WO2024257715A1 - 2‐m‐ヒドロキシフェニル酢酸の製造方法及び2‐m‐ヒドロキシフェニル酢酸 - Google Patents
2‐m‐ヒドロキシフェニル酢酸の製造方法及び2‐m‐ヒドロキシフェニル酢酸 Download PDFInfo
- Publication number
- WO2024257715A1 WO2024257715A1 PCT/JP2024/020996 JP2024020996W WO2024257715A1 WO 2024257715 A1 WO2024257715 A1 WO 2024257715A1 JP 2024020996 W JP2024020996 W JP 2024020996W WO 2024257715 A1 WO2024257715 A1 WO 2024257715A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxybenzaldehyde
- reaction
- hydroxyphenylacetic acid
- producing
- wittig
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/18—Preparation of ethers by reactions not forming ether-oxygen bonds
- C07C41/30—Preparation of ethers by reactions not forming ether-oxygen bonds by increasing the number of carbon atoms, e.g. by oligomerisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/03—Ethers having all ether-oxygen atoms bound to acyclic carbon atoms
- C07C43/14—Unsaturated ethers
- C07C43/178—Unsaturated ethers containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/42—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by hydrolysis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/26—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing hydroxy groups
- C07C47/27—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing hydroxy groups containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/30—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing six-membered aromatic rings
Definitions
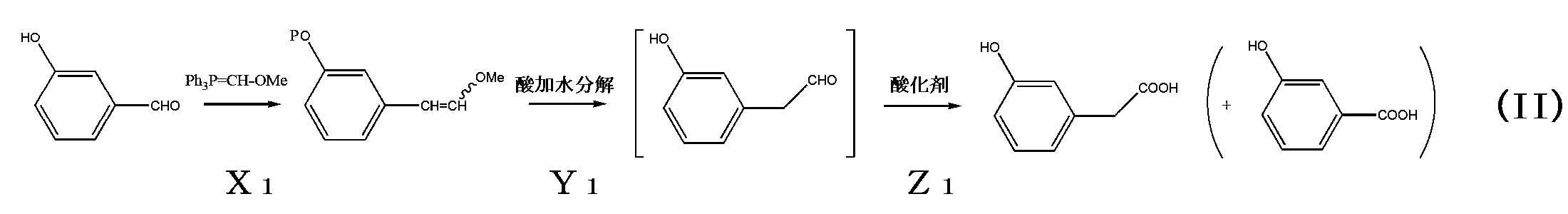
- the present invention relates to a method for producing 2-m-hydroxyphenylacetic acid and 2-m-hydroxyphenylacetic acid.
- 2-m-Hydroxyphenylacetic acid is at least a very useful chemical product as a starting material for preparations for preventing or treating allergic inflammatory skin diseases or skin barrier dysfunction (Patent Document 1), a chemical substance for preserving the freshness of plants and/or for promoting their continued growth (Patent Document 2), and methyl carboxymethylphenylaminocarboxypropylphosphonate (also known as Nahlsgen (registered trademark)), an anti-aging agent added to cosmetics and the like due to its activity as a GGT inhibitor (Patent Document 3).
- 2-m-hydroxyphenylacetic acid is thought to be a substance that can be obtained by, for example, using a CN (cyanide) source (NaCN or KCN), converting the methyl group of m-crezol to a halogen to obtain m-hydroxyphenyl halide, which is then converted to m-hydroxyphenyl cyanide and then hydrolyzing it.
- CN cyanide
- KCN KCN
- TMSCN trimethylsilyl cyanide
- TMSCN trimethylsilyl cyanide
- Non-Patent Document 2 The possibility of producing 2-m-hydroxyphenylacetic acid using the Willgerodt reaction has also been disclosed in the past (Non-Patent Document 2).
- a sulfur (S)-based reactant is used.
- Non-Patent Document 2 Even if the sulfur-based reactant disclosed in Non-Patent Document 2 is used, it is difficult to adopt from an industrial point of view because it requires equipment to deal with the odor of sulfur-based gas and the number of steps required to obtain 2-m-hydroxyphenylacetic acid is large.
- the inventors have been working diligently on research into a method for producing 2-m-hydroxyphenylacetic acid, based on the concerns of improving safety and reducing the number of steps compared to the above-mentioned known techniques.
- m-hydroxybenzaldehyde or an alkali metal salt of said m-hydroxybenzaldehyde as the starting material and using the Wittig reaction from among the many reactions that can be used may enable the production of 2-m-hydroxyphenylacetic acid safely and with fewer steps, and have therefore continued their research.
- the inventors therefore attempted to react m-hydroxybenzaldehyde as a starting material with a Wittig reaction agent represented by the following general formula (I), which is a 2-Methoxymethyl triphenylphosphazene derivative, in an amount that greatly exceeds the reaction equivalent of the Wittig reaction.
- a Wittig reaction agent represented by the following general formula (I), which is a 2-Methoxymethyl triphenylphosphazene derivative
- a portion of the Wittig reagent can substitute a phosphinium group for the proton of the hydroxy group of the starting material m-hydroxybenzaldehyde.
- Another portion of the Wittig reagent can react with the aldehyde group of the starting material to produce an enol derivative by increasing the carbon number by one, a one-carbon homologation reaction.
- the Wittig reaction is a non-aqueous reaction, it is incompatible to use a starting material having a hydroxyl group.
- the above-mentioned action (a) i.e., the replacement of the proton of the hydroxyl group with a phosphinium group derived from the Wittig reactant, makes it possible to use a starting material having a hydroxyl group, which was previously considered incompatible.
- the Wittig reactant H 3 C—O—CH ⁇ PPh 3 is not limited to being used as a preformed compound.
- the Wittig reactant H 3 C—O—CH ⁇ PPh 3 can be generated by reacting (methoxymethyl)triphenylphosphonium chloride (H 3 C—O—CH 2 -P + (Cl ⁇ )Ph 3 ) with a strong base (e.g., potassium tert-butoxide) in-situ in a solvent (e.g., toluene or tetrahydrofuran) commonly used in the Wittig reaction.
- a strong base e.g., potassium tert-butoxide
- solvent e.g., toluene or tetrahydrofuran
- H 3 C—O—CH ⁇ PPh 3 as a Wittig reactant also includes the Wittig reaction of the Wittig reactant that can be generated from a raw material compound with the starting material m-hydroxybenzaldehyde.
- the upper limit of the amount of the Wittig reactant introduced is not particularly limited. From the viewpoint of the economic disadvantage of introducing an unnecessarily large amount, the prevention or suppression of the influence of side reactions, and/or the optimization of the accompanying purification step, it is a preferred embodiment to set the amount of the Wittig reactant to 3 times or less (more preferably 2.5 times or less) the reaction equivalent amount.
- the enol derivative is easily hydrolyzed under acidic conditions. Therefore, it is possible to produce 2-(m-hydroxyphenyl)acetaldehyde, which is an additional carbon atom from the starting material m-hydroxybenzaldehyde. However, it has been confirmed that 2-(m-hydroxyphenyl)acetaldehyde is an unstable compound that is very difficult to isolate.
- a method for producing 2-m-hydroxyphenylacetic acid according to the present invention comprises a first step of producing m-hydroxystyryl methyl ether by reacting m-hydroxybenzaldehyde with a Wittig reactant represented by general formula (I) in an amount of 2 molar equivalents or more relative to the m-hydroxybenzaldehyde, a second step of producing 2-(m-hydroxyphenyl)acetaldehyde by acid hydrolysis of the m-hydroxystyryl methyl ether, and a third step of treating the 2-(m-hydroxyphenyl)acetaldehyde with an oxidizing agent.
- the method for producing 2-m-hydroxyphenylacetic acid comprises carrying out the second step and the third step in situ.
- the following chemical reaction formula (III) is a reaction formula when a sodium (Na) salt of m-hydroxybenzaldehyde, which is an example of an alkali metal salt of m-hydroxybenzaldehyde, is used.
- a sodium (Na) salt of m-hydroxybenzaldehyde is used as a starting material, the Wittig reactant does not substantially need to play the role (a) of the above-mentioned two roles, namely, roles (a) and (b).
- the present inventors have found that by introducing the Wittig reactant in an amount necessary to play the role (b), in other words, to play the carbon-increasing reaction, it is possible to produce an effect similar to that of the first step in the method for producing 2-m-hydroxyphenylacetic acid when m-hydroxybenzaldehyde is used as a starting material.
- the sodium (Na) salt of m-hydroxybenzaldehyde is explained as an example of an alkali metal salt of m-hydroxybenzaldehyde, but the alkali metal salt of m-hydroxybenzaldehyde is not limited to the sodium (Na) salt of m-hydroxybenzaldehyde.
- the potassium (K) salt of m-hydroxybenzaldehyde can also achieve the same results as the sodium (Na) salt.
- the amount of the Wittig reactant introduced to realize the step (reaction step) shown in X2 of the above-mentioned chemical reaction formula (III) with high accuracy may be equal to or more than the reaction equivalent to the alkali metal salt of m-hydroxybenzaldehyde.
- the upper limit of the amount introduced is not particularly limited. However, from the viewpoint of the economic disadvantage of introducing an unnecessarily large amount, prevention or suppression of the influence of side reactions, and/or optimization of the accompanying purification step, it is a preferred embodiment to make the amount less than twice the reaction equivalent of the Wittig reactant (more preferably, 1.5 times or less).
- Another method for producing 2-m-hydroxyphenylacetic acid of the present invention comprises a first step of producing m-hydroxystyryl methyl ether by reacting an alkali metal salt of m-hydroxybenzaldehyde with a Wittig reactant represented by general formula (I) in an amount equal to or greater than the reaction amount of the alkali metal salt, a second step of producing 2-(m-hydroxyphenyl)acetaldehyde by acid hydrolysis of the m-hydroxystyryl methyl ether, and a third step of treating the 2-(m-hydroxyphenyl)acetaldehyde with an oxidizing agent.
- the second step and the third step of the method for producing 2-m-hydroxyphenylacetic acid are carried out in situ.
- the amount of the Wittig reactant when reacting the alkali metal salt with the Wittig reactant represented by the above general formula (I) is sufficient as long as it is equal to or greater than the reaction amount. In other words, it is possible to suppress the amount of the Wittig reactant to be lower than when m-hydroxybenzaldehyde is used as a starting material.
- the 2-m-hydroxyphenylacetic acid composition of the present invention contains less than 0.1 wt% m-hydroxybenzaldehyde.
- this 2-m-hydroxyphenylacetic acid composition by adopting the above-described method for producing 2-m-hydroxyphenylacetic acid, it is possible to realize a high-purity 2-m-hydroxyphenylacetic acid in which the amount of m-hydroxybenzaldehyde, which is the starting material, is reduced to less than 0.1 wt%.
- this 2-m-hydroxyphenylacetic acid composition is highly safe because it does not contain any toxic CN source reactant or any sulfur-based reactant that requires odor control.
- 2-m-hydroxyphenylacetic acid composition means a composition that may contain, in addition to 2-m-hydroxyphenylacetic acid, other substances (for example, m-hydroxybenzaldehyde or an alkali metal salt of said m-hydroxybenzaldehyde as a starting material, or by-products generated in the production process of said 2-m-hydroxyphenylacetic acid), and the composition of the "2-m-hydroxyphenylacetic acid composition” is not particularly limited.
- m-hydroxystyryl methyl ether can be produced with high certainty from the starting material. Furthermore, by carrying out the second step (corresponding to the acid hydrolysis step) and the third step (corresponding to the oxidation step) in this production method in situ, 2-m-hydroxyphenylacetic acid can be produced with high certainty in which a carbon-increasing reaction is realized from the starting material. In addition, according to this production method, 2-m-hydroxyphenylacetic acid can be produced safely and efficiently without using any toxic CN source reactants or sulfur-based reactants that require odor control.
- 2-m-hydroxyphenylacetic acid composition of the present invention it is possible to realize highly pure 2-m-hydroxyphenylacetic acid in which the amount of m-hydroxybenzaldehyde, the starting material, present, in other words the remaining amount of said m-hydroxybenzaldehyde, is reduced to less than 0.1 wt%. Furthermore, this 2-m-hydroxyphenylacetic acid composition is highly safe because it does not contain any toxic CN source reactants or sulfur-based reactants that require odor control.
- FIG. 1 shows a HPLC (high performance liquid chromatography) diagram of the intermediate stage of the first step immediately after completion of the dropwise addition of the starting material m-hydroxybenzaldehyde to the reaction mixture under a specified temperature condition after the preparation of the Wittig reactant in Example 1.
- FIG. 1 is a HPLC diagram after the first step (Wittig reaction) in Example 1.
- FIG. 2 is a HPLC diagram after the second step (acid hydrolysis reaction) in Example 1.
- FIG. 1 is an HPLC diagram after the third step (oxidation reaction) in Example 1.
- 1 is a diagram showing a proton nuclear magnetic resonance ( 1 H NMR) spectrum of the purified product obtained after the third step (oxidation reaction) in Example 1.
- FIG. 2 is a diagram showing a C-13 nuclear magnetic resonance ( 13 C NMR) spectrum of the purified product obtained after the third step (oxidation reaction) in Example 1.
- FIG. 2 is an IR spectrum (infrared absorption spectrum) of the purified product obtained after the third step (oxidation reaction) in Example 1.
- This embodiment describes a method for producing 2-m-hydroxyphenylacetic acid when the starting material is m-hydroxybenzaldehyde.
- reaction steps (A) and (B) are carried out in a solvent (e.g., toluene or tetrahydrofuran) commonly used in the Wittig reaction. Therefore, the reaction steps (A) and (B) are carried out in situ, so to speak.
- a solvent e.g., toluene or tetrahydrofuran
- reaction step (A) i.e., the step of producing the Wittig reactant
- the starting material m-hydroxybenzaldehyde is introduced into a container (e.g., a flask) that contains the Wittig reactant, thereby realizing the production of m-hydroxystyryl methyl ether based on an in-situ Wittig reaction.
- a container e.g., a flask
- reaction steps (A) and (B) in-situ as in this embodiment, commercially available raw material compounds can be used, and there is no need to isolate the Wittig reactant, making it possible to produce m-hydroxystyryl methyl ether very efficiently and with little waste in terms of yield.
- the amount of the Wittig reactant produced in the above-mentioned reaction step (A) is an amount that greatly exceeds the reaction equivalent in the Wittig reaction with respect to the starting material, more specifically, 2 molar equivalents or more, or if the amounts of the above-mentioned raw material compound and strong base are adjusted to such an amount.
- reducing the amount of the starting material (m-hydroxybenzaldehyde) remaining after the reaction step (B) as much as possible leads to ensuring that the production of m-hydroxybenzoic acid, an unnecessary and difficult-to-isolate by-product, is prevented after the third step described below.
- reaction step (B) corresponds to the first step in this embodiment (corresponding to the reaction step of X1 in the chemical reaction formula (II) described above).
- the organic layer is obtained by separating the reaction mixture containing m-hydroxystyryl methyl ether obtained in the first step (including unreacted compounds) with hydrochloric acid having a concentration of several wt %.
- the solvent from the first step is distilled off under reduced pressure, and a solution of an ester such as ethyl acetate is added and stirred, and the solid precipitated under cooling is filtered.
- the solution of an ester such as ethyl acetate is then distilled off from the obtained organic layer under reduced pressure to obtain a viscous solid, which is the crude product of m-hydroxystyryl methyl ether.
- the viscous solid is a mixture of the crude product of m-hydroxystyryl methyl ether and a portion of the by-product of the Wittig reaction produced in the above-mentioned first step (i.e., a viscous mixture).
- the second step is a step in which the viscous mixture containing m-hydroxystyryl methyl ether obtained in the first step is diluted with, for example, an organic solvent (e.g., acetonitrile), and then hydrochloric acid is added to the container containing the mixture to cause an acid hydrolysis reaction.
- an organic solvent e.g., acetonitrile
- hydrochloric acid is used for the acid hydrolysis reaction in this embodiment, at least some of the effects of this embodiment can be achieved even if dilute sulfuric acid, methylsulfonic acid, p-toluenesulfonic acid, or the like is used instead of hydrochloric acid.
- the subsequent third step (corresponding to the reaction step of Z1 in chemical reaction formula (II) already explained) is carried out without taking out the reaction mixture (typically, 2- (m-hydroxyphenyl)acetaldehyde), i.e., in situ.
- the reaction mixture typically, 2- (m-hydroxyphenyl)acetaldehyde
- the third step is a step of adding an oxidizing agent to the reaction system in which the second step described above has been carried out while the temperature is appropriately controlled.
- the oxidation reaction of 2-(m-hydroxyphenyl)acetaldehyde with the oxidizing agent produces a reaction mixture containing 2-m-hydroxyphenylacetic acid.
- the organic solvent is removed from the filtrate obtained by filtering the solid from the reaction mixture under reduced pressure, and then a solution of an ester such as ethyl acetate and sodium hydroxide (NaOH) are added and stirred to obtain an alkaline aqueous solution through liquid separation.
- the organic layer is then obtained by adding a solution of the ester and concentrated hydrochloric acid to the aqueous solution and separating the liquids.
- the ester solution is removed from the organic layer under reduced pressure to obtain a crude product of 2-m-hydroxyphenylacetic acid, which is a viscous solid.
- the crude product is heated under reflux with a large amount of an organic solvent (e.g., toluene), then filtered, and the resulting filtrate is cooled to precipitate a light brown solid substance.
- This light brown solid substance is purified 2-m-hydroxyphenylacetic acid.
- the above-mentioned third step which uses an oxidizing agent, is preferably carried out at a temperature between -10°C and 10°C. If the third step is carried out at a temperature below -10°C, the conversion of the generated 2-(m-hydroxyphenyl)acetaldehyde to the desired 2-m-hydroxyphenylacetic acid in situ is extremely slow and industrially impractical, whereas if the third step is carried out at a temperature above 10°C, the oxygen radical attacks the methylene carbon, which is the carbon that has increased in carbon, causing a cleavage reaction and then providing an oxidation reaction, which can result in the problem of giving m-hydroxybenzoic acid (the same compound as the oxidation product of the starting material).
- the type of oxidizing agent capable of realizing the reaction in the above-mentioned third step is not limited as long as it satisfies the following requirements (x) and (y).
- the oxidizing agent used in the third step is at least one selected from the group consisting of peroxides including hydroxyperoxide and Oxone (registered trademark) ( 2KHSO5.KHSO4.K2SO4 ).
- Oxone registered trademark
- the use of Oxone is particularly preferred as it most reliably produces the effects of this embodiment.
- m-hydroxystyryl methyl ether can be produced with high certainty from the starting material m-hydroxybenzaldehyde. In other words, the amount of the starting material remaining after the first step can be reduced with high certainty.
- 2-m-hydroxyphenylacetic acid can be produced with high certainty. It is also noteworthy that by adopting the method for producing 2-m-hydroxyphenylacetic acid of this embodiment, 2-m-hydroxyphenylacetic acid can be produced safely and efficiently without using any toxic CN source reactant or any sulfur-based reactant that requires odor control, and without going through multiple steps.
- the amount of the remaining starting material can be reduced with a high degree of certainty. Therefore, the amount of m-hydroxybenzaldehyde contained in the 2-m-hydroxyphenylacetic acid composition produced by this production method can be reduced to less than 0.1 wt%.
- Second Embodiment A method for producing 2-m-hydroxyphenylacetic acid in this embodiment, which is different from that in the first embodiment, will be described below.
- This embodiment describes a method for producing 2-m-hydroxyphenylacetic acid when the starting material is an alkali metal salt of m-hydroxybenzaldehyde.
- chemical reaction formula (III) is a reaction formula when sodium (Na) salt of m-hydroxybenzaldehyde, which is an example of an alkali metal salt of m-hydroxybenzaldehyde, is used.
- Na sodium
- m-hydroxybenzaldehyde which is an example of an alkali metal salt of m-hydroxybenzaldehyde
- a pre-existing alkali metal salt of m-hydroxybenzaldehyde may be used as the starting material, but one possible embodiment is to generate an alkali metal salt of m-hydroxybenzaldehyde from a raw compound of an alkali metal salt of m-hydroxybenzaldehyde.
- an alkali metal salt of m-hydroxybenzaldehyde is used as a starting material.
- a synthesis example of a sodium salt of m-hydroxybenzaldehyde, which is a representative example of an alkali metal salt of m-hydroxybenzaldehyde, from the raw material compound m-hydroxybenzaldehyde is as follows.
- the amount of the Wittig reactant introduced may be equal to or greater than the reaction equivalent of the alkali metal salt of m-hydroxybenzaldehyde (sodium salt in the above example). This is because in the starting material of this embodiment, the protons of the hydroxyl groups have already been replaced with sodium groups, and therefore the influence of the hydroxyl groups during the Wittig reaction is substantially eliminated.
- the subsequent second step in this embodiment (corresponding to the acid hydrolysis reaction step of Y2 in chemical reaction formula (III) as already explained) is the same as the second step in the first embodiment (corresponding to the acid hydrolysis reaction step of Y1 in chemical reaction formula (II) as already explained).
- the subsequent third step in this embodiment (corresponding to the oxidation reaction step of Z2 in chemical reaction formula (III) as already explained) is the same as the third step in the first embodiment (corresponding to the acid hydrolysis reaction step of Z1 in chemical reaction formula (II) as already explained).
- 2-m-hydroxyphenylacetic acid can be produced with high accuracy by carrying out the second and third steps in situ, as in the first embodiment. It is also noteworthy that by adopting the method for producing 2-m-hydroxyphenylacetic acid of this embodiment, 2-m-hydroxyphenylacetic acid can be produced safely and efficiently without using any toxic CN source reactants or sulfur-based reactants that require odor control, as in the first embodiment, without going through multiple steps.
- Example 1 Method for producing 2-m-hydroxyphenylacetic acid when m-hydroxybenzaldehyde is used as the starting material
- the first reaction mixture (including unreacted compounds) in the first step was cooled to about 10°C, and the organic layer was separated using 100 g of ice and 200 g of 5% aqueous hydrochloric acid. After that, the organic layer was separated using aqueous hydrochloric acid and water, and the organic solvent toluene was distilled off under reduced pressure, and 100 mL of ethyl acetate was added and stirred at a temperature of about 0°C. As a result, a viscous mixture (first mixture) containing a crude product of m-hydroxystyryl methyl ether and a by-product of the Wittig reaction was obtained.
- Figure 1 is an HPLC (high performance liquid chromatography) diagram of the first step immediately after the dropwise addition of the starting material m-hydroxybenzaldehyde to the reaction mixture under a specified temperature condition after the preparation of the Wittig reactant in this embodiment is completed.
- Figure 2 is an HPLC diagram after the first step (Wittig reaction) in this embodiment.
- P1 in the diagram is the Wittig reactant
- Q1 is the starting material (m-hydroxybenzaldehyde).
- R1 is a by-product of the Wittig reaction
- S1 is m-hydroxystyryl methyl ether, which is the product of the Wittig reaction.
- T1 is toluene, which is the solvent.
- HPLC diagrams other than Figure 1 common symbols are used for common compounds unless otherwise specified.
- the Q1 peak which indicates the starting material
- the Q1 peak becomes smaller, and after the first step in this embodiment, the Q1 peak becomes so small that it is barely visible. Therefore, as described above, by reacting 2 or more molar equivalents of the Wittig reactant (e.g., about 2.4 molar equivalents) with the starting material m-hydroxybenzaldehyde in the Wittig reaction, it is possible to reduce the remaining amount of starting material with a high degree of certainty.
- the Wittig reactant e.g., about 2.4 molar equivalents
- FIG. 3 shows the HPLC results after the second step (acid hydrolysis reaction) in this example. Note that U1 in the figure is 2-(m-hydroxyphenyl)acetaldehyde, and V1 is the remaining ethyl acetate.
- reaction mixture (second mixture) containing 2-(m-hydroxyphenyl)acetaldehyde was not taken out, i.e., the subsequent third step (corresponding to the reaction step of Z1 in chemical reaction formula (II) described above) was carried out in situ.
- toluene in an amount about 10 times the volume of the crude product and a small amount of activated carbon were added to the container containing the crude product, and the crude product was heated to reflux and then filtered.
- the filtrate obtained by the filtration was cooled to obtain 19.65 g of 2-m-hydroxyphenylacetic acid, a light brown solid substance.
- the yield in this example was about 51.6%.
- FIG 4 shows the HPLC trace of the above-mentioned purified light brown solid material after the third step (oxidation reaction) in this example.
- W1 in the figure is 2-m-hydroxyphenylacetic acid.
- Fig. 5 is a proton nuclear magnetic resonance ( 1 H NMR) spectrum of the purified product obtained after the third step (oxidation reaction) in this example.
- Fig. 6 is a C-13 nuclear magnetic resonance ( 13 C NMR) spectrum of the purified product obtained after the third step (oxidation reaction) in this example.
- Fig. 7 is an IR spectrum (infrared absorption spectrum) of the purified product obtained after the third step (oxidation reaction) in this example.
- the purified 2-m-hydroxyphenylacetic acid contains almost no m-hydroxybenzaldehyde, which is the starting material. More specifically, it was revealed that the 2-m-hydroxyphenylacetic acid composition contains less than 0.1 wt% of m-hydroxybenzaldehyde, which is the starting material.
- Example 2 [Method for producing 2-m-hydroxyphenylacetic acid when the starting material is an alkali metal salt of m-hydroxybenzaldehyde]
- the amount of starting material in this example is about 40% by molar ratio compared to Example 1. Therefore, as described above, in this example, the amount of Wittig reactant introduced is a relatively small amount, more than the reaction equivalent amount (more specifically, a small excess amount of the reaction equivalent amount; a typical example is about 1.2 equivalents) relative to the alkali metal salt of m-hydroxybenzaldehyde.
- the amount of the compound used in each step was adjusted to about 40% of the amount of the compound in Example 1, and then each treatment (including purification treatment) described in the second embodiment was performed.
- each treatment (including purification treatment) described in the second embodiment was performed.
- a reaction similar to that described in Example 1 was performed, and 7.76 g of 2-m-hydroxyphenylacetic acid, a light brown solid substance, was obtained.
- the yield in this example was about 51.0%.
- various HPLC analyses, proton nuclear magnetic resonance ( 1 H NMR) spectrum analyses, C-13 nuclear magnetic resonance ( 13 C NMR) spectrum analyses, and IR spectrum analyses were performed in the same manner as in Example 1, and the results were similar to those in Example 1.
- the method for producing 2-m-hydroxyphenylacetic acid of the present invention and the 2-m-hydroxyphenylacetic acid of the present invention can be widely used as a useful chemical substance or its production method for materials with a variety of applications (e.g., functional materials or intermediates thereof used in various pharmaceuticals or cosmetics, etc.).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description

(a)ウィッティヒ反応剤の一部が、出発材であるm‐ヒドロキシベンズアルデヒドのヒドロキシ基のプロトンをホスフィニウム基に置換し得る。
(b)ウィッティヒ反応剤の他の一部が、出発材のアルデヒド基と反応させることにより炭素数を1つ増やす、増炭反応(One‐carbon Homologation Reaction)を実現したエノール誘導体を生成することができる。
一般式(I)
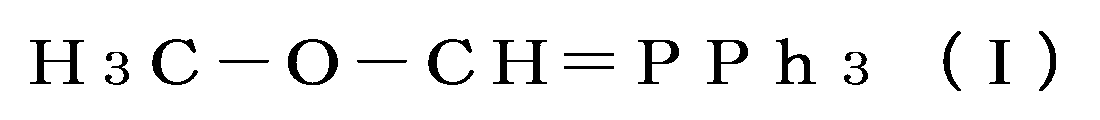

一般式(I)

なお、本願において、「2‐m‐ヒドロキシフェニル酢酸組成物」とは、2‐m‐ヒドロキシフェニル酢酸に加えて、その他の物質(例えば、出発材としてのm‐ヒドロキシベンズアルデヒド又は該m‐ヒドロキシベンズアルデヒドのアルカリ金属塩、あるいは該2‐m‐ヒドロキシフェニル酢酸の製造過程において生成される副生成物)を含有し得る組成物という意味であり、該「2‐m‐ヒドロキシフェニル酢酸組成物」の組成は特に限定されない。
以下に、本実施形態における2‐m‐ヒドロキシフェニル酢酸の製造方法、及び該2‐m‐ヒドロキシフェニル酢酸について説明する。
本実施形態は、出発材をm‐ヒドロキシベンズアルデヒドとしたときの2‐m‐ヒドロキシフェニル酢酸を製造する方法を説明する。
(A)市販されている、原料化合物の(メトキシメチル)トリフェニルホスホニウムクロリド(H3C‐O‐CH2‐P+(Cl-)Ph3)と、強塩基(例えば、カリウムtert‐ブトキシド)とを反応させて該ウィッティヒ反応剤を生成する工程
(B)(A)反応に続く、m‐ヒドロキシベンズアルデヒドと該ウィッティヒ反応剤とのウィッティヒ反応を生じさせることによってm‐ヒドロキシスチリルメチルエーテルを生成させる工程
(x)2‐(m‐ヒドロキシフェニル)アセトアルデヒドにおけるアルデヒド基をカルボキシル基へ導く反応を実現し得る酸化剤
(y)該アルデヒド基とは異なる官能基に対する副次的反応を生じさせ難い酸化剤
以下に、本実施形態における第1の実施形態とは異なる2‐m‐ヒドロキシフェニル酢酸の製造方法について説明する。
本実施形態は、出発材をm‐ヒドロキシベンズアルデヒドのアルカリ金属塩としたときの2‐m‐ヒドロキシフェニル酢酸を製造する方法を説明する。
上述のとおり、本実施形態においては、m‐ヒドロキシベンズアルデヒドのアルカリ金属塩を出発材として採用する。原料化合物であるm‐ヒドロキシベンズアルデヒドから、代表的なm‐ヒドロキシベンズアルデヒドのアルカリ金属塩の例である、m‐ヒドロキシベンズアルデヒドのナトリウム塩の合成例は、次のとおりである。
上述のとおり合成されたm‐ヒドロキシベンズアルデヒドナトリウム(Na)塩は、第1の実施形態と同様に、ウィッティヒ反応剤であるH3C‐O‐CH=PPh3との反応(ウィッティヒ反応)によって、m‐ヒドロキシスチリルメチルエーテルを生成させる(本実施形態における第1工程)。
以下の各実施例を通じて上述の実施形態を具体的に説明するが、該実施例の記載によって本発明及び該実施形態の範囲は限定されない。
[出発材をm‐ヒドロキシベンズアルデヒドとしたときの2‐m‐ヒドロキシフェニル酢酸を製造方法について]
本実施例においては、有機溶媒(トルエン)300mLが収容されたフラスコに、原料化合物の(メトキシメチル)トリフェニルホスホニウムクロリド206g(0.6mol)とカリウムtert‐ブトキシド74.1g(0.66mol)とを、窒素ガスを流しながら反応系内を5℃以下になるように温度制御された状態で反応させる。その結果、ウィッティヒ反応剤であるH3C‐O‐CH=PPh3が生成される。
その後、上述のウィッティヒ反応剤の生成が行われた反応系内に、該反応系内が20℃以下になるように温度制御された状態でm‐ヒドロキシベンズアルデヒド30.6g(0.251mol)を添加し、室温下において撹拌した。なお、本実施例においては、上述のウィッティヒ反応剤の生成工程において生成されたウィッティヒ反応剤は、出発材であるm‐ヒドロキシベンズアルデヒドの約2.4モル等量である。
その後、上述の第1混合物を、アセトニトリル300mlを用いて希釈した後、該第1混合物を収容する容器に、濃塩酸5gに対して水5gを添加した塩酸を加えて、室温下において撹拌することにより、酸加水分解反応(既に説明した、化学反応式(II)のY1の反応工程に相当)を生じさせた。
上述のとおり、in‐situで、0℃以上5℃以下の温度条件下において、該第2混合物を収容する容器内に、酸化剤であるオキソン(登録商標)(2KHSO5.KHSO4.K2SO4)153.7gを加えて撹拌することにより、2‐m‐ヒドロキシフェニル酢酸を含む反応混合物(第3混合物)を得た。
[出発材をm‐ヒドロキシベンズアルデヒドのアルカリ金属塩としたときの2‐m‐ヒドロキシフェニル酢酸の製造方法について]
その後、本実施例においては、上述のウィッティヒ反応剤の生成が行われた反応系内に、該反応系内が20℃以下になるように温度制御された状態で、第2の実施形態において説明した出発材であるm‐ヒドロキシベンズアルデヒドナトリウム(Na)塩14.4g(0.10mol)を添加し、室温下において撹拌した。
Claims (6)
- m‐ヒドロキシベンズアルデヒドと、前記m‐ヒドロキシベンズアルデヒドに対して2モル等量以上の一般式(I)で表されるウィッティヒ反応剤とを反応させることによってm‐ヒドロキシスチリルメチルエーテルを生成する第1工程と、
前記m‐ヒドロキシスチリルメチルエーテルの酸加水分解反応によって2‐(m‐ヒドロキシフェニル)アセトアルデヒドを生成する第2工程と、
前記2‐(m‐ヒドロキシフェニル)アセトアルデヒドを、酸化剤を用いて処理する第3工程と、を含み、
前記第2工程と前記第3工程を、in‐situで行う、
2‐m‐ヒドロキシフェニル酢酸の製造方法。
一般式(I)

- m‐ヒドロキシベンズアルデヒドのアルカリ金属塩と、前記アルカリ金属塩に対して反応等量以上の一般式(I)で表されるウィッティヒ反応剤とを反応させることによってm‐ヒドロキシスチリルメチルエ‐テルを生成する第1工程と、
前記m‐ヒドロキシスチリルメチルエーテルの酸加水分解反応によって2‐(m‐ヒドロキシフェニル)アセトアルデヒドを生成する第2工程と、
前記2‐(m‐ヒドロキシフェニル)アセトアルデヒドを、酸化剤を用いて処理する第3工程と、を含み、
前記第2工程と前記第3工程を、in‐situで行う、
2‐m‐ヒドロキシフェニル酢酸の製造方法。
一般式(I)

- 前記m‐ヒドロキシベンズアルデヒドと、前記m‐ヒドロキシベンズアルデヒドに対して2モル等量超の前記ウィッティヒ反応剤とを反応させる、
請求項1に記載の2‐m‐ヒドロキシフェニル酢酸の製造方法。 - 前記第3工程を、-10℃以上10℃以下において行う、
請求項1又は請求項2に記載の2‐m‐ヒドロキシフェニル酢酸の製造方法。 - 前記酸化剤が、ヒドロキシペルオキシドを含む過酸化物及びオキソン(登録商標)(2KHSO5.KHSO4.K2SO4)の群から選択される少なくとも1種である、
請求項1又は請求項2に記載の2‐m‐ヒドロキシフェニル酢酸の製造方法。 - 2‐m‐ヒドロキシフェニル酢酸組成物が、m‐ヒドロキシベンズアルデヒドを、0.1wt%未満含む、
2‐m‐ヒドロキシフェニル酢酸。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202480006888.1A CN120476101A (zh) | 2023-06-14 | 2024-06-10 | 2-间羟基苯乙酸的制造方法及2-间羟基苯乙酸 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023-097577 | 2023-06-14 | ||
| JP2023097577A JP7625643B2 (ja) | 2023-06-14 | 2023-06-14 | 2‐m‐ヒドロキシフェニル酢酸の製造方法及び2‐m‐ヒドロキシフェニル酢酸 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024257715A1 true WO2024257715A1 (ja) | 2024-12-19 |
Family
ID=93852123
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/020996 Ceased WO2024257715A1 (ja) | 2023-06-14 | 2024-06-10 | 2‐m‐ヒドロキシフェニル酢酸の製造方法及び2‐m‐ヒドロキシフェニル酢酸 |
Country Status (3)
| Country | Link |
|---|---|
| JP (2) | JP7625643B2 (ja) |
| CN (1) | CN120476101A (ja) |
| WO (1) | WO2024257715A1 (ja) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60116646A (ja) * | 1983-11-16 | 1985-06-24 | ビーチヤム・グループ・ピーエルシー | アラキドン酸類似体、その製法及びその用途 |
| WO2022127321A1 (zh) * | 2020-12-18 | 2022-06-23 | 四川大学 | 一种新型中间体及其制备方法和应用 |
-
2023
- 2023-06-14 JP JP2023097577A patent/JP7625643B2/ja active Active
-
2024
- 2024-06-10 WO PCT/JP2024/020996 patent/WO2024257715A1/ja not_active Ceased
- 2024-06-10 CN CN202480006888.1A patent/CN120476101A/zh active Pending
-
2025
- 2025-01-22 JP JP2025008994A patent/JP2025066782A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60116646A (ja) * | 1983-11-16 | 1985-06-24 | ビーチヤム・グループ・ピーエルシー | アラキドン酸類似体、その製法及びその用途 |
| WO2022127321A1 (zh) * | 2020-12-18 | 2022-06-23 | 四川大学 | 一种新型中间体及其制备方法和应用 |
Non-Patent Citations (3)
| Title |
|---|
| OTT ARNOLD C., LEONARD A. MATTANO, GERALD H. COLEMAN: "Preparation of Hydroxyphenylalkanoic Acids by the Willgerodt Reaction", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, vol. 68, no. 12, 1 December 1946 (1946-12-01), pages 2633 - 2634, XP093248124, ISSN: 0002-7863, DOI: 10.1021/ja01216a061 * |
| SUSANNE H. KLEINFELD; ELINA WEGELIUS; DIETER HOPPE: "(−)‐Sparteine‐Mediated Asymmetric Cyclocarbolithiation of Alkenes Combined with a Stereospecific retro‐[1,4]‐Brook Rearrangement", HELVETICA CHIMICA ACTA, VERLAG HELVETICA CHIMICA ACTA., HOBOKEN, USA, vol. 82, no. 12, 21 December 1999 (1999-12-21), Hoboken, USA, pages 2413 - 2424, XP071267281, ISSN: 0018-019X, DOI: 10.1002/(SICI)1522-2675(19991215)82:12<2413::AID-HLCA2413>3.0.CO;2-F * |
| WEBB, K.S. RUSZKAY, S.J.: "Oxidation of Aldehydes with Oxone@? in Aqueous Acetone", TETRAHEDRON, ELSEVIER SIENCE PUBLISHERS, AMSTERDAM, NL, vol. 54, no. 3-4, 15 January 1998 (1998-01-15), AMSTERDAM, NL , pages 401 - 410, XP004106631, ISSN: 0040-4020, DOI: 10.1016/S0040-4020(97)10299-X * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN120476101A (zh) | 2025-08-12 |
| JP2025066782A (ja) | 2025-04-23 |
| JP2024179066A (ja) | 2024-12-26 |
| JP7625643B2 (ja) | 2025-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Marsden et al. | Aerobic oxidation of aldehydes under ambient conditions using supported gold nanoparticle catalysts | |
| HU198437B (en) | Process for producing mono- or bis-carbonyl-compounds | |
| Fringuelli et al. | Easy and environmentally friendly uncatalyzed synthesis of β-hydroxy arylsulfides by thiolysis of 1, 2-epoxides in water | |
| JP5070936B2 (ja) | 2−ヒドロキシ−4−(メチルチオ)酪酸またはそのエステルの製造方法およびその中間体 | |
| EP2543658B1 (en) | Method for producing a-acyloxycarbonyl compound and novel a-acyloxycarbonyl compound | |
| JP7625643B2 (ja) | 2‐m‐ヒドロキシフェニル酢酸の製造方法及び2‐m‐ヒドロキシフェニル酢酸 | |
| JP7718088B2 (ja) | 3-(メタ)アクリロイルスルホランの製造方法 | |
| JP2830210B2 (ja) | α,β―不飽和ケトン類の合成法 | |
| JP5664779B2 (ja) | ヘキサフルオロアセトンまたはその水和物の製造法 | |
| JP2678784B2 (ja) | アダマンタントリオール類の製造方法 | |
| JP3855570B2 (ja) | 4−アセチルテトラヒドロピランの製法 | |
| JP2007238517A (ja) | ジスルフィド化合物の製造方法 | |
| JPH02200653A (ja) | 1級アルコールからアルデヒドの製造法 | |
| EP1583730A1 (en) | Novel process for preparing a 5-hydroxy-3-oxo-hexanoic acid derivative | |
| JP2002212149A (ja) | フッ化テトラアルキルアンモニウムの製造方法、およびそれを用いたβ−ヒドロキシケトンの製造方法 | |
| EP0278845A2 (fr) | Procédé de préparation d'hydroxybiphenyles | |
| KR100792004B1 (ko) | 4-브로모-3,5-디플루오로 페닐아세테이트의 합성방법 | |
| CN114907196A (zh) | 通过芳基取代邻二醇氧化裂解制备羰基化合物的方法 | |
| JPH03287576A (ja) | キノリン酸の製造法 | |
| Badri et al. | Facile, mild and selective silica sulfuric acid catalyzed oxidation of benzylalcohols to benzaldehyde derivatives by potassium peroxodisulfate | |
| SU1625866A1 (ru) | Способ получени 5-хлорпентановой кислоты | |
| FR2643633A1 (fr) | Procede d'hydroxylation de phenols et d'ethers de phenols | |
| CN120794829A (zh) | 一种1-烷基-4-甲基-2,3-二醇-1,4-丁二酮的制备工艺 | |
| JP4294130B2 (ja) | α,β−不飽和ケトン化合物の製造方法 | |
| KR100570279B1 (ko) | 코엔자임 Qn의 중간체 및 그 중간체의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24823331 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202480006888.1 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202517072811 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202480006888.1 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202517072811 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |