.714:0.186:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液15.00gを1.9minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは2.0であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを10minかけて滴下した。TEOS添加終了後に22.30mass%のアンモニア溶液62.77gを8.1minで添加(第二の中和工程)した。第二中和工程後のpHは8.8であった。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.8であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は2.84であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)も2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該実施例4に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[実施例5]
実施例5として、第一中和工程に用いるアンモニア水溶液の添加量および添加時間を51.00gおよび6.5min、第一の中和工程後のpHを3.0、第二の中和工程に用いるアンモニア溶液の添加量および添加時間を27.24gおよび3.5minとした以外は実施例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該実施例5に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化鉄磁性粒子についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
実施例5として、第一中和工程に用いるアンモニア水溶液の添加量および添加時間を51.00gおよび6.5min、第一の中和工程後のpHを3.0、第二の中和工程に用いるアンモニア溶液の添加量および添加時間を27.24gおよび3.5minとした以外は実施例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.8であった。当該実施例5に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化鉄磁性粒子についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[実施例6]
実施例6として、第一中和工程に用いるアンモニア水溶液の添加量および添加時間を53.00gおよび6.8min、第一の中和工程後のpHを4.0、第二の中和工程に用いるアンモニア溶液の添加量および添加時間を24.78gおよび3.2minとした以外は実施例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程後の反応液pH、および前記20hの撹拌をしている間の反応液のpHは8.8であった。当該実施例5に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化鉄磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表1に併せて示す。
実施例6として、第一中和工程に用いるアンモニア水溶液の添加量および添加時間を53.00gおよび6.8min、第一の中和工程後のpHを4.0、第二の中和工程に用いるアンモニア溶液の添加量および添加時間を24.78gおよび3.2minとした以外は実施例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、第二の中和工程後の反応液pH、および前記20hの撹拌をしている間の反応液のpHは8.8であった。当該実施例5に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化鉄磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表1に併せて示す。
[実施例7]
1L反応槽にて、純水746.26gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液102.73g、純度98%硝酸アルミニウム(III)9水和物10.74g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.02gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.680:0.220:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液51.59gを60minで添加(第一の中和工程)し、滴下終了後に10min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは3.0であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを5minかけて滴下した。TEOS添加終了後に22.30mass%のアンモニア溶液27.09gを14minで添加(第二の中和工程)した。第二中和工程後のpHは8.8であった。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.8であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、アルミニウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は2.84であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、アルミニウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該実施例7に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
得られた置換型ε酸化鉄磁性粒子粉につき、上述した方法により電波吸収特性を測定した。その結果、周波数が50GHzから100GHzまでの範囲での圧粉体の最大吸収周波数は80.1GHz、単位厚みあたりの透過減衰量は4.1dB/mmであった。
1L反応槽にて、純水746.26gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液102.73g、純度98%硝酸アルミニウム(III)9水和物10.74g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.02gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.680:0.220:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液51.59gを60minで添加(第一の中和工程)し、滴下終了後に10min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは3.0であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを5minかけて滴下した。TEOS添加終了後に22.30mass%のアンモニア溶液27.09gを14minで添加(第二の中和工程)した。第二中和工程後のpHは8.8であった。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.8であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、アルミニウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は2.84であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、アルミニウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該実施例7に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ0%であった。この値は後述する比較例4および5により得られた置換型ε酸化磁性粒子粉についてのそれらよりも優れたものである。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
得られた置換型ε酸化鉄磁性粒子粉につき、上述した方法により電波吸収特性を測定した。その結果、周波数が50GHzから100GHzまでの範囲での圧粉体の最大吸収周波数は80.1GHz、単位厚みあたりの透過減衰量は4.1dB/mmであった。
[比較例4]
比較例4として、第二の中和工程を実施しない以外は実施例6と同様の手順で、置換型ε酸化鉄粉体を得た。なお、TEOS添加終了時点での反応液のpH、および前記20hの撹拌をしている間の反応液のpHは4.0であった。当該比較例4に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ87.5%と、実施例1~7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
比較例4として、第二の中和工程を実施しない以外は実施例6と同様の手順で、置換型ε酸化鉄粉体を得た。なお、TEOS添加終了時点での反応液のpH、および前記20hの撹拌をしている間の反応液のpHは4.0であった。当該比較例4に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ87.5%と、実施例1~7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
[比較例5]
比較例5として、第一の中和工程に用いるアンモニア水溶液の添加量および添加時間を57.00gおよび7.3min、第一の中和工程終了時のpHを6.0とした以外は比較例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点での反応液のpH、および前記20hの撹拌をしている間の反応液のpHは6.0であった。当該比較例5に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ36.9%と、実施例1~7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
以上の結果から、第二の中和工程を設けないと、α相の含有率の低い置換型ε酸化鉄磁性粒子粉が得られないことが判る。また、出発物質の鉄原料として硝酸塩および硫酸塩の何れを用いても、ほぼ同じ結果が得られている。
比較例5として、第一の中和工程に用いるアンモニア水溶液の添加量および添加時間を57.00gおよび7.3min、第一の中和工程終了時のpHを6.0とした以外は比較例4と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点での反応液のpH、および前記20hの撹拌をしている間の反応液のpHは6.0であった。当該比較例5に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ36.9%と、実施例1~7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
以上の結果から、第二の中和工程を設けないと、α相の含有率の低い置換型ε酸化鉄磁性粒子粉が得られないことが判る。また、出発物質の鉄原料として硝酸塩および硫酸塩の何れを用いても、ほぼ同じ結果が得られている。
[実施例8]
中間の熟成工程における撹拌時間を10minとし、テトラエトキシシラン(TEOS)を5minかけて添加した以外は、実施例5と同様の手順により置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点での反応液のpHは3.0であり、前記20hの撹拌をしている間の反応液のpHは8.6であった。
得られた置換型ε酸化鉄磁性粒子粉のα相の含有率は0%であり、上述した方法により電波吸収特性を測定した。その結果、周波数が50GHzから100GHzまでの範囲での圧粉体の最大吸収周波数は67.2GHz、単位厚みあたりの透過減衰量は4.6dB/mmであった。
中間の熟成工程における撹拌時間を10minとし、テトラエトキシシラン(TEOS)を5minかけて添加した以外は、実施例5と同様の手順により置換型ε酸化鉄磁性粒子粉を得た。なお、TEOS添加終了時点での反応液のpHは3.0であり、前記20hの撹拌をしている間の反応液のpHは8.6であった。
得られた置換型ε酸化鉄磁性粒子粉のα相の含有率は0%であり、上述した方法により電波吸収特性を測定した。その結果、周波数が50GHzから100GHzまでの範囲での圧粉体の最大吸収周波数は67.2GHz、単位厚みあたりの透過減衰量は4.6dB/mmであった。
[比較例6]
5L反応槽にて、純水3688.56gに、Fe濃度11.58mass%の硫酸第二鉄(III)溶液527.22g、Ga濃度11.55mass%の硝酸Ga(III)溶液71.61g、純度97%硝酸コバルト(II)6水和物9.57g、Ti濃度15.1mass%の硫酸チタン(IV)10.11gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.714:0.186:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液388.91gを10minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは8.6であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)794.40gを10minかけて滴下した。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.6であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該比較例6に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ4.9%と、実施例8と比較して高い値であった。また組成の化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
5L反応槽にて、純水3688.56gに、Fe濃度11.58mass%の硫酸第二鉄(III)溶液527.22g、Ga濃度11.55mass%の硝酸Ga(III)溶液71.61g、純度97%硝酸コバルト(II)6水和物9.57g、Ti濃度15.1mass%の硫酸チタン(IV)10.11gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Ga:Co:Ti=1.714:0.186:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液388.91gを10minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは8.6であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)794.40gを10minかけて滴下した。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.6であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該比較例6に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ4.9%と、実施例8と比較して高い値であった。また組成の化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
[実施例9]
硫酸第二鉄(III)溶液101.50g、および硝酸アルミニウム(III)9水和物11.7gを使用した以外は実施例7と同様の手順により、置換型ε酸化鉄粉体を得た。なお、TEOS添加終了時点での反応液のpHは3.0であり、前記20hの撹拌をしている間の反応液のpHは8.6であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.660:0.240:0.050:0.050である。組成の化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
硫酸第二鉄(III)溶液101.50g、および硝酸アルミニウム(III)9水和物11.7gを使用した以外は実施例7と同様の手順により、置換型ε酸化鉄粉体を得た。なお、TEOS添加終了時点での反応液のpHは3.0であり、前記20hの撹拌をしている間の反応液のpHは8.6であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.660:0.240:0.050:0.050である。組成の化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
[比較例7]
1L反応槽にて、純水746.36gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液101.51g、純度98%硝酸Al(III)9水和物11.72g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.12gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.660:0.240:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液78.68gを10minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは8.6であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを10minかけて滴下した。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.6であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該比較例7に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ8.7%と、追加実施例2と比較して高い値であった。また化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
1L反応槽にて、純水746.36gに、Fe濃度11.65mass%の硫酸第二鉄(III)溶液101.51g、純度98%硝酸Al(III)9水和物11.72g、純度97%硝酸コバルト(II)6水和物1.91g、Ti濃度15.1mass%の硫酸チタン(IV)2.12gを大気雰囲気中、撹拌羽根により機械的に撹拌しながら溶解した(手順1)。この溶解液のpHは約1であった。この仕込み溶液中の金属イオンのモル比は、Fe:Al:Co:Ti=1.660:0.240:0.050:0.050である。
大気雰囲気中、この仕込み溶解液を30℃の条件下で、撹拌羽根により機械的に撹拌しながら、22.30mass%のアンモニア水溶液78.68gを10minで添加(第一の中和工程)し、滴下終了後に30min間撹拌を続けて生成した沈殿物の熟成を行った(中間の熟成工程)。その際、沈殿物を含むスラリーのpHは8.6であった(手順2)。
手順2で得られたスラリーを撹拌しながら、大気中30℃で、純度95.0mass%のテトラエトキシシラン(TEOS)158.88gを10minかけて滴下した。その後20hそのまま撹拌し続け、シリコン化合物の化学反応生成物で沈殿物を被覆した(手順3)。前記20hの撹拌をしている間の反応液のpHは8.6であった。なお、この条件で下では、pH2.0以上7.0以下の分散液に添加したテトラエトキシシランに含まれるSi元素の量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比、S1/(F+M)は0であり、分散液に滴下するテトラエトキシシランに含まれるSi元素の全量と、原料溶液中に含まれる鉄、ガリウム、コバルト、チタンイオンの量とのモル比S2/(F+M)は2.84である。
その後は実施例1と同様の手順で置換型ε酸化鉄磁性粒子粉を得た。当該比較例7に係る置換型ε酸化鉄粉体についてXRD測定を行い、α相の含有率を求めたところ8.7%と、追加実施例2と比較して高い値であった。また化学分析、磁気特性および電波吸収特性の評価を行った。測定結果を表2に併せて示す。
[比較例8]
本比較例は、実施例7の組成比の従来製法に対応する実験例である。
添加する金属塩の量をFe濃度11.65mass%の硫酸第二鉄(III)溶液102.73g、純度98%硝酸Al(III)9水和物10.74gとした以外は追加比較例2と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。
なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.6であった。当該比較例に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ8.1%と実施例7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。
本比較例は、実施例7の組成比の従来製法に対応する実験例である。
添加する金属塩の量をFe濃度11.65mass%の硫酸第二鉄(III)溶液102.73g、純度98%硝酸Al(III)9水和物10.74gとした以外は追加比較例2と同様の手順で、置換型ε酸化鉄磁性粒子粉を得た。
なお、TEOS添加終了時点の反応液pH、および前記20hの撹拌をしている間の反応液のpHはいずれも8.6であった。当該比較例に係る置換型ε酸化鉄磁性粒子粉についてXRD測定を行い、α相の含有率を求めたところ8.1%と実施例7と比較して高い値であった。また組成の化学分析および磁気特性の評価を行った。測定結果を表2に併せて示す。

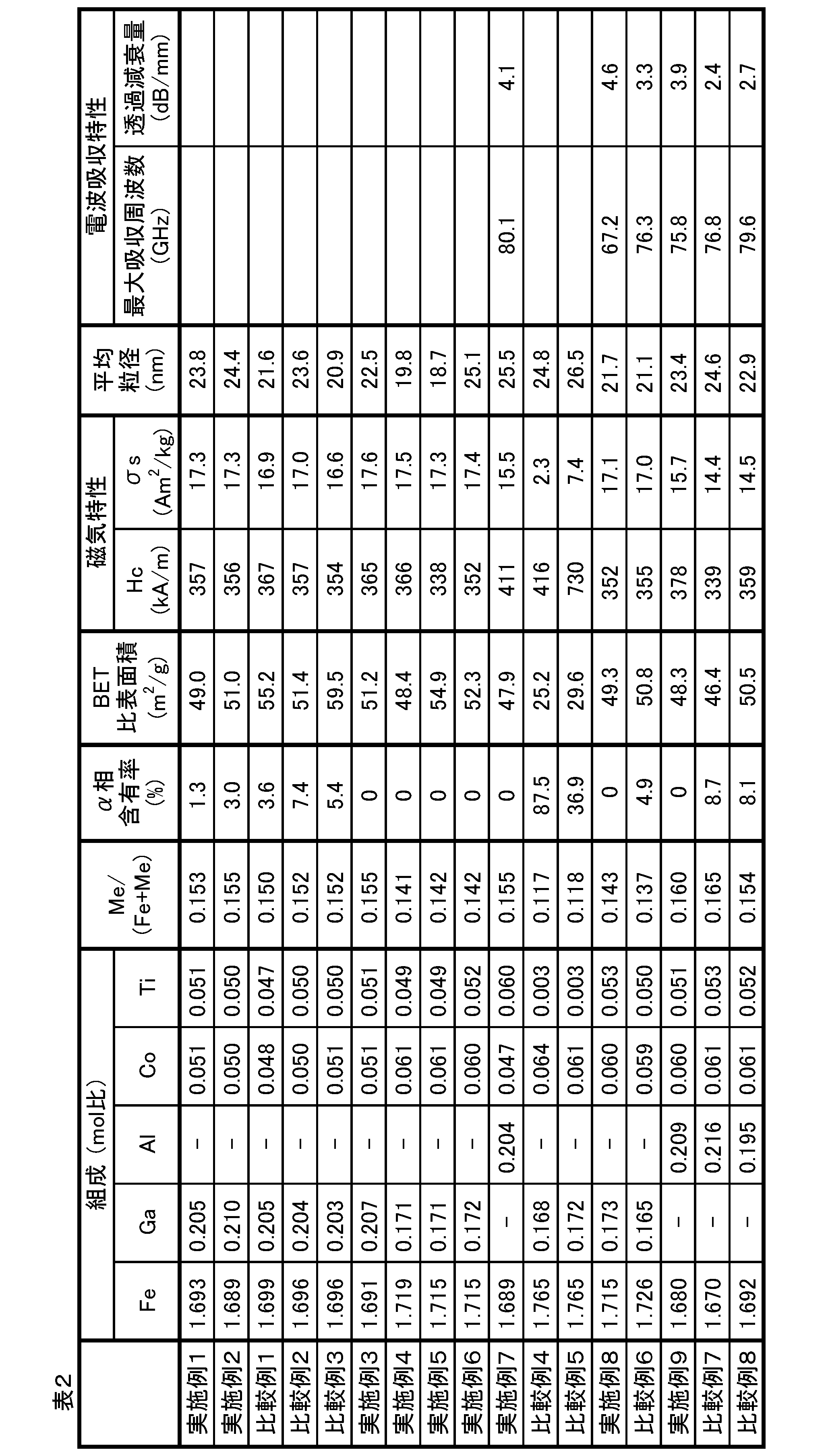
Claims (11)
- ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉であって、前記置換型ε酸化鉄磁性粒子粉に含まれるFeのモル数をFe、Feサイトを置換した全金属元素のモル数をMeとしたとき、Me/(Fe+Me)で定義される他の金属元素によるFeの置換量が0.08以上0.17以下であり、かつ、X線回折法により測定されるαタイプの鉄系酸化物の含有率が3%以下である、置換型ε酸化鉄磁性粒子粉。
- 前記のFeサイトを一部置換する他の金属元素がCo、Tiならびに、GaおよびAlから選ばれる1種以上である、請求項1に記載の置換型ε酸化鉄磁性粒子粉。
- 請求項1または2に記載の置換型ε酸化鉄磁性粒子粉からなる圧粉体。
- 請求項1または2に記載の置換型ε酸化鉄磁性粒子粉が樹脂またはゴムに分散されてなる電波吸収体。
- 原料溶液として3価の鉄イオンと前記Feサイトを一部置換する金属のイオンを含む酸性の水溶液を用い、前記の原料溶液にアルカリを添加してpH8.0以上10.0以下まで中和し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水 酸化物の混合物を含む分散液を得る中和工程と、
前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物を含む前記の分散液に、加水分解基を持つシリコン化合物を添加するシリコン化合物添加工程と、
前記の置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物と前記のシリコン化合物を含む分散液を、pH8.0以上10.0以下で保持し、置換金属元素を含むオキシ水酸化鉄もしくはオキシ水酸化鉄と置換金属元素の水酸化物の混合物に前記シリコン化合物の化学反応生成物を被覆する熟成工程と、
を含む、ε-Fe2O3のFeサイトの一部を他の金属元素で置換したεタイプの鉄系酸化物を主として含む置換型ε酸化鉄磁性粒子粉の製造方法であって、
前記の加水分解基を持つシリコン化合物の添加を、前記の中和工程において分散液のpHが2.0以上7.0以下になった時点で開始し、
pH2.0以上7.0以下の分散液に添加する前記のシリコン化合物のモル数をS1、原料溶液中に含まれるFeイオンのモル数をF、置換金属元素イオンの全モル数をMとしたとき、S1/(F+M)が0.01以上10.0以下であり、かつ、
前記のシリコン化合物の全添加モル数をS2としたとき、S2/(F+M)が0.50以上10.0以下である、
置換型ε酸化鉄磁性粒子粉の製造方法。 - 前記の中和工程におけるアルカリ添加と、前記のシリコン化合物添加工程におけるシリコン化合物の添加を、いずれも連続的に行う、請求項5に記載の置換型ε酸化鉄磁性粒子粉の製造方法。
- 前記の中和工程におけるアルカリ添加を連続的に、前記のシリコン化合物添加工程におけるシリコン化合物の添加を間歇的に行う、請求項5に記載の置換型ε酸化鉄磁性粒子粉の製造方法。
- 前記の中和工程におけるアルカリ添加と、前記のシリコン化合物添加工程におけるシリコン化合物の添加を、いずれも間歇的に行う、請求項5に記載の置換型ε酸化鉄磁性粒子粉の製造方法。
- 前記の中和工程におけるアルカリ添加を間歇的に、前記のシリコン化合物添加工程におけるシリコン化合物の添加を連続的に行う、請求項5に記載の置換型ε酸化鉄磁性粒子粉の製造方法。
- 前記のFeサイトを一部置換する他の金属元素がCo、Tiならびに、GaおよびAlから選ばれる1種以上である、請求項3に記載の置換型ε酸化鉄磁性粒子粉の製造方法。
- 請求項1~3のいずれか一項に記載の置換型ε酸化鉄磁性粒子粉を圧縮成形して圧粉体を得る、圧粉体の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202080018538.9A CN113508442B (zh) | 2019-03-05 | 2020-02-28 | 置换型ε氧化铁磁性粒子粉、置换型ε氧化铁磁性粒子粉的制造方法、压粉体、压粉体的制造方法和电波吸收体 |
| US17/433,672 US20220089456A1 (en) | 2019-03-05 | 2020-02-28 | Substitution-type epsilon-iron oxide magnetic particle powder, method for producing substitution-type epsilon-iron oxide magnetic particle powder, green compact, method for producing green compact, and electromagnetic wave |
| EP20766384.0A EP3937194A4 (en) | 2019-03-05 | 2020-02-28 | SUBSTITUTION-LIKE EPSILONE IRON OXIDE MAGNETIC PARTICLE POWDER, METHOD FOR PRODUCTION OF A SUBSTITUTION-LIKE EPSILONE IRON OXIDE MAGNETIC PARTICLE POWDER, GREEN LING, METHOD FOR PRODUCTION OF A GREEN LING AND ABSORBER OF ELECTROMAGNETIC WAVES |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019040063 | 2019-03-05 | ||
| JP2019-040063 | 2019-03-05 | ||
| JP2019-180329 | 2019-09-30 | ||
| JP2019180329A JP7509524B2 (ja) | 2019-03-05 | 2019-09-30 | 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020179659A1 true WO2020179659A1 (ja) | 2020-09-10 |
Family
ID=72338605
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/008278 Ceased WO2020179659A1 (ja) | 2019-03-05 | 2020-02-28 | 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20220089456A1 (ja) |
| EP (1) | EP3937194A4 (ja) |
| WO (1) | WO2020179659A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016130208A (ja) * | 2015-01-09 | 2016-07-21 | Dowaエレクトロニクス株式会社 | 鉄系酸化物磁性粒子粉およびその製造方法並びに塗料および磁気記録媒体 |
| JP2018092691A (ja) * | 2016-11-30 | 2018-06-14 | 富士フイルム株式会社 | ε−酸化鉄型強磁性粉末およびその製造方法ならびにε−酸化鉄型強磁性粉末含有組成物 |
| JP2019003970A (ja) * | 2017-06-09 | 2019-01-10 | 富士フイルム株式会社 | 磁性粉、磁性粉の製造方法、及び磁気記録媒体 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4859791B2 (ja) * | 2006-09-01 | 2012-01-25 | 国立大学法人 東京大学 | 電波吸収材料用の磁性結晶および電波吸収体 |
| JP5445843B2 (ja) * | 2007-05-31 | 2014-03-19 | 国立大学法人 東京大学 | 磁性酸化鉄粒子、磁性体、および電波吸収体 |
| JP5071902B2 (ja) * | 2008-02-20 | 2012-11-14 | 国立大学法人 東京大学 | 電波吸収材料および当該電波吸収材料を用いた電波吸収体 |
| WO2017018407A1 (ja) * | 2015-07-27 | 2017-02-02 | Dowaエレクトロニクス株式会社 | 鉄系酸化物磁性粒子粉の製造方法 |
| WO2018062478A1 (ja) * | 2016-09-30 | 2018-04-05 | Dowaエレクトロニクス株式会社 | イプシロン型鉄酸化物磁性粒子及びその製造方法、磁性粒子から構成される磁性粉ならびに磁性塗料および磁気記録媒体 |
-
2020
- 2020-02-28 EP EP20766384.0A patent/EP3937194A4/en active Pending
- 2020-02-28 WO PCT/JP2020/008278 patent/WO2020179659A1/ja not_active Ceased
- 2020-02-28 US US17/433,672 patent/US20220089456A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016130208A (ja) * | 2015-01-09 | 2016-07-21 | Dowaエレクトロニクス株式会社 | 鉄系酸化物磁性粒子粉およびその製造方法並びに塗料および磁気記録媒体 |
| JP2018092691A (ja) * | 2016-11-30 | 2018-06-14 | 富士フイルム株式会社 | ε−酸化鉄型強磁性粉末およびその製造方法ならびにε−酸化鉄型強磁性粉末含有組成物 |
| JP2019003970A (ja) * | 2017-06-09 | 2019-01-10 | 富士フイルム株式会社 | 磁性粉、磁性粉の製造方法、及び磁気記録媒体 |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP3937194A4 * |
| TOWATA, ATSUYA: "Influence of ε-Fe2O3 Synthesis Conditions on Particle Properties in Silica Xerogels", JOURNAL OF THE SOCIETY OF POWDER TECHNOLOGY, vol. 54, no. 9, 10 September 2017 (2017-09-10), Kyoto, pages 569 - 575, XP009529771, ISSN: 0386-6157, DOI: 10.4164/sptj.54.569 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3937194A4 (en) | 2022-12-14 |
| US20220089456A1 (en) | 2022-03-24 |
| EP3937194A1 (en) | 2022-01-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10622127B2 (en) | Iron-based oxide magnetic particle powder, method for producing same, coating material, and magnetic recording medium | |
| JP6676493B2 (ja) | 鉄系酸化物磁性粒子粉の製造方法 | |
| JP5966064B1 (ja) | 鉄系酸化物磁性粒子粉および鉄系酸化物磁性粒子粉の製造方法 | |
| JP6480715B2 (ja) | 鉄系酸化物磁性粒子粉の前駆体およびそれを用いた鉄系酸化物磁性粒子粉の製造方法 | |
| WO2016047559A1 (ja) | 鉄系酸化物磁性粒子粉および鉄系酸化物磁性粒子粉の製造方法 | |
| WO2016111224A1 (ja) | 鉄系酸化物磁性粒子粉およびその製造方法並びに塗料および磁気記録媒体 | |
| TWI732445B (zh) | 鐵系氧化物磁性粉及其製造方法 | |
| JP7509524B2 (ja) | 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 | |
| JP2020167365A (ja) | 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 | |
| TWI761154B (zh) | 鐵系氧化物磁性粉,以及使用該磁性粉而得的壓粉體及電波吸收體 | |
| WO2020179659A1 (ja) | 置換型ε酸化鉄磁性粒子粉、置換型ε酸化鉄磁性粒子粉の製造方法、圧粉体、圧粉体の製造方法および電波吸収体 | |
| US20220162089A1 (en) | Substituted epsilon-iron oxide magnetic particle powder, production method for substituted epsilon-iron oxide magnetic particle powder, green compact, production method for green compact, and electromagnetic wave absorber | |
| JP7497258B2 (ja) | 置換型ε酸化鉄磁性粒子粉および置換型ε酸化鉄磁性粒子粉の製造方法 | |
| WO2021200481A1 (ja) | 鉄系酸化物磁性粉およびその製造方法 | |
| JP7659408B2 (ja) | 鉄系酸化物磁性粉の製造方法 | |
| JP2021150448A (ja) | 鉄系酸化物磁性粉の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20766384 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020766384 Country of ref document: EP Effective date: 20211005 |