WO2020196228A1 - 硬化性組成物、及び硬化物 - Google Patents
硬化性組成物、及び硬化物 Download PDFInfo
- Publication number
- WO2020196228A1 WO2020196228A1 PCT/JP2020/012224 JP2020012224W WO2020196228A1 WO 2020196228 A1 WO2020196228 A1 WO 2020196228A1 JP 2020012224 W JP2020012224 W JP 2020012224W WO 2020196228 A1 WO2020196228 A1 WO 2020196228A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polymer
- meth
- weight
- reactive silyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/336—Polymers modified by chemical after-treatment with organic compounds containing silicon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/14—Homopolymers or copolymers of esters of esters containing halogen, nitrogen, sulfur, or oxygen atoms in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
- C08L71/02—Polyalkylene oxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2312/00—Crosslinking
Definitions
- the present invention relates to a curable composition and a cured product.
- An organic polymer having a hydroxyl group or a hydrolyzable group on a silicon atom and a silicon-containing group capable of forming a siloxane bond (hereinafter referred to as "reactive silyl group”) is known as a moisture-reactive polymer. It is contained in many industrial products such as adhesives, sealants, coating materials, paints, and adhesives, and is used in a wide range of fields.
- a polyoxyalkylene polymer having a reactive silyl group and a (meth) acrylic acid ester polymer having a reactive silyl group are blended for use in applications such as a highly weather resistant sealing material and an elastic adhesive.
- Curable compositions are known (see, for example, Patent Documents 1 to 4).
- the cured product obtained from a curable composition obtained by blending a polyoxyalkylene polymer having a reactive silyl group and a (meth) acrylic acid ester polymer having a reactive silyl group exhibits high strength, while exhibiting high strength. In some cases, the elongation was not sufficient.
- the present invention has a polyoxyalkylene polymer having a reactive silyl group and a (meth) acrylic acid having a reactive silyl group, which can form a cured product showing high elongation while having high strength.
- An object of the present invention is to provide a curable composition containing an ester-based polymer.
- the present inventors have found that the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton in one molecule of the polyoxyalkylene polymer, and (meth ) Acrylic acid ester-based polymer (B)
- the average number of reactive silyl groups at the end of the polymer skeleton and the average number of reactive silyl groups inside the polymer skeleton per molecule are set within specific ranges.
- the present invention has the following general formula (1): -SiR 1 a X 3-a (1)
- R 1 represents a substituted or unsubstituted hydrocarbon group having 1 to 20 carbon atoms.
- X represents a hydroxyl group or a hydrolyzable group.
- A is 0, 1 or 2.
- R 2 represents an unsubstituted hydrocarbon group having 1 to 20 carbon atoms.
- Y represents a hydroxyl group or a hydrolyzable group.
- R 2 or Y are 0, 1 or 2.
- R 2 or Y When present, they include a (meth) acrylic acid ester-based polymer (B), which has a reactive silyl group represented by (which may be the same or different.), And a polyoxyalkylene-based polymer (.
- the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton in one molecule is 0.80 or more, and the terminal of the polymer skeleton in one molecule of the (meth) acrylic acid ester-based polymer (B)
- the average number of reactive silyl groups bonded to is 0.30 or more and 1.00 or less, and the reactive silyl bonded to the inside of the polymer skeleton in one molecule of the (meth) acrylic acid ester-based polymer (B).
- the present invention relates to a curable composition having an average number of groups of 0 or more and less than 0.30.
- the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton in one molecule of the polyoxyalkylene polymer (A) is 0.85 or more.
- the average number of reactive silyl groups bonded to the ends of the polymer skeleton in one molecule of the (meth) acrylic acid ester-based polymer (B) is 0.85 or more and 1.00 or less.
- the average number of reactive silyl groups bonded to the inside of the polymer skeleton in one molecule of the (meth) acrylic acid ester-based polymer (B) is 0 or more and less than 0.15.
- the weight ratio of the polyoxyalkylene polymer (A): (meth) acrylic acid ester polymer (B) is 80:20 to 50:50.
- the proportion of methyl methacrylate in the constituent monomer units of the (meth) acrylic acid ester-based polymer (B) is 35% by weight or more and 85% by weight or less.
- the total proportion of alkyl methacrylate in the constituent monomer units of the (meth) acrylic acid ester-based polymer (B) is 70% by weight or more and 85% by weight or less, and the alkyl has 1 carbon atom. It is an unsubstituted alkyl group of ⁇ 4.
- the present invention also relates to a cured product obtained by curing the curable composition.
- a polyoxyalkylene polymer having a reactive silyl group and a (meth) acrylic acid ester-based polymer having a reactive silyl group capable of forming a cured product showing high elongation while having high strength can be provided.
- the curable composition of the present invention contains a polyoxyalkylene polymer (A) having a reactive silyl group. Since the polyoxyalkylene polymer (A) has the reactive silyl group, it exhibits curability based on the hydrolysis and dehydration condensation reaction of the reactive silyl group.
- the polyoxyalkylene polymer (A) has a polymer skeleton of polyoxyalkylene and a terminal structure bonded to the end of the polymer skeleton.
- the polymer skeleton refers to a polymer main chain composed of oxyalkylene repeating units.
- the polymer skeleton of the polyoxyalkylene polymer (A) may be a linear one or a branched chain one.
- the linear polymer skeleton is preferable because the cured product of the curable composition has high elongation, and the branched-chain polymer skeleton is preferable because the cured product of the curable composition has high strength.
- the linear polymer skeleton can be formed by using an initiator having two hydroxyl groups in one molecule in the polymerization method for forming the polymer skeleton, and the branched polymer skeleton can be formed. It can be formed by using an initiator having three or more hydroxyl groups in one molecule.
- the polymer skeleton is a polymer skeleton composed of only a plurality of oxyalkylene repeating units linked to each other, or is derived from an initiator used during polymerization in addition to the plurality of oxyalkylene repeating units. It is preferable that the polymer skeleton contains a structure and is composed only of these.
- the oxyalkylene repeating unit refers to a repeating unit constituting a polyether, for example, an oxyalkylene unit having 2 to 6 carbon atoms, preferably 2 to 4 carbon atoms.
- the polymer skeleton of the polyoxyalkylene is not particularly limited, and for example, polyoxyethylene, polyoxypropylene, polyoxybutylene, polyoxytetramethylene, polyoxyethylene-polyoxypropylene copolymer, polyoxypropylene-polyoxy. Butylene copolymer and the like can be mentioned. Polyoxypropylene is preferred. As the polymer skeleton, only one type may be used, or two or more types may be used in combination.
- the terminal structure refers to a site that does not contain the oxyalkylene repeating unit constituting the polymer skeleton of polyoxyalkylene and is bonded to the end of the polymer skeleton.
- the terminal structure is preferably bonded to an oxyalkylene unit located at the end of the polymer skeleton via an oxygen atom. Further, it is preferable that the reactive silyl group contained in the polyoxyalkylene polymer (A) is contained in the terminal structure.
- the polyoxyalkylene polymer (A) has a reactive silyl group in a specific ratio. That is, the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton in one molecule of the polyoxyalkylene polymer (A) is 0.80 or more.
- the cured product of the curable composition of the present invention has high strength. It is possible to achieve both high growth.
- the numerical value of the average ratio can be determined by the method described in the examples. In addition to the methods described in Examples, the numerical value of the average ratio can also be calculated from the results of GPC measurement and NMR measurement of the polyoxyalkylene polymer (A).
- the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton refers to the number of reactive silyl groups contained in one terminal structure of the polymer skeleton on average, and is contained in one molecule of the polymer.
- the number of terminals of the polymer skeleton in one molecule of the polymer is 2 when all the polymer skeletons are linear, and 3 or more when all the polymer skeletons are branched chains. It can also be between 2 and 3 if the polymer backbone is a mixture of linear and branched chains.
- the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton is 0.80 or more, but 0.85 or more because the curable composition of the present invention can exhibit higher strength after curing. Is preferable, 0.90 or more is preferable, and 0.95 or more is more preferable. Further, 1.0 or more is preferable, 1.1 or more is more preferable, 1.3 or more is further preferable, and 1.5 or more is particularly preferable. Further, the ratio is preferably 5 or less, and more preferably 3 or less.
- the reactive silyl group contained in the polyoxyalkylene polymer (A) has the following general formula (1): -SiR 1 a X 3-a (1) It is represented by.
- R 1 represents a substituted or unsubstituted hydrocarbon group having 1 to 20 carbon atoms.
- the carbon number is preferably 1 to 10, the carbon number 1 to 8 is more preferable, the carbon number 1 to 6 is further preferable, the carbon number 1 to 3 is more preferable, and the carbon number 1 or 2 is particularly preferable.
- the hydrocarbon group has a substituent, the substituent is not particularly limited, and examples thereof include a halogen group such as a chloro group, an alkoxy group such as a methoxy group, and an amino group such as an N, N-diethylamino group. ..
- Aryl groups such as phenyl group, toluyl group and 1-naphthyl group; aralkyl groups such as benzyl group and the like can be mentioned.
- R 1 it is preferably a substituted or unsubstituted alkyl group, more preferably a methyl group, an ethyl group, a chloromethyl group or a methoxymethyl group, still more preferably a methyl group or a methoxymethyl group, and particularly preferably a methyl group.
- R 1 only one type of group may be used, or two or more types of groups may be used in combination.
- X represents a hydroxyl group or a hydrolyzable group.
- Examples of X include hydroxyl groups, hydrogens, halogens, alkoxy groups, acyloxy groups, ketoximate groups, amino groups, amide groups, acid amide groups, aminooxy groups, mercapto groups, alkenyloxy groups and the like.
- the above-mentioned alkoxy group and the like may have a substituent.
- An alkoxy group is preferable, a methoxy group, an ethoxy group, an n-propoxy group, and an isopropoxy group are more preferable, a methoxy group and an ethoxy group are further preferable, and a methoxy group is particularly preferable, because the hydrolyzability is mild and easy to handle.
- X only one type of group may be used, or two or more types of groups may be used in combination.
- a in the formula (1) is 0, 1 or 2. It is preferably 0 or 1. It is more preferably 1 in terms of the balance between the curability of the curable composition and the physical properties of the cured product.
- Examples of the reactive silyl group represented by the general formula (1) include a trimethoxysilyl group, a triethoxysilyl group, a tris (2-propenyloxy) silyl group, a triacetoxysilyl group, a methyldimethoxysilyl group, and a methyldi.
- Ethoxysilyl group dimethoxyethylsilyl group, (chloromethyl) dimethoxysilyl group, (chloromethyl) diethoxysilyl group, (methoxymethyl) dimethoxysilyl group, (methoxymethyl) diethoxysilyl group, (N, N-diethylaminomethyl) ) Dimethoxysilyl group, (N, N-diethylaminomethyl) diethoxysilyl group and the like.
- a trimethoxysilyl group, a (chloromethyl) dimethoxysilyl group, and a (methoxymethyl) dimethoxysilyl group are more preferable.
- a methyldimethoxysilyl group, a methyldiethoxysilyl group, and a triethoxysilyl group are more preferable. Further, a trimethoxysilyl group, a triethoxysilyl group and a methyldimethoxysilyl group are more preferable because they are easy to produce. Of these, the methyldimethoxysilyl group is most preferable.
- the terminal structure having a reactive silyl group is not particularly limited, but as a typical one, the terminal represented by the following general formula (3) or (4) is represented. The structure can be mentioned.
- R 3 represents a direct bond or a divalent hydrocarbon group having 1 to 4 carbon atoms
- R 4 represents hydrogen or an alkyl group having 1 to 6 carbon atoms.
- the oxygen at the left end indicates oxygen in the oxyalkylene unit located at the end of the polymer skeleton.
- R 1 , X, and a are the same as those described above for equation (1).
- the R 3 preferably a hydrocarbon group having 1 to 3 carbon atoms, more preferably a hydrocarbon group having 1 to 2 carbon atoms.
- a hydrocarbon group an alkylene group is preferable, and a methylene group, an ethylene group, a propylene group, and a butylene group can be used. Methylene groups are particularly preferred.
- the alkyl group include hydrogen, methyl group, ethyl group, propyl group, butyl group and the like.
- R 4 hydrogen, a methyl group and an ethyl group are preferable, and hydrogen and a methyl group are more preferable.
- a methyl group is preferable as R 4.
- R 5 is a direct bond or a divalent linking group having 1 to 6 carbon atoms.
- R 6 is hydrogen or a hydrocarbon group having 1 to 10 carbon atoms.
- n is an integer from 1 to 10.
- the oxygen at the left end indicates oxygen in the oxyalkylene unit located at the end of the polymer skeleton formed by connecting a plurality of oxyalkylene units.
- R 1 , R 3 , R 4 , X, and a are the same as those described above for equations (1) and (3).
- R 5 may be a divalent organic group having 1 to 6 carbon atoms.
- the organic group is preferably a hydrocarbon group or a hydrocarbon group containing an oxygen atom.
- the number of carbon atoms is preferably 1 to 4, more preferably 1 to 3, and even more preferably 1 to 2. It is preferably CH 2 OCH 2 , CH 2 O, and CH 2 , and more preferably CH 2 OCH 2 .
- a hydrogen atom and a methyl group are particularly preferable, and a hydrogen atom is most preferable.
- the terminal structure represented by the general formula (4) represents one terminal structure bonded to one terminal of the polymer skeleton. Although two or more reactive silyl groups are shown in formula (4), formula (4) does not show two or more ends, but two or more reactive in one terminal structure. It indicates that a silyl group is present. Further, the formula (4) does not include a polymer skeleton composed of an oxyalkylene repeating unit. That is, the n structures in parentheses in the formula (4) do not correspond to the oxyalkylene repeating unit in the polymer skeleton.
- the number average molecular weight of the polyoxyalkylene polymer (A) is not particularly limited, but is preferably 3,000 to 100,000, more preferably 3,000 to 50,000 in terms of polystyrene-equivalent molecular weight in GPC. , More preferably 3,000 to 30,000.
- the number average molecular weight is 3,000 or more, the relative amount of the reactive silyl group with respect to the entire polymer is in an appropriate range, which is desirable from the viewpoint of production cost. Further, when the number average molecular weight is 100,000 or less, it is easy to achieve a desired viscosity from the viewpoint of workability.
- the number average molecular weight can be determined by GPC measurement in terms of polystyrene.
- the molecular weight distribution (Mw / Mn) of the polyoxyalkylene polymer (A) is not particularly limited, but it is preferably narrow because it enables low viscosity. Specifically, it is preferably less than 2.0, more preferably 1.6 or less, further preferably 1.5 or less, and particularly preferably 1.4 or less. Further, from the viewpoint of improving various mechanical properties such as improving the durability and elongation of the cured product, 1.2 or less is preferable.
- the molecular weight distribution (Mw / Mn) can be calculated from the number average molecular weight and the weight average molecular weight obtained in terms of polystyrene by GPC measurement.
- the polyoxyalkylene-based polymer (A) is a reaction having reactivity with the olefin group after introducing an olefin group into the hydroxyl group-terminated polyoxyalkylene-based polymer (C) by utilizing the reactivity of the hydroxyl group. It can be produced by reacting a compound containing a reactive silyl group to introduce a reactive silyl group.
- the polymer skeleton of the polyoxyalkylene polymer can be formed by polymerizing an epoxy compound with an initiator having a hydroxyl group by a conventionally known method, whereby the hydroxyl-terminated polyoxyalkylene polymer (C) can be formed. Is obtained.
- the specific polymerization method is not particularly limited, but since a hydroxyl group-terminated polymer having a small molecular weight distribution (Mw / Mn) can be obtained, a polymerization method using a composite metal cyanide complex catalyst such as a zinc hexacyanocobaltate glyme complex. Is preferable.
- the initiator having a hydroxyl group is not particularly limited, and for example, ethylene glycol, propylene glycol, glycerin, pentaerythritol, low molecular weight polyoxypropylene glycol, low molecular weight polyoxypropylene triol, allyl alcohol, and low molecular weight polyoxypropylene.
- examples thereof include organic compounds having one or more hydroxyl groups, such as monoallyl ether and low molecular weight polyoxypropylene monoalkyl ether.
- the epoxy compound is not particularly limited, and examples thereof include alkylene oxides such as ethylene oxide and propylene oxide, and glycidyl ethers such as methyl glycidyl ether and butyl glycidyl ether. Propylene oxide is preferable.
- reaction with alkali metal salts In introducing an olefin group into the hydroxyl group-terminated polyoxyalkylene polymer (C), first, an alkali metal salt is allowed to act on the hydroxyl group-terminated polyoxyalkylene polymer (C) to make the terminal hydroxyl group a metaloxy group. It is preferable to convert to. Further, a composite metal cyanide complex catalyst can be used instead of the alkali metal salt. As a result, the metaloxy group-terminated polyoxyalkylene polymer (D) is formed.
- the alkali metal salt is not particularly limited, and examples thereof include sodium hydroxide, sodium alkoxide, potassium hydroxide, potassium alkoxide, lithium hydroxide, lithium alkoxide, cesium hydroxide, and cesium alkoxide. From the viewpoint of ease of handling and solubility, sodium hydroxide, sodium methoxide, sodium methoxide, sodium tert-butoxide, potassium hydroxide, potassium methoxide, potassium ethoxide, potassium tert-butoxide are preferable, and sodium methoxide and sodium tert are preferable. -Butoxide is more preferred. Sodium methoxide is particularly preferred in terms of availability, and sodium tert-butoxide is particularly preferred in terms of reactivity.
- the alkali metal salt may be subjected to the reaction in a state of being dissolved in a solvent.
- the amount of the alkali metal salt used is not particularly limited, but the molar ratio of the hydroxyl-terminated polyoxyalkylene polymer (C) to the hydroxyl group is preferably 0.5 or more, more preferably 0.6 or more, and 0. 7 or more is more preferable, and 0.8 or more is even more preferable.
- the molar ratio is preferably 1.2 or less, more preferably 1.1 or less.
- the alkali metal salt is used to convert the hydroxyl group of the hydroxyl-terminated polyoxyalkylene polymer (C) into a metaloxy group, but in order to efficiently proceed with this conversion reaction, water or polyoxy is used. It is preferable to remove a substance having a hydroxyl group other than the alkylene polymer from the reaction system in advance. For removal, a known method may be used, and for example, heat evaporation, vacuum evaporation, spray vaporization, thin film evaporation, azeotropic evaporation and the like can be used.
- the temperature at which the alkali metal salt is allowed to act can be appropriately set by those skilled in the art, but is preferably 50 ° C. or higher and 150 ° C. or lower, and more preferably 110 ° C. or higher and 145 ° C. or lower.
- the time for the alkali metal salt to act is preferably 10 minutes or more and 5 hours or less, and more preferably 30 minutes or more and 3 hours or less.
- reaction with electrophile (E) By allowing the electrophile (E) having an olefin group to act on the metaloxy group-terminated polyoxyalkylene polymer (D) obtained as described above, the metaloxy group has a structure containing an olefin group. Can be converted to. As a result, a polyoxyalkylene polymer (F) having an olefin group in the terminal structure is formed.
- the electrophilic agent (E) having an olefin group is particularly limited as long as it is a compound capable of reacting with the metaloxy group of the polyoxyalkylene polymer (D) and introducing an olefin group into the polyoxyalkylene polymer.
- an organic halide having an olefin group (E1), an epoxy compound having an olefin group (E2), and the like can be mentioned.
- the organic halide (E1) having an olefin group which is one aspect of the electrophile (E), reacts with the metaloxy group by a halogen substitution reaction to form an ether bond, thereby forming a polyoxyalkylene-based weight.
- a structure containing an olefin group can be introduced as the terminal structure of the coalescence.
- organic halide (E1) having an olefin group are not particularly limited, but vinyl chloride, allyl chloride, methallyl chloride, vinyl bromide, allyl bromide, methallyl bromide, vinyl iodide, allyl iodide, Examples include metallyl iodide. Allyl chloride and methallyl chloride are preferable because of ease of handling. Further, since the average ratio of the number of reactive silyl groups to the number of terminals of the polymer skeleton described above is improved, metallyl chloride, metallic bromide, and metallic iodide are preferable.
- the amount of the organic halide (E1) having an olefin group added is not particularly limited, but the molar ratio of the organic halide (E1) to the hydroxyl group of the polyoxyalkylene polymer (C) is 0.7 or more. Preferably, 1.0 or more is more preferable. The molar ratio is preferably 5.0 or less, more preferably 2.0 or less.
- the temperature at which the organic halide having an olefin group (E1) is reacted with the metaloxy group-terminated polyoxyalkylene polymer (D) is preferably 50 ° C. or higher and 150 ° C. or lower, and 110 ° C. or higher and 140 ° C. or lower. Is more preferable.
- the reaction time is preferably 10 minutes or more and 5 hours or more, and more preferably 30 minutes or more and 3 hours or less.
- a structure containing an olefin group and a hydroxyl group can be introduced as the terminal structure of the alkylene polymer.
- the ring-opening addition reaction one molecule or two or more molecules of the epoxy compound (E2) are used for one metaloxy group by adjusting the amount of the epoxy compound (E2) used for the metaloxy group and the reaction conditions. ) Can be added.
- the epoxy compound (E2) having the olefin group is not limited, but the following general formula (6):
- R 5 and R 6 are the same groups as R 5 and R 6 described above for the general formula (4), respectively.
- epoxy compound (E2) having an olefin group are not particularly limited, but allyl glycidyl ether, metallicyl glycidyl ether, glycidyl acrylate, glycidyl methacrylate, and butadiene monooxide are preferable from the viewpoint of reaction activity, and allyl glycidyl ether is preferable. Especially preferable.
- the amount of the epoxy compound (E2) having an olefin group added can be any amount in consideration of the amount of the olefin group introduced into the polymer and the reactivity.
- the molar ratio of the epoxy compound (E2) to the hydroxyl group of the polyoxyalkylene polymer (C) is preferably 0.2 or more, more preferably 0.5 or more.
- the molar ratio is preferably 5.0 or less, more preferably 2.0 or less.
- the reaction temperature when the epoxy compound (E2) having an olefin group is subjected to a cycloaddition reaction with the metaloxy group-terminated polyoxyalkylene polymer (D) is preferably 60 ° C. or higher and 150 ° C. or lower, preferably 110. More preferably, it is °C or more and 140 °C or less.
- the epoxy compound (E2) having an olefin group is allowed to act on the metaloxy group-terminated polyoxyalkylene polymer (D) as described above, a new metaloxy group is generated by opening the ring of the epoxy group. Therefore, after the epoxy compound (E2) is allowed to act, the above-mentioned organic halide having an olefin group (E1) can be continuously acted on.
- the organic halide (E1) having an olefin group used in this embodiment the same compound as described above can be used, and the amount and reaction temperature thereof are the same as described above. This method is preferable because the amount of the olefin group introduced into the polymer and the amount of the reactive silyl group introduced can be further increased.
- the polyoxyalkylene polymer (F) having an olefin group in the terminal structure obtained as described above is subjected to a hydrosilylation reaction with a hydrosilane compound (G) having a reactive silyl group, whereby the polymer is reactive silyl.
- the group can be introduced.
- the polyoxyalkylene polymer (A) having a reactive silyl group can be produced.
- the hydrosilylation reaction has the advantages that it can be easily carried out, the amount of the reactive silyl group introduced can be easily adjusted, and the physical properties of the obtained polymer are stable.
- hydrosilane compound (G) having a reactive silyl group examples include trichlorosilane, dichloromethylsilane, chlorodimethylsilane, dichlorophenylsilane, (chloromethyl) dichlorosilane, (dichloromethyl) dichlorosilane, and bis (chloromethyl).
- Halosilanes such as chlorosilane, (methoxymethyl) dichlorosilane, (dimethoxymethyl) dichlorosilane, bis (methoxymethyl) chlorosilane; trimethoxysilane, triethoxysilane, dimethoxymethylsilane, diethoxymethylsilane, dimethoxyphenylsilane, ethyl Dimethoxysilane, methoxydimethylsilane, ethoxydimethylsilane, (chloromethyl) methylmethoxysilane, (chloromethyl) dimethoxysilane, (chloromethyl) diethoxysilane, bis (chloromethyl) methoxysilane, (methoxymethyl) methylmethoxysilane, (Methoxymethyl) dimethoxysilane, bis (methoxymethyl) methoxysilane, (methoxymethyl) diethoxysilane, (ethoxymethyl) diethoxysi
- Alkoxysilanes Alkoxysilanes; Asyloxysilanes such as diacetoxymethylsilane and diacetoxyphenylsilane; Ketoximatesilanes such as bis (dimethylketoximate) methylsilane and bis (cyclohexylketoximate) methylsilane, triisopropeniloxisilane , (Chloromethyl) diisopropenyloxysilane, (methoxymethyl) diisopropenyloxysilane and other isopropenyloxysilanes (deacetone type) and the like.
- the amount of the hydrosilane compound (G) having a reactive silyl group may be appropriately set in consideration of the amount of the olefin group contained in the polyoxyalkylene polymer (F).
- the molar ratio of the hydrosilane compound (G) to the olefin group of the polyoxyalkylene polymer (F) is preferably 0.05 or more and 10 or less, and 0.3 or more and 3 or less from the viewpoint of reactivity. More preferred.
- the molar ratio is more preferably 0.5 or more, and particularly preferably 0.7 or more, in that the modulus value of the cured product of the obtained polyoxyalkylene polymer (A) can be increased.
- the molar ratio is more preferably 2.5 or less, and particularly preferably 2 or less.
- the hydrosilylation reaction is preferably carried out in the presence of a hydrosilylation catalyst in order to promote the reaction.
- a hydrosilylation catalyst metals such as cobalt, nickel, iridium, platinum, palladium, rhodium, and ruthenium, and complexes thereof and the like are known, and these can be used.
- the hydrosilylation reaction can be carried out without using a solvent, but for the purpose of uniformly dissolving the polyoxyalkylene polymer (F), the hydrosilane compound (G), and the hydrosilylation catalyst, and the reaction.
- an organic solvent may be added.
- the temperature conditions for the hydrosilylation reaction are not particularly limited and can be appropriately set by those skilled in the art, but the reaction under heating conditions is preferable for the purpose of lowering the viscosity of the reaction system and improving the reactivity, specifically.
- the reaction at 50 ° C. to 150 ° C. is more preferable, and the reaction at 70 ° C. to 120 ° C. is further preferable.
- the reaction time may be appropriately set, but it is preferable to adjust the reaction time together with the temperature conditions so that the unintended condensation reaction between the polymers does not proceed.
- the reaction time is preferably 30 minutes or more and 5 hours or less, and more preferably 3 hours or less.
- hydrosilylation reaction may be carried out in the presence of an orthocarboxylic acid trialkyl ester. This makes it possible to suppress thickening during the hydrosilylation reaction and improve the storage stability of the obtained polymer.
- orthocarboxylic acid trialkyl ester examples include trimethyl orthoformate, triethyl orthoformate, trimethyl orthoacetate, and triethyl orthoacetate. Preferred are trimethyl orthoformate and trimethyl orthoacetate.
- the amount used is not particularly limited, but is preferably about 0.1 to 10 parts by weight, preferably about 0.1 to 10 parts by weight, based on 100 parts by weight of the polyoxyalkylene polymer (A). About 3 parts by weight is more preferable.
- a method of introducing a reactive silyl group by forming a urethane bond can also be applied. Also by this method, the polyoxyalkylene polymer (A) having a reactive silyl group can be produced.
- the compound (H) having a reactive silyl group and an isocyanate group in one molecule includes an isocyanate group capable of a urethanization reaction with a hydroxyl group of the polyoxyalkylene polymer (C) and a reactive silyl group.
- the compound is not particularly limited as long as it is contained in the molecule, but specific examples thereof include (3-isocyanatepropyl) trimethoxysilane, (3-isocyanatepropyl) dimethoxymethylsilane, (3-isocyanatepropyl) triethoxysilane, and (3-isocyanatepropyl) triethoxysilane.
- Examples thereof include 3-isocyanatepropyl) diethoxymethylsilane, (isocyanatemethyl) trimethoxysilane, (isocyanatemethyl) triethoxysilane, (isocyanatemethyl) dimethoxymethylsilane, and (isocyanatemethyl) diethoxymethylsilane.
- the amount of the compound (H) having a reactive silyl group and an isocyanate group in one molecule may be appropriately determined in consideration of the amount of the hydroxyl group of the polyoxyalkylene polymer (C).
- the molar ratio of the isocyanate group of the compound (H) to the hydroxyl group of the polyoxyalkylene polymer (C) is preferably 0.5 or more, more preferably 0.8 or more, and 0.9. The above is more preferable.
- the upper limit of the amount of compound (H) used is not particularly limited, and the molar ratio is preferably 1 or less, but may exceed 1.
- the excess compound (H) may be removed by a treatment such as devolatile under reduced pressure after the urethanization reaction, or may be reacted with an active hydrogen group-containing compound or the like to form another compound. It may be converted or may remain in the polyoxyalkylene polymer (A).
- the compound (H) remaining in the polyoxyalkylene polymer (A), or a derivative thereof, can act as a silane coupling agent.
- the urethanization reaction may be carried out without using the urethanization catalyst, but may be carried out in the presence of the urethanization catalyst for the purpose of improving the reaction rate or the reaction rate.
- urethanization catalysts include Polyurethanes: Chemistry and Technology, Part I, Table 30, Chapter 4, Sanders and Frisch, Interscience Publicly known catalysts such as Polyurethane 19 and New.
- a catalyst can be used. Specific examples thereof include, but are not limited to, organic tin compounds, bismuth compounds, base catalysts such as organic amines, and the like.
- a catalyst having low activity with respect to the reactive silyl group is preferable, and from this viewpoint, dibutyltinbis (isononyl-3-mercaptopropionate), dibutyltinbis (isooctyl mercaptopropionate), dibutyltinbis (isooctylthioate).
- a tin catalyst containing a sulfur atom, such as glycolate) is particularly preferred.
- the amount of the urethanization catalyst added can be appropriately set by those skilled in the art, but from the viewpoint of reaction activity, 1 to 1000 ppm is preferable, and 10 to 100 ppm is more preferable with respect to 100 parts by weight of the polyoxyalkylene polymer (C). In this range, in addition to obtaining sufficient reaction activity, the obtained polyoxyalkylene polymer (A) retains good physical properties such as heat resistance, weather resistance, hydrolysis resistance, and storage stability. it can.
- the urethanization reaction can be carried out without using a solvent, but for the purpose of uniformly dissolving the polyoxyalkylene polymer (C), the compound (H), and the urethanization catalyst, and the reaction system.
- an organic solvent may be added.
- the temperature of the urethanization reaction can be appropriately set by those skilled in the art, but is preferably 50 ° C. or higher and 120 ° C. or lower, and more preferably 70 ° C. or higher and 100 ° C. or lower.
- the reaction time may be appropriately set, but it is preferable to adjust the reaction time together with the temperature conditions so that the unintended condensation reaction between the polymers does not proceed.
- the reaction time is preferably 15 minutes or more and 5 hours or less, and more preferably 30 minutes or more and 3 hours or less.
- a silyl group reactive in one molecule and a mercaptan with respect to the polyoxyalkylene polymer (F) having an olefin group in the terminal structure As yet another method for producing the polyoxyalkylene polymer (A), a silyl group reactive in one molecule and a mercaptan with respect to the polyoxyalkylene polymer (F) having an olefin group in the terminal structure.
- a method in which a compound (I) having a group is allowed to act to form a sulfide bond by adding a mercaptan group to an olefin group to introduce a reactive silyl group can also be applied. Also by this method, the polyoxyalkylene polymer (A) having a reactive silyl group can be produced.
- Examples of the compound (I) having a reactive silyl group and a mercaptan group in one molecule include a mercaptan group capable of an addition reaction to an olefin group of the polyoxyalkylene polymer (F) and a reactive silyl group.
- the compound is not particularly limited as long as it is a compound contained in the molecule, but specific examples thereof include (3-mercaptopropyl) methyldimethoxysilane, (3-mercaptopropyl) trimethoxysilane, and (3-mercaptopropyl) methyldiethoxysilane.
- Examples thereof include (3-mercaptopropyl) triethoxysilane, (mercaptomethyl) methyldimethoxysilane, (mercaptomethyl) trimethoxysilane, (mercaptomethyl) methyldiethoxysilane, and (mercaptomethyl) triethoxysilane.
- the amount of the compound (I) having a reactive silyl group and a mercaptan group in one molecule may be appropriately determined in consideration of the amount of the olefin group contained in the polyoxyalkylene polymer (F).
- the molar ratio of the mercaptan group of the compound (I) to the olefin group of the polyoxyalkylene polymer (F) is preferably 0.5 or more, more preferably 0.8 or more, and 0. 0.9 or more is more preferable.
- the upper limit of the amount of compound (I) used is not particularly limited, and the molar ratio is preferably 1 or less, but may exceed 1.
- the excess compound (I) may be removed by a treatment such as vacuum devolatilization after the addition reaction, or may be reacted with an unsaturated group-containing compound or the like to convert it into another compound. Alternatively, it may remain in the polyoxyalkylene polymer (A).
- the compound (I) remaining in the polyoxyalkylene polymer (A), or a derivative thereof, can act as a silane coupling agent.
- the addition reaction of the mercaptan group to the olefin group may be carried out without using a radical initiator, but it is carried out in the presence of a radical initiator for the purpose of improving the reaction rate or the reaction rate. May be good.
- a radical initiator conventionally known ones can be used. Specific examples thereof include, but are not limited to, an azo-based initiator and a peroxide-based initiator.
- a catalyst having low activity with respect to the reactive silyl group is preferable, and from this viewpoint, 2,2'-azobis (isobutyronitrile) (AIBN) and 2,2'-azobis (2).
- Azo-based initiators such as -methylbutyronitrile) (V-59) and 2,2'-azobis (1-methylcyclohexanecarbonitrile) (V-40) are particularly preferred.
- the amount of the radical initiator added can be appropriately set by those skilled in the art, but from the viewpoint of reaction activity, it is preferably 0.01 to 10 parts by weight, preferably 0.1 to 10 parts by weight, based on 100 parts by weight of the polyoxyalkylene polymer (F). 10 parts by weight is more preferable.
- the radical initiator may be used in a state of being dissolved in an organic solvent.
- the temperature of the addition reaction can be appropriately set by those skilled in the art, but is preferably 50 ° C. or higher and 120 ° C. or lower, and more preferably 70 ° C. or higher and 100 ° C. or lower.
- the reaction time may be appropriately set, but it is preferable to adjust the reaction time together with the temperature conditions so that the unintended condensation reaction between the polymers does not proceed.
- the reaction time is preferably 15 minutes or more and 10 hours or less, and more preferably 30 minutes or more and 6 hours or less.
- the curable composition of the present invention further contains a (meth) acrylic acid ester-based polymer (B) having a reactive silyl group.
- the reactive silyl group contained in the (meth) acrylic acid ester polymer (B) has the following general formula (2): -SiR 2 b Y 3-b (2) It is represented by.
- R 2 represents an unsubstituted hydrocarbon group having 1 to 20 carbon atoms.
- the carbon number is preferably 1 to 10, the carbon number 1 to 8 is more preferable, the carbon number 1 to 6 is further preferable, the carbon number 1 to 3 is more preferable, and the carbon number 1 or 2 is particularly preferable.
- the R 2 for example, a methyl group, an ethyl group, n- propyl group, an isopropyl group, n- butyl group, tert- butyl group, n- hexyl, 2-ethylhexyl group, n- alkyl groups such as dodecyl group; Unsaturated hydrocarbon groups such as vinyl group, isopropenyl group and allyl group; cycloalkyl group such as cyclohexyl group; aryl group such as phenyl group, toluyl group and 1-naphthyl group; aralkyl group such as benzyl group and the like. ..
- R 2 it is preferably an alkyl group or an aryl group, more preferably a methyl group, an ethyl group or a phenyl group, still more preferably a methyl group or an ethyl group, and particularly preferably a methyl group.
- R 2 only one type of group may be used, or two or more types of groups may be used in combination.
- Y represents a hydroxyl group or a hydrolyzable group.
- Examples of Y include hydroxyl groups, hydrogens, halogens, alkoxy groups, acyloxy groups, ketoximate groups, amino groups, amide groups, acid amide groups, aminooxy groups, mercapto groups, alkenyloxy groups and the like.
- the above-mentioned alkoxy group and the like may have a substituent.
- An alkoxy group is preferable, a methoxy group, an ethoxy group, an n-propoxy group, and an isopropoxy group are more preferable, a methoxy group and an ethoxy group are further preferable, and a methoxy group is particularly preferable, because the hydrolyzability is mild and easy to handle.
- Y only one type of group may be used, or two or more types of groups may be used in combination.
- B in equation (2) is 0, 1 or 2. It is preferably 0 or 1. It is more preferably 1 in terms of the balance between the curability of the curable composition and the physical properties of the cured product, and more in that the curability of the composition and the resilience of the cured product can be further enhanced. It is preferably 0.
- Examples of the reactive silyl group represented by the general formula (2) include a trimethoxysilyl group, a triethoxysilyl group, a tris (2-propenyloxy) silyl group, a triacetoxysilyl group, a methyldimethoxysilyl group, and a methyldi.
- Ethoxysilyl group ethyldimethoxysilyl group, ethyldiethoxysilyl group, n-propyldimethoxysilyl group, n-hexyldimethoxysilyl group, phenyldimethoxysilyl group, phenyldiethoxysilyl group, methyldiisopropenoxysilyl group, methyldi Examples thereof include a phenoxysilyl group and a dimethylmethoxysilyl group.
- a methyldimethoxysilyl group is more preferable from the viewpoint of achieving both storage stability and curability of the curable composition, and trimethoxysilyl can further enhance the curability of the composition and the resilience of the cured product. Groups are more preferred.
- reactive silyl group of the polyoxyalkylene polymer (A) and the reactive silyl group of the (meth) acrylic acid ester polymer (B) may be the same or different.
- the bond positions and the number of reactive silyl groups of the (meth) acrylic acid ester-based polymer (B) satisfy specific conditions. That is, in one molecule of the (meth) acrylic acid ester-based polymer (B), the average number of reactive silyl groups bonded to the ends of the polymer skeleton is 0.30 or more and 1.00 or less, and the polymer skeleton. The average number of reactive silyl groups bonded to the inside of is 0 or more and less than 0.30.
- the average number of the former is the average number of reactive silyl groups bonded to the end of the polymer skeleton among the reactive silyl groups contained in one molecule of the polymer (B), and the average number of the latter is , The average number of reactive silyl groups bonded to the inside of the polymer skeleton among the reactive silyl groups contained in one molecule of the polymer (B).
- the curable composition of the present invention has high strength and high strength after curing. It is possible to achieve both growth.
- the numerical value of the average number can be calculated by the method described in the examples. In addition to the methods described in Examples, the numerical value of the average number can also be calculated from the results of GPC measurement and NMR measurement of the (meth) acrylic acid ester-based polymer (B).
- the reactive silyl group bonded to the terminal of the polymer skeleton is a reactive silyl group bonded to the terminal of the polymer skeleton formed by linking a plurality of monomer units. Point to that.
- a reactive silyl group attached to the terminal can be suitably formed, for example, by a method using a chain transfer agent described later, for example, without limitation.
- the (meth) acrylic acid ester-based polymer (B) having an average number of reactive silyl groups bonded to the ends of 1.00 or less in the present invention is, for example, a reactive silyl group with respect to one terminal of the polymer skeleton. It can be formed by adopting the method of introducing.
- the (meth) acrylic acid ester-based polymer (B) in the present invention is a polymer having a reactive silyl group at one end of the polymer skeleton but not having a reactive silyl group at the other end. Is preferable.
- the (meth) acrylic acid ester-based polymer (B) in the present invention preferably does not contain a (meth) acrylic acid ester-based polymer having a reactive silyl group at each of the two terminals of such a polymer skeleton. ..
- the reactive silyl group bonded to the inside of the polymer skeleton is a plurality of monomer units constituting the polymer skeleton except for the monomer unit located at the end of the polymer skeleton described above.
- a reactive silyl group attached to either Such a reactive silyl group may be described as being attached to the polymer as a side chain.
- Such an internally bonded reactive silyl group is not limited, but can be suitably formed by, for example, a method of copolymerizing a monomer having a reactive silyl group described later. In this case, the average number of reactive silyl groups bonded to the inside of the polymer skeleton in one polymer molecule can be adjusted by adjusting the amount of the monomer having the reactive silyl group used.
- the average number of reactive silyl groups bonded to the ends of the polymer skeleton in one molecule of the (meth) acrylic acid ester-based polymer (B) is 0.30 or more and 1.00 or less, but the curability of the present invention Since the strength of the cured product of the composition can be further improved, it is preferably 0.65 or more and 1.00 or less, more preferably 0.83 or more and 1.00 or less, and further preferably 0.85 or more and 1.00 or less. , 0.90 or more and 1.00 or less is particularly preferable.
- the average number of reactive silyl groups bonded to the inside of the polymer skeleton is 0 or more and less than 0.30, that is, bonded to the inside.
- the reactive silyl group may not be present (ie, the average number may be 0).
- the average number is preferably less than 0.20 because the strength and elongation of the cured product of the curable composition of the present invention can be improved together. , Less than 0.15, more preferably less than 0.05.
- the average number of reactive silyl groups contained in one molecule of the (meth) acrylic acid ester-based polymer (B) (that is, the average number of reactive silyl groups bonded to the ends of the polymer skeleton described above, and the polymer.
- the total number of reactive silyl groups bonded to the inside of the skeleton) is limited to the range of 0.30 or more and less than 1.30 depending on the above-mentioned conditions, but is preferably 0.65 or more and 1.20 or less. , More preferably 0.85 or more and 1.05 or less.
- the (meth) acrylic acid ester-based monomer constituting the main chain of the (meth) acrylic acid ester-based polymer (B) is not particularly limited, and various types can be used. Specifically, methyl (meth) acrylate, ethyl (meth) acrylate, n-propyl (meth) acrylate, isopropyl (meth) acrylate, n-butyl (meth) acrylate, isobutyl (meth) acrylate.
- Examples of the monomer unit other than the above include acrylic acids such as acrylic acid and methacrylic acid; amide groups such as N-methylolacrylamide and N-methylolmethacrylicamide, epoxy groups such as glycidylacrylate and glycidylmethacrylate, and diethylaminoethylacrylate. , Diethylaminoethyl methacrylate, etc., and monomers containing nitrogen-containing groups.
- the (meth) acrylic acid ester-based polymer (B) a polymer obtained by copolymerizing a (meth) acrylic acid ester-based monomer and a vinyl-based monomer copolymerizable therewith is used. You can also do it.
- the vinyl-based monomer is not particularly limited, and is, for example, a styrene-based monomer such as styrene, vinyltoluene, ⁇ -methylstyrene, chlorostyrene, styrenesulfonic acid and a salt thereof; perfluoroethylene, perfluoropropylene, etc.
- Fluorine-containing vinyl monomers such as vinylidene fluoride; silicon-containing vinyl monomers such as vinyltrimethoxysilane and vinyltriethoxysilane; maleic anhydride, maleic acid, monoalkyl and dialkyl esters of maleic acid; fumal Acids, monoalkyl esters and dialkyl esters of fumaric acid; maleimide-based monomers such as maleimide, methylmaleimide, ethylmaleimide, propylmaleimide, butylmaleimide, hexylmaleimide, octylmaleimide, dodecylmaleimide, stearylmaleimide, phenylmaleimide, cyclohexylmaleimide, etc.
- Nitrile group-containing vinyl-based monomers such as acrylonitrile and maleimide nitrile; amide group-containing vinyl-based monomers such as acrylamide and methacrylicamide; vinyl acetate, vinyl propionate, vinyl pivalate, vinyl benzoate, vinyl laurate Vinyl ester-based monomers such as; alkenyl-based monomers such as ethylene and propylene; conjugated diene-based monomers such as butadiene and isoprene; vinyl chloride, vinylidene chloride, allyl chloride, allyl alcohol and the like. It is also possible to use a plurality of these as copolymerization components.
- methyl methacrylate is preferably contained as a monomer constituting the main chain of the (meth) acrylic acid ester-based polymer (B), and the (meth) acrylic acid ester-based polymer (B) is used.
- the proportion of methyl methacrylate in all the constituent monomer units is preferably 35% by weight or more and 85% by weight or less.
- the weight of all monomer units includes only the weight of the monomer and does not include the weight of the chain transfer agent and the radical initiator.
- the proportion of methyl methacrylate in the (meth) acrylic acid ester-based polymer (B) is within the above range, the strength of the cured product of the composition according to the present invention can be further improved.
- the proportion of methyl methacrylate is more preferably 60% by weight or more and 85% by weight or less, and more preferably 70% by weight or more and 85% by weight or less.
- the total proportion of alkyl methacrylate among all the monomer units constituting the (meth) acrylic acid ester-based polymer (B) is 70% by weight or more and 85% by weight or less.
- the alkyl of the alkyl methacrylate is an unsubstituted alkyl group having 1 to 4 carbon atoms (specifically, a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, tert).
- the weight of all monomer units includes only the weight of the monomer and does not include the weight of the chain transfer agent and the radical initiator.
- the proportion of alkyl methacrylate in the (meth) acrylic acid ester-based polymer (B) is within the above range, the strength of the cured product of the composition according to the present invention can be further improved.
- the ratio of the alkyl methacrylate is more preferably 75% by weight or more and 85% by weight or less.
- the monomer composition of the (meth) acrylic acid ester-based polymer (B) is generally selected by those skilled in the art depending on the application and purpose, but in applications that require strength such as an adhesive, it is used.
- the (meth) acrylic acid ester polymer (B) preferably has a relatively high glass transition temperature (Tg), preferably 0 ° C. or higher and 200 ° C. or lower, and more preferably 20 ° C. or higher and 100 ° C. or lower. Is good. Tg is obtained from the following Fox formula.
- the method for synthesizing the (meth) acrylic acid ester-based polymer (B) is not particularly limited, and examples thereof include known methods.
- the radical polymerization method is preferable from the viewpoint of versatility of the monomer and ease of control of the polymerization reaction.
- the radical polymerization method can be roughly divided into a “free radical polymerization method” and a “living radical polymerization method".
- the “free radical polymerization method” is a method for polymerizing a monomer using an azo compound, a peroxide or the like as a polymerization initiator, and is a simple polymerization method. According to the “free radical polymerization method”, it is also possible to obtain a polymer having a functional group at the end of the polymer skeleton by using a chain transfer agent having a specific functional group. On the other hand, in the "living radical polymerization method", the polymer growth end grows under specific reaction conditions without causing a side reaction such as a termination reaction.
- a polymer having an arbitrary molecular weight, a narrow molecular weight distribution, and a low viscosity can be obtained, and a constituent unit amount derived from a monomer having a specific functional group. It is possible to introduce the body unit at almost any position on the polymer. Details of these polymerization methods are disclosed in paragraphs [0086] to [0094] of International Publication No. 2012/20560 and [0061] to [0068] of JP-A-2014-114434.
- a method for obtaining an acrylic polymer by using a metallocene catalyst as shown in JP-A-2001-040037 and a thiol compound having at least one reactive silyl group in the molecule As a polymerization method other than the above, a method for obtaining an acrylic polymer by using a metallocene catalyst as shown in JP-A-2001-040037 and a thiol compound having at least one reactive silyl group in the molecule.
- a vinyl-based monomer as shown in JP-A-57-502171, JP-A-59-006207, and JP-A-60-511992 is used in a stirring tank type reactor. It is also possible to use a high-temperature continuous polymerization method or the like in which continuous polymerization is carried out using.
- the method for introducing the reactive silyl group into the (meth) acrylic acid ester polymer is not particularly limited, and for example, the following method can be used.
- (Iv) A method of copolymerizing a compound having a polymerizable unsaturated group and a reactive silyl group together with the above-mentioned monomer. Using this method, reactive silyl groups tend to be randomly introduced inside the polymer backbone.
- (V) A method for polymerizing a (meth) acrylic acid ester-based polymer using a mercaptosilane compound having a reactive silyl group as a chain transfer agent. Using this method, reactive silyl groups can be introduced at the ends of the polymer backbone.
- Vi A method in which a compound having a polymerizable unsaturated group and a reactive functional group (V group) is copolymerized, and then the reactive silyl group and the compound having a functional group that reacts with the V group are reacted. Specifically, a method of copolymerizing 2-hydroxyethyl acrylate and then reacting with an isocyanate silane having a reactive silyl group, or a method of copolymerizing glycidyl acrylate and then reacting with an aminosilane compound having a reactive silyl group. An example of how to make it.
- (Vii) A method for introducing a reactive silyl group by modifying the terminal functional group of the (meth) acrylic acid ester-based polymer synthesized by the living radical polymerization method.
- a functional group can be easily introduced at the end of the polymer skeleton, and by modifying this, a reactive silyl group can be introduced at the end of the polymer skeleton.
- Examples of the silicon compound that can be used to introduce a reactive silyl group into the (meth) acrylic acid ester-based polymer by the above method include the following compounds.
- Examples of the compound having a polymerizable unsaturated group and a reactive silyl group used in the method (iv) include 3- (trimethoxysilyl) propyl (meth) acrylate and 3- (dimethoxymethylsilyl) propyl (meth) acrylate.
- Examples of the mercaptosilane compound having a reactive silyl group used in the method (v) include (3-mercaptopropyl) trimethoxysilane, (3-mercaptopropyl) dimethoxymethylsilane, (3-mercaptopropyl) triethoxysilane, and (3. Examples thereof include mercaptomethyl) trimethoxysilane, (mercaptomethyl) dimethoxymethylsilane, and (mercaptomethyl) triethoxysilane.
- Examples of the compound having a functional group that reacts with the reactive silyl group and the V group used in the method (vi) include (3-isocyanuppropyl) trimethoxysilane, (3-isocyanvopropyl) dimethoxymethylsilane, and (3-isocyanabylpropyl).
- Silanesilane compounds such as (Isocyanicmethyl) trimethoxysilane, (Isocyanicmethyl) triethoxysilane, (Isocyanicmethyl) dimethoxymethylsilane, (Isocyanicmethyl) diethoxymethylsilane; (3-glycidoxypropyl) ) Trimethoxysilane, (3-glycidoxypropyl) triethoxysilane, (3-glycidoxypropyl) dimethoxymethylsilane, (glycidoxymethyl) trimethoxysilane, (glycidoxymethyl) triethoxysilane, ( Epoxysilane compounds such as glycidoxymethyl) dimethoxymethylsilane, (glycidoxymethyl) diethoxymethylsilane; (3-aminopropyl) trimethoxysilane, (3-aminopropyl) triethoxysilane, (3-aminopropyl) ) Dimethoxymethylsilane,
- any modification reaction can be used.
- a method using a compound having a functional group and a reactive silicon group capable of reacting with the terminally reactive group obtained by polymerization, or a terminal reactivity is used.
- a method of introducing a double bond at the end of the polymer skeleton using a compound having a functional group capable of reacting with a group and a double bond, and introducing a reactive silyl group into the terminal by hydrosilylation or the like can be used.
- the number average molecular weight of the (meth) acrylic acid ester-based polymer (B) is not particularly limited, but the polystyrene-equivalent molecular weight measured by GPC is preferably 500 to 100,000, more preferably 1,000 to 10,000, and 1 , 200-3,000 are particularly preferable.
- the number average molecular weight of the (meth) acrylic acid ester-based polymer (B) is within the above range, it is easy to form a cured product showing sufficient strength and elongation, and a desirable viscosity is achieved from the viewpoint of workability. Cheap.
- the method for blending the polyoxyalkylene polymer and the (meth) acrylic acid ester polymer is JP-A-59-122541, JP-A-63-112642, JP-A-6-172631, JP-A-11-116763. It is proposed in the publication. In addition, a method of polymerizing a (meth) acrylic acid ester-based monomer in the presence of a polyoxypropylene-based polymer having a reactive silyl group can be used. This production method is specifically disclosed in JP-A-59-78223, JP-A-60-228516, JP-A-60-228517 and the like. Also in the present invention, the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B) can be blended by the same method, but the present invention is not limited thereto.
- the weight ratio of the polyoxyalkylene polymer (A): (meth) acrylic acid ester polymer (B) in the curable composition of the present invention is not particularly limited, but is preferably 90:10 to 40:60, and is preferably 80. : 20 to 50:50 is more preferable, 80:20 to 55:45 is even more preferable, and 75:25 to 65:35 is particularly preferable.
- the curable composition of the present invention tends to achieve a desired viscosity from the viewpoint of workability, and tends to achieve high strength and high elongation after curing.
- the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B) are compatible with each other.
- both polymers can be configured to be compatible with each other.
- the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B) of the present invention only one kind may be used, or two or more kinds may be used in combination.
- the curable composition of the present invention hydrolyzes and dehydrates and condenses the reactive silyl groups of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B), that is, a curing reaction.
- a silanol condensation catalyst for the purpose of facilitating, it is preferable to contain a silanol condensation catalyst.
- silanol condensation catalyst conventionally known ones can be used, and specifically, organotin compounds, carboxylic acid metal salts, amine compounds, carboxylic acids, alkoxy metals, inorganic acids and the like can be used.
- organic tin compound examples include dibutyltin dilaurate, dibutyltin dioctanoate, dibutyltin bis (butylmaleate), dibutyltin diacetate, dibutyltin oxide, dibutyltin bis (acetylacetonate), dibutyltin oxide and silicate compounds.
- dioctyltin diacetate dioctyltin dilaurate
- dioctyltin bis (ethylmalate) dioctyltin bis (octylmaleate)
- dioctyltin bis (acetylacetate) Nart dioctyltin compounds are preferred.
- metal carboxylate salt examples include tin carboxylate, bismuth carboxylate, titanium carboxylate, zirconium carboxylate, iron carboxylate, and the like.
- carboxylic acid group the following carboxylic acids and various metals can be combined.
- amine compounds include amines such as octylamine, 2-ethylhexylamine, laurylamine, stearylamine, etc .; pyridine, 1,8-diazabicyclo [5,4,0] undecene-7 (DBU), 1, Nitrogen-containing heterocyclic compounds such as 5-diazabicyclo [4,3,0] nonen-5 (DBN); guanidines such as guanidine, phenylguanidine, diphenylguanidine; butylbiguanide, 1-o-tolylbiguanide and 1- Biganides such as phenylbiguanide; amino group-containing silane coupling agents; ketimine compounds and the like can be mentioned.
- amines such as octylamine, 2-ethylhexylamine, laurylamine, stearylamine, etc .
- DBU 1,8-diazabicyclo [5,4,0] undecene-7
- DBN Nitrogen-
- carboxylic acid examples include acetic acid, propionic acid, butyric acid, 2-ethylhexanoic acid, lauric acid, stearic acid, oleic acid, linoleic acid, neodecanoic acid, and versatic acid.
- the alkoxy metal include titanium compounds such as tetrabutyl titanate titanium tetrakis (acetylacetonate) and diisopropoxytitanium bis (ethylacetatete), aluminum tris (acetylacetonate), and diisopropoxyaluminum ethylacetate.
- titanium compounds such as tetrabutyl titanate titanium tetrakis (acetylacetonate) and diisopropoxytitanium bis (ethylacetatete), aluminum tris (acetylacetonate), and diisopropoxyaluminum ethylacetate.
- aluminum compounds such as, and zirconium compounds such as zirconium tetrakis (acetylacetonate).
- silanol condensation catalysts fluorine anion-containing compounds, photoacid generators and photobase generators can also be used.
- the silanol condensation catalyst may be used in combination with two or more different catalysts. For example, by using the amine compound and the carboxylic acid in combination, the effect of improving the reactivity may be obtained.
- the blending amount of the silanol condensation catalyst is preferably 0.001 to 20 parts by weight with respect to 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). Further, 0.01 to 15 parts by weight is more preferable, and 0.01 to 10 parts by weight is particularly preferable. If the blending amount of the silanol condensation catalyst is less than 0.001 part by weight, the reaction rate may be insufficient. On the other hand, if the blending amount of the silanol condensation catalyst exceeds 20 parts by weight, the reaction rate is too fast, so that the usable time of the composition is shortened, which tends to deteriorate workability and storage stability.
- silanol condensation catalysts may exude to the surface of the cured product or contaminate the surface of the cured product after the curable composition is cured.
- the amount of the silanol condensation catalyst used may be set to 0.01 to 3.0 parts by weight, the surface condition of the cured product can be kept good while ensuring the curability.
- additives In the curable composition of the present invention, as other additives, a silicon compound, an adhesive-imparting agent, a plasticizer, a solvent, a diluent, a silicate, a filler, an antioxidant, an antioxidant, a light stabilizer, and ultraviolet rays. Even if absorbents, property modifiers, tackifier resins, epoxy group-containing compounds, photocurable substances, oxygen curable substances, surface improvers, epoxy resins, other resins, flame retardants, and foaming agents are added. good.
- various additives may be added to the curable composition of the present invention as necessary for the purpose of adjusting various physical properties of the curable composition or the cured product. Examples of such additives include curability regulators, radical inhibitors, metal deactivators, ozone deterioration inhibitors, phosphorus peroxide decomposing agents, lubricants, pigments, fungicides and the like. Be done.
- fillers can be added to the composition of the present invention.
- fillers heavy calcium carbonate, collagen carbonate, magnesium carbonate, silica soil, clay, talc, titanium oxide, fumed silica, precipitated silica, crystalline silica, molten silica, silicic acid anhydride, hydrous silicic acid, Examples thereof include carbon black, ferric oxide, fine aluminum powder, zinc oxide, active zinc white, PVC powder, PMMA powder, glass fiber and filament.
- the above filler may be used alone or in combination of two or more.
- the amount of the filler used is preferably 1 to 300 parts by weight, particularly 10 to 300 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). 250 parts by weight is preferable.
- Organic balloons and inorganic balloons may be added for the purpose of reducing the weight (lower specific gravity) of the composition.
- the balloon is a spherical filler with a hollow inside, and the material of this balloon is an inorganic material such as glass, silas, and silica, and an organic material such as phenol resin, urea resin, polystyrene, and saran. Materials can be mentioned. Only one type of the balloon may be used, or two or more types may be mixed and used.
- the amount of the balloon used is preferably 0.1 to 100 parts by weight, particularly 1 by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). Up to 20 parts by weight is preferable.
- An adhesiveness-imparting agent can be added to the composition of the present invention.
- a silane coupling agent and a reaction product of the silane coupling agent can be added.
- silane coupling agent examples include 3-aminopropyltrimethoxysilane, 3-aminopropylmethyldimethoxysilane, N- (2-aminoethyl) -3-aminopropyltrimethoxysilane, and N- (2-aminoethyl).
- the amount of the adhesive-imparting agent used is preferably 0.1 to 20 parts by weight with respect to 100 parts by weight in total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). In particular, 0.5 to 10 parts by weight is preferable.
- plasticizer can be added to the composition of the present invention.
- specific examples of the plasticizer include phthalate compounds such as dibutylphthalate, diisononylphthalate (DINP), diheptylphthalate, di (2-ethylhexyl) phthalate, diisodecylphthalate (DIDP), and butylbenzylphthalate; bis (2-ethylhexyl).
- a polymer plasticizer can be used.
- the polymer plasticizer include a vinyl polymer; a polyester plasticizer; a polyether polyol such as polyethylene glycol and polypropylene glycol having a number average molecular weight of 500 or more, and the hydroxy group of these polyether polyols is an ester group or an ether group.
- Polyethers such as derivatives converted into the above; polystyrenes; polybutadiene, polybutene, polyisobutylene, butadiene-acrylonitrile, polychloroprene and the like can be mentioned.
- the amount of the plasticizer used is preferably 5 to 150 parts by weight, preferably 10 to 120 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). By weight is more preferred, especially 20 to 100 parts by weight. If it is less than 5 parts by weight, the effect as a plasticizer will not be exhibited, and if it exceeds 150 parts by weight, the mechanical strength of the cured product will be insufficient.
- the plasticizer may be used alone or in combination of two or more.
- Solvents or diluents can be added to the compositions of the present invention.
- the solvent and diluent are not particularly limited, but aliphatic hydrocarbons, aromatic hydrocarbons, alicyclic hydrocarbons, halogenated hydrocarbons, alcohols, esters, ketones, ethers and the like can be used.
- the boiling point of the solvent is preferably 150 ° C. or higher, more preferably 200 ° C. or higher, and particularly preferably 250 ° C. or higher, due to the problem of air pollution when the composition is used indoors. ..
- the above solvent or diluent may be used alone or in combination of two or more.
- a sagging inhibitor may be added to the composition of the present invention in order to prevent sagging and improve workability.
- the sagging inhibitor is not particularly limited, and examples thereof include polyamide waxes; hydrogenated castor oil derivatives; and metal soaps such as calcium stearate, aluminum stearate, and barium stearate. These anti-sauce agents may be used alone or in combination of two or more.
- the amount of the sagging inhibitor used is preferably 0.1 to 20 parts by weight with respect to 100 parts by weight in total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B).
- Antioxidants can be used in the compositions of the present invention.
- the use of antioxidants can enhance the weather resistance of the cured product.
- examples of the antioxidant include hindered phenols, monophenols, bisphenols, and polyphenols. Specific examples of the antioxidant are also described in JP-A-4-283259 and JP-A-9-194731.
- the amount of the antioxidant used is preferably 0.1 to 10 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). In particular, 0.2 to 5 parts by weight is preferable.
- a light stabilizer can be used in the composition of the present invention.
- the use of a light stabilizer can prevent photooxidation deterioration of the cured product.
- Examples of the light stabilizer include benzotriazole-based compounds, hindered amine-based compounds, and benzoate-based compounds, but hindered amine-based compounds are particularly preferable.
- the amount of the light stabilizer used is preferably 0.1 to 10 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). In particular, 0.2 to 5 parts by weight is preferable.
- An ultraviolet absorber can be used in the composition of the present invention.
- the use of an ultraviolet absorber can enhance the surface weather resistance of the cured product.
- Examples of the ultraviolet absorber include benzophenone-based, benzotriazole-based, salicylate-based, substituted trill-based and metal chelate-based compounds, but benzotriazole-based compounds are particularly preferable, and commercially available names such as tinubin P, tinubin 213, tinubin 234 and tinubin 326, Examples thereof include chinubin 327, chinubin 328, chinubin 329, and chinubin 571 (all manufactured by BASF).
- the amount of the ultraviolet absorber used is preferably 0.1 to 10 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). In particular, 0.2 to 5 parts by weight is preferable.
- a physical property adjusting agent for adjusting the tensile properties of the produced cured product may be added to the curable composition of the present invention.
- the physical property adjusting agent is not particularly limited, and for example, alkylalkoxysilanes such as phenoxytrimethylsilane, methyltrimethoxysilane, dimethyldimethoxysilane, trimethylmethoxysilane, and n-propyltrimethoxysilane; diphenyldimethoxysilane and phenyltrimethoxysilane.
- Allylalkoxysilanes such as: dimethyldiisopropenoxysilane, methyltriisopropenoxysilane, alkylisopropenoxysilane such as ⁇ -glycidoxypropylmethyldiisopropenoxysilane; tris (trimethylsilyl) borate, tris (triethyl) Examples thereof include trialkylsilylborates such as silyl) borate; silicone varnishes; polysiloxanes and the like.
- the physical property adjusting agent may be used alone or in combination of two or more.
- a compound that produces a compound having a monovalent silanol group in the molecule by hydrolysis has an action of lowering the modulus of the cured product without aggravating the stickiness of the surface of the cured product.
- compounds that produce trimethylsilanol are preferred.
- Compounds that produce compounds having a monovalent silanol group in the molecule by hydrolysis are derivatives of alcohols such as hexanol, octanol, phenol, trimethylolpropane, glycerin, pentaerythritol, and sorbitol, and are silane monomono by hydrolysis. Silicon compounds that produce oars can be mentioned.
- the amount of the physical property adjusting agent used is preferably 0.1 to 10 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). In particular, 0.5 to 5 parts by weight is preferable.
- a tackifier resin can be added to the composition of the present invention for the purpose of enhancing the adhesiveness and adhesion to the base material, or if necessary.
- the tackifying resin is not particularly limited, and a commonly used resin can be used.
- terpen-based resins aromatic-modified terpen resins, hydrocarbon-modified terpen resins, terpen-phenol resins, phenol resins, modified phenol resins, xylene-phenol resins, cyclopentadiene-phenol resins, kumaron inden resins, and rosin-based resins.
- the amount of the tackifier resin used is preferably 2 to 100 parts by weight, preferably 5 to 50 parts by weight, based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). It is more preferably parts by weight, and even more preferably 5 to 30 parts by weight.
- a compound containing an epoxy group can be used.
- the compound having an epoxy include epoxidized unsaturated fats and oils, epoxidized unsaturated fatty acid esters, alicyclic epoxy compounds, compounds shown in epichlorohydrin derivatives, and mixtures thereof.
- Epoxy butyl steerate and the like The epoxy compound is preferably used in the range of 0.5 to 50 parts by weight with respect to a total of 100 parts by weight of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). ..
- a photocurable substance can be used in the composition of the present invention.
- a photocurable material When a photocurable material is used, a film of a photocurable substance is formed on the surface of the cured product, and the stickiness of the cured product and the weather resistance of the cured product can be improved.
- Many compounds of this type are known, such as organic monomers, oligomers, resins, and compositions containing them, and typical ones include one or several acrylic or methacrylic unsaturated groups.
- An unsaturated acrylic compound, vinyl polysilicate dermatates, an azide resin, or the like, which is a monomer, an oligomer, or a mixture thereof, can be used.
- the amount of the photocurable substance used is preferably 0.1 to 20 parts by weight based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). More preferably, 0.5 to 10 parts by weight.
- An oxygen-curable substance can be used in the composition of the present invention.
- An example of an unsaturated compound that can react with oxygen in the air is an oxygen-curable substance that reacts with oxygen in the air to form a cured film near the surface of the cured product, which causes stickiness on the surface and dust on the surface of the cured product. It acts to prevent the adhesion of dust and dirt.
- Specific examples of the oxygen-curable substance include dry oils such as diene oil and linseed oil, and various alkyd resins obtained by modifying the compounds; acrylic polymers and epoxy resins modified with the dry oils.
- Silicon resin 1,2-polybutadiene, 1,4-polybutadiene, C5-C8 diene polymers obtained by polymerizing or copolymerizing diene compounds such as butadiene, chloroprene, isoprene, 1,3-pentadiene, etc.
- diene compounds such as butadiene, chloroprene, isoprene, 1,3-pentadiene, etc.
- liquid polymers These may be used alone or in combination of two or more.
- the amount of the oxygen-curable substance used is preferably 0.1 to 20 parts by weight based on 100 parts by weight of the total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B). More preferably, 0.5 to 10 parts by weight.
- the oxygen-curable substance is preferably used in combination with the photo-curable substance.
- Epoxy resin can be used in combination with the composition of the present invention.
- the composition to which the epoxy resin is added is particularly preferable as an adhesive, particularly an adhesive for outer wall tiles.
- the epoxy resin include bisphenol A type epoxy resins and novolac type epoxy resins.
- a curing agent that cures the epoxy resin can be used in combination with the composition of the present invention.
- the epoxy resin curing agent that can be used is not particularly limited, and a generally used epoxy resin curing agent can be used.
- the amount used is preferably in the range of 0.1 to 300 parts by weight with respect to 100 parts by weight of the epoxy resin.
- the curable composition of the present invention can be prepared as a one-component type in which all the compounding components are compounded and sealed in advance and cured by the moisture in the air after construction, and separately as a curing agent, a silanol condensation catalyst, It is also possible to prepare a two-component type in which components such as a filler, a plasticizer, and water are blended and the compounding material and the resin composition are mixed before use. From the viewpoint of workability, the one-component type is preferable.
- the moist-containing compounding components are either dehydrated and dried in advance before use, or dehydrated by decompression or the like during compounding kneading. Is preferable.
- the amount of the dehydrating agent, particularly the silicon compound capable of reacting with water such as vinyltrimethoxysilane, is 100 parts by weight in total of the polyoxyalkylene polymer (A) and the (meth) acrylic acid ester polymer (B).
- the range of 0.1 to 20 parts by weight is preferable, and the range of 0.5 to 10 parts by weight is more preferable.
- the curable composition of the present invention is an adhesive, a sealing material for buildings, ships, automobiles, roads, etc., an adhesive, a waterproofing material, a coating film waterproofing material, a molding agent, a vibration-proofing material, a vibration-damping material, and a soundproofing material.
- the cured product obtained by curing the curable composition of the present invention is excellent in flexibility and adhesiveness, and therefore can be suitably used as a sealing material or an adhesive.
- the curable composition of the present invention includes electric / electronic component materials such as a solar cell backside sealing material, electrical / electronic components such as an insulating coating material for electric wires / cables, an electrical insulating material for an apparatus, and an acoustic insulating material.
- electric / electronic component materials such as a solar cell backside sealing material
- electrical / electronic components such as an insulating coating material for electric wires / cables, an electrical insulating material for an apparatus, and an acoustic insulating material.
- the curable composition of the present invention is an adhesive for interior panels, an adhesive for exterior panels, an adhesive for tiles, an adhesive for stones, an adhesive for ceiling finishing, an adhesive for floor finishing, and an adhesive for wall finishing.
- Adhesives vehicle panel adhesives, electrical / electronic / precision equipment assembly adhesives, leather, textiles, fabrics, paper, board and rubber bonding adhesives, reactive post-bridge pressure sensitive adhesives, direct It can also be used as a glazing sealant, a multi-layer glass sealant, an SSG method sealant, a building working joint sealant, a civil engineering material, and a bridge material. Furthermore, it can also be used as an adhesive material such as an adhesive tape or an adhesive sheet.
- the number average molecular weight in the examples is the GPC molecular weight measured under the following conditions.
- Liquid transfer system Tosoh HLC-8220GPC Column: Tosoh TSKgel SuperH series Solvent: THF Molecular weight: Polystyrene conversion Measurement temperature: 40 ° C
- the polyoxyalkylene-based weight is adopted. Calculated as the sum of the molar ratio of the reacted epoxy compound (E2) to the hydroxyl group of the coalescence (C) and the molar ratio of the reacted organic halide (E1) to the hydroxyl group of the polyoxyalkylene polymer (C). it can.
- the "reactive silyl group introduction rate" is determined by measuring 1 H NMR for the polyoxyalkylene-based polymer (A) having a reactive silyl group and using the integrated value of the signal indicating each group.
- the "average number of reactive silyl groups bonded to the inside of the skeleton" is referred to as "the number of terminally bonded silyl groups” and “the number of internally bonded silyl groups", respectively.
- Number of end-bonded silyl groups (number average molecular weight of polymer / total number of solids during polymer synthesis) x [(total number of copies of each reactive silyl group-containing chain transfer agent used / molecular weight of the chain transfer agent)]
- Number of internally bonded silyl groups (number average molecular weight of polymer / total number of solids during polymer synthesis) ⁇ [(total number of copies of each reactive silyl group-containing monomer / molecular weight of the monomer)]
- Linear polyoxypropylene (A-5) having a silyl group number / terminal number ratio of 0.75, a number average molecular weight of 28,000, and a methyldimethoxysilyl group at the end by reacting at ° C. for 2 hours.
- the blending amount is the number of parts by weight with respect to 100 parts by weight of each polymer mixture as the base polymer.
- the curable composition was filled in a mold having a thickness of about 5 mm using a spatula at 23 ° C. and a relative humidity of 50%, and the time when the surface was flattened was defined as the curing start time.
- the surface was touched with a spatula, and the curability was measured by setting the time until the composition did not adhere to the spatula as the skinning time.
- the curable composition was packed into a 3 mm thick sheet-like formwork at 23 ° C. and 50% relative humidity. After curing at 23 ° C. and a relative humidity of 50% for 3 days, it was cured in a dryer at 50 ° C. for 4 days to obtain a sheet-like cured product. The obtained cured product was punched into a No. 3 dumbbell mold according to JIS K 6251 to obtain a test piece. Using the obtained test piece, a tensile test (tensile speed 200 mm / min) was performed using an autograph at 23 ° C. and 50% relative humidity, and 50% elongation stress, 100% elongation stress, and fracture stress were performed. , And elongation at break was measured.
- the number of terminally bonded silyl groups is 0.30 or more and 1.00 or less with respect to the polyoxyalkylene polymer (A) having a silyl group number / terminal number ratio of 0.80 or more.
- the cured products of the compositions (each example) containing the polymer mixture containing the (meth) acrylic acid ester-based polymer (B) having an internally bonded silyl group number of 0 or more and less than 0.30 are the same.
- a composition containing a polymer mixture obtained by mixing a (meth) acrylic acid ester-based polymer having an internally bonded silyl group number of 0.30 or more with respect to the polymer (A) (Comparative Examples 1-1, 1-2, Compared to the cured products of 1-3, 1-4, 2-1, 2-2, 3-1, 3-2, 4-1), 4-2, and 5-1), the stress at break and the stress at break It can be seen that both growth improved.
- the number of terminally bonded silyl groups is 0.30 or more and 1.00 or less and the number of internally bonded silyl groups is 0 or more with respect to the polyoxyalkylene polymer having a silyl group number / terminal number ratio of less than 0.80.
- the cured products of the compositions (Comparative Examples 1-5, 2-3, and 3-3) containing the polymer mixture obtained by mixing the (meth) acrylic acid ester-based polymer (B) of less than 0.30 are the same.
- a composition containing a polymer mixture obtained by mixing a (meth) acrylic acid ester-based polymer having an internally bonded silyl group number of 0.30 or more with respect to a polyoxyalkylene-based polymer (Comparative Examples 1-6, 1-7). It can be seen that, as compared with the cured products of 2-4, 2-5 and 3-4), the elongation at break is improved, but the stress at break is reduced.
- Example 4-1 compared with the case where the ratio of the number of silyl groups / the number of terminals of the polymer (A) is 0.80 (Example 4-1). It can be seen that when the ratio is 0.85 or more (Example 4-2), the effect of improving the stress at break is strongly exhibited.
- Table 8 show a (meth) acrylic acid ester-based polymer showing various internal bond silyl groups with respect to the polyoxyalkylene-based polymer (A) having a silyl group number / terminal number ratio of 1.50. This is the result of using a polymer mixture mixed with B), but when the number of internally bonded silyl groups of the (meth) acrylic acid ester polymer is 0 or more and less than 0.30 (each example), the cured product is It can be seen that it shows high breaking stress and breaking elongation.
- Examples 6-1 to 6-6 when the number of internally bonded silyl groups is less than 0.20 (Examples 6-1 to 6-6), the effect is strongly exhibited, and when it is less than 0.15 (Examples 6-1 to 6-5). ), And when it is less than 0.05 (Examples 6-1 to 6-4), it is particularly strongly exerted.
- Comparative Example 6-1 in Table 8 is the result of using a (meth) acrylic acid ester-based polymer in which both the number of terminal-bonded silyl groups and the number of internal-bonded silyl groups are 0, and Comparative Example 6-2 is the result.
- This is the result of using a (meth) acrylic acid ester polymer having 0 terminal-bonded silyl groups and 1 or more internal-bonded silyl groups, but the cured product has low breaking stress and breaking elongation. You can see that. From this, it can be seen that the (meth) acrylic acid ester-based polymer needs to have a reactive silyl group bonded to the end of the polymer skeleton in order to exert the effect of the present invention.
- Example 6-1 and Example 7-1 when the number of terminal-bonded silyl groups is 0.65 or more (Example 6-1 and Example 7-1), the effect is strongly exhibited, and when it is 0.85 or more (Example 6-1). It can be seen that it is exerted even more strongly.
- Comparative Example 2-1 and Comparative Example 2-2 in Table 8 show a (meth) acrylic acid ester with respect to the polyoxyalkylene polymer (A) having a silyl group number / terminal number ratio of 1.50.
- This is the result of using a polymer mixture in which a (meth) acrylic acid ester-based polymer having an internal bond silyl group number of 0.30 or more of the based polymer is mixed.
- Examples 6-1 and Examples As shown in 7-1, even if the number of terminal-bonded silyl groups of the (meth) acrylic acid ester-based polymer is increased from less than 0.85 to 0.85 or more, the stress at break of the cured product does not improve.
- the results shown in Table 10 show that the polyoxyalkylene polymer (A) having a silyl group number / terminal number ratio of 0.80 or more has various (meth) acrylic acid ester-based polymers that satisfy the requirements of the present invention. This is the result of using a polymer mixture in which the polymer (B) is mixed.
- the (meth) acrylic acid ester-based polymer (B) having an end-bonded silyl group number of 0.30 or more and 1.00 or less and an internally bonded silyl group of 0 or more and less than 0.30 is used in combination (each example).
- the number of terminally bonded silyl groups is 0.30 or more and 1.00 or less and the internal bond is relative to the polyoxyalkylene polymer (A) having a silyl group number / terminal number ratio of 0.80 or more.
- a composition containing a polymer mixture containing a (meth) acrylic acid ester-based polymer (B) having a silyl group number of 0 or more and less than 0.30 has a curable composition showing high breaking stress and breaking elongation after curing. It can be seen that it can be suitably used as a product.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Polyethers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
-SiR1 aX3-a (1)
(式中、R1は炭素数1~20の置換または無置換の炭化水素基を表す。Xは水酸基または加水分解性基を表す。aは、0、1または2である。R1又はXが複数存在するとき、それらは同じでもよく、異なっていてもよい。)で表される反応性シリル基を有するポリオキシアルキレン系重合体(A)、及び、下記一般式(2):
-SiR2 bY3-b (2)
(式中、R2は炭素数1~20の無置換の炭化水素基を表す。Yは水酸基または加水分解性基を表す。bは、0、1または2である。R2又はYが複数存在するとき、それらは同じでもよく、異なっていてもよい。)で表される反応性シリル基を有する(メタ)アクリル酸エステル系重合体(B)、を含み、ポリオキシアルキレン系重合体(A)1分子における重合体骨格の末端の数に対する反応性シリル基の数の平均比率が0.80以上であり、(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の末端に結合した反応性シリル基の平均個数が0.30以上1.00以下であり、かつ、(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の内部に結合した反応性シリル基の平均個数が0以上0.30未満である、硬化性組成物に関する。好ましくは、ポリオキシアルキレン系重合体(A)1分子における重合体骨格の末端の数に対する反応性シリル基の数の平均比率が0.85以上である。好ましくは、(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の末端に結合した反応性シリル基の平均個数が0.85以上1.00以下である。好ましくは、(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の内部に結合した反応性シリル基の平均個数が0以上0.15未満である。好ましくは、ポリオキシアルキレン系重合体(A):(メタ)アクリル酸エステル系重合体(B)の重量比が80:20~50:50である。好ましくは、(メタ)アクリル酸エステル系重合体(B)の構成単量体単位のうちメタクリル酸メチルが占める割合が、35重量%以上85重量%以下である。好ましくは、(メタ)アクリル酸エステル系重合体(B)の構成単量体単位のうちメタクリル酸アルキルの合計が占める割合が、70重量%以上85重量%以下であり、前記アルキルが炭素数1~4の非置換アルキル基である。
また本発明は前記硬化性組成物を硬化させた硬化物にも関する。
(ポリオキシアルキレン系重合体(A))
本発明の硬化性組成物は、反応性シリル基を有するポリオキシアルキレン系重合体(A)を含有する。ポリオキシアルキレン系重合体(A)は当該反応性シリル基を有することで、反応性シリル基の加水分解及び脱水縮合反応に基づく硬化性を示す。
-SiR1 aX3-a (1)
で表される。
式中、R3は、直接結合、又は炭素数1~4の2価の炭化水素基を表し、R4は水素または炭素数1~6のアルキル基を表す。左端の酸素は、重合体骨格の末端に位置するオキシアルキレン単位中の酸素を示す。R1、X、及びaは、式(1)について上述したものと同じである。

次にポリオキシアルキレン系重合体(A)を製造する方法について説明する。ポリオキシアルキレン系重合体(A)は、水酸基末端ポリオキシアルキレン系重合体(C)に対し、水酸基の反応性を利用してオレフィン基を導入した後、該オレフィン基との反応性を有する反応性シリル基含有化合物を反応させて反応性シリル基を導入することで製造できる。
ポリオキシアルキレン系重合体の重合体骨格は、従来公知の方法によって、水酸基を有する開始剤にエポキシ化合物を重合させることで形成することができ、これによって水酸基末端ポリオキシアルキレン系重合体(C)が得られる。具体的な重合方法としては特に限定されないが、分子量分布(Mw/Mn)の小さい水酸基末端重合体が得られることから、亜鉛ヘキサシアノコバルテートグライム錯体等の複合金属シアン化物錯体触媒を用いた重合方法が好ましい。
水酸基末端ポリオキシアルキレン系重合体(C)に対しオレフィン基を導入するにあたっては、まず、水酸基末端ポリオキシアルキレン系重合体(C)に対しアルカリ金属塩を作用させて末端の水酸基をメタルオキシ基に変換することが好ましい。また、アルカリ金属塩の代わりに、複合金属シアン化物錯体触媒を用いることもできる。以上によって、メタルオキシ基末端ポリオキシアルキレン系重合体(D)が形成される。
以上のようにして得られたメタルオキシ基末端ポリオキシアルキレン系重合体(D)に対し、オレフィン基を有する求電子剤(E)を作用させることで、メタルオキシ基を、オレフィン基を含む構造に変換することができる。これにより、末端構造中にオレフィン基を有するポリオキシアルキレン系重合体(F)が形成される。
Z-R3-C(R4)=CH2 (5)
で表すことができる。式中、R3及びR4は、それぞれ、一般式(3)について上述したR3及びR4と同じ基である。Zは、ハロゲン原子を表す。当該有機ハロゲン化物(E1)を反応させて得られた末端構造中にオレフィン基を有するポリオキシアルキレン系重合体(F)に対して、後に説明する反応性シリル基の導入を行うと、前記一般式(3)で表される末端構造が形成され得る。
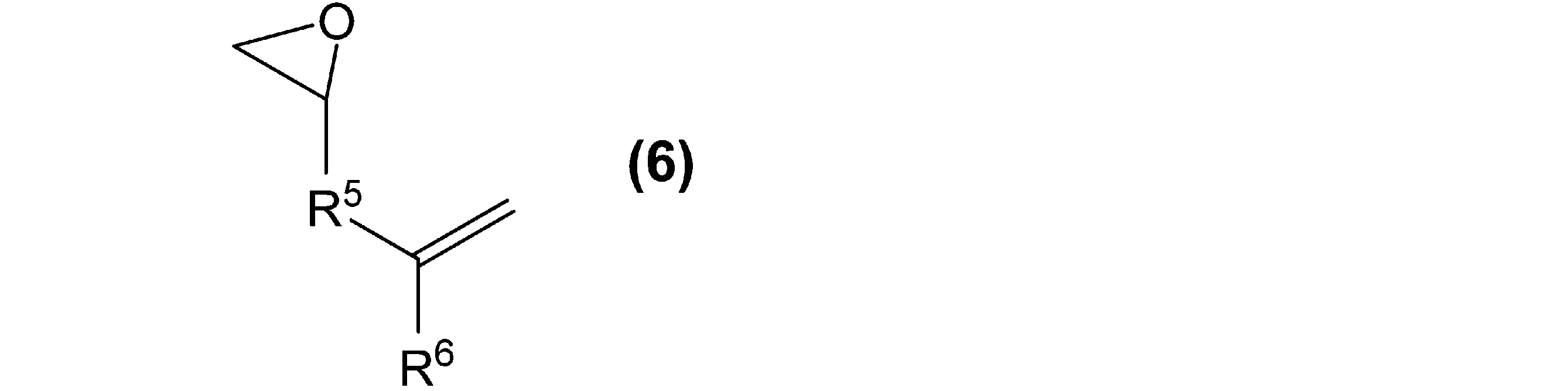
以上によって得られた末端構造中にオレフィン基を有するポリオキシアルキレン系重合体(F)に対し、反応性シリル基を有するヒドロシラン化合物(G)をヒドロシリル化反応させることで、重合体に反応性シリル基を導入することができる。これにより、反応性シリル基を有するポリオキシアルキレン系重合体(A)が製造され得る。ヒドロシリル化反応には、簡便に実施できることに加え、反応性シリル基の導入量の調整が容易であり、また、得られる重合体の物性が安定している利点がある。
本発明の硬化性組成物は、反応性シリル基を有する(メタ)アクリル酸エステル系重合体(B)をさらに含有する。
-SiR2 bY3-b (2)
で表される。
1/(Tg(K))=Σ(Mi/Tgi)
(式中、Miは重合体を構成する単量体i成分の重量分率、Tgiは単量体iのホモポリマーのガラス転移温度(K)を表す。)
(iv)重合性不飽和基と反応性シリル基を有する化合物を、上述の単量体とともに共重合する方法。この方法を用いると反応性シリル基は重合体骨格の内部にランダムに導入される傾向がある。
(v)連鎖移動剤として、反応性シリル基を有するメルカプトシラン化合物を使用して(メタ)アクリル酸エステル系重合体を重合する方法。この方法を用いると、反応性シリル基を重合体骨格の末端に導入することができる。
(vi)重合性不飽和基と反応性官能基(V基)を有する化合物を、共重合した後、反応性シリル基とV基に反応する官能基を有する化合物を反応させる方法。具体的には、アクリル酸2-ヒドロキシエチルを共重合した後、反応性シリル基を有するイソシアネートシランを反応させる方法や、アクリル酸グリシジルを共重合した後、反応性シリル基を有するアミノシラン化合物を反応させる方法などが例示できる。
(vii)リビングラジカル重合法によって合成した(メタ)アクリル酸エステル系重合体の末端官能基を変性して、反応性シリル基を導入する方法。リビングラジカル重合法は重合体骨格の末端に官能性基を導入しやすく、これを変性することで重合体骨格の末端に反応性シリル基を導入することができる。
本発明の硬化性組成物は、ポリオキシアルキレン系重合体(A)及び(メタ)アクリル酸エステル系重合体(B)が有する反応性シリル基を加水分解・脱水縮合させる反応、即ち硬化反応を促進する目的で、シラノール縮合触媒を含有することが好ましい。
本発明の硬化性組成物には、その他の添加剤として、シリコン化合物、接着性付与剤、可塑剤、溶剤、希釈剤、シリケート、充填剤、タレ防止剤、酸化防止剤、光安定剤、紫外線吸収剤、物性調整剤、粘着付与樹脂、エポキシ基を含有する化合物、光硬化性物質、酸素硬化性物質、表面性改良剤、エポキシ樹脂、その他の樹脂、難燃剤、発泡剤を添加しても良い。また、本発明の硬化性組成物には、硬化性組成物又は硬化物の諸物性の調整を目的として、必要に応じて各種添加剤を添加してもよい。このような添加物の例としては、例えば、硬化性調整剤、ラジカル禁止剤、金属不活性化剤、オゾン劣化防止剤、リン系過酸化物分解剤、滑剤、顔料、防かび剤等が挙げられる。
本発明の組成物には、種々の充填剤を配合することができる。充填剤としては、重質炭酸カルシウム、膠質炭酸カルシウム、炭酸マグネシウム、ケイソウ土、クレー、タルク、酸化チタン、ヒュームドシリカ、沈降性シリカ、結晶性シリカ、溶融シリカ、無水ケイ酸、含水ケイ酸、カーボンブラック、酸化第二鉄、アルミニウム微粉末、酸化亜鉛、活性亜鉛華、PVC粉末、PMMA粉末、ガラス繊維およびフィラメント等が挙げられる。上記充填剤は1種類のみで使用しても良いし、2種類以上混合使用しても良い。
本発明の組成物には、接着性付与剤を添加することができる。接着性付与剤としては、シランカップリング剤、シランカップリング剤の反応物を添加することができる。
本発明の組成物には、可塑剤を添加することができる。可塑剤の具体例としては、ジブチルフタレート、ジイソノニルフタレート(DINP)、ジヘプチルフタレート、ジ(2-エチルヘキシル)フタレート、ジイソデシルフタレート(DIDP)、ブチルベンジルフタレートなどのフタル酸エステル化合物;ビス(2-エチルヘキシル)-1,4-ベンゼンジカルボキシレートなどのテレフタル酸エステル化合物;1,2-シクロヘキサンジカルボン酸ジイソノニルエステルなどの非フタル酸エステル化合物;アジピン酸ジオクチル、セバシン酸ジオクチル、セバシン酸ジブチル、コハク酸ジイソデシル、アセチルクエン酸トリブチルなどの脂肪族多価カルボン酸エステル化合物;オレイン酸ブチル、アセチルリシノール酸メチルなどの不飽和脂肪酸エステル化合物;アルキルスルホン酸フェニルエステル;リン酸エステル化合物;トリメリット酸エステル化合物;塩素化パラフィン;アルキルジフェニル、部分水添ターフェニルなどの炭化水素系油;プロセスオイル;エポキシ化大豆油、エポキシステアリン酸ベンジルなどのエポキシ可塑剤、などをあげることができる。
本発明の組成物には溶剤または希釈剤を添加することができる。溶剤及び希釈剤としては、特に限定されないが、脂肪族炭化水素、芳香族炭化水素、脂環族炭化水素、ハロゲン化炭化水素、アルコール、エステル、ケトン、エーテルなどを使用することができる。溶剤または希釈剤を使用する場合、組成物を屋内で使用した時の空気への汚染の問題から、溶剤の沸点は、150℃以上が好ましく、200℃以上がより好ましく、250℃以上が特に好ましい。上記溶剤または希釈剤は単独で用いてもよく、2種以上併用してもよい。
本発明の組成物には、必要に応じてタレを防止し、作業性を良くするためにタレ防止剤を添加しても良い。また、タレ防止剤としては特に限定されないが、例えば、ポリアミドワックス類;水添ヒマシ油誘導体類;ステアリン酸カルシウム、ステアリン酸アルミニウム、ステアリン酸バリウム等の金属石鹸類等が挙げられる。これらタレ防止剤は単独で用いてもよく、2種以上併用してもよい。
本発明の組成物には、酸化防止剤(老化防止剤)を使用することができる。酸化防止剤を使用すると硬化物の耐候性を高めることができる。酸化防止剤としてはヒンダードフェノール系、モノフェノール系、ビスフェノール系、ポリフェノール系が例示できる。酸化防止剤の具体例は特開平4-283259号公報や特開平9-194731号公報にも記載されている。
本発明の組成物には、光安定剤を使用することができる。光安定剤を使用すると硬化物の光酸化劣化を防止できる。光安定剤としてベンゾトリアゾール系、ヒンダードアミン系、ベンゾエート系化合物等が例示できるが、特にヒンダードアミン系が好ましい。
本発明の組成物には、紫外線吸収剤を使用することができる。紫外線吸収剤を使用すると硬化物の表面耐候性を高めることができる。紫外線吸収剤としてはベンゾフェノン系、ベンゾトリアゾール系、サリチレート系、置換トリル系及び金属キレート系化合物等が例示できるが、特にベンゾトリアゾール系が好ましく、市販名チヌビンP、チヌビン213、チヌビン234、チヌビン326、チヌビン327、チヌビン328、チヌビン329、チヌビン571(以上、BASF製)が挙げられる。
本発明の硬化性組成物には、必要に応じて生成する硬化物の引張特性を調整する物性調整剤を添加しても良い。物性調整剤としては特に限定されないが、例えば、フェノキシトリメチルシラン、メチルトリメトキシシラン、ジメチルジメトキシシラン、トリメチルメトキシシラン、n-プロピルトリメトキシシラン等のアルキルアルコキシシラン類;ジフェニルジメトキシシラン、フェニルトリメトキシシランなどのアリールアルコキシシラン類;ジメチルジイソプロペノキシシラン、メチルトリイソプロペノキシシラン、γ-グリシドキシプロピルメチルジイソプロペノキシシラン等のアルキルイソプロペノキシシラン;トリス(トリメチルシリル)ボレート、トリス(トリエチルシリル)ボレートなどのトリアルキルシリルボレート類;シリコーンワニス類;ポリシロキサン類等が挙げられる。前記物性調整剤を用いることにより、本発明の組成物を硬化させた時の硬度を上げたり、逆に硬度を下げ、破断伸びを出したりし得る。上記物性調整剤は単独で用いてもよく、2種以上併用してもよい。
本発明の組成物には、基材への接着性や密着性を高める目的、あるいはその他必要に応じて粘着付与樹脂を添加できる。粘着付与樹脂としては、特に制限はなく通常使用されているものを使うことが出来る。
本発明の組成物においてはエポキシ基を含有する化合物を使用できる。エポキシを有する化合物としてはエポキシ化不飽和油脂類、エポキシ化不飽和脂肪酸エステル類、脂環族エポキシ化合物類、エピクロルヒドリン誘導体に示す化合物及びそれらの混合物等が例示できる。具体的には、エポキシ化大豆油、エポキシ化あまに油、ビス(2-エチルヘキシル)-4,5-エポキシシクロヘキサン-1,2-ジカーボキシレート(E-PS)、エポキシオクチルステアレ-ト、エポキシブチルステアレ-ト等が挙げられる。エポキシ化合物は、ポリオキシアルキレン系重合体(A)と(メタ)アクリル酸エステル系重合体(B)との合計100重量部に対して0.5~50重量部の範囲で使用するのがよい。
本発明の組成物には光硬化性物質を使用できる。光硬化性物資を使用すると硬化物表面に光硬化性物質の皮膜が形成され、硬化物のべたつきや硬化物の耐候性を改善できる。この種の化合物には有機単量体、オリゴマー、樹脂或いはそれらを含む組成物等多くのものが知られており、代表的なものとしては、アクリル系又はメタクリル系不飽和基を1ないし数個有する単量体、オリゴマー或いはそれ等の混合物である不飽和アクリル系化合物、ポリケイ皮酸ビニル類あるいはアジド化樹脂等が使用できる。
本発明の組成物には酸素硬化性物質を使用することができる。酸素硬化性物質には空気中の酸素と反応し得る不飽和化合物を例示でき、空気中の酸素と反応して硬化物の表面付近に硬化皮膜を形成し表面のべたつきや硬化物表面へのゴミやホコリの付着を防止するなどの作用をする。酸素硬化性物質の具体例には、キリ油、アマニ油などで代表される乾性油や、該化合物を変性して得られる各種アルキッド樹脂;乾性油により変性されたアクリル系重合体、エポキシ系樹脂、シリコン樹脂;ブタジエン、クロロプレン、イソプレン、1,3-ペンタジエンなどのジエン系化合物を重合または共重合させてえられる1,2-ポリブタジエン、1,4-ポリブタジエン、C5~C8ジエンの重合体などの液状重合体などが挙げられる。これらは単独で用いてもよく、2種以上併用してもよい。
本発明の組成物にはエポキシ樹脂を併用することができる。エポキシ樹脂を添加した組成物は特に接着剤、殊に外壁タイル用接着剤として好ましい。エポキシ樹脂としてはビスフェノールA型エポキシ樹脂類またはノボラック型エポキシ樹脂などが挙げられる。
本発明の硬化性組成物は、すべての配合成分を予め配合密封保存し、施工後空気中の湿気により硬化する1成分型として調製することも可能であり、硬化剤として別途、シラノール縮合触媒、充填材、可塑剤、水等の成分を配合しておき、該配合材と樹脂組成物を使用前に混合する2成分型として調製することもできる。作業性の点からは、1成分型が好ましい。
本発明の硬化性組成物は、粘着剤、建造物・船舶・自動車・道路などのシーリング材、接着剤、防水材、塗膜防水材、型取剤、防振材、制振材、防音材、発泡材料、塗料、吹付材として使用することができる。本発明の硬化性組成物を硬化して得られる硬化物は、柔軟性および接着性に優れることから、シーリング材または接着剤として好適に使用することができる。
送液システム:東ソー製HLC-8220GPC
カラム:東ソー製TSKgel SuperHシリーズ
溶媒:THF
分子量:ポリスチレン換算
測定温度:40℃
「ポリオキシアルキレン系重合体骨格の1つの末端に存在する、反応性シリル基を導入可能な基(例えばオレフィン基)の数」に対し、「反応性シリル基導入率」を掛け合わせることによって算出した。
「ポリオキシアルキレン系重合体骨格の1つの末端に存在する、反応性シリル基を導入可能な基の数」は、例えば、ポリオキシアルキレン系重合体(C)が有する水酸基に対してオレフィン基を有するエポキシ化合物(E2)を反応させた後、オレフィン基を有する有機ハロゲン化物(E1)を反応させてポリオキシアルキレン系重合体(A)を製造する方法を採用した場合は、ポリオキシアルキレン系重合体(C)が有する水酸基に対する、反応したエポキシ化合物(E2)のモル比と、ポリオキシアルキレン系重合体(C)が有する水酸基に対する、反応した有機ハロゲン化物(E1)のモル比の合計として算出できる。
「反応性シリル基導入率」は、反応性シリル基を有するポリオキシアルキレン系重合体(A)について1H NMRを測定して、各基を示すシグナルの積分値を利用して、式:(反応性シリル基のモル数)/(反応性シリル基のモル数と、反応性シリル基が導入されずに残留した、反応性シリル基を導入可能な基(例えば末端オレフィン基)のモル数と、前記反応性シリル基を導入可能な基が変性した基(例えば末端オレフィン基が内部異性化した内部オレフィン基)のモル数の合計)に基づいて算出した。
なお、以下では、「ポリオキシアルキレン系重合体骨格の末端の数に対する反応性シリル基の数の平均比率」を、「シリル基数/末端数の比率」として言及する。
反応性シリル基含有単量体および反応性シリル基含有連鎖移動剤の使用量から計算される重合体中の反応性シリル基濃度、およびGPCによって測定した重合体の数平均分子量によって算出した。具体的には、下記式を用いて算出した。なお、以下では、「(メタ)アクリル酸エステル系重合体1分子において重合体骨格の末端に結合した反応性シリル基の平均個数」及び「(メタ)アクリル酸エステル系重合体1分子において重合体骨格の内部に結合した反応性シリル基の平均個数」を、それぞれ、「末端結合シリル基数」及び「内部結合シリル基数」として言及する。
末端結合シリル基数 = (重合体の数平均分子量/重合体合成時の固形分部数合計)×[(各反応性シリル基含有連鎖移動剤の使用部数/該連鎖移動剤の分子量)の合計]
内部結合シリル基数 = (重合体の数平均分子量/重合体合成時の固形分部数合計)×[(各反応性シリル基含有単量体の部数/該単量体の分子量)の合計]
数平均分子量約4,500のポリオキシプロピレンジオールを開始剤とし、亜鉛ヘキサシアノコバルテートグライム錯体触媒にて、下記分子量に達するまでプロピレンオキシドの重合を行い、数平均分子量が約28,000の直鎖型水酸基末端ポリオキシプロピレン(C-1)を得た。
数平均分子量約4,500のポリオキシプロピレンジオールを開始剤とし、亜鉛ヘキサシアノコバルテートグライム錯体触媒にて、下記分子量に達するまでプロピレンオキシドの重合を行い、数平均分子量が約15,000の直鎖型水酸基末端ポリオキシプロピレン(C-2)を得た。
数平均分子量約4,500のポリオキシプロピレントリオールを開始剤とし、亜鉛ヘキサシアノコバルテートグライム錯体触媒にて、下記分子量に達するまでプロピレンオキシドの重合を行い、数平均分子量が約26,000の分岐型水酸基末端ポリオキシプロピレン(C-3)を得た。
数平均分子量約4,500のポリオキシプロピレンジオールを開始剤とし、亜鉛ヘキサシアノコバルテートグライム錯体触媒にて、下記分子量に達するまでプロピレンオキシドの重合を行い、数平均分子量が約21,000の直鎖型水酸基末端ポリオキシプロピレン(C-4)を得た。
重合体(C-1)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-2-メチル-1-プロペンを添加して末端の水酸基をメタリル基に変換した。次に、得られたメタリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体溶液(白金換算で3質量%濃度のイソプロピルアルコール溶液)100ppm、硫黄溶液(0.25重量%濃度のヘキサン溶液)100ppmおよび(メトキシメチル)ジメトキシシラン2.3重量部を加え、100℃にて、メタリル基の残存量が反応開始前の1%未満となるまで反応を継続した。減圧脱揮により、余剰の(メトキシメチル)ジメトキシシラン等を除くことにより、シリル基数/末端数の比率が0.95、数平均分子量が28,000の、末端に(メトキシメチル)ジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-1)を得た。
重合体(C-1)の水酸基に対して1.0当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.0当量のアリルグリシジルエーテルを140℃で添加して2時間反応させてアリル基と水酸基を含む構造を導入し、さらに1.5当量の3-クロロ-1-プロペンを添加して水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)36ppmおよび(メトキシメチル)ジメトキシシラン2.1重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が1.50、数平均分子量が28,000の、末端に(メトキシメチル)ジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-2)を得た。
重合体(C-1)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-1-プロペンを添加して末端の水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)36ppmおよび(メトキシメチル)ジメトキシシラン1.1重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が0.75、数平均分子量が28,000の、末端に(メトキシメチル)ジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-3)を得た。
重合体(C-1)の水酸基に対して1.0当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.0当量のアリルグリシジルエーテルを140℃で添加して2時間反応させてアリル基と水酸基を含む構造を導入し、さらに1.5当量の3-クロロ-1-プロペンを添加して水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)36ppmおよびメチルジメトキシシラン1.7重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が1.50、数平均分子量が28,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-4)を得た。
重合体(C-1)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-1-プロペンを添加して末端の水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)36ppmおよびメチルジメトキシシラン0.95重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が0.75、数平均分子量が28,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-5)を得た。
重合体(C-1)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-1-プロペンを添加して末端の水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)50ppmおよびトリメトキシシラン1.1重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が0.80、数平均分子量が28,000の、末端にトリメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-6)を得た。
重合体(C-1)の水酸基に対して1.0当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.0当量のアリルグリシジルエーテルを140℃で添加して2時間反応させてアリル基と水酸基を含む構造を導入し、さらに1.5当量の3-クロロ-1-プロペンを添加して水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)50ppmおよびトリメトキシシラン1.9重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が1.50、数平均分子量が28,000の、末端にトリメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-7)を得た。
重合体(C-2)100重量部に対して、メルカプト錫系触媒(日東化成製U-360)50ppm、および(イソシアネートメチル)ジメトキシメチルシラン3.1重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が0.90、数平均分子量が15,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-8)を得た。
重合体(C-3)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-2-メチル-1-プロペンを添加して末端の水酸基をメタリル基に変換した。次に、得られたメタリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体溶液(白金換算で3質量%濃度のイソプロピルアルコール溶液)100ppm、硫黄溶液(0.25重量%濃度のヘキサン溶液)100ppmおよび(メトキシメチル)ジメトキシシラン2.4重量部を加え、100℃にて、メタリル基の残存量が反応開始前の1%未満となるまで反応を継続した。減圧脱揮により、余剰の(メトキシメチル)ジメトキシシラン等を除くことにより、シリル基数/末端数の比率が0.95、数平均分子量が26,000の、末端に(メトキシメチル)ジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-9)を得た。
重合体(C-2)の水酸基に対して1.0当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.0当量のアリルグリシジルエーテルを140℃で添加して2時間反応させてアリル基と水酸基を含む構造を導入し、さらに1.5当量の3-クロロ-1-プロペンを添加して水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体(白金換算で3質量%のイソプロピルアルコール溶液)36ppmおよびメチルジメトキシシラン3.4重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が1.60、数平均分子量が15,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-10)を得た。
重合体(C-2)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-1-プロペンを添加して末端の水酸基をアリル基に変換した。次に、得られたアリル基末端ポリオキシプロピレン100重量部に対して、(3-メルカプトプロピル)メチルジメトキシシラン6.7重量部と2,2’-アゾビス(2-メチルブチロニトリル)0.15重量部を加えて90℃とし、2時間後と4時間後に2,2’-アゾビス(2-メチルブチロニトリル)0.15重量部ずつを加え、6時間反応を継続した。減圧脱揮により、余剰の(3-メルカプトプロピル)メチルジメトキシシラン等を除くことにより、シリル基数/末端数の比率が0.95、数平均分子量が15,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-11)を得た。
重合体(C-1)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-2-メチル-1-プロペンを添加して末端の水酸基をメタリル基に変換した。次に、得られたメタリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体溶液(白金換算で3質量%濃度のイソプロピルアルコール溶液)100ppm、硫黄溶液(0.25重量%濃度のヘキサン溶液)100ppmおよびメチルジメトキシシラン2.3重量部を加え、100℃にて、メタリル基の残存量が反応開始前の1%未満となるまで反応を継続した。減圧脱揮により、余剰のメチルジメトキシシラン等を除くことにより、シリル基数/末端数の比率が0.95、数平均分子量が28,000の、末端にメチルジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-12)を得た。
重合体(C-4)の水酸基に対して1.2当量のナトリウムメトキシドのメタノール溶液を添加して140℃でメタノールを留去し、1.6当量の3-クロロ-2-メチル-1-プロペンを添加して末端の水酸基をメタリル基に変換した。次に、得られたメタリル基末端ポリオキシプロピレン100重量部に対して、白金ジビニルジシロキサン錯体溶液(白金換算で3質量%濃度のイソプロピルアルコール溶液)100ppm、硫黄溶液(0.25重量%濃度のヘキサン溶液)100ppmおよび(メトキシメチル)ジメトキシシラン3.0重量部を加え、100℃にて、メタリル基の残存量が反応開始前の1%未満となるまで反応を継続した。減圧脱揮により、余剰の(メトキシメチル)ジメトキシシラン等を除くことにより、シリル基数/末端数の比率が0.95、数平均分子量が21,000の、末端に(メトキシメチル)ジメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-13)を得た。
重合体(C-1)100重量部に対して、メルカプト錫系触媒(日東化成製U-360)50ppm、および3-イソシアネートプロピルトリメトキシシラン(モメンティブ・パフォーマンス・マテリアルズ社製SilquestA-Link35)2.1重量部を加え、90℃で2時間反応させることにより、シリル基数/末端数の比率が0.85、数平均分子量が28,000の、末端にトリメトキシシリル基を有する直鎖状のポリオキシプロピレン(A-14)を得た。
攪拌機を備えた四口フラスコにイソブチルアルコール57.2重量部を入れ、窒素雰囲気下、105℃まで昇温した。そこに、メタクリル酸メチル78.3重量部、アクリル酸ブチル6.7重量部、メタクリル酸ステアリル15.0重量部、(3-メルカプトプロピル)メチルジメトキシシラン10.0重量部、および2,2’-アゾビス(2-メチルブチロニトリル)0.35重量部をイソブチルアルコール16.3重量部に溶解した混合溶液を3.5時間かけて滴下した。さらに105℃で2時間重合を行い、数平均分子量が1,950、重量平均分子量が3,400である(メタ)アクリル酸エステル系重合体(B-1)のイソブチルアルコール溶液(固形分60重量%)を得た。該重合体(B-1)において末端結合シリル基数は0.98、内部結合シリル基数は0であった。
合成例3-1と同様の手順で、表1に記載の各重合処方に従って各種単量体、連鎖移動剤、ラジカル開始剤、及び溶剤を使用し、(メタ)アクリル酸エステル系重合体(B-2)~(B-20)のイソブチルアルコール溶液を得た。得られた各重合体(B)について、末端結合シリル基数、内部結合シリル基数、数平均分子量、重量平均分子量、及び固形分濃度を表1に示した。
合成例2-1で得られた重合体(A-1)と、合成例3-1で得られた重合体(B-1)のイソブチルアルコール溶液とを、固形分比(質量比)70/30で混合した。得られた混合液をロータリーエバポレーターを用いて110℃に加熱し、減圧条件下でイソブチルアルコールの減圧留去を行い、固形分濃度99%以上で、重合体(A-1)/重合体(B-1)の質量比が70/30である重合体混合物を得た。この重合体混合物の23℃における粘度は、105Pa・sであった。
合成例4-1と同様の手順で、表2に記載の各重合体種類及び混合物組成に従って重合体(A)と重合体(B)から重合体混合物を得た。これら重合体混合物の23℃における粘度を表2に示した。

表3~表10に示した各組成に従って、各合成例で得た重合体混合物と、次に示す各種添加剤のうち充填剤及びタレ防止剤を混合して十分混合した後、3本ペイントロールに3回通して分散させ、主剤を作製した。その後、脱水剤、接着性付与剤、シラノール縮合触媒を添加して十分混合し、自転公転ミキサーを用いて均一に混錬脱泡して、各硬化性組成物を作製した。作製した各硬化性組成物を用い、23℃、相対湿度50%の恒温恒湿雰囲気下にて各種試験体を作製し、各種評価を行った。
全ての実施例及び比較例の組成物物性の評価において、次に示す添加剤を使用した。配合量は、ベースポリマーである各重合体混合物100重量部に対する重量部数である。
充填剤:
(i) 脂肪酸処理沈降炭酸カルシウム(白艶華CCR、白石工業(株)製)、50重量部
(ii) 重質炭酸カルシウム(ホワイトンSB赤、白石カルシウム(株)製)、50重量部
タレ防止剤:脂肪酸アマイドワックス(ディスパロン#6500、楠本化成(株))、0.5重量部
脱水剤:ビニルトリメトキシシラン(A-171、Momentive(株)製)、3重量部
接着性付与剤:3-(N-2-アミノエチルアミノ)プロピルトリメトキシシラン(A-1120、Momentive(株)製)、3重量部
シラノール縮合触媒:以下のいずれか1種(各表に種類と添加量を記載)
(iii) 50%フェニルグアニジン溶液(N-ブチルベンゼンスルホンアミドの50重量パーセント溶液、日本カーバイド(株)製)
(iv) ジオクチル錫ジラウレート(U-810、日東化成(株)製)
(v) ジブチル錫ビス(アセトアセテート)(U-220H、日東化成(株)製)
作製した硬化性組成物の硬化性、及びダンベル物性を下記の方法にて測定した。その結果も、同じく表3~表10に示した。
23℃、相対湿度50%下で、硬化性組成物を厚さ約5mmの型枠にスパチュラを用いて充填し、表面を平面状に整えた時間を硬化開始時間とした。表面をスパチュラで触り、スパチュラに組成物が付着しなくなるまでの時間を皮張り時間として硬化性の測定を行った。
23℃、相対湿度50%下で、硬化性組成物を3mm厚のシート状型枠に充填した。23℃、相対湿度50%下で3日間硬化させた後、50℃乾燥機内で4日間養生し、シート状硬化物を得た。得られた硬化物をJIS K 6251に従って3号ダンベル型に打ち抜き試験片を得た。得られた試験片を用い、23℃、相対湿度50%下で、オートグラフを用いて引張試験(引張速度200mm/分)を行い、50%伸長時応力、100%伸長時応力、破断時応力、及び破断時伸びを測定した。

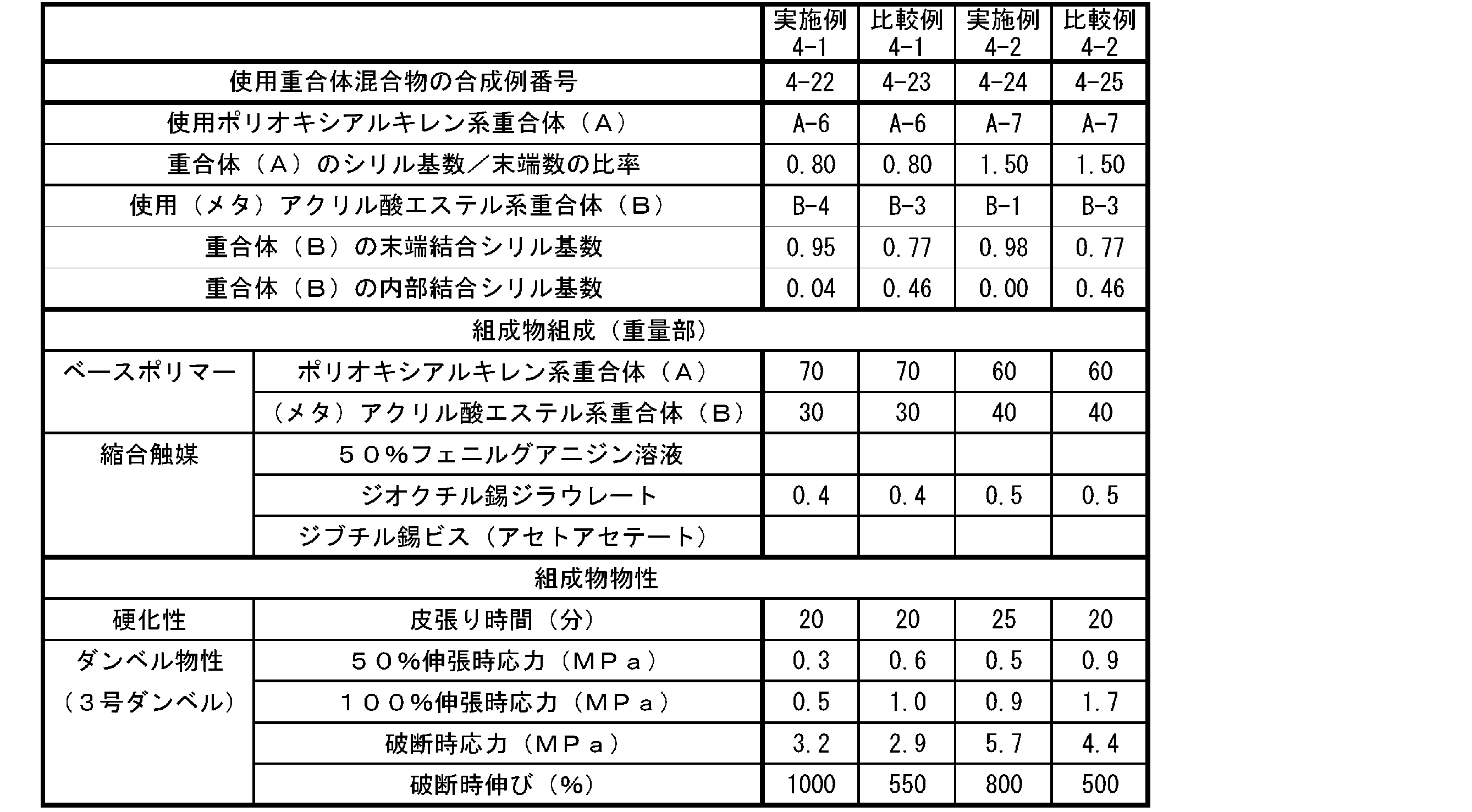



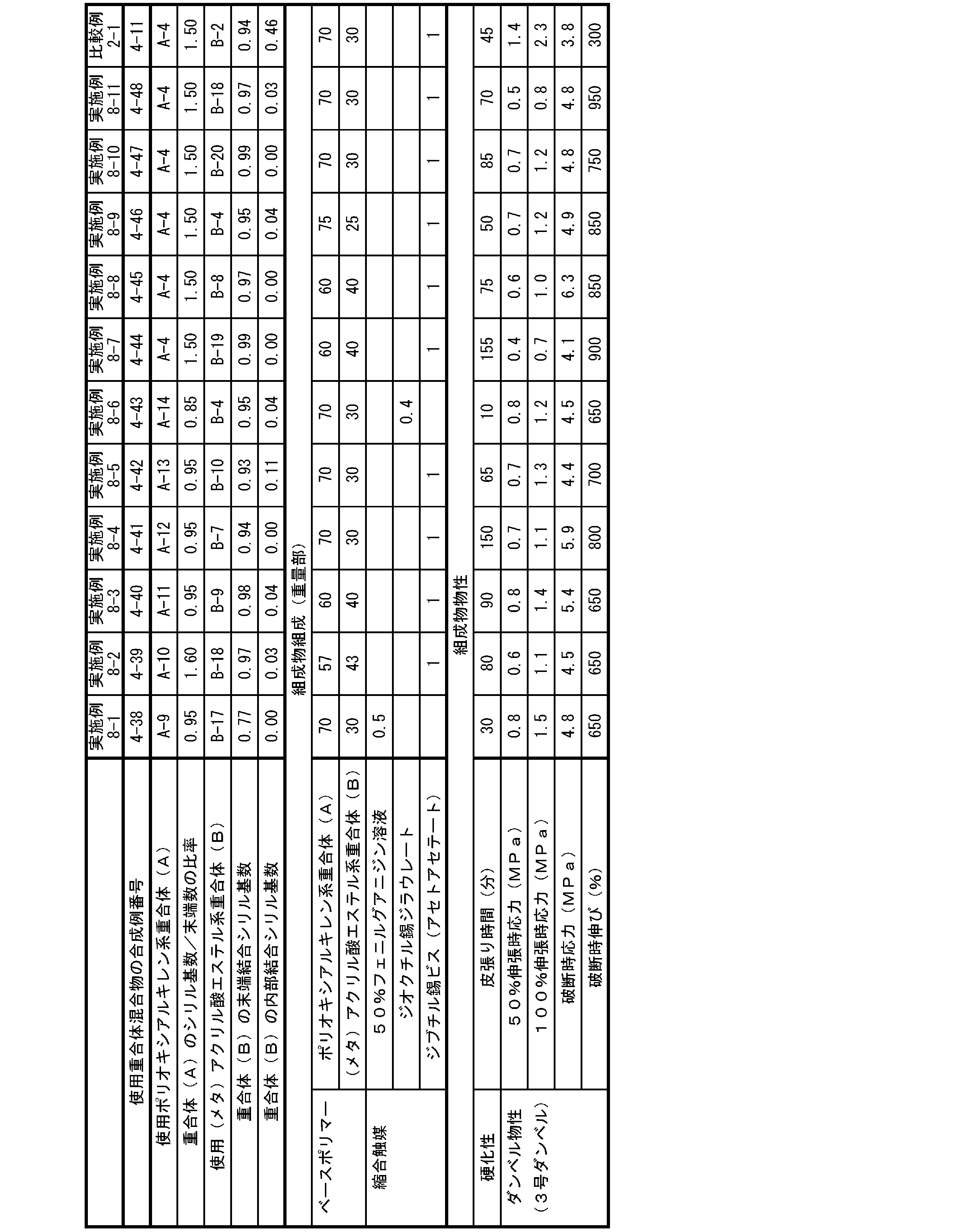
Claims (8)
- 下記一般式(1):
-SiR1 aX3-a (1)
(式中、R1は炭素数1~20の置換または無置換の炭化水素基を表す。Xは水酸基または加水分解性基を表す。aは、0、1または2である。R1又はXが複数存在するとき、それらは同じでもよく、異なっていてもよい。)で表される反応性シリル基を有するポリオキシアルキレン系重合体(A)、及び
下記一般式(2):
-SiR2 bY3-b (2)
(式中、R2は炭素数1~20の無置換の炭化水素基を表す。Yは水酸基または加水分解性基を表す。bは、0、1または2である。R2又はYが複数存在するとき、それらは同じでもよく、異なっていてもよい。)で表される反応性シリル基を有する(メタ)アクリル酸エステル系重合体(B)、を含み、
ポリオキシアルキレン系重合体(A)1分子における重合体骨格の末端の数に対する反応性シリル基の数の平均比率が0.80以上であり、
(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の末端に結合した反応性シリル基の平均個数が0.30以上1.00以下であり、かつ、(メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の内部に結合した反応性シリル基の平均個数が0以上0.30未満である、硬化性組成物。 - ポリオキシアルキレン系重合体(A)1分子における重合体骨格の末端の数に対する反応性シリル基の数の平均比率が0.85以上である、請求項1に記載の硬化性組成物。
- (メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の末端に結合した反応性シリル基の平均個数が0.85以上1.00以下である、請求項1又は2に記載の硬化性組成物。
- (メタ)アクリル酸エステル系重合体(B)1分子において重合体骨格の内部に結合した反応性シリル基の平均個数が0以上0.15未満である、請求項1~3のいずれか1項に記載の硬化性組成物。
- ポリオキシアルキレン系重合体(A):(メタ)アクリル酸エステル系重合体(B)の重量比が80:20~50:50である、請求項1~4のいずれか1項に記載の硬化性組成物。
- (メタ)アクリル酸エステル系重合体(B)の構成単量体単位のうちメタクリル酸メチルが占める割合が、35重量%以上85重量%以下である、請求項1~5のいずれか1項に記載の硬化性組成物。
- (メタ)アクリル酸エステル系重合体(B)の構成単量体単位のうちメタクリル酸アルキルの合計が占める割合が、70重量%以上85重量%以下であり、前記アルキルが炭素数1~4の非置換アルキル基である、請求項1~6のいずれか1項に記載の硬化性組成物。
- 請求項1~7のいずれか1項に記載の硬化性組成物を硬化させた硬化物。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202080022598.8A CN113795547B (zh) | 2019-03-28 | 2020-03-19 | 固化性组合物及固化物 |
| EP20777726.9A EP3950832B1 (en) | 2019-03-28 | 2020-03-19 | Curable composition and cured product |
| JP2021509296A JP7461338B2 (ja) | 2019-03-28 | 2020-03-19 | 硬化性組成物、及び硬化物 |
| US17/481,520 US12152143B2 (en) | 2019-03-28 | 2021-09-22 | Curable composition and cured product |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-062765 | 2019-03-28 | ||
| JP2019062765 | 2019-03-28 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/481,520 Continuation US12152143B2 (en) | 2019-03-28 | 2021-09-22 | Curable composition and cured product |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020196228A1 true WO2020196228A1 (ja) | 2020-10-01 |
Family
ID=72610527
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/012224 Ceased WO2020196228A1 (ja) | 2019-03-28 | 2020-03-19 | 硬化性組成物、及び硬化物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US12152143B2 (ja) |
| EP (1) | EP3950832B1 (ja) |
| JP (1) | JP7461338B2 (ja) |
| CN (1) | CN113795547B (ja) |
| WO (1) | WO2020196228A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022135913A (ja) * | 2021-03-03 | 2022-09-15 | Agc株式会社 | 硬化性組成物及びその硬化物 |
| JP2022143083A (ja) * | 2021-03-17 | 2022-10-03 | 株式会社カネカ | 有機重合体の製造方法、並びに、有機重合体、硬化性組成物、及び硬化物 |
| US12378406B2 (en) * | 2020-03-31 | 2025-08-05 | Kaneka Corporation | Mixture of polyoxyalkylene polymers and curable composition |
| WO2026042692A1 (ja) * | 2024-08-20 | 2026-02-26 | 株式会社カネカ | 硬化性組成物 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023215360A1 (en) * | 2022-05-04 | 2023-11-09 | Dow Silicones Corporation | Silicone-polyether copolymer, sealants comprising same, and related methods |
Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57502171A (ja) | 1982-01-21 | 1982-12-09 | ||
| JPS596207A (ja) | 1982-06-15 | 1984-01-13 | エス・シ−・ジヨンソン・アンド・サン・インコ−ポレ−テツド | バルク重合方法とポリマ−生成物 |
| JPS5978223A (ja) | 1982-10-27 | 1984-05-07 | Kanegafuchi Chem Ind Co Ltd | 重合体の製造方法 |
| JPS59122541A (ja) | 1982-12-28 | 1984-07-16 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPS60228517A (ja) | 1984-04-26 | 1985-11-13 | Kanegafuchi Chem Ind Co Ltd | 新規重合体の製造法 |
| JPS60228516A (ja) | 1984-04-26 | 1985-11-13 | Kanegafuchi Chem Ind Co Ltd | 新規重合体の製造法 |
| JPS63112642A (ja) | 1986-10-29 | 1988-05-17 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH03160053A (ja) | 1989-11-16 | 1991-07-10 | Kanegafuchi Chem Ind Co Ltd | 室温硬化性組成物 |
| JPH04283259A (ja) | 1991-03-11 | 1992-10-08 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH06172631A (ja) | 1992-12-04 | 1994-06-21 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH09194731A (ja) | 1996-01-23 | 1997-07-29 | Asahi Glass Co Ltd | 硬化性組成物 |
| JPH11116763A (ja) | 1997-07-28 | 1999-04-27 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JP2001040037A (ja) | 1999-07-30 | 2001-02-13 | Soken Chem & Eng Co Ltd | 反応性アクリル系重合体、硬化性アクリル系重合体、硬化性組成物、硬化体およびこれらの用途 |
| JP2003313418A (ja) | 2002-04-19 | 2003-11-06 | Kanegafuchi Chem Ind Co Ltd | 硬化性樹脂組成物 |
| WO2004090035A1 (ja) * | 2003-04-10 | 2004-10-21 | Kaneka Corporation | 硬化性組成物 |
| JP2004346146A (ja) * | 2003-05-21 | 2004-12-09 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JP2006274094A (ja) * | 2005-03-30 | 2006-10-12 | Kaneka Corp | 放熱シート用組成物およびそれを硬化させてなる放熱シート |
| WO2012020560A1 (ja) | 2010-08-10 | 2012-02-16 | 株式会社カネカ | 硬化性組成物 |
| JP2012214755A (ja) * | 2011-03-31 | 2012-11-08 | Kaneka Corp | 硬化性組成物 |
| WO2014024963A1 (ja) * | 2012-08-10 | 2014-02-13 | 株式会社カネカ | 湿分硬化性組成物 |
| JP2014114434A (ja) | 2012-11-14 | 2014-06-26 | Kaneka Corp | 硬化性組成物 |
| WO2016002907A1 (ja) * | 2014-07-02 | 2016-01-07 | 株式会社カネカ | 硬化性組成物およびその硬化物 |
| WO2016035718A1 (ja) * | 2014-09-01 | 2016-03-10 | 株式会社カネカ | 硬化性組成物 |
| JP2017066349A (ja) | 2015-10-02 | 2017-04-06 | 株式会社カネカ | 硬化性組成物 |
| JP2017125098A (ja) * | 2016-01-12 | 2017-07-20 | セメダイン株式会社 | 光硬化性接着剤の硬化方法、硬化物、光硬化性接着剤、及び製品 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5355848B2 (ja) | 2006-06-23 | 2013-11-27 | 積水フーラー株式会社 | 硬化性組成物、シーリング材および接着剤 |
| WO2009133811A1 (ja) * | 2008-05-02 | 2009-11-05 | 株式会社カネカ | 室温硬化性組成物およびその硬化物 |
| WO2011152002A1 (ja) | 2010-06-03 | 2011-12-08 | 株式会社カネカ | 湿気硬化型反応性ホットメルト接着剤組成物 |
| WO2012057092A1 (ja) * | 2010-10-27 | 2012-05-03 | 株式会社カネカ | 硬化性組成物 |
| US9505879B2 (en) * | 2012-05-31 | 2016-11-29 | Kaneka Corporation | Polymer having terminal structure including plurality of reactive silicon groups, method for manufacturing same, and use for same |
| WO2014192914A1 (ja) * | 2013-05-30 | 2014-12-04 | 株式会社カネカ | 硬化性組成物およびその硬化物 |
| CN105246979B (zh) * | 2013-05-30 | 2018-12-14 | 株式会社钟化 | 固化性组合物 |
| JP6527589B2 (ja) * | 2015-10-02 | 2019-06-05 | 株式会社カネカ | 硬化性組成物 |
-
2020
- 2020-03-19 CN CN202080022598.8A patent/CN113795547B/zh active Active
- 2020-03-19 EP EP20777726.9A patent/EP3950832B1/en active Active
- 2020-03-19 WO PCT/JP2020/012224 patent/WO2020196228A1/ja not_active Ceased
- 2020-03-19 JP JP2021509296A patent/JP7461338B2/ja active Active
-
2021
- 2021-09-22 US US17/481,520 patent/US12152143B2/en active Active
Patent Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57502171A (ja) | 1982-01-21 | 1982-12-09 | ||
| JPS596207A (ja) | 1982-06-15 | 1984-01-13 | エス・シ−・ジヨンソン・アンド・サン・インコ−ポレ−テツド | バルク重合方法とポリマ−生成物 |
| JPS5978223A (ja) | 1982-10-27 | 1984-05-07 | Kanegafuchi Chem Ind Co Ltd | 重合体の製造方法 |
| JPS59122541A (ja) | 1982-12-28 | 1984-07-16 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPS60228517A (ja) | 1984-04-26 | 1985-11-13 | Kanegafuchi Chem Ind Co Ltd | 新規重合体の製造法 |
| JPS60228516A (ja) | 1984-04-26 | 1985-11-13 | Kanegafuchi Chem Ind Co Ltd | 新規重合体の製造法 |
| JPS63112642A (ja) | 1986-10-29 | 1988-05-17 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH03160053A (ja) | 1989-11-16 | 1991-07-10 | Kanegafuchi Chem Ind Co Ltd | 室温硬化性組成物 |
| JPH04283259A (ja) | 1991-03-11 | 1992-10-08 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH06172631A (ja) | 1992-12-04 | 1994-06-21 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JPH09194731A (ja) | 1996-01-23 | 1997-07-29 | Asahi Glass Co Ltd | 硬化性組成物 |
| JPH11116763A (ja) | 1997-07-28 | 1999-04-27 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JP2001040037A (ja) | 1999-07-30 | 2001-02-13 | Soken Chem & Eng Co Ltd | 反応性アクリル系重合体、硬化性アクリル系重合体、硬化性組成物、硬化体およびこれらの用途 |
| JP2003313418A (ja) | 2002-04-19 | 2003-11-06 | Kanegafuchi Chem Ind Co Ltd | 硬化性樹脂組成物 |
| WO2004090035A1 (ja) * | 2003-04-10 | 2004-10-21 | Kaneka Corporation | 硬化性組成物 |
| JP2004346146A (ja) * | 2003-05-21 | 2004-12-09 | Kanegafuchi Chem Ind Co Ltd | 硬化性組成物 |
| JP2006274094A (ja) * | 2005-03-30 | 2006-10-12 | Kaneka Corp | 放熱シート用組成物およびそれを硬化させてなる放熱シート |
| WO2012020560A1 (ja) | 2010-08-10 | 2012-02-16 | 株式会社カネカ | 硬化性組成物 |
| JP2012214755A (ja) * | 2011-03-31 | 2012-11-08 | Kaneka Corp | 硬化性組成物 |
| WO2014024963A1 (ja) * | 2012-08-10 | 2014-02-13 | 株式会社カネカ | 湿分硬化性組成物 |
| JP2014114434A (ja) | 2012-11-14 | 2014-06-26 | Kaneka Corp | 硬化性組成物 |
| WO2016002907A1 (ja) * | 2014-07-02 | 2016-01-07 | 株式会社カネカ | 硬化性組成物およびその硬化物 |
| WO2016035718A1 (ja) * | 2014-09-01 | 2016-03-10 | 株式会社カネカ | 硬化性組成物 |
| JP2017066349A (ja) | 2015-10-02 | 2017-04-06 | 株式会社カネカ | 硬化性組成物 |
| JP2017125098A (ja) * | 2016-01-12 | 2017-07-20 | セメダイン株式会社 | 光硬化性接着剤の硬化方法、硬化物、光硬化性接着剤、及び製品 |
Non-Patent Citations (2)
| Title |
|---|
| SAUNDERSFRISCH: "Polyurethanes: Chemistry and Technology", INTERSCIENCE PUBLISHERS, pages: 1963 |
| See also references of EP3950832A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12378406B2 (en) * | 2020-03-31 | 2025-08-05 | Kaneka Corporation | Mixture of polyoxyalkylene polymers and curable composition |
| JP2022135913A (ja) * | 2021-03-03 | 2022-09-15 | Agc株式会社 | 硬化性組成物及びその硬化物 |
| JP7722155B2 (ja) | 2021-03-03 | 2025-08-13 | Agc株式会社 | 硬化性組成物及びその硬化物 |
| JP2022143083A (ja) * | 2021-03-17 | 2022-10-03 | 株式会社カネカ | 有機重合体の製造方法、並びに、有機重合体、硬化性組成物、及び硬化物 |
| WO2026042692A1 (ja) * | 2024-08-20 | 2026-02-26 | 株式会社カネカ | 硬化性組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3950832A1 (en) | 2022-02-09 |
| CN113795547A (zh) | 2021-12-14 |
| JP7461338B2 (ja) | 2024-04-03 |
| EP3950832A4 (en) | 2022-12-07 |
| EP3950832B1 (en) | 2025-10-22 |
| US12152143B2 (en) | 2024-11-26 |
| CN113795547B (zh) | 2023-06-23 |
| US20220002541A1 (en) | 2022-01-06 |
| JPWO2020196228A1 (ja) | 2020-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105246979B (zh) | 固化性组合物 | |
| JP6527589B2 (ja) | 硬化性組成物 | |
| JP6088790B2 (ja) | 硬化性組成物およびその硬化物 | |
| JP7461338B2 (ja) | 硬化性組成物、及び硬化物 | |
| JP6682227B2 (ja) | 硬化性組成物 | |
| US12234322B2 (en) | Polyoxyalkylene polymer and curable composition | |
| CN104350084A (zh) | 具有有多个反应性硅基的末端结构的聚合物及其制造方法和利用 | |
| WO2015133564A1 (ja) | 硬化性組成物 | |
| JP2014234396A (ja) | 室温硬化性組成物およびその硬化物 | |
| JP2012214755A (ja) | 硬化性組成物 | |
| JP2014001358A (ja) | 硬化性組成物 | |
| WO2022163562A1 (ja) | ポリオキシアルキレン系重合体の混合物及び硬化性組成物 | |
| WO2022163563A1 (ja) | ポリオキシアルキレン系重合体及びその混合物 | |
| US12378406B2 (en) | Mixture of polyoxyalkylene polymers and curable composition | |
| JP7356247B2 (ja) | 硬化性組成物、及び硬化物 | |
| JP2021055012A (ja) | 硬化性組成物 | |
| JP2022049666A (ja) | 硬化性組成物 | |
| JP2023138378A (ja) | 硬化性組成物 | |
| WO2024224831A1 (ja) | 結晶性ポリオキシアルキレン系重合体およびそれを含む硬化性組成物 | |
| WO2023171425A1 (ja) | ポリオキシアルキレン系重合体の混合物および硬化性組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20777726 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021509296 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020777726 Country of ref document: EP Effective date: 20211028 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2020777726 Country of ref document: EP |