WO2024253831A1 - Non-coordinated alkylaluminum free anion modified alumoxanes and methods thereof - Google Patents
Non-coordinated alkylaluminum free anion modified alumoxanes and methods thereof Download PDFInfo
- Publication number
- WO2024253831A1 WO2024253831A1 PCT/US2024/030225 US2024030225W WO2024253831A1 WO 2024253831 A1 WO2024253831 A1 WO 2024253831A1 US 2024030225 W US2024030225 W US 2024030225W WO 2024253831 A1 WO2024253831 A1 WO 2024253831A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- mao
- composition
- compound
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/12—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing organo-metallic compounds or metal hydrides
- B01J31/14—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing organo-metallic compounds or metal hydrides of aluminium or boron
- B01J31/143—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing organo-metallic compounds or metal hydrides of aluminium or boron of aluminium
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/52—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides selected from boron, aluminium, gallium, indium, thallium or rare earths
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/64003—Titanium, zirconium, hafnium or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/64082—Tridentate ligand
- C08F4/64141—Dianionic ligand
- C08F4/64158—ONO
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65908—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an ionising compound other than alumoxane, e.g. (C6F5)4B-X+
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65916—Component covered by group C08F4/64 containing a transition metal-carbon bond supported on a carrier, e.g. silica, MgCl2, polymer
Definitions
- TITLE Non-Coordinated Alkylaluminum Free Anion Modified Alumoxanes and Methods Thereof Inventors: Lubin Luo; Jo-Ann M. Canich; Alexander V. Zabula; Xuan Ye CROSS REFERENCE TO RELATED APPLICATIONS [0001]
- the present disclosure relates to aluminoxane compositions substantially or completely undetectable hydrocarbyl aluminum content, methods of forming such alumoxane compositions, catalyst systems having the alumoxane compositions, and methods of polymerizing olefins using catalyst systems having the alumoxane compositions.
- BACKGROUND [0003] Olefin polymerization catalysts are of great use in industry. Hence, there is interest in finding new catalyst systems that increase the commercial usefulness of the catalyst and allow the production of polymers having improved properties. In order to polymerize monomers to form polyolefins, catalysts are activated to provide an active site on the catalyst and promote polymerization of the monomers.
- MAO Active methylaluminoxane
- TMA trimethylaluminum
- MAO has become the aluminum co- catalyst (also called an activator) of choice in the industry. It is available commercially in the form of 10 to 30 wt% solutions in an aromatic diluent, typically toluene.
- WO 2009/029857 shows dimethylaluminum cation (AlMe 2 + ) formation from MAO upon treatment of MAO with a Lewis base, e.g., tetrahydrofuran, in a toluene solution.
- a Lewis base e.g., tetrahydrofuran
- Lewis base stabilized dialkylaluminum cation such as AlMe 2 + can also be derived from non-MAO sources and used as metallocene catalyst activators; see for example Klosin et al., WO 2000/011006, and Organometallics, 2000, v.19, pp.4684-4686; US 9090720 shows a metallocene with dimethoxy leaving groups ethylenebisindenylzirconium dimethoxide (EtInd 2 Zr(OMe) 2 ) extracts AlMe 2 + from MAO to form a [EtInd 2 Zr( ⁇ -OMe) 2 AlMe 2 ] + species, which are slowly alkylated to form fully activated species [EtInd 2 Zr( ⁇ -Me) 2 AlMe 2 ] + , as a strong evidence of AlMe 2 + activation from MAO.
- EtInd 2 Zr(OMe) 2 ethylenebisindenylzirconium dimethoxid
- the fully activated [EtInd 2 Zr( ⁇ -Me) 2 AlMe 2 ] + species is similar to other MAO activated metallocenes that also form the metallocene-dialkylaluminum cation species, for example, [Cp 2 Zr( ⁇ -Me) 2 AlMe 2 ] + or [Cp 2 Ti( ⁇ -Me) 2 AlMe 2 ] + , such as examples in Babushkin and Brintzinger, J. Am. Chem. Soc., 2002, v.124, pp.12869-12873, and Sarzotti et al., J. Polymer Sci.
- the coordinated TMA is believed to be in equilibrium with free TMA because the attempt to physically remove all free TMA results in the loss of both free and coordinated TMA and the formation of the more thermally stable MAO gel, which become much less useful due to its insolubility that becomes unsupportable to form supported finished catalysts dominantly used in both gas- and slurry-phase polymerizations or not possible to use in solution polymerization.
- the equilibrium is shown in Scheme 1 using the Sinn’s fresh MAO formula as an attempt to help understanding of the gelation process.
- CGC constrained-geometry-complex
- the present disclosure relates to an active aluminoxane composition with undetectable or low free alkylaluminum content, methods of forming such an active aluminoxane composition, catalyst systems comprising such an active aluminoxane composition, and methods of polymerizing olefins using catalyst systems comprising such an active aluminoxane composition.
- the aluminoxane composition with undetectable or low free aluminum alkyl content is an electron withdrawing group modified aluminoxane composition containing about 8.5 mol% or less of THF extractable alkylaluminum, based on total aluminum content of the aluminoxane composition.
- a method of making a alumoxane composition with undetectable or lower in free trialkylaluminum includes introducing an alumoxane with an electron withdrawing compound containing at least one electron withdrawing group to form an electron withdrawing group modified alumoxane composition.
- the method includes introducing a hydrocarbyl aluminum compound with an oxygen source at a temperature of about -60°C to about -5°C to form the alumoxane composition.
- the aluminoxane composition with undetectable or low free trialkylaluminum content is a methylaluminoxane (MAO) composition formed by contacting an electron withdrawing compound capable of reducing the THF extractable total trimethylaluminum (TMA) in a unsupported or a supported MAO composition to 8.5 mol% or lower, based on total aluminum content of the MAO composition.
- MAO methylaluminoxane
- a catalyst system comprises a pre-catalyst compound and an MAO composition with undetectable or low free trialkylaluminum content, wherein the MAO composition with undetectable or low free TMA content includes an MAO, an electron withdrawing group-containing hydrocarbyl aluminum compound, and about 8.5 mol% or less of THF extractable total trialkylaluminum, based on total aluminum content of the MAO composition.
- the methods of forming the active MAO composition with undetectable or low free TMA content comprise a method of in-situ conversion of the THF extractable TMA in the unsupported or supported MAO to AlMe 2 X, a compound capable of serving as a coordinated and free TMA equilibrium blocking agent (so-called a TEB agent), with a compound containing at least one electron withdrawing group X, so-called an electron withdrawing compound.
- a TEB agent a compound capable of serving as a coordinated and free TMA equilibrium blocking agent
- the methods of forming the active MAO composition with undetectable or low free TMA content comprise the in-situ conversion of the majority or all of the THF extractable TMA in the MAO composition to a TEB agent AlMe 2 X through bringing into contact of an electron withdrawing compound containing at least one electron withdrawing group X and an unsupported or supported MAO composition, wherein X is a fluorine atom or a perfluorinated aryloxy group.
- an alumoxane composition with undetectable or low free trialkylaluminum content includes an alumoxane, a TEB agent AlR 2 X, wherein R is C 1 to C 10 hydrocarbyl group and the two R can be the same or different, and about 2 wt% Al or less as free or dimeric trihydrocarbyl aluminum compound AlR 3 , based on total aluminum content of the alumoxane.
- a catalyst system comprises a pre-catalyst compound and a alumoxane composition with undetectable or low free trialkylaluminum content, wherein the aluminoxane composition with undetectable or low free trialkylaluminum content comprises an alumoxane, a TEB agent AlR2X, where R is C1 to C10 hydrocarbyl group and the two R can be the same or different, and about 2 wt% Al or less as free or dimeric hydrocarbyl aluminum compounds AlR3, based on total aluminum content of the alumoxane.
- the methods of forming the supported or solid MAO composition with undetectable or low free TMA content comprise a pre-formed AlR2X treatment and a free TMA removal process, e.g., a filtration or decantation step.
- the electron withdrawing compound used to form the TEB agent comprises at least one Si-F unit.
- methods of polymerizing olefins include using catalyst systems. BRIEF DESCRIPTION OF THE DRAWINGS [0022]
- FIG. 1 is a graph illustrating ethylene uptake of post-metallocene Complex 6 finished catalyst using an inventive activator, according to an embodiment. [0023] FIG.
- FIGs.3A-B illustrate 1 H NMR spectra of an inventive activator (3A) showing less THF extractable TMA(THF) and more AlMe 2 + (THF) 2 with inert species SiMe4 and [(NHAlMe) 3 ] 2 vs. regular MAO (4B), both with toluene solvent as the reference, according to an embodiment.
- FIGs.4A-B illustrate 1 H NMR spectra of a 30% commercial MAO solution after KF treatment; with 4A) showing the upper solution phases after 2, 4, 7, and 10 mol% KF treatment, respectively; and with 4B) showing the final K + (F-MAO)- clathrate phase (b) and the non-treated solution MAO (a) for comparison.
- FIG. 4A illustrates 1 H NMR spectra of a 30% commercial MAO solution after KF treatment; with 4A) showing the upper solution phases after 2, 4, 7, and 10 mol% KF treatment, respectively; and with 4B) showing the final K + (F-MAO)- clathrate phase (b) and the non-treated solution MAO (a) for comparison.
- FIG.4A-B illustrate 1 H NMR spectra of a 30% commercial MAO solution after KF treatment; with 4A) showing the upper solution phases after 2, 4, 7, and 10 mol% KF treatment, respectively; and with 4B) showing the final K + (F-MAO)- cla
- FIG. 5 is a graph illustrating the solution ethylene-butadiene copolymerization activities of three Group 3 post-metallocenes Complex 36, 34, and 35 activated with the inventive TMA free MAO (TF-MAO), respectively to compare with the regular MAO solution and the perfluoraromatic boron/Al i Bu 2 H activator systems, according to an embodiment.
- TF-MAO TMA free MAO
- An “olefin,” alternatively referred to as “alkene,” is a linear, branched, or cyclic compound of carbon and hydrogen having at least one double bond.
- a polymer or copolymer when referred to as comprising an olefin, the olefin present in such polymer or copolymer is the polymerized form of the olefin.
- a copolymer when a copolymer is said to have an "ethylene" content of 35 wt% to 55 wt%, it is understood that the mer unit in the copolymer is derived from ethylene in the polymerization reaction and said derived units are present at 35 wt% to 55 wt%, based upon the weight of the copolymer.
- a “polymer” has two or more of the same or different mer units.
- a “homopolymer” is a polymer having mer units that are the same.
- a “copolymer” is a polymer having two or more mer units that are different from each other.
- a “terpolymer” is a polymer having three mer units that are different from each other. Accordingly, the definition of copolymer, as used herein, includes terpolymers and the like. “Different” as used to refer to mer units indicates that the mer units differ from each other by at least one atom or are different isomerically.
- ethylene polymer or "ethylene copolymer” is a polymer or copolymer comprising at least 50 mol% ethylene derived units
- a "propylene polymer” or “propylene copolymer” is a polymer or copolymer comprising at least 50 mole% propylene derived units, and so on.
- Ethylene shall be considered an ⁇ -olefin.
- metalocene refers to a catalyst compound containing two substituted or unsubstituted cyclopentadienyl moieties, bridging or non-bridging together, where the two cyclopentadienyl moieties bind directly to the transition metal center having at least two leaving groups when the metal center is charge neutral or having at least one leaving group and an optional weak donor when it bears a positive charge;
- half-metallocene refers to a catalyst compound containing one substituted or unsubstituted cyclopentadienyl moiety and a heteroatom containing ligand, bridging or non-bridging together, where the cyclopentadienyl moiety and at least one of the heteroatom on the heteroatom containing ligand bind directly to the transition metal center having at least two leaving groups when the metal center is charge neutral or having at least one leaving group and an optional weak donor when the metal center bears a positive charge, including so-called “constrained
- post-metallocene refers to a catalyst compound containing no cyclopentadienyl moiety but ligands with hetero-atoms, e.g., N, O, P, B, S, and the like, directly binding to the catalyst metal center having at least two leaving groups when it is charge neutral or having at least one leaving group and an optional weak donor when it bears a positive charge.
- aluminoxane and “alumoxane” are used interchangeably to refer to the composition made from the reaction of a trialkylaluminum, e.g., C 1 -C 10 trialkylaluminum or the mixture thereof, with an oxygen source, which may or may not include the coordinated and free trialkylaluminum.
- the term “MAO” can refer to the MAO composition that includes MAO, coordinated TMA, free TMA, and gel, e.g., species in Scheme 1, but can sometimes refer to just the MAO main molecule only, e.g., (Al 4 O 3 Me 6 ) 4 , without the coordinated TMA and free TMA.
- Al-alkyl or alkylaluminum means compounds containing at least one Al-alkyl (Al-R, where R is a C 1 to C 12 hydrocarbyl group) unit and may be coordinated to or not coordinated to the main aluminoxane structure.
- Al-R Al-alkyl
- R is a C 1 to C 12 hydrocarbyl group
- Such a compound if not coordinated to the main aluminoxane structure is also called a non-coordinated aluminumalkyl or a free aluminumalkyl, which may coordinate to each other to form a dimer, for example, the AlMe3 dimer in Scheme 1.
- Non-coordinated alkylaluminum or “free alkylaluminum” has the same meaning to represent an aluminum compound having at least one alkyl group, e.g., Me, Et, iBu, Oct, in the form of either monomer or dimer that is not chemically bound to the aluminoxane structure.
- the free alkylaluminum can exchange with the coordinated alkylaluminum on the aluminoxane structure to become coordinated, the regeneration of the free alkylaluminum from the originally coordinated alkylaluminum maintains the free alkylaluminum concentration under the same conditions.
- aluminoxane alumoxane
- alkylaluminoxane alkylaluminoxane
- alkylalumoxane alkylaluminoxane
- alkylalumoxane usually used to represent free alkylaluminum.
- TMA usually used to represent free alkylaluminum, e.g., TMA means free TMA.
- Free or “free of” means undetectable with the current analytical methods, such as NMR spectroscopy or a conventional wet titration method.
- Low in means 2 wt% or 2 mol% or less based on total same element in the system, e.g., low in free TMA means the Al weight (or mol) of the free TMA content is 2 wt% (or mol%) or less based on the total Al weight (or mol) in the MAO composition.
- free or “free of” includes the “low in description, e.g., TMA free MAO may indicate the Al weight or molar number of the free TMA content is 2 wt% or 2 mol% or less based on the total Al content in MAO.
- the term “undetectable” means a species quantification result from an analytical method, e.g., an NMR measurement method or a chemical titration method, is zero or near zero.
- the terms “anion modified alkylaluminoxane”, “anion modified aluminoxane”, “electron-withdrawing group modified alkylaluminoxane”, “electron-withdrawing group modified aluminoxane”, and “F-MAO” have the similar meaning and are used interchangeably.
- the term “electron withdrawing group” (EWG) may refer to an atom or a group X capable of withdrawing electron from the atom where X is directly bonded to, as defined in organic chemistry.
- EWC electron withdrawing compound
- (NH 4 ) 2 SiF 6 , SiF 4 , HOC 6 F 5 and the like can be used to react with AlMe 3 , AlEt 3 , AlOct 3 to form AlMe 2 F, AlEt 2 F, AlOct 2 F, AlMe 2 (OC 6 F 5 ), AlEt 2 (OC 6 F 5 ), and AlOct 2 (OC 6 F 5 ), respectively, either in-situ in an MAO composition or ex-situ then adding to an MAO composition; KF, NaF, K(OC 6 F 5 ), Na(OC 6 F 5 ) and the like can be used to react with AlMe 2 Cl, AlMe 2 Br to form AlMe 2 F, AlMe 2 (OC 6 F 5 ), respectively, which can then be separated from the byproduct metal salts, such as KCl or NaCl, before adding to an aluminoxane composition.
- TEB coordinated and free TMA equilibrium blocking agent
- Such a compound is cable of replacing the coordinated TMA in an MAO composition therefore to eliminate or limit the conversion of coordinated TMA to free TMA while maintaining the capability of providing AlR 2 + as the active site (e.g., Scheme 4).
- the TEB can form in-situ or preform through bringing into contact of a so-called electron withdrawing compound as defined above with either the AlR 3 component in an aluminoxane composition or a neat AlR 3 or AlR 2 Y compound, where Y is a non-fluorine halide, such as Cl, Br.
- Y is a non-fluorine halide, such as Cl, Br.
- Cn means hydrocarbon(s) having n carbon atom(s) per molecule, wherein n is a positive integer.
- hydrocarbon means a class of compounds containing hydrogen bound to carbon, and encompasses (i) saturated hydrocarbon compounds, (ii) unsaturated hydrocarbon compounds, and (iii) mixtures of hydrocarbon compounds (saturated and/or unsaturated), including mixtures of hydrocarbon compounds having different values of n.
- a “C m -C y ” group or compound refers to a group or compound comprising carbon atoms at a total number thereof in the range from m to y.
- a C 1 -C 50 alkyl group refers to an alkyl group comprising carbon atoms at a total number thereof in the range from 1 to 50.
- hydrocarbyl radical hydrocarbyl group
- hydrocarbyl group hydrocarbyl
- hydrocarbyl hydrocarbyl
- Hydrocarbyls may be C 1 -C 100 radicals that may be linear, branched, or cyclic, and when cyclic, aromatic or non-aromatic.
- radicals include, but are not limited to, alkyl groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, and the like, aryl groups, such as phenyl, benzyl, naphthalenyl, and the like.
- alkyl groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl, cyclopropyl, cyclobut
- substituted means that at least one hydrogen atom has been replaced with at least one non-hydrogen group, such as a hydrocarbyl group, a heteroatom, or a heteroatom containing group, such as halide (such as Br, Cl, F or I) or at least one functional group such as -NR* 2 , -OR*, -SeR*, -TeR*, -PR* 2 , -AsR* 2 , -SbR* 2 , -SR*, -BR* 2 , -SiR* 3 , -GeR* 3 , -SnR* 3 , -PbR* 3 , where each R* is independently a hydrocarbyl or halocarbyl radical, and two or more R* may join together to form a substituted or un
- substituted hydrocarbyl means a hydrocarbyl radical in which at least one hydrogen atom of the hydrocarbyl radical has been substituted with at least one heteroatom (such as halide, e.g., Br, Cl, F or I) or heteroatom-containing group (such as a functional group, e.g., -NR* 2 , -OR*, -SeR*, -TeR*, -PR* 2 , -AsR* 2 , -SbR* 2 , -SR*, -BR* 2 , -SiR* 3 , -GeR* 3 , -SnR* 3 , -PbR* 3 , where each R* is independently a hydrocarbyl or halocarbyl radical, and two or more R* may join together to form a substituted or unsubstituted completely saturated, partially unsaturated, or aromatic cyclic or polycyclic ring structure), or
- aryl or "aryl group” means an aromatic ring and the substituted variants thereof, such as phenyl, 2-methyl-phenyl, xylyl, 4-bromo-xylyl.
- heteroaryl means an aryl group where a ring carbon atom (or two or three ring carbon atoms) has been replaced with a heteroatom, such as N, O, or S.
- aromatic also refers to pseudoaromatic heterocycles which are heterocyclic substituents that have similar properties and structures (nearly planar) to aromatic heterocyclic ligands, but are not by definition aromatic; likewise, the term aromatic also refers to substituted aromatics.
- substituted aromatic means an aromatic group having 1 or more hydrogen groups replaced by a hydrocarbyl, substituted hydrocarbyl, heteroatom or heteroatom containing group.
- a "substituted phenolate” is a phenolate group where at least one, two, three, four or five hydrogen atoms in the 2, 3, 4, 5, and/or 6 positions has been replaced with at least one non-hydrogen group, such as a hydrocarbyl group, a heteroatom or heteroatom-containing group, such as halogen (such as Br, Cl, F or I) or at least one functional group such as -NR* 2 , -OR*, -SeR*, -TeR*, -PR* 2 , -AsR* 2 , -SbR* 2 , -SR*, -BR* 2 , -SiR* 3 , -GeR* 3 , -SnR* 3 , -PbR* 3 ,
- a "substituted phenolate" group in the catalyst compounds described herein is represented by the formula: where R 18 is hydrogen, C 1 -C 40 hydrocarbyl (such as C 1 -C 40 alkyl) or C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom-containing group, E 17 is oxygen, sulfur, or NR 17 , and each of R 17 , R 19 , R 20 , and R 21 is independently selected from hydrogen, C 1 -C 40 hydrocarbyl (such as C 1 -C 40 alkyl) or C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom- containing group, or two or more of R 18 , R 19 , R 20 , and R 21 are joined together to form a C 4 -C 62 cyclic or polycyclic ring structure, or a combination thereof, and the wavy line shows where the substituted phenolate group forms bonds to the rest of the catalyst compound.
- alkyl substituted phenolate is a phenolate group where at least one, two, three, four or five hydrogen atoms in the 2, 3, 4, 5, and/or 6 positions has been replaced with at least one alkyl group, such as a C 1 to C 40 , alternately C 2 to C 20 , alternately C 3 to C 12 alkyl, such as methyl, ethyl, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, adamantyl and the like including their substituted analogues.
- alkyl group such as a C 1 to C 40 , alternately C 2 to C 20 , alternately C
- An "aryl substituted phenolate” is a phenolate group where at least one, two, three, four or five hydrogen atoms in the 2, 3, 4, 5, and/or 6 positions has been replaced with at least one aryl group, such as a C 1 to C 40 , alternately C 2 to C 20 , alternately C 3 to C 12 aryl group, such as phenyl, 4-fluorophenyl, 2-methylphenyl, 2-propylphenyl, 2,6-dimethylphenyl, mesityl, 2-ethylphenyl, naphthalenyl, and the like including their substituted analogues.
- aryl group such as a C 1 to C 40 , alternately C 2 to C 20 , alternately C 3 to C 12 aryl group, such as phenyl, 4-fluorophenyl, 2-methylphenyl, 2-propylphenyl, 2,6-dimethylphenyl, mesityl, 2-ethylphenyl, n
- ring atom means an atom that is part of a cyclic ring structure.
- a benzyl group has six ring atoms and tetrahydrofuran has 5 ring atoms.
- a heterocyclic ring also referred to as a heterocycle, is a ring having a heteroatom in the ring structure as opposed to a “heteroatom-substituted ring” where a hydrogen on a ring atom is replaced with a heteroatom.
- tetrahydrofuran is a heterocyclic ring and 4-N,N-dimethylamino-phenyl is a heteroatom substituted ring.
- a substituted heterocyclic ring means a heterocyclic ring having 1 or more hydrogen groups replaced by a hydrocarbyl, substituted hydrocarbyl, heteroatom or heteroatom containing group.
- a substituted hydrocarbyl ring means a ring comprised of carbon and hydrogen atoms having 1 or more hydrogen groups replaced by a hydrocarbyl, substituted hydrocarbyl, heteroatom or heteroatom containing group.
- the term “substituted” means that a hydrogen group has been replaced with a hydrocarbyl group, a heteroatom or heteroatom-containing group, such as halogen (such as Br, Cl, F or I) or at least one functional group such as -NR* 2 , -OR*, -SeR*, -TeR*, -PR* 2 , -AsR* 2 , -SbR* 2 , -SR*, -BR* 2 , -SiR* 3 , -GeR* 3 , -SnR* 3 , -PbR* 3 , and the like, where each R* is independently hydrogen, a hydrocarbyl or halocarbyl radical, and two or more R* may join together to form a substituted or unsubstituted completely saturated, partially unsaturated, or aromatic cyclic
- a tertiary hydrocarbyl group possesses a carbon atom bonded to three other carbon atoms.
- tertiary hydrocarbyl groups are also referred to as tertiary alkyl groups.
- tertiary hydrocarbyl groups include tert-butyl, 2-methylbutan-2-yl, 2-methylhexan-2-yl, 2-phenylpropan-2-yl, 2-cyclohexylpropan-2-yl, 1-methylcyclohexyl, 1-adamantyl, bicyclo[2.2.1]heptan-1-yl and the like.
- Tertiary hydrocarbyl groups can be illustrated by the formula: , wherein R A , R B and R C are independently hydrocarbyl groups or substituted hydrocarbyl groups that may optionally be bonded to one another, and the wavy line shows where the tertiary hydrocarbyl group forms bonds to other groups.
- a cyclic tertiary hydrocarbyl group is defined as a tertiary hydrocarbyl group that forms at least one alicyclic (non-aromatic) ring. Cyclic tertiary hydrocarbyl groups are also referred to as alicyclic tertiary hydrocarbyl groups.
- cyclic tertiary hydrocarbyl groups are also referred to as cyclic tertiary alkyl groups or alicyclic tertiary alkyl groups.
- cyclic tertiary hydrocarbyl groups include 1-adamantyl, 1-methylcyclohexyl, 1-methylcyclopentyl, 1-methylcyclooctyl, 1-methylcyclodecyl, 1-methylcyclododecyl, bicycle[3.3.1]nonan-1-yl, bicyclo[2.2.1]heptan-1- yl, bicyclo[2.3.3]hexan-1-yl, bicycle[1.1.1]pentan-1-yl, bicycle[2.2.2]octan-1-yl, and the like.
- Cyclic tertiary hydrocarbyl groups can be illustrated by Formula (B): wherein R A is a hydrocarbyl group or substituted hydrocarbyl group, each R D is independently hydrogen or a hydrocarbyl group or substituted hydrocarbyl group, w is an integer from 1 to about 30, and R A , and one or more R D , and or two or more R D may optionally be bonded to one another to form additional rings.
- R A is a hydrocarbyl group or substituted hydrocarbyl group
- each R D is independently hydrogen or a hydrocarbyl group or substituted hydrocarbyl group
- w is an integer from 1 to about 30
- R A and one or more R D , and or two or more R D may optionally be bonded to one another to form additional rings.
- a cyclic tertiary hydrocarbyl group contains more than one alicyclic ring, it can be referred to as polycyclic tertiary hydrocarbyl group or if the hydrocarbyl group is an alkyl group, it may be referred to as a polycyclic tertiary alkyl group.
- alkyl radical and “alkyl” are used interchangeably throughout this disclosure.
- alkyl radical is defined to be C1-C100 alkyls that may be linear, branched, or cyclic.
- radicals can include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, and the like including their substituted analogues.
- Substituted alkyl radicals are radicals in which at least one hydrogen atom of the alkyl radical has been substituted with at least a non-hydrogen group, such as a hydrocarbyl group, a heteroatom or heteroatom-containing group, such as halogen (such as Br, Cl, F or I) or at least one functional group such as -NR* 2 , -OR*, -SeR*, -TeR*, -PR* 2 , -AsR* 2 , -SbR* 2 , -SR*, -BR* 2 , -SiR* 3 , -GeR* 3 , -SnR* 3 , -PbR* 3 , and the like, where each R* is independently hydrogen, a hydrocarbyl or halocarbyl radical, and two or more R* may join together to form a substituted or unsubstituted completely saturated, partially unsaturated, or aromatic cyclic or polycyclic ring
- isomers of a named alkyl, alkenyl, alkoxide, or aryl group exist (e.g., n-butyl, iso-butyl, sec-butyl, and tert-butyl)
- reference to an alkyl, alkenyl, alkoxide, or aryl group without specifying a particular isomer e.g., butyl
- expressly discloses all isomers e.g., n-butyl, iso-butyl, sec-butyl, and tert-butyl).
- Mn is number average molecular weight
- Mw is weight average molecular weight
- Mz is z average molecular weight
- wt% is weight percent
- mol% is mole percent.
- Molecular weight distribution also referred to as polydispersity index (PDI)
- PDI polydispersity index
- Me is methyl
- Et is ethyl
- MAO MAO
- TMS trimethylsilyl
- Oct is octyl
- Bu is butyl
- iPr is isopropyl
- Bn is benzyl (i.e., CH 2 Ph)
- THF also referred to as thf
- RT room temperature (and is 23 °C unless otherwise indicated)
- tol is toluene
- Cp is cyclopentadienyl
- NMR nuclear magnetic resonance
- TMA is trimethylaluminum.
- a catalyst system is a combination of at least one pre-catalyst compound, an activator, an optional coactivator, and an optional support material.
- catalyst system means the unactivated catalyst complex (pre- catalyst) together with an activator and, optionally, a coactivator.
- pre- catalyst unactivated catalyst complex
- coactivator When it is used to describe such a pair after activation, it means the activated complex and the activator or other charge- balancing moiety.
- the catalyst compound may be neutral as in a pre-catalyst, or a charged species with a counter ion as in an activated catalyst system.
- catalyst systems are described as including neutral stable forms of the components, it is well understood by one of ordinary skill in the art, that the ionic form of the component is the form that reacts with the monomers to produce polymers.
- a polymerization catalyst system is a catalyst system that can polymerize monomers to polymer.
- catalyst compounds and activators represented by formulae herein embrace both neutral and ionic forms of the catalyst compounds and activators.
- the catalyst may be described as a catalyst, a catalyst precursor, a pre-catalyst compound, catalyst compound or a transition metal compound, and these terms are used interchangeably.
- anionic ligand is a negatively charged ligand which donates one or more pairs of electrons to a metal ion.
- anionic donor is used interchangeably with “anionic ligand”.
- anionic donors may include, but are not limited to, methyl, chloride, fluoride, alkoxide, aryloxide, alkyl, alkenyl, thiolate, carboxylate, amido, benzyl, hydrido, amidinate, amidate, and phenyl. Two anionic donors may be joined to form a dianionic group.
- a “neutral Lewis base” or “neutral donor group” is an uncharged (neutral) group which donates one or more pairs of electrons to a metal ion.
- neutral Lewis bases include ethers, thioethers, amines, phosphines, ethyl ether, tetrahydrofuran, dimethylsulfide, triethylamine, pyridine, alkenes, alkynes, alenes, and carbenes.
- Lewis bases may be joined together to form bidentate or tridentate Lewis bases.
- phenolate donors can include Ph-O-, Ph-S-, and Ph-N(R ⁇ )- groups, where R ⁇ is hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom-containing group, and Ph is optionally substituted phenyl.
- R ⁇ is hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom-containing group, and Ph is optionally substituted phenyl.
- a method of making an electron withdrawing group modified alumoxane includes introducing an alumoxane with an electron withdrawing group to form an alumoxane.
- a method of making a low trialkylaluminum alumoxane includes introducing an alumoxane with an electron-withdrawing compound to form a strong electron-withdrawing atom or group modified alumoxane. The method includes introducing a hydrocarbyl aluminum compound with an oxygen source at a temperature of about -60°C to about 0°C to form the alumoxane.
- an electron withdrawing group modified alumoxane includes an alumoxane, an electron withdrawing group-containing hydrocarbyl aluminum compound, and about 2 wt% Al or less from free or dimeric hydrocarbyl aluminum compounds, based on total aluminum content of the alumoxane composition.
- a catalyst system comprises a catalyst compound and an electron withdrawing group modified alumoxane.
- the electron withdrawing group modified alumoxane includes an alumoxane, an electron withdrawing group-containing hydrocarbyl aluminum compound, and about 2 wt% Al or less of from free or dimeric hydrocarbyl aluminum compounds, based on total aluminum content of the alumoxane composition.
- methods of polymerizing olefins include using catalyst systems.
- the presence of the electron withdrawing group containing content of the treated alumoxane provides more aluminum cations associated with the alumoxane.
- the ratio of strong electron withdrawing atoms to hydrocarbyl aluminum compounds is about 1:1, the amount of free hydrocarbyl aluminum compound (such as trimethylaluminum) present in a catalyst system can be reduced or eliminated, which provides reduced side reaction of oxygen-containing catalyst compounds and/or nitrogen-containing catalyst compounds with aluminum in the catalyst system.
- the presence of strong electron withdrawing atoms reduces formation of the stable hydrocarbyl aluminum dimers (e.g., dimeric Al(CH 3 ) 3 ) that would otherwise form in the absence of strong electron withdrawing atoms.
- the presence of strong electron withdrawing atoms instead forms more active aluminum cations.
- the presence of an electron withdrawing compound in the treated alumoxane likewise reduces or eliminates side reactions of oxygen-containing catalyst compounds and/or nitrogen-containing catalyst compounds, promoting improved catalyst activity and lifetime.
- hydrocarbyl aluminum compounds having an electron withdrawing atom can be formed in-situ upon forming the alumoxane.
- an electron withdrawing compound can provide multiple electron withdrawing atoms (e.g., (NH 4 ) 2 SiF 6 ) to multiple available aluminum atoms, which promotes atom economy. Many such electron withdrawing compounds are commercially available and relatively inexpensive. Use of such electron withdrawing compounds reduces the cost and atom economy of forming overall catalyst systems which likewise improves cost and atom economy of polymers produced from such catalyst systems.
- alumoxanes of the present disclosure can be supported or unsupported with one or more support particles (e.g., silica).
- support particles e.g., silica
- fluorinated supports are known, it has been discovered that the fluorine atoms are not completely converted to aluminum- fluorine-type alumoxane. Instead, upon calcination of a fluorinated-support, a significant amount of HF and SiF4 gases are formed which has been shown to be difficult to control.
- alumoxanes and methods of the present disclosure do not form HF and/or SiF 4 gases due to the very strong bond strength of aluminum-fluorine atoms.
- the present disclosure relates to TMA free active MAO composition, methods of forming such an MAO composition, catalyst systems having TMA free active MAO, and methods of polymerizing olefins using catalyst systems having TMA free active MAO, wherein TMA free MAO means that the free TMA content in MAO is zero or near zero while the MAO active sites capable of providing AlMe 2 + are maintained or increased through the treatment of a so-called coordinated and free TMA equilibrium blocking agent (TEB agent).
- TMA free MAO means that the free TMA content in MAO is zero or near zero while the MAO active sites capable of providing AlMe 2 + are maintained or increased through the treatment of a so-called coordinated and free TMA equilibrium blocking agent (TEB agent).
- TMA free MAO means that the free TMA content in MAO is zero or near zero while the MAO active sites capable of providing AlMe 2 + are maintained or increased through the treatment of a so-called coordinated and free TMA equilibrium blocking agent (TEB agent).
- TEB agent coordinated and free TMA
- the TMA free active MAO composition with maintained or improved activity is made possible, without being bound by theory, where either the fluorinated silica or the electron withdrawing compound (e.g., (NH 4 ) 2 SiF 6 ), both containing Si-F units, are capable of converting free TMA in MAO to AlMe 2 F, which then replaces the coordinated TMA in MAO to form the new active site that not only blocks coordinated TMA to free TMA equilibrium, but also is capable of releasing more AlMe 2 + for pre-catalyst ionization as well as reducing the ion-pair interaction to increase the individual active molecule activity due to the F atom electron withdrawing effect (Scheme 4): Scheme 4 [0080] Without being bound by theory, the replacement of coordinated TMA with AlMe 2 F converts the equilibrium of Scheme 1 (Scheme 5 a) to Scheme 5 b therefore efficiently blocks the free and coordinated TMA equilibrium, presumably due to the presence of
- a method of making a TMA free MAO includes the treatment of MAO, in a solution or a supported form, with an electron withdrawing compound capable of converting the total TMA (free and coordinated TMA) to AlMe 2 F as the major derivative and an optional minor non-fluorinated inert aluminumalkyl derivative, depending on the electron withdrawing compound structure in use.
- a method of making a TMA free MAO composition in solution or supported form includes introducing a solution or supported MAO composition containing free TMA and coordinated TMA and an electron withdrawing compound containing at least one strong electron withdrawing atom or group X capable of converting free TMA to AlMe 2 X to form a modified MAO composition with undetectable or low free TMA content.
- the method includes introducing a hydrocarbyl aluminum compound with an oxygen source optionally in a support at a temperature of about -60°C to about 0°C to form a regular MAO composition before the fluorination treatment.
- a TMA free MAO composition includes an electron withdrawing group modified MAO in solution form or supported form with about 8.5 mol% or less of THF extractable trihydrocarbyl aluminum compounds, based on total aluminum content of the MAO.
- a catalyst system comprises a catalyst compound and a TMA free MAO composition in solution or supported form, wherein the TMA free MAO composition includes an electron withdrawing group modified MAO in solution form or supported form with about 8.5 mol% or less of THF extractable trihydrocarbyl aluminum compounds, based on total aluminum content of the MAO.
- methods of making the TMA free supported MAO composition include the treatment of the supported MAO with an electron withdrawing compound and a trialkylaluminum, following a filtration step to remove the excess free TMA.
- methods of making the TMA free supported MAO composition include the treatment of the support with an electron withdrawing compound before the MAO supportation, but with the reactive fluorine atoms on the support adjusted to match the total TMA in the MAO later loaded in the support to obtain a TMA free supported MAO composition.
- methods of polymerizing olefins include using catalyst systems.
- an electron withdrawing group modified alkylaluminum species can be formed and coordinated with the MAO, to form a more reactive aluminum species of the MAO, which has been discovered to promote catalyst activity and catalyst lifetime. Without being bound by theory, it is believed the presence of the electron withdrawing group content of the treated MAO provides more aluminum cations associated with the MAO as shown in Scheme 4.
- the ratio of strong electron withdrawing atoms or groups to hydrocarbyl aluminum compounds is about 1:1, the amount of free trihydrocarbyl aluminum compound (such as trimethylaluminum) present in a catalyst system can be reduced or eliminated, which therefore reduces the decomposition reactions of oxygen-containing catalyst compounds and/or nitrogen-containing catalyst compounds with the oxygen or nitrogen reactive trihydrocarbyl aluminum in the catalyst system.
- the presence of fluorine atoms converts the most reactive primary trihydrocarbyl aluminum (e.g., dimeric form of AlMe3) to a less active secondary dihydrocarbyl aluminum (e.g., as Al(CH 3 ) 2 F) therefore to reduce or eliminate the formation of the primary trihydrocarbyl aluminum as the equilibrium reaction of coordinated AlMe 2 F in the MAO composition shown in Scheme 5 that would otherwise form in the absence of fluorine atoms as the equilibrium reaction of coordinated TMA in regular MAO shown in Scheme 5.
- the presence of fluorine atoms instead forms more aluminum cations and more active ion-pair due to weaker ion-pair interaction, as shown in Scheme 4.
- the presence of fluorine in the treated MAO likewise reduces or eliminates the chance of forming free TMA in the regular MAO free TMA and coordinated TMA equilibrium and thus reduces or eliminates the side reactions of oxygen-containing catalyst compounds and/or nitrogen-containing catalyst compounds, promoting improved catalyst activity and lifetime.
- a secondary aluminumalkyl such as AlMe 2 F can also be formed ex- situ and added to a supported MAO composition following a free TMA removal step, such as a filtration and wash step.
- a free TMA removal step such as a filtration and wash step.
- adding a secondary aluminumalkyl such as AlMe 2 F to a solution MAO is less preferred because of the challenging free TMA removal in the solution system.
- MAO of the present disclosure can be supported or unsupported with one or more support particles (e.g., silica).
- support particles e.g., silica
- fluorinated supports are known, it has been discovered that the support fluorination processes, e.g., those described in WO 2000/12565, generate equipment corrosive HF and SiF 4 gases as well as are difficult to obtain an accurate fluorine loading due to uncontrollable F loss.
- MAO and methods of the present disclosure do not form HF and/or SiF4 gases due to the very strong bond strength of aluminum-strong electron withdrawing atoms or groups therefore the fluorine loading can be more accurately controlled.
- Alumoxanes are oligomeric compounds containing —Al(R)—O— or —Al(R)2— O— subunits, where R is an alkyl group, typically a C 1 to C 12 alkyl group, such as the inactive MAO gel shown in Scheme 1.
- alumoxanes examples include methylalumoxane (MAO), modified methylalumoxane (MMAO), ethylalumoxane, triethylalumoxane, triisobutylalumoxane, tetraethyldialumoxane, and di-isobutylalumoxane.
- MAO methylalumoxane
- MMAO modified methylalumoxane
- ethylalumoxane triethylalumoxane
- triisobutylalumoxane triisobutylalumoxane
- tetraethyldialumoxane tetraethyldialumoxane
- di-isobutylalumoxane di-isobutylalumoxane.
- Unsupported MAO may refer to either solution MAO, such as the commercial MAO solution products produced by W. R.
- Unsupported solid MAO can be prepared through the removal of the solvent of a solution MAO product, and form controlled particle sizes with different solid formation methods, e.g., spray-drying.
- Methods for preparing MAO and modified MAO such as the methods described in U.S. Pat. No.4,542,199 and Chen and Marks, 100 Chem. Rev.1391 (2000).
- MAO can also be modified for different purposes, e.g., increasing activity, solubility. Examples of useful MAO include MAO from TMA with an oxygenate (e.g., W. R.
- Active MAO is formed from the contact of largely excess TMA with an oxygen source (such as water, metal salt coordinated water, CO 2 , methylacylic acid, benzoic acid, or other reactive oxygen containing organics) under suitable reaction conditions.
- an oxygen source such as water, metal salt coordinated water, CO 2 , methylacylic acid, benzoic acid, or other reactive oxygen containing organics
- Active MAO of the present disclosure can be prepared in-situ by contacting the hydocarbyl aluminum compound with an oxygen source, e.g., TMA with water in an aliphatic or aromatic diluent, at a temperature of less than 0°C to ⁇ 60°C, such as ⁇ 10°C to ⁇ 50°C, such as ⁇ 15°C to ⁇ 30°C.
- Supported MAO of the present disclosure can be prepared by conventional methods such as bringing into contact of a pre-formed MAO solution with a support (e.g., silica).
- the solution MAO can be added to a solid support or a support slurry or a reverse addition following by optional heating to form the supported MAO.
- Supported MAO of the present disclosure can also be prepared in-situ by contacting the hydrocarbyl aluminum compound with an oxygen source loaded in a support.
- a support material e.g., silica
- water pre-loaded in a support material e.g., silica
- a support material e.g., silica
- optional cooling can be added to a TMA solution cooled to a temperature of less than 0°C to ⁇ 60°C, such as ⁇ 10°C to ⁇ 50°C, such as ⁇ 15°C to ⁇ 30°C following a heating process as described in U.S.11,161,922; or a non-hydrolytic organic oxygenate can be mixed with TMA under cooling, e.g., at a temperature of less than 0°C to ⁇ 60°C, such as ⁇ 10°C to ⁇ 50°C, such as ⁇ 15°C to ⁇ 30°C to form a pre-MAO composition and then mix a support (
- suitable diluents for forming a support slurry are capable of dissolving MAO to ensure a good MAO distribution inside the pores of the support, such as diluents such as toluene, benzene, or xylenes.
- suitable diluents are materials in which the reactants, e.g., the hydrocarbyl aluminum such as TMA, the non-hydrolytic organic oxygenate, and the derivatives of the two reagents, are at least partially soluble and which are liquid at reaction temperatures.
- Suitable aromatic diluents can include toluene, benzene, or xylenes.
- the active alumoxane composition (such as MAO) can be exclusively formed with trimethylaluminum (TMA), but other aluminumalkyl compounds can be used to modify the MAO.
- TMA trimethylaluminum
- the hydrocarbyl aluminum compounds used for alumoxane modification can be alkylaluminum compounds such as a trialkylaluminum compound.
- the alkyl substituents can be alkyl groups of up to 10 carbon atoms, such as octyl, isobutyl, ethyl or methyl.
- suitable hydrocarbyl aluminum compounds may include trimethylaluminum, triethylaluminum, tripropylalumiuum, tri-n-butylaluminum, tri-isobutyl-aluminum, tri(2-methylpentyl)aluminum, trihexylaluminum, tri-n-octylaluminum, and tri-n-decylaluminum.
- hydrocarbyl aluminum compounds are trimethylaluminum and tri-n-octylaluminum.
- hydrocarbyl aluminum compounds are represented by the formula R 3 Al where each R is independently a hydrocarbon containing between 1 and 30 carbon atoms.
- the hydrocarbyl aluminum compound is one or more of trialkylaluminum mixtures, e.g., dimethylethylaluminum or methyldiethylaluminum from AlMe3 and AlEt3 mixture, diethylisobutylaluminum or ethyldiisobutylaluminum from AlEt3 and AliBu3 mixture, and the like.
- Oxygen Sources are any oxygen sources in which one or more oxygen atoms is able to react with the hydrocarbyl aluminum compound to form a new Al—O bond.
- the oxygen source may be or include water, such as pure water or water in a metal salt hydrate.
- the oxygen source can be one or more hydroxy or carbonyl containing compounds for example an alcohol, CO or CO 2 , an acetone, or a carboxylic acid.
- the oxygen source is one or more of carbon dioxide, a carboxylic acid, a ketone, an aldehyde, an ester, an anhydride, an alcohol, or combination thereof.
- the oxygen source is represented by the formula R 1 R 2 C ⁇ CR 3 CO 2 H wherein each of R 1 and R 2 is independently hydrogen, alkyl, alkenyl, aryl or heteroatom containing group and R 3 is alkyl, alkenyl, aryl or heteroatom containing group.
- the oxygen source includes in the hydrocarbyl aluminum compound, e.g., the reaction product of TMA with an alcohol, a ketone, an ester, or an organic acid.
- hydrocarbyl aluminum compounds which include an oxygen source include dimethyl aluminum methoxide, dimethyl aluminum ethoxide, dimethyl aluminum isopropoxide, dimethyl aluminum n-butoxide, dimethyl aluminum isobutoxide, pentamethyldialuminum-t-butoxide, tetramethyldialuminumdi-t- butoxide, pentamethyldialuminum-i-propoxide, tetramethyldialuminumdi-i-propoxide, or the mixture of the listed compounds and the like.
- the starting charging molar ratio of Al:O where O is the active oxygen in the active oxygen containing compound, can be 100:1, 60:1, 30:1, 10:1, 1:1, or 0.9:1 to form the desired MAO compositions with or without excess free hydrocarbyl aluminum compounds.
- the molar ratio of Al:O can be about 0.9:1 to about 100:1, such as about 1:1 to about 10:1, alternatively about 10:1 to about 60:1, such as about 30:1 to about 60:1.
- the oxygen source is one or more of carbon dioxide, a carboxylic acid, an ester, an anhydride, an alcohol or combination thereof.
- the oxygen source is one or more of carbon dioxide, a carboxylic acid, an ester, an anhydride and an alcohol or combination thereof, optionally containing water.
- the oxygen source is R 1 R 2 C ⁇ CR 3 CO 2 H wherein each of R 1 and R 2 is independently hydrogen, alkyl, alkenyl, aryl or heteroatom containing group and R 3 is alkyl, alkenyl, aryl or heteroatom containing group.
- the oxygen source is methacrylic acid.
- the oxygen source is a hydrocarbylboroxine as described in Welborn, US 5,001,244.
- TMA can react with hydrolytic compounds such as an alcohol ROH to rapidly form AlMe (3-n) (OR) n (n ⁇ 3) with n and the OR position depending on the ROH reactivity, steric hindrance, and the reaction conditions.
- hydrolytic compounds such as an alcohol ROH to rapidly form AlMe (3-n) (OR) n (n ⁇ 3) with n and the OR position depending on the ROH reactivity, steric hindrance, and the reaction conditions.
- Small R groups e.g., MeOH, EtOH, and t BuOH are MAO poison because, without being bound by theory, the small R groups convert both the free and coordinated TMA to form very stable oxygen bridging structures and the RO- group is a strong electron donating group that destabilizes the MAO anion (Scheme 6): Scheme 6 .
- Sterically hindered alcohols such as 3,5-di-t-butyl-4-hydroxytoluene (BHT) form terminal OR groups but may need largely excess for near TMA free system, without being bound by theory, due to the equilibrium in Scheme 7: Scheme 7 .
- BHT 3,5-di-t-butyl-4-hydroxytoluene
- Ijpeij et al., U.S. 7,956,140 uses BHT:Al from 0.5:1 to 2 ratio for MAO treatment to provide the activation of CGC catalyst precursors containing nitrogen donor ligands.
- Such a system may have a large amount of neutral BHT in the system that is not desired in some end products since MAO is usually used in a largely excess amount to ensure efficient activation and the amount of the coordinated TMA (the active site) may also decrease due to the coordinated TMA to free TMA equilibrium (Scheme 1).
- the TEB agent can then replace the coordinated TMA (which becomes free TMA) therefore to eliminate the coordinated TMA to free TMA equilibrium as well as to provide more AlMe 2 + for pre-catalyst ionization and more dispersed MAO anion charge to weaken the active ion-pair interaction due to the introduction of the strong electron withdrawing atoms on the MAO anions as shown in Scheme 4, therefore, to increase the system’s activity.
- the total TMA in MAO to TEB agent conversion is therefore a much more efficient method to remove the free TMA in MAO while maintaining or improving the activation efficiency to obtain a system that is suitable to activate pre-catalysts built with ligands containing TMA reactive hetero-atom donors, e.g., N, O, S, and/or P donors in the ligands for post-metallocene and CGC half-metallocene pre-catalysts.
- TMA reactive hetero-atom donors e.g., N, O, S, and/or P donors in the ligands for post-metallocene and CGC half-metallocene pre-catalysts.
- the inorganic fluorine containing compound having the Formula (I) is selected from NH 4 BF 4 , (NH 4 ) 2 SiF 6 , NH 4 PF 6 , NH 4 F, (NH 4 ) 2 TaF 7 , NH 4 NbF 4 , (NH 4 ) 2 GeF 6 , (NH 4 ) 2 SmF 6 , (NH 4 ) 2 TiF 6 , (NH 4 ) 2 ZrF 6 , MoF 6 , ReF 6 , GaF 3 , SO 2 ClF, F 2 , SiF 4 , SF 6 , ClF3, ClF5, BrF5, IF7, NF3, HF, BF3, B(OC6F5)3, AlF3, Al(OC6F5)3, NHF2 and NH4HF2.
- R is a C 1 to C 10 hydrocarbyl group
- M is a group 13 or 14 element
- X is an electron withdrawing atom or group
- u is the valence state of element M.
- the organic fluorine compound having the Formula (II) is selected from Me 3 SiF, Me 2 SiF 2 , MeSiF 3 , Et 3 SiF, Et 2 SiF 2 , EtSiF 3 , Ph 3 SiF, Ph 2 SiF 2 , PhSiF 3 , Me 3 CF, Me 2 CF 2 , MeCF 3 , Et 3 CF, Et 2 CF 2 , EtCF 3 , Ph 3 CF, Ph 2 CF 2 , PhCF 3 , Me 2 BF, MeBF 2 , MeAlF 2 , Et 2 BF, EtBF 2 , EtAlF 2 , Ph 2 BF, PhBF 2 , Me 3 Si(OC 6 F 5 ), Me 2 Si(OC 6 F 5 ) 2 , MeSi(OC 6 F 5 ) 3 , Me 3 C(OC 6 F 5 ), Ph 3 C(OC 6 F 5 ), Me 2 B(OC 6 F 5 ), MeB(OC 6 F 5 ) 2 ,
- the amount of the electron withdrawing compound relative to trihydrocarbyl aluminum compound (e.g., TMA) in MAO can be controlled such that after the TEB agent forms and replaces the coordinated trihydrocarbyl aluminum compound (e.g., coordinated TMA) in MAO, resulting in little or no free trihydrocarbyl aluminum compound (or dimer thereof) remained.
- TMA trihydrocarbyl aluminum compound
- a ratio of the active strong electron withdrawing atom (e.g., F) or group (e.g., C6F5O-) number of the strong electron withdrawing compound to hydrocarbyl aluminum compound is about 1.5:1 to about 1:1.5, such as about 1.3:1 to about 1:1.3, such as about 1.2:1 to about 1:1.2, such as about 1.1:1 to about 1:1.1, such as about 1.05:1 to about 1:1.05.
- the ratio is a molar ratio or alternatively is based on the number of the strong electron withdrawing atoms or groups in the strong electron withdrawing compound relative to moles of the hydrocarbyl aluminum compound, e.g., (NH 4 )SiF 6 to Al(CH 3 ) 3 would be a 8:1 molar ratio but a 6:6 ratio (i.e., 1:1 ratio) based on the number of fluorine atoms in the strong electron withdrawing compound relative to moles of the hydrocarbyl aluminum compound, plus a 2:2 ratio (i.e., 1:1 ratio) of TMA to TMA reactive NH4 + that forms an inert compound presumably with a formula of (Al 3 Me 3 N 3 H 3 ) 2 .
- the amount of free trihydrocarbyl aluminum compound (or dimer thereof) is determined after formation of the solution, solid, or supported MAO composition.
- a sample of unsupported MAO or supported MAO produced or obtained commercially can be treated with tetrahydrofuran (THF) to convert both free or coordinated TMA in MAO to a TMA-THF adduct, THF-MAO-adducts, as well as an AlMe 2 + - THF 2 adduct as shown in Scheme 8: Scheme 8 .
- THF tetrahydrofuran
- the relative amount of each adduct can be determined by nuclear magnetic resonance (NMR) spectroscopy.
- an amount of the strong electron withdrawing compound can be introduced to the MAO based on a predetermined ratio of the electron withdrawing group of the strong electron withdrawing to the free hydrocarbyl aluminum in MAO.
- a ratio of the strong electron withdrawing atoms or groups in the strong electron withdrawing compound to total trihydrocarbyl aluminum compound in MAO is about 1.5:1 to about 1:1.5, such as about 1.3:1 to about 1:1.3, such as about 1.2:1 to about 1:1.2, such as about 1.1:1 to about 1:1.1, such as about 1.05:1 to about 1:1.05.
- the ratio is a molar ratio or alternatively is based on the number of strong electron withdrawing atoms or groups in the strong electron withdrawing compound relative to moles of the trihydrocarbyl aluminum compound (e.g., (NH 4 ) 2 SiF 6 to Al(CH 3 ) 3 would be a 8:1 molar ratio but a 6:6 ratio based on the number of fluorine atoms in the strong electron withdrawing compound relative to moles of the trihydrocarbyl aluminum compound, plus a 2:2 ratio of TMA to TMA reactive NH 4 + that forms an inert compound presumably with a formula of (Al 3 Me 3 N 3 H 3 ) 2 ).
- the trihydrocarbyl aluminum compound e.g., (NH 4 ) 2 SiF 6 to Al(CH 3 ) 3 would be a 8:1 molar ratio but a 6:6 ratio based on the number of fluorine atoms in the strong electron withdrawing compound relative to moles of the trihydrocarbyl
- the TEB agent formation reaction (of electron withdrawing compound with free hydrocarbyl aluminum compound (or dimer thereof)) can proceed at any suitable temperature, such as about 0°C to about 100°C, such as about 10°C to about 30°C, such as about 20°C, such as ambient temperature.
- the reaction can proceed neat (e.g., solid-solid) or can proceed using any suitable diluent.
- a diluent can be an organic diluent, such as an aliphatic diluent or an aromatic diluent.
- Aromatic diluent may include benzene, toluene, or xylenes.
- an MAO (unsupported or supported) can have an amount of Al from free hydrocarbyl aluminum compound of about 2 wt% or less, such as about 1.5 wt% or less, such as about 1 wt% or less, such as about 0.5 wt% or less, such as about 0.25 wt% or less, such as about 0.1 wt% to about 2 wt%, such as about 0.1 wt% to about 1.5 wt%, such as about 0.2 wt% to about 1 wt%, such as about 0.3 wt% to about 0.7 wt%, based on total aluminum content of the MAO.
- the TEB agent is formed before adding to the solid or supported MAO composition, following a free TMA removal step, e.g., a filtration or decant step to remove free TMA without the requirement of TMA quantification based on the chemistry of Scheme 9 below: Scheme 9 .
- a free TMA removal step e.g., a filtration or decant step to remove free TMA without the requirement of TMA quantification based on the chemistry of Scheme 9 below: Scheme 9 .
- Preparation methods for pre-formed TEB agents include but not limited to: 1) bringing into contact of AlR 3 , where R is a C 1 to C 8 hydrocarbyl group or the mixture there of, such as Me, Et, i Bu, Oct, and preferably the Me group, with a strong electron withdrawing compound, to form in-situ the AlR 2 X compound as the major product.
- AlR 2 Y is selected from AlMe 2 Cl, AlMe 2 Br, AlMe 2 I, AlEt 2 Cl, AlEt 2 Br, AlEt 2 I, Al i Bu 2 Cl, Al i Bu 2 Br, Al i Bu 2 I, AlOct 2 Cl, AlOct 2 Br, AlOct 2 I, AlMe 2 CN, AlEt2 C N, Al i Bu 2 CN, AlOct 2 CN, and the like; and MXu is selected from LiF, NaF, KF, MgF 2 , CaF 2 , BaF 2 , LiOC 6 F 5 , NaOC 6 F 5 , KOC 6 F 5 , Mg(OC 6 F 5 ) 2 , Ca(OC 6 F 5 ) 2 , Ba(OC 6 F 5 ) 2 , and the like.
- ClMgOC 6 F 5 and the like may also be used, e.g., AlMe 2 Cl + ClMgOC 6 F 5 to form AlMe 2 OC 6 F 5 + MgCl 2 .
- An MgCl 2 agglomerator such as dioxolane can be used to oligomerize MgCl2 for better solid separation form the desired AlMe 2 F or AlMe 2 OC 6 F 5 product.
- Optional Support Materials and the Derived Trihydrocarbyl Aluminum-free Supported Alumoxanes may include a support material.
- a support material can be contacted with a pre-formed solution alumoxane, e.g., a commercial solution MAO, to form the supported MAO, followed by contacting the supported MAO with a strong electron withdrawing compound to form the TEB agent in-situ or a pre- formed TEB agent of the present disclosure.
- a support material can be contacted with TMA free MAO of the present disclosure to form a supported activator followed by contacting the supported activator with a pre-catalyst compound.
- a pre-catalyst compound is contacted with a TMA free MAO to form a solution catalyst system followed by contacting the catalyst system with a support material to form a supported catalyst system.
- a supported material can be loaded with an oxygen source, e.g., water, followed by adding the oxygen source loaded support, in a solid form or slurry for, with or without cooling, to a cold TMA solution with optional heating to form the supported MAO composition, and then followed by the treatment of either an in-situ formed TEB agent through contacting the supported MAO with a strong electron withdrawing compound with optional AlR3 and optional filtration/wash step or a pre-formed TEB agent with necessary filtration/wash step to form the TMA free supported MAO before contacting a pre-catalyst to form the finished catalyst.
- the support material can be a porous support material, for example, talc, and inorganic oxides.
- the support material can be an inorganic oxide.
- the inorganic oxide can be in a finely divided form.
- Suitable inorganic oxide materials for use in catalyst systems herein may include groups 2, 4, 13, and 14 metal oxides, such as silica, alumina, and mixtures thereof.
- Other inorganic oxides that may be employed either alone or in combination with the silica, or alumina can be magnesia, titania, zirconia.
- Other suitable support materials can be employed, for example, finely divided functionalized polyolefins, such as finely divided polyethylene.
- suitable supports may include magnesia, titania, zirconia, montmorillonite, phyllosilicate, zeolites, talc, clays. Also, combinations of these support materials may be used, for example, silica-chromium, silica-alumina, silica-titania. In at least one embodiment, the support material is selected from Al 2 O 3 , ZrO 2 , SiO 2 , SiO 2 /Al 2 O 3 , SiO 2 /TiO 2 , silica clay, silicon oxide/clay, or mixtures thereof.
- the support material such as an inorganic oxide, can have a surface area of about 10 m /g to about 800 m /g, pore volume of about 0.1 cm 3 /g to about 4.0 cm 3 /g and average particle size of about 3 ⁇ m to about 300 ⁇ m.
- the surface area of the support material can be of about 50 m 2 /g to about 500 m /g, pore volume of about 0.5 cm /g to about 3.5 cmVg and average particle size of about 10 ⁇ m to about 200 ⁇ m.
- the surface area of the support material can be about 100 m 2 /g to about 400 m 2 /g, pore volume of about 0.8 cm 3 /g to about 3.0 cm 3 /g and average particle size can be about 5 ⁇ m to about 100 ⁇ m.
- the average pore size of the support material useful in the present disclosure can be of about 50 ⁇ to about 1000 ⁇ , such as about 60 ⁇ to about 500 ⁇ , and such as about 75 ⁇ to about 350 ⁇ .
- suitable silicas can be the silicas marketed under the tradenames of DAVISONTM 952 or DAVISONTM 955 (Davison Chemical Division of W.R. Grace and Company). In other embodiments, DAVISONTM 948 is used.
- a silica can be ES-70, ES70X, ES757. PD17062, PD16042, PD16043, or PD 14024 silica (Ecovyst.
- Sipemat 310 Sipemat 50 (Evonik) that has been calcined, for example, at 200°C, 400°C, 600°C, or 875°C.
- the support material should be dry, that is, free or substantially free of absorbed water for pre-formed MAO supportation but can be uncalcined for in-situ MAO supportation when water is used as the oxygen source. Drying of the support material can be affected by heating or calcining at about 100°C to about 1000°C, such as at least about 600°C. When the support material is silica, it is heated to at least 200°C, such as about 200°C to about 850°C, and such as at about 600°C; and for a time of about 30 minutes to about 100 hours, about 4 hours to about 72 hours, or about 24 hours to about 60 hours.
- the calcined support material must have at least some reactive hydroxyl (OH) groups to produce supported catalyst systems of the present disclosure.
- the calcined support material is then contacted with at least one polymerization catalyst including at least one catalyst compound and an activator.
- the support material having reactive surface groups, such as hydroxyl groups, is slurried in a non-polar diluent and the resulting slurry is contacted with a pre-catalyst compound in solid form or solution form and MAO in any sequence when with a pre-formed TMA free MAO and with preformed regular MAO first and then with a pre-catalyst compound after the supported MAO is treated with an in-situ formed TEB agent or a pre-formed TEB agent and other necessary steps to obtain the TMA free supported MAO.
- the slurry of the support material is first contacted with the activator (e.g., TMA free MAO) for a period of time of about 0.5 hour to about 24 hours, about 2 hours to about 16 hours, or about 4 hours to about 8 hours.
- the solution or solid form of the pre-catalyst compound is then contacted with the supported activator.
- the supported catalyst system is generated in-situ.
- the slurry of the TMA free supported MAO is first contacted with the pre-catalyst compound for a period of time of about 0.5 hour to about 24 hours, about 1 hour to about 16 hours, or about 2 hours to about 8 hours.
- Suitable non-polar diluents are materials in which all of the reactants used herein, e.g., the activator and the pre-catalyst compound, are at least partially soluble and which are liquid at polymerization temperatures.
- Non-polar diluents for in-situ MAO supportation can be alkanes, such as isopentane, hexane, isohexane, n-heptane, octane, nonane, and decane, although a variety of other materials including cycloalkanes, such as cyclohexane, aromatics, such as benzene, toluene, and ethylbenzene, may also be employed, whereas for pre-formed MAO supportation, aromatics, such as benzene, toluene, and ethylbenzene, can be used.
- alkanes such as isopentane, hexane, isohexane, n-heptane, octane, nonane, and decane
- cycloalkanes such as cyclohexane
- aromatics such as benzene, toluene,
- the supported activator is a supported TMA free MAO (TF-MAO), which is a silica (e.g., ES70 silica calcined at 400°C) supported MAO with undetectable or low free TMA after the total TMA in MAO is partially or completely converted to a TEB agent to form a coordinated TEB agent on the main MAO structure with an optional step of free TMA removal.
- TF-MAO supported TMA free MAO
- silica e.g., ES70 silica calcined at 400°C
- Embodiments of the present disclosure include methods for preparing a catalyst system including contacting in an organic diluent the unsupported MAO (TMA free solution) or supported MAO (TMA free support) with at least one pre-catalyst compound having a Group 3 through Group 12 metal atom or lanthanide metal atom.
- a TMA free solution MAO is first brought into contact with at least one pre-catalyst compound before contacting the support.
- the unsupported MAO or supported MAO is heated prior to contact with the catalyst compound.
- the unsupported MAO or supported MAO can be solvated or slurried in an organic diluent and the resulting mixture is contacted with a solution of at least one catalyst compound.
- the catalyst compound can also be added as a solid to the mixture of the organic diluent and the MAO.
- the mixture of the MAO is contacted with the catalyst compound for a period of time of about 0.02 hours to about 24 hours, such as about 0.1 hour to about 1 hour, about 0.2 hours to about 0.6 hours, about 2 hours to about 16 hours, or about 4 hours to about 8 hours.
- the mixture of the catalyst compound and the MAO may be heated to a temperature of about 0°C to about 70°C, such as about 23°C to about 60°C, for example room temperature. Contact times may be about 0.02 hours to about 24 hours, such as about 0.1 hours to about 1 hour, about 0.2 hours to about 0.6 hours, about 2 hours to about 16 hours, or about 4 hours to about 8 hours.
- Suitable organic diluents are materials in which some or all of the reactants used herein, e.g., the MAO and the catalyst compound, are at least partially soluble (or in the case of the solid support, suspended) and which are liquid at reaction temperatures.
- Non-limiting example diluents are non-cyclic alkanes with formula CnH(2n+2) where n is 4 to 30, such as isobutane, butane, isopentane, hexane, n-heptane, octane, nonane, decane and the like, and cycloalkanes with formula CnH(2n-2) where n is 5 to 30, such as cyclopentane, methylcyclopentane, cyclohexane, methylcyclohexane and mixtures thereof.
- Aromatic diluent can include benzene, toluene, or xylenes.
- the diluent can be charged into a reactor, followed by an MAO. Catalyst can then be charged into the reactor, such as a solution of catalyst in an organic diluent or as a solid.
- the mixture can be stirred at a temperature, such as room temperature. Additional diluent may be added to the mixture to form a mixture having a desired consistency, such as a slurry having from about 2 cc/g of silica to about 20 cc/g silica, such as about 4 cc/g.
- the diluent can then be removed. Removing diluent dries the mixture and may be performed under a vacuum atmosphere, purged with inert atmosphere, heating of the mixture, or combinations thereof.
- any suitable temperature can be used that evaporates the aliphatic diluent. It is to be understood that reduced pressure under vacuum will lower the boiling point of the aliphatic diluent depending on the pressure of the reactor. Diluent removal temperatures can be about 10°C to about 200°C, such as about 60°C to about 140°C, such as about 60°C to about 120°C, for example about 80°C or less, such as about 70°C or less.
- removing diluent includes applying heat, applying vacuum, and applying nitrogen purged from bottom of the vessel by bubbling nitrogen through the mixture. The mixture is dried.
- the present disclosure provides a catalyst system comprising a catalyst compound having a metal atom.
- the catalyst compound can be a metallocene catalyst compound.
- the metal can be a Group 3 through Group 12 metal atom, such as Group 3 through Group 10 metal atoms, or lanthanide Group atoms.
- the catalyst compound having a Group 3 through Group 12 metal atom can be monodentate or multidentate, such as bidentate, tridentate, or tetradentate, where a heteroatom of the catalyst, such as phosphorous, oxygen, nitrogen, or sulfur is chelated to the metal atom of the catalyst.
- a heteroatom of the catalyst such as phosphorous, oxygen, nitrogen, or sulfur is chelated to the metal atom of the catalyst.
- Non- limiting examples include bis(phenolate)s.
- the Group 3 through Group 12 metal atom is selected from Group 5, Group 6, Group 8, or Group 10 metal atoms.
- a Group 3 through Group 10 metal atom is selected from Cr, Sc, Ti, Zr, Hf, V, Nb, Ta, Mn, Re, Fe, Ru, Os, Co, Rh, Ir, and Ni.
- a metal atom is selected from Groups 4, 5, and 6 metal atoms. In at least one embodiment, a metal atom is a Group 4 metal atom selected from Ti, Zr, or Hf.
- the oxidation state of the metal atom can range from 0 to +7, for example +1, +2, +3, +4, or +5, for example +2, +3 or +4.
- a catalyst compound of the present disclosure can be a chromium or chromium- based catalyst.
- Chromium-based catalysts include chromium oxide (CrO 3 ) and silylchromate catalysts. Chromium catalysts have been the subject of much development in the area of continuous fluidized-bed gas-phase polymerization for the production of polyethylene polymers.
- Mono-Cp catalyst precursor compounds useful in the present disclosure have one cyclopentadienyl (Cp) ligand (which includes ligands that are isolobal to cyclopentadienyl) and at least one polar atom in at least one non-Cp ligand, bridging or unbridging to the Cp ligand, directly bonded to the pre-catalyst metal center.
- Cp cyclopentadienyl
- the mono-Cp pre-catalyst compound of the present disclosure is represented by formula (MC-I): where Cp is independently a substituted or unsubstituted cyclopentadienyl ligand or substituted or unsubstituted ligand isolobal to cyclopentadienyl such as indenyl, fluorenyl, tetrahydro- s-indaccny I and tetrahydro-as-indecenyl.
- M is a group 4 transition metal, such as Hf, Ti, or Zr.
- G is a heteroatom group represented by the formula JR* Z where J is N, P, O or S, and R* is a linear, branched, or cyclic C 1 -C 20 hydrocarby l.
- z is 1 or 2.
- T is a bridging group, y is 0 or 1.
- J is N
- R* is methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, cyclooctyl, cyclododecyl, decyl, undecyl, dodecyl, adamantyl or an isomer thereof.
- Exemplary JR* Z groups include t-butyl amido and cyclododecylamido.
- tire bridging group T examples include CH 2 , CH 2 CH 2 . SiMe 2 . SiPh 2 . SiMePh, Si(CH 2 ) 3 , Si(CH 2 ) 4 , O. S. NPh. PPh, NMe, PMe, NEt. NPr, NBu, PEt, PPr. Me 2 SiOSiMe 2 , and PBu.
- T is represented by the formula or (ER d 2 ) 2 , where E is C, Si, or Gc, and each R d is, independently, hydrogen, halogen, C 1 to C 20 hydrocarbyl (such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, or dodecyl) or a C
- C 1 to C 20 hydrocarbyl such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, or dodecyl
- to C20 substituted hydrocarbyl or two R
- Each X is independently selected from the group consisting of hydrocarbyl radicals having from 1 to 20 carbon atoms, aryls, hydrides, amides, alkoxides, sulfides, phosphides, halides, dienes, amines, phosphines, ethers, and a combination thereof, (two Xs may form a part of a fused ring or a ring system), such as each X is independently selected from halides, aryls and C 1 to C 5 alkyl groups, such as each X is a phenyl, methyl, ethyl, propyl, butyl, pentyl, or chloro group.
- MC-I is selected from: dimethylsilandiyl (2,3,4,5-tetramethylcyclopentadienyl)(cyclododecylamido)M(R) 2 : dimethylsilandiyl (2,3,4,5-tetramethylcyclopentadienyl)(cycloundecylamido)M(R) 2 ; dimethylsilandiyl (2,3,4,5-tetramethylcyclopentadienyl)(cyclodecylamido)M(R) 2 ; dimethylsilandiyl (2,3,4,5-tetramethylcyclopentadienyl)(t-butylamido)M(R) 2 ; dimethylsilandiyl (cyclopentadienyl)(l-adamantylamido)M(R) 2 ; dimethylsilandiyl (3-tertbutylcyclopentadienyl)(1-adamantylamido)M
- M is Ti and each R is methyl.
- R is methyl.
- the mono-Cp pre-catalyst compounds can also include compounds having the structure represented by formula (MC-II) preferably having C s or pseudo-C s symmetry: wherein: M is zirconium; L 1 is a unsubstituted fluorenyl, heterocyclopentapentalenyl, or heterofluorenyl, or a substituted fluorenyl, heterocyclopentapentalenyl, or heterofluorenyl ligand with one or more symmetric or pseudo symmetric substituents, each substituent group being, independently, a radical group which is a hydrocarbyl, substituted hydrocarbyl, halocarbyl, substituted halocarbyl, silylcarbyl or germylcarbyl, and optionally two or more adjacent substituents may join to form a substituted or unsubstituted, saturated, partially unsaturated or aromatic, cyclic or polycyclic substituent; G is a bridging group; J is a hetero
- L 1 is fluorenyl or substituted fluorenyl, such as fluorenyl, 2,7-dimethylfluorenyl, 2,7-diethylfluorenyl, 2,7-dipropylfluorenyl, 2,7-dibutylfluorenyl, 2,7-diphenylfluorenyl, 2,7-dichlorofluorenyl, 2,7-dibromofluorenyl, 3,6-dimethylfluorenyl, 3,6-diethylfluorenyl, 3,6-dipropylfluorenyl, 3,6-dibutylfluorenyl, 3,6-diphenylfluorenyl, 3,6-dichlorofluorenyl, 3,6-dibromofluorenyl or 1,1,4,4,7,7,10,10-octamethyl- octahydrodibenzofluoren
- G is methylene, dimethylmethylene, diphenylmethylene, dimethylsilylene, methylphenylsilylene, diphenylsilylene, di(4-triethylsilylphenyl)silylene, ethylene, such as diphenylmethylene, diphenylsilylene, methylphenylsilylene, and dimethylsilylene; such as dimethylsilylene.
- Suitable J can be nitrogen.
- R’ is hydrocarbyl or halocarbyl, such as C3-C20 hydrocarbyl, such as all isomers (including cyclics and polycyclics) of propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, benzyl, phenyl and substituted phenyl, such as tert-butyl, neopentyl, benzyl, phenyl, diisopropylphenyl, adamantyl, norbornyl, cyclohexyl, cyclooctyl, cyclodecyl, and cyclododecyl, such as tert-butyl, adamant-1-yl, norborn-2-yl, cyclohexyl, cyclooctyl, and cyclododecyl
- X is hydrocarbyl or halo, such as methyl, benzyl, floro or chloro, such as methyl or chloro; w is zero (L’ being absent); M is zirconium.
- polar donor(s) include but are not limited to:
- the mono-metallocene pre-catalyst compound may also be selected from: dimethylsilyl (tetramethylcyclopentadienyl)(cyclododecylamido)titanium dimethyl; dimethylsilyl (tetramethylcyclopentadienyl)(cyclododecylamido)titanium dichloride; dimethylsilyl (tetramethylcyclopentadienyl)(t-butylamido)titanium dimethyl; or dimethylsilyl (tetramethylcyclopentadienyl)(t-butylamido)titanium dichloride.
- pre-catalysts of the present disclosure can be “post- metallocene” catalysts having an oxygen and/or nitrogen atom(s).
- a catalyst of the present disclosure can be a metal complex having: a metal selected from groups 3-10 or lanthanide metals, and a tridentate, mono- or di-anionic ligand containing one or two anionic donor groups and two or one neutral Lewis base donor, where the one or two neutral Lewis base donors is covalently bonded between the two anionic donors, and where the metal-ligand complex features a pair of 4-, 5-, 6-, 7-, or 8-membered metallocycle rings or a pair of mixed membered metallocycle rings, such as a mix of 4- and 5-membered rings, a mix of 5- and 6- membered rings, a mix of 6- and 7-membered rings, a mix of 5- and 7-membered rings, or a mix of 7- and -8 membered rings.
- the catalyst complexes of the present disclosure include a metal selected from groups 3-10 or lanthanide metals of the Periodic Table of the Elements, a tridentate dianionic ligand containing two anionic donor groups and a neutral heterocyclic Lewis base donor, wherein the heterocyclic donor is covalently bonded between the two anionic donors.
- the dianionic, tridentate ligand features a central heterocyclic donor group and two phenolate donors and the tridentate ligand coordinates to the metal center to form two eight-membered rings.
- the heterocyclic Lewis base donor of the catalyst compound features a nitrogen or oxygen donor atom.
- heterocyclic groups include derivatives of pyridine, pyrazine, pyrimidine, triazine, thiazole, imidazole, thiophene, oxazole, thiazole, furan, and substituted variants of thereof.
- the heterocyclic Lewis base lacks hydrogen(s) in the position alpha to the donor atom.
- a heterocyclic Lewis base donor includes pyridine, 3-substituted pyridines, and 4-substituted pyridines.
- the anionic donors of the tridentate dianionic ligand may be arylthiolates, phenolates, or anilides. In some embodiments, anionic donors are phenolates.
- the tridentate dianionic ligand coordinates to the metal center to form a complex that may lack a mirror plane of symmetry. In some embodiments, the tridentate dianionic ligand coordinates to the metal center to form a complex that has a two-fold rotation axis of symmetry; when determining the symmetry of the bis(phenolate) complexes only the metal and dianionic tridentate ligand are considered (i.e., ignore remaining ligands).
- Catalyst compounds of the present disclosure can be bis(aryl phenolate)pyridine complexes.
- Bis(aryl phenolate)pyridine complexes may have a tridentate bis(aryl phenolate)pyridine ligand that is coordinated to a group 4 transition metal with the formation of two eight-membered rings.
- a bis(aryl phenolate)pyridine complexes includes transition metal complexes of a dianionic, tridentate ligand that features a central neutral donor group and two phenolate donors, where the tridentate ligands coordinate to the metal center to form two eight-membered rings, for example, the post-metallocene catalyst can be an 8-8 catalyst.
- the central neutral donor it is advantageous for the central neutral donor to be a heterocyclic group.
- heterocyclic group it is advantageous for the heterocyclic group to lack hydrogens in the position alpha to the heteroatom.
- phenolates may also be advantageous for the phenolates to be substituted with one or more cyclic tertiary alkyl substituents.
- the use of cyclic tertiary alkyl substituted phenolates can improve the ability of these catalysts to produce high molecular weight polymer.
- bis(phenolate) ligands can be tridentate dianionic ligands that coordinate to the metal M in such a fashion that a pair of 8-membered metallocycle rings are formed.
- the bis(phenolate) ligands wrap around the metal to form a complex with a 2-fold rotation axis, thus giving the complexes C 2 symmetry.
- the C 2 geometry and the 8-membered metallocycle rings are features of these complexes that make them effective catalyst components for the production of polyolefins, particularly isotactic poly(alpha olefins). If the ligands were coordinated to the metal in such a manner that the complex had mirror-plane (Cs) symmetry, then the catalyst would be expected to produce only atactic poly(alpha olefins); these symmetry-reactivity concepts are summarized by Bercaw, J. E. (2009) in Macromolecules, v.42, pp. 8751-8762.
- the pair of 8-membered metallocycle rings of the catalyst compounds is also a notable feature that is advantageous for temperature stability, and isoselectivity of monomer enchainment.
- Related group 4 complexes featuring smaller 6-membered metallocycle rings are known (Macromolecules 2009, v.42, pp.8751-8762) to form mixtures of C 2 and C s symmetric complexes when used in olefin polymerizations and are thus not well suited to the production of highly isotactic poly(alpha olefins).
- Bis(phenolate) ligands that contain oxygen donor groups can be substituted with alkyl, substituted alkyl, aryl, or other groups. It can be advantageous that each phenolate group be substituted in the ring position that is adjacent to the oxygen donor atom.
- substitution at the position adjacent to the oxygen donor atom can be an alkyl group containing 1-20 carbon atoms.
- the substitution at the position next to the oxygen donor atom can be a non-aromatic cyclic alkyl group with one or more five- or six-membered rings. Substitution at the position next to the oxygen donor atom can be a cyclic tertiary alkyl group.
- substitution at the position next to the oxygen donor atom is adamantan-1-yl or substituted adamantan-1-yl.
- the neutral heterocyclic Lewis base donor is covalently bonded between the two anionic donors (e.g., between two phenolate groups) via linker groups that join the heterocyclic Lewis base to the anionic donors.
- the “linker groups” are indicated by (A 3 A 2 ) and (A 2’ A 3’ ) in Formula (PM-I).
- the choice of each linker group may affect the catalyst performance, such as the tacticity of the poly(alpha olefin) produced.
- Each linker group can be a C2-C40 divalent group that is two-atoms in length.
- One or both linker groups may independently be phenylene, substituted phenylene, heteroaryl, vinylene, or a non-cyclic two- carbon long linker group.
- the alkyl substituents on the phenylene group may be chosen to optimize catalyst performance.
- one or both phenylenes may be unsubstituted or may be independently substituted with C1 to C20 alkyl, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl, eicosyl, or an isomer thereof, such as isopropyl, etc.
- C1 to C20 alkyl such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl,
- a catalyst compound is represented by Formula (PM-I): wherein: M is a group 3, 4, 5, or 6 transition metal or a lanthanide (such as Hf, Zr or Ti); E and E' are each independently O, S, or NR 9 , where R 9 is independently hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, or a heteroatom-containing group, such as O, such as both E and E' are O; Q is group 14, 15, or 16 atom that forms a dative bond to metal M, such as Q is C, O, S or N, such as Q is C, N, or O, such as Q is N; A 1 QA 1’ are part of a heterocyclic Lewis base containing 4 to 40 non-hydrogen atoms that links A 2 to A 2’ via a 3-atom bridge with Q being the central atom of the 3-atom bridge (A 1 QA 1’ combined with the curved line joining A 1 and A 1’ represents the heterocycl
- the metal, M is selected from group 3, 4, 5, or 6 elements, such as group 4.
- the metal, M is zirconium or hafnium.
- the donor atom Q of the neutral heterocyclic Lewis base can be nitrogen, carbon, or oxygen. In some embodiments, Q is nitrogen.
- Non-limiting examples of neutral heterocyclic Lewis base groups include derivatives of pyridine, pyrazine, pyrimidine, triazine, thiazole, imidazole, thiophene, oxazole, thiazole, furan, and substituted variants of thereof.
- Heterocyclic Lewis base groups can include derivatives of pyridine, pyrazine, thiazole, and imidazole.
- Each of A 1 and A 1’ of the heterocyclic Lewis base (of Formula (PM-I)) is independently C, N, or C(R 22 ), where R 22 is selected from hydrogen, C 1 -C 20 hydrocarbyl, and C 1 -C 20 substituted hydrocarbyl.
- each of A 1 and A 1' is carbon.
- Q is carbon
- each of A 1 and A 1’ can be selected from nitrogen and C(R 22 ).
- the heterocyclic Lewis base (of Formula (PM-I)) represented by A 1 QA 1’ combined with the curved line joining A 1 and A 1’ can be selected from the following, with each R 23 group selected from hydrogen, heteroatoms, C 1 -C 20 alkyls, C 1 -C 20 alkoxides, C 1 -C 20 amides, and C 1 -C 20 substituted alkyls.
- the heterocyclic Lewis base (of Formula (PM-I)) represented by A 1 QA 1’ combined with the curved line joining A 1 and A 1’ is a six membered ring containing zero or one ring heteroatoms or a five membered ring containing zero, one two or three ring heteroatoms.
- the heterocyclic Lewis base (of Formula (PM-I)) represented by A 1 QA 1’ combined with the curved line joining A 1 and A 1’ is not a six membered ring containing two or more ring heteroatoms.
- Q is C, N or O, such as Q is N.
- each of A 1 and A 1' is independently carbon, nitrogen, or C(R 22 ), with R 22 selected from hydrogen, C 1 -C 20 hydrocarbyl, substituted C 1 -C 20 hydrocarbyl. In some embodiments, each of A 1 and A 1’ is carbon.
- a 1 QA 1’ of Formula (PM-I) is part of a heterocyclic Lewis base, such as a pyridine, pyrazine, pyrimidine, triazine, thiazole, imidazole, thiophene, oxazole, thiazole, furan, or a substituted variant of thereof.
- a 1 QA 1’ are part of a heterocyclic Lewis base containing 2 to 20 non-hydrogen atoms that links A 2 to A 2’ via a 3-atom bridge with Q being the central atom of the 3-atom bridge.
- each A 1 and A 1' is a carbon atom and the A 1 QA 1’ fragment forms part of a pyridine, pyrazine, pyrimidine, triazine, thiazole, imidazole, thiophene, oxazole, thiazole, furan, or a substituted variant of thereof group, or a substituted variant thereof.
- Q is carbon and each of A 1 and A 1' is N or C(R 22 ), where R 22 is selected from hydrogen, C 1 -C 20 hydrocarbyl, substituted C 1 -C 20 hydrocarbyl, a heteroatom or a heteroatom-containing group.
- R 22 is selected from hydrogen, C 1 -C 20 hydrocarbyl, substituted C 1 -C 20 hydrocarbyl, a heteroatom or a heteroatom-containing group.
- the A 1 QA 1’ fragment forms part of a cyclic carbene, N-heterocyclic carbene, cyclic amino alkyl carbene, or a substituted variant thereof.
- [0176] is a divalent group containing 2 to 20 non-hydrogen atoms that links A 1' to the E'-bonded aryl group via a 2-atom bridge, where the is a linear alkyl or forms part of a cyclic group (such as an optionally substituted ortho-phenylene group, or ortho- arylene group or, or a substituted variant thereof.
- M is Zr or Hf
- Q is nitrogen
- both A 1 and A 1’ are carbon
- both E and E ’ are oxygen
- both R 1 and R 1’ are C 4 -C 20 cyclic tertiary alkyls.
- M is Zr or Hf
- Q is nitrogen
- both A 1 and A 1’ are carbon
- both E and E ’ are oxygen
- both R 1 and R 1’ are adamantan-1-yl or substituted adamantan-1-yl.
- M is Zr or Hf
- Q is nitrogen
- both A 1 and A 1’ are carbon
- both E and E ’ are oxygen
- both R 1 and R 1’ are C 6 -C 20 aryls.
- a catalyst compound is represented by Formula (PM-II): wherein: M is a group 3, 4, 5, or 6 transition metal or a lanthanide (such as a group 4 transition metal that is Hf, Zr or Ti); E and E' are each independently O, S, or NR 9 , where R 9 is independently hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, or a heteroatom-containing group, such as O, such as both E and E' are O; each L is independently a Lewis base; each X is independently an anionic ligand; n is 1, 2 or 3; m is 0, 1, or 2; n+m is not greater than 4; each of R 1 , R 2 , R 3 , R 4 , R 1' , R 2' , R 3' , and R 4' is independently hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, a heteroatom or a
- E and E’ are each selected from oxygen or NR 9 , where R 9 is independently hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, or a heteroatom-containing group. In some embodiments, E and E’ are oxygen. When E and/or E’ are NR 9 , R 9 can be selected from C 1 to C 20 hydrocarbyls, alkyls, or aryls.
- E and E’ are each selected from O, S, or N(alkyl) or N(aryl), where the alkyl can be a C 1 to C 20 alkyl, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, octyl, nonyl, decyl, undecyl, dodecyl, and the like, and aryl is a C 6 to C 40 aryl group, such as phenyl, naphthalenyl, benzyl, methylphenyl, and the like. [0182] In some embodiments, and are independently a divalent hydrocarbyl group, such as C 1 to C 12 hydrocarbyl group.
- each phenolate group when E and E’ are oxygen, each phenolate group can be substituted in the position that is next to the oxygen atom (i.e., R 1 and R 1’ in Formula (PM-I) and (PM-II)).
- each of R 1 and R 1' is independently a C 1 -C 40 hydrocarbyl, a C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom-containing group, such as each of R 1 and R 1' is independently a non-aromatic cyclic alkyl group with one or more five- or six-membered rings (such as cyclohexyl, cyclooctyl, adamantyl, or 1-methylcyclohexyl, or substituted adamantyl), such as a non-aromatic cyclic tertiary alkyl group (such as 1-methylcyclohexyl, adamantyl, or substituted adamantyl).
- a non-aromatic cyclic alkyl group with one or more five- or six-membered rings such as cyclohexyl, cyclooctyl, adamantyl, or 1-methylcyclohexyl, or substitute
- each of R 1 and R 1' is independently a tertiary hydrocarbyl group. In other embodiments of Formula (PM-I) or (PM-II), each of R 1 and R 1' is independently a cyclic tertiary hydrocarbyl group. In other embodiments of the catalyst compound of Formula (PM-I) or (PM-II), each of R 1 and R 1' is independently a polycyclic tertiary hydrocarbyl group. [0185] In some embodiments of the catalyst compound of Formula (PM-I) or (PM-II), each of R 1 and R 1' is independently a tertiary hydrocarbyl group.
- each of R 1 and R 1' is independently a cyclic tertiary hydrocarbyl group. In other embodiments of the catalyst compound of Formula (PM-I) or (PM-II), each of R 1 and R 1' is independently a polycyclic tertiary hydrocarbyl group.
- the linker groups i.e., in Formula (PM-I)
- R 7 and R 7’ positions of Formula (PM-II) may be hydrogen or C 1 to C 20 alkyl, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl, eicosyl, or an isomer thereof, such as isopropyl, etc.
- C 1 to C 20 alkyl such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetrade
- R 7 and R 7’ positions of Formula (PM-II) may be a C 1 to C 20 alkyl, such as for both R 7 and R 7’ to be a C 1 to C 3 alkyl.
- M is a group 4 metal, such as Hf or Zr.
- each of E and E' is O.
- each of R 1 , R 2 , R 3 , R 4 , R 1' , R 2' , R 3' , and R 4' is independently hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, a heteroatom or a heteroatom-containing group, or one or more of R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 1' and R 2' , R 2’ and R 3' , R 3' and R 4' may be joined to form one or more substituted hydrocarbyl rings, unsubstituted hydrocarbyl rings, substituted heterocyclic rings, or unsubstituted heterocyclic rings each having 5, 6, 7, or 8 ring atoms, and where substitutions on the ring can join to form additional rings, such as hydrogen, methyl, ethyl, propyl, butyl, pentyl, hexy
- each of R 1 , R 2 , R 3 , R 4 , R 1' , R 2' , R 3' , and R 4' is independently selected from methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl, eicosyl, heneicosyl, docosyl, tricosyl, tetracosyl, pentacosyl, hexacosyl, heptacosyl, octacosyl, nonacosyl, triacontyl, phenyl, substitute
- each of R 4 and R 4' is independently hydrogen or a C 1 to C 3 hydrocarbyl, such as methyl, ethyl or propyl.
- R 9 is hydrogen, C 1 -C 40 hydrocarbyl, C 1 -C 40 substituted hydrocarbyl, or a heteroatom-containing group, such as hydrogen, methyl, ethyl, propyl, butyl, pentyl, hexyl, or an isomer thereof.
- R 9 is C 1 to C 6 alkyl (such as methyl, ethyl, propyl, or butyl), phenyl, 2-methylphenyl, 2,6-dimethylphenyl, or 2,4,6-trimethylphenyl.
- each X is, independently, selected from the group consisting of hydrocarbyl radicals having from 1 to 20 carbon atoms (such as alkyls or aryls), hydrides, amides, alkoxides, sulfides, phosphides, halides, alkyl sulfonates, and a combination thereof, (two or more X’s may form a part of a fused ring or a ring system), such as each X is independently selected from halides, aryls, and C 1 to C 5 alkyl groups, such as each X is independently a hydrido, dimethylamido, diethylamido, methyltrimethylsilyl, neopentyl, phenyl, benzyl, methyl, ethyl, propyl, butyl, pentyl, fluoro, iodo, bromo,
- each X may be, independently, a halide, a hydride, an alkyl group, or an alkenyl group.
- each L is a Lewis base, independently, selected from the group consisting of ethers, thio-ethers, amines, nitriles, imines, pyridines, halocarbons, and phosphines, such as ethers, thioethers, or a combination thereof, optionally two or more L’s may form a part of a fused ring or a ring system, such as each L is independently selected from ether or thioether groups, such as each L is an ethyl ether, tetrahydrofuran, dibutyl ether, or dimethylsulfide group.
- each of R 1 and R 1’ is independently cyclic tertiary alkyl groups.
- n is 1, 2 or 3, such as 2.
- m is 0, 1 or 2, such as 0.
- each of R 1 and R 1' is not hydrogen.
- M is Hf or Zr, each of E and E' is O; each of R 1 and R 1’ is independently a C 1 -C 40 hydrocarbyl, a substituted C 1 -C 40 hydrocarbyl, a heteroatom or a heteroatom-containing group, each of R 2 , R 3 , R 4 , R 2' , R 3' , and R 4' is independently hydrogen, C 1 -C 20 hydrocarbyl, substituted C 1 -C 20 hydrocarbyl, a heteroatom or a heteroatom-containing group, or one or more of R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 1' and R 2' , R 2’ and R 3' , R 3' and R 4' may be joined to form one or more substituted hydrocarbyl rings, unsubstituted hydrocarbyl rings, substituted heterocyclic rings, or unsubstituted heterocyclic
- each of R 5 , R 6 , R 7 , R 8 , R 5' , R 6' , R 7' , R 8' , R 10 , R 11 and R 12 is independently hydrogen, C 1 -C 40 hydrocarbyl, substituted C 1 -C 40 hydrocarbyl, a heteroatom or a heteroatom-containing group, or one or more adjacent R groups may be joined to form one or more substituted hydrocarbyl rings, unsubstituted hydrocarbyl rings, substituted heterocyclic rings, or unsubstituted heterocyclic rings each having 5, 6, 7, or 8 ring atoms, and where substitutions on the ring can join to form additional rings.
- each of R 5 , R 6 , R 7 , R 8 , R 5' , R 6' , R 7' , R 8' , R 10 , R 11 and R 12 is independently hydrogen, methyl, ethyl, propyl, butyl, pentyl, hexyl, or an isomer thereof.
- each of R 5 , R 6 , R 7 , R 8 , R 5' , R 6' , R 7' , R 8 ', R 10 , R 11 and R 12 is independently selected from methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl, eicosyl, heneicosyl, docosyl, tricosyl, tetracosyl, pentacosyl, hexacosyl, heptacosyl, octacosyl, nonacosyl, triacontyl, phen
- M is Hf or Zr, each of E and E' is O; each of R 1 and R 1' is independently a C 1 -C 40 hydrocarbyl, a substituted C 1 -C 40 hydrocarbyl, a heteroatom or a heteroatom-containing group; each of R 1 , R 2 , R 3 , R 4 , R 1' , R 2' , R 3' , and R 4' is independently hydrogen, C 1 -C 20 hydrocarbyl, substituted C 1 -C 20 hydrocarbyl, a heteroatom or a heteroatom-containing group, or one or more of R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 1' and R 2' , R 2’ and R 3' , R 3' and R 4' may be joined to form one or more substituted hydrocarbyl rings, unsubstituted hydrocarbyl rings, substituted heterocyclic rings, or unsubstituted
- M is Zr or Hf
- both E and E ’ are oxygen
- both R 1 and R 1’ are C 4 -C 20 cyclic tertiary alkyls.
- M is Zr or Hf
- both E and E ’ are oxygen
- both R 1 and R 1’ are adamantan-1-yl or substituted adamantan-1-yl.
- M is Zr or Hf
- both E and E ’ are oxygen
- each of R 1 , R 1’ , R 3 and R 3’ are adamantan-1-yl or substituted adamantan-1-yl.
- M is Zr or Hf
- both E and E ’ are oxygen
- both R 1 and R 1’ are C 4 -C 20 cyclic tertiary alkyls
- both R 7 and R 7’ are C 1 -C 20 alkyls.
- a catalyst compound is one or more of: dimethylzirconium[2',2'''-(pyridine-2,6-diyl)bis(3-adamantan-1-yl)-5-(tert-butyl)-[1,1'- biphenyl]-2-olate)], dimethylhafnium[2',2'''-(pyridine-2,6-diyl)bis(3-adamantan-1-yl)-5-(tert- butyl)-[1,1'-biphenyl]-2-olate)], dimethylzirconium[6,6'-(pyridine-2,6- diylbis(benzo[b]thiophene-3,2-diyl))bis(2-adamantan-1-yl)-4-methylphenolate)], dimethylhafnium[6,6'-(pyridine-2,6-diylbis(benzo[b]thiophene-3,2-diyl)))
- a pre-catalyst compound is one or more of the Group 3 based pre-catalysts:
- TMA free (or trihydrocarbyl aluminum free) solution alumoxane (e.g., MAO), solid alumoxane (e.g., MAO), or supported alumoxane (e.g., MAO) can also be used for bis-Cp metallocene pre-catalyst compounds not having polar donors for the purpose of polymer property control, e.g., for regulating the polymer molecule weight and molecular weight distribution through limiting the free aluminum alkyls in the system that may cause chain transfer from the catalytic metal center to the free aluminum alkyls.
- Metallocene pre-catalyst compounds as used herein include metallocenes comprising Group 3 to Group 10 metal complexes, preferably, Group 4 to Group 6 metal complexes, for example, Group 4 metal complexes.
- the metallocene catalyst compound of catalyst systems of the present disclosure may be unbridged metallocene catalyst compounds represented by the formula (BC-I): Cp A Cp B M’X’n (BC-I) wherein each Cp A and Cp B is independently selected from cyclopentadienyl ligands and ligands isolobal to cyclopentadienyl, one or both Cp A and Cp B may contain heteroatoms, and one or both Cp A and Cp B may be substituted by one or more R’’ groups.
- M’ is selected from Groups 3 through 12 atoms and lanthanide Group atoms.
- X’ is an anionic leaving group.
- n is 0 or an integer from 1 to 4.
- R’’ is selected from alkyl, lower alkyl, substituted alkyl, heteroalkyl, alkenyl, lower alkenyl, substituted alkenyl, heteroalkenyl, alkynyl, lower alkynyl, substituted alkynyl, heteroalkynyl, alkoxy, lower alkoxy, aryloxy, alkylthio, lower alkylthio, arylthio, aryl, substituted aryl, heteroaryl, aralkyl, aralkylene, alkaryl, alkarylene, haloalkyl, haloalkenyl, haloalkynyl, heteroalkyl, heterocycle, heteroaryl, a heteroatom-containing group, hydrocarbyl, lower hydrocarbyl, substituted
- each Cp A and Cp B is independently selected from cyclopentadienyl, indenyl, fluorenyl, cyclopentaphenanthreneyl, benzindenyl, fluorenyl, octahydrofluorenyl, cyclooctatetraenyl, cyclopentacyclododecene, phenanthrindenyl, 3,4-benzofluorenyl, 9-phenylfluorenyl, 8-H-cyclopent[a]acenaphthylenyl, 7-H-dibenzofluorenyl, indeno[1,2-9]anthrene, thiophenoindenyl, thiophenofluorenyl, and hydrogenated versions thereof.
- the metallocene catalyst compound may be a bridged metallocene catalyst compound represented by the formula (BC-II): Cp A (A)Cp B M’X’n (BC-II) wherein each Cp A and Cp B is independently selected from cyclopentadienyl ligands and ligands isolobal to cyclopentadienyl.
- Cp A and Cp B may contain heteroatoms, and one or both Cp A and Cp B may be substituted by one or more R’’ groups.
- M’ is selected from Groups 3 through 12 atoms and lanthanide Group atoms.
- X’ is an anionic leaving group.
- n is 0 or an integer from 1 to 4.
- (A) is selected from divalent alkyl, divalent lower alkyl, divalent substituted alkyl, divalent heteroalkyl, divalent alkenyl, divalent lower alkenyl, divalent substituted alkenyl, divalent heteroalkenyl, divalent alkynyl, divalent lower alkynyl, divalent substituted alkynyl, divalent heteroalkynyl, divalent alkoxy, divalent lower alkoxy, divalent aryloxy, divalent alkylthio, divalent lower alkylthio, divalent arylthio, divalent aryl, divalent substituted aryl, divalent heteroaryl, divalent aralkyl, divalent aralkylene, divalent alkaryl, divalent alkarylene, divalent haloalkyl, divalent haloalkenyl, divalent haloalkynyl, divalent heteroalkyl, divalent heterocycle, divalent heteroaryl, a divalent heteroatom-containing group,
- R’’ is selected from alkyl, lower alkyl, substituted alkyl, heteroalkyl, alkenyl, lower alkenyl, substituted alkenyl, heteroalkenyl, alkynyl, lower alkynyl, substituted alkynyl, heteroalkynyl, alkoxy, lower alkoxy, aryloxy, alkylthio, lower alkylthio, arylthio, aryl, substituted aryl, heteroaryl, aralkyl, aralkylene, alkaryl, alkarylene, haloalkyl, haloalkenyl, haloalkynyl, heteroalkyl, heterocycle, heteroaryl, a heteroatom-containing group, hydrocarbyl, lower hydrocarbyl, substituted hydrocarbyl, heterohydrocarbyl, silyl, boryl, phosphino, phosphine, amino, amine, germanium, ether, and thioether.
- each of Cp A and Cp B is independently selected from cyclopentadienyl, n-propylcyclopentadienyl, indenyl, pentamethylcyclopentadienyl, tetramethylcyclopentadienyl, and n-butylcyclopentadienyl.
- (A) may be CR’2 or SiR’2, where each R’ is independently hydrogen or C 1 -C 20 hydrocarbyl.
- two or more different pre-catalyst compounds are present in the catalyst system used herein.
- two or more different pre-catalyst compounds are present in the reaction zone where the process(es) described herein occur. It may be preferable to use the same activator for the transition metal compounds, however, two different activators, such as TMA free supported or unsupported MAO from the current disclosure and a strong Lewis acid activator (e.g., trisperfluoroaromatic boranes) or non- or weak-coordinating anion activator (e.g., N,N-dimethylanalenium or trityl tetrakisperfluoroaromatic borates), can be used in combination.
- a strong Lewis acid activator e.g., trisperfluoroaromatic boranes
- non- or weak-coordinating anion activator e.g., N,N-dimethylanalenium or trityl tetrakisperfluoroaromatic borates
- transition metal compounds contain an X group which is not a hydride, hydrocarbyl, or substituted hydrocarbyl
- the MAO can be contacted with the transition metal compounds prior to addition of the non-coordinating anion activator.
- the two transition metal compounds may be used in any ratio.
- molar ratios of (A) transition metal compound to (B) transition metal compound fall within the range of (A:B) 1:1000 to 1000:1, alternatively 1:100 to 500:1, alternatively 1:10 to 200:1, alternatively 1:1 to 100:1, and alternatively 1:1 to 75:1, and alternatively 5:1 to 50:1.
- useful mole percents are 10% to 99.9% A to 0.1% to 90% B, alternatively 25% to 99% A to 0.5% to 50% B, alternatively 50% to 99% A to 1% to 25% B, and alternatively 75% to 99% A to 1% to 10% B.
- the leaving groups of the pre-catalysts described above are preferably pre-alkylated, such as methylated, ethylated, benzylated, or trimethylsilylmethylenated because the alkylation agent in MAO, e.g., free TMA, has been removed significantly.
- non-alkylated pre-catalysts may still be used either with a mild alkylation agent, such as high carbon trialkylaluminum (e.g., trioctylaluminum) or a secondary aluminumalkyl (e.g., AlMe 2 BHT or AlEt 2 BHT) or without a mild alkylation agent if the solution, solid, or supported MAO system has a low TMA or trihydrocarbylaluminum residue enough for the alkylation of the pre-catalyst.
- a mild alkylation agent such as high carbon trialkylaluminum (e.g., trioctylaluminum) or a secondary aluminumalkyl (e.g., AlMe 2 BHT or AlEt 2 BHT) or without a mild alkylation agent if the solution, solid, or supported MAO system has a low TMA or trihydrocarbylaluminum residue enough for the alkylation of the pre-catalyst.
- the present disclosure also relates to polymerization processes where monomer (e.g., ethylene; propylene), and optionally a one or more comonomers, are contacted with catalyst systems made from one of the methods described in this disclosure in a single polymerization reactor or multiple polymerization reactors in sequence following corresponding polymerization processes to obtain desired polymer products including single- phasic polymers or copolymers and multiple-phasic copolymers, such as in a single reactor for solution-, slurry-, and gas-phase polymerization and copolymerization, such as in multiple reactors for solution-, slurry-, and gas-phase sequential copolymerization.
- monomer e.g., ethylene; propylene
- a one or more comonomers are contacted with catalyst systems made from one of the methods described in this disclosure in a single polymerization reactor or multiple polymerization reactors in sequence following corresponding polymerization processes to obtain desired polymer products including single- phasic polymers or copo
- the pre-catalyst compound and activator may be combined in any suitable order.
- the pre-catalyst compound and activator may be combined prior to contacting with the monomer, e.g., the finished catalyst system can form first by combing a pre-catalyst compound and a TMA free silica supported MAO before it is fed into the polymerization reactor to contact with monomers.
- the pre-catalyst compound and activator may be introduced into the polymerization reactor separately, wherein the pre-catalyst compound and activator subsequently react to form the active catalyst.
- Monomers may include substituted or unsubstituted C 2 to C 40 alpha olefins, such as C 2 to C 20 alpha olefins, such as C 2 to C 12 alpha olefins, such as ethylene, propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene and isomers thereof.
- the monomer includes ethylene and an optional comonomer including one or more C 3 to C 40 olefins, such as C 4 to C 20 olefins, such as C 6 to C 12 olefins.
- the C 3 to C 40 olefin monomers may be linear, branched, or cyclic.
- the C 3 to C 40 cyclic olefins may be strained or unstrained, monocyclic or polycyclic, and may optionally include heteroatoms and or one or more functional groups.
- the monomer includes propylene and an optional comonomer including one or more ethylene or C 4 to C 40 olefins, such as C 4 to C 20 olefins, such as C 6 to C 12 olefins.
- the C 4 to C 40 olefin monomers may be linear, branched, or cyclic.
- the C 4 to C 40 cyclic olefins may be strained or unstrained, monocyclic or polycyclic, and may optionally include heteroatoms and or one or more functional groups.
- Exemplary C 2 to C 40 olefin monomers and optional comonomers may include ethylene, propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, ethylidenenorbornene, vinylnorbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof, such as hexene, heptene, he
- Polymerization processes of the present disclosure can be carried out in any suitable manner. Any suitable suspension, homogeneous, bulk, solution, slurry, or gas phase polymerization process can be used. Such processes can be run in a batch, semi-batch, or continuous mode. Homogeneous polymerization processes and slurry processes can be employed. (A homogeneous polymerization process is defined to be a process where at least 90 wt% of the product is soluble in the reaction media.) A homogeneous polymerization process can be a bulk homogeneous process.
- a bulk process is defined to be a process where monomer concentration in all feeds to the reactor is 70 volume % or more.
- no diluent is present or added in the reaction medium, (except for the small amounts used as the carrier for the catalyst system or other additives, or amounts found with the monomer; e.g., propane in propylene).
- the process is a slurry process.
- slurry polymerization process means a polymerization process where a supported catalyst is employed and monomers are polymerized on the supported catalyst particles. At least 95 wt% of polymer products derived from the supported catalyst are in granular form as solid particles (not dissolved in the diluent).
- Suitable diluents for polymerization may include non-coordinating, inert liquids.
- Examples of diluents for polymerization may include straight and branched-chain hydrocarbons, such as isobutane, butane, pentane, isopentane, hexanes, isohexane, heptane, octane, dodecane, and mixtures thereof; cyclic and alicyclic hydrocarbons, such as cyclohexane, cycloheptane, methylcyclohexane, methylcycloheptane, and mixtures thereof, such as can be found commercially (e.g., IsoparTM); perhalogenated hydrocarbons, such as perfluorinated C4 to C10 alkanes, chlorobenzene, and aromatic and alkylsubstituted aromatic compounds, such as benzene, toluene, mesitylene, and xylene.
- Suitable diluents may also include liquid olefins which may act as monomers or comonomers including ethylene, propylene, 1-butene, 1-hexene, 1-pentene, 3-methyl-1-pentene, 4-methyl-1-pentene, 1-octene, 1-decene, and mixtures thereof.
- aliphatic hydrocarbon diluents are used as the diluent, such as isobutane, butane, pentane, isopentane, hexanes, isohexane, heptane, octane, dodecane, and mixtures thereof; cyclic and alicyclic hydrocarbons, such as cyclohexane, cycloheptane, methylcyclohexane, methylcycloheptane, and mixtures thereof.
- the diluent is not aromatic, such as aromatics are present in the diluent at less than 1 wt%, such as less than 0.5 wt%, such as 0 wt% based upon the weight of the diluents.
- a feedstream to the reactor has a feed concentration of the monomers and comonomers for the polymerization is 60 vol% diluent or less, such as 40 vol% or less, such as 20 vol% or less, based on the total volume of the feedstream.
- the polymerization is run in a bulk process.
- Polymerizations can be run at any temperature and or pressure suitable to obtain the desired polymers.
- Suitable temperatures for solution polymerization include a temperature of about 50°C to about 200°C, such as about 60°C to about 180°C, such as about 65°C to about 160°C, such as about 80°C to about 150°C, such as about 85°C to about 140°C.
- Suitable temperatures for slurry- or gas-phase polymerization include a temperature of about 50°C to about 120°C, such as about 60°C to about 110°C, such as about 65°C to about 100°C, such as about 70°C to about 85°C, such as about 75°C to about 80°C.
- Polymerizations can be run at a pressure of about 0.1 MPa to about 25 MPa, such as about 0.45 MPa to about 6 MPa, or about 0.5 MPa to about 4 MPa.
- the run time of the reaction can be up to about 300 minutes, such as about 5 minutes to about 250 minutes, such as about 10 minutes to about 120 minutes, such as about 20 minutes to about 90 minutes, such as about 30 minutes to about 60 minutes.
- the run time may be the average residence time of the reactor.
- the run time of the reaction is up to about 45 minutes.
- the run time may be the average residence time of the reactor.
- hydrogen is present in the polymerization reactor at a partial pressure of about 0.001 psig to about 50 psig (0.007 kPa to 345 kPa), such as about 0.01 psig to about 25 psig (0.07 kPa to 172 kPa), such as about 0.1 psig to about 10 psig (0.7 kPa to 70 kPa).
- the hydrogen content is about 0.0001 ppm to about 2,000 ppm, such as about 0.0001 ppm to about 1,500 ppm, such as about 0.0001 ppm to about 1,000 ppm, such as about 0.0001 ppm to about 500 ppm.
- hydrogen can be present at zero ppm.
- MAO can be present at zero mol%, alternately the MAO can be present at a molar ratio of aluminum to transition metal less than 500:1, such as less than 300:1, such as less than 100:1, such as less than 1:1.
- Catalyst productivity is a measure of how many grams of polymer (P) are produced using a polymerization catalyst comprising W g of catalyst (cat), over a period of time of T hours; and may be expressed by the following formula: P/(T x W) and expressed in units of gPgcat -1 hr -1 .
- catalyst activity is a measure of how active the catalyst is and is reported as the mass of product polymer (P) produced per mole of catalyst (cat) used (kgP/molcat) for solution MAO derived catalyst system or as the mass of product polymer (P) produced per mass of catalyst (cat) used (kgP/gcat or gP/gcat) for solution, solid, or supported MAO derived catalyst systems.
- Catalyst activity may also be expressed over a period of time T of hours and reported as the mass of product polymer (P) produced per mole or millimole of catalyst (cat) used and expressed in units of gPmmolcat -1 hr -1 for solution or solid MAO as the activator and in units of kgPgcat -1 hr -1 for solution, solid, or supported MAO as the activator.
- a solution catalyst system with TMA free solution MAO as the activator has a catalyst activity of greater than about 10 to 1,000 kgPgcat -1 hr -1 , such as greater than about 20 kgPgcat -1 hr -1 , such as greater than about 30 kgPgcat -1 hr -1 ; such as about 100 kgPgcat -1 hr -1 to about 300 kgPgcat -1 hr -1 ;
- a TMA free supported MAO derived finished catalyst system used in slurry- or gas-phase polymerization has a catalyst activity of greater than about 3 to 30 kgPgcat -1 hr -1 , such as about 4 kgPgcat -1 hr -1 to about 20 kgPgcat -1 hr -1 , such as about 6 kgPgcat -1 hr -1 to about 15 kgPgcat- 1 hr -1 , such as about 8 kgPgcat -1 hr -1 to
- the catalyst residence time in a reactor can be about 10 minutes to about 120 minutes, such as about 20 minutes to about 90 minutes, such as about 30 minutes to about 60 minutes; for solution polymerization, the catalyst residence time in a reactor can be about 10 minutes to about 120 minutes, such as about 20 minutes to about 90 minutes, such as about 30 minutes to about 60 minutes; for supported catalyst polymerization, e.g., slurry- or gas-phase polymerization, the catalyst residence time in a reactor can be about 10 minutes to about 240 minutes, such as about 30 minutes to about 120 minutes, such as about 60 minutes to about 90 minutes; and for the self-supported MAO derived catalyst system; the residence time in a slurry- or gas-phase polymerization reactor is similar to the supported catalyst system.
- the polymerization 1) is conducted at temperatures of about 0°C to about 300°C (such as about 25°C to about 250°C, such as about 50°C to about 160°C, such as about 80°C to about 140°C); 2) is conducted at a pressure of atmospheric pressure to about 10 MPa (such as about 0.35 MPa to about 10 MPa, such as about 0.45 MPa to about 6 MPa, such as about 0.5 MPa to about 4 MPa); 3) is conducted without an aliphatic hydrocarbon diluent, such as in a gas phase reactor, or with the monomer also serves as a diluent, such as in a slurry reactor with propylene as the monomer and diluent, or in an aliphatic hydrocarbon diluent, such as isobutane, butane, pentane, isopentane, hexanes, isohexane, heptane, octane, do
- the catalyst system used in the polymerization includes more than one pre-catalyst compound.
- a "reaction zone” also referred to as a “polymerization zone” is a vessel where polymerization takes place, for example a stirred-tank reactor or a loop reactor. When multiple reactors are used in a continuous polymerization process, each reactor is considered as a separate polymerization zone. For a multi-stage polymerization in a batch polymerization process, each polymerization stage is considered as a separate polymerization zone. In at least one embodiment, the polymerization occurs in one reaction zone or multiple reaction zones. Room temperature is 23°C unless otherwise noted.
- additives may also be used in the polymerization, as desired, such as one or more scavengers, hydrogen, aluminum alkyls, or chain transfer agents such as higher alkyl modified MAO, a compound represented by the formula AlR3 or ZnR2 (where each R is, independently, a C 1 -C 8 aliphatic radical, such as methyl, ethyl, propyl, butyl, pentyl, hexyl octyl or an isomer thereof) or a combination thereof, such as diethyl zinc, MAO, trimethylaluminum, triisobutylaluminum, trioctylaluminum, or a combination thereof.
- scavengers hydrogen, aluminum alkyls, or chain transfer agents
- a compound represented by the formula AlR3 or ZnR2 where each R is, independently, a C 1 -C 8 aliphatic radical, such as methyl, ethyl, propyl, butyl,
- Polymers that may be prepared include polyethylene, polypropylene, homopolymers of C 4 -C 20 olefins, copolymers of C 4 -C 20 olefins, copolymers of ethylene with C 3 -C 20 olefins, copolymers of propylene with C 4 -C 20 olefins, terpolymers of C 4 -C 20 olefins, terpolymers of ethylene and propylene with C 4 -C 20 olefins, and terpolymers of ethylene and propylene with 5-ethylidene-2-norbornene.
- the processes described herein may be used to produce polymers such as HDPE, MDPE, LDPE, or LLDPE with butene, hexene, or octene as the comonomer to turn the polymer density; such as iPP, sPP, or aPP from different stereo- or regio-regulation on the pre-catalysts; such as random copolymer plastomers from propylene rich ethylene copolymers with ethylene content not more than 30% or ethylene rich propylene copolymers with propylene content not more that 30%, ethylene propylene elastomers (rubber)s, i.e., EP rubbers, with ethylene and propylene close to 50:50, such as 30:70, 40:60, 50:50, 60:40, or 70:30, such as ethylene-butadiene copolymers from solution polymerization; such as impact copolymers, e.g., reactor made iPP-EPR, iPP-E
- the melt index (MI) for PE based polymers is from 0.01 to about 50 g/10 min, such as 0.1 to 10g/10 min, such as 0.5 to 5 g/10 min, such as 1-2 g/10 min; or the melt flow rate or mass flow rate (MFR) for PP based polymers is from 0.01 to 2000 g/10 min, such as 0.05 to 1000 g/10 min, such as 0.1 to 500 g/10 min, such as 0.5 to 100g/10 min, such as 2-50g/10 min, with the measurement method described in the similar standards ASTM D1238 and ISO 1133.
- the weight average molecular weight Mw of the polymer products is in the range of 10k to 2000k measured by GPC methods, such as 50k to 1000k, such as 60k to 500k, such as 100k to 300k; and the molecular weight distribution (MWD) or polydispersity index (PDI) is 1.5 to 30, such as 2 to 10, such as 2.5 to 9, which can have single modal or multiple modal distributions, for example, bimodal distributions from a two-stage polymerization process in two different reaction zones or from a one-stage polymerization process in one reaction zone with a catalyst system containing two different pre-catalyst compounds.
- GPC methods such as 50k to 1000k, such as 60k to 500k, such as 100k to 300k
- MWD molecular weight distribution
- PDI polydispersity index
- the comonomer distribution in the polymer products can be a conventional distribution, i.e., the comonomer incorporation is getting less with the increase of Mw; can be a flat distribution, i.e., similar incorporation with different molecular weight compositions; or can be a broad orthogonal comonomer distribution, i.e., the comonomer incorporation is getting more with the increase of Mw.
- EXPERIMENTAL [0239] In studies to improve upon prior technology (e.g., US 6,368,999), the fluorinated silica support containing Si-F units were found to react with free TMA in MAO to become more active supported MAO.
- Si-F units e.g., (NH 4 ) 2 SiF 6
- Example 1 Quantification of Total THF Extractable TMA Contents in a Commercial MAO Solution The total TMA content including the coordinated and free TMA in either a supported or unsupported MAO composition can be quantified through a THF solvent treatment to convert both the coordinated and free TMA to AlMe 3 (THF) as the major product and AlMe 2 (THF) 2 + as a minor product according to Scheme 8 reaction.
- THF AlMe 3
- THF AlMe 2
- the total TMA is therefore the sum of AlMe 3 (THF) and AlMe 2 (THF) 2 + (converted back to TMA in calculation) and can be quantified with the 1 H NMR methods below with the solvent toluene as the internal standard for solution MAO or with an added inert compound as the internal standard for ether a solution MAO, a solid MAO, or a supported MAO.
- the MAO formula without coordinated TMA is Al1O0.78Me1.44 based on the COA to give a Mw 61.1.
- AlMe 2 + is counted as TMA because it is generated from coordinated TMA.
- the clathrate phase from the sample with complete conversion of regular MAO to ionic MAO was also analyzed with 1 H NMR spectroscopy in THF-d8 NMR solvent and the spectrum of the Al-Me region is shown in Fig.4B with the mother MAO solution for comparison, which shows the absence of AlMe 2 + species for the ionic MAO, meaning all coordinated TMA is removed by KF as a confirmation that the coordinated TMA is the source of AlMe 2 + .
- the total Al% is 13.8 wt% and the total TMA (free + coordinated) is 5.33 wt% based on Table 1.
- the total TMA can be converted to 2.00 wt% Al to give 14.5 mol%.
- Example 3 (NH4)2SiF6 Treatment of a Commercial MAO Solution
- This example uses (NH 4 ) 2 SiF 6 as the electron withdrawing compound to convert the majority of total TMA into AlMe 2 F.
- the NMR reaction stoichiometry studies suggest that 1 eq. of (NH 4 ) 2 SiF 6 is able to consume 8 eq. TMA to form 6 eq. AlMe 2 F and a species having 2 eq. N-H and 2 eq.
- the THF extractable TMA (AlMe 3 (THF)) is reduced from an integral of 78.96 (3B) to 37.94 (3A) and the AlMe 2 (THF) 2 + species integral is increased from 7.98 (3B) to 15.23 (3A).
- the THF extractable TMA is believed to be from the coordinated AlMe 2 F according to the reaction below (Scheme 13), which shows AlMe 2 F as a poor free TMA source if no donor is around due to the strong electron withdrawing group F that doesn’t favor the F bonded Al to become coordinately unsaturated (left reaction).
- Scheme 13 also helps understanding of the observed EWC treated MAO chemistry that a monodentate donor such as THF can either replace the AlMe3 form from the AlMe 2 of the coordinated AlMe 2 F with a nearby Me directly bonded to the Al of the coordinated AlMe 2 F to form the THF coordinated TMA (AlMe3(THF) in FIG.
- a monodentate donor such as THF
- Examples 4-1, 4-2, 4-3 Small Scale Solution Propylene Polymerizations [0257] Polymerization Reagents: Pre-catalyst solutions were made using a given transition metal complex dissolved in toluene (ExxonMobil Chemical-anhydrous, stored under N2) (98%), typically at a concentration of 0.5 mmol/L. Activation of the complexes was performed using various methylaluminoxes (MAO) including commercial methylalumoxane (cMAO, 10 wt% in toluene, Albemarle Corp. - control) and F-MAO. Complex 6 can be prepared as described in US 11,254,763.
- MAO methylaluminoxes
- cMAO commercial methylalumoxane
- Complex 6 can be prepared as described in US 11,254,763.
- MAOs were typically used as a 0.2 wt% toluene solution. Micromoles of MAO reported below are based on the micromoles of aluminum in MAO, which has a formula weight of 58.0 grams/mole. [0259] Solvents, polymerization grade toluene and/or isohexanes were supplied by ExxonMobil Chemical Co.
- Polymerization grade propylene was purified by passage through a series of columns: 2,250 cc OXICLEAR cylinder from Labclear followed by a 2,250 cc column packed with 3 ⁇ molecular sieves (8-12 mesh; Aldrich Chemical Company), then two 500 cc columns in series packed with 5 ⁇ molecular sieves (8-12 mesh; Aldrich Chemical Company), then a 500 cc column packed with SELEXSORB CD (BASF), and finally a 500 cc column packed with SELEXSORB COS (BASF).
- toluene, MAO, propylene (1.0 ml unless otherwise listed in the tables) and comonomer (if used) were added via syringe.
- the reactor was then heated to process temperature (typically 70°C or 100°C unless otherwise mentioned) while stirring at 800 RPM.
- process temperature typically 70°C or 100°C unless otherwise mentioned
- the pre-catalyst solution was added via syringe with the reactor at process conditions.
- the reactor temperature was monitored and typically maintained within +/ ⁇ 1°C. Polymerizations were halted by addition of approximately 50 psi of an air gas mixture or CO 2 gas to the autoclaves for approximately 30 seconds.
- the polymerizations were quenched based on a predetermined pressure loss of approximately 8 psi unless specified differently (max quench value in psi) or for a maximum of 30 minutes polymerization time unless specified differently.
- the reactors were then cooled and vented.
- the polymers were isolated after solvent removal in-vacuo. Actual quench times are reported. Quench times less than maximum reaction times indicate the reaction quenched with uptake. Yields reported include total weight of polymer and residual catalyst.
- Catalyst activity is reported as grams of polymer per mmol complex per hour of reaction time (gP/mmol cat•hr). Propylene homopolymerization examples including characterization are summarized in Table 1 below. [0263] Small Scale Polymer Characterization.
- polymer sample solutions were prepared by dissolving polymer in 1,2,4-trichlorobenzene (TCB, 99+% purity) containing 2,6-di-tert-butyl-4-methylphenol (BHT, Sigma-Aldrich, 99%) at 165°C in a shaker oven for approximately 3 hours.
- the typical concentration of polymer in solution was from 0.1 to 0.9 mg/ml with a BHT concentration of 1.25 mg BHT/ml of TCB. Samples were cooled to 135°C for testing.
- ELSD evaporative light scattering detector
- samples were measured by Gel Permeation Chromatography using a Symyx Technology GPC equipped with dual wavelength infrared detector and calibrated using polystyrene standards (Polymer Laboratories: Polystyrene Calibration Kit S-M-10: Mp (peak Mw) between 580 and 3,039,000).
- Samples 250 ⁇ L of a polymer solution in TCB were injected into the system) were run at an eluent flow rate of 2.0 ml/minute (135°C sample temperatures, 165°C oven/columns) using three Polymer Laboratories: PLgel 10 ⁇ m Mixed-B 300 x 7.5mm columns in series. No column spreading corrections were employed.
- DSC Differential Scanning Calorimetry
- the reactor was heated to 100°C, stirred at 225 rpm, and then pressurized with ethylene (250 psi, Sigma, 99.5%). The reactor was re-pressurized when the pressure dropped below 240 psi during the first hour. After 4 hours the reactor was cooled and then depressurized. The polymer products were isolation from each vial by precipitating and washing with acetone and methanol. Then the solids were filtered out and washed with copious acetone and methanol. The polymer samples were then dried in a 50°C vacuum oven for 18 hours.
- TF-MAO to the pre-catalyst metal
- the examples here used a post-metallocene (Complex 6) to test on a TMA free supported MAO (TF-sMAO) and compare to the regular supported MAO (sMAO).
- Example 8-12 Catalyst Preparation Procedure 2.04 g silica and 12 g toluene in a 20 mL vial Slowly add MAO solution 2.8 g (14 mmol Al, based on 7.0 mmol/g silica charge); heated to 100°C for 4 hours.
- 1-hexene was fed into the reactor as a ratio to ethylene flow (0.1 g/g).
- Hydrogen was fed to the reactor as a ratio to ethylene flow (0.5 mg/g).
- the hydrogen and ethylene ratios were measured by on-line GC analysis. Polymerizations were halted after 1 hour by venting the reactor, cooling to room temperature then exposing to air. The salt was removed by washing with water two times; the polymer was isolated by filtration, briefly washed with acetone, and dried in air for at least two days.
- hydrocarbyl aluminum compounds having strong electron withdrawing atoms or groups can be formed in-situ upon forming the MAO.
- the phrases, unless otherwise specified, "consists essentially of” and “consisting essentially of” do not exclude the presence of other steps, elements, or materials, whether or not, specifically mentioned in this specification, so long as such steps, elements, or materials, do not affect the basic and novel characteristics of the present disclosure, additionally, they do not exclude impurities and variances normally associated with the elements and materials used.
- consists essentially of and “consisting essentially of” do not exclude the presence of other steps, elements, or materials, whether or not, specifically mentioned in this specification, so long as such steps, elements, or materials, do not affect the basic and novel characteristics of the present disclosure, additionally, they do not exclude impurities and variances normally associated with the elements and materials used.
- ranges from any lower limit may be combined with any upper limit to recite a range not explicitly recited, as well as ranges from any lower limit may be combined with any other lower limit to recite a range not explicitly recited, in the same way, ranges from any upper limit may be combined with any other upper limit to recite a range not explicitly recited.
- ranges from any upper limit may be combined with any other upper limit to recite a range not explicitly recited.
- within a range includes every point or individual value between its end points even though not explicitly recited. Thus, every point or individual value may serve as its own lower or upper limit combined with any other point or individual value or any other lower or upper limit, to recite a range not explicitly recited.
- compositions, an element or a group of elements are preceded with the transitional phrase “comprising,” it is understood that we also contemplate the same composition or group of elements with transitional phrases “consisting essentially of,” “consisting of,” “selected from the group of consisting of,” or “is” preceding the recitation of the composition, element, or elements and vice versa.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Abstract
Description

















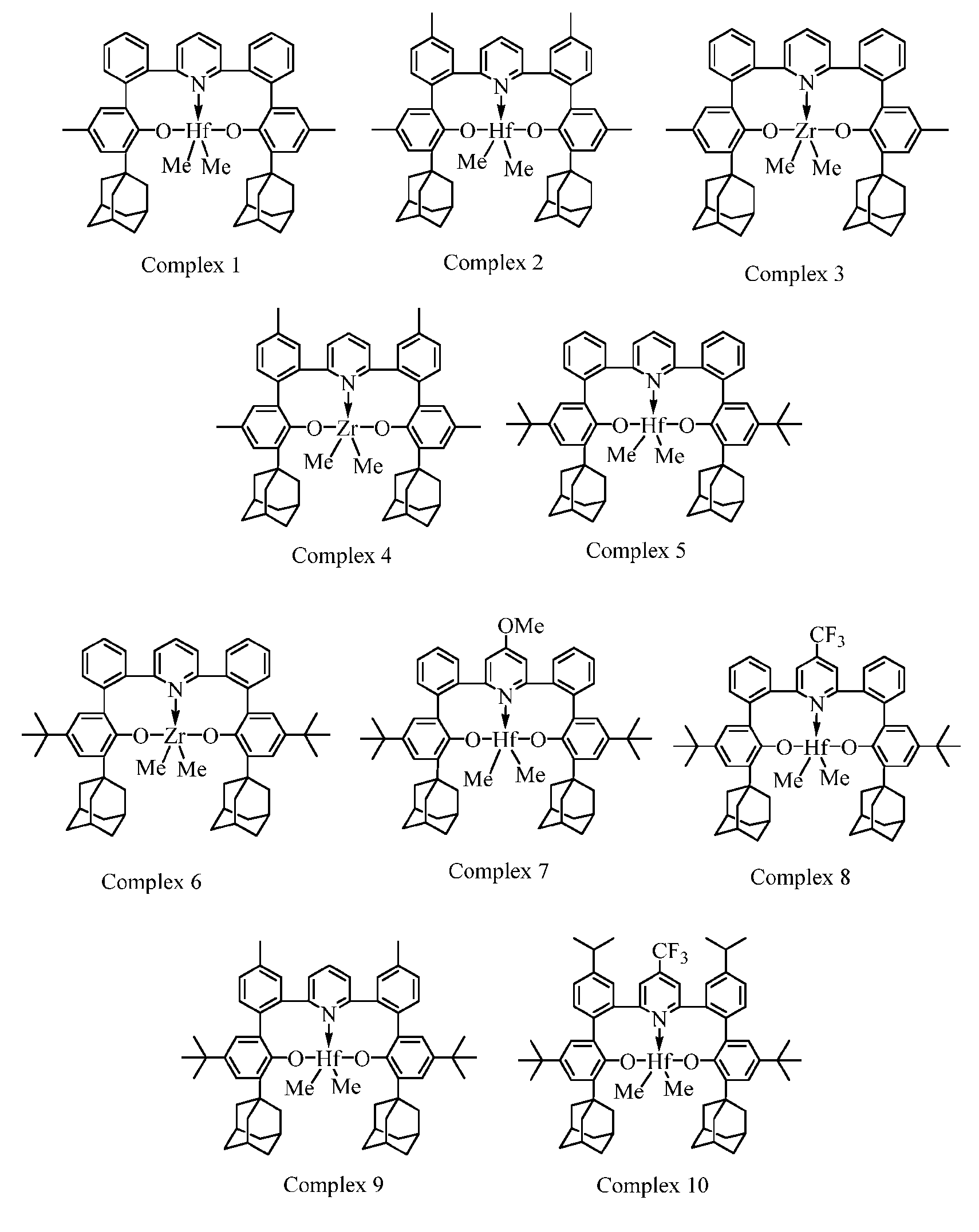



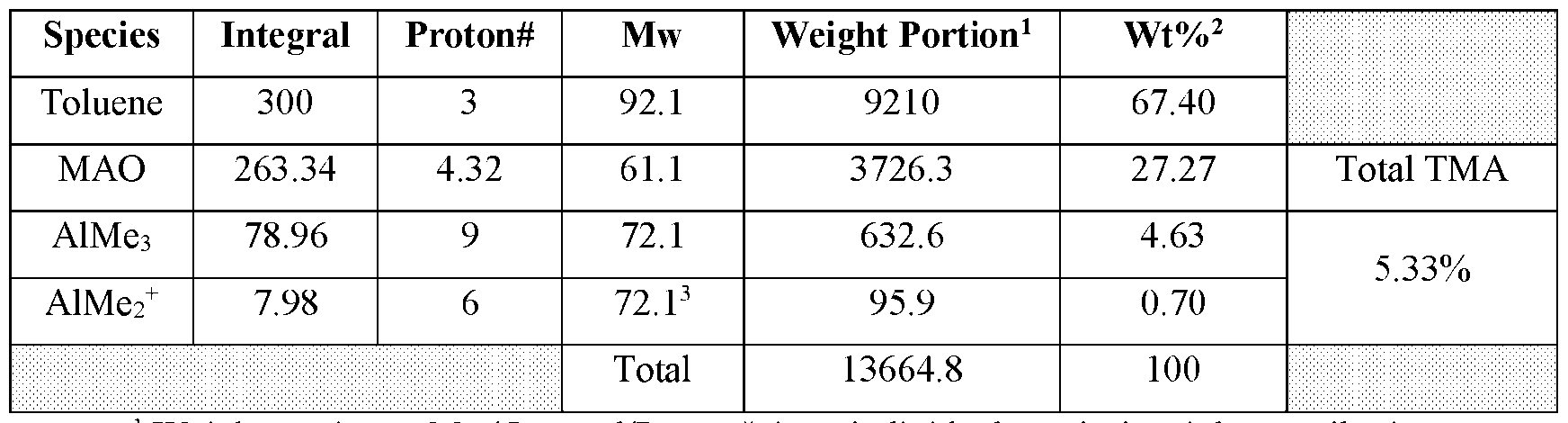





Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202480044515.3A CN121487971A (en) | 2023-06-06 | 2024-05-20 | Anionic modified aluminumoxanes without noncoordinated alkyl aluminum and their methods |
| EP24733405.5A EP4724505A1 (en) | 2023-06-06 | 2024-05-20 | Non-coordinated alkylaluminum free anion modified alumoxanes and methods thereof |
| KR1020267000313A KR20260017476A (en) | 2023-06-06 | 2024-05-20 | Anion-modified alumoxane without non-coordinated alkyl aluminum and method thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363506543P | 2023-06-06 | 2023-06-06 | |
| US63/506,543 | 2023-06-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024253831A1 true WO2024253831A1 (en) | 2024-12-12 |
Family
ID=91581173
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/030225 Ceased WO2024253831A1 (en) | 2023-06-06 | 2024-05-20 | Non-coordinated alkylaluminum free anion modified alumoxanes and methods thereof |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4724505A1 (en) |
| KR (1) | KR20260017476A (en) |
| CN (1) | CN121487971A (en) |
| WO (1) | WO2024253831A1 (en) |
Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4542199A (en) | 1981-07-09 | 1985-09-17 | Hoechst Aktiengesellschaft | Process for the preparation of polyolefins |
| US5001244A (en) | 1988-06-22 | 1991-03-19 | Exxon Chemical Patents Inc. | Metallocene, hydrocarbylaluminum and hydrocarbylboroxine olefin polymerization catalyst |
| US5547675A (en) | 1989-09-13 | 1996-08-20 | Exxon Chemical Patents Inc. | Modified monocyclopentadienyl transition metal/alumoxane catalyst system for polymerization of olefins |
| US5621126A (en) | 1987-01-30 | 1997-04-15 | Exxon Chemical Patents Inc. | Monocyclopentadienyl metal compounds for ethylene-α-olefin-copolymer production catalysts |
| WO2000011006A1 (en) | 1998-08-18 | 2000-03-02 | The Dow Chemical Company | Metalloid salt catalyst/activators |
| WO2000012565A1 (en) | 1998-08-26 | 2000-03-09 | Exxon Chemical Patents Inc. | Highly active supported catalyst compositions |
| US6175409B1 (en) | 1999-04-02 | 2001-01-16 | Symyx Technologies, Inc. | Flow-injection analysis and variable-flow light-scattering methods and apparatus for characterizing polymers |
| US6260407B1 (en) | 1998-04-03 | 2001-07-17 | Symyx Technologies, Inc. | High-temperature characterization of polymers |
| US6294388B1 (en) | 1998-04-03 | 2001-09-25 | Symyx Technologies, Inc. | Indirect calibration of polymer characterization systems |
| US6406632B1 (en) | 1998-04-03 | 2002-06-18 | Symyx Technologies, Inc. | Rapid characterization of polymers |
| US6436292B1 (en) | 1999-04-02 | 2002-08-20 | Symyx Technologies, Inc. | Parallel high-performance liquid chromatography with post-separation treatment |
| US6491816B2 (en) | 1999-04-02 | 2002-12-10 | Symyx Technologies, Inc. | Apparatus for parallel high-performance liquid chromatography with serial injection |
| US6833417B2 (en) | 2002-12-31 | 2004-12-21 | Univation Technologies, Llc | Processes for transitioning between chrome-based and mixed polymerization catalysts |
| US6841630B2 (en) | 2002-12-31 | 2005-01-11 | Univation Technologies, Llc | Processes for transitioning between chrome-based and mixed polymerization catalysts |
| US20050143254A1 (en) * | 2003-12-31 | 2005-06-30 | Sangokoya Samuel A. | Haloaluminoxane compositions, their preparation, and their use in catalysis |
| US6989344B2 (en) | 2002-12-27 | 2006-01-24 | Univation Technologies, Llc | Supported chromium oxide catalyst for the production of broad molecular weight polyethylene |
| US7202313B2 (en) | 2003-03-28 | 2007-04-10 | Union Carbide Chemicals & Plastics Technology Corporation | Chromium-based catalysts in mineral oil for production of polyethylene |
| WO2009029857A1 (en) | 2007-08-29 | 2009-03-05 | Albemarle Corporation | Aluminoxane catalyst activators derived from dialkylaluminum cation precursor agents, processes for making same, and use thereof in catalysts and polymerization of olefins |
| US20090124486A1 (en) | 2007-11-08 | 2009-05-14 | Holtcamp Matthew W | Halogen substituted heterocyclic heteroatom containing ligands-alumoxane activation of metallocenes |
| US20110010938A1 (en) | 2008-02-27 | 2011-01-20 | Univation Technologies, Llc | Modified Chromium-Based Catalysts and Polymerization Processes for Using the Same |
| US7915357B2 (en) | 2005-07-27 | 2011-03-29 | Univation Technologies, Llc | Blow molding polyethylene resins |
| US7956140B2 (en) | 2004-03-17 | 2011-06-07 | Dsm Ip Assets B.V. | Polymerization catalyst comprising amidine ligand |
| US8101691B2 (en) | 2008-12-22 | 2012-01-24 | Univation Technologies, Llc | Systems and methods for fabricating polymers |
| US8129484B2 (en) | 2005-07-27 | 2012-03-06 | Univation Technologies, Llc | Blow molding polyethylene resins |
| US20130253155A1 (en) | 2010-11-22 | 2013-09-26 | Albemarie Corporation | Activator Compositions, Their Preparation, and Their Use In Catalysis |
| US9090720B2 (en) | 2011-03-09 | 2015-07-28 | Albemarle Corporation | Aluminoxane catalyst activators containing carbocation agents, and use thereof in polyolefin catalysts |
| WO2016171810A1 (en) * | 2015-04-20 | 2016-10-27 | Exxonmobil Chemical Patents Inc. | Supported catalyst systems and processes for use thereof |
| US20180142046A1 (en) | 2015-06-05 | 2018-05-24 | Exxonmobil Chemical Patents Inc. | Catalyst System Comprising Supported Alumoxane and Unsupported Alumoxane Particles |
| US20190127499A1 (en) | 2017-10-31 | 2019-05-02 | Exxonmobil Chemical Patents Inc. | Toluene Free Silica Supported Single-Site Metallocene Catalysts from In-situ Supported Alumoxane Formation in Aliphatic Solvents |
| US20190153135A1 (en) | 2012-04-27 | 2019-05-23 | W. R. Grace & Co.-Conn. | Activator Compositions, Their Preparation, And Their Use In Catalysts |
| US11161922B2 (en) | 2017-10-31 | 2021-11-02 | Exxonmobil Chemical Patents Inc. | Toluene free silica supported single-site metallocene catalysts from in-situ supported MAO formation in aliphatic solvents |
| US11254763B2 (en) | 2019-02-12 | 2022-02-22 | Exxonmobil Chemical Patents Inc. | Transition metal bis(phenolate) complexes and their use as catalysts for olefin polymerization |
-
2024
- 2024-05-20 WO PCT/US2024/030225 patent/WO2024253831A1/en not_active Ceased
- 2024-05-20 KR KR1020267000313A patent/KR20260017476A/en active Pending
- 2024-05-20 EP EP24733405.5A patent/EP4724505A1/en active Pending
- 2024-05-20 CN CN202480044515.3A patent/CN121487971A/en active Pending
Patent Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4542199A (en) | 1981-07-09 | 1985-09-17 | Hoechst Aktiengesellschaft | Process for the preparation of polyolefins |
| US5621126A (en) | 1987-01-30 | 1997-04-15 | Exxon Chemical Patents Inc. | Monocyclopentadienyl metal compounds for ethylene-α-olefin-copolymer production catalysts |
| US5001244A (en) | 1988-06-22 | 1991-03-19 | Exxon Chemical Patents Inc. | Metallocene, hydrocarbylaluminum and hydrocarbylboroxine olefin polymerization catalyst |
| US5547675A (en) | 1989-09-13 | 1996-08-20 | Exxon Chemical Patents Inc. | Modified monocyclopentadienyl transition metal/alumoxane catalyst system for polymerization of olefins |
| US6454947B1 (en) | 1998-04-03 | 2002-09-24 | Symyx Technologies, Inc. | Overlaid injection for rapid characterizations of polymers |
| US6491823B1 (en) | 1998-04-03 | 2002-12-10 | Symyx Technologies, Inc. | Targeted separation protocols for rapid characterizations of polymers |
| US6260407B1 (en) | 1998-04-03 | 2001-07-17 | Symyx Technologies, Inc. | High-temperature characterization of polymers |
| US6294388B1 (en) | 1998-04-03 | 2001-09-25 | Symyx Technologies, Inc. | Indirect calibration of polymer characterization systems |
| US6475391B2 (en) | 1998-04-03 | 2002-11-05 | Symyx Technologies, Inc. | Rapid characterization of polymers for combinatorial, analytical and process control applications |
| US6406632B1 (en) | 1998-04-03 | 2002-06-18 | Symyx Technologies, Inc. | Rapid characterization of polymers |
| US6461515B1 (en) | 1998-04-03 | 2002-10-08 | Symyx Technologies, Inc. | Parallel liquid chromatography for analyzing combinatorial libraries of non-biological polymers |
| WO2000011006A1 (en) | 1998-08-18 | 2000-03-02 | The Dow Chemical Company | Metalloid salt catalyst/activators |
| WO2000012565A1 (en) | 1998-08-26 | 2000-03-09 | Exxon Chemical Patents Inc. | Highly active supported catalyst compositions |
| US6667272B2 (en) | 1998-08-26 | 2003-12-23 | Exxonmobil Chemical Patents Inc. | Highly active supported catalyst compositions |
| US6368999B1 (en) | 1998-08-26 | 2002-04-09 | Exxon Mobil Chemical Patents Inc. | Highly active supported catalyst compositions |
| US6175409B1 (en) | 1999-04-02 | 2001-01-16 | Symyx Technologies, Inc. | Flow-injection analysis and variable-flow light-scattering methods and apparatus for characterizing polymers |
| US6491816B2 (en) | 1999-04-02 | 2002-12-10 | Symyx Technologies, Inc. | Apparatus for parallel high-performance liquid chromatography with serial injection |
| US6436292B1 (en) | 1999-04-02 | 2002-08-20 | Symyx Technologies, Inc. | Parallel high-performance liquid chromatography with post-separation treatment |
| US8420754B2 (en) | 2002-12-27 | 2013-04-16 | Univation Technologies, Llc | Production of polyethylene |
| US6989344B2 (en) | 2002-12-27 | 2006-01-24 | Univation Technologies, Llc | Supported chromium oxide catalyst for the production of broad molecular weight polyethylene |
| US7504463B2 (en) | 2002-12-27 | 2009-03-17 | Univation Technologies, Llc | Production of broad molecular weight polyethylene |
| US7563851B2 (en) | 2002-12-27 | 2009-07-21 | Univation Technologies, Llc | Production of broad molecular weight polyethylene |
| US6833417B2 (en) | 2002-12-31 | 2004-12-21 | Univation Technologies, Llc | Processes for transitioning between chrome-based and mixed polymerization catalysts |
| US6841630B2 (en) | 2002-12-31 | 2005-01-11 | Univation Technologies, Llc | Processes for transitioning between chrome-based and mixed polymerization catalysts |
| US7202313B2 (en) | 2003-03-28 | 2007-04-10 | Union Carbide Chemicals & Plastics Technology Corporation | Chromium-based catalysts in mineral oil for production of polyethylene |
| US7193100B2 (en) | 2003-12-31 | 2007-03-20 | Albemarle Corporation | Haloaluminoxane compositions, their preparation, and their use in catalysis |
| US20050143254A1 (en) * | 2003-12-31 | 2005-06-30 | Sangokoya Samuel A. | Haloaluminoxane compositions, their preparation, and their use in catalysis |
| US7355058B2 (en) | 2003-12-31 | 2008-04-08 | Albemarle Corporation | Haloaluminoxane compositions, their preparation, and their use in catalysis |
| US7956140B2 (en) | 2004-03-17 | 2011-06-07 | Dsm Ip Assets B.V. | Polymerization catalyst comprising amidine ligand |
| US7915357B2 (en) | 2005-07-27 | 2011-03-29 | Univation Technologies, Llc | Blow molding polyethylene resins |
| US8129484B2 (en) | 2005-07-27 | 2012-03-06 | Univation Technologies, Llc | Blow molding polyethylene resins |
| US8575284B2 (en) | 2007-08-29 | 2013-11-05 | Albemarle Corporation | Aluminoxane catalyst activators derived from dialkylaluminum cation precursor agents, processes for making same, and use thereof in catalysts and polymerization of olefins |
| WO2009029857A1 (en) | 2007-08-29 | 2009-03-05 | Albemarle Corporation | Aluminoxane catalyst activators derived from dialkylaluminum cation precursor agents, processes for making same, and use thereof in catalysts and polymerization of olefins |
| US20090124486A1 (en) | 2007-11-08 | 2009-05-14 | Holtcamp Matthew W | Halogen substituted heterocyclic heteroatom containing ligands-alumoxane activation of metallocenes |
| US20110010938A1 (en) | 2008-02-27 | 2011-01-20 | Univation Technologies, Llc | Modified Chromium-Based Catalysts and Polymerization Processes for Using the Same |
| US8101691B2 (en) | 2008-12-22 | 2012-01-24 | Univation Technologies, Llc | Systems and methods for fabricating polymers |
| US20130253155A1 (en) | 2010-11-22 | 2013-09-26 | Albemarie Corporation | Activator Compositions, Their Preparation, and Their Use In Catalysis |
| US9090720B2 (en) | 2011-03-09 | 2015-07-28 | Albemarle Corporation | Aluminoxane catalyst activators containing carbocation agents, and use thereof in polyolefin catalysts |
| US20190153135A1 (en) | 2012-04-27 | 2019-05-23 | W. R. Grace & Co.-Conn. | Activator Compositions, Their Preparation, And Their Use In Catalysts |
| WO2016171810A1 (en) * | 2015-04-20 | 2016-10-27 | Exxonmobil Chemical Patents Inc. | Supported catalyst systems and processes for use thereof |
| US20180142046A1 (en) | 2015-06-05 | 2018-05-24 | Exxonmobil Chemical Patents Inc. | Catalyst System Comprising Supported Alumoxane and Unsupported Alumoxane Particles |
| US20190127499A1 (en) | 2017-10-31 | 2019-05-02 | Exxonmobil Chemical Patents Inc. | Toluene Free Silica Supported Single-Site Metallocene Catalysts from In-situ Supported Alumoxane Formation in Aliphatic Solvents |
| US11021552B2 (en) | 2017-10-31 | 2021-06-01 | Exxonmobil Chemical Patents Inc. | Toluene free silica supported single-site metallocene catalysts from in-situ supported alumoxane formation in aliphatic solvents |
| US11161922B2 (en) | 2017-10-31 | 2021-11-02 | Exxonmobil Chemical Patents Inc. | Toluene free silica supported single-site metallocene catalysts from in-situ supported MAO formation in aliphatic solvents |
| US11254763B2 (en) | 2019-02-12 | 2022-02-22 | Exxonmobil Chemical Patents Inc. | Transition metal bis(phenolate) complexes and their use as catalysts for olefin polymerization |
Non-Patent Citations (9)
| Title |
|---|
| BABUSHKINBRINTZINGER, J. AM. CHEM. SOC., vol. 124, 2002, pages 12869 - 12873 |
| BERCAW, J. E., MACROMOLECULES, vol. 42, 2009, pages 8751 - 8762 |
| BRYLIAKOVTALSIBOCHMANN, ORGANOMETALLICS, vol. 23, 2004, pages 149 - 152 |
| CHENMARKS, 100 CHEM. REV., vol. 1391, 2000 |
| KILPATRICK ALEXANDER F. ET AL: "Slurry-Phase Ethylene Polymerization Using Pentafluorophenyl- and Pentafluorophenoxy-Modified Solid Polymethylaluminoxanes", ORGANOMETALLICS, vol. 37, no. 1, 8 January 2018 (2018-01-08), pages 156 - 164, XP093195141, ISSN: 0276-7333, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/acs.organomet.7b00846> DOI: 10.1021/acs.organomet.7b00846 * |
| LUOJAINHARLAN: "American Chemical Society", 5 April 2017, PRIESTLEY MEDALIST SYMPOSIUM IN HONOR OF TOBIN J. MARKS, article "INOR 1169" |
| ORGANOMETALLICS, vol. 19, 2000, pages 4684 - 4686 |
| SARZOTTI ET AL., J. POLYMER SCI. A, vol. 45, 2007, pages 1677 - 1690 |
| SINN ET AL.: "Metalorg. Cat. for Synth. & Polym", 1999, SPRINGER-VERLAG, article "Formation, Structure, and Mechanism of Oligomeric Methylaluminoxane", pages: 105 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4724505A1 (en) | 2026-04-15 |
| KR20260017476A (en) | 2026-02-05 |
| CN121487971A (en) | 2026-02-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107636029B (en) | Catalyst compositions comprising fluorinated supports and methods of use thereof | |
| US7842763B2 (en) | Catalyst compositions and polyolefins for extrusion coating applications | |
| RU2382793C2 (en) | Dual metallocene catalyst for producing film resins with good machine direction (md) elmendorf tear strength | |
| ES2638094T3 (en) | Polymerization catalysts to produce polymers with low levels of long chain branching | |
| CN112088172A (en) | Process for preparing non-coordinating anionic activators in aliphatic and cycloaliphatic hydrocarbon solvents | |
| CN103732604B (en) | Comprise the highly active catalyst composition of the silyl-bridged metallocene with large volume substituting group | |
| JP2005538215A (en) | Method for producing fluorination catalyst | |
| JP2002528572A (en) | Compounds comprising transition metal cations linked to aluminoxate anions and their use as catalyst components | |
| AU2003282528A1 (en) | Synthesis of polymerization catalyst components | |
| BR112013018910B1 (en) | catalyst composition, olefin polymerization process comprising said catalyst composition and compound | |
| CN103596686B (en) | Comprise the catalyst system of multiple non-coordinating anion activator and use its polymerization process | |
| CN110431160A (en) | Supported catalyst systems and methods of use thereof | |
| CN107531840B (en) | Supported catalyst compositions having improved flow characteristics and preparation thereof | |
| CN1172960C (en) | Process for producing highly productive supported ionic catalysts for gas phase polymerization reactions | |
| WO2024253831A1 (en) | Non-coordinated alkylaluminum free anion modified alumoxanes and methods thereof | |
| US7332551B2 (en) | Partially fluorinated naphthyl-based borates | |
| CN111356705A (en) | Polymerization process | |
| KR20260017477A (en) | Cationically modified alumoxane without uncoordinated alkyl aluminum and method thereof | |
| RU2510646C2 (en) | Metallocene compound, catalyst composition including it and method of olefin polymerisation, applying it | |
| CN110557948A (en) | Hafnium complexes; supported hafnium complexes; methods of forming polymers using such complexes | |
| CN108290975B (en) | Production of Polyolefins with Internal Unsaturation Using Metallocene Catalyst Systems | |
| CN110312741B (en) | Hafnocene catalyst compounds and methods of use thereof | |
| CN121620541A (en) | Solution catalyst system and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24733405 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2025571428 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025571428 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020267000313 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020267000313 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024733405 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024733405 Country of ref document: EP Effective date: 20260107 |
|
| ENP | Entry into the national phase |
Ref document number: 2024733405 Country of ref document: EP Effective date: 20260107 |
|
| ENP | Entry into the national phase |
Ref document number: 2024733405 Country of ref document: EP Effective date: 20260107 |
|
| ENP | Entry into the national phase |
Ref document number: 2024733405 Country of ref document: EP Effective date: 20260107 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020267000313 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2024733405 Country of ref document: EP Effective date: 20260107 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024733405 Country of ref document: EP |