WO2022050419A1 - iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物 - Google Patents
iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物 Download PDFInfo
- Publication number
- WO2022050419A1 WO2022050419A1 PCT/JP2021/032734 JP2021032734W WO2022050419A1 WO 2022050419 A1 WO2022050419 A1 WO 2022050419A1 JP 2021032734 W JP2021032734 W JP 2021032734W WO 2022050419 A1 WO2022050419 A1 WO 2022050419A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- h1foo
- cells
- ips
- protein
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4738—Cell cycle regulated proteins, e.g. cyclin, CDC, INK-CCR
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/95—Fusion polypeptide containing a motif/fusion for degradation (ubiquitin fusions, PEST sequence)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/602—Sox-2
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/603—Oct-3/4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/604—Klf-4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/606—Transcription factors c-Myc
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/11—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells
- C12N2506/115—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells from monocytes, from macrophages
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1307—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from adult fibroblasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18811—Sendai virus
- C12N2760/18841—Use of virus, viral particle or viral elements as a vector
- C12N2760/18843—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Definitions
- the present invention relates to an iPS cell quality improving agent, a method for producing iPS cells, iPS cells, and a composition for producing iPS cells.
- iPS cells induced pluripotent stem cells; also called “induced pluripotent stem cells” or “induced pluripotent stem cells” are produced from somatic cells by introducing Oct3 / 4, Sox2, Klf4 and c-Myc.
- Non-Patent Document 1, Patent Document 1 This can be achieved by reprogramming the transcriptional network of parental cells and the epigenetic signature.
- iPS cells bring various benefits to basic research, pharmaceutical innovation, and regenerative medicine. However, it is still a serious problem that the generated iPS cell group has a heterogeneous quality as compared with the embryonic stem cell (ES cell) cell group.
- ES cells the variation in properties among cells is small, and almost any cell can be differentiated into a target cell.
- iPS cells cells that cannot be differentiated into target cells often appear due to large variations in properties among cells. It is important for basic research and clinical purposes to obtain a cell group of iPS cells that show high quality for all iPS cells without variation among cells.
- Patent Document 2 when a predetermined amount of Oct3 / 4 gene, Klf4 gene, c-Myc gene and Sox2 gene is introduced into a somatic cell a predetermined number of times, the production efficiency and stability of iPS cells can be improved. It is stated to that effect.
- Patent Document 3 in addition to the Oct3 / 4 gene or its gene product, the Sox2 gene or its gene product, the Klf4 gene or its gene product, and the c-Myc gene or its gene product, the Prdm14 gene or its gene product, Esrrb It is described that by introducing a gene or a gene product thereof and a All4a gene or a gene product thereof into a somatic cell, iPS cells having excellent quality can be efficiently produced in a short period of time.
- Patent Document 4 describes the Oct3 / 4 gene or its gene product, the Sox2 gene or its gene product, the Klf4 gene or its gene product, and the c-Myc gene or its gene product, as well as the Jard2 variant gene or its gene product.
- the linker histone H1 family binds to the linker DNA and gives rise to a higher chromatin structure in order to control gene expression.
- Members of the linker histone H1 family include histones H1a, H1b, H1c, H1d, H1e, H1foo, H1x, H1.0, H1t, H1T2, HILS1.
- Most members of the linker histone family consist of somatic linker histones that condense chromatin. Therefore, such a structure generally suppresses the overall gene transcription activity (Non-Patent Documents 2 and 3).
- the present inventors When inducing iPS cells from somatic cells, the present inventors have introduced the H1foo gene, which is a member of the linker histone H1 family, in addition to the above genes, so that the iPS cells are of high quality and have little variation in quality. (Patent Document 5).
- Japanese Patent No. 4183742 Japanese Unexamined Patent Publication No. 2011-004674 Japanese Unexamined Patent Publication No. 2014-217344 Japanese Unexamined Patent Publication No. 2014-217345 International Publication No. 2017/010080
- the method for producing iPS cells described in Patent Document 5 can produce iPS cells having high quality and less variation in quality as compared with the conventional method.
- the number of colonies obtained after iPS cell induction is similar to that of the conventional method. Therefore, the present invention is an iPS cell quality improving agent capable of increasing the number of colonies obtained after iPS cell induction, a method for producing iPS cells, an iPS cell produced by such a production method, and an iPS cell for producing iPS cells.
- An object of the present invention is to provide a composition.
- the present inventors have controlled the time and amount of H1foo protein expressed from the H1foo gene introduced into somatic cells in the cells. , It was found that the number of colonies after iPS cell induction was significantly increased, and the present invention was completed.
- a control sequence capable of controlling at least one of the intracellular abundance and the existence time of the H1foo protein expressed from the H1foo gene when the H1foo gene is introduced into the cell.
- An iPS cell quality improving agent containing a polynucleotide having a polynucleotide.
- the iPS cell quality improving agent according to [1] wherein the polynucleotide is inserted into an expression vector in a state in which the H1foo gene can be expressed in the cell into which the polynucleotide is introduced.
- the iPS cell quality improving agent according to [2] wherein the expression vector is a Sendai virus vector.
- the control sequence contains a nucleotide sequence encoding a destabilizing domain, and the destabilizing domain is a domain that promotes degradation of a fusion protein containing the destabilizing domain by the proteasome.
- the quality of the iPS cell according to any one of [1] to [3], which is linked to the H1foo gene so that the fusion protein of the destabilizing domain and the H1foo protein can be expressed. Improvement agent.
- the iPS cell quality improving agent according to any one of [1] to [4], wherein the control sequence contains a promoter sequence that controls transcription of the H1foo gene in response to a chemical stimulus. ..
- iPS cell quality improving agent containing a fusion protein of an H1foo protein and a destabilizing domain, wherein the destabilizing domain is a domain that promotes degradation of the fusion protein by the proteasome. Quality improver.
- Nuclear reprogramming material and A method for producing an iPS cell which comprises a step of introducing the iPS cell quality improving agent according to any one of [1] to [6] into a somatic cell.
- the nuclear reprogramming substance is a gene of the Oct gene family, a gene of the Sox gene family, a gene of the Klf gene family, a gene of the Myc gene family, a gene of the Lin gene family, and a Nanog gene, and their gene products.
- the nuclear reprogramming substance is one of [7] to [9], which is an Oct3 / 4 gene, a Sox2 gene, a Klf4 gene, L-Myc or c-Myc, and a gene product thereof.
- the method for producing an iPS cell according to the above method is an Oct3 / 4 gene, a Sox2 gene, a Klf4 gene, L-Myc or c-Myc, and a gene product thereof.
- the nuclear reprogramming substance is selected from the group consisting of a gene of the Oct gene family, a gene of the Sox gene family, a gene of the Klf gene family, a gene of the Myc gene family, a gene of the Lin gene family, and a Nanog gene.
- [12] The method for producing an iPS cell according to [11], wherein the expression vector is a Sendai virus vector.
- [13] Including the step of introducing a nuclear reprogramming substance and the H1foo gene into somatic cells.
- a method for producing an iPS cell wherein the H1foo protein expressed from the H1foo gene introduced into the somatic cell is controlled at least one of the existence time and the abundance in the somatic cell.
- the nuclear reprogramming substance is selected from the group consisting of a gene of the Oct gene family, a gene of the Sox gene family, a gene of the Klf gene family, a gene of the Myc gene family, a gene of the Lin gene family, and a Nanog gene.
- a composition for producing iPS cells which comprises a nuclear reprogramming substance and the quality improving agent for iPS cells according to any one of [1] to [6].
- a method for producing an iPS cell which comprises a step of introducing a substance having the above into the somatic cell.
- the present invention provides an agent for improving the quality of iPS cells, a method for producing iPS cells, iPS cells produced by such a production method, and a composition for producing iPS cells. According to the present invention, the number of colonies obtained after iPS cell induction can be increased. Moreover, many of the iPS cell groups produced in this way are of high quality as iPS cells such as high pluripotency.
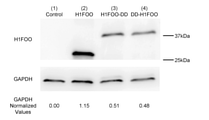
- FIG. 1 The structures of three Sendai virus vectors (SeV18 + H1FOO / TS15 ⁇ F, SeV18 + H1FOO-DD / TS15 ⁇ F, SeV18 + DD-H1FOO / TS15 ⁇ F) containing the H1FOO gene used in the examples are shown.
- One of the three Sendai virus vectors shown in FIG. 1 was introduced into human skin fibroblasts, and the expression levels of H1FOO (2), H1FOO-DD fusion protein (3), and DD-H1FOO fusion protein (4) were expressed in Western. The result of comparison by blotting is shown.
- "(1) Control” is a "(a) SeV18 + H1FOO / TS15 ⁇ F" shown in FIG.
- ALP alkaline phosphatase
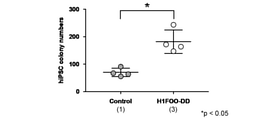
- Prime-type iPS cells use any of the Sendai virus vectors (a) to (c) shown in FIG. 1 together with a Sendai virus vector containing an Oct3 / 4 gene, a Sox2 gene, a Klf4 gene, and an L-Myc gene.
- ALP-positive colonies in prime-type iPS cells prepared from human peripheral blood mononuclear cells is shown.
- Prime-type iPS cells were prepared using SeV18 + H1FOO-DD together with a Sendai virus vector containing an Oct3 / 4 gene, a Sox2 gene, a Klf4 gene, and an L-Myc gene.
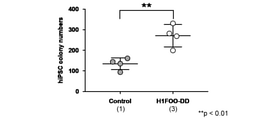
- the number of ALP-positive colonies in naive iPS cells prepared from human skin fibroblasts is shown.
- Naive iPS cells were generated using SeV18 + H1FOO-DD together with a Sendai virus vector containing the Oct3 / 4, Sox2 gene, Klf4 gene, and L-Myc gene.
- the number of ALP-positive colonies in naive iPS cells prepared from human peripheral blood mononuclear cells is shown.
- Naive iPS cells were generated using SeV18 + H1FOO-DD together with a Sendai virus vector containing the Oct3 / 4, Sox2 gene, Klf4 gene, and L-Myc gene.
- the results of comparative evaluation of the variation in gene expression level between (1) Control-iPS and (3) H1FOO-DD-iPS in prime type iPS cells prepared from human skin fibroblasts are shown.
- the results of comparative evaluation of the variation in gene expression level between (1) Control-iPS and (3) H1FOO-DD-iPS in naive iPS cells prepared from human skin fibroblasts are shown.
- Control-iPS and (3) H1FOO-DD-iPS evaluated albumin secretion by ELISA are shown.
- a measurement example is shown in which the proportion of cells positive for both PDGFRA and AMPEP in cells induced to differentiate from naive iPS cells using a primitive endoderm differentiation-inducing medium is measured by flow cytometry.
- the left figure is (1) a measurement example of Control-iPS, and the right figure is a measurement example of (3) H1FOO-DD-iPS.
- HDF Human skin fibroblast (TIG) 120; H9 ESC: H9 ES cell; H1FOO OE HDF: Cell on the second day after introducing H1FOO-DD into human skin fibroblast; OSKL day2: Human skin fibroblast 2nd day cells after introduction of nuclear reprogramming substance (OCT4 / SOX2 / KLF4 / LMYC); OSKLH day2: Nuclear reprogramming substance (OCT4 / SOX2 / KLF4 / LMYC) and H1FOO-DD into human skin fibroblasts. The cells on the second day after the introduction.
- the term “comprise” means that components other than the target component may be included.
- the term “consist of” means that it does not contain any component other than the component of interest.
- the term “consistentially of” does not include components other than the target component in a mode that exerts a special function (such as a mode in which the effect of the invention is completely lost). means.
- the term “comprise” includes the “consist of” mode and the “essentially constist of” mode.
- Proteins polypeptides
- peptides polynucleotides (DNA, RNA), vectors, and cells
- isolated means a state isolated from the natural state.
- the proteins (polypeptides), peptides, polynucleotides (DNA, RNA), vectors, and cells described herein are isolated proteins (isolated polypeptides), isolated peptides, simplex. It can be an isolated polynucleotide (isolated DNA, isolated RNA), an isolated vector, and an isolated cell.
- gene means a polynucleotide containing at least one open reading frame encoding a particular protein.
- the gene may contain only exons, as well as one or more of the introns, 5'UTRs, and 3'UTRs.
- the term "functionally linked" as used with respect to a polynucleotide means that the first base sequence is located close enough to the second base sequence and the first base sequence is the second base sequence or the second base sequence. It means that it can affect the area under the control of.
- functionally ligating a polynucleotide to a promoter means that the polynucleotide is ligated to be expressed under the control of the promoter.
- the gene is usually functionally linked to the promoter.
- the “expressible state” means a state in which the polynucleotide can be transcribed in the cell into which the polynucleotide has been introduced.
- the “expression vector” refers to a vector containing a target polynucleotide, which is provided with a system for making the target polynucleotide expressible in the cell into which the vector has been introduced.
- the present invention provides an agent for improving the quality of iPS cells.
- the iPS cell quality improving agent of the present embodiment is at least one of the H1foo gene and the H1foo protein expressed from the H1foo gene in the cell when the H1foo gene is introduced into the cell.
- the iPS cell quality improving agent of the present embodiment can improve the quality of somatic cell-derived iPS cells by introducing it into somatic cells together with a nuclear reprogramming substance described later.
- the "quality of iPS cells” includes high expression of undifferentiated markers (Nanog, Tra-1-60, ALP, etc.) in iPS cells, high embryoid body formation ability, and embryo-like formation from iPS cells.
- Various properties such as uniformity of body size, low abnormal methylation, high ability to form chimera in mouse iPS cells, high ability to differentiate into differentiated cells (eg, myocardial cells), and the above among iPS cells.
- the uniformity of various properties such as the above can be mentioned.
- the iPS cell quality improving agent of the present embodiment By introducing the iPS cell quality improving agent of the present embodiment into somatic cells together with the nuclear reprogramming substance, various properties of the iPS cells as described above are improved as compared with the case where only the nuclear reprogramming substance is introduced. Or, it has the effect of improving the uniformity of various properties between iPS cells.
- the iPS cell quality improving agent of the present embodiment has effects such as improvement of the ability to generate prime type iPS cells expressing ALP and improvement of the ability to generate naive type iPS cells expressing ALP.
- the iPS cell quality improving agent of the present embodiment is introduced into somatic cells together with a nuclear reprogramming substance to improve gene expression homogeneity and DNA methylation homogeneity in the generated iPS cells.
- the differentiated cells are not particularly limited, but are, for example, endometrial cells, mesophyll cells, ectodermal cells, myocardial cells, hepatocytes, renal cells, muscle cells, fibroblasts, nerve cells, and immune cells (lymph). Spheres, etc.), vascular cells, eye cells (retinal pigment epithelial cells, etc.), blood cells (giant nuclei, erythrocytes, etc.), other tissue cells, and precursor cells thereof.
- H1foo gene means a polynucleotide encoding an H1foo protein.
- the species from which the H1foo gene is derived is not particularly limited and may be appropriately selected depending on the intended purpose. Examples thereof include arbitrary mammals such as humans, mice, rats, cows, sheep, horses and monkeys. ..
- the sequence information of the H1foo gene can be obtained from a known database. For example, in GenBank, it can be obtained with accession number BC047943 (human) or BC137916 (mouse).
- the nucleotide sequence of the human H1foo gene of the accession number is shown in SEQ ID NO: 1, and the amino acid sequence of the human H1foo protein encoded by the nucleotide sequence is shown in SEQ ID NO: 2.
- the nucleotide sequence of the mouse H1foo gene of the accession number is shown in SEQ ID NO: 3, and the amino acid sequence of the mouse H1foo protein is shown in SEQ ID NO: 4.
- H1FOO when the alphabet "H1FOO" is written in all capital letters, it means a human H1foo gene or H1foo protein. When described as "H1foo", it includes the H1foo gene or H1foo protein of all species including humans.
- the H1foo gene is not limited to the wild-type H1foo gene, and may include mutations (either deletion, substitution, insertion, and addition, or a combination thereof).
- the number of mutations is not particularly limited as long as it has a nucleotide sequence encoding a protein having H1foo activity.
- the term "H1foo activity" refers to at least one of the functions of the wild-type H1foo protein.
- Examples of the H1foo activity include an activity of binding to a linker DNA that connects nucleosomes.
- the H1foo activity is more preferably an activity that binds to a linker DNA that connects nucleosomes and keeps the linker DNA region loose.
- H1foo gene examples include the following (a) to (g).
- A Wild-type H1foo gene (eg, a polynucleotide consisting of a nucleotide sequence represented by SEQ ID NO: 1 or 3)
- B Polynucleotide consisting of a nucleotide sequence encoding a wild-type H1foo protein (eg, a protein consisting of the amino acid sequence represented by SEQ ID NO: 2 or 4)
- c Amino acid sequence of the wild-type H1foo protein (eg, sequence) In the amino acid sequence represented by No.
- the amino acid sequence of the polynucleotide (d) wild-type H1foo protein encoding a protein consisting of an amino acid sequence in which one or more amino acids are mutated and having H1foo activity.
- a polynucleotide encoding a protein consisting of an amino acid sequence having 70% or more sequence identity and having H1foo activity with the amino acid sequence represented by SEQ ID NO: 2 or 4)
- (e) A nucleotide of a wild-type H1foo gene (e) A nucleotide of a wild-type H1foo gene.
- a polynucleotide (f) wild encoding a protein in a sequence (eg, a nucleotide sequence represented by SEQ ID NO: 1 or SEQ ID NO: 3) consisting of a nucleotide sequence in which one or more nucleotides are mutated and having H1foo activity.
- a poly comprising a nucleotide sequence of a type H1foo gene (eg, a nucleotide sequence represented by SEQ ID NO: 1 or SEQ ID NO: 3) and a nucleotide sequence having 70% or more sequence identity and encoding a protein having H1foo activity.
- Nucleotide A poly that encodes a protein that hybridizes with wild-type H1foo gene (eg, a polynucleotide containing the nucleotide sequence represented by SEQ ID NO: 1 or SEQ ID NO: 3) under stringent conditions and has H1foo activity.
- the "mutation” may be any of deletion, substitution, addition, and insertion, and may be a combination thereof.
- the “plurality” is not particularly limited as long as the resulting protein has H1foo activity, but for example, 2 to 30 are exemplified, preferably 2 to 20 and more preferably 2 to 10. Preferably, 2 to 5 are even more preferred, and 2 or 3 are particularly preferred.
- the “plurality” is not particularly limited as long as the resulting polynucleotide encodes a protein having H1foo activity, but for example, 2 to 60 are exemplified, and 2 to 50 are preferable.
- sequence identity is not particularly limited as long as it is 70% or more, but 80% or more is preferable, 85% or more is more preferable, and 90% or more, 95% or more, 96%. More preferably, 97% or more, 98% or more, or 99% or more.
- Sequence identity between amino acid sequences or nucleotide sequences can be obtained by juxtaposing two amino acid sequences or nucleotide sequences with gaps in the parts corresponding to insertions and deletions so that the corresponding amino acids or nucleotides match most.
- sequence identity between amino acid sequences or nucleotide sequences can be determined by using various homology search software known in the art.
- sequence identity value of an amino acid sequence or nucleotide sequence can be obtained by calculation based on the alignment obtained by the known homology search software BLASTP or BLASTN.
- the “stringent condition” includes, for example, the condition described in Molecular Cloning-A LABORATORY MANUAL THIRD EDITION (Sambrook et al., Cold Spring Harbor Laboratory Press).
- ⁇ SSC composition of 20 ⁇ SSC: 3M sodium chloride, 0.3M citrate solution, pH 7.0
- 5 ⁇ Denhardt solution composition of 100 ⁇ Denhardt solution: 2% by mass, bovine serum albumin, 2% by mass
- Ficoll 2% by weight polyvinylpyrrolidone
- 0.5% by weight SDS 0.5% by weight SDS
- 0.1 mg / mL salmon sperm DNA 50% by volume formamide in a hybridization buffer at 42-70 ° C. for several hours to overnight.
- the conditions for hybridization by performing incubation are exemplified.
- the washing buffer used for washing after incubation include a 1 ⁇ SSC solution containing 0.1% by mass SDS, and more preferably a 0.1 ⁇ SSC solution containing 0.1% by mass SDS.
- the degenerate codons used are somatic cells that use the quality improving agent for iPS cells of the present embodiment and have a high codon usage frequency.
- codons frequently used in human cells are used. That is, it is preferably optimized for human codons.
- codons frequently used in mouse cells are used. That is, it is preferably optimized for mouse codons.
- H1foo protein may refer to a fusion protein of a destabilizing domain and H1foo protein described later, or an H1foo protein region in the fusion protein.
- the control sequence is a nucleotide sequence capable of controlling at least one of the intracellular abundance and the existence time of the H1foo protein expressed from the H1foo gene when the H1foo gene is introduced into the cell.
- the control sequence is not particularly limited as long as it can control either the intracellular abundance or the existence time of the H1foo protein.
- the control sequence includes, for example, a nucleotide sequence encoding a destabilizing domain.
- the control sequence includes, for example, a promoter sequence that controls transcription of the H1foo gene in response to a chemical stimulus.
- the term "Destabilized Domain” refers to a domain that promotes the degradation of a fusion protein containing the domain by the proteasome. That is, by linking the destabilizing domain to the N-terminal or C-terminal of the desired protein to obtain a fusion protein containing the destabilizing domain, the degradation of the fusion protein containing the desired protein by the proteasome is promoted.
- the destabilizing domain is not particularly limited as long as it promotes the decomposition of the fusion protein containing the domain, and known destabilizing domains can be used. Examples of the destabilizing domain include a destabilizing domain derived from FKBP12 (Banaszynski et al., Cell.
- destabilizing domain derived from ecDHFR (Iwamoto et al). ., Chem Biol. 2010 Sep 24; 17 (9): 981-8.) And the like.
- destabilizing domain derived from FKBP12 include those having the amino acid sequence set forth in SEQ ID NO: 14, and examples of the nucleotide sequence encoding the amino acid sequence include the nucleotide sequence set forth in SEQ ID NO: 13. ..
- the fusion protein of the desired protein and the destabilizing domain may contain a polypeptide between the desired protein and the destabilizing domain.
- Degradation of fusion proteins containing these destabilizing domains is carried out by membrane-permeable small molecule compounds called Sheald1 (in the case of destabilizing domains derived from FKBP12) or Guard (in the case of destabilizing domains derived from ecDHFR).
- Sheald1 in the case of destabilizing domains derived from FKBP12
- Guard in the case of destabilizing domains derived from ecDHFR
- stabilizing compound membrane-permeable small molecule compounds
- the polynucleotide encoding these destabilizing domains hereinafter referred to as "destabilizing domain gene" and the stabilizing compound shall be used by PreteoTuner TM Standard System (Clontech), PreteoTuner TM Guard System (Clontech) and the like. Can be done.
- the destabilizing domain can also be said to be a degron sequence.
- degrons examples include mTOR degron (US Patent Application Publication No. 2009/0215169), dihydrofolate reductase (DHFR) degron (US Patent Application Publication No. 2012/0178168), PEST (International Publication No. 99/54348), TetR. Examples include, but are not limited to, degron (International Publication No. 2007/032555) and auxin-inducible degron (AID) (International Publication No. 2010/125620).
- the destabilizing domain gene may be linked to the H1foo gene so that the fusion protein of the destabilizing domain and the H1foo protein can be expressed.
- the H1foo protein is expressed as a fusion protein with a destabilizing domain. Therefore, the H1foo protein is rapidly degraded by the proteasome after expression. That is, by expressing the H1foo protein as a fusion protein with the destabilizing domain, the abundance of the H1foo protein in the cell can be controlled to be small.
- the expression vector for the H1foo protein can be used as the expression vector for the fusion protein. It can be controlled within a certain period after the introduction. In this way, by controlling the H1foo protein to be expressed at a certain period from the introduction of the nuclear reprogramming substance to the completion of reprogramming and to be present at a relatively low level thereafter, high-quality iPS cells can be produced at a high quality. It can be manufactured with efficiency.
- the fusion protein containing the destabilizing domain can control the intracellular abundance and abundance time by adding a stabilizing compound.
- a stabilizing compound is added at an arbitrary period, and the stabilizing compound is removed after the lapse of the arbitrary period. By doing so, the abundance and time of existence of the H1foo protein may be controlled.
- the polynucleotide contained in the quality improving agent for iPS cells of the present embodiment may be in a form in which the H1foo gene is inserted into the expression vector in a state in which it can be expressed in the cell into which it is introduced.
- the destabilizing domain gene is linked to the replication-related protein gene so that a fusion protein of the destabilizing domain and a protein required for replication of the expression vector (hereinafter referred to as “replication-related protein”) can be expressed. It may have been done.
- "Replication-related protein gene” means a polynucleotide encoding a replication-related protein.
- the replication-related protein expressed as a fusion protein with the destabilizing domain is rapidly degraded by the proteasome after expression.
- the replication-related protein to be used as the fusion protein with the destabilizing domain is not particularly limited and may be appropriately selected depending on the type of expression vector.
- replication-related proteins include nucleocapsid protein (N), phosphorylated protein (P), matrix protein (M), and large protein (L). Among these, P protein is preferable.
- the destabilizing domain gene may be ligated to any two or more of the plurality of replication-related protein genes.
- control sequence may be linked to at least one of the 5'end and 3'end of the H1foo gene or replication-related protein gene.
- the control sequence may be linked to either the 5'end or the 3'end of the protein gene, or may be linked to both the 5'end and the 3'end.
- the control sequence is linked to the H1foo gene or replication-related protein gene so that the destabilizing domain gene and the H1foo gene or replication-related protein gene do not undergo a frameshift (ie, in-frame).
- a frameshift ie, in-frame
- the nucleotide sequence set forth in SEQ ID NO: 15 encodes a fusion protein (SEQ ID NO: 16) in which the destabilizing domain derived from FKBP12 is added to the C-terminus of the H1foo protein.
- SEQ ID NO: 16 the nucleotide sequence when the destabilizing domain gene derived from FKBP12 is linked to the 5'end of the H1foo gene is shown in SEQ ID NO: 17.
- the nucleotide sequence set forth in SEQ ID NO: 17 encodes a fusion protein (SEQ ID NO: 18) in which the destabilizing domain derived from FKBP12 is added to the N-terminus of the H1foo protein.
- the control sequence may be linked to both the H1foo gene and the replication-related protein gene.
- the control sequence may include a promoter sequence that controls transcription of the H1foo gene in response to external stimuli.
- the external stimulus include chemical substances, heat, light, pH, osmotic pressure and the like.
- a promoter that controls transcription of the H1foo gene in response to an external stimulus (hereinafter referred to as “external stimulus responsive promoter”) is placed upstream of the H1foo gene so as to control transcription of the H1foo gene.
- the external stimulus-responsive promoter is not particularly limited, and known promoters can be used. Examples of the external stimulus-responsive promoter include a tetracycline-responsive promoter (eg, tetracycline-responsive factor: TRE).
- the iPS cell quality improving agent of the present embodiment contains a polynucleotide having the above H1foo gene and the above control sequence.
- the polynucleotide is one in which the H1foo gene is inserted into an expression vector in a state in which it can be expressed under the control of a control sequence.
- the "expression vector capable of expressing the H1foo gene" may be one that expresses the H1foo protein, or may be one that expresses a fusion protein of the H1foo protein and the destabilizing domain.
- the expression vector contains, if necessary, a promoter that controls the expression of the H1foo gene or the H1foo gene and the destabilizing domain gene (hereinafter collectively referred to as “H1foo gene and the like”) in addition to the H1foo gene and the control sequence. It is preferable to be.
- the promoter is arranged upstream of the H1foo gene or the like so as to control the expression of the H1foo gene or the like.
- the control sequence is an external stimulus-responsive promoter, it is preferable that the expression vector does not contain another promoter that controls the expression of the H1foo gene.
- promoters include SR ⁇ promoter, SV40 initial promoter, retrovirus LTR, CMV (cytomegalovirus) promoter, RSV (rous sarcoma virus) promoter, HSV-TK (herpes simplex virus thymidine kinase) promoter, EF1 ⁇ promoter, and metallothioneine. Examples include promoters and heat shock promoters.
- an enhancer for the IE gene of human CMV may be used together with a promoter.
- a CAG promoter including a cytomegalovirus enhancer, a chicken ⁇ -actin promoter and a poly A signal site of the ⁇ -globin gene
- the stimulus-responsive promoter mentioned in the above-mentioned "control sequence" section may be used.
- the expression vector may optionally contain an enhancer, a poly A addition signal, a marker gene, an origin of replication, a gene encoding a protein that binds to the origin of replication and controls replication, and the like.
- the marker gene refers to a gene that enables selection and selection of cells by introducing the marker gene into cells.
- Specific examples of the marker gene include a drug resistance gene, a fluorescent protein gene, a luminescent enzyme gene, a color-developing enzyme gene, and the like. These may be used alone or in combination of two or more.
- the drug resistance gene examples include neomycin resistance gene, tetracycline resistance gene, canamycin resistance gene, zeosin resistance gene, hyglomycin resistance gene, puromycin resistance gene and the like.
- Specific examples of the fluorescent protein gene include a green fluorescent protein (GFP) gene, a yellow fluorescent protein (YFP) gene, a red fluorescent protein (RFP) gene, and the like.
- Specific examples of the luminescent enzyme gene include a luciferase gene and the like.
- Specific examples of the color-developing enzyme gene include ⁇ -galactosidase gene, ⁇ -glucuronidase gene, alkaline phosphatase gene and the like.
- the expression vector may contain an enhancer gene or a repressor gene that binds to the promoter in response to the stimulus.
- the expression vector may contain genes such as reverse tetracycline-regulated transactivating factor (rtTA) or tetracycline-regulated transactivating factor (rTA). These enhancer genes or repressor genes may be contained in an expression vector different from the expression vector having the H1foo gene.
- the type of expression vector is not particularly limited, and a known expression vector can be used.
- Examples of the expression vector include an episomal vector, an artificial chromosome vector, a plasmid vector, a virus vector and the like.
- the episomal vector is a vector capable of autonomous replication outside the chromosome. Specific means of using episomal vectors are disclosed in Yu et al., Science, 324, 797-801 (2009). For example, an episomal vector in which loxP sequences are arranged in the same direction on the 5'side and the 3'side of the vector element required for replication of the episomal vector can be used. Since episomal vectors can replicate autonomously extrachromosomally, they can provide stable expression in host cells without being integrated into the genome, but once iPS cells are established, the vector is rapidly removed. Is desirable.
- the autonomous replication ability of the episomal vector can be lost, and the vector can be lost. Can be shed from iPS cells at an early stage.
- the episomal vector examples include a vector containing a sequence required for autonomous replication derived from EBV, SV40, etc. as a vector element.
- the vector element required for autonomous replication is an origin of replication and a gene encoding a protein that binds to the origin of replication and controls replication.
- EBV the origin of replication oriP.
- EBNA-1 gene the origin of replication ori and the SV40LT gene can be mentioned.
- the artificial chromosome vector examples include a YAC (Yeast artificial chromosome) vector, a BAC (Bacterial artificial chromosome) vector, and a PAC (P1-derived artificial chromosome) vector.
- YAC yeast artificial chromosome
- BAC Bacillus chromosome
- PAC P1-derived artificial chromosome
- the plasmid vector is not particularly limited as long as it is a plasmid vector that can be expressed in the somatic cell to be introduced.
- a generally used plasmid vector for expressing animal cells can be used.
- the plasmid vector for animal cell expression include pA1-11, pXT1, pRc / CMV, pRc / RSV, pcDNAI / Neo and the like.
- Virus vectors include retrovirus (including lentivirus) vector, adenovirus vector, adeno-associated virus vector, Sendai virus vector, herpesvirus vector, vaccinia virus vector, poxvirus vector, poliovirus vector, sylvis virus vector, and loved. Examples thereof include a virus vector, a paramixovirus vector, an orthomixovirus vector, and the like.
- retrovirus including lentivirus
- adenovirus vector adeno-associated virus vector
- Sendai virus vector Sendai virus vector
- herpesvirus vector vaccinia virus vector
- poxvirus vector poxvirus vector
- poliovirus vector poliovirus vector
- sylvis virus vector sylvis virus vector
- Virus vectors include retrovirus (including lentivirus) vector, adenovirus vector, adeno-associated virus vector, Sendai virus vector, herpesvirus vector, vaccinia virus vector, poxvirus vector, poliovirus vector, s
- Sendai virus vector is preferable.
- Sendai virus is one of the Mononegavirales viruses and belongs to the genus Paramyxoviridae; Paramyxovirus, Morbilivirus, Rubulavirus, and Pnemovirus, which is a genus of viruses, including a single protein chain. It contains the RNA of the virus (antisense strand against the sense strand) as a genome. Negative-strand RNA is also called negative-strand RNA.
- Sendai viral vectors are non-chromosomally integrated viral vectors that are expressed in the cytoplasm. Therefore, there is no risk that the transgene will be integrated into the host chromosome. Therefore, it is highly safe and it is possible to remove the vector from the introduced cells after achieving the purpose.
- the Sendai virus vector is a complex consisting of an infectious virus particle, a virus core, a complex of a virus genome and a virus protein, or a non-infectious virus particle, and is a gene to be loaded by introduction into a cell. Includes a complex capable of expressing.
- a ribonucleoprotein core portion of a virus consisting of the Sendai virus genome and Sendai virus proteins (NP, P, and L proteins) that bind to it can express the transgene in the cell by introducing it into the cell. Yes (International Publication No. 00/70055).
- the introduction into cells may be carried out by appropriately using a transfection reagent or the like. Therefore, such a ribonucleoprotein (RNP) is also included in the Sendai viral vector.
- RNP ribonucleoprotein
- the Sendai virus genome consists of NP (nucleocapsid) gene, P (phospho) gene, M (matrix) gene, F (fusion) gene, and HN (hemogglutinin / neuraminidase) in order from the 3'end to the 5'end. ) Gene and L (large) gene are included. Of these, Sendai virus can sufficiently function as a vector if it has an NP gene, a P gene, and an L gene, can replicate the genome in a cell, and can express the loaded gene. Since Sendai virus has minus-strand RNA in its genome, the 3'side of the genome is upstream and the 5'side is downstream, which is the opposite of the usual case.
- accession numbers of the base sequence database (GenBank) of each of the above genes of Sendai virus are, for example, M29343, M30202, M30203, M30204, M51331, M555565, M69046, X17218 for the NP gene; M30202, M30203 for the P gene.
- HN gene D26475, M12397, M30202, M30203, M30204, M69046, X00586, X02808, X56131; , X58886.
- a plurality of strains of Sendai virus are known, and there are genes consisting of sequences other than those exemplified above depending on the strains.
- Sendai viral vectors having viral genes derived from any of these genes are also useful as Sendai viral vectors.
- the Sendai virus vector has 90% or more, preferably 95% or more, 96% or more, 97% or more, 98% or more, or 99% or more sequence identity with the coding sequence of any of the above viral genes. It may contain a base sequence having.
- the Sendai virus vector is, for example, 90% or more, preferably 95% or more, 96% or more, 97% or more, 98% or more, or 99 with the amino acid sequence encoded by the coding sequence of any of the above viral genes. It may contain a base sequence encoding an amino acid sequence having% or more identity.
- the Sendai virus vector has, for example, 10 or less, preferably 9 or less, 8 or less, 7 or less, 6 or less, 5 in the amino acid sequence encoded by the coding sequence of any of the above viral genes. Within, 4 or less, 3 or less, 2 or less, or an amino acid sequence in which one amino acid is substituted, inserted, deleted, and / or added, which retains the function of each gene product. It may contain the encoding base sequence.
- sequence to which the database accession number such as the base sequence and amino acid sequence described in the present specification is referred to refers to the sequence as of the filing date of the present application, and is specified as the sequence as of the filing date of the present application.
- sequence at each point in time can be identified by referring to the database revision history.
- the Sendai virus vector used in this embodiment may be a derivative.
- Derivatives of the Sendai virus vector include a virus in which the viral gene is modified so as not to impair the gene transfer ability of the Sendai virus, a virus in which the virus gene is chemically modified, and the like.
- the Sendai virus may be derived from a natural strain, a wild strain, a mutant strain, a laboratory passage strain, an artificially constructed strain, or the like.
- Z strain can be mentioned (Medical Journal of Osaka University Vol.6, No.1, March 1955 p1-15). That is, the virus may be a viral vector having a structure similar to that of a virus isolated from nature, or a virus artificially modified by genetic recombination, as long as the desired function can be achieved.
- any gene possessed by the wild-type virus may be mutated or deleted. It is also possible to use incomplete viruses such as DI particles (J. Virol. 68: 8413-8417, 1994).
- a virus having a mutation or deletion in at least one gene encoding the envelope protein or the outer shell protein of the virus can be preferably used.
- a viral vector is, for example, a viral vector that can replicate the genome in infected cells but cannot form infectious viral particles.
- a virus vector lacking in transmission ability is highly safe because there is no concern that the infection will spread to the surroundings.
- a viral vector that does not contain at least one gene encoding an enveloped protein or spike protein such as F and / or HN, or a combination thereof can be used (International Publication No. 00/70055, International Publication No. 00/70055). 00/70070, Li, H.-O.et al., J. Virol.
- the proteins required for genome replication are encoded in genomic RNA
- the genome can be amplified in infected cells.
- a defective virus for example, a defective gene product or a protein capable of complementing it is externally supplied to virus-producing cells (International Publication No. 00/70055, International Publication No. 00/70070, Li, H.-O.et al., J. Virol. 74 (14) 6564-6569 (2000)).
- the viral vector is recovered as an RNP (eg, an RNP consisting of N, L, P proteins, and genomic RNA), the vector can be produced without complementing the enveloped protein.
- Suitable Sendai virus vectors include, for example, G69E, T116A and A183S mutations in the M protein, A262T, G264 and K461G mutations in the HN protein, L511F mutations in the P protein, and N1197S and K1795E mutations in the L protein. It may be an F gene-deficient Sendai virus vector having a gene (for example, Z stripe), or may be a vector in which a mutation of TS 7, TS 12, TS 13, TS 14, or TS 15 is further introduced into this vector. Specifically, SeV18 + / TS ⁇ F (International Publication No. 2010/008054, International Publication No.
- TS ⁇ F has G69E, T116A and A183S mutations in the M protein, A262T, G264R and K461G mutations in the HN protein, L511F mutations in the P protein, and N1197S and K1795E mutations in the L protein, and has the F gene. It means to delete.
- Suitable Sendai virus vectors include, for example, a vector in which a degron sequence is added to a P protein so that it can be easily removed from infected cells (International Publication No. 2016/125364).
- Deglon includes mTOR Deglon (US Patent Application Publication No. 2009/0215169), Dihydrofolate Reductase (DHFR) Deglon (US Patent Application Publication No. 2012/0178168), PEST (International Publication No. 99/54348), TetR Deglon (US Patent Application Publication No. 99/54348). International Publication No. 2007/032555) and auxin-inducible degron (AID) (International Publication No. 2010/125620), but are not limited to these.
- DD-tag which is a kind of mTOR degron, is added to the C-terminal of P protein is preferably used.
- Reconstruction of the recombinant Sendai virus vector having the polynucleotide to be introduced can be carried out by using a known method. Specifically, (a) a cDNA encoding Sendai virus genomic RNA (minus strand) or its complementary strand (plus strand) is used to express viral proteins (N, P, and L) required for virus particle formation. It can be produced by the step of transferring in (b) and the step of collecting the culture supernatant containing the generated virus. The viral protein required for particle formation may be expressed from transcribed viral genomic RNA or may be supplied to trans from other than genomic RNA.
- expression plasmids encoding N, P, and L proteins can be introduced and supplied into cells. If the viral gene required for particle formation is deleted in the genomic RNA, the viral gene can be separately expressed in virus-producing cells to complement the particle formation.
- a vector in which the DNA encoding the protein or genomic RNA is ligated downstream of an appropriate promoter that functions in the host cell is introduced into the host cell.
- the transcribed genomic RNA is replicated in the presence of viral proteins to form infectious viral particles.
- the defective protein or another viral protein capable of complementing its function can be expressed in virus-producing cells.
- Sendai virus can be carried out by using the following known methods (International Publication No. 97/16539; International Publication No. 97/16538; International Publication No. 00/70055; International Publication No. 97. 00/70070; International Publication No. 01/18223; International Publication No. 03/025570; International Publication No. 2005/071092; International Publication No. 2006/137517; International Publication No. 2007/083644; International Publication No. 2008 / No. 007581; Hasan, MK et al., J. Gen. Virol. 78: 2813-2820, 1997, Kato, A. et al., 1997, EMBO J.
- the H1foo gene or the like may be located at any position of the Sendai virus genomic RNA (minus strand) as long as each gene of the Sendai virus is not destroyed.
- the H1foo gene or the like may be on the 3'side of the NP gene, may be between the NP gene and the P gene, may be between the P gene and the M gene, and may be between the M gene and the F gene. It may be between the F gene and the HN gene (if it does not have the F gene, it may be between the M gene and the HN gene), it may be between the HN gene and the L gene, and it may be on the 5'side of the L gene. There may be.
- the H1foo gene or the like In order to increase the expression level of the H1foo gene or the like, it is preferable to place the H1foo gene or the like on the 3'side.
- the H1foo gene or the like is placed on the 3'side of the Sendai virus genomic RNA (minus strand) (for example, 3'side of the NP gene)
- the expression level of the H1foo gene or the like increases most.
- a Sendai virus vector it is preferable to place the H1foo gene or the like on the 3'side of all the genes of Sendai virus. That is, it is preferable to place the H1foo gene or the like on the 3'side most.
- virus particles obtained by using packaging cells may be used.
- a packaging cell is a cell into which a gene encoding a structural protein of a virus has been introduced, and when a recombinant viral vector in which the target gene is incorporated into the cell is introduced, recombinant virus particles incorporating the target gene are produced.
- the packaging cells are not particularly limited and can be appropriately selected depending on the intended purpose.
- packaging cells based on HEK293 cells derived from human kidneys or NIH3T3 cells derived from mouse fibroblasts; PLAT-E cells designed to express ecotropic virus-derived enveloped glycoproteins; PLAT-A cells designed to express PLAT-A cells; and PLAT-GP cells designed to express enveloped glycoproteins derived from bullous stomatitis virus.
- the packaging cell is preferably PLAT-A cell, PLAT-GP cell or the like in terms of host directivity.
- the method for introducing the viral vector into the packaging cells is not particularly limited, and can be appropriately selected depending on the intended purpose. For example, a lipofection method, an electroporation method, a calcium phosphate method and the like can be mentioned.
- the expression vector may contain a nuclear reprogramming substance described later.
- the nuclear reprogramming substance may be one kind or two or more kinds.
- a base sequence encoding a self-cleaving peptide such as a 2A peptide or an IRES (Internal Ribozyme Entry) between the H1foo gene or the like and the nuclear reprogramming substance is used.
- Site An array or the like may be interposed. By interposing these sequences, a plurality of proteins can be independently expressed from one promoter.
- the quality improver of the present embodiment is a Sendai virus vector carrying a polynucleotide encoding a fusion protein of H1foo protein and a destabilizing domain in an expressible form.
- the quality improver of the present embodiment is a Sendai virus vector carrying a polynucleotide encoding a fusion protein of H1foo protein and a destabilizing domain derived from FKBP12 in an expressible form.
- the fusion protein containing H1FOO include a protein containing the amino acid sequence set forth in SEQ ID NO: 16 or SEQ ID NO: 18.
- Specific examples of the polynucleotide encoding these proteins include polynucleotides containing the nucleotide sequence set forth in SEQ ID NO: 15 or SEQ ID NO: 17.
- the present invention provides an iPS cell quality improving agent comprising a fusion protein of H1foo protein and destabilizing domain.
- the destabilizing domain is a domain that induces the degradation of the fusion protein by the proteasome.
- the H1foo protein is a protein produced by transcription and translation from the H1foo gene described in the above " ⁇ 1st embodiment>" and has H1foo activity.
- the organism species from which the H1foo protein is derived is not particularly limited and may be appropriately selected depending on the intended purpose. Examples thereof include arbitrary mammals such as humans, mice, rats, cows, sheep, horses and monkeys. ..
- the amino acid sequence of the human H1foo protein is exemplified in SEQ ID NO: 2. Further, the amino acid sequence of the mouse H1foo protein is exemplified in SEQ ID NO: 4.
- the H1foo protein is not limited to the wild-type H1foo protein, and may include mutations (either deletion, substitution, insertion, and addition, or a combination thereof).
- the number of mutations is not particularly limited as long as it is a protein having H1foo activity.
- H1foo protein examples include the following (a) to (c).
- A Wild-type H1foo protein (eg, a polypeptide consisting of the amino acid sequence represented by SEQ ID NO: 2 or SEQ ID NO: 4)
- B A protein (c) consisting of an amino acid sequence in which one or more amino acids are mutated in the amino acid sequence of the wild-type H1foo protein (eg, the amino acid sequence represented by SEQ ID NO: 2 or 4) and having H1foo activity.
- a protein consisting of an amino acid sequence of a wild-type H1foo protein for example, an amino acid sequence represented by SEQ ID NO: 2 or 4) and an amino acid sequence having 70% or more sequence identity and having H1foo activity.
- the "mutation” may be any of deletion, substitution, addition, and insertion, and may be a combination thereof.
- the "plurality” is not particularly limited as long as the resulting protein has H1foo activity, but for example, 2 to 30 are exemplified, preferably 2 to 20 and more preferably 2 to 10. Preferably, 2 to 5 are even more preferred, and 2 or 3 are particularly preferred.
- the sequence identity is not particularly limited as long as it is 70% or more, but 80% or more is preferable, 85% or more is more preferable, 90% or more, 95% or more, 96% or more, 97%. As mentioned above, 98% or more, or 99% or more is more preferable.
- the destabilizing domain is the same as that described in the above “ ⁇ 1st embodiment>", and the same as those exemplified in the above “ ⁇ 1st embodiment>” are exemplified.
- the fusion protein of the H1foo protein and the destabilizing domain is one in which the destabilizing domain is linked to at least one of the N-terminal and the C-terminal of the H1foo protein.
- the destabilizing domain may be ligated to either the N-terminus or the C-terminus of the H1foo protein, or may be ligated to both the N-terminus and the C-terminus.
- Fusion proteins of the H1foo protein and the destabilizing domain can be prepared using an expression vector comprising a polynucleotide in which the destabilizing domain gene is linked to at least one of the 5'end or 3'end of the H1foo gene.
- the fusion protein can be obtained by introducing the expression vector into an appropriate cell, culturing the cell, and isolating / purifying the fusion protein from the culture supernatant or the cultured cell or cell.
- the fusion protein of the H1foo protein and the destabilizing domain may further have a protein transfer domain (PTD).
- PTDs protein transfer domains
- those using cell transit domains of proteins such as AntP derived from Drosophila, TAT derived from HIV, and VP22 derived from HSV have been developed. Having a PTD allows the fusion protein to be introduced into cells without the use of protein transfer reagents.
- the fusion protein of the H1foo protein and the destabilizing domain is rapidly degraded by the proteasome because it has the destabilizing domain when introduced into somatic cells.
- the fusion protein containing the destabilizing domain can control the intracellular abundance and abundance time by adding the stabilizing compound. Therefore, it is possible to control the abundance and time of existence of H1foo protein in cells.
- the fusion protein of the H1foo protein and the destabilizing domain is intracellular, for example, only at an arbitrary time immediately after the introduction. By controlling the presence of iPS cells, high-quality iPS cells can be produced.
- the cells are reprogrammed by being introduced into somatic cells together with a nuclear reprogramming substance instead of the polynucleotide having the H1foo gene and the control sequence. It is also possible to use a substance having a function of suppressing the spontaneous immune response in the cell at the same time when it is started. Suppression of the innate immune response means suppression of the expression of innate immune response markers such as IFIT1 and IFNA as a specific example, and more specifically, the expression of FKBP1A in the cell was introduced only by a nuclear reprogramming substance.
- the increase is 2 times or more, preferably 2 to 20 times, more preferably 5 to 15 times, as compared with the case.
- the time when cell reprogramming is started means 2 to 15 days, preferably 3 to 6 days after the introduction of the nuclear reprogramming substance.
- the substance having a function of suppressing the innate immune response is not particularly limited as long as it can suppress the expression of the innate immune response marker.
- Examples of the substance having a function of suppressing the natural immune response include siRNA or antisense RNA against the natural immune response marker gene, a transcriptional inhibitor of the natural immune response marker gene, FKBP1A, FKBP1A gene, transcription promoting factor of FKBP1A and the like. However, it is not limited to these.
- the present invention provides a method for producing iPS cells. It comprises a step of introducing a nuclear reprogramming substance and a quality improving agent for iPS cells according to the above embodiment into somatic cells.
- the method for producing iPS cells of the present embodiment provides a method for producing iPS cells. It comprises a step of introducing a nuclear reprogramming substance and a quality improving agent for iPS cells according to the above embodiment into somatic cells.
- the iPS cell quality improving agent may be either the first embodiment or the second embodiment described in the above-mentioned "[iPS cell quality improving agent]".
- the quality improving agents for iPS cells of both the first embodiment and the second embodiment may be used in combination.
- nuclear reprogramming substance means a substance (group) capable of inducing a somatic cell into an iPS cell by introducing it into the somatic cell.
- the nuclear reprogramming substance is not particularly limited as long as it is a substance (group) capable of inducing iPS cells from somatic cells.
- the nuclear reprogramming substance may be any substance such as a gene (including a form incorporated in an expression vector), a gene product thereof, or a small molecule compound.
- “Gene product” means an mRNA transcribed from a gene and a protein translated from that mRNA.
- the gene product used as a nuclear reprogramming substance may be mRNA, protein, or both.
- a gene that is a nuclear reprogramming substance means a polynucleotide that encodes a protein that is a nuclear reprogramming substance.
- the nuclear reprogramming substance is a gene or a gene product thereof, the Oct gene family gene, the Sox gene family gene, the Klf gene family gene, the Myc gene family gene, the Lin gene family gene, and the Nanog gene, and them.
- At least one selected from the group consisting of genetic products of International Publication No. 2007/69666; Patent No. 5696282; Science, 2007, 318: 1917-1920.
- at least one selected from the group consisting of a gene of the Oct gene family, a gene of the Sox gene family, a gene of the Klf gene family, a gene of the Myc gene family, and a gene product of these genes is mentioned. ..
- a nuclear reprogramming substance consisting of genes of the Oct gene family (B) A combination of two nuclear reprogramming substances consisting of a gene of the Oct gene family and a gene of the Sox gene family; (C) A combination of two nuclear reprogramming substances consisting of a gene of the Oct gene family and a gene of the Klf gene family; (D) A combination of two nuclear reprogramming substances consisting of a gene of the Oct gene family and a Nanog gene; (E) A combination of three nuclear reprogramming substances consisting of a gene of the Oct gene family, a gene of the Sox gene family, and a gene of the Klf gene family; (F) A combination of three nuclear reprogramming substances consisting of a gene of the Oct gene family, a gene of the Klf gene family, and a gene of the Myc gene family.
- G A combination of four nuclear reprogramming substances consisting of a gene of the Oct gene family, a gene of the Sox gene family, a gene of the Klf gene family and a gene of the Myc gene family; and (h) a gene of the Oct gene family and a Sox gene.
- a combination of four nuclear reprogrammers consisting of a family of genes, a Lin gene family of genes, and a Nanog gene.
- the Sox2 gene can be replaced with the Sox1 gene, the Sox3 gene, the Sox15 gene, the Sox17 gene, or the Sox18 gene.
- the Klf4 gene can be replaced with the Klf1 gene, the Klf2 gene, or the Klf5 gene.
- the c-Myc gene can be replaced with a T58A (active mutant) gene, an N-Myc gene, or an L-Myc gene.
- Oct3 / 4 gene, Sox2 gene, Klf4 gene, c-Myc gene, Nanog gene, Lin28 gene see Cell Research (2008) 600-603.
- Oct3 / 4 gene, Sox2 gene, Klf4 gene, c-Myc gene, SV40LT gene see also Stem Cells, 26, 1998-2005 (2008).
- Oct3 / 4 gene, Klf4 gene see Nature 454: 646-650 (2008), Cell Stem Cell, 2: 525-528 (2008)
- Oct3 / 4 gene, c-Myc gene see Nature 454: 646-650 (2008)
- 16) Oct3 / 4 gene, Sox2 gene see Nature, 451, 141-146 (2008), International Publication No.
- a member gene of another Oct gene family (for example, Oct1A, Oct6, etc.) can be used instead of the Oct3 / 4 gene.
- a member gene of another Sox gene family (for example, the Sox7 gene) can also be used.
- a member gene of another Lin gene family (for example, the Lin28b gene) can also be used.
- one or more nuclear reprogramming substances selected from the group consisting of the Fbx15 gene, the Eras gene, the ECAT15-2 gene, the Tcl1 gene, and the ⁇ -catenin gene may be combined, and / Or from the group consisting of ECAT1 gene, Esg1 gene, Dnmt3L gene, ECAT8 gene, Gdf3 gene, Mybl2 gene, ECAT15-1 gene, Fthl17 gene, All4 gene, Rex1 gene, UTF1 gene, Stella gene, Stat3 gene, and Grb2 gene. It is also possible to combine one or more selected nuclear reprogramming substances. These combinations are specifically described in WO 2007/69666.
- Preferred nuclear reprogramming agents are selected from the group consisting of Oct3 / 4 gene, Sox2 gene, Klf4 gene, c-Myc gene (or L-Myc gene), Lin28 gene, Nanog gene, and gene products of these genes. At least one is exemplified. Preferably, two or more combinations selected from the group consisting of Oct3 / 4 gene, Sox2 gene, Klf4 gene, c-Myc gene (or L-Myc gene), Lin28 gene, Nanog gene, and their gene products, and more. It is preferably a combination of three or more.
- nuclear reprogramming substances that are preferably introduced includes (1) Oct3 / 4 gene or its gene product, Sox2 gene or its gene product, Klf4 gene or its gene product, and (2) Oct3. / 4 gene or its gene product, Sox2 gene or its gene product, Klf4 gene or its gene product, and c-Myc gene or its gene product, (3) Oct3 / 4 gene or its gene product, Sox2 gene or its gene product , Klf4 gene or its gene product, and L-Myc gene or its gene product.
- a product and a combination of the L-Myc gene or a gene product thereof are preferred.
- the combination of Oct3 / 4 gene, Sox2 gene, and Klf4 gene, and the combination of Oct3 / 4 gene, Sox2 gene, Klf4 gene, and L-Myc gene are more preferable.
- the nuclear reprogramming substance is a gene or a gene product thereof
- the species from which the gene is derived is not particularly limited and may be appropriately selected depending on the intended purpose. For example, humans, mice, rats, cattle and sheep. , Horses, monkeys and any other mammals.
- the cDNA sequence information of each of the above nuclear reprogramming substances can be obtained from a known database.
- GenBank described in International Publication No. 2007/069666
- the Nanog gene is described in the above publication under the name "ECAT4".
- mouse and human cDNA sequence information of four particularly preferable genes are described below.
- the cDNA sequence of the human Oct3 / 4 gene registered in the above GenBank accession number is shown in SEQ ID NO: 5, and the amino acid sequence of the human Oct3 / 4 protein is shown in SEQ ID NO: 6.
- the cDNA sequence of the human Sox2 gene is shown in SEQ ID NO: 7, and the amino acid sequence of the human Sox2 protein is shown in SEQ ID NO: 8.
- the cDNA sequence of the human Klf4 gene is shown in SEQ ID NO: 9, and the amino acid sequence of the human Klf4 protein is shown in SEQ ID NO: 10.
- the cDNA sequence of the human L-Myc gene is shown in SEQ ID NO: 11, and the amino acid sequence of the human L-Myc protein is shown in SEQ ID NO: 12.
- the cDNA of each of the above nuclear reprogramming substances can be easily obtained from the cells of the organism from which the sequence is derived by using a known method such as PCR method, based on the above cDNA sequence information or the sequence information registered in a known database. Can be isolated in.
- nucleotide sequences are not limited to the nucleotide sequences of wild-type genes, and may contain mutations as long as they have a nuclear reprogramming effect.
- Each nucleotide sequence of each of the above genes may be only a protein coding sequence, or may contain other portions (for example, intron, 5'UTR, 3'UTR, etc.).
- the nuclear reprogramming substance is a protein encoded by each of the above genes
- their amino acid sequences are not limited to the amino acid sequences of wild-type proteins, and may contain mutations as long as they have a nuclear reprogramming effect. ..
- nuclear reprogramming action means an action of inducing a somatic cell into an iPS cell by introducing it into the somatic cell.
- nuclear reprogramming substance is a gene or mRNA
- examples of the nuclear reprogramming substance include the following (a) to (g).
- A Wild-type gene or wild-type mRNA exemplified as the above-mentioned nuclear reprogramming substance.
- B A polynucleotide consisting of a nucleotide sequence encoding a wild-type protein exemplified as the nuclear reprogramming substance
- c An amino acid sequence in which one or more amino acids are mutated in the wild-type protein exemplified as the nuclear reprogramming substance.
- Polynucleotide comprising and encoding a protein having a nuclear reprogramming effect (d) consisting of an amino acid sequence having 70% or more sequence identity with the amino acid sequence of the wild-type protein exemplified as the nuclear reprogramming substance and in the nuclear initial stage Polynucleotide encoding a protein having a chemogenic effect (e)
- a nucleotide sequence of a wild-type gene exemplified as the above-mentioned nuclear reprogramming substance one or more nucleotides are mutated nucleotide sequences and have a nuclear reprogramming effect.
- Polynucleotide encoding the protein (f) A protein consisting of the nucleotide sequence of the wild-type gene exemplified as the above-mentioned nuclear reprogramming substance and a nucleotide sequence having 70% or more sequence identity and having a nuclear reprogramming action.
- Encoding polynucleotide (g) A polynucleotide encoding a protein that hybridizes with the nucleotide sequence of a wild-type gene exemplified as the above-mentioned nuclear reprogramming substance under stringent conditions and has a nuclear reprogramming action.
- the "mutation” may be any of deletion, substitution, addition, and insertion, and may be a combination thereof.
- the above (c) and “plurality” are not particularly limited as long as the resulting protein has nuclear reprogramming activity, but for example, 2 to 30 are exemplified, preferably 2 to 20 and 2 to 10. More preferably, 2 to 5 are even more preferable, and 2 or 3 are particularly preferable.
- the "plurality” is not particularly limited as long as the resulting polynucleotide encodes a protein having a nuclear reprogramming action, but for example, 2 to 60 are exemplified, and 2 to 50 are preferable.
- the sequence identity is not particularly limited as long as it is 70% or more, but 80% or more is preferable, 85% or more is more preferable, and 90% or more, 95% or more, 96%. More preferably, 97% or more, 98% or more, or 99% or more.
- the "stringent condition” is the same as the condition exemplified in the above-mentioned "[iPS cell quality improving agent]", ⁇ first embodiment>, (H1foo gene) ". Can be mentioned.
- the degenerate codons used are somatic cells using the iPS cell quality improving agent of the present embodiment having a high codon usage frequency.
- codons frequently used in human cells are used. That is, it is preferably optimized for human codons.
- codons frequently used in mouse cells are used. That is, it is preferably optimized for mouse codons.
- the nuclear reprogramming substance is a gene
- the gene is integrated into an expression vector.
- the expression vector include the same as those mentioned in the above-mentioned "[quality improver for iPS cells], (polynucleotide)".
- the Sendai virus vector is preferable as the expression vector.
- an expression vector is used, only one kind of nuclear reprogramming substance may be incorporated into one expression vector, or two or more kinds of nuclear reprogramming substances may be incorporated.
- the expression vector contains two or more kinds of nuclear reprogramming substances, a base sequence encoding a self-cleaving peptide such as 2A peptide, an IRES sequence, etc. are interposed between two or more kinds of genes which are nuclear reprogramming substances. You may let me.
- nuclear reprogramming substance When the nuclear reprogramming substance is a protein, examples of the nuclear reprogramming substance include the following (A) to (C).
- B) The amino acid sequence of the wild protein exemplified as the nuclear reprogramming substance consists of an amino acid sequence in which one or more amino acids are mutated, and the nuclear initial stage.
- Protein with activating activity C
- the "mutation” may be any of deletion, substitution, addition, and insertion, and may be a combination thereof.
- the “plurality” is not particularly limited as long as the resulting protein has nuclear reprogramming activity, but for example, 2 to 30 are exemplified, preferably 2 to 20 and 2 to 10. Is more preferable, 2 to 5 pieces are more preferable, and 2 or 3 pieces are particularly preferable.
- the sequence identity is not particularly limited as long as it is 70% or more, but 80% or more is preferable, 85% or more is more preferable, 90% or more, 95% or more, 96% or more, 97%. As mentioned above, 98% or more, or 99% or more is more preferable.
- a protein transfer domain may be linked for introduction into somatic cells.
- the nuclear reprogramming substance is a gene or a gene product thereof, an embodiment using only the gene, an embodiment using only the gene product, or an embodiment using both the gene and the gene product thereof may be used. May be.
- the nuclear reprogramming substance is a gene or a gene product thereof and two or more kinds of "genes or gene products thereof" are used in combination, a gene product is used for one gene and a gene is used for another gene. May be.
- the nuclear reprogramming substance is preferably a gene or a combination of genes exemplified above, and more preferably in the form of an expression vector.
- somatic cell The somatic cells are not particularly limited and can be appropriately selected depending on the intended purpose.
- somatic cells include fetal somatic cells, mature somatic cells and the like.
- Specific examples of mature somatic cells include tissue stem cells (somatic stem cells) such as mesenchymal stem cells, hematopoietic stem cells, adipose tissue-derived stromal cells, adipose tissue-derived stromal stem cells, nerve stem cells, and sperm stem cells; tissue precursor cells. ;
- tissue stem cells such as mesenchymal stem cells, hematopoietic stem cells, adipose tissue-derived stromal cells, adipose tissue-derived stromal stem cells, nerve stem cells, and sperm stem cells; tissue precursor cells.
- Already differentiated cells such as fibroblasts, epithelial cells, lymphocytes, muscle cells; etc.
- the species from which somatic cells are derived is not particularly limited and can be appropriately selected according to the purpose.
- Species from which somatic cells are derived include, for example, any mammal such as humans, mice, rats, cows, sheep, horses, monkeys.
- the organism from which the somatic cells are derived and the organism from which the nuclear reprogramming substance is derived do not have to be the same, but are preferably the same.
- the organism species from which the H1foo gene or H1foo protein contained in the quality improving agent for iPS cells of the above embodiment is derived and the organism species from which somatic cells are derived may not be the same, but are preferably the same.
- the individual from which somatic cells are derived there are no particular restrictions on the individual from which somatic cells are derived, and it can be appropriately selected according to the purpose.
- the individual subject to the regenerative medicine or the individual subject to the regenerative medicine and the MHC Other individuals of the same type or substantially the same type are preferable.
- the fact that the types of MHC are substantially the same means that when cells obtained by inducing differentiation from somatic cell-derived iPS cells are transplanted into an individual, the transplanted cells can be obtained by using an immunosuppressive agent or the like. It means that the types of MHC match to the extent that they can be engrafted.
- the somatic cell may be a recombinant cell into which an allogeneic gene has been introduced in order to facilitate selection of iPS cells.
- recombinant cells include recombinant cells in which at least one of a reporter gene and a drug resistance gene is incorporated into a locus of a gene that is specifically highly expressed in a pluripotent cell.
- genes specifically highly expressed in differentiated pluripotent cells include the Fbx15 gene, Nanog gene, Oct3 / 4 gene, and the like.
- the reporter gene include a green fluorescent protein (GFP) gene, a luciferase gene, a ⁇ -galactosidase gene and the like.
- the drug resistance gene include a blastocidin gene, a hygromycin gene, a puromycin resistance gene, and a neomycin resistance gene.
- the culture conditions for somatic cells are not particularly limited and can be appropriately selected depending on the intended purpose.
- the culture temperature is about 37 ° C.
- the CO 2 concentration is about 2% to 5%
- the O 2 concentration is about 5% to 20%, and the like.
- the medium used for culturing somatic cells is not particularly limited and can be appropriately selected depending on the intended purpose.
- minimum essential medium (MEM), Dulbecco modified medium (DMEM), RPMI1640 medium, 199 medium, F12 medium and the like containing 5% by mass to 20% by mass of serum can be mentioned.
- what is simply expressed as "%" for the concentration means "volume%".
- the method for introducing the nuclear reprogramming substance and the quality improving agent for iPS cells of the above embodiment is not particularly limited and may be appropriately selected depending on the intended purpose. be able to. Considering the ease of introduction into somatic cells, it is preferable to introduce the nuclear reprogramming substance into somatic cells in the form of a gene or in the form of mRNA of the gene, rather than introducing it into somatic cells in the form of protein. , It is more preferable to introduce it into somatic cells in the form of a gene.
- the nuclear reprogramming substance is preferably introduced into somatic cells in the form of an expression vector.
- the polynucleotide contained in the quality improving agent is preferably in the form of an expression vector.
- the method for introducing the expression vector into somatic cells is not particularly limited and can be appropriately selected according to the purpose.
- Examples of the method for introducing the expression vector into somatic cells include a lipofection method, a microinjection method, a DEAE dextran method, a gene gun method, an electroporation method, and a calcium phosphate method.
- examples of the method for infecting somatic cells with the viral vector include the polybrene method.
- a Sendai virus vector When a Sendai virus vector is used as an expression vector, a nuclear reprogramming substance or a quality improving agent, or a Sendai virus vector carrying a nuclear reprogramming substance and a quality improving agent is prepared by using the vector (Sendai virus particles) as a medium containing somatic cells. By adding to the virus and infecting the somatic cell, the virus is introduced into the somatic cell.
- the dose of the Sendai virus vector can be adjusted as appropriate, and is, for example, multiplicity of infection (MOI) of 0.1 or more, preferably 0.3 or more, 0.5 or more, 1 or more, 2 or more, or 3 or more.
- MOI multiplicity of infection
- Sendai virus vector is in the form of RNP, it can be introduced into cells by a method such as electroporation, lipofection, or microinjection.
- the method for introducing mRNA (messenger RNA) into somatic cells is not particularly limited, and a known method can be appropriately selected and used.
- mRNA can be introduced into somatic cells using a commercially available RNA transfection reagent such as Lipofectamine (registered trademark) MessengerMAX (manufactured by Life Technologies).
- the method for introducing the protein into somatic cells is not particularly limited, and a known method can be appropriately selected and used. Examples of such a method include a method using a protein-introducing reagent, a method using a protein-introduced domain (PTD) fusion protein, a microinjection method, and the like.
- a known method can be appropriately selected and introduced into somatic cells.
- BioPOTER registered trademark
- Protein Delivery Reagent manufactured by Gene Therapy Systmes
- Pro-JectTM Protein Transfection Regent manufactured by PIERCE
- cationic lipids Lipid-based Project-1 (manufactured by PIERCE).
- Targeting Systems Penetratin Peptide (Qbiogene) and Protein Kit (Active Motif) based on membrane-permeable peptides
- GenomONE Ishihara Sangyo
- HVJ envelope inactivated Sendai virus
- a suitable solvent for example, a buffer solution such as PBS, HEPES
- an introduction reagent was added, and the mixture was incubated at room temperature for about 5 to 15 minutes to form a complex, which was replaced with a serum-free medium. Incubate at 37 ° C. for 1 to several hours. Then, the medium is removed and replaced with a serum-containing medium.
- PTD examples include the same as those mentioned in the above-mentioned "[quality improver for iPS cells], ⁇ second embodiment>, (fusion protein)".
- PTD protein introduction reagent
- the microinjection method is a method in which a protein solution is put into a glass needle having a tip diameter of about 1 ⁇ m and punctured and introduced into cells, and the protein can be reliably introduced into cells.
- a nuclear reprogramming substance in the form of a protein together with CPP such as polyarginine and TAT have been developed in mice and humans, and these methods are used. It can also be done (Cell Stem Cell, 4: 381-384 (2009)).
- the number of introductions of the nuclear reprogramming substance and the quality improving agent into the somatic cells may be once or twice or more.
- the introduction time is not particularly limited and can be appropriately selected according to the purpose. All of the nuclear reprogramming substances and quality improvers may be introduced at the same time, or some or all of them may be introduced at different times. It is preferable to introduce all the nuclear reprogramming substances and quality improving agents into somatic cells at the same time.
- the number of introductions is preferably once.
- the amount of the nuclear reprogramming substance introduced into the somatic cell is not particularly limited as long as the nucleus of the somatic cell can be reprogrammed, and all the genes or the gene product thereof to be used are not particularly limited. May be introduced in equal amounts, or may be introduced in different amounts.
- an Oct3 / 4 gene may be used in a large amount (for example, about 3 times) with respect to the Sox2 gene, Klf4 gene, or c-Myc gene or L-Myc gene. (PNAS 106 (31): 12759-12764 (2009), J.Biol.Chem.287 (43): 36273-36282 (2012)).
- the contact of the low molecular weight compound with the somatic cells is carried out by using the low molecular weight compound in an aqueous or non-aqueous solvent at an appropriate concentration.
- Medium that is lysed and suitable for culturing somatic cells eg, minimal essential medium (MEM) containing about 5-20% fetal bovine serum, Dulbecco modified Eagle's medium (DMEM), RPMI1640 medium, 199 medium, F12 medium, etc.
- MEM minimal essential medium
- DMEM Dulbecco modified Eagle's medium
- RPMI1640 199 medium
- F12 medium F12 medium
- the low molecular weight compound solution is added to the medium so that the concentration of the low molecular weight compound is sufficient for nuclear reprogramming to occur in the somatic cells and the cytotoxicity is not observed, and the cells are cultured for a certain period of time.
- the concentration of the small molecule compound which is the nuclear reprogramming substance varies depending on the type of the small molecule compound used, but can be appropriately selected in the range of about 0.1 nM to about 100 nM.
- the contact period is not particularly limited as long as it is sufficient for the nuclear reprogramming of cells to be achieved, but it is usually sufficient to allow the cells to coexist in the medium until the appearance of positive colonies.
- the method for producing iPS cells of the present embodiment may include, if necessary, other steps in addition to the above-mentioned introduction step.
- the other steps are not particularly limited as long as the effects of the present invention are not impaired, and can be appropriately selected depending on the intended purpose.
- a step of culturing somatic cells into which a nuclear reprogramming substance and a quality improving agent have been introduced hereinafter, also simply referred to as “introduced cells”
- introduction cell culture step hereinafter, also simply referred to as “introduced cell culture step”
- the introduced cell culture step is a step of culturing the introduced cells.
- the culture conditions for the introduced cells are not particularly limited, and commonly used stem cell culture conditions can be used.
- examples of the culture conditions include conditions suitable for ES cell culture. Examples of such conditions include a culture temperature of about 37 ° C., a CO 2 concentration of about 2% to 5%, and an O 2 concentration of about 5% to 20%.
- the medium used for culturing the introduced cells is not particularly limited and can be appropriately selected depending on the intended purpose. In the case of mouse cells, Leukemia Inhibitory Factor (LIF) is added as a differentiation inhibitor to a normal medium and cultured.
- LIF Leukemia Inhibitory Factor
- LIF or basic fibroblast growth factor (bFGF) and / or stem cell factor (SCF) is added depending on the culture conditions.
- bFGF basic fibroblast growth factor
- SCF stem cell factor
- bFGF basic fibroblast growth factor
- MEFs fibroblasts
- STO cells and the like are usually often used, but SNL cells (McMahon, AP & Bradley, A. Cell 62, 1073-1085 (1990)) and the like are often used for the induction of iPS cells.
- Co-culture with feeder cells may be started before the introduction of the nuclear reprogramming substance and the quality improving agent, at the time of the introduction, or after the introduction (for example, 1 to 10 days later). good.
- the period of the above-mentioned introduced cell culture step is not particularly limited and can be appropriately selected according to the purpose.
- the time and amount of H1foo protein present in the cells are determined by the control sequence contained in the quality improving agent. You can control at least one of them. Therefore, the existence time or amount of the H1foo protein can be appropriately controlled, and as a result, high-quality iPS cells can be obtained.
- the present invention provides a method for producing an iPS cell, which comprises the step of introducing a nuclear reprogramming substance and an H1foo gene into a somatic cell.
- the H1foo protein expressed from the H1foo gene introduced into the somatic cell is controlled at least one of the presence time and the abundance in the somatic cell.
- the production method of the present embodiment includes a step of introducing a nuclear reprogramming substance and an H1foo gene into somatic cells.
- nuclear reprogramming material (Nuclear reprogramming material)
- the nuclear reprogramming material is the same as that described in the above “ ⁇ 1st embodiment>”.
- Preferred nuclear reprogramming substances include those similar to those mentioned in the above “ ⁇ 1st embodiment>”.
- H1foo gene The H1foo gene is the same as that described in the above-mentioned "[Quality improving agent for iPS cells], ⁇ first embodiment> (H1foo gene)".
- Preferred H1foo genes include those similar to those mentioned in the above-mentioned "[quality improver for iPS cells], ⁇ first embodiment> (H1foo gene)".
- the method for producing iPS cells of the present embodiment at least one of the abundance time and abundance of the H1foo protein expressed from the H1foo gene introduced into the somatic cell is controlled in the somatic cell.
- the method for controlling the time and amount of the H1foo protein present in somatic cells is not particularly limited, and known methods can be used.
- a method for controlling the abundance time and abundance of the H1foo protein preferably, a method using the control sequence described in "[quality improving agent for iPS cells], ⁇ first embodiment> (control sequence)" can be mentioned. ..
- control sequence control sequence
- a method in which a destabilizing domain gene is ligated to the H1foo gene and expressed as a fusion protein of the H1foo protein and the destabilizing domain when introduced into a somatic cell can be mentioned.
- the fusion protein has an destabilizing domain, so that it is rapidly degraded by the proteasome after being produced intracellularly.
- the time from the fusion gene of the H1foo gene and the destabilizing domain gene to the transcription and translation of the fusion protein to produce it and then to decompose it into the proteasome is, for example, 6 hours or less, preferably 4 hours. Within, more preferably within 3 hours, and particularly preferably within 2 hours.
- the time and amount of the fusion protein may be controlled by adding a stabilizing substance. For example, by adding a stabilizing substance to the medium of the introduced cells, or by recovering the cells from the medium and transferring the cells to a medium containing no stabilizing substance, the fusion protein is allowed to be present in the cells at a desired time. be able to.
- the intracellular abundance of the fusion protein can be controlled by adjusting the addition amount of the stabilizing substance.
- a stimulus-responsive promoter may be linked upstream of the H1foo gene so that the H1foo gene is expressed under the control of the stimulus-responsive promoter when introduced into somatic cells.
- the presence time and amount of the H1foo protein can be controlled by giving the introduced cells a "stimulation" in which the stimulus-responsive promoter responds.
- the stimulus-responsive promoter is a tetracycline-responsive promoter, tetracycline or a derivative thereof (doxycycline or the like) can be used to control the expression timing and expression level of the H1foo gene, and as a result, cells introducing the H1foo protein. It is possible to control the time and amount of existence within.
- the method for producing iPS cells of the present embodiment may include, if necessary, other steps in addition to the above-mentioned introduction step.
- the other steps are not particularly limited as long as the effects of the present invention are not impaired, and can be appropriately selected depending on the intended purpose.
- Examples of other steps include an introduced cell culture step as in the above “ ⁇ 1st embodiment>”.
- the introduced cell culture step can be performed in the same manner as the method described in the above " ⁇ 1st embodiment>".
- the method for producing iPS cells of the present embodiment may include a step of controlling at least one of the abundance amount and the abundance time of the H1foo protein in the introduced cells (hereinafter, referred to as "H1foo protein control step").
- the abundance and timing of the H1foo protein can be controlled, for example, by using a stabilizing substance when the H1foo protein is expressed as a fusion protein with a destabilizing domain. Further, for example, when the H1foo gene is expressed under the control of a stimulus-responsive promoter, the abundance and timing of the H1foo protein are controlled by giving the stimulus that the responsive promoter responds to the introduced cells. Can be done.
- the abundance and timing of H1foo protein in the introduced cells is 50% or less, preferably 30% or less, more preferably 30% or less, more preferably 30% or less of the maximum expression level by the time the somatic cell reprogramming is completed when iPS cells are produced. It is controlled to be 20% or less. More specifically, the H1foo protein introduced into the cell is controlled to be 50% or less, preferably 30% or less, more preferably 20% or less 24 hours after the maximum expression in the cell. Will be done. Alternatively, the duration of expression of the H1foo gene after introduction of the nuclear reprogramming substance into somatic cells may be within 24 hours after introduction. Alternatively, the abundance of H1foo protein in the cells into which the nuclear reprogramming substance has been introduced is less than about 50% of the maximum abundance of H1foo protein after the introduction at a time about 5 days after the introduction. May be good.
- At least one of the intracellular presence time and the abundance amount of the H1foo protein can be appropriately controlled. As a result, high quality iPS cells can be obtained.
- the present invention provides iPS cells produced by the method for producing iPS cells according to the above embodiment.
- IPS cells are pluripotent stem cells derived from somatic cells, and have pluripotency for differentiation and self-renewal ability.
- Pluripotency means that it can differentiate into all three germ layer lines.
- the self-replicating ability means the ability to proliferate while maintaining an undifferentiated state.
- the cells produced by the method for producing iPS cells of the above embodiment are iPS cells can be confirmed by a known method. For example, it can be confirmed whether the cells are iPS cells by the expression of undifferentiated markers, the ability to form embryoid bodies, and / or the ability to form chimeras.
- the cell produced by the method for producing an iPS cell according to the above embodiment may be an iPS cell containing a nuclear reprogramming substance and a quality improving agent for the iPS cell according to the above embodiment.
- the cell produced by the method for producing an iPS cell of the above embodiment may be a cell containing a nuclear reprogramming substance, an H1foo gene, and a control sequence.
- the cell produced by the method for producing an iPS cell of the above embodiment may be a cell containing a nuclear reprogramming substance and a fusion gene of an H1foo gene and a control sequence (eg, a destabilizing domain gene). ..
- the cell produced by the method for producing an iPS cell according to the above embodiment may be a cell containing a nuclear reprogramming substance and the H1foo gene functionally linked to a stimulus-responsive promoter.
- the iPS cells produced by the production method of the above embodiment are higher quality iPS cells than the iPS cells produced by the conventional method because the abundance time and abundance amount of the H1foo protein are appropriately controlled in the introduced cells. Is.
- the iPS cells of the present embodiment have an improved number of colonies expressing Tra-1-60.
- the ability to generate naive iPS cells is improved.
- the naive type iPS cells can be confirmed by the fact that the colonies have a naive type-specific dome shape and that the naive type-specific genes are highly expressed.
- the ability to differentiate into target cells for example, cardiomyocytes, primitive endoderm cells
- the cell population of iPS cells produced by the production method of the above embodiment can be a uniform cell population with no variation in gene expression.
- composition for producing iPS cells provides a composition for producing iPS cells.
- the composition for producing iPS cells of the present embodiment contains a nuclear reprogramming substance and a quality improving agent for iPS cells of the above embodiment.
- the nuclear reprogramming substance is the same as that described in the above "[Method for producing iPS cells] ⁇ First Embodiment>".
- Preferred nuclear reprogramming substances include those similar to those mentioned in the above " ⁇ 1st embodiment>”.
- the quality improving agent for iPS cells is the same as that described in the above "[Quality improving agent for iPS cells]”.
- Preferred quality improvers include those similar to those mentioned in the above-mentioned "[iPS cell quality improver]”.
- the amounts of the nuclear reprogramming substances and the quality improving agents are not particularly limited and may be the same amount or different amounts.
- the amount of the Oct3 / 4 gene is the Sox2 gene, the Klf4 gene, or the c-Myc.
- the amount may be large, for example, about 3 times the amount of the gene or the L-Myc gene.
- the composition for producing iPS cells of the present embodiment may contain other components in addition to the nuclear reprogramming substance and the quality improving agent.
- the other components are not particularly limited, and examples thereof include a buffer, a gene transfer reagent, and a protein transfer reagent.
- the present invention provides, as another embodiment, a method for improving the quality of iPS cells, which comprises the step of introducing the iPS cell quality improving agent of the above embodiment into somatic cells.
- the present invention provides the use of a fusion gene of an H1foo gene and a destabilizing domain gene in the production of a quality improving agent for iPS cells.
- the present invention provides, as another embodiment, the use of fusion proteins of H1foo proteins with destabilizing domains in the production of quality improvers for iPS cells.
- the present invention provides, as another embodiment, the use of the H1foo gene linked downstream of a stimulus-responsive promoter in the production of quality improvers for iPS cells.
- the present invention provides, as another embodiment, the use of a nuclear reprogramming agent and a fusion gene of the H1foo gene and a destabilizing domain in the production of a composition for producing iPS cells.
- the present invention provides, as another embodiment, the use of a nuclear reprogramming agent and a fusion protein of H1foo protein and destabilizing domain in the production of compositions for producing iPS cells.
- the present invention provides, as another embodiment, the use of a nuclear reprogramming agent and the H1foo gene linked downstream of a stimulus-responsive promoter in the production of compositions for iPS cell production.
- the present invention provides, as another embodiment, a kit for producing iPS cells, which comprises a nuclear reprogramming substance and a quality improving agent for iPS cells according to the above embodiment.
- the nuclear reprogramming substance and the quality improving agent may be divided into individual containers, may be collected in one container, or may be collected in any number of containers. It may also contain a gene transfer reagent, a protein transfer reagent, packaging cells, a buffer, a diluent and the like.
- Example 1 Construction of Sendai virus vector containing H1FOO gene H1FOO cDNA (Teranishi, T., et al.) Using Sendai virus vector (SeV18; manufactured by ID Pharma Co., Ltd.) and ProteoTuner TM Sheld System (manufactured by Clontech). Three Sendai virus vectors were prepared, including Rapid replacement of somatic linker histones with the oocyte-specific linker histone H1foo in nuclear transfer. Developmental Biology 266, 76-86 (2004).) (Fig. 1).
- SeV18 + H1FOO / TS15 ⁇ F is a vector that does not contain the destabilized domein (DD) of the ProteoTuner TM Steeld System.
- SeV18 + H1FOO-DD / TS15 ⁇ F is a vector containing a sequence in which a DD coding sequence is added to the 3'end of the H1FOO cDNA.
- the vector (b) expresses a fusion protein in which DD is added to the C-terminal of H1FOO, and the degradation of H1FOO is promoted.
- (C) SeV18 + DD-H1FOO / TS15 ⁇ F is a vector containing a sequence in which a DD coding sequence is added to the 5'end of the H1FOO cDNA.
- the vector (c) expresses a fusion protein in which DD is added to the N-terminal of H1FOO, and the degradation of H1FOO is promoted.
- Example 2 Western blotting of H1FOO protein
- H1FOO protein Each of the above Sendai virus vectors (a) to (c) containing the H1FOO gene was transferred from human skin fibroblasts (TIG120 strain, Tokyo Metropolitan Institute of Gerontology, Kondo et al. , Exp Cell Res. 1995 Oct; 220 (2): 501-4), respectively, introduced with infection multiplicity (MOI) 3 and cultured for 5 days, then each cell was collected and anti-H1FOO antibody (HPA037992; Sigma- Western blotting was performed using Aldrich). The results are shown in FIG. In FIG.
- (1) to (4) are (1) H1FOO-free Control, (2) H1FOO (vector (a)), (3) H1FOO-DD (vector (b)), and (4).
- the results in each human skin fibroblast into which each vector of DD-H1FOO (vector (c)) has been introduced are shown.
- the expression level of H1FOO protein was remarkably high in (2) H1FOO.
- (3) H1FOO-DD and (4) DD-H1FOO the expression level of the H1FOO-DD fusion protein or the DD-H1FOO fusion protein was decreased.
- a Sendai virus vector containing the Azami-Green gene was used instead of the H1FOO gene in the vector of the above (a).
- Example 3 Preparation and culture conditions of prime-type human iPS cells The preparation of human iPS cells is described in the literature (Ban, H., Nishishita, N., Fusaki, N., Tabata, T., Saeki, K., Shikamura.
- iPS cells were prepared from fibroblasts (TIG120 strain) and peripheral blood mononuclear cells (product number: CTL-UP1, Lot: HHU20140120, Cellular Technology Ltd.) (each with infection multiplicity 0.1 to 0.3). Infected cells).
- iPS cells Control-iPS
- iPS cells were prepared using the Sendai virus vector containing the Azami-Green gene instead of the H1FOO gene in the vector (a) above. This experiment was conducted according to the guidelines for genetic recombination experiments at Keio University and Kyoto University.
- the human iPS cells prepared as described above are cultured in StemFit AK02N (manufactured by Ajinomoto Co., Inc.) culture medium (hereinafter referred to as "prime type human iPS cell culture medium") coated with iMatrix-511 (manufactured by Nippi Co., Ltd.). Cultured and maintained on a dish.
- the culture medium of prime-type human iPS cells was changed every 2 days, and the cells were passaged every 5 to 7 days using StemPro Accutase (manufactured by Gibco).
- Example 4 Preparation and culture conditions of naive human iPS cells The preparation of human iPS cells is described in the literature (Ban, H. et al., Proc Natl Acad Sci USA 108, 14234-9, 2011; Seki, T. et. Al., Nature Protocols 7, 718-728, 2012; Liu, X et al., Nature Methods 14, 1055-1062, 2017). Human skin using any of the Sendai virus vectors (a) to (c) prepared in Example 1 above, together with a Sendai virus vector containing an Oct3 / 4 gene, a Sox2 gene, a Klf4 gene, and an L-Myc gene.
- Naive iPS cells were generated from fibroblasts and human peripheral blood mononuclear cells (each cell was infected with an infection multiplicity of 0.1 to 0.3). Being naive iPS cells means that the colonies have a naive-specific dome shape and that they highly express naive-specific genes (KLF17, TFCP2L1, PRDM14, and DPPA3). The gene was confirmed by quantitative PCR and immunostaining).
- iPS cells (Control-iPS) were prepared using the Sendai virus vector containing the Azami-Green gene instead of the H1FOO gene in the vector (a) above. This experiment was carried out according to the guidelines for genetic recombination experiments at Keio University and Kyoto University.
- the human iPS cells prepared as described above are NDiff227 (manufactured by Takara Bio), CHIR99021 (manufactured by Sigma-Aldrich), PD0325901 (manufactured by Sigma-Aldrich), Go6983 (manufactured by Sigma-Aldrich), and Human recombinant.
- NDiff227 manufactured by Takara Bio
- CHIR99021 manufactured by Sigma-Aldrich
- PD0325901 manufactured by Sigma-Aldrich
- Go6983 manufactured by Sigma-Aldrich
- Human recombinant Human recombinant.
- the culture medium of naive human iPS cells was replaced every 2 days, and the cells were subcultured using StemPro Accutase (manufactured by Gibco) every 3 to 4 days. Culturing and subculture were performed in a hypoxic environment with an oxygen concentration of 5% using an incubator capable of adjusting the oxygen concentration.
- Example 5 Comparison of iPS cell establishment efficiency by measuring the number of ALP-positive iPS cell colonies According to the method described in Example 3 above, prime-type (1) Control-iPS, (2) H1FOO from human skin fibroblasts. After preparing -iPS, (3) H1FOO-DD-iPS, and (4) DD-H1FOO-iPS, they were cultured in a prime human iPS cell culture medium for 15 days, and the number of ALP-positive colonies was measured. ALP is known as a stem cell marker.
- Prime-type (1) Control-iPS and (3) H1FOO-DD-iPS cells were prepared from human peripheral blood mononuclear cells, and prime-type human iPS cell culture was performed. The cells were cultured in the solution for 15 days, and the number of ALP-positive colonies was measured. Further, in order to compare the establishment efficiency of naive iPS cells, naive (1) Control-iPS and (3) H1FOO from human skin fibroblasts and human peripheral blood mononuclear cells according to the method described in Example 4 above. -DD-iPS cells were prepared and cultured in a naive human iPS cell culture medium for 15 days, and the number of ALP-positive colonies was measured.
- FIGS. 4A-4C The results are shown in FIGS. 4A-4C.
- FIG. 4A primary human iPS cells established from human peripheral blood mononuclear cells
- FIG. 4B naive human iPS cells established from human skin fibroblasts
- FIG. 4C established from human peripheral blood mononuclear cells.
- H1FOO-DD improves the establishment efficiency of human iPS cells regardless of the original cell type and the type of iPS cells to be established.
- Example 6 Comparison of variation in gene expression level Prime-type (1) Control-iPS and (3) H1FOO-DD-iPS were prepared from human skin fibroblasts according to the method described in Example 3 above. .. After culturing in a prime-type human iPS cell culture medium for about 210 days (28 passages), 8 clones each of (1) and (3) were selected and RNA-seq was performed. Based on the RNA-seq data analysis results, we obtained the number of genes with a mean absolute error (MAE) greater than 2.0 (large variation) in the 8 clones of (1). rice field. In the same manner, the number of genes having a MAE greater than 2.0 was obtained in the 8 clones of (3). The results are shown in FIG. 5A.
- MAE mean absolute error
- naive type (1) Control-iPS and (3) H1FOO-DD-iPS were prepared from human skin fibroblasts.
- the cells were cultured in naive human iPS cell culture medium for about 190 days (45 passages), and 8 clones of each of (1) and (3) were selected for RNA-seq.
- the number of genes having a MAE greater than 2.0 was obtained in each of the 8 clones (1) and (3). The results are shown in FIG. 5B.
- H1FOO-DD-iPS has about half the number of genes whose expression levels vary widely among clones as compared with (1) Control-iPS. was suppressed.
- Example 7 Comparison of variations in DNA methylation DNA of (1) Control-iPS 8 clones and (3) H1FOO-DD-iPS 8 clones of prime-type human iPS cells selected in Example 6 above.
- DNA methylation was analyzed using a methylation array (Infinium Human Mechanism 450K BeadChip Kit, Illumina). Based on the analysis results by the DNA methylation array, the number of methylation probes having a mean absolute error (MAE) of more than 2.0 (large variation) was obtained in the 8 clones of (1). In the same manner, the number of genes having a MAE greater than 2.0 was obtained in the 8 clones of (3). The results are shown in FIG. 6A.
- H1FOO-DD-iPS has about half the number of methylated probes that vary widely among clones compared to (1) Control-iPS. It was suppressed.
- Control-iPS and (3) H1FOO-DD-iPS were prepared from human peripheral blood mononuclear cells according to the method described in Example 3 above. .. Three clones of (1) and (3) were selected and cultured in a myocardial differentiation-inducing medium to induce differentiation into cardiomyocytes (Tohyama, S., et al. (2013). Cell Stem Cell 12). (1): 127-137.). Cells were collected 10 days after the start of culture in the myocardial differentiation-inducing medium. The expression of TNNT2 in the recovered cells was detected using a flow cytometer, and the proportion of TNN2-positive cells was calculated. TNNT2 is a representative cardiomyocyte marker. Each clone was subjected to a differentiation induction test into cardiomyocytes 6 times.
- FIG. 7A shows an example of measuring the proportion (%) of TNNT2-positive cells by a flow cytometer.
- the left figure is (1) a measurement example of Control-iPS, and the right figure is a measurement example of (3) H1FOO-DD-iPS.
- (1) In Control-iPS only about 6% of all cells were TNNT2-positive, and differentiation into cardiomyocytes was poor.
- FIG. 7B shows the percentage of TNNT2-positive cells in each clone.
- (3) H1FOO-DD-iPS the proportion of TNNT2-positive cells was higher than that in (1) Control-iPS. Further, it was confirmed that (3) H1FOO-DD-iPS has less variation between each cardiomyocyte differentiation induction test than (1) Control-iPS, and can induce cardiomyocyte differentiation more stably. These results indicate that H1FOO-DD-iPS has excellent cardiomyocyte differentiation potential and can differentiate cardiomyocytes with higher homogeneity.
- Control-iPS and (3) H1FOO-DD-iPS were prepared from human skin fibroblasts according to the method described in Example 3 above. Eight clones each of (1) and (3) were selected and induced to differentiate into three germ layer components by STEMdiff Trilineage differentiation kit (Cat: ST-05230, VERITAS). On the 5th day from the start of differentiation induction, cells were collected. In the recovered cells, the expression of markers related to endoderm was semi-comprehensively evaluated by qPCR using a TaqMan hPSC Scorecard kit (Cat: A15876, Thermo Fisher SCIENTIFIC).
- Example 10 Comparison of hepatocyte differentiation potential (1)
- prime type (1) Control-iPS and (3) H1FOO-DD-iPS were prepared from human peripheral blood mononuclear cells.
- Ten clones of each of (1) and (3) were selected to induce differentiation into hepatocytes (a type of terminally differentiated cells of the endoderm system) (Kajiwara, M., et al. (2012). Proc. Natl Acad Sci USA 109 (31): 12538-12543 .; Takebe, T., et al. (2017). Cell Rep 21 (10): 2661-2670.).
- ASGR1 which is one of the representative mature hepatocyte markers, was quantitatively evaluated by qRT-PCR.
- Example 11 Comparison of hepatocyte differentiation potential (2) According to the method described in Example 3 above, prime type (1) Control-iPS and (3) H1FOO-DD-iPS were prepared from human peripheral blood mononuclear cells. Ten clones of each of (1) and (3) were selected to induce differentiation into hepatocytes in the same manner as in Example 11. On the 21st day from the start of differentiation induction, the culture supernatant was collected. In the collected culture supernatant, the amount of secreted albumin was quantitatively evaluated by ELISA. Since mature hepatocytes produce and secrete albumin, albumin is used as a marker for mature hepatocytes, similar to ASGR1.
- Example 12 Comparison of primitive endoderm differentiation potential Naive type (1) Control-iPS and (3) H1FOO from human skin fibroblasts and human peripheral blood mononuclear cells according to the method described in Example 4 above. -DD-iPS was produced. For each of (1) and (3), 6 clones (derived from human skin fibroblasts: 4 clones, derived from human peripheral blood mononuclear cells: 2 clones) were selected, cultured in a primitive endoderm differentiation-inducing medium, and cultured in the primitive endoderm. Induction of differentiation into germ layers was performed (International Publication No. 2019/093340). Cells were harvested 3 days after the start of culture in the primitive endoderm differentiation-inducing medium.
- PDGFRA and ANPEP are typical primitive endoderm markers.
- Each clone was subjected to a differentiation induction test into a primitive endoderm three times.
- the primordial endoderm differentiation-inducing medium was 25 ng / mL FGF4 (Peprotech), 1 ⁇ g / mL Heparin (Wako), 10 ng / mL BMP4 (R & D), and 10 ng / mL PDGFRA (Peprotech) in N2B27 medium (NDiff227).
- FIG. 11A shows an example of measuring the ratio (%) of PDGFRA-positive cells and ANPEP-positive cells by a flow cytometer.
- the figure on the left is (1) a measurement example of Control-iPS.
- the proportion of cells positive for both PDGFRA and ANPEP was about 19% of all cells.
- the figure on the right is a measurement example of (3) H1FOO-DD-iPS.
- the proportion of cells positive for both PDGFRA and ANPEP was about 37% of all cells.
- FIG. 11B shows the percentage of cells positive for both PDGFRA and ANPEP in each clone.
- (3) H1FOO-DD-iPS the proportion of positive cells was higher than that in (1) Control-iPS. Further, it was confirmed that (3) H1FOO-DD-iPS has less variation among clones than (1) Control-iPS, and can induce differentiation of primitive endoderm more stably. These results indicate that H1FOO-DD-iPS has excellent primordial endoderm differentiation potential and is capable of differentiating into more homogeneous primordial endoderm.
- Example 13 Verification of metabolic function Naive type (1) Control-iPS and (3) H1FOO-DD- from human skin fibroblasts and human peripheral blood mononuclear cells according to the method described in Example 4 above. iPS was produced. Six clones of each of (1) and (3) were selected, and an extracellular flux analyzer (Agilent Searose XF96 Extracellular Flux Analyzer, Prime Tech Co., Ltd.) was used to perform spare respiratory capacity (electron transport chain function). Evaluation) was measured.
- extracellular flux analyzer Align Searose XF96 Extracellular Flux Analyzer, Prime Tech Co., Ltd.
- Control-iPS and (3) H1FOO-DD-iPS were prepared from human skin fibroblasts or human peripheral blood mononuclear cells.
- OCR oxygen consumption rate
- ECAR extracellular acidification rate
- the results are shown in FIG. 12B.
- the X-axis shows EPAR and reflects anaerobic metabolic capacity.
- the Y-axis indicates OCR and reflects aerobic metabolic capacity.
- Each plot shows the mean value measured 6 times with one clone.
- H1FOO-DD-iPS as compared with (1) Control-iPS, the plot was present in the upper right corner as a whole, and both ECAR and OCR were in a high state.
- Example 14 Verification of presence / absence of X-chromosome activation
- one X-chromosome is inactivated in female-derived somatic cells and prime-type iPS / ES cells.
- both X chromosomes are activated in the naive type, especially in the pre-implantation epiblast.
- mRNA-FISH was performed on each of the four naive clones (1) Control-iPS and (3) H1FOO-DD-iPS, and the expression of the three markers was compared and verified.
- naive types Control-iPS and (3) H1FOO-DD-iPS were prepared from human skin fibroblasts and human peripheral blood mononuclear cells. Four clones each of (1) and (3) were selected, and the expression of UTX, HUWE1, and XIST was confirmed by mRNA-FISH (Sahakyan, A., et al. (2017). Cell Stem Cell 20 (1). ): 87-101.). Since a large number of polyploid cells are generated in the currently obtained naive human iPS cells, it is necessary to select cells having a normal number of chromosomes for correct evaluation. UTX is expressed with or without X-chromosome activation.
- HUWE1 expressed when the X chromosome is activated
- XIST originally involved in X-chromosome inactivation, but known to be expressed on both bilateral X chromosomes in pre-implantation epiblasts. Expression was examined by mRNA-FISH.
- FIG. 13A shows an example of a fluorescence microscope image of mRNA-FISH.
- the figure on the left is an example of HUWEI +/+ XIST +/ +.
- the figure on the right is an example of HUWEI +/- XIST ⁇ / ⁇ .
- FIG. 13B shows the results of summarizing the expression patterns of HUWE1 and XIST for 100 cells of each clone.
- the numbers below the bar graph indicate the clone Nos.
- Control-iPS and (3) H1FOO-DD-iPS. Is shown.
- Clone No. Nos. 1 and 2 are derived from skin fibroblasts, and clone No. 1 and 2 are derived from skin fibroblasts.
- 5 and 6 are derived from human peripheral blood mononuclear cells.
- the phenotype closest to the pre-implantation epiblast is HUWE1 +/+ and XIST +/+, and the phenotype equivalent to it is HUWE1 +/+ and XIST +/-.
- Example 15 Comparison of FKBP1A expression level H1FOO-DD was introduced into human fibroblasts (TIG120) using the Sendai virus vector of (c) prepared in Example 1 (H1FOO OE HDF).
- the Oct3 / 4, Sox2 gene, Klf4 gene, and L-Myc gene were introduced into human fibroblasts (TIG120) using a Sendai virus vector to prepare iPS cells (OSKL).
- OSKL Sendai virus vector of (c) prepared in Example 1 above, together with the Sendai virus vector containing the Oct3 / 4 gene, Sox2 gene, Klf4 gene, and L-Myc gene, to human fibroblasts (TIG120).
- Each of the above genes was introduced to prepare H1FOO-DD-iPS (OSKLH).
- the H1FOO OE HDF, OSKL, and OSKLH prepared above were cultured, and the cells on the second day after the introduction of each of the genes were collected. Then, the expression level of FKBP1A was measured by qRT-PCR. As a control, the expression level of FKBP1A was similarly measured for human fibroblasts (HDF) and H9 ES cells (H9 ESC). The expression level of FKBP1A was standardized by the expression level of GAPDH.
- HDF is human skin fibroblast
- H9 ESC is H9 ES cell
- H1FOO OE HDF is a cell cultured for 2 days after preparation of H1FOO OE HDF
- OSKL day2 is a cell cultured for 2 days after preparation of OSKL
- OSKLH day2 is The cells cultured for 2 days after the preparation of OSKLH.
- the expression level of FKBP1A is shown as a relative expression level when the expression level of OSKLH day2 is 1.0. As shown in FIG. 14, the expression level of FKBP1A was remarkably high only in OSKLH day2.
- the expression level of FKBP1A was about 10 times higher than that in other cells. This result indicates that the expression level of FKBP1A is increased in the cells in the early stage of induction of nuclear reprogramming by introducing H1FOO-DD together with the nuclear reprogramming substance. Furthermore, although the results are omitted, it was confirmed that FKBP1A suppresses the expression of IFIT1 and IFNA, which are innate immune response expression markers. Therefore, it is presumed that the expression of the innate immune response marker is suppressed by introducing H1FOO-DD together with the nuclear reprogramming substance. These results suggest that suppression of the expression of innate immune response markers may contribute to improving the quality of iPS cells.
- an iPS cell quality improving agent capable of producing high quality iPS cells, a method for producing iPS cells, iPS cells produced by such a production method, and a composition for producing iPS cells.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Cell Biology (AREA)
- Toxicology (AREA)
- Virology (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Developmental Biology & Embryology (AREA)
- Transplantation (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
H1foo遺伝子と、前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得る制御配列と、を有するポリヌクレオチドを含む、iPS細胞の品質改善剤。
Description
本発明は、iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物に関する。
本願は、2020年9月4日に、日本に出願された特願2020-149447号に基づき優先権を主張し、その内容をここに援用する。
本願は、2020年9月4日に、日本に出願された特願2020-149447号に基づき優先権を主張し、その内容をここに援用する。
iPS細胞(induced pluripotent stem cell;「人工多能性幹細胞」又は「誘導多能性幹細胞」とも呼ばれる。)は、Oct3/4、Sox2、Klf4及びc-Mycを導入することによって、体細胞から産生することができる(非特許文献1、特許文献1)。これは、親体細胞の転写ネットワーク及びエピジェネティックシグネチャー(epigenetic signature)をリプログラミングすることによって達成できる。iPS細胞は、基礎研究、医薬の革新、及び再生医療にさまざまな利益をもたらす。しかし、生成したiPS細胞の細胞群が、胚性幹細胞(ES細胞)の細胞群と比較して不均一な品質(heterogeneous quality)であることは、依然として深刻な問題である。例えば、ES細胞の場合、細胞毎の性質のばらつきが小さく、おおむねどの細胞も目的とする細胞へ分化させることができる。これに対し、iPS細胞は、細胞毎の性質のばらつきが大きく、目的とする細胞に分化させることができない細胞がしばしば出現する。細胞毎のばらつきがなく、いずれのiPS細胞も高品質を示すiPS細胞の細胞群を得ることは、基礎研究及び臨床目的のために重要である。
iPS細胞の細胞群の品質が不均一であるという問題を解決するために、多くの試みがなされてきた。例えば、特許文献2には、所定量のOct3/4遺伝子、Klf4遺伝子、c-Myc遺伝子及びSox2遺伝子を、体細胞に所定回数導入すると、iPS細胞の製造効率及び安定性を向上させることができる旨が記載されている。特許文献3には、Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、Klf4遺伝子又はその遺伝子産物、及びc-Myc遺伝子又はその遺伝子産物に加えて、Prdm14遺伝子又はその遺伝子産物、Esrrb遺伝子又はその遺伝子産物、及びSall4a遺伝子又はその遺伝子産物を体細胞に導入することにより、品質面に優れたiPS細胞を効率良く、短期間で製造することができる旨が記載されている。特許文献4には、Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、Klf4遺伝子又はその遺伝子産物、及びc-Myc遺伝子又はその遺伝子産物に加えて、Jarid2変異体遺伝子又はその遺伝子産物を体細胞に導入することにより、品質面に優れたiPS細胞を効率良く、短期間で製造することができる旨が記載されている。しかし、iPS細胞の品質にはまだ改善の余地があった。そこで、より高品質で、且つ品質のばらつきがより少ないiPS細胞の製造方法の開発が求められていた。
リンカーヒストンH1ファミリーは、リンカーDNAに結合し、遺伝子発現を制御するために、高次クロマチン構造を生じさせる。リンカーヒストンH1ファミリーのメンバーには、ヒストンH1a、H1b、H1c、H1d、H1e、H1foo、H1x、H1.0、H1t、H1T2、HILS1が含まれている。リンカーヒストンファミリーの大部分のメンバーは、クロマチンを凝縮する体細胞リンカーヒストンからなる。したがって、かかる構造は、一般的に全体的な遺伝子転写活性を抑制する(非特許文献2、3)。本発明者らは、体細胞からiPS細胞を誘導する際に、上記の遺伝子に加えて、リンカーヒストンH1ファミリーのメンバーであるH1foo遺伝子を導入することにより、高品質で品質のばらつきが少ないiPS細胞を製造できることを見出している(特許文献5)。
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell126, 663-676 (2006)
Steinbach, O.C., Wolffe, A.P. & Rupp, R.A. Somatic linker histones cause loss of mesodermal competence in Xenopus. Nature 389, 395-399 (1997)
Hebbar, P.B. & Archer, T.K. Altered histone H1 stoichiometry and an absence of nucleosome positioning on transfected DNA. The Journal of biological chemistry 283, 4595-4601 (2008)
特許文献5に記載のiPS細胞の製造方法では、従来の方法に比べて、高品質で品質のばらつきが少ないiPS細胞を製造することができる。しかしながら、iPS細胞誘導後に得られるコロニー数は、従来の方法と同程度である。
そこで、本発明は、iPS細胞誘導後に得られるコロニー数を増加させることが可能な、iPS細胞の品質改善剤、iPS細胞の製造方法、かかる製造方法により製造されるiPS細胞、及びiPS細胞製造用組成物を提供することを課題とする。
そこで、本発明は、iPS細胞誘導後に得られるコロニー数を増加させることが可能な、iPS細胞の品質改善剤、iPS細胞の製造方法、かかる製造方法により製造されるiPS細胞、及びiPS細胞製造用組成物を提供することを課題とする。
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、体細胞に導入したH1foo遺伝子から発現されるH1fooタンパク質の、細胞内における存在時期及び存在量のいずれかを制御することにより、iPS細胞誘導後のコロニー数が有意に増加することを見出し、本発明を完成させるに至った。
本発明は以下の態様を含む。
[1]H1foo遺伝子と、前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得る制御配列と、を有するポリヌクレオチドを含む、iPS細胞の品質改善剤。
[2]前記ポリヌクレオチドが、これを導入する細胞内で、前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されているものである、[1]に記載のiPS細胞の品質改善剤。
[3]前記発現ベクターが、センダイウイルスベクターである、[2]に記載のiPS細胞の品質改善剤。
[4]前記制御配列が、不安定化ドメインをコードするヌクレオチド配列を含み、前記不安定化ドメインは、前記不安定化ドメインを含む融合タンパク質のプロテアソームによる分解を促進するドメインであり、前記制御配列が、前記不安定化ドメインと前記H1fooタンパク質との融合タンパク質を発現し得るように、前記H1foo遺伝子に連結されている、[1]~[3]のいずれか1つに記載のiPS細胞の品質改善剤。
[5]前記制御配列が、化学的刺激に応答して、前記H1foo遺伝子の転写を制御するプロモーター配列を含む、[1]~[4]のいずれか1つに記載のiPS細胞の品質改善剤。
[6]H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む、iPS細胞の品質改善剤であって、前記不安定化ドメインは、前記融合タンパク質のプロテアソームによる分解を促進するドメインである、iPS細胞の品質改善剤。
[7]核初期化物質と、
[1]~[6]のいずれか1つに記載のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む、iPS細胞の製造方法。
[8]前記iPS細胞が、プライム型またはナイーブ型iPS細胞である、[7]に記載のiPS細胞の製造方法。
[9]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子、並びに、それらの遺伝子産物からなる群から選択される少なくとも1つを含む、[7]または[8]に記載のiPS細胞の製造方法。
[10]前記核初期化物質が、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、L-Myc若しくはc-Myc、およびそれらの遺伝子産物である、[7]~[9]のいずれか1つに記載のiPS細胞の製造方法。
[11]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子が導入される細胞内で前記少なくとも1つの遺伝子を発現可能な状態で発現ベクターに挿入されている、[7]~[9]のいずれか1つに記載のiPS細胞の製造方法。
[12]前記発現ベクターが、センダイウイルスベクターである、[11]に記載のiPS細胞の製造方法。
[13]核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含み、
前記体細胞に導入された前記H1foo遺伝子から発現されるH1fooタンパク質は、前記体細胞内における存在時期及び存在量の少なくとも一方が制御される、iPS細胞の製造方法。
[14]前記H1foo遺伝子が、前記H1foo遺伝子が導入される細胞内で前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されている、[13]に記載のiPS細胞の製造方法。
[15]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子を発現可能な発現ベクターにコードされている、[13]又は[14]に記載のiPS細胞の製造方法。
[16]前記発現ベクターが、センダイウイルスベクターである、[14]又は[15]に記載のiPS細胞の製造方法。
[17][7]~[16]のいずれか1つに記載のiPS細胞の製造方法により製造されるiPS細胞。
[18]核初期化物質と、[1]~[6]のいずれか1つに記載のiPS細胞の品質改善剤と、を含む、iPS細胞製造用組成物。
[19]核初期化物質と、前記核初期化物質とともに体細胞に導入されてから2~15日目までのいずれかで、前記体細胞の初期化を誘導するとともに、自然免疫を抑制する機能を有する物質とを前記体細胞に導入する工程を含む、iPS細胞の製造方法。
[1]H1foo遺伝子と、前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得る制御配列と、を有するポリヌクレオチドを含む、iPS細胞の品質改善剤。
[2]前記ポリヌクレオチドが、これを導入する細胞内で、前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されているものである、[1]に記載のiPS細胞の品質改善剤。
[3]前記発現ベクターが、センダイウイルスベクターである、[2]に記載のiPS細胞の品質改善剤。
[4]前記制御配列が、不安定化ドメインをコードするヌクレオチド配列を含み、前記不安定化ドメインは、前記不安定化ドメインを含む融合タンパク質のプロテアソームによる分解を促進するドメインであり、前記制御配列が、前記不安定化ドメインと前記H1fooタンパク質との融合タンパク質を発現し得るように、前記H1foo遺伝子に連結されている、[1]~[3]のいずれか1つに記載のiPS細胞の品質改善剤。
[5]前記制御配列が、化学的刺激に応答して、前記H1foo遺伝子の転写を制御するプロモーター配列を含む、[1]~[4]のいずれか1つに記載のiPS細胞の品質改善剤。
[6]H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む、iPS細胞の品質改善剤であって、前記不安定化ドメインは、前記融合タンパク質のプロテアソームによる分解を促進するドメインである、iPS細胞の品質改善剤。
[7]核初期化物質と、
[1]~[6]のいずれか1つに記載のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む、iPS細胞の製造方法。
[8]前記iPS細胞が、プライム型またはナイーブ型iPS細胞である、[7]に記載のiPS細胞の製造方法。
[9]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子、並びに、それらの遺伝子産物からなる群から選択される少なくとも1つを含む、[7]または[8]に記載のiPS細胞の製造方法。
[10]前記核初期化物質が、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、L-Myc若しくはc-Myc、およびそれらの遺伝子産物である、[7]~[9]のいずれか1つに記載のiPS細胞の製造方法。
[11]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子が導入される細胞内で前記少なくとも1つの遺伝子を発現可能な状態で発現ベクターに挿入されている、[7]~[9]のいずれか1つに記載のiPS細胞の製造方法。
[12]前記発現ベクターが、センダイウイルスベクターである、[11]に記載のiPS細胞の製造方法。
[13]核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含み、
前記体細胞に導入された前記H1foo遺伝子から発現されるH1fooタンパク質は、前記体細胞内における存在時期及び存在量の少なくとも一方が制御される、iPS細胞の製造方法。
[14]前記H1foo遺伝子が、前記H1foo遺伝子が導入される細胞内で前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されている、[13]に記載のiPS細胞の製造方法。
[15]前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子を発現可能な発現ベクターにコードされている、[13]又は[14]に記載のiPS細胞の製造方法。
[16]前記発現ベクターが、センダイウイルスベクターである、[14]又は[15]に記載のiPS細胞の製造方法。
[17][7]~[16]のいずれか1つに記載のiPS細胞の製造方法により製造されるiPS細胞。
[18]核初期化物質と、[1]~[6]のいずれか1つに記載のiPS細胞の品質改善剤と、を含む、iPS細胞製造用組成物。
[19]核初期化物質と、前記核初期化物質とともに体細胞に導入されてから2~15日目までのいずれかで、前記体細胞の初期化を誘導するとともに、自然免疫を抑制する機能を有する物質とを前記体細胞に導入する工程を含む、iPS細胞の製造方法。
本発明により、iPS細胞の品質改善剤、iPS細胞の製造方法、かかる製造方法により製造されるiPS細胞、及びiPS細胞製造用組成物が提供される。本発明によれば、iPS細胞誘導後に得られるコロニー数を増加させることができ、
かつそのように製造されたiPS細胞群の中には高い分化多能性等のiPS細胞としての品質が高いものが多く含まれるものである。
かつそのように製造されたiPS細胞群の中には高い分化多能性等のiPS細胞としての品質が高いものが多く含まれるものである。
「を含む」(comprise)という用語は、対象となる構成要素以外の構成要素を含んでいてもよいことを意味する。「からなる」(consist of)という用語は、対象となる構成要素以外の構成要素を含まないことを意味する。「から本質的になる」(consist essentially of)という用語は、対象となる構成要素以外の構成要素を特別な機能を発揮する態様(発明の効果を完全に喪失させる態様など)では含まないことを意味する。本明細書において、「を含む」(comprise)と記載する場合、「からなる」(consist of)態様、及び「から本質的になる」(essentially consist of)態様を包含する。
タンパク質(ポリペプチド)、ペプチド、ポリヌクレオチド(DNA、RNA)、ベクター、及び細胞は、単離されたものであり得る。「単離された」とは、天然状態から分離された状態を意味する。本明細書に記載されるタンパク質(ポリペプチド)、ペプチド、ポリヌクレオチド(DNA、RNA)、ベクター、及び細胞は、単離されたタンパク質(単離されたポリペプチド)、単離されたペプチド、単離されたポリヌクレオチド(単離されたDNA、単離されたRNA)、単離されたベクター、及び単離された細胞であり得る。
「遺伝子」という用語は、特定のタンパク質をコードする少なくとも1つのオープンリーディングフレームを含むポリヌクレオチドを意味する。遺伝子は、エクソンのみを含んでもよく、エクソン、並びにイントロン、5’UTR、及び3’UTRのいずれか1つ以上を含んでもよい。
ポリヌクレオチドに関して用いる「機能的に連結」という用語は、第一の塩基配列が第二の塩基配列に十分に近くに配置され、第一の塩基配列が第二の塩基配列又は第二の塩基配列の制御下の領域に影響を及ぼしうることを意味する。例えば、ポリヌクレオチドがプロモーターに機能的に連結するとは、当該ポリヌクレオチドが、当該プロモーターの制御下で発現するように連結されていることを意味する。遺伝子の上流にプロモーターが配置される場合、通常、前記遺伝子は前記プロモーターに機能的に連結される。
「発現可能な状態」とは、ポリヌクレオチドが導入された細胞内で、該ポリヌクレオチドが転写され得る状態にあることをいう。
「発現ベクター」とは、対象ポリヌクレオチドを含むベクターであって、該ベクターを導入した細胞内で、対象ポリヌクレオチドを発現可能な状態にするシステムを備えたベクターをいう。
「発現ベクター」とは、対象ポリヌクレオチドを含むベクターであって、該ベクターを導入した細胞内で、対象ポリヌクレオチドを発現可能な状態にするシステムを備えたベクターをいう。
[iPS細胞の品質改善剤]
<第1実施形態>
一実施形態において、本発明は、iPS細胞の品質改善剤を提供する。本実施形態のiPS細胞の品質改善剤は、H1foo遺伝子と、前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得るヌクレオチド配列(以下、「制御配列」という。)と、を有するポリヌクレオチドを含む。
<第1実施形態>
一実施形態において、本発明は、iPS細胞の品質改善剤を提供する。本実施形態のiPS細胞の品質改善剤は、H1foo遺伝子と、前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得るヌクレオチド配列(以下、「制御配列」という。)と、を有するポリヌクレオチドを含む。
本実施形態のiPS細胞の品質改善剤は、後述する核初期化物質とともに体細胞に導入することにより、体細胞から誘導されるiPS細胞の品質を改善することができる。「iPS細胞の品質」としては、iPS細胞における、未分化マーカー(Nanog、Tra-1-60、ALPなど)発現が高いこと、胚様体形成能が高いこと、iPS細胞から形成される胚様体サイズの均一性、異常メチル化が低いこと、マウスiPS細胞におけるキメラ形成能が高いこと、分化細胞(例、心筋細胞)への分化能が高いこと等の各種性質、及びiPS細胞間における前記のような各種性質の均一性等が挙げられる。本実施形態のiPS細胞の品質改善剤は、核初期化物質とともに体細胞に導入することにより、核初期化物質のみを導入した場合と比較して、前記のようなiPS細胞の各種性質の向上、又はiPS細胞間の各種性質の均一性の向上等の効果を奏する。特に、本実施形態のiPS細胞の品質改善剤は、ALPを発現するプライム型iPS細胞の生成能の向上、ALPを発現するナイーブ型iPS細胞の生成能の向上等の効果を奏する。また、本実施形態のiPS細胞の品質改善剤は、核初期化物質とともに体細胞に導入することにより、生成したiPS細胞において、遺伝子発現の均一性の向上、DNAメチル化の均一性の向上、目的とする細胞への分化能の向上、及びそれらの性質の均一性の向上、均一性の高い分化細胞集団に分化する能力の向上、及びナイーブ型表現型(代謝機能、HUWE1/XIST発現パターン等)の発現向上等の効果を奏する。分化細胞における均一性は、例えば、分化細胞マーカーの発現量のばらつきを調べることにより、確認することができる。分化細胞としては、特に限定されないが、例えば、内胚葉系細胞、中胚葉系細胞、外胚葉系細胞、心筋細胞、肝細胞、腎細胞、筋細胞、線維芽細胞、神経細胞、免疫細胞(リンパ球等)、血管細胞、眼細胞(網膜色素上皮細胞等)、血液細胞(巨核球、赤血球等)、その他各組織細胞、及びそれらの前駆細胞等が挙げられる。
(H1foo遺伝子)
本明細書において、「H1foo遺伝子」とは、H1fooタンパク質をコードするポリヌクレオチドを意味する。H1foo遺伝子が由来する生物種としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。上記H1foo遺伝子の配列情報は、公知のデータベースから得ることができる。例えば、GenBankでは、アクセッション番号BC047943(ヒト)、又はBC137916(マウス)で入手することができる。前記アクセッション番号のヒトのH1foo遺伝子のヌクレオチド配列を配列番号1に示し、当該ヌクレオチド配列にコードされるヒトのH1fooタンパク質のアミノ酸配列を配列番号2に示す。前記アクセッション番号のマウスのH1foo遺伝子のヌクレオチド配列を配列番号3に示し、マウスのH1fooタンパク質のアミノ酸配列を配列番号4に示す。本明細書においては、「H1FOO」とアルファベットを全て大文字で記載した場合には、ヒトのH1foo遺伝子又はH1fooタンパク質を指す。「H1foo」と記載した場合には、ヒトを含む全ての生物種のH1foo遺伝子又はH1fooタンパク質を包含する。
本明細書において、「H1foo遺伝子」とは、H1fooタンパク質をコードするポリヌクレオチドを意味する。H1foo遺伝子が由来する生物種としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。上記H1foo遺伝子の配列情報は、公知のデータベースから得ることができる。例えば、GenBankでは、アクセッション番号BC047943(ヒト)、又はBC137916(マウス)で入手することができる。前記アクセッション番号のヒトのH1foo遺伝子のヌクレオチド配列を配列番号1に示し、当該ヌクレオチド配列にコードされるヒトのH1fooタンパク質のアミノ酸配列を配列番号2に示す。前記アクセッション番号のマウスのH1foo遺伝子のヌクレオチド配列を配列番号3に示し、マウスのH1fooタンパク質のアミノ酸配列を配列番号4に示す。本明細書においては、「H1FOO」とアルファベットを全て大文字で記載した場合には、ヒトのH1foo遺伝子又はH1fooタンパク質を指す。「H1foo」と記載した場合には、ヒトを含む全ての生物種のH1foo遺伝子又はH1fooタンパク質を包含する。
H1foo遺伝子は、野生型のH1foo遺伝子に限定されず、変異(欠失、置換、挿入、及び付加のいずれか、又はこれらの組み合わせ)を含むものであってもよい。H1foo活性を有するタンパク質をコードするヌクレオチド配列を有するものであれば、変異の数は特に限定されない。なお、本明細書において、「H1foo活性」とは、野生型のH1fooタンパク質が有する機能の少なくとも1つを指す。H1foo活性としては、例えば、ヌクレオソーム間を繋ぐリンカーDNAに結合する活性が挙げられる。H1foo活性は、より好ましくは、ヌクレオソーム間を繋ぐリンカーDNAに結合し、且つ当該リンカーDNA領域が緩んだ状態を維持する活性である。
H1foo遺伝子としては、例えば、以下の(a)~(g)が挙げられる。
(a)野生型のH1foo遺伝子(例、配列番号1又は3で表されるヌクレオチド配列からなるポリヌクレオチド)
(b)野生型のH1fooタンパク質(例、配列番号2又は4で表されるアミノ酸配列からなるタンパク質)をコードするヌクレオチド配列からなるポリヌクレオチド
(c)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(d)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)と70%以上の配列同一性を有するアミノ酸配列からなり且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(e)野生型のH1foo遺伝子のヌクレオチド配列(例、配列番号1又は配列番号3で表されるヌクレオチド配列)において、1又は複数個のヌクレオチドが変異したヌクレオチド配列からなり、且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(f)野生型のH1foo遺伝子のヌクレオチド配列(例、配列番号1又は配列番号3で表されるヌクレオチド配列)と、70%以上の配列同一性を有するヌクレオチド配列からなり、且つH1foo活性を有するタンパク質をコードするポリヌクレオチド
(g)野生型のH1foo遺伝子(例、配列番号1又は配列番号3で表されるヌクレオチド配列を含むポリヌクレオチド)とストリンジェントな条件でハイブリダイズし、且つH1foo活性を有するタンパク質をコードするポリヌクレオチド
(a)野生型のH1foo遺伝子(例、配列番号1又は3で表されるヌクレオチド配列からなるポリヌクレオチド)
(b)野生型のH1fooタンパク質(例、配列番号2又は4で表されるアミノ酸配列からなるタンパク質)をコードするヌクレオチド配列からなるポリヌクレオチド
(c)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(d)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)と70%以上の配列同一性を有するアミノ酸配列からなり且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(e)野生型のH1foo遺伝子のヌクレオチド配列(例、配列番号1又は配列番号3で表されるヌクレオチド配列)において、1又は複数個のヌクレオチドが変異したヌクレオチド配列からなり、且つH1foo活性を有するタンパク質、をコードするポリヌクレオチド
(f)野生型のH1foo遺伝子のヌクレオチド配列(例、配列番号1又は配列番号3で表されるヌクレオチド配列)と、70%以上の配列同一性を有するヌクレオチド配列からなり、且つH1foo活性を有するタンパク質をコードするポリヌクレオチド
(g)野生型のH1foo遺伝子(例、配列番号1又は配列番号3で表されるヌクレオチド配列を含むポリヌクレオチド)とストリンジェントな条件でハイブリダイズし、且つH1foo活性を有するタンパク質をコードするポリヌクレオチド
上記(c)及び(e)において、「変異」は、欠失、置換、付加、及び挿入のいずれであってもよく、これらの組合せであってもよい。
上記(c)において、「複数個」は、結果として生じるタンパク質がH1foo活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(e)において、「複数個」は、結果として生じるポリヌクレオチドがH1foo活性を有するタンパク質をコードする限り特に限定されないが、例えば、2~60個が例示され、2~50個が好ましく、2~40個又は2~30個がより好ましく、2~20個又は2~10個がさらに好ましく、2~5個又は2個若しくは3個が特に好ましい。
上記(d)及び(f)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。アミノ酸配列同士又はヌクレオチド配列同士の配列同一性は、2つのアミノ酸配列又はヌクレオチド配列を、対応するアミノ酸又はヌクレオチドが最も多く一致するように、挿入及び欠失に当たる部分にギャップを入れながら並置し、得られたアラインメント中のギャップを除くアミノ酸配列全体又はヌクレオチド配列全体に対する一致したアミノ酸又はヌクレオチドの割合として求められる。アミノ酸配列同士又はヌクレオチド配列同士の配列同一性は、当該技術分野で公知の各種相同性検索ソフトウェアを用いて求めることができる。例えば、アミノ酸配列又はヌクレオチド配列の配列同一性の値は、公知の相同性検索ソフトウェアBLASTP又はBLASTNにより得られたアライメントを元にした計算によって得ることができる。
上記(g)において、「ストリンジェントな条件」とは、例えば、Molecular Cloning-A LABORATORY MANUAL THIRD EDITION (Sambrookら、Cold Spring Harbor Laboratory Press)に記載の条件が挙げられる。例えば、6×SSC(20×SSCの組成:3M塩化ナトリウム、0.3Mクエン酸溶液、pH7.0)、5×デンハルト溶液(100×デンハルト溶液の組成:2質量%ウシ血清アルブミン、2質量%フィコール、2質量%ポリビニルピロリドン)、0.5質量%のSDS、0.1mg/mLサケ精子DNA、及び50容量%フォルムアミドからなるハイブリダイゼーションバッファー中で、42~70℃で数時間から一晩インキュベーションを行うことによりハイブリダイズさせる条件が例示される。インキュベーション後の洗浄の際に用いる洗浄バッファーとしては、好ましくは0.1質量%SDS含有1×SSC溶液、より好ましくは0.1質量%SDS含有0.1×SSC溶液が挙げられる。
上記(c)において、「複数個」は、結果として生じるタンパク質がH1foo活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(e)において、「複数個」は、結果として生じるポリヌクレオチドがH1foo活性を有するタンパク質をコードする限り特に限定されないが、例えば、2~60個が例示され、2~50個が好ましく、2~40個又は2~30個がより好ましく、2~20個又は2~10個がさらに好ましく、2~5個又は2個若しくは3個が特に好ましい。
上記(d)及び(f)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。アミノ酸配列同士又はヌクレオチド配列同士の配列同一性は、2つのアミノ酸配列又はヌクレオチド配列を、対応するアミノ酸又はヌクレオチドが最も多く一致するように、挿入及び欠失に当たる部分にギャップを入れながら並置し、得られたアラインメント中のギャップを除くアミノ酸配列全体又はヌクレオチド配列全体に対する一致したアミノ酸又はヌクレオチドの割合として求められる。アミノ酸配列同士又はヌクレオチド配列同士の配列同一性は、当該技術分野で公知の各種相同性検索ソフトウェアを用いて求めることができる。例えば、アミノ酸配列又はヌクレオチド配列の配列同一性の値は、公知の相同性検索ソフトウェアBLASTP又はBLASTNにより得られたアライメントを元にした計算によって得ることができる。
上記(g)において、「ストリンジェントな条件」とは、例えば、Molecular Cloning-A LABORATORY MANUAL THIRD EDITION (Sambrookら、Cold Spring Harbor Laboratory Press)に記載の条件が挙げられる。例えば、6×SSC(20×SSCの組成:3M塩化ナトリウム、0.3Mクエン酸溶液、pH7.0)、5×デンハルト溶液(100×デンハルト溶液の組成:2質量%ウシ血清アルブミン、2質量%フィコール、2質量%ポリビニルピロリドン)、0.5質量%のSDS、0.1mg/mLサケ精子DNA、及び50容量%フォルムアミドからなるハイブリダイゼーションバッファー中で、42~70℃で数時間から一晩インキュベーションを行うことによりハイブリダイズさせる条件が例示される。インキュベーション後の洗浄の際に用いる洗浄バッファーとしては、好ましくは0.1質量%SDS含有1×SSC溶液、より好ましくは0.1質量%SDS含有0.1×SSC溶液が挙げられる。
上記(b)~(e)において、縮重コドンは、本実施形態のiPS細胞の品質改善剤を使用する体細胞において、コドン使用頻度が高いものが用いられることが好ましい。例えば、ヒトの体細胞に用いる場合には、ヒト細胞において使用頻度の高いコドンが用いられていることが好ましい。すなわち、ヒトコドンに最適化されていることが好ましい。また、マウスの体細胞に用いる場合には、マウス細胞において使用頻度の高いコドンが用いられていることが好ましい。すなわち、マウスコドンに最適化されていることが好ましい。
本明細書において、「H1fooタンパク質」は、後述する不安定化ドメインとH1fooタンパク質との融合タンパク質、又は当該融合タンパク質中のH1fooタンパク質領域を指す場合がある。
(制御配列)
制御配列は、H1foo遺伝子が細胞に導入された場合に、H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得るヌクレオチド配列である。制御配列は、H1fooタンパク質の細胞内での存在量及び存在時期のいずれかを制御し得るものであれば、制御方法は特に限定されない。制御配列としては、例えば、不安定化ドメインをコードするヌクレオチド配列が挙げられる。あるいは、制御配列としては、例えば、化学的刺激に応答して、前記H1foo遺伝子の転写を制御するプロモーター配列が挙げられる。
制御配列は、H1foo遺伝子が細胞に導入された場合に、H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得るヌクレオチド配列である。制御配列は、H1fooタンパク質の細胞内での存在量及び存在時期のいずれかを制御し得るものであれば、制御方法は特に限定されない。制御配列としては、例えば、不安定化ドメインをコードするヌクレオチド配列が挙げられる。あるいは、制御配列としては、例えば、化学的刺激に応答して、前記H1foo遺伝子の転写を制御するプロモーター配列が挙げられる。
本明細書において、「不安定化ドメイン」(Destabilized Domain:DD)とは、当該ドメインを含む融合タンパク質のプロテアソームによる分解を促進するドメインを指す。すなわち、所望のタンパク質のN末端又はC末端に不安定化ドメインを連結し、不安定化ドメインを含む融合タンパク質とすることにより、所望タンパク質を含む融合タンパク質のプロテアソームによる分解が促進される。
不安定化ドメインは、当該ドメインを含む融合タンパク質の分解を促進するものであれば、特に限定されず、公知のものを用いることができる。不安定化ドメインとしては、例えば、FKBP12由来の不安定化ドメイン(Banaszynski et al., Cell. 2006 Sep 8;126(5):995-1004.)、ecDHFR由来の不安定化ドメイン(Iwamoto et al., Chem Biol. 2010 Sep 24;17(9):981-8.)等が挙げられる。FKBP12由来の不安定化ドメインとしては、例えば、配列番号14に記載のアミノ酸配列を有するものが例示され、前記アミノ酸配列をコードするヌクレオチド配列としては、配列番号13に記載のヌクレオチド配列が例示される。なお、所望タンパク質と不安定化ドメインの融合タンパク質は、所望タンパク質と不安定化ドメインとの間にポリペプチドが含まれていてもよい。
これらの不安定化ドメインを含む融合タンパク質の分解は、Shield1(FKBP12由来の不安定化ドメインの場合)、又はGuard(ecDHFR由来の不安定化ドメインの場合)と呼ばれる膜透過性の低分子化合物(以下、「安定化化合物」という。)により抑制することができる。これらの不安定化ドメインをコードするポリヌクレオチド(以下、「不安定化ドメイン遺伝子」という。)及び安定化化合物は、PreteoTunerTM Shield System(Clontech)、PreteoTunerTM Guard System(Clontech)等により利用することができる。
不安定化ドメインは、デグロン配列であるということもできる。デグロンの例としては、mTORデグロン(米国特許出願公開第2009/0215169号)、ジヒドロ葉酸レダクターゼ(DHFR)デグロン(米国特許出願公開第2012/0178168号)、PEST(国際公開第99/54348)、TetRデグロン(国際公開第2007/032555号)、およびオーキシン誘導性デグロン(auxin-inducible degron,AID)(国際公開第2010/125620号)などが挙げられるが、これらに限定されない。
不安定化ドメインは、当該ドメインを含む融合タンパク質の分解を促進するものであれば、特に限定されず、公知のものを用いることができる。不安定化ドメインとしては、例えば、FKBP12由来の不安定化ドメイン(Banaszynski et al., Cell. 2006 Sep 8;126(5):995-1004.)、ecDHFR由来の不安定化ドメイン(Iwamoto et al., Chem Biol. 2010 Sep 24;17(9):981-8.)等が挙げられる。FKBP12由来の不安定化ドメインとしては、例えば、配列番号14に記載のアミノ酸配列を有するものが例示され、前記アミノ酸配列をコードするヌクレオチド配列としては、配列番号13に記載のヌクレオチド配列が例示される。なお、所望タンパク質と不安定化ドメインの融合タンパク質は、所望タンパク質と不安定化ドメインとの間にポリペプチドが含まれていてもよい。
これらの不安定化ドメインを含む融合タンパク質の分解は、Shield1(FKBP12由来の不安定化ドメインの場合)、又はGuard(ecDHFR由来の不安定化ドメインの場合)と呼ばれる膜透過性の低分子化合物(以下、「安定化化合物」という。)により抑制することができる。これらの不安定化ドメインをコードするポリヌクレオチド(以下、「不安定化ドメイン遺伝子」という。)及び安定化化合物は、PreteoTunerTM Shield System(Clontech)、PreteoTunerTM Guard System(Clontech)等により利用することができる。
不安定化ドメインは、デグロン配列であるということもできる。デグロンの例としては、mTORデグロン(米国特許出願公開第2009/0215169号)、ジヒドロ葉酸レダクターゼ(DHFR)デグロン(米国特許出願公開第2012/0178168号)、PEST(国際公開第99/54348)、TetRデグロン(国際公開第2007/032555号)、およびオーキシン誘導性デグロン(auxin-inducible degron,AID)(国際公開第2010/125620号)などが挙げられるが、これらに限定されない。
不安定化ドメイン遺伝子は、不安定化ドメインとH1fooタンパク質との融合タンパク質を発現し得るように、H1foo遺伝子に連結されていてもよい。これにより、本実施形態のiPS細胞の品質改善剤を細胞に導入した場合に、H1fooタンパク質は、不安定化ドメインとの融合タンパク質として発現する。そのため、H1fooタンパク質は、発現後すみやかにプロテアソームにより分解される。すなわち、不安定化ドメインとの融合タンパク質としてH1fooタンパク質を発現させることにより、H1fooタンパク質の細胞内での存在量が少なくなるように制御可能である。さらに、前記融合タンパク質の発現ベクターとして、遺伝子を宿主染色体に組み込まずに細胞質内で遺伝子を発現し、遺伝子発現後に細胞から速やかに消失するベクターを用いることにより、H1fooタンパク質の存在時期を当該発現ベクター導入後からの一定期間内に制御可能である。このように、H1fooタンパク質が、核初期化物質の導入から初期化完了までの一定期間に発現し、それ以降は比較的低いレベルで存在するように制御することにより、品質の高いiPS細胞を高効率で作製することができる。
また、不安定化ドメインを含む融合タンパク質は、安定化化合物の添加により、細胞内での存在量及び存在時期を制御することもできる。例えば、本実施形態のiPS細胞の品質改善剤を、後述する核初期化物質とともに体細胞に導入した後、安定化化合物を任意の期間に添加し、前記任意の期間経過後に安定化化合物を除去することにより、H1fooタンパク質の存在量及び存在時期を制御してもよい。
また、本実施形態のiPS細胞の品質改善剤に含まれるポリヌクレオチドは、H1foo遺伝子がこれを導入する細胞内で発現可能な状態で発現ベクターに挿入されている形態であってもよい。不安定化ドメイン遺伝子は、不安定化ドメインと当該発現ベクターの複製に必要なタンパク質(以下、「複製関連タンパク質」という。)との融合タンパク質を発現し得るように、当該複製関連タンパク質遺伝子に連結されていてもよい。「複製関連タンパク質遺伝子」は、複製関連タンパク質をコードするポリヌクレオチドを意味する。不安定化ドメインとの融合タンパク質として発現された複製関連タンパク質は、発現後すみやかにプロテアソームにより分解される。そのため、当該複製関連タンパク質量の低下により、発現ベクターの複製が阻害される。これにより、間接的に、H1fooタンパク質の細胞内での存在時期及び存在量を制御することができる。
不安定化ドメインとの融合タンパク質とする複製関連タンパク質は、特に限定されず、発現ベクターの種類に応じて適宜選択すればよい。例えば、発現ベクターが、センダイウイルスベクターである場合、複製関連タンパク質としては、ヌクレオカプシドタンパク質(N)、リン酸化タンパク質(P)、マトリクスタンパク質(M)、ラージタンパク質(L)が挙げられる。これらの中でも、Pタンパク質が好ましい。
発現ベクターが、複数の複製関連タンパク質遺伝子を含む場合、前記複数の複製関連タンパク質遺伝子のいずれか2つ以上に、不安定化ドメイン遺伝子を連結してもよい。
不安定化ドメインとの融合タンパク質とする複製関連タンパク質は、特に限定されず、発現ベクターの種類に応じて適宜選択すればよい。例えば、発現ベクターが、センダイウイルスベクターである場合、複製関連タンパク質としては、ヌクレオカプシドタンパク質(N)、リン酸化タンパク質(P)、マトリクスタンパク質(M)、ラージタンパク質(L)が挙げられる。これらの中でも、Pタンパク質が好ましい。
発現ベクターが、複数の複製関連タンパク質遺伝子を含む場合、前記複数の複製関連タンパク質遺伝子のいずれか2つ以上に、不安定化ドメイン遺伝子を連結してもよい。
制御配列が不安定化ドメイン遺伝子配列を含む場合、制御配列は、H1foo遺伝子又は複製関連タンパク質遺伝子の5’末端及び3’末端の少なくとも一方に連結すればよい。制御配列は、前記タンパク質遺伝子の5’末端及び3’末端のいずれに連結してもよく、5’末端及び3’末端の両方に連結してもよい。制御配列と、H1foo遺伝子又は複製関連タンパク質遺伝子との連結は、不安定化ドメイン遺伝子及びH1foo遺伝子若しくは複製関連タンパク質遺伝子に、フレームシフトが起こらないように(すなわち、インフレームで)行う。一例として、H1foo遺伝子の3’末端にFKBP12由来の不安定化ドメイン遺伝子を連結した場合のヌクレオチド配列を配列番号15に示す。配列番号15に記載のヌクレオチド配列は、H1fooタンパク質のC末端にFKBP12由来の不安定化ドメインが付加された融合タンパク質(配列番号16)をコードする。一例として、H1foo遺伝子の5’末端にFKBP12由来の不安定化ドメイン遺伝子を連結した場合のヌクレオチド配列を配列番号17に示す。配列番号17に記載のヌクレオチド配列は、H1fooタンパク質のN末端にFKBP12由来の不安定化ドメインが付加された融合タンパク質(配列番号18)をコードする。
制御配列は、H1foo遺伝子と複製関連タンパク質遺伝子との両方に連結してもよい。
制御配列は、H1foo遺伝子と複製関連タンパク質遺伝子との両方に連結してもよい。
制御配列は、外部刺激に応答して、H1foo遺伝子の転写を制御するプロモーター配列を含んでいてもよい。外部刺激としては、例えば、化学物質、熱、光、pH、浸透圧等が挙げられる。外部刺激に応答してH1foo遺伝子の転写を制御するプロモーター(以下、「外部刺激応答性プロモーター」という。)は、H1foo遺伝子の転写を制御するように、H1foo遺伝子の上流に配置される。
外部刺激応答性プロモーターは、特に限定されず、公知のものを用いることができる。外部刺激応答性プロモーターとしては、例えば、テトラサイクリン応答性プロモーター(例、テトラサイクリン応答因子:TRE)が挙げられる。
外部刺激応答性プロモーターは、特に限定されず、公知のものを用いることができる。外部刺激応答性プロモーターとしては、例えば、テトラサイクリン応答性プロモーター(例、テトラサイクリン応答因子:TRE)が挙げられる。
(ポリヌクレオチド)
本実施形態のiPS細胞の品質改善剤は、上記H1foo遺伝子と上記制御配列とを有するポリヌクレオチドを含む。前記ポリヌクレオチドは、H1foo遺伝子が制御配列の制御のもとに発現可能な状態で発現ベクターに挿入されているものである。
なお、「H1foo遺伝子を発現可能な発現ベクター」は、H1fooタンパク質を発現するものであってもよく、H1fooタンパク質と不安定化ドメインとの融合タンパク質を発現するものであってもよい。
本実施形態のiPS細胞の品質改善剤は、上記H1foo遺伝子と上記制御配列とを有するポリヌクレオチドを含む。前記ポリヌクレオチドは、H1foo遺伝子が制御配列の制御のもとに発現可能な状態で発現ベクターに挿入されているものである。
なお、「H1foo遺伝子を発現可能な発現ベクター」は、H1fooタンパク質を発現するものであってもよく、H1fooタンパク質と不安定化ドメインとの融合タンパク質を発現するものであってもよい。
発現ベクターは、必要に応じて、H1foo遺伝子及び制御配列に加えて、H1foo遺伝子又はH1foo遺伝子及び不安定化ドメイン遺伝子(以下、まとめて「H1foo遺伝子等」という。)の発現を制御するプロモーターを含んでいることが好ましい。発現ベクターにおいて、当該プロモーターは、H1foo遺伝子等の発現を制御するように、H1foo遺伝子等の上流に配置される。但し、前記制御配列が、外部刺激応答性プロモーターである場合、発現ベクターは、H1foo遺伝子の発現を制御する別のプロモーターを含まないことが好ましい。
プロモーターとしては、例えば、SRαプロモーター、SV40初期プロモーター、レトロウイルスのLTR、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーター、EF1αプロモーター、メタロチオネインプロモーター、ヒートショックプロモーター等が挙げられる。また、ヒトCMVのIE遺伝子のエンハンサーをプロモーターと共に用いてもよい。一例としては、CAGプロモーター(サイトメガロウイルスエンハンサーとニワトリβ-アクチンプロモーターとβ-グロビン遺伝子のポリAシグナル部位を含む)を用いることができる。
また、上記「制御配列」の項で挙げた刺激応答性プロモーターを用いてもよい。
また、上記「制御配列」の項で挙げた刺激応答性プロモーターを用いてもよい。
発現ベクターは、プロモーターの他に、所望によりエンハンサー、ポリA付加シグナル、マーカー遺伝子、複製開始点、複製開始点に結合して複製を制御するタンパク質をコードする遺伝子等を含んでいてもよい。マーカー遺伝子とは、該マーカー遺伝子を細胞に導入することにより、細胞の選別や選択を可能とするような遺伝子をいう。マーカー遺伝子の具体例としては、薬剤耐性遺伝子、蛍光タンパク質遺伝子、発光酵素遺伝子、発色酵素遺伝子等が挙げられる。これらは、1種単独で使用してもよいし、2種以上を併用してもよい。薬剤耐性遺伝子の具体例としては、ネオマイシン耐性遺伝子、テトラサイクリン耐性遺伝子、カナマイシン耐性遺伝子、ゼオシン耐性遺伝子、ハイグロマイシン耐性遺伝子、ピューロマイシン耐性遺伝子などが挙げられる。蛍光タンパク質遺伝子の具体例としては、緑色蛍光タンパク質(GFP)遺伝子、黄色蛍光タンパク質(YFP)遺伝子、赤色蛍光タンパク質(RFP)遺伝子などが挙げられる。発光酵素遺伝子の具体例としては、ルシフェラーゼ遺伝子などが挙げられる。発色酵素遺伝子の具体例としては、βガラクトシターゼ遺伝子、βグルクロニダーゼ遺伝子、アルカリホスファターゼ遺伝子等が挙げられる。
プロモーターとして、刺激応答性プロモーターを用いた場合には、当該刺激に応答してプロモーターに結合するエンハンサー遺伝子又はリプレッサー遺伝子を、発現ベクターが含んでいてもよい。例えば、テトラサイクリン応答性プロモーターの場合には、リバーステトラサイクリン制御性トランス活性化因子(rtTA)又はテトラサイクリン制御性トランス活性化因子(rTA)等の遺伝子を、発現ベクターが含んでいてもよい。これらのエンハンサー遺伝子又はリプレッサー遺伝子は、H1foo遺伝子を有する発現ベクターとは別の発現ベクターに、含まれていてもよい。
プロモーターとして、刺激応答性プロモーターを用いた場合には、当該刺激に応答してプロモーターに結合するエンハンサー遺伝子又はリプレッサー遺伝子を、発現ベクターが含んでいてもよい。例えば、テトラサイクリン応答性プロモーターの場合には、リバーステトラサイクリン制御性トランス活性化因子(rtTA)又はテトラサイクリン制御性トランス活性化因子(rTA)等の遺伝子を、発現ベクターが含んでいてもよい。これらのエンハンサー遺伝子又はリプレッサー遺伝子は、H1foo遺伝子を有する発現ベクターとは別の発現ベクターに、含まれていてもよい。
発現ベクターの種類は、特に限定されず、公知の発現ベクターを用いることができる。発現ベクターとしては、例えば、エピソーマルベクター、人工染色体ベクター、プラスミドベクター、ウイルスベクター等が挙げられる。
エピソーマルベクターは、染色体外で自律複製可能なベクターである。エピソーマルベクターを用いる具体的手段は、Yu et al., Science, 324, 797-801 (2009) に開示されている。例えば、エピソーマルベクターの複製に必要なベクター要素の5’側及び3’側にloxP配列を同方向に配置したエピソーマルベクターを使用することができる。エピソーマルベクターは染色体外で自律複製可能なため、ゲノムに組み込まれなくとも宿主細胞内での安定な発現を提供し得るが、いったんiPS細胞が樹立された後は、当該ベクターは速やかに除かれることが望ましい。エピソーマルベクターの複製に必要なベクター要素を2つのloxP配列で挟み、これにCreリコンビナーゼを作用させて当該ベクター要素を切り出すことにより、エピソーマルベクターの自律複製能を喪失させることができ、該ベクターを早期にiPS細胞から脱落させることができる。
エピソーマルベクターとしては、例えば、EBV、SV40等に由来する自律複製に必要な配列をベクター要素として含むベクターが挙げられる。自律複製に必要なベクター要素としては、具体的には、複製開始点、及び複製開始点に結合して複製を制御するタンパク質をコードする遺伝子であり、例えば、EBVにあっては複製開始点oriPとEBNA-1遺伝子、SV40にあっては複製開始点oriとSV40LT遺伝子が挙げられる。
人工染色体ベクターとしては、YAC(Yeast artificial chromosome)ベクター、BAC(Bacterial artificial chromosome)ベクター、PAC(P1-derived artificial chromosome)ベクター等が挙げられる。
プラスミドベクターとしては、導入対象となる体細胞内で発現可能なプラスミドベクターであれば、特に限定されない。導入対象となる体細胞が哺乳動物である場合は、動物細胞発現用プラスミドベクターとして、一般的に用いられているものを用いることができる。動物細胞発現用プラスミドベクターとしては、例えば、pA1-11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neo等が挙げられる。
ウイルスベクターとしては、レトロウイルス(レンチウイルスを含む)ベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター、センダイウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、ポックスウイルスベクター、ポリオウイルスベクター、シルビスウイルスベクター、ラブドウイルスベクター、パラミクソウイルスベクター、オルソミクソウイルスベクター等が挙げられる。
本明細書において、ウイルスベクターとは、当該ウイルスに由来するゲノム核酸を有し、該核酸に導入遺伝子を組み込むことにより、該遺伝子を発現させることができるベクターを意味する。
本明細書において、ウイルスベクターとは、当該ウイルスに由来するゲノム核酸を有し、該核酸に導入遺伝子を組み込むことにより、該遺伝子を発現させることができるベクターを意味する。
ウイルスベクターとしては、センダイウイルスベクターが好ましい。センダイウイルスは、モノネガウイルス目(Mononegavirales)ウイルスの1つでパラミクソウイルス科(Paramyxoviridae;Paramyxovirus,Morbillivirus,Rubulavirus,およびPnemovirus等の属を含む)に属し、一本のマイナス鎖(ウイルス蛋白質をコードするセンス鎖に対するアンチセンス鎖)のRNAをゲノムとして含んでいる。マイナス鎖RNAはネガティブ鎖RNAとも呼ばれる。センダイウイルスベクターは、染色体非組み込み型ウイルスベクターであり、ベクターは細胞質中で発現される。そのため、導入遺伝子が宿主の染色体に組み込まれる危険性がない。従って安全性が高く、目的達成後に導入細胞からベクターを除去することが可能である。
センダイウイルスベクターには、感染性ウイルス粒子の他、ウイルスコア、ウイルスゲノムとウイルス蛋白質との複合体、または非感染性ウイルス粒子などからなる複合体であって、細胞に導入することにより搭載する遺伝子を発現する能力を持つ複合体が含まれる。例えば、センダイウイルスゲノムとそれに結合するセンダイウイルス蛋白質(NP、P、およびL蛋白質)からなるリボヌクレオ蛋白質(ウイルスのコア部分)は、細胞に導入することにより該細胞内で導入遺伝子を発現することができる(国際公開第00/70055号)。細胞への導入は、適宜トランスフェクション試薬等を用いて行えばよい。したがって、このようなリボヌクレオ蛋白質(RNP)も、センダイウイルスベクターに包含される。
センダイウイルスのゲノムは、3’端から5’端に向けて順に、NP(ヌクレオキャプシド)遺伝子、P(ホスホ)遺伝子、M(マトリックス)遺伝子、F(フュージョン)遺伝子、HN(赤血球凝集素/ノイラミニダーゼ)遺伝子、及びL(ラージ)遺伝子が含まれている。このうち、センダイウイルスは、NP遺伝子、P遺伝子、およびL遺伝子があればベクターとして十分機能でき、細胞中でゲノムを複製し、搭載されている遺伝子を発現させることができる。センダイウイルスは、マイナス鎖RNAをゲノムに持つことから、通常とは逆で、ゲノムの3’側が上流にあたり、5’側が下流にあたる。
センダイウイルスの上記各遺伝子の塩基配列のデータベース(GenBank)のアクセッション番号は、例えばNP遺伝子については、M29343,M30202,M30203,M30204,M51331,M55565,M69046,X17218;P遺伝子については、M30202,M30203,M30204,M55565,M69046,X00583,X17007,X17008;M遺伝子については、D11446,K02742,M30202,M30203,M30204,M69046,U31956,X00584,X53056;F遺伝子については、D00152,D11446,D17334,D17335,M30202,M30203,M30204,M69046,X00152,X02131;HN遺伝子については、D26475,M12397,M30202,M30203,M30204,M69046,X00586,X02808,X56131;L遺伝子については、D00053,M30202,M30203,M30204,M69040,X00587,X58886を参照することにより特定することができる。
但し、センダイウイルスには複数の株が知られており、株の違いにより上記に例示した以外の配列からなる遺伝子も存在する。これらのいずれかの遺伝子に由来するウイルス遺伝子を持つセンダイウイルスベクターもまた、センダイウイルスベクターとして有用である。例えば、センダイウイルスベクターは、上記のいずれかのウイルス遺伝子のコード配列と、90%以上、好ましくは95%以上、96%以上、97%以上、98%以上、または99%以上の配列同一性を持つ塩基配列を含み得る。また、センダイウイルスベクターは、例えば、上記のいずれかのウイルス遺伝子のコード配列がコードするアミノ酸配列と、90%以上、好ましくは95%以上、96%以上、97%以上、98%以上、または99%以上の同一性を持つアミノ酸配列をコードする塩基配列を含み得る。また、センダイウイルスベクターは、例えば、上記のいずれかのウイルス遺伝子のコード配列がコードするアミノ酸配列において、10個以内、好ましくは9個以内、8個以内、7個以内、6個以内、5個以内、4個以内、3個以内、2個以内、または1個のアミノ酸が置換、挿入、欠失、および/または付加されたアミノ酸配列であって、各遺伝子産物の機能を保持するポリペプチドをコードする塩基配列を含み得る。
但し、センダイウイルスには複数の株が知られており、株の違いにより上記に例示した以外の配列からなる遺伝子も存在する。これらのいずれかの遺伝子に由来するウイルス遺伝子を持つセンダイウイルスベクターもまた、センダイウイルスベクターとして有用である。例えば、センダイウイルスベクターは、上記のいずれかのウイルス遺伝子のコード配列と、90%以上、好ましくは95%以上、96%以上、97%以上、98%以上、または99%以上の配列同一性を持つ塩基配列を含み得る。また、センダイウイルスベクターは、例えば、上記のいずれかのウイルス遺伝子のコード配列がコードするアミノ酸配列と、90%以上、好ましくは95%以上、96%以上、97%以上、98%以上、または99%以上の同一性を持つアミノ酸配列をコードする塩基配列を含み得る。また、センダイウイルスベクターは、例えば、上記のいずれかのウイルス遺伝子のコード配列がコードするアミノ酸配列において、10個以内、好ましくは9個以内、8個以内、7個以内、6個以内、5個以内、4個以内、3個以内、2個以内、または1個のアミノ酸が置換、挿入、欠失、および/または付加されたアミノ酸配列であって、各遺伝子産物の機能を保持するポリペプチドをコードする塩基配列を含み得る。
本明細書に記載した塩基配列およびアミノ酸配列などのデータベースアクセッション番号が参照された配列は、本願出願日における配列を参照するものであって、本願出願日時点における配列として特定される。各時点での配列はデータベースのリビジョンヒストリーを参照することにより特定することができる。
本実施形態において用いられるセンダイウイルスベクターは誘導体であってもよい。センダイウイルスベクターの誘導体には、センダイウイルスによる遺伝子導入能を損なわないように、ウイルス遺伝子が改変されたウイルス、および化学修飾されたウイルス等が含まれる。
センダイウイルスは、天然株、野生株、変異株、ラボ継代株、および人為的に構築された株などに由来してもよい。例えばZ株が挙げられる(Medical Journal of Osaka University Vol.6, No.1, March 1955 p1-15)。つまり、当該ウイルスは、目的とする機能を達成できる限り、天然から単離されたウイルスと同様の構造を持つウイルスベクターであっても、遺伝子組み換えにより人為的に改変したウイルスであってもよい。例えば、野生型ウイルスが持ついずれかの遺伝子に変異や欠損があるものであってよい。また、DI粒子(J.Virol.68:8413-8417,1994)などの不完全ウイルスを用いることも可能である。例えば、ウイルスのエンベロープ蛋白質または外殻蛋白質をコードする少なくとも1つの遺伝子に変異または欠損を有するウイルスを好適に用いることができる。このようなウイルスベクターは、例えば感染細胞においてはゲノムを複製することはできるが、感染性ウイルス粒子を形成できないウイルスベクターである。このような伝搬能欠損型のウイルスベクターは、周囲に感染を拡大する懸念がないので安全性が高い。例えば、Fおよび/またはHNなどのエンベロープ蛋白質またはスパイク蛋白質をコードする少なくとも1つの遺伝子、あるいはそれらの組み合わせが含まれていないウイルスベクターを用いることができる(国際公開第00/70055号、国際公開第00/70070号、Li,H.-O.et al., J. Virol. 74(14) 6564-6569 (2000))。ゲノム複製に必要な蛋白質(例えばNP、P、およびL蛋白質)をゲノムRNAにコードしていれば、感染細胞においてゲノムを増幅することができる。欠損型ウイルスを製造するには、例えば、欠損している遺伝子産物またはそれを相補できる蛋白質をウイルス産生細胞において外来的に供給する(国際公開第00/70055号、国際公開第00/70070号、Li, H.-O.et al., J. Virol. 74(14) 6564-6569 (2000))。ウイルスベクターをRNP(例えばN、L、P蛋白質、およびゲノムRNAからなるRNP)として回収する場合は、エンベロープ蛋白質を相補することなくベクターを製造することができる。
好適なセンダイウイルスベクターとしては、例えば、M蛋白質にG69E,T116A及びA183Sの変異を、HN蛋白質にA262T,G264及びK461Gの変異を、P蛋白質にL511F変異を、そしてL蛋白質にN1197S及びK1795E変異を持つF遺伝子欠失型センダイウイルスベクター(例えばZ strain)であってよく、このベクターにさらにTS 7、TS 12、TS 13、TS 14、またはTS 15の変異を導入したベクターであってもよい。具体的には、SeV18+/TSΔF(国際公開第2010/008054号、国際公開第2003/025570号)、SeV(PM)/TSΔF、および、これらにさらにTS 7、TS 12、TS 13、TS 14、またはTS 15の変異を導入したベクターなどが挙げられるが、これらに限定されない。
「TSΔF」は、M蛋白質にG69E,T116A及びA183Sの変異を、HN蛋白質にA262T,G264R及びK461Gの変異を、P蛋白質にL511F変異を、そしてL蛋白質にN1197S及びK1795E変異を持ち、F遺伝子を欠失することをいう。
「TSΔF」は、M蛋白質にG69E,T116A及びA183Sの変異を、HN蛋白質にA262T,G264R及びK461Gの変異を、P蛋白質にL511F変異を、そしてL蛋白質にN1197S及びK1795E変異を持ち、F遺伝子を欠失することをいう。
好適なセンダイウイルスベクターとしては、例えば、Pタンパク質にデグロン配列を付加して、感染細胞から容易に除去することができるようにしたベクターが挙げられる(国際公開第2016/125364号)。デグロンとしてはmTORデグロン(米国特許出願公開第2009/0215169号)、ジヒドロ葉酸レダクターゼ(DHFR)デグロン(米国特許出願公開第2012/0178168号)、PEST(国際公開第99/54348号)、TetRデグロン(国際公開第2007/032555号)、およびオーキシン誘導性デグロン(auxin-inducible degron:AID)(国際公開第2010/125620号)などが挙げられるが、これらに限定されない。例えば、mTORデグロンの1種であるDD-tagをPタンパク質のC末端に付加したベクターが好適に用いられる。
導入対象のポリヌクレオチド(H1foo遺伝子、あるいはH1foo遺伝子及び制御配列)を持つ組換えセンダイウイルスベクターの再構成は公知の方法を利用して行うことができる。
具体的には、(a)センダイウイルスゲノムRNA(マイナス鎖)またはその相補鎖(プラス鎖)をコードするcDNAを、ウイルス粒子形成に必要なウイルス蛋白質(N、P、およびL)を発現する細胞で転写させる工程、(b)生成したウイルスを含む培養上清を回収する工程、により製造することができる。粒子形成に必要なウイルス蛋白質は、転写させたウイルスゲノムRNAから発現されてもよいし、ゲノムRNA以外からトランスに供給されてもよい。例えば、N、P、およびL蛋白質をコードする発現プラスミドを細胞に導入して供給することができる。ゲノムRNAにおいて粒子形成に必要なウイルス遺伝子が欠損している場合は、そのウイルス遺伝子をウイルス産生細胞で別途発現させ、粒子形成を相補することもできる。ウイルス蛋白質やRNAゲノムを細胞内で発現させるためには、該蛋白質やゲノムRNAをコードするDNAを宿主細胞で機能する適当なプロモーターの下流に連結したベクターを宿主細胞に導入する。転写されたゲノムRNAは、ウイルス蛋白質の存在下で複製され、感染性ウイルス粒子が形成される。エンベロープ蛋白質などの遺伝子を欠損する欠損型ウイルスを製造する場合は、欠損する蛋白質またはその機能を相補できる他のウイルス蛋白質などをウイルス産生細胞において発現させることもできる。
具体的には、(a)センダイウイルスゲノムRNA(マイナス鎖)またはその相補鎖(プラス鎖)をコードするcDNAを、ウイルス粒子形成に必要なウイルス蛋白質(N、P、およびL)を発現する細胞で転写させる工程、(b)生成したウイルスを含む培養上清を回収する工程、により製造することができる。粒子形成に必要なウイルス蛋白質は、転写させたウイルスゲノムRNAから発現されてもよいし、ゲノムRNA以外からトランスに供給されてもよい。例えば、N、P、およびL蛋白質をコードする発現プラスミドを細胞に導入して供給することができる。ゲノムRNAにおいて粒子形成に必要なウイルス遺伝子が欠損している場合は、そのウイルス遺伝子をウイルス産生細胞で別途発現させ、粒子形成を相補することもできる。ウイルス蛋白質やRNAゲノムを細胞内で発現させるためには、該蛋白質やゲノムRNAをコードするDNAを宿主細胞で機能する適当なプロモーターの下流に連結したベクターを宿主細胞に導入する。転写されたゲノムRNAは、ウイルス蛋白質の存在下で複製され、感染性ウイルス粒子が形成される。エンベロープ蛋白質などの遺伝子を欠損する欠損型ウイルスを製造する場合は、欠損する蛋白質またはその機能を相補できる他のウイルス蛋白質などをウイルス産生細胞において発現させることもできる。
また、センダイウイルスの製造は、以下の公知の方法を利用して実施することができる(国際公開第97/16539号;国際公開第97/16538号;国際公開第00/70055号;国際公開第00/70070号;国際公開第01/18223号;国際公開第 03/025570号;国際公開第2005/071092号;国際公開第2006/137517号;国際公開第2007/083644号;国際公開第2008/007581号;Hasan, M.K. et al., J. Gen. Virol. 78: 2813-2820, 1997、Kato, A. et al., 1997, EMBO J. 16: 578-587及びYu, D. et al., 1997, Genes Cells 2: 457-466;Durbin, A.P. et al., 1997, Virology 235: 323-332;Whelan, S. P. et al., 1995, Proc. Natl. Acad. Sci. USA 92: 8388-8392;Schnell. M.J. et al., 1994, EMBO J. 13: 4195-4203;Radecke, F. et al., 1995, EMBO J. 14: 5773-5784;Lawson, N.D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481;Garcin, D. et al., 1995, EMBO J. 14: 6087-6094;Kato, A. et al.,1996, Genes Cells 1: 569-579;Baron, M.D. and Barrett, T., 1997, J. Virol. 71: 1265-1271;Bridgen, A. and Elliott, R.M., 1996, Proc. Natl. Acad. Sci. USA 93: 15400-15404;Tokusumi, T. et al. Virus Res. 2002: 86; 33-38、Li,H.-O.et al., J. Virol. 2000: 74; 6564-6569)。
センダイウイルスベクターを用いる場合、H1foo遺伝子等は、センダイウイルスの各遺伝子を破壊しない限り、センダイウイルスゲノムRNA(マイナス鎖)のいずれの位置にあってもよい。例えば、H1foo遺伝子等は、NP遺伝子よりも3’側にあってもよく、NP遺伝子とP遺伝子の間にあってもよく、P遺伝子とM遺伝子の間にあってもよく、M遺伝子とF遺伝子の間にあってもよく、F遺伝子とHN遺伝子の間にあってもよく(F遺伝子を有しない場合は、M遺伝子とHN遺伝子の間)、HN遺伝子とL遺伝子の間にあってもよく、L遺伝子よりも5’側にあってもよい。H1foo遺伝子等の発現量を増加させるためには、H1foo遺伝子等をより3’側に配置することが好ましい。例えば、センダイウイルスゲノムRNA(マイナス鎖)上で、H1foo遺伝子等を最も3’側に配置した場合(例えば、NP遺伝子よりも3’側)、H1foo遺伝子等の発現量が最も増加する。センダイウイルスベクターを用いる場合、H1foo遺伝子等をセンダイウイルスの全ての遺伝子よりも3’側に配置することが好ましい。すなわち、H1foo遺伝子等を最も3’側に配置することが好ましい。これにより、H1fooタンパク質の発現量と、不安定化ドメインにより促進されるH1fooタンパク質の分解量とのバランスが適度に維持されやすくなる。そのため、より品質の高いiPS細胞を高効率で作製することができる。また、センダイウイルスを導入対象細胞に感染させることで、より品質の高いiPS細胞を高効率で作製することができる。
発現ベクターとして、ウイルスベクターを用いる場合には、パッケージング細胞を用いて得られたウイルス粒子を用いてもよい。パッケージング細胞は、ウイルスの構造タンパク質をコードする遺伝子を導入した細胞であり、該細胞に目的遺伝子を組み込んだ組換えウイルスベクターを導入すると、該目的遺伝子を組み込んだ組換えウイルス粒子を産生する。パッケージング細胞は、特に限定されず、目的に応じて適宜選択することができる。例えば、ヒト腎臓由来のHEK293細胞又はマウス線維芽細胞由来のNIH3T3細胞をベースとしたパッケージング細胞;Ecotropic virus由来エンベロープ糖タンパク質を発現するよう設計されているPLAT-E細胞;Amphotropic virus由来エンベロープ糖タンパク質を発現するよう設計されているPLAT-A細胞;及び水疱性口内炎ウイルス由来エンベロープ糖タンパク質を発現するよう設計されているPLAT-GP細胞などが挙げられる。ウイルスベクターの導入対象がヒト体細胞である場合には、パッケージング細胞としては、宿主指向性の点で、PLAT-A細胞、PLAT-GP細胞等が好ましい。パッケージング細胞へのウイルスベクターの導入方法は、特に限定されず、目的に応じて適宜選択することができる。例えば、リポフェクション法、エレクトロポレーション法、リン酸カルシウム法等が挙げられる。
発現ベクターは、後述する核初期化物質を含んでいてもよい。発現ベクターが、核初期化物質を含む場合、当該核初期化物質は、1種であってもよく、2種以上であってもよい。発現ベクターが、H1foo遺伝子等とともに、核初期化物質を有する場合、H1foo遺伝子等と核初期化物質との間に、2Aペプチドなどの自己切断型ペプチドをコードする塩基配列、又はIRES(Internal Ribozyme Entry Site)配列等を介在させてもよい。これらの配列を介在させることにより、1つのプロモーターから、複数のタンパク質を独立して発現させることができる。
好ましい態様において、本実施形態の品質改善剤は、H1fooタンパク質と不安定化ドメインとの融合タンパク質をコードするポリヌクレオチドを発現可能な形態で搭載したセンダイウイルスベクターである。さらなる好ましい態様において、本実施形態の品質改善剤は、H1fooタンパク質とFKBP12由来の不安定化ドメインとの融合タンパク質をコードするポリヌクレオチドを発現可能な形態で搭載したセンダイウイルスベクターである。H1FOOを含む前記融合タンパク質の具体例としては、配列番号16又は配列番号18に記載のアミノ酸配列を含むタンパク質が挙げられる。これらのタンパク質をコードするポリヌクレオチドの具体例としては、配列番号15又は配列番号17に記載のヌクレオチド配列を含むポリヌクレオチドが挙げられる。
<第2実施形態>
一実施形態において、本発明は、H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む、iPS細胞の品質改善剤を提供する。前記不安定化ドメインは、前記融合タンパク質のプロテアソームによる分解を誘導するドメインである。
一実施形態において、本発明は、H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む、iPS細胞の品質改善剤を提供する。前記不安定化ドメインは、前記融合タンパク質のプロテアソームによる分解を誘導するドメインである。
(H1fooタンパク質)
H1fooタンパク質は、上記「<第1実施形態>」で説明したH1foo遺伝子から転写及び翻訳されて生じるタンパク質であり、H1foo活性を有する。H1fooタンパク質が由来する生物種としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。ヒトのH1fooタンパク質のアミノ酸配列を配列番号2に例示する。また、マウスのH1fooタンパク質のアミノ酸配列を配列番号4に例示する。
H1fooタンパク質は、上記「<第1実施形態>」で説明したH1foo遺伝子から転写及び翻訳されて生じるタンパク質であり、H1foo活性を有する。H1fooタンパク質が由来する生物種としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。ヒトのH1fooタンパク質のアミノ酸配列を配列番号2に例示する。また、マウスのH1fooタンパク質のアミノ酸配列を配列番号4に例示する。
H1fooタンパク質は、野生型のH1fooタンパク質に限定されず、変異(欠失、置換、挿入、及び付加のいずれか、又はこれらの組合せ)を含むものであってもよい。H1foo活性を有するタンパク質であれば、変異の数は特に限定されない。
H1fooタンパク質としては、例えば、以下の(a)~(c)が挙げられる。
(a)野生型のH1fooタンパク質(例、配列番号2又は配列番号4で表されるアミノ酸配列からなるポリペプチド)
(b)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり、且つH1foo活性を有するタンパク質
(c)野生型のH1fooタンパク質のアミノ酸配列(例えば、配列番号2又は4で表されるアミノ酸配列)と、70%以上の配列同一性を有するアミノ酸配列からなり、且つH1foo活性を有するタンパク質
(a)野生型のH1fooタンパク質(例、配列番号2又は配列番号4で表されるアミノ酸配列からなるポリペプチド)
(b)野生型のH1fooタンパク質のアミノ酸配列(例、配列番号2又は4で表されるアミノ酸配列)において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり、且つH1foo活性を有するタンパク質
(c)野生型のH1fooタンパク質のアミノ酸配列(例えば、配列番号2又は4で表されるアミノ酸配列)と、70%以上の配列同一性を有するアミノ酸配列からなり、且つH1foo活性を有するタンパク質
上記(b)において、「変異」は、欠失、置換、付加、及び挿入のいずれであってもよく、これらの組合せであってもよい。
上記(b)において、「複数個」は、結果として生じるタンパク質がH1foo活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(c)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
上記(b)において、「複数個」は、結果として生じるタンパク質がH1foo活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(c)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
(不安定化ドメイン)
不安定化ドメインは、上記「<第1実施形態>」で説明したものと同様であり、上記「<第1実施形態>」で例示したものと同様のものが例示される。
不安定化ドメインは、上記「<第1実施形態>」で説明したものと同様であり、上記「<第1実施形態>」で例示したものと同様のものが例示される。
(融合タンパク質)
H1fooタンパク質と不安定化ドメインとの融合タンパク質は、H1fooタンパク質のN末端及びC末端の少なくとも一方に不安定化ドメインが連結されたものである。不安定化ドメインは、H1fooタンパク質のN末端及びC末端のいずれに連結されていてもよく、N末端及びC末端の両方に連結されていてもよい。
H1fooタンパク質と不安定化ドメインとの融合タンパク質は、H1fooタンパク質のN末端及びC末端の少なくとも一方に不安定化ドメインが連結されたものである。不安定化ドメインは、H1fooタンパク質のN末端及びC末端のいずれに連結されていてもよく、N末端及びC末端の両方に連結されていてもよい。
H1fooタンパク質と不安定化ドメインとの融合タンパク質は、H1foo遺伝子の5’末端又は3’末端の少なくとも一方に不安定化ドメイン遺伝子が連絡されたポリヌクレオチドを含む、発現ベクターを用いて製造することができる。当該融合タンパク質は、当該発現ベクターを、適切な細胞に導入し、当該細胞を培養し、その培養上清又は培養菌体若しくは細胞から、融合タンパク質を単離・精製することにより得ることができる。
H1fooタンパク質と不安定化ドメインとの融合タンパク質は、さらに、タンパク質導入ドメイン(PTD)を有していてもよい。PTDとしては、ショウジョウバエ由来のAntP、HIV由来のTAT、HSV由来のVP22等のタンパク質の細胞通過ドメインを用いたものが開発されている。PTDを有することにより、タンパク質導入試薬を用いることなく、融合タンパク質を細胞に導入することができる。
H1fooタンパク質と不安定化ドメインとの融合タンパク質は、体細胞に導入した場合に、不安定化ドメインを有するために、すみやかにプロテアソームにより分解される。なお、不安定化ドメインを含む融合タンパク質は、安定化化合物の添加により、細胞内での存在量及び存在時期を制御することができる。したがって、細胞内におけるH1fooタンパク質の存在量及び存在時期を制御することができる。本実施形態のiPS細胞の品質改善剤を、後述する核初期化物質とともに体細胞に導入した後、H1fooタンパク質と不安定化ドメインとの融合タンパク質が、例えば、導入直後の任意の時間のみ細胞内に存在するよう制御することにより、品質の高いiPS細胞を製造することができる。
本発明のiPS細胞の品質改善剤、及びiPS細胞の製造方法においては、上記H1foo遺伝子と制御配列とを有するポリヌクレオチドの代わりに、核初期化物質とともに体細胞に導入され、細胞の初期化が開始された際に、同時に当該細胞内の自然免疫応答を抑制する機能を有する物質を用いることもできる。自然免疫応答の抑制とは、具体例としてはIFIT1、IFNA等の自然免疫応答マーカーの発現抑制を意味し、さらに具体的には当該細胞内でFKBP1Aの発現を、核初期化物質のみを導入した場合に比べて2倍以上、好ましくは2~20倍、さらに好ましくは5~15倍に増加させることを意味する。ここで、細胞の初期化が開始された際とは、核初期化物質導入から2~15日、好ましくは3~6日目をいう。このような初期化工程を行うことにより、iPS細胞の樹立効率が高くなるだけでなく、製造されたiPS細胞群の中には高い分化多能性等のiPS細胞としての品質が高いものが多く含まれるものとなるため、他家iPS細胞の製造方法としても有効であるが、自家iPS細胞の製造時で細胞株として樹立せずバルクで扱うような場合に特に大きな効果を有するものである。
自然免疫応答を抑制する機能を有する物質は、自然免疫応答マーカーの発現を抑制し得る限り、特に限定されない。自然免疫応答を抑制する機能を有する物質としては、例えば、自然免疫応答マーカー遺伝子に対するsiRNA若しくはアンチセンスRNA、自然免疫応答マーカー遺伝子の転写阻害因子、FKBP1A、FKBP1A遺伝子、FKBP1Aの転写促進因子等が挙げられるが、これらに限定されない。
自然免疫応答を抑制する機能を有する物質は、自然免疫応答マーカーの発現を抑制し得る限り、特に限定されない。自然免疫応答を抑制する機能を有する物質としては、例えば、自然免疫応答マーカー遺伝子に対するsiRNA若しくはアンチセンスRNA、自然免疫応答マーカー遺伝子の転写阻害因子、FKBP1A、FKBP1A遺伝子、FKBP1Aの転写促進因子等が挙げられるが、これらに限定されない。
[iPS細胞の製造方法]
<第1実施形態>
一実施形態において、本発明は、iPS細胞の製造方法を提供する。核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む。
<第1実施形態>
一実施形態において、本発明は、iPS細胞の製造方法を提供する。核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む。
≪導入工程≫
本実施形態のiPS細胞の製造方法は、iPS細胞の製造方法を提供する。核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む。
本実施形態のiPS細胞の製造方法は、iPS細胞の製造方法を提供する。核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を体細胞に導入する工程を含む。
(iPS細胞の品質改善剤)
iPS細胞の品質改善剤は、上記「[iPS細胞の品質改善剤]」で説明した第1実施形態及び第2実施形態のいずれであってもよい。また、第1実施形態及び第2実施形態の両方のiPS細胞の品質改善剤を併用してもよい。
iPS細胞の品質改善剤は、上記「[iPS細胞の品質改善剤]」で説明した第1実施形態及び第2実施形態のいずれであってもよい。また、第1実施形態及び第2実施形態の両方のiPS細胞の品質改善剤を併用してもよい。
(核初期化物質)
本明細書において、「核初期化物質」とは、体細胞に導入することにより、当該体細胞をiPS細胞に誘導することができる物質(群)を意味する。核初期化物質は、体細胞からiPS細胞を誘導することができる物質(群)であれば、特に限定されない。核初期化物質は、遺伝子(発現ベクターに組み込まれた形態を含む)若しくはその遺伝子産物、又は低分子化合物等のいかなる物質であってもよい。、「遺伝子産物」とは、遺伝子から転写されるmRNA、及び当該mRNAから翻訳されるタンパク質を意味する。核初期化物質として用いる遺伝子産物は、mRNAであってもよく、タンパク質であってもよく、その両方であってもよい。核初期化物質である遺伝子とは、核初期化物質であるタンパク質をコードするポリヌクレオチドを意味する。
本明細書において、「核初期化物質」とは、体細胞に導入することにより、当該体細胞をiPS細胞に誘導することができる物質(群)を意味する。核初期化物質は、体細胞からiPS細胞を誘導することができる物質(群)であれば、特に限定されない。核初期化物質は、遺伝子(発現ベクターに組み込まれた形態を含む)若しくはその遺伝子産物、又は低分子化合物等のいかなる物質であってもよい。、「遺伝子産物」とは、遺伝子から転写されるmRNA、及び当該mRNAから翻訳されるタンパク質を意味する。核初期化物質として用いる遺伝子産物は、mRNAであってもよく、タンパク質であってもよく、その両方であってもよい。核初期化物質である遺伝子とは、核初期化物質であるタンパク質をコードするポリヌクレオチドを意味する。
核初期化物質が遺伝子又はその遺伝子産物の場合、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子、並びに、それらの遺伝子産物からなる群から選択される少なくとも1つが挙げられる(国際公開第2007/69666号;特許第5696282号;Science, 2007, 318:1917-1920)。これらの中でも、好ましくは、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリー遺伝子、及びMyc遺伝子ファミリーの遺伝子、及びこれらの遺伝子の遺伝子産物からなる群より選択される少なくとも1つが挙げられる。
これらのファミリーの遺伝子及びその組み合わせの具体例を以下に列挙する。なお、以下においては遺伝子の名称のみを記載するが、その遺伝子産物を用いる場合も含まれる。
(a)Oct遺伝子ファミリーの遺伝子からなる1種の核初期化物質;
(b)Oct遺伝子ファミリーの遺伝子及びSox遺伝子ファミリーの遺伝子からなる2種の核初期化物質の組み合わせ;
(c)Oct遺伝子ファミリーの遺伝子及びKlf遺伝子ファミリーの遺伝子からなる2種の核初期化物質の組み合わせ;
(d)Oct遺伝子ファミリーの遺伝子及びNanog遺伝子からなる2種の核初期化物質の組み合わせ;
(e)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、及びKlf遺伝子ファミリーの遺伝子からなる3種の核初期化物質の組み合わせ;
(f)Oct遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子及びMyc遺伝子ファミリーの遺伝子からなる3種の核初期化物質の組み合わせ。
(g)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子及びMyc遺伝子ファミリーの遺伝子からなる4種の核初期化物質の組み合わせ;並びに
(h)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる4種の核初期化物質の組み合わせ。
(b)Oct遺伝子ファミリーの遺伝子及びSox遺伝子ファミリーの遺伝子からなる2種の核初期化物質の組み合わせ;
(c)Oct遺伝子ファミリーの遺伝子及びKlf遺伝子ファミリーの遺伝子からなる2種の核初期化物質の組み合わせ;
(d)Oct遺伝子ファミリーの遺伝子及びNanog遺伝子からなる2種の核初期化物質の組み合わせ;
(e)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、及びKlf遺伝子ファミリーの遺伝子からなる3種の核初期化物質の組み合わせ;
(f)Oct遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子及びMyc遺伝子ファミリーの遺伝子からなる3種の核初期化物質の組み合わせ。
(g)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子及びMyc遺伝子ファミリーの遺伝子からなる4種の核初期化物質の組み合わせ;並びに
(h)Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる4種の核初期化物質の組み合わせ。
より具体的には、以下の組み合わせが例示されるが、これらに限定されない。以下の組合せにおいて、Sox2遺伝子は、Sox1遺伝子、Sox3遺伝子、Sox15遺伝子、Sox17遺伝子、又はSox18遺伝子で置換可能である。Klf4遺伝子は、Klf1遺伝子、Klf2遺伝子、又はKlf5遺伝子で置換可能である。c-Myc遺伝子は、T58A(活性型変異体)遺伝子、N-Myc遺伝子、又はL-Myc遺伝子で置換可能である。
(1)Oct3/4遺伝子、Klf4遺伝子、c-Myc遺伝子
(2)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子
(3)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、Fbx15遺伝子、Nanog遺伝子、Eras遺伝子、ECAT15-2遺伝子、TclI遺伝子、β-catenin(活性型変異体S33Y)
(4)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、SV40 Large T antigen(以下、SV40LT)遺伝子
(5)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV16 E6遺伝子
(6)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV16 E7遺伝子
(7)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV6 E6遺伝子、HPV16 E7遺伝子
(8)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、Bmil遺伝子
(上記(1)~(8)の組み合わせについては、国際公開第2007/069666号を参照(但し、上記(2)の組み合わせにおいて、Sox2遺伝子からSox18遺伝子への置換、Klf4遺伝子からKlf1遺伝子若しくはKlf5遺伝子への置換については、Nature Biotechnology, 26, 101-106 (2008)を参照)。「Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子」の組み合わせについては、Cell, 126, 663-676 (2006)、Cell, 131, 861-872 (2007) 等も参照。「Oct3/4遺伝子、Sox2遺伝子、Klf2(又はKlf5)遺伝子、c-Myc遺伝子」の組み合わせについては、Nat. Cell Biol., 11, 197-203 (2009) も参照。「Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、SV40LT遺伝子」の組み合わせについては、Nature, 451, 141-146 (2008)も参照。)
(9)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子(Nature Biotechnology, 26, 101-106 (2008)を参照)
(10)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子(Science, 318, 1917-1920 (2007)を参照)
(11)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子、hTERT遺伝子、SV40LT遺伝子(Stem Cells, 26, 1998-2005 (2008)を参照)
(12)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、Nanog遺伝子、Lin28遺伝子(Cell Research (2008) 600-603を参照)
(13)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、SV40LT遺伝子(Stem Cells, 26, 1998-2005 (2008)も参照)
(14)Oct3/4遺伝子、Klf4遺伝子(Nature 454:646-650 (2008)、Cell Stem Cell, 2:525-528(2008))を参照)
(15)Oct3/4遺伝子、c-Myc遺伝子(Nature 454:646-650 (2008)を参照)
(16)Oct3/4遺伝子、Sox2遺伝子(Nature, 451, 141-146 (2008), 国際公開第2008/118820号を参照)
(17)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子(国際公開第2008/118820号を参照)
(18)Oct3/4遺伝子、Sox2遺伝子、Lin28遺伝子(国際公開第2008/118820号を参照)
(19)Oct3/4遺伝子、Sox2遺伝子、c-Myc遺伝子、Esrrb遺伝子(Essrrb遺伝子はEsrrg遺伝子で置換可能である。Nat. Cell Biol., 11, 197-203 (2009) を参照)
(20)Oct3/4遺伝子、Sox2遺伝子、Esrrb遺伝子(Nat. Cell Biol., 11, 197-203 (2009) を参照)
(21)Oct3/4遺伝子、Klf4遺伝子、L-Myc遺伝子
(22)Oct3/4遺伝子、Nanog遺伝子
(23)Oct3/4遺伝子
(24)Oct3/4遺伝子、Klf4遺伝子、c-Myc遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子、SV40LT遺伝子(Science, 324: 797-801 (2009)を参照)
(1)Oct3/4遺伝子、Klf4遺伝子、c-Myc遺伝子
(2)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子
(3)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、Fbx15遺伝子、Nanog遺伝子、Eras遺伝子、ECAT15-2遺伝子、TclI遺伝子、β-catenin(活性型変異体S33Y)
(4)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、SV40 Large T antigen(以下、SV40LT)遺伝子
(5)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV16 E6遺伝子
(6)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV16 E7遺伝子
(7)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、HPV6 E6遺伝子、HPV16 E7遺伝子
(8)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、Bmil遺伝子
(上記(1)~(8)の組み合わせについては、国際公開第2007/069666号を参照(但し、上記(2)の組み合わせにおいて、Sox2遺伝子からSox18遺伝子への置換、Klf4遺伝子からKlf1遺伝子若しくはKlf5遺伝子への置換については、Nature Biotechnology, 26, 101-106 (2008)を参照)。「Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子」の組み合わせについては、Cell, 126, 663-676 (2006)、Cell, 131, 861-872 (2007) 等も参照。「Oct3/4遺伝子、Sox2遺伝子、Klf2(又はKlf5)遺伝子、c-Myc遺伝子」の組み合わせについては、Nat. Cell Biol., 11, 197-203 (2009) も参照。「Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、hTERT遺伝子、SV40LT遺伝子」の組み合わせについては、Nature, 451, 141-146 (2008)も参照。)
(9)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子(Nature Biotechnology, 26, 101-106 (2008)を参照)
(10)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子(Science, 318, 1917-1920 (2007)を参照)
(11)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子、hTERT遺伝子、SV40LT遺伝子(Stem Cells, 26, 1998-2005 (2008)を参照)
(12)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、Nanog遺伝子、Lin28遺伝子(Cell Research (2008) 600-603を参照)
(13)Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子、SV40LT遺伝子(Stem Cells, 26, 1998-2005 (2008)も参照)
(14)Oct3/4遺伝子、Klf4遺伝子(Nature 454:646-650 (2008)、Cell Stem Cell, 2:525-528(2008))を参照)
(15)Oct3/4遺伝子、c-Myc遺伝子(Nature 454:646-650 (2008)を参照)
(16)Oct3/4遺伝子、Sox2遺伝子(Nature, 451, 141-146 (2008), 国際公開第2008/118820号を参照)
(17)Oct3/4遺伝子、Sox2遺伝子、Nanog遺伝子(国際公開第2008/118820号を参照)
(18)Oct3/4遺伝子、Sox2遺伝子、Lin28遺伝子(国際公開第2008/118820号を参照)
(19)Oct3/4遺伝子、Sox2遺伝子、c-Myc遺伝子、Esrrb遺伝子(Essrrb遺伝子はEsrrg遺伝子で置換可能である。Nat. Cell Biol., 11, 197-203 (2009) を参照)
(20)Oct3/4遺伝子、Sox2遺伝子、Esrrb遺伝子(Nat. Cell Biol., 11, 197-203 (2009) を参照)
(21)Oct3/4遺伝子、Klf4遺伝子、L-Myc遺伝子
(22)Oct3/4遺伝子、Nanog遺伝子
(23)Oct3/4遺伝子
(24)Oct3/4遺伝子、Klf4遺伝子、c-Myc遺伝子、Sox2遺伝子、Nanog遺伝子、Lin28遺伝子、SV40LT遺伝子(Science, 324: 797-801 (2009)を参照)
上記(1)~(24)の組み合わせにおいて、Oct3/4遺伝子に代えて、他のOct遺伝子ファミリーのメンバー遺伝子(例えばOct1A、Oct6など)を用いることもできる。Sox2遺伝子(又はSox1遺伝子、Sox3遺伝子、Sox15遺伝子、Sox17遺伝子、Sox18遺伝子)に代えて、他のSox遺伝子ファミリーのメンバー遺伝子(例えばSox7遺伝子など)を用いることもできる。Lin28遺伝子に代えて、他のLin遺伝子ファミリーのメンバー遺伝子(例えばLin28b遺伝子など)を用いることもできる。
上記(1)~(24)の組み合わせには該当しないが、それらのいずれかにおける構成要素をすべて含み、且つ任意の他の物質をさらに含む組み合わせも、本発明における「核初期化物質」の範疇に含まれ得る。核初期化の対象となる体細胞が上記(1)~(24)の組み合わせのいずれかにおける構成要素の一部を、核初期化のために十分なレベルで内在的に発現している条件下にあっては、当該構成要素を除いた残りの構成要素のみの組み合わせもまた、本発明における「核初期化物質」の範疇に含まれ得る。
上記の核初期化物質に加えて、Fbx15遺伝子、ERas遺伝子、ECAT15-2遺伝子、Tcl1遺伝子、及びβ-catenin遺伝子からなる群から選ばれる1種以上の核初期化物質を組み合わせてもよく、及び/又はECAT1遺伝子、Esg1遺伝子、Dnmt3L遺伝子、ECAT8遺伝子、Gdf3遺伝子、Mybl2遺伝子、ECAT15-1遺伝子、Fthl17遺伝子、Sall4遺伝子、Rex1遺伝子、UTF1遺伝子、Stella遺伝子、Stat3遺伝子、及びGrb2遺伝子からなる群から選ばれる1種以上の核初期化物質を組み合わせることもできる。これらの組み合わせについては国際公開第2007/69666号に具体的に説明されている。
好ましい核初期化物質としては、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子(又はL-Myc遺伝子)、Lin28遺伝子、Nanog遺伝子、及びこれらの遺伝子の遺伝子産物からなる群から選択される少なくとも1つが例示される。好ましくは、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子(又はL-Myc遺伝子)、Lin28遺伝子、Nanog遺伝子、それらの遺伝子産物からなる群から選択される2つ以上の組み合わせ、より好ましくは3つ以上の組み合わせである。このうち、少なくとも導入されることが好ましい核初期化物質の組み合わせとしては、(1)Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、及びKlf4遺伝子又はその遺伝子産物、(2)Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、Klf4遺伝子又はその遺伝子産物、及びc-Myc遺伝子又はその遺伝子産物、(3)Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、Klf4遺伝子又はその遺伝子産物、及びL-Myc遺伝子又はその遺伝子産物が挙げられる。中でも、Oct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、及びKlf4遺伝子又はその遺伝子産物の組み合わせ、並びにOct3/4遺伝子又はその遺伝子産物、Sox2遺伝子又はその遺伝子産物、Klf4遺伝子又はその遺伝子産物、及びL-Myc遺伝子又はその遺伝子産物の組み合せが好ましい。中でも、Oct3/4遺伝子、Sox2遺伝子、及びKlf4遺伝子の組み合わせ、並びにOct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子の組み合わせがより好ましい。
核初期化物質が遺伝子又はその遺伝子産物の場合、かかる遺伝子が由来する生物種としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。
上記の各核初期化物質のcDNA配列情報は、公知のデータベースから得ることができる。例えば、国際公開第2007/069666号に記載のGenBankにおけるアクセッション番号を参照してもよい。Nanog遺伝子は前記公報中では「ECAT4」との名称で記載されている。
上記の各核初期化物質のうち、特に好ましい4つの遺伝子(Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、L-Myc遺伝子)のマウス及びヒトcDNA配列情報を、以下に記載する。
遺伝子名 マウス ヒト
Oct3/4 NM_013633 NM_002701
Sox2 NM_011443 NM_003106
Klf4 NM_010637 NM_004235
L-Myc NM_008506 NM_001033081
遺伝子名 マウス ヒト
Oct3/4 NM_013633 NM_002701
Sox2 NM_011443 NM_003106
Klf4 NM_010637 NM_004235
L-Myc NM_008506 NM_001033081
上記GenBankアクセッション番号に登録されるヒトのOct3/4遺伝子のcDNA配列を配列番号5に示し、ヒトのOct3/4タンパク質のアミノ酸配列を配列番号6に示す。ヒトのSox2遺伝子のcDNA配列を配列番号7に示し、ヒトのSox2タンパク質のアミノ酸配列を配列番号8に示す。ヒトのKlf4遺伝子のcDNA配列を配列番号9に示し、ヒトのKlf4タンパク質のアミノ酸配列を配列番号10に示す。ヒトのL-Myc遺伝子のcDNA配列を配列番号11に示し、ヒトのL-Mycタンパク質のアミノ酸配列を配列番号12に示す。
また、上記の各核初期化物質のうち、国際公開第2007/069666号にGenBankアクセッション番号が記載されていない遺伝子のマウス及びヒトcDNA配列情報を以下に記載する。
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
上記各核初期化物質のcDNAは、上記のcDNA配列情報又は公知のデータベースに登録された配列情報に基づき、当該配列が由来する生物の細胞から、PCR法等の公知の方法を用いて、容易に単離することができる。
核初期化物質が、上記各遺伝子又はそのmRNAである場合、それらのヌクレオチド配列は、野生型遺伝子のヌクレオチド配列に限定されず、核初期化作用を有する限り、変異を含んでいてもよい。上記各遺伝子の各ヌクレオチド配列は、タンパク質コード配列のみであってもよく、それ以外の部分(例えば、イントロン、5’UTR、3’UTR等)を含んでいてもよい。
核初期化物質が、上記各遺伝子にコードされるタンパク質である場合、それらのアミノ酸配列は、野生型タンパク質のアミノ酸配列に限定されず、核初期化作用を有する限り、変異を含んでいてもよい。
本明細書において、「核初期化作用」とは、体細胞に導入することにより、当該体細胞をiPS細胞に誘導する作用を意味する。
核初期化物質が、上記各遺伝子にコードされるタンパク質である場合、それらのアミノ酸配列は、野生型タンパク質のアミノ酸配列に限定されず、核初期化作用を有する限り、変異を含んでいてもよい。
本明細書において、「核初期化作用」とは、体細胞に導入することにより、当該体細胞をiPS細胞に誘導する作用を意味する。
核初期化物質が遺伝子又はmRNAである場合、核初期化物質としては、例えば、以下の(a)~(g)が挙げられる。
(a)上記核初期化物質として例示した野生型遺伝子又は野生型mRNA
(b)上記核初期化物質として例示した野生型タンパク質をコードするヌクレオチド配列からなるポリヌクレオチド
(c)上記核初期化物質として例示した野生型タンパク質において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(d)上記核初期化物質として例示した野生型タンパク質のアミノ酸配列と70%以上の配列同一性を有するアミノ酸配列からなり且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(e)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列において、1又は複数個のヌクレオチドが変異したヌクレオチド配列からなり、且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(f)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列と、70%以上の配列同一性を有するヌクレオチド配列からなり、且つ核初期化作用を有するタンパク質をコードするポリヌクレオチド
(g)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列とストリンジェントな条件でハイブリダイズし、且つ核初期化作用を有するタンパク質をコードするポリヌクレオチド
(a)上記核初期化物質として例示した野生型遺伝子又は野生型mRNA
(b)上記核初期化物質として例示した野生型タンパク質をコードするヌクレオチド配列からなるポリヌクレオチド
(c)上記核初期化物質として例示した野生型タンパク質において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(d)上記核初期化物質として例示した野生型タンパク質のアミノ酸配列と70%以上の配列同一性を有するアミノ酸配列からなり且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(e)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列において、1又は複数個のヌクレオチドが変異したヌクレオチド配列からなり、且つ核初期化作用を有するタンパク質、をコードするポリヌクレオチド
(f)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列と、70%以上の配列同一性を有するヌクレオチド配列からなり、且つ核初期化作用を有するタンパク質をコードするポリヌクレオチド
(g)上記核初期化物質として例示した野生型遺伝子のヌクレオチド配列とストリンジェントな条件でハイブリダイズし、且つ核初期化作用を有するタンパク質をコードするポリヌクレオチド
上記(c)及び(e)において、「変異」は、欠失、置換、付加、及び挿入のいずれであってもよく、これらの組合せであってもよい。
上記(c)、「複数個」は、結果として生じるタンパク質が核初期化活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(e)において、「複数個」は、結果として生じるポリヌクレオチドが核初期化作用を有するタンパク質をコードする限り特に限定されないが、例えば、2~60個が例示され、2~50個が好ましく、2~40個又は2~30個がより好ましく、2~20個又は2~10個がさらに好ましく、2~5個又は2個若しくは3個が特に好ましい。
上記(d)及び(f)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
上記(g)において、「ストリンジェントな条件」とは、上記「[iPS細胞の品質改善剤]」、<第1実施形態>、(H1foo遺伝子)」の項で例示した条件と、同様の条件が挙げられる。
上記(c)、「複数個」は、結果として生じるタンパク質が核初期化活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(e)において、「複数個」は、結果として生じるポリヌクレオチドが核初期化作用を有するタンパク質をコードする限り特に限定されないが、例えば、2~60個が例示され、2~50個が好ましく、2~40個又は2~30個がより好ましく、2~20個又は2~10個がさらに好ましく、2~5個又は2個若しくは3個が特に好ましい。
上記(d)及び(f)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
上記(g)において、「ストリンジェントな条件」とは、上記「[iPS細胞の品質改善剤]」、<第1実施形態>、(H1foo遺伝子)」の項で例示した条件と、同様の条件が挙げられる。
上記(b)~(e)において、縮重コドンは、本実施形態のiPS細胞の品質改善剤を使用する体細胞において、コドン使用頻度が高いものが用いられていることが好ましい。例えば、ヒトの体細胞に用いる場合には、ヒト細胞において使用頻度の高いコドンが用いられていることが好ましい。すなわち、ヒトコドンに最適化されていることが好ましい。また、マウスの体細胞に用いる場合には、マウス細胞において使用頻度の高いコドンが用いられていることが好ましい。すなわち、マウスコドンに最適化されていることが好ましい。
核初期化物質が遺伝子である場合、当該遺伝子は、発現ベクターに組み込まれていることが好ましい。発現ベクターとしては、上記「[iPS細胞の品質改善剤]、(ポリヌクレオチド)」の項で挙げたものと同様のものが挙げられる。中でも、発現ベクターとしては、センダイウイルスベクターが好ましい。発現ベクターを用いる場合、1つの発現ベクターに、1種の核初期化物質のみを組み込んでもよく、2種以上の核初期化物質を組み込んでもよい。発現ベクターが、2種以上の核初期化物質を含む場合、核初期化物質である2種以上の遺伝子間に、2Aペプチドなどの自己切断型ペプチドをコードする塩基配列や、IRES配列等を介在させてもよい。
核初期化物質がタンパク質である場合、核初期化物質としては、例えば、以下の(A)~(C)が挙げられる。
(A)上記核初期化物質として例示した野生型タンパク質
(B)上記核初期化物質として例示した野生型タンパク質のアミノ酸配列において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり、かつ核初期化活性を有するタンパク質
(C)上記核初期化物質として例示した野生型タンパク質と、70%以上の配列同一性を有するアミノ酸配列からなり、かつ核初期化活性を有するタンパク質
(A)上記核初期化物質として例示した野生型タンパク質
(B)上記核初期化物質として例示した野生型タンパク質のアミノ酸配列において、1又は複数個のアミノ酸が変異したアミノ酸配列からなり、かつ核初期化活性を有するタンパク質
(C)上記核初期化物質として例示した野生型タンパク質と、70%以上の配列同一性を有するアミノ酸配列からなり、かつ核初期化活性を有するタンパク質
上記(B)において、「変異」は、欠失、置換、付加、及び挿入のいずれであってもよく、これらの組合せであってもよい。
上記(B)において、「複数個」は、結果として生じるタンパク質が核初期化活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(C)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
上記(B)において、「複数個」は、結果として生じるタンパク質が核初期化活性を有する限り特に限定されないが、例えば、2~30個が例示され、2~20個が好ましく、2~10個がより好ましく、2~5個がさらに好ましく、2個又は3個が特に好ましい。
上記(C)において、配列同一性は、70%以上である限り、特に限定されないが、80%以上が好ましく、85%以上がより好ましく、90%以上、95%以上、96%以上、97%以上、98%以上、又は99%以上がさらに好ましい。
核初期化物質がタンパク質である場合、体細胞への導入のために、タンパク質導入ドメイン(PTD)を連結してもよい。
核初期化物質が遺伝子又はその遺伝子産物である場合、遺伝子のみを用いる態様であってもよいし、その遺伝子産物のみを用いる態様であってもよいし、遺伝子とその遺伝子産物の両方を用いる態様であってもよい。核初期化物質が遺伝子又はその遺伝子産物であって、かつ、2種類以上の「遺伝子又はその遺伝子産物」を併用する場合、ある遺伝子については遺伝子産物を用い、他の遺伝子については遺伝子を用いる態様であってもよい。
核初期化物質は、上記例示した遺伝子又は遺伝子の組合せであることが好ましく、発現ベクターの形態であることがより好ましい。
核初期化物質は、上記例示した遺伝子又は遺伝子の組合せであることが好ましく、発現ベクターの形態であることがより好ましい。
(体細胞)
体細胞は、特に限定されず、目的に応じて適宜選択することができる。体細胞としては、例えば、胎児期の体細胞、成熟した体細胞等が挙げられる。成熟した体細胞の具体例としては、間葉系幹細胞、造血幹細胞、脂肪組織由来間質細胞、脂肪組織由来間質幹細胞、神経幹細胞、精子幹細胞などの組織幹細胞(体性幹細胞);組織前駆細胞;線維芽細胞、上皮細胞、リンパ球、筋肉細胞等の既に分化した細胞;等が挙げられる。
体細胞は、特に限定されず、目的に応じて適宜選択することができる。体細胞としては、例えば、胎児期の体細胞、成熟した体細胞等が挙げられる。成熟した体細胞の具体例としては、間葉系幹細胞、造血幹細胞、脂肪組織由来間質細胞、脂肪組織由来間質幹細胞、神経幹細胞、精子幹細胞などの組織幹細胞(体性幹細胞);組織前駆細胞;線維芽細胞、上皮細胞、リンパ球、筋肉細胞等の既に分化した細胞;等が挙げられる。
体細胞が由来する生物種は、特に限定されず、目的に応じて適宜選択することができる。体細胞が由来する生物種としては、例えば、ヒト、マウス、ラット、ウシ、ヒツジ、ウマ、サルなどの任意の哺乳動物が挙げられる。体細胞が由来する生物種と、核初期化物質が由来する生物種とは、同じでなくてもよいが、同じであることが好ましい。上記実施形態のiPS細胞の品質改善剤に含まれるH1foo遺伝子又はH1fooタンパク質が由来する生物種と、体細胞が由来する生物種は同じでなくてもよいが、同じであることが好ましい。
体細胞が由来する個体としては、特に制限はなく、目的に応じて適宜選択することができる。ただし、本実施形態の方法で製造したiPS細胞を再生医療用途に用いる場合には、拒絶反応の観点から、当該再生医療の対象となる個体自身、又は当該再生医療の対象となる個体とMHCの型が同一若しくは実質的に同一の他個体が好ましい。ここで、上記MHCの型が実質的に同一とは、上記体細胞由来のiPS細胞から分化誘導して得られた細胞を個体に移植した場合に、免疫抑制剤などの使用により、移植細胞が生着可能な程度にMHCの型が一致していることをいう。
体細胞は、iPS細胞の選択を容易にするために、他家遺伝子を導入した組換え体細胞であってもよい。組換え体細胞の具体例としては、分化多能性細胞において特異的に高発現する遺伝子の遺伝子座に、レポーター遺伝子、及び薬剤耐性遺伝子の少なくともいずれかを組み込んだ組換え体細胞が挙げられる。分化多能性細胞において特異的に高発現する遺伝子としては、例えば、Fbx15遺伝子、Nanog遺伝子、Oct3/4遺伝子、などが挙げられる。レポーター遺伝子としては、例えば、緑色蛍光タンパク質(GFP)遺伝子、ルシフェラーゼ遺伝子、β-ガラクトシダーゼ遺伝子などが挙げられる。上記薬剤耐性遺伝子としては、ブラストシジン遺伝子、ハイグロマイシン遺伝子、ピューロマイシン耐性遺伝子、ネオマイシン耐性遺伝子などが挙げられる。
体細胞の培養条件は、特に限定されず、目的に応じて適宜選択することができる。例えば、培養温度は、約37℃、CO2濃度は約2%~5%、O2濃度は約5%~20%などが挙げられる。体細胞の培養に用いる培地は、特に限定されず、目的に応じて適宜選択することができる。例えば、5質量%~20質量%の血清を含む、最小必須培地(MEM)、ダルベッコ改変培地(DMEM)、RPMI1640培地、199培地、F12培地などが挙げられる。本明細書において、濃度について単に「%」と表記するものは「容量%」を意味する。
(体細胞への導入)
核初期化物質と、上記実施形態のiPS細胞の品質改善剤(以下、単に「品質改善剤」という。)を、体細胞に導入する方法は、特に限定されず、目的に応じて適宜選択することができる。体細胞への導入の容易さを考慮すると、核初期化物質は、タンパク質の形態で体細胞へ導入するよりも、遺伝子の形態、又は該遺伝子のmRNAの形態で体細胞へ導入する方が好ましく、遺伝子の形態で体細胞に導入することがより好ましい。特に、核初期化物質は、発現ベクターの形態で体細胞に導入することが好ましい。同様に、品質改善剤に含まれるポリヌクレオチドも、発現ベクターの形態であることが好ましい。
核初期化物質と、上記実施形態のiPS細胞の品質改善剤(以下、単に「品質改善剤」という。)を、体細胞に導入する方法は、特に限定されず、目的に応じて適宜選択することができる。体細胞への導入の容易さを考慮すると、核初期化物質は、タンパク質の形態で体細胞へ導入するよりも、遺伝子の形態、又は該遺伝子のmRNAの形態で体細胞へ導入する方が好ましく、遺伝子の形態で体細胞に導入することがより好ましい。特に、核初期化物質は、発現ベクターの形態で体細胞に導入することが好ましい。同様に、品質改善剤に含まれるポリヌクレオチドも、発現ベクターの形態であることが好ましい。
発現ベクターを体細胞に導入する方法は、特に限定されず、目的に応じて適宜選択することができる。発現ベクターの体細胞への導入方法としては、例えば、リポフェクション法、マイクロインジェクション法、DEAEデキストラン法、遺伝子銃法、エレクトロポレーション法、リン酸カルシウム法などが挙げられる。発現ベクターがウイルスベクターである場合には、ウイルスベクターを体細胞へ感染させる方法としては、例えば、ポリブレン法が挙げられる。
発現ベクターとしてセンダイウイルスベクターを用いる場合、核初期化物質若しくは品質改善剤、或いは核初期化物質及び品質改善剤を搭載したセンダイウイルスベクターは、該ベクター(センダイウイルス粒子)を、体細胞を含む培地に添加して該体細胞に感染させることにより、該体細胞内に導入される。センダイウイルスベクターの用量は適宜調節することができるが、例えば、感染多重度(MOI)0.1以上、好ましくは0.3以上、0.5以上、1以上、2以上、又は3以上であり、かつ100以下、好ましくは90以下、80以下、70以下、60以下、50以下、40以下、30以下、20以下、10以下、又は5以下で感染させることができる。好ましくは、MOI 0.3~100、より好ましくはMOI 0.5~50、MOI 1~40、MOI 1~30、MOI 2~30、又はMOI 3~30で感染させる。
センダイウイルスベクターがRNPの形態である場合には、例えば、エレクトロポレーション、リポフェクション、マイクロインジェクションなどの手法によって細胞内に導入することができる。
センダイウイルスベクターがRNPの形態である場合には、例えば、エレクトロポレーション、リポフェクション、マイクロインジェクションなどの手法によって細胞内に導入することができる。
核初期化物質がmRNAである場合、mRNA(メッセンジャーRNA)を体細胞に導入する方法は、特に限定されず、公知の方法を適宜選択して用いることができる。例えば、mRNAは、Lipofectamine(登録商標)MessengerMAX(Life Technologies社製)などの市販のRNAトランスフェクション試薬などを用いて体細胞に導入することができる。
核初期化物質がタンパク質である場合、タンパク質を体細胞に導入する方法は、特に限定されず、公知の方法を適宜選択して用いることができる。そのような方法としては、例えば、タンパク質導入試薬を用いる方法、タンパク質導入ドメイン(PTD)融合タンパク質を用いる方法、マイクロインジェクション法などが挙げられる。品質改善剤が、H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む場合も、公知の方法を適宜選択して、体細胞に導入することができる。
タンパク質導入試薬としては、カチオン性脂質をベースとしたBioPOTER(登録商標)Protein Delivery Reagent(Gene Therapy Systmes社製)及びPro-JectTM Protein Transfection Reagent(PIERCE社製);脂質をベースとしたProfect-1(Targeting Systems社製);膜透過性ペプチドをベースとしたPenetratin Peptide(Q biogene社製)及びChariot Kit(Active Motif社製);HVJエンベロープ(不活化センダイウイルス)を利用したGenomONE(石原産業社製)等が市販されている。タンパク質の導入はこれらの試薬に添付のプロトコールに従って行うことができるが、一般的な手順は以下の通りである。タンパク質を適当な溶媒(例えば、PBS、HEPES等の緩衝液)に希釈し、導入試薬を加えて室温で5~15分間程度インキュベートして複合体を形成させ、これを無血清培地に交換した細胞に添加して37℃で1時間~数時間インキュベートする。その後、培地を除去して血清含有培地に交換する。
PTDとしては、上記「[iPS細胞の品質改善剤]、<第2実施形態>、(融合タンパク質)」の項で挙げたものと同様のものが挙げられる。PTDを用いる場合も、タンパク質導入試薬を添加しない以外は上記と同様にして行うことができる。
マイクロインジェクション法は、先端径1μm程度のガラス針にタンパク質溶液を入れ、細胞に穿刺導入する方法であり、確実に細胞内にタンパク質を導入することができる。以前より、マウスおよびヒトにおいて、タンパク質の形態の核初期化物質をポリアルギニンやTAT等のCPP(cell penetrating peptide)と共に導入してiPS細胞を樹立する方法が開発されており、これらの手法を用いることもできる(Cell Stem Cell, 4:381-384 (2009))。
本実施形態のiPS細胞の製造方法において、核初期化物質及び品質改善剤の体細胞への導入回数は、1回であってもよいし、2回以上であってもよい。その導入時期は、特に限定されず、目的に応じて適宜選択することができる。核初期化物質及び品質改善剤の全てを同時期に導入してもよいし、一部又は全てを異なる時期に導入してもよい。核初期化物質及び品質改善剤は、全て同時期に体細胞に導入することが好ましい。導入回数は、1回であることが好ましい。
核初期化物質が遺伝子又はその遺伝子産物の場合、核初期化物質の体細胞への導入量としては、かかる体細胞の核を初期化し得る限り特に制限されず、用いる全ての遺伝子又はその遺伝子産物を等量ずつ導入してもよいし、異なる量で導入してもよい。遺伝子又はその遺伝子産物として、遺伝子を用いる場合の例としては、Oct3/4遺伝子を、Sox2遺伝子、Klf4遺伝子、又はc-Myc遺伝子若しくはL-Myc遺伝子に対して多量(例えば約3倍量)となるように導入する方法が挙げられる(PNAS 106(31):12759-12764 (2009)、J.Biol.Chem.287(43): 36273-36282 (2012))。
本実施形態のiPS細胞の製造方法において用いる核初期化物質が低分子化合物である場合、かかる低分子化合物の体細胞への接触は、その低分子化合物を適当な濃度で水性もしくは非水性溶媒に溶解し、体細胞の培養に適した培地(例えば、約5~20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地など)中に、その低分子化合物の濃度が体細胞において核初期化が起こるのに十分で且つ細胞毒性がみられない範囲となるようにその低分子化合物溶液を添加して、細胞を一定期間培養することにより実施することができる。核初期化物質である低分子化合物の濃度は用いるその低分子化合物の種類によって異なるが、約0.1nM~約100nMの範囲で適宜選択することができる。接触期間は細胞の核初期化が達成されるのに十分な時間であれば特に制限はないが、通常は陽性コロニーが出現するまで培地に共存させておけばよい。
≪その他の工程≫
本実施形態のiPS細胞の製造方法は、上記導入工程に加えて、必要に応じて更にその他の工程を含み得る。その他の工程としては、本発明の効果を損なわない限り特に制限はなく、目的に応じて適宜選択することができる。例えば、核初期化物質及び品質改善剤を導入した体細胞(以下、単に「導入細胞」とも記載する。)を培養する工程(以下、単に「導入細胞培養工程」とも表示する。)などが挙げられる。
本実施形態のiPS細胞の製造方法は、上記導入工程に加えて、必要に応じて更にその他の工程を含み得る。その他の工程としては、本発明の効果を損なわない限り特に制限はなく、目的に応じて適宜選択することができる。例えば、核初期化物質及び品質改善剤を導入した体細胞(以下、単に「導入細胞」とも記載する。)を培養する工程(以下、単に「導入細胞培養工程」とも表示する。)などが挙げられる。
(導入細胞培養工程)
導入細胞培養工程は、導入細胞を培養する工程である。導入細胞の培養条件としては、特に限定されず、通常用いられる幹細胞の培養条件を用いることができる、例えば、培養条件としては、ES細胞の培養に適した条件を挙げることができる。かかる条件としては、例えば、培養温度は約37℃、CO2濃度は約2%~5%、O2濃度は約5%~20%などが挙げられる。導入細胞の培養に用いる培地は、特に限定されず、目的に応じて適宜選択することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、培養条件に応じてLIFもしくは塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加する。通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞(MEF)の共存下で培養される。MEFとしては、通常STO細胞等がよく使われるが、iPS細胞の誘導には、SNL細胞(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。フィーダー細胞との共培養は、核初期化物質及び品質改善剤の導入より前から開始してもよいし、該導入時から、あるいは該導入より後(例えば1~10日後)から開始してもよい。
導入細胞培養工程は、導入細胞を培養する工程である。導入細胞の培養条件としては、特に限定されず、通常用いられる幹細胞の培養条件を用いることができる、例えば、培養条件としては、ES細胞の培養に適した条件を挙げることができる。かかる条件としては、例えば、培養温度は約37℃、CO2濃度は約2%~5%、O2濃度は約5%~20%などが挙げられる。導入細胞の培養に用いる培地は、特に限定されず、目的に応じて適宜選択することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、培養条件に応じてLIFもしくは塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加する。通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞(MEF)の共存下で培養される。MEFとしては、通常STO細胞等がよく使われるが、iPS細胞の誘導には、SNL細胞(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。フィーダー細胞との共培養は、核初期化物質及び品質改善剤の導入より前から開始してもよいし、該導入時から、あるいは該導入より後(例えば1~10日後)から開始してもよい。
上記の導入細胞培養工程の期間としては、特に制限はなく、目的に応じて適宜選択することができる。
本実施形態のiPS細胞の製造方法では、核初期化物質及び品質改善剤が体細胞に導入された後、品質改善剤に含まれる制御配列により、H1fooタンパク質の細胞内での存在時期及び存在量の少なくとも一方を制御することができる。そのため、H1fooタンパク質の存在時期又は存在量を適切に制御することができ、その結果、品質の高いiPS細胞を得ることができる。
<第2実施形態>
一実施形態において、本発明は、核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含む、iPS細胞の製造方法を提供する。前記体細胞に導入された前記H1foo遺伝子から発現されるH1fooタンパク質は、前記体細胞内における存在時期及び存在量の少なくとも一方が制御される。
一実施形態において、本発明は、核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含む、iPS細胞の製造方法を提供する。前記体細胞に導入された前記H1foo遺伝子から発現されるH1fooタンパク質は、前記体細胞内における存在時期及び存在量の少なくとも一方が制御される。
≪導入工程≫
本実施形態の製造方法は、核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含む。
本実施形態の製造方法は、核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含む。
(核初期化物質)
核初期化物質は、上記「<第1実施形態>」で説明したものと同様である。好ましい核初期化物質も、上記「<第1実施形態>」で挙げたものと同様のものが挙げられる。
核初期化物質は、上記「<第1実施形態>」で説明したものと同様である。好ましい核初期化物質も、上記「<第1実施形態>」で挙げたものと同様のものが挙げられる。
(H1foo遺伝子)
H1foo遺伝子は、上記「[iPS細胞の品質改善剤]、<第1実施形態>(H1foo遺伝子)」で説明したものと同様である。好ましいH1foo遺伝子も、上記「[iPS細胞の品質改善剤]、<第1実施形態>(H1foo遺伝子)」で挙げたものと同様のものが挙げられる。
H1foo遺伝子は、上記「[iPS細胞の品質改善剤]、<第1実施形態>(H1foo遺伝子)」で説明したものと同様である。好ましいH1foo遺伝子も、上記「[iPS細胞の品質改善剤]、<第1実施形態>(H1foo遺伝子)」で挙げたものと同様のものが挙げられる。
本実施形態のiPS細胞の製造方法において、体細胞に導入されたH1foo遺伝子から発現されるH1fooタンパク質は、体細胞内における存在時期及び存在量の少なくとも一方が制御される。前記H1fooタンパク質の体細胞内での存在時期及び存在量を制御する方法は、特に限定されず、公知の方法を用いることができる。
H1fooタンパク質の存在時期及び存在量を制御する方法としては、好ましくは、「[iPS細胞の品質改善剤]、<第1実施形態>(制御配列)」で挙げた制御配列を用いる方法が挙げられる。例えば、H1foo遺伝子に不安定化ドメイン遺伝子を連結し、体細胞に導入されたときに、H1fooタンパク質と不安定化ドメインとの融合タンパク質として発現させる方法が挙げられる。この場合、当該融合タンパク質は、不安定化ドメインを有することにより、細胞内で生成後、プロテアソームによりすみやかに分解される。H1foo遺伝子と不安定化ドメイン遺伝子との融合遺伝子から、融合タンパク質が転写及び翻訳されて生成し、その生成後にプロテアソームに分解されるまでの時間は、例えば、6時間以内であり、好ましくは4時間以内であり、さらに好ましくは3時間以内であり、特に好ましくは2時間以内である。
融合タンパク質の存在時期及び存在量は、安定化物質の添加により制御してもよい。例えば、導入細胞の培地に安定化物質を添加したり、当該培地から細胞を回収して安定化物質を含まない培地に移したりすることにより、所望の時期に、融合タンパク質を細胞内に存在させることができる。また、安定化物質の添加量を調整することにより、融合タンパク質の細胞内での存在量を制御することができる。
融合タンパク質の存在時期及び存在量は、安定化物質の添加により制御してもよい。例えば、導入細胞の培地に安定化物質を添加したり、当該培地から細胞を回収して安定化物質を含まない培地に移したりすることにより、所望の時期に、融合タンパク質を細胞内に存在させることができる。また、安定化物質の添加量を調整することにより、融合タンパク質の細胞内での存在量を制御することができる。
あるいは、H1foo遺伝子の上流に刺激応答性プロモーターを連結し、体細胞に導入されたときに、H1foo遺伝子が当該刺激応答性プロモーターの制御下で発現されるようにしてもよい。この場合、導入細胞に、当該刺激応答性プロモーターが応答する「刺激」を与えることにより、H1fooタンパク質の存在時期及び存在量を制御することができる。刺激応答性プロモーターがテトラサイクリン応答性プロモーターである場合には、テトラサイクリン又はその誘導体(ドキシサイクリン等)を用いて、H1foo遺伝子の発現時期及び発現量を制御することができ、その結果としてH1fooタンパク質の導入細胞内での存在時期及び存在量を制御することができる。
(体細胞への導入)
核初期化物質及びH1foo遺伝子を体細胞に導入する方法としては、上記「<第1実施形態>」で挙げた方法と同様の方法が挙げられる。
核初期化物質及びH1foo遺伝子を体細胞に導入する方法としては、上記「<第1実施形態>」で挙げた方法と同様の方法が挙げられる。
<他の工程>
本実施形態のiPS細胞の製造方法は、上記導入工程に加えて、必要に応じて更にその他の工程を含み得る。その他の工程としては、本発明の効果を損なわない限り特に制限はなく、目的に応じて適宜選択することができる。その他の工程としては、例えば、上記「<第1実施形態>」と同様に、導入細胞培養工程などが挙げられる。導入細胞培養工程は、上記「<第1実施形態>」で挙げた方法と同様に行うことができる。
本実施形態のiPS細胞の製造方法は、上記導入工程に加えて、必要に応じて更にその他の工程を含み得る。その他の工程としては、本発明の効果を損なわない限り特に制限はなく、目的に応じて適宜選択することができる。その他の工程としては、例えば、上記「<第1実施形態>」と同様に、導入細胞培養工程などが挙げられる。導入細胞培養工程は、上記「<第1実施形態>」で挙げた方法と同様に行うことができる。
本実施形態のiPS細胞の製造方法は、導入細胞におけるH1fooタンパク質の存在量及び存在時期の少なくとも一方を制御する工程(以下、「H1fooタンパク質制御工程」という。)を含んでいてもよい。H1fooタンパク質の存在量及び存在時期の制御は、例えば、H1fooタンパク質が不安定化ドメインとの融合タンパク質として発現する場合には、安定化物質を用いて行うことができる。また、例えば、H1foo遺伝子が刺激応答性プロモーターの制御下で発現される場合には、当該応答性プロモーターが応答する刺激を導入細胞に与えることにより、H1fooタンパク質の存在量及び存在時期を制御することができる。
導入細胞内でのH1fooタンパク質の存在量及び存在時期は、iPS細胞を製造する際に、体細胞の初期化が完了するまでに最大発現量の50%以下、好ましくは30%以下、さらに好ましくは20%以下になるように制御される。さらに具体的には、細胞に導入されたH1fooタンパク質が、細胞内での発現が最大になってから24時間後に50%以下、好ましくは30%以下、さらに好ましくは20%以下となるように制御される。あるいは、核初期化物質の体細胞への導入後のH1foo遺伝子の発現持続期間は、前記導入後24時間以内であってもよい。あるいは、核初期化物質を導入した細胞におけるH1fooタンパク質の存在量は、前記導入後5日程度経過した時期、前記導入後のH1fooタンパク質の最大存在量と比較して、約50%未満であってもよい。
本実施形態のiPS細胞の製造方法では、H1fooタンパク質の細胞内での存在時期及び存在量の少なくとも一方を適切に制御することができる。その結果、品質の高いiPS細胞を得ることができる。
[iPS細胞]
一実施形態において、本発明は、上記実施形態のiPS細胞の製造方法により製造されるiPS細胞を提供する。
一実施形態において、本発明は、上記実施形態のiPS細胞の製造方法により製造されるiPS細胞を提供する。
iPS細胞は、体細胞から誘導される多能性幹細胞であり、分化多能性及び自己複製能を有する。分化多能性とは、三胚葉系列すべてに分化できることを意味する。また、前記自己複製能とは、未分化状態を保持したまま増殖できる能力を意味する。
上記実施形態のiPS細胞の製造方法により製造された細胞が、iPS細胞であるか否かの確認は、公知の方法により行うことができる。例えば、未分化マーカーの発現、胚様体形成能、及び/又はキメラ形成能等により、iPS細胞であるかを確認することができる。
上記実施形態のiPS細胞の製造方法により製造された細胞は、核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を含むiPS細胞であってもよい。上記実施形態のiPS細胞の製造方法により製造された細胞は、核初期化物質と、H1foo遺伝子と、制御配列と、を含む細胞であってもよい。上記実施形態のiPS細胞の製造方法により製造された細胞は、核初期化物質と、H1foo遺伝子と制御配列(例、不安定化ドメイン遺伝子)との融合遺伝子と、を含む細胞であってもよい。上記実施形態のiPS細胞の製造方法により製造された細胞は、核初期化物質と、刺激応答性プロモーターに機能的に連結されたH1foo遺伝子とを含む細胞であってもよい。
上記実施形態の製造方法で製造されたiPS細胞は、導入細胞において、H1fooタンパク質の存在時期及び存在量を適切に制御されるため、従来の方法で製造されたiPS細胞よりも品質の高いiPS細胞である。特に、本実施形態のiPS細胞は、Tra-1-60を発現するコロニー形成数が向上している。また、ナイーブ型iPS細胞の生成能が向上している。ナイーブ型iPS細胞であることは、コロニーがナイーブ型特異的なドーム型の形状をしていることと、ナイーブ型に特異的な遺伝子を高発現していることにより確認することができる。また、目的細胞(例えば、心筋細胞、原始内胚葉細胞)への分化能が向上している。さらに、上記実施形態の製造方法で製造されたiPS細胞の細胞集団は、遺伝子発現にばらつきのない均一な細胞集団であり得る。
[iPS細胞製造用組成物]
一実施形態において、本発明は、iPS細胞製造用組成物を提供する。本実施形態のiPS細胞製造用組成物は、核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を含む。
一実施形態において、本発明は、iPS細胞製造用組成物を提供する。本実施形態のiPS細胞製造用組成物は、核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を含む。
核初期化物質は、上記「[iPS細胞の製造方法]<第1実施形態>」で説明したものと同様である。好ましい核初期化物質も、上記「<第1実施形態>」で挙げたものと同様のものが挙げられる。
iPS細胞の品質改善剤は、上記「[iPS細胞の品質改善剤]」で説明したものと同様である。好ましい品質改善剤も、上記「[iPS細胞の品質改善剤]」で挙げたものと同様のものが挙げられる。
iPS細胞の品質改善剤は、上記「[iPS細胞の品質改善剤]」で説明したものと同様である。好ましい品質改善剤も、上記「[iPS細胞の品質改善剤]」で挙げたものと同様のものが挙げられる。
本実施形態のiPS細胞製造用組成物において、各核初期化物質及び品質改善剤の量は、特に限定されず、全て等量であってもよく、それぞれ異なる量であってもよい。例えば、核初期化物質として、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c-Myc遺伝子若しくはL-Myc遺伝子を用いる場合、Oct3/4遺伝子の量が、Sox2遺伝子、Klf4遺伝子、又はc-Myc遺伝子若しくはL-Myc遺伝子に対して多量、例えば約3倍量となるようにしてもよい。
本実施形態のiPS細胞製造用組成物は、核初期化物質及び品質改善剤に加えて、他の成分を含んでいてもよい。他の成分としては、特に限定されないが、例えば、バッファー、遺伝子導入試薬、タンパク質導入試薬等が挙げられる。
[他の実施形態]
本発明は、他の実施形態として、上記実施形態のiPS細胞の品質改善剤を体細胞に導入する工程を含む、iPS細胞の品質の改善方法を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、H1foo遺伝子と不安定化ドメイン遺伝子との融合遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、H1fooタンパク質と不安定化ドメインとの融合タンパク質の使用を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、刺激応答性プロモーターの下流に連結されたH1foo遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及びH1foo遺伝子と不安定化ドメインとの融合遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及びH1fooタンパク質と不安定化ドメインとの融合タンパク質の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及び刺激応答性プロモーターの下流に連結されたH1foo遺伝子の使用を提供する。
本発明は、他の実施形態として、核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を含むiPS細胞製造用キットを提供する。核初期化物質、及び品質改善剤は、個別の容器に分けられていてもよく、1つの容器にまとめられていてもよく、任意の数ごとに容器にまとめられていてもよい。また、遺伝子導入試薬、タンパク質導入試薬、パッケージング細胞、バッファー、希釈液等を含んでいてもよい。
本発明は、他の実施形態として、上記実施形態のiPS細胞の品質改善剤を体細胞に導入する工程を含む、iPS細胞の品質の改善方法を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、H1foo遺伝子と不安定化ドメイン遺伝子との融合遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、H1fooタンパク質と不安定化ドメインとの融合タンパク質の使用を提供する。
本発明は、他の実施形態として、iPS細胞の品質改善剤の製造における、刺激応答性プロモーターの下流に連結されたH1foo遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及びH1foo遺伝子と不安定化ドメインとの融合遺伝子の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及びH1fooタンパク質と不安定化ドメインとの融合タンパク質の使用を提供する。
本発明は、他の実施形態として、iPS細胞製造用組成物の製造における、核初期化物質、及び刺激応答性プロモーターの下流に連結されたH1foo遺伝子の使用を提供する。
本発明は、他の実施形態として、核初期化物質と、上記実施形態のiPS細胞の品質改善剤と、を含むiPS細胞製造用キットを提供する。核初期化物質、及び品質改善剤は、個別の容器に分けられていてもよく、1つの容器にまとめられていてもよく、任意の数ごとに容器にまとめられていてもよい。また、遺伝子導入試薬、タンパク質導入試薬、パッケージング細胞、バッファー、希釈液等を含んでいてもよい。
以下に実施例により本発明を詳細に説明するが、本発明はこれらの実施例に限定されるものではない。以下の全ての実験は、慶應義塾大学および京都大学の動物及びDNA実験指針にしたがって行い、慶應義塾大学および京都大学の倫理委員会によって承認され、アメリカ国立衛生研究所の実験動物の管理と使用に関する指針に準拠している。
[実施例1]H1FOO遺伝子を含むセンダイウイルスベクターの構築
センダイウイルスベクター(SeV18;IDファーマ社製)とProteoTunerTM Shield System(Clontech社製)を用いて、H1FOO cDNA(Teranishi, T., et al. Rapid replacement of somatic linker histones with the oocyte-specific linker histone H1foo in nuclear transfer. Developmental Biology 266, 76-86 (2004).)を含む、3種のセンダイウイルスベクターを作製した(図1)。
(a)SeV18+H1FOO/TS15ΔFは、ProteoTunerTM Shield Systemの不安定化ドメイン(Destabilized domein:DD)を含まないベクターである。
(b)SeV18+H1FOO-DD/TS15ΔFは、H1FOO cDNAの3’末端にDDコード配列を付加した配列を含むベクターである。(b)のベクターは、H1FOOのC末端にDDが付加された融合タンパク質を発現し、H1FOOの分解が促進される。
(c)SeV18+DD-H1FOO/TS15ΔFは、H1FOO cDNAの5’末端にDDコード配列を付加した配列を含むベクターである。(c)のベクターは、H1FOOのN末端にDDが付加された融合タンパク質を発現し、H1FOOの分解が促進される。
センダイウイルスベクター(SeV18;IDファーマ社製)とProteoTunerTM Shield System(Clontech社製)を用いて、H1FOO cDNA(Teranishi, T., et al. Rapid replacement of somatic linker histones with the oocyte-specific linker histone H1foo in nuclear transfer. Developmental Biology 266, 76-86 (2004).)を含む、3種のセンダイウイルスベクターを作製した(図1)。
(a)SeV18+H1FOO/TS15ΔFは、ProteoTunerTM Shield Systemの不安定化ドメイン(Destabilized domein:DD)を含まないベクターである。
(b)SeV18+H1FOO-DD/TS15ΔFは、H1FOO cDNAの3’末端にDDコード配列を付加した配列を含むベクターである。(b)のベクターは、H1FOOのC末端にDDが付加された融合タンパク質を発現し、H1FOOの分解が促進される。
(c)SeV18+DD-H1FOO/TS15ΔFは、H1FOO cDNAの5’末端にDDコード配列を付加した配列を含むベクターである。(c)のベクターは、H1FOOのN末端にDDが付加された融合タンパク質を発現し、H1FOOの分解が促進される。
上記(a)~(c)のベクター中の各配列を以下の配列番号に示す。
DDコード配列:配列番号13
H1FOO-DDコード配列:配列番号15
DD-H1FOOコード配列:配列番号17
また、上記ベクター(a)~(c)の作製に用いたプライマー配列を表1に示す。
DDコード配列:配列番号13
H1FOO-DDコード配列:配列番号15
DD-H1FOOコード配列:配列番号17
また、上記ベクター(a)~(c)の作製に用いたプライマー配列を表1に示す。

[実施例2]H1FOOタンパク質のウエスタンブロッティング
H1FOO遺伝子を含む上記(a)~(c)の各センダイウイルスベクターをヒト皮膚線維芽細胞(TIG120株、東京都老人総合研究所より譲渡、Kondo et al., Exp Cell Res. 1995 Oct;220(2):501-4)に感染多重度(MOI)3でそれぞれ導入し、5日間培養した後、各細胞を回収し、抗H1FOO抗体(HPA037992;Sigma-Aldrich社製)を用いて、ウエスタンブロッティングを行った。
結果を図2に示す。図2中、(1)~(4)は、(1)H1FOOを含まないControl、(2)H1FOO(ベクター(a))、(3)H1FOO-DD(ベクター(b))、及び(4)DD-H1FOO(ベクター(c))の各ベクターをそれぞれ導入した各ヒト皮膚線維芽細胞における結果をそれぞれ示す。5日後においてなお、(2)H1FOOでは、H1FOOタンパク質の発現量が著明に高かった。一方、(3)H1FOO-DD、及び(4)DD-H1FOOにおいては、H1FOO-DD融合タンパク質又はDD-H1FOO融合蛋白質の発現量が低下していた。
(1)H1FOOを含まないControlには、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクターを用いた。
H1FOO遺伝子を含む上記(a)~(c)の各センダイウイルスベクターをヒト皮膚線維芽細胞(TIG120株、東京都老人総合研究所より譲渡、Kondo et al., Exp Cell Res. 1995 Oct;220(2):501-4)に感染多重度(MOI)3でそれぞれ導入し、5日間培養した後、各細胞を回収し、抗H1FOO抗体(HPA037992;Sigma-Aldrich社製)を用いて、ウエスタンブロッティングを行った。
結果を図2に示す。図2中、(1)~(4)は、(1)H1FOOを含まないControl、(2)H1FOO(ベクター(a))、(3)H1FOO-DD(ベクター(b))、及び(4)DD-H1FOO(ベクター(c))の各ベクターをそれぞれ導入した各ヒト皮膚線維芽細胞における結果をそれぞれ示す。5日後においてなお、(2)H1FOOでは、H1FOOタンパク質の発現量が著明に高かった。一方、(3)H1FOO-DD、及び(4)DD-H1FOOにおいては、H1FOO-DD融合タンパク質又はDD-H1FOO融合蛋白質の発現量が低下していた。
(1)H1FOOを含まないControlには、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクターを用いた。
[実施例3]プライム型ヒトiPS細胞の作製と培養条件
ヒトiPS細胞の作製は、文献(Ban, H., Nishishita, N., Fusaki, N., Tabata, T., Saeki, K., Shikamura, M., Takada, N., Inoue, M., Hasegawa, M., Kawamata, S., Nishikawa, S., Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors, Proc Natl Acad Sci U S A 108, 14234-9, 2011およびSeki, T., Yuasa, S., Fukuda, K., Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus, Nature Protocols 7, 718-728, 2012)に記載されたプロトコールにしたがって行った。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(a)~(c)のいずれかのセンダイウイルスベクターを用いて、ヒト皮膚線維芽細胞(TIG120株)および末梢血単核球(品番:CTL-UP1、Lot:HHU20140120、Cellular Technology Ltd.)からプライム型iPS細胞を作製した(感染多重度0.1~0.3で各細胞に感染させた)。コントロールとして、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクター用いて、iPS細胞(Control-iPS)を作製した。なお、本実験は慶應義塾大学および京都大学の遺伝子組み換え実験指針にしたがって行った。
ヒトiPS細胞の作製は、文献(Ban, H., Nishishita, N., Fusaki, N., Tabata, T., Saeki, K., Shikamura, M., Takada, N., Inoue, M., Hasegawa, M., Kawamata, S., Nishikawa, S., Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors, Proc Natl Acad Sci U S A 108, 14234-9, 2011およびSeki, T., Yuasa, S., Fukuda, K., Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus, Nature Protocols 7, 718-728, 2012)に記載されたプロトコールにしたがって行った。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(a)~(c)のいずれかのセンダイウイルスベクターを用いて、ヒト皮膚線維芽細胞(TIG120株)および末梢血単核球(品番:CTL-UP1、Lot:HHU20140120、Cellular Technology Ltd.)からプライム型iPS細胞を作製した(感染多重度0.1~0.3で各細胞に感染させた)。コントロールとして、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクター用いて、iPS細胞(Control-iPS)を作製した。なお、本実験は慶應義塾大学および京都大学の遺伝子組み換え実験指針にしたがって行った。
上記のように作製したヒトiPS細胞は、StemFit AK02N(味の素社製)培養液(以下、「プライム型ヒトiPS細胞培養液」という。)中で、iMatrix-511(ニッピ社製)でコーティングした培養皿上で培養及び維持した。プライム型ヒトiPS細胞の培養液は2日毎に交換し、細胞は5~7日毎にStemPro Accutase(Gibco社製)を用いて継代した。
[実施例4]ナイーブ型ヒトiPS細胞の作製と培養条件
ヒトiPS細胞の作製は、文献(Ban, H. et al., Proc Natl Acad Sci U S A 108, 14234-9, 2011; Seki, T. et al., Nature Protocols 7, 718-728, 2012; Liu, X et al., Nature Methods 14, 1055-1062, 2017)に記載されたプロトコールにしたがって行った。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(a)~(c)のいずれかのセンダイウイルスベクターを用いて、ヒト皮膚線維芽細胞およびヒト末梢血単核球からナイーブ型iPS細胞を作製した(感染多重度0.1~0.3で各細胞に感染させた)。ナイーブ型iPS細胞であることは、コロニーがナイーブ型特異的なドーム型の形状をしていることと、ナイーブ型に特異的な遺伝子を高発現していること(KLF17、TFCP2L1、PRDM14、およびDPPA3遺伝子について、定量的PCRおよび免疫染色法にて確認)により確認した。コントロールとして、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクター用いて、iPS細胞(Control-iPS)を作製した。本実験は慶應義塾大学および京都大学の遺伝子組み換え実験指針にしたがって行った。
ヒトiPS細胞の作製は、文献(Ban, H. et al., Proc Natl Acad Sci U S A 108, 14234-9, 2011; Seki, T. et al., Nature Protocols 7, 718-728, 2012; Liu, X et al., Nature Methods 14, 1055-1062, 2017)に記載されたプロトコールにしたがって行った。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(a)~(c)のいずれかのセンダイウイルスベクターを用いて、ヒト皮膚線維芽細胞およびヒト末梢血単核球からナイーブ型iPS細胞を作製した(感染多重度0.1~0.3で各細胞に感染させた)。ナイーブ型iPS細胞であることは、コロニーがナイーブ型特異的なドーム型の形状をしていることと、ナイーブ型に特異的な遺伝子を高発現していること(KLF17、TFCP2L1、PRDM14、およびDPPA3遺伝子について、定量的PCRおよび免疫染色法にて確認)により確認した。コントロールとして、上記(a)のベクターにおいてH1FOO遺伝子に代えてAzami-Green遺伝子を含むセンダイウイルスベクター用いて、iPS細胞(Control-iPS)を作製した。本実験は慶應義塾大学および京都大学の遺伝子組み換え実験指針にしたがって行った。
上記のように作製したヒトiPS細胞は、NDiff227(タカラバイオ社製)に、CHIR99021(Sigma-Aldrich社製)、PD0325901(Sigma-Aldrich社製)、Go6983(Sigma-Aldrich社製)、及びHuman recombinant leukemia inhibitory factor(富士フィルム和光純薬社製)を添加した培養液(以下、「ナイーブ型ヒトiPS細胞培養液」という。)中で、放射線照射後マウス胎児線維芽細胞を播種した培養皿上で培養及び維持した。ナイーブ型ヒトiPS細胞の培養液は2日毎に交換し、細胞は3~4日毎にStemPro Accutase(Gibco社製)を用いて継代した。酸素濃度を調節できるインキュベーターを用いて酸素濃度5%の低酸素環境下で培養および継代を行った。
[実施例5]ALP陽性iPS細胞コロニー数の計測によるiPS細胞樹立効率の比較
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、(2)H1FOO-iPS、(3)H1FOO-DD-iPS、及び(4)DD-H1FOO-iPSを作製後、プライム型ヒトiPS細胞培養液中で15日間培養し、ALP陽性のコロニー数を測定した。ALPは幹細胞マーカーとして知られている。
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、(2)H1FOO-iPS、(3)H1FOO-DD-iPS、及び(4)DD-H1FOO-iPSを作製後、プライム型ヒトiPS細胞培養液中で15日間培養し、ALP陽性のコロニー数を測定した。ALPは幹細胞マーカーとして知られている。
結果を図3に示す。(3)H1FOO-DD-iPSおよび(4)DD-H1FOO-iPSは、(1)Control-iPSに比べ、ALP陽性細胞コロニー数が有意に増加することが示された。
次に元の細胞種による効果の違いを検証するためヒト末梢血単核球からプライム型の(1)Control-iPSおよび(3)H1FOO-DD-iPS細胞を作製し、プライム型ヒトiPS細胞培養液中で15日間培養し、ALP陽性のコロニー数を測定した。更にナイーブ型iPS細胞の樹立効率を比較するため、上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞およびヒト末梢血単核球からナイーブ型の(1)Control-iPSおよび(3)H1FOO-DD-iPS細胞をそれぞれ作製し、ナイーブ型ヒトiPS細胞培養液中で15日間培養し、ALP陽性のコロニー数を測定した。
結果を図4A~4Cに示す。図4A(ヒト末梢血単核球から樹立したプライム型ヒトiPS細胞)、図4B(ヒト皮膚線維芽細胞から樹立したナイーブ型ヒトiPS細胞)、及び図4C(ヒト末梢血単核球から樹立したナイーブ型ヒトiPS細胞)のいずれにおいても、(3)H1FOO-DD-iPSでは、(1)Control-iPSに比べ、図3の結果と同様にALP陽性細胞コロニー数が有意に増加することが示された。
以上の結果より、元の細胞種および樹立するiPS細胞の種類を問わず、H1FOO-DDはヒトiPS細胞の樹立効率を改善することが示された。
以上の結果より、元の細胞種および樹立するiPS細胞の種類を問わず、H1FOO-DDはヒトiPS細胞の樹立効率を改善することが示された。
[実施例6]遺伝子発現量のばらつきの比較
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。プライム型ヒトiPS細胞培養液中で約210日間(28継代)培養し、(1)及び(3)について各8クローンを選択して、RNA-seqを行った。RNA-seqのデータ解析結果に基づき、(1)の8クローンにおいて、遺伝子の発現量の平均絶対誤差(mean absolute error: MAE)が2.0よりも大きい(ばらつきが大きい)遺伝子の数を得た。同様の方法で、(3)の8クローンにおいても、MAEが2.0よりも大きい遺伝子の数を得た。その結果を図5Aに示した。
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。プライム型ヒトiPS細胞培養液中で約210日間(28継代)培養し、(1)及び(3)について各8クローンを選択して、RNA-seqを行った。RNA-seqのデータ解析結果に基づき、(1)の8クローンにおいて、遺伝子の発現量の平均絶対誤差(mean absolute error: MAE)が2.0よりも大きい(ばらつきが大きい)遺伝子の数を得た。同様の方法で、(3)の8クローンにおいても、MAEが2.0よりも大きい遺伝子の数を得た。その結果を図5Aに示した。
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。ナイーブ型ヒトiPS細胞培養液中で約190日間(45継代)培養し、(1)及び(3)について各8クローンを選択して、RNA-seqを行った。上記プライム型ヒトiPS細胞と同様の方法で、(1)及び(3)の各8クローンにおいて、MAEが2.0よりも大きい遺伝子の数を得た。その結果を図5Bに示した。
プライム型及びナイーブ型のいずれのヒトiPS細胞においても、(3)H1FOO-DD-iPSは、(1)Control-iPSと比較して、クローン間で発現量のばらつきの大きい遺伝子の数が半分程度に抑制されていた。これらの結果は、H1FOO-DD-iPSでは、グローバルな遺伝子発現がクローン間でより均一であることを示す。
[実施例7]DNAメチル化のばらつきの比較
上記実施例6で選択した、プライム型ヒトiPS細胞の(1)Control-iPSの8クローン及び(3)H1FOO-DD-iPSの8クローンについて、DNAメチル化アレイ(Infinium Human Methylation 450K BeadChip Kit、Illumina)を用いて、DNAメチル化の解析を行った。DNAメチル化アレイによる解析結果に基づき、(1)の8クローンにおいて、平均絶対誤差(mean absolute error: MAE)が2.0よりも大きい(ばらつきが大きい)メチル化プローブの数を得た。同様の方法で、(3)の8クローンにおいても、MAEが2.0よりも大きい遺伝子の数を得た。その結果を図6Aに示した。
上記実施例6で選択した、プライム型ヒトiPS細胞の(1)Control-iPSの8クローン及び(3)H1FOO-DD-iPSの8クローンについて、DNAメチル化アレイ(Infinium Human Methylation 450K BeadChip Kit、Illumina)を用いて、DNAメチル化の解析を行った。DNAメチル化アレイによる解析結果に基づき、(1)の8クローンにおいて、平均絶対誤差(mean absolute error: MAE)が2.0よりも大きい(ばらつきが大きい)メチル化プローブの数を得た。同様の方法で、(3)の8クローンにおいても、MAEが2.0よりも大きい遺伝子の数を得た。その結果を図6Aに示した。
上記実施例6で選択した、ナイーブ型ヒトiPS細胞の(1)Control-iPSの8クローン及び(3)H1FOO-DD-iPSの8クローンについて、DNAメチル化アレイを用いて、DNAメチル化の解析を行った。上記プライム型ヒトiPS細胞と同様の方法で、(1)及び(3)の各8クローンにおいて、MAEが2.0よりも大きいメチル化プローブの数を得た。その結果を図6Bに示した。
プライム型及びナイーブ型のいずれのヒトiPS細胞においても、(3)H1FOO-DD-iPSは、(1)Control-iPSと比較して、クローン間でばらつきの大きいメチル化プローブの数が半分程度に抑制されていた。これらの結果は、H1FOO-DD-iPSでは、DNAメチル化がクローン間でより均一であることを示す。
[実施例8]心筋細胞分化能の比較
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各3クローンを選択して、心筋分化誘導培地で培養し、心筋細胞への分化誘導を行った(Tohyama, S., et al. (2013). Cell Stem Cell 12(1): 127-137.)。心筋分化誘導培地での培養開始から10日目に、細胞を回収した。回収した細胞におけるTNNT2の発現を、フローサイトメーターを用いて検出し、TNN2陽性細胞の割合を算出した。TNNT2は、代表的な心筋細胞マーカーである。各クローンについて、それぞれ6回ずつ心筋細胞への分化誘導試験を行った。
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各3クローンを選択して、心筋分化誘導培地で培養し、心筋細胞への分化誘導を行った(Tohyama, S., et al. (2013). Cell Stem Cell 12(1): 127-137.)。心筋分化誘導培地での培養開始から10日目に、細胞を回収した。回収した細胞におけるTNNT2の発現を、フローサイトメーターを用いて検出し、TNN2陽性細胞の割合を算出した。TNNT2は、代表的な心筋細胞マーカーである。各クローンについて、それぞれ6回ずつ心筋細胞への分化誘導試験を行った。
図7Aは、フローサイトメーターによるTNNT2陽性細胞の割合(%)の測定例を示す。左図は(1)Control-iPSの測定例であり、右図は(3)H1FOO-DD-iPSの測定例である。(1)Control-iPSでは、全細胞の約6%のみTNNT2陽性であり、心筋細胞への分化が不良であった。(3)H1FOO-DD-iPSでは、全細胞の約95%がTNNT2強陽性であり、良好な心筋分化能を示した。
図7Bは、各クローンにおけるTNNT2陽性細胞の割合(%)を示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、TNNT2陽性細胞の割合が高かった。また、(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、各心筋細胞分化誘導試験間でばらつきが小さく、より安定して心筋細胞を分化誘導できることが確認された。これらの結果は、H1FOO-DD-iPSが、優れた心筋細胞分化能を有し、且つより均一性の高い心筋細胞を分化できることを示す。
[実施例9]内胚葉分化能の比較
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各8クローンを選択して、STEMdiff Trilineage diffrerentiation kit(Cat:ST-05230、VERITAS)による三胚葉成分への分化誘導を行った。分化誘導開始から5日目に、細胞を回収した。回収した細胞において、TaqMan hPSC Scorecard kit(Cat:A15876、ThermoFisher SCIENTIFIC)を用いて半網羅的に内胚葉に関連するマーカーの発現をqPCRで評価した。
上記実施例3に記載の方法に従って、ヒト皮膚線維芽細胞からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各8クローンを選択して、STEMdiff Trilineage diffrerentiation kit(Cat:ST-05230、VERITAS)による三胚葉成分への分化誘導を行った。分化誘導開始から5日目に、細胞を回収した。回収した細胞において、TaqMan hPSC Scorecard kit(Cat:A15876、ThermoFisher SCIENTIFIC)を用いて半網羅的に内胚葉に関連するマーカーの発現をqPCRで評価した。
結果を図8に示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、Algorithmic scoreが高かった。また、(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、各クローン間でのばらつきが小さく、より安定して内胚葉を分化誘導できることが確認された。これらの結果は、H1FOO-DD-iPSが、優れた内胚葉分化能を有し、且つより均一性の高い内胚葉に分化できることを示す。
[実施例10]肝細胞分化能の比較(1)
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各10クローンを選択して、肝細胞(内胚葉系の終末分化細胞の一種)への分化誘導を行った(Kajiwara, M., et al. (2012). Proc Natl Acad Sci U S A 109(31): 12538-12543.; Takebe, T., et al. (2017). Cell Rep 21(10): 2661-2670.)。分化誘導開始から21日目に、細胞を回収した。回収した細胞において、代表的な成熟肝細胞マーカーの一つであるASGR1をqRT-PCRで定量評価した。
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各10クローンを選択して、肝細胞(内胚葉系の終末分化細胞の一種)への分化誘導を行った(Kajiwara, M., et al. (2012). Proc Natl Acad Sci U S A 109(31): 12538-12543.; Takebe, T., et al. (2017). Cell Rep 21(10): 2661-2670.)。分化誘導開始から21日目に、細胞を回収した。回収した細胞において、代表的な成熟肝細胞マーカーの一つであるASGR1をqRT-PCRで定量評価した。
結果を図9に示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、全体的にAGSR1の発現量が高く、平均値(Median)及び中央値(Average)が高かった。また、(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、各クローン間でのばらつき(STDVP、CV)が小さく、より安定して肝細胞を分化誘導できることが確認された。これらの結果は、H1FOO-DD-iPSが、優れた肝細胞分化能を有し、且つより均一性の高い肝細胞に分化できることを示す。
[実施例11]肝細胞分化能の比較(2)
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各10クローンを選択して、実施例11と同様に、肝細胞への分化誘導を行った。分化誘導開始から21日目に、培養上清を回収した。回収した培養上清において、ELISAにより分泌Albuminの量を定量評価した。成熟肝細胞はAlbuminを産生して分泌するため、AlbuminはASGR1と同様に、成熟肝細胞のマーカーとして使用される。
上記実施例3に記載の方法に従って、ヒト末梢血単核球からプライム型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各10クローンを選択して、実施例11と同様に、肝細胞への分化誘導を行った。分化誘導開始から21日目に、培養上清を回収した。回収した培養上清において、ELISAにより分泌Albuminの量を定量評価した。成熟肝細胞はAlbuminを産生して分泌するため、AlbuminはASGR1と同様に、成熟肝細胞のマーカーとして使用される。
結果を図10に示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、全体的にAlbuminの分泌量が高く、中央値(Median)が高かった。また、(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、各クローン間でのばらつき(STDVP、CV)が小さく、より安定して肝細胞を分化誘導できることが確認された。これらの結果は、H1FOO-DD-iPSが、優れた肝細胞分化能を有し、且つより均一性の高い肝細胞に分化できることを示す。
[実施例12]原始内胚葉分化能の比較
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞およびヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各6クローン(ヒト皮膚線維芽細胞由来:4クローン、ヒト末梢血単核球由来:2クローン)を選択して、原始内胚葉分化誘導培地で培養し、原始内胚葉への分化誘導を行った(国際公開第2019/093340号)。原始内胚葉分化誘導培地での培養開始から3日目に、細胞を回収した。回収した細胞におけるPDGFRA及びANPEPの発現を、フローサイトメーターを用いて検出し、PDGFRA陽性細胞及びANPEP陽性細胞の割合を算出した。PDGFRA及びANPEPは、代表的な原始内胚葉マーカーである。各クローンについて、それぞれ3回ずつ原始内胚葉への分化誘導試験を行った。
原始内胚葉分化誘導培地は、N2B27培地(NDiff227)に、25ng/mLのFGF4(Peprotech)、1μg/mLのHeparin(Wako)、10ng/mLのBMP4(R&D)、10ng/mLのPDGFRA(Peprotech)、1μMのXAV939(Wako)、3μMのA83-01(Wako)、及び0.1μMのRetinoic Acid(Sigma)を添加して、調製した。分化誘導開始後2日目に、10ng/mLのIL-6(R&D)を添加した。
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞およびヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各6クローン(ヒト皮膚線維芽細胞由来:4クローン、ヒト末梢血単核球由来:2クローン)を選択して、原始内胚葉分化誘導培地で培養し、原始内胚葉への分化誘導を行った(国際公開第2019/093340号)。原始内胚葉分化誘導培地での培養開始から3日目に、細胞を回収した。回収した細胞におけるPDGFRA及びANPEPの発現を、フローサイトメーターを用いて検出し、PDGFRA陽性細胞及びANPEP陽性細胞の割合を算出した。PDGFRA及びANPEPは、代表的な原始内胚葉マーカーである。各クローンについて、それぞれ3回ずつ原始内胚葉への分化誘導試験を行った。
原始内胚葉分化誘導培地は、N2B27培地(NDiff227)に、25ng/mLのFGF4(Peprotech)、1μg/mLのHeparin(Wako)、10ng/mLのBMP4(R&D)、10ng/mLのPDGFRA(Peprotech)、1μMのXAV939(Wako)、3μMのA83-01(Wako)、及び0.1μMのRetinoic Acid(Sigma)を添加して、調製した。分化誘導開始後2日目に、10ng/mLのIL-6(R&D)を添加した。
図11Aは、フローサイトメーターによるPDGFRA陽性細胞及びANPEP陽性細胞の割合(%)の測定例を示す。左図は(1)Control-iPSの測定例である。PDGFRA及びANPEPの両方が陽性である細胞の割合は、全細胞の約19%であった。
右図は(3)H1FOO-DD-iPSの測定例である。PDGFRA及びANPEPの両方が陽性である細胞の割合は、全細胞の約37%であった。
右図は(3)H1FOO-DD-iPSの測定例である。PDGFRA及びANPEPの両方が陽性である細胞の割合は、全細胞の約37%であった。
図11Bは、各クローンにおける、PDGFRA及びANPEPの両方が陽性である細胞の割合(%)を示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、陽性細胞の割合が高かった。また、(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、各クローン間でのばらつきが小さく、より安定して原始内胚葉を分化誘導できることが確認された。これらの結果は、H1FOO-DD-iPSが、優れた原始内胚葉分化能を有し、且つより均一性の高い原始内胚葉に分化できることを示す。
[実施例13]代謝機能の検証
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞及びヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各6クローンを選択して、細胞外フラックスアナライザー(Agilent Seahorse XF96 Extracellular Flux Analyzer、プライムテック株式会社)を用いて、予備呼吸能(Spare respiratory capacity;電子伝達系の機能評価)を測定した。
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞及びヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各6クローンを選択して、細胞外フラックスアナライザー(Agilent Seahorse XF96 Extracellular Flux Analyzer、プライムテック株式会社)を用いて、予備呼吸能(Spare respiratory capacity;電子伝達系の機能評価)を測定した。
結果を図12Aに示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、予備呼吸能が高かった。
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞又はヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各6クローンを選択して、細胞外フラックスアナライザー(Agilent Seahorse XF96 Extracellular Flux Analyzer、プライムテック株式会社)を用いて、酸素消費速度(OCR、電子伝達系の機能評価)及び細胞外酸性化速度(ECAR、解糖系の機能評価)を測定した。
結果を図12Bに示す。X軸はECARを示し、嫌気性代謝能を反映する。Y軸はOCRを示し、好気性代謝能を反映する。各プロットは、1つのクローンで6回測定した平均値を示す。(3)H1FOO-DD-iPSでは、(1)Control-iPSと比較して、全体にプロットが右上に存在し、ECAR及びOCRのいずれも高い状態にあった。これらの結果は、H1FOO-DD-iPSが、よりナイーブ型の代謝能を有することを示す。
[実施例14]X染色体活性化の有無の検証
女性由来の体細胞及びプライム型iPS/ES細胞では原則として片方のX染色体は不活性化されている。一方、ナイーブ型、特に着床前エピブラストでは、両方のX染色体が活性化型となっていることが知られている。これについて検証するため、ナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSの各4クローンについて、mRNA-FISHを行い、3つのマーカーの発現を比較検証した。
女性由来の体細胞及びプライム型iPS/ES細胞では原則として片方のX染色体は不活性化されている。一方、ナイーブ型、特に着床前エピブラストでは、両方のX染色体が活性化型となっていることが知られている。これについて検証するため、ナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSの各4クローンについて、mRNA-FISHを行い、3つのマーカーの発現を比較検証した。
上記実施例4に記載の方法に従って、ヒト皮膚線維芽細胞及びヒト末梢血単核球からナイーブ型の(1)Control-iPS、及び(3)H1FOO-DD-iPSを作製した。(1)及び(3)について各4クローンを選択して、UTX、HUWE1、及びXISTの発現をmRNA-FISHにより確認した(Sahakyan, A., et al. (2017). Cell Stem Cell 20(1): 87-101.)。
現在得られるナイーブ型ヒトiPS細胞は多倍体細胞が多数発生するため、正しい評価のために染色体数が正常な細胞を選ぶ必要がある。UTXはX染色体活性化の有無に関係なく発現している。そこで、UTXを2つ発現している細胞を正常な細胞として選んだ。次いで、HUWE1(X染色体が活性化していると発現する)、及びXIST(本来はX染色体不活性化に関与するが、着床前エピブラストでは両側X染色体に発現することが知られている)の発現を、mRNA-FISHにより調べた。
現在得られるナイーブ型ヒトiPS細胞は多倍体細胞が多数発生するため、正しい評価のために染色体数が正常な細胞を選ぶ必要がある。UTXはX染色体活性化の有無に関係なく発現している。そこで、UTXを2つ発現している細胞を正常な細胞として選んだ。次いで、HUWE1(X染色体が活性化していると発現する)、及びXIST(本来はX染色体不活性化に関与するが、着床前エピブラストでは両側X染色体に発現することが知られている)の発現を、mRNA-FISHにより調べた。
図13Aに、mRNA-FISHの蛍光顕微鏡画像の例を示す。左図は、HUWEI+/+XIST+/+の例である。右図は、HUWEI+/-XIST-/-の例である。
図13Bに、各クローンの100細胞について、HUWE1及びXISTの発現パターンをまとめた結果を示す。図13B中、棒グラフの下の数字は、(1)Control-iPS、及び(3)H1FOO-DD-iPSにおけるクローンNo.を示す。クローンNo.1及び2は皮膚線維芽細胞由来であり、クローンNo.5及び6はヒト末梢血単核球由来である。
最も着床前エピブラストに近い表現型はHUWE1+/+且つXIST+/+であり、それに準ずる表現型はHUWE1+/+且つXIST+/-である(1)Control-iPSでは、プライム型に近い表現型を示す細胞が多数を占めたクローンがあった(クローン1、2)。一方、(3)H1FOO-DD-iPSでは、全てのクローンにおいて、ナイーブ型の表現型を示す細胞が多数を占めた。これらの結果は、H1FOO-DD-iPSが、よりナイーブ型に近い表現型を有することを示す。
最も着床前エピブラストに近い表現型はHUWE1+/+且つXIST+/+であり、それに準ずる表現型はHUWE1+/+且つXIST+/-である(1)Control-iPSでは、プライム型に近い表現型を示す細胞が多数を占めたクローンがあった(クローン1、2)。一方、(3)H1FOO-DD-iPSでは、全てのクローンにおいて、ナイーブ型の表現型を示す細胞が多数を占めた。これらの結果は、H1FOO-DD-iPSが、よりナイーブ型に近い表現型を有することを示す。
[実施例15]FKBP1Aの発現量の比較
ヒト線維芽細胞(TIG120)に、実施例1で作製した(c)のセンダイウイルスベクターを用いて、H1FOO-DDを導入した(H1FOO OE HDF)。ヒト線維芽細胞(TIG120)に、センダイウイルスベクターを用いて、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を導入し、iPS細胞を作製した(OSKL)。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(c)のセンダイウイルスベクターを用いて、ヒト線維芽細胞(TIG120)に、前記各遺伝子を導入し、H1FOO-DD-iPSを作製した(OSKLH)。
ヒト線維芽細胞(TIG120)に、実施例1で作製した(c)のセンダイウイルスベクターを用いて、H1FOO-DDを導入した(H1FOO OE HDF)。ヒト線維芽細胞(TIG120)に、センダイウイルスベクターを用いて、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を導入し、iPS細胞を作製した(OSKL)。Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、及びL-Myc遺伝子を含むセンダイウイルスベクターと共に、上記実施例1で作製した(c)のセンダイウイルスベクターを用いて、ヒト線維芽細胞(TIG120)に、前記各遺伝子を導入し、H1FOO-DD-iPSを作製した(OSKLH)。
上記で作製したH1FOO OE HDF、OSKL、及びOSKLHを培養し、前記各遺伝子の導入から2日目の細胞を回収した。次いで、qRT-PCRにより、FKBP1Aの発現量を測定した。コントロールとして、ヒト線維芽細胞(HDF)、及びH9 ES細胞(H9 ESC)についても同様に、FKBP1Aの発現量を測定した。FKBP1Aの発現量は、GAPDHの発現量で標準化した。
結果を図14に示す。図中、HDFはヒト皮膚線維芽細胞、H9 ESCはH9 ES細胞、H1FOO OE HDFはH1FOO OE HDFの作製後2日間培養した細胞、OSKL day2はOSKLの作製後2日間培養した細胞、OSKLH day2はOSKLHの作製後2日間培養した細胞を示す。FKBP1Aの発現量は、OSKLH day2の発現量を1.0としたときの相対発現量として示した。
図14に示すように、FKBP1Aの発現量は、OSKLH day2のみで、顕著に高かった。OSKLH day2では、他の細胞と比較して、FKBP1Aの発現量が10倍程度高かった。この結果は、核初期化物質とともにH1FOO-DDを導入することにより、核初期化誘導初期の細胞において、FKBP1Aの発現量が上昇することを示す。さらに、結果は割愛するが、FKBP1Aは、自然免疫応答発現マーカーであるIFIT1、IFNAの発現を抑制することが確認された。そのため、核初期化物質とともにH1FOO-DDを導入することにより、自然免疫応答マーカーの発現が抑制されると推測される。これらの結果から、自然免疫応答マーカーの発現抑制が、iPS細胞の品質向上に寄与する可能性が示唆された。
図14に示すように、FKBP1Aの発現量は、OSKLH day2のみで、顕著に高かった。OSKLH day2では、他の細胞と比較して、FKBP1Aの発現量が10倍程度高かった。この結果は、核初期化物質とともにH1FOO-DDを導入することにより、核初期化誘導初期の細胞において、FKBP1Aの発現量が上昇することを示す。さらに、結果は割愛するが、FKBP1Aは、自然免疫応答発現マーカーであるIFIT1、IFNAの発現を抑制することが確認された。そのため、核初期化物質とともにH1FOO-DDを導入することにより、自然免疫応答マーカーの発現が抑制されると推測される。これらの結果から、自然免疫応答マーカーの発現抑制が、iPS細胞の品質向上に寄与する可能性が示唆された。
本発明によれば、品質が良好なiPS細胞を製造可能な、iPS細胞の品質改善剤、iPS細胞の製造方法、かかる製造方法により製造されるiPS細胞、及びiPS細胞製造用組成物が提供される。
以上、本発明の好ましい実施形態を説明および図示してきたが、これらは本発明を例示するものであり、限定的なものとみなされるべきではないことを理解すべきである。本発明の精神または範囲から逸脱することなく、追加、省略、置換、およびその他の変更を行うことができる。したがって、本発明は、前述の説明によって限定されるものとはみなされず、添付の請求項の範囲によってのみ限定される。
Claims (19)
- H1foo遺伝子と、
前記H1foo遺伝子が細胞に導入された場合に、前記H1foo遺伝子から発現されるH1fooタンパク質の細胞内での存在量及び存在時期の少なくとも一方を制御し得る制御配列と、
を有するポリヌクレオチドを含む、iPS細胞の品質改善剤。 - 前記ポリヌクレオチドが、これを導入する細胞内で、前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されているものである、請求項1に記載のiPS細胞の品質改善剤。
- 前記発現ベクターが、センダイウイルスベクターである、請求項2に記載のiPS細胞の品質改善剤。
- 前記制御配列が、不安定化ドメインをコードするヌクレオチド配列を含み、前記不安定化ドメインは、前記不安定化ドメインを含む融合タンパク質のプロテアソームによる分解を促進するドメインであり、
前記制御配列が、前記不安定化ドメインと前記H1fooタンパク質との融合タンパク質を発現し得るように、前記H1foo遺伝子に連結されている、
請求項1~3のいずれか一項に記載のiPS細胞の品質改善剤。 - 前記制御配列が、化学的刺激に応答して、前記H1foo遺伝子の転写を制御するプロモーター配列を含む、請求項1~4のいずれか一項に記載のiPS細胞の品質改善剤。
- H1fooタンパク質と不安定化ドメインとの融合タンパク質を含む、iPS細胞の品質改善剤であって、
前記不安定化ドメインは、前記融合タンパク質のプロテアソームによる分解を促進するドメインである、
iPS細胞の品質改善剤。 - 核初期化物質と、
請求項1~6のいずれか一項に記載のiPS細胞の品質改善剤と、
を体細胞に導入する工程を含む、iPS細胞の製造方法。 - 前記iPS細胞が、プライム型またはナイーブ型iPS細胞である、請求項7に記載のiPS細胞の製造方法。
- 前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子、並びに、それらの遺伝子産物からなる群から選択される少なくとも1つを含む、
請求項7または8に記載のiPS細胞の製造方法。 - 前記核初期化物質が、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、L-Myc若しくはc-Myc、およびそれらの遺伝子産物である、
請求項7~9のいずれか一項に記載のiPS細胞の製造方法。 - 前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、
前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子が導入される細胞内で前記少なくとも1つの遺伝子を発現可能な状態で発現ベクターに挿入されている、
請求項7~9のいずれか一項に記載のiPS細胞の製造方法。 - 前記発現ベクターが、センダイウイルスベクターである、請求項11に記載のiPS細胞の製造方法。
- 核初期化物質と、H1foo遺伝子と、を体細胞に導入する工程を含み、
前記体細胞に導入された前記H1foo遺伝子から発現されるH1fooタンパク質は、前記体細胞内における存在時期及び存在量の少なくとも一方が制御される、
iPS細胞の製造方法。 - 前記H1foo遺伝子が、前記H1foo遺伝子が導入される細胞内で前記H1foo遺伝子を発現可能な状態で発現ベクターに挿入されている、請求項13に記載のiPS細胞の製造方法。
- 前記核初期化物質が、Oct遺伝子ファミリーの遺伝子、Sox遺伝子ファミリーの遺伝子、Klf遺伝子ファミリーの遺伝子、Myc遺伝子ファミリーの遺伝子、Lin遺伝子ファミリーの遺伝子、及びNanog遺伝子からなる群より選択される少なくとも1つの遺伝子であり、
前記少なくとも1つの遺伝子が、前記少なくとも1つの遺伝子を発現可能な発現ベクターにコードされている、
請求項13又は14に記載のiPS細胞の製造方法。 - 前記発現ベクターが、センダイウイルスベクターである、請求項14又は15に記載のiPS細胞の製造方法。
- 請求項7~16のいずれか一項に記載のiPS細胞の製造方法により製造されるiPS細胞。
- 核初期化物質と、
請求項1~6のいずれか一項に記載のiPS細胞の品質改善剤と、
を含む、iPS細胞製造用組成物。 - 核初期化物質と、前記核初期化物質とともに体細胞に導入されてから2~15日目までのいずれかで、前記体細胞の初期化を誘導するとともに、自然免疫を抑制する機能を有する物質とを前記体細胞に導入する工程を含む、iPS細胞の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202180054397.0A CN116018402A (zh) | 2020-09-04 | 2021-09-06 | iPS细胞的品质改善剂、iPS细胞的制备方法、iPS细胞、及iPS细胞制备用组合物 |
| JP2022547004A JP7808298B2 (ja) | 2020-09-04 | 2021-09-06 | iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物 |
| US18/024,646 US20230220025A1 (en) | 2020-09-04 | 2021-09-06 | Quality Improving Agent for IPS Cells, Method of Producing IPS Cells, IPS Cells, and Composition for Producing IPS Cells |
| EP21864470.6A EP4209593A4 (en) | 2020-09-04 | 2021-09-06 | IPS CELL QUALITY IMPROVING AGENT, METHOD FOR PRODUCING IPS CELLS, IPS CELLS, AND COMPOSITION FOR PRODUCING IPS CELLS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020149447 | 2020-09-04 | ||
| JP2020-149447 | 2020-09-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022050419A1 true WO2022050419A1 (ja) | 2022-03-10 |
Family
ID=80491146
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/032734 Ceased WO2022050419A1 (ja) | 2020-09-04 | 2021-09-06 | iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20230220025A1 (ja) |
| EP (1) | EP4209593A4 (ja) |
| JP (1) | JP7808298B2 (ja) |
| CN (1) | CN116018402A (ja) |
| WO (1) | WO2022050419A1 (ja) |
Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997016538A1 (en) | 1995-10-31 | 1997-05-09 | Dnavec Research Inc. | (-)-strand rna virus vector having autonomously replicating activity |
| WO1997016539A1 (en) | 1995-11-01 | 1997-05-09 | Dnavec Research Inc. | Recombinant sendai virus |
| WO1999054348A1 (en) | 1998-04-17 | 1999-10-28 | Clontech Laboratories, Inc. | Rapidly degrading gfp-fusion proteins and methods of use |
| WO2000070055A1 (en) | 1999-05-18 | 2000-11-23 | Dnavec Research Inc. | Rnp originating in paramyxovirus |
| WO2000070070A1 (en) | 1999-05-18 | 2000-11-23 | Dnavec Research Inc. | Paramyxoviridae virus vector defective in envelope gene |
| WO2001018223A1 (en) | 1999-09-06 | 2001-03-15 | Dnavec Research Inc. | Paramyxoviruses having modified transcription initiation sequence |
| WO2003025570A1 (fr) | 2001-09-18 | 2003-03-27 | Dnavec Research Inc. | Methode permettant d'examiner un vecteur de virus a arn a brin negatif |
| WO2005071092A1 (ja) | 2004-01-22 | 2005-08-04 | Dnavec Research Inc. | サイトメガロウイルスエンハンサーおよびニワトリβ-アクチンプロモーターを含むハイブリッドプロモーターを利用したマイナス鎖RNAウイルスベクターの製造方法 |
| WO2006137517A1 (ja) | 2005-06-24 | 2006-12-28 | Dnavec Corporation | 幼少個体への遺伝子導入用ベクター |
| WO2007032555A1 (ja) | 2005-09-15 | 2007-03-22 | Japan Science And Technology Agency | テトラサイクリン系抗生物質によるタンパク質分解制御法 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| WO2007083644A1 (ja) | 2006-01-17 | 2007-07-26 | Dnavec Corporation | 新規タンパク質発現系 |
| WO2008007581A1 (en) | 2006-07-13 | 2008-01-17 | Dnavec Corporation | Non-replicating paramyxoviridae virus vector |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| US20090215169A1 (en) | 2007-02-09 | 2009-08-27 | Wandless Thomas J | Method for regulating protein function in cells using synthetic small molecules |
| WO2010008054A1 (ja) | 2008-07-16 | 2010-01-21 | ディナベック株式会社 | 染色体非組み込み型ウイルスベクターを用いてリプログラムされた細胞を製造する方法 |
| WO2010125620A1 (ja) | 2009-04-30 | 2010-11-04 | 国立大学法人大阪大学 | 哺乳類細胞におけるタンパク質分解誘導方法 |
| JP2011004674A (ja) | 2009-06-26 | 2011-01-13 | Fujitsu Ltd | 誘導多能性幹細胞(iPS細胞)の製造方法 |
| JP2014217345A (ja) | 2013-05-10 | 2014-11-20 | 学校法人 埼玉医科大学 | 人工多能性幹細胞製造用組成物、及び人工多能性幹細胞の製造方法 |
| JP2014217344A (ja) | 2013-05-10 | 2014-11-20 | 学校法人 埼玉医科大学 | 人工多能性幹細胞製造用組成物、及び人工多能性幹細胞の製造方法 |
| JP5696282B2 (ja) | 2009-06-10 | 2015-04-08 | 国立大学法人京都大学 | 人工多能性幹細胞樹立効率改善剤 |
| JP2015519898A (ja) * | 2012-05-21 | 2015-07-16 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 合成自己複製RNAによるヒトiPS細胞の作製 |
| WO2016125364A1 (ja) | 2015-02-02 | 2016-08-11 | 株式会社Idファーマ | 改良されたマイナス鎖rnaウイルスベクター |
| WO2017010080A1 (ja) | 2015-07-10 | 2017-01-19 | Heartseed株式会社 | 高品質なiPS細胞の製造方法 |
| WO2019093340A1 (ja) | 2017-11-07 | 2019-05-16 | 国立大学法人京都大学 | ナイーブ型多能性幹細胞からの原始内胚葉誘導方法 |
| JP2020511529A (ja) * | 2017-03-03 | 2020-04-16 | オブシディアン セラピューティクス, インコーポレイテッド | 免疫療法のためのcd19組成物及び方法 |
| JP2020149447A (ja) | 2019-03-14 | 2020-09-17 | 富士ゼロックス株式会社 | 表示情報生成装置及びプログラム |
| WO2021145128A1 (ja) * | 2020-01-16 | 2021-07-22 | 国立研究開発法人理化学研究所 | 変異型klfタンパク質、及び誘導多能性幹細胞の製造方法 |
-
2021
- 2021-09-06 WO PCT/JP2021/032734 patent/WO2022050419A1/ja not_active Ceased
- 2021-09-06 CN CN202180054397.0A patent/CN116018402A/zh active Pending
- 2021-09-06 JP JP2022547004A patent/JP7808298B2/ja active Active
- 2021-09-06 US US18/024,646 patent/US20230220025A1/en active Pending
- 2021-09-06 EP EP21864470.6A patent/EP4209593A4/en active Pending
Patent Citations (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997016538A1 (en) | 1995-10-31 | 1997-05-09 | Dnavec Research Inc. | (-)-strand rna virus vector having autonomously replicating activity |
| WO1997016539A1 (en) | 1995-11-01 | 1997-05-09 | Dnavec Research Inc. | Recombinant sendai virus |
| WO1999054348A1 (en) | 1998-04-17 | 1999-10-28 | Clontech Laboratories, Inc. | Rapidly degrading gfp-fusion proteins and methods of use |
| WO2000070055A1 (en) | 1999-05-18 | 2000-11-23 | Dnavec Research Inc. | Rnp originating in paramyxovirus |
| WO2000070070A1 (en) | 1999-05-18 | 2000-11-23 | Dnavec Research Inc. | Paramyxoviridae virus vector defective in envelope gene |
| WO2001018223A1 (en) | 1999-09-06 | 2001-03-15 | Dnavec Research Inc. | Paramyxoviruses having modified transcription initiation sequence |
| WO2003025570A1 (fr) | 2001-09-18 | 2003-03-27 | Dnavec Research Inc. | Methode permettant d'examiner un vecteur de virus a arn a brin negatif |
| WO2005071092A1 (ja) | 2004-01-22 | 2005-08-04 | Dnavec Research Inc. | サイトメガロウイルスエンハンサーおよびニワトリβ-アクチンプロモーターを含むハイブリッドプロモーターを利用したマイナス鎖RNAウイルスベクターの製造方法 |
| WO2006137517A1 (ja) | 2005-06-24 | 2006-12-28 | Dnavec Corporation | 幼少個体への遺伝子導入用ベクター |
| WO2007032555A1 (ja) | 2005-09-15 | 2007-03-22 | Japan Science And Technology Agency | テトラサイクリン系抗生物質によるタンパク質分解制御法 |
| JP4183742B1 (ja) | 2005-12-13 | 2008-11-19 | 国立大学法人京都大学 | 誘導多能性幹細胞の製造方法 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| WO2007083644A1 (ja) | 2006-01-17 | 2007-07-26 | Dnavec Corporation | 新規タンパク質発現系 |
| WO2008007581A1 (en) | 2006-07-13 | 2008-01-17 | Dnavec Corporation | Non-replicating paramyxoviridae virus vector |
| US20120178168A1 (en) | 2007-02-09 | 2012-07-12 | The Board Of Trustees Of The Leland Stanford Junior University | Method for regulating protein function in cells using synthetic small molecules |
| US20090215169A1 (en) | 2007-02-09 | 2009-08-27 | Wandless Thomas J | Method for regulating protein function in cells using synthetic small molecules |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| WO2010008054A1 (ja) | 2008-07-16 | 2010-01-21 | ディナベック株式会社 | 染色体非組み込み型ウイルスベクターを用いてリプログラムされた細胞を製造する方法 |
| WO2010125620A1 (ja) | 2009-04-30 | 2010-11-04 | 国立大学法人大阪大学 | 哺乳類細胞におけるタンパク質分解誘導方法 |
| JP5696282B2 (ja) | 2009-06-10 | 2015-04-08 | 国立大学法人京都大学 | 人工多能性幹細胞樹立効率改善剤 |
| JP2011004674A (ja) | 2009-06-26 | 2011-01-13 | Fujitsu Ltd | 誘導多能性幹細胞(iPS細胞)の製造方法 |
| JP2015519898A (ja) * | 2012-05-21 | 2015-07-16 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 合成自己複製RNAによるヒトiPS細胞の作製 |
| JP2014217344A (ja) | 2013-05-10 | 2014-11-20 | 学校法人 埼玉医科大学 | 人工多能性幹細胞製造用組成物、及び人工多能性幹細胞の製造方法 |
| JP2014217345A (ja) | 2013-05-10 | 2014-11-20 | 学校法人 埼玉医科大学 | 人工多能性幹細胞製造用組成物、及び人工多能性幹細胞の製造方法 |
| WO2016125364A1 (ja) | 2015-02-02 | 2016-08-11 | 株式会社Idファーマ | 改良されたマイナス鎖rnaウイルスベクター |
| WO2017010080A1 (ja) | 2015-07-10 | 2017-01-19 | Heartseed株式会社 | 高品質なiPS細胞の製造方法 |
| JP2020511529A (ja) * | 2017-03-03 | 2020-04-16 | オブシディアン セラピューティクス, インコーポレイテッド | 免疫療法のためのcd19組成物及び方法 |
| WO2019093340A1 (ja) | 2017-11-07 | 2019-05-16 | 国立大学法人京都大学 | ナイーブ型多能性幹細胞からの原始内胚葉誘導方法 |
| JP2020149447A (ja) | 2019-03-14 | 2020-09-17 | 富士ゼロックス株式会社 | 表示情報生成装置及びプログラム |
| WO2021145128A1 (ja) * | 2020-01-16 | 2021-07-22 | 国立研究開発法人理化学研究所 | 変異型klfタンパク質、及び誘導多能性幹細胞の製造方法 |
Non-Patent Citations (48)
| Title |
|---|
| "GenBank", Database accession no. BC137916 |
| AKIRA KUNITOMI, YUASA SHINSUKE, SUGIYAMA FUMIHIRO, SAITO YUKI, SEKI TOMOHISA, KUSUMOTO DAI, KASHIMURA SHIN, TAKEI MAKOTO, TOHYAMA : "H1foo Has a Pivotal Role in Qualifying Induced Pluripotent Stem Cells", STEM CELL REPORTS, vol. 6, no. 6, 14 June 2016 (2016-06-14), United States , pages 825 - 833, XP055509242, ISSN: 2213-6711, DOI: 10.1016/j.stemcr.2016.04.015 * |
| ANONYMOUS: "iPS cell production kit CytoTune -iPS", NACALAI.CO, February 2019 (2019-02-01), pages 1 - 7, XP055907436, Retrieved from the Internet <URL:https://www.nacalai.co.jp/products/entry/d012011.html> * |
| BAN, H. ET AL., PROC NATL ACAD SCI U S A, vol. 108, 2011, pages 14234 - 9 |
| BAN, H.NISHISHITA, N.FUSAKI, N.TABATA, T.SAEKI, K.SHIKAMURA, M.TAKADA, N.INOUE, M.HASEGAWA, M.KAWAMATA, S.: "Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors", PROC NATL ACAD SCI U S A, vol. 108, 2011, pages 14234 - 9, XP002684868, DOI: 10.1073/PNAS.1103509108 |
| BANASZYNSKI ET AL., CELL, vol. 126, no. 5, 8 September 2006 (2006-09-08), pages 995 - 1004 |
| BARON, M. D.BARRETT, T., J. VIROL., vol. 71, 1997, pages 1265 - 1271 |
| BRIDGEN, A.ELLIOTT, R. M., PROC. NATL. ACAD. SCI. USA, vol. 93, 1996, pages 15400 - 15404 |
| CELL RESEARCH, 2008, pages 600 - 603 |
| CELL STEM CELL, vol. 2, 2008, pages 525 - 528 |
| CELL STEM CELL, vol. 4, 2009, pages 381 - 384 |
| CELL, vol. 126, 2006, pages 663 - 676 |
| CELL, vol. 131, 2007, pages 861 - 872 |
| DURBIN, A. P. ET AL., VIROLOGY, vol. 235, 1997, pages 323 - 332 |
| HASAN, M. K. ET AL., J. GEN. VIROL., vol. 78, 1997, pages 2813 - 2820 |
| HEBBAR, P.B.ARCHER, T.K.: "Altered histone H1 stoichiometry and an absence of nucleosome positioning on transfected DNA", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 283, 2008, pages 4595 - 4601 |
| IWAMOTO ET AL., CHEM BIOL., vol. 17, no. 9, 24 September 2010 (2010-09-24), pages 981 - 8 |
| J. BIOL. CHEM., vol. 287, no. 43, 2012, pages 36273 - 36282 |
| J. VIROL., vol. 68, 1994, pages 8413 - 8417 |
| KAJIWARA, M. ET AL., PROC NATL ACAD SCI U S A, vol. 109, no. 31, 2012, pages 12538 - 12543 |
| KATO, A. ET AL., EMBO J., vol. 16, 1997, pages 578 - 587 |
| KATO, A. ET AL., GENES CELLS, vol. 1, 1996, pages 569 - 579 |
| KONDO ET AL., EXP CELL RES., vol. 220, no. 2, October 1995 (1995-10-01), pages 501 - 4 |
| LI, H.-O. ET AL., J. VIROL., vol. 74, no. 14, 2000, pages 6564 - 6569 |
| LIU, X ET AL., NATURE METHODS, vol. 14, 2017, pages 1055 - 1062 |
| MCMAHON, A. P.BRADLEY, A., CELL, vol. 62, 1990, pages 1073 - 1085 |
| MEDICAL JOURNAL OF OSAKA UNIVERSITY, vol. 6, no. 1, March 1955 (1955-03-01), pages 1 - 15 |
| NAT. CELL BIOL., vol. 11, 2009, pages 197 - 203 |
| NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 101 - 106 |
| NATURE, vol. 454, 2008, pages 646 - 650 |
| PNAS, vol. 106, no. 31, 2009, pages 12759 - 12764 |
| RADECKE, F. ET AL., EMBO J., vol. 14, 1995, pages 6087 - 6094 |
| SAHAKYAN, A. ET AL., CELL STEM CELL, vol. 20, no. 1, 2017, pages 87 - 101 |
| SCHNELL. M. J. ET AL., EMBO J., vol. 13, 1994, pages 4195 - 4203 |
| SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| See also references of EP4209593A4 |
| SEKI, T. ET AL., NATURE PROTOCOLS, vol. 7, 2012, pages 718 - 728 |
| SEKI, T.YUASA, S.FUKUDA, K.: "Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus", NATURE PROTOCOLS, vol. 7, 2012, pages 718 - 728, XP055223860, DOI: 10.1038/nprot.2012.015 |
| STEINBACH, O.C.WOLFFE, A.P.RUPP, R.A.: "Somatic linker histones cause loss of mesodermal competence in Xenopus", NATURE, vol. 389, 1997, pages 395 - 399 |
| STEM CELLS, vol. 26, 2008, pages 1998 - 2005 |
| TAKAHASHI, K.YAMANAKA, S.: "Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors", CELL, vol. 126, 2006, pages 663 - 676 |
| TAKEBE, T. ET AL., CELL REP, vol. 21, no. 10, 2017, pages 2661 - 2670 |
| TERANISHI, T. ET AL.: "Rapid replacement of somatic linker histones with the oocyte-specific linker histone Hlfoo in nuclear transfer", DEVELOPMENTAL BIOLOGY, vol. 266, 2004, pages 76 - 86 |
| TOHYAMA, S. ET AL., CELL STEM CELL, vol. 12, no. 1, 2013, pages 127 - 137 |
| TOKUSUMI, T. ET AL., VIRUS RES., vol. 86, 2002, pages 33 - 38 |
| WHELAN, S. P. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 8388 - 8392 |
| YU ET AL., SCIENCE, vol. 324, 2009, pages 797 - 801 |
| YU, D. ET AL., GENES CELLS, vol. 2, 1997, pages 457 - 466 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116018402A (zh) | 2023-04-25 |
| JPWO2022050419A1 (ja) | 2022-03-10 |
| EP4209593A1 (en) | 2023-07-12 |
| US20230220025A1 (en) | 2023-07-13 |
| EP4209593A4 (en) | 2024-10-09 |
| JP7808298B2 (ja) | 2026-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101685209B1 (ko) | 유도된 다능성 줄기 세포의 효율적인 확립 방법 | |
| JP5827220B2 (ja) | 人工多能性幹細胞の樹立効率改善方法 | |
| CN101855350B (zh) | 核重编程序方法 | |
| CN102190731B (zh) | 用人工转录因子诱导产生多能干细胞 | |
| US20110003365A1 (en) | Method of preparing induced pluripotent stem cells deprived of reprogramming gene | |
| CN105916980B (zh) | 用于改进诱导多能干细胞的效率的方法 | |
| JP6927578B2 (ja) | 高品質なiPS細胞の製造方法 | |
| JP5773393B2 (ja) | 効率的な人工多能性幹細胞の樹立方法 | |
| JP7808298B2 (ja) | iPS細胞の品質改善剤、iPS細胞の製造方法、iPS細胞、及びiPS細胞製造用組成物 | |
| HK40096086A (en) | Quality improving agent for ips cells, method of producing ips cells, ips cells, and composition for producing ips cells | |
| WO2011055851A1 (en) | Method of efficiently establishing induced pluripotent stem cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21864470 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022547004 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021864470 Country of ref document: EP Effective date: 20230404 |