WO2022158458A1 - 改質硫化物固体電解質及びその製造方法 - Google Patents
改質硫化物固体電解質及びその製造方法 Download PDFInfo
- Publication number
- WO2022158458A1 WO2022158458A1 PCT/JP2022/001664 JP2022001664W WO2022158458A1 WO 2022158458 A1 WO2022158458 A1 WO 2022158458A1 JP 2022001664 W JP2022001664 W JP 2022001664W WO 2022158458 A1 WO2022158458 A1 WO 2022158458A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- solid electrolyte
- sulfide solid
- hydrocarbon group
- general formula
- modified
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0561—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of inorganic materials only
- H01M10/0562—Solid materials
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B17/00—Sulfur; Compounds thereof
- C01B17/22—Alkali metal sulfides or polysulfides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/14—Sulfur, selenium, or tellurium compounds of phosphorus
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B1/00—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors
- H01B1/06—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of other non-metallic substances
- H01B1/10—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of other non-metallic substances sulfides
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B13/00—Apparatus or processes specially adapted for manufacturing conductors or cables
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/82—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by IR- or Raman-data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/021—Physical characteristics, e.g. porosity, surface area
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0065—Solid electrolytes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0065—Solid electrolytes
- H01M2300/0068—Solid electrolytes inorganic
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0065—Solid electrolytes
- H01M2300/0068—Solid electrolytes inorganic
- H01M2300/008—Halides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a modified sulfide solid electrolyte and a method for producing the same.
- a sulfide solid electrolyte has been conventionally known as a solid electrolyte used in a solid electrolyte layer, and improvement in ionic conductivity is first desired for the sulfide solid electrolyte.
- a method for producing a composite solid electrolyte has been proposed in which the surface of a sulfide-based solid electrolyte is coated with a predetermined halogenated hydrocarbon compound as a coating material (see, for example, Patent Document 1). ).
- Patent Document 2 discloses that the affinity between the active material and the sulfide solid electrolyte used for the negative electrode, the positive electrode, etc.
- the ester compound binds to the surface of the conductive sulfide or is adsorbed to improve the cycle characteristics of a solid battery
- the sulfide solid electrolyte comprises a step of wet pulverizing a slurry containing a lithium ion conductive sulfide, an organic solvent, and an ester compound.
- the present invention has been made in view of such circumstances, and even if it is a sulfide solid electrolyte with a large specific surface area, it is excellent in coating aptitude when coated as a paste, and a battery that is excellent in efficiency.
- An object of the present invention is to provide a modified sulfide solid electrolyte capable of exhibiting performance and a method for producing the same.
- Another object of the present invention is to provide an electrode mixture and a lithium ion battery that exhibit excellent battery performance.
- the modified sulfide solid electrolyte according to the present invention is A sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, and an epoxy compound, A modified sulfide solid electrolyte having a peak at 2800 to 3000 cm -1 in an infrared absorption spectrum by FT-IR analysis (ATR method), is.
- the method for producing a modified sulfide solid electrolyte according to the present invention comprises: mixing a sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, an epoxy compound, and an organic solvent; A method for producing a modified sulfide solid electrolyte, comprising removing the organic solvent; is.
- the modified sulfide solid electrolyte according to the present invention is A sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, and an epoxy compound, modified sulfide solid electrolyte, is.
- the electrode mixture according to the present invention is an electrode mixture containing the modified sulfide solid electrolyte according to the present invention and an electrode active material; is. Further, the lithium ion battery according to the present invention is A lithium ion battery containing at least one of the modified sulfide solid electrolyte according to the present invention and the electrode mixture according to the present invention, is.
- the present invention it is possible to provide a modified sulfide solid electrolyte that is excellent in coating aptitude when applied as a paste and can efficiently exhibit excellent battery performance, and a method for producing the same. Further, according to the present invention, it is possible to provide an electrode mixture and a lithium ion battery that exhibit excellent battery performance.
- FIG. 4 shows CV curves of the modified sulfide solid electrolytes of Examples 23 and 24 and the sulfide solid electrolyte of Comparative Example 1.
- FIG. 4 shows CV curves of the modified sulfide solid electrolytes of Examples 23 and 24 and the sulfide solid electrolyte of Comparative Example 1.
- this embodiment An embodiment of the present invention (hereinafter sometimes referred to as "this embodiment") will be described below.
- the upper and lower numerical values of the numerical ranges of “more than”, “less than”, and “to” are numerical values that can be arbitrarily combined, and the numerical values in the examples are used as the upper and lower numerical values. can also
- Patent Documents 1 to 3 the technology is used to improve the ion conductivity, increase the affinity between the active material used for the negative electrode, positive electrode, etc. when manufacturing a lithium ion battery and the sulfide solid electrolyte to improve the cycle characteristics.
- the challenge is to improve battery performance, such as improving the
- a paste is prepared by mixing a solid electrolyte, other predetermined components, and a solvent, and the paste is applied to form a separator layer. , to form an electrode mixture layer.
- a solid electrolyte that constitutes these layers, and it is effective to use a solid electrolyte with a large specific surface area to improve the density.
- a sulfide solid electrolyte having a large specific surface area of 10 m 2 /g or more has a high viscosity when made into a paste, and not only does the coating aptitude decrease remarkably, but also a large amount of solvent is required to reduce the viscosity of the paste. is required, the drying time is prolonged, and the battery performance is significantly lowered due to the decrease in density.
- Patent Documents 1 to 3 many studies have been conducted on the subject of improving ion conductivity and battery performance, but under the circumstances where practical use of lithium ion batteries is progressing rapidly, attention is paid to mass production. Then, the inventors noticed that no study has been made on a method for improving the performance in the manufacturing process, such as the coating suitability of the paste. The inventors of the present invention have followed the technique of coating the surface of the sulfide solid electrolyte disclosed in Patent Documents 1 to 3 with some kind of compound, and have focused on the compound to be coated on the surface and continued intensive research.
- a sulfide solid electrolyte having a surface area as large as 10 m 2 /g or more by attaching lithium halide to the surface, it has excellent coating suitability when coated as a paste, and can be efficiently applied.
- the present inventors have found that a sulfide solid electrolyte capable of exhibiting excellent battery performance can be obtained.
- Lithium halide has hitherto been used as a raw material for sulfide solid electrolytes, but by adhering it to the surface of sulfide solid electrolytes, the specific surface area is as large as 10 m 2 /g or more. It is a surprising fact that even a sulfide solid electrolyte has an effect of being excellent in coating aptitude when coated as a paste, which has not been recognized at all so far.
- solid electrolyte means an electrolyte that remains solid at 25°C under a nitrogen atmosphere.
- the "sulfide solid electrolyte” obtained by the production method of the present embodiment is a solid electrolyte containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms and having ionic conductivity attributable to lithium atoms.
- Sulfide solid electrolyte includes both a crystalline sulfide solid electrolyte having a crystal structure and an amorphous sulfide solid electrolyte.
- a crystalline sulfide solid electrolyte is a solid electrolyte in which peaks derived from the solid electrolyte are observed in the X-ray diffraction pattern in powder X-ray diffraction (XRD) measurement. It is a material that does not matter whether or not there is a peak derived from the raw material.
- the crystalline sulfide solid electrolyte includes a crystal structure derived from the solid electrolyte, and even if part of the crystal structure is derived from the solid electrolyte, the entire crystal structure is derived from the solid electrolyte. It is a thing. If the crystalline sulfide solid electrolyte has the X-ray diffraction pattern as described above, part of it contains an amorphous sulfide solid electrolyte (also referred to as a "glass component"). It is acceptable. Therefore, crystalline sulfide solid electrolytes include so-called glass ceramics obtained by heating an amorphous solid electrolyte (glass component) to a crystallization temperature or higher.
- the amorphous sulfide solid electrolyte means a halo pattern in which no peaks other than peaks derived from the material are substantially observed in the X-ray diffraction pattern in powder X-ray diffraction (XRD) measurement. It means that the presence or absence of a peak derived from the raw material of the solid electrolyte does not matter.
- XRD powder X-ray diffraction
- the modified sulfide solid electrolyte according to the first form of the present embodiment is A sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, and an epoxy compound, A modified sulfide solid electrolyte having a peak at 2800 to 3000 cm -1 in an infrared absorption spectrum by FT-IR analysis (ATR method), is.
- Sulfide solid electrolytes containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms are obtained by conventional methods, for example, lithium sulfide, diphosphorus pentasulfide, lithium halides, elemental halogens, etc., as raw materials. are typically exemplified by sulfide solid electrolytes. That is, the modified sulfide solid electrolyte of the present embodiment contains a sulfide solid electrolyte having a large BET specific surface area of 10 m 2 /g or more according to the conventional method, and an epoxy compound.
- a conventional sulfide solid electrolyte with a large BET specific surface area of 10 m 2 /g or more in order to achieve a predetermined battery performance, the content is contained at a content necessary to ensure the density of the solid electrolyte in the layer.
- the coating performance of the applied paste was remarkably deteriorated, and it was extremely difficult to efficiently form the positive electrode, the negative electrode, and the electrolyte layer.
- the sulfide solid electrolyte of the present embodiment is dramatically improved in coatability by containing a conventional sulfide solid electrolyte and an epoxy compound. It should be called a "modified sulfide solid electrolyte".
- the modified sulfide solid electrolyte according to the first embodiment has a peak at 2800 to 3000 cm ⁇ 1 in infrared absorption spectrum by FT-IR analysis (ATR method). Since this peak is not detected from the sulfide solid electrolyte, it is considered to be a peak derived from the epoxy compound contained in the modified sulfide solid electrolyte.
- the peak detected at 2800 to 3000 cm -1 is the alkyl chain in the epoxy compound. It is derived from C—H stretching vibration. As described above, the peak is a peak that is not detected in the sulfide solid electrolyte by the conventional method, and is a peak that appears due to the inclusion of the epoxy compound, and the epoxy compound is assumed to exist while maintaining its structure. Conceivable.
- the epoxy compound adheres to the surface of the sulfide solid electrolyte while maintaining its structure. It is speculated that This is because, according to the examples described later, the peak is clearly detected, so it is considered that the peak exists in an easily detectable manner.
- the modified sulfide solid electrolyte of the present embodiment has a lower oil absorption than a sulfide solid electrolyte that does not contain an epoxy compound. Confirmed. It is natural to think that the reduction in oil absorption is due to the epoxy compound adhering to the surface of the sulfide solid electrolyte and blocking at least some of the pores of the sulfide solid electrolyte. It is generally known that oil absorption is related to improvement of coatability in the same way as specific surface area. It is presumed that the adhesion of the epoxy compound to the surface of the sulfide solid electrolyte reduces the oil absorption and improves the coatability.
- the details of the mode of adhesion that is, whether it is physical adhesion or chemical adhesion, are also unknown.
- heteroatoms such as oxygen atoms have the property of easily bonding with lithium atoms, halogen atoms, etc.
- the oxygen atoms contained in the epoxy compound bond with the lithium atoms, halogen atoms, etc. constituting the sulfide solid electrolyte, It is likely to adhere to the surface, ie chemical adhesion.
- the modified sulfide solid electrolyte of the present embodiment even if the adhesion is any of the above, if an epoxy compound is attached to the surface, the oil absorption is easily reduced, and as a result, the coatability is improved. Therefore, it is considered that the battery performance can be improved.
- the modified sulfide solid electrolyte according to the third aspect of the present embodiment has a peak of 0.0 to 5.0 ppm derived from the alkyl chain in the 1 H-NMR spectrum.
- the modified sulfide solid electrolyte according to the present embodiment has this peak, and it is considered that the epoxy compound exists while maintaining its structure in the modified sulfide solid electrolyte.
- the modified sulfide solid electrolyte according to the fourth form of the present embodiment includes, as the epoxy compounds, epoxy compound 1 represented by general formula (1), poxy compound 2 represented by general formula (2) and general formula ( It is specified that at least one compound selected from poxy compounds 3 shown in 3) is used. A detailed description of the epoxy compounds represented by formulas (1) to (3) will be given later.
- the modified sulfide solid electrolyte of the present embodiment if it contains an epoxy compound, it has excellent coatability. , excellent coatability is easily obtained, and the coatability itself is improved.
- an epoxy compound having a certain molecular size or more as represented by the general formulas (1) to (3) it is easy to obtain the effect of reducing oil absorption and improve coatability. It is thought that it can be made easier.
- the content of the epoxy compound is 0.03 parts by mass or more and 25 parts by mass or less with respect to 100 parts by mass of the sulfide solid electrolyte. It is.
- the content of the epoxy compound is within the above range, it is possible to moderately disperse and adhere to the surface of the sulfide solid electrolyte to reduce the oil absorption and maintain the lithium ion conductivity appropriately. The aptitude is likely to be improved, and efficient and excellent battery performance is likely to be exhibited. If the usage amount of the epoxy compound used in producing the modified sulfide solid electrolyte is known, the "content of the epoxy compound" is the usage amount as it is.
- the epoxy compound has a molecular weight of 60 or more.
- the effect of reducing the oil absorption can be easily obtained, and the coatability can be easily improved.
- the epoxy compound 1 in the fourth aspect is the general formula (1), wherein X 11 is a monovalent hydrocarbon having 3 to 24 carbon atoms or a monovalent halogenated hydrocarbon group having 3 to 24 carbon atoms, wherein X 12 and X 13 are hydrogen atoms.
- X 11 is a monovalent hydrocarbon having 3 to 24 carbon atoms or a monovalent halogenated hydrocarbon group having 3 to 24 carbon atoms
- X 12 and X 13 are hydrogen atoms.
- those defined in the seventh form adhere to the surface of the sulfide solid electrolyte and easily reduce oil absorption, thereby improving coatability. It is easy to perform and it is easy to express excellent battery performance efficiently.
- the epoxy compound 2 in the fourth aspect is a group represented by the general formula (2a) in which X 21 is the general formula (2). and X 22 and X 23 are hydrogen atoms, and in the general formula (2a), R 21 is a divalent hydrocarbon group having 1 to 4 carbon atoms, and R 22 is a monovalent hydrocarbon group having 2 to 24 carbon atoms It is a compound that is a hydrocarbon group.
- the epoxy compounds 2 represented by the general formula (2) those defined in the eighth form adhere to the surface of the sulfide solid electrolyte and easily reduce oil absorption, thereby improving coatability. It is easy to perform and it is easy to express excellent battery performance efficiently.
- the epoxy compound 3 in the fourth aspect is the general formula (3), wherein X 31 is a group represented by the general formula (3a) and X 32 and X 33 are hydrogen atoms, and in the general formula (3a), R 31 is a divalent hydrocarbon group having 1 to 4 carbon atoms, and R 32 is a divalent hydrocarbon group having 2 to 8 carbon atoms a hydrocarbon group, X 34 and X 36 are groups represented by general formula (3b), X 35 is a monovalent hydrocarbon group having 1 to 4 carbon atoms, and in general formula (3b), R It is a compound in which 34 to R 36 are monovalent hydrocarbon groups having 1 to 4 carbon atoms.
- those defined in the ninth form adhere to the surface of the sulfide solid electrolyte and easily reduce oil absorption, thereby improving coatability. It is easy to perform and it is easy to express excellent battery performance efficiently.
- the epoxy compound 3 in the fourth form is a group represented by the general formula (3) in which X 31 is represented by the general formula (3a) and X 22 and X 23 are hydrogen atoms, and in general formula (3a), R 31 is a divalent hydrocarbon group having 1 to 4 carbon atoms, R 32 is a single bond, and X 34 to X 36 is a compound in which 36 is a monovalent hydrocarbon group having 1 to 8 carbon atoms.
- epoxy compounds 3 represented by the general formula (3) those defined in the tenth form adhere to the surface of the sulfide solid electrolyte, as with the compound defined in the ninth form, It is easy to reduce the oil absorption amount, which makes it easy to improve the coating suitability, and it is easy to efficiently express excellent battery performance.
- a method for producing a modified sulfide solid electrolyte according to the eleventh form of the present embodiment includes: mixing a sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, an epoxy compound, and an organic solvent; removing the organic solvent; That's what it means.
- the modified sulfide solid electrolyte of the present embodiment is not particularly limited in its production method as long as it contains an epoxy compound.
- the epoxy compound can be present so as to adhere to the surface of the sulfide solid electrolyte, so it is excellent in coatability and efficient.
- the modified sulfide solid electrolyte of the present embodiment can be produced more efficiently.
- the organic solvent used in the method for producing the eleventh aspect is an aliphatic hydrocarbon solvent or an alicyclic hydrocarbon solvent. , aromatic hydrocarbon solvents, ester solvents, nitrile solvents and ether solvents.
- the electrode mixture according to the thirteenth form of the present embodiment includes the modified sulfide solid electrolyte or the like of the first form and an electrode active material, That's what it means.
- the lithium ion battery according to the fourteenth form of the present embodiment includes at least one of the modified sulfide solid electrolyte or the like of the first form and the electrode active material of the thirteenth form, That's what it means.
- the modified sulfide solid electrolyte of the present embodiment has excellent coating aptitude when applied as a paste, and can efficiently exhibit excellent battery performance. Therefore, since the electrode mixture containing the modified sulfide solid electrolyte of the present embodiment also has excellent coating suitability, a lithium ion battery can be efficiently produced, and the obtained lithium ion battery is excellent. It has battery performance.
- the modified sulfide solid electrolyte according to the fifteenth form of the present embodiment is A sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom, and an epoxy compound, That's what it means.
- the modified sulfide solid electrolyte having such a configuration also has the same effects as the modified sulfide solid electrolyte of the first embodiment, that is, it has excellent coating suitability when coated as a paste, and is excellent in efficiency. This is effective in that it is possible to express a good battery performance.
- the modified sulfide solid electrolyte of the first form has a peak at 2800 to 3000 cm -1 in the infrared absorption spectrum by FT-IR analysis (ATR method) in the modified sulfide solid electrolyte of the fifteenth form. It can also be said that it is defined as having. However, the peak is a peak derived from the epoxy compound contained in the modified sulfide solid electrolyte as described above, and the peak is expressed by containing the epoxy compound. .
- the electrode mixture according to the sixteenth form of the present embodiment is comprising the modified sulfide solid electrolyte of the fifteenth form and an electrode active material, That's what it means.
- the lithium ion battery according to the seventeenth form of the present embodiment includes at least one of the modified sulfide solid electrolyte etc. of the fifteenth form and the electrode active material of the sixteenth form, That's what it means.
- the electrode mixture and the lithium ion battery are the same as the electrode mixture of the thirteenth embodiment and the lithium ion battery of the fourteenth embodiment.
- the modified sulfide solid electrolyte of the present embodiment includes a sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms, and an epoxy compound, It has a peak at 2800 to 3000 cm ⁇ 1 in an infrared absorption spectrum obtained by FT-IR analysis (ATR method). Since this peak is not detected in a sulfide solid electrolyte containing no epoxy compound, it is a peak derived from an epoxy compound.
- FT-IR analysis refers to analysis using a Fourier transform infrared spectrophotometer, meaning measurement employing a total reflection measurement method (ATR method), and the infrared absorption spectrum is FT-IR It is the spectrum measured under the following conditions using the apparatus.
- Measurement method total reflection measurement method (ATR method) Measurement wavenumber range: 650 to 4000 cm -1 Light source: Grover lamp (SiC) Detector: DTGS detector Resolution: 4 cm -1 Measurement time: 1 second/time Accumulated times: 256 times
- the peak at 2800 to 3000 cm ⁇ 1 in the infrared absorption spectrum is derived from C—H stretching vibration.
- the peak is considered to be derived from the epoxy compound, and more specifically derived from the C—H bond of the epoxy compound.
- the epoxy compound has an alkyl chain
- the peak is derived from the C—H bond (C—H stretching vibration) of the alkyl chain in the epoxy compound. Since the peak is clearly detected in the modified sulfide solid electrolyte of the present embodiment, the epoxy compound is contained in a manner that is easy to detect, that is, it exists so as to adhere to the surface of the sulfide solid electrolyte. It is considered that It is believed that such presence can reduce the oil absorption of the sulfide solid electrolyte, resulting in excellent coatability.
- the modified sulfide solid electrolyte of the present embodiment preferably has a 0.0 to 5.0 ppm peak derived from the alkyl chain of the epoxy compound in the 1 H-NMR spectrum.
- peaks in 1 H-NMR spectra are measured under the following conditions using a nuclear magnetic resonance spectrometer (NMR spectrometer). Observation nucleus: 1 H Resonance frequency: 500MHz Probe: 5mm ⁇ TCI cryoprobe Measurement temperature: 25°C Accumulated times: 16 times
- the modified sulfide solid electrolyte of the present embodiment has a peak of 2800 to 3000 cm ⁇ 1 in the infrared absorption spectrum, and the peak is clearly detected, that is, the sulfide solid electrolyte It is thought that it exists so as to adhere to the surface of the The detection of the peak derived from the alkyl chain in the 1 H-NMR spectrum also means that it exists so as to adhere to the surface of the sulfide solid electrolyte, as in the infrared absorption spectrum. It is considered that work suitability can be obtained.
- a modified sulfide solid electrolyte obtained by mixing a sulfide solid electrolyte and an epoxy compound in an organic solvent is added to a solvent such as toluene to form a slurry.
- a solvent such as toluene
- GC/MS method gas chromatography mass spectrometry
- no epoxy compounds were detected.
- 1 H-NMR measurement a chemical shift of a group (such as an alkyl group) derived from the epoxy compound is detected. From this phenomenon, in the modified sulfide solid electrolyte, the epoxy compound strongly adheres to the surface of the sulfide solid electrolyte, and this adhesion reduces the oil absorption and is considered to have excellent coatability. .
- the epoxy compound adheres to the surface of the sulfide solid electrolyte, it may adhere so as to cover the entire surface of the sulfide solid electrolyte, or may adhere to a part thereof.
- the epoxy compound is not particularly limited as long as it is a compound having an epoxy ring, and from the viewpoint of more efficiently improving the coating suitability and improving the battery performance, it is represented by the following general formulas (1) to (3).
- Epoxy compounds 1 to 3 are preferably mentioned, respectively.
- Epoxy compound 1 is a compound represented by the following general formula (1).
- X 11 to X 13 are each independently a hydrogen atom, a halogen atom, a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group, and at least one of X 11 to X 13 is a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group.
- the monovalent hydrocarbon groups of X 11 to X 13 include monovalent aliphatic hydrocarbon groups, alicyclic hydrocarbon groups and aromatic hydrocarbon groups, and aliphatic hydrocarbon groups and alicyclic hydrocarbon groups.
- a hydrogen group is preferred, and an aliphatic hydrocarbon group is more preferred.
- Preferred examples of the aliphatic hydrocarbon group include alkyl groups and alkenyl groups, with alkyl groups being preferred.
- the number of carbon atoms in the aliphatic hydrocarbon group is preferably 1 or more, more preferably 2 or more, still more preferably 4 or more, and even more preferably 6 or more, and the upper limit is preferably 24 or less, more preferably is 20 or less, more preferably 16 or less.
- alkenyl group it is 2 or more, preferably 4 or more, and the upper limit is preferably 24 or less, more preferably 20 or less, and still more preferably 16 or less.
- the aliphatic hydrocarbon groups of X 11 to X 13 may be linear or branched, preferably linear. Also, the aliphatic hydrocarbon group may be partially substituted with a hydroxyl group or the like. When multiple of X 11 to X 13 are aliphatic hydrocarbon groups, the multiple aliphatic hydrocarbon groups may be the same or different.
- Preferred examples of the monovalent alicyclic hydrocarbon groups of X 11 to X 13 include cycloalkyl groups and cycloalkenyl groups, with cycloalkyl groups being preferred.
- the number of carbon atoms in the alicyclic hydrocarbon group is 3 or more, preferably 4 or more, and the upper limit is preferably 12 or less, more preferably 8 or less, and still more preferably 6 or less.
- the alicyclic hydrocarbon groups of X 11 to X 13 may be partially substituted with a hydroxyl group, the above monovalent aliphatic hydrocarbon group (eg, alkyl group, alkenyl group), or the like. Further, when a plurality of X 11 to X 13 are alicyclic hydrocarbon groups, the plurality of alicyclic hydrocarbon groups may be the same or different.
- the monovalent aromatic hydrocarbon groups of X 11 to X 13 include phenyl group, naphthyl group, biphenyl group, diphenylmethyl group, trityl group, anthranyl group, perylenyl group, pyrenyl group and the like.
- the monovalent aromatic hydrocarbon groups of X 11 to X 13 may be partially substituted with hydroxyl groups, the above monovalent aliphatic hydrocarbon groups (eg, alkyl groups, alkenyl groups), and the like. Also. When multiple of X 11 to X 13 are aromatic hydrocarbon groups, the multiple aromatic hydrocarbon groups may be the same or different.
- Examples of the monovalent halogenated hydrocarbon groups for X 11 to X 13 include groups in which the hydrocarbon groups exemplified above as the hydrocarbon groups for X 11 to X 13 are partially substituted with halogen atoms.
- the hydrocarbon group substituted with a halogen atom is preferably an aliphatic hydrocarbon group or an alicyclic hydrocarbon group, more preferably an aliphatic hydrocarbon group, and an alkyl group. More preferred.
- the halogen atoms contained in the monovalent halogenated hydrocarbon groups of X 11 to X 13 are preferably fluorine, chlorine, bromine and iodine, more preferably fluorine, chlorine and bromine, and still more preferably fluorine.
- the number of halogen atoms in the halogenated hydrocarbon group varies depending on the number of carbon atoms in the hydrocarbon group, so it cannot be said unconditionally, but it is preferably 1 or more, more preferably 2 or more, still more preferably 3 or more, and still more preferably 1 or more. It is preferably 6 or more, and the upper limit is preferably 16 or less, more preferably 12 or less, and still more preferably 9 or less.
- the halogen atoms of X 11 to X 13 are preferably fluorine, chlorine, bromine and iodine, more preferably fluorine, chlorine and bromine, and still more preferably fluorine, like the halogen atoms that can be contained in the halogenated hydrocarbon group.
- At least one of X 11 to X 13 is a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group, that is, one, two or three of X 11 to X 13 are monovalent hydrocarbon groups or It may be a monovalent halogenated hydrocarbon group, and from the viewpoint of improving coatability, one is preferably a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group.
- X 11 is a monovalent hydrocarbon group having 1 to 24 carbon atoms or a monovalent halogenated hydrocarbon group having 1 to 24 carbon atoms
- X 12 and X 13 are preferably hydrogen atoms
- the hydrocarbon group is preferably an aliphatic hydrocarbon group, more preferably an alkyl group or an alkenyl group, and still more preferably an alkyl group, as described above.
- the number of carbon atoms is also as described above, and when it is an alkyl group, it is preferably 1 or more, more preferably 2 or more, still more preferably 4 or more, and even more preferably 6 or more, and the upper limit is preferably 24. Below, more preferably 20 or less, still more preferably 16 or less.
- Epoxy compound 2 is a compound represented by the following general formula (2).
- X 21 to X 23 are each independently a hydrogen atom, a halogen atom, a monovalent hydrocarbon group, a monovalent halogenated hydrocarbon group or a group represented by general formula (2a).
- X 21 to X 23 is a group represented by general formula (2a).
- R 21 is a divalent hydrocarbon group and R 22 is a hydrogen atom, a halogen atom, a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group.
- the monovalent hydrocarbon groups for X 21 to X 23 include the same hydrocarbon groups, preferably an aliphatic hydrocarbon group or an alicyclic hydrocarbon group, more preferably an aliphatic hydrocarbon group.
- the aliphatic hydrocarbon groups alkyl groups and alkenyl groups are more preferred, and alkyl groups are even more preferred.
- Examples of the monovalent halogenated hydrocarbon groups for X 21 to X 23 include those exemplified as the monovalent halogenated hydrocarbon groups for X 11 to X 13 above .
- Examples of the monovalent hydrocarbon group include those exemplified.
- At least one of X 21 to X 23 is a group represented by general formula (2a). That is, one, two or three of X 21 to X 23 may be groups represented by general formula (2a), preferably one.
- the divalent hydrocarbon for R 21 is a hydrocarbon obtained by removing one hydrogen atom from the above monovalent hydrocarbon groups for X 21 to X 23 A hydrogen group is mentioned. Therefore, the divalent hydrocarbon of R 21 includes a divalent aliphatic hydrocarbon group, an alicyclic hydrocarbon group and an aromatic hydrocarbon group, and an aliphatic hydrocarbon group and an alicyclic hydrocarbon group. is preferred, and an aliphatic hydrocarbon group is more preferred. As the divalent aliphatic hydrocarbon group, an alkylene group and an alkenylene group are preferable, and an alkylene group is more preferable.
- the number of carbon atoms in the alkylene group is preferably 1 or more, and the upper limit is preferably 8 or less, more preferably 6 or less, still more preferably 4 or less, and still more preferably 2 or less.
- the number of carbon atoms is preferably 2 or more, and the upper limit is the same as that of the alkylene group.
- the monovalent hydrocarbon represented by R 22 is the same as the monovalent hydrocarbon group represented by X 21 to X 23 , that is, a monovalent aliphatic group hydrocarbon groups, alicyclic hydrocarbon groups and aromatic hydrocarbon groups, with aliphatic hydrocarbon groups and aromatic hydrocarbon groups being preferred.
- the aliphatic hydrocarbon group is preferably an alkyl group or an alkenyl group, more preferably an alkyl group, and the aliphatic hydrocarbon group may be linear or branched.
- the number of carbon atoms is preferably 1 or more, more preferably 2 or more, still more preferably 4 or more, and the upper limit is preferably 24 or less, more preferably 16 or less, and still more preferably. is 12 or less, more preferably 10 or less.
- the aromatic hydrocarbon group is preferably a phenyl group, a biphenyl group, a diphenylmethyl group, or a trityl group, more preferably a phenyl group, a diphenylmethyl group, or a trityl group, and still more preferably a phenyl group or a trityl group.
- these aromatic hydrocarbon groups may be substituted with halogen atoms, hydroxyl groups, monovalent aliphatic hydrocarbon groups (alkyl groups, alkenyl groups), and the like.
- Examples of the monovalent halogenated hydrocarbon group for R 22 include groups in which the above monovalent hydrocarbon group for R 22 is partially substituted with a halogen atom.
- the halogen atom contained in the monovalent halogenated hydrocarbon represented by R 22 and the halogen atom represented by R 22 are preferably fluorine, chlorine, bromine and iodine, more preferably fluorine, chlorine and bromine, and still more preferably fluorine.
- X 21 is a group represented by the general formula (2a)
- X 22 and X 23 are hydrogen atoms
- R 21 is a divalent hydrocarbon group having 1 to 8 carbon atoms
- R 22 is preferably a monovalent hydrocarbon group having 1 to 24 carbon atoms.
- the monovalent hydrocarbon group is as described above.
- the monovalent hydrocarbon group of R 22 is a phenyl group, it is preferably substituted with a monovalent aliphatic hydrocarbon group, preferably with an alkyl group.
- the alkyl group preferably has 1 or more carbon atoms, more preferably 2 or more carbon atoms, and still more preferably 4 or more carbon atoms, and the upper limit is preferably 24 or less, more preferably 12 or less, and still more preferably 8 or less. Preferably it is 6 or less.
- the aliphatic hydrocarbon group may be linear or branched, preferably branched.
- R 22 is preferably an aliphatic hydrocarbon group having a quaternary carbon atom, particularly preferably a tert-butyl group.
- Epoxy compound 3 is a compound represented by the following general formula (3).
- X 31 to X 33 are each independently a hydrogen atom, a halogen atom, a monovalent hydrocarbon group, a monovalent halogenated hydrocarbon group, or a group represented by general formula (3a).
- X 31 to X 33 is a group represented by general formula (3a).
- R 31 and R 32 are each independently a single bond or a divalent hydrocarbon group
- X 34 to X 36 are each independently a hydrogen atom, a halogen atom or a monovalent hydrocarbon group.
- R 33 to R 36 are each independently a hydrogen atom, a halogen atom, a monovalent hydrocarbon group or a monovalent halogenated hydrocarbon group.
- Examples of the monovalent hydrocarbon groups for X 31 to X 33 include the monovalent aliphatic hydrocarbon groups, alicyclic hydrocarbon groups and aromatic Examples thereof include the same hydrocarbon groups, preferably an aliphatic hydrocarbon group or an alicyclic hydrocarbon group, more preferably an aliphatic hydrocarbon group.
- alkyl groups and alkenyl groups are more preferred, and alkyl groups are even more preferred.
- Examples of the monovalent halogenated hydrocarbon groups for X 31 to X 33 include those exemplified as the monovalent halogenated hydrocarbon groups for X 11 to X 13 above .
- Examples of the monovalent hydrocarbon group include those exemplified.
- At least one of X 31 to X 33 is a group represented by general formula (3a). That is, one, two or three of X 31 to X 33 may be groups represented by general formula (3a), preferably one.
- the divalent hydrocarbons represented by R 31 and R 32 include one hydrogen atom from the above monovalent hydrocarbon groups represented by X 31 to X 33 . Included are hydrocarbon groups that have been removed. Therefore, the divalent hydrocarbon of R 31 includes a divalent aliphatic hydrocarbon group, an alicyclic hydrocarbon group, an aromatic hydrocarbon group, an aliphatic hydrocarbon group, an alicyclic hydrocarbon group is preferred, and an aliphatic hydrocarbon group is more preferred. As the divalent aliphatic hydrocarbon group, an alkylene group and an alkenylene group are preferable, and an alkylene group is more preferable.
- the number of carbon atoms in the alkylene group is preferably 1 or more, and the upper limit is preferably 8 or less, more preferably 6 or less, still more preferably 4 or less, and still more preferably 2 or less.
- the number of carbon atoms is preferably 2 or more, and the upper limit is the same as that of the alkylene group.
- the monovalent hydrocarbon groups and monovalent halogenated hydrocarbon groups for X 34 to X 36 are the same as those exemplified as the above monovalent hydrocarbon groups and halogenated hydrocarbon groups for X 31 to X 33 . mentioned.
- R 33 in —OR 33 of X 34 to X 36 , the monovalent hydrocarbon group of R 34 to R 36 in general formula (3b), and the monovalent halogenated hydrocarbon group of X 34 to X 36 The same as the monovalent hydrocarbon group and the monovalent halogenated hydrocarbon group can be mentioned.
- X 31 is a group represented by the general formula (3a)
- X 32 and X 33 are hydrogen atoms
- R 31 is a divalent hydrocarbon group having 1 to 8 carbon atoms
- R 32 is a divalent hydrocarbon group having 1 to 8 carbon atoms
- X 34 and X 36 are groups represented by general formula (3b) and X 35 is a monovalent hydrocarbon group having 1 to 24 carbon atoms
- R 34 to R 36 are preferably monovalent hydrocarbon groups having 1 to 24 carbon atoms.
- the divalent hydrocarbon groups of R 31 and R 32 are as described above, and the number of carbon atoms in the divalent hydrocarbon group of R 31 is preferably 1 or more, and the upper limit is preferably It is 8 or less, more preferably 6 or less, even more preferably 4 or less, and even more preferably 2 or less.
- the number of carbon atoms in the divalent hydrocarbon group of R 32 is preferably 1 or more, more preferably 2 or more, and the upper limit is preferably 8 or less, more preferably 6 or less, and still more preferably 4 or less.
- the monovalent hydrocarbon groups of X 35 and R 34 to R 36 are as described above.
- the number of carbon atoms in the monovalent hydrocarbon group of X 35 is preferably 1 or more, and the upper limit is preferably It is 24 or less, more preferably 12 or less, still more preferably 8 or less, even more preferably 4 or less, and particularly preferably 2 or less.
- the number of carbon atoms in the monovalent hydrocarbon groups of R 34 to R 36 is also the same as the number of carbon atoms in the monovalent hydrocarbon group of X 35 .
- X 31 is a group represented by the general formula (3a), X 22 and X 23 are hydrogen atoms, and in the general formula (3a),
- R 31 is a divalent hydrocarbon group having 1 to 8 carbon atoms
- R 32 is a single bond
- X 34 to X 36 are monovalent hydrocarbon groups having 1 to 24 carbon atoms.
- the divalent hydrocarbon group for R 31 is as described above. It is more preferably 6 or less, still more preferably 4 or less, and even more preferably 2 or less.
- the monovalent hydrocarbon groups of X 34 to X 36 are as described above, and the number of carbon atoms in the monovalent hydrocarbon groups of X 34 and X 36 is preferably 1 or more, and the upper limit is preferably It is 24 or less, more preferably 12 or less, still more preferably 8 or less, even more preferably 4 or less, and particularly preferably 2 or less.
- the number of carbon atoms in the monovalent hydrocarbon group of X 35 is preferably 1 or more, more preferably 2 or more, still more preferably 4 or more, and the upper limit is preferably 24 or less, more preferably 12 or less, and further It is preferably 8 or less, and even more preferably 6 or less.
- the hydrocarbon group for X 35 may be linear or branched, preferably branched, preferably an aliphatic hydrocarbon group having a quaternary carbon atom, particularly tert -Butyl groups are preferred.
- the epoxy compound contained in the modified sulfide solid electrolyte of the present embodiment preferably has a molecular weight of 60 or more, more preferably 70 or more, and the upper limit is preferably 400 or less, more preferably 380 or less, and even more preferably 350 or less.
- the content of the epoxy compound may vary depending on the type of epoxy compound used, it cannot be generalized, but it is preferably 0.03 parts by mass or more, more preferably 0.05 parts by mass, with respect to 100 parts by mass of the sulfide solid electrolyte. Part by mass, more preferably 0.1 part by mass or more, still more preferably 0.5 part by mass, and the upper limit is preferably 25 parts by mass or less, more preferably 20 parts by mass or less.
- the sulfide solid electrolyte that can be used in the present embodiment contains lithium atoms, sulfur atoms, phosphorus atoms, and halogen atoms, and can be used without particular limitation as long as it has a BET specific surface area of 10 m 2 /g or more. , a commercially available product can be used as it is, or it can be used after being manufactured. A method for producing a sulfide solid electrolyte that can be used in the present embodiment will be described.
- the sulfide solid electrolyte that can be used in the present embodiment is produced, for example, by mixing two or more raw materials selected from compounds containing at least one atom of a lithium atom, a sulfur atom, a phosphorus atom, and a halogen atom. obtained by the method.
- the compound that can be used as a raw material contains at least one atom of a lithium atom, a sulfur atom, a phosphorus atom, and a halogen atom.
- Compounds that can be used as raw materials other than the above include, for example, compounds containing at least one atom selected from the above four atoms and containing atoms other than the four atoms, more specifically lithium oxide, Lithium compounds such as lithium hydroxide and lithium carbonate; alkali metal sulfides such as sodium sulfide, potassium sulfide, rubidium sulfide, and cesium sulfide; silicon sulfide, germanium sulfide, boron sulfide, gallium sulfide, tin sulfide (SnS, SnS2 ), sulfide metal sulfides such as aluminum and zinc sulfide; phosphoric acid compounds such as sodium phosphate and lithium phosphate; aluminum halides, silicon halides, germanium halides, arsenic halides, selenium halides, tin halides, antimony halides, metal halides such as telluri
- halogen atoms chlorine, bromine and iodine atoms are preferable, and bromine and iodine atoms are more preferable, from the viewpoint of obtaining a sulfide solid electrolyte having high ion conductivity more easily.
- these atoms may be used singly or in combination. That is, taking lithium halide as an example, lithium bromide may be used alone, lithium iodide may be used alone, or lithium bromide and lithium iodide may be used in combination. .
- the compounds that can be used as raw materials include, among the above , lithium sulfide ; F 2 ), chlorine (Cl 2 ), bromine (Br 2 ), and iodine (I 2 ); lithium halides such as lithium fluoride, lithium chloride, lithium bromide, and lithium iodide; Phosphorus pentasulfide is preferable among phosphorus, chlorine (Cl 2 ), bromine (Br 2 ), and iodine (I 2 ) are preferable among simple halogens, and lithium chloride, lithium bromide, and lithium iodide are preferable among lithium halides. preferable.
- Combinations of compounds that can be used as raw materials include, for example, a combination of lithium sulfide, diphosphorus pentasulfide and a lithium halide, and a combination of lithium sulfide, diphosphorus pentasulfide and an elemental halogen.
- Lithium iodide and lithium chloride are preferable, and chlorine, bromine and iodine are preferable as simple halogens.
- the lithium sulfide is preferably in the form of particles.
- the average particle size (D 50 ) of the lithium sulfide particles is preferably 10 ⁇ m or more and 2000 ⁇ m or less, more preferably 30 ⁇ m or more and 1500 ⁇ m or less, and even more preferably 50 ⁇ m or more and 1000 ⁇ m or less.
- the average particle size (D 50 ) is the particle size that reaches 50% of the whole when the particle size distribution integrated curve is drawn, and the particle size is accumulated sequentially from the smallest particle size, and the volume distribution is , for example, the average particle size that can be measured using a laser diffraction/scattering particle size distribution analyzer.
- the solid raw materials exemplified above those having an average particle size approximately equal to that of the lithium sulfide particles are preferable, that is, those having an average particle size within the same range as the lithium sulfide particles. preferable.
- the ratio of lithium sulfide to the total of lithium sulfide and diphosphorus pentasulfide is from the viewpoint of obtaining higher chemical stability
- the PS 4 fraction is From the viewpoint of obtaining improved high ionic conductivity, it is preferably 60 mol% or more, more preferably 65 mol% or more, and still more preferably 68 mol% or more, and the upper limit is preferably 80 mol% or less, more preferably 78 mol% or less. More preferably, it is 76 mol % or less.
- the content of lithium sulfide and diphosphorus pentasulfide with respect to the total of these is preferably 60 mol% or more, more preferably is 65 mol % or more, more preferably 70 mol % or more, and the upper limit is preferably 100 mol % or less, more preferably 90 mol % or less, and still more preferably 80 mol % or less.
- the total of lithium bromide and lithium iodide is preferably 1 mol% or more, more preferably 20 mol% or more, still more preferably 40 mol% or more, still more preferably 50 mol% or more, and the upper limit is preferably 99 mol% or less, more preferably 90 mol%. Below, more preferably 80 mol % or less, still more preferably 70 mol % or less.
- the total number of moles of lithium sulfide and phosphorus pentasulfide excluding the same number of moles of lithium sulfide as the number of moles of the halogen simple substance is preferably in the range of 60 to 90%, more preferably in the range of 65 to 85%.
- the content of elemental halogen with respect to the total amount of lithium sulfide, phosphorus pentasulfide, and elemental halogen is 1 to 50 mol%. is preferred, 2 to 40 mol% is more preferred, 3 to 25 mol% is still more preferred, and 3 to 15 mol% is even more preferred.
- the content of elemental halogen ( ⁇ mol%) and the content of lithium halide ( ⁇ mol%) relative to the total amount are as follows: It preferably satisfies the formula (1), more preferably satisfies the following formula (2), further preferably satisfies the following formula (3), and even more preferably satisfies the following formula (4). 2 ⁇ 2 ⁇ + ⁇ 100 (1) 4 ⁇ 2 ⁇ + ⁇ 80 (2) 6 ⁇ 2 ⁇ + ⁇ 50 (3) 6 ⁇ 2 ⁇ + ⁇ 30 (4)
- Mixing of two or more raw materials selected from compounds containing at least one atom selected from a lithium atom, a sulfur atom, a phosphorus atom and a halogen atom can be carried out using, for example, a mixer. Moreover, it can also be carried out using a stirrer, a pulverizer, or the like. This is because the raw materials can be mixed even when a stirrer is used, and the raw materials are pulverized when a pulverizer is used, but mixing also occurs at the same time. That is, the sulfide solid electrolyte used in the present embodiment is prepared by stirring, mixing, pulverizing, or It can also be said that the processing can be performed by combining any of these.
- the stirrer and mixer include, for example, a mechanical stirring mixer that is equipped with stirring blades in the reaction vessel and capable of stirring (mixing by stirring, which can also be referred to as stirring and mixing).
- mechanical stirring mixers include high-speed stirring mixers and double-arm mixers.
- the high-speed stirring mixer includes a vertical shaft rotary mixer, a horizontal shaft rotary mixer, and the like, and either type of mixer may be used.
- the shape of the stirring impeller used in the mechanical stirring mixer includes blade type, arm type, anchor type, paddle type, full zone type, ribbon type, multi-blade type, double arm type, shovel type, twin blade type, Flat blade type, C type blade type, etc., and from the viewpoint of promoting the reaction of raw materials more efficiently, shovel type, flat blade type, C type blade type, anchor type, paddle type, full zone type, etc. are preferable. Anchor type, paddle type and full zone type are more preferred.
- the rotation speed of the stirring blades may be appropriately adjusted according to the volume and temperature of the fluid in the reaction vessel, the shape of the stirring blades, etc., and is not particularly limited, but is usually 5 rpm or more and 400 rpm or less. From the viewpoint of promoting the reaction of the raw materials more efficiently, the rotation speed is preferably 10 rpm or more and 300 rpm or less, more preferably 15 rpm or more and 250 rpm or less, and even more preferably 20 rpm or more and 200 rpm or less.
- the temperature conditions for mixing using a mixer are not particularly limited, and are usually -30 to 120°C, preferably -10 to 100°C, more preferably 0 to 80°C, and still more preferably 10 to 60°C. is.
- the mixing time is usually 0.1 to 500 hours, preferably 1 to 450 hours, more preferably 10 to 425 hours, still more preferably 20 to 400 hours, from the viewpoint of making the dispersion state of the raw materials more uniform and promoting the reaction. hours, more preferably 40 to 375 hours.
- a method of performing mixing accompanied by pulverization using a pulverizer is a method that has been conventionally employed as a solid-phase method (mechanical milling method).
- a medium-type pulverizer using a pulverizing medium can be used.
- Media-type pulverizers are broadly classified into container-driven pulverizers and medium-agitation pulverizers. Examples of the container-driven pulverizer include a stirring tank, a pulverizing tank, or a combination of these, such as a ball mill and a bead mill.
- medium agitating pulverizers include impact pulverizers such as cutter mills, hammer mills and pin mills; tower pulverizers such as tower mills; stirring tank pulverizers such as attritors, aquamizers and sand grinders; circulation tank-type pulverizers such as pearl mills; circulation tube-type pulverizers; annular-type pulverizers such as coball mills; continuous dynamic pulverizers; Among them, ball mills and bead mills exemplified as container-driven pulverizers are preferred, and planetary-type pulverizers are particularly preferred, in view of the ease of adjusting the particle size of the resulting sulfide.
- impact pulverizers such as cutter mills, hammer mills and pin mills
- tower pulverizers such as tower mills
- stirring tank pulverizers such as attritors, aquamizers and sand grinders
- circulation tank-type pulverizers
- pulverizers can be appropriately selected according to the desired scale, etc.
- container-driven pulverizers such as ball mills and bead mills can be used.
- other types of pulverizers may be used.
- a wet pulverizer capable of coping with wet pulverization is preferable.
- wet pulverizers include wet bead mills, wet ball mills, wet vibration mills, and the like.
- a wet bead mill used as a is preferred.
- dry pulverizers such as dry medium pulverizers such as dry bead mills, dry ball mills and dry vibration mills, and dry non-medium pulverizers such as jet mills can also be used.
- a flow-type pulverizer that is capable of circulating and operating as necessary.
- a pulverizer that circulates between a pulverizer (pulverization mixer) for pulverizing slurry and a temperature holding tank (reaction vessel).
- the size of the beads and balls used in the ball mill and bead mill may be appropriately selected according to the desired particle size, throughput, etc.
- the diameter of the beads is usually 0.05 mm ⁇ or more, preferably 0.1 mm ⁇ or more It is more preferably 0.3 mm ⁇ or more, and the upper limit is usually 5.0 mm ⁇ or less, preferably 3.0 mm ⁇ or less, and more preferably 2.0 mm ⁇ or less.
- the diameter of the ball is usually 2.0 mm ⁇ or more, preferably 2.5 mm ⁇ or more, more preferably 3.0 mm ⁇ or more, and the upper limit is usually 20.0 mm ⁇ or less, preferably 15.0 mm ⁇ or less, more preferably 10.0 mm ⁇ or less.
- Materials include, for example, metals such as stainless steel, chrome steel and tungsten carbide; ceramics such as zirconia and silicon nitride; and minerals such as agate.
- the number of revolutions varies depending on the scale of the treatment and cannot be generalized. It is usually 1,000 rpm or less, preferably 900 rpm or less, more preferably 800 rpm or less, still more preferably 700 rpm or less.
- the pulverization time varies depending on the scale of the treatment and cannot be generalized. hours, and the upper limit is usually 100 hours or less, preferably 72 hours or less, more preferably 48 hours or less, and even more preferably 36 hours or less.
- the size and material of the medium (beads, balls) to be used, the number of rotations of the rotor, time, etc., it is possible to perform mixing, stirring, pulverization, or a combination of any of these treatments.
- the particle size of the sulfide can be adjusted.
- solvent In the above mixing, a solvent can be added to and mixed with the above raw materials.
- the solvent various solvents that are widely called organic solvents can be used.
- solvent it is possible to widely employ solvents that have been conventionally used in the production of solid electrolytes.
- hydrocarbon solvents such as aliphatic hydrocarbon solvents, alicyclic hydrocarbon solvents, and aromatic hydrocarbon solvents Solvents can be mentioned.
- Aliphatic hydrocarbons include, for example, hexane, pentane, 2-ethylhexane, heptane, octane, decane, undecane, dodecane, and tridecane
- alicyclic hydrocarbons include cyclohexane, methylcyclohexane, and the like.
- aromatic hydrocarbon solvents include benzene, toluene, xylene, mesitylene, ethylbenzene, tert-butylbenzene, trifluoromethylbenzene, nitrobenzene and the like.
- solvents containing atoms other than carbon atoms and hydrogen atoms such as heteroatoms such as nitrogen atoms, oxygen atoms, sulfur atoms, and halogen atoms, are also included.
- Such a solvent has the property of easily forming a complex with a compound containing a lithium atom, a phosphorus atom, a sulfur atom and a halogen atom used as a raw material (hereinafter, such a solvent is referred to as a "complexing agent").
- sulfide solid electrolyte It is also referred to as sulfide solid electrolyte.), and has the property of making it easier for halogen atoms to remain within the structure of the sulfide solid electrolyte, which is useful in that higher ionic conductivity can be obtained.
- a complexing agent include, for example, ether solvents, ester solvents, alcohol solvents, aldehyde solvents, and ketone solvents containing an oxygen atom as a heteroatom.
- Ether solvents include dimethyl ether, diethyl ether, tert-butyl methyl ether, dimethoxymethane, dimethoxyethane, diethylene glycol dimethyl ether (diglyme), triethylene oxide glycol dimethyl ether (triglyme), and aliphatic ethers such as diethylene glycol and triethylene glycol; Alicyclic ethers such as ethylene oxide, propylene oxide, tetrahydrofuran, tetrahydropyran, dimethoxytetrahydrofuran, cyclopentyl methyl ether, dioxane; heterocyclic ethers such as furan, benzofuran, benzopyran; methylphenyl ether (anisole), ethylphenyl ether, dibenzyl Aromatic ethers such as ether and diphenyl ether are preferred.
- ester solvents include methyl formate, ethyl formate, methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate; methyl propionate, ethyl propionate, dimethyl oxalate, diethyl oxalate, dimethyl malonate, diethyl malonate, succinic acid; Aliphatic esters such as dimethyl and diethyl succinate; Alicyclic esters such as methyl cyclohexanecarboxylate, ethyl cyclohexanecarboxylate, and dimethyl cyclohexanedicarboxylate; methyl pyridinecarboxylate, methyl pyrimidinecarboxylate, acetolactone, propiolactone, butyrolactone , valerolactone; and aromatic esters such as methyl benzoate, ethyl benzoate, dimethyl phthalate, diethyl phthalate, but
- Alcohol solvents such as ethanol and butanol; aldehyde solvents such as formaldehyde, acetaldehyde and dimethylformamide; and ketone solvents such as acetone and methyl ethyl ketone are also preferred.
- Solvents containing a nitrogen atom as a heteroatom include solvents containing a nitrogen atom-containing group such as an amino group, an amide group, a nitro group, or a nitrile group.
- solvents having an amino group include aliphatic amines such as ethylenediamine, diaminopropane, dimethylethylenediamine, diethylethylenediamine, dimethyldiaminopropane, tetramethyldiaminomethane, tetramethylethylenediamine (TMEDA), and tetramethyldiaminopropane (TMPDA); cyclopropanediamine, cyclohexanediamine, bisaminomethylcyclohexane, etc.; heterocyclic amines, such as isophoronediamine, piperazine, dipiperidylpropane, dimethylpiperazine; Aromatic amines such as dimethylnaphthalenediamine, dimethylphenylenediamine, te
- Preferred solvents containing halogen atoms as heteroatoms include dichloromethane, chlorobenzene, trifluoromethylbenzene, chlorobenzene, chlorotoluene, bromobenzene and the like.
- Preferred examples of solvents containing sulfur atoms include dimethylsulfoxide and carbon disulfide.
- the amount of solvent used is preferably 100 mL or more, more preferably 200 mL or more, still more preferably 250 mL or more, and even more preferably 300 mL or more, relative to 1 kg of the total amount of raw materials. It is 3000 mL or less, more preferably 2500 mL or less, still more preferably 2000 mL or less, and even more preferably 1550 mL or less. When the amount of the solvent used is within the above range, the raw materials can be efficiently reacted.
- the mixing is performed using a solvent, it may include drying the fluid (usually a slurry) obtained by the mixing after the mixing.
- a complexing agent is used as a solvent, the complexing agent is removed from the complex containing the complexing agent.
- a sulfide solid electrolyte is obtained by removing the agent and the solvent, or by removing the solvent when a solvent other than the complexing agent is used. The resulting sulfide solid electrolyte exhibits ionic conductivity due to lithium atoms.
- Drying can be performed on the fluid obtained by mixing at a temperature depending on the type of solvent. For example, it can be carried out at a temperature equal to or higher than the boiling point of the complexing agent. In addition, it is usually dried at 5 to 100° C., preferably 10 to 85° C., more preferably 15 to 70° C., still more preferably about room temperature (23° C.) (for example, room temperature about ⁇ 5° C.) under reduced pressure using a vacuum pump or the like. (Vacuum drying) to volatilize the complexing agent and optionally used solvent.
- Drying may be performed by filtering the fluid using a glass filter or the like, solid-liquid separation by decantation, or solid-liquid separation using a centrifugal separator or the like.
- a solvent other than the complexing agent is used, a sulfide solid electrolyte can be obtained by solid-liquid separation.
- drying under the above temperature conditions may be performed to remove the complexing agent incorporated in the complex.
- the fluid in solid-liquid separation, the fluid is transferred to a container, and after sulfide (or a complex (which can also be referred to as a precursor of a sulfide solid electrolyte) if a complexing agent is included) is precipitated, the supernatant is It is easy to perform decantation to remove the complexing agent and solvent, and filtration using a glass filter having a pore size of about 10 to 200 ⁇ m, preferably 20 to 150 ⁇ m.
- Drying may be performed after mixing and before hydrogen treatment, which will be described later, or after hydrogen treatment.
- the sulfide solid electrolyte obtained by performing the above mixing is basically an amorphous sulfide solid electrolyte (glass component) unless mixing is performed by pulverizing using a pulverizer to the extent that it crystallizes, for example. .
- the sulfide solid electrolyte obtained by the above mixing may be an amorphous sulfide solid electrolyte (glass component) or a crystalline sulfide solid electrolyte. can be selected.
- the amorphous sulfide solid electrolyte obtained by the above mixing can be heated to obtain a crystalline sulfide solid electrolyte.
- an amorphous component (glass component) is formed on the surface.
- the sulfide solid electrolyte containing an amorphous component includes an amorphous sulfide solid electrolyte and a crystalline sulfide solid electrolyte having an amorphous component formed on its surface. Also included are electrolytes.
- a crystalline sulfide solid electrolyte Further heating may be included when producing a crystalline sulfide solid electrolyte.
- an amorphous sulfide solid electrolyte (glass component) is obtained by the above mixing, a crystalline sulfide solid electrolyte is obtained by heating, and a crystalline sulfide solid electrolyte is obtained. In this case, a crystalline sulfide solid electrolyte with improved crystallinity can be obtained.
- a complexing agent is used as a solvent for mixing, a complex containing the complexing agent is formed. After removal, a sulfide solid electrolyte is obtained, which can be amorphous or crystalline depending on the heating conditions.
- the heating temperature is determined according to the structure of the crystalline sulfide solid electrolyte obtained by heating the amorphous sulfide solid electrolyte.
- the amorphous sulfide solid electrolyte is subjected to differential thermal analysis (DTA) using a differential thermal analysis apparatus (DTA apparatus) under a temperature rising condition of 10 ° C./min, and the lowest temperature side
- DTA differential thermal analysis
- the temperature is preferably 5°C or less, more preferably 10°C or less, and still more preferably 20°C or less, and the lower limit is not particularly limited.
- the peak top temperature of the exothermic peak observed on the lowest temperature side may be about ⁇ 40° C. or higher.
- the heating temperature for obtaining the amorphous sulfide solid electrolyte depends on the structure of the crystalline sulfide solid electrolyte to be obtained, and cannot be generally defined, but is usually preferably 135° C. or less. 130° C. or lower is more preferable, and 125° C. or lower is even more preferable.
- the lower limit is not particularly limited, it is preferably 90° C. or higher, more preferably 100° C. or higher, and still more preferably 105° C. or higher.
- the heating temperature may be determined according to the structure of the crystalline sulfide solid electrolyte. It is preferable that the heating temperature is higher than the above heating temperature for obtaining a solid solid electrolyte. Differential thermal analysis (DTA) is performed under temperature conditions, and the temperature of the peak top of the exothermic peak observed on the lowest temperature side is preferably 5 ° C. or higher, more preferably 10 ° C. or higher, and still more preferably 20 ° C. or higher. The temperature may be within the range, and the upper limit is not particularly limited, but may be about 40°C or less.
- DTA Differential thermal analysis
- the heating temperature for obtaining a crystalline sulfide solid electrolyte varies depending on the composition and structure of the obtained crystalline sulfide solid electrolyte, and cannot be generally defined, but is usually preferably 130° C. or higher. , more preferably 135° C. or higher, more preferably 140° C. or higher, and although the upper limit is not particularly limited, it is preferably 600° C. or lower, more preferably 550° C. or lower, and still more preferably 500° C. or lower.
- the heating time is not particularly limited as long as the desired amorphous sulfide solid electrolyte or crystalline sulfide solid electrolyte can be obtained. More preferably, it is 30 minutes or more, and even more preferably 1 hour or more.
- the upper limit of the heating time is not particularly limited, but is preferably 24 hours or less, more preferably 10 hours or less, still more preferably 5 hours or less, and even more preferably 3 hours or less.
- the heating is preferably performed in an inert gas atmosphere (for example, a nitrogen atmosphere or an argon atmosphere) or a reduced pressure atmosphere (especially in a vacuum). It may be an inert gas atmosphere containing hydrogen at a certain concentration, for example, the concentration of hydrogen in the hydrogen treatment described later. This is because deterioration (for example, oxidation) of the crystalline sulfide solid electrolyte can be prevented.
- the heating method is not particularly limited, and examples thereof include a method using a hot plate, a vacuum heating device, an argon gas atmosphere furnace, and a firing furnace. Industrially, a horizontal dryer having a heating means and a feeding mechanism, a horizontal vibrating fluidized dryer, or the like can be used, and the drying apparatus may be selected according to the amount of heat to be processed.
- the sulfide solid electrolyte used in this embodiment may be a commercially available product or a manufactured product.
- the sulfide solid electrolyte obtained by the above method is an amorphous (glass component), crystalline sulfide solid electrolyte containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms, and is preferably Used as a sulfide solid electrolyte.
- the specific surface area of the sulfide solid electrolyte used in this embodiment has a BET specific surface area of 10 m 2 /g or more. Despite having such a large specific surface area, the modified sulfide solid electrolyte of the present embodiment has excellent coating aptitude when coated as a paste, and efficiently exhibits excellent battery performance. The higher the BET specific surface area of the sulfide solid electrolyte, the more superior the effect can be shown. From such a viewpoint, the BET specific surface area is preferably 12 m 2 /g or more, more preferably 15 m 2 /g or more, and even more preferably 20 m 2 /g or more.
- the upper limit is not particularly limited from the same viewpoint, it is practically 100 m 2 /g or less, preferably 75 m 2 /g or less, more preferably 50 m 2 /g or less.
- the BET specific surface area is a specific surface area measured using krypton as an adsorbate in accordance with JIS Z 8830:2013 (Method for measuring specific surface area of powder (solid) by gas adsorption).
- the amorphous sulfide solid electrolyte obtained by the above method contains lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms, and typical examples include Li 2 SP 2 S 5 -LiI, composed of lithium sulfide, phosphorus sulfide and lithium halide such as Li 2 SP 2 S 5 -LiCl, Li 2 SP 2 S 5 -LiBr, Li 2 SP 2 S 5 -LiI -LiBr; a solid electrolyte further containing other atoms such as oxygen atoms and silicon atoms, such as Li 2 SP 2 S 5 —Li 2 O—LiI, Li 2 S — SiS 2 —P 2 S 5 —LiI, etc.
- Solid electrolytes are preferred. From the viewpoint of obtaining higher ionic conductivity, Li 2 SP 2 S 5 -LiI, Li 2 SP 2 S 5 -LiCl, Li 2 SP 2 S 5 -LiBr, Li 2 SP 2 S Solid electrolytes composed of lithium sulfide such as 5 -LiI-LiBr, phosphorus sulfide and lithium halide are preferred.
- the type of atoms forming the amorphous sulfide solid electrolyte can be confirmed by, for example, an ICP emission spectrometer.
- the shape of the amorphous sulfide solid electrolyte is not particularly limited, but may be, for example, particulate.
- the average particle size (D 50 ) of the particulate amorphous sulfide solid electrolyte can be, for example, within the range of 0.01 ⁇ m to 500 ⁇ m and 0.1 to 200 ⁇ m.
- the crystal structure of the crystalline sulfide solid electrolyte obtained by the production method of the present embodiment is preferably the thiolysicone region II type crystal structure among the above, because higher ion conductivity can be obtained.
- the “thiolysicone region II type crystal structure” is a Li 4-x Ge 1-x P x S 4 system thio-LISICON Region II type crystal structure, Li 4-x Ge 1-x P x S 4 -based thio-LISICON Region II type and similar crystal structures.
- the crystalline sulfide solid electrolyte obtained by the production method of the present embodiment may have the thiolysicone region II type crystal structure described above, or may have the main crystal. From the viewpoint of obtaining high ionic conductivity, it is preferable to have it as a main crystal.
- the crystalline sulfide solid electrolyte obtained by the production method of the present embodiment does not contain crystalline Li 3 PS 4 ( ⁇ -Li 3 PS 4 ) from the viewpoint of obtaining higher ionic conductivity. is preferred.
- the Li 4-x Ge 1-x P x S 4 -based thiolysicone region II Diffraction peaks of the (thio-LISICON Region II) type crystal
- the thiolysicone region II type crystal structure when the thiolysicone region II type crystal structure is obtained in the present embodiment, it preferably does not contain crystalline Li 3 PS 4 ( ⁇ -Li 3 PS 4 ).
- a crystal structure basically having a structural framework of these Li 7 PS 6 is also referred to as an aldirodite-type crystal structure. These peak positions may be shifted within a range of ⁇ 0.5°.
- the shape of the crystalline sulfide solid electrolyte is not particularly limited, but may be, for example, particulate.
- the average particle size (D 50 ) of the particulate crystalline sulfide solid electrolyte can be, for example, within the range of 0.01 ⁇ m to 500 ⁇ m and 0.1 to 200 ⁇ m.
- the modified sulfide solid electrolyte of the present embodiment has a BET specific surface area of 10 m 2 /g or more and has a large specific surface area. From the viewpoint that the higher the BET specific surface area of the sulfide solid electrolyte, the more superior the effect can be shown, the BET specific surface area is preferably 12 m 2 /g or more, and 15 m 2 /g or more. are more preferable, and those of 20 m 2 /g or more are even more preferable.
- the upper limit is not particularly limited from the same viewpoint, it is practically 100 m 2 /g or less, preferably 75 m 2 /g or less, more preferably 50 m 2 /g or less. Even if the lithium halide adheres to the surface, the BET specific surface area of the sulfide solid electrolyte is not greatly affected, and the BET specific surface area of the sulfide solid electrolyte used in this embodiment and the modified sulfide The BET specific surface area of the solid electrolyte is substantially the same.
- the modified sulfide solid electrolyte naturally has a BET specific surface area of 10 m 2 /g or more.
- the oil absorption of the modified sulfide solid electrolyte of the present embodiment is small, usually less than 0.9 mL/g, due to the effect of the lithium halide adhering to the surface, although the BET specific surface area is large as described above. , and further becomes 0.85 mL/g or less and less than 0.80 mL/g.
- the modified sulfide solid electrolyte of the present embodiment has a large BET specific surface area, it has a small oil absorption. Since the paste has excellent coating suitability and does not require the use of a solvent or the like to suppress an increase in paste viscosity, excellent battery performance can be easily obtained.
- the oil absorption is measured by taking 1 g of the modified sulfide solid electrolyte as a sample, adding one drop of butyl butyrate in a mortar or the like and stirring with a spatula until the sample becomes a paste. The total amount of butyl butyrate added repeatedly was taken as the oil absorption (mL/g).
- "paste” is defined in "7.2 Measurement” of JIS K5101-13-1:2004 (Pigment test method-Part 13: Oil absorption-Section 1: Refined linseed oil method) It means "a state that can be spread without cracking or crumbling, and that adheres lightly to the measuring plate.”
- the ionic conductivity of the modified sulfide solid electrolyte of the present embodiment is usually 0.5 mS/cm or more, and furthermore, 1.0 mS/cm or more, 1.5 mS/cm or more, 2.0 mS/cm or more. , 2.5 mS/cm or more, and has extremely high ionic conductivity, so that a lithium battery having excellent battery performance can be obtained.
- the modified sulfide solid electrolyte of the present embodiment is excellent in coatability and can be used for battery production without using a solvent or the like, so that it can efficiently exhibit excellent battery performance. Moreover, since it has high ionic conductivity and excellent battery performance, it is suitably used for batteries.
- the modified sulfide solid electrolyte of the present embodiment may be used for the positive electrode layer, the negative electrode layer, or the electrolyte layer. In addition, each layer can be manufactured by a well-known method.
- the above battery preferably uses a current collector, and known current collectors can be used.
- a layer coated with Au or the like can be used, such as Au, Pt, Al, Ti, or Cu, which reacts with the solid electrolyte.
- the method for producing a modified sulfide solid electrolyte of the present embodiment includes a sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms; an epoxy compound; and an organic solvent, and removing the organic solvent. According to the production method of the present embodiment, it is possible to efficiently produce the modified sulfide solid electrolyte of the present embodiment. It is preferable to manufacture by the manufacturing method of.
- the sulfide solid electrolyte having a BET specific surface area of 10 m 2 /g or more and containing lithium atoms, sulfur atoms, phosphorus atoms and halogen atoms used in the production method of the present embodiment is the modified sulfide solid of the present embodiment.
- the same sulfide solid electrolytes described as may be used in the electrolyte are employed. Therefore, as the sulfide solid electrolyte, a commercially available product may be used, or a product manufactured by the above-described sulfide solid electrolyte method may be used.
- Examples of the organic solvent used in the production method of the present embodiment include the solvents described as being usable in the method for producing the sulfide solid electrolyte. Aspects of promoting mixing of a sulfide solid electrolyte and an epoxy compound to efficiently obtain a modified sulfide solid electrolyte containing the sulfide solid electrolyte and an epoxy compound, and adhesion of the epoxy compound to the sulfide solid electrolyte. From the viewpoint of promoting , aromatic hydrocarbon solvents are more preferred. In the production method of the present embodiment, as the organic solvent, one of these can be used alone, or a combination of two or more of them can be used.
- the method of mixing the sulfide solid electrolyte, the epoxy compound, and the organic solvent can be performed by the same method as "mixing" in the method of producing the sulfide solid electrolyte. can.
- the removal of the organic solvent can be carried out by the same method as “drying” in the method for producing the sulfide solid electrolyte. Further, in the manufacturing method of the present embodiment, “heating” in the method of manufacturing the sulfide solid electrolyte may be performed.
- modified sulfide solid electrolyte has a BET specific surface area of 10 m 2 /g or more, lithium atoms, It includes a sulfide solid electrolyte containing sulfur atoms, phosphorus atoms and halogen atoms, and an epoxy compound.
- such a modified sulfide solid electrolyte in another form also has excellent coating suitability when coated as a paste, like the modified sulfide solid electrolyte of the present embodiment described above, and This is effective in that excellent battery performance can be expressed efficiently.
- the BET specific surface area, the epoxy compound, the sulfide solid electrolyte and the method for producing the same, and the method for producing the modified sulfide solid electrolyte in the modified sulfide solid electrolyte of another form are the modified sulfide solid electrolyte of the present embodiment. is the same as described for
- the electrode mixture of the present embodiment includes the modified sulfide solid electrolyte of the present embodiment described above and an electrode active material. and a material.
- Electrode active material As the electrode active material, a positive electrode active material and a negative electrode active material are employed depending on whether the electrode mixture is used for a positive electrode or a negative electrode.
- positive electrode active material in relation to the negative electrode active material, atoms employed as atoms that exhibit ionic conductivity, preferably lithium atoms, as long as they can promote the battery chemical reaction accompanied by movement of lithium ions.
- positive electrode active materials capable of intercalating and deintercalating lithium ions include oxide-based positive electrode active materials and sulfide-based positive electrode active materials.
- sulfide-based positive electrode active materials include titanium sulfide (TiS 2 ), molybdenum sulfide (MoS 2 ), iron sulfide (FeS, FeS 2 ), copper sulfide (CuS), nickel sulfide (Ni 3 S 2 ), and the like.
- Niobium selenide (NbSe 3 ) or the like can also be used in addition to the positive electrode active material described above.
- a positive electrode active material can be used individually by 1 type or in combination of multiple types.
- an atom employed as an atom that expresses ionic conductivity preferably a metal capable of forming an alloy with a lithium atom, an oxide thereof, an alloy of the metal and a lithium atom, etc., preferably a lithium atom
- a metal capable of forming an alloy with a lithium atom, an oxide thereof, an alloy of the metal and a lithium atom, etc. preferably a lithium atom
- Any material can be used without particular limitation as long as it can promote the battery chemical reaction accompanied by the movement of lithium ions caused by .
- the negative electrode active material capable of intercalating and deintercalating lithium ions any known negative electrode active material in the field of batteries can be employed without limitation.
- negative electrode active materials include metals capable of forming an alloy with metal lithium or metal lithium, such as metal lithium, metal indium, metal aluminum, metal silicon, metal tin, oxides of these metals, and metals with these metals.
- metals capable of forming an alloy with metal lithium or metal lithium such as metal lithium, metal indium, metal aluminum, metal silicon, metal tin, oxides of these metals, and metals with these metals.
- An alloy with metal lithium and the like can be mentioned.
- the electrode active material used in this embodiment may have a coating layer on which the surface is coated.
- Materials for forming the coating layer include ionic conductors such as nitrides and oxides of atoms, preferably lithium atoms, which exhibit ionic conductivity in the sulfide solid electrolyte, or composites thereof.
- lithium nitride (Li 3 N) a conductor having a lysicon-type crystal structure such as Li 4-2x Zn x GeO 4 having a main structure of Li 4 GeO 4 , and a Li 3 PO 4 -type skeleton conductors having a thiolysicone crystal structure such as Li 4-x Ge 1-x P x S 4 , conductors having a perovskite crystal structure such as La 2/3-x Li 3x TiO 3 , LiTi 2 Conductors having a NASICON-type crystal structure such as (PO 4 ) 3 are included.
- Li 3 N lithium nitride
- a conductor having a lysicon-type crystal structure such as Li 4-2x Zn x GeO 4 having a main structure of Li 4 GeO 4
- a Li 3 PO 4 -type skeleton conductors having a thiolysicone crystal structure such as Li 4-x Ge 1-x P x S 4
- Lithium titanates such as Li y Ti 3-y O 4 (0 ⁇ y ⁇ 3 ) and Li 4 Ti 5 O 12 ( LTO); Lithium metal oxide, also Li2O - B2O3 - P2O5 system, Li2O - B2O3 - ZnO system , Li2O - Al2O3 - SiO2 - P2O5 - TiO 2 -based oxide-based conductors, and the like.
- An electrode active material having a coating layer is obtained, for example, by depositing a solution containing various atoms constituting the material forming the coating layer on the surface of the electrode active material, and then heating the electrode active material after deposition to preferably 200° C. or higher and 400° C. or lower. It is obtained by firing at
- the solution containing various atoms for example, a solution containing alkoxides of various metals such as lithium ethoxide, titanium isopropoxide, niobium isopropoxide and tantalum isopropoxide may be used.
- alcoholic solvents such as ethanol and butanol
- aliphatic hydrocarbon solvents such as hexane, heptane and octane
- aromatic hydrocarbon solvents such as benzene, toluene and xylene
- the above adhesion may be performed by immersion, spray coating, or the like.
- the firing temperature is preferably 200° C. or higher and 400° C. or lower, more preferably 250° C. or higher and 390° C. or lower, from the viewpoint of improving production efficiency and battery performance, and the firing time is usually about 1 minute to 10 hours. and preferably 10 minutes to 4 hours.
- the coverage of the coating layer is preferably 90% or more, more preferably 95% or more, still more preferably 100%, based on the surface area of the electrode active material, that is, the entire surface is preferably covered.
- the thickness of the coating layer is preferably 1 nm or more, more preferably 2 nm or more, and the upper limit is preferably 30 nm or less, more preferably 25 nm or less.
- the thickness of the coating layer can be measured by cross-sectional observation with a transmission electron microscope (TEM), and the coverage rate is the thickness of the coating layer, the elemental analysis value, the BET specific surface area, can be calculated from
- the electrode composite material of the present embodiment may contain other components such as a conductive material and a binder in addition to the modified sulfide solid electrolyte and the electrode active material. That is, in the method of manufacturing the electrode composite material of the present embodiment, other components such as a conductive material and a binder may be used in addition to the modified sulfide solid electrolyte and the electrode active material. Other components such as a conductive agent and a binder are added to the modified sulfide solid electrolyte and the electrode active material in mixing the modified sulfide solid electrolyte and the electrode active material. A mixture may be used.
- artificial graphite, graphite carbon fiber, resin-baked carbon, pyrolytic vapor-grown carbon, coke, mesocarbon microbeads, furfuryl alcohol resin-baked carbon are used from the viewpoint of improving battery performance by improving electronic conductivity.
- polyacene, pitch-based carbon fiber, vapor-grown carbon fiber, natural graphite, and non-graphitizable carbon are used from the viewpoint of improving battery performance by improving electronic conductivity.
- the binder is not particularly limited as long as it can impart functions such as binding properties and flexibility.
- examples include fluorine-based polymers such as polytetrafluoroethylene and polyvinylidene fluoride, butylene rubber, and styrene-butadiene rubber.
- Various resins such as thermoplastic elastomers, acrylic resins, acrylic polyol resins, polyvinyl acetal resins, polyvinyl butyral resins, and silicone resins are exemplified.
- the compounding ratio (mass ratio) of the electrode active material and the modified sulfide solid electrolyte in the electrode mixture is preferably 99.5:0.5 to 40 in consideration of improving battery performance and manufacturing efficiency. :60, more preferably 99:1 to 50:50, still more preferably 98:2 to 60:40.
- the content of the conductive material in the electrode mixture is not particularly limited. It is at least 1.5% by mass, more preferably at least 1.5% by mass, and the upper limit is preferably 10% by mass or less, preferably 8% by mass or less, and more preferably 5% by mass or less.
- the content of the binder in the electrode mixture is not particularly limited, but considering the improvement of battery performance and production efficiency, it is preferably 1% by mass or more, more preferably. is 3% by mass or more, more preferably 5% by mass or more, and the upper limit is preferably 20% by mass or less, preferably 15% by mass or less, and further preferably 10% by mass or less.
- the lithium ion battery of the present embodiment contains at least one selected from the modified sulfide solid electrolyte of the present embodiment and the electrode mixture, and the modified sulfide solid electrolyte of another form and the above A lithium ion battery containing at least one selected from an electrode mixture.
- the lithium ion battery of the present embodiment includes either the modified sulfide solid electrolyte of the present embodiment, an electrode mixture containing the same, a modified sulfide solid electrolyte of another form, or an electrode mixture containing the same.
- an electrode mixture containing the same There are no particular restrictions on the configuration as long as it contains a lithium ion battery, as long as it has the configuration of a widely used lithium ion battery.
- the lithium ion battery of the present embodiment preferably includes, for example, a positive electrode layer, a negative electrode layer, an electrolyte layer, and a current collector.
- the electrode mixture of the present embodiment is preferably used for the positive electrode layer and the negative electrode layer, and the modified sulfide solid electrolyte of the present embodiment and another modified sulfide solid electrolyte are used as the electrolyte layer. preferably used.
- a known current collector may be used.
- a layer coated with Au or the like can be used, such as Au, Pt, Al, Ti, or Cu, which reacts with the solid electrolyte.
- amorphous sulfide solid electrolyte was heated at 140° C. under vacuum for 2 hours to obtain a crystalline sulfide solid electrolyte 1 (heating temperature for obtaining a crystalline sulfide solid electrolyte (this 140° C. in the example) is sometimes referred to as the “crystallization temperature”).
- the BET specific surface areas of the obtained amorphous sulfide solid electrolyte and crystalline sulfide solid electrolyte were measured, both were 40 m 2 /g.
- Production Example 2 Production of Sulfide Solid Electrolyte 2 Into a reactor with a stirring blade (capacity: 500 mL), 30.0 g of the sulfide solid electrolyte powder obtained in Production Example 1 and 470 g of toluene were charged under a nitrogen atmosphere. After rotating the stirring blade, a bead mill capable of circulating microbeads ("UAM-015 (model number)", manufactured by Hiroshima Metal & Machinery Co., Ltd.) was used under predetermined conditions (bead material: zirconia, bead diameter: 0.005 mm).
- Production Example 3 Production of Sulfide Solid Electrolyte 3 Into a reactor with a stirring blade (capacity: 500 mL), 30.0 g of the sulfide solid electrolyte powder obtained in Production Example 1 and 470 g of toluene were charged under a nitrogen atmosphere. After rotating the stirring blade, a bead mill capable of circulating microbeads ("UAM-015 (model number)", manufactured by Hiroshima Metal & Machinery Co., Ltd.) was used under predetermined conditions (bead material: zirconia, bead diameter: 0.005 mm).
- Example 1 3 g of the crystalline sulfide solid electrolyte 1 obtained in Production Example 1 was weighed and added to Schlenk (capacity: 100 mL) with a stirrer under a nitrogen atmosphere, and 22 g of toluene was added and stirred to form a slurry fluid. did. 0.05 g of butylene oxide as an epoxy compound (5 parts by mass with respect to 100 parts by mass of the crystalline sulfide solid electrolyte) is added to the slurry-like fluid, and stirred for 10 minutes. Toluene was distilled off by vacuum drying to obtain a modified sulfide solid electrolyte.
- the obtained modified sulfide solid electrolyte was measured for oil absorption and ionic conductivity according to the following methods. Also, the rate of decrease in oil absorption was calculated according to the following method. Table 1 shows the measurement results and calculation results.
- FT-IR analysis (ATR method) was conducted according to the following method, and it was confirmed that the infrared absorption spectrum had a peak at 2800 to 3000 cm ⁇ 1 . Further, 1 H-NMR measurement was carried out based on the method described below, and it was also confirmed to have a peak of 0.0 to 5.0 ppm derived from the alkyl chain.
- Example 1 a modified sulfide solid electrolyte was prepared in the same manner as in Example 1, except that the type of crystalline sulfide solid electrolyte and the type and amount of epoxy compound used were as shown in Table 1. was made. The obtained modified sulfide solid electrolyte was measured for oil absorption and ionic conductivity according to the following methods. Also, the rate of decrease in oil absorption was calculated according to the following method. Table 1 shows the measurement results and calculation results.
- FT-IR analysis (ATR method) was conducted according to the following method, and it was confirmed that the infrared absorption spectrum had a peak at 2800 to 3000 cm ⁇ 1 .
- 1 H-NMR measurement was performed based on the method described below, and it was also confirmed that there was a peak of 0.0 to 5.0 ppm derived from the alkyl chain.
- Comparative Examples 1-3 The sulfide solid electrolytes 1 to 3 obtained in Production Examples 1 to 3 were measured for oil absorption and ionic conductivity according to the following methods. Also, the rate of decrease in oil absorption was calculated according to the following method. Table 1 shows the measurement results and calculation results. The oil absorptions of sulfide solid electrolytes 1 to 3 were 1.03 mL/g, 0.93 mL/g and 0.66 mL/g, respectively. Further, when FT-IR analysis (ATR method) was performed based on the following method, no peak could be confirmed at 2800 to 3000 cm -1 in the infrared absorption spectrum. Further, when 1 H-NMR measurement was performed based on the method described below, no peaks from 0.0 to 5.0 ppm originating from alkyl chains could be confirmed.
- ATR method FT-IR analysis
- the oil absorption reduction rate of Example 1 is calculated by setting the oil absorption amount of the sulfide solid electrolyte 1 as the oil absorption amount A and the oil absorption amount of the modified sulfide solid electrolyte of Example 1 as the oil absorption amount B. do.
- Reduction rate of oil absorption (oil absorption A - oil absorption B) / oil absorption A x 100 (%)
- the ionic conductivity was measured as follows. A circular pellet having a diameter of 10 mm (cross-sectional area S: 0.785 cm 2 ) and a height (L) of 0.1 to 0.3 cm was molded from the sulfide solid electrolyte to obtain a sample. Electrode terminals were taken from the top and bottom of the sample, and measurement was performed at 25° C. by the AC impedance method (frequency range: 1 MHz to 100 Hz, amplitude: 10 mV) to obtain a Cole-Cole plot.
- AC impedance method frequency range: 1 MHz to 100 Hz, amplitude: 10 mV
- the real part Z' ( ⁇ ) at the point where -Z'' ( ⁇ ) is the minimum is the bulk resistance R ( ⁇ ) of the electrolyte, and according to the following formula, ion Conductivity ⁇ (S/cm) was calculated.
- the measured ionic conductivity was evaluated according to the following criteria. A. 2.5 mS/cm or more B. 0.5 mS/cm or more and less than 2.5 mS/cm C.I. Less than 0.5 mS/cm
- Measurement device FR-IR spectrometer “VERTEX70v (model number)”, manufactured by Bruker Measurement method: Total reflection measurement method (ATR method) Measurement wavenumber range: 650 to 4000 cm -1 Light source: Grover lamp (SiC) Detector: DTGS detector Resolution: 4 cm -1 Measurement time: 1 second/time Accumulated times: 256 times Measurement conditions: Using a diamond prism, irradiation at an incident angle of 45°
- the modified sulfide solid electrolyte of the present embodiment has an oil absorption evaluation of A or B, and the reduction rate of oil absorption is all 15% or more, so the specific surface area is 10 m It was confirmed that the oil absorption was small and the coating suitability was excellent, although the oil absorption was as large as 2 /g or more. It was also confirmed that the ionic conductivity was also high in A or B evaluation.
- the sulfide solid electrolytes of Comparative Examples 1 to 3 which were not mixed with an epoxy compound and did not contain an epoxy compound, were the sulfide solid electrolytes 1 to 3 produced in Production Examples 1 to 3, respectively, and were conventional sulfide solid electrolytes. It is the electrolyte itself.
- the sulfide solid electrolytes 1 and 2 of Comparative Examples 1 and 2 having a specific surface area of 10 m 2 /g or more were evaluated as C in terms of oil absorption, and it was confirmed that they were inferior in coatability.
- the sulfide solid electrolyte 3 of Comparative Example 3 was evaluated as A in both the oil absorption amount and the ionic conductivity, and it was confirmed that there was little need for modification. That is, the method for producing a modified sulfide solid electrolyte of the present embodiment can reduce oil absorption and improve coating suitability for a material having a large specific surface area of 10 m 2 /g or more. It has been found to be suitable.
- Example 20 The modified sulfide solid electrolytes obtained in the above examples were examined below in order to confirm whether or not the epoxy compound adhered to the sulfide solid electrolytes.
- the content of 1,2-epoxyoctadecane in Example 6 was 1% by mass (the amount of 1,2-epoxyoctadecane used was 1 part by mass with respect to 100 parts by mass of the sulfide solid electrolyte).
- Toluene was added to the resulting modified sulfide solid electrolyte to form a slurry (slurry concentration: 12% by mass), and the slurry was allowed to stand for 12 hours.
- a supernatant liquid produced by sedimentation of the sulfide solid electrolyte was sampled and analyzed by gas chromatography mass spectrometry (GC/MS method).
- the charged liquid (1 mass% toluene solution of 1,2-epoxyoctadecane) was also analyzed in the same manner as the above supernatant liquid, and the peak area of 1,2-epoxyoctadecane in the charged liquid was set to 1.
- the peak area of 1,2-epoxyoctadecane remaining in the supernatant the closer the peak area of the supernatant is to 1, the more the epoxy compound is liberated from the sulfide solid electrolyte and dissolved in toluene. means.
- no epoxy compounds were detected in the supernatant, so it is considered that all the epoxy compounds adhered to the sulfide solid electrolyte.
- the sedimented sulfide solid electrolyte was washed by repeating three times a process of adding toluene to the sedimented sulfide solid electrolyte, stirring the mixture, leaving the mixture at rest for 12 hours, and removing the supernatant. After washing, the sulfide solid electrolyte obtained by drying toluene was dissolved in heavy methanol and subjected to 1 H-NMR measurement by the above method. Shift detected.
- Example 21 Except that the content of 1,2-epoxyoctadecane in Example 6 was 3% by mass (the amount of 1,2-epoxyoctadecane used was 3 parts by mass with respect to 100 parts by mass of the sulfide solid electrolyte) obtained a modified sulfide solid electrolyte in the same manner as in Example 6.
- the obtained modified sulfide solid electrolyte was measured for oil absorption in accordance with the measurement of oil absorption described above. Also, the rate of decrease in oil absorption was calculated according to the following method. Table 1 shows the measurement results and calculation results.
- the supernatant liquid of the obtained modified sulfide solid electrolyte was analyzed by gas chromatography mass spectrometry (GC/MS method) in the same manner as in Example 20. According to the analysis, although some epoxy compounds were detected from the supernatant (peak area: 0.25), it was confirmed that the epoxy compounds adhered to the sulfide solid electrolyte.
- 1 H-NMR measurement was performed on the precipitated powder in the same manner as in Example 20, and a chemical shift of a group (such as an alkyl group) derived from the epoxy compound was detected.
- Example 22 Except that the content of 1,2-epoxyoctadecane in Example 6 was 10% by mass (the amount of 1,2-epoxyoctadecane used was 10 parts by mass with respect to 100 parts by mass of the sulfide solid electrolyte) obtained a modified sulfide solid electrolyte in the same manner as in Example 6.
- the obtained modified sulfide solid electrolyte was measured for oil absorption in accordance with the measurement of oil absorption described above. Also, the rate of decrease in oil absorption was calculated according to the following method. Table 1 shows the measurement results and calculation results.
- the supernatant liquid of the obtained modified sulfide solid electrolyte was analyzed by gas chromatography mass spectrometry (GC/MS method) in the same manner as in Example 20. According to the analysis, although some epoxy compounds were detected in the supernatant (peak area: 0.43), it was confirmed that the epoxy compounds adhered to the sulfide solid electrolyte. In addition, 1 H-NMR measurement was performed on the precipitated powder in the same manner as in Example 20, and a chemical shift of a group (such as an alkyl group) derived from the epoxy compound was detected.
- a group such as an alkyl group
- the epoxy compound is contained in the modified sulfide solid electrolyte so as to adhere to the sulfide solid electrolyte, and most of it is in the modified sulfide solid electrolyte even after washing with toluene. confirmed to remain. 1 H-NMR measurement confirmed that the epoxy compound remained in the modified sulfide solid electrolyte.
- Example 23 a modified sulfide solid electrolyte was prepared in the same manner as in Example 1, except that the type of crystalline sulfide solid electrolyte and the type and amount of epoxy compound used were as shown in Table 2. was made. The obtained modified sulfide solid electrolyte was measured for oil absorption and ionic conductivity based on the above methods. Also, the rate of decrease in oil absorption was calculated based on the above method. Table 2 shows the measurement results and calculation results.
- Comparative example 4 For each of the sulfide solid electrolytes 4 obtained in Production Example 4, the oil absorption and ionic conductivity were measured according to the following methods. Also, the rate of decrease in oil absorption was calculated based on the above method. Table 2 shows the measurement results and calculation results. The oil absorption of the sulfide solid electrolyte 4 was 1.06 mL/g. Further, when FT-IR analysis (ATR method) was performed based on the above method, no peak could be confirmed at 2800 to 3000 cm ⁇ 1 in the infrared absorption spectrum. In addition, when 1 H-NMR measurement was performed based on the above method, no peaks from 0.0 to 5.0 ppm originating from the alkyl chain could be confirmed.
- ATR method FT-IR analysis
- the electrolyte for the above separator was synthesized under the following conditions. 20.5 g of L 2 S, 33.1 g of P 2 S 5 , 10.0 g of LiI, and 6.5 g of LiBr were added to a 1 L reaction vessel equipped with a stirring blade under nitrogen atmosphere. After rotating the stirring blade, 630 g of toluene was introduced and the slurry was stirred for 10 minutes.
- the reaction vessel was connected to a circulating bead mill (“Star Mill LMZ015 (trade name)”, manufactured by Ashizawa Finetech Co., Ltd., zirconia bead material: zirconia, bead diameter: 0.5 mm ⁇ , amount of beads used: 456 g).
- a pulverization treatment was performed for a period of time (pump flow rate: 650 mL/min, bead mill peripheral speed: 12 m/s, mill jacket temperature: 45°C).
- the obtained slurry was dried at room temperature (25° C.) under vacuum and then heated (80° C.) to obtain a white powder of an amorphous solid electrolyte.
- the obtained crystalline solid electrolyte had an average particle size (D 50 ) of 4.5 ⁇ m and an ionic conductivity of 5.0 mS/cm.
- InLi foil having a layered structure, "/" means between layers.
- 10 mm ⁇ ⁇ 0.1 mm / Li 9 mm ⁇ ⁇ 0.1 mm
- 08 mm/SUS 10 mm ⁇ 0.1 mm
- the cell was fixed by four screws sandwiching an insulator so as not to cause a short circuit between the measurement powder (1) and the InLi foil, and the screws were fixed with a torque of 8 N ⁇ m to obtain a measurement cell.
- the obtained measurement cell was connected to a measuring instrument (“VSP-3 (model number)” manufactured by Biologic) to obtain a CV curve under the following conditions.
- Measurement temperature 25°C Sweep speed: 0.1mV/s
- Potential measurement range Open circuit voltage (+2.7V) ⁇ +5.0V ⁇ +2.7V Number of cycles: 5 times
- the modified sulfide solid electrolyte of the present embodiment has an oil absorption evaluation of A or B, and the reduction rate of oil absorption is all 17% or more. Although the surface area was as large as 10 m 2 /g or more, it was confirmed that the oil absorption was small and the coatability was excellent. It was also confirmed that the ionic conductivity was also high in A or B evaluation.
- FIG. 1 shows the CV curve of the first cycle. According to the CV curve in FIG. 1, it can be confirmed that the modified sulfide solid electrolytes of Examples 23 and 24 show a decrease in the oxidation current. I found it possible. On the other hand, it was found that the sulfide solid electrolyte of Comparative Example 1 was inferior to those of Examples 23 and 24 in the effect of suppressing the oxidation reaction.
- the modified sulfide solid electrolyte of the present embodiment even if it is a sulfide solid electrolyte with a large specific surface area, has excellent coating aptitude when coated as a paste, and can efficiently exhibit excellent battery performance. It is.
- the modified sulfide solid electrolyte of the present embodiment has high ionic conductivity, it is used for batteries, especially for information-related equipment and communication equipment such as personal computers, video cameras, and mobile phones. It is suitably used for a battery that is
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Organic Chemistry (AREA)
- Materials Engineering (AREA)
- General Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Physics & Mathematics (AREA)
- Conductive Materials (AREA)
- Secondary Cells (AREA)
- Battery Electrode And Active Subsutance (AREA)
Abstract
Description
従来、このような用途に用いられる電池において可燃性の有機溶媒を含む電解液が用いられていたため、短絡時の温度上昇を抑制する安全装置の取付、短絡防止のための構造、材料面での改善が必要となる。これに対して、電解液を固体電解質にかえて、電池を全固体化することで、電池内に可燃性の有機溶媒を用いず、安全装置の簡素化が図れ、製造コスト、生産性に優れることから、電解液を固体電解質層に換えた電池の開発が行われている。
表面を被覆する技術として、例えば、特許文献2には、リチウムイオン電池を製造する際の負極、正極等に用いられる活物質と硫化物固体電解質の親和性を高めてサイクル特性を向上させるため、当該固体電解質の表面にC=O結合を有する化合物、S=O結合を有する化合物により被膜を形成した固体電解質組成物が開示されている。また、特許文献3には、リチウム元素、リン元素及び硫黄元素を含み、かつカルボン酸とアルコールとのエステル化合物も含有する硫化物固体電解質において、当該エステル化合物が当該伝導性硫化物の表面に結合ないし吸着しており、固体電池のサイクル特性を向上し得ること、また当該硫化物固体電解質が、リチウムイオン伝導性硫化物と有機溶媒とエステル化合物とを含むスラリーを湿式粉砕する工程を有する製造方法により得られることが開示されている。このように、近年リチウムイオン電池の実用化に向けて、単に硫化物固体電解質自体のイオン伝導度を向上させるだけにとどまらず、それ以外の性能の向上に対する要望が多様化している。そして、そのような要望に対応するため、表面を被覆する技術は適用されている。
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含み、
FT-IR分析(ATR法)による赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有する改質硫化物固体電解質、
である。
また、本発明に係る改質硫化物固体電解質の製造方法は、
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、有機溶媒と、を混合すること、
前記有機溶媒を除去すること、を含む改質硫化物固体電解質の製造方法、
である。
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含む、
改質硫化物固体電解質、
である。
上記本発明に係る改質硫化物固体電解質と、電極活物質と、を含む電極合材、
である。
また、本発明に係るリチウムイオン電池は、
上記本発明に係る改質硫化物固体電解質及び上記本発明に係る電極合材の少なくとも一方を含む、リチウムイオン電池、
である。
本発明者らは、上記の課題を解決すべく鋭意検討した結果、下記の事項を見出し、本発明を完成するに至った。
特許文献1~3のように、硫化物固体電解質の表面に何らかの化合物を被覆させる技術は従来から存在している。特許文献1~3では、当該技術を用いて、イオン伝導度を向上させる、リチウムイオン電池を製造する際の負極、正極等に用いられる活物質と硫化物固体電解質の親和性を高めてサイクル特性を向上させる、といった電池性能の向上を課題としている。
本発明者らは、特許文献1~3に開示される硫化物固体電解質の表面に何らかの化合物を被覆させる技術を踏襲しながら、表面に被覆させる化合物に着目し鋭意研究を続けてきたところ、比表面積として10m2/g以上と大きいものである硫化物固体電解質であっても、その表面にハロゲン化リチウムを付着させることにより、ペーストとして塗工する際の塗工適性に優れ、かつ効率的に優れた電池性能を発現し得る硫化物固体電解質となり得ることを見出すに至った。ハロゲン化リチウムは、これまで硫化物固体電解質の原料として用いられてきたものであるが、これを硫化物固体電解質の表面に付着させることで、比表面積として10m2/g以上と大きいものである硫化物固体電解質であってもペーストとして塗工する際の塗工適性に優れるという効果が得られることは、これまで全く認知されていない、驚くべき事象である。
上記結晶性及び非晶性の区別は、本実施形態において、硫化物固体電解質、改質硫化物固体電解質のいずれにも適用される。
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含み、
FT-IR分析(ATR法)による赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有する改質硫化物固体電解質、
である。
本実施形態の改質硫化物固体電解質は、付着が上記のいずれであったとしても、その表面にエポキシ化合物が付着していれば、吸油量が低減しやすく、その結果として塗工適性は向上し、電池性能の向上を図ることができると考えられる。
エポキシ化合物の含有量が上記範囲内にあると、適度に硫化物固体電解質の表面に分散して付着して吸油量を低減するとともに、リチウムイオン伝導度を適正に保つことができるため、塗工適性が向上しやすく、また効率的に優れた電池性能を発現しやすくなる。「エポキシ化合物の含有量」は、改質硫化物固体電解質を製造する際に用いるエポキシ化合物の使用量が分かれば、当該使用量がそのまま含有量となる。
既述のように、エポキシ化合物は一定以上の分子の大きさを有するものを採用することで、吸油量の低減効果が得られやすく、塗工適性を向上させやすく得る。
一般式(1)で示されるエポキシ化合物1の中でも、第七の形態に規定されるものは、硫化物固体電解質の表面に付着して、吸油量を低減させやすく、これにより塗工適性が向上しやすく、また効率的に優れた電池性能を発現しやすくなる。
一般式(2)で示されるエポキシ化合物2の中でも、第八の形態に規定されるものは、硫化物固体電解質の表面に付着して、吸油量を低減させやすく、これにより塗工適性が向上しやすく、また効率的に優れた電池性能を発現しやすくなる。
一般式(3)で示されるエポキシ化合物3の中でも、第九の形態に規定されるものは、硫化物固体電解質の表面に付着して、吸油量を低減させやすく、これにより塗工適性が向上しやすく、また効率的に優れた電池性能を発現しやすくなる。
一般式(3)で示されるエポキシ化合物3の中でも、第十の形態に規定されるものは、上記第九の形態に規定される化合物と同様に、硫化物固体電解質の表面に付着して、吸油量を低減させやすく、これにより塗工適性が向上しやすく、また効率的に優れた電池性能を発現しやすくなる。
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、有機溶媒と、を混合すること、
前記有機溶媒を除去すること、を含む、
というものである。
既述のように、本実施形態の改質硫化物固体電解質は、エポキシ化合物を含有するものとなれば、その製造方法は特に制限されるものではないが、本実施形態の第十一の形態に係る改質硫化物固体電解質の製造方法によれば、その特徴からみてエポキシ化合物が硫化物固体電解質の表面に付着するように存在する者となり得るため、塗工適性に優れ、効率的に優れた電池性能を発現するものとなり、そのような本実施形態の改質硫化物固体電解質をより効率的に製造することが可能となる。
有機溶媒として、前記溶媒を用いることにより、エポキシ化合物の硫化物固体電解質の表面への付着を促進することができ、塗工適性を向上させやすくなる。
というものである。
また、本実施形態の第十四の形態に係るリチウムイオン電池は、前記第一の形態の改質硫化物固体電解質等及び前記第十三の形態の電極活物質の少なくとも一方を含む、
というものである。
BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含む、
というものである。
このような構成を有する改質硫化物固体電解質も、前記第一の形態の改質硫化物固体電解質と同様の効果、すなわちペーストとして塗工する際の塗工適性に優れ、かつ効率的に優れた電池性能を発現し得るという効果を奏するものである。前記第一の形態の改質硫化物固体電解質は、第十五の形態の改質硫化物固体電解質において、FT-IR分析(ATR法)による赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有すると規定されるものである、ともいえる。しかし、当該ピークは、既述のように改質硫化物固体電解質に含まれるエポキシ化合物に由来するピークであり、当該ピークはエポキシ化合物を含むことにより発現するものであることを示すものだからである。
前記第十五の形態の改質硫化物固体電解質と、電極活物質と、を含む、
というものである。
また、本実施形態の第十七の形態に係るリチウムイオン電池は、前記第十五の形態の改質硫化物固体電解質等及び前記第十六の形態の電極活物質の少なくとも一方を含む、
というものである。
これらの電極合材及びリチウムイオン電池については、前記第十三の形態の電極合材、及び前記第十四の形態のリチウムイオン電池に対する説明と同様である。
本実施形態の改質硫化物固体電解質は、BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含み、FT-IR分析(ATR法)による赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有するものである。当該ピークは、エポキシ化合物を含まない硫化物固体電解質では検出されないことから、エポキシ化合物に由来するピークである。
測定方法:全反射測定法(ATR法)
測定波数範囲:650~4000cm-1
光源:グローバー灯(SiC)
検出器:DTGS検出器
分解能:4cm-1
測定時間:1秒/回
積算回数:256回
本実施形態の改質硫化物固体電解質は、当該ピークが明確に検出されることから、エポキシ化合物は検出されやすい態様で含まれている、すなわち硫化物固体電解質の表面に付着するようにして存在するものと考えられる。そして、そのように存在することで、硫化物固体電解質の吸油量を低減させることができ、優れた塗工適性が得られるものと考えられる。
本明細書において、1H-NMRスペクトルによるピークは、核磁気共鳴装置(NMR装置)を用いて、以下の諸条件のもとで測定したものである。
観測核:1H
共鳴周波数:500MHz
プローブ:5mmφ TCIクライオプローブ
測定温度:25℃
積算回数:16回
この事象より、改質硫化物固体電解質において、エポキシ化合物は硫化物固体電解質の表面に強く付着しており、当該付着により吸油量が低減し、塗工適性が優れたものとなるものと考えられる。
エポキシ化合物1は、下記一般式(1)で示される化合物である。
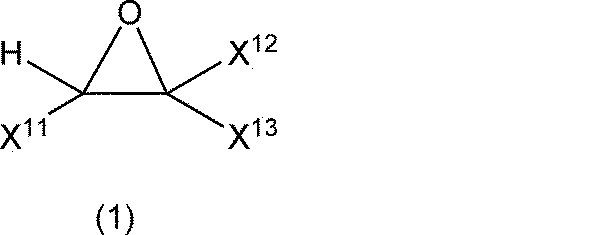
脂肪族炭化水素基としては、アルキル基、アルケニル基が好ましく挙げられ、アルキル基が好ましい。脂肪族炭化水素基の炭素数は、アルキル基の場合は好ましくは1以上、より好ましくは2以上、更に好ましくは4以上、より更に好ましくは6以上であり、上限として好ましくは24以下、より好ましくは20以下、更に好ましくは16以下である。またアルケニル基の場合は、2以上、好ましくは4以上であり、上限として好ましくは24以下、より好ましくは20以下、更に好ましくは16以下である。
X11~X13の脂肪族炭化水素基は、直鎖状、分岐状のいずれであってもよく、直鎖状が好ましい。また、脂肪族炭化水素基は、水酸基等により一部が置換されていてもよい。X11~X13の複数が脂肪族炭化水素基の場合、複数の脂肪族炭化水素基は同じでも異なっていてもよい。
X11~X13の脂環族炭化水素基は、水酸基、上記1価の脂肪族炭化水素基(例えば、アルキル基、アルケニル基)等により一部が置換されていてもよい。また、X11~X13の複数が脂環族炭化水素基の場合、複数の脂環族炭化水素基は同じでも異なっていてもよい。
X11~X13の1価の芳香族炭化水素基は、水酸基、上記1価の脂肪族炭化水素基(例えば、アルキル基、アルケニル基)等により一部が置換されていてもよい。また。X11~X13の複数が芳香族炭化水素基である場合、複数の芳香族炭化水素基は同じでも異なっていてもよい。
ハロゲン化炭化水素基中のハロゲン原子の数は、炭化水素基の炭素数に応じてかわるため一概にはいえないが、好ましくは1以上、より好ましくは2以上、更に好ましくは3以上、より更に好ましくは6以上であり、上限として好ましくは16以下、より好ましくは12以下、更に好ましくは9以下である。
エポキシ化合物2は、下記一般式(2)で示される化合物である。

2価の脂肪族炭化水素基としては、アルキレン基、アルケニレン基が好ましく、アルキレン基がより好ましい。アルキレン基の場合の炭素数としては、好ましくは1以上であり、上限として好ましくは8以下、より好ましくは6以下、更に好ましくは4以下、より更に好ましくは2以下である。アルケニレン基の場合は、炭素数は好ましくは2以上であり、上限はアルキレン基と同じである。
脂肪族炭化水素基としては、アルキル基、アルケニル基が好ましく、アルキル基がより好ましく、脂肪族炭化水素基は直鎖状でも分岐状でもよい。また、脂肪族炭化水素基がアルキル基の場合、炭素数は好ましくは1以上、より好ましくは2以上、更に好ましくは4以上であり、上限として好ましくは24以下、より好ましくは16以下、更に好ましくは12以下であり、より更に好ましくは10以下である。
また、R22の1価の炭化水素基がフェニル基の場合、1価の脂肪族炭化水素基で置換されていることが好ましく、アルキル基で置換されていることが好ましい。アルキル基としては、炭素数は好ましくは1以上、より好ましくは2以上、更に好ましくは4以上であり、上限として好ましくは24以下、より好ましくは12以下、更に好ましくは8以下であり、より更に好ましくは6以下である。この場合、脂肪族炭化水素基は直鎖状、分岐状のいずれであってもよく、分岐状であることが好ましい。R22としては、これらの中でも第四級炭素原子を有する脂肪族炭化水素基が好ましく、特にtert-ブチル基が好ましい。
エポキシ化合物3は、下記一般式(3)で示される化合物である。

2価の脂肪族炭化水素基としては、アルキレン基、アルケニレン基が好ましく、アルキレン基がより好ましい。アルキレン基の場合の炭素数としては、好ましくは1以上であり、上限として好ましくは8以下、より好ましくは6以下、更に好ましくは4以下、より更に好ましくは2以下である。アルケニレン基の場合は、炭素数は好ましくは2以上であり、上限はアルキレン基と同じである。
X34~X36の-OR33におけるR33、一般式(3b)のR34~R36の1価の炭化水素基、1価のハロゲン化炭化水素基についても、上記X34~X36の1価の炭化水素基、1価のハロゲン化炭化水素基と同じものが挙げられる。
この場合、R31及びR32の2価の炭化水素基は既述の通りであるが、R31の2価の炭化水素基の炭素数としては、好ましくは1以上であり、上限として好ましくは8以下、より好ましくは6以下、更に好ましくは4以下、より更に好ましくは2以下である。また、R32の2価の炭化水素基の炭素数としては、好ましくは1以上、より好ましくは2以上であり、上限として好ましくは8以下、より好ましくは6以下、更に好ましくは4以下である。X35、R34~R36の1価の炭化水素基は既述の通りであるが、X35の1価の炭化水素基の炭素数としては、好ましくは1以上であり、上限として好ましくは24以下、より好ましくは12以下、更に好ましくは8以下、より更に好ましくは4以下、特に好ましくは2以下である。また、R34~R36の1価の炭化水素基の炭素数も、X35の1価の炭化水素基の炭素数と同じである。
この場合、R31の2価の炭化水素基は既述の通りであるが、R31の2価の炭化水素基の炭素数としては、好ましくは1以上であり、上限として好ましくは8以下、より好ましくは6以下、更に好ましくは4以下、より更に好ましくは2以下である。X34~X36の1価の炭化水素基は既述の通りであるが、X34、X36の1価の炭化水素基の炭素数としては、好ましくは1以上であり、上限として好ましくは24以下、より好ましくは12以下、更に好ましくは8以下、より更に好ましくは4以下、特に好ましくは2以下である。また、X35の1価の炭化水素基の炭素数としては、好ましくは1以上、より好ましくは2以上、更に好ましくは4以上であり、上限として好ましくは24以下、より好ましくは12以下、更に好ましくは8以下であり、より更に好ましくは6以下である。またこの場合、X35の炭化水素基は直鎖状、分岐状のいずれであってもよく、分岐状であることが好ましく、第四級炭素原子を有する脂肪族炭化水素基が好ましく、特にtert-ブチル基が好ましい。
次に、本実施形態の改質硫化物固体電解質を形成する硫化物固体電解質について説明する。本実施形態で用いられ得る硫化物固体電解質は、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含み、BET比表面積が10m2/g以上のものであれば特に制限なく用いることが可能であり、市販品をそのまま用いることもできるし、製造して用いることもできる。
本実施形態において用いられ得る硫化物固体電解質を製造して用いる場合について、その製造方法を説明する。本実施形態において用いられ得る硫化物固体電解質は、例えば、リチウム原子、硫黄原子、リン原子及びハロゲン原子の少なくとも一の原子を含む化合物から選ばれる二種以上の原料を混合することを含む、製造方法により得られる。
原料としては、リチウム原子、硫黄原子、リン原子、ハロゲン原子の少なくとも一つの原子を含む化合物から選ばれる二種以上の化合物を採用し得る。
原料として用い得る化合物は、リチウム原子、硫黄原子、リン原子、ハロゲン原子の少なくとも一つの原子を含むものであり、より具体的には、硫化リチウム;フッ化リチウム、塩化リチウム、臭化リチウム、ヨウ化リチウム等のハロゲン化リチウム;ヨウ化ナトリウム、フッ化ナトリウム、塩化ナトリウム、臭化ナトリウム等のハロゲン化ナトリウムなどのハロゲン化アルカリ金属;三硫化二リン(P2S3)、五硫化二リン(P2S5)等の硫化リン;各種フッ化リン(PF3、PF5)、各種塩化リン(PCl3、PCl5、P2Cl4)、各種臭化リン(PBr3、PBr5)、各種ヨウ化リン(PI3、P2I4)等のハロゲン化リン;フッ化チオホスホリル(PSF3)、塩化チオホスホリル(PSCl3)、臭化チオホスホリル(PSBr3)、ヨウ化チオホスホリル(PSI3)、二塩化フッ化チオホスホリル(PSCl2F)、二臭化フッ化チオホスホリル(PSBr2F)等のハロゲン化チオホスホリル;などの上記四種の原子から選ばれる少なくとも二種の原子からなる原料、フッ素(F2)、塩素(Cl2)、臭素(Br2)、ヨウ素(I2)等のハロゲン単体、好ましくは臭素(Br2)、ヨウ素(I2)が代表的に挙げられる。
また、同様の観点から、原料に用い得る化合物としては、上記の中でも、硫化リチウム;三硫化二リン(P2S3)、五硫化二リン(P2S5)等の硫化リン;フッ素(F2)、塩素(Cl2)、臭素(Br2)、ヨウ素(I2)等のハロゲン単体;フッ化リチウム、塩化リチウム、臭化リチウム、ヨウ化リチウム等のハロゲン化リチウム;が好ましく、硫化リンの中でも五硫化二リンが好ましく、ハロゲン単体の中でも塩素(Cl2)、臭素(Br2)、ヨウ素(I2)が好ましく、ハロゲン化リチウムの中でも塩化リチウム、臭化リチウム、ヨウ化リチウムが好ましい。
硫化リチウム粒子の平均粒径(D50)は、10μm以上2000μm以下であることが好ましく、30μm以上1500μm以下であることがより好ましく、50μm以上1000μm以下であることがさらに好ましい。本明細書において、平均粒径(D50)は、粒子径分布積算曲線を描いた時に粒子径の最も小さい粒子から順次積算して全体の50%に達するところの粒子径であり、体積分布は、例えば、レーザー回折/散乱式粒子径分布測定装置を用いて測定することができる平均粒径のことである。また、上記の原料として例示したもののうち固体の原料については、上記硫化リチウム粒子と同じ程度の平均粒径を有するものが好ましい、すなわち上記硫化リチウム粒子の平均粒径と同じ範囲内にあるものが好ましい。
2≦2α+β≦100…(1)
4≦2α+β≦80 …(2)
6≦2α+β≦50 …(3)
6≦2α+β≦30 …(4)
リチウム原子、硫黄原子、リン原子及びハロゲン原子の少なくとも一つの原子を含む化合物から選ばれる二種以上の原料を混合は、例えば当該原料を混合機を用いて行うことができる。また、撹拌機、粉砕機等を用いて行うこともできる。
撹拌機を用いても原料の混合は起こり得るし、粉砕機を用いると原料の粉砕が生じることとなるが、同時に混合も生じるからである。すなわち、本実施形態で用いられる硫化物固体電解質は、リチウム原子、硫黄原子、リン原子及びハロゲン原子の少なくとも一つの原子を含む化合物から選ばれる二種以上の原料を、撹拌、混合、粉砕、又はこれらのいずれかを組合せた処理により行うことができる、ともいえる。
媒体式粉砕機は、容器駆動式粉砕機、媒体撹拌式粉砕機に大別される。容器駆動式粉砕機としては、撹拌槽、粉砕槽、あるいはこれらを組合せたボールミル、ビーズミル等が挙げられる。また、媒体撹拌式粉砕機としては、カッターミル、ハンマーミル、ピンミル等の衝撃式粉砕機;タワーミルなどの塔型粉砕機;アトライター、アクアマイザー、サンドグラインダー等の撹拌槽型粉砕機;ビスコミル、パールミル等の流通槽型粉砕機;流通管型粉砕機;コボールミル等のアニュラー型粉砕機;連続式のダイナミック型粉砕機;一軸又は多軸混練機などの各種粉砕機が挙げられる。中でも、得られる硫化物の粒径の調整のしやすさ等を考慮すると、容器駆動式粉砕機として例示したボールミル、ビーズミルが好ましく、中でも遊星型のものが好ましい。
湿式粉砕機としては、湿式ビーズミル、湿式ボールミル、湿式振動ミル等が代表的に挙げられ、粉砕操作の条件を自由に調整でき、より小さい粒径のものに対応しやすい点で、ビーズを粉砕メディアとして用いる湿式ビーズミルが好ましい。また、乾式ビーズミル、乾式ボールミル、乾式振動ミル等の乾式媒体式粉砕機、ジェットミル等の乾式非媒体粉砕機等の乾式粉砕機を用いることもできる。
また、材質としては、例えば、ステンレス、クローム鋼、タングステンカーバイド等の金属;ジルコニア、窒化ケイ素等のセラミックス;メノウ等の鉱物が挙げられる。
また、この場合の粉砕時間としては、その処理する規模に応じてかわるため一概にはいえないが、通常0.5時間以上、好ましくは1時間以上、より好ましくは5時間以上、更に好ましくは10時間以上であり、上限としては通常100時間以下、好ましくは72時間以下、より好ましくは48時間以下、更に好ましくは36時間以下である。
上記の混合にあたり、上記の原料に、溶媒を加えて混合することができる。溶媒としては、広く有機溶媒と称される各種溶媒等を用いることができる。
例えば、アミノ基を有する溶媒としては、エチレンジアミン、ジアミノプロパン、ジメチルエチレンジアミン、ジエチルエチレンジアミン、ジメチルジアミノプロパン、テトラメチルジアミノメタン、テトラメチルエチレンジアミン(TMEDA)、テトラメチルジアミノプロパン(TMPDA)等の脂肪族アミン;シクロプロパンジアミン、シクロヘキサンジアミン、ビスアミノメチルシクロヘキサン等の脂環式アミン;イソホロンジアミン、ピペラジン、ジピペリジルプロパン、ジメチルピペラジン等の複素環式アミン;フェニルジアミン、トリレンジアミン、ナフタレンジアミン、メチルフェニレンジアミン、ジメチルナフタレンジアミン、ジメチルフェニレンジアミン、テトラメチルフェニレンジアミン、テトラメチルナフタレンジアミン等の芳香族アミンが好ましく挙げられる。
アセトニトリル、アクリロニトリル等のニトリル溶媒;ジメチルホルムアミド、ニトロベンゼン等の窒素原子を含む溶媒も好ましく挙げられる。
また、硫黄原子を含む溶媒としては、ジメチルスルホキシド、二硫化炭素等が好ましく挙げられる。
溶媒を用いて混合を行った場合は、混合を行った後、混合により得られた流体(通常、スラリー)を乾燥することを含んでもよい。溶媒として錯化剤を用いた場合は、錯化剤を含む錯体から当該錯化剤を除去することにより、錯化剤と溶媒とを併用した場合は、錯化剤を含む錯体から当該錯化剤を除去し、かつ溶媒を除去することにより、また錯化剤以外の溶媒を用いた場合は当該溶媒を除去することにより、硫化物固体電解質が得られる。得られた硫化物固体電解質は、リチウム原子に起因するイオン伝導度を発現するものである。
また、通常5~100℃、好ましくは10~85℃、より好ましくは15~70℃、より更に好ましくは室温(23℃)程度(例えば室温±5℃程度)で真空ポンプ等を用いて減圧乾燥(真空乾燥)して、錯化剤及び必要に応じて用いられる溶媒を揮発させて行うことができる。
固液分離は、具体的には、流体を容器に移し、硫化物(あるいは錯化剤を含む場合は錯体(硫化物固体電解質の前駆体とも称し得るものである。)が沈殿した後に、上澄みとなる錯化剤、溶媒を除去するデカンテーション、また例えばポアサイズが10~200μm程度、好ましくは20~150μmのガラスフィルターを用いたろ過が容易である。
上記混合を行って得られる硫化物固体電解質は、例えば結晶化する程度に粉砕機を用いて粉砕による混合を行わない限り、基本的には非晶性の硫化物固体電解質(ガラス成分)となる。
硫化物固体電解質としては、結晶性の硫化物固体電解質の粉末の粒径を調整するために、例えば後述する粉砕等の処理を施した結果、その表面に非晶性の成分(ガラス成分)が形成した結晶性の硫化物固体電解質も含まれ得る。よって、非晶性成分を含む硫化物固体電解質には、非晶性の硫化物固体電解質、また結晶性の硫化物固体電解質であって、その表面に非晶性の成分が形成した硫化物固体電解質も含まれる。
結晶性の硫化物固体電解質を製造する場合、さらに加熱することを含んでもよい。上記混合することにより非晶性の硫化物固体電解質(ガラス成分)が得られた場合は、加熱することにより結晶性の硫化物固体電解質が得られ、また結晶性の硫化物固体電解質が得られた場合は、より結晶化度を向上させた結晶性の硫化物固体電解質が得られる。
また、混合を行う際に溶媒として錯化剤を用いた場合は、錯化剤を含む錯体が形成しているが、上記の乾燥を行わずに加熱することによっても、錯体より錯化剤を除去し、硫化物固体電解質が得られ、加熱の条件によって、非晶性のものとすることもできるし、結晶性のものとすることもできる。
加熱の方法は、特に制限されるものではないが、例えば、ホットプレート、真空加熱装置、アルゴンガス雰囲気炉、焼成炉を用いる方法等を挙げることができる。また、工業的には、加熱手段と送り機構を有する横型乾燥機、横型振動流動乾燥機等を用いることもでき、加熱する処理量に応じて選択すればよい。
既述のように、本実施形態で用いられる硫化物固体電解質は、市販品を用いても、製造したものを用いてもよい。
上記の方法により得られる硫化物固体電解質は、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む、非晶性(ガラス成分)、結晶性の硫化物固体電解質であり、好適に本実施形態における硫化物固体電解質として用いられる。
本実施形態で用いられる硫化物固体電解質の比表面積はBET比表面積が10m2/g以上のものである。本実施形態の改質硫化物固体電解質は、このように比表面積が大きいものであるにもかかわらず、ペーストとして塗工する際の塗工適性に優れ、かつ効率的に優れた電池性能を発現するという効果を奏するものであり、硫化物固体電解質のBET比表面積は高ければ高いほど、当該効果の優位性をより示すことが可能となる。このような観点から、BET比表面積は12m2/g以上のものが好ましく、15m2/g以上のものがより好ましく、20m2/g以上のものが更に好ましい。同様の観点から上限としては特に制限はないが、現実的には100m2/g以下、好ましくは75m2/g以下、より好ましくは50m2/g以下である。
本明細書において、BET比表面積は、JIS Z 8830:2013(ガス吸着による粉体(固体)の比表面積測定方法)に準拠し、吸着質としてクリプトンを用いて測定される比表面積である。
上記の方法により得られる非晶性硫化物固体電解質としては、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含み、代表的なものとしては、例えば、Li2S-P2S5-LiI、Li2S-P2S5-LiCl、Li2S-P2S5-LiBr、Li2S-P2S5-LiI-LiBr等の、硫化リチウム、硫化リン及びハロゲン化リチウムとから構成される固体電解質;さらに酸素原子、珪素原子等の他の原子を含む、例えば、Li2S-P2S5-Li2O-LiI、Li2S-SiS2-P2S5-LiI等の固体電解質が好ましく挙げられる。より高いイオン伝導度を得る観点から、Li2S-P2S5-LiI、Li2S-P2S5-LiCl、Li2S-P2S5-LiBr、Li2S-P2S5-LiI-LiBr等の硫化リチウムと硫化リンとハロゲン化リチウムとから構成される固体電解質が好ましく挙げられる。
非晶性硫化物固体電解質を構成する原子の種類は、例えば、ICP発光分光分析装置により確認することができる。
上記の方法により得られる結晶性硫化物固体電解質は、非晶性固体電解質を結晶化温度以上に加熱して得られる、いわゆるガラスセラミックスであってもよく、その結晶構造としては、Li3PS4結晶構造、Li4P2S6結晶構造、Li7PS6結晶構造、Li7P3S11結晶構造、2θ=20.2°近傍及び23.6°近傍にピークを有する結晶構造(例えば、特開2013-16423号公報)等が挙げられる。
なお、これらのピーク位置については、±0.5°の範囲内で前後していてもよい。
本実施形態の改質硫化物固体電解質は、BET比表面積が10m2/g以上であり、大きい比表面積を有するものである。硫化物固体電解質のBET比表面積は高ければ高いほど、当該効果の優位性をより示すことが可能となる観点から、BET比表面積は12m2/g以上のものが好ましく、15m2/g以上のものがより好ましく、20m2/g以上のものが更に好ましい。同様の観点から上限としては特に制限はないが、現実的には100m2/g以下、好ましくは75m2/g以下、より好ましくは50m2/g以下である。
ハロゲン化リチウムがその表面に付着しても、硫化物固体電解質のBET比表面積には大きな影響を及ぼすことはなく、本実施形態で用いられる硫化物固体電解質のBET比表面積と、改質硫化物固体電解質のBET比表面積とは、実質的に同じである。よって、硫化物固体電解質としては、BET比表面積が10m2/g以上のものを用いれば、自ずと改質硫化物固体電解質のBET比表面積も10m2/g以上のものとなる。
本明細書において、吸油量は、改質硫化物固体電解質1gを試料とし、乳鉢等において酪酸ブチルを1滴添加してはスパチュラで撹拌する操作を行い、試料がペースト状になるまで当該操作を繰り返し、添加した酪酸ブチルの合計量を吸油量(mL/g)とした。ここで、「ペースト状」は、JIS K5101-13-1:2004(顔料試験方法-第13部:吸油量-第1節:精製あまに油法)の「7.2 測定」に規定される「割れたり,ぼろぼろになったりせず広げることができ,かつ,測定板に軽く付着する程度のもの」の状態を意味する。
本実施形態の改質硫化物固体電解質は、塗工適性に優れ、溶媒等を用いなくても電池の製造に供することができることから、効率的に優れた電池性能を発現し得るものである。また、イオン伝導度が高く、優れた電池性能を有しているため、電池に好適に用いられる。
本実施形態の改質硫化物固体電解質は、正極層に用いてもよく、負極層に用いてもよく、電解質層に用いてもよい。なお、各層は、公知の方法により製造することができる。
本実施形態の改質硫化物固体電解質の製造方法は、BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、有機溶媒と、を混合すること、前記有機溶媒を除去すること、を含む、製造方法である。本実施形態の製造方法によれば、上記本実施形態の改質硫化物固体電解質を効率的に製造することが可能である、すなわち、本実施形態の改質硫化物固体電解質は、本実施形態の製造方法により製造することが好ましい。
本実施形態の製造方法において、有機溶媒としては、これらの中から単独で、又は複数種を組合わせて用いることができる。
また、本実施形態の製造方法においては、上記硫化物固体電解質を製造する方法における「加熱」することを行ってもよい。
本実施形態の別形態の改質硫化物固体電解質(上記本実施形態の第十五の形態に係る改質硫化物固体電解質)は、BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含む、というものである。既述のように、このような別形態の改質硫化物固体電解質も、上記の本実施形態の改質硫化物固体電解質と同様に、ペーストとして塗工する際の塗工適性に優れ、かつ効率的に優れた電池性能を発現し得るという効果を奏するものである。
本実施形態の電極合材は、上記の本実施形態の改質硫化物固体電解質と、電極活物質と、を含む電極合材、また上記の別形態の改質硫化物固体電解質と、電極活物質と、を含む電極合材である。
電極活物質としては、電極合材が正極、負極のいずれに用いられるかに応じて、各々正極活物質、負極活物質が採用される。
硫化物系正極活物質としては、硫化チタン(TiS2)、硫化モリブデン(MoS2)、硫化鉄(FeS、FeS2)、硫化銅(CuS)、硫化ニッケル(Ni3S2)等が挙げられる。
また、上記正極活物質の他、セレン化ニオブ(NbSe3)等も使用可能である。
正極活物質は、一種単独で、又は複数種を組み合わせて用いることが可能である。
このような負極活物質としては、例えば、金属リチウム、金属インジウム、金属アルミ、金属ケイ素、金属スズ等の金属リチウム又は金属リチウムと合金を形成し得る金属、これら金属の酸化物、またこれら金属と金属リチウムとの合金等が挙げられる。
被覆層を形成する材料としては、硫化物固体電解質においてイオン伝導度を発現する原子、好ましくはリチウム原子の窒化物、酸化物、又はこれらの複合物等のイオン伝導体が挙げられる。具体的には、窒化リチウム(Li3N)、Li4GeO4を主構造とする、例えばLi4-2xZnxGeO4等のリシコン型結晶構造を有する伝導体、Li3PO4型の骨格構造を有する例えばLi4-xGe1-xPxS4等のチオリシコン型結晶構造を有する伝導体、La2/3-xLi3xTiO3等のペロブスカイト型結晶構造を有する伝導体、LiTi2(PO4)3等のNASICON型結晶構造を有する伝導体等が挙げられる。
また、LiyTi3-yO4(0<y<3)、Li4Ti5O12(LTO)等のチタン酸リチウム、LiNbO3、LiTaO3等の周期表の第5族に属する金属の金属酸リチウム、またLi2O-B2O3-P2O5系、Li2O-B2O3-ZnO系、Li2O-Al2O3-SiO2-P2O5-TiO2系等の酸化物系の伝導体等が挙げられる。
ここで、各種原子を含む溶液としては、例えばリチウムエトキシド、チタンイソプロポキシド、ニオブイソプロポキシド、タンタルイソプロポキシド等の各種金属のアルコキシドを含む溶液を用いればよい。この場合、溶媒としては、エタノール、ブタノール等のアルコール系溶媒、ヘキサン、ヘプタン、オクタン等の脂肪族炭化水素溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素溶媒等を用いればよい。
また、上記の付着は、浸漬、スプレーコーティング等により行えばよい。
被覆層の厚さは、透過型電子顕微鏡(TEM)による断面観察により、被覆層の厚さを測定することができ、被覆率は、被覆層の厚さと、元素分析値、BET比表面積と、から算出することができる。
本実施形態の電極合材は、上記の改質硫化物固体電解質、電極活物質の他、例えば導電材、結着剤等のその他成分を含んでもよい。すなわち、本実施形態の電極合材の製造方法は、上記の改質硫化物固体電解質、電極活物質の他、例えば導電材、結着剤等のその他成分を用いてもよい。導電剤、結着剤等のその他成分は、上記の改質硫化物固体電解質と、電極活物質と、を混合することにおいて、これらの改質硫化物固体電解質及び電極活物質に、さらに加えて混合して用いればよい。
導電材としては、電子伝導性の向上により電池性能を向上させる観点から、人造黒鉛、黒鉛炭素繊維、樹脂焼成炭素、熱分解気相成長炭素、コークス、メソカーボンマイクロビーズ、フルフリルアルコール樹脂焼成炭素、ポリアセン、ピッチ系炭素繊維、気相成長炭素繊維、天然黒鉛、難黒鉛化性炭素等の炭素系材料が挙げられる。
結着剤としては、結着性、柔軟性等の機能を付与し得るものであれば特に制限はなく、例えば、ポリテトラフルオロエチレン、ポリフッ化ビニリデン等のフッ素系ポリマー、ブチレンゴム、スチレン-ブタジエンゴム等の熱可塑性エラストマー、アクリル樹脂、アクリルポリオール樹脂、ポロビニルアセタール樹脂、ポリビニルブチラール樹脂、シリコーン樹脂等の各種樹脂が例示される。
また、結着剤を含有する場合、電極合材中の結着剤の含有量は特に制限はないが、電池性能を向上させ、かつ製造効率を考慮すると、好ましくは1質量%以上、より好ましくは3質量%以上、更に好ましくは5質量%以上であり、上限として好ましくは20質量%以下、好ましくは15質量%以下、更に好ましくは10質量%以下である。
本実施形態のリチウムイオン電池は、上記の本実施形態の改質硫化物固体電解質及び上記の電極合材から選ばれる少なくとも一方を含む、また上記の別形態の改質硫化物固体電解質及び上記の電極合材から選ばれる少なくとも一方を含む、リチウムイオン電池である。
撹拌子入りシュレンク(容量:100mL)に、窒素雰囲気下、硫化リチウム0.59g、五硫化二リン0.95g、臭化リチウム0.19g、ヨウ化リチウム0.28gを導入した。撹拌子を回転させた後、錯化剤のテトラメチルエチレンジアミン(TMEDA)20mLを加え、12時間撹拌を継続し、得られた錯体含有物を真空下で乾燥(室温:23℃)して粉末の錯体を得た。次いで、錯体の粉末を真空下で120℃で加熱を2時間行い、非晶性の硫化物固体電解質を得た。更に、非晶性の硫化物固体電解質を真空下で140℃で加熱を2時間行い、結晶性の硫化物固体電解質1を得た(結晶性の硫化物固体電解質を得るための加熱温度(本例では140℃)を「結晶化温度」と称することがある。)。
得られた非晶性の硫化物固体電解質、結晶性の硫化物固体電解質のBET比表面積を測定したところ、いずれも40m2/gであった。
撹拌翼付き反応槽(容量:500mL)に、窒素雰囲気下で製造例1で得られた硫化物固体電解質の粉末30.0g、トルエン470gを投入した。撹拌翼を回転させた後、循環運転可能な微小ビーズ対応ビーズミル(「UAM-015(型番)」、株式会社広島メタル&マシナリー製)を用いて所定条件(ビーズ材質:ジルコニア、ビーズ直径:0.1mmφ、ビーズ使用量:391g、ポンプ流量150mL/min、周速8m/s、ミルジャケット温度20℃)で30分間の粉砕処理行った。得られたスラリーを真空下で乾燥(室温:23℃)して非晶性の固体電解質の白色粉末を得た。この白色粉末を160℃で2時間結晶化を行うことで、結晶性の硫化物固体電解質2を得た。得られた結晶性の硫化物固体電解質2のBET比表面積を測定したところ、10m2/gであった。
撹拌翼付き反応槽(容量:500mL)に、窒素雰囲気下で製造例1で得られた硫化物固体電解質の粉末30.0g、トルエン470gを投入した。撹拌翼を回転させた後、循環運転可能な微小ビーズ対応ビーズミル(「UAM-015(型番)」、株式会社広島メタル&マシナリー製)を用いて所定条件(ビーズ材質:ジルコニア、ビーズ直径:0.05mmφ、ビーズ使用量:391g、ポンプ流量150mL/min、周速8m/s、ミルジャケット温度20℃)で30分間の第一粉砕処理行った。次いで、周速を12.5m/sに変更し10分間の第二粉砕処理を行った。得られたスラリーを真空下で乾燥(室温:23℃)して非晶性の固体電解質の白色粉末を得た。この白色粉末を160℃で2時間結晶化を行うことで、結晶性の硫化物固体電解質3を得た。得られた結晶性の硫化物固体電解質3のBET比表面積を測定したところ、8m2/gであった。
窒素雰囲気のグローブボックス内にて、モル比がLi2S:P2S5:LiBr:LiCl=47.5:12.5:15.0:25.0であり、合計110.0gとなるように計量したものを、ガラス容器内で振とうして粗混合した。
窒素雰囲気下で、粗混合した原料を、脱水トルエン(和光純薬製)1140mL及び脱水イソブチロニトリル(キシダ化学製)7mLの混合溶媒中に分散させたスラリー(スラリー濃度:10質量%)を、循環運転可能なビーズミル(「LMZ015(型番)」、アシザワ・ファインテック社製)を用いて、1時間混合粉砕した(ビーズ材質:ジルコニア、ビーズ直径:0.5mmφ、ビーズ使用量:456g、ポンプ流量:500mL/min、周速:12m/s)。次いで、スラリーを減圧乾燥して得られた原料混合物30.0gを、エチルベンゼン(和光純薬社製)300mLに分散させたスラリーを、減圧乾燥して溶媒を留去したものを、窒素雰囲気下のグローブボックス内の電気炉(「F-1404-A(型番)」、東京硝子器械株式会社製)を用いて380℃で1時間以上保持して焼成し、アルジロダイト型結晶構造を有する結晶性の硫化物固体電解質4を得た。得られた結晶性の硫化物固体電解質4のBET比表面積を測定したところ、12m2/gであった。
撹拌子入りシュレンク(容量:100mL)に、窒素雰囲気下、製造例1で得られた結晶性の硫化物固体電解質1を3g秤量して加え、トルエン22gを加えて撹拌し、スラリー状の流体とした。当該スラリー状の流体に、更にエポキシ化合物としてブチレンオキサイドを0.05g(結晶性硫化物固体電解質100質量部に対して5質量部)の割合となるような量で加え、10分撹拌した後、真空乾燥によりトルエンを留去し、改質硫化物固体電解質を得た。
得られた改質硫化物固体電解質について、下記の方法に基づき、吸油量、イオン伝導度を測定した。また下記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、第1表に示す。また、下記の方法に基づき、FT-IR分析(ATR法)を行ったところ、赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有することが確認された。また、下記の方法に基づき、1H-NMR測定を行ったところ、アルキル鎖に由来する0.0~5.0ppmのピークを有することも確認された。
実施例1において、結晶性の硫化物固体電解質の種類、エポキシ化合物の種類及び使用量を、第1表に示されるものとした以外は、実施例1と同様にして、改質硫化物固体電解質を作製した。
得られた改質硫化物固体電解質について、下記の方法に基づき、吸油量、イオン伝導度を測定した。また下記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、第1表に示す。また、下記の方法に基づき、FT-IR分析(ATR法)を行ったところ、赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有することが確認された。また、下記の方法に基づき、1H-NMR測定を行ったところ、アルキル鎖に由来する0.0~5.0ppmのピークを有することも確認された。
上記製造例1~3で得られた各々硫化物固体電解質1~3について、下記の方法に基づき、吸油量、イオン伝導度を測定した。また下記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、第1表に示す。硫化物固体電解質1~3の吸油量は、各々1.03mL/g、0.93mL/g、0.66mL/gであった。
また、下記の方法に基づき、FT-IR分析(ATR法)を行ったところ、赤外線吸収スペクトルにおいて、2800~3000cm-1においてピークは確認できなかった。また、下記の方法に基づき、1H-NMR測定を行ったところ、アルキル鎖に由来する0.0~5.0ppmのピークも確認できなかった。
実施例及び比較例で得られた固体電解質1gを試料とし、メノウ乳鉢において、スポイトを用いて酪酸ブチルを1滴添加してはスパチュラで撹拌する操作を行い、試料がペースト状になるまで当該操作を繰り返し、添加した酪酸ブチルの合計量を吸油量(mL/g)とした。測定した吸油量について、以下の基準で評価とした。
A.0.8mL/g未満
B.0.8mL/g以上0.9mL/g未満
C.0.9mL/g以上
上記(吸油量の測定)と同様にして、製造例1~4で得られた硫化物固体電解質1~4の吸油量を測定した。硫化物固体電解質1~4の吸油量Aと、上記(吸油量の測定)による実施例及び比較例で得られた硫化物固体電解質の吸油量Bと、を用いて、以下の式により算出される数値を吸油量の減少率とした。ここで、吸油量Aは、実施例及び比較例で用いられた硫化物固体電解質1~4のいずれかの吸油量を用いることとする。例えば、実施例1の吸油量の減少率は、硫化物固体電解質1の吸油量を吸油量Aとして、実施例1の改質硫化物固体電解質の吸油量を吸油量Bとして、減少率を計算する。
吸油量の減少率=(吸油量A-吸油量B)/吸油量A×100(%)
本実施例において、イオン伝導度の測定は、以下のようにして行った。
硫化物固体電解質から、直径10mm(断面積S:0.785cm2)、高さ(L)0.1~0.3cmの円形ペレットを成形して試料とした。その試料の上下から電極端子を取り、25℃において交流インピーダンス法により測定し(周波数範囲:1MHz~100Hz、振幅:10mV)、Cole-Coleプロットを得た。高周波側領域に観測される円弧の右端付近で、-Z’’(Ω)が最小となる点での実数部Z’(Ω)を電解質のバルク抵抗R(Ω)とし、以下式に従い、イオン伝導度σ(S/cm)を計算した。
R=ρ(L/S)
σ=1/ρ
測定したイオン伝導度について、以下の基準で評価した。
A.2.5mS/cm以上
B.0.5mS/cm以上2.5mS/cm未満
C.0.5mS/cm未満
測定装置:FR-IR分光計「VERTEX70v(型番)」、Bruker社製
測定方法:全反射測定法(ATR法)
測定波数範囲:650~4000cm-1
光源:グローバー灯(SiC)
検出器:DTGS検出器
分解能:4cm-1
測定時間:1秒/回
積算回数:256回
測定条件:ダイヤモンドプリズムを用い、入射角45°で照射
核磁気共鳴装置(NMR装置):AVANCE III HD(BEUKER社製)
観測核:1H
共鳴周波数:500MHz
プローブ:5mmφ TCIクライオプローブ
測定温度:25℃
積算回数:16回
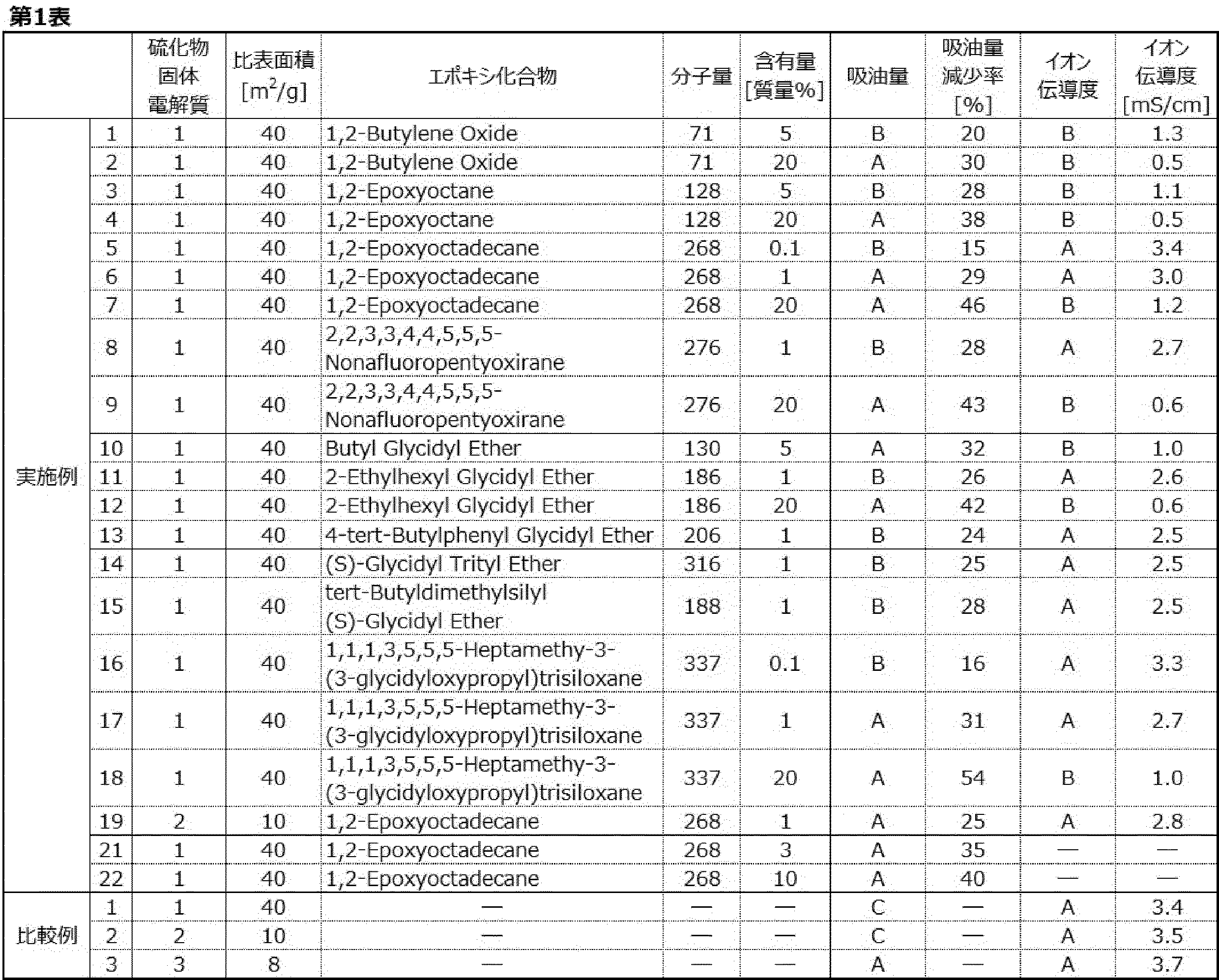
一方、エポキシ化合物と混合せず、エポキシ化合物を含まない比較例1~3の硫化物固体電解質は、各々製造例1~3で作製した硫化物固体電解質1~3であり、従来の硫化物固体電解質そのものである。比較例1及び2の比表面積が10m2/g以上の硫化物固体電解質1及び2は、吸油量の点でC評価となり、塗工適性に劣るものであることが確認された。また、比較例3の硫化物固体電解質3については、吸油量、イオン伝導度の評価のいずれもA評価であり、改質の必要性が乏しいものであることが確認された。すなわち、本実施形態の改質硫化物固体電解質の製造方法は、比表面積が10m2/g以上と大きいものについて、吸油量を低減させて、塗工適性を向上させる効果を発現し得るため、好適であることが確認された。
上記実施例で得られた改質硫化物固体電解質について、エポキシ化合物が硫化物固体電解質に付着しているか否かを確認するため、以下検証した。
まず、実施例6の1,2-エポキシオクタデカンを1質量%の含有量(硫化物固体電解質100質量部に対して1,2-エポキシオクタデカンの使用量を1質量部の割合の使用量)として得られた改質硫化物固体電解質について、トルエンを加えてスラリー状にした後(スラリー濃度:12質量%)、12時間静置した。硫化物固体電解質が沈降することで生じた上澄み液を採取し、ガスクロマトグラフィー質量分析法(GC/MS法)により分析した。本分析における定量は、仕込液(1,2-エポキシオクタデカンの1質量%トルエン溶液)も上記上澄み液と同様にして分析し、当該仕込液中の1,2-エポキシオクタデカンのピーク面積を1とし、上記上澄み液中に残存した1,2-エポキシオクタデカンのピーク面積と比較した(上澄み液のピーク面積が1に近いほど、エポキシ化合物が硫化物固体電解質から遊離して、トルエンに溶けだしたことを意味する。)。当該分析によると、上澄み液からはエポキシ化合物は検出されなかったことから、エポキシ化合物は硫化物固体電解質に全て付着したと考えられる。
ガスクロマトグラフ:6890B(Agient社製)
分析カラム:HP-5ms(Agilent社製)
GCオーブン昇温条件:初期温度 50℃
50℃~300℃ 10℃/分で昇温
300℃で5分保持
サンプル注入量:1μL
実施例6の1,2-エポキシオクタデカンを3質量%の含有量(硫化物固体電解質100質量部に対して1,2-エポキシオクタデカンの使用量を3質量部の割合の使用量)とした以外は実施例6と同様にして改質硫化物固体電解質を得た。得られた改質硫化物固体電解質について、上記吸油量の測定に従い、吸油量を測定した。また下記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、結果を第1表に示す。また、得られた改質硫化物固体電解質について、実施例20と同様にして上澄み液をガスクロマトグラフィー質量分析法(GC/MS法)により分析した。当該分析によると、上澄み液からはエポキシ化合物は若干検出(ピーク面積:0.25)されたものの、エポキシ化合物は硫化物固体電解質に付着していることが確認された。
また、沈降した粉末についても、実施例20と同様に1H-NMR測定を行ったところ、エポキシ化合物に由来する基(アルキル基等)のケミカルシフトが検出された。
実施例6の1,2-エポキシオクタデカンを10質量%の含有量(硫化物固体電解質100質量部に対して1,2-エポキシオクタデカンの使用量を10質量部の割合の使用量)とした以外は実施例6と同様にして改質硫化物固体電解質を得た。得られた改質硫化物固体電解質について、上記吸油量の測定に従い、吸油量を測定した。また下記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、結果を第1表に示す。また、得られた改質硫化物固体電解質について、実施例20と同様にして上澄み液をガスクロマトグラフィー質量分析法(GC/MS法)により分析した。当該分析によると、上澄み液からはエポキシ化合物は若干検出(ピーク面積:0.43)されたものの、エポキシ化合物は硫化物固体電解質に付着していることが確認された。
また、沈降した粉末についても、実施例20と同様に1H-NMR測定を行ったところ、エポキシ化合物に由来する基(アルキル基等)のケミカルシフトが検出された。
以上実施例20~22の結果から、改質硫化物固体電解質においてエポキシ化合物は硫化物固体電解質に付着するように含まれており、トルエンによる洗浄を行ってもほとんどが改質硫化物固体電解質に残留することが確認された。また、1H-NMR測定から、エポキシ化合物は改質硫化物固体電解質に残留していることが確認された。
実施例1において、結晶性の硫化物固体電解質の種類、エポキシ化合物の種類及び使用量を、第2表に示されるものとした以外は、実施例1と同様にして、改質硫化物固体電解質を作製した。
得られた改質硫化物固体電解質について、上記の方法に基づき、吸油量、イオン伝導度を測定した。また上記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、第2表に示す。
上記製造例4で得られた各々硫化物固体電解質4について、下記の方法に基づき、吸油量、イオン伝導度を測定した。また上記の方法に基づき、吸油量の減少率を算出した。測定結果及び算出結果について、第2表に示す。硫化物固体電解質4の吸油量は、1.06mL/gであった。
また、上記の方法に基づき、FT-IR分析(ATR法)を行ったところ、赤外線吸収スペクトルにおいて、2800~3000cm-1においてピークは確認できなかった。また、上記の方法に基づき、1H-NMR測定を行ったところ、アルキル鎖に由来する0.0~5.0ppmのピークも確認できなかった。
不可逆容量を評価するため、下記のCV測定用セルを用いた。
実施例で得られた硫化物固体電解質及びデンカブラック粒状品(粒径:35nm、デンカ株式会社製)を合計100mg(硫化物固体電解質:デンカブラック(質量比)=85:15)を乳鉢を用いて10分間混合し、測定用粉体(1)を得た。
直径10mmの電池セルに、セパレーター層用の電解質100mgを加え、SUS製金型で10MPa/cm2で120°ずつ回転させながら3回プレスした後、測定用粉体(1)を50mg加え、20MPa/cm2で120°ずつ回転させながら3回プレスした。次いで、上記測定用粉体(1)の逆側から、20MPa/cm2で120°ずつ回転させながら3回プレスした。
1L撹拌翼付き反応容器に、窒素雰囲気下でL2Sを20.5g、P2S5を33.1g、LiIを10.0g、LiBrを6.5g添加した。撹拌翼を回転させた後、トルエン630gを導入し、このスラリーを10分間撹拌した。循環運転可能なビーズミル(「スターミルLMZ015(商品名)」、アシザワファインテック株式会社製、ジルコニア製ビーズ材質:ジルコニア、ビーズ直径:0.5mmφ、ビーズ使用量:456g)に反応容器を接続し、45時間の粉砕処理(ポンプ流量:650mL/min、ビーズミル周速:12m/s、ミルジャケット温度:45℃)を行った。
得られたスラリーを真空下で室温乾燥(25℃)した後、加熱(80℃)を行い非晶性の固体電解質の白色粉末を得た。さらに、得られた白色粉末を真空下で195℃の加熱を2時間行うことにより、結晶性固体電解質の白色粉末を得た。結晶性固体電解質のXRDスペクトルでは2θ=20.2°、23.6°に結晶化ピークが検出され、チオリシコンリージョンII型結晶構造を有していることを確認した。また、得られた結晶性固体電解質の平均粒径(D50)は4.5μm、イオン伝導度は5.0mS/cmであった。
測定温度:25℃
掃引速度:0.1mV/s
電位測定範囲:開回路電圧(+2.7V)→+5.0V→+2.7V
サイクル数:5回

Claims (17)
- BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含み、
FT-IR分析(ATR法)による赤外線吸収スペクトルにおいて、2800~3000cm-1にピークを有する、
改質硫化物固体電解質。 - 前記ピークが、前記エポキシ化合物におけるアルキル鎖のC-H伸縮振動に由来するものである、請求項1記載の改質硫化物固体電解質。
- 1H-NMRスペクトルにおいて、前記アルキル鎖に由来する0.0~5.0ppmのピークを有する、請求項2に記載の改質硫化物固体電解質。
- 前記エポキシ化合物が、下記一般式(1)で示されるエポキシ化合物1、一般式(2)で示されるエポキシ化合物2及び一般式(3)で示されるエポキシ化合物3から選ばれる少なくとも一種の化合物である、請求項1~3のいずれか1項に記載の改質硫化物固体電解質。

(一般式(1)において、X11~X13は、各々独立に水素原子、ハロゲン原子、1価の炭化水素基又は1価のハロゲン化炭化水素基であり、X11~X13の少なくとも一つは1価の炭化水素基又は1価のハロゲン化炭化水素基である。
一般式(2)において、X21~X23は、各々独立に水素原子、ハロゲン原子、1価の炭化水素基、1価のハロゲン化炭化水素基又は一般式(2a)で示される基であり、X21~X23の少なくとも一つは一般式(2a)で示される基である。一般式(2a)において、R21は2価の炭化水素基であり、R22は水素原子、ハロゲン原子、1価の炭化水素基又は1価のハロゲン化炭化水素基である。
一般式(3)において、X31~X33は、各々独立に水素原子、ハロゲン原子、1価の炭化水素基、1価のハロゲン化炭化水素基、一般式(3a)で示される基であり、X31~X33の少なくとも一つは一般式(3a)で示される基である。一般式(3a)において、R31及びR32は各々独立に単結合又は2価の炭化水素基であり、X34~X36は、各々独立に水素原子、ハロゲン原子、1価の炭化水素基、1価のハロゲン化炭化水素基、-OR33又は一般式(3b)で示される基である。R33~R36は、各々独立に水素原子、ハロゲン原子、1価の炭化水素基又は1価のハロゲン化炭化水素基である。) - 前記エポキシ化合物の含有量が、前記硫化物固体電解質100質量部に対して0.03質量部以上25質量部以下である請求項1~4のいずれか1項に記載の改質硫化物固体電解質。
- 前記エポキシ化合物の分子量が、60以上である、請求項1~5のいずれか1項に記載の改質硫化物固体電解質。
- 前記エポキシ化合物1が、前記一般式(1)において、X11が炭素数1~24の1価の炭化水素基又は炭素数1~24の1価のハロゲン化炭化水素基であり、X12及びX13が水素原子である化合物である、請求項4~6のいずれか1項に記載の改質硫化物固体電解質。
- 前記エポキシ化合物2が、前記一般式(2)において、X21が一般式(2a)で示される基であり、X22及びX23が水素原子であり、一般式(2a)において、R21が炭素数1~8の2価の炭化水素基であり、R22が炭素数1~24の1価の炭化水素基である化合物である、請求項4~7のいずれか1項に記載の改質硫化物固体電解質。
- 前記エポキシ化合物3が、前記一般式(3)において、X31が一般式(3a)で示される基であり、X32及びX33が水素原子であり、一般式(3a)において、R31が炭素数1~8の2価の炭化水素基であり、R32が炭素数1~8の2価の炭化水素基であり、X34及びX36が一般式(3b)で示される基であり、X35が炭素数1~24の1価の炭化水素基であり、一般式(3b)において、R34~R36が炭素数1~24の1価の炭化水素基である化合物である、請求項4~8のいずれか1項に記載の改質硫化物固体電解質。
- 前記エポキシ化合物3が、前記一般式(3)において、X31が一般式(3a)で示される基であり、X22及びX23が水素原子であり、一般式(3a)において、R31が炭素数1~8の2価の炭化水素基であり、R32が単結合であり、X34~X36が炭素数1~24の1価の炭化水素基である化合物である、請求項4~8のいずれか1項に記載の改質硫化物固体電解質。
- BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、有機溶媒と、を混合すること、
前記有機溶媒を除去すること、を含む改質硫化物固体電解質の製造方法。 - 前記有機溶媒が、脂肪族炭化水素溶媒、脂環族炭化水素溶媒、芳香族炭化水素溶媒、エーテル溶媒、エステル溶媒及びニトリル溶媒から選ばれる少なくとも一種の溶媒である、請求項11に記載の改質硫化物固体電解質の製造方法。
- 請求項1~10のいずれか1項に記載の改質硫化物固体電解質と、電極活物質と、を含む電極合材。
- 請求項1~10のいずれか1項に記載の改質硫化物固体電解質及び請求項13に記載の電極合材の少なくとも一方を含む、リチウムイオン電池。
- BET比表面積が10m2/g以上であり、リチウム原子、硫黄原子、リン原子及びハロゲン原子を含む硫化物固体電解質と、エポキシ化合物と、を含む、
改質硫化物固体電解質。 - 請求項15に記載の改質硫化物固体電解質と、電極活物質と、を含む電極合材。
- 請求項15に記載の改質硫化物固体電解質及び請求項16に記載の電極合材の少なくとも一方を含む、リチウムイオン電池。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22742580.8A EP4283633A4 (en) | 2021-01-19 | 2022-01-18 | MODIFIED SULFIDE SOLID ELECTROLYTE AND PROCESS FOR PRODUCING THE SAME |
| US18/272,166 US20240105985A1 (en) | 2021-01-19 | 2022-01-18 | Modified sulfide solid electrolyte and method for producing same |
| CN202280009816.3A CN116745868A (zh) | 2021-01-19 | 2022-01-18 | 改性硫化物固体电解质及其制造方法 |
| JP2022576696A JP7818535B2 (ja) | 2021-01-19 | 2022-01-18 | 改質硫化物固体電解質及びその製造方法 |
| KR1020237023540A KR20230133288A (ko) | 2021-01-19 | 2022-01-18 | 개질 황화물 고체 전해 질 및 그 제조 방법 |
| JP2025176101A JP2025188267A (ja) | 2021-01-19 | 2025-10-20 | 改質硫化物固体電解質及びその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021006663 | 2021-01-19 | ||
| JP2021-006663 | 2021-01-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022158458A1 true WO2022158458A1 (ja) | 2022-07-28 |
Family
ID=82549404
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/001664 Ceased WO2022158458A1 (ja) | 2021-01-19 | 2022-01-18 | 改質硫化物固体電解質及びその製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20240105985A1 (ja) |
| EP (1) | EP4283633A4 (ja) |
| JP (2) | JP7818535B2 (ja) |
| KR (1) | KR20230133288A (ja) |
| CN (1) | CN116745868A (ja) |
| TW (1) | TW202236729A (ja) |
| WO (1) | WO2022158458A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20240104011A (ko) | 2022-12-27 | 2024-07-04 | 도요타 지도샤(주) | 정극층, 정극 및 고체 전지 |
| WO2024147411A1 (en) * | 2023-01-06 | 2024-07-11 | Samsung Electro-Mechanics Co., Ltd. | All-solid-state battery |
| KR20250084855A (ko) | 2023-12-04 | 2025-06-11 | 도요타지도샤가부시키가이샤 | 고체 전지 및 고체 전지의 제조 방법 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022186155A1 (ja) * | 2021-03-05 | 2022-09-09 | 三井金属鉱業株式会社 | 固体電解質並びにそれを含む電極合剤及び電池 |
| EP4303183A4 (en) * | 2021-03-05 | 2024-10-02 | Mitsui Mining & Smelting Co., Ltd. | METHOD FOR PRODUCING A SULFIDE SOLID ELECTROLYTE |
| CN119096307A (zh) * | 2022-03-30 | 2024-12-06 | 出光兴产株式会社 | 改性硫化物固体电解质及其制造方法和电极复合材料及锂离子电池 |
| CN116190663B (zh) * | 2023-04-18 | 2023-07-21 | 蔚来电池科技(安徽)有限公司 | 二次电池和装置 |
| KR20250094398A (ko) * | 2023-12-18 | 2025-06-25 | 포스코홀딩스 주식회사 | 고체 전해질 및 이를 포함하는 전고체 전지 |
| KR20250150752A (ko) * | 2024-04-12 | 2025-10-21 | 현대자동차주식회사 | 기계적 물성이 우수한 리튬이차전지용 황화물계 고체전해질 및 이의 제조방법 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012226854A (ja) * | 2011-04-15 | 2012-11-15 | Daiso Co Ltd | 非水電解質二次電池 |
| JP2013016423A (ja) | 2011-07-06 | 2013-01-24 | Toyota Motor Corp | 硫化物固体電解質材料、リチウム固体電池、および、硫化物固体電解質材料の製造方法 |
| JP2018521173A (ja) * | 2015-06-24 | 2018-08-02 | クアンタムスケイプ コーポレイション | 複合電解質 |
| JP2019199394A (ja) * | 2018-05-18 | 2019-11-21 | トヨタ自動車株式会社 | 硫化物系固体電解質、当該硫化物系固体電解質の製造方法、及び、全固体電池の製造方法 |
| WO2020105736A1 (ja) * | 2018-11-22 | 2020-05-28 | 出光興産株式会社 | 硫化物固体電解質及びその処理方法 |
| JP2020173992A (ja) * | 2019-04-11 | 2020-10-22 | トヨタ自動車株式会社 | 硫化物固体電解質、硫化物固体電解質の製造方法、電極体および全固体電池 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6709065B2 (ja) | 2016-02-19 | 2020-06-10 | 富士フイルム株式会社 | 固体電解質組成物、全固体二次電池用シート及び全固体二次電池、並びに、全固体二次電池用シート及び全固体二次電池の製造方法 |
| JP7176937B2 (ja) | 2018-11-21 | 2022-11-22 | トヨタ自動車株式会社 | 複合固体電解質の製造方法 |
| WO2020203231A1 (ja) | 2019-03-29 | 2020-10-08 | 三井金属鉱業株式会社 | 硫化物固体電解質 |
-
2022
- 2022-01-18 EP EP22742580.8A patent/EP4283633A4/en active Pending
- 2022-01-18 US US18/272,166 patent/US20240105985A1/en active Pending
- 2022-01-18 CN CN202280009816.3A patent/CN116745868A/zh active Pending
- 2022-01-18 KR KR1020237023540A patent/KR20230133288A/ko active Pending
- 2022-01-18 JP JP2022576696A patent/JP7818535B2/ja active Active
- 2022-01-18 WO PCT/JP2022/001664 patent/WO2022158458A1/ja not_active Ceased
- 2022-01-19 TW TW111102233A patent/TW202236729A/zh unknown
-
2025
- 2025-10-20 JP JP2025176101A patent/JP2025188267A/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012226854A (ja) * | 2011-04-15 | 2012-11-15 | Daiso Co Ltd | 非水電解質二次電池 |
| JP2013016423A (ja) | 2011-07-06 | 2013-01-24 | Toyota Motor Corp | 硫化物固体電解質材料、リチウム固体電池、および、硫化物固体電解質材料の製造方法 |
| JP2018521173A (ja) * | 2015-06-24 | 2018-08-02 | クアンタムスケイプ コーポレイション | 複合電解質 |
| JP2019199394A (ja) * | 2018-05-18 | 2019-11-21 | トヨタ自動車株式会社 | 硫化物系固体電解質、当該硫化物系固体電解質の製造方法、及び、全固体電池の製造方法 |
| WO2020105736A1 (ja) * | 2018-11-22 | 2020-05-28 | 出光興産株式会社 | 硫化物固体電解質及びその処理方法 |
| JP2020173992A (ja) * | 2019-04-11 | 2020-10-22 | トヨタ自動車株式会社 | 硫化物固体電解質、硫化物固体電解質の製造方法、電極体および全固体電池 |
Non-Patent Citations (3)
| Title |
|---|
| KANNO ET AL., JOURNAL OF THE ELECTROCHEMICAL SOCIETY, vol. 148, no. 7, 2001, pages A742 - 746 |
| See also references of EP4283633A4 |
| SOLID STATE IONICS, vol. 177, 2006, pages 2721 - 2725 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20240104011A (ko) | 2022-12-27 | 2024-07-04 | 도요타 지도샤(주) | 정극층, 정극 및 고체 전지 |
| JP2024093769A (ja) * | 2022-12-27 | 2024-07-09 | トヨタ自動車株式会社 | 正極層、正極及び固体電池 |
| JP7764357B2 (ja) | 2022-12-27 | 2025-11-05 | トヨタ自動車株式会社 | 正極層、正極及び固体電池 |
| WO2024147411A1 (en) * | 2023-01-06 | 2024-07-11 | Samsung Electro-Mechanics Co., Ltd. | All-solid-state battery |
| KR20250084855A (ko) | 2023-12-04 | 2025-06-11 | 도요타지도샤가부시키가이샤 | 고체 전지 및 고체 전지의 제조 방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116745868A (zh) | 2023-09-12 |
| KR20230133288A (ko) | 2023-09-19 |
| TW202236729A (zh) | 2022-09-16 |
| US20240105985A1 (en) | 2024-03-28 |
| EP4283633A1 (en) | 2023-11-29 |
| EP4283633A4 (en) | 2025-07-09 |
| JP2025188267A (ja) | 2025-12-25 |
| JP7818535B2 (ja) | 2026-02-20 |
| JPWO2022158458A1 (ja) | 2022-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7818535B2 (ja) | 改質硫化物固体電解質及びその製造方法 | |
| JP7324849B2 (ja) | 電極合材及びその製造方法 | |
| KR20230079084A (ko) | 황화물 고체 전해질 유리 세라믹스 및 그 제조 방법 | |
| JP2023152783A (ja) | 硫化物固体電解質の製造方法 | |
| JP7744375B2 (ja) | 改質硫化物固体電解質及びその製造方法 | |
| JP2023152966A (ja) | 硫化物固体電解質、その製造方法、電極合材及びリチウムイオン電池 | |
| WO2023276860A1 (ja) | 改質硫化物固体電解質粉体の製造方法 | |
| US20240063425A1 (en) | Method for producing a crystalline solid electrolyte, a crystalline solid electrolyte, and an electrode combined material and a lithium ion battery using it | |
| US20250253333A1 (en) | Modified sulfide solid electrolyte and production method for same, and electrode combined material and lithium-ion battery | |
| EP4339970B1 (en) | Sulfide solid electrolyte composition, electrode mixture containing same, and method for producing sulfide solid electrolyte composition | |
| WO2024024825A1 (ja) | 改質硫化物固体電解質及びその製造方法並びに電極合材及びリチウムイオン電池 | |
| JP7751485B2 (ja) | 硫化物固体電解質の製造方法及び電極合材の製造方法 | |
| KR20230015357A (ko) | 전극 합재 및 그 제조 방법 | |
| US20230335788A1 (en) | Method for producing atomization sulfide solid electrolyte, atomization sulfide solid electrolyte, electrode mixture and lithium ion battery | |
| JP7742365B2 (ja) | 固体電解質の製造方法 | |
| KR20260020073A (ko) | 결정성 황화물 고체 전해질 | |
| KR20260020931A (ko) | 결정성 황화물 고체 전해질 | |
| WO2024024824A1 (ja) | 改質硫化物固体電解質及びその製造方法並びに電極合材及びリチウムイオン電池 | |
| WO2025037517A1 (ja) | 結晶性硫化物固体電解質 | |
| WO2025173702A1 (ja) | 硫化物固体電解質及び硫化物固体電解質の製造方法 | |
| WO2026054035A1 (ja) | 酸硫化物固体電解質の製造方法、酸硫化物固体電解質前駆体及び酸硫化物固体電解質 | |
| JP2023168318A (ja) | 硫化物固体電解質の製造方法及び硫化物固体電解質 | |
| KR20240171087A (ko) | 황화물 고체 전해질 유리 세라믹스 및 그 제조 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22742580 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022576696 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280009816.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18272166 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022742580 Country of ref document: EP Effective date: 20230821 |