WO2022004859A1 - 経口用医薬組成物及びその製造方法 - Google Patents
経口用医薬組成物及びその製造方法 Download PDFInfo
- Publication number
- WO2022004859A1 WO2022004859A1 PCT/JP2021/025019 JP2021025019W WO2022004859A1 WO 2022004859 A1 WO2022004859 A1 WO 2022004859A1 JP 2021025019 W JP2021025019 W JP 2021025019W WO 2022004859 A1 WO2022004859 A1 WO 2022004859A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- pharmaceutical composition
- acid
- active ingredient
- pharmaceutically active
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0056—Mouth soluble or dispersible forms; Suckable, eatable, chewable coherent forms; Forms rapidly disintegrating in the mouth; Lozenges; Lollipops; Bite capsules; Baked products; Baits or other oral forms for animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0087—Galenical forms not covered by A61K9/02 - A61K9/7023
- A61K9/0095—Drinks; Beverages; Syrups; Compositions for reconstitution thereof, e.g. powders or tablets to be dispersed in a glass of water; Veterinary drenches
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention relates to an oral pharmaceutical composition for administering a pharmaceutically active ingredient having a pH-dependent dissolution profile and a method for producing the same.
- the pharmaceutical composition for oral administration of the pharmaceutically active ingredient exhibits certain dissolving characteristics in order to achieve stable absorption of the pharmaceutically active ingredient by the target gastrointestinal tract.
- the pH in the digestive tract is not constant, and in particular, the pH in the stomach varies greatly among individuals, and even within the same individual, there are large fluctuations depending on conditions such as physical condition and dietary content. Therefore, especially in the case of a pharmaceutically active ingredient having a pH-dependent dissolution profile, it is extremely difficult to administer it orally and stably absorb it.
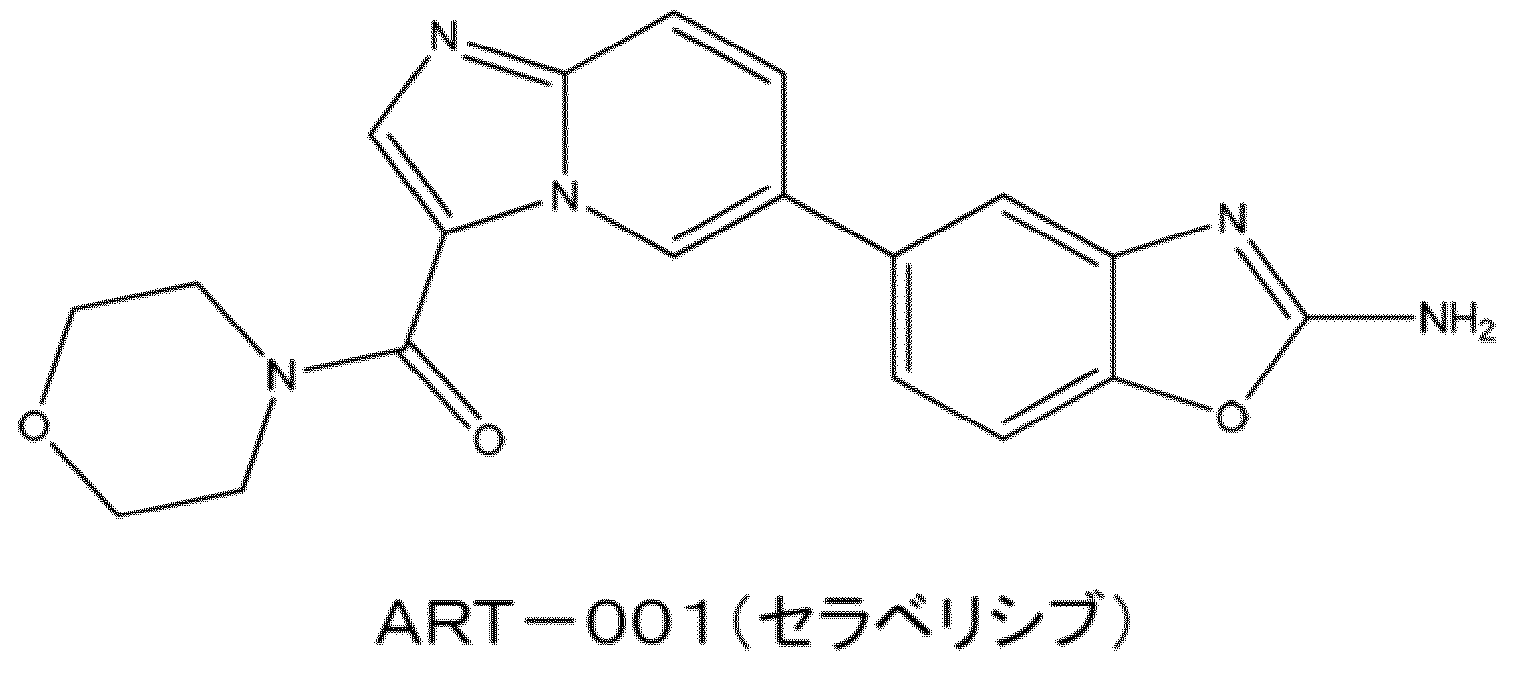
- ART-001 As an example of a pharmaceutically active ingredient having such a pH-dependent dissolution profile, the compound ART-001 (compound name: Cerabellicive) currently under development by the applicant can be mentioned.
- the chemical formula of ART-001 is shown below.
- ART-001 is currently undergoing clinical trials as a pharmaceutical active ingredient showing a therapeutic effect on diseases such as vascular malformations.
- Non-Patent Documents It has been reported that ART-001 has a pH-dependent dissolution profile in which solubility is extremely reduced in neutral to basic solutions while exhibiting high solubility in acidic solutions. 1). ART-001 showed a high degree of variability in pharmacokinetics and limited therapeutic effect in the clinical phase I study conducted in the past in patients with advanced solid tumor (Non-Patent Document 2). ART-001 also showed remarkable pharmacokinetic variability in pharmacokinetic studies in healthy adults (Non-Patent Document 1).
- the present invention has been made in view of the above problems, and is a pharmaceutical composition for orally administering a pharmaceutically active ingredient having a pH-dependent dissolution profile, and achieves excellent absorption efficiency regardless of the pH in the gastrointestinal tract. It is an object of the present invention to provide a pharmaceutical composition which can be used.
- the present inventors have formulated a pharmaceutically active ingredient having such a pH-dependent dissolution profile together with an inorganic or organic acid and a hydrophilic polymer, thereby being excellent regardless of the pH in the gastrointestinal tract.
- the present invention has been completed by finding that a pharmaceutical composition capable of achieving the absorption efficiency can be obtained.
- the gist of the present invention relates to, for example, the following.
- An oral pharmaceutical composition comprising a pharmaceutically active ingredient having a pH-dependent dissolution profile, an inorganic or organic acid, and a hydrophilic polymer.
- the oral pharmaceutical composition according to Item 1 wherein the pharmaceutically active ingredient is an aromatic compound having a ⁇ -conjugated system.
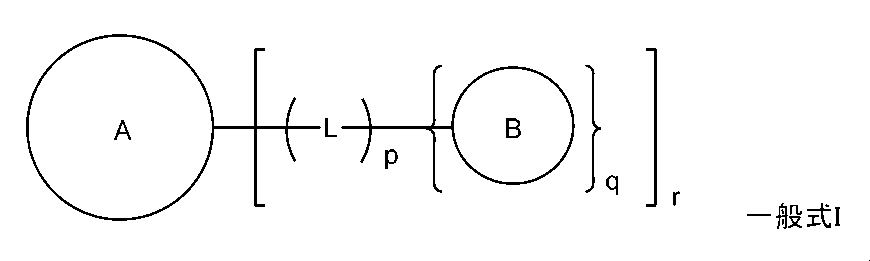
- the oral pharmaceutical composition according to Item 1 or 2 wherein the pharmaceutically active ingredient is a compound having a structure represented by the following general formula I. (During the ceremony, Ring A represents a 5- to 16-membered monocyclic or fused bicyclic or tricyclic aromatic hydrocarbon group or aromatic heterocyclic group which may have one or more substituents.
- Ring B represents a 5- to 12-membered monocyclic or fused bicyclic aromatic hydrocarbon group or aromatic heterocyclic group which may have one or more substituents.
- L represents a divalent or trivalent linking group which may have one or more substituents.
- p represents an integer of 0 or 1 and represents q represents an integer of 1 or 2.
- r represents an integer of 1 or 2.
- the pharmaceutically active ingredient is one or more compounds selected from ART-001 (ceravericive), alperiticib, afatinib, gefitinib, bosutinib, alectinib, palbociclib, tacericib, and copanricib, Item 1 to.
- the oral pharmaceutical composition according to any one of 3.
- the ratio of the solubility of the pharmaceutically active ingredient at pH 2.5 to the solubility at pH 6 is 50% or less, 30% or less, 20% or less, 10% or less, or 5% or less.
- the inorganic or organic acid is selected from phosphoric acid, hydrochloric acid, citric acid, malic acid, tartaric acid, ascorbic acid, fumaric acid, succinic acid, aspartic acid, lactic acid, acetic acid, glutamic acid, and adipic acid 1
- the hydrophilic polymer is selected from polyvinyl alcohol, povidone, hypromellose, copolyvidone, hydroxypropyl cellulose, polyvinyl alcohol-polyethylene glycol graft copolymer, hypromellose phthalate ester, and hypromellose acetate succinic acid ester 1 Item 6.
- the oral pharmaceutical composition according to any one of Items 1 to 6, which is a seed or two or more kinds of polymers.
- the content of the pharmaceutically active ingredient is 2% by mass or more, 5% by mass or more, 10% by mass or more, or 15% by mass or more, and 40% by mass or less, or 35% by mass or less, or 30. Item 6.
- the content ratio of the inorganic or organic acid to 1 part by mass of the pharmaceutical active ingredient is 0.1 part by mass or more, 0.3 part by mass or more, 0.5 part by mass or more, and 10 parts by mass or less.
- the content ratio of the hydrophilic polymer to 1 part by mass of the pharmaceutical active ingredient is 0.02 parts by mass or more, 0.05 parts by mass or more, 0.1 parts by mass or more, and 5 parts by mass or less, or Item 6.
- the oral pharmaceutical composition according to any one of Items 1 to 10 which is a dry syrup agent.
- the oral composition of the present invention when a pharmaceutically active ingredient having a pH-dependent dissolution profile is orally administered, it is possible to achieve excellent absorption efficiency regardless of the pH in the gastrointestinal tract. .. Further, according to one aspect of the oral composition of the present invention, it is possible to obtain stable pharmacokinetics with suppressed variation among individuals as compared with the case of administration using a conventional dosage form such as a capsule or a tablet. It will be possible.
- composition of the present invention relates to a pharmaceutical composition for orally administering a pharmaceutically active ingredient having a pH-dependent dissolution profile (appropriately referred to as "the pharmaceutical composition of the present invention").
- the pharmaceutical composition of the present invention contains an inorganic or organic acid and a hydrophilic polymer in addition to the pharmaceutically active ingredient having such a pH-dependent dissolution profile.
- the pharmaceutical composition of the present invention having such a composition, when a pharmaceutically active ingredient having a pH-dependent dissolution profile is orally administered, it is possible to achieve excellent absorption efficiency regardless of the pH in the gastrointestinal tract. ..
- an animal administration test was carried out as shown in Examples described later.
- conventional capsules and tablets were also used in human administration tests.
- the pharmaceutical composition of the present invention contains a pharmaceutically active ingredient having a pH-dependent dissolution profile.
- the "pharmaceutical active ingredient” means an ingredient contained in a pharmaceutical product that exhibits physiological activity for a desired indication.
- the indication and physiological activity of the pharmaceutically active ingredient in the present disclosure are not limited, and examples thereof include a pharmaceutically active ingredient that exerts its medicinal effect by being orally administered to a subject and absorbed in the gastrointestinal tract (for example, in the stomach).
- the "pH-dependent dissolution profile” means that the solubility varies depending on the pH.
- the terms “dissolution profile” and “solubility” in the present disclosure mean solubility in an aqueous medium.
- aqueous medium means water or various aqueous solutions.
- Aqueous digestive juices eg saliva, gastric juice, etc.
- aqueous body fluids other than digestive fluids blood, lymph, tissue fluid, ascites, etc.
- the pharmaceutically active ingredient having a pH-dependent dissolution profile in the present disclosure is not limited to this, but exhibits high solubility under acidic pH conditions, while dissolving as the pH rises. It is a compound that has reduced properties and shows almost no solubility under neutral to alkaline pH conditions. More specifically, when comparing the solubility of the pharmaceutically active ingredient in an aqueous solvent near room temperature (for example, 30 ° C.), a neutral pH condition (for example, pH 6) for solubility under an acidic pH condition (for example, pH 2.5). ), The solubility ratio in) is usually 50% or less, and among them, compounds having a solubility ratio of 30% or less, 20% or less, or 10% or less, particularly 5% or less are included.
- examples of the pharmaceutically active ingredient in the pharmaceutical composition of the present invention include compounds having a basic nitrogen-containing heterocycle and compounds having a basic functional group, for example, an unsubstituted or substituted amino group. ..
- the type of the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is not limited as long as it has a pH-dependent dissolution profile, and examples thereof include the pharmaceutically active ingredient capable of forming a stable crystal structure.
- examples of such pharmaceutically active ingredients include aromatic compounds having a ⁇ -conjugated system and capable of causing ⁇ - ⁇ stacking.
- ⁇ - ⁇ stacking means a phenomenon in which two or more aromatic compound molecules are stabilized by ⁇ - ⁇ interaction in a state of being stacked on a plane (stacking).
- Aromatic compounds that can cause ⁇ - ⁇ stacking are generally capable of forming stable crystal structures and have low solubility in aqueous media. In particular, by applying the present invention to such aromatic compounds, a remarkable effect of improving the dissolution profile can be obtained.
- examples of the pharmaceutically active ingredient in the pharmaceutical composition of the present invention include aromatic compounds having a large ⁇ -conjugated system, for example, compounds having a fused aromatic ring and / or a plurality of linked aromatic rings. Since such compounds are liable to cause ⁇ - ⁇ stacking, which in turn is likely to crystallize and cause deterioration of solubility, the effect of improving the dissolution profile by applying the pharmaceutical composition of the present invention becomes more remarkable.
- examples of such a pharmaceutically active ingredient include, but are not limited to, compounds having a structure represented by the following general formula I.
- ring A may have one or more substituents, a 5- to 16-membered monocyclic or fused bicyclic or tricyclic aromatic hydrocarbon group or aromatic complex. Represents a cyclic group.
- ring B may have one or more substituents, a 5- to 12-membered monocyclic or fused bicyclic aromatic hydrocarbon group or aromatic heterocyclic group. Represents.
- the aromatic hydrocarbon group includes, but is not limited to, a group selected from the following group. (Note that the following is indicated by the name of a monovalent group. When r is 2, the ring A becomes a divalent group obtained by removing one more hydrogen atom from the following monovalent group.) .. -(Monocyclic) phenyl group. -(Condensation bicycle) indenyl group, naphthyl group, azrenyl group. -(Condensation tricycle) anthrasenyl group, phenanthonasenyl group, fluorenyl group.
- the aromatic heterocyclic group is one or more heteros selected from a nitrogen atom, an oxygen atom, and a sulfur atom. Contains atoms.
- the type of the aromatic heterocyclic group is not limited to these, and examples thereof include a group selected from the following groups (note that the following are indicated by the names of monovalent groups; n is 2). In some cases, ring A becomes a divalent group obtained by further removing one hydrogen from the following monovalent group).
- -(Monocyclic) pyrrolyl group pyrazolyl group, imidazolyl group, triazolyl group, furanyl group, thiophenyl group, oxazolyl group, isoxazolyl group, thiazolyl group, oxadiazolyl group, thiadiazolyl group, pyridinyl group, pyridazinyl group, pyrimidinyl group, pyrazinyl group, Triazinyl group, pyranyl group.
- -(Condensation tricycle) carbazolyl group dibenzofuranyl group, acridinyl group, phenazinyl group, phenoxadinyl group, phenothiadinyl group, phenoxatiynyl group.
- L represents a divalent or trivalent linking group which may have one or more substituents. Specifically, when q is 1, L is a divalent linking group, and when q is 2, L is a trivalent linking group.
- the type of linking group is not particularly limited, but is preferably a group that can be conjugated with ring A and / or ring B.
- examples of the linking group of L include, but are not limited to, a group selected from the following group.
- -Alkyl group (methyl group, ethyl group, propyl group (n-propyl group, isopropyl group), butyl group (n-butyl group, sec-butyl group, isobutyl group, tert-butyl group), pentyl group, hexyl group, A divalent or trivalent group obtained by removing one or two hydrogen atoms from a heptyl group, an octyl group, a nonyl group, a decyl group), an alkenyl group, or an alkynyl group.
- the number of carbon atoms of the group is not particularly limited, but is usually 1 to 10, preferably 1 to 7, and more preferably 1 to 5.
- -Carbonyl group A divalent or trivalent group, an amide group obtained by further removing one or two hydrogen atoms from an azo group, a diazo group, or a (primary, secondary, or tertiary) amino group.
- -Sulfide group sulfinyl group, sulfonyl group.
- -Oxy group dioxy group.
- linking groups for example, an alkoxy group, an alkenyloxy group, a carboxy group, a sulfinyloxy group, a sulfonyloxy group, a carbonylamino group. Included in the example of linking groups. Further, the connecting positions of the linking group and the rings A and B are not particularly limited and are arbitrary.
- p is an integer of 0 or 1.
- ring A and ring B are connected by a single bond.
- q is an integer of 1 or 2.
- the plurality of rings B may be the same as each other or may be different from each other.
- r is an integer of 1 or 2.
- the plurality of B, L, p, and q may be the same as each other or may be different from each other.
- the substituent which the aromatic hydrocarbon group or the aromatic heterocyclic group of the ring A and the ring B may have, and the substituent which the linking group L may have may be used as the substituent.
- substituents include, but are not limited to, groups selected from the following groups.
- -Halogen atom hydroxyl group, carboxyl group, nitro group, cyano group, thiol group, sulfinic acid group, sulfonic acid group, amino group, amide group, imino group, imide group.
- -Hydrocarbon group hydrocarbon oxy group, hydrocarbon carbonyl group (acyl group), hydrocarbon oxycarbonyl group, hydrocarbon carbonyloxy group, hydrocarbon substituted amino group, hydrocarbon substituted aminocarbonyl group, hydrocarbon carbonyl substituted amino group , Hydrocarbon substituted thiol group, hydrocarbon sulfonyl group, hydrocarbon oxysulfonyl group, hydrocarbon sulfonyloxy group.
- the "hydrocarbon” group may be an aliphatic or aromatic, or a combination thereof.
- Aliphatic hydrocarbons may be chained or cyclic, or may be a combination thereof.
- the chain hydrocarbon may be linear or branched.
- the cyclic hydrocarbon may be a monocyclic type or a compound ring type, and in the case of the compound ring type, it may be a condensed ring type, a bridged ring type, a spiro ring type, or a combination thereof.
- the "hydrocarbon” group may be saturated or unsaturated, in other words, it may contain one or more carbon-carbon double bonds.
- the "hydrocarbon” group is a concept including an alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, a cycloalkenyl group, a cycloalkynyl group, an aryl group and the like.
- the number of carbon atoms of the "hydrocarbon” group is not particularly limited, but in the case of a chain hydrocarbon group, it is usually 1 to 20, preferably 1 to 15, more preferably 1 to 12, and in the case of a cyclic hydrocarbon group. , Usually 3 to 20, preferably 4 to 15, and even more preferably 5 to 12.
- one or more hydrogen atoms of the "hydrocarbon” group may be substituted with any substituent, and one or more carbon atoms of the hydrocarbon may have a valence. It may be replaced with any suitable heteroatom.
- the type of heteroatom is not limited, and examples thereof include a nitrogen atom, an oxygen atom, a sulfur atom, a phosphorus atom, and a silicon atom.
- heterocyclic oxy group heterocyclic carbonyl group, heterocyclic oxycarbonyl group, heterocyclic carbonyloxy group, heterocyclic amino group, heterocyclic aminocarbonyl group, heterocyclic carbonyl substituted amino group, heterocyclic substituted thiol Group, heterocyclic sulfonyl group, heterocyclic oxysulfonyl group, heterocyclic sulfonyloxy group.
- the "heterocyclic” group may be saturated or unsaturated, in other words, it may contain one or more carbon-carbon double bonds. In the case of unsaturated, it may or may not have aromaticity. Further, the "heterocyclic” group may be a monocyclic type or a compound ring type, and in the case of a compound ring type, it may be a fused ring type, a bridged ring type or a spiro ring type. The number of ring-constituting atoms of the "heterocyclic” group is not particularly limited, but is usually 3 or more, 4 or more, or 5 or more, and usually 20 or less, 15 or less, or 12 or less.
- heteroatom contained in the "heterocycle” group is not limited, and examples thereof include a nitrogen atom, an oxygen atom, a sulfur atom, a phosphorus atom, and a silicon atom.
- the number of heteroatoms is also not particularly limited, but is usually 1 to 8, preferably 1 to 5, and more preferably 1 to 3.
- heterocyclic When the "heterocyclic" group contains two or more heteroatoms, these heteroatoms may be the same or different from each other.
- a functional group in which any group of the above-mentioned substituents is further substituted with one or more of the above-mentioned substituents as long as its valence and physicochemical properties allow It shall be included in the "substituent" in the present invention.
- the number thereof is not particularly limited as long as its valence and physicochemical properties allow.
- these substituents may be the same or different from each other.
- the number of the substituents may be one, but may be two or more. good.
- these substituents may be the same or different from each other.
- the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is selected from the compounds listed in Table 1 below.
- the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is selected from cerabellisive, alpericib, afatinib, gefitinib, bosutinib, alectinib, palbociclib, tacericib, and copanricib.
- the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is cerabellisive or alpericive.
- the pharmaceutical composition of the present invention may contain any one of these pharmaceutically active ingredients alone, or may contain two or more of them in any combination and ratio.
- the pharmaceutically active ingredient in the pharmaceutical composition of the present invention does not maintain a stable crystal structure in the pharmaceutical composition of the present invention and is in a partially amorphous state.
- the pharmaceutically active ingredient is an aromatic compound capable of causing ⁇ - ⁇ stacking
- such an aromatic compound causes ⁇ - ⁇ stacking. It does not exist, and each molecule exists in a separated state. The method for incorporating the pharmaceutically active ingredient in the pharmaceutical product in an amorphous state will be described later.
- the content of the pharmaceutically active ingredient in the pharmaceutical composition of the present invention may be appropriately selected according to various conditions such as the type of the pharmaceutically active ingredient, indication, dosage form, administration route and the like.
- the content of the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is, for example, 2% by mass or more, 5% by mass or more, 10% by mass or more, or 15% by mass or more, and for example. It is 40% by mass or less, 35% by mass or less, 30% by mass or less, or 25% by mass or less.
- the pharmaceutical composition of the present invention is one or two kinds having no pH-dependent dissolution profile in addition to the pharmaceutically active ingredient having a pH-dependent dissolution profile within the range not deviating from the gist of the present invention. It may contain the above additional pharmaceutically active ingredients.
- the pharmaceutical composition of the present invention contains an inorganic or organic acid.
- the inorganic or organic “acid” means an inorganic or organic compound (so-called Bronsted acid) capable of donating a proton (H +).
- Bronsted acid an inorganic or organic compound capable of donating a proton (H +).
- the acid in the pharmaceutical composition of the present invention is not limited, but the acid dissociation constant (pKa value) is, for example, 4.2 or less, 3.1 or less, or 2.2 or less. It is an acid.
- the acid dissociation constant (pKa value) in the present disclosure means an acid dissociation constant in water, and in the case of a polyvalent acid, one hydrogen ion is released. It shall mean the acid dissociation constant (so-called pKa 1 value) related to the reaction.
- An acid having an acid dissociation constant (pKa value) of not less than or equal to the upper limit facilitates protonation of the basic pharmaceutically active ingredient and solubilizes the pharmaceutically active ingredient having a pH-dependent dissolution profile.
- the acid in the pharmaceutical composition of the present invention is an acid having a molecular mass within a predetermined range.
- the molecular mass of the acid is not limited, but is usually 30 or more and 200 or less.
- Specific examples of the acid in the pharmaceutical composition of the present invention are not limited, but according to one embodiment, phosphoric acid, hydrochloric acid, citric acid, malic acid, tartaric acid, ascorbic acid, fumaric acid, succinic acid, and the like. Examples thereof include aspartic acid, lactic acid, acetic acid, glutamic acid and adipic acid. According to one aspect, specific examples of the acid include phosphoric acid, hydrochloric acid, citric acid, malic acid, tartaric acid and the like. According to one aspect, specific examples of the acid in the pharmaceutical composition of the present invention include phosphoric acid, citric acid, and tartaric acid.
- the pharmaceutical composition of the present invention may contain any one of these acids alone, or may contain two or more of them in any combination and ratio.
- the acid in the pharmaceutical composition of the present invention is a compound having two or more acidic functional groups.
- compounds having two or more acidic functional groups are not limited, but are not limited to compounds having two or more hydroxyl groups (for example, phosphoric acid) and compounds having two or more carboxyl groups (for example, citric acid and apple). Acids, tartrate acids, etc.), compounds having one or more hydroxyl groups and one or more carboxyl groups (eg, lactic acid, etc.) and the like can be mentioned.
- the acid content in the pharmaceutical composition of the present invention is not limited, but is, for example, 5% by mass or more, 10% by mass or more, or 15% by mass or more, and for example. It is 50% by mass or less, 45% by mass or less, or 40% by mass or less.
- the mass ratio of the inorganic or organic acid to the pharmaceutically active ingredient in the pharmaceutical composition of the present invention is, for example, 0.1 mass by mass of the inorganic or organic acid with respect to 1 part by mass of the pharmaceutically active ingredient. It is 10 parts by mass or more, 0.3 parts by mass or more, 0.5 parts by mass or more, and for example, 10 parts by mass or less, 7.5 parts by mass or less, or 5 parts by mass or less.
- the pharmaceutical composition of the present invention may contain one or more unit-priced inorganic or organic acids in addition to the inorganic or organic acids as long as the gist of the present invention is not deviated.
- Hydrophilic polymer The pharmaceutical composition of the present invention contains a hydrophilic polymer.
- a hydrophilic polymer intervenes between the molecules of the pharmaceutically active ingredient to prevent its reaggregation and crystallization. By doing so, it is presumed that the dissolution profile is stabilized regardless of pH.
- the "hydrophilic polymer” means a polymer having a property of being soluble in water at room temperature. Specifically, it means a polymer having a viscosity of 1 to 10% aqueous solution of 1 mPa ⁇ s or more under the conditions of 20 ° C. and normal pressure.
- the hydrophilic polymer in the pharmaceutical composition of the present invention is not limited, but examples thereof include those having a carboxyl group, a hydroxyl group, a sulfo group, an amide group and the like as hydrophilic substituents. ..
- Such hydrophilic polymers can prevent their reaggregation and crystallization through hydrophobic interactions between the molecules of the pharmaceutically active ingredient, which in turn can contribute to the stabilization of its dissolution profile.
- hydrophilic polymer in the pharmaceutical composition of the present invention are not limited, but according to one embodiment, polyvinyl alcohol, povidone, hypromellose, copolyvidone, hydroxypropyl cellulose, polyvinyl alcohol / polyethylene glycol graft. Examples thereof include polymers, hypromellose phthalates, hypromellose acetates and succinates. According to a specific embodiment, specific examples of the hydrophilic polymer in the pharmaceutical composition of the present invention include polyvinyl alcohol, povidone, hypromellose, hydroxypropyl cellulose and the like.
- the pharmaceutical composition of the present invention may contain any one of these hydrophilic polymers alone, or may contain two or more of them in any combination and ratio.
- the content of the hydrophilic polymer in the pharmaceutical composition of the present invention is not limited, but is, for example, 1% by mass or more, 2% by mass or more, or 3% by mass or more, and also. For example, 15% by mass or less, 12.5% by mass or less, or 10% by mass or less.
- the mass ratio of the hydrophilic polymer to the pharmaceutically active ingredient is, for example, usually 0.02 parts by mass or more, or 0.05 parts by mass or more, based on 1 part by mass of the pharmaceutically active ingredient. , Or 0.1 parts by mass or more, and usually 5 parts by mass or less, 3 parts by mass or less, or 2 parts by mass or less.
- the pharmaceutical composition of the present invention may contain one or more unit-priced inorganic or organic acids in addition to the inorganic or organic acids as long as the gist of the present invention is not deviated.
- the pharmaceutical composition of the present invention may further contain other components.
- other components include, but are not limited to, excipients such as mannitol, erythritol, powdered reduced malt sugar water candy, crystalline cellulose, and corn starch; crospovidone, croscarmellose sodium, and sodium starch glycolate.
- Disintegrants such as partially pregelatinized starch, carmellose calcium, low substitution hydroxypropyl cellulose; sweeteners such as sucralose, aspartame, acesulfame potassium, saccharin sodium, monoammonium glycyrrhizinate, taumatin; fluidizers, colorants, fragrances, etc.
- Other additives such as.
- the pharmaceutical composition of the present invention may contain any one of these components alone, or may contain two or more of them in any combination and ratio.
- the ingredients that can be used in the pharmaceutical composition of the present invention see, for example, University of the Sciences in Philadelphia, "Remington: The Sciences and Practice and Practice, Lippincot, 20th EDIT, etc.” It can be taken into consideration as appropriate.
- Dosage form of the product is not limited, and any dosage form that can be orally administered is arbitrary. Examples thereof include various oral dosage forms described in the Japanese Pharmacopoeia, that is, tablets, capsules, granules, powders, oral solutions, syrups, oral jellies and the like. These dosage forms may be appropriately selected depending on the pharmaceutically active ingredient and the type of indication.
- the pharmaceutical composition of the present invention is in the form of a dry syrup.
- dry syrup is a subclass of "syrup” described in the Japanese Pharmacopoeia, and is a granule that can be reconstituted into a syrup by adding water at the time of administration and dissolving or suspending at the time of use. It means a dry solid agent in the form of a powder or a powder.
- Such a dry syrup preparation is a pharmaceutical active ingredient (for example, ART-001, alpericive, etc.) that is mainly administered to children in terms of its storage stability, portability, ease of administration, and the like. It is advantageous in some cases.
- the method for producing the pharmaceutical composition of the present invention is not limited, and any known method according to the dosage form may be used.
- a pharmaceutically active ingredient, an inorganic or organic acid, and a hydrophilic polymer, and optionally other ingredients may be mixed using a known pharmaceutical method, and granulation or the like may be appropriately performed. It is sufficient to add the processing of.
- the raw material mixture may be granulated by using a known granulation method such as a dry granulation method or a wet granulation method.
- the granulated product of the raw material mixture obtained by a known granulation method may be compressed into tablets, and the obtained tablets may be coated as necessary.
- the raw material mixture may be directly tableted by a direct tableting method without going through a granulation step.
- the raw materials are a pharmaceutically active ingredient, an inorganic or organic acid, and a hydrophilic polymer, and optionally other ingredients. It can be produced by a method of processing by the following steps (appropriately referred to as "the production method of the present invention"). -A step of mixing the raw materials in an arbitrary mixing order. -A step of wet granulating the mixture. -A step of drying the wet granulated product.
- raw material components other than the pharmaceutical active ingredient may be added later to the raw material mixture after wet granulation, if desired.
- the solvent used at the time of granulation is not particularly limited, and any known solvent can be used. Examples include water, ethanol, methanol, acetone, dichloromethane and the like. Among them, water, ethanol and the like can be mentioned from the viewpoint of handling. Any one of these solvents may be used alone, or two or more of them may be used in any combination and ratio. Such a solvent and each raw material component are mixed and subjected to a granulation step.
- the granulation method to be used is not particularly limited, and any known granulation method can be used.
- the granulation method is a fluidized bed granulation method.
- the wet granulator is not particularly limited, and any known granulator can be used depending on the solvent and the granulation method used. Examples include a cylindrical extrusion granulator, a rolling fluidized bed granulator, a spray drying granulator, a fluidized bed granulator, a stirring granulator and the like.
- the granulator is a rolling fluidized bed granulator, a fluidized bed granulator, or the like.
- a step of wet or dry crushing each raw material component a step of sieving each raw material component or raw material mixture, a step of coating the granulated product with a predetermined coating material, etc.
- steps well known in the pharmaceutical field may be added.
- the pharmaceutical composition of the present invention is orally administered to a subject for the purpose of treating / preventing / controlling a disease or condition according to the type of the pharmaceutically active ingredient.
- the administration target of the pharmaceutical composition of the present invention is not particularly limited, and examples thereof include humans, mammals other than humans, and other animals.
- the dose and frequency of administration of the pharmaceutical composition of the present invention are not particularly limited, but may be appropriately adjusted according to, for example, the type and activity of the pharmaceutical active ingredient, the species to be administered, age, body weight, condition and the like.
- two or more kinds of pharmaceutical compositions of the present invention containing two or more kinds of pharmaceutical active ingredients individually may be administered to the same subject simultaneously or continuously, and the pharmaceutical composition of the present invention containing a certain pharmaceutical active ingredient may be administered.
- the product and another pharmaceutical composition containing another pharmaceutical active ingredient may be administered to the same subject simultaneously or continuously.
- the first pharmaceutical agent having pH-dependent solubility is administered.
- the pharmaceutical composition of the present invention in which the active ingredient is formulated is combined with a conventional pharmaceutical composition in which a second pharmaceutical active ingredient whose solubility does not depend on pH is formulated, and these are simultaneously or continuously targeted. It may be administered.
- the pharmaceutical composition administered in combination with the pharmaceutical composition of the present invention may be orally administered, or may be administered by another route.
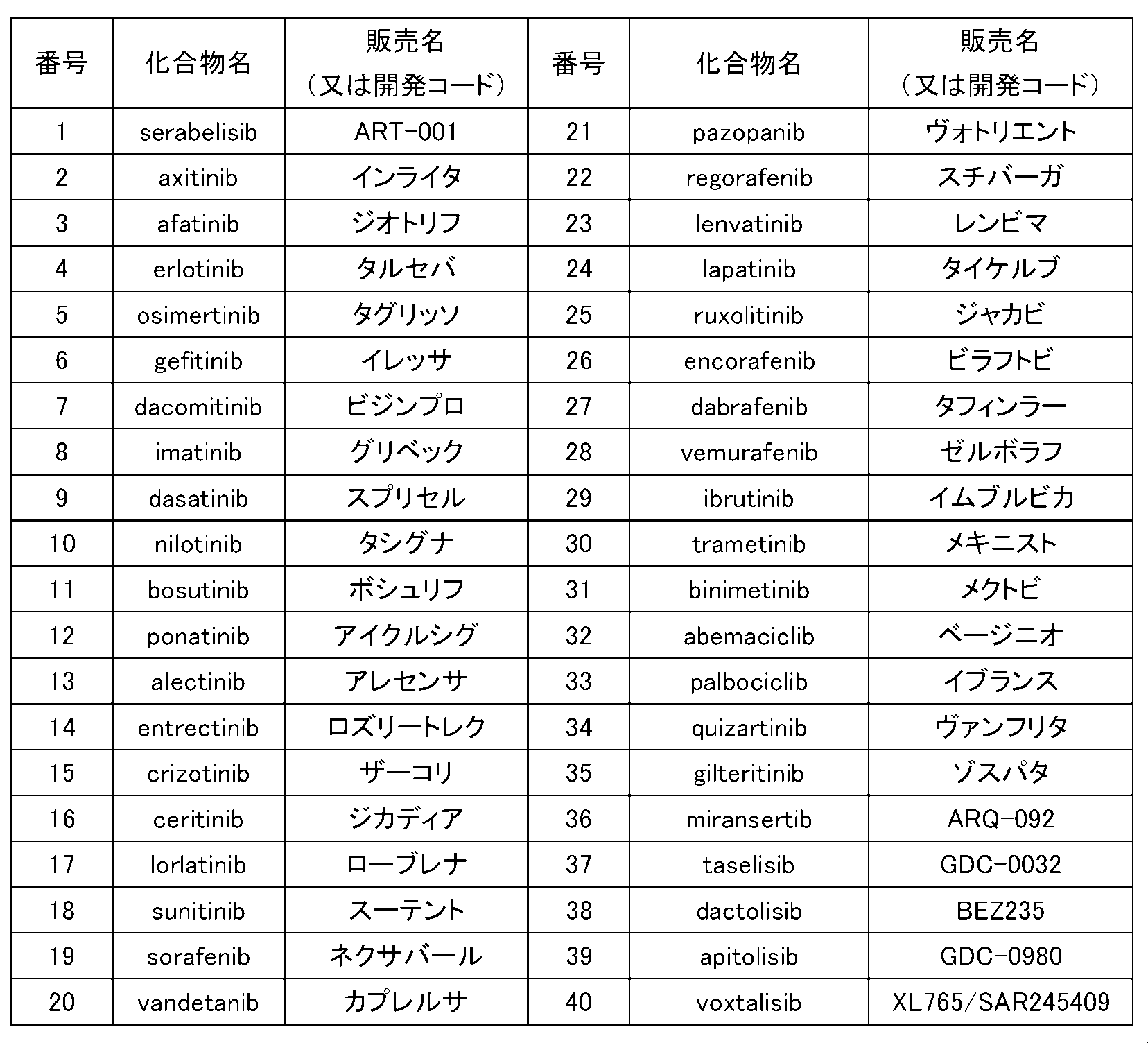
- ART-001 exhibits extremely high solubility under low pH conditions (eg pH 2.5 or less) at room temperature (RT) (solubility 312 mcg / mL to about 10,000 mcg / mL).
- RT room temperature
- solubility 312 mcg / mL to about 10,000 mcg / mL.
- high pH conditions for example, pH 6 or higher
- solubility 1.3 mcg / mL: 100% solubility under low pH conditions Then about 4%). From this, it can be seen that ART-001 has a highly pH-dependent dissolution profile.
- -Formulation B (1) 270 g of D-mannitol (Bussan Food Science Co., Ltd.) and 30 g of crystalline cellulose (KG-1000, Asahi Kasei Co., Ltd.) passed through a 42M sieve were mixed. (2) 75 g of phosphoric acid (Domestic Chemical Co., Ltd.) and 25 g of popidone (PLASSONE K-25, ASHLAND) were dissolved in 600 g of purified water. (3) 100 g of ART-001 (Carbogen Amcis AG) was added to the solution of (2) above and dispersed.
- ART-001 Carbogen Amcis AG
- -Formulation C (1) 48 g of crystalline cellulose (KG-1000, Asahi Kasei Co., Ltd.) and 32 g of polyvinyl alcohol (partially saponified product) (Gosenol EG-05PW, Mitsubishi Chemical Co., Ltd.) were mixed. (2) 160 g of D-mannitol (Busan Food Science Co., Ltd.) passed through a 42M sieve was mixed with the mixture of the above (1). (3) 120 g of phosphoric acid (Taipei Chemical Industry Co., Ltd.) and 40 g of popidone (PLASSONE K-25, ASHLAND) were dissolved in 960 g of purified water.
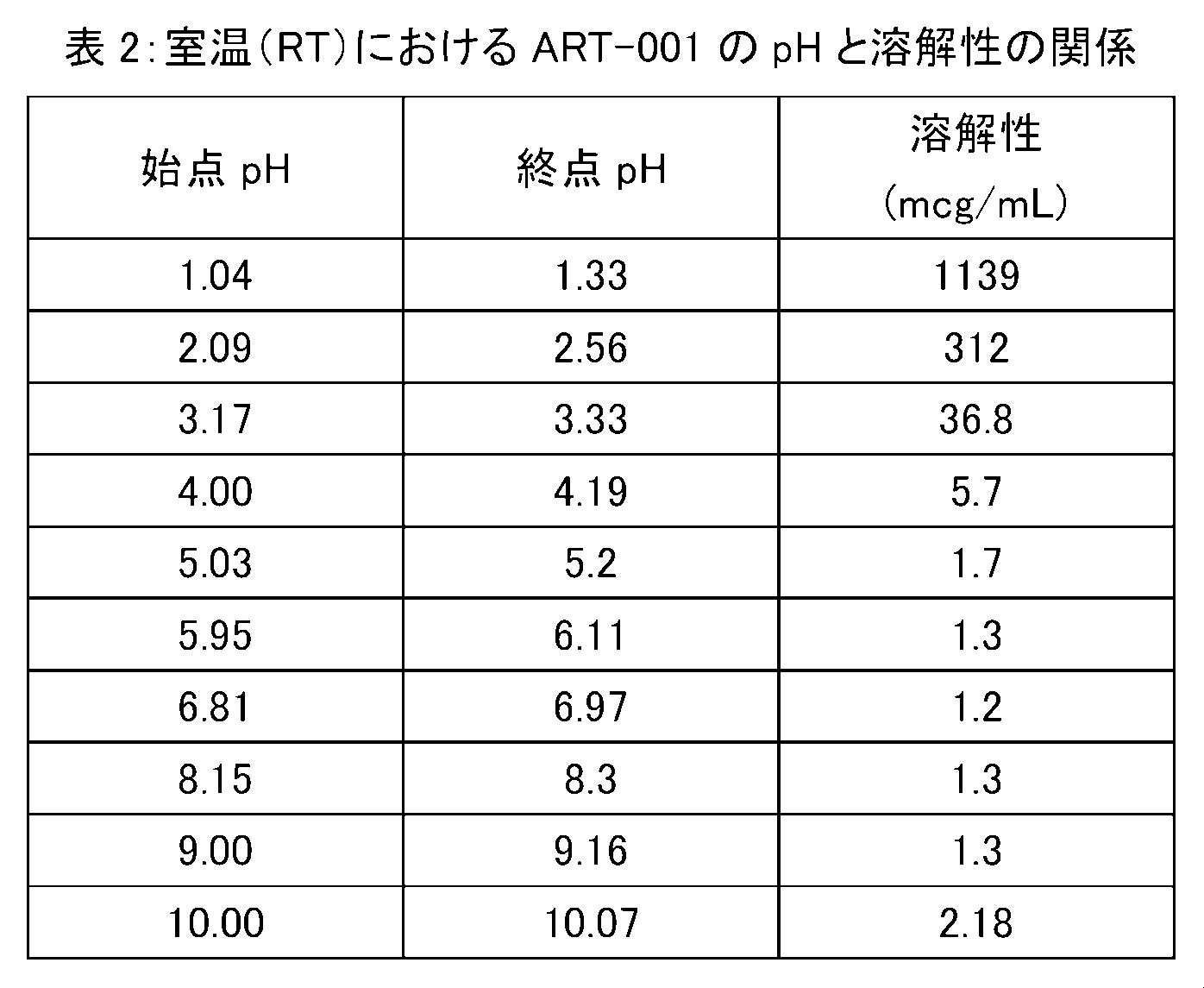
- ART-001 dry syrup preparation The dry syrup preparation of ART-001 prepared in the above procedure was administered to dogs according to the procedure described in detail below, and the blood pharmacokinetics of ART-001 was examined.
- a preparation in which ART-001 was suspended in a 0.5% methylcellulose (MC; METOROSE, SM-100 manufactured by Shin-Etsu Chemical Co., Ltd.) aqueous solution was also prepared, and similarly administered to dogs, ART- The blood dynamics of 001 were examined.
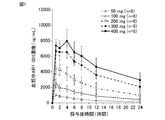
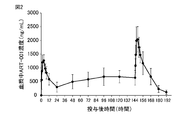
- the dry syrup preparation of ART-001 was dissolved in 100 mL of water at doses of 50 mg, 100 mg, 200 mg, 300 mg, or 400 mg as ART-001, respectively, and each was simply administered to 6 healthy adult males. It was orally administered once. Blood was collected over time from before administration to 48 hours after administration, and the plasma concentration of ART-001 (ng / mL) was measured by HPLC. In the repeated administration test, the dry syrup preparation of ART-001 was dissolved in 100 mL of water at a dose of 100 mg as ART-001 and orally administered to 6 healthy adult males once a day for 7 days over time. Blood was collected in.
- the dry syrup preparation corresponding to the pharmaceutical composition of the present invention can be used as a conventional capsule or tablet when used for administration of ART-001, which is an example of a pharmaceutically active ingredient having a pH-dependent dissolution profile.
- ART-001 which is an example of a pharmaceutically active ingredient having a pH-dependent dissolution profile.
- the tight and stable pharmacokinetics obtained by such a dry syrup preparation facilitates dose selection for obtaining blood exposure with expected efficacy, and also enables avoidance of side effects due to high exposure dose.
- the present invention is widely applicable in the pharmaceutical field, especially for administering various pharmaceutically active ingredients having a pH-dependent dissolution profile, and its utility value is extremely high.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Zoology (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
[項1]pH依存性の溶解プロファイルを有する医薬活性成分と、無機又は有機の酸と、親水性ポリマーとを含む経口用医薬組成物。
[項2]医薬活性成分が、π共役系を有する芳香族化合物である、項1に記載の経口用医薬組成物。
[項3]医薬活性成分が、下記一般式Iで表される構造を有する化合物である、項1又は2に記載の経口用医薬組成物。
環Aは、1又は2以上の置換基を有していてもよい、5~16員の単環式又は縮合二環若しくは三環式の芳香族炭化水素基又は芳香族複素環式基を表し、
環Bは、1又は2以上の置換基を有していてもよい、5~12員の単環式又は縮合二環式の芳香族炭化水素基又は芳香族複素環式基を表し、
Lは、1又は2以上の置換基を有していてもよい、二価又は三価の連結基を表し、
pは、0又は1の整数を表し、
qは、1又は2の整数を表し、
rは、1又は2の整数を表す。)
[項4]医薬活性成分が、ART-001(セラベリシブ)、アルペリシブ、アファチニブ、ゲフィチニブ、ボスチニブ、アレクチニブ、パルボシクリブ、タセリシブ、及びコパンリシブから選択される1種又は2種以上の化合物である、項1~3の何れか一項に記載の経口用医薬組成物。
[項5]医薬活性成分のpH2.5での溶解性に対するpH6での溶解性の比率が、50%以下、又は30%以下、又は20%以下、又は10%以下、又は5%以下である、項1~4の何れか一項に記載の経口用医薬組成物。
[項6]無機又は有機の酸が、リン酸、塩酸、クエン酸、リンゴ酸、酒石酸、アスコルビン酸、フマル酸、コハク酸、アスパラギン酸、乳酸、酢酸、グルタミン酸、及びアジピン酸から選択される1種又は2種以上の酸である、項1~5の何れか一項に記載の経口用医薬組成物。
[項7]親水性ポリマーが、ポリビニルアルコール、ポビドン、ヒプロメロース、コポリビドン、ヒドロキシプロピルセルロース、ポリビニルアルコール・ポリエチレングリコール・グラフトコポリマー、ヒプロメロースフタル酸エステル、及びヒプロメロース酢酸エステルコハク酸エステルから選択される1種又は2種以上のポリマーである、項1~6の何れか一項に記載の経口用医薬組成物。
[項8]医薬活性成分の含有率が、2質量%以上、又は5質量%以上、又は10質量%以上、又は15質量%以上、また、40質量%以下、又は35質量%以下、又は30質量%以下、又は25質量%以下である、項1~7の何れか一項に記載の経口用医薬組成物。
[項9]医薬活性成分1質量部に対する無機又は有機の酸の含有割合が、0.1質量部以上、又は0.3質量部以上、又は0.5質量部以上、また、10質量部以下、又は7.5質量部以下、又は5質量部以下の範囲である、項1~8の何れか一項に記載の経口用医薬組成物。
[項10]医薬活性成分1質量部に対する親水性ポリマーの含有割合が、0.02質量部以上、又は0.05質量部以上、又は0.1質量部以上、また、5質量部以下、又は3質量部以下、又は2質量部以下の範囲である、項1~9の何れか一項に記載の経口用医薬組成物。
[項11]ドライシロップ剤である、項1~10の何れか一項に記載の経口用医薬組成物。
[項12]項1~11の何れか一項に記載の経口用医薬組成物を製造する方法であって、医薬活性成分、無機又は有機の酸、及び親水性ポリマーを含む混合物を湿式造粒し、乾燥することを含む方法。
[項13]項12に記載の方法により製造される経口用医薬組成物。
本発明の一態様は、pH依存性の溶解プロファイルを有する医薬活性成分を経口投与するための医薬組成物(適宜「本発明の医薬組成物」と称する。)に関する。本発明の医薬組成物は、斯かるpH依存性の溶解プロファイルを有する医薬活性成分に加えて、無機又は有機の酸と、親水性ポリマーとを含有する。
本発明の医薬組成物は、pH依存性の溶解プロファイルを有する医薬活性成分を含有する。

・(単環)フェニル基。
・(縮合二環)インデニル基、ナフチル基、アズレニル基。
・(縮合三環)アントラセニル基、フェナントナセニル基、フルオレニル基。
・(縮合二環)インデニル基、インドリル基、イソインドリル基、インドリジニル基、インダゾリル基、ベンゾイミダゾリル基、アザインドリル基、アザインダゾリル基、ピラゾロピリミジニル基、プリニル基、ベンゾフラニル基、イソベンゾフラニル基、ベンゾチオフェニル基、ベンゾオキサゾリル基、ベンゾチアゾリル基、ベンゾイソオキサゾリル基、ベンゾイソチアゾリル基、キノリニル基、イソキノリニル基、キノリジニル基、キノキサリニル基、フタラジニル基、キナゾリニル基、ナフチリジニル基、ピリドピリミジニル基、ピリドピラジニル基、ベンゾピラニル基。
・(縮合三環)カルバゾリル基、ジベンゾフラニル基、アクリジニル基、フェナジニル基、フェノキサジニル基、フェノチアジニル基、フェノキサチイニル基。
・カルボニル基。
・アゾ基、ジアゾ基、(一級、二級、又は三級)アミノ基から更に1つ又は2つの水素原子を取り去って得られる二価又は三価の基、アミド基。
・スルフィド基、スルフィニル基、スルホニル基。
・オキシ基、ジオキシ基。
また、前記の連結基と環A及び環Bとの連結位置も特に制限されず、任意である。
本発明の医薬組成物は、無機又は有機の酸を含有する。本開示において無機又は有機の「酸」とは、プロトン(H+)を供与し得る無機又は有機の化合物(いわゆるブレンステッド酸)を意味する。理論に束縛されるものではないが、本発明の医薬組成物中では、斯かる酸が医薬活性成分の分子間に介在して存在することにより、その溶解プロファイルがpHによらず安定化するものと推測される。
本発明の医薬組成物は、親水性ポリマーを含有する。理論に束縛されるものではないが、本発明の医薬組成物中では、前記の酸に加えて、斯かる親水性ポリマーが医薬活性成分の分子間に介在してその再凝集や結晶化を防止することにより、その溶解プロファイルがpHによらず安定化するものと推測される。
本発明の医薬組成物は、更にその他の成分を含有していてもよい。その他の成分の具体例としては、制限されるものではないが、マンニトール、エリスリトール、粉末還元麦芽糖水アメ、結晶セルロース、トウモロコシデンプン等の賦形剤;クロスポビドン、クロスカルメロースナトリウム、デンプングリコール酸ナトリウム、部分アルファー化デンプン、カルメロースカルシウム、低置換度ヒドロキシプロピルセルロース等の崩壊剤;スクラロース、アスパルテーム、アセスルファムカリウム、サッカリンナトリウム、グリチルリチン酸モノアンモニウム、タウマチン等の甘味剤;流動化剤、着色剤、香料等のその他の添加物が挙げられる。これらの成分は、本発明の医薬組成物の剤形等に応じて、適宜選択して使用することが可能である。なお、本発明の医薬組成物は、これらの成分のうち何れか1種を単独で含んでいてもよく、2種以上を任意の組み合わせ及び比率で含んでいてもよい。なお、本発明の医薬組成物に使用可能な成分の詳細については、例えばUniversity of the Sciences in Philadelphia, “Remington: The Science and Practice of Pharmacy, 20th EDITION”, Lippincott Williams & Wilkins, 2000等の記載を適宜参酌することができる。
本発明の医薬組成物の剤形は限定されず、経口投与可能な剤形であれば任意である。例としては、日本薬局方に記載される各種の経口投与剤形、即ち錠剤、カプセル剤、顆粒剤、散剤、経口液剤、シロップ剤、経口ゼリー剤等が挙げられる。これらの剤形は、医薬活性成分や適応の種類に応じて、適宜選択すればよい。
本発明の医薬組成物を製造する方法は制限されず、剤形に応じた任意の公知の方法により製造すればよい。例えば、目的とする剤形に応じて、医薬活性成分、無機又は有機の酸、及び親水性ポリマー、並びに任意により用いられる他の成分を、公知の製剤手法を用いて混合し、適宜造粒等の処理を加えればよい。例えば顆粒製剤の場合には、乾式造粒法、湿式造粒法等の公知の造粒法を用いて原料混合物を造粒すればよい。錠剤の場合には、公知の造粒法によって得られた原料混合物の造粒物を圧縮して錠剤化し、得られた錠剤に必要に応じてコーティングを施せばよい。或いは、造粒工程を経ずに原料混合物を直接打錠法により直接打錠して製造してもよい。
・前記原料を任意の混合順で混合する工程。
・前記混合物を湿式造粒する工程。
・前記湿式造粒物を乾燥する工程。
本発明の医薬組成物は、医薬活性成分の種類に応じた疾患や状態の治療・予防・調節等の目的で、対象に対して経口投与される。本発明の医薬組成物の投与対象は特に制限されないが、例としてはヒト、ヒト以外の哺乳類、その他の動物等が挙げられる。本発明の医薬組成物の投与量や投与回数も特に制限されないが、例えば医薬活性成分の種類や活性、投与対象の種、年齢、体重、状態等に応じて、適宜調整すればよい。また、2種以上の医薬活性成分を個別に含む2種以上の本発明の医薬組成物を、同一対象に同時に又は連続して投与してもよく、ある医薬活性成分を含む本発明の医薬組成物と、別の医薬活性成分を含む別の医薬組成物を、同一対象に同時に又は連続して投与してもよい。特に、pH依存溶解性を有する第1の医薬活性成分と、溶解性がpHに依存しない第2の医薬活性成分とを同一対象に投与する場合には、pH依存溶解性を有する第1の医薬活性成分を製剤化した本発明の医薬組成物と、溶解性がpHに依存しない第2の医薬活性成分を製剤化した従来の医薬組成物とを組み合わせて、これらを対象に同時に又は連続して投与してもよい。なお、本発明の医薬組成物との組み合わせで投与される医薬組成物は、経口投与されるものであってもよいが、別の経路で投与されるものであってもよい。
様々なpH環境下におけるART-001の溶解プロファイルを、以下の手順により試験した。即ち、6規定塩酸又は10規定水酸化ナトリウム水溶液を用いて、pHを1~10の範囲内で種々調整した、50mMリン酸緩衝液(pH1、2、7、及び8)、クエン酸緩衝液(pH3、4、5、及び6)、ホウ酸緩衝液(pH9)、及び重炭酸緩衝液(pH10)に対して、過剰量のART-001を加え、室温下で攪拌した。溶解状態が平衡に到達した時点で一定量のサンプルを採取し、1000rpm、10分間の遠心分離を行ったのち、上清を採取しHPLCにて測定した。溶解始点及び溶解終点のpHも同時に測定し、記録した。
以下の手順によりART-001のドライシロップ製剤を調製した。
(1)42Mの篩を通したD-マンニトール(物産フードサイエンス(株))270g及び結晶セルロース(KG-1000、旭化成(株))30gを混合した。
(2)リン酸(国産化学(株))75g及びポピドン(PLASDONE K-25、ASHLAND)25gを精製水600gに溶解させた。
(3)ART-001(Carbogen Amcis AG)100gを前記(2)の溶液に加えて分散させた。
(4)前記(1)の混合物を流動層造粒乾燥機(FD-MP-01D/SFP型、(株)パウレック)に投入し、前記(3)の分散液を噴霧して造粒した。
(5)前記(4)で得られた造粒物を、60℃で乾燥した。
(6)前記(5)で得られた乾燥後の造粒物を30Mの篩により整粒した。
(7)前記(6)で得られた整粒物0.25gとクエン酸水和物(国産化学(株))0.15gを混合し、本発明の医薬組成物に該当する目的のドライシロップ製剤を得た。
(1)42Mの篩を通したD-マンニトール(物産フードサイエンス(株))270g及び結晶セルロース(KG-1000、旭化成(株))30gを混合した。
(2)リン酸(国産化学(株))75g及びポピドン(PLASDONE K-25、ASHLAND)25gを精製水600gに溶解させた。
(3)ART-001(Carbogen Amcis AG)100gを前記(2)の溶液に加えて分散させた。
(4)前記(1)の混合物を流動層造粒乾燥機(FD-MP-01D/SFP型、(株)パウレック)に投入し、前記(3)の分散液を噴霧して造粒した。
(5)前記(4)で得られた造粒物を、60℃で乾燥した。
(6)前記(5)で得られた乾燥後の造粒物を30Mの篩により整粒した。
(7)前記(6)で得られた整粒物0.25g、酒石酸(L(+)-酒石酸、国産化学(株))0.0583g及びL-グルタミン酸塩酸塩(富士フィルム和光純薬(株))0.035gを混合し、本発明の医薬組成物に該当する目的のドライシロップ製剤を得た。
(1)結晶セルロース(KG-1000、旭化成(株))48g及びポリビニルアルコール(部分けん化物)(ゴーセノール EG-05PW、三菱ケミカル(株))32gを混合した。
(2)42Mの篩を通したD-マンニトール(物産フードサイエンス(株))160gを前記(1)の混合物と混合した。
(3)リン酸(太平化学産業(株))120g及びポピドン(PLASDONE K-25、ASHLAND)40gを精製水960gに溶解させた。
(4)ART-001(Carbogen Amcis AG)160gを前記(3)の溶液に加えて分散させた。
(5)前記(2)の混合物を流動層造粒乾燥機(FD-MP-01D/SFP型、(株)パウレック)に投入し、前記(4)の分散液を噴霧して造粒した。
(6)前記(5)で得られた造粒物を、60℃で乾燥した。
(7)前記(6)で得られた乾燥後の造粒物を30Mの篩により整粒し、本発明の医薬組成物に該当する目的のドライシロップ製剤を得た。
前記手順で調製したART-001のドライシロップ製剤を、以下に詳述する手順でイヌに投与し、ART-001の血中薬物動態を検討した。なお、対照として、0.5%メチルセルロース(MC;信越化学工業株式会社製METOLOSE、SM-100)水溶液にART-001を懸濁させた製剤も調製し、同様にイヌに投与して、ART-001の血中動態を検討した。
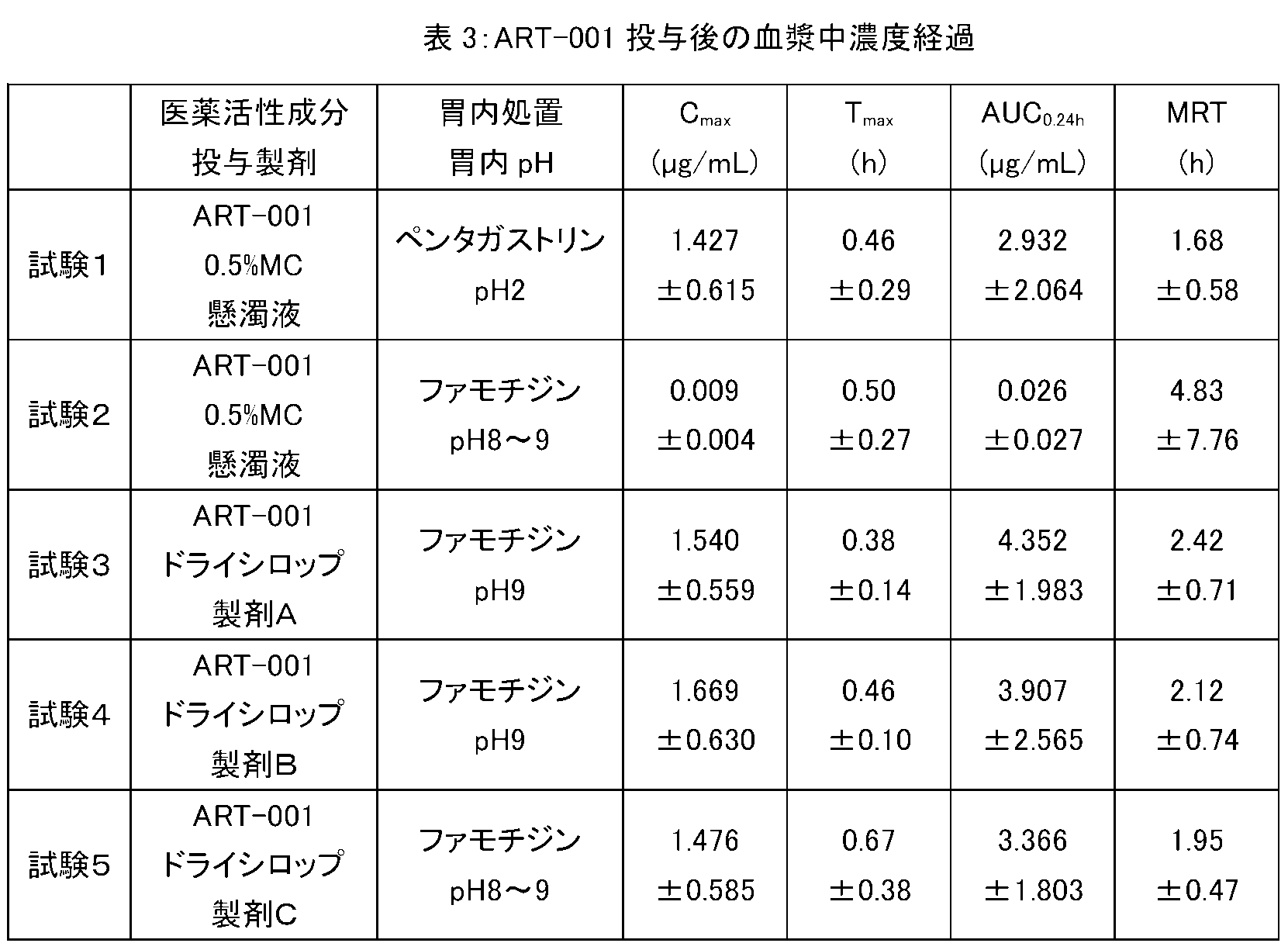
前記手順で調製したART-001のドライシロップ製剤(本発明の医薬組成物)を、健康成人男性に単回投与又は反復投与し、その後のART-001の血中薬物動態を検討した。


Claims (13)
- pH依存性の溶解プロファイルを有する医薬活性成分と、無機又は有機の酸と、親水性ポリマーとを含む経口用医薬組成物。
- 医薬活性成分が、π共役系を有する芳香族化合物である、請求項1に記載の経口用医薬組成物。
- 医薬活性成分が、下記一般式Iで表される構造を有する化合物である、請求項1又は2に記載の経口用医薬組成物。

環Aは、1又は2以上の置換基を有していてもよい、5~16員の単環式又は縮合二環若しくは三環式の芳香族炭化水素基又は芳香族複素環式基を表し、
環Bは、1又は2以上の置換基を有していてもよい、5~12員の単環式又は縮合二環式の芳香族炭化水素基又は芳香族複素環式基を表し、
Lは、1又は2以上の置換基を有していてもよい、二価又は三価の連結基を表し、
pは、0又は1の整数を表し、
qは、1又は2の整数を表し、
rは、1又は2の整数を表す。) - 医薬活性成分が、ART-001(セラベリシブ)、アルペリシブ、アファチニブ、ゲフィチニブ、ボスチニブ、アレクチニブ、パルボシクリブ、タセリシブ、及びコパンリシブから選択される1種又は2種以上の化合物である、請求項1~3の何れか一項に記載の経口用医薬組成物。
- 医薬活性成分のpH2.5での溶解性に対するpH6での溶解性の比率が50%以下である、請求項1~4の何れか一項に記載の経口用医薬組成物。
- 無機又は有機の酸が、リン酸、塩酸、クエン酸、リンゴ酸、酒石酸、アスコルビン酸、フマル酸、コハク酸、アスパラギン酸、乳酸、酢酸、グルタミン酸、及びアジピン酸から選択される1種又は2種以上の酸である、請求項1~5の何れか一項に記載の経口用医薬組成物。
- 親水性ポリマーが、ポリビニルアルコール、ポビドン、ヒプロメロース、コポリビドン、ヒドロキシプロピルセルロース、ポリビニルアルコール・ポリエチレングリコール・グラフトコポリマー、ヒプロメロースフタル酸エステル、及びヒプロメロース酢酸エステルコハク酸エステルから選択される1種又は2種以上のポリマーである、請求項1~6の何れか一項に記載の経口用医薬組成物。
- 医薬活性成分の含有率が2~40質量%の範囲である、請求項1~7の何れか一項に記載の経口用医薬組成物。
- 医薬活性成分1質量部に対する無機又は有機の酸の含有割合が0.1~10質量部の範囲である、請求項1~8の何れか一項に記載の経口用医薬組成物。
- 医薬活性成分1質量部に対する親水性ポリマーの含有割合が0.02~5質量部の範囲である、請求項1~9の何れか一項に記載の経口用医薬組成物。
- ドライシロップ剤である、請求項1~10の何れか一項に記載の経口用医薬組成物。
- 請求項1~11の何れか一項に記載の経口用医薬組成物を製造する方法であって、医薬活性成分、無機又は有機の酸、及び親水性ポリマーを含む混合物を湿式造粒し、乾燥することを含む方法。
- 請求項12に記載の方法により製造される経口用医薬組成物。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020227041888A KR20230034207A (ko) | 2020-07-02 | 2021-07-01 | 경구용 의약 조성물 및 그 제조 방법 |
| JP2022529745A JP7149449B2 (ja) | 2020-07-02 | 2021-07-01 | 経口用医薬組成物及びその製造方法 |
| CA3182166A CA3182166A1 (en) | 2020-07-02 | 2021-07-01 | Oral pharmaceutical composition and method for producing same |
| US18/013,616 US20230364063A1 (en) | 2020-07-02 | 2021-07-01 | Oral pharmaceutical composition and method for producing same |
| CN202180044261.1A CN115989022A (zh) | 2020-07-02 | 2021-07-01 | 口服用药物组合物及其制备方法 |
| AU2021299348A AU2021299348A1 (en) | 2020-07-02 | 2021-07-01 | Oral pharmaceutical composition and method for producing same |
| EP21834289.7A EP4176902A4 (en) | 2020-07-02 | 2021-07-01 | ORAL PHARMACEUTICAL COMPOSITION AND METHOD FOR MANUFACTURING SAME |
| JP2022152235A JP2022174317A (ja) | 2020-07-02 | 2022-09-26 | 経口用医薬組成物及びその製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020114978 | 2020-07-02 | ||
| JP2020-114978 | 2020-07-02 | ||
| JP2021011901 | 2021-01-28 | ||
| JP2021-011901 | 2021-01-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022004859A1 true WO2022004859A1 (ja) | 2022-01-06 |
Family
ID=79316406
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/025019 Ceased WO2022004859A1 (ja) | 2020-07-02 | 2021-07-01 | 経口用医薬組成物及びその製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20230364063A1 (ja) |
| EP (1) | EP4176902A4 (ja) |
| JP (2) | JP7149449B2 (ja) |
| KR (1) | KR20230034207A (ja) |
| CN (1) | CN115989022A (ja) |
| AU (1) | AU2021299348A1 (ja) |
| CA (1) | CA3182166A1 (ja) |
| WO (1) | WO2022004859A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023239337A1 (en) * | 2022-06-08 | 2023-12-14 | Abdi Ibrahim Ilac Sanayi Ve Ticaret Anonim Sirketi | A pharmaceutical composition comprising palbociclib |
| WO2024128311A1 (ja) * | 2022-12-16 | 2024-06-20 | ARTham Therapeutics株式会社 | 脈管奇形用医薬組成物及び脈管奇形の治療方法 |
| US12324807B2 (en) | 2018-06-01 | 2025-06-10 | Cornell University | Combination therapy for PI3K-associated disease or disorder |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024242504A1 (ko) * | 2023-05-25 | 2024-11-28 | 주식회사 삼양홀딩스 | 팔보시클립의 경구용 조성물 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004518676A (ja) * | 2000-12-20 | 2004-06-24 | シャイア ラボラトリーズ,インコーポレイテッド | 最小化pH依存性溶解プロフィールを有する徐放性薬学的剤形 |
| JP2009504796A (ja) * | 2005-08-22 | 2009-02-05 | ノバルティス アクチエンゲゼルシャフト | pH依存性薬剤化合物、pH調整剤および遅延剤を含む医薬組成物 |
| JP2009504795A (ja) * | 2005-08-22 | 2009-02-05 | ノバルティス アクチエンゲゼルシャフト | 1−(4−クロロアニリノ)−4−(4−ピリジルメチル)フタラジンおよびpH調整剤を含む固体医薬組成物 |
| WO2016175230A1 (ja) * | 2015-04-28 | 2016-11-03 | アステラス製薬株式会社 | 経口投与用医薬組成物 |
| JP2017002034A (ja) * | 2015-06-04 | 2017-01-05 | ファイザー・インク | パルボシクリブの固形剤形 |
| JP2019510059A (ja) * | 2016-03-29 | 2019-04-11 | シェンチェン ファーマシン シーオー.,エルティーディー. | パルボシクリブの医薬製剤およびその調製方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013071272A1 (en) * | 2011-11-11 | 2013-05-16 | Intellikine, Llc | Kinase inhibitor polymorphs |
| TWI791053B (zh) * | 2017-10-10 | 2023-02-01 | 美商亞雷生物製藥股份有限公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之結晶形式及其醫藥組合物 |
-
2021
- 2021-07-01 CN CN202180044261.1A patent/CN115989022A/zh active Pending
- 2021-07-01 KR KR1020227041888A patent/KR20230034207A/ko active Pending
- 2021-07-01 US US18/013,616 patent/US20230364063A1/en active Pending
- 2021-07-01 WO PCT/JP2021/025019 patent/WO2022004859A1/ja not_active Ceased
- 2021-07-01 JP JP2022529745A patent/JP7149449B2/ja active Active
- 2021-07-01 EP EP21834289.7A patent/EP4176902A4/en active Pending
- 2021-07-01 AU AU2021299348A patent/AU2021299348A1/en active Pending
- 2021-07-01 CA CA3182166A patent/CA3182166A1/en active Pending
-
2022
- 2022-09-26 JP JP2022152235A patent/JP2022174317A/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004518676A (ja) * | 2000-12-20 | 2004-06-24 | シャイア ラボラトリーズ,インコーポレイテッド | 最小化pH依存性溶解プロフィールを有する徐放性薬学的剤形 |
| JP2009504796A (ja) * | 2005-08-22 | 2009-02-05 | ノバルティス アクチエンゲゼルシャフト | pH依存性薬剤化合物、pH調整剤および遅延剤を含む医薬組成物 |
| JP2009504795A (ja) * | 2005-08-22 | 2009-02-05 | ノバルティス アクチエンゲゼルシャフト | 1−(4−クロロアニリノ)−4−(4−ピリジルメチル)フタラジンおよびpH調整剤を含む固体医薬組成物 |
| WO2016175230A1 (ja) * | 2015-04-28 | 2016-11-03 | アステラス製薬株式会社 | 経口投与用医薬組成物 |
| JP2017002034A (ja) * | 2015-06-04 | 2017-01-05 | ファイザー・インク | パルボシクリブの固形剤形 |
| JP2019510059A (ja) * | 2016-03-29 | 2019-04-11 | シェンチェン ファーマシン シーオー.,エルティーディー. | パルボシクリブの医薬製剤およびその調製方法 |
Non-Patent Citations (11)
| Title |
|---|
| C. PATEL , S. SANKOH , Y. SHOU , C. GRIFFIN , K. VENKATAKRISHNAN: "PII-110 Clinical biopharmaceutic assessments of the sources of absorption variability for the anticancer agent TAK-117, an inhibitor of phosphoinositide 3-kinase-a (PI3Kx)", CLINICAL PHARMACOLOGY AND THERAPEUTICS, vol. 101, no. S1, 1 February 2017 (2017-02-01), pages S82, XP055897162, ISSN: 1532-6535, DOI: 10.1002/cpt.570 * |
| CLINICAL CANCER RESEARCH, vol. 23, no. 17, 2017, pages 5218 - 5224 |
| CLINICAL PHARMACOLOGY IN DRUG DEVELOPMENT, vol. 8, no. 5, 2019, pages 637 - 646 |
| FDACENTER FOR DRUG EVALUATION AND RESEARCH, MULTI-DISCIPLINE |
| FDACENTER FOR DRUG EVALUATION AND RESEARCH, PRODUCT QUALITY REVIEW(S, 24 May 2019 (2019-05-24) |
| JURIC ET AL., CLIN. CANCER RES., vol. 23, no. 17, 2017, pages 5015 - 5023 |
| PATEL CHIRAG G., RANGACHARI LAKSHMI, PATTI MARK, GRIFFIN CELINA, SHOU YAPING, VENKATAKRISHNAN KARTHIK: "Characterizing the Sources of Pharmacokinetic Variability for TAK-117 (Serabelisib), an Investigational Phosphoinositide 3-Kinase Alpha Inhibitor: A Clinical Biopharmaceutics Study to Inform Development Strategy", CLINICAL PHARMACOLOGY IN DRUG DEVELOPMENT, vol. 8, no. 5, 1 July 2019 (2019-07-01), GB , pages 637 - 646, XP055897147, ISSN: 2160-763X, DOI: 10.1002/cpdd.613 * |
| PATEL ET AL., CLIN. PHARMACOL. DRUG DEV, vol. 8, no. 5, 2019, pages 637 - 646 |
| PATEL ET AL., CLIN. PHARMACOL. DRUG DEV., vol. 8, no. 5, 2019, pages 637 - 646 |
| See also references of EP4176902A4 |
| UNIVERSITY OF THE SCIENCES: "Remington: The Science and Practice of Pharmacy", 2000, LIPPINCOTT WILLIAMS & WILKINS |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12324807B2 (en) | 2018-06-01 | 2025-06-10 | Cornell University | Combination therapy for PI3K-associated disease or disorder |
| WO2023239337A1 (en) * | 2022-06-08 | 2023-12-14 | Abdi Ibrahim Ilac Sanayi Ve Ticaret Anonim Sirketi | A pharmaceutical composition comprising palbociclib |
| WO2024128311A1 (ja) * | 2022-12-16 | 2024-06-20 | ARTham Therapeutics株式会社 | 脈管奇形用医薬組成物及び脈管奇形の治療方法 |
| JP7536987B2 (ja) | 2022-12-16 | 2024-08-20 | ARTham Therapeutics株式会社 | 脈管奇形用医薬組成物及び脈管奇形の治療方法 |
| EP4635499A1 (en) | 2022-12-16 | 2025-10-22 | ARTham Therapeutics Inc. | Pharmaceutical composition for vascular malformations and method for curing vascular malformations |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3182166A1 (en) | 2022-01-06 |
| TW202216117A (zh) | 2022-05-01 |
| JP2022174317A (ja) | 2022-11-22 |
| EP4176902A4 (en) | 2024-07-31 |
| AU2021299348A1 (en) | 2023-02-02 |
| CN115989022A (zh) | 2023-04-18 |
| JP7149449B2 (ja) | 2022-10-06 |
| KR20230034207A (ko) | 2023-03-09 |
| JPWO2022004859A1 (ja) | 2022-01-06 |
| EP4176902A1 (en) | 2023-05-10 |
| US20230364063A1 (en) | 2023-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7149449B2 (ja) | 経口用医薬組成物及びその製造方法 | |
| JP6339089B2 (ja) | 非晶質トルバプタンを含有する経口投与懸濁剤 | |
| CN114306245A (zh) | 无定形固体分散体的药物组合物及其制备方法 | |
| JP2025134801A (ja) | オラパリブの溶解度及び生体利用効率が改善された組成物 | |
| AU2007268772B2 (en) | Oral composition comprising 3-[5-[4-(cyclopentyloxy) -2-hydroxybenzoyl]-2-[(3-hydroxy-1,2-benzisoxazol-6- yl)methoxy]phenyl]propionic acid or salt thereof | |
| CN108135915A (zh) | 片剂 | |
| JP2025038168A (ja) | ベンズイミダゾール誘導体化合物を含有する口腔内崩壊錠およびその調整方法 | |
| CA2648538C (en) | Oral rapid release pharmaceutical formulation for pyridylmethylsulfinyl-benzimidazoles | |
| AU2018294561B2 (en) | New oral formulations of belinostat | |
| WO2011078821A1 (en) | Effervescent tablet and granule formulation comprising cefixime | |
| RU2840493C1 (ru) | Фармацевтическая композиция для перорального применения и способ ее получения | |
| WO2020111089A1 (ja) | 医薬組成物 | |
| JP2026067959A (ja) | 経口用医薬組成物及びその製造方法 | |
| HK40089965A (zh) | 口服用药物组合物及其制备方法 | |
| JPWO2011081118A1 (ja) | 経口投与用医薬組成物 | |
| CN109316451B (zh) | 治疗高血压和相关疾病的口服固体制剂 | |
| WO2014007780A1 (en) | Orally-disintegrating formulations of dexketoprofen | |
| EP2682104A1 (en) | Orally-disintegrating formulations of dexketoprofen | |
| JPWO2009069643A1 (ja) | 3−{5−[4−(シクロペンチルオキシ)−2−ヒドロキシベンゾイル]−2−[(3−ヒドロキシ−1,2−ベンズイソオキサゾール−6−イル)メトキシ]フェニル}プロピオン酸またはその塩を含有する固体分散体またはその組成物 | |
| JP2019073488A (ja) | アプレピタントを有効成分とする医薬錠剤 | |
| TR202022248A2 (tr) | Rebami̇pi̇d i̇çeren geli̇şti̇ri̇lmi̇ş granül formülasyonlari | |
| JP2019073445A (ja) | アプレピタントを有効成分とする医薬組成物 | |
| WO2016107605A1 (zh) | 药物组合物及其制备方法 | |
| CN121774936A (zh) | 固态组合物、布洛芬的溶出性改善剂及溶出性改善方法 | |
| BRPI0607372B1 (pt) | Medicamento destinado à administração oral, compreendendo um inibidor da ciclooxigenase-2, e método de preparação do mesmo |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21834289 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022529745 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3182166 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202217071869 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2021299348 Country of ref document: AU Date of ref document: 20210701 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022134743 Country of ref document: RU Ref document number: 2021834289 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2021834289 Country of ref document: EP Effective date: 20230202 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2022134743 Country of ref document: RU |