WO2022102565A1 - 4-ボロノ-l-フェニルアラニン及びその中間体の製造方法 - Google Patents
4-ボロノ-l-フェニルアラニン及びその中間体の製造方法 Download PDFInfo
- Publication number
- WO2022102565A1 WO2022102565A1 PCT/JP2021/040947 JP2021040947W WO2022102565A1 WO 2022102565 A1 WO2022102565 A1 WO 2022102565A1 JP 2021040947 W JP2021040947 W JP 2021040947W WO 2022102565 A1 WO2022102565 A1 WO 2022102565A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- linear
- mmol
- branched
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
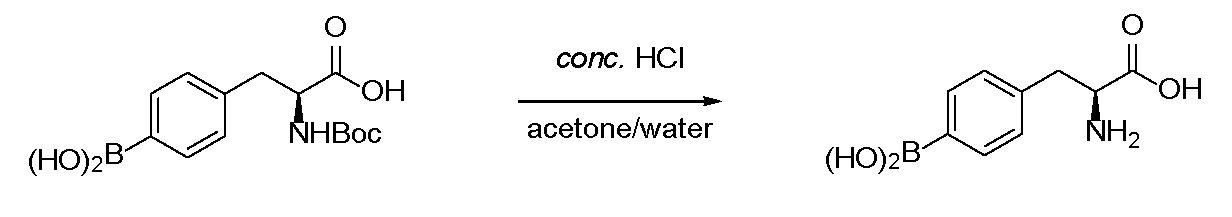
- the present invention relates to a method for producing 4-borono-L-phenylalanine and its intermediate.
- Boron neutron capture therapy is attracting attention as a treatment method for cancer.
- a boron compound containing a boron 10 isotope ( 10 B) is taken up by cancer cells, irradiated with low-energy neutron rays (for example, thermal neutrons), and locally caused by a nuclear reaction occurring inside the cells. It is a therapeutic method that destroys cancer cells. In this therapeutic method, it is important to selectively accumulate a boron compound containing 10B in cells of cancer tissue in order to enhance the therapeutic effect. Therefore, it is necessary to develop a boron compound that is selectively incorporated into cancer cells. Is required.
- a boron-containing compound having a boron atom or a boron atom group introduced into the basic skeleton has been synthesized.
- Drugs used in actual clinical practice include 4-borono-L-phenylalanine (L-BPA) and mercaptoundecahydrododecaborate (BSH).
- L-BPA 4-borono-L-phenylalanine
- BSH mercaptoundecahydrododecaborate
- 4-Borono-L-Phenylalanine is incorporated into LAT1, a type of amino acid transporter, as a mimic of phenylalanine. Since the expression of LAT1 is enhanced in cancer cells, L-BPA is likely to accumulate and is used for the treatment of cancer by utilizing its property (see, for example, Non-Patent Document 1).
- Patent Document 1 As a method for producing 4-borono-L-phenylalanine, a method using boronotoluene as a raw material has been proposed (see Patent Document 1).
- An object of the present invention is to provide a new method for producing 4-borono-L-phenylalanine.
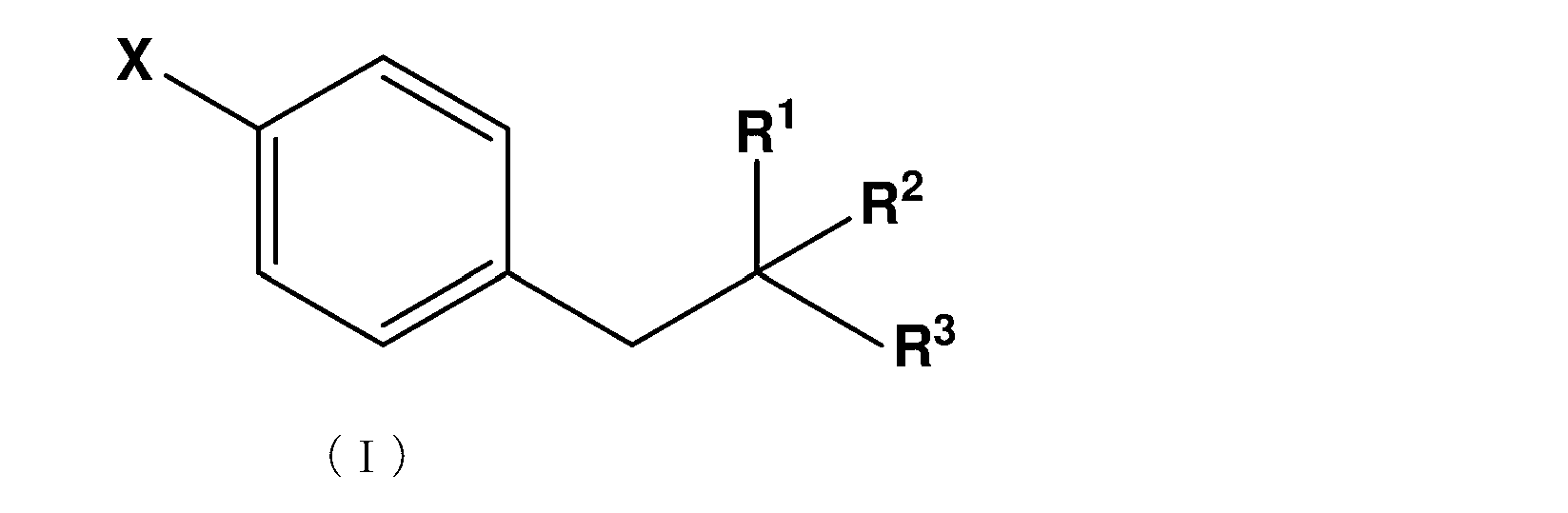
- X represents Cl, F, Br or I and represents R 1 represents NR 11 R 12 , R 11 represents H, or an amino protecting group, and R 12 represents an amino protecting group.
- R 2 represents COOR 21 and R 21 represents H, or a linear or branched C1 to C10 alkyl or benzyl group.
- R 3 represents H or COOR 31 and R 31 represents a linear or branched C1-C10 alkyl or benzyl group; R4 represents either boronic acid (B (OH) 2 ), boronic acid ester or boronic acid amide;
- the first step in which the compound represented by the formula (I) reacts with the linear or branched C1-C10 alkyl magnesium halide in the presence of the metal halide; and the compound obtained in the first step.
- a production method comprising a second step of reacting with a borate ester or borate amide; [2] The production method according to [1], wherein the metal halide is lithium chloride, and the alkylmagnesium halide is isopropylmagnesium chloride or sec-butylmagnesium chloride. [3] The X represents Br or I, and represents Br or I. The production method according to [1] or [2], wherein R 3 is COOR 31 and R 31 represents a linear or branched alkyl group of C1 to C10. [4] The X represents Br or I, and represents Br or I. The R 2 represents COOH. The production method according to any one of [1] to [3], wherein R 3 represents H.
- both the first reaction step and the second reaction step include a step of using at least one selected from the group consisting of an ether solvent and a hydrocarbon solvent as a solvent.
- the manufacturing method according to any one.
- [6] The production method according to any one of [1] to [5], wherein both the first reaction step and the second reaction step are carried out at a reaction temperature in the range of ⁇ 78 ° C. to 0 ° C.
- a compound represented by the formula (I), a mixture of a metal halide and an alkylmagnesium halide and a borate ester represented by B (OR) 3 (R is a linear or branched C1 to C10 alkyl group, The production method according to any one of [1] to [6], wherein the reaction between the first step and the second step is promoted by mixing the phenyl group or the benzyl group).
- R is a linear or branched C1 to C10 alkyl group
- the production method according to any one of [1] to [6] wherein the reaction between the first step and the second step is promoted by mixing the phenyl group or the benzyl group).
- the production method according to [7] wherein the amount ratio of the mixture of the metal halide and the alkylmagnesium halide and the boric acid ester is 1: 0.5 to 2 in an equivalent ratio.
- a method for producing 4-borono-L-phenylalanine which comprises a step of
- the novel production method of the present invention can reduce by-products and increase the yield thereof, especially in the production of 4-borono-L-phenylalanine.
- the compound when a compound having an asymmetric carbon is represented, the compound may be racemic, L-form, or D-form unless otherwise specified.
- the B atom when 4-borono-L-phenylalanine or an intermediate (precursor) thereof has a B atom, the B atom contains 10 B and 11 B only, unless otherwise specified. Any of these is acceptable.
- Typical existing methods for producing 4-borono-L-phenylalanine use boronotoluene as a raw material, and since B addition is in the early stages of the production process, in particular, not only 11 B atoms but also 10 B. When atoms are used, the cost of 10 B is lost in the subsequent manufacturing process, which increases the manufacturing cost. Further, a photobromination reaction using light as an active source when synthesizing a precursor is included. The raw material for the bromination reaction does not dissolve in the solvent, and by-products other than the target compound are produced, which causes a problem that the overall yield is lowered.
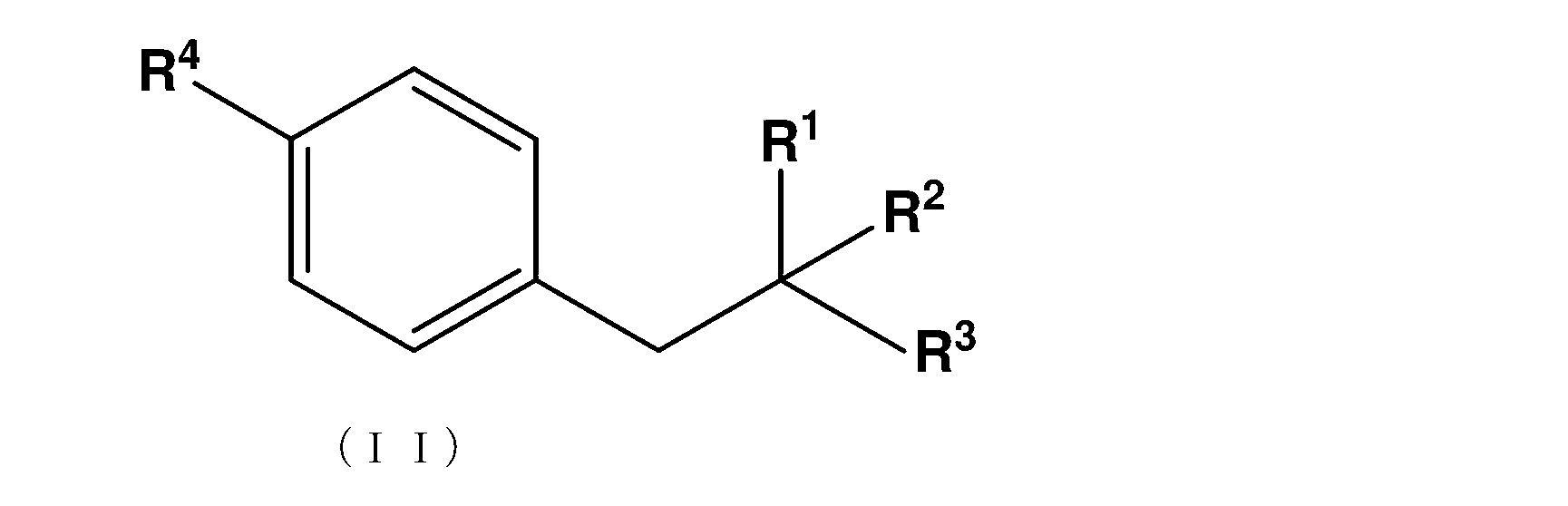
- the novel method for producing 4-borono-L-phenylalanine is a compound that is an intermediate between the compound represented by the following formula (I) and 4-borono-L-phenylalanine represented by the formula (II). Includes the process of manufacturing.
- the compound represented by the following general formula (II) may be particularly referred to as an intermediate or a precursor.
- X represents Cl, F, Br or I and represents R 1 represents NR 11 R 12 , R 11 represents H, a protecting group for an amino group, and R 12 represents a protecting group for an amino group.
- R 2 represents COOR 21 and R 21 represents H, or a linear or branched C1 to C10 alkyl or benzyl group.
- R 3 represents H or COOR 31 and R 31 represents a linear or branched C1-C10 alkyl or benzyl group; R4 represents either boronic acid (B (OH) 2 ), boronic acid ester or boronic acid amide;
- the first step in which the compound represented by the formula (I) reacts with the linear or branched C1-C10 alkyl magnesium halide in the presence of the metal halide; and the compound obtained in the first step.
- the second step in which boric acid ester or boric acid amide reacts with the boric acid ester.
- the amino-protecting group may be any group that protects an amino acid, and is not limited, but is preferably an acyl-based protecting group, an alkyl-protecting group, a carbamate-based protecting group, or the like. ..
- examples of the acyl-based protecting group include an acetyl group, a pivaloyl group, a benzoyl group and the like
- examples of the alkyl-protecting group include a benzyl group
- examples of the carbamate-based protecting group include a tert-butoxycarbonyl group and a benzyl group. Examples thereof include an oxycarbonyl group and a 9-fluorenylmethyloxycarbonyl group.
- an acyl-based protecting group or a carbamate-based protecting group is used.
- borate ester or borate amide is not limited, but is preferably B (OR) 3 , B (NR) 3 , B (OR) 2 (NR), B (OR).
- (NR) 2 R refers to a compound represented by a linear or branched C1-C10 alkyl group, phenyl group, or benzyl group).
- B (OR) 3 is particularly preferably used.
- any alkyl group having 1 to 10 carbon atoms may be used, but linear or branched alkyl of C1 to C8 is preferable.
- a group more preferably a linear or branched C1-C6 alkyl group.
- these groups include, but are not limited to, a methyl group, an ethyl group, an isopropyl group, a butyl group, and the like.
- the compound represented by the above formula (I) can be synthesized using, for example, a commercially available acetamide malonic acid diester such as diethyl acetamide malonate as a starting material.
- a commercially available acetamide malonic acid diester such as diethyl acetamide malonate
- an alcohol such as ethanol, an ether solvent, or an aprotonic polar solvent such as DMF is used as a solvent, and a metal alkoxide exemplified by lithium or sodium is added dropwise to prepare an enolate of an acetamide malonic acid diester.
- the corresponding halogenated benzyl bromide for example 4-iodobenzyl bromide, 4-bromobenzyl bromide, 4-chlorobenzyl bromide, etc.
- the product can also be obtained, for example, by adjusting the pH of the reaction solution to near neutral, filtering the solid if it precipitates, and drying it under reduced pressure. Alternatively, it can be obtained by extracting the crude product and then purifying it by column chromatography or the like.
- the following method can be mentioned. It is synthesized using diethyl acetamide malonate as a starting material. For example, alcohol such as ethanol is added, and sodium ethoxide is added dropwise to this solution at room temperature. After completion of the dropping, stir under reflux for 30 minutes to 1 hour, cool under an ice bath, add halogenated benzyl bromide such as 4-iodobenzyl bromide, 4-bromobenzyl bromide, or 4-chlorobenzyl bromide, and add 1 more. The reaction is carried out under reflux for an hour to 10 hours. It can be obtained by cooling under an ice bath, adjusting the pH to 6-7, filtering and washing the precipitated solid, and drying under reduced pressure.
- halogenated benzyl bromide such as 4-iodobenzyl bromide, 4-bromobenzyl bromide, or 4-chlorobenzyl bromide
- R 2 represents COOR 21
- R 3 represents H or COOR 31
- R 21 represents H or a linear or branched C1 to C10 alkyl or benzyl group
- Reference numeral 31 represents a linear or branched C1 to C10 alkyl group or benzyl group.
- any alkyl group having 1 to 10 carbon atoms may be used. It is preferably a linear or branched C1 to C8 alkyl group, and more preferably a linear or branched C1 to C6 alkyl group. Examples of these groups include, but are not limited to, a methyl group, an ethyl group, an isopropyl group, a butyl group, and the like.
- R4 represents any of the groups of boronic acid (B (OH) 2 ), boronic acid ester, or boronic acid amide, but the group of boronic acid ester or boronic acid amide in this definition.
- B (OH) 2 boronic acid
- boronic acid ester or boronic acid amide
- R 41 represents a linear or branched alkyl group of C1 to C10.
- any alkyl group having 1 to 10 carbon atoms may be used in the same manner as described above. It is preferably a linear or branched C1 to C8 alkyl group, and more preferably a linear or branched C1 to C6 alkyl group. Examples of these groups include, but are not limited to, a methyl group, an ethyl group, an isopropyl group, a butyl group and the like. Further, in the cyclic structure referred to here, not only the O atom but also the N atom may be present.
- pinacol 2,2-dimethyl-1,3-propanediol, N-methyldiethanolamine, 1,8-diaminonaphthalene, N-methyliminodiacetic acid, 1,1,1-trishydroxymethylethane.
- an ester or ester analog composed of atom B and any selected from the group consisting of catechol.
- boronic acid pinacole ester boronic acid MIDA ester
- boronic acid 1,3-propanediol ester boronic acid neopentyl glycol ester
- boronic acid catechol ester boronic acid pinandiol ester, boron.
- the metal constituting the metal halide is preferably an alkali metal such as lithium, sodium, potassium, rubidium, and cesium.
- the metal halide include lithium halide such as lithium chloride, lithium bromide and lithium iodide, sodium chloride, sodium bromide and sodium iodide such as sodium iodide. Of these, lithium chloride is particularly preferably used.
- the alkylmagnesium halide is not particularly limited as long as it contains a linear or branched C1 to C10 alkyl group.
- any alkyl group having 1 to 10 carbon atoms may be used, but linear or branched alkyl of C1 to C8 is preferable.
- alkyl magnesium halide examples include methyl magnesium chloride, ethyl magnesium chloride, butyl magnesium chloride (for example, sec-butyl magnesium chloride), hexyl magnesium chloride, isopropyl magnesium chloride, methyl magnesium bromide, and ethyl magnesium bromide.
- Butylmagnesium bromide (sec-butylmagnesium bromide, etc.), hexylmagnesium bromide, isopropylmagnesium bromide, etc. are preferably used.
- X is preferably a compound represented by Br or I.
- R 2 is COOH and R 3 is H. Further, it is also preferable that R 2 is COOR 21 (where R 21 is a linear or branched alkyl group of C1 to C10), and R 3 is H.
- both the first reaction step and the second reaction step include a step of using at least one selected from the group consisting of an ether solvent and a hydrocarbon solvent as a solvent. It is more preferable that the step uses at least one selected from the group consisting of an ether solvent and a hydrocarbon solvent as the solvent.
- the ether solvent include, but are not limited to, diethyl ether, diisopropyl ether, tetrahydrofuran (THF), 2-methyltetrahydrofuran, dioxane, cyclopentyl methyl ether, glyme, diglyme and the like.
- the hydrocarbon solvent is not limited, and examples thereof include aromatic hydrocarbon solvents such as toluene, xylene, and benzene.
- aromatic hydrocarbon solvents such as toluene, xylene, and benzene.
- tetrahydrofuran or toluene is particularly preferably used, and these can also be used in combination.
- Both the first reaction step and the second reaction step are carried out at a reaction temperature in the range of ⁇ 78 ° C. to 0 ° C.
- the reaction temperature is preferably in the range of ⁇ 60 ° C. to 0 ° C., more preferably in the range of ⁇ 50 ° C. to ⁇ 10 ° C.
- the reaction times of the first reaction step and the second reaction step are, respectively, 10 minutes to 72 hours, more preferably 20 minutes to 60 hours, and further preferably 30 minutes to 48 hours. Further, a space may be provided between the first reaction step and the second reaction step.
- the reaction between the first step and the second step can be advanced.
- the amount ratio of the mixture of the metal halide and the alkylmagnesium halide and B (OR) 3 is an equivalent ratio, preferably 1: 0.5 to 2, more preferably 1: 0.7 to 1.5. More preferably, it can be 1: 0.8 to 1.3.
- the crude product of the formula (II) obtained in the above steps is subjected to the extraction, washing and purification steps as it is or arbitrarily, and is subjected to the next deprotection step.
- the extraction, cleaning and purification steps typically include the following steps: That is, the pH of the reaction product containing the crude product of the formula (II) can be adjusted to a temperature of ⁇ 78 ° C. or higher, preferably around room temperature (20 ° C.), and the pH can be adjusted arbitrarily.
- Inorganic acids and organic acids can be used as the pH adjuster, hydrochloric acid, sulfuric acid, hydrobromic acid, hydroiodic acid, etc. can be used as the inorganic acids, and citric acid, acetic acid, trifluoroacetic acid, etc. are exemplified as the organic acids. Will be done.
- the pH is preferably adjusted to 1 to 5, more preferably 1 to 3.
- An organic solvent can be mixed with the reaction mixture, and the organic phase can be recovered and washed.
- the organic solvent used here is, for example, an ester-based solvent such as ethyl acetate, an ether-based solvent such as methyl tert-butyl ether, ketones, a halogen-based solvent and the like.
- the washing can be performed with saturated saline or the like. It can be dried, filtered to remove impurities and the filtrate concentrated under reduced pressure.
- alcohols, ethers, ketones, aprotonic polar solvents, water, halogen-based solvents and the like may be added to the reaction mixture to precipitate a solid, which may be obtained by filtration.
- water can be added to the organic phase to adjust the basicity
- the target substance can be transferred to the aqueous phase
- the aqueous phase can be washed with an organic solvent.
- organic solvent exemplified here include ester-based solvents such as ethyl acetate, ether-based solvents such as methyl tert-butyl ether, alcohol-based solvents such as 1-butyl alcohol and isobutyl alcohol, ketones, halogen-based solvents and the like.
- the aqueous phase can be acidified to precipitate a solid, and the desired product can be obtained.
- the cleaning solvent used here is not limited, and examples thereof include ether-based, alcohols, halogen-based, ketones, aprotic polar solvents, water and the like.
- the solid thus obtained can be dried to obtain a compound represented by the formula (II) with high purity.
- the obtained solid can be recrystallized to further increase the purity.
- the obtained compound represented by the formula (II) can be deprotected to produce 4-borono-L-phenylalanine.
- Deprotection follows a conventional method, but can be performed by, for example, hydrolysis, catalytic hydrogenation, or decarboxylation.
- the compound when it is a racemate, it can be used as it is, or for example, the optical purity of the L-form can be increased in order to obtain a preferable compound for use in boron neutron capture therapy.
- a known method may be appropriately used for the optical division of the L-4-boronophenylalanine derivative.
- the compound represented by the formula (II) is optically divided ( ⁇ ) through a hydrolysis step and an esterification step.
- a simplified method including a simplified step using an acylase via a hydrolysis step from the compound represented by the formula (II) can also be adopted.
- R 3 is a compound of H
- the step of using an acylase or the like can be omitted.
- acylases can be used as the above acylases.
- the optical resolution treatment using the above acylase can be carried out in a reaction temperature range of 30 to 60 ° C. and a pH range of 7.0 to 9.0 with a reaction time of 24 to 60 hours. From the viewpoint of reaction yield and suppression of impurities, it is preferable to carry out the reaction at a reaction temperature of 50 ° C., a pH of 8.0 and a reaction time of 48 hours.
- the method for producing the novel compound of the present invention is not limited, but is thus particularly preferably provided by being incorporated into the process of the method for producing 4- borono -L-phenylalanine, and particularly includes 10B. It is provided by incorporating it into the process of the method for producing 4-borono-L-phenylalanine.
- such a compound can be obtained in good condition with high yield and high purity.
- the expensive loss of 10 B can be reduced and the method of the present invention can be conveniently used.
- X represents Cl, F, Br or I and represents R 1 represents NR 11 R 12 , R 11 represents H, or an amino protecting group, and R 12 represents an amino protecting group.
- R 2 represents COOR 21 and R 21 represents H, or a linear or branched C1 to C10 alkyl or benzyl group.
- R 3 represents H or COOR 31 and R 31 represents a linear or branched C1-C10 alkyl or benzyl group; R4 represents either boronic acid (B (OH) 2 ), boronic acid ester or boronic acid amide;
- the first step in which the compound represented by the formula (I) reacts with the linear or branched C1-C10 alkyl magnesium halide in the presence of the metal halide; and the compound obtained in the first step.
- a production method comprising a second step of reacting with a borate ester or borate amide; [2] The production method according to [1], wherein the metal halide is lithium chloride, and the alkylmagnesium halide is isopropylmagnesium chloride or sec-butylmagnesium chloride. [3] The X represents Br or I, and represents Br or I. The production method according to [1] or [2], wherein R 3 is COOR 31 and R 31 represents a linear or branched alkyl group of C1 to C10. [4] The X represents Br or I, and represents Br or I. The production method according to [1] or [2], wherein R 3 represents H.
- the X represents Br or I, and represents Br or I.
- R 4 is boronic acid (B (OH) 2 ).
- R 1 represents NR 11 R 12
- R 11 represents H, butoxycarbonyl, or benzyl
- R 12 represents butoxycarbonyl, or benzyl, any one of [1] to [6].
- the X represents Br or I, and represents Br or I.
- the R 2 represents COOH.
- both the first reaction step and the second reaction step include a step of using at least one selected from the group consisting of an ether solvent and a hydrocarbon solvent as a solvent.
- [10] The production method according to [9], wherein the solvent is tetrahydrofuran, toluene, or a combination thereof.
- the compound represented by the formula (I) contains a mixture of a metal halide and an alkylmagnesium halide and a borate ester represented by B (OR) 3 (R is a linear or branched C1 to C10 alkyl group, The production method according to any one of [1] to [11], wherein the reaction between the first step and the second step is promoted by mixing the phenyl group or the benzyl group). [13] The production method according to any one of [1] to [12], wherein the borate ester represented by B (OR) 3 is triethyl borate or tributyl borate.
- the obtained white powder was suspended in 5 mL of hexane, washed, filtered off and dried under reduced pressure to obtain 18.5 g of a white powder of 4-bromomethylphenylboric acid pinacol ester. The yield was 52%.
- the obtained white powder was identified by 1 H-NMR (400 MHz), and it was confirmed that this was 4-bromomethylphenylborate pinacol ester. 1 In the measurement of the 1 H-NMR spectrum, deuterated chloroform was used as a solvent and tetramethylsilane was used as an internal standard substance. The obtained data are shown below.
- the obtained orange amorphous substance was identified by 1 H-NMR (400 MHz), and this was diethyl2-acetamide-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolane-). It was confirmed that it was 2-yl) benzyl) malonate.
- deuterated chloroform was used as a solvent and tetramethylsilane was used as an internal standard substance. The obtained data are shown below.
- the precipitate was separated by filtration, acetone was distilled off under reduced pressure, diluted with 50 mL of ethyl acetate, and washed with water and saturated brine. The solvent was distilled off under reduced pressure to obtain 0.40 g of a white powder of 4- (2-acetamide-3-ethoxy-2- (ethoxycarbonyl) -3-oxopropyl) phenylboronic acid. The yield was 99%.
- the obtained white powder was identified by 1 H-NMR (400 MHz), and it was determined that this was 4- (2-acetamide-3-ethoxy-2- (ethoxycarbonyl) -3-oxopropyl) phenylboronic acid. confirmed. 1 In the measurement of the 1 H-NMR spectrum, deuterated chloroform + deuterated methanol was used as a solvent, and tetramethylsilane was used as an internal standard substance. The obtained data are shown below.
- the obtained white powder was identified by 1 H-NMR (400 MHz), and it was confirmed that this was 2-acetamide-3- [4- (dihydroxyboryl) phenyl] propionic acid.
- 1 In the measurement of the 1 H-NMR spectrum, deuterated methanol was used as the solvent and methanol was used as the internal standard substance. The obtained data are shown below.
- the obtained white powder was identified by 1 H-NMR (400 MHz), and it was confirmed that this was (2S) -2-amino-3- [4- (dihydroxyboryl-10B) phenyl] propionic acid.
- 2S 2-amino-3- [4- (dihydroxyboryl-10B) phenyl] propionic acid.
- TSP sodium 3-trimethylsilylpropionate-d4
- Example 1 (4- (2-Acetamide-3-ethoxy-2- (ethoxycarbonyl) -3-oxopropyl) phenyl)
- boronic acid Under a nitrogen atmosphere, anhydrous tetrahydrofuran (2.0 mL) was added to diethyl 2-acetamide-2- (4-iodobenzyl) malonate (433 mg, 1.00 mmol) obtained in Reference Production Example 2 to prepare a solution (0.5 M). bottom.
- An isopropylmagnesium chloride-lithium chloride complex (1.29 M tetrahydrofuran solution, 2.2 eq., 1.7 mL, 2.2 mmol) at ⁇ 20 ° C. was added dropwise to this solution.
- the yield after using the boric acid ester is about 70%, and the boric acid ester is reacted with diethyl2-acetamido-2- (4-iodobenzyl) malonate in an equal amount or more.
- the loss of the B atom used was suppressed, and it was confirmed that this was an excellent method. Further adjustment of the amount of boric acid ester can further suppress the loss of 10 B atoms.
- Example 1 The 4- (2-acetamido-3-ethoxy-2- (ethoxycarbonyl) -3-oxopropyl) phenylboronic acid obtained in Example 1 was finally (2S) -2-amino-according to a conventional method. 3- [4- (Dihydroxyboronic) phenyl] propanoic acid can be obtained. Specifically, the same method as in Reference Production Example 1 can also be adopted.
- Example 2 Production of (S)-(4- (3-tert-butoxy) -2-((tert-butoxycarbonyl) amino) -3-oxopropyl) phenyl) boronic acid Under a nitrogen atmosphere, tert-butyl (S) -2-((tert-butoxycarbonyl) amino) -3- (4-iodophenyl) propanoate (6.71 g, 15.0 mmol) and anhydrous toluene / anhydrous tetrahydrofuran mixed solution ( 1 / 1,75 mL) was added.
- Example 4 Production of (S)-(4- (2- (bis (tert-butoxycarbonyl) amino) -3- (tert-butoxy) -3-oxopropyl) phenyl) boronic acid Under a nitrogen atmosphere, anhydrous tetrahydrofuran (2.1 mL) was added to tert-butyl (S) -2- (bis (tert-butoxycarbonyl) amino) -3- (4-iodophenyl) propanoate (566 mg, 1.03 mmol). rice field.
- Example 6 Comparing Example 6 and Comparative Example 2, it was confirmed that in Example 6 the reaction time was shorter and the yield was improved in both the halogen / magnesium exchange reaction and the boration reaction. The time-yield advantage of the technique was shown.
- tributyl borate (1.84 g, 8.00 mmol) was added dropwise at the same temperature.
- 3% hydrochloric acid (20 mL) was added dropwise so as not to exceed 10 ° C.
- concentrated hydrochloric acid was added dropwise so as not to exceed 10 ° C. to adjust the pH to 1.
- extraction by adding ethyl acetate (30 mLx2) the organic phase was washed with saturated brine (30 mL). This was dried over anhydrous sodium sulfate, filtered, and the obtained filtrate was concentrated under reduced pressure.
- Example 9 The method of Example 9 had a high yield and good results.
- the liquid was separated and the aqueous phase was extracted with methyl t-butyl ether (80 mL).
- the organic phase and the MTBE phase obtained earlier were combined, water (100 mL) was added, and the pH was adjusted to 12.8 by adding an 8M aqueous sodium hydroxide solution while cooling in an ice bath.
- the liquid was separated, and 3% hydrochloric acid was added to the obtained aqueous phase to adjust the pH to 10-11.
- 1-butanol 60 mLx2
- concentrated hydrochloric acid was added dropwise to adjust the pH to about 3, and a solid was precipitated.
- the solid could be efficiently stirred without precipitating during the exchange reaction, and the target product could be efficiently obtained with 58% as the standard of 10 B.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
[1]
下記式(I)で表される化合物から式(II)で表される化合物を製造する方法であって、


Xは、Cl、F、Br又はIを表わし、
R1は、NR11R12を表し、R11は、H、又はアミノ基の保護基を表し、R12は、アミノ基の保護基を表し、
R2は、COOR21を表し、R21は、H、又は直鎖状又は分岐状のC1~C10のアルキル基又はベンジル基を表し、
R3は、H又はCOOR31を表し、R31は、直鎖状又は分岐状のC1~C10アルキル基又はベンジル基を表す;
R4は、ボロン酸(B(OH)2)、ボロン酸エステル又はボロン酸アミドのいずれかを表わす;
式(I)で表される化合物が、金属ハロゲン化物の存在下において、直鎖状又は分岐状のC1~C10アルキルマグネシウムハロゲン化物と反応する第一工程;及び
前記第一の工程で得られる化合物とホウ酸エステル又はホウ酸アミドとが反応する第二工程;が含まれる、製造方法。
[2]
前記金属ハロゲン化物が、塩化リチウムであり、前記アルキルマグネシウムハロゲン化物が、イソプロピルマグネシウム塩化物又はsec-ブチルマグネシウム塩化物である、[1]に記載の製造方法。
[3]
前記Xは、Br又はIを表わし、
前記R3は、COOR31であり、R31は、直鎖状又は分岐状のC1~C10のアルキル基を表す
[1]又は[2]に記載の製造方法。
[4]
前記Xは、Br又はIを表わし、
前記R2は、COOHを表し、
前記R3は、Hを表す
[1]~[3]のいずれか1項に記載の製造方法。
[5]
前記第一反応工程及び第二反応工程がいずれも、溶媒として、エーテル系溶媒及び炭化水素系溶媒からなる群より選択される少なくとも1種を使用する工程を含む、[1]~[4]のいずれか1項に記載の製造方法。
[6]
前記第一反応工程及び第二反応工程がいずれも、-78℃から0℃の範囲の反応温度で行われる、[1]~[5]のいずれか1項に記載の製造方法。
[7]
式(I)で表される化合物に、金属ハロゲン化物・アルキルマグネシウムハロゲン化物の混合物とB(OR)3で表されるホウ酸エステル(Rは直鎖状又は分岐状のC1~C10アルキル基、フェニル基、又はベンジル基)とを混合することで、前記第一工程と第二工程の反応を進める、[1]~[6]のいずれか1項に記載の製造方法。
[8]
金属ハロゲン化物とアルキルマグネシウムハロゲン化物の混合物とホウ酸エステルとの量比が、当量比で、1:0.5~2である、[7]に記載の製造方法。
[9]
[1]~[8]のいずれか1項の工程で得られた前記式(II)の化合物を、さらに脱保護する工程を含む、4-ボロノ-L-フェニルアラニンの製造方法。
本発明において、新規な4-ボロノ-L-フェニルアラニンの製造方法は、下記式(I)で表される化合物から式(II)で表される4-ボロノ-L-フェニルアラニンの中間体となる化合物を製造する工程を含む。本明細書において、下記一般式(II)で示される化合物を特に中間体又は前駆体と表すことがある。


Xは、Cl、F、Br又はIを表わし、
R1は、NR11R12を表し、R11は、H、アミノ基の保護基を表し、R12は、アミノ基の保護基を表し、
R2は、COOR21を表し、R21は、H、又は直鎖状又は分岐状のC1~C10のアルキル基又はベンジル基を表し、
R3は、H又はCOOR31を表し、R31は、直鎖状又は分岐状のC1~C10アルキル基又はベンジル基を表す;
R4は、ボロン酸(B(OH)2)、ボロン酸エステル又はボロン酸アミドのいずれかを表わす;
式(I)で表される化合物が、金属ハロゲン化物の存在下において、直鎖状又は分岐状のC1~C10アルキルマグネシウムハロゲン化物と反応する第一工程;及び
前記第一の工程で得られる化合物とホウ酸エステル又はホウ酸アミドとが反応する第二工程。
このうち、R4は、特にボロン酸(B(OH)2)、あるいは、鎖状又は環状構造のボロン酸エステルが好ましく、ボロン酸が最も好ましい。
すなわち、前記式(II)の粗生成物を含む反応生成物を、-78℃以上の温度、好ましくは、室温(20℃)前後とし、任意に、pH調節することができる。pH調節剤としては無機酸や有機酸が使用でき、無機酸としては塩酸、硫酸、臭化水素酸、ヨウ化水素酸などが使用でき、有機酸ではクエン酸、酢酸、トリフルオロ酢酸などが例示される。pHは、好ましくは1~5、より好ましくは1~3に調整されることが好ましい。
反応混合物に有機溶媒を混合し、有機相を回収して洗浄することができる。ここで用いられる有機溶媒は、例えば、酢酸エチルなどのエステル系、メチルtert-ブチルエーテルなどのエーテル系、ケトン類、ハロゲン系溶媒等である。洗浄は、飽和食塩水等で行うことができる。
これを乾燥させ、濾過して不純物を除去し、濾液を減圧濃縮することができる。もしくは反応混合物にアルコール類、エーテル系、ケトン類、非プロトン性極性溶媒、水、ハロゲン系溶媒等を加え固体を析出させ、濾過して取得することもできる。
もしくは、有機相に水を加えて塩基性に調整して目的物を水相に移し、有機溶媒で水相を洗浄することもできる。ここで例示される有機溶媒は、例えば、酢酸エチルなどのエステル系、メチルtert-ブチルエーテルなどのエーテル系、1‐ブチルアルコールやイソブチルアルコールなどのアルコール系、ケトン類、ハロゲン系溶媒等である。次に水相を酸性にして固体を析出させ、目的物を取得することもできる。
[1]
下記式(I)で表される化合物から式(II)で表される化合物を製造する方法であって、
Xは、Cl、F、Br又はIを表わし、
R1は、NR11R12を表し、R11は、H、又はアミノ基の保護基を表し、R12は、アミノ基の保護基を表し、
R2は、COOR21を表し、R21は、H、又は直鎖状又は分岐状のC1~C10のアルキル基又はベンジル基を表し、
R3は、H又はCOOR31を表し、R31は、直鎖状又は分岐状のC1~C10アルキル基又はベンジル基を表す;
R4は、ボロン酸(B(OH)2)、ボロン酸エステル又はボロン酸アミドのいずれかを表わす;
式(I)で表される化合物が、金属ハロゲン化物の存在下において、直鎖状又は分岐状のC1~C10アルキルマグネシウムハロゲン化物と反応する第一工程;及び
前記第一の工程で得られる化合物とホウ酸エステル又はホウ酸アミドとが反応する第二工程;が含まれる、製造方法。
[2]
前記金属ハロゲン化物が、塩化リチウムであり、前記アルキルマグネシウムハロゲン化物が、イソプロピルマグネシウム塩化物又はsec-ブチルマグネシウム塩化物である、[1]に記載の製造方法。
[3]
前記Xは、Br又はIを表わし、
前記R3は、COOR31であり、R31は、直鎖状又は分岐状のC1~C10のアルキル基を表す
[1]又は[2]に記載の製造方法。
[4]
前記Xは、Br又はIを表わし、
前記R3は、Hを表す
[1]又は[2]に記載の製造方法。
[5]
前記Xは、Br又はIを表わし、
R2は、COOR21を表し、R21は、H、又はメチル基、エチル基、イソプロピル基、又はブチル基を表す、[1]~[4]のいずれかに記載の製造方法。
[6]
前記R4は、ボロン酸(B(OH)2)である、[1]~[5]のいずれか1項に記載の製造方法。
[7]
前記R1は、NR11R12を表し、R11は、H、ブトキシカルボニル、又はベンジルを表し、R12は、ブトキシカルボニル、又はベンジルを表す、[1]~[6]のいずれか1項に記載の製造方法。
[8]
前記Xは、Br又はIを表わし、
前記R2は、COOHを表し、
前記R3は、Hを表す、[1]~[7]のいずれかに記載の製造方法。
[9]
前記第一反応工程及び第二反応工程がいずれも、溶媒として、エーテル系溶媒及び炭化水素系溶媒からなる群より選択される少なくとも1種を使用する工程を含む、[1]~[8]のいずれか1項に記載の製造方法。
[10]
前記溶媒が、テトラヒドロフラン又はトルエン、又はこれらの組み合わせである、[9]に記載の製造方法。
[11]
前記第一反応工程及び第二反応工程がいずれも、-78℃から0℃の範囲の反応温度で行われる、[1]~[10]のいずれか1項に記載の製造方法。
[12]
式(I)で表される化合物に、金属ハロゲン化物・アルキルマグネシウムハロゲン化物の混合物とB(OR)3で表されるホウ酸エステル(Rは直鎖状又は分岐状のC1~C10アルキル基、フェニル基、又はベンジル基)とを混合することで、前記第一工程と第二工程の反応を進める、[1]~[11]のいずれか1項に記載の製造方法。
[13]
前記B(OR)3で表されるホウ酸エステルが、ホウ酸トリエチル又はホウ酸トリブチルである、[1]~[12]のいずれか1項に記載の製造方法。
[14]
金属ハロゲン化物とアルキルマグネシウムハロゲン化物の混合物とホウ酸エステルとの量比が、当量比で、1:0.5~2である、[12]又は[13]に記載の製造方法。
[15]
前記式(II)で表される化合物中のB原子が、10Bを含む、[1]~[14]のいずれか1項に記載の製造方法。
[16]
前記B(OR)3で表されるホウ酸エステルが、10Bを含む、[12]~[14]のいずれか1項に記載の製造方法。
[17]
[1]~[16]のいずれか1項の工程で得られた前記式(II)の化合物を、さらに脱保護する工程を含む、4-ボロノ-L-フェニルアラニンの製造方法。
(4-ブロモメチルフェニルホウ酸ピナコールエステルの合成)
ピナコール14.2g(120mmol)と4-メチルフェニルホウ酸16.3g(120mmol)を酢酸エチル240mLに溶かした溶液を90℃で2時間加熱還流した。この溶液を25℃に冷却して、n-ヘキサン240mLを加えて希釈した。次に臭素酸ナトリウム40.6g(269mmol)を水138mlに溶かした溶液を加え、これに亜硫酸水素ナトリウム28.8g(276mmol)の水溶液278mLを内温35℃以下に保つように12分間かけて滴下した後、室温で4時間撹拌した。反応液に酢酸エチル20mLを加えて分液し、有機相を5%Na2S2O3水溶液で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下に溶媒を留去した。粗生成物の白色粉末32.8gを得た。得られた白色粉末をヘキサン5mLに懸濁させて洗浄し、次いでろ別して減圧乾燥し、4-ブロモメチルフェニルホウ酸ピナコールエステルの白色粉末18.5gを得た。収率は52%であった。
20%ナトリウムエトキシド5.10g(15.0mmol)をエタノール19.5mLで希釈した溶液に、アセトアミドマロン酸ジエチル3.04g(14.0mmol)を加え、25℃で30分間撹拌した。次に4-ブロモメチルフェニルホウ酸ピナコールエステル2.97g(10.0mmol)を加え、100℃で12時間加熱還流した。この溶液を25℃に冷却して、3N塩酸を3.3mL加えた。減圧下でエタノールを留去したのち、酢酸エチル95mLで希釈し、飽和食塩水で洗浄した。無水硫酸マグネシウムで乾燥した後、減圧下に溶媒を留去して、粗生成物のオレンジ色オイル4.60gを得た。これをシリカゲルカラムクロマトグラフィー(溶出溶媒;n-ヘキサン:酢酸エチル=4:1)に付し、ジエチル2-アセトアミド-2-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ベンジル)マロネートのオレンジ色無定形物質3.45gを得た。収率は80%であった。
(4-(2-アセトアミド-3-エトキシ-2-(エトキシカルボニル)-3-オキソプロピル)フェニルボロン酸の合成)
得られたジエチル2-アセトアミド-2-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ベンジル)マロネート0.5g(1.15mmol)をアセトン40mLに溶かした溶液に、過ヨウ素酸ナトリウム0.74g(3.45mmol)、酢酸アンモニウム0.27g(3.45mmol)及び水20mLを加え、25℃で48時間撹拌した。析出物をろ別し、減圧下でアセトンを留去した後、酢酸エチル50mLで希釈し、水、飽和食塩水で洗浄した。減圧下に溶媒を留去して、4-(2-アセトアミド-3-エトキシ-2-(エトキシカルボニル)-3-オキソプロピル)フェニルボロン酸の白色粉末0.40gを得た。収率は99%であった。
4-(2-アセトアミド-3-エトキシ-2-(エトキシカルボニル)-3-オキソプロピル)フェニルボロン酸21.6g(61.7mmol)、5%水酸化ナトリウム水溶液179mLを85.0-91.0℃で4時間撹拌した。43-53℃に冷却し、3N塩酸90mLを加え、さらに85.0-90.0℃で1時間撹拌した。45℃に冷却し、反応液を減圧濃縮した。残留物に水32mLを加えて20℃で30分間撹拌し、析出した結晶をろ過して、水20mLで洗浄した。結晶を45℃で減圧乾燥し、2-アセトアミド-3-[4-(ジヒドロキシボリル)フェニル]プロパン酸の白色粉末11.5gを得た。収率は74.6%であった。
水酸化ナトリウム1.84g(46.0mmol)を水58mLに溶かし、50℃で2-アセトアミド-3-[4-(ジヒドロキシボリル)フェニル]プロパン酸11.5g(46.0mmol)を加えて撹拌した。この溶液に濃塩酸を加えてpH8.0に調整し、次にアシラーゼ(天野エンザイム社製、アシラーゼ「アマノ」)0.575gを加えて撹拌した。濃塩酸を加えて反応液のpH8.0を調整しながら,50℃で48時間撹拌した。反応液を25℃に冷却し,濃塩酸を加えてpH6.4に調整した。5℃で1時間静置したのち、水23mLを加えて希釈し,析出した結晶をろ過し,水23mL、エタノール12mLで洗浄した。結晶を減圧乾燥し、(2S)-2-アミノ-3-[4-(ジヒドロキシボリル)フェニル]プロパン酸の白色粉末4.54gを得た。収率は47.5%であった。
ジエチル2-アセトアミド-2-(4-ヨードベンジル)マロネートの製造
アセトアミドマロン酸ジエチル(10.0g,46.0mmol)にエタノール(75mL)を加え溶液(0.6M)とした。この溶液に室温下ナトリウムエトキシド(20%エタノール溶液,18.8g,55.2mmol)を20分間かけて滴下した。滴下終了後、還流下45分間攪拌した後、氷浴下で冷却し、4-ヨードベンジルブロミド(15.0g,50.5mmol)を加え、さらに還流下4時間反応させた。氷浴下で冷却し、2N塩酸を滴下してpHを6~7に調整し、析出した固体を濾過した。得た固体を水、ヘキサンで順次洗浄し、減圧下乾燥させることでジエチル2-アセトアミド-2-(4-ヨードベンジル)マロネート(15.1g,34.9mmol,75%)を黄色固体として得た。
1H NMR (500MHz,CDCl3):δ 7.59(d,J=5.0Hz,2H,ArH),6.75(d,J=5.0Hz,2H,ArH),4.27(q,J=5.0Hz,4H,CH2),3.59(s,2H,CH2),2.03(s,3H,CH3),1.30(t,J=5.0Hz,6H,CH3).
(4-(2-アセトアミド-3-エトキシ-2-(エトキシカルボニル)-3-オキソプロピル)フェニル)ボロン酸の製造
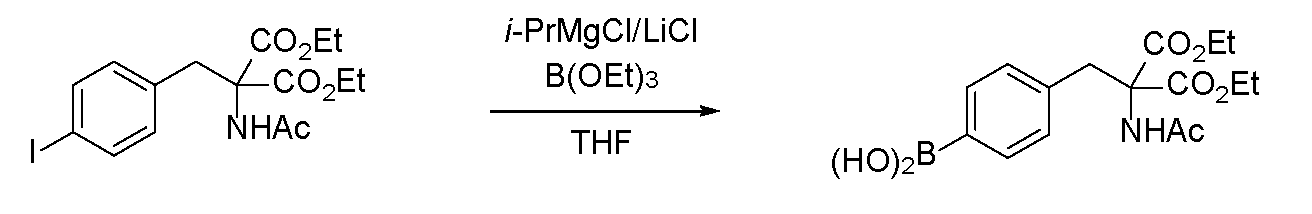
1H NMR(400MHz,DMSO-d6):7.96(s,2H,OH),7.68(d,J=8.0Hz,2H,ArH),6.94(d,J=8.0Hz,2H,ArH),4.15(q,J=8.0Hz,4H,CH2),3.43(s,1H,NH),1.94(s,3H,CH3),1.17(t,J=8.0Hz,6H,CH3).
(S)-(4-(3-tert-ブトキシ)-2-((tert-ブトキシカルボニル)アミノ)-3-オキソプロピル)フェニル)ボロン酸の製造

1H NMR(500MHz,DMSO-d6):δ7.95(s,2H,OH),7.69(d,J=10.0Hz,2H,ArH),7.18(d,J=10.0Hz,2H,ArH),7.14(d,J=5.0Hz,1H,NH),4.02(m,1H,CH),2.93(dd,J=5.0,15.0Hz,1H,CH2),2.84(dd,J=10.0,15.0Hz,1H,CH2),1.34(s,18H,CH3).
(2S)-2-アミノ-3-[4-(ジヒドロキシボリル)フェニル]プロパン酸の製造

1H NMR(D2O+DCl):7.77(d,J=8.0Hz,2H,ArH),7.38(d,J=8.0Hz,2H,ArH),4.42(dd,J=6.0,7.6Hz,1H,CH),3.40(dd,J=6.0,14.8Hz,1H,CH2),3.27(dd,J=7.6,14.8Hz,1H,CH2).
(S)-(4-(2-(ビス(tert-ブトキシカルボニル)アミノ)-3-(tert-ブトキシ)-3-オキソプロピル)フェニル)ボロン酸の製造
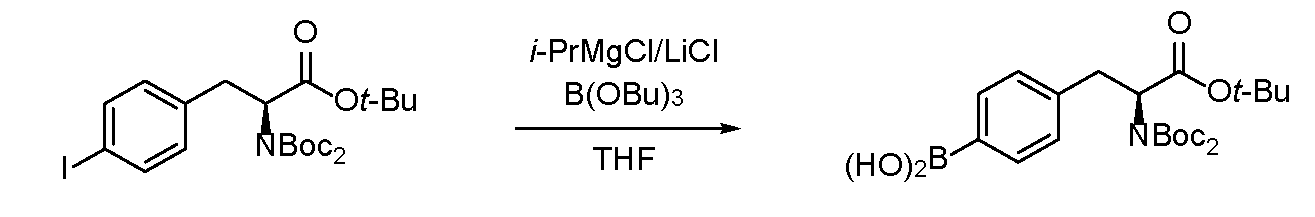
1H NMR(500MHz,CDCl3):δ7.58(dd,J=5.0,10.0Hz,2H,ArH),6.93(dd,J=5.0,10.0Hz,2H,ArH),5.00(dd,J=5.0,10.0Hz,1H,CH),3.33(dd,J=5.0,15.0Hz,1H,CH2),3.15(dd,J=10.0,15.0Hz,1H,CH2),1.47(s,9H,CH3),1.42(s,18H,CH3).
(2S)-2-アミノ-3-[4-(ジヒドロキシボリル)フェニル]プロパン酸の製造

1H NMR(500MHz,D2O):δ7.77(d,J=10.0Hz,2H,ArH),7.37(d,J=10.0Hz,2H,ArH),4.34(dd,J=5.0,10.0Hz,1H,CH),3.38(dd,J=5.0,15.0Hz,1H,CH2),3.25(dd,J=10.0,15.0Hz,1H,CH2).
(S)-(4-(3-(ベンジルオキシ)-2-(ジベンジルアミノ)-3-オキソプロピル)フェニル)ボロン酸の製造

1H NMR(500MHz,CDCl3):δ8.10(s,2H,OH),7.40-7.12(m,19H,ArH),5.26(d,J=15.0Hz,1H,CH2),5.15(d,J=15.0Hz,1H,CH2),3.96(d,J=15.0Hz,2H,CH2),3.80(t,J=5.0Hz,1H,CH),3.57(d,J=15.0Hz,2H,CH2),3.23(dd,J=5.0,15.0Hz,1H,CH2),3.11(d,J=5.0,15.0Hz,1H,CH2).
(S)-(4-(3-(ベンジルオキシ)-2-(ジベンジルアミノ)-3-オキソプロピル)フェニル)ボロン酸の製造

(S)-3-(4-ボロノフェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパン酸の製造

1H NMR(500MHz,acetone-d6):δ7.81(d,J=10.0Hz,2H,ArH),7.28(d,J=10.0Hz,2H,ArH),7.11(brs,1H,OH),6.01(d,J=10.0Hz,1H,NH),4.45(m,1H,CH),3.22(dd,J=5.0,15.0Hz,1H,CH2),3.03(dd,J=10.0,15.0Hz,1H,CH2),1.36(s,9H,CH3).
窒素雰囲気下、水素化ナトリウム(60% oil suspension,121mg,3.02mmol)、ホウ酸トリブチル(2.09g,9.06mmol)に(S)-2-((tert-ブトキシカルボニル)アミノ)-3-(4-ヨードフェニル)プロパン酸(1.18g,3.02mmol)を加えた。氷浴下塩化tert-ブチルマグネシウム(0.88Mテトラヒドロフラン溶液,27mL,24mmol)を20℃を超えないよう10分かけて滴下した。室温に昇温し、24時間攪拌後、氷浴下3%塩酸を加えて反応を停止し、pH1にした。Tert-ブチルメチルエーテル(30mL)を加え2回抽出した後、有機相を飽和食塩水(30mL)で洗浄した。これを無水硫酸ナトリウムにて乾燥させ濾過後、得られた濾液を減圧濃縮した。得られた粗生成物の1H NMR測定したところ、原料回収がほとんどであった。
(S)-2-アミノ-3-(4-ボロノフェニル)プロパン酸の製造
(S)-3-(4-ボロノフェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパン酸の製造
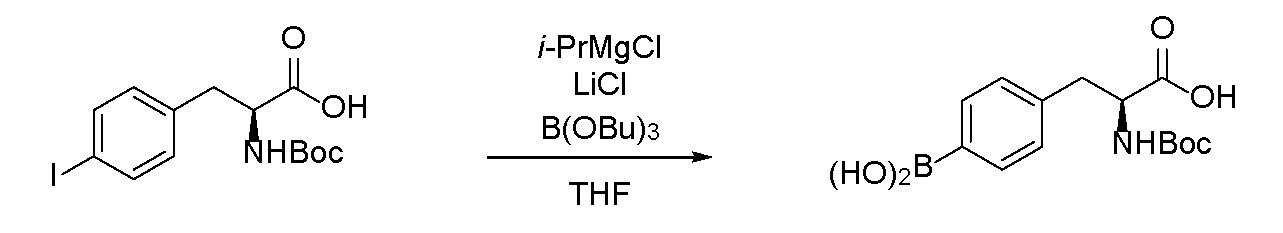
(S)-3-(4-ボロノフェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパン酸の製造

(S)-3-(4-ボロノフェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパン酸の製造
窒素雰囲気下(S)-2-((tert-ブトキシカルボニル)アミノ)-3-(4-ヨードフェニル)プロパン酸(12.5g,32.0mmol)にテトラヒドロフラン(64mL)を溶液とした。この溶液を-35℃下まで冷却し、塩化イソプロピルマグネシウム(2.0M テトラヒドロフラン溶液、64mL,128mmol)を45分かけて-30℃を超えないように滴下した。-30℃下3時間攪拌していくことで固体が析出して固まり、攪拌が全くできない状況となった。
Claims (9)
- 下記式(I)で表される化合物から式(II)で表される化合物を製造する方法であって、


Xは、Cl、F、Br又はIを表わし、
R1は、NR11R12を表し、R11は、H、又はアミノ基の保護基を表し、R12は、アミノ基の保護基を表し、
R2は、COOR21を表し、R21は、H、あるいは直鎖状又は分岐状のC1~C10のアルキル基又はベンジル基を表し、
R3は、H又はCOOR31を表し、R31は、直鎖状又は分岐状のC1~C10アルキル基又はベンジル基を表す;
R4は、ボロン酸(B(OH)2)、ボロン酸エステル又はボロン酸アミドのいずれかを表わす;
式(I)で表される化合物が、金属ハロゲン化物の存在下において、直鎖状又は分岐状のC1~C10アルキルマグネシウムハロゲン化物と反応する第一工程;及び
前記第一の工程で得られる化合物とホウ酸エステル又はホウ酸アミドとが反応する第二工程;が含まれる、製造方法。 - 前記金属ハロゲン化物が、塩化リチウムであり、前記アルキルマグネシウムハロゲン化物が、イソプロピルマグネシウム塩化物又はsec-ブチルマグネシウム塩化物である、請求項1記載の製造方法。
- 前記Xは、Br又はIを表わし、
前記R3は、COOR31であり、R31は、直鎖状又は分岐状のC1~C10のアルキル基を表す
請求項1又は2に記載の製造方法。 - 前記Xは、Br又はIを表わし、
前記R2は、COOHを表し、
前記R3は、Hを表す
請求項1~3のいずれか1項に記載の製造方法。 - 前記第一工程及び第二工程がいずれも、溶媒として、エーテル系溶媒、及び炭化水素系溶媒からなる群より選択される少なくとも1種を使用する工程を含む、請求項1~4のいずれか1項に記載の製造方法。
- 前記第一工程及び第二工程がいずれも、-78℃から0℃の範囲の反応温度で行われる、請求項1~5のいずれか1項に記載の製造方法。
- 式(I)で表される化合物に、金属ハロゲン化物・アルキルマグネシウムハロゲン化物と一般式B(OR)3のホウ酸エステル(Rは直鎖状又は分岐状のC1~C10のアルキル基、フェニル基、又はベンジル基)とを混合することで、前記第一工程と第二工程の反応を進めさせる、請求項1~6のいずれか1項に記載の製造方法。
- 金属ハロゲン化物とアルキルマグネシウムハロゲン化物の混合物とホウ酸エステルとの量比が、当量比で、1:0.5~2である、請求項7に記載の製造方法。
- 請求項1~8のいずれか1項の工程で得られた前記式(II)の化合物を、さらに脱保護する工程を含む、4-ボロノ-L-フェニルアラニンの製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020237009241A KR20230107791A (ko) | 2020-11-13 | 2021-11-08 | 4-보로노-l-페닐알라닌 및 그 중간체의 제조 방법 |
| EP21891811.8A EP4223762A4 (en) | 2020-11-13 | 2021-11-08 | PROCESS FOR PRODUCING 4-BORONO-L-PHENYLALANINE AND AN INTERMEDIATE THEREOF |
| AU2021377857A AU2021377857A1 (en) | 2020-11-13 | 2021-11-08 | Method for producing 4-borono-l-phenylalanine and intermediate thereof |
| CN202180062978.9A CN116113636A (zh) | 2020-11-13 | 2021-11-08 | 4-硼-l-苯基丙氨酸及其中间体的制造方法 |
| US18/252,305 US12612421B2 (en) | 2020-11-13 | 2021-11-08 | Method for producing 4-borono-L-phenylalanine and intermediate thereof |
| JP2022561897A JPWO2022102565A1 (ja) | 2020-11-13 | 2021-11-08 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020189349 | 2020-11-13 | ||
| JP2020-189349 | 2020-11-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022102565A1 true WO2022102565A1 (ja) | 2022-05-19 |
Family
ID=81601285
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/040947 Ceased WO2022102565A1 (ja) | 2020-11-13 | 2021-11-08 | 4-ボロノ-l-フェニルアラニン及びその中間体の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP4223762A4 (ja) |
| JP (1) | JPWO2022102565A1 (ja) |
| KR (1) | KR20230107791A (ja) |
| CN (1) | CN116113636A (ja) |
| AU (1) | AU2021377857A1 (ja) |
| TW (1) | TWI896809B (ja) |
| WO (1) | WO2022102565A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115466279A (zh) * | 2022-10-19 | 2022-12-13 | 重庆波克底科技开发有限责任公司 | 一种4-二羟10硼基-l-苯丙氨酸的制备方法 |
| CN115925732A (zh) * | 2022-12-16 | 2023-04-07 | 重庆高硼生物科技有限公司 | 4-硼酸基-l-苯丙氨酸的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008214319A (ja) | 2007-03-07 | 2008-09-18 | Stella Chemifa Corp | フェニルホウ素誘導体およびそれを用いたp−ボロノフェニルアラニンの製造方法 |
| JP2018525398A (ja) * | 2015-08-14 | 2018-09-06 | 南京中硼▲聯▼康医▲療▼科技有限公司Neuboron Medtech Ltd. | L−bpaの調製方法 |
| WO2019163738A1 (en) * | 2018-02-20 | 2019-08-29 | Otsuka Pharmaceutical Co., Ltd. | Method of 4-boronophenylalanine production |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2609396T3 (es) * | 2013-10-28 | 2017-04-20 | Taiwan Biotech Co., Ltd. | Procedimiento para preparar 4-borono-L-fenilalanina |
-
2021
- 2021-11-08 CN CN202180062978.9A patent/CN116113636A/zh active Pending
- 2021-11-08 EP EP21891811.8A patent/EP4223762A4/en active Pending
- 2021-11-08 WO PCT/JP2021/040947 patent/WO2022102565A1/ja not_active Ceased
- 2021-11-08 JP JP2022561897A patent/JPWO2022102565A1/ja active Pending
- 2021-11-08 KR KR1020237009241A patent/KR20230107791A/ko active Pending
- 2021-11-08 AU AU2021377857A patent/AU2021377857A1/en active Pending
- 2021-11-12 TW TW110142148A patent/TWI896809B/zh active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008214319A (ja) | 2007-03-07 | 2008-09-18 | Stella Chemifa Corp | フェニルホウ素誘導体およびそれを用いたp−ボロノフェニルアラニンの製造方法 |
| JP2018525398A (ja) * | 2015-08-14 | 2018-09-06 | 南京中硼▲聯▼康医▲療▼科技有限公司Neuboron Medtech Ltd. | L−bpaの調製方法 |
| WO2019163738A1 (en) * | 2018-02-20 | 2019-08-29 | Otsuka Pharmaceutical Co., Ltd. | Method of 4-boronophenylalanine production |
Non-Patent Citations (5)
| Title |
|---|
| ANONYMOUS: "[Special Lecture] Development of Turbo Grignard Reagents ", REAGENTS-FUJI FILM WAKO PURE CHEMICAL INDUSTRIES, 1 June 2013 (2013-06-01), XP055931687, Retrieved from the Internet <URL:https://labchem-wako.fujifilm.com/jp/siyaku-blog/010871.html> [retrieved on 20220615] * |
| KOPP FELIX, WUNDERLICH STEFAN, KNOCHEL PAUL: "Halogen–magnesium exchange on unprotected aromatic and heteroaromatic carboxylic acids", CHEMICAL COMMUNICATIONS, ROYAL SOCIETY OF CHEMISTRY, UK, no. 20, 1 January 2007 (2007-01-01), UK , pages 2075 - 2077, XP055779521, ISSN: 1359-7345, DOI: 10.1039/B618923G * |
| KRASOVSKIY A, KNOCHEL P: "A LICL-MEDIATED BR/MG EXCHANGE REACTION FOR THE PREPARATION OF FUNCTIONALIZED ARYL- AND HETEROARYLMAGNESIUM COMPOUNDS FROM ORGANIC BROMIDES", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, VERLAG CHEMIE, vol. 43, 21 June 2004 (2004-06-21), pages 3333 - 3336, XP002294441, ISSN: 1433-7851, DOI: 10.1002/anie.200454084 * |
| See also references of EP4223762A4 |
| WONGTHAI P ET AL., CANCER SCI, vol. 106, no. 3, March 2015 (2015-03-01), pages 279 - 86 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115466279A (zh) * | 2022-10-19 | 2022-12-13 | 重庆波克底科技开发有限责任公司 | 一种4-二羟10硼基-l-苯丙氨酸的制备方法 |
| CN115925732A (zh) * | 2022-12-16 | 2023-04-07 | 重庆高硼生物科技有限公司 | 4-硼酸基-l-苯丙氨酸的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI896809B (zh) | 2025-09-11 |
| KR20230107791A (ko) | 2023-07-18 |
| US20230416280A1 (en) | 2023-12-28 |
| EP4223762A4 (en) | 2025-01-01 |
| CN116113636A (zh) | 2023-05-12 |
| TW202229229A (zh) | 2022-08-01 |
| AU2021377857A1 (en) | 2023-06-22 |
| EP4223762A1 (en) | 2023-08-09 |
| JPWO2022102565A1 (ja) | 2022-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3874306B2 (ja) | (s)−3−(アミノメチル)−5−メチルヘキサン酸を製造する方法 | |
| US20110275855A1 (en) | Separation of an enantiomer mixture of (r)- and (s)-3-amino-1-butanol | |
| WO2022102565A1 (ja) | 4-ボロノ-l-フェニルアラニン及びその中間体の製造方法 | |
| MX2008014759A (es) | Sintesis del acido (s)-(+)-3-(aminometil)-5-metil hexanoico. | |
| JPH0623150B2 (ja) | 光学活性3−ヒドロキシブタン酸類の製造方法 | |
| WO2015037460A1 (ja) | 光学活性な3-(ビフェニル-4-イル)-2-[(t-ブトキシカルボニル)アミノ]プロパン-1-オールの製造方法 | |
| HUT68255A (en) | Process for the preparation of beta-phenylisoserine derivatives and use thereof | |
| WO2003042180A9 (en) | Process for producing optically active oxoheptenoic acid ester | |
| US20100286442A1 (en) | Novel method for preparing pregabalin | |
| JP2008214319A (ja) | フェニルホウ素誘導体およびそれを用いたp−ボロノフェニルアラニンの製造方法 | |
| US5817867A (en) | Reduction and resolution methods for the preparation of compounds useful as intermediates for preparing taxanes | |
| EP1008590B1 (en) | Process for preparing optically active oxazolidinone derivatives | |
| US12612421B2 (en) | Method for producing 4-borono-L-phenylalanine and intermediate thereof | |
| JP2021070660A (ja) | 光学活性な2−ピロリドン化合物の製造方法 | |
| JP7662640B2 (ja) | 光学活性な化合物の製造方法 | |
| CA2184500A1 (en) | Synthesis of compounds with predetermined chirality | |
| JPH08109158A (ja) | 光学活性l−アミノ酸の製法及び新規の光学活性l−アミノ酸 | |
| JP2016183132A (ja) | シクロヘキサンジカルボン酸モノエステル化合物の製造方法 | |
| CN111556861A (zh) | 茉莉酸酯化合物的制备方法 | |
| HK40088032A (zh) | 4-硼-l-苯基丙氨酸及其中间体的制造方法 | |
| EP1078919B1 (en) | Synthesis of alpha-amino-alpha',alpha'-dihaloketones and process for the preparation of beta)-amino acid derivatives by the use of the same | |
| JP2825608B2 (ja) | 光学活性トレオ―3―アミノ―2―ヒドロキシペンタン酸及びその製造法 | |
| JP4829418B2 (ja) | 光学活性なハロヒドリン誘導体およびその使用方法 | |
| JP2546067B2 (ja) | メタンジホスホン酸化合物の製造方法 | |
| JPH1160543A (ja) | 1−アミノテトラリン−2−オールの光学分割方法と分割の過程で生じるジアステレオマー塩 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21891811 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022561897 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18252305 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2021891811 Country of ref document: EP Effective date: 20230503 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021377857 Country of ref document: AU Date of ref document: 20211108 Kind code of ref document: A |