WO2024190892A1 - 抗体製剤 - Google Patents
抗体製剤 Download PDFInfo
- Publication number
- WO2024190892A1 WO2024190892A1 PCT/JP2024/010151 JP2024010151W WO2024190892A1 WO 2024190892 A1 WO2024190892 A1 WO 2024190892A1 JP 2024010151 W JP2024010151 W JP 2024010151W WO 2024190892 A1 WO2024190892 A1 WO 2024190892A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formulation
- pharmaceutical composition
- aqueous pharmaceutical
- bispecific antibody
- heavy chain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/46—Hybrid immunoglobulins
Definitions
- the present disclosure relates to a pharmaceutical composition (hereinafter sometimes abbreviated as “pharmaceutical composition of the present invention”) containing an antibody, in particular a bispecific antibody capable of specifically binding to PD-1 and CD3, respectively, as an active ingredient, and a formulation thereof (hereinafter sometimes abbreviated as “formulation of the present invention”).
- pharmaceutical composition of the present invention containing an antibody, in particular a bispecific antibody capable of specifically binding to PD-1 and CD3, respectively, as an active ingredient, and a formulation thereof (hereinafter sometimes abbreviated as “formulation of the present invention”).
- PD-1 is an immunosuppressive receptor belonging to the immunoglobulin family, and is a molecule that has the function of suppressing immune activation signals of T cells activated by stimulation from antigen receptors. From the analysis of PD-1 knockout mice, it is known that this PD-1 signal plays an important role in suppressing autoimmune diseases such as autoimmune dilated cardiomyopathy, lupus-like syndrome, autoimmune encephalomyelitis, systemic lupus erythematosus, graft-versus-host disease, type I diabetes, and rheumatoid arthritis. Therefore, it has been pointed out that substances that enhance PD-1 signals could be used as preventive or therapeutic agents for autoimmune diseases.
- autoimmune diseases such as autoimmune dilated cardiomyopathy, lupus-like syndrome, autoimmune encephalomyelitis, systemic lupus erythematosus, graft-versus-host disease, type I diabetes, and rheumatoid arthritis. Therefore, it has been
- Bispecific antibodies that recognize PD-1 have been known to date as substances that enhance PD-1 signals (Patent Documents 1 to 5). These bispecific antibodies are made by genetically linking the antigen recognition site of an antibody that recognizes CD3, a member of the T cell receptor complex, with the antigen recognition site of an antibody that recognizes PD-1, and have the effect of enhancing the inhibitory signal of PD-1 against the T cell receptor complex by increasing the frequency with which PD-1 is located in the vicinity of the T cell receptor complex.
- the pharmaceutical composition of the present invention is characterized in that it contains as an active ingredient an anti-PD-1/CD3 bispecific antibody in a concentration range of about 0.01 mg/mL to about 200 mg/mL, and contains acetate buffer, trehalose, L-methionine, polysorbate 80, and sodium chloride as one embodiment of the formulation additives.
- an anti-PD-1/CD3 bispecific antibody in a concentration range of about 0.01 mg/mL to about 200 mg/mL, and contains acetate buffer, trehalose, L-methionine, polysorbate 80, and sodium chloride as one embodiment of the formulation additives.
- Patent Document 6 relates to a formulation that can minimize the immunosuppressive effect in target tissues by administering anti-CD3 antibodies via non-systemic administration, such as intravenous injection, in order to achieve target tissue-specific immunomodulation.
- formulations include an oral formulation containing an anti-CD3 antibody with a unit dose of 0.1 to 10 mg as the active ingredient and sodium acetate trihydrate, sodium chloride, polysorbate 80, trehalose, and methionine as formulation additives, and a subcutaneous formulation containing 2 mg/mL anti-CD3 antibody as the active ingredient and sodium acetate trihydrate, sodium chloride, and polysorbate 80 as formulation additives.
- Patent Document 7 discloses a formulation containing 150 mg/mL anti-OX40L antibody as an active ingredient, and acetate buffer, trehalose, polysorbate 20, and methionine as formulation additives.
- trehalose and methionine are used as isotonicity agents or stabilizers, but there is no suggestion whatsoever of further adding sodium chloride, which can also function as an isotonicity agent or stabilizer, in the presence of trehalose and methionine.
- polysorbate 20 which is included in a formulation example that exhibits favorable performance, in place of polysorbate 80.
- Patent document 8 discloses a formulation containing 150 mg/mL anti-GM-CSF-R ⁇ antibody as the active ingredient and acetate buffer, sodium chloride, polysorbate 80, and trehalose as formulation additives, but makes no suggestion whatsoever about the addition of L-methionine.
- the pH is usually set to one pH difference from the pI value of the antibody, taking into consideration the colloidal stability of the antibody.
- Patent Document 8 is intended to be applied to preparations that achieve a pH in the range of 5.5 to 7.5, which is close to the pI value of the antibody, and the formulation composition is not inherently applicable to pharmaceutical compositions with other pH values (for example, the pharmaceutical composition of the present invention having a pH of 4.7 to 5.3).
- Patent document 9 discloses a formulation containing approximately 45-55 mg/mL adalimumab as the active ingredient and acetate buffer, sodium chloride, polysorbate 80 and trehalose as formulation additives, but indicates that the formulation contains substantially no or no free amino acids, or contains no more than 0.1 mM free amino acids, and further discloses that the presence of free amino acids, including L-methionine, significantly worsens the stability of the antibody (antibody fragmentation at pH 5.0 or less). Furthermore, while a less complex formulation employing fewer ingredients would be desirable, it is suggested that the antioxidant properties of trehalose are highly promising, and therefore no additional antioxidants are required.
- Patent documents 10 and 11 disclose a formulation containing 20 mg/mL anti-EGFR antibody as the active ingredient and acetate buffer, sodium chloride, polysorbate 80 and trehalose as formulation additives, but the concentration of sodium chloride is equivalent to 100 mM.
- the concentration of sodium chloride is approximately 34.2 mM or less, and at a concentration equivalent to 100 mM, significant formation of aggregates occurs and the formulation cannot be applied. Furthermore, there is no suggestion whatsoever about the addition of L-methionine.
- Patent Documents 12 and 13 disclose a formulation containing an acetate buffer, polysorbate 80, trehalose, methionine and DTPA to deal with visible fine particles that are prominent in IgG4 isotype antibodies, but there is no suggestion that this is applicable to the anti-PD-1/CD3 bispecific antibody of the present invention, which is an IgG1 isotype antibody (DOI(JaLC): 10.15083/00006491), which has different physical properties. Furthermore, while the concentration of polysorbate 80 in the formulations in these documents is 0.02% (w/v), there is no suggestion that an additional concentration of approximately 0.1% (w/v) is optimal or necessary for the pharmaceutical composition of the present invention from the standpoint of suppressing aggregation.
- Patent document 14 discloses a formulation containing an anti-TIGIT antibody as an active ingredient, and as one embodiment of the formulation, an acetate buffer, trehalose, sodium chloride, and polysorbate 80 as additives; however, in practice, the addition of sodium chloride caused a rapid decrease in the monomer purity of the antibody, and it is therefore preferable not to include it; and, after comparing polysorbate 80 concentrations of 0.02% and 0.04%, it was found that a lower concentration of 0.02% was preferable in terms of the rapid decrease in monomer purity.
- sodium chloride is added to the pharmaceutical composition of the present invention, and a polysorbate 80 concentration of approximately 0.1% (w/v) is more preferable.
- the objective of the present invention is to provide a pharmaceutical composition and a formulation thereof that has a single composition and can be used for both subcutaneous and intravenous administration of an antibody, particularly an anti-PD-1/CD3 bispecific antibody, that is used in a wide concentration range.
- aqueous pharmaceutical composition for subcutaneous or intravenous administration comprising an antibody (preferably, an anti-PD-1/CD3 bispecific antibody) as an active ingredient, and further comprising a buffer (preferably, an acetate buffer), trehalose, an antioxidant (preferably, L-methionine), polysorbate 80, and sodium chloride, or a formulation thereof;
- a buffer preferably, an acetate buffer
- trehalose preferably, an antioxidant
- polysorbate 80 preferably, and sodium chloride
- sodium chloride sodium chloride
- An aqueous pharmaceutical composition for subcutaneous or intravenous administration which is biologically equivalent to the aqueous pharmaceutical composition or a formulation thereof described in the preceding paragraph ⁇ 1>, and ⁇ 3> use of the aqueous pharmaceutical composition or a formulation thereof described in the preceding paragraph ⁇ 1> or ⁇ 2> in the prevention, inhibition of progression of symptoms, inhibition of recurrence, and/or treatment of autoimmune disease, graft-versus-host disease (GVHD), or blood cancer
- the pharmaceutical composition of the present invention relating to the anti-PD-1/CD3 bispecific antibody or a preparation thereof will be a new drug for preventing, inhibiting the progression of symptoms, inhibiting recurrence, and/or treating autoimmune diseases, graft-versus-host disease (GVHD), or blood cancer.
- GVHD graft-versus-host disease
- the heavy chain constant region constituting anti-PD-1/CD3 bispecific antibody B (described in detail below) has an amino acid sequence in each of the heavy chain and light chain that constitutes the antigen-binding site that specifically binds to PD-1.
- FIG. 1 shows the amino acid sequences of the heavy chain constant regions of the heavy and light chains that constitute the antigen-binding site that specifically binds to CD3, among the heavy chain constant regions that constitute anti-PD-1/CD3 bispecific antibody B.
- FIG. 1 shows the relationship between the binding activity (%) of antibody ⁇ (described in detail below) to PD-1 (relative activity assuming the activity of a standard antibody ⁇ as 100%) and the oxidation degree (%) of the 101st methionine residue (hereinafter sometimes abbreviated as "Met101") present in the complementarity determining region of the antibody to PD-1 (the proportion of antibody ⁇ in which the 101st methionine residue is oxidized).
- the present invention relates to antibodies, in particular to antibodies that have binding specificities for two different antigen molecules or epitopes in a single molecule, i.e., bispecific antibodies.
- the anti-PD-1/CD3 bispecific antibodies of the present invention are (i) a heavy chain and a light chain that form an antigen-binding site that specifically binds to PD-1, and (ii) a heavy chain and a light chain that form an antigen-binding site that specifically binds to CD3, wherein: (a) the heavy chain of the preceding paragraph (i) consists of any one of the amino acid sequences selected from SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 and SEQ ID NO:5; (b) the heavy chain of (ii) above consists of the amino acid sequence of SEQ ID NO:6; and (c) the light chains of (i) and (ii) above both consist of the amino acid sequence of SEQ ID NO:7.
- heavy and light chains constituting an antigen-binding site that specifically binds to PD-1 refers to a heavy and light chain complex in which the heavy and light chains associate with each other via disulfide bonds to form an antigen-binding site that specifically binds to PD-1.
- an “antigen-binding site” is the minimum unit for an antibody to have binding activity to its antigen, and “specifically binds to PD-1” is used to mean a characteristic in which an antibody can directly bind to PD-1 with a binding activity having an affinity (dissociation constant (Kd value)) of at least 1x10-5 M, preferably 1x10-7 M, more preferably 1x10-9 M or higher, and does not substantially bind to at least other receptor members belonging to the so-called CD28 family of receptors, such as CD28, CTLA-4, and ICOS.
- Kd value affinity
- CD28 family of receptors such as CD28, CTLA-4, and ICOS.
- “heavy and light chains constituting an antigen-binding site that specifically binds to CD3” refers to a heavy and light chain complex in which the heavy and light chains associate with each other via disulfide bonds to form an antigen-binding site that specifically binds to CD3.
- “specifically binds to CD3” is used to mean a characteristic of being able to directly bind to CD3 with a binding activity having an affinity (dissociation constant (Kd value)) of at least 1x10-5 M, preferably 1x10-7 M, more preferably 1x10-9 M or higher, and not substantially binding to other proteins.
- anti-PD-1/CD3 bispecific antibody A the antibody to which the pharmaceutical composition or formulation of the present invention is applied is preferably an anti-PD-1/CD3 bispecific antibody (hereinafter sometimes abbreviated as "antibody ⁇ ") in which the heavy chain constituting the antigen-binding site that specifically binds to PD-1 is composed of the amino acid sequence of SEQ ID NO:5.
- Anti-PD-1/CD3 bispecific antibody A can be produced according to the method disclosed in WO2019/156199, and corresponds to anti-PD-1/CD3 bispecific antibody clones PD1-1(Bi) to PD1-5(Bi), of which antibody ⁇ corresponds to clone PD1-5(Bi).
- anti-PD-1/CD3 bispecific antibody B an anti-PD-1/CD3 bispecific antibody in which the heavy chain constant region of the heavy chain constituting the antigen-binding site that specifically binds to PD-1 of antibody ⁇ is replaced with a heavy chain constant region consisting of any one of the amino acid sequences selected from SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12, and SEQ ID NO:13, and the heavy chain constant region of the heavy chain constituting the antigen-binding site that specifically binds to CD3 is replaced with a heavy chain constant region consisting of any one of the amino acid sequences selected from SEQ ID NO:14, SEQ ID NO:15, SEQ ID NO:16, SEQ ID NO:17, SEQ ID NO:18, and SEQ ID NO:19.
- Anti-PD-1/CD3 bispecific antibody B can be produced according to the heavy chain constant region of the heavy chain constituting the antigen-binding site that specifically binds to
- the isotype of both anti-PD-1/CD3 bispecific antibody A and anti-PD-1/CD3 bispecific antibody B is IgG1.
- compositions of the present invention comprise the anti-PD-1/CD3 bispecific antibody at any concentration between about 0.01 and about 200 mg/mL (e.g., about 0.01 mg/mL, about 0.02 mg/mL, about 0.03 mg/mL, about 0.04 mg/mL, about 0.05 mg/mL, about 0.06 mg/mL, about 0.07 mg/mL, about 0.08 mg/mL, about 0.09 mg/mL, about 0.1 mg/mL, about 0.2 mg/mL, about 0.3 mg/mL, about 0.4 mg/mL, about 0.5 mg/mL, about 0.6 mg/mL, about 0.7 mg/mL, about 0.8 mg/mL, about 0.9 mg/mL, about 10 mg/mL, about 15 mg/mL, about 16 mg/mL, about 17 mg/mL, about 18 mg/mL, about 19 mg/mL, about 20 mg/mL, about 21 mg/mL, about 22 mg/mL, about 23 mg/mL, about
- the anti-PD-1/CD3 bispecific antibody applied to the aqueous pharmaceutical composition of the present invention (preferably for intravenous administration) having a volume per unit formulation of, for example, 5 mL may be any amount between about 0.05 and about 1000 mg (preferably, about 0.5 to about 750 mg, more preferably, about 0.5 to about 6.0 mg, about 25 to about 125 mg or about 400 to about 750 mg, even more preferably, about 2.5 mg, about 5 mg/mL, about 50 mg/mL or about 500 mg/mL), while the volume per unit formulation of, for example, 1
- the amount of the anti-PD-1/CD3 bispecific antibody applied to the 5 mL aqueous pharmaceutical composition (preferably for subcutaneous administration) will be 1/5 of the amount of the antibody applied to the 5 mL aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6 mL, about 0.5 mL, about 0.4 mL, and about 0.2 mL,
- the term "pharmaceutical composition” refers to a mixture of an active ingredient and one or more other additives
- aqueous pharmaceutical composition refers to a pharmaceutical composition that is substantially dissolved in an aqueous solvent.
- the preferred form of the pharmaceutical composition of the present invention is an aqueous pharmaceutical composition.
- formulation refers to a pharmaceutical composition in a form suitable for an appropriately selected route or method of administration.
- “about” means that the indicated numerical value may vary within a range of 10% below or above the indicated numerical value, and “to” indicating a range means that the lower and upper numerical values are included.
- the aqueous pharmaceutical composition of the present invention contains a buffer to stabilize its pH. Any buffer is acceptable as long as it is sufficiently compatible with the pH stability of the aqueous pharmaceutical composition of the present invention and does not affect the stability of the anti-PD-1/CD3 bispecific antibody, but an acetate buffer is preferred.
- the acetate buffer concentration in the aqueous pharmaceutical composition of the present invention may be any concentration between about 7.3 and about 73.0 mM (e.g., about 7.3 mM, about 10 mM, about 14.6 mM, about 15 mM, about 18 mM, about 20 mM, about 20.07 mM, about 21 mM, about 22 mM, about 25 mM, about 30 mM, about 35 mM, about 40 mM, about 43.8 mM, about 50 mM, about 55 mM, about 56 mM, about 57 mM, about 58 mM, about 59 mM, about 60 mM, about 61 mM, about 62 mM, about 63 mM, about 64 mM, about 65 mM, about 66 mM, about 67 mM, about 68 mM, about 69 mM, about 70 mM, about 71 mM, about 72 mM
- the acetate buffer can be prepared by a known method of adding acetic acid to a sodium acetate solution (e.g., an aqueous solution of sodium acetate anhydrous, sodium acetate dihydrate, or sodium acetate trihydrate) to obtain a desired pH.
- a sodium acetate solution e.g., an aqueous solution of sodium acetate anhydrous, sodium acetate dihydrate, or sodium acetate trihydrate
- the acetate buffer to be used in an aqueous pharmaceutical composition having a volume of 5 mL per unit formulation is composed of (i) any amount of sodium acetate trihydrate equivalent to about 3.4 to about 34 mg (preferably, about 6.8 to about 20.4 mg, more preferably, about 8.4 to about 10.2 mg), and (ii) any amount of glacial acetic acid corresponding to the amount of sodium acetate trihydrate such that the pH of the acetate buffer solution is about 4.9 to 5.1, and in this case, the acetate buffer in a preferred embodiment is composed of about 9.4 mg (specifically, about 9.35 mg) of sodium acetate trihydrate and about 1.9 mg of glacial acetic acid.
- the acetate buffer applied to an aqueous pharmaceutical composition having a volume of, for example, about 1 mL is composed of about 1/5 of the amounts of sodium acetate trihydrate and glacial acetic acid applied to the 5 mL aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6 mL, about 0.5 mL, about 0.4 mL and about 0.2 mL, respectively, it is composed of about 2/5, about 4/25, about 3/25, about 1/10, about 2/25 and about 1/25 of the amounts of sodium acetate trihydrate and glacial acetic acid applied to the 5 mL aqueous pharmaceutical composition, respectively.
- the buffering agent in the aqueous pharmaceutical composition of the present invention also includes a buffering agent having pH buffering properties equivalent to those of the acetate buffering agent described above.
- the aqueous pharmaceutical composition of the present invention has any pH (e.g., about 4.7, about 4.8, about 4.9, about 5.0, about 5.1, about 5.2 or about 5.3) in the range of about 4.7 to about 5.3 (i.e., in the range of about 5.0 ⁇ 0.3), preferably has any pH in the range of about 4.9 to about 5.1 (i.e., in the range of about 5.0 ⁇ 0.1), and more preferably has a pH of about 5.0.
- any pH e.g., about 4.7, about 4.8, about 4.9, about 5.0, about 5.1, about 5.2 or about 5.3
- any pH in the range of about 4.9 to about 5.3 i.e., in the range of about 5.0 ⁇ 0.3
- preferably has any pH in the range of about 4.9 to about 5.1 i.e., in the range of about 5.0 ⁇ 0.1
- more preferably has a pH of about 5.0 preferably has a pH of about 5.0.
- the aqueous pharmaceutical composition of the present invention contains a saccharide mainly for adjusting its osmotic pressure. Any saccharide is acceptable as long as it can sufficiently adjust the osmotic pressure of the aqueous pharmaceutical composition of the present invention and does not affect the stabilization of the anti-PD-1/CD3 bispecific antibody, but trehalose is preferred, and trehalose may be added in the form of a hydrate (e.g., monohydrate, preferably dihydrate) or anhydrous.
- a saccharide mainly for adjusting its osmotic pressure. Any saccharide is acceptable as long as it can sufficiently adjust the osmotic pressure of the aqueous pharmaceutical composition of the present invention and does not affect the stabilization of the anti-PD-1/CD3 bispecific antibody, but trehalose is preferred, and trehalose may be added in the form of a hydrate (e.g., monohydrate, preferably dihydrate) or anhydrous.
- the concentration of trehalose in the aqueous pharmaceutical composition of the present invention may be any concentration between about 5 and about 286.1 mM (e.g., about 5 mM, about 10 mM, about 20 mM, about 30 mM, about 40 mM, about 50 mM, about 60 mM, about 70 mM, about 80 mM, about 90 mM, about 100 mM, about 110 mM, about 120 mM, about 130 mM, about 140 mM, about 150 mM, about 160 mM, about 170 mM, about 172 mM, about 180 mM, about 182 mM, about 182.3 mM, about 182.38 mM, about 182.4 mM, about 190 mM, about 193 mM, about 194 mM, about 195 mM, about 196 mM, about 197 mM, about 198 mM, about 199 mM, about 200 mM, about
- the trehalose to be applied to an aqueous pharmaceutical composition having a volume per unit dosage form of, for example, 5 mL is present in any amount of about 9.46 to about 541.2 mg as its dihydrate (preferably, about 18.9 to about 393.3 mg, more preferably, about 325 to about 365.1 mg, and even more preferably, about 345 mg); on the other hand, when the saccharide in the aqueous pharmaceutical composition of the present invention is applied to an aqueous pharmaceutical composition having a volume per unit dosage form of, for example, 1 mL (preferably, The amount of trehalose applied to the 5 mL aqueous pharmaceutical composition (for subcutaneous administration or for intravenous administration) is 1/5 of the amount of trehalose dihydrate applied to the 5 mL aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6
- the sugars in the aqueous pharmaceutical composition of the present invention also include sugars that have the same osmotic pressure adjusting ability as trehalose.
- the aqueous pharmaceutical composition of the present invention contains an antioxidant to suppress oxidation of the anti-PD-1/CD3 bispecific antibody or the resulting decrease in pharmacological activity.
- Any antioxidant is acceptable as long as it can suppress the oxidation of the anti-PD-1/CD3 bispecific antibody or the resulting decrease in pharmacological activity to the desired extent, but L-methionine or DTPA (Diethylene Triamine Pentaacetic Acid) is preferred, and L-methionine is more preferred.
- the concentration thereof may be any concentration between about 5 and about 30.2 mM (e.g., about 5 mM, about 8 mM, about 10 mM, about 10.05 mM, about 12 mM, about 15 mM, about 20 mM, about 25 mM, about 30 mM, or about 30.2 mM), preferably any concentration between about 5 and about 20 mM, more preferably any concentration between about 5 and about 15 mM, even more preferably any concentration between about 8 and about 12 mM, and most preferably about 10 mM, and more specifically about 10.05 mM.
- concentration between about 5 and about 30.2 mM e.g., about 5 mM, about 8 mM, about 10 mM, about 10.05 mM, about 12 mM, about 15 mM, about 20 mM, about 25 mM, about 30 mM, or about 30.2 mM
- any concentration between about 5 and about 20 mM more preferably any concentration between about 5 and
- the antioxidant in the aqueous pharmaceutical composition of the present invention is L-methionine
- the L-methionine applied to an aqueous pharmaceutical composition (preferably for intravenous administration) having a volume per unit dosage form of, for example, 5 mL is any amount between about 3.75 and about 22.5 mg (preferably, about 3.75 to about 15.0 mg, more preferably, about 3.75 to about 11.25 mg, even more preferably, about 6 to about 9 mg, and most preferably, about 7.5 mg)
- the L-methionine applied to an aqueous pharmaceutical composition having a volume per unit dosage form of, for example, 1 mL is any amount between about 3.75 and about 22.5 mg (preferably, about 3.75 to about 15.0 mg, more preferably, about 3.75 to about 11.25 mg, even more preferably, about 6 to about 9 mg, and most preferably, about 7.5 mg).
- the amount of L-methionine applied to the pharmaceutical composition is 1/5 of the amount of L-methionine applied to the 5 mL volume of the aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6 mL, about 0.5 mL, about 0.4 mL, and about 0.2 mL, respectively, the amounts are about 2/5, about 4/25, about 3/25, about 1/10, about 2/25, and about 1/25 of the amount of L-methionine applied to the 5 mL volume of the aqueous pharmaceutical composition.
- the antioxidants in the aqueous pharmaceutical composition of the present invention also include those that have antioxidant activity against the anti-PD-1/CD3 bispecific antibody equivalent to that of the L-methionine.
- the aqueous pharmaceutical composition of the present invention contains a surfactant primarily to inhibit aggregation of the anti-PD-1/CD3 bispecific antibody. Any surfactant capable of inhibiting aggregation of the anti-PD-1/CD3 bispecific antibody is acceptable, but polysorbate 80 is preferred.
- the surfactant may be any concentration between about 0.01 and about 0.5% (w/v) (e.g., about 0.01% (w/v), about 0.02% (w/v), about 0.04% (w/v), about 0.05% (w/v), about 0.06% (w/v), about 0.08% (w/v), about 0.1% (w/v), about 0.12% (w/v), about 0.18% (w/v), about 0.19% (w/v), about 0.21% (w/v), about 0.26% (w/v), about 0.28% (w/v), about 0.29% (w/v), about 0.31% (w/v), about 0.32% (w/v), about 0.33% (w/v), about 0.34% (w/v), about 0.35% (w/v), about 0.36% (w/v), about 0.37% (w/v), about 0.38% (w/v), about 0.39% (w/v), about 0.41% (w/v), about 0.42% (w/v), about 0.46%
- the surfactant in the aqueous pharmaceutical composition of the present invention is polysorbate 80, and the polysorbate 80 applied to an aqueous pharmaceutical composition having a volume per unit preparation of, for example, 5 mL (preferably for intravenous administration) is any amount between about 0.5 and about 25 mg (preferably, about 2.5 to about 15 mg, more preferably, about 4 to about 6 mg, and even more preferably, about 5 mg), while the polysorbate 80 applied to an aqueous pharmaceutical composition having a volume per unit preparation of, for example, 1 mL (preferably for subcutaneous administration) is any amount between about 0.5 and about 25 mg (preferably, about 2.5 to about 15 mg, more preferably, about 4 to about 6 mg, and even more preferably, about 5 mg).
- the amount of polysorbate 80 applied to the 5 mL aqueous pharmaceutical composition is 1/5 of the amount of polysorbate 80 applied to the 5 mL aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6 mL, about 0.5 mL, about 0.4 mL, and about 0.2 mL, respectively, the amounts of polysorbate 80 applied to the 5 mL aqueous pharmaceutical composition are about 2/5, about 4/25, about 3/25, about 1/10, about 2/25, and about 1/25, respectively.
- Surfactants in the aqueous pharmaceutical composition of the present invention include those that have the same ability to inhibit aggregation of anti-PD-1/CD3 bispecific antibodies as polysorbate 80.
- the aqueous pharmaceutical composition of the present invention contains a salt for stabilization (inhibition of aggregation).
- a salt for stabilization is acceptable, but preferably sodium chloride is used, and the concentration may be any concentration between about 0.5 and about 34.2 mM (e.g., about 0.5 mM, about 1 mM, about 2 mM, about 3 mM, about 4 mM, about 5 mM, about 6 mM, about 7 mM, about 8 mM, about 9 mM, about 10 mM, about 11 mM, about 12 mM, about 13 mM, about 14 mM, about 15 mM, about 16 mM, about 17 mM, about 18 mM, etc.).
- the aqueous pharmaceutical composition of the present invention preferably has a conductivity of about 2.6 to about 3.1 mS/cm (e.g., about 2.6 mS/cm, about 2.7 mS/cm, about 2.8 mS/cm, about 2.9 mS/cm, about 3.0 mS/cm or about 3.1 mS/cm), and more preferably has a conductivity of about 2.8 mS/cm.
- the amount of sodium chloride applied to an aqueous pharmaceutical composition having a volume per unit dosage form of, for example, 5 mL is any amount between about 0.15 and about 10.0 mg (preferably, about 0.29 to about 7.5 mg, more preferably, about 5.8 to about 7.5 mg, and even more preferably, about 6.8 mg), and when the salt in the aqueous pharmaceutical composition of the present invention is used in an aqueous pharmaceutical composition having a volume per unit dosage form of, for example, 1 mL (preferably, for intravenous administration), the amount of sodium chloride applied to the aqueous pharmaceutical composition is any amount between about 0.15 and about 10.0 mg (preferably, about 0.29 to about 7.5 mg, more preferably, about 5.8 to about 7.5 mg, and even more preferably, about 6.8 mg).
- the amount of sodium chloride applied to the 5 mL aqueous pharmaceutical composition is 1/5 of the amount applied to the 5 mL aqueous pharmaceutical composition, and when the volumes are, for example, about 2 mL, about 0.8 mL, about 0.6 mL, about 0.5 mL, about 0.4 mL, and about 0.2 mL, respectively, the amount of sodium chloride applied to the 5 mL aqueous pharmaceutical composition is about 2/5, about 4/25, about 3/25, about 1/10, about 2/25, and about 1/25, respectively.
- the salts in the aqueous pharmaceutical composition of the present invention include those that have a stabilizing ability equivalent to that of sodium chloride.
- the amount of each additive contained in the aqueous pharmaceutical composition of the present invention may be the amount of each of the aforementioned additives corresponding to a unit volume (e.g., 1 mL), and an example of such an amount may include 1.87 mg/mL sodium acetate trihydrate, 0.38 mg/mL acetic acid, 69 mg/mL trehalose dihydrate, 1.5 mg/mL L-methionine, 1.0 mg/mL polysorbate 80, and 1.36 mg/mL sodium chloride.
- aqueous pharmaceutical composition of the present invention is, for example, (A) (a) about 0.1 to about 1.2 mg/mL or about 5 to about 25 mg/mL (preferably about 1 mg/mL or about 10 mg/mL, respectively) of an anti-PD-1/CD3 bispecific antibody (preferably antibody ⁇ ) as an active ingredient, and further comprising: (b) a buffering agent (preferably, an acetate buffer of about 7.3 to about 73.0 mM (preferably, about 14.6 to about 43.8 mM, more preferably, about 18 to about 22 mM, even more preferably, about 20 mM, more specifically, about 20.07 mM) or a buffering agent having a pH buffering capacity equivalent to that of the acetate buffering agent); (c) about 5 to about 286.1 mM (preferably, about 10 to about 207.9 mM, more preferably, about 172 to about 193 mM, even more preferably, about 182 mM, and more specifically, about 18
- one embodiment of the aqueous pharmaceutical composition for subcutaneous administration is, for example, (D) (a) about 80 to about 150 mg/mL (preferably, about 100 mg/mL) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising: (b) a buffering agent (preferably, an acetate buffer of about 7.3 to about 73.0 mM (preferably, about 14.6 to about 43.8 mM, more preferably, about 18 to about 22 mM, even more preferably, about 20 mM, more specifically, about 20.07 mM) or a buffering agent having a pH buffering capacity equivalent to that of the acetate buffering agent); (c) about 5 to about 286.1 mM (preferably, about 10 to about 207.9 mM, more preferably, about 172 to about 193 mM, even more preferably, about 182 mM, and more specifically, about 18
- a buffering agent preferably, an acetate buffer of about
- aqueous pharmaceutical composition of the present invention is, for example, (A1)(a) about 0.1 to about 1.2 mg/mL or about 5 to about 25 mg/mL (preferably about 0.5 mg/mL, about 1 mg/mL, or about 10 mg/mL, respectively) of an anti-PD-1/CD3 bispecific antibody (preferably antibody ⁇ ) as an active ingredient, and further comprising: (b) about 7.3 to about 73.0 mM acetate buffer; (c) about 5 to about 286.1 mM trehalose; (d) about 5 to about 30.2 mM L-methionine; (e) about 0.01 to about 0.5% (w/v) polysorbate 80, and (f) about 0.5 to about 34.2 mM sodium chloride, and a pH of about 4.7 to about 5.3, preferably (B1)(a) about 0.1 to about 1.2 mg/mL or about 5 to about 25 mg/mL (preferably about 0.5 mg/mL, about 1 mg/m
- another embodiment of the aqueous pharmaceutical composition for subcutaneous administration is, for example, (D1)(a) about 80 to about 150 mg/mL (preferably, about 100 mg/mL) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising: (b) about 7.3 to about 73.0 mM acetate buffer; (c) about 5 to about 286.1 mM trehalose; (d) about 5 to about 30.2 mM L-methionine; (e) about 0.01 to about 0.5% (w/v) polysorbate 80, and (f) about 0.5 to about 34.2 mM sodium chloride, and a pH of about 4.7 to about 5.3, preferably (E1)(a) about 80 to about 150 mg/mL (preferably, about 100 mg/mL) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising:
- aqueous pharmaceutical composition of the present invention for example, when the volume per unit preparation is about 5 mL, (A2)(a) about 0.5 to about 6 mg or about 25 to about 125 mg (preferably about 2.5 mg, about 5 mg, or about 50 mg, respectively) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising: (b) an acetate buffer comprising (b1) about 3.4 to about 34.0 mg of sodium acetate trihydrate, and (b2) glacial acetic acid in an amount corresponding to the amount of sodium acetate trihydrate in (b1) above, such that the pH is about 4.7 to about 5.3; (c) about 9.46 to about 541.2 mg of trehalose dihydrate; (d) about 3.75 to about 22.5 mg L-methionine; (e) about 0.5 to about 25.0 mg of polysorbate 80; and (f) about 0.15 to about 10.0 mg of sodium chloride, and a pH of about 4.7 to about
- the volume per unit formulation may be selected according to the dosage per administration.
- the volume per unit formulation may be selected from about 1 mL, about 2 mL, about 3 mL, about 4 mL, about 6 mL, about 8 mL, about 10 mL, and about 20 mL.
- the amounts of the anti-PD-1/CD3 bispecific antibody and each additive are about 1/5, about 2/5, about 3/5, about 4/5, about 6/5, about 8/5, about 2 times, and about 4 times the amounts of the respective amounts when the volume per unit formulation described in the preceding paragraphs (A2) to (C2) is about 5 mL.
- (D2) (a) about 0.1 to about 1.2 mg/mL or about 5 to about 25 mg/mL (preferably about 0.5 mg/mL, about 1 mg/mL, or about 10 mg/mL, respectively) of an anti-PD-1/CD3 bispecific antibody (preferably antibody ⁇ ) as an active ingredient, and further comprising: (b) an acetate buffer comprising (b1) about 0.68 to about 6.8 mg/mL of sodium acetate trihydrate, and (b2) an amount of acetic acid that provides a pH of about 4.7 to about 5.3, corresponding to the amount of sodium acetate trihydrate in (b1); (c) about 1.9 to about 108.3 mg/mL of trehalose dihydrate; (d) about 0.75 to about 4.5 mg/mL L-methionine; (e) about 0.1 to about 5.0 mg/mL of polysorbate 80, and (f) about 0.03 to about 2.0 mg/mL of an anti-PD-1/CD3 bispecific antibody (preferably antibody ⁇ ) as
- another embodiment of the aqueous pharmaceutical composition for subcutaneous administration is, for example, when the volume per unit preparation is about 1 mL, (A3)(a) about 80 to about 150 mg (preferably, about 100 mg) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising: (b) an acetate buffer comprising (b1) about 0.68 to about 6.8 mg of sodium acetate trihydrate, and (b2) glacial acetic acid in an amount corresponding to the amount of sodium acetate trihydrate in (b1) above, such that the pH is about 4.7 to about 5.3; (c) about 1.9 to about 108.3 mg of trehalose dihydrate; (d) about 0.75 to about 4.5 mg of L-methionine; (e) about 0.1 to about 5.0 mg of polysorbate 80; and (f) about 0.03 to about 2.0 mg of sodium chloride, and a pH of about 4.7 to about
- the volume per unit formulation may be selected from, for example, about 0.1 mL, about 0.2 mL, about 0.4 mL, about 0.5 mL, about 0.6 mL, about 0.8 mL, and about 1.2 mL.
- the amounts of the anti-PD-1/CD3 bispecific antibody and each additive are about 1/10, about 1/5, about 2/5, about 1/2, about 3/5, about 4/5, and about 6/5 times the amount of each additive when the volume per unit formulation described in the preceding paragraphs (A3) to (C3) is about 1 mL.
- still another embodiment of the aqueous pharmaceutical composition for subcutaneous administration is, for example, (D3)(a) about 80 to about 150 mg/mL (preferably, about 100 mg/mL) of an anti-PD-1/CD3 bispecific antibody (preferably, antibody ⁇ ) as an active ingredient, and further comprising: (b) an acetate buffer comprising (b1) about 0.68 to about 6.8 mg/mL of sodium acetate trihydrate, and (b2) an amount of acetic acid that provides a pH of about 4.7 to about 5.3, corresponding to the amount of sodium acetate trihydrate in (b1); (c) about 1.9 to about 108.3 mg/mL of trehalose dihydrate; (d) about 0.75 to about 4.5 mg/mL L-methionine; (e) about 0.1 to about 5.0 mg/mL of polysorbate 80, and (f) about 0.03 to about 2.0 mg/mL of sodium chloride, and a pH of about
- the aqueous pharmaceutical composition of the present invention includes an aqueous pharmaceutical composition for intravenous or subcutaneous administration that has a bioavailability equivalent to any of the pharmaceutical compositions listed in (A)-(F), (A1)-(F1), (A2)-(F2), and (A3)-(F3) above in a bioequivalence test and contains the anti-PD-1/CD3 bispecific antibody of the present invention (preferably antibody ⁇ ) as an active ingredient.
- the pharmaceutical composition includes those that contain the same additives as any of the pharmaceutical compositions listed in (A)-(F), (A1)-(F1), (A2)-(F2), and (A3)-(F3) above, but have a different composition ratio or one or more additives that are different.
- a pharmaceutical composition containing at least trehalose, L-methionine, polysorbate 80, and sodium chloride there are (i) a pharmaceutical composition containing at least trehalose, L-methionine, polysorbate 80, and sodium chloride, (ii) a pharmaceutical composition containing at least acetate buffer, trehalose, polysorbate 80, and sodium chloride, and (iii) a pharmaceutical composition containing at least trehalose, polysorbate 80, and sodium chloride.
- bioequivalence can be determined by bioequivalence testing as described in the "Guideline for Bioequivalence Testing of Generic Drugs" established by the Ministry of Health, Labour and Welfare in Notification No. 1124005 of the Pharmaceutical and Food Safety Bureau dated November 24, 2006, or in subsequent revisions.
- the crossover method is used as a general rule, and when blood is used as a sample, the AUCt (AUC (area under the blood concentration-time curve) up to the final sampling time t) and Cmax (maximum blood concentration) in a single-dose test are used as bioequivalence determination parameters.
- the test method is appropriately selected, and the bioequivalence parameters of the product to be tested and the reference product are statistically processed. If the parameters are within the specified range, the test product can be determined to be biologically equivalent to the reference product.
- the acceptable range of bioequivalence is 0.80 to 1.25 when expressed as the ratio of the population means of the parameters of the test product and the reference product when AUC and Cmax are log-normally distributed, and the test product and the reference product are determined to be biologically equivalent when the 90% confidence interval of the difference in the mean logarithmic values of the bioequivalence parameters of the test product and the reference product is within the range of log(0.80) to log(1.25).
- the two products are determined to be bioequivalent. However, this regulation applies only when a total of 20 subjects (10 subjects per group) or more are used.
- the formulations consisting of the pharmaceutical compositions listed above in (A) to (F), (A1) to (F1), (A2) to (F2), and (A3) to (F3) respectively correspond to the "reference formulation.”
- the aqueous pharmaceutical composition of the present invention is preferably sterilized before production.
- the composition can be produced, for example, by diluting or dissolving a weighed amount of the antibody of the present invention and each additive in water for injection (e.g., sterilized purified water and distilled water for injection) and sterilizing the solution by filtration.
- the pharmaceutical composition also includes one prepared by reconstituting a lyophilized product of the antibody of the present invention and each additive with water for injection.
- the formulation of the present invention can take a suitable form for either intravenous or subcutaneous administration.
- intravenous administration it can be enclosed in a vial or ampule container, the size of which can be selected according to the total liquid volume of the aqueous pharmaceutical composition. Even if it is enclosed in a vial or ampule container, it may be collected in a syringe in the medical field and then administered subcutaneously.
- the pharmaceutical composition can be provided as a prefilled syringe formulation in which the pharmaceutical composition is prefilled in a cleaned and sterilized syringe.
- the pharmaceutical composition of the present invention can also be provided in a form in which the pharmaceutical composition is filled and sealed in a cleaned and sterilized cartridge, and the cartridge is further filled in an injector pen, an autoinjector, or a needleless device (e.g., MediJector and BioJector (registered trademark)).
- aqueous pharmaceutical composition of the present invention relating to an anti-PD-1/CD3 bispecific antibody or a formulation thereof is useful for preventing, inhibiting the progression of symptoms, inhibiting recurrence, and/or treating autoimmune diseases, graft-versus-host disease (GVHD), or blood cancer.
- GVHD graft-versus-host disease
- autoimmune diseases that can be prevented, inhibited in progression of symptoms, inhibited in recurrence, and/or treated with the aqueous pharmaceutical composition of the present invention or a formulation thereof include Behcet's disease, systemic lupus erythematosus, chronic discoid lupus erythematosus, multiple sclerosis (systemic sclerosis, progressive systemic sclerosis), scleroderma, polymyositis, dermatomyositis, periarteritis nodosa (polyarteritis nodosa, microscopic polyangiitis), aortitis syndrome (Takayasu's arteritis), malignant rheumatoid arthritis, rheumatoid arthritis, juvenile idiopathic arthritis, spondyloarthritis, mixed connective tissue disease, Sjogren's syndrome, Adult Still's disease, vasculitis, allergic granulomatous vascu
- ulcerative colitis Crohn's disease
- celiac disease ankylosing spondylitis
- severe asthma chronic urticaria
- transplant immunity familial Mediterranean fever
- eosinophilic sinusitis eosinophilic sinusitis
- dilated cardiomyopathy eosinophilic sinusitis
- inclusion body myositis eosinophilic sinusitis
- autoimmune diseases that can be prevented, inhibited in progression of symptoms, inhibited in recurrence, and/or treated with the aqueous pharmaceutical composition of the present invention or a formulation thereof include psoriasis, psoriatic arthritis, rheumatoid arthritis, Crohn's disease, ulcerative colitis, and myasthenia gravis.
- examples of blood cancers that can be prevented, inhibited in progression of symptoms, inhibited in recurrence, and/or treated with the aqueous pharmaceutical composition of the present invention or a formulation thereof include multiple myeloma, malignant lymphoma (e.g., non-Hodgkin's lymphoma (e.g., B-cell non-Hodgkin's lymphoma and T/NK-cell non-Hodgkin's lymphoma (e.g., precursor T-cell lymphoblastic lymphoma, chronic T-lymphocytic leukemia, T-cell large granular lymphocytic leukemia, large granular NK cell leukemia, rapidly progressive NK cell leukemia, peripheral T-cell lymphoma, peripheral T-cell lymphoma, non-specific type) , unclassifiable peripheral T-cell lymphoma, angioimmunoblastic T-cell lymphoma, anaplastic large cell (CD30 positive)
- Preferred examples of blood cancers that can be prevented, inhibited from progressing, inhibited from recurrence, and/or treated with the aqueous pharmaceutical composition of the present invention or a formulation thereof include peripheral T-cell lymphoma and cutaneous T-cell lymphoma.
- treatment means, for example, curing or improving a certain disease or its symptoms
- prevention means preventing the onset of a certain disease or symptom before it occurs or delaying it for a certain period of time
- suppression of progression of symptoms means suppressing the progression or worsening of symptoms to stop the progression of the pathology.
- prevention also includes suppression of recurrence.
- suppression of recurrence means preventing the recurrence of a certain disease or symptom or reducing the possibility of recurrence.
- the present invention provides, for example, the following embodiments [1] to [95], [1-1] to [1-3], and [2-1] to [2-3].
- An aqueous pharmaceutical composition or a formulation thereof for subcutaneous or intravenous administration comprising an antibody (preferably, an anti-PD-1/CD3 bispecific antibody) as an active ingredient, and further comprising a buffer (preferably, an acetate buffer), trehalose, an antioxidant (preferably, L-methionine), polysorbate 80, and sodium chloride;
- the anti-PD-1/CD3 bispecific antibody is about 0.01 mg/mL, about 0.
- [1-1] A prefilled syringe or cartridge prefilled with the aqueous pharmaceutical composition for subcutaneous administration according to any one of the preceding items [1] to [91], [93] and [95]; [1-2] An injector pen, an autoinjector, or a needleless device loaded with the cartridge according to the preceding item [1-1]; [1-3] A vial or ampoule container containing the aqueous pharmaceutical composition (preferably for intravenous administration) according to any one of the above items [1] to [95];
- KD diffusion interaction parameter
- Tagg aggregation onset temperature
- solubility was measured for each of the 30 samples. Accelerated and storage tests were also conducted, and the concentration and turbidity of each were measured, as well as the amount of remaining monomer by SEC-HPLC.
- the diffusion interaction parameters were measured and analyzed at 25°C for each antibody concentration (1.25, 2.5, 5.0, 10 and 15 mg/mL) of each sample, and calculated by plotting the diffusion coefficient ( ⁇ m 2 /s) for each formulation.
- the aggregation onset temperature was measured and analyzed by heating each sample from 25 to 80°C for an antibody concentration of 1 mg/mL. By plotting the particle size (nm) for each formulation, the value at which the average particle size increased by 10% from the average particle size at 25°C was determined as the aggregation onset temperature.
- a plate reader (DynaPro PlateReaderII (WYATT Technology)) and analysis software (Dynamics V7) were used to measure the diffusion interaction parameters and aggregation onset temperature.
- each 2 mg/mL sample was reacted with a solvent containing 10 ammonium sulfate concentrations ranging from 1.1 M to 2.0 M in 0.1 M intervals, and then centrifuged at 15,000 g for 30 minutes. The absorbance of the supernatant after centrifugation was measured at 280 nm. The solubility (%) of each sample was plotted against the ammonium sulfate concentration, and the ammonium sulfate concentration at 50% solubility was compared.
- a plate reader IFINITE 200 PRO M PLEX (TECAN) was used to measure absorbance.
- the samples were stored at 4°C and 20°C for 0 and 4 weeks, respectively.
- the stored samples were evaluated for concentration, turbidity, and SEC-HPLC analysis.
- the samples used for measurement were the supernatants obtained after centrifugation at 15,000 g for 30 minutes.
- the concentration was calculated based on the absorbance at 280 nm (path length 2 mm).
- the turbidity was calculated based on the absorbance at 500 nm (path length 1 cm).
- SEC-HPLC was performed using an Alliance HPLC system (Waters) under the conditions shown in Table 3.
- the accelerated test consisted of shaking at 500 rpm for three days at 20°C. Evaluation was carried out in the same manner as the storage test.
- sample 4 (20 mM acetate buffer (pH 5.0)
- sample 5 (20 mM acetate buffer (pH 5.0)
- sample 22 (20 mM histidine buffer (pH 6.0)
- 20 mM shown as the acetate buffer concentration is an abbreviated notation, and the correct value is 20.07 mM.
- Tables 8 to 11 Tables 27, Tables 33, Tables 38, Tables 45 and 51, as well as the corresponding notations in Examples 1 to 10.
- Example 2 Evaluation of additives (1) To each of the formulations of sample numbers 4, 5 and 22 selected in the evaluation of Example 1, sugars (sucrose, trehalose, D-mannitol and sorbitol) were added to prepare 24 types of formulations S1 to S24 as shown in Table 8.
- sugars sucrose, trehalose, D-mannitol and sorbitol
- the diffusion interaction parameter (KD) and aggregation onset temperature (Tagg) were measured for 24 samples.
- the diffusion interaction parameters were measured and analyzed at 25°C for each antibody concentration (1, 2, 4, 8, and 10 mg/mL) of each sample, and calculated by plotting the diffusion coefficient ( ⁇ m 2 /s) for each formulation.
- the aggregation onset temperature was measured and analyzed by heating each sample from 25 to 80°C for an antibody concentration of 1 mg/mL.
- the particle size (nm) was plotted for each formulation, and the value at which the average particle size increased by 10% from the average particle size at 25°C was determined as the aggregation onset temperature.
- the KD values calculated for each sample are shown in Table 9 in descending order.
- the aggregation onset temperature (Tagg) measured for each sample is shown in Table 10 in descending order.
- samples S3 (20 mM acetate buffer (pH 5.0), 285 mM trehalose), S5 (20 mM acetate buffer (pH 5.0), 285 mM D-mannitol) and S23 (20 mM histidine buffer (pH 6.0), 285 mM sorbitol) were selected as formulations to proceed to the next evaluation. Note that, because acetate buffers containing sodium chloride all had low scores, formulations that did not contain sodium chloride were selected.
- Example 3 Evaluation of additives (2) Fifteen types of samples, E1 to E15, were prepared by adding polysorbate (PS) (polysorbate 80 (PS80) and polysorbate 20 (PD20)) to the formulations of samples S3, S5, and S23 selected in the evaluation of Example 2, as shown in Table 11.
- PS polysorbate
- PD20 polysorbate 20
- the 15 samples (1 mg/mL) were sterilized using a 0.22 ⁇ m filter and dispensed into vials, after which storage tests, accelerated tests, and freeze-thaw tests were performed on each.
- the samples were stored at 20°C and 40°C for four weeks. The stored samples were visually evaluated and evaluated for concentration, turbidity and SEC-HPLC analysis.
- the samples used for evaluation were the supernatants obtained by centrifugation at 15,000 g for 30 minutes. The concentration was calculated based on the absorbance at 280 nm (path length 2 mm). The turbidity was calculated based on the absorbance at 500 nm (path length 1 cm).
- the SEC-HPLC analysis was carried out under the conditions shown in Table 2.
- the accelerated test consisted of shaking at 500 rpm for three days at 20°C.
- the freeze-thaw test consisted of freezing (-80°C for 10 minutes, then 25°C for 10 minutes) repeated three times.
- the results of the storage test are shown in Table 12, the results of the storage test (at 40°C for 4 weeks) are shown in Table 13, the results of the accelerated test (shaking at 20°C for 3 days) are shown in Table 14, and the results of the freeze-thaw test (at -80°C for 10 minutes, at 25°C for 10 minutes) are shown in Table 15.
- Samples containing 100 mg/mL antibody ⁇ were prepared for each of the acetate formulations (20 mM acetate buffer (pH 5.0), 285 mM trehalose, 0.1% PS80) and histidine formulations (20 mM histidine buffer (pH 6.0), 0.1% PS80) selected for evaluation in Example 3.
- the samples were sterilized using a 0.22 ⁇ m filter and dispensed in 1 mL portions into sterilized vials. After subjecting each sample to the conditions shown in Table 16, an evaluation test was conducted on the stability of each sample.
- the samples subjected to each of the conditions shown in the above table were evaluated by (a) pH measurement, (b) antibody concentration measurement, (c) turbidity measurement, (d) SEC-HPLC (size exclusion chromatography) analysis, (e) SDS-PAGE analysis, (f) IE-HPLC (ion exchange chromatography) analysis, (g) viscosity measurement, (h) FIA (flow imaging analysis), and (i) sliding resistance measurement.
- a pH meter LAQUA F-74 (HORIBA)
- a microsample measurement electrode Micro Tou pH electrode (HORIBA)
- the turbidity of the acetic acid formulation sample increased by 20% or more after storage at 40°C
- the histidine formulation sample increased by 20% or more after storage at 25°C and 40°C.
- the turbidity of the acetic acid formulation sample increased by about 50% after 12 weeks of storage at 40°C.
- the turbidity of the histidine formulation sample increased by 90% or more after storage at 5°C, 25°C, and 40°C, compared to the initial sample.
- the histidine formulation sample After storage at 25°C for four weeks, the histidine formulation sample showed a 3% increase in acidic molecular species and a 3% decrease in neutral molecular species.
- the acetic acid formulation sample After storage at 40°C for four weeks, the acetic acid formulation sample showed a 15% increase in acidic molecular species, a 19% decrease in neutral molecular species, and a 5% increase in basic molecular species.
- the histidine formulation sample also showed a 15% increase in acidic molecular species and a 15% decrease in neutral molecular species.
- the acetic acid formulation sample After 12 weeks of storage at 25°C, the acetic acid formulation sample showed a 9% increase in acidic molecular species and a 9% decrease in neutral molecular species.
- the histidine formulation sample also showed an 8% increase in acidic molecular species and an 8% decrease in neutral molecular species.
- the acetic acid formulation sample After 12 weeks of storage at 40°C, the acetic acid formulation sample showed a 33% increase in acidic molecular species, a 37% decrease in neutral molecular species, and a 4% increase in basic molecular species.
- the histidine formulation sample also showed a 32% increase in acidic molecular species and a 32% decrease in neutral molecular species.
- the initial viscosity of the samples was 4.41 mPa ⁇ s for the acetic acid formulation and 4.2 mPa ⁇ s for the histidine formulation, both of which were very low even at a concentration of 100 mg/mL.
- the sliding resistance values were very low (7.66 N and 7.46 N) for both the acetic acid formulation and the histidine formulation.
- Table 21 shows the particle concentrations obtained by subtracting the particle concentration of the formulation solution alone from the particle concentration of each antibody-containing formulation before and after the stability evaluation test.
- the acetic acid formulation sample had the highest particle concentration when stored for 12 weeks at 40°C, while the histidine formulation sample had the highest particle concentration when stored for 12 weeks at 25°C.
- particle concentrations in the histidine formulation were tens to thousands of times higher than in the acetic acid formulation.
- Example 5 Evaluation of additives (3) The acetic acid formulation (20 mM acetate buffer (pH 5.0), 285 mM trehalose, 0.1% PS80) selected based on the evaluation in Example 4 was further subjected to optimization studies.
- the salt concentration was examined to ensure stability of the conductivity.
- N1 to N8 Eight types of samples, N1 to N8, were prepared by adding sodium chloride to the selected acetic acid formulation (20 mM acetate buffer (pH 5.0), 285 mM trehalose, 0.1% PS80) as shown in Table 22.
- the eight samples were sterilized using a 0.22 ⁇ m filter and dispensed into sterilized vials at 1 or 5 mL each, and stability evaluation was performed under the conditions shown in Table 23.
- the color index was calculated as (fluorescence intensity value) / (UV280 nm absorption area value) x 100.
- the ratio of the color index to the standard ((color index of sample) / (color index of standard)) was used as an index value showing the amount of colored bodies in the sample.
- Binding activity to PD-1 and CD3 was measured by surface plasmon resonance (SPR).
- SPR surface plasmon resonance
- a 500 ⁇ L sample was measured at a flow rate of 0.1 mL/min using a flow imaging device, and aggregates with a size of 2 ⁇ m or more were evaluated.
- Peptide map analysis was performed using LC-MS.
- Storage test (a) The percentage of high molecular weight species (HMWS) before and after the storage test, evaluated by SEC-HPLC analysis, is shown in Table 27. At any concentration, no significant increase in HMWS was observed after 12 weeks of storage at 5°C.
- HMWS At 25°C, no increase in HMWS was observed in the formulation with an antibody concentration of 20 mg/mL, but a linear increase in HMWS was observed in the formulation with an antibody concentration of 100 mg/mL. On the other hand, no difference in behavior due to conductivity was observed. At 40°C, HMWS increased at any concentration, but in the formulation with 20 mg/mL, HMWS tended to increase with increasing conductivity. It was confirmed that the formulation was stable at 5°C and 25°C.
- Example 6 Stability evaluation (2) Since Example 5 suggested that the antibody ⁇ of the present invention may be less stable at low concentrations, the stability in the low concentration range was evaluated.
- Samples L1 and L2 containing 1 and 50 mg/mL of antibody ⁇ , respectively, in an acetic acid formulation (20 mM acetate buffer (pH 5.0), 285 mM trehalose, 0.1% PS80, 1.25 mg/mL sodium chloride) were prepared as shown in Table 33.
- the samples stored under each condition were subjected to (a) SEC-HPLC analysis, (b) IE-HPLC analysis, and (c) measurement of binding activity to PD-1 and CD3.
- the SEC-HPLC analysis was performed under the conditions shown in Table 24, and the IE-HPLC analysis was performed under the conditions shown in Table 25.
- the binding activity to PD-1 and CD3 was measured by SPR.
- (a) The percentage of HMWS before and after storage, as evaluated by SEC-HPLC, is shown in Table 35. In all formulations, HMWS increased significantly after long-term storage at 40°C, but accumulation of HMWS was more significant in L1, which had a low antibody concentration. This suggests that the possibility of aggregation increases under low concentration conditions.
- Example 7 Evaluation of additives (4)
- Example 6 suggests that long-term storage causes the oxidation of the antibody ⁇ of the present invention, resulting in a significant decrease in binding activity to PD-1 and CD3, in particular, in binding activity to PD-1.
- antioxidants L-methionine or DTPA
- acetic acid formulation (20 mM acetate buffer (pH 5.0), 193 mM trehalose, 0.1% PS80, 1.15 mg/mL sodium chloride).
- each of the eight samples was sterilized using a 0.22 ⁇ m filter and dispensed into sterilized vials at 1 mL each, and stability was evaluated in the same manner as in Table 34.
- the samples stored under the conditions shown in Table 34 were subjected to (a) SEC-HPLC analysis, (b) IE-HPLC analysis, (c) RP-HPLC analysis, (d) measurement of binding activity to PD-1 and CD3, and (e) peptide map analysis.
- the SEC-HPLC analysis was performed under the conditions shown in Table 24, the IE-HPLC analysis was performed under the conditions shown in Table 25, and the RP-HPLC analysis was performed under the conditions shown in Table 26.
- Table 42 a peptide map was used to analyze the causes of the decrease in PD-1 binding activity during long-term storage at 40°C.
- Table 44 shows the degree of oxidation (%) of the methionine residue (Met101) present in the complementarity determining region for PD-1. It was confirmed that Met101 was highly oxidized when the antibody was stored at a concentration of 1 mg/mL at 40°C.
- the addition of an antioxidant dramatically suppressed the oxidation of Met101, and furthermore, dramatically suppressed the decrease in binding activity to PD-1. It was therefore confirmed that the addition of an antioxidant is extremely useful for improving the stability of antibody ⁇ .
- Example 8 Stability test (3) In the studies of Examples 1 to 7, conditions were found under which the antibody ⁇ of the present invention can be stably stored. In order to set more stable conditions, the concentration range of sugar was determined.
- Example 9 Stability test (4) In the studies of Examples 1 to 8, conditions were found under which the antibody ⁇ of the present invention can be stably stored. Taking these conditions into consideration, a stability test was carried out under conditions that were deemed optimal.
- Samples F1 and F2 containing 1 mg/mL or 100 mg/mL of antibody ⁇ , respectively, in an acetate formulation (20 mM acetate buffer (pH 5.0), 182.38 mM trehalose, 0.1% PS80, 1.36 mg/mL sodium chloride, 10 mM L-methionine) were prepared as shown in Table 51.
- the samples stored under each condition were subjected to (a) SEC-HPLC, (b) IE-HPLC analysis, (c) isoelectric focusing (iCIEF), (d) RP-HPLC, and (e) measurement of binding activity to PD-1 and CD3.
- SEC-HPLC analysis was performed under the conditions shown in Table 24.
- IE-HPLC analysis was performed under the conditions shown in Table 25.
- iCIEF was performed using a capillary isoelectric focusing apparatus.
- RP-HPLC was performed under the conditions shown in Table 26.
- Measurement of binding activity to PD-1 and CD3 was performed by SPR.
- (a) The percentage of HMWS before and after storage evaluated by SEC-HPLC is shown in Table 53. At a concentration of 100 mg/mL, an increase in HMWS was observed at 25°C and 40°C, but no significant increase was observed at 5°C. It was considered stable at 5°C.
- samples L1 to L4 were prepared as shown in Table 59, containing 0.01 mg/mL to 1.0 mg/mL of antibody ⁇ in an acetate formulation (20 mM acetate buffer (pH 5.0), 182.38 mM trehalose, 0.1% PS80, 1.36 mg/mL sodium chloride, 10 mM L-methionine).
- Example 10 Formulation preparation method A method for preparing the aqueous pharmaceutical composition of the present invention is described below using sample F2 of Example 9 as an example. Note that aqueous pharmaceutical compositions of the present invention having different antibody concentrations from those of sample F2 can also be prepared in the same manner.
- (a) Prepare formulation preparation 1 to contain 20 mM acetate buffer (pH 5.0), 182.38 mM trehalose, 1.36 mg/mL sodium chloride, and 10 mM L-methionine.
- Formulation preparation 2 is prepared to contain 20 mM acetate buffer (pH 5.0), 182.38 mM trehalose, 2% PS80, 1.36 mg/mL sodium chloride, and 10 mM L-methionine.
- formulation preparation 3 to contain 20 mM acetate buffer (pH 5.0), 182.38 mM trehalose, 0.1% PS80, 1.36 mg/mL sodium chloride, and 10 mM L-methionine.
- the process solution obtained from the virus filtration step is replaced in solution composition with the formulation preparation solution 1 using an ultrafiltration/diafiltration membrane.
- the process solution containing antibody ⁇ the solution composition of which has been replaced with formulation preparation solution 1, is concentrated to approximately 140 mg/mL using an ultrafiltration/diafiltration membrane.
- formulation preparation solution 2 containing a high concentration of PS80 is added to adjust the PS80 concentration to 0.1%.
- the process solution containing antibody ⁇ adjusted to the desired PS80 concentration is added with formulation preparation solution 3 to dilute the antibody to the desired antibody concentration of 100 mg/mL.
- the pharmaceutical composition or preparation thereof of the present invention which contains an anti-PD-1/CD3 bispecific antibody as an active ingredient, is useful for preventing, inhibiting the progression of, or treating autoimmune diseases, graft-versus-host disease (GVHD), or blood cancer.
- GVHD graft-versus-host disease
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Transplantation (AREA)
- Biochemistry (AREA)
- Neurology (AREA)
- Inorganic Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pain & Pain Management (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
<1> 抗体(好ましくは、抗PD-1/CD3二重特異性抗体)を有効成分として含み、さらに、緩衝剤(好ましくは、酢酸緩衝剤)、トレハロース、酸化防止剤(好ましくは、L-メチオニン)、ポリソルベート80および塩化ナトリウムを含む、皮下投与用もしくは静脈内投与用水性医薬組成物またはその製剤、
<2> 前項<1>記載の水性医薬組成物またはその製剤と生物学的同等性を有する皮下投与用もしくは静脈内投与用水性医薬組成物またはその製剤、ならびに
<3> 前項<1>もしくは<2>記載の水性医薬組成物またはその製剤の自己免疫疾患もしくは移植片対宿主病(GVHD)あるいは血液がんの予防、症状進展抑制、再発抑制および/または治療における使用。
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖から構成される。ここで、
(a)前項(i)の重鎖は、配列番号1、配列番号2、配列番号3、配列番号4および配列番号5から選択される何れか一つのアミノ酸配列からなり、
(b)前項(ii)の重鎖は、配列番号6のアミノ酸配列からなり、ならびに
(c)前項(i)および前項(ii)の軽鎖はともに、配列番号7のアミノ酸配列からなる。
(A)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニン、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である医薬組成物、
(B)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である医薬組成物、ならびに、
(C)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である医薬組成物である。なお、これら(A)~(C)の水性医薬組成物は、静脈内投与および皮下投与の何れにおいても使用できる。
(D)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニン、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である医薬組成物、
(E)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である医薬組成物、ならびに、
(F)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20 mM、より具体的には、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182 mM、より具体的には、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10 mM、より具体的には、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、および
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23 mM、より具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは約5.0)である医薬組成物である。
(A1)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約7.3~約73.0 mMの酢酸緩衝剤、
(c)約5~約286.1 mMのトレハロース、
(d)約5~約30.2 mMのL-メチオニン、
(e)約0.01~約0.5 %(w/v)のポリソルベート80、および
(f)約0.5~約34.2 mMの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(B1)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、および
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(C1)(a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分(好ましくは、抗体α)として含み、さらに、
(b)約20.1 mM(具体的には、約20.07 mM)の酢酸緩衝剤、
(c)約182.4 mM(具体的には、約182.38 mM)のトレハロース、
(d)約10.1 mM(具体的には、約10.05 mM)のL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、および
(f)約23.3 mM(具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約5.0である医薬組成物である。なお、これら(A1)~(C1)の水性医薬組成物も、静脈内投与および皮下投与の何れにおいても使用できる。
(D1)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約7.3~約73.0 mMの酢酸緩衝剤、
(c)約5~約286.1 mMのトレハロース、
(d)約5~約30.2 mMのL-メチオニン、
(e)約0.01~約0.5 %(w/v)のポリソルベート80、および
(f)約0.5~約34.2 mMの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(E1)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、および
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(F1)(a)約100 mg/mLの抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約20.1 mM(具体的には、約20.07 mM)の酢酸緩衝剤、
(c)約182.4 mM(具体的には、約182.38 mM)のトレハロース、
(d)約10.1 mM(具体的には、約10.05 mM)のL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、および
(f)約23.3 mM(具体的には、約23.27 mM)の塩化ナトリウムを含み、pHが約5.0である医薬組成物である。
(A2)(a)約0.5~約6 mgまたは約25~約125 mg(各々好ましくは、約2.5 mg、約5 mgまたは約50 mg)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約3.4~約34.0 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約9.46~約541.2 mgのトレハロース二水和物、
(d)約3.75~約22.5 mgのL-メチオニン、
(e)約0.5~約25.0 mgのポリソルベート80、ならびに
(f)約0.15~約10.0 mgの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(B2)(a)約0.5~約6 mgまたは約25~約125 mg(各々好ましくは、約2.5 mg、約5 mgまたは約50 mg)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約6.8~約20.4 mg(好ましくは、約8.4~約10.2 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約18.9~約393.3 mg(好ましくは、約325~約365.1 mg)のトレハロース二水和物、
(d)約3.75~約15.0 mg(好ましくは、約3.75~11.25 mg)のL-メチオニン、
(e)約2.5~約15.0 mg(好ましくは、約4~6 mg)のポリソルベート80、ならびに
(f)約0.29~約7.5 mg(好ましくは、約5.8~約7.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(C2)(a)約2.5 mg、約5 mgまたは約50 mgの抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約9.4 mg(具体的には、約9.35 mg)の酢酸ナトリウム三水和物および約1.9 mgの氷酢酸からなる酢酸緩衝剤、
(c)約345 mgのトレハロース二水和物、
(d)約7.5 mgのL-メチオニン、
(e)約5.0 mgのポリソルベート80、ならびに
(f)約6.8 mgの塩化ナトリウムを含み、pHが約5.0である医薬組成物である。なお、これら(A2)~(C2)の水性医薬組成物も、静脈内投与および皮下投与の何れにおいても使用できる。
(D2)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mg/mLの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mg/mLのトレハロース二水和物、
(d)約0.75~約4.5 mg/mLのL-メチオニン、
(e)約0.1~約5.0 mg/mLのポリソルベート80、ならびに
(f)約0.03~約2.0 mg/mLの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(E2)(a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mL(好ましくは、約65.1~約72.6 mg/mL)のトレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(F2)(a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である医薬組成物である。なお、これら(D2)~(F2)の水性医薬組成物も、同様に、静脈内投与および皮下投与の何れにおいても使用できる。
(A3)(a)約80~約150 mg(好ましくは、約100 mg)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mgのトレハロース二水和物、
(d)約0.75~約4.5 mgのL-メチオニン、
(e)約0.1~約5.0 mgのポリソルベート80、ならびに
(f)約0.03~約2.0 mgの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(B3)(a)約80~約150 mg(好ましくは、約100 mg)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg(好ましくは、約1.68~約2.05 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mgの(好ましくは、約65.1~約72.6 mg)トレハロース二水和物、
(d)約0.75~約3.0 mg(好ましくは、約0.75~約2.25 mg)のL-メチオニン、
(e)約0.5~約3.0 mgのポリソルベート80、ならびに
(f)約0.06~約1.5 mg(好ましくは、約1.15~約1.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(C3)(a)約100 mgの抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約1.9 mg(具体的には、約1.87 mg)の酢酸ナトリウム三水和物および約0.4 mg(具体的には、約0.38 mg)の氷酢酸からなる酢酸緩衝剤、
(c)約69 mgのトレハロース二水和物、
(d)約1.5 mgのL-メチオニン、
(e)約1.0 mgのポリソルベート80、ならびに
(f)約1.4 mg(具体的には、約1.36 mg)の塩化ナトリウムを含み、pHが約5.0である医薬組成物である。
(D3)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mg/mLの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mg/mLのトレハロース二水和物、
(d)約0.75~約4.5 mg/mLのL-メチオニン、
(e)約0.1~約5.0 mg/mLのポリソルベート80、ならびに
(f)約0.03~約2.0 mg/mLの塩化ナトリウムを含み、pHが約4.7~約5.3である医薬組成物であり、好ましくは、
(E3)(a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mLの(好ましくは、約65.1~約72.6 mg/mL)トレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である医薬組成物であり、より好ましくは、
(F3)(a)約100 mg/mLの抗PD-1/CD3二重特異性抗体(好ましくは、抗体α)を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である医薬組成物である。
抗PD-1/CD3二重特異性抗体にかかる本発明の水性医薬組成物またはその製剤は、自己免疫疾患もしくは移植片対宿主病(GVHD)あるいは血液がんの予防、症状進展抑制、再発抑制および/または治療に有用である。
[1] 抗体(好ましくは、抗PD-1/CD3二重特異性抗体)を有効成分として含み、さらに、緩衝剤(好ましくは、酢酸緩衝剤)、トレハロース、酸化防止剤(好ましくは、L-メチオニン)、ポリソルベート80および塩化ナトリウムを含む、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[2] 抗PD-1/CD3二重特異性抗体が、約0.01~約200 mg/mL(好ましくは、約0.1~約150 mg/mL(好ましくは、約0.1~約1.2 mg/mL、約5~約25 mg/mLまたは約80~約150 mg/mL))である、前項[1]記載の水性医薬組成物またはその製剤;
[3] 抗PD-1/CD3二重特異性抗体が、約0.01 mg/mL、約0.02 mg/mL、約0.03 mg/mL、約0.04 mg/mL、約0.05 mg/mL、約0.06 mg/mL、約0.07 mg/mL、約0.08 mg/mL、約0.09 mg/mL、約0.1 mg/mL、約0.2 mg/mL、約0.3 mg/mL、約0.4 mg/mL、約0.5 mg/mL、約0.6 mg/mL、約0.7 mg/mL、約0.8 mg/mL、約0.9 mg/mL、約1 mg/mL、約1.1 mg/mL、約1.2 mg/mL、約1.3 mg/mL、約1.4 mg/mL、約1.5 mg/mL、約2 mg/mL、約5 mg/mL、約10 mg/mL、約15 mg/mL、約20 mg/mL、約25 mg/mL、約30 mg/mL、約40 mg/mL、約50 mg/mL、約60 mg/mL、約70 mg/mL、約80 mg/mL、約90 mg/mL、約100 mg/mL、約110 mg/mL、約120 mg/mL、約130 mg/mL、約140 mg/mL、約150 mg/mL、約160 mg/mL、約170 mg/mL、約180 mg/mL、約190 mg/mLまたは約200 mg/mLである、前項[1]記載の水性医薬組成物またはその製剤;
[4] 抗PD-1/CD3二重特異性抗体が、約0.5 mg/mL、約1 mg/mL、約10 mg/mLまたは約100 mg/mLである、前項[1]記載の水性医薬組成物またはその製剤;
[5] 酢酸緩衝剤が、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM)である、前項[1]~[4]の何れか一項記載の水性医薬組成物またはその製剤;
[6] 酢酸緩衝剤が、約7.3 mM、約10 mM、約14.6 mM、約15 mM、約18 mM、約20 mM、約20.07 mM、約21 mM、約22 mM、約25 mM、約30 mM、約35 mM、約40 mM、約43.8 mM、約45 mM、約50 mM、約55 mM、約60 mM、約70 mMまたは約73.0 mMである、前項[1]~[4]の何れか一項記載の水性医薬組成物またはその製剤;
[7] 酢酸緩衝剤が、約20.07 mMである、前項[1]~[4]の何れか一項記載の水性医薬組成物またはその製剤;
[8] 酢酸緩衝剤が、酢酸ナトリウム三水和物および酢酸の組み合わせからなる、前項[5]~[7]の何れか一項記載の水性医薬組成物またはその製剤;
[9] トレハロースが、約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM)である、前項[1]~[8]の何れか一項記載の水性医薬組成物またはその製剤;
[10] トレハロースが、約5 mM、約10 mM、約20 mM、約30 mM、約40 mM、約50 mM、約60 mM、約70 mM、約80 mM、約90 mM、約100 mM、約110 mM、約120 mM、約130 mM、約140 mM、約150 mM、約160 mM、約170 mM、約172 mM、約180 mM、約182 mM、約182.3 mM、約182.38 mM、約182.4 mM、約190 mM、約193 mM、約200 mM、約207.9 mM、約210 mM、約220 mM、約225 mM、約230 mM、約240 mM、約250 mM、約260 mM、約270 mM、約280 mM、約285 mM、約286 mMまたは約286.1 mMである、前項[1]~[8]の何れか一項記載の水性医薬組成物またはその製剤;
[11] トレハロースが、約182.38 mMである、前項[1]~[8]の何れか一項記載の水性医薬組成物またはその製剤;
[12] トレハロースが、トレハロース二水和物である、前項[1]~[8]の何れか一項記載の水性医薬組成物またはその製剤;
[13] L-メチオニンが、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM)である、前項[1]~[12]の何れか一項記載の水性医薬組成物またはその製剤;
[14] L-メチオニンが、約5 mM、約10 mM、約10.05 mM、約15 mM、約20 mM、約25 mM、約30 mMまたは約30.2 mMである、前項[1]~[12]の何れか一項記載の水性医薬組成物またはその製剤;
[15] L-メチオニンが、約10.05 mMである、前項[1]~[12]の何れか一項記載の水性医薬組成物またはその製剤;
[16] ポリソルベート80が、約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、さらに好ましくは、約0.08~約0.12 %(w/v))である、前項[1]~[15]の何れか一項記載の水性医薬組成物またはその製剤;
[17] ポリソルベート80が、約0.01 %(w/v)、約0.02 %(w/v)、約0.04 %(w/v)、約0.05 %(w/v)、約0.06 %(w/v)、約0.08 %(w/v)、約0.1 %(w/v)、約0.12 %(w/v)、約0.15 %(w/v)、約0.2 %(w/v)、約0.25 %(w/v)、約0.3 %(w/v)、約0.35 %(w/v)、約0.4 %(w/v)、約0.45 %(w/v)または約0.5 %(w/v)である、前項[1]~[15]の何れか一項記載の水性医薬組成物またはその製剤;
[18] ポリソルベート80が、約0.1 %(w/v)である、前項[1]~[15]の何れか一項記載の水性医薬組成物またはその製剤;
[19] 塩化ナトリウムが、約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM)である、前項[1]~[18]の何れか一項記載の水性医薬組成物またはその製剤;
[20] 塩化ナトリウムが、約0.5 mM、約1 mM、約2 mM、約3 mM、約4 mM、約5 mM、約6 mM、約7 mM、約8 mM、約9 mM、約10 mM、約11 mM、約12 mM、約13 mM、約14 mM、約15 mM、約16 mM、約17 mM、約18 mM、約19 mM、約19.6 mM、約20 mM、約21 mM、約22 mM、約23 mM、約23.27 mM、約24.0 mM、約25 mM、約25.7 mM、約30 mMまたは約34.2 mMである、前項[1]~[18]の何れか一項記載の水性医薬組成物またはその製剤;
[21] 塩化ナトリウムが、約23.27 mMである、前項[1]~[18]の何れか一項記載の水性医薬組成物またはその製剤;
[22] 水性医薬組成物(好ましくは、静脈内投与用)の単位製剤当たりの容量が約5 mLである、前項[1]~[21]の何れか一項記載の水性医薬組成物またはその製剤;
[23] 前項[22]記載の水性医薬組成物に適用される酢酸緩衝剤が、(i)約3.4~約34 mgの酢酸ナトリウム三水和物、および(ii)前項(i)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)となる分量の氷酢酸からなる、前項[22]記載の水性医薬組成物またはその製剤;
[24] 前項[22]記載の水性医薬組成物に適用される酢酸緩衝剤が、(i)約6.8~約20.4 mg(好ましくは、約8.4~約10.2 mg)の酢酸ナトリウム三水和物、および(ii)前項(i)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)となる分量の氷酢酸からなる、前項[22]記載の水性医薬組成物またはその製剤;
[25] 前項[22]記載の水性医薬組成物に適用される酢酸緩衝剤が、約9.4 mg(具体的には、約9.35 mg)の酢酸ナトリウム三水和物および約1.9 mgの氷酢酸からなる、前項[22]記載の水性医薬組成物またはその製剤;
[26] トレハロースがトレハロース二水和物である、前項[22]~[25]の何れか一項記載の水性医薬組成物またはその製剤;
[27] トレハロース二水和物が、約9.46~約541.2 mg(好ましくは、約18.9~約393.3 mg、より好ましくは、約325~約365.1 mg、さらにより好ましくは、約345 mg)である、前項[26]記載の水性医薬組成物またはその製剤;
[28] 酸化防止剤がL-メチオニンである、前項[22]~[27]の何れか一項記載の水性医薬組成物またはその製剤;
[29] L-メチオニンが、約3.75~約22.5 mg(好ましくは、約3.75~約15.0 mg、より好ましくは、約3.75~約11.25 mg、さらにより好ましくは、約7.5 mg)である、前項[28]記載の水性医薬組成物またはその製剤;
[30] ポリソルベート80が、約0.5~約25 mg(好ましくは、約2.5~約15 mg、より好ましくは、約5 mg)である、前項[22]~[29]の何れか一項記載の水性医薬組成物またはその製剤;
[31] 塩化ナトリウムが、約0.15~約10.0 mg(好ましくは、約0.29~約7.5 mg、より好ましくは、約5.8~約7.5 mg、さらにより好ましくは、約6.8 mg)である、前項[22]~[30]の何れか一項記載の水性医薬組成物またはその製剤;
[32] 水性医薬組成物の単位製剤当たりの容量が約2 mLであり、各添加物の分量が、前項[23]~[31]の何れか一または二以上に記載の各添加物の分量の約2/5量である、前項[1]~[21]の何れか一項記載の水性医薬組成物またはその製剤;
[33] 水性医薬組成物(好ましくは、皮下投与用)の単位製剤当たりの容量が約1 mLである、前項[1]~[21]の何れか一項記載の水性医薬組成物またはその製剤;
[34] 前項[33]記載の水性医薬組成物に適用される酢酸緩衝剤が、(i)約0.68~約6.8 mgの酢酸ナトリウム三水和物、および(ii)前項(i)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)となる分量の氷酢酸からなる、前項[33]記載の水性医薬組成物またはその製剤;
[35] 前項[33]記載の水性医薬組成物に適用される酢酸緩衝剤が、(i)約1.36~約4.08 mg(好ましくは、約1.68~約2.05 mg)の酢酸ナトリウム三水和物、および(ii)前項(i)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)となる分量の氷酢酸からなる、前項[33]記載の水性医薬組成物またはその製剤;
[36] 前項[33]記載の水性医薬組成物に適用される酢酸緩衝剤が、約1.9 mg(具体的には、約1.87 mg)の酢酸ナトリウム三水和物および約0.4 mg(具体的には、約0.38 mg)の氷酢酸からなる、前項[33]記載の水性医薬組成物またはその製剤;
[37] トレハロースがトレハロース二水和物である、前項[33]~[36]の何れか一項記載の水性医薬組成物またはその製剤;
[38] トレハロース二水和物が、約1.9~約108.3 mg(好ましくは、約3.8~約78.7 mg、より好ましくは、約65.1~約72.6 mg、さらにより好ましくは、約69 mg)である、前項[37]記載の水性医薬組成物またはその製剤;
[39] 酸化防止剤がL-メチオニンである、前項[33]~[38]の何れか一項記載の水性医薬組成物またはその製剤;
[40] L-メチオニンが、約0.75~約4.5 mg(好ましくは、約0.75~約3.0 mg、より好ましくは、約0.75~約2.25 mg、さらにより好ましくは、約1.5 mg)である、前項[39]記載の水性医薬組成物またはその製剤;
[41] ポリソルベート80が、約0.1~約5.0 mg(好ましくは、約0.5~約3.0 mg、より好ましくは、約1.0 mg)である、前項[33]~[40]の何れか一項記載の水性医薬組成物またはその製剤;
[42] 塩化ナトリウムが、約0.03~約2.0 mg(好ましくは、約0.06~約1.5 mg、より好ましくは、約1.15~約1.5 mg、さらにより好ましくは、約1.4 mg(具体的には、約1.36 mg))である、前項[33]~[41]の何れか一項記載の水性医薬組成物またはその製剤;
[43] 当該水性医薬組成物のpHが、約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、前項[1]~[42]の何れか一項記載の水性医薬組成物またはその製剤;
[44] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約7.3~約73.0 mMの酢酸緩衝剤、
(c)約5~約286.1 mMのトレハロース、
(d)約5~約30.2 mMのL-メチオニン、
(e)約0.01~約0.5 %(w/v)のポリソルベート80、ならびに
(f)約0.5~約34.2 mMの塩化ナトリウムを含み、pHが約4.7~約5.3である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[45] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、ならびに
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[46] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約20.07 mMの酢酸緩衝剤、
(c)約182.38 mMのトレハロース、
(d)約10.05 mMのL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、ならびに
(f)約23.27 mMの塩化ナトリウムを含み、pHが約5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[47] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約7.3~約73.0 mMの酢酸緩衝剤、
(c)約5~約286.1 mMのトレハロース、
(d)約5~約30.2 mMのL-メチオニン、
(e)約0.01~約0.5 %(w/v)のポリソルベート80、ならびに
(f)約0.5~約34.2 mMの塩化ナトリウムを含み、pHが約4.7~約5.3である、皮下投与用の水性医薬組成物またはその製剤;
[48] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、ならびに
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用の水性医薬組成物またはその製剤;
[49] (a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約20.07 mMの酢酸緩衝剤、
(c)約182.38 mMのトレハロース、
(d)約10.05 mMのL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、ならびに
(f)約23.27 mMの塩化ナトリウムを含み、pHが約5.0である、皮下投与用の水性医薬組成物またはその製剤;
[50] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mg/mLの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mg/mLのトレハロース二水和物、
(d)約0.75~約4.5 mg/mLのL-メチオニン、
(e)約0.1~約5.0 mg/mLのポリソルベート80、ならびに
(f)約0.03~約2.0 mg/mLの塩化ナトリウムを含み、pHが約4.7~約5.3である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[51] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mL(好ましくは、約65.1~約72.6 mg/mL)のトレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[52] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[53] (a)約0.5~約6 mgまたは約25~約125 mg(各々好ましくは、約2.5 mg、約5 mgまたは約50 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約3.4~約34.0 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約9.46~約541.2 mgのトレハロース二水和物、
(d)約3.75~約22.5 mgのL-メチオニン、
(e)約0.5~約25.0 mgのポリソルベート80、ならびに
(f)約0.15~約10.0 mgの塩化ナトリウムを含み、pHが約4.7~約5.3であり、単位製剤当たりの容量が約5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[54] (a)約0.5~約6 mgまたは約25~約125 mg(各々好ましくは、約2.5 mg、約5 mgまたは約50 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約6.8~約20.4 mg(好ましくは、約8.4~約10.2 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約18.9~約393.3 mg(好ましくは、約325~約365.1 mg)のトレハロース二水和物、
(d)約3.75~約15.0 mg(好ましくは、約3.75~約11.25 mg)のL-メチオニン、
(e)約2.5~約15.0 mg(好ましくは、約4~6 mg)のポリソルベート80、ならびに
(f)約0.29~約7.5 mg(好ましくは、約5.8~約7.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[55] (a)約2.5 mg、約5 mgまたは約50 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約9.4 mg(具体的には、約9.35 mg)の酢酸ナトリウム三水和物および約1.9 mgの氷酢酸からなる酢酸緩衝剤、
(c)約345 mgのトレハロース二水和物、
(d)約7.5 mgのL-メチオニン、
(e)約5.0 mgのポリソルベート80、ならびに
(f)約6.8 mgの塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[56] (a)約0.2~約2.4 mgまたは約10~約50 mg(各々好ましくは、約1 mg、約2 mgまたは約20 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約13.6 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約3.8~約216.6 mgのトレハロース二水和物、
(d)約1.5~約9.0 mgのL-メチオニン、
(e)約0.2~約10.0 mgのポリソルベート80、ならびに
(f)約0.06~約4.0 mgの塩化ナトリウムを含み、pHが約4.7~約5.3であり、単位製剤当たりの容量が約2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[57] (a)約0.2~約2.4 mgまたは約10~約50 mg(各々好ましくは、約1 mg、約2 mgまたは約20 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約2.72~約8.16 mg(好ましくは、約3.36~約4.1 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約7.6~約157.4 mg(好ましくは、約130.2~約145.2 mg)のトレハロース二水和物、
(d)約1.5~約6.0 mg(好ましくは、約1.5~約4.5 mg)のL-メチオニン、
(e)約1.0~約6.0 mgのポリソルベート80、ならびに
(f)約0.12~約3.0 mg(好ましくは、約2.3~約3.0 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[58] (a)約1 mg、約2 mgまたは約20 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約3.7 mg(具体的には、約3.74 mg)の酢酸ナトリウム三水和物および約0.8 mg(具体的には、約0.76 mg)の氷酢酸からなる酢酸緩衝剤、
(c)約138 mgのトレハロース二水和物、
(d)約3.0 mgのL-メチオニン、
(e)約2.0 mgのポリソルベート80、ならびに
(f)約2.7 mg(具体的には、約2.72 mg)の塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[59] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mg/mLの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mg/mLのトレハロース二水和物、
(d)約0.75~約4.5 mg/mLのL-メチオニン、
(e)約0.1~約5.0 mg/mLのポリソルベート80、ならびに
(f)約0.03~約2.0 mg/mLの塩化ナトリウムを含み、pHが約4.7~約5.3である、皮下投与用の水性医薬組成物またはその製剤;
[60] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mL(好ましくは、約65.1~約72.6 mg/mL)のトレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用の水性医薬組成物またはその製剤;
[61] (a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である、皮下投与用の水性医薬組成物またはその製剤;
[62] (a)約80~約150 mg(好ましくは、約100 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約0.68~約6.8 mgの酢酸ナトリウム三水和物、および(b2)前記(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.7~約5.3となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約1.9~約108.3 mgのトレハロース二水和物、
(d)約0.75~約4.5 mgのL-メチオニン、
(e)約0.1~約5.0 mgのポリソルベート80、ならびに
(f)約0.03~約2.0 mgの塩化ナトリウムを含み、pHが約4.7~約5.3であり、単位製剤当たりの容量が約1 mLである、皮下投与用の水性医薬組成物またはその製剤;
[63] (a)約80~約150 mg(好ましくは、約100 mg)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg(好ましくは、約1.68~約2.05 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg(好ましくは、約65.1~約72.6 mg)のトレハロース二水和物、
(d)約0.75~約3.0 mg(好ましくは、約0.75~約2.25 mg)のL-メチオニン、
(e)約0.5~約3.0 mgのポリソルベート80、ならびに
(f)約0.06~約1.5 mg(好ましくは、約1.15~約1.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約1 mLである、皮下投与用の水性医薬組成物またはその製剤;
[64] (a)約100 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg(具体的には、約1.87 mg)の酢酸ナトリウム三水和物および約0.4 mg(具体的には、約0.38 mg)の氷酢酸からなる酢酸緩衝剤、
(c)約69 mgのトレハロース二水和物、
(d)約1.5 mgのL-メチオニン、
(e)約1.0 mgのポリソルベート80、ならびに
(f)約1.4 mg(具体的には、約1.36 mg)の塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約1 mLである、皮下投与用の水性医薬組成物またはその製剤;
[65] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニン、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、ならびに
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[66] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)であり、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v)である)のポリソルベート80、ならびに
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[67] (a)約0.1~約1.2 mg/mLまたは約5~約25 mg/mL(各々好ましくは、約0.5 mg/mL、約1 mg/mLまたは約10 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、ならびに
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[68] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニン、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、ならびに
(f)約0.5~約34.2 mMの塩化ナトリウム(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)を含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、皮下投与用の水性医薬組成物またはその製剤;
[69] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、ならびに
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは約5.0)である、皮下投与用の水性医薬組成物またはその製剤;
[70] (a)約80~約150 mg/mL(好ましくは、約100 mg/mL)の抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)緩衝剤(好ましくは、約7.3~約73.0 mM(好ましくは、約14.6~約43.8 mM、より好ましくは、約18~約22 mM、さらにより好ましくは、約20.07 mM)の酢酸緩衝剤または当該酢酸緩衝剤と同等のpH緩衝性能を有する緩衝剤)、
(c)約5~約286.1 mM(好ましくは、約10~約207.9 mM、より好ましくは、約172~約193 mM、さらにより好ましくは、約182.38 mM)のトレハロース、
(d)酸化防止剤(好ましくは、約5~約30.2 mM(好ましくは、約5~約20 mM、より好ましくは、約5~約15 mM、さらにより好ましくは、約10.05 mM)のL-メチオニンまたは当該L-メチオニンと同等の酸化防止能を有する酸化防止剤)、
(e)約0.01~約0.5 %(w/v)(好ましくは、約0.05~約0.3 %(w/v)、より好ましくは、約0.08~約0.12 %(w/v)、さらに好ましくは、約0.1 %(w/v))のポリソルベート80、ならびに
(f)約0.5~約34.2 mM(好ましくは、約1.0~約25.7 mM、より好ましくは、約19.6~約25.7 mM、さらにより好ましくは、約23.27 mM)の塩化ナトリウムを含み、pHが約4.7~約5.3(好ましくは、約4.9~約5.1、より好ましくは、約5.0)である、皮下投与用の水性医薬組成物またはその製剤;
[71] 当該抗PD-1/CD3二重特異性抗体が、WO2019/156199により特定される特許出願明細書に開示された当該抗PD-1/CD3二重特異性抗体である、前項[1]~[70]の何れか一項記載の水性医薬組成物またはその製剤;
[72] 当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(a)前項(i)の重鎖が、配列番号1、配列番号2、配列番号3、配列番号4および配列番号5から選択される何れか一つのアミノ酸配列からなり、
(b)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(c)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、前項[1]~[70]の何れか一項記載の水性医薬組成物またはその製剤;
[73] PD-1に特異的に結合する抗原結合部位を構成する重鎖が、配列番号5のアミノ酸配列からなる、前項[72]記載の水性医薬組成物またはその製剤;
[74] 前項[72]または[73]記載のPD-1に特異的に結合する抗原結合部位を構成する重鎖の重鎖定常領域が、配列番号8、配列番号9、配列番号10、配列番号11、配列番号12および配列番号13から選択される何れか一つのアミノ酸配列からなる重鎖定常領域に置換され、前項[72]記載のCD3に特異的に結合する抗原結合部位を構成する重鎖の重鎖定常領域が、配列番号14、配列番号15、配列番号16、配列番号17、配列番号18および配列番号19から選択される何れか一つのアミノ酸配列からなる重鎖定常領域に置換された、前項[71]~[73]の何れか一項記載の水性医薬組成物またはその製剤;
[75] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、ならびに
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[76] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約20.07 mMの酢酸緩衝剤、
(c)約182.38 mMのトレハロース、
(d)約10.05 mMのL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、ならびに
(f)約23.27 mMの塩化ナトリウムを含み、pHが約5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[77] (a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約14.6~約43.8 mM(好ましくは、約18~約22 mM)の酢酸緩衝剤、
(c)約10~約207.9 mM(好ましくは、約172~約193 mM)のトレハロース、
(d)約5~約20 mM(好ましくは、約5~約15 mM)のL-メチオニン、
(e)約0.05~約0.3 %(w/v)(好ましくは、約0.08~約0.12 %(w/v))のポリソルベート80、ならびに
(f)約1.0~約25.7 mM(好ましくは、約19.6~約25.7 mM)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[78] (a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約20.07 mMの酢酸緩衝剤、
(c)約182.38 mMのトレハロース、
(d)約10.05 mMのL-メチオニン、
(e)約0.1 %(w/v)のポリソルベート80、ならびに
(f)約23.27 mMの塩化ナトリウムを含み、pHが約5.0である、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[79] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mL(好ましくは、約65.1~約72.6 mg/mL)のトレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[80] (a)約0.5 mg/mL、約1 mg/mLまたは約10 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[81] (a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg/mL(好ましくは、約1.68~約2.05 mg/mL)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg/mL(好ましくは、約65.1~約72.6 mg/mL)のトレハロース二水和物、
(d)約0.75~約3.0 mg/mL(好ましくは、約0.75~約2.25 mg/mL)のL-メチオニン、
(e)約0.5~約3.0 mg/mL(好ましくは、約0.8~約1.2 mg/mL)のポリソルベート80、ならびに
(f)約0.06~約1.5 mg/mL(好ましくは、約1.15~約1.5 mg/mL)の塩化ナトリウムを含み、pHが約4.9~約5.1である、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、および
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[82](a)約100 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg/mL(具体的には、約1.87 mg/mL)の酢酸ナトリウム三水和物および約0.4 mg/mL(具体的には、約0.38 mg/mL)の酢酸からなる酢酸緩衝剤、
(c)約69 mg/mLのトレハロース二水和物、
(d)約1.5 mg/mLのL-メチオニン、
(e)約1.0 mg/mLのポリソルベート80、ならびに
(f)約1.4 mg/mL(具体的には、約1.36 mg/mL)の塩化ナトリウムを含み、pHが約5.0である、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[83] (a)約2.5 mg、約5 mgまたは約50 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約6.8~約20.4 mg(好ましくは、約8.4~約10.2 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約18.9~約393.3 mg(好ましくは、約325~約365.1 mg)のトレハロース二水和物、
(d)約3.75~約15.0 mg(好ましくは、約3.75~約11.25 mg)のL-メチオニン、
(e)約2.5~約15.0 mg(好ましくは、約4~6 mg)のポリソルベート80、ならびに
(f)約0.29~約7.5 mg(好ましくは、約5.8~約7.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[84] (a)約2.5 mg、約5 mgまたは約50 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約9.4 mg(具体的には、約9.35 mg)の酢酸ナトリウム三水和物および約1.9 mgの氷酢酸からなる酢酸緩衝剤、
(c)約345 mgのトレハロース二水和物、
(d)約7.5 mgのL-メチオニン、
(e)約5.0 mgのポリソルベート80、ならびに
(f)約6.8 mgの塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[85](a)約1 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約2.72~約8.16 mg(好ましくは、約3.36~約4.1 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約7.6~約157.4 mg(好ましくは、約130.2~約145.2 mg)のトレハロース二水和物、
(d)約1.5~約6.0 mg(好ましくは、約1.5~約4.5 mg)のL-メチオニン、
(e)約1.0~約6.0 mgのポリソルベート80、ならびに
(f)約0.12~約3.0 mg(好ましくは、約2.3~約3.0 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[86](a)約1 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約3.7 mg(具体的には、約3.74 mg)の酢酸ナトリウム三水和物および約0.8 mg(具体的には、約0.76 mg)の氷酢酸からなる酢酸緩衝剤、
(c)約138 mgのトレハロース二水和物、
(d)約3.0 mgのL-メチオニン、
(e)約2.0 mgのポリソルベート80、ならびに
(f)約2.7 mg(具体的には、約2.72 mg)の塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[87] (a)約100 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)約1.36~約4.08 mg(好ましくは、約1.68~約2.05 mg)の酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが約4.9~約5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)約3.8~約78.7 mg(好ましくは、約65.1~約72.6 mg)のトレハロース二水和物、
(d)約0.75~約3.0 mg(好ましくは、約0.75~約2.25 mg)のL-メチオニン、
(e)約0.5~約3.0 mgのポリソルベート80、ならびに
(f)約0.06~約1.5 mg(好ましくは、約1.15~約1.5 mg)の塩化ナトリウムを含み、pHが約4.9~約5.1であり、単位製剤当たりの容量が約1 mLである、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、および
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[88](a)約100 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)約1.9 mg(具体的には、約1.87 mg)の酢酸ナトリウム三水和物および約0.4 mg(具体的には、約0.38 mg)の氷酢酸からなる酢酸緩衝剤、
(c)約69 mgのトレハロース二水和物、
(d)約1.5 mgのL-メチオニン、
(e)約1.0 mgのポリソルベート80、ならびに
(f)約1.4 mg(具体的には、約1.36 mg)の塩化ナトリウムを含み、pHが約5.0であり、単位製剤当たりの容量が約1 mLである、皮下投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤;
[89] 導電率が、約2.6~約3.1 mS/cmである、前項[1]~[88]の何れか一項記載の水性医薬組成物またはその製剤;
[90] 抗体(好ましくは、抗PD-1/CD3二重特異性抗体)を有効成分として含み、さらに、緩衝剤(好ましくは、酢酸緩衝剤)、トレハロース、酸化防止剤(好ましくは、L-メチオニン)、ポリソルベート80を含み、導電率が約2.6~約3.1 mS/cmである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤;
[91] 導電率が、約2.8 mS/cmである、前項[1]~[90]の何れか一項記載の水性医薬組成物またはその製剤;
[92] 前項[75]、[76]、[79]、[80]、[83]~[86]および[89]~[91]の何れか一項記載の水性医薬組成物またはその製剤と生物学的同等性を示し、同項記載の抗PD-1/CD3二重特異性抗体を有効成分として含有する、静脈内投与用の水性医薬組成物またはその製剤;
[93] 前項[75]~[91]の何れか一項記載の水性医薬組成物またはその製剤と生物学的同等性を示し、同項記載の抗PD-1/CD3二重特異性抗体を有効成分として含有する、皮下投与用の水性医薬組成物またはその製剤;
[94] 生物学的同等性が、以下の(i)および(ii)の差の90%信頼区間がlog(0.80)~log(1.25)の範囲にある関係である、前項[92]記載の水性医薬組成物またはその製剤:
(i)その投与による生物学的同等性判定パラメータの対数値の平均値、および
(ii)前項[75]、[76]、[79]、[80]、[83]~[86]および[89]~[91]の何れか一項記載の水性医薬組成物またはその製剤の投与による生物学的同等性判定パラメータの対数値の平均値;
[95] 生物学的同等性が、以下の(i)および(ii)の差の90%信頼区間がlog(0.80)~log(1.25)の範囲にある関係である、前項[93]記載の水性医薬組成物またはその製剤:
(i)その投与による生物学的同等性判定パラメータの対数値の平均値、および
(ii)前項[75]~[91]の何れか一項記載の水性医薬組成物またはその製剤の投与による生物学的同等性判定パラメータの対数値の平均値;
[1-2] 前項[1-1]記載のカートリッジが充填されたインジェクターペン、オートインジェクターまたは無針装置;
[1-3] 前項[1]~[95]の何れか一項記載の水性医薬組成物(好ましくは、静脈内投与用)が封入されたバイアル容器またはアンプル容器;
[2-2] 自己免疫疾患が、乾癬、乾癬性関節炎、関節リウマチ、クローン病、潰瘍性大腸炎または重症筋無力症である、前項[2-1]記載の使用;ならびに
[2-3] 血液がんが、末梢性T細胞リンパ腫または皮膚T細胞性リンパ腫である、前項[2-1]記載の使用。
本発明にかかる抗PD-1/CD3二重特異性抗体である抗体αに最適な処方の基本条件の探索を行った。表1および表2の条件で、抗体濃度が各々20 mg/mLとなるよう、試料B1~B30の30種を調製した。




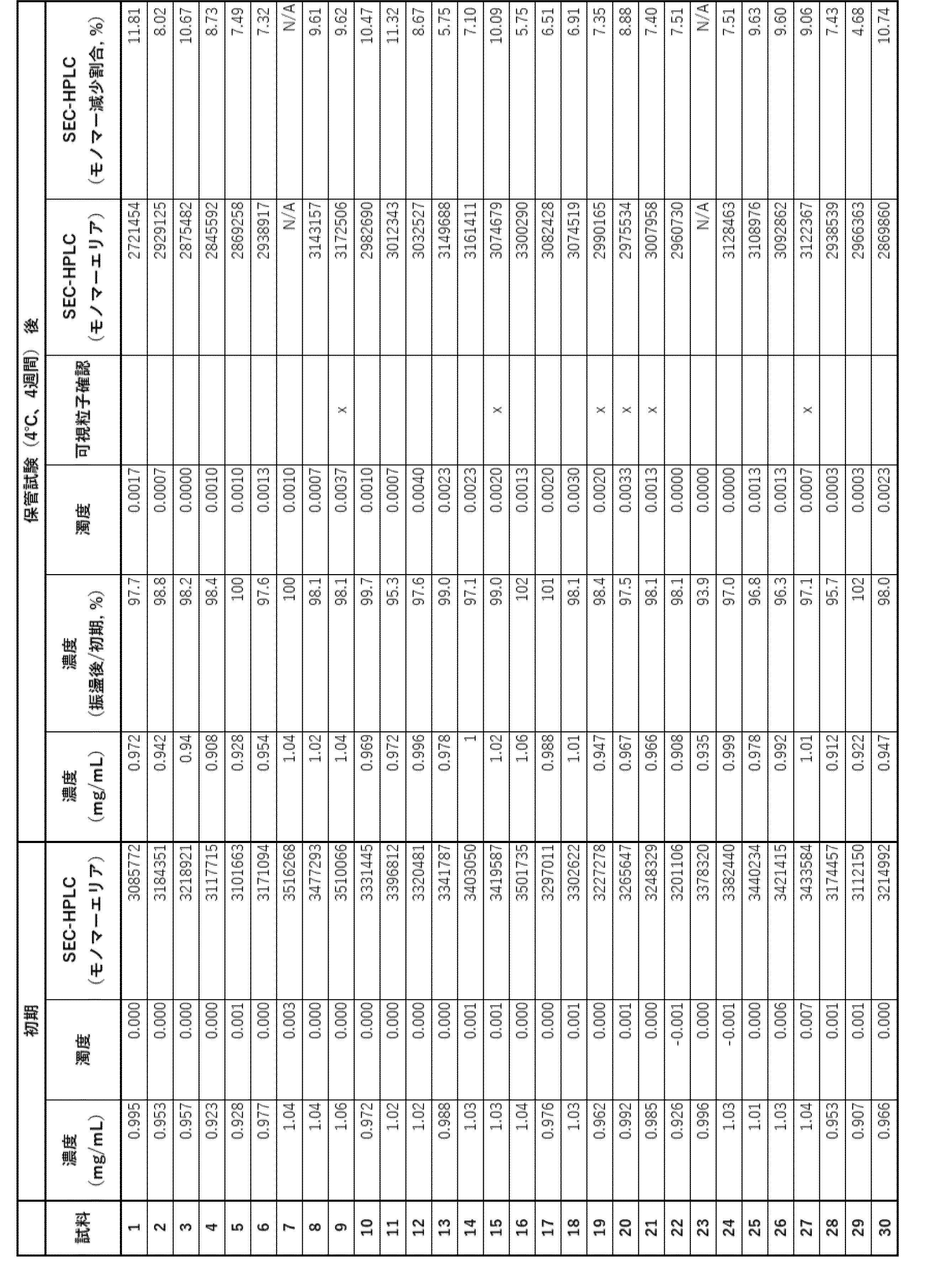

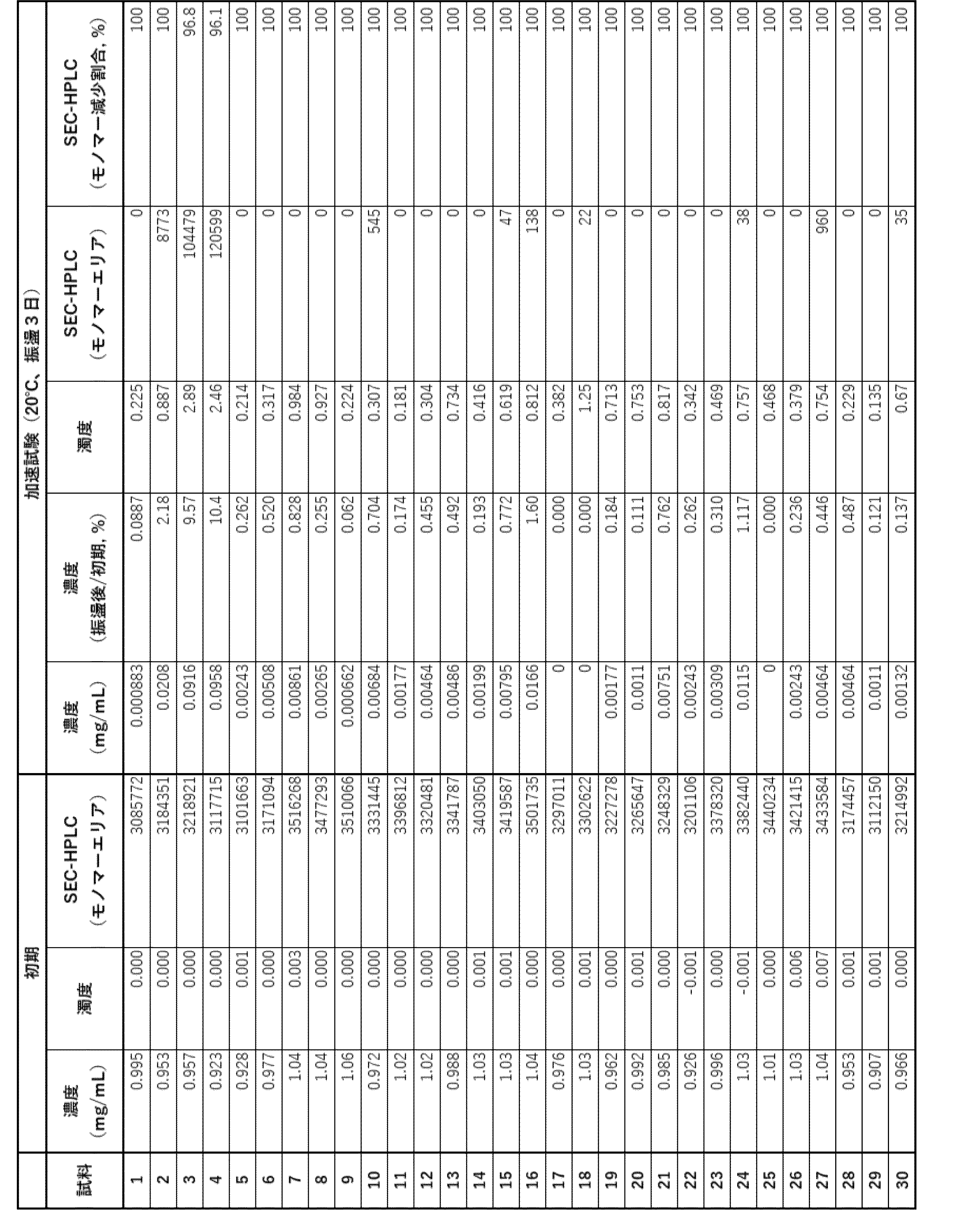
実施例1の評価にて選択された試料番号4、5および22の各処方に、糖(スクロース、トレハロース、D-マンニトールおよびソルビトール)を各々添加した処方S1~S24の24種を表8のとおり調製した。



実施例2の評価にて選択された試料S3、S5およびS23の各処方に、ポリソルベート(PS)(ポリソルベート80(PS80)およびポリソルベート20(PD20))を各々添加した試料E1~E15の15種を表11のとおり調製した。

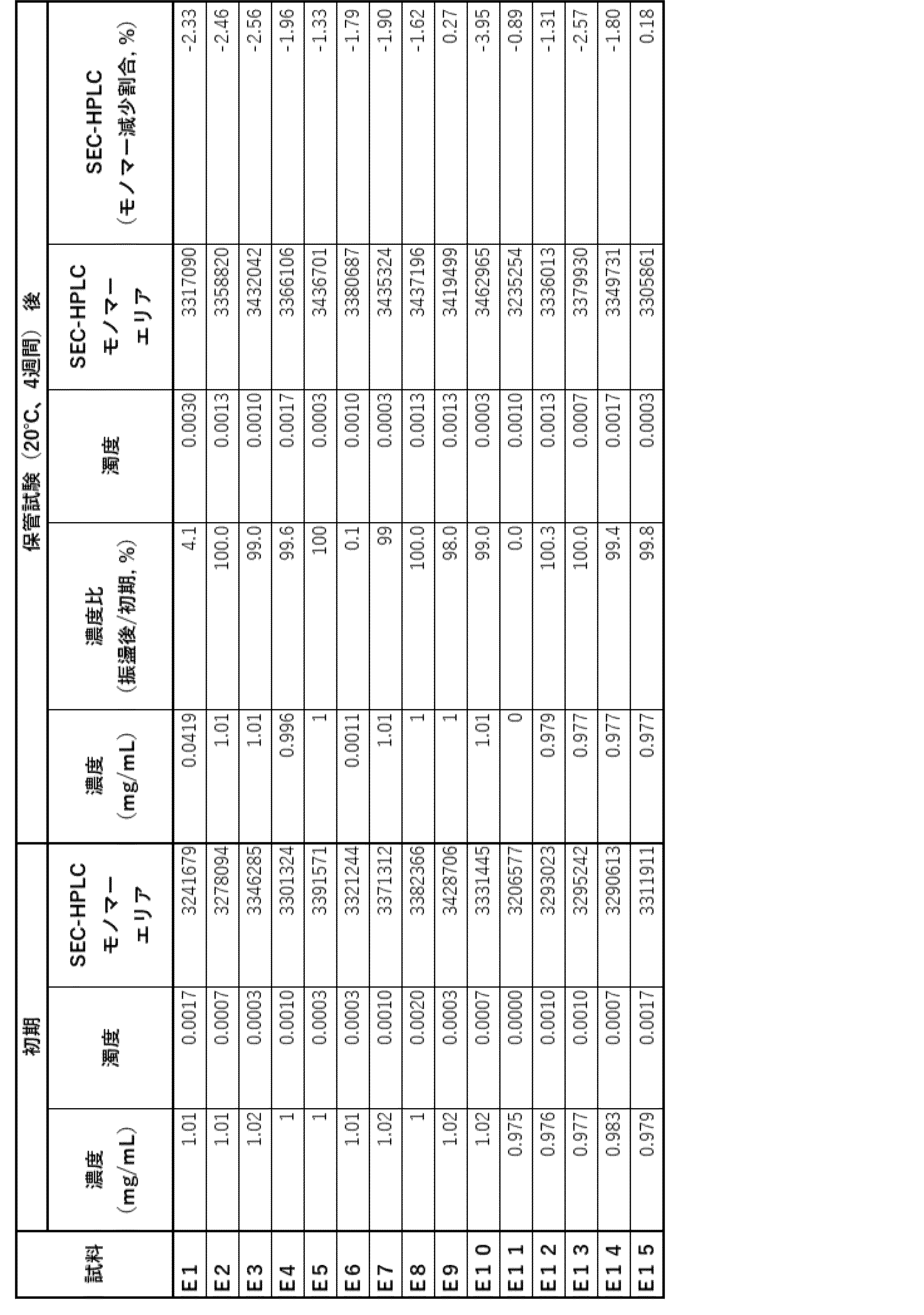


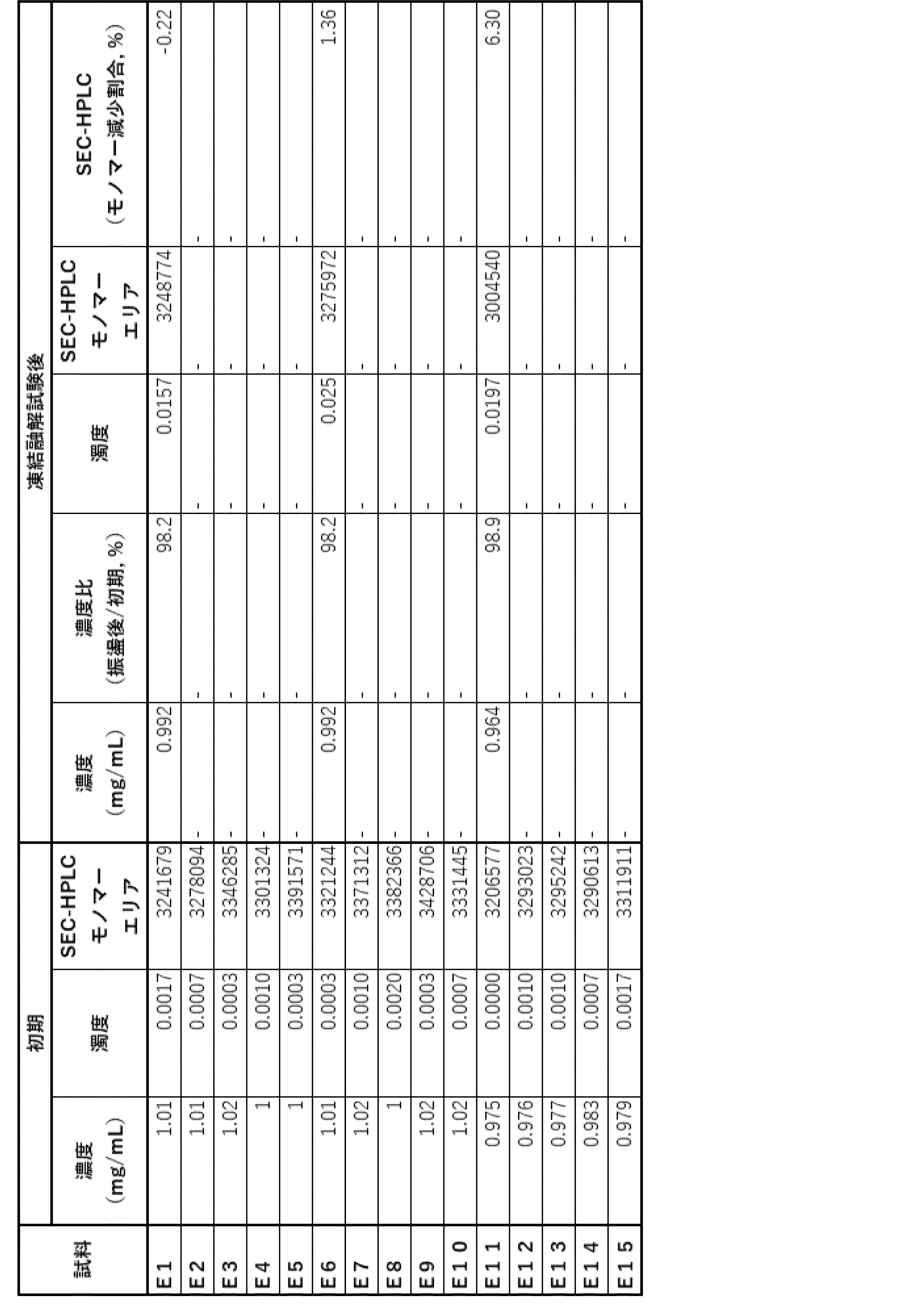

(a)pH測定には、pHメーター(LAQUA F-74(HORIBA))および微量試料測定用電極(マイクロ Tou pH 電極(HORIBA))が使用された。
(b)抗体濃度は、各試料を15,000 g、30分間遠心した上清について、280 nmの吸光度を1 cmまたは2 mmの光路長で測定された。なお、各試料について、吸光度が0.1~1.0になるよう希釈して測定され、抗体の吸光係数ε280 = 1.51(mg/mL)-1cm-1から濃度が算出された。なお、測定には、分光光度計(BioSpectormeter(Eppendorf))が使用された。
(c)濁度測定においては、光路長1 cmで500 nmの吸光度を測定した。なお、測定には、分光光度計が使用された。
(d)SEC-HPLC解析は、表3に示した条件で実施された。
(e)SDS-PAGE解析は、各試料を15,000 g、30分間遠心分離した上清を用い、還元下(100 mM DTT)および非還元下(-DTT)において、2 μgの試料を、4~12% Bis-Trisゲル上で100 V、約30分間電気泳動して実施された。
(f)IE-HPLC解析は、表17に示す条件で実施された。

(h)摺動抵抗測定:測定試料として、15,000 g、30分間遠心分離した上清を用いた。29G staked 1mL COP syringeに試料を400 μLずつ充填し、5時間程度静置した。オートグラフを用い、6 mL/分でシリンジを押し出し、摺動抵抗値が測定された。なお、摺動抵抗測定は、初期状態の試料についてのみ実施した。
(i)FIAにおいては、フローイメージング装置を用いて、150 μLの試料を流速0.05 mL/分で測定し、1 μm以上のサイズの凝集体について評価された。
安定性評価試験前後の試料について、目視での状態確認が実施された。初期状態の試料では、酢酸処方の試料およびヒスチジン処方の試料ともに黄色を呈し、4週間および12週間保管ならびに3日間振とうした各条件下において、明らかな色の変化は見られなかった。ヒスチジン処方の試料に関して、5℃下での4週間保管後ならびに5℃および25℃下で各々12週間保管した後において微粒子が確認された。また、酢酸処方およびヒスチジン処方の試料ともに、40℃下での4週間保管後において微粒子は見られなかったが、全体が白く濁っていた。
(a)安定性評価試験後のすべての試料において、顕著なpHの変化は認められなかった。
(b)安定性試験後のすべての試料において、大きな濃度の変化は見られなかった。
(c)安定性試験前後の濁度の測定結果を表18に示す。

(d)安定性試験前後のSEC-HPLC解析の測定結果を表19に示す。

(e)安定性試験前後のSDS-PAGE解析から、還元条件および非還元条件の何れによっても、初期状態および各試料において、標品と同様のバンドが観察され、精製、濃縮などの試料調製で分解が生じていないことが確認された。
(f)安定性試験前後のIE-HPLC分析の結果を表20に示す。

(g)安定性評価試験前後での粘度測定の結果、何れの処方も、安定性評価試験前後において有意な粘度の変化は見られなかった。
(h)初期状態の試料での摺動抵抗値測定の結果、酢酸処方およびヒスチジン処方の試料の何れにおいても、非常に低い摺動抵抗値(7.66 Nおよび7.46 N)であった。
(i)安定性評価試験前後の抗体を含む各処方の粒子濃度から、処方液のみの粒子濃度を引いた粒子濃度を表21に示す。

実施例4の評価に基づき選択された酢酸処方(20 mM酢酸緩衝液(pH 5.0)、285 mM トレハロース、0.1 % PS80)について、さらに最適化検討を実施した。





(e)FIAにおいては、フローイメージング装置を用いて、500 μLの試料を流速0.1 mL/分で測定し、2 μm以上のサイズの凝集体が評価された。
(f)ペプチドマップ解析は、LC-MSを用いて実施された。
(保管試験)
(a)SEC-HPLC解析にて評価した、保管試験前後での高分子種(HMWS)の割合を表27に示す。いずれの濃度においても、5℃下での12週間保管では、HMWSの顕著な増加は認められなかった。25℃下においては、抗体濃度が20 mg/mLの処方では、HMWSの増加は認められなかったものの、100 mg/mLの処方では、HMWSの直線的な増加傾向が認められた。一方、導電率による挙動の差は認められなかった。40℃下では、いずれの濃度においてもHMWSは増加したが、20 mg/mLの処方では、導電率の上昇に伴い、HMWSが高まる傾向が認められた。5℃および25℃下では、安定な処方であることが確認された。
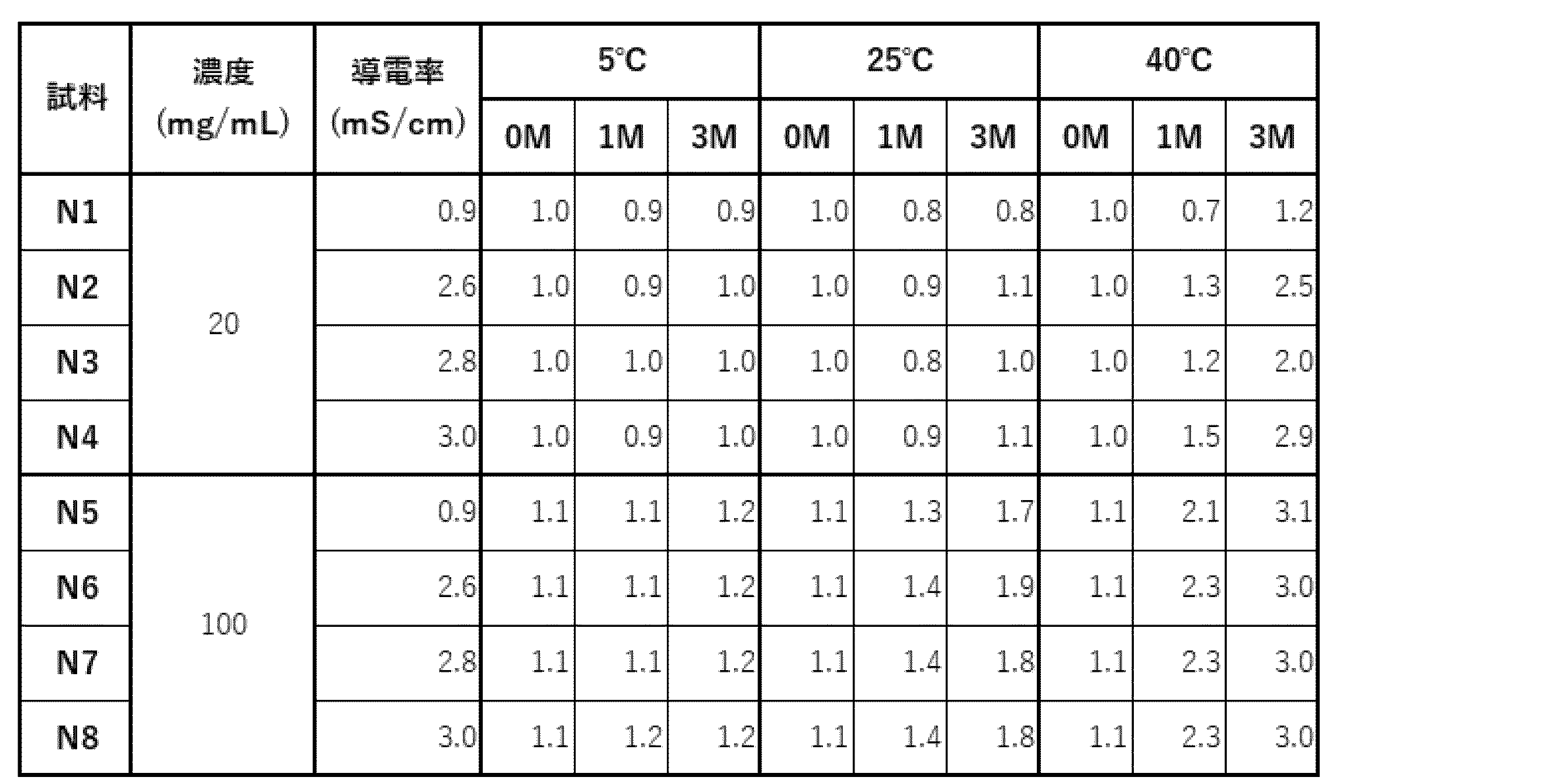

(c)RP-HPLC解析にて評価した、保管試験前後での着色体の割合を表29に示す。
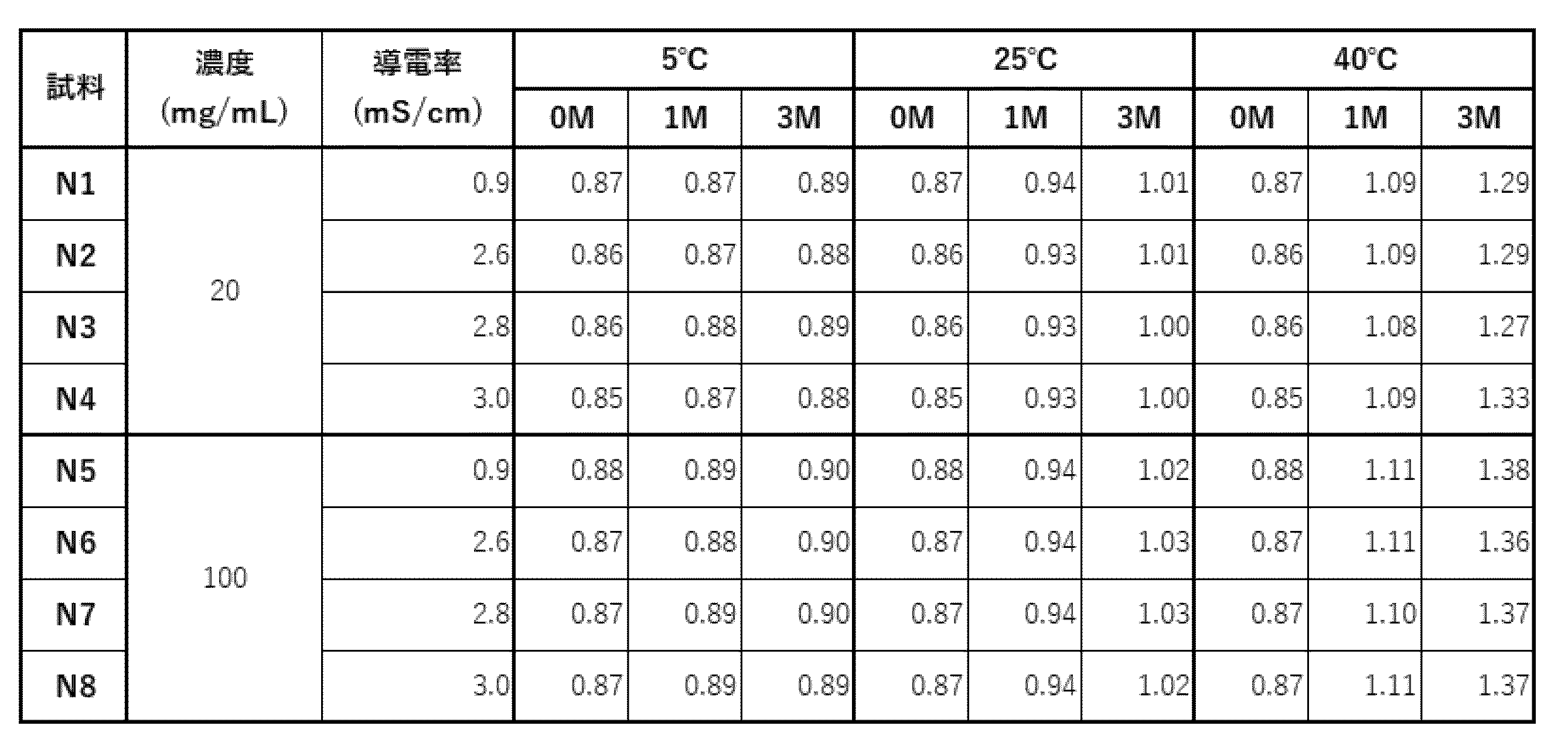
(d)SPRにて評価した、保管試験前後でのPD-1への結合活性の結果を表30に、CD3結合活性の結果を表31に各々示す。各表中の数値は、抗体αの標準品に対する比活性(%)を表す。5℃および25℃下では、PD-1、CD3ともに結合活性の低下は認められなかった。一方、40℃下では、PD-1への結合活性の低下が認められ、さらに、抗体濃度が20 mg/mLにおいては、その結合活性の低下は顕著であった。

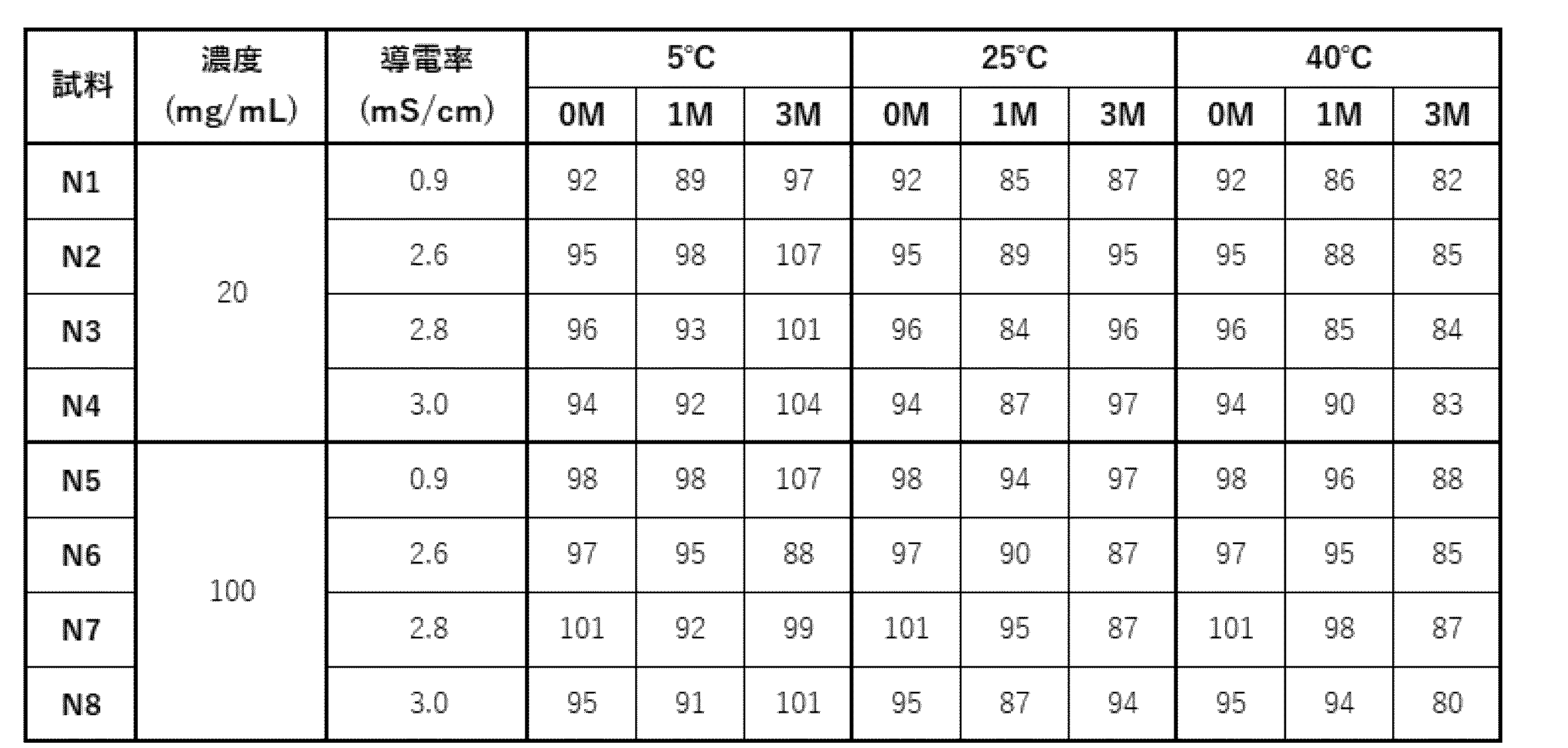
(振とう試験)
(a)SEC-HPLCにて、振とう試験前後におけるHMWSの割合を評価したが、すべての試料および条件において、HMWSの顕著な増加は認められなかった。
(b)IE-HPLCにて、振とう試験前後における酸性分子種の割合を評価したが、すべての試料および条件において、酸性分子種の顕著な増加は認められなかった。
(c)RP-HPLCにて、振とう試験前後における着色体の割合を評価したが、すべての試料および条件において、着色体の顕著な増加は認められなかった。
(d)SPRにて、振とう試験前後におけるPD-1およびCD3に対する結合活性を各々評価したが、試料間において結合活性の低下に有意な差は認められなかった。
(e)FIAにて、試料N5およびN8の振とう試験後における粒子数を評価したが、何れについても、振とうストレスによる微粒子数および最大粒子径の増加は認められなかった。
(凍結融解)
(a)SEC-HPLCにて、凍結融解試験前後でのHMWSの割合を評価したが、すべての試料においてHMWSの割合に大きな変化はなく、凍結融解ストレスに安定であることが確認された。
(b)IE-HPLCにて、凍結融解試験前後での酸性分子種の割合を評価したが、すべての試料において酸性分子種の割合に大きな変化はなく、同様に、凍結融解ストレスに安定であることが確認された。
(c)RP-HPLCにて、凍結融解試験前後での着色体の割合を評価したが、すべての試料において着色体の割合にも大きな変化は認められなかった。
(d)SPRにて、凍結融解試験前後でのPD-1およびCD3への結合活性を各々評価したが、すべての試料において結合活性に大きな変化は認められなかった。
(e)FIAにて、試料N5およびN8の凍結融解試験後における粒子数を各々評価したが、何れについても凍結融解ストレスによる微粒子数および最大粒子径の増加は認められなかった。また、評価した試料間に有意な差も認められなかった。
SEC-HPLCにて、試料N8のシリンジ通液試験前後におけるHMWSの割合(%)を評価したが、HMWSの生成は認められなかった。
表30に示したように、40℃下での長期保管におけるPD-1への結合活性の低下について、ペプチドマップによる要因解析を行った。代表例として、PD-1に対する相補性決定領域に存在する101番目のメチオニン残基(以下、「Met101」と略記することがある。)への酸化度(%)を表32に示す。5℃および25℃下では、Met101の酸化度は低値を示しており、試料間差は認められなかった。また、抗体濃度が100 mg/mLの場合に比べ、20 mg/mLの場合において顕著な酸化が認められた。図4に示すように、Met101の酸化とPD-1結合活性の低下には相関関係があった。このような酸化が、結合活性低下の原因である可能性が認められた。

本発明にかかる抗体αが、低濃度において、安定性が低下する可能性が実施例5において示唆されたため、低濃度域での安定性を評価した。


(a)SEC-HPLCにて評価した、保管前後でのHMWSの割合を表35に示す。何れの処方においても、40℃下での長期保管においてHMWSが大幅に上昇したが、抗体が低濃度のL1において、HMWSの蓄積はより顕著であった。低濃度条件で凝集の可能性が高まることが示唆された。



長期保管の影響により、本発明にかかる抗体αの酸化が進行し、PD-1およびCD3への結合活性の低下、特に、PD-1への結合活性の低下が顕著であることが実施例6において示唆された。

(a)SEC-HPLC解析にて評価した、保管前後でのHMWSの割合(%)を表39に示す。抗酸化剤未添加でも、5℃および25℃では変化はなかった。一方で、L-メチオニンを10 mMまたはDTPAを20 μM添加した試料について、40℃下で12週間保管した場合、HMWSの生成が劇的に抑制された。L-メチオニンまたはDTPAのHMWSの生成抑制効果は、抗体濃度1および20 mg/mLにおいても同様に認められた。


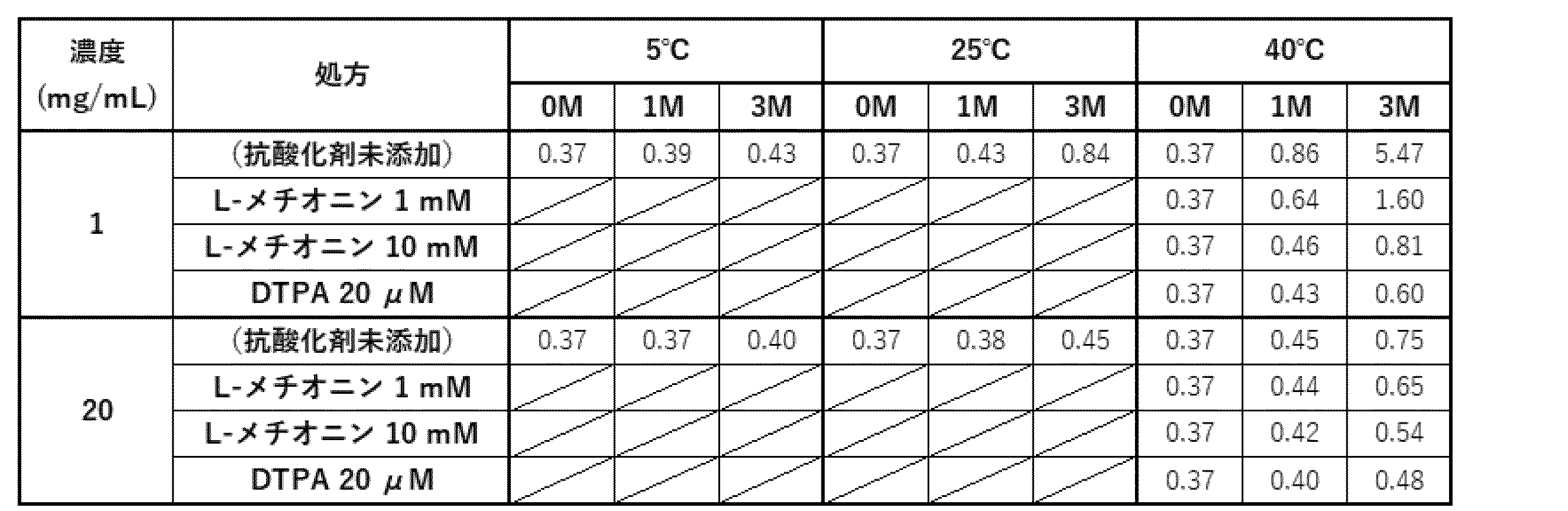



実施例1~7までの検討において、本発明にかかる抗体αが、安定に保管され得る条件を見出した。さらに安定な条件を設定するため、糖の濃度範囲を決定することとした。


(a)IE-HPLCにて評価した、保管前後での酸性分子種の割合(%)を表47に示す。何れの処方においても、25℃および40℃下での長期保管において酸性分子種が上昇した。通常の保管温度となる5℃下では安定であった。処方間で差は認められなかった。
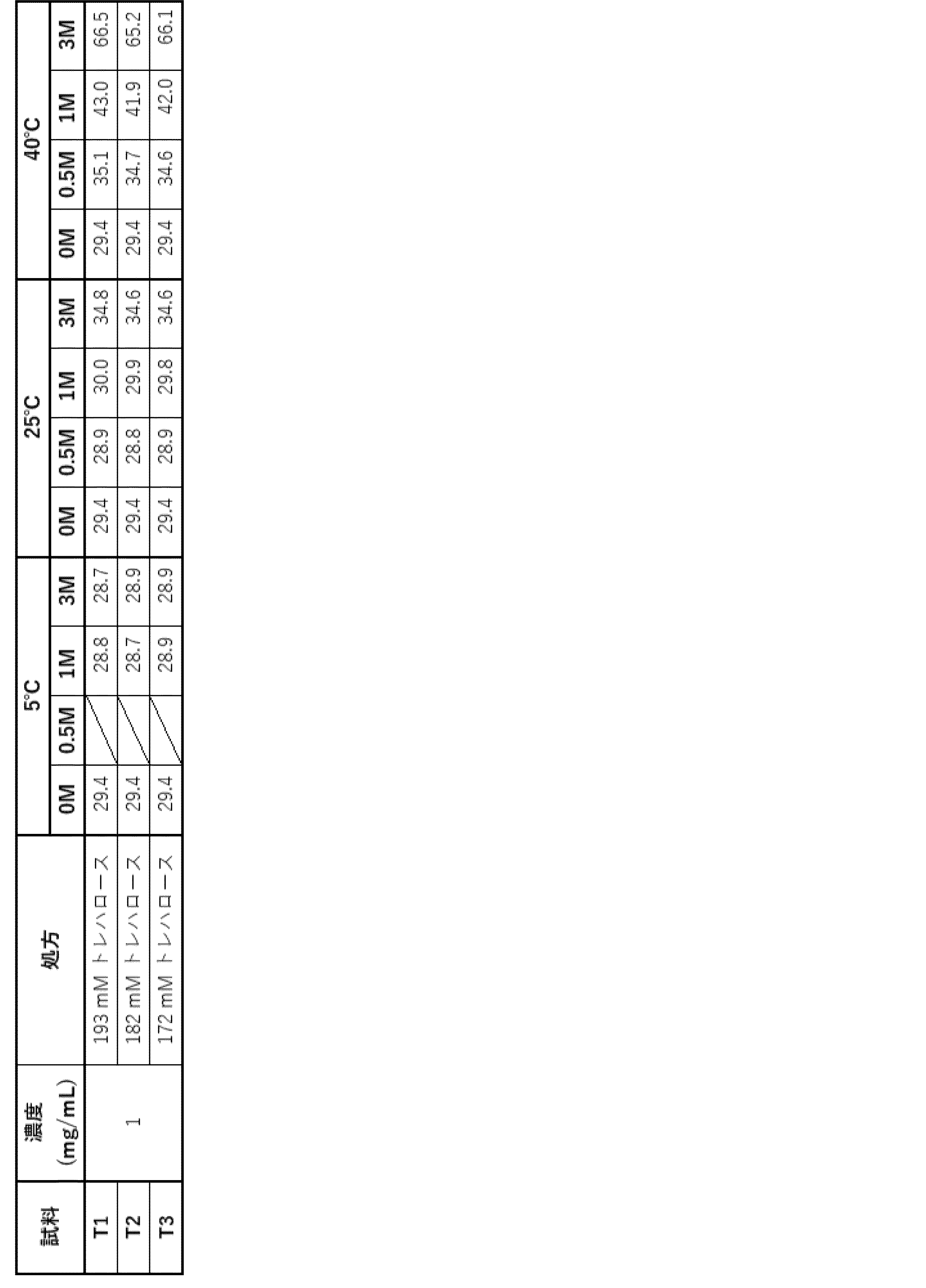



実施例1~8までの検討において、本発明にかかる抗体αが、安定に保管され得る条件を見出した。それらを総合して最適と考えられた条件の安定性試験を実施した。


(a)SEC-HPLCにて評価した、保管前後でのHMWSの割合を表53に示す。100 mg/mLの濃度においては、25℃および40℃下の条件でHMWSの増加が認められたが、5℃下では顕著な増加は認められなかった。5℃下では安定であると考えられた。
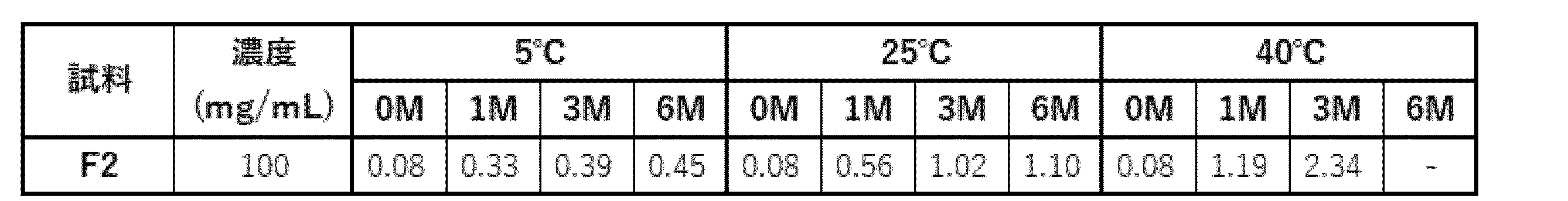







(a)SEC-HPLCにて評価した、保管前後でのHMWSの割合を表61に示す。0.5 mg/mLおよび1.0 mg/mLの濃度において、5、25および40 ℃の条件下では、HMWSの割合の増加が認められなかった。





本発明の水性医薬組成物を調製する方法について、実施例9の試料F2を例に、以下に示す。なお、同試料F2と抗体濃度が異なる本発明の水性医薬組成物についても、同様に調製することができる。
(a)処方調製液1を、20 mM酢酸緩衝液(pH 5.0)、182.38 mMトレハロース、1.36 mg/mL 塩化ナトリウムおよび10 mM L-メチオニンとなるように調製する。
(b)処方調製液2を、20 mM酢酸緩衝液(pH 5.0)、182.38 mMトレハロース、2 % PS80、1.36 mg/mL 塩化ナトリウムおよび10 mM L-メチオニンとなるように調製する。
(c)処方調製液3を、20 mM酢酸緩衝液(pH 5.0)、182.38 mMトレハロース、0.1 % PS80、1.36 mg/mL 塩化ナトリウムおよび10 mM L-メチオニンとなるように調製する。
(d)抗体αの製造工程のうち、ウイルスろ過工程から得られた工程液を、限外ろ過/透析ろ過膜で処方調製液1に溶液組成を置換する。
(e)前項(d)において、溶液組成を処方調製液1に置換した、抗体αを含む工程液を、限外ろ過/透析ろ過膜で約140 mg/mLまで濃縮する。
(f)前項(e)において濃縮した抗体αを含む工程液に、高濃度のPS80を含む処方調製液2を添加し、PS80濃度が0.1%となるように調製する。
(g)前項(f)において、目的のPS80濃度に調整した抗体αを含む工程液に、処方調製液3を添加し、抗体濃度が目的の100 mg/mLとなるように希釈する。
Claims (12)
- (a)0.1~1.2 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)18~22 mMの酢酸緩衝剤、
(c)172~193 mMのトレハロース、
(d)5~15 mMのL-メチオニン、
(e)0.05~0.3 %(w/v)のポリソルベート80、および
(f)19.6~25.7 mMの塩化ナトリウムを含み、pHが4.9~5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - 抗PD-1/CD3二重特異性抗体が、0.5 mg/mLまたは1 mg/mLである、請求項1記載の水性医薬組成物またはその製剤。
- (a)0.5 mg/mLまたは1 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)20.07 mMの酢酸緩衝剤、
(c)182.38 mMのトレハロース、
(d)10.05 mMのL-メチオニン、
(e)0.1 %(w/v)のポリソルベート80、ならびに
(f)23.27 mMの塩化ナトリウムを含み、pHが5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - (a)0.1~1.2 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)1.68~2.05 mg/mLの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが4.9~5.1となる分量の酢酸からなる酢酸緩衝剤、
(c)65.1~72.6 mg/mLのトレハロース二水和物、
(d)0.75~2.25 mg/mLのL-メチオニン、
(e)0.5~3.0 mg/mLのポリソルベート80、ならびに
(f)1.15~1.5 mg/mLの塩化ナトリウムを含み、pHが4.9~5.1である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - 抗PD-1/CD3二重特異性抗体が、0.5 mg/mLまたは1 mg/mLである、請求項4記載の水性医薬組成物またはその製剤。
- (a)0.5 mg/mLまたは1 mg/mLの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)1.87 mg/mLの酢酸ナトリウム三水和物および0.38 mg/mLの酢酸からなる酢酸緩衝剤、
(c)69 mg/mLのトレハロース二水和物、
(d)1.5 mg/mLのL-メチオニン、
(e)1.0 mg/mLのポリソルベート80、ならびに
(f)1.36 mg/mLの塩化ナトリウムを含み、pHが5.0である、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - (a)0.5~6 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)8.4~10.2 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが4.9~5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)325~365.1 mgのトレハロース二水和物、
(d)3.75~11.25 mgのL-メチオニン、
(e)2.5~15.0 mgのポリソルベート80、ならびに
(f)5.8~7.5 mgの塩化ナトリウムを含み、pHが4.9~5.1であり、単位製剤当たりの容量が5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - 抗PD-1/CD3二重特異性抗体が、2.5 mgまたは5 mgである、請求項7記載の水性医薬組成物またはその製剤。
- (a)2.5 mgまたは5 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)9.35 mgの酢酸ナトリウム三水和物および1.9 mgの氷酢酸からなる酢酸緩衝剤、
(c)345 mgのトレハロース二水和物、
(d)7.5 mgのL-メチオニン、
(e)5.0 mgのポリソルベート80、ならびに
(f)6.8 mgの塩化ナトリウムを含み、pHが5.0であり、単位製剤当たりの容量が5 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - (a)0.2~2.4 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)(b1)3.36~4.1 mgの酢酸ナトリウム三水和物、および(b2)前項(b1)の酢酸ナトリウム三水和物の分量に対応して、pHが4.9~5.1となる分量の氷酢酸からなる酢酸緩衝剤、
(c)130.2~145.2 mgのトレハロース二水和物、
(d)1.5~4.5 mgのL-メチオニン、
(e)1.0~6.0 mgのポリソルベート80、ならびに
(f)2.3~3.0 mgの塩化ナトリウムを含み、pHが4.9~5.1であり、単位製剤当たりの容量が2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。 - 抗PD-1/CD3二重特異性抗体が、1 mgである、請求項10記載の水性医薬組成物またはその製剤。
- (a)1 mgの抗PD-1/CD3二重特異性抗体を有効成分として含み、さらに、
(b)3.74 mgの酢酸ナトリウム三水和物および0.76 mgの氷酢酸からなる酢酸緩衝剤、
(c)138 mgのトレハロース二水和物、
(d)3.0 mgのL-メチオニン、
(e)2.0 mgのポリソルベート80、ならびに
(f)2.72 mgの塩化ナトリウムを含み、pHが5.0であり、単位製剤当たりの容量が2 mLである、皮下投与用もしくは静脈内投与用の水性医薬組成物またはその製剤であって、
ここで、当該抗PD-1/CD3二重特異性抗体が、
(i)PD-1に特異的に結合する抗原結合部位を構成する重鎖および軽鎖、ならびに
(ii)CD3に特異的に結合する抗原結合部位を構成する重鎖および軽鎖からなり、
(α)前項(i)の重鎖が、配列番号5のアミノ酸配列からなり、
(β)前項(ii)の重鎖が、配列番号6のアミノ酸配列からなり、ならびに
(γ)前項(i)および前項(ii)の軽鎖がともに、配列番号7のアミノ酸配列からなる当該二重特異性抗体である、当該水性医薬組成物またはその製剤。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024534766A JP7548475B1 (ja) | 2023-03-16 | 2024-03-15 | 抗体製剤 |
| EP24770994.2A EP4681730A1 (en) | 2023-03-16 | 2024-03-15 | Antibody preparation |
| AU2024237533A AU2024237533A1 (en) | 2023-03-16 | 2024-03-15 | Antibody preparation |
| CN202480016597.0A CN120752054A (zh) | 2023-03-16 | 2024-03-15 | 抗体制剂 |
| KR1020257029539A KR20250156121A (ko) | 2023-03-16 | 2024-03-15 | 항체 제제 |
| JP2024126120A JP2024153869A (ja) | 2023-03-16 | 2024-08-01 | 抗体製剤 |
| MX2025010452A MX2025010452A (es) | 2023-03-16 | 2025-09-04 | Formulacion de anticuerpos |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023041761 | 2023-03-16 | ||
| JP2023-041761 | 2023-03-16 | ||
| JP2023154195 | 2023-09-21 | ||
| JP2023-154195 | 2023-09-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024190892A1 true WO2024190892A1 (ja) | 2024-09-19 |
Family
ID=92755353
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/010151 Ceased WO2024190892A1 (ja) | 2023-03-16 | 2024-03-15 | 抗体製剤 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202502380A (ja) |
| WO (1) | WO2024190892A1 (ja) |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003011911A1 (fr) | 2001-07-31 | 2003-02-13 | Ono Pharmaceutical Co., Ltd. | Substance specifique pour pd-1 |
| WO2004072286A1 (ja) | 2003-01-23 | 2004-08-26 | Ono Pharmaceutical Co., Ltd. | ヒトpd−1に対し特異性を有する物質 |
| WO2011161226A2 (en) | 2010-06-25 | 2011-12-29 | F. Hoffmann-La Roche Ag | Novel antibody formulation |
| WO2013022091A1 (ja) | 2011-08-11 | 2013-02-14 | 小野薬品工業株式会社 | Pd-1アゴニストからなる自己免疫疾患治療剤 |
| JP2017516846A (ja) | 2014-05-23 | 2017-06-22 | アレス トレーディング ソシエテ アノニム | 液体医薬組成物 |
| CN107987161A (zh) | 2016-10-26 | 2018-05-04 | 泰州迈博太科药业有限公司 | 一种抗egfr单克隆抗体制剂 |
| WO2019156199A1 (ja) | 2018-02-09 | 2019-08-15 | 小野薬品工業株式会社 | 二重特異性抗体 |
| JP2019526627A (ja) | 2016-08-29 | 2019-09-19 | ティジアーナ ライフ サイエンシズ パブリック リミティド カンパニー | 抗−cd3抗体製剤 |
| JP2020509025A (ja) | 2017-03-01 | 2020-03-26 | メドイミューン・リミテッドMedImmune Limited | モノクローナル抗体製剤 |
| WO2021020416A1 (ja) | 2019-07-30 | 2021-02-04 | 小野薬品工業株式会社 | 二重特異性抗体 |
| WO2021210662A1 (ja) * | 2020-04-17 | 2021-10-21 | 小野薬品工業株式会社 | タンパク質製剤原薬の着色除去方法 |
| WO2022152245A1 (zh) | 2021-01-14 | 2022-07-21 | 上海君实生物医药科技股份有限公司 | 抗tigit抗体药物组合物及其用途 |
| JP2022553058A (ja) * | 2019-10-25 | 2022-12-21 | アムジェン インコーポレイテッド | 低タンパク質濃度のタンパク質損失を最小限に抑えるための組成物及び方法 |
| WO2023031969A1 (en) | 2021-09-03 | 2023-03-09 | Dr. Reddy’S Laboratories Limited | A method of improving stability of immune check point inhibitors |
| WO2023031970A1 (en) | 2021-09-03 | 2023-03-09 | Dr. Reddy’S Laboratories Limited | A pharmaceutical formulation of immune check point inhibitors |
| CN115869395A (zh) | 2021-09-26 | 2023-03-31 | 泰州迈博太科药业有限公司 | 一种抗体-缓冲液体系 |
-
2024
- 2024-03-15 WO PCT/JP2024/010151 patent/WO2024190892A1/ja not_active Ceased
- 2024-03-15 TW TW113109681A patent/TW202502380A/zh unknown
Patent Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003011911A1 (fr) | 2001-07-31 | 2003-02-13 | Ono Pharmaceutical Co., Ltd. | Substance specifique pour pd-1 |
| WO2004072286A1 (ja) | 2003-01-23 | 2004-08-26 | Ono Pharmaceutical Co., Ltd. | ヒトpd−1に対し特異性を有する物質 |
| WO2011161226A2 (en) | 2010-06-25 | 2011-12-29 | F. Hoffmann-La Roche Ag | Novel antibody formulation |
| WO2013022091A1 (ja) | 2011-08-11 | 2013-02-14 | 小野薬品工業株式会社 | Pd-1アゴニストからなる自己免疫疾患治療剤 |
| JP2017516846A (ja) | 2014-05-23 | 2017-06-22 | アレス トレーディング ソシエテ アノニム | 液体医薬組成物 |
| JP2019526627A (ja) | 2016-08-29 | 2019-09-19 | ティジアーナ ライフ サイエンシズ パブリック リミティド カンパニー | 抗−cd3抗体製剤 |
| CN107987161A (zh) | 2016-10-26 | 2018-05-04 | 泰州迈博太科药业有限公司 | 一种抗egfr单克隆抗体制剂 |
| JP2020509025A (ja) | 2017-03-01 | 2020-03-26 | メドイミューン・リミテッドMedImmune Limited | モノクローナル抗体製剤 |
| WO2019156199A1 (ja) | 2018-02-09 | 2019-08-15 | 小野薬品工業株式会社 | 二重特異性抗体 |
| WO2021020416A1 (ja) | 2019-07-30 | 2021-02-04 | 小野薬品工業株式会社 | 二重特異性抗体 |
| JP2022553058A (ja) * | 2019-10-25 | 2022-12-21 | アムジェン インコーポレイテッド | 低タンパク質濃度のタンパク質損失を最小限に抑えるための組成物及び方法 |
| WO2021210662A1 (ja) * | 2020-04-17 | 2021-10-21 | 小野薬品工業株式会社 | タンパク質製剤原薬の着色除去方法 |
| WO2022152245A1 (zh) | 2021-01-14 | 2022-07-21 | 上海君实生物医药科技股份有限公司 | 抗tigit抗体药物组合物及其用途 |
| WO2023031969A1 (en) | 2021-09-03 | 2023-03-09 | Dr. Reddy’S Laboratories Limited | A method of improving stability of immune check point inhibitors |
| WO2023031970A1 (en) | 2021-09-03 | 2023-03-09 | Dr. Reddy’S Laboratories Limited | A pharmaceutical formulation of immune check point inhibitors |
| CN115869395A (zh) | 2021-09-26 | 2023-03-31 | 泰州迈博太科药业有限公司 | 一种抗体-缓冲液体系 |
Non-Patent Citations (2)
| Title |
|---|
| MINISTRY OF HEALTH, LABOUR AND WELFARE, 24 November 2006 (2006-11-24) |
| See also references of EP4681730A1 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202502380A (zh) | 2025-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102397713B1 (ko) | 안정한 액체 약제학적 제제 | |
| AU2013276112B2 (en) | Antibody formulation | |
| EP3991745A1 (en) | Formulations containing anti-cd47/pd-l1 bispecific antibody and preparation method therefor and use thereof | |
| EP2830658B1 (en) | Stable igg4 binding agent formulations | |
| ES2783448T3 (es) | Métodos de tratamiento del lupus eritematoso sistémico utilizando un anticuerpo de dominio dirigido contra CD28 | |
| TW201021831A (en) | Method and formulation for reducing aggregation of a macromolecule under physiological conditions | |
| KR20220124208A (ko) | 프로그램된 세포 사멸 수용체1 항체 제제 및 그 용도 | |
| JP7848114B2 (ja) | 抗cd20抗体製剤及びcd20陽性疾患の治療のための抗cd20抗体の使用 | |
| JP7791813B2 (ja) | 高濃縮タンパク質製剤中の粘度低減剤としてのカンファースルホン酸およびそのカチオン性賦形剤との組み合わせ | |
| EP4094777A1 (en) | Recombinant fully human anti-tigit monoclonal antibody preparations, preparation method therefor and use thereof | |
| CN109498565B (zh) | 一种稳定的抗pd-1抗体制剂及其用途 | |
| JP7548475B1 (ja) | 抗体製剤 | |
| WO2024190892A1 (ja) | 抗体製剤 | |
| WO2024025440A1 (en) | Pharmaceutical composition of anti-cd20 antibody and use thereof | |
| KR20240100493A (ko) | 항-cd22 항체의 수성 제제 및 이의 용도 | |
| KR20230020449A (ko) | 레빌리맙의 수성 약학적 조성물 | |
| US11459399B2 (en) | Pharmaceutical compositions of a HER2/neu antibody and use of the same | |
| RU2824627C2 (ru) | Фармацевтическая композиция анти-cd20 антитела и ее применение | |
| HK40093464A (zh) | 稳定的药物制剂 | |
| TR2025000822T2 (tr) | Anti̇-cd20 anti̇kor farmasöti̇k bi̇leşi̇mi̇ ve bunun kullanimi | |
| US20230053747A1 (en) | Pharmaceutical Compositions of a HER2/neu Antibody and Use of the Same | |
| WO2025185555A1 (en) | Pharmaceutical compositions comprising ox40 targeting antibodies and uses thereof | |
| JP2024526773A (ja) | B7‐h3抗体の医薬組成物及びその使用 | |
| WO2026064732A1 (en) | Anti-pd-1 antibody-attenuated il-2 immunoconjugate formulations | |
| CN120239616A (zh) | 抗体制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024534766 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24770994 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2024237533 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202480016597.0 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 1020257029539 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202547084134 Country of ref document: IN |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112025019072 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2024237533 Country of ref document: AU Date of ref document: 20240315 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 202480016597.0 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024770994 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 202547084134 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257029539 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2024770994 Country of ref document: EP Effective date: 20251016 |
|
| ENP | Entry into the national phase |
Ref document number: 2024770994 Country of ref document: EP Effective date: 20251016 |
|
| ENP | Entry into the national phase |
Ref document number: 2024770994 Country of ref document: EP Effective date: 20251016 |
|
| ENP | Entry into the national phase |
Ref document number: 2024770994 Country of ref document: EP Effective date: 20251016 |
|
| ENP | Entry into the national phase |
Ref document number: 2024770994 Country of ref document: EP Effective date: 20251016 |