WO2022209558A1 - 銅粒子及びその製造方法 - Google Patents
銅粒子及びその製造方法 Download PDFInfo
- Publication number
- WO2022209558A1 WO2022209558A1 PCT/JP2022/009029 JP2022009029W WO2022209558A1 WO 2022209558 A1 WO2022209558 A1 WO 2022209558A1 JP 2022009029 W JP2022009029 W JP 2022009029W WO 2022209558 A1 WO2022209558 A1 WO 2022209558A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- copper
- particles
- ions
- carboxylate
- copper particles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/10—Metallic powder containing lubricating or binding agents; Metallic powder containing organic material
- B22F1/102—Metallic powder coated with organic material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/05—Metallic powder characterised by the size or surface area of the particles
- B22F1/054—Nanosized particles
- B22F1/0545—Dispersions or suspensions of nanosized particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/05—Metallic powder characterised by the size or surface area of the particles
- B22F1/054—Nanosized particles
- B22F1/056—Submicron particles having a size above 100 nm up to 300 nm
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F9/00—Making metallic powder or suspensions thereof
- B22F9/16—Making metallic powder or suspensions thereof using chemical processes
- B22F9/18—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds
- B22F9/24—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds starting from liquid metal compounds, e.g. solutions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B23—MACHINE TOOLS; METAL-WORKING NOT OTHERWISE PROVIDED FOR
- B23K—SOLDERING OR UNSOLDERING; WELDING; CLADDING OR PLATING BY SOLDERING OR WELDING; CUTTING BY APPLYING HEAT LOCALLY, e.g. FLAME CUTTING; WORKING BY LASER BEAM
- B23K35/00—Rods, electrodes, materials, or media, for use in soldering, welding, or cutting
- B23K35/02—Rods, electrodes, materials, or media, for use in soldering, welding, or cutting characterised by mechanical features, e.g. shape
- B23K35/0222—Rods, electrodes, materials, or media, for use in soldering, welding, or cutting characterised by mechanical features, e.g. shape for use in soldering or brazing
- B23K35/0244—Powders, particles or spheres; Preforms made therefrom
- B23K35/025—Pastes, creams or slurries
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B23—MACHINE TOOLS; METAL-WORKING NOT OTHERWISE PROVIDED FOR
- B23K—SOLDERING OR UNSOLDERING; WELDING; CLADDING OR PLATING BY SOLDERING OR WELDING; CUTTING BY APPLYING HEAT LOCALLY, e.g. FLAME CUTTING; WORKING BY LASER BEAM
- B23K35/00—Rods, electrodes, materials, or media, for use in soldering, welding, or cutting
- B23K35/22—Rods, electrodes, materials, or media, for use in soldering, welding, or cutting characterised by the composition or nature of the material
- B23K35/24—Selection of soldering or welding materials proper
- B23K35/30—Selection of soldering or welding materials proper with the principal constituent melting at less than 1550°C
- B23K35/302—Cu as the principal constituent
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22C—ALLOYS
- C22C1/00—Making non-ferrous alloys
- C22C1/04—Making non-ferrous alloys by powder metallurgy
- C22C1/0425—Copper-based alloys
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2201/00—Treatment under specific atmosphere
- B22F2201/10—Inert gases
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2301/00—Metallic composition of the powder or its coating
- B22F2301/10—Copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2304/00—Physical aspects of the powder
- B22F2304/05—Submicron size particles
- B22F2304/054—Particle size between 1 and 100 nm
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2998/00—Supplementary information concerning processes or compositions relating to powder metallurgy
- B22F2998/10—Processes characterised by the sequence of their steps
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2999/00—Aspects linked to processes or compositions used in powder metallurgy
Definitions
- the present invention relates to copper particles used as a raw material for conductive or bonding pastes and a method for producing the same.
- a method of printing a conductive paste or conductive ink containing copper particles of a bonding material as a conductive filler on a substrate is widely known.
- the copper particles are prepared in a slurry state, mixed with an organic substance, and used as a bonding material such as a conductive paste or a conductive ink.
- a method has been developed to easily form wiring on a substrate by directly applying such conductive ink or conductive paste to the substrate using an inkjet printer, screen printer, offset printer, or the like. has been put into practical use.
- a typical method for producing nano-sized or submicron-sized particles is the liquid phase reduction method.
- the raw material is dissolved in a solvent to prepare a solution containing metal ions, and the metal ions are reduced in the presence of a dispersant to suppress aggregation of the particles, thereby precipitating the metal particles.
- a method for producing copper particles is disclosed in which a reducing agent is applied in the absence of the agent. In this method, by adding a hydrazine compound or sodium borohydride or the like in a pH range of 8 to 14 via a copper-nitrilotriacetic acid complex, the average particle size of the primary particles is 0.01 ⁇ m or more and 0.3 ⁇ m or less. making particles.
- Patent Document 2 after adjusting the pH of the copper salt aqueous solution to 12 or more using sodium hydroxide before adding the hydrazine-based reducing agent, reducing sugar is added, and then the hydrazine-based reducing agent is added. describes a method for producing copper particles with a particle size of 0.5 ⁇ m to 3.1 ⁇ m.
- Patent Document 3 after adding an aqueous sodium hydroxide solution to an aqueous copper sulfate solution to generate cupric oxide, a reducing agent such as glucose is added, and then a hydrazine-based reducing agent is added. , describes a method for producing copper particles having an average particle size of 0.1 to 10 ⁇ m.
- metal fine particles such as gold, silver, copper, platinum, or palladium, the surface of which is coated with a protective agent, wherein the protective agent is selected from at least one of an amine compound and a carboxylic acid compound, and the metal is Metal fine particles are disclosed in which the total content of alkali metals, halogens, sulfur, and phosphorus contained in the fine particles is less than 0.1 mass% with respect to the mass of the metal fine particles (Patent Document 4 ( See claims 1, 4 and 8, paragraph [0055], paragraph [0098], paragraph [0099]).
- 0.1 mass% is 1000 ppm by mass.
- metal nuclei are deposited by reduction from a metal compound dispersed in a solid state in a liquid phase containing a reducing agent and a protective agent, and the metal nuclei are aggregated and coated with a protective agent.
- impurities such as alkali metals, halogens, sulfur and phosphorus contained in the metal fine particles are removed to purify the metal fine particles.
- the metal compound used as the raw material preferably does not contain impurities such as alkali metals, halogens, sulfur and phosphorus, such as metal oxides and noble metal oxides.
- Example 5 shown in Patent Document 4 bis(acetylacetonato)copper is used as a raw material, bis(2-ethylhexyl)amine is used as a protective agent and a reducing agent, and dodecylamine is used as a protective agent.
- a mixed solution of these was heated to 220° C. to reduce Cu(C 5 H 7 O 2 ) 2 to obtain a dispersion of Cu metal fine particles coated with dodecylamine and bis(2-ethylhexyl)amine. there is Cu metal microparticles are obtained by purifying this dispersion.
- Patent Document 4 when the impurity elements contained in the Cu metal fine particles obtained in Example 5 and their contents were measured by ion chromatography or ICP fluorescence spectroscopy, the Cl component was 0.005 mass% (50 mass ppm), and halogens other than Cl, alkali metals, sulfur, and phosphorus were not detected.
- bonding material particles having organic protection formed on the surface of copper nanoparticles and a method for producing the same have been disclosed (see Patent Document 5 (claims 1 and 4, paragraph [0012] and paragraph [0015]).
- the bonding material particles have a BET specific surface area of 3.5 m 2 /g or more and 8 m 2 /g or less, a BET diameter converted from the specific surface area of 80 nm or more and 200 nm or less, and the organic protective
- the film is contained in the range of 0.5% by mass or more and 2.0% by mass or less with respect to 100% by mass of the bonding material particles.
- a pH adjuster is added to an aqueous dispersion of copper citrate at room temperature to adjust the pH to pH 3 or more and less than pH 7, and the pH-adjusted copper citrate is placed in an inert gas atmosphere.
- a hydrazine compound is added to and mixed with the aqueous dispersion of the above, and the mixture is heated to a temperature of 60 ° C. or higher and 80 ° C. or lower in an inert gas atmosphere and held for 1.5 hours or more and 2.5 hours or less to obtain citric acid. Copper is reduced to generate copper nanoparticles, and an organic protective film is formed on the surfaces of the copper nanoparticles.
- the nano-sized or submicron-sized copper particles obtained by the liquid phase reduction method shown in Patent Documents 1 to 3 have the oxidation-reduction potential of copper ions and the hydrazine compound or sodium borohydride, which are the driving force for oxidation-reduction. Due to the difference in oxidation-reduction potential of such a reducing agent, metal impurities with a concentration of 10 ppm by mass or more and on the order of several tens of ppm by mass were sometimes included.
- Patent Document 3 When the industrial copper sulfate shown in Patent Document 3 is used as a copper raw material, it tends to contain metal elements such as nickel, iron, lead, and silver as metal impurities. Since the redox potential of these metal elements is higher than the redox potential of hydrazine compounds and sodium borohydride, these metal elements precipitate when copper ions are reduced to copper, and the concentration is 10 ppm by mass or more. metal impurities in the copper particles.
- Patent Document 1 when reduction is performed in an alkaline region as in Patent Documents 1 and 2, metal impurities are likely to precipitate, and further, sodium borohydride is used as a reducing agent as in Patent Document 1, When sodium hydroxide is used as a pH adjuster, there is a problem that sodium is likely to precipitate, resulting in the production of copper particles containing metal impurities at a concentration of 10 ppm by mass or more.
- Patent Document 4 As an evaluation of the copper particles of Example 5 shown in Patent Document 4, it is stated that alkali metals are not detected as contained elements in the copper particles, but Patent Document 4 describes metal impurities other than alkali metals. There is a high probability that the copper particles contain metals other than alkali metals for the following reasons. That is, the aliphatic amine compound used as a reducing agent in the method shown in Patent Document 4, bis(2-ethylhexyl)amine in Example 5, exhibits coordinative adsorption to metal fine particles and electron-donating properties. By having an alkyl group of , the electron density of the lone pair on the nitrogen atom increases, resulting in a high reducing property. For this reason, there is a problem that metal elements other than alkali metals, such as metal elements such as iron and nickel, which can become metal impurities from the viewpoint of oxidation-reduction potential, precipitate as impurities in metal compounds.
- metal elements other than alkali metals such as metal elements such as
- the lower limit of the BET diameter is set to 80 nm. was sometimes difficult to form.
- the method for producing particles for a bonding material disclosed in Patent Document 5 that is, in the method for producing copper particles, when the pH of the mixed solution is in the range of pH 6 to 7, which is close to neutrality, the heating temperature of the mixed solution exceeds 75°C. In this case, there is a problem that the impurity concentration does not become less than 10 ppm by mass.
- Such copper particles containing metal impurities at a concentration of 10 mass ppm or more are excellent in electrical conductivity and heat transfer properties of copper wiring when used as a wiring material. There is concern about the deterioration of the characteristics of other members due to the diffusion of For example, when a conductive paste containing copper particles is applied to a ceramic base material such as a MLCC (multilayer ceramic capacitor), metal impurities diffuse into the ceramic base material, possibly impairing the insulation of the base material. .
- MLCC multilayer ceramic capacitor
- An object of the present invention is to provide copper particles that do not impair the properties of members and a method for producing the same.
- the present inventors used copper carboxylate as a source of copper, used a hydrazine compound as a reducing agent, and in an acidic region without using sodium hydroxide, at a predetermined temperature, a predetermined time, a predetermined oxidation
- copper carboxylate is reduced with a hydrazine compound having a reduction potential
- the surface of the particles is reduced to an organic compound derived from copper carboxylate without using a dispersant and a surface protective agent for suppressing aggregation between particles and/or oxidation of particles.
- a first aspect of the present invention is a copper particle in which the surface of a core particle made of metallic copper is coated with an organic protective film composed of an organic molecule derived from copper carboxylate, and the oxidation-reduction potential contained in the copper particle is
- the total concentration of impurities of metals less noble than copper is less than 10 mass ppm
- the average particle diameter of the copper particles in the state of primary particles is in the range of more than 50 nm and 200 nm or less
- the copper particles are subjected to time-of-flight secondary
- the amount of CuC 2 O 2 H - ions detected is in the range of 0.02 times or more the amount of Cu + ions detected when analyzed using the next ion mass spectrometry (TOF-SIMS), Copper particles in which the amount of C 2 H 3 O 2 - ions detected is 0.02 times or more the amount of Cu + ions detected.
- a second aspect of the present invention is a method for producing the copper particles of the first aspect without using a dispersant and a surface protective agent for suppressing aggregation between particles and/or oxidation of particles,
- a third aspect of the present invention is an invention based on the second aspect, wherein the copper carboxylate is sparingly soluble in water and is selected from the group consisting of copper carboxylates having 4 to 8 carbon atoms. It is a method for producing copper particles that are one or two or more copper salts.
- a fourth aspect of the present invention is an invention based on the second aspect, and is a method for producing copper particles, wherein the pH adjuster is ammonium carboxylate.
- the copper particles of the first aspect of the present invention since the core particle surface is coated with an organic protective film composed of organic molecules derived from copper carboxylate, the copper particles have excellent oxidation resistance during storage. Especially in the copper particles shown in Patent Document 4, since the particle surface is coated with a protective agent containing additives such as amine compounds and carboxylic acid compounds, there is a high risk of contamination with metal impurities.
- the copper particles of aspect 1 are characterized by a low risk of contamination with metal impurities because the particle surfaces are coated with an organic protective film derived from copper carboxylate.
- the total concentration of impurities of metals whose oxidation-reduction potential is less noble than that of copper is less than 10 ppm by mass, when copper particles are used as a wiring material or a bonding material, diffusion of the metal impurities will not impair the characteristics of other members. There is no
- the average particle size of the primary particles of the copper particles is in the range of more than 50 nm and 200 nm or less, when used as a wiring material or a bonding material, the copper wiring has excellent conductivity and heat transfer properties, and forms an electronic circuit.
- the reaction area of the copper particles is large and the reactivity by heating is high, so that the copper particles can be sintered at a relatively low temperature.
- the copper particles of the first aspect of the present invention have a detected amount of CuC 2 O 2 H ⁇ ions when the copper particles are analyzed using time-of-flight secondary ion mass spectrometry (TOF-SIMS).
- TOF-SIMS time-of-flight secondary ion mass spectrometry
- the range of 0.02 times or more the amount of Cu + ions detected, and the range of the amount of C 2 H 3 O 2 ⁇ ions detected is 0.02 times or more the amount of Cu + ions detected. Therefore, the amount of the organic protective film is sufficient to protect the copper particles. This organic protective film prevents the surfaces of the copper particles from being oxidized and prevents the copper particles from aggregating with each other.
- the supply source of copper ions is not a water-soluble complex in the liquid, but a metal compound called carboxylic acid copper as a copper ion supply source. Since the copper ions slightly eluted from acid copper and the hydrazine compound, which is a reducing agent, are sequentially reacted, it is possible to reduce the copper ion concentration in the liquid even in the acidic region, and the average particle size of the primary particles is reduced to 50 nm. Copper particles in the range of more than 200 nm or less can be obtained.
- the organic molecule which is a non-metallic portion that constitutes the copper carboxylate, coats the surface of the core particle of the copper particle as a surface protective film, as shown in Patent Document 4, it is possible to prevent the aggregation of the copper particle. There is no need to use a dispersant or a surface protective agent, and there is no contamination of impurities caused by the surface protective agent, so that the impurity concentration of the copper particles can be further reduced.
- the organic molecule is present on the surface of the copper particles, the dissolution of the core particles is suppressed, and the copper ions are less likely to become copper (II) hydroxide and are less likely to precipitate as copper (II) hydroxide. can be produced in high yield. Furthermore, the obtained copper particles are excellent in oxidation resistance during storage.
- the pH of the aqueous dispersion of copper carboxylate is adjusted to an acidic range of 3 or more and less than 6, and a hydrazine compound having an oxidation-reduction potential of -0.7 V to -0.5 V is used at a temperature of 60 ° C. to 75 ° C.
- the oxidation-reduction potential of the copper ion and the oxidation-reduction potential of the hydrazine compound do not contain a pH value close to neutralization of pH 6-7, as described in Patent Document 5.
- the deposition of metals less noble than copper is suppressed, and the total concentration of impurities of metals less noble than copper becomes less than 10 ppm by mass.
- one or more copper salts selected from the group consisting of copper carboxylates having 4 to 8 carbon atoms and which are sparingly soluble in water is. Since the copper carboxylate is sparingly soluble in water, when the copper carboxylate is mixed with water, the copper carboxylate does not dissolve, resulting in an aqueous dispersion of the copper carboxylate.
- the particle size can be changed, and copper particles having an average primary particle size of more than 50 nm and 200 nm or less can be obtained. Also, by using two or more kinds of copper salts, it is possible to adjust the particle size distribution.
- an acidic region is created by using ammonium carboxylate without using, for example, sodium hydroxide containing sodium, which causes residual metal impurities, as a pH adjuster. Therefore, metal impurities in the obtained copper particles can be further reduced.
- FIG. 10 is a photograph of the copper particles of Example 5 taken with a scanning electron microscope.
- FIG. 10 is a photograph of the copper particles of Example 10 taken with a scanning electron microscope.
- FIG. 10 is a photograph of the copper particles of Example 16 taken with a scanning electron microscope.
- a copper particle 10 of this embodiment the surface of a core particle 11 made of metallic copper is covered with an organic protective film 12 composed of organic molecules derived from copper carboxylate.
- the copper particles 10 have a total concentration of impurities of metals having oxidation-reduction potentials less noble than that of copper contained in the particles of less than 10 ppm by mass.
- the total concentration of these impurities is preferably less than 1 mass ppm. If the total concentration of the impurities is 10 ppm by mass or more, when copper particles are used as a wiring material, diffusion of the metal impurities may impair the characteristics of other members. For example, there is a risk that impurities may contaminate a substrate or the like on which wiring is provided, impairing the insulating properties of the substrate or the like.
- copper carboxylate one or more copper salts selected from the group consisting of copper carboxylates having 4 to 8 carbon atoms and which are sparingly soluble in water are preferably used.
- copper benzoate (14 carbon atoms) or the like can also be used.
- Carboxylic acid copper having less than 4 carbon atoms is easily dissolved in water. When copper carboxylate is dissolved in water, copper particles are formed when an aqueous solution of a hydrazine compound is added. do not have.
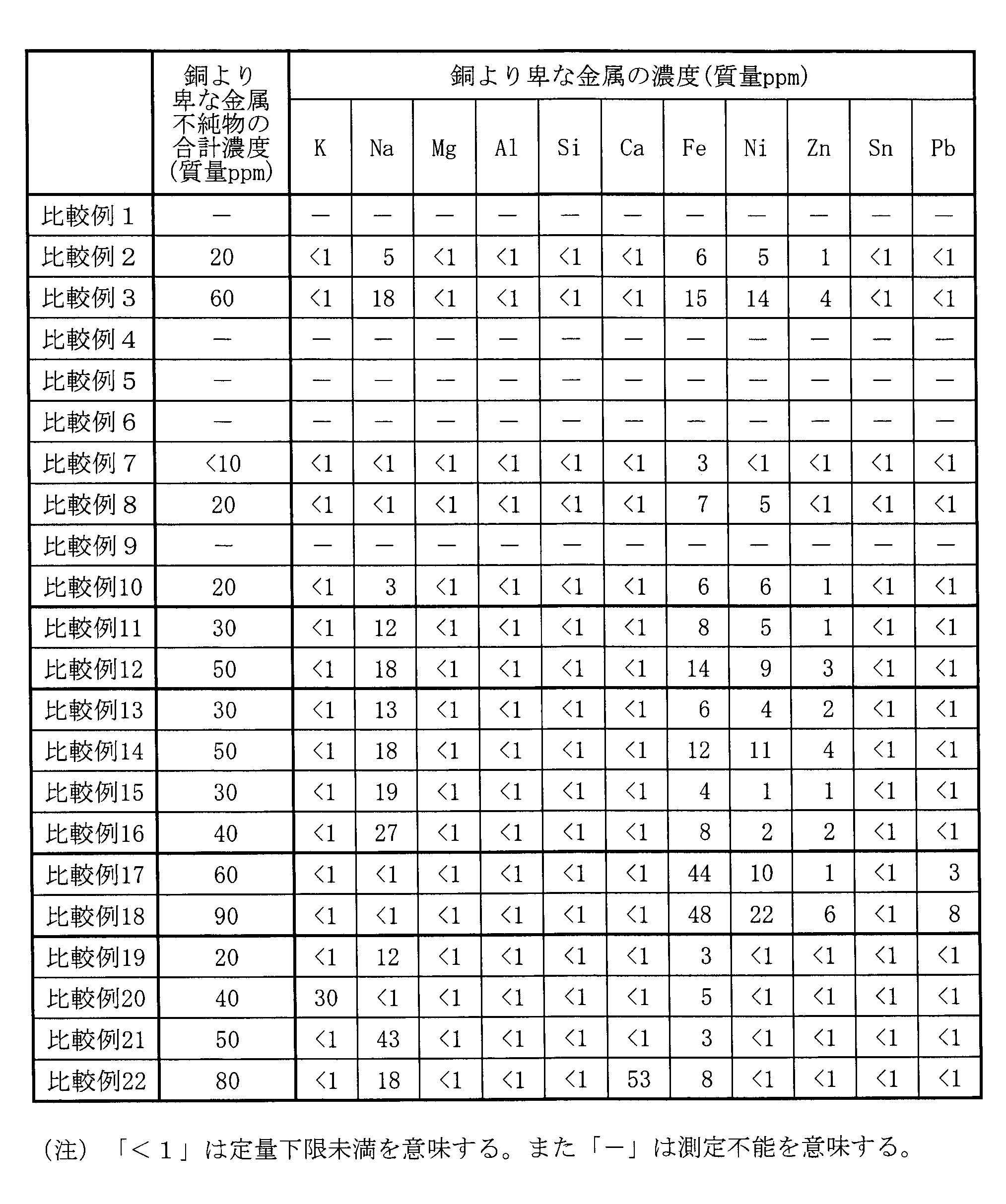
- Examples of metals contained in the copper particles 10 whose oxidation-reduction potential is less noble than that of copper include not only alkali metals such as potassium and sodium shown in Patent Document 4, but also alkaline earth metals other than alkali metals, transition metals, and the like. .
- the concentrations of these metals are measured by ICP-MS (Inductively Coupled Plasma Mass Spectrometry).
- ICP-MS Inductively Coupled Plasma Mass Spectrometry
- the copper particles 10 have an average particle size in the range of more than 50 nm and 200 nm in the state of primary particles.
- the lower limit of the average particle diameter of the primary particles is set to 80 nm in terms of BET diameter. Therefore, when forming a wiring pattern with a paste made using this particle, it is possible to form a fine wiring pattern with a narrow pitch.
- the copper particles of the present embodiment have a value exceeding the lower limit of 50 nm. It becomes possible to form easily. If the average particle size is 50 nm or less, there is a problem that the viscosity increases with a predetermined composition when producing a paste using copper particles.
- the average particle size of the primary particles is preferably in the range of more than 50 nm and less than 80 nm from the viewpoint of easily forming a fine wiring pattern with a narrow pitch.
- the average particle size of the primary particles is obtained by the following method. First, using a scanning electron microscope (SEM), the magnification is determined according to the size of the copper particles, and an SEM image of the copper particles is photographed. It is preferable to photograph in the range of 10000 times to 50000 times. Next, the SEM image is analyzed using image analysis software, the Heywood diameter is obtained for 300 or more particles per sample, and the arithmetic mean value of the Heywood diameters is taken as the average particle diameter of the primary particles.
- SEM scanning electron microscope
- the organic protective film 12 is composed of organic molecules derived from copper carboxylate.
- the organic protective film 12 covers the surfaces of the core particles 11 made of metallic copper and serves to prevent the core particles 11 from being oxidized during storage from manufacture to paste.
- the coating amount of the organic protective film is preferably analyzed by analyzing the copper particles using time-of-flight secondary ion mass spectrometry (TOF-SIMS). In this analysis method, it is detected as a peak value when copper particles are analyzed.
- TOF-SIMS time-of-flight secondary ion mass spectrometry
- the presence or absence of the organic protective film is measured by CuC 2 O 2 H - ions and C 2 H 3 O 2 - ions.
- the detected amount of CuC 2 O 2 H ⁇ ions is in the range of 0.02 times or more the detected amount of Cu + ions.
- the core particles 11 are coated so that the detected amount of C 2 H 3 O 2 ⁇ ions is in the range of 0.02 times or more the detected amount of Cu + ions.
- the detected amount of CuC 2 O 2 H - ions relative to the detected amount of Cu + ions and the detected amount of C 2 H 3 O 2 - ions relative to the detected amount of Cu + ions were determined by the coating derived from copper carboxylate present on the surface of the core particles. It represents the quantity of things.
- the amount of the organic protective film is sufficient to protect the core particles 11 . If it is less than the lower limit of the above range, the coating amount of the organic protective film becomes small, and the surfaces of the core particles become active to oxidize the copper particles.
- a pH adjuster is added to an aqueous dispersion of copper carboxylate to adjust the pH of the aqueous dispersion to an acidic range of 3 or more and less than 6, and the pH-adjusted copper carboxylate
- the mixed liquid is stirred at 60° C. under an inert gas atmosphere. C. to 75.degree. C. and held for 1.5 to 2.5 hours to reduce the copper carboxylate and obtain a copper particle dispersion in which copper particles are dispersed.
- this carboxylic acid copper is obtained by placing an aqueous carboxylate solution and a copper electrolyte solution in a reaction tank in an air atmosphere, stirring them at a temperature of 60° C. to 80° C. and reacting them. Powdery high-purity copper carboxylate obtained by obtaining a copper suspension, washing, solid-liquid separation, and drying the solid content may also be used.
- the aqueous carboxylate solution is prepared by dissolving sodium salts or ammonium salts of carboxylic acids such as citric acid, phthalic acid, benzoic acid and tartaric acid in pure water such as ion-exchanged water and distilled water.
- powdery copper carboxylate is put into pure water such as ion-exchanged water or distilled water at room temperature, stirred so as to be uniformly dispersed, and the concentration is adjusted to 25% by mass or more and 40% by mass or less.
- An aqueous dispersion of copper carboxylate is obtained.
- a pH adjuster is added to the aqueous dispersion of copper carboxylate to adjust the pH of the aqueous dispersion to an acidic range of 3 or more and less than 6.
- ammonium carboxylate containing no metal component is preferred.
- the reason why the pH is adjusted to pH 3 or more and less than 6 by the pH adjuster is that when the pH is less than 3, the elution of copper ions from the copper carboxylate is slow, the reaction is difficult to proceed quickly, and it is difficult to obtain the target particles.
- the pH is 6 or more
- copper carboxylate is reduced with a hydrazine compound
- eluted copper ions tend to become copper(II) hydroxide and tend to precipitate, making it impossible to produce copper particles at a high yield.
- the reducing power of hydrazine becomes stronger and the reaction proceeds more easily, so the average particle size of the primary particles increases.
- a preferable pH is 4 or more and 5 or less.
- an aqueous solution of a hydrazine compound having an oxidation-reduction potential in the range of ⁇ 0.7 V to ⁇ 0.5 V was added as a reducing agent to the pH-adjusted aqueous dispersion of copper carboxylate under an atmosphere. Add and mix to obtain a mixed solution.
- Hydrazine-based reducing agents such as hydrazine monohydrate are known to react differently in the acidic range and the alkaline range.
- the oxidation-reduction potential means the potential difference with respect to the standard hydrogen electrode (NHE).
- the oxidation-reduction potential E(V) is represented by the following formula (1) based on the pH value.
- the mixed solution is heated to a temperature of 60° C. or more and 75° C. or less in an inert gas atmosphere and held for 1.5 hours or more and 2.5 hours or less to convert the copper carboxylate.
- Core particles are formed by reduction, and an organic protective film derived from copper carboxylate is formed on the surface of the core particles to prepare a dispersion of copper particles having a desired particle size.
- the purpose of heating and holding in an inert gas atmosphere is to prevent oxidation of the core particles. If the heating temperature of the mixture is less than 60° C., the reducing power of copper carboxylate is too low to complete the reduction reaction.
- the temperature exceeds 75°C or the holding time exceeds 2.5 hours the amount of copper ions eluted from the copper carboxylate increases and the reaction rate increases, so metal impurities of 10 ppm by mass or more are included. Along with this, the coating amount of the organic protective film decreases. Furthermore, it becomes difficult to control the particle size of the primary particles, and it becomes difficult to obtain an average particle size of the primary particles in the range of 200 nm or less.
- the holding time is less than 1.5 hours, the desired particles cannot be obtained because the copper carboxylate is not completely reduced. If the time exceeds 2.5 hours, the disappearance of fine particles and the growth of coarse particles occur so as to alleviate the free energy caused by the difference in particle size, and thus grain growth occurs. Primary particles are not obtained.
- a preferred heating temperature is 65° C. or higher and 70° C. or lower, and a preferred holding time is 2 hours or longer and 2.5 hours or shorter.
- a hydrazine compound has advantages such as not producing a residue after a reduction reaction when reducing copper carboxylate under an acidic condition, relatively high safety, and easy handling.
- the hydrazine compound include hydrazine monohydrate, anhydrous hydrazine, hydrazine hydrochloride, and hydrazine sulfate. Among these, hydrazine monohydrate is preferred because it is desirable that it does not contain components such as sulfur and chlorine that can be impurities.
- a hydrazine compound as a reducing agent when added to and mixed with an acidic liquid having a pH of less than 6 to generate core particles in the liquid, a component derived from carboxylate ions generated from copper carboxylate forms the surface of the core particles. It coats quickly and suppresses the dissolution of the core particles.
- the acidic liquid having a pH of less than 6 is preferably kept at a temperature of 60° C. or higher and 70° C. or lower so that the reduction reaction proceeds easily.
- the target copper particles having the organic protective film formed on the surface of the core particles described above can be obtained. Since the surfaces of the core particles of the copper particles are coated with an organic protective film, the particles can be prevented from being oxidized even if they are stored in the atmosphere until they are used as a paste for wiring or bonding.
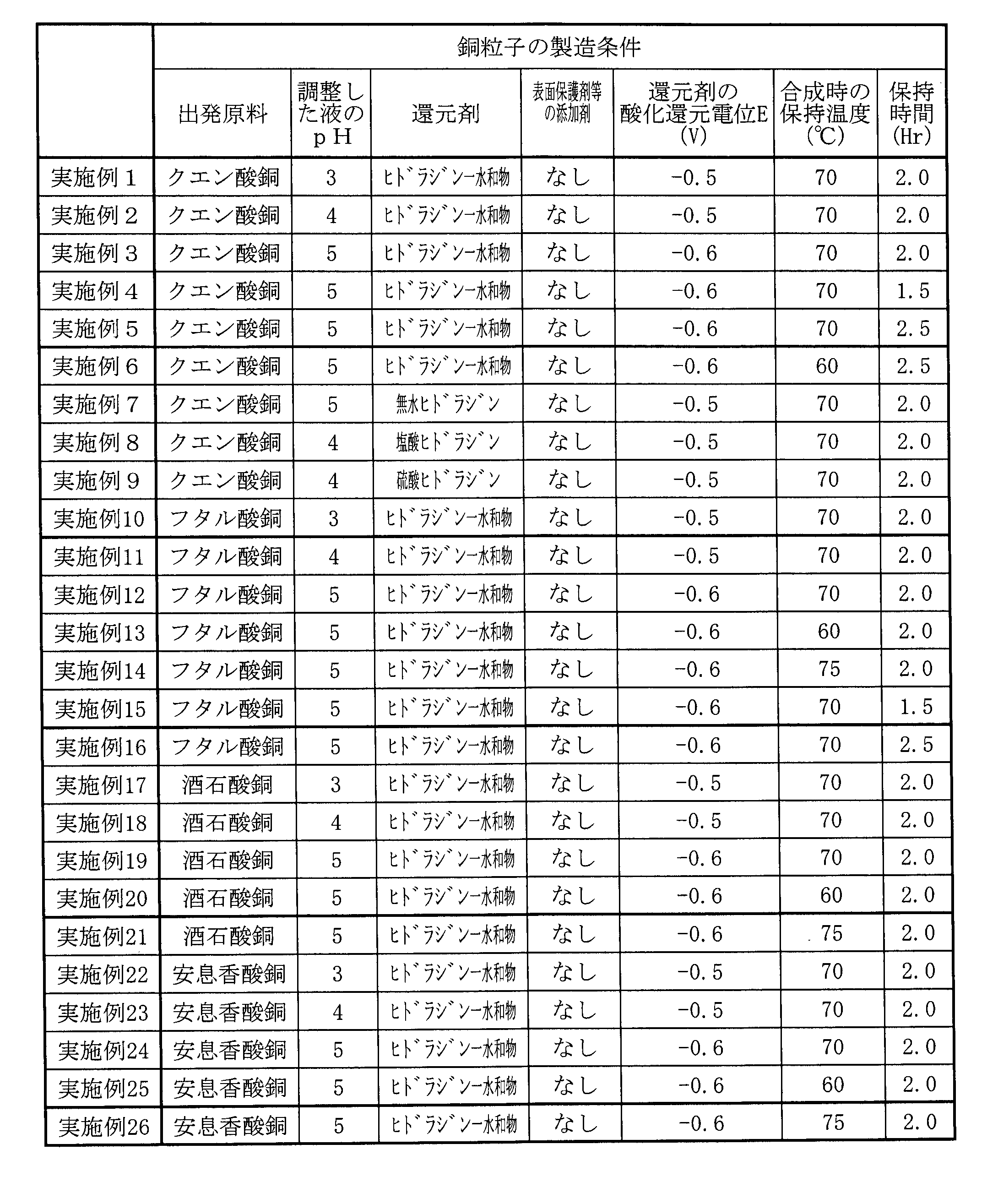
- the type and number of carbon atoms of copper carboxylates used in Examples and Comparative Examples the total concentration of impurities of metals having oxidation-reduction potentials less noble than that of copper contained in the copper carboxylates, and the impurity concentration of each metal were determined. , as shown in Table 1 below. In addition, at the bottom of Table 1, the method for producing each copper carboxylate is shown. The total concentrations of impurities of metals with oxidation-reduction potentials less noble than that of copper shown in Table 1 are approximate values.
- Example 1 First, copper citrate shown in Table 1 was prepared as a starting raw material, copper carboxylate. This copper citrate was added to deionized water at room temperature and stirred using a stirring blade to prepare an aqueous dispersion of copper citrate having a concentration of 30% by mass. Next, an aqueous solution of ammonium citrate was added as a pH adjuster to the aqueous dispersion of copper citrate to adjust the pH of the aqueous dispersion to 3.
- the pH-adjusted liquid is brought to a temperature of 50° C., and the oxidation-reduction potential, which is 1.2 equivalents capable of reducing copper ions, is -0.5 V as a reducing agent in the pH-adjusted liquid in a nitrogen gas atmosphere.
- hydrazine monohydrate aqueous solution (2-fold dilution) was added at once, and mixed uniformly using a stirring blade.
- the mixture of the aqueous dispersion and the reducing agent was heated to a holding temperature of 70° C. under a nitrogen gas atmosphere and held at 70° C. for 2 hours. Particles generated in the heated liquid were collected by solid-liquid separation using a centrifuge. The collected particles were dried by a vacuum drying method to produce copper particles of Example 1.
- Example 1 and Examples 2 to 26 described below are shown in Table 2 below, and the production conditions for the copper particles of Comparative Examples 1 to 22 are shown in Table 3 below.
- Examples 2 to 26, Comparative Examples 1 to 3, 6 to 16 Using copper carboxylate which is the same as or different from copper citrate which is the starting material of Example 1, the adjusted pH value is the same as or different from that in Example 1, and the type of reducing agent and oxidation-reduction potential are the same as or different from those in Example 1.
- the holding temperature and the holding time during the synthesis of the copper particles were the same as in Example 1 or changed.
- Copper particles of Examples 2 to 26 and Comparative Examples 1 to 3 and 6 to 16 were produced in the same manner as in Example 1 except for the above. Scanning electron microscope photographs of the copper particles obtained in Examples 5, 10 and 16 among these copper particles are shown in FIGS. 3, 4 and 5, respectively.
- Example 4 Hydrazine monohydrate as the reducing agent in Example 1 was changed to ammonium formate, the redox potential of this reducing agent was changed to 0.3 V, and the pH value was changed to 5. The holding temperature and holding time during the synthesis of Example 1 were not changed. Otherwise, copper particles of Comparative Example 4 were produced in the same manner as in Example 1.
- Example 19 The pH value of Example 1 was changed to 5, the oxidation-reduction potential of the reducing agent was changed to -0.6 V, the holding temperature during synthesis was unchanged, and the holding time was changed to 1.5 hours. Furthermore, 5% by mass of methyl cellulose was added to the particles as a surface protective agent for the core particles. Otherwise, copper particles of Comparative Example 19 were produced in the same manner as in Example 1.
- Comparative Example 20 Methylcellulose, which is a surface protective agent for the core particles used in Comparative Example 19, was changed to polyethylene glycol, and 5% by mass was added to the particles. Otherwise, copper particles of Comparative Example 20 were produced in the same manner as in Comparative Example 19.
- Methylcellulose which is the surface protective agent for the core particles used in Comparative Example 19, was changed to polyvinyl alcohol, and added in an amount of 5% by mass based on the particles. Otherwise, copper particles of Comparative Example 21 were produced in the same manner as in Comparative Example 19.
- the total concentration of metal impurities whose oxidation-reduction potential is less noble than that of copper is the total concentration of each metal impurity.
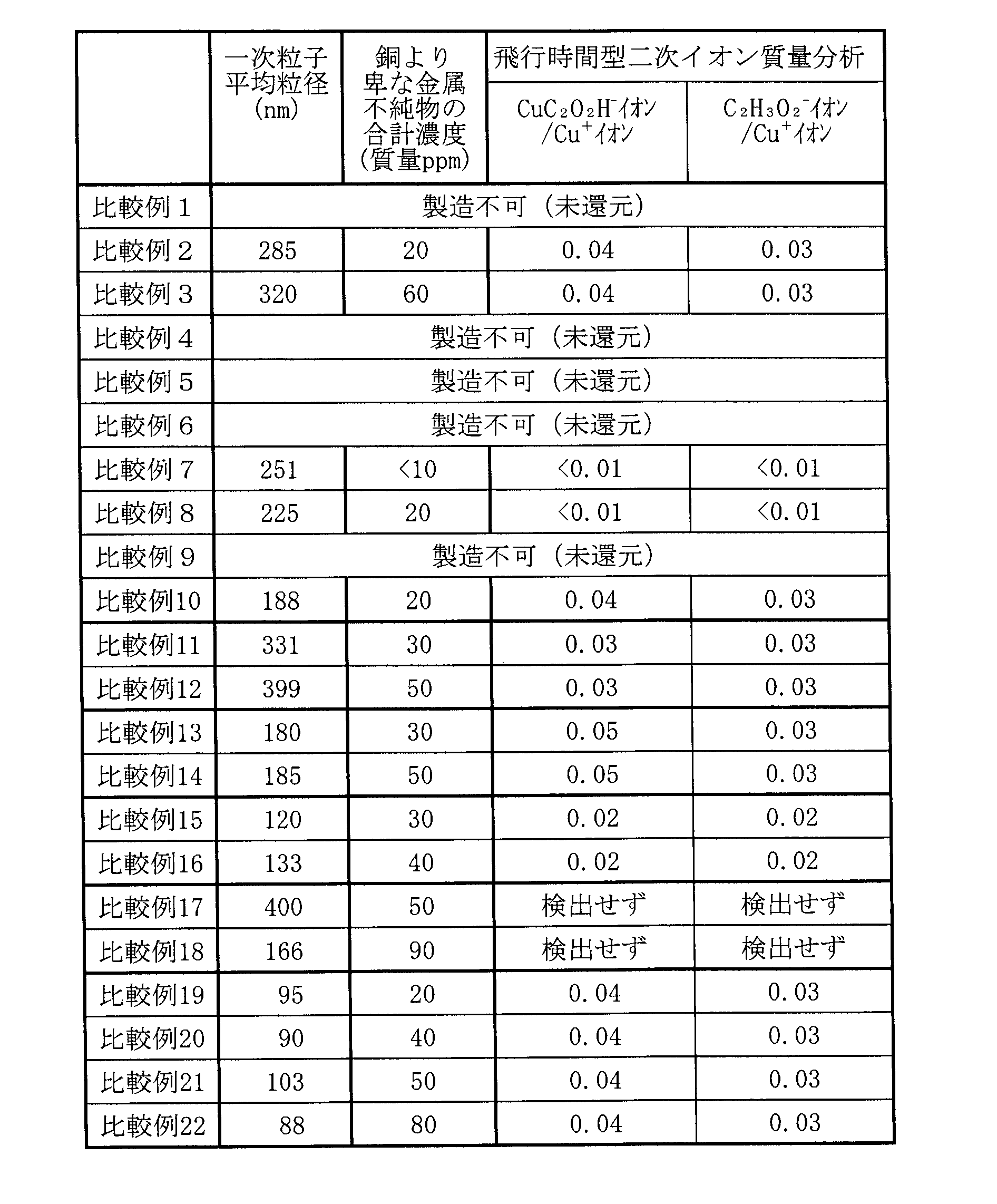
- Tables 5 and 7 below show the total concentration of impurities of metals whose oxidation-reduction potential is less noble than that of copper and the concentration of each metal impurity. For Comparative Examples 1, 4 to 6, and 9, which could not be synthesized due to insufficient reduction, the values in the above items are not listed.
- Comparative Example 2 the pH of the copper citrate aqueous dispersion was adjusted to 8, and a reducing agent of -0.8 V was added under alkaline conditions, so grain growth occurred in the liquid and the average particle size of the primary particles was 285 nm.
- the total metal impurity concentration of the obtained copper particles was as high as 20 ppm by mass.
- Comparative Example 3 the pH of the aqueous dispersion of copper citrate was adjusted to 10, and a reducing agent of -1.0 V was added under strong alkalinity. 320 nm, and the total metal impurity concentration of the obtained copper particles was as high as 60 ppm by mass.
- Comparative Example 6 -0.6 V hydrazine-hydrate was used as a reducing agent for copper citrate, and the mixture was heated at 70°C, but the holding time was too short at 1.0 hours. Reduction of copper citrate was not completed, and copper particles could not be produced.
- Comparative Example 11 the pH of the aqueous dispersion of copper phthalate was adjusted to 8 and a reducing agent of ⁇ 0.9 V was used, so the mixed solution was heated to 70° C. and held for 2.0 hours. Grain growth occurred in 1, and the average particle diameter of the primary particles became 331 nm, and the total metal impurity concentration of the obtained copper particles was as high as 30 ppm by mass.
- Comparative Example 12 the pH of the copper phthalate aqueous dispersion was adjusted to 10 and a reducing agent of ⁇ 1.0 V was used. Grain growth occurred in 1, and the average particle diameter of the primary particles became 399 nm, and the total metal impurity concentration of the obtained copper particles was as high as 50 ppm by mass.
- Comparative Example 13 the aqueous dispersion of copper tartrate was adjusted to pH 8 and a reducing agent of ⁇ 0.9 V was used, so the mixture was heated to 70° C. and held for 2.0 hours.
- the total metal impurity concentration of the copper particles was as high as 30 mass ppm.
- Comparative Example 14 the aqueous dispersion of copper tartrate was adjusted to pH 10 and a reducing agent of ⁇ 1.0 V was used, so the mixture was heated to 70° C. and held for 2.0 hours.
- the total metal impurity concentration of the copper particles was as high as 50 mass ppm.
- Comparative Example 15 the aqueous dispersion of copper benzoate was adjusted to pH 8 and a reducing agent of ⁇ 0.9 V was used.
- the total metal impurity concentration of the copper particles was as high as 30 mass ppm.
- Comparative Example 16 the aqueous dispersion of copper benzoate was adjusted to pH 10 and a reducing agent of ⁇ 1.0 V was used.
- the total metal impurity concentration of the copper particles was as high as 40 mass ppm.
- hydrazine-hydrate was used as the reducing agent. Since the oxidation-reduction potential of this reducing agent was -0.9 V and the copper ammine complex was used under acidity with a pH value of 5, the mixed solution was heated to 70°C and held for 2.0 hours. Grain growth occurred at 100.degree. C., the average particle diameter of the primary particles was 400 nm, and the total metal impurity concentration of the obtained copper particles was as high as 50 mass ppm. The amount of CuC 2 O 2 H - ions and C 2 H 3 O 2 - ions detected with respect to the amount of Cu + ions detected was also examined, but none of them were detected because the component derived from copper carboxylate was not contained. rice field.
- hydrazine-hydrate was used as the reducing agent. Since the oxidation-reduction potential of this reducing agent was -1.0 V and the copper ammine complex was used in an acidic environment with a pH value of 5, the mixed solution was heated to 70°C and held for 2.0 hours. Grain growth occurred at 100.degree. C., the average particle diameter of the primary particles was 166 nm, and the total metal impurity concentration of the obtained copper particles was as high as 90 mass ppm. The amount of CuC 2 O 2 H - ions and C 2 H 3 O 2 - ions detected with respect to the amount of Cu + ions detected was also examined, but none of them were detected because the component derived from copper carboxylate was not contained. rice field.
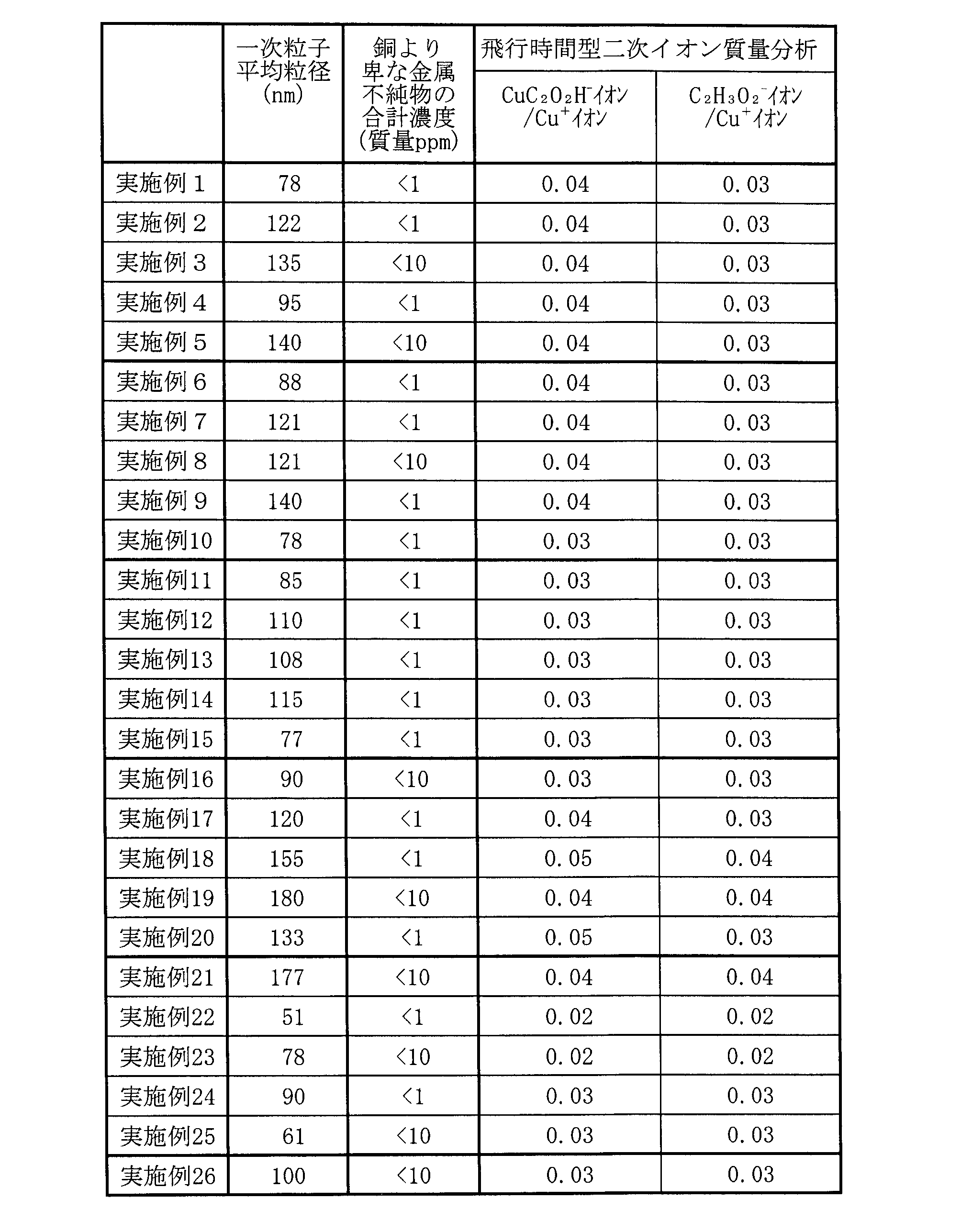
- Examples 1 to 26 a reducing agent was added and mixed under an acidic condition of pH 3 or more and less than pH 6, a hydrazine compound was used as the reducing agent, and the holding temperature during heating of the synthesis liquid was 60° C. or more and 75° C. or less. and the retention time was set to 1.5 hours or more and 2.5 hours or less, so that the average particle size of the primary particles of the copper particles was in the range of 51 nm (Example 22) to 180 nm (Example 19).
- the total concentration of impurities containing metals having oxidation-reduction potentials lower than that of copper was less than 10 ppm by mass.
- the copper particles of the present invention can be used as fine-pitch lead-free wiring or bonding particles, and the wiring paste or bonding paste obtained by using the wiring particles or bonding particles as a raw material can be used for fine electronic parts. It can be suitably used for mounting.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nanotechnology (AREA)
- Mechanical Engineering (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Manufacture Of Metal Powder And Suspensions Thereof (AREA)
- Powder Metallurgy (AREA)
Abstract
Description
また、この接合材料用粒子の製造方法では、室温のクエン酸銅の水分散液にpH調整剤を加えてpH3以上pH7未満にpH調整し、不活性ガス雰囲気下でこのpH調整したクエン酸銅の水分散液にヒドラジン化合物を添加混合し、不活性ガス雰囲気下でこの混合液を60℃以上80℃以下の温度に加熱し1.5時間以上2.5時間以下保持することにより、クエン酸銅を還元して銅ナノ粒子を生成させ、この銅ナノ粒子の表面に有機保護膜を形成している。
図1に示すように、この実施形態の銅粒子10では、金属銅からなるコア粒子11の表面がカルボン酸銅由来の有機分子により構成された有機保護膜12で被覆される。この銅粒子10は、粒子に含まれる酸化還元電位が銅より卑な金属の不純物の合計濃度が10質量ppm未満である。この不純物の合計濃度は、1質量ppm未満であることが好ましい。上記不純物の合計濃度が10質量ppm以上になると、銅粒子を配線材料として用いた場合に、金属不純物の拡散により他部材の特性を損なうおそれがある。例えば配線が施される基板等を不純物が汚染して基板等の絶縁性を損なうおそれがある。
本実施形態の銅粒子の製造方法では、カルボン酸銅の水分散液にpH調整剤を加えてこの水分散液のpHを3以上6未満の酸性領域に調整し、このpH調整したカルボン酸銅の水分散液に、酸化還元電位が-0.7V~-0.5Vの範囲にあるヒドラジン化合物の水溶液を添加混合して混合液を得た後、不活性ガス雰囲気下、前記混合液を60℃~75℃の温度に加熱し、1.5時間~2.5時間保持することにより、カルボン酸銅を還元して銅粒子が分散した銅粒子分散液を得る。
ここで、酸化還元電位とは標準水素電極(NHE)に対する電位差の意味である。酸化還元電位が-0.7V未満では、銅との酸化還元電位差が大きくなり10質量ppm以上の金属不純物が含まれてしまう不具合があり、-0.5Vを超えると銅との酸化還元電位差が小さくなるため、カルボン酸銅の還元が完了しない不具合がある。好ましい酸化還元電位の範囲は-0.6V~-0.5Vである。
酸化還元電位E(V)は、pH値に基づいて、以下の式(1)で表される。
(酸性域)N2H5 + = N2 + 5H+ + 4e-
(アルカリ域)N2H4 + 4OH- = N2 + 4H2O + 4e-
酸化還元電位E(V):-0.23 -0.075×pH (1)
例えば、pHが3であるときには、上記式(1)は[-0.23 -0.075×3]となり、酸化還元電位は、-0.455Vとなる。なお、後述する実施例及び比較例における酸化還元電位は、小数点以下第二位を四捨五入して、例えば-0.455Vは-0.5Vで示している。

先ず、出発原料であるカルボン酸銅として、表1に示すクエン酸銅を用意した。このクエン酸銅を室温のイオン交換水に入れ、撹拌羽根を用いて撹拌し、濃度30質量%のクエン酸銅の水分散液を調製した。次いで、このクエン酸銅の水分散液にpH調整剤としてのクエン酸アンモニウム水溶液を加えて、上記水分散液のpHが3になるように調整した。次に、pH調整した液を50℃の温度にし、窒素ガス雰囲気下で、pH調整した液に還元剤として、銅イオンを還元できる1.2倍当量分である酸化還元電位が-0.5Vのヒドラジン一水和物水溶液(2倍希釈)を一気に添加し、撹拌羽を用いて均一に混合した。更に、目標とする銅粒子を合成するために、上記水分散液と上記還元剤との混合液を窒素ガス雰囲気下で保持温度の70℃まで昇温し、70℃で2時間保持した。遠心分離機を用いて、加熱保持した液中に生成した粒子を固液分離して回収した。回収した粒子を減圧乾燥法で乾燥し、実施例1の銅粒子を製造した。

実施例1の出発原料であるクエン酸銅と同一又は異なるカルボン酸銅を用い、調整したpH値を実施例1と同一又は変更し、還元剤の種類及び酸化還元電位を実施例1と同一又は変更し、銅粒子の合成時の保持温度とその保持時間を実施例1と同一又は変更した。それ以外は実施例1と同様にして、実施例2~26、比較例1~3、6~16の銅粒子を製造した。これらの銅粒子の中で、実施例5、実施例10及び実施例16で得られた各銅粒子を走査型電子顕微鏡で撮影した写真図を、図3、図4及び図5にそれぞれ示す。
実施例1の還元剤であるヒドラジン一水和物をギ酸アンモニウムに変更し、この還元剤の酸化還元電位を0.3Vに変更し、pH値を5に変更した。実施例1の合成時の保持温度及びその保持時間は変更しなかった。それ以外は実施例1と同様にして、比較例4の銅粒子を製造した。
実施例1の還元剤であるヒドラジン一水和物をギ酸に変更し、この還元剤の酸化還元電位を-0.2Vに変更した。実施例1の合成時の保持温度は変更せずに、その保持時間を1.5時間に変更した。それ以外は実施例1と同様にして、比較例5の銅粒子を製造した。
実施例1のカルボン酸銅の代わりに、銅アンミン錯体を用いて、pH値を5に変更し、実施例1と同一の還元剤を用いて、還元剤の酸化還元電位を-0.9Vに変更した。それ以外は実施例1と同様にして、比較例17の銅粒子を製造した。
実施例1のカルボン酸銅の代わりに、銅アンミン錯体を用いて、pH値を5に変更し、実施例1と同一の還元剤を用いて、還元剤の酸化還元電位を-1.0Vに変更した。それ以外は実施例1と同様にして、比較例18の銅粒子を製造した。
実施例1のpH値を5に変更し、還元剤の酸化還元電位を-0.6Vに変更し、合成時の保持温度は変更せずに、その保持時間を1.5時間に変更した。更にコア粒子の表面保護剤として、メチルセルロースを粒子に対して5質量%添加した。それ以外は実施例1と同様にして、比較例19の銅粒子を製造した。
比較例19で使用したコア粒子の表面保護剤であるメチルセルロースを、ポリエチレングリコールに変更し、粒子に対して5質量%添加した。それ以外は比較例19と同様にして、比較例20の銅粒子を製造した。
比較例19で使用したコア粒子の表面保護剤であるメチルセルロースを、ポリビニルアルコールに変更し、粒子に対して5質量%添加した。それ以外は比較例19と同様にして、比較例21の銅粒子を製造した。
比較例19で使用したコア粒子の表面保護剤であるメチルセルロースを、ゼラチンに変更し、粒子に対して5質量%添加した。それ以外は比較例19と同様にして、比較例22の銅粒子を製造した。
実施例1~26及び比較例1~22の中で、銅粒子を製造することができた例における(1)銅粒子の一次粒子の平均粒径、(2)酸化還元電位が銅より卑な金属の不純物の合計濃度、及び(3)銅粒子を飛行時間型二次イオン質量分析法(TOF-SIMS)を用いて分析したときのCu+イオンの検出量に対するCuC2O2H-イオンとC2H3O2 -イオンの各検出量を、上述した方法でそれぞれ求めた。それらの結果を以下の表4及び表6にそれぞれ示す。ここで、酸化還元電位が銅より卑な金属不純物の合計濃度は、各金属不純物を合計した濃度である。酸化還元電位が銅より卑な金属の不純物の合計濃度と各金属不純物の濃度を、以下の表5及び表7に示す。また還元不十分で合成できなかった比較例1、比較例4~6及び比較例9については、上記項目における値を記載していない。

11 コア粒子
12 有機保護膜
Claims (4)
- 金属銅からなるコア粒子の表面がカルボン酸銅由来の有機分子により構成された有機保護膜で被覆された銅粒子において、
前記銅粒子に含まれる酸化還元電位が銅より卑な金属の不純物の合計濃度が10質量ppm未満であり、
前記銅粒子が一次粒子の状態でその平均粒径が50nmを超え200nm以下の範囲にあり、
前記銅粒子を飛行時間型二次イオン質量分析法(TOF-SIMS)を用いて分析したときに、CuC2O2H-イオンの検出量が、Cu+イオンの検出量に対して0.02倍以上の範囲にあって、C2H3O2 -イオンの検出量が、Cu+イオンの検出量に対して0.02倍以上の範囲にあることを特徴とする銅粒子。 - 粒子間での凝集及び/又は粒子の酸化を抑制するための分散剤及び表面保護剤を用いずに、請求項1に記載された銅粒子を製造する方法であって、
カルボン酸銅の水分散液にpH調整剤を加えて前記水分散液のpHを3以上6未満の酸性領域に調整する工程と、
前記pH調整したカルボン酸銅の水分散液に酸化還元電位が-0.7V~-0.5Vの範囲にあるヒドラジン化合物の水溶液を添加混合して混合液を得る工程と、
不活性ガス雰囲気下、前記混合液を60℃~75℃の温度に加熱し、1.5時間~2.5時間保持することにより、前記カルボン酸銅を還元して銅粒子が分散した銅粒子分散液を得る工程と
を含むことを特徴とする銅粒子の製造方法。 - 前記カルボン酸銅が、水に難溶性であって、炭素数が4~8であるカルボン酸銅からなる群より選ばれた1種又は2種以上の銅塩である請求項2記載の銅粒子の製造方法。
- 前記pH調整剤がカルボン酸アンモニウムである請求項2記載の銅粒子の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22779783.4A EP4316698A4 (en) | 2021-03-29 | 2022-03-03 | Copper particles and method for producing same |
| CN202280020923.6A CN116981527A (zh) | 2021-03-29 | 2022-03-03 | 铜粒子及其制造方法 |
| US18/283,015 US20240165699A1 (en) | 2021-03-29 | 2022-03-03 | Copper particles and method for producing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021054554A JP7121884B1 (ja) | 2021-03-29 | 2021-03-29 | 銅粒子及びその製造方法 |
| JP2021-054554 | 2021-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022209558A1 true WO2022209558A1 (ja) | 2022-10-06 |
Family
ID=82929855
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/009029 Ceased WO2022209558A1 (ja) | 2021-03-29 | 2022-03-03 | 銅粒子及びその製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20240165699A1 (ja) |
| EP (1) | EP4316698A4 (ja) |
| JP (1) | JP7121884B1 (ja) |
| CN (1) | CN116981527A (ja) |
| TW (1) | TWI912489B (ja) |
| WO (1) | WO2022209558A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116174705A (zh) * | 2023-03-03 | 2023-05-30 | 嘉庚创新实验室 | 铜复合颗粒及其制备方法、铜导电浆料和铜膜 |
| CN116586627A (zh) * | 2023-05-18 | 2023-08-15 | 南京工业大学 | 一种蛇莓形状纳米铜颗粒及其制备方法与应用 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2024080436A (ja) * | 2022-12-02 | 2024-06-13 | Jx金属株式会社 | 銅粉 |
| KR20250156147A (ko) * | 2023-04-26 | 2025-10-31 | 엠. 테크닉 가부시키가이샤 | 구리 입자의 제조방법 |
| WO2026043474A1 (en) * | 2024-08-20 | 2026-02-26 | Hewlett-Packard Development Company, L.P. | Binder fluid for three-dimensional printing |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05331508A (ja) * | 1992-05-29 | 1993-12-14 | Murata Mfg Co Ltd | 銅粉末の製造方法 |
| JP2638271B2 (ja) | 1990-09-06 | 1997-08-06 | 住友金属工業株式会社 | 銅微粉末の製造方法 |
| JP3934869B2 (ja) | 2000-10-10 | 2007-06-20 | 三井金属鉱業株式会社 | 回路形成用銅微粉末 |
| JP2009084614A (ja) * | 2007-09-28 | 2009-04-23 | Dowa Electronics Materials Co Ltd | 銅粉およびその製造方法、銅ペースト、積層セラミックコンデンサ、並びに銅粉判定方法 |
| JP2010116625A (ja) * | 2008-10-17 | 2010-05-27 | Mitsubishi Materials Corp | 金属ナノ粒子の合成方法 |
| CN102581294A (zh) * | 2012-03-26 | 2012-07-18 | 沈阳化工大学 | 一种纳米金属铜粉体的制备方法 |
| JP2013072091A (ja) | 2011-09-26 | 2013-04-22 | Hitachi Cable Ltd | 金属微粒子およびその製造方法、金属微粒子を含む金属ペースト、並びに金属ペーストから形成される金属被膜 |
| JP2014221927A (ja) * | 2013-05-13 | 2014-11-27 | 国立大学法人東北大学 | 銅微粒子およびその製造方法 |
| JP2016069716A (ja) * | 2014-10-01 | 2016-05-09 | 協立化学産業株式会社 | 被覆銅粒子及びその製造方法 |
| JP2017115119A (ja) | 2015-12-24 | 2017-06-29 | 三星電子株式会社Samsung Electronics Co.,Ltd. | 重合体、補償フィルム、光学フィルム及び表示装置 |
| JP2019077926A (ja) * | 2017-10-25 | 2019-05-23 | 株式会社村田製作所 | 複合銅粒子、銅インク、および、複合銅粒子の製造方法 |
| JP2020059914A (ja) | 2018-10-04 | 2020-04-16 | 三菱マテリアル株式会社 | 接合材料用粒子及びその製造方法、接合用ペースト及びその調製方法並びに接合体の製造方法 |
| JP2020090725A (ja) * | 2020-02-14 | 2020-06-11 | 協立化学産業株式会社 | 複合粒子、銅ペースト組成物、および導電体 |
| JP2021054554A (ja) | 2019-09-27 | 2021-04-08 | 株式会社神戸製鋼所 | 吊り荷の水平移動補助装置およびこれを備えたクレーン、吊り荷の水平移動方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5898400B2 (ja) * | 2007-11-05 | 2016-04-06 | 住友金属鉱山株式会社 | 銅微粒子とその製造方法及び銅微粒子分散液 |
| JP5117420B2 (ja) * | 2008-03-27 | 2013-01-16 | 古河電気工業株式会社 | 銅微粒子分散水溶液の製造方法及び銅微粒子分散水溶液の保管方法 |
| KR20170122208A (ko) * | 2015-02-27 | 2017-11-03 | 히타치가세이가부시끼가이샤 | 구리 함유 입자, 도체 형성 조성물, 도체의 제조 방법, 도체 및 장치 |
| US20220040759A1 (en) * | 2018-10-04 | 2022-02-10 | Mitsubishi Materials Corporation | Particles for joining material and production method thereof, joining paste and preparation method thereof, and production method of joined body |
-
2021
- 2021-03-29 JP JP2021054554A patent/JP7121884B1/ja active Active
-
2022
- 2022-03-03 US US18/283,015 patent/US20240165699A1/en not_active Abandoned
- 2022-03-03 WO PCT/JP2022/009029 patent/WO2022209558A1/ja not_active Ceased
- 2022-03-03 EP EP22779783.4A patent/EP4316698A4/en not_active Withdrawn
- 2022-03-03 CN CN202280020923.6A patent/CN116981527A/zh active Pending
- 2022-03-16 TW TW111109562A patent/TWI912489B/zh active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2638271B2 (ja) | 1990-09-06 | 1997-08-06 | 住友金属工業株式会社 | 銅微粉末の製造方法 |
| JPH05331508A (ja) * | 1992-05-29 | 1993-12-14 | Murata Mfg Co Ltd | 銅粉末の製造方法 |
| JP3934869B2 (ja) | 2000-10-10 | 2007-06-20 | 三井金属鉱業株式会社 | 回路形成用銅微粉末 |
| JP2009084614A (ja) * | 2007-09-28 | 2009-04-23 | Dowa Electronics Materials Co Ltd | 銅粉およびその製造方法、銅ペースト、積層セラミックコンデンサ、並びに銅粉判定方法 |
| JP2010116625A (ja) * | 2008-10-17 | 2010-05-27 | Mitsubishi Materials Corp | 金属ナノ粒子の合成方法 |
| JP2013072091A (ja) | 2011-09-26 | 2013-04-22 | Hitachi Cable Ltd | 金属微粒子およびその製造方法、金属微粒子を含む金属ペースト、並びに金属ペーストから形成される金属被膜 |
| CN102581294A (zh) * | 2012-03-26 | 2012-07-18 | 沈阳化工大学 | 一种纳米金属铜粉体的制备方法 |
| JP2014221927A (ja) * | 2013-05-13 | 2014-11-27 | 国立大学法人東北大学 | 銅微粒子およびその製造方法 |
| JP2016069716A (ja) * | 2014-10-01 | 2016-05-09 | 協立化学産業株式会社 | 被覆銅粒子及びその製造方法 |
| JP2017115119A (ja) | 2015-12-24 | 2017-06-29 | 三星電子株式会社Samsung Electronics Co.,Ltd. | 重合体、補償フィルム、光学フィルム及び表示装置 |
| JP2019077926A (ja) * | 2017-10-25 | 2019-05-23 | 株式会社村田製作所 | 複合銅粒子、銅インク、および、複合銅粒子の製造方法 |
| JP2020059914A (ja) | 2018-10-04 | 2020-04-16 | 三菱マテリアル株式会社 | 接合材料用粒子及びその製造方法、接合用ペースト及びその調製方法並びに接合体の製造方法 |
| JP2021054554A (ja) | 2019-09-27 | 2021-04-08 | 株式会社神戸製鋼所 | 吊り荷の水平移動補助装置およびこれを備えたクレーン、吊り荷の水平移動方法 |
| JP2020090725A (ja) * | 2020-02-14 | 2020-06-11 | 協立化学産業株式会社 | 複合粒子、銅ペースト組成物、および導電体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4316698A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116174705A (zh) * | 2023-03-03 | 2023-05-30 | 嘉庚创新实验室 | 铜复合颗粒及其制备方法、铜导电浆料和铜膜 |
| CN116586627A (zh) * | 2023-05-18 | 2023-08-15 | 南京工业大学 | 一种蛇莓形状纳米铜颗粒及其制备方法与应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI912489B (zh) | 2026-01-21 |
| JP7121884B1 (ja) | 2022-08-19 |
| EP4316698A4 (en) | 2025-01-29 |
| US20240165699A1 (en) | 2024-05-23 |
| CN116981527A (zh) | 2023-10-31 |
| JP2022151975A (ja) | 2022-10-12 |
| TW202302248A (zh) | 2023-01-16 |
| EP4316698A1 (en) | 2024-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7121884B1 (ja) | 銅粒子及びその製造方法 | |
| JP4489389B2 (ja) | 微粒銀粉の製造方法 | |
| JP5377483B2 (ja) | 微小金属粒子含有組成物及びその製造方法 | |
| EP2208559A1 (en) | Silver microparticle-containing composition, process for production of the composition, process for production of the silver microparticle, and paste containing the silver microparticle | |
| JP5949654B2 (ja) | 銀粉およびその製造方法 | |
| JP6168837B2 (ja) | 銅微粒子およびその製造方法 | |
| JP4428085B2 (ja) | 銅微粒子の製造方法 | |
| CN103079726A (zh) | 银粒子及其制备方法 | |
| JP2018104724A (ja) | 銀コート銅粉の製造方法 | |
| JP2017039991A (ja) | 銀コート銅粉とその製造方法、及びそれを用いた導電性ペースト | |
| WO2020017564A1 (ja) | 球状銀粉の製造方法 | |
| EP4516431A1 (en) | Spherical silver powder, method for producing spherical silver powder, apparatus for producing spherical silver powder, and conductive paste | |
| JP2017206751A (ja) | ニッケル粉末の製造方法 | |
| TW201631603A (zh) | 覆銀銅粉及其製造方法 | |
| TW201338893A (zh) | 銀粉 | |
| JP2018199844A (ja) | 錫コート銅粉の製造方法および導電性ペーストの製造方法 | |
| JP2021147684A (ja) | 銅および酸化銅含有微粒子及びその製造方法 | |
| JP2021055142A (ja) | 銀被覆金属粉末およびその製造方法並びに導電性塗料 | |
| KR102940155B1 (ko) | 은팔라듐 합금 분말 및 그의 이용 | |
| KR20200061193A (ko) | 단분산 은 분말의 제조방법 | |
| US12031196B2 (en) | Metal powder | |
| JP7065676B2 (ja) | 銀被覆金属粉末およびその製造方法、銀被覆金属粉末を含む導電性ペースト、並びに導電性ペーストを用いた導電膜の製造方法 | |
| JP2024012999A (ja) | 銅粒子および銅粒子の製造方法 | |
| JP7600782B2 (ja) | ニッケル粒子、ニッケル粒子の表面処理方法およびニッケル粉末の製造方法 | |
| JP7596754B2 (ja) | ニッケル粒子の表面処理方法およびニッケル粉末の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22779783 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280020923.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18283015 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022779783 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022779783 Country of ref document: EP Effective date: 20231030 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2022779783 Country of ref document: EP |