WO2021164538A1 - 一种多靶点酪氨酸激酶抑制剂 - Google Patents
一种多靶点酪氨酸激酶抑制剂 Download PDFInfo
- Publication number
- WO2021164538A1 WO2021164538A1 PCT/CN2021/074809 CN2021074809W WO2021164538A1 WO 2021164538 A1 WO2021164538 A1 WO 2021164538A1 CN 2021074809 W CN2021074809 W CN 2021074809W WO 2021164538 A1 WO2021164538 A1 WO 2021164538A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cancer
- hydrogen
- ethyl
- following groups
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- ZJUXJQSYXBYFFO-UHFFFAOYSA-N CN1CCN(Cc(cc2)ccc2C(O)=O)CC1 Chemical compound CN1CCN(Cc(cc2)ccc2C(O)=O)CC1 ZJUXJQSYXBYFFO-UHFFFAOYSA-N 0.000 description 1
- QQPSMIFQTBSALU-UHFFFAOYSA-N CN1CCN(Cc(cc2)ccc2C(OCC[n]2ncc(-c3cc(nccc4Oc(ccc(NC(C5(CC5)C(Nc(cc5)ccc5F)=O)=O)c5)c5F)c4[s]3)c2)=O)CC1 Chemical compound CN1CCN(Cc(cc2)ccc2C(OCC[n]2ncc(-c3cc(nccc4Oc(ccc(NC(C5(CC5)C(Nc(cc5)ccc5F)=O)=O)c5)c5F)c4[s]3)c2)=O)CC1 QQPSMIFQTBSALU-UHFFFAOYSA-N 0.000 description 1
- NWPRHHFLSFLQDM-UHFFFAOYSA-N OCC[n]1ncc(-c2cc3nccc(Oc(ccc(NC(C4(CC4)C(Nc(cc4)ccc4F)=O)=O)c4)c4F)c3[s]2)c1 Chemical compound OCC[n]1ncc(-c2cc3nccc(Oc(ccc(NC(C4(CC4)C(Nc(cc4)ccc4F)=O)=O)c4)c4F)c3[s]2)c1 NWPRHHFLSFLQDM-UHFFFAOYSA-N 0.000 description 1
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system having sulfur as a ring hetero atom, e.g. ticlopidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- the invention belongs to the technical field of medicine, and specifically relates to a multi-target tyrosine kinase inhibitor with 2-substituted pyrazolylpyrido[3,2-b]thiophene as the core and its use in the medical field application.
- HGF hepatocyte growth factor
- c-Met The receptor tyrosine kinase c-Met is the only known receptor for hepatocyte growth factor (HGF), and the HGF/c-Met pathway plays an important role in the process of embryo formation. HGF and c-Met are abnormally expressed in a variety of tumors. The excessive activation of c-Met can promote tumor cell growth and survival, the formation of new vascular system, and invasion and metastasis.
- HGF and/or Met abnormal expression of HGF and/or Met has been found in many patients with different tumors, including glioma, melanoma, hepatocellular carcinoma, lung cancer, pancreatic cancer, prostate cancer, ovarian cancer, breast cancer, Gastric cancer, renal cell carcinoma, etc., and usually accompanied by a poor prognosis.
- the formation of the new vascular system is considered to be an important step in the occurrence and development of tumors.
- many drugs target the angiogenic factor VEGF or its receptor VEGFR, and inhibit tumor growth by inhibiting the angiogenesis of tumor cells, such as bevacizumab and sedative.
- Nitinib, sorafenib and other drugs have the above-mentioned pharmacological effects.
- the anti-cancer effect of this type of drug has been significantly reduced after several weeks of its efficacy.
- Mechanism studies have found that the hypoxia-inducible factor HIF- ⁇ induces high expression of Met, leading to tolerance to the inhibition of the VEGF/VEGFR pathway.
- VEGFR inhibitors such as sunitinib
- VEGF monoclonal antibodies can increase the invasion and migration ability of tumor cells, and these tumor cells are accompanied by higher levels of Met expression. Therefore, simultaneous inhibition of VEGFR and c-Met may inhibit tumors better, and also reduce tumor cell invasion and metastasis.
- c-kit kinase is also an important anti-tumor therapeutic target.
- About 80% of gastrointestinal stromal tumors (GIST) have c-kit activating mutations.
- GIST gastrointestinal stromal tumors
- c-kit mutations are also accompanied.
- Chinese patent application CN108530464A involves a new compound td32-4(N-3-fluoro-4-((2-(1-(2-hydroxyethyl)-1H-pyrazol-4-yl)thieno[ 3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dimethylamide), which has a positive effect on VEGFR/c-Met/c-
- the compound shows good biological activity, the extremely poor solubility of td32-4 limits the further development of the compound as a good targeted antitumor drug. It is mainly reflected in the following aspects: 1
- 2 The difference in the absorption of poorly soluble drugs between individuals It is huge, and there are greater risks and difficulties in the preclinical/clinical safety evaluation of the drug; 3
- 3 The absorption of the drug is saturated when the drug is administered in a large dose, no matter how to increase the dose, the blood drug concentration still cannot be increased.
- the present invention modifies the structure of td32-4 and proposes a series of compounds represented by the following formula (I), which can not only solve the solubility problem of td32-4, but also maintain its potent anti-tumor activity in vivo.
- formula (I) a series of compounds represented by the following formula (I)
- the present invention relates to a compound or a pharmaceutically acceptable salt, isomer, or racemate thereof, characterized in that the compound has a structure represented by the general formula (I):
- R1 is optionally selected from the following substituents: C3-C8 alkyl acyl substituted by carboxyl, substituted or unsubstituted phosphono,
- R2 is optionally selected from the following groups: hydrogen, halogen, C1-C8 alkyl substituted with R5, C6-C12 aryl substituted with R5;
- R3 and R4 are optionally selected from the following groups: hydrogen, C1-C6 alkyl substituted by R5, or R3 and R4 constitute a five- to twelve-membered aliphatic heterocyclic ring;
- R5 is optionally selected from the following groups: hydrogen, 1-3 halogens, hydroxyl, C1-C6 alkoxy, C6-C12 aryl;
- R1 can be selected from 3-carboxypropionyl, 4-carboxybutyryl, 5-carboxyvaleryl, 6-carboxyhexanoyl,
- R2 is optionally selected from the following groups: hydrogen, halogen, C1-C8 alkyl substituted with R5, C6-C12 aryl substituted with R5;
- R3 and R4 are optionally selected from the following groups: hydrogen, C1-C6 alkyl substituted by R5, or R3 and R4 constitute a five- to twelve-membered aliphatic heterocyclic ring;
- R5 is optionally selected from the following groups: hydrogen, 1-3 halogens, hydroxyl, C1-C6 alkoxy, C6-C12 aryl; further, when Hour,
- R2 is optionally selected from the following groups: hydrogen, fluorine, chlorine, bromine, methyl, -CH2F, -CHF2, -CF3, ethyl, propyl, isopropyl, cyclopropyl, butyl, isobutyl, benzyl Group, 4-hydroxybenzyl, phenyl, 4-fluorophenyl;
- R3 and R4 are optionally selected from the following groups: hydrogen, methyl, ethyl, 2,2-dimethoxyethyl, propyl, butyl, or R3 and R4 constitute morpholine, pyridine, pyrrole, piperidine, Piperidinylpiperidine, N-methylpiperidine;
- R2 is preferably selected from the following groups: hydrogen, methyl, -CH2F, -CHF2, -CF3, ethyl, propyl, isopropyl, isobutyl, benzyl, 4-hydroxybenzyl, phenyl;
- R3 and R4 are preferably selected from the following groups: hydrogen, methyl, ethyl, 2,2-dimethoxyethyl, or R3 and R4 constitute morpholine or N-methylpiperidine;
- R3 and R4 are optionally selected from the following groups: hydrogen, methyl, ethyl, 2,2-dimethoxyethyl, propyl, butyl, or R3 and R4 constitute morpholine, pyridine, pyrrole, piperidine, Piperidinyl piperidine, N-methyl piperidine.
- Another object of the present invention is to provide a pharmaceutical composition containing the compound of general formula (I) or a pharmaceutically acceptable salt, isomer, or racemate thereof as an active ingredient.
- Another object of the present invention is to provide an application of a compound containing the general formula (I) or a pharmaceutically acceptable salt, isomer, or racemate thereof in the preparation of a drug for treating tumors, the tumors comprising : Cervical cancer, seminoma, testicular lymphoma, prostate cancer, ovarian cancer, lung cancer, rectal cancer, breast cancer, skin squamous cell carcinoma, colon cancer, liver cancer, pancreatic cancer, stomach cancer, esophageal cancer, thyroid cancer, and / Or bladder transitional epithelial cancer, leukemia.
- the compound provided by the present invention obtains significantly improved solubility by modifying the structure of td32-4, and overcomes the existing problems of td32-4 in pharmacokinetic research, safety evaluation, analytical method development, formulation research, etc. difficulty.
- due to the extremely poor water solubility of td32-4 its oral bioavailability in rat pharmacokinetic studies is extremely poor, which limits the further development of this compound.
- the compound of the present invention has been verified by experiments that after oral administration into the body, it can be converted into td32-4 to exert pharmacological effects. Compared with direct oral administration of td32-4, the oral bioavailability is greatly improved, especially at high doses (200mg ⁇ kg -1 ) at the time of administration. It has been verified that the compound of the present invention has a strong tumor inhibitory effect when administered orally in animals, while td32-4 has no obvious effect on tumor suppression when the same dose is directly administered orally.
- Figure 1 The plasma concentration-time curve of td32-4 administered by intravenous injection of 5 mg ⁇ kg -1 and oral administration of 10 mg ⁇ kg -1 and 200 mg ⁇ kg -1.
- FIG. 2 The plasma concentration-time curve of td32-4P3 and the td32-4 and td32-4 transformed into the body by direct oral administration, the doses are both 10mg ⁇ kg -1 .
- FIG. 3 The plasma concentration-time curve of td32-4P3 and td32-4 and td32-4 transformed into td32-4 and td32-4 directly orally.
- the doses are both 200mg ⁇ kg -1 .
- Figure 4 The plasma concentration-time curve of td32-4P10 and the td32-4 and td32-4 transformed in vivo by direct oral administration, the doses are both 10mg ⁇ kg -1 .
- Figure 5 The plasma concentration-time curve of td32-4P10 and the td32-4 and td32-4 transformed in vivo by direct oral administration, the doses are both 200mg ⁇ kg -1 .
- FIG. 7 Tumor suppressive activity data of td32-4P10 in nude mouse MV-4-11 mouse xenograft model (50mpk dose).
- Figure 8 The tumor suppressor activity data of td32-4P10 in the nude mouse H358 mouse xenograft model (50mpk dose).
- FIG. 9 Tumor inhibitory activity data of td32-4P10 in a nude mouse H82 mouse xenograft model (50mpk dose).
- the reagent materials of the present invention are all commercially available products.
- Td32-4 (chemical name: N-3-fluoro-4-((2-(1-(2-hydroxyethyl)-1H-pyrazol-4-yl)thieno[3,2-b]pyridine -7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dimethylamide) (5g, 8.7mmol), 4-DMAP (0.21g, 1.7mmol), Succinic anhydride (4.34g, 43.5mmol) was dissolved in 50mL DMF, and the reaction was heated at 100°C for 8 hours.
- the reaction solution was poured into 500 mL of water, stirred to precipitate solids, filtered with suction, and the filter cake was washed with 200 mL of water. After the filter cake is dried, heat and stir the product with 20% (w%) sodium carbonate aqueous solution to gradually dissolve, add 5% activated carbon and reflux for 1 hour, suction and filter while hot, and adjust the pH of the filtrate to 1 ⁇ 2 with 2mol/L dilute hydrochloric acid , A large amount of white solid precipitated, filtered with suction and dried in an oven at 55°C to obtain 4.82 g of white solid, yield: 82.1%, HPLC purity: 98.0%, LC-MS: 675.4 [MH] - .
- td32-4 (5g, 8.7mmol) and triethylamine (1.05g, 10.4mmol) were added to 60mL of tetrahydrofuran, cooled to 0°C, and the tetrahydrofuran solution of phosphorus oxychloride was added dropwise.
- the reaction system was at 0°C. ⁇ 5°C and continue to stir and react for 3 hours.
- the above reaction liquid was added dropwise to 100 mL of ice water, and a solid was precipitated with stirring. Suction filtration, wash the filter cake with water until the filtrate becomes neutral, and dry the sample.
- N,N-dimethylglycine (1.79g, 17.4mmol)
- 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (abbreviation EDCI, 4g , 20.9mmol)
- 4-dimethylaminopyridine (abbreviated DMAP, 1.59g, 13.0mmol) was added to 60mL of anhydrous tetrahydrofuran, after 30 minutes of reaction, td32-4 (5g, 8.7mmol) was added to the reaction system, transfer To room temperature, react for 24 hours.
- reaction solution was filtered, 200mL 1mol/L of dilute hydrochloric acid solution was added to the filtrate, stirred, filtered by suction, the filtrate was adjusted to pH 10 with saturated sodium carbonate solution, a large amount of white solid was precipitated, filtered by suction, the filter cake was washed with 100mL of water, and the filter cake was dried Got crude.
- the crude product was dissolved in 100 mL of 2mol/L dilute hydrochloric acid, filtered to remove insolubles, the filtrate was adjusted to pH 10 with saturated sodium carbonate, a large amount of white solid was precipitated, filtered with suction, the filter cake was washed with 100 mL of water, and the filter cake was dried.
- the solid was dissolved in 80 mL of anhydrous acetone, 2 mL of concentrated hydrochloric acid was added, and the mixture was stirred overnight at room temperature.
- the product hydrochloride was precipitated and filtered with suction.
- the filter cake was washed with 50 mL of anhydrous acetone, and the filter cake was dried in an oven at 55°C. Obtained 3.75 g of white solid, yield: 65.3%, HPLC purity: 98.8%, LC-MS: 661.3 [M+H] + .
- Boc-glycine (3.04g, 17.4mmol), 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (abbreviation EDCI, 4g, 20.9mmol), 4-Dimethylaminopyridine (abbreviation DMAP, 1.59g, 13.0mmol) was added to 60mL of anhydrous tetrahydrofuran, after 30 minutes of reaction, td32-4 (5g, 8.7mmol) was added to the reaction system, transferred to room temperature, reaction 24 Hour.
- EDCI 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride
- DMAP 4-Dimethylaminopyridine
- reaction solution was filtered, 200mL 1mol/L dilute hydrochloric acid solution was added to the filtrate, a white solid was precipitated out with stirring, filtered with suction, and the solid was dried.
- A is very easy to dissolve 1g of solute can be dissolved in less than 1mL of solvent
- B is soluble 1g of solute can be dissolved in 1 ⁇ 10mL
- C dissolves 1g of solute can be dissolved in 10 ⁇ 30mL
- D is slightly soluble 1g of solute can be dissolved in 30 ⁇ 100mL
- E slightly soluble 1g of solute can be dissolved in 100 ⁇ 1000mL
- F very slightly soluble 1g of solute can be dissolved in 1000 ⁇ 10000mL
- G is almost insoluble or insoluble 1g solute does not dissolve in 10000mL solvent
- the compound of the present invention introduces a hydrophilic functional group into the td32-4 structure through an ester bond to increase the solubility of the compound. After being taken orally into the body, it needs to be transformed into td32-4 under the physical and chemical environment of the body to exert its pharmacological effects.
- some representative compounds were selected to carry out in vivo pharmacokinetic experiments in SD rats and compared with direct oral administration of td32-4. The experiment was conducted with high (200mg ⁇ kg -1 ) and low (10 mg ⁇ kg -1 ) doses.
- DMA dimethylacetamide
- test product weigh an appropriate amount of the test product into a glass bottle. Add PEG400, vortex and mix. Ultrasound to get a clear solution.
- the SD rats were weighed, and the dosage was calculated based on the body weight.
- the plasma concentration data of 10 mg ⁇ kg -1 and 200 mg ⁇ kg -1 are as follows. By calculation, it can be concluded that at low concentrations, the oral bioavailability of compound td32-4 reaches 83.96%, but it drops sharply at high concentrations , Only 18.6%. It is likely that the compound accumulates and precipitates in the gastrointestinal tract at high concentrations, resulting in the inability to be absorbed into the blood circulation. The results are shown in Figure 1.
- td32-4P10 when td32-4P10 is orally administered at a dose of 200 mg ⁇ kg -1 , it can be converted into td32-4 in the body, and the exposure of the converted td32-4 (the area under the drug-time curve AUC) is higher than High-dose oral td32-4.
- the compound in the application of the present invention can exhibit significantly better pharmacokinetic properties than td32-4 in vivo, especially in large doses. (200mg ⁇ kg -1 ), its oral bioavailability is greatly improved, and the blood concentration of td32-4 is increased.
- the compound of the present invention can solve the problem of poor medicinal properties of td32-4 compounds to a certain extent.
- the animal model can be selected as a gastric cancer cell MKN45 xenograft model.
- Test method 5 ⁇ 10 6 MKN-45 gastric cancer cells were inoculated under the fat pad of the right rib of nude mice, and tumor formation was observed. Two weeks after the inoculation, the tumor-bearing mice were divided into groups, and td32-4 and td32-4 were administered respectively. td32-4P3, td32-4P6, td32-4P9, td32-4P10, orally administered, the dosage is 100mg ⁇ kg -1 , once a day for 14 days. Measure and record the tumor volume with vernier calipers.
- the present invention modifies the structure of the compound td32-4 and introduces hydrophilic structural fragments, which can significantly improve the water solubility of the compound.
- the compound can be converted into td32-4 after oral absorption and exert anti-tumor activity. After verification of pharmacokinetic experiments, it can overcome the inherent shortcoming of poor oral absorption of td32-4. It exhibits potent tumor suppressor activity in tumor-bearing mice and is expected to be developed as a new multi-target anti-tumor drug.
- the compound td32-4P10 was selected for in vivo activity evaluation, and the tumor suppressor activity data of td32-4P10 in nude mice MV-4-11, H358 and H82 mouse xenograft models were tested respectively.
- the test method is the same as above, and the dosage is shown in Table 3.
- the tumor volume change was measured at the same time; during the administration period, the drug caused the weight change of the nude mice.
- the tumor weight was counted, and the anatomical image of the tumor after the end of the administration was recorded.
- the test results are shown in Table 3, Figures 7, 8 and 9.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
本发明涉及一种多靶点酪氨酸激酶抑制剂,其为具有式(I)所示的蛋白激酶抑制活性的化合物,所述化合物体内代谢转化成抑制肿瘤生长的活性小分子td32-4,从而发挥抗肿瘤作用。与td32-4相比,本发明化合物具有显著提高的溶解度和口服生物利用度,在动物体内实验中表现出强效抗肿瘤活性。本发明涉及式(I)化合物的合成方法与其在制药领域的应用。
Description
本发明属于医药技术领域,具体地说,涉及一种以2-取代吡唑基吡啶并[3,2-b]噻吩为母核的多靶点酪氨酸激酶抑制剂及其在医药领域的应用。
细胞内许多重要的生命活动都与蛋白质的磷酸化密切相关,例如细胞的增殖、分化与凋亡。在肿瘤细胞中广泛存在蛋白激酶活性异常的现象,随着医药科技的进步,越来越多的蛋白激酶已经成为行之有效的药物靶点,激酶抑制剂药物成为肿瘤靶向治疗领域研究不可或缺的策略。
受体酪氨酸激酶c-Met是肝细胞生长因子(HGF)的唯一已知受体,HGF/c-Met通路在胚胎形成过程中具有重要作用。HGF、c-Met在多种肿瘤中均发现异常表达现象,c-Met的过度激活能促进肿瘤细胞生长和存活,新血管系统的生成,以及侵袭和转移。
临床上,在许多不同肿瘤病人体内均发现了HGF和(或)Met的异常表达现象,包括神经胶质瘤、黑色素瘤、肝细胞癌、肺癌、胰腺癌、前列腺癌、卵巢癌、乳腺癌、胃癌、肾细胞癌等,而且通常伴随着预后不良。
新血管系统的生成被认为是肿瘤发生发展的重要步骤,目前有许多药物靶向血管生成因子VEGF或其受体VEGFR,通过抑制肿瘤细胞的血管生成来抑制肿瘤生长,例如贝伐单抗、舒尼替尼、索拉菲尼等药物均有上述药理作用。但是临床上发现,这类药物在发挥药效数周后,抗癌效果明显下降。机制研究发现,低氧诱导因子HIF-α诱导Met的高表达,导致机体对VEGF/VEGFR通路的抑制产生耐受。最新研究表明,一些VEGFR抑制剂(如舒尼替尼)以及VEGF单抗,能够增加肿瘤细胞的侵袭和迁移能力,而这些肿瘤细胞中伴随着更高水平的Met表达。因此,同时抑制VEGFR和c-Met可能对肿瘤的抑制更好,也会减少肿瘤细胞侵袭和转移的发生。
c-kit激酶也是一个重要的抗肿瘤治疗靶标,在胃肠道间质瘤(GIST)中,约有80%存在c-kit激活型突变。在黑色素瘤、系统性肥大细胞增多症、急性髓性细胞白血病中,也伴随c-kit的突变。
中国专利申请CN108530464A中涉及一种新的化合物td32-4(N-3-氟-4-((2-(1-(2-羟乙基)-1H-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-N-(4-氟苯基)环丙烷-1,1-二甲酰胺),其对VEGFR/c-Met/c-Kit三个靶点表现出强效抑制活性,同时在多个肿瘤细胞模型中表现出强效的抗增殖活性。在低剂量(10mg·kg
-1)给药剂量下,表现出良好的口服生物利用度(F=71.6%)。尽管该化合物表现出良好 的生物活性,但是td32-4极差的溶解性限制了该化合物被进一步开发为良好的靶向抗肿瘤药。主要体现在以下几个方面:①化合物水溶性极差,小剂量时可以溶液形式给药,能够达到较好的吸收,但是在治疗剂量增大(>60mg·kg
-1)时,以固体制剂/混悬溶液形式给药,药物在动物/人体内无法有效释放,血药浓度极低,口服生物利用度很差(F=3%~15%);②难溶性药物在个体间的吸收差异巨大,在进行药物的临床前/临床安全性评价时存在较大风险与困难;③大剂量给药时,药物具有吸收饱和现象,无论如何提高给药量,血药浓度仍然无法提升。
为解决上述问题,本发明对td32-4进行结构修饰,提出以下列式(I)为代表的系列化合物,既能够解决td32-4的溶解度难题,又保持其强效的体内抗肿瘤活性,有望开发成为新一代VEGFR/c-Met/c-Kit多靶点激酶抑制剂药物。

发明内容
本发明涉及一种化合物或其药学可接受的盐、异构体、消旋体,其特征在于所述化合物具有通式(I)所示的结构:

其中,
R1任选自以下取代基:羧基取代的C3~C8烷基酰基、取代或未取代的膦酰基、

R2任选自以下基团:氢、卤素、R5取代的C1~C8烷基、R5取代的C6~C12芳基;
R3、R4任选自以下基团:氢、R5取代的C1~C6烷基,或R3、R4构成五元~十二元脂肪杂环;
R5任选自以下基团:氢、1~3个卤素、羟基、C1~C6烷氧基、C6~C12芳基;
进一步地,R1可选自3-羧基丙酰基、4-羧酸丁酰基、5-羧基戊酰基、6-羧基己 酰基、

再进一步地,当
 时,
时,
R2任选自以下基团:氢、卤素、R5取代的C1~C8烷基、R5取代的C6~C12芳基;
R3、R4任选自以下基团:氢、R5取代的C1~C6烷基,或R3、R4构成五元~十二元脂肪杂环;
R5任选自以下基团:氢、1~3个卤素、羟基、C1~C6烷氧基、C6~C12芳基;进一步地,当
 时,
时,
R2任选自以下基团:氢、氟、氯、溴、甲基、-CH2F、-CHF2、-CF3、乙基、丙基、异丙基、环丙基、丁基、异丁基、苄基、4-羟基苄基、苯基、4-氟苯基;
R3、R4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或R3、R4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶;
进一步地,
 时,
时,
R2优选自以下基团:氢、甲基、-CH2F、-CHF2、-CF3、乙基、丙基、异丙基、异丁基、苄基、4-羟基苄基、苯基;
R3、R4优选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基,或R3、R4构成吗啉、N-甲基哌啶;
当
 时,
时,
R3、R4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或R3、R4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶。
在本发明的具体实施方式中,优选的具有式I的化合物如下:



本发明的另一个目的是提供一种含有通式(I)所述化合物或其药学上可接受的盐、异构体、消旋体,作为活性成分制备的药物组合物。
本发明的又一目的是提供一种含有通式(I)所述化合物或其药学上可接受的盐、异构体、消旋体在用于制备治疗肿瘤药物中的应用,所述肿瘤包括:宫颈癌、精原细胞瘤、睾丸淋巴瘤、前列腺癌、卵巢癌、肺癌、直肠癌、乳腺癌、皮肤鳞状细胞癌、结肠癌、肝癌、胰腺癌、胃癌、食管癌、甲状腺癌、和/或膀胱移行上皮癌、白血病。
本发明提供的化合物通过对td32-4的结构进行修饰,获得明显提高的溶解度,克服了对td32-4进行药代动力学研究、安全性评价、分析方法的开发、制剂处方的研究等存在的困难。除此之外,由于td32-4水溶性极差,其在大鼠药代动力学研究中表现出的口服生物利用度极差,限制了该化合物的进一步开发。本发明的化合物通过实验验证,在口服进入体内后,可转化为td32-4发挥药理作用,同时和直接口服td32-4相比,大大提高了口服生物利用度,尤其是在高剂量(200mg·kg
-1)给药时。经验证,本发明化合物在动物体内经口服给药,具有强效的肿瘤抑制作用,而td32-4在同等剂量直接口服给药时对肿瘤抑制没有明显的作用。
图1 td32-4在5mg·kg
-1静脉注射给药和10mg·kg
-1、200mg·kg
-1口服给药的血药浓度-时间曲线。
图2 td32-4P3及其体内转化而成的td32-4、td32-4直接口服给药的血药浓度-时间曲线,剂量均为10mg·kg
-1。
图3 td32-4P3及其体内转化而成的td32-4、td32-4直接口服给药的血药浓度-时间曲线,剂量均为200mg·kg
-1。
图4 td32-4P10及其体内转化而成的td32-4、td32-4直接口服给药的血药浓度-时间曲线,剂量均为10mg·kg
-1。
图5 td32-4P10及其体内转化而成的td32-4、td32-4直接口服给药的血药浓度-时间曲线,剂量均为200mg·kg
-1。
图6 td32-4P3、td32-4P6、td32-4P9、td32-4P10的小鼠体内肿瘤抑制作用。
图7 td32-4P10在裸鼠MV-4-11小鼠异种移植模型中肿瘤抑制活性数据(50mpk剂量)。A)给药期间,肿瘤体积的变化情况;B)给药期间,药物引起裸鼠体重变化情况;C)给药结束后肿瘤重量统计;D)给药结束后肿瘤解剖图像。
图8 td32-4P10在裸鼠H358小鼠异种移植模型中肿瘤抑制活性数据(50mpk剂量)。A)给药期间,肿瘤体积的变化情况;B)给药期间,药物引起裸鼠体重变化情况;C)给药结束后肿瘤重量统计;D)给药结束后肿瘤解剖图像。
图9 td32-4P10在裸鼠H82小鼠异种移植模型中肿瘤抑制活性数据(50mpk剂量)。A)给药期间,肿瘤体积的变化情况;B)给药期间,药物引起裸鼠体重变化情况;C)给药结束后肿瘤重量统计;D)给药结束后肿瘤解剖图像。
以下将结合部分实施例进一步地说明本发明的技术方案,下述实施例不构成对本发明的任何限制。
本发明的试剂材料均为市售产品。
实施例1
化合物4-(2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙氧基)-4-氧代丁酸(td32-4P1)的合成

将td32-4(化学名:N-3-氟-4-((2-(1-(2-羟乙基)-1H-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-N-(4-氟苯基)环丙烷-1,1-二甲酰胺)(5g,8.7mmol),4-DMAP(0.21g,1.7mmol),丁二酸酐(4.34g,43.5mmol)溶于50mL DMF中,100℃加热反应8小时。将反应液倒入500mL水中,搅拌析出固体,抽滤,200mL水洗涤滤饼。滤饼烘干后,将产物与20%(w%)碳酸钠水溶液加热搅拌,逐渐溶解,加入5%活性炭回流1小时,趁热抽滤,滤液用2mol/L稀盐酸调节pH至1~2,析出大量白色固体,抽滤,55℃烘箱中干燥,得白色固体4.82g,收率:82.1%,HPLC纯度:98.0%,LC-MS:675.4[M-H]
-。
实施例2
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基磷酸酯(td32-4P2)的合成

室温下将td32-4(5g,8.7mmol)与三乙胺(1.05g,10.4mmol)加入60mL四氢呋喃中,冷却至0℃,滴加三氯氧磷的四氢呋喃溶液,滴毕,反应体系于0~5℃继续搅拌反应3小时。将上述反应液滴加至100mL冰水中,搅拌析出固体。抽滤,水洗滤饼至滤液成中性,烘干样品。滤饼烘干后,将产物与20%(w%)碳酸钠水溶液加热搅拌,逐渐溶解,加入5%活性炭回流1小时,趁热抽滤,滤液用2mol/L稀盐酸调节pH至1~2,析出大量白色固体,抽滤,55℃烘箱中干燥,得白色固体3.56g,收率:62.5%,HPLC纯度:96.1%,LC-MS:654.1[M-H]
-。
实施例3
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基二甲基甘氨酸酯(td32-4P3)的合成

0℃条件下,将N,N-二甲基甘氨酸(1.79g,17.4mmol)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(缩写EDCI,4g,20.9mmol)、4-二甲氨基吡啶(缩写DMAP,1.59g,13.0mmol)加入到60mL的无水四氢呋喃中,反应30分钟后,将td32-4(5g,8.7mmol)加入反应体系,转移至室温,反应24小时。反应液过滤,向滤液中加入200mL 1mol/L的稀盐酸溶液,搅拌,抽滤,滤液用饱和碳酸钠溶液调节pH至10,析出大量白色固体,抽滤,100mL水洗滤饼,滤饼烘干得粗品。
将粗品溶于2mol/L的稀盐酸100mL中,过滤除去不溶物,滤液再用饱和碳酸钠调节pH至10,析出大量白色固体,抽滤,100mL水洗滤饼,滤饼烘干。将固体溶于80mL无水丙酮中,加入浓盐酸2mL,室温搅拌过夜,析出产品盐酸盐,抽滤,滤饼用50mL无水丙酮洗涤,滤饼于55℃烘箱中干燥。得白色固体3.75g,收率:65.3%,HPLC纯度:98.8%,LC-MS:661.3[M+H]
+。
实施例4
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基二乙基甘氨酸酯(td32-4P4)的合成

以N,N-二乙基甘氨酸(2.28g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体4.49g,收率:75.1%,HPLC纯度:97.9%,LC-MS:689.3[M+H]
+。
实施例5
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基2-吗啉乙酸酯(td32-4P5)的合成

以2-吗啉基乙酸(2.523g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体5.02g,收率:82.2%,HPLC纯度:96.3%,LC-MS:703.4[M+H]
+。
实施例6
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基甘氨酸酯(td32-4P6)的合成

1)化合物6A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)甘氨酸酯的合成
0℃条件下,将Boc-甘氨酸(3.04g,17.4mmol)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(缩写EDCI,4g,20.9mmol)、4-二甲氨基吡啶(缩写DMAP,1.59g,13.0mmol)加入到60mL的无水四氢呋喃中,反应30分钟后,将td32-4(5g,8.7mmol)加入反应体系,转移至室温,反应24小时。反应液过滤,向滤液中加入200mL 1mol/L的稀盐酸溶液,搅拌析出白色固体,抽滤,烘干固体。中间体粗品用水/乙醇(V/V=1/1)体系重结晶处理,得到中间体6A,白色固体4.81g,收率:75.5%。
2)化合物td32-4P6的合成
将中间体6A,溶于30mL无水丙酮中,加入浓盐酸2mL,40℃反应5小时,产生白色沉淀,抽滤,用20mL无水丙酮洗涤沉淀,真空干燥得到td32-4P6的盐酸盐。白色固体3.97g,收率:95.1%,HPLC纯度:98.5%,LC-MS:632.3[M+H]
+。
实施例7
2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基丙氨酸酯(td32-4P7)

1)化合物7A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)丙氨酸酯的合成
以Boc-丙氨酸(3.29g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例 6中6A的合成方法,得到化合物7A,白色固体5.21g,收率:80.2%。
化合物td32-4P7的合成
以化合物7A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基丙氨酸酯(td32-4P7)盐酸盐。白色固体4.29g,收率:90.1%,HPLC纯度:98.1%,LC-MS:647.3[M+H]
+。
实施例8
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基亮氨酸酯(td32-4P8)的合成

1)化合物8A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)亮氨酸酯的合成
以Boc-亮氨酸(4.02g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物8A,白色固体4.47g,收率:65.1%。
2)化合物td32-4P8的合成
以化合物8A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基亮氨酸酯(td32-4P8)。白色固体3.66g,收率:89.2%,HPLC纯度:99.0%,LC-MS:689.8[M+H]
+。
实施例9
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基异亮氨酸酯(td32-4P9)的合成

1)化合物9A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)异亮氨酸酯的合成
以Boc-异亮氨酸(4.02g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施 例6中6A的合成方法,得到化合物9A,白色固体4.88g,收率:71.2%。
2)化合物td32-4P9的合成
以化合物9A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基异亮氨酸酯(td32-4P9)。白色固体3.85g,收率:85.7%,HPLC纯度:97.3%,LC-MS:689.7[M+H]
+。
实施例10
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基缬氨酸酯(td32-4P10)的合成

1)化合物10A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)缬氨酸酯的合成
以Boc-缬氨酸(3.77g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物10A,白色固体5.33g,收率:79.1%。
2)化合物td32-4P10的合成
以化合物10A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基缬氨酸酯(td32-4P10)。白色固体4.46g,收率:91.1%,HPLC纯度:97.2%,LC-MS:675.8[M+H]
+。
实施例11
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基甲硫氨酸酯(td32-4P11)的合成

1)化合物11A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)甲硫氨酸酯的合成
以Boc-甲硫氨酸(4.33g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施 例6中6A的合成方法,得到化合物11A,白色固体4.64g,收率:66.1%。
2)化合物td32-4P11的合成
以化合物11A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-甲硫氨酸酯(td32-4P11)。白色固体4.03g,收率:94.3%,HPLC纯度:97.3%,LC-MS:707.9[M+H]
+。
实施例12
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-苯丙氨酸酯(td32-4P12)的合成

1)化合物12A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)苯丙氨酸酯的合成
以Boc-苯丙氨酸(4.61g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物12A,白色固体5.07g,收率:70.8%。
2)化合物td32-4P12的合成
以化合物12A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-苯丙氨酸酯(td32-4P12)的盐酸盐。白色固体4.38g,收率:93.6%,HPLC纯度:98.1%,LC-MS:723.8[M+H]
+。
实施例13
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基脯氨酸酯(td32-4P13)的合成

1)化合物13A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)脯氨酸酯的合成
以Boc-脯氨酸(3.74g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例 6中6A的合成方法,得到化合物13A,白色固体6.72g,收率:77.4%。
2)化合物td32-4P13的合成
以化合物13A为原料,参考实施例6中td32-4P6合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-脯氨酸酯(td32-4P13)盐酸盐。白色固体4.27g,收率:89.4%,HPLC纯度:95.1%,LC-MS:673.6[M+H]
+。
实施例14
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基天冬酰胺酯(td32-4P15)的合成

1)化合物14A的合成
2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)天冬酰胺酯
以Boc-天冬酰胺(4.03g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物14A,白色固体4.05g,收率:58.9%。2)化合物td32-4P14的合成
以化合物14A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基天冬酰胺酯(td32-4P14)盐酸盐。白色固体3.17g,收率:85.3%,HPLC纯度:97.9%,LC-MS:691.1[M+H]
+。
实施例15
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基谷氨酰胺酯(td32-4P15)的合成

1)化合物15A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)谷氨酰胺酯的合成
以Boc-谷氨酰胺(4.28g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施 例6中6A的合成方法,得到化合物15A,白色固体3.90g,收率:55.8%。
2)化合物td32-4P15的合成
以化合物15A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-谷氨酰胺酯(td32-4P15)盐酸盐。白色固体2.99g,收率:83.3%,HPLC纯度:99.0%,LC-MS:704.6[M+H]
+。
实施例16
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基色氨酸酯(td32-4P16)的合成

1)化合物16A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(叔丁氧羰基)色氨酸酯的合成
以Boc-色氨酸(5.29g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物16A,白色固体4.89g,收率:65.3%。
2)化合物td32-4P16的合成
以化合物16A为原料,参考实施例7中td32-4P7的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基L-色氨酸酯(td32-4P16)盐酸盐。白色固体3.48g,收率:76.7%,HPLC纯度:94.6%,LC-MS:762.7[M+H]
+。
实施例17
2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(2,2-二甲氧乙基)甘氨酸酯(td32-4P17)

1)化合物17A:2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基) 噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(2,2-二甲氧乙基)(叔丁氧羰基)甘氨酸酯的合成
以N-Boc-N-2,2-二甲氧乙基甘氨酸(4.57g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例6中6A的合成方法,得到化合物17A,白色固体5.50g,收率:77.1%。
2)化合物td32-4P17的合成
以化合物17A为原料,参考实施例6中td32-4P6的合成方法,得到化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基(2,2-二甲氧乙基)甘氨酸酯(td32-4P17)盐酸盐。白色固体4.53g,收率:89.3%,HPLC纯度:93.5%,LC-MS:721.9[M+H]
+。
实施例18
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基N,N-二甲基丙氨酸酯(td32-4P18)的合成

以N,N-二甲基丙氨酸(2.03g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体5.86g,收率:76.1%,HPLC纯度:93.3%,LC-MS:675.3[M+H]
+。
实施例19
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基4-(吗啉基甲基)苯甲酸酯(td32-4P19)的合成

以4-(吗啉基甲基)苯甲酸(3.84g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体4.50g,收率:66.5%,HPLC纯度:95.9%,LC-MS:780.1[M+H]
+。
实施例20
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基3-(吗啉基甲基)苯甲酸酯(td32-4P20)的合成

以3-(吗啉基甲基)苯甲酸(3.84g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体4.68g,收率:69.1%,HPLC纯度:95.2%,LC-MS:779.9[M+H]
+。
实施例21
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基4-((甲基哌嗪-1-基)甲基)苯甲酸酯(td32-4P21)的合成

以4-((甲基哌嗪-1-基)甲基)苯甲酸(4.07g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体4.14g,收率:60.1%,HPLC纯度:96.4%,LC-MS:793.0[M+H]
+。
实施例22
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基3-((甲基哌嗪-1-基)甲基)苯甲酸酯(td32-4P22)的合成

以3-((甲基哌嗪-1-基)甲基)苯甲酸(4.07g,17.4mmol)、td32-4(5g,8.7mmol) 为原料,参照实施例3的合成方法,得到白色固体3.91g,收率:56.8%,HPLC纯度:93.9%,LC-MS:792.9[M+H]
+。
实施例23
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基4-(吡咯-1-基甲基)苯甲酸酯(td32-4P23)的合成

以4-(吡咯-1-基甲基)苯甲酸(3.57g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体3.67g,收率:55.4%,HPLC纯度:96.6%,LC-MS:763.9[M+H]
+。
实施例24
化合物2-(4-(7-(2-氟-4-(1-((4-氟苯基)氨甲酰基)环丙烷-1-甲酰胺)苯氧基)噻吩并[3,2-b]吡啶-2-基)-1H-吡唑基-1-基)乙基3-(吡咯-1-基甲基)苯甲酸酯(td32-4P24)的合成

以3-(吡咯-1-基甲基)苯甲酸(3.57g,17.4mmol)、td32-4(5g,8.7mmol)为原料,参照实施例3的合成方法,得到白色固体3.04g,收率:45.8%,HPLC纯度:97.5%,LC-MS:763.8[M+H]
+。
实施例25溶解度测试
对实施例1-24所合成的系列化合物进行溶解度测试(水),溶解度测试方法参考2015版《中国药典》。具体试验操作与评价标准如下:
称取研磨成细粉状的化合物粉末适量(1mg~10mg),置于25℃±2℃,一定容量的溶剂中,每隔5分钟强力振摇30秒;观察30分钟内的溶解情况,如无目视可见的溶质颗粒或液滴时,即视为完全溶解。
| A极易溶解 | 溶质1g能在溶剂不到1mL中溶解 |
| B易溶 | 溶质1g能在1~10mL溶剂中溶解 |
| C溶解 | 溶质1g能在10~30mL溶剂中溶解 |
| D略溶 | 溶质1g能在30~100mL溶剂中溶解 |
| E微溶 | 溶质1g能在100~1000mL溶剂中溶解 |
| F极微溶解 | 溶质1g能在1000~10000mL溶剂中溶解 |
| G几乎不溶或不溶 | 溶质1g在10000mL溶剂中不溶解 |
化合物水中溶解性测试结果如表1所示:
表1化合物在水中的溶解度测试
| Compd. | Result | Compd. | Result | Compd. | Result |
| td32-4P1 | F | td32-4P10 | C | td32-4P19 | F |
| td32-4P2 | F | td32-4P11 | E | td32-4P20 | F |
| td32-4P3 | C | td32-4P12 | F | td32-4P21 | D |
| td32-4P4 | C | td32-4P13 | E | td32-4P22 | D |
| td32-4P5 | C | td32-4P14 | D | td32-4P23 | D |
| td32-4P6 | C | td32-4P15 | D | td32-4P24 | D |
| td32-4P7 | D | td32-4P16 | F | td32-4 | G |
| td32-4P8 | E | td32-4P17 | E | ||
| td32-4P9 | D | td32-4P18 | D |
实验结果表明,本发明涉及的化合物水溶解性均明显好于td32-4,能够解决td32-4低溶解度引起的口服吸收差等问题,有望开发为新型多靶点酪氨酸激酶抑制剂抗肿瘤药物。
实施例26体内转化实验
本发明涉及的化合物将亲水性官能团通过酯键的方式引入到td32-4结构中来增加化合物的溶解度。在口服进入体内后,需要在体内物理、化学环境下转化为td32-4发挥药理作用。本实施例选择部分代表性化合物,进行SD大鼠体内药动学实验研究,并与直接口服td32-4作比对。实验选用高(200mg·kg
-1)、低(10mg·kg
-1)两种剂量进行。
实验材料:
SD雄性鼠,SPF级。上海西普尔-必凯实验动物有限公司。其它试剂均为市售产品。
1)溶液配制:
静脉溶媒:DMA+PEG400+Saline
称取供试品5mg至玻璃瓶中。加入1mL的DMA(二甲基乙酰胺),涡旋振荡,使固体物质完全溶解。加入1.5mL的PEG400,涡旋振荡,混匀。加入2.5mL的注射用生理盐水,涡旋振荡,混匀,使用过滤膜(PALL,Nylon,0.45μm)过滤,得无色澄清溶液。供试品以5mg·kg
-1(1mg·mL
-1)剂量经尾静脉注射给予实验动物(n=3)。
口服溶媒:PEG400
称取供试品适量至玻璃瓶中。加入PEG400,涡旋振荡,混匀。超声得澄清溶液。供试品以10mg·kg
-1和200mg·kg
-1剂量经灌胃给予实验动物(n=3)。
2)试验方法:
给药前称重SD大鼠,根据体重,计算给药量。通过静脉注射或灌胃口服给药。按0,0.083,0.25,0.5,1,2,4,8,12,24小时间隔,经颌下静脉或其他合适方式采血,每个样品采集约0.20mL,EDTA-K2抗凝,采集后放置冰上。并于1小时之内离心分离血浆(离心条件:6800g,6分钟,2-8℃)。血浆样本在分析前存放时则放于-80℃冰箱内。使用LC-MS/MS进样分析各样品的血药浓度。通过不同时间点的血药浓度数据,运用Phoenix WinNonlin7.0计算药代动力学参数,提供AUC0-t、AUC0-∞、MRT0-∞、Cmax、Tmax、和T1/2等参数及其平均值和标准差。本发明涉及的化合物,静脉给药4小时候后全部转化为td32-4,其数据视为100%生物利用度,用作计算口服利用度。
3)代表性实验结果如下:
化合物td32-4:
在10mg·kg
-1和200mg·kg
-1的血药浓度数据如下,通过计算可以得出,在低浓度下,化合物td32-4的口服生物利用度达83.96%,但在高浓度下急剧下降,仅18.6%。很可能是高浓度下化合物在胃肠道聚集沉淀,导致无法吸收入血液循环,结果见图1所示。
化合物td32-4P3低浓度:
如图2所示,td32-4P3在按照10mg·kg
-1剂量口服给药时,在体内能够转化成td32-4,且由td32-4P3体内转化的td32-4的暴露量(药时曲线下面积AUC)与直接口服化合物td32-4接近,表明td32-4P3在体内快速转化为td32-4。按照td32-4的暴露量计算其口服生物利用度F=70.2%。
化合物td32-4P3高浓度:
如图3所示,化合物td32-4P3在按照200mg·kg
-1剂量口服给药时,在体内能够转化成td32-4,转化得到的td32-4的暴露量(药时曲线下面积AUC)高于直接高剂量口服化合物td32-4。td32-4P3在体内18小时几乎完全转化为td32-4,按 照td32-4的暴露量计算口服生物利用度F=60.1%,远高于直接口服td32-4的F=18.6%。
化合物td32-4P10低浓度:
如图4所示,td32-4P10在按照10mg·kg
-1剂量口服给药时,在体内能够转化成td32-4,转化而来的td32-4的暴露量(药时曲线下面积AUC)与直接口服td32-4接近。td32-4P3在体内快速转化为td32-4。按照转化而来的td32-4的暴露量计算口服生物利用度F=48.9%。
化合物td32-4P10高浓度:
如图5所示,td32-4P10在按照200mg·kg
-1剂量口服给药时,在体内能够转化成td32-4,转化成的td32-4的暴露量(药时曲线下面积AUC)高于高剂量口服td32-4。td32-4P10在体内18小时几乎完全转化为td32-4。按照转化成的td32-4的暴露量计算口服生物利用度F=44.8%。(直接口服td32-4的F=18.6%)
实验结果汇总如下(表2):
表2:部分化合物的口服生物利用度(按照血浆中td32-4计算)
| Compd. | F%(10mpk) | F%(200mpk) | Compd. | F%(10mpk) | F%(200mpk) |
| td32-4 | 83.4% | 18.6% | td32-4P10 | 48.9% | 44.8% |
| td32-4P1 | 50.2% | 20.1% | td32-4P13 | 48.7% | 19.9% |
| td32-4P2 | 45.1% | 15.3% | td32-4P19 | 45.1% | 20.0% |
| td32-4P3 | 70.2% | 60.1% | td32-4P21 | 38.9% | 17.3% |
| td32-4P5 | 66.9% | 45.6% | td32-4P22 | 41.1% | 21.2% |
| td32-4P6 | 71.3% | 55.7% | td32-4P23 | 30.8% | 25.1% |
| td32-4P9 | 45.1% | 41.9% | td32-4P24 | 35.1% | 22.6% |
通过代表性化合物td32-4P3、td32-4P10的药代动力学实验数据可以看出,本发明申请中的化合物能够在体内表现出明显优于td32-4的药动学性质,特别是在大剂量(200mg·kg
-1)时,其口服生物利用度大幅度提高,提高了td32-4的血药浓度。本发明的化合物通过改善了化合物的溶解度,能够一定程度上解决td32-4类化合物药性差的难题。
实施例27体内药效学测试
1.选用化合物(td32-4、td32-4P3、td32-4P6、td32-4P9、td32-4P10)进行单浓 度体内活性评价,动物模型选能用胃癌细胞MKN45异种移植瘤模型。
试验方法:5×10
6个MKN-45胃癌细胞接种于裸鼠右侧肋部脂肪垫下,观察成瘤情况,接种两周后,对荷瘤小鼠进行分组,分别给药td32-4和td32-4P3、td32-4P6、td32-4P9、td32-4P10,口服给药,给药剂量为100mg·kg
-1,每天给药一次共14天。用游标卡尺测量肿瘤体积并记录。
实验结果表明td32-4P3/9/10在裸鼠模型中均表现出强效的肿瘤抑制作用。td32-4P6表现出中等抑制活性,而相同剂量下,由于溶解度限制了生物利用度,td32-4并未表现出明显活性,数据结果见图6所示。
本发明对化合物td32-4进行结构修饰,引入亲水结构片段,能够显著提高化合物的水溶性,实验证明化合物经口服吸收后,能够转化为td32-4,发挥抗肿瘤活性。经过药代动力学实验验证,能够克服td32-4固有的口服吸收差的缺陷。在荷瘤小鼠中表现出强效的肿瘤抑制活性,有望开发成为新型多靶点抗肿瘤药物。
2.选用化合物td32-4P10进行体内活性评价,分别测试了td32-4P10在裸鼠MV-4-11、H358和H82小鼠异种移植模型中肿瘤抑制活性数据。试验方法同上,给药剂量如表3所示。在给药期间,同时测量肿瘤体积的变化情况;给药期间,药物引起裸鼠体重变化情况。在给药结束后对肿瘤重量统计,记录给药结束后肿瘤解剖图像。试验结果如表3、图7、8和9所示。
表3化合物td32-4P10体内活性评价

Claims (9)
- 一种多靶点酪氨酸激酶抑制剂,是一种化合物或其药学可接受的盐、异构体、消旋体,其特征在于所述化合物具有通式(I)所示的结构:
 其中,R 1任选自以下取代基:羧基取代的C 3~C 8烷基酰基、取代或未取代的膦酰基、
其中,R 1任选自以下取代基:羧基取代的C 3~C 8烷基酰基、取代或未取代的膦酰基、 R 2任选自以下基团:氢、卤素、R 5取代的C 1~C 8烷基、R 5取代的C 6~C 12芳基;R 3、R 4任选自以下基团:氢、R 5取代的C 1~C 6烷基,或R 3、R 4构成五元~十二元脂肪杂环;其中,R 5任选自以下基团:氢、1~3个卤素、羟基、C 1~C 6烷氧基、C 6~C 12芳基。
R 2任选自以下基团:氢、卤素、R 5取代的C 1~C 8烷基、R 5取代的C 6~C 12芳基;R 3、R 4任选自以下基团:氢、R 5取代的C 1~C 6烷基,或R 3、R 4构成五元~十二元脂肪杂环;其中,R 5任选自以下基团:氢、1~3个卤素、羟基、C 1~C 6烷氧基、C 6~C 12芳基。 - 如权利要求1所述的多靶点酪氨酸激酶抑制剂,其特征在于:R 1选自3-羧基丙酰基、4-羧酸丁酰基、5-羧基戊酰基、6-羧基己酰基或

- 如权利要求1所述的多靶点酪氨酸激酶抑制剂,其特征在于:R 1为时,
 R 2任选自以下基团:氢、卤素、R 5取代的C 1~C 8烷基、R 5取代的C 6~C 12芳基;R 3、R 4任选自以下基团:氢、R 5取代的C 1~C 6烷基,或R 3、R 4构成五元~十二元脂肪杂环;其中,R 5任选自以下基团:氢、1~3个卤素、羟基、C 1~C 6烷氧基、或C 6~C 12芳基。
R 2任选自以下基团:氢、卤素、R 5取代的C 1~C 8烷基、R 5取代的C 6~C 12芳基;R 3、R 4任选自以下基团:氢、R 5取代的C 1~C 6烷基,或R 3、R 4构成五元~十二元脂肪杂环;其中,R 5任选自以下基团:氢、1~3个卤素、羟基、C 1~C 6烷氧基、或C 6~C 12芳基。 - 如权利要求3所述的多靶点酪氨酸激酶抑制剂,其特征在于:时,
 R 2任选自以下基团:氢、氟、氯、溴、甲基、-CH 2F、-CHF 2、-CF 3、乙基、丙基、异丙基、环丙基、丁基、异丁基、苄基、4-羟基苄基、苯基、4-氟苯基;R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或 R 3、R 4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶。
R 2任选自以下基团:氢、氟、氯、溴、甲基、-CH 2F、-CHF 2、-CF 3、乙基、丙基、异丙基、环丙基、丁基、异丁基、苄基、4-羟基苄基、苯基、4-氟苯基;R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或 R 3、R 4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶。 - 如权利要求3所述的多靶点酪氨酸激酶抑制剂,其特征在于:时,
 R 2选自以下基团:氢、甲基、-CH 2F、-CHF 2、-CF 3、乙基、丙基、异丙基、异丁基、苄基、4-羟基苄基、苯基;R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基,或R 3、R 4构成吗啉、N-甲基哌啶。
R 2选自以下基团:氢、甲基、-CH 2F、-CHF 2、-CF 3、乙基、丙基、异丙基、异丁基、苄基、4-羟基苄基、苯基;R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基,或R 3、R 4构成吗啉、N-甲基哌啶。 - 如权利要求1所述的多靶点酪氨酸激酶抑制剂,其特征在于:
 时,
时, R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或R 3、R 4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶。
R 3、R 4任选自以下基团:氢、甲基、乙基、2,2-二甲氧基乙基、丙基、丁基,或R 3、R 4构成吗啉、吡啶、吡咯、哌啶、哌啶基哌啶、N-甲基哌啶。 - 如权利要求1所述的多靶点酪氨酸激酶抑制剂,包括以下化合物:


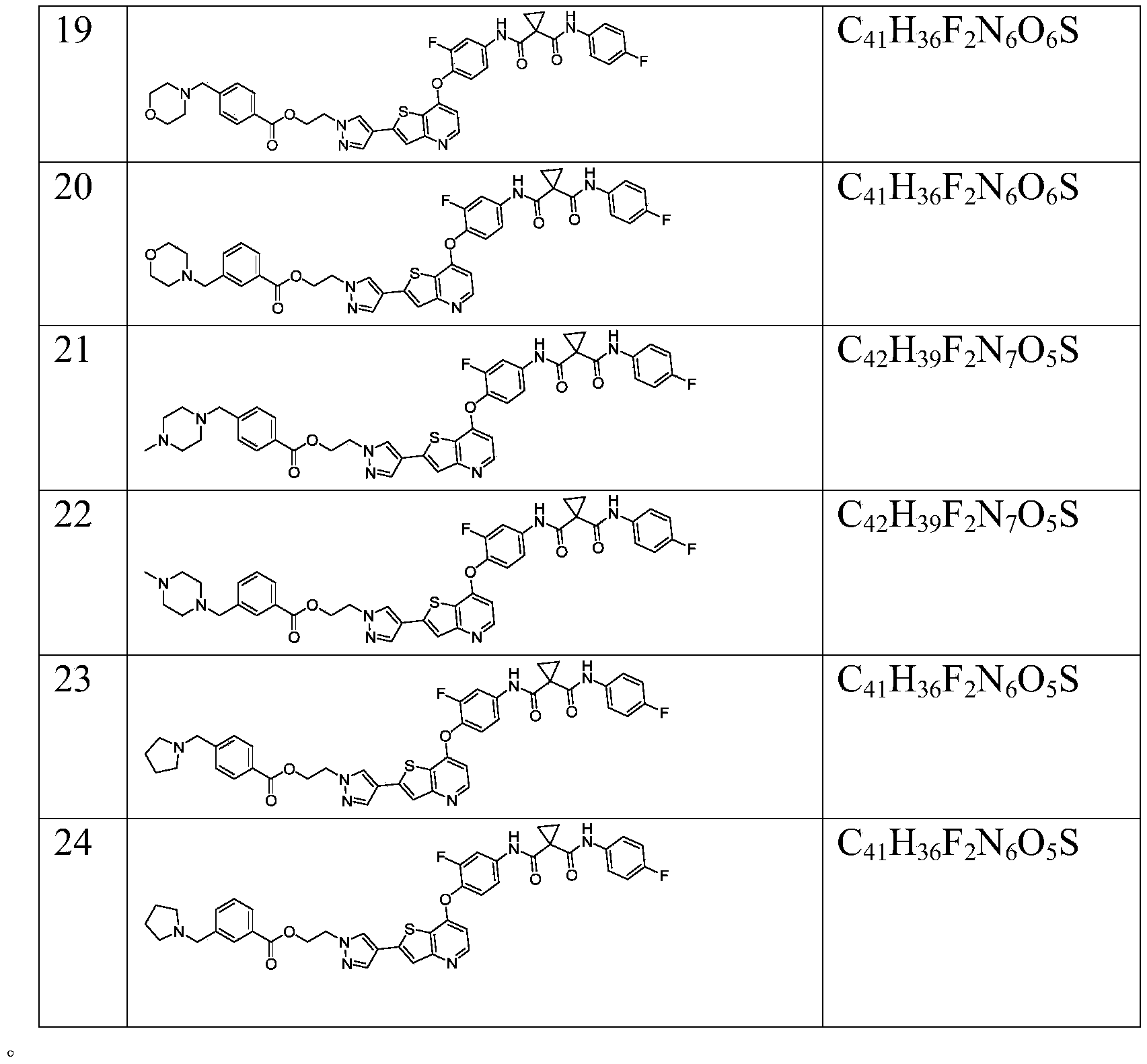
- 一种药物组合物,含有如权利要求1~7任意一项所述的多靶点酪氨酸激酶抑制剂。
- 权利要求1~7任一项所述的多靶点酪氨酸激酶抑制剂在用于制备治疗肿瘤药物中的应用,所述肿瘤包括:宫颈癌、精原细胞瘤、睾丸淋巴瘤、前列腺癌、卵巢癌、肺癌、直肠癌、乳腺癌、皮肤鳞状细胞癌、结肠癌、肝癌、胰腺癌、胃癌、食管癌、甲状腺癌、和/或膀胱移行上皮癌、白血病。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/795,516 US12391706B2 (en) | 2020-02-18 | 2021-02-02 | Multi-target tyrosine kinase inhibitor |
| EP21757014.2A EP4108666A4 (en) | 2020-02-18 | 2021-02-02 | MULTI-TARGETED TYROSINE KINASE INHIBITOR |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010099747.2A CN113336768B (zh) | 2020-02-18 | 2020-02-18 | 一种多靶点酪氨酸激酶抑制剂 |
| CN202010099747.2 | 2020-02-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021164538A1 true WO2021164538A1 (zh) | 2021-08-26 |
Family
ID=77391832
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2021/074809 Ceased WO2021164538A1 (zh) | 2020-02-18 | 2021-02-02 | 一种多靶点酪氨酸激酶抑制剂 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US12391706B2 (zh) |
| EP (1) | EP4108666A4 (zh) |
| CN (1) | CN113336768B (zh) |
| WO (1) | WO2021164538A1 (zh) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114354789B (zh) * | 2021-12-27 | 2023-08-29 | 深圳海王医药科技研究院有限公司 | 一种同时测定卡博替尼类似物及其有关物质的方法 |
| CN114644642B (zh) * | 2022-04-06 | 2023-05-12 | 深圳海王医药科技研究院有限公司 | 一种噻吩并吡啶化合物的晶型a、制备方法及其药物组合物 |
| CN115177618A (zh) * | 2022-07-26 | 2022-10-14 | 广州市第一人民医院(广州消化疾病中心、广州医科大学附属市一人民医院、华南理工大学附属第二医院) | Flt3抑制剂在制备治疗急性髓系白血病药物中的应用 |
| CN118846073B (zh) * | 2024-06-25 | 2026-01-30 | 深圳海王医药科技研究院有限公司 | 一种酪氨酸激酶抑制剂的新应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008041053A2 (en) * | 2005-05-20 | 2008-04-10 | Methylgene, Inc. | Inhibitors of vegf receptor and hgf receptor signaling |
| CN108530464A (zh) | 2017-03-02 | 2018-09-14 | 深圳海王医药科技研究院有限公司 | 一种多靶点激酶抑制剂 |
| CN109384799A (zh) * | 2018-11-12 | 2019-02-26 | 深圳海王医药科技研究院有限公司 | 一种多靶点激酶抑制剂化合物的晶型a及制备方法和含有其的药物组合物 |
-
2020
- 2020-02-18 CN CN202010099747.2A patent/CN113336768B/zh active Active
-
2021
- 2021-02-02 EP EP21757014.2A patent/EP4108666A4/en active Pending
- 2021-02-02 US US17/795,516 patent/US12391706B2/en active Active
- 2021-02-02 WO PCT/CN2021/074809 patent/WO2021164538A1/zh not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008041053A2 (en) * | 2005-05-20 | 2008-04-10 | Methylgene, Inc. | Inhibitors of vegf receptor and hgf receptor signaling |
| CN108530464A (zh) | 2017-03-02 | 2018-09-14 | 深圳海王医药科技研究院有限公司 | 一种多靶点激酶抑制剂 |
| CN109384799A (zh) * | 2018-11-12 | 2019-02-26 | 深圳海王医药科技研究院有限公司 | 一种多靶点激酶抑制剂化合物的晶型a及制备方法和含有其的药物组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN113336768A (zh) | 2021-09-03 |
| EP4108666A4 (en) | 2024-03-13 |
| US12391706B2 (en) | 2025-08-19 |
| CN113336768B (zh) | 2022-08-19 |
| EP4108666A1 (en) | 2022-12-28 |
| US20230102146A1 (en) | 2023-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021164538A1 (zh) | 一种多靶点酪氨酸激酶抑制剂 | |
| ES2552386T3 (es) | Composiciones de {3-[5-(4-cloro-fenil)-1H-pirrol[2,3-b]piridin-3-carbonil]-2,4-difluoro-fenil}-amida del ácido propano-1-sulfónico y usos de las mismas | |
| ES2954451T3 (es) | Composiciones y usos de derivados de amidina | |
| JP6612200B2 (ja) | 抗炎症性の置換シクロブテンジオン化合物のコリン塩 | |
| US8476320B2 (en) | Formulations for parenteral administration of amino-substituted (E)-2, 6-dialkoxystyryl 4-substituted benzylsulfones | |
| PT1663194E (pt) | Utilizaão de saha para o tratamento de mesotelioma | |
| US10323035B2 (en) | Co-crystal of a CDK inhibitor and an MEK inhibitor and process of preparation thereof | |
| US20200376004A1 (en) | Amorphous onapristone compositions and methods of making the same | |
| KR20200044873A (ko) | 안구 제약학적 조성물 | |
| US8063109B2 (en) | Formulations for parenteral administration of (e)-2, 6-dialkoxystyryl 4-substituted benzylsulfones | |
| CN105246483A (zh) | 用于γ-谷氨酰循环调节的方法和组合物 | |
| CN102665716B (zh) | 用于治疗唐氏综合征的方法和药物组合物 | |
| BR112019003973A2 (pt) | inibição de atividade olig2 | |
| WO2019219019A1 (zh) | 一种kor受体激动剂药物组合物 | |
| US10266523B2 (en) | Crystaline forms of N-[6-(cis-2,6-dimethylmorpholine-4-yl)pyridine-3-yl]-2-Methyl-4′-(trifluoromethoxy) [1,1′-biphenyl]-3-Methanamide monophosphate, and process of preparation thereof | |
| ES2965179T3 (es) | Derivado polimérico soluble en agua de venetoclax | |
| CN114573582B (zh) | 1,2,3,4-四氢吡啶并[2,3-d]嘧啶类化合物及其制备方法和应用 | |
| CN119235877B (zh) | 一种钌(iii)酸盐的组合物及其应用 | |
| CN110078729A (zh) | 一种水溶性氟苯尼考前药及其制备方法 | |
| CN105732642B (zh) | 一种cdk抑制剂和mek抑制剂的共晶及其制备方法 | |
| CN110934868A (zh) | 难溶性复合物、药物组合物及其应用 | |
| EP4631496A1 (en) | Solid dispersion, preparation method therefor, and use thereof | |
| US20240368088A1 (en) | Ep4 antagonist compound as well as salt, polymorph and use thereof | |
| ES3039287T3 (en) | Crystal of pyrazolo[3,4-d]pyrimidine | |
| CN102079717A (zh) | 一种二元酯酸的精氨酸盐化合物及其制备方法和药物应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21757014 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021757014 Country of ref document: EP Effective date: 20220919 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 17795516 Country of ref document: US |