WO2025022019A1 - Procédé de diagnostic prédictif d'une pathologie ou d'un état pathologique - Google Patents
Procédé de diagnostic prédictif d'une pathologie ou d'un état pathologique Download PDFInfo
- Publication number
- WO2025022019A1 WO2025022019A1 PCT/EP2024/071489 EP2024071489W WO2025022019A1 WO 2025022019 A1 WO2025022019 A1 WO 2025022019A1 EP 2024071489 W EP2024071489 W EP 2024071489W WO 2025022019 A1 WO2025022019 A1 WO 2025022019A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- training
- microorganisms
- subject
- data
- diagnosis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/689—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for bacteria
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Definitions
- the present application relates to a method, in particular in vitro, for diagnosing or predictively diagnosing a pathology or a pathological condition from a biological sample taken from a subject.
- the invention relates to a method, in particular in vitro, for predictively diagnosing a pathology of the digestive system or an extra-digestive pathology of a subject from the analysis of the microbiota present in a biological sample taken from the digestive system, and/or outside the digestive system such as the vagina and/or in the stools of a subject.
- the invention relates to a method for diagnosing necrotizing ulcerative enterocolitis (NUE) in premature newborns from a biological sample taken from their stools.
- NUE necrotizing ulcerative enterocolitis
- the subject of the present invention is the predictive diagnosis of premature delivery from a biological sample taken from the vagina of a pregnant woman. The present method is therefore in the field of diagnosis, predictive diagnosis, particularly in vitro, and personalized medicine.
- preterm birth is a major cause of morbidity and mortality in newborns.
- a proportion of spontaneous preterm births appear to result from an inflammatory reaction following an infection of the genital tract.
- a large proportion of preterm births remain without an identified cause, without clinical signs.
- clinicians currently have no reliable tool to predict the risk of preterm birth.
- Patent EP 3161167 describes a method for assessing a risk of premature delivery based on the detection, in a vaginal or cervical sample obtained by swabbing from a pregnant woman, of the quantity of the following bacteria: Vimonas micra, Ureaplasma urealyticum or Ureaplasma parvum, Atopobium vaginae, Peptoniphilus lacrimalis, Megasphaera cerevisiae and Parvibacter caecicola, compared to a reference level.
- the quantification of the bacteria is carried out by amplification of a small region of ribosomal DNA (16S rDNA) by quantitative polymerase chain reaction (qPCR).
- Patent EP 2 972 308 B9 describes a serum or plasma peptide biomarker, produced by human cells, and not by the microbiota, the detection of which is used in a method for assessing a risk of premature birth.
- International application WO 2020/227053 describes a method for determining the risk of premature birth comprising determining the abundance of Saccharibacteria TM7-H1 and optionally BVAB1, Sneathia amnii and Prevotella in a vaginal sample from a pregnant woman, from the nucleotide sequence of a small portion of the 16S rDNA of the microorganisms.
- rRNA ribosomal RNA
- 16S rDNA for prokaryotic microorganisms, such as bacteria and archaea
- 18S rDNA for eukaryotes, including yeasts
- the method involves the simultaneous quantification by qPCR amplification of specific small fragments of DNA from each of the following 10 microorganisms: Lactobacillus crispatus, Lactobacillus iners, Weissella koreensis, Bacteroides fragilis, Prevotella bivia, Prevotella amnii, Prevotella salivae, Ureaplasma urealyticum, Ureaplasma parvum, Gardnerella vaginalis.
- necrotizing enterocolitis is the most common life-threatening gastrointestinal emergency encountered by preterm infants in neonatal intensive care units. It is defined as ulcerative inflammation of the intestinal wall.
- Current clinical practice for diagnosing NEC is based on clinical, radiological and haematological findings constituting the Bell criteria, according to a recent review (D'Angelo et al., 2018).
- the clinical signs of early NEC are often very subtle and may initially manifest as feeding intolerance and nonspecific symptoms (malaise, bradycardia) before gastrointestinal symptoms become evident.
- T1D type I diabetes
- T1D affects children and young adults. In the short term, it is responsible for a significant deterioration in quality of life since affected subjects must constantly adapt their insulin intake (subcutaneously) to blood sugar, food intake and energy expenditure. In the medium and long term, chronic hyperglycemia leads to multi-organ alterations, particularly nervous and vascular.
- T1D has been increasing continuously since at least 1988. In France, it is 18 per 100,000 in those under 15 years of age, over the period 2013-2015, i.e. a prevalence of around 1.3 per 1,000.
- the incidence of diabetes in young people is increasing by 3 to 4% per year, at the same time as the age of onset is decreasing (Gale E 2002).
- Immune activation is multifactorial and depends partly on the HLA system and postnatal infectious events. There is thus a familial aggregation of cases, an association with other autoimmune diseases, and a possible link with certain viral agents, notably group B coxsackieviruses.
- a first method called "metabarcoding” makes it possible to determine the taxa present in a sample thanks to their genetic signature, unique for each taxa.
- the idea is to have a DNA fragment present in all the taxa to be analyzed and which constitutes a genetic marker.
- This marker is a DNA fragment framed by highly conserved regions and therefore the most “universal” possible, and which, once sequenced, shows variations in genetic sequences between different taxa.
- this method often includes the amplification of fragments of a size between 300 and 470 base pairs of the V3 and/or V4 regions of the gene expressing the 16S rRNA.
- biases are likely to be generated during the amplification step carried out by PCR and can alter the vision of the real diversity of the microbiota. Indeed, it is known that the primers used that cannot be "universal" to amplify nucleotide sequences will favor the amplification of the sequences of certain microorganisms to the detriment of others, resulting in a possibly erroneous abundance of microorganisms or even the non-detection of certain microorganisms. In addition, the short length of the sequenced DNA fragments provides only a low taxonomic resolution, not allowing the description of microbial communities at the species level.
- This method advantageously comprises the use of all the microorganisms identified in the microbiota of a subject by an artificial intelligence model to establish a diagnosis or a predictive diagnosis of a pathology or pathological condition.
- the present invention thus has as its first subject a method, in particular in vitro, for diagnosis or predictive diagnosis of a pathology or pathological condition in a subject, from at least one biological sample taken from the subject and containing microorganisms, said method comprising the following steps: a) sequencing, from the nucleic acid isolated from the subject's sample, the nucleotide sequences corresponding to at least one sequence of interest selected from the group consisting of: a fragment of a gene expressing 16S ribosomal RNA (rRNA), a fragment of a gene expressing 18S rRNA, a fragment of 16S rRNA, a fragment of 18S rRNA, b) from the sequencing of step a), determination of the identity and relative abundance of the microorganisms present in said sample without any preselection, c) determination of the predictive diagnosis of said pathology or pathological condition by a model artificial intelligence model from at least the abundances of the identities obtained in step b), said artificial intelligence model having previously been trained on the basis of a
- the phenotype label assigned to each training subject depends on the purpose of the method according to the invention and the type of data used for training.
- the labeled data set includes at least two different states for the phenotypes and in particular antinomic states: a positive phenotype associated with a diagnosis/diagnosis positive predictive and a negative phenotype associated with a diagnosis/negative predictive diagnosis.
- the training subject phenotype can be classified as "not affected” or "affected” by the pathology or pathological condition or "healthy” and “sick", these types of classification being synonymous.
- the training subject phenotype can be classified as "having developed” or “not having developed” the pathology or pathological condition or "with appearance” or “without appearance” of the pathology or pathological condition, these types of classification being synonymous.
- the invention has the advantage of training the artificial intelligence model more efficiently by using the identity of all the microorganisms identified in the labeled data set.
- the absence of a step of pre-selection of the identity of microorganisms in the labeled data set for training the artificial intelligence model makes it possible to preserve all the diversity and individual variability of the microbiotas and all the associated microbial interactions in the context of a specific pathology or pathological state.
- the method according to the invention has the advantage of restricting to a minimum (or even of not applying any restriction) the exclusion of the identities of the microorganisms from the data from step b) transmitted to the artificial intelligence model during step c), making it possible to preserve as much as possible the microbial diversity present in the subject's sample.
- the selection of the identities sent to the artificial intelligence model is in no way done on the basis of a relative abundance that is too low in the subject's sample or their absence of known involvement in the pathology or pathological condition, but only on the basis of their presence in the training data set.
- the data set is sufficiently large and exhaustive, no identity of microorganisms is excluded from the data transmitted to the artificial intelligence model to carry out step c).
- microbiota analysis techniques exist, they do not allow for a precise characterization between microbiotas and pathologies, the risk of developing said pathologies, or the evolution of the latter. Thus, the most likely result would have been to obtain a large number of false positive or false negative diagnoses.
- the method of the invention takes into account as the identity of each microorganism the classification by taxonomic rank, this rank preferably being the species of the microorganism. No preselection is carried out during the identification, in particular on the basis of their relative abundance and/or their known involvement in the diagnosis or predictive diagnosis.
- the microorganisms of the labeled data set as well as those of step b) are identified at the level of the same taxonomic rank.
- This rank is notably chosen from the phylum to the species, and is preferably the species.
- the identity of each microorganism corresponds to the most confident taxonomic rank, which can be a species, a genus, a family, an order, a class or a phylum.
- the identities of the microorganisms will not all have the same rank. This aspect advantageously allows to preserve the maximum exhaustiveness of the labeled dataset when training the intelligence model.
- “Most confident taxonomic rank” means the most precise taxonomic rank obtainable from the nucleotide sequence or set of nucleotide sequences used to identify a microorganism. Obtaining the most confident rank depends on several factors, described in detail below.
- the diversity of microbiotas given to the artificial intelligence model during its training can be ensured by using data from training subjects of multinational origins, in particular multi-continental, in particular from all continents.
- the training subjects are divided into different groups of geographical origin.
- the distribution of subjects in the different groups is as representative as possible of geographical diversity.
- the labeled data set comprises at least one determined clinical data item, where each training subject profile comprises a value for the or each determined clinical data item, and where step c) comprises providing the artificial intelligence model with the corresponding value of the subject for the or each determined clinical data item.
- the method according to the invention thus has the advantage, from a simple sample of vaginal microbiota during pregnancy, in the 1st trimester and/or in the 2nd trimester and/or 3rd trimester, and its sequencing, of predicting with high certainty the occurrence of a premature birth or a full-term birth.
- the method of the invention allows the predictive diagnosis of the occurrence of premature birth, the accuracy of which can reach 88%. Such a degree of reliability is unmatched among the methods for diagnosing premature birth to date.
- the method according to the invention also has the advantage, from a simple sample of microbiota in the stools of a subject, and its sequencing, of determining with high certainty the development of a disease of the digestive system or an extra-digestive disease.
- This approach can advantageously be used in the context of personalized medicine to evaluate the relevance of more precise clinical monitoring and/or the use of therapeutic treatment.
- the method of the invention allows a reliable prediction of ulcerative necrotizing enterocolitis with an accuracy of up to 94.9%. Such a degree of reliability is very useful for identifying premature newborns at risk, strengthening monitoring and allowing rapid therapeutic responses avoiding possible serious health problems. To this end, the method of the invention allows early and very effective diagnosis of ECUN and equally effective distinction of unaffected infants.
- the method is intended for the predictive diagnosis of type I diabetes in a child.
- the method according to the invention in a similar manner, also makes it possible to reliably predict the occurrence of type I diabetes (T1D), with an accuracy of up to 73.6% in particular.
- T1D type I diabetes
- the method of the invention thus makes it possible to identify early on children at high risk of developing autoimmunity and then diabetes, which would allow a therapeutic revolution towards personalized preventive medicine to avoid the disabling consequences of the pathology.
- the method aims at a predictive diagnosis of neonatal sepsis in an infant.
- the method according to the invention also makes it possible to reliably predict the occurrence of sepsis, with an accuracy of up to 92.3%.
- the method of the invention thus makes it possible to identify premature newborns at risk, to strengthen monitoring and to adapt treatment to the profile of these bacteria involved in the pathology.
- the invention also relates to a method for training an artificial intelligence model intended to obtain a diagnosis or a predictive diagnosis, said method using a labeled data set comprising profiles of training subjects, where each training subject profile comprises the identity and relative abundance of all the microorganisms identified in at least one sample of said training subject without any preselection, and where each profile is labeled with the phenotype of the training subject from which it originates.
- the training method according to the invention makes it possible to obtain a more reliable and more precise artificial intelligence model in these predictions, for the aforementioned reasons.
- the data obtained using the training process therefore also make it possible to have a precise mapping of the microorganisms associated with the presence of a state which could lead to a pathology or a pathological state, and of the microorganisms associated with the absence of a state leading to a pathology or a pathological state, on the other hand.
- the method according to the invention also has the advantage of not increasing the number of obstetric examinations on pregnant women carried out during pregnancy, since the vaginal sample can be recovered during an examination already scheduled.
- the method according to the invention advantageously makes it possible to carry out early therapeutic interventions in order to prevent the development or the worst complications of an extra-digestive pathology based on the analysis of the intestinal or fecal microbiome of a subject.
- the present invention also relates to a computer program product comprising executable instructions, which when executed on a computer allow the implementation of step c) of determining the diagnosis/predictive diagnosis of the method according to the invention.
- the characteristics previously and subsequently described in relation to the artificial intelligence model apply mutatis mutandis to the present subject.
- the computer program product comprises instructions enabling the predictive diagnosis of premature delivery in a subject.
- the computer program product comprises instructions enabling the predictive diagnosis of ECUN in a subject.
- the computer program product comprises instructions enabling the predictive diagnosis of type I diabetes in a subject.
- the computer program product comprises instructions enabling the predictive diagnosis of sepsis in a subject.
- the invention also relates to the use of a computer program product according to the invention for the diagnosis/predictive diagnosis of a pathology or a pathological condition.
- the characteristics previously and subsequently described in relation to the diagnostic/predictive diagnosis method according to the invention apply mutatis mutandis to the present subject.
- the invention finally relates to the management or treatment of a subject whose diagnosis or positive diagnosis of a pathology or pathological condition has been determined as positive using the diagnostic/predictive diagnostic method of the invention.
- Said treatment may be a curative treatment or a prophylactic treatment depending on the situation.
- the management may be enhanced clinical monitoring, particularly in the context of the predictive diagnosis of premature birth.
- the present invention thus has as its first object a method, in particular in vitro, for diagnosis or predictive diagnosis of a pathology or a pathological state in a subject, from at least one biological sample taken from the subject and containing microorganisms.
- diagnosis is meant in the invention the determination of the presence or absence of a pathology or pathological condition in a subject.
- a positive diagnosis is understood in the invention as corresponding to the determination of the presence of the pathology or pathological condition in the subject.
- a negative diagnosis is understood as corresponding to the determination of the absence of the pathology or pathological condition in the subject.
- predictive diagnosis is meant in the invention the determination of the risk of developing/occurring/appearing a pathology or the occurrence of a pathological condition in a subject not presenting any symptoms.
- the positive predictive diagnosis is understood in the present invention as a high risk of appearing the pathology or the pathological condition.
- a negative predictive diagnosis is understood in the present invention as a low risk of appearing the pathology or the pathological condition.
- a positive diagnosis/predictive diagnosis may be considered determined when the associated certainty is greater than 50%, preferably a certainty greater than or equal to 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or equal to 100%.
- a negative diagnosis/predictive diagnosis may be considered determined when the associated certainty is greater than 50%, preferably a certainty greater than or equal to 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or equal to 100%.
- “Pathology” means a disease, a biological imbalance or discomfort.
- the pathology corresponds in particular to a digestive pathology, an extra-digestive pathology or a pathology of the newborn, in particular enterocolitis of the type, more particularly ulcerative necrotizing enterocolitis (ECIIN).
- Enterocolitis of the type, more particularly ulcerative necrotizing enterocolitis (ECIIN).
- Ulcerative necrotizing enterocolitis means a disease characterized by inflammation and necrosis of the intestinal mucosa. Even more particularly, among said digestive pathologies, we can cite: digestive cancers, that is to say affecting at least one of the organs of the digestive system, chronic inflammatory diseases, such as in particular Crohn’s disease, ulcerative colitis, irritable bowel syndrome and celiac disease.
- the pathology is advantageously either a pathology of the organ from which the biological sample is taken, or a pathology of another organ in the environment from which the sample is taken.
- Extra-digestive pathology means a condition or pathology that does not directly affect an organ of the digestive system but one of the consequences of which is likely to directly or indirectly affect the microbiota of the digestive system and vice versa.
- extra-digestive or non-digestive conditions and pathologies for which a predictive diagnosis can be carried out by a method according to the invention we can cite: diabetes, sepsis, obesity, cardiovascular diseases, metabolic diseases, liver diseases, kidney diseases, urogenital diseases, pulmonary diseases, joint diseases, muscle diseases, inflammatory diseases, asthma, allergies, arthritis, neurodegenerative diseases (Parkinson's, Alzheimer's, etc.), psychiatric diseases, behavioral diseases, all types of cancer for all types of organs.
- pathological condition means a state of alteration of the functions, morphology or health of an organ or organism, the cause of which is known or unknown, and which is characterized by the presence or absence of one or more signs.
- a pathological condition includes, in particular, premature delivery.
- condition or pathology of the digestive system is meant a condition or pathology affecting at least one organ selected from: the mouth, the salivary glands, the pharynx, the esophagus, the stomach, the pancreas, the liver, the gallbladder, the bile duct, the small intestine and the large intestine.
- the large intestine includes the ascending colon, the transverse colon, the sigmoid colon and the rectum.
- said pathology is an intestinal pathology.
- Premature delivery means delivery occurring before the start of the 37th week of amenorrhea.
- said pathology is a digestive pathology of a subject chosen from: children, infants (children beyond their first month of life and up to the age of 24 or 30 months) and newborns (children under 28 days according to the definition of the World Health Organization), said newborns being born at term, i.e. between the 37th week and the end of the 40th week of amenorrhea, or premature, i.e. born before the 37th week of amenorrhea.
- the term “subject” means an animal or a human being, the animal being in particular a mammal.
- the stage of development of the subject is chosen from: adult (from 18 years), adolescent (12 - 17 years), child (2 - 11 years), infant (28 days - 23 months), newborn (0 - 27 days) and premature newborn ( ⁇ 37 weeks of amenorrhea).
- the subject is a pregnant woman, a newborn, an infant or a human child.
- biological sample means any sample from the subject containing microorganisms.
- said biological sample is chosen from: a sample from the digestive system, a sample of excretions, in particular a sample of stool from the subject, a vaginal sample, a cervical sample, a skin sample, and any other biological sample containing microorganisms.
- sample collection is carried out in particular in a conventional and well-known manner by a specialist.
- a given biological sample comprises a community of microorganisms designated by the term “microbiota”.
- the sample may correspond to the grouping of several samples taken from various areas of a sampling region in the subject, in order to attempt to obtain the maximum diversity of microorganisms.
- “Microorganism” means any unicellular or multicellular microorganism such as, but not limited to, bacteria, archaea, viruses, unicellular eukaryotes such as yeasts, etc.
- microbiota hosted by a human subject we can distinguish the skin microbiota, the mucosal microbiota, the pulmonary microbiota, the oral microbiota, the vaginal microbiota, the urinary microbiota, and the microbiotas of the digestive system (oral or salivary microbiota, stomach microbiota, small intestine microbiota, colonic microbiota, anal microbiota).
- the microbiota present in the stools, or fecal microbiota corresponds to all the microorganisms found in the stools following transit through the digestive system of a subject, which may reflect the intestinal microbiota in the broad sense with a closer proximity to the colonic microbiota.
- microbiome refers to all the genomes carrying the genes hosted by the microorganisms constituting the microbiota.
- the microbiome can also be considered as the set of microorganisms including their genomes in a particular biological environment such as the colon.
- “Digestive system” means the set of organs of multicellular animals that receives food, digests it to extract nutrients, and excretes waste in the form of fecal matter.
- the organs of the human digestive system include: the mouth, salivary glands, pharynx, esophagus, stomach, pancreas, liver, gallbladder, bile duct, small intestine, and large intestine.
- the large intestine includes the ascending colon, transverse colon, sigmoid colon, and rectum.
- “Excretion” means unusable or toxic waste that is excreted by the subject such as urine, feces, or stool, or secretion products such as bile or saliva.
- Step a) corresponds to the sequencing of the nucleic acid of the microorganisms present in the biological sample(s), said nucleic acid having been previously isolated from the sample.
- nucleic acid means all nucleic acid molecules present in the biological sample, in particular deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), including respectively the genes expressing 16S ribosomal RNA (rRNA) and/or those expressing 18S rRNA, in particular rRNA and even more particularly 16S rRNA and 18S rRNA.
- rRNA ribosomal RNA
- 18S rRNA in particular rRNA and even more particularly 16S rRNA and 18S rRNA.
- 16S rRNA-expressing gene means the DNA nucleotide sequence comprising the nucleotide sequence encoding the 16S rRNA.
- a gene expressing a 16S rRNA is also referred to as "16S rDNA”.
- a “gene expressing 18S rRNA” means the DNA nucleotide sequence comprising the DNA nucleotide sequence encoding 18S rRNA.
- a gene expressing 18S rRNA is also referred to as “18S rDNA”.
- any commercial nucleic acid extraction kit can be used. It should be noted that the yield (quantity of nucleic acids) of the kits as well as the quality of the nucleic acids can vary depending on the type of sample. It is generally necessary to compare the efficiency of the kits to select the most efficient one.
- the extraction can be carried out manually or using an automaton.
- extraction processes for which the reagents are produced directly in the laboratory.
- extraction protocol standards aimed at homogenizing nucleic acid extraction procedures worldwide. In particular, in the context of ECUN, the H protocol published by the IHMS (International Human Microbiome Standards) can be used for DNA extraction from newborn stools: (see IHMS (human-microbiome.org)).
- the method comprises the isolation of the nucleic acid from a plurality of microorganisms present in said biological sample, in particular from all of the microorganisms.
- the isolated nucleic acid is then sequenced in order to obtain the nucleotide sequences corresponding to at least one sequence of interest chosen from the group consisting of: a fragment of a gene expressing 16S rRNA, a fragment of a gene expressing 18S rRNA, a fragment of 16S rRNA and a fragment of 18S rRNA (hereinafter referred to as "sequences of interest").
- sequences of interest a fragment of a gene expressing 16S rRNA, a fragment of a gene expressing 18S rRNA, a fragment of 16S rRNA and a fragment of 18S rRNA.
- the aim of the sequencing step is to recover all of the sequences corresponding to at least one sequence of interest.
- set of sequences means the set of sequences that the sequencing method can obtain. The key point here is that there is no discrimination of certain sequences of interest among those found in the sample, no preselection is carried out.
- the analysis uses the entire sequencing data.
- nucleotide sequences corresponding to at least one sequence of interest chosen from the group consisting of: a fragment of a gene expressing 16S rRNA and a fragment of a gene expressing 18S rRNA are obtained.
- “Sequencing” means any known method for determining the nucleotide sequence of a nucleic acid. Among these methods, direct metagenomic sequencing known as “shotgun” is preferred, and is notably described in the document Quince C, et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017 Sep 12;35(9):833-844.
- this type of sequencing involves the fragmentation of the isolated nucleic acid into fragments whose size varies depending on the sequencing platform used (typically from 200 to 550 bp on average for the Illumina® platform and from a few dozen bases to > 100,000 bp for the Nanopore® platform), which are subsequently linked to adapters (here also specific to the platform used) for the preparation of the sequencing library.
- the libraries obtained are then sequenced using a high-throughput sequencing platform (typically Illumina® or Nanopore®).
- the sequences obtained are then filtered to remove poor quality sequences and sequences corresponding to the subject's genome, according to well-established principles in the technical field.
- the filtered sequences are then organized for identification, as seen in detail below.
- Illumina® sequencing data from gene capture approaches by hybridization is also preferred and notably described in the document Comtet-Marre, Sophie & Chakoory, Oshma & Peyret, Pierre, (2022), Targeted 16S rRNA Gene Capture by Hybridization and Bioinformatic Analysis. Briefly, the isolated nucleic acid is fragmented and linked to sequencing adapters in a manner similar to the "shotgun" method. In parallel, oligonucleotide probes, in particular biotinylated, complementary to the sequences of interest are synthesized and then hybridized with the sequencing libraries.
- the complexes formed are captured, in particular using magnetic beads coated with streptavidin, and amplified by PCR using primers complementary to the adapters.
- the captured and amplified fragments are sequenced with a high-throughput sequencing platform, then filtered, as previously described.
- the filtered sequences are then organized.
- said method comprises a preliminary step of specific isolation of the nucleic acid from a plurality of microorganisms present in said biological sample.
- Sequencing can also be of the "amplicon sequencing" or "metabarcoding" type, notably described in the document Durazzi, F., Sala, C., Castellani, G. et al.
- a “fragment” of a nucleotide sequence means a fragment of at least 20% of the length of that sequence.
- a “fragment of at least 20%” means a fragment of at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, at least 99% or 100% of the nucleotide sequence in question.
- the sequenced 16S rDNA and/or 16S rRNA fragment of microorganisms belongs in particular to prokaryotes. Additionally, the 18S rDNA and/or 18S rRNA fragment also belongs to eukaryotes and micro-eukaryotes.
- step b) The purpose of step b) is to identify all the microorganisms present in the sample from the sequencing of step a) as well as their relative abundance, and to provide relevant input data to the artificial intelligence model for determining the diagnosis.
- “set of microorganisms” means all the microorganisms identifiable according to the sequencing method used. Identifying all the microorganisms present in the sample and providing this set (without the identities absent from the training set) to the artificial intelligence model makes it possible to preserve the maximum individual variability of the subject as well as the associated microbial interactions in the context of a specific pathology or pathological condition and to ensure a personalized diagnosis/predictive diagnosis.
- the method comprises organizing the sequenced sequences to reconstruct the nucleotide sequence of at least one part of a gene expressing 16S rRNA and/or of a gene expressing 18S rRNA.
- step b) comprises in particular firstly a step of organizing the sequences obtained in step a) by aligning them with known sequences of microorganisms present in a database.
- Said known sequences comprise at least said sequence of interest selected for the greatest number of known microorganisms, in order to determine direct correspondences or to reconstruct sequences of new microorganisms and/or to obtain longer sequences in order to increase the reliability of the identity of the microorganisms present in the biological sample of the subject.
- the organization is done in particular by direct correspondence.
- the organization can be done by direct correspondence and/or reconstruction.
- the determined set of microorganisms is in particular selected from those available in online databases, in particular public ones.
- the SILVA database https://arb-silva.de.
- Another example of databases is the “Greengenes” database (https://greengenes.secondgenome.com/). The person skilled in the art can thus easily determine whether a given nucleotide sequence comes from a known or unknown microorganism, or from a human or animal subject.
- the method according to the invention comprises a step of reconstructing at least part of the sequence of the gene expressing the 16S rRNA and/or the sequence of the gene expressing the 18S rRNA of the microorganisms present in the biological sample.
- the reconstructable length depends on the sequenced length of the fragment of the sequence of interest and the sequencing effort, i.e. the number of readings generated during sequencing (sequencing depth).
- At least 70% of the length of the gene expressing the 16S rRNA and/or at least 70% of the length of the 16S rRNA is reconstructed.
- An increase in the size of the reconstructed part allows for greater precision in determining the identity of the microorganism, making it possible to go as far as the taxonomic rank of the species.
- the length of a 16S rDNA gene being approximately 1500 base pairs on average, a nucleotide sequence of at least 70% of the length of the gene comprises approximately 1050 base pairs, on average.
- the invention uses all of the metagenomic data of the microbiota which then allow the reconstruction of complete sequences of interest and a precise affiliation of the microorganisms of the microbial community at the genus or species level, or even the identification of new microorganisms.
- the organization step is notably followed by a classification step by taxonomic ranks of the correspondences and/or reconstructions making it possible to determine the identity of the microorganisms present in the subject's biological sample.
- identity determination we mean the identification of microorganisms, following a nomenclature, organized into hierarchical categories (classification by taxonomic ranks), in other words in taxonomic ranks, these categories consist of belonging to the domain of life (least precise rank) to the definition of the species (most precise rank).
- the taxonomic ranks of interest extend from the phylum to the species.
- the taxonomic classification is carried out by comparing each reconstructed sequence of interest or whose correspondence is attributed with 16S rDNA sequences and/or 18S rDNA sequences contained in databases.
- SILVA database Among the public databases that can be used, we can notably cite again the SILVA database.
- the most confident taxonomic rank that can be identified depends on several parameters including the type of sequencing, the sequencing parameters, the determined set of microorganisms used for the alignment (see below), etc.
- the invention thus has the advantage of taking into account each determined identification. There is thus no preselection carried out, making it possible to preserve all the diversity of the subject's sample. This exhaustiveness contributes to obtaining a diagnosis/predictive diagnosis of higher quality than with the methods of the prior art.
- the same taxonomic rank among the taxonomic ranks of interest is preserved for all the sequences.
- the most precise taxonomic rank among the taxonomic ranks of interest for each sequence is determined. This second aspect allows better identification of the microbial diversity of the sample, and ensures a more reliable diagnosis.
- [112]By “determination of the relative abundance” is meant the determination for each of the microorganisms considered for the method according to the invention, of the abundance of the microorganism relative to the total abundance of the microorganisms considered for the method according to the invention. The determination of the abundance depends on the sequencing method used, and is well known to those skilled in the art.
- an artificial intelligence model previously trained on the basis of a labeled data set determines the diagnosis/predictive diagnosis on the basis of the data obtained in step b).
- the artificial intelligence model can also take as input at least one clinical data of the subject, as will be seen in detail later.
- the artificial intelligence model thus presents an internal structure reflecting the relationship between on the one hand (1) the relative abundance of microorganisms within the sample, as well as optionally at least one clinical data of the subject, and on the other hand (2) the diagnosis/predictive diagnosis of the pathology or pathological state.
- the artificial intelligence model is a supervised learning model and corresponds in particular to a classification model, a deep learning model, a neural network (NN), a deep neural network, a decision tree, a K-nearest neighbors model (KNN), a random forest (RF), a naive Bayesian classification (NB), an “Extreme gradient boosting” algorithm (XGBoost), a logistic regression or a support vector machine (SVM).
- the artificial intelligence model is a deep neural network with an input layer composed of neurons equivalent to the number of features in the training data, followed by one or more hidden layers and an output layer which gives the result of the diagnosis/predictive diagnosis.
- the invention thus also relates to a method for training an artificial intelligence model intended to obtain a diagnosis or a predictive diagnosis, said method using a labeled data set.
- the labeled data set or training set comprises profiles of training subjects.
- the training subjects belong to the same species as the subject whose sample(s) are analyzed in the method of the invention.
- the training subjects advantageously come from various nations, and in particular from various continents. Parity between the sex types of the subjects in the training set is also advantageous, depending of course on the pathology or of the pathological state considered.
- the principle of the invention is to preserve all the diversity of the microbiota of each of the training subjects, so that the artificial intelligence model can determine all the possible relationships, independently of any bias introduced by the knowledge at a given time.
- the results obtained following the training give an excellent accuracy of prediction of diagnosis/predictive diagnosis of the physiological or pathological state for which the artificial intelligence model was trained.
- the inventors were thus able to show that microorganisms with a very low relative abundance, generally excluded from the training for this reason, proved to be very relevant for determining the predictive diagnosis of pathologies and pathological states. What could previously be considered noise is demonstrated here as a discriminating point.
- the training subjects may in particular be specifically recruited for this purpose, or may come from one or more databases, in particular public ones, and more particularly from the most exhaustive and diversified subject cohort databases available. These databases include in particular raw sequencing data from one or more samples from each subject, and optionally at least one clinical data from each subject.
- the training subjects are notably divided into two groups, namely a training group and a test group.
- the training group is used to train the artificial intelligence model, and the test group is used to qualify its performance.
- the training group represents 80% of all training subjects, and the test group 20%.
- the training subject profiles each comprise the identity and relative abundance of the identified microorganisms present in at least one sample of the training subject, as well as optionally at least one clinical data of the training subject.
- the relative abundances are notably obtained by implementing steps a) and b) described above on samples of subjects, or the single step b) on sequencing data of samples of subjects.
- the identities of the microorganisms can be restricted for training to the same given taxonomic rank so that all of the microorganisms are identified at the level of the same rank, starting from the phylum and up to the species.
- no preselection is carried out on the identified microorganisms, notably on the basis of their relative abundance and/or their known involvement in the diagnosis or the predictive diagnosis. According to one embodiment preferred, no restriction on taxonomic rank is made, and the most confident taxonomic rank is retained for all identities.
- the sample(s) of each training subject are in particular taken during the same trimester, and typically during the 1st , 2nd or 3rd trimester, or even the same month.
- [123]Learning supervision is achieved by labeling the profiles of training subjects with their phenotype.
- the subjects are classified into at least two phenotypes, and preferably into two opposing phenotypes.
- the phenotypes of the subjects are notably affected/not affected by the pathology/pathological state.
- Concerning the predictive diagnosis the phenotypes of the subjects are notably with appearance/without appearance of the pathology or pathological state.
- the training set includes a balanced number of each phenotype, or a greater proportion of positive phenotype.
- the data of the training subjects are notably normalized.
- This normalization is in particular of the min-max type on the entire training set.
- This type of normalization corresponds to a linear transformation of the features in a uniform range, while preserving all the distance ratios of the original data. This is done to prevent the numerical values of the larger features (abundances of microorganisms) from surpassing those of the smaller numerical features, thus minimizing the bias in the discrimination of pathological states.
- the main objective is to ensure the comparability of the data across microbial samples or groups of samples, such as those classified as diseased or healthy. Indeed, the large variability of the sizes of the databases and the sequencing depth induces strong dependencies among the abundances of the different taxa.
- data normalization ensures that all features (taxa) in the data contribute equally to the learning process, although not all features are equally important for the classification decision.
- At least one clinical datum is used in the input data in addition to the data relating to microorganisms, it is of course relevant to the pathology or pathological condition for which the diagnosis/predictive diagnosis is carried out.
- at least one clinical datum is meant one, two, three, four, five, six, seven, eight, nine, ten or more than ten clinical data characteristic of the subject.
- the clinical data may belong to the subject himself or to his mother.
- it may be used in particular: at least one of the following data:
- ethnicity is meant a group of people who are brought together by a certain number of characteristics.
- the characteristic “ethnicity” is notably chosen from the group consisting of: “African-American”, “American-Indian”, “Black”, “White”, “Caucasian”, “Hispanic”, “Asian”, “Multi-ethnicity”.
- categorical data such as gender and mode of birth in the case of newborns
- categorical variable which is converted to 1.
- Continuous data actual age, birth weight and gestational age in the case of newborns
- bins contiguous intervals
- the clinical data "day of life” is discretized into intervals with an increasing step of 9 (from 0 to 99 days) and 99 (100 to 499 days).
- a time step of 1 could also be considered over the first 3 weeks of life when the pathology most frequently appears.
- the clinical data "weight" is discretized into intervals with an increasing step of 99 (from 500 to 2899 grams).
- the weight of the children can also be followed if necessary by interval of 9 throughout the first 3 weeks of life until the possible appearance of the pathology.
- Gestational age at birth can be converted into factors due to the limited number of values.
- the duration of gestation may in particular be expressed in number of weeks of gestation or designated by the period at which the biological sample is taken.
- This period is notably chosen from: the first trimester of pregnancy, the second trimester of pregnancy, the third trimester of pregnancy.
- the age of the pregnant woman in a method according to the invention, can be defined in number of years or by her belonging to an age group. More particularly, the age of the pregnant woman can be attributed to one of the following two groups: “less than 35 years” and “equal to or greater than 35 years”.
- the artificial intelligence model comprises at least 500 microorganism identity abundance entries, in particular at least 600 entries, in particular at least 700 entries, in particular at least 1000 entries, particularly at least 1300 entries.
- the artificial intelligence model comprises at least 10 determined clinical data inputs, in particular at least 20, particularly at least 30, in particular at least 40.
- the artificial intelligence model comprises at least 600 entries of abundance of identities of microorganisms and optionally at least 10, in particular at least 15, entries of determined clinical data.
- the artificial intelligence model comprises at least 1000, in particular at least 1300, entries of abundance of identities of microorganisms and optionally at least 40, in particular at least 45, entries of determined clinical data.
- the artificial intelligence model comprises at least 1000, in particular at least 1300, entries of abundance of identities of microorganisms and optionally at least 40 entries of determined clinical data.
- the artificial intelligence model comprises at least 600, in particular at least 1300, microorganism abundance entries and optionally at least 40 determined clinical data entries.
- first signatures positive diagnosis/predictive diagnosis
- second signatures negative diagnosis/predictive diagnosis
- signature is meant a set of identities of microorganisms. This method also allows the discovery of new microorganisms.
- a first signature of microorganisms associated with a diagnosis of the appearance and/or development of ECUN, in particular obtained by a method according to the invention is characterized in particular by the presence of microorganisms of the species:
- a first signature associated with a high probability of premature delivery is characterized in particular by the presence of microorganisms of the genus:
- Prevotella in particular Prevotella bivia
- Gardnerella in particular Gardnerella vaginalis
- a second signature associated with a plurality of microorganisms statistically associated with a diagnosis of absence of ECUN is characterized in particular by the presence of microorganisms of several species of Lactobacillus associated with non-ECUN cases. Indeed, these microorganisms were discovered as present or present in greater quantity in the biological samples statistically associated with a prediction of absence of ECUN.
- the second signature associated with a diagnosis of absence of ECUN may comprise other microorganisms, such as: the genera Bifidobacterium, Bacteroides, the species Bifidobacterium longum, Bacteroides fragilis, Lactobacillus casei.
- a second signature associated with a high probability of delivery at term (more than 70%), notably obtained by a method according to the invention, is characterized in particular by the presence of microorganisms of the Christensenellaceae family and of the genus:
- Lactobacillus in particular Lactobacillus crispatus.
- the diagnosis/predictive diagnosis is determined from the identities and abundances of microorganisms determined during step b). Of these data obtained in step b), only those of the microorganisms absent from the training set are purified. In this sense, the larger the training set, the more likely it is that it will be exhaustive, and that no purification will be carried out in the data obtained in step b). However, in the event that a sample from a subject was discovered to include a microorganism identity that was not present in the training set, it is possible a posteriori to re-train the artificial intelligence model with this new input. It is thus possible to obtain a continuous enrichment of the artificial intelligence model, and therefore a continually improved accuracy of the predictions.
- step b) The data retained from step b) following the exclusion of microorganisms absent from the training data set are in particular normalized. This normalization is in particular of the min-max type on the basis of the training set.
- the diagnosis/diagnosis obtained in step c) may in particular be associated with a certainty/confidence index, typically ranging from 0 to 1, reflecting the probability of correspondence.
- the artificial intelligence model can determine a positive diagnosis of a pathology with a confidence index of 0.8, indicating that there is an 80% chance that the analyzed microbiota is associated with this pathology.
- the artificial intelligence model can determine a negative diagnosis with a confidence index of 0.8, indicating that there is an 80% chance that the analyzed microbiota is not associated with the pathology and therefore a 20% chance that it is.
- the method according to the invention may comprise a step d) of compiling several diagnoses/predictive diagnoses for a final determination of the diagnosis/predictive diagnosis.
- At least two biological samples from the subject are used, in particular at least three.
- at least two biological samples is meant two, three, four, five, six, seven, eight, nine, ten or more than ten biological samples from the same subject.
- the samples can be taken at the same time, or at different times.
- step d) comprises the compilation of the diagnosis/predictive diagnosis obtained in step c) for each sample and the final determination of the diagnosis/predictive diagnosis.
- the diagnosis/predictive diagnosis can be considered positive/negative if more than 50% of the result of steps c) correspond to this state.
- step d) when the samples are taken at the same time, step d) makes it possible to reinforce a first diagnosis determined in the first step c), in particular to overcome a potential undesired selection of microorganisms by the choice of the sampling area in a sampling region.
- the samples are in particular taken in different areas of the same sampling region, in order to ensure the exhaustiveness of the representation of the microorganisms in the subject's sampling region.
- step d) when the samples are taken at different times, step d) makes it possible to monitor the changes in the subject's microbiota and in particular the change in their phenotype (from sick to healthy following treatment, or from healthy to sick), allowing a clinician to confirm a curative effect or to take the necessary measures in the event of the appearance of a pathology or pathological state.
- Figure 1 shows an overview of the steps followed for an embodiment of the method for diagnosing a pathology according to the invention from the identification of microorganisms and their abundance in the sample of a subject, followed by a step of predicting the diagnosis/predictive diagnosis using the trained and optimized DNN model.
- Figure 2 illustrates the steps of an example of training a deep neural network model according to the invention and the adjustment of its hyperparameters allowing the optimization of the prediction of the diagnosis/predictive diagnosis.
- Figure 3 illustrates the prediction performance obtained by the deep neural network model based on the input data provided.
- the input data are the data from direct metagenomic sequencing (Fettweis cohort) processed by RiboTaxa or by MetaPhlAn3.
- MetaPhlAn3 uses the high-quality reads from direct metagenomic sequencing to compare them to a reference genome database of microorganisms available at: segatalab.cibio.unitn.it/data/Pasolli_et_al.html and determine the taxonomic composition of the analyzed microbiota (from domain to species) and the relative abundances of the identified microorganisms (TSV file).
- Figure 4 shows the performance of deep neural network models trained on data from direct metagenomic sequencing and genus-level metabarcoding.
- Figure 5 represents the final structure of an artificial intelligence model (trained deep neural network model) according to the invention optimized to predict ECUN.
- Figure 6 shows the true positive rate (on the ordinate) as a function of the false positive rate (on the abscissa) in the context of predicting the occurrence of ECUN, where the AUC is equal to 0.987.
- Figure 7 shows the accuracy (ordinate) versus sensitivity (abscissa) in predicting the occurrence of ECUN, where the AUC is equal to 0.992.
- Figure 8 shows the 20 input features of the trained deep neural network model contributing most to the prediction of ECU N or non-ECU N phenotypes summarized by the SHAP explainer.
- Figure 9 illustrates the analysis of the longitudinal follow-up of samples following the prediction of the deep neural network model trained in the context of predicting the occurrence of NEC.
- the unlabeled circle on the left represents the actual phenotype of the infant. Samples from infants without pathology are indicated in dark gray and samples from NEC infants in light gray. Each labeled circle represents a sample collected from each of the infants and the numbers inside the circles correspond to the day of collection (in days of life).
- the color of these circles represents the phenotype predicted by the neural network according to the same color code as the unlabeled circles.
- the single square represents the samples that were reclassified into the “control” group and the double square represents the samples that were reclassified into the “NEC” group.
- Figure 14 shows the 20 input features of the trained deep neural network model contributing most to the prediction of T1D or non-T1D phenotypes summarized by the SHAP explainer.
- Figure 15 represents the longitudinal analysis approach of the predictions made on the set of samples of children who had at least 3 samples in the “sepsis” test set. The final phenotype of the child is determined by the phenotypic group having the greatest number of samples of the same condition.
- Figure 16 shows the 20 input features of the trained deep neural network model contributing the most to the prediction of sepsis phenotypes summarized by the SHAP explainer. Examples
- the inventors collected raw microbiota sequencing data and associated clinical data from patient cohorts established as part of studies of different pathologies and pathological conditions: preterm birth (PB), necrotizing enterocolitis (NCE), sepsis and type 1 diabetes (T1D).
- PB preterm birth
- NCE necrotizing enterocolitis
- T1D type 1 diabetes
- the first step consisted in selecting relevant scientific publications that had made these data available. A search by precise keywords was carried out in the PubMed and Google Scholar publication databases. The microbiota sequencing data had to have been obtained by direct metagenomic sequencing, known as “shotgun”. Only prospective studies with samples taken before the onset of the pathology or pathological state, allowing a predictive diagnosis, were retained. In addition, the inclusion of control subjects was required.
- the shotgun metagenomic data were processed with the RiboTaxa bioinformatics chain (Chakoory et al., 2022) to obtain the taxonomic profiles of the microbiota (identification of microorganisms at all taxonomic ranks and associated relative abundances).
- the RiboTaxa approach consists of reconstructing 16S and 18S rDNA sequences using reference databases, here, the SILVA SSU 138.1 NR99 database (Quast et al., 2013), then allowing identification of microorganisms down to the species level. RiboTaxa performs quality control of the raw reads, reconstruction of the 16 and 18S rDNA sequences, determination of their relative abundance and the identity of the microorganisms.
- the C parameter “insert_stddev” represents the standard deviation of the size distribution of inserts of paired-end reads.
- Parameters B and C were estimated using the script “mean_size.py”, available at: gist.github.com/timoast/af73c0e9fac00187ee49.
- the architecture of the deep neural network consists of an input layer whose number of neurons depends on the number of input characteristics (number of microorganisms identified and number and nature of clinical data), hidden layers whose number and the number of corresponding neurons are determined during training and optimization of the model, and an output layer containing 2 neurons, one for a “pathology/pathological state” output, the other for a “no pathology/pathological state” output.
- the rectified linear unit activation function (ReLLI) was used for all hidden layers. Activation functions play an important role in training neural networks by providing the nonlinearity needed for the model to learn complex representations.
- the neuron dropout technique on each hidden layer was also employed to mitigate overfitting of the neural network, which leads to poor generalization of the model and reduced performance on new data.
- Neuron dropout is a learning method that involves randomly removing neurons during model training, with the removed nodes being excluded from subsequent steps.
- the output layer activation function uses the Softmax function to assign a value based on a probability between 0 and 1 to each class (pathology/disease state, no pathology/disease state). This value allows the model to make a ‘risk of pathology’ or ‘no risk of pathology’ decision.
- [182]Different values of other hyperparameters were tested.
- the number of epochs (number of times the entire dataset is propagated through the neural network) was varied from 1 to 40.
- the cross-entropy loss between the target value and the predicted value was optimized over the epochs with learning rates, ranging from 0.0001 to 0.01.
- the number of hidden layers was varied from 1 to 3 and the number of neurons in the first hidden layer from 32 to 512 with an increasing step size of 32. To facilitate model convergence, the number of neurons in the hidden layers was set to half that of the previous layer.
- the performance of the optimized deep neural network model was estimated on the test data (20% of the entire data set) by comparing the phenotype predicted by the model and the phenotype observed in the subject. For example, if the model correctly classifies a sample from a subject with a pathology or disease state, it is considered a true positive (TP), otherwise it is a false negative (FN). On the other hand, if the model correctly classifies a sample from a subject without a pathology or disease state, it is considered a true negative (TN), otherwise it is a false positive (FP).
- model performance was measured using several metrics: accuracy (total number of correct predictions over the total number of subjects), sensitivity (rate of subjects with the pathology correctly predicted by the model or true positive rate), specificity (rate of subjects without the pathology correctly predicted by the model or true negative rate), area under the curve (AUC) of the receiver operating characteristic (ROC)/AUROC, and precision-recall AUC (PR-AUC). [185]Accuracy is calculated as follows:
- AUROC corresponds to the area under the ROC curve which shows the sensitivity (rate of true positives) as a function of the specificity (rate of true negatives).
- the PR-AIIC measures the sensitivity over the precision (ratio of TPs to the total number of TPs and FPs).
- AUCs were calculated using the scikit-learn package (Pedregosa et al., 2011) and plotted using matplotlib (Hunter, 2007) (v3.1).
- the 95% confidence intervals (CIs) of the AUCs were estimated using the bootstrap method (Efron and Tibshirani, 1994) with 1,000 iterations.
- ROC curves and the Sankey plot were generated using matplotlib and plotly (v5.15.0), respectively.
- SHAP SHapley Additive exPlanations
- Models can be interpreted by calculating the importance of input data related to the classification performance of the model.
- the importance of input elements was calculated using SHAP.
- SHAP's DeepExplainer function is a method for decomposing the output of a deep neural network (prediction) by assigning contribution values to each data of the neural network input. This function allows highlighting the input data with the most weight in predicting a phenotype.
- x is the original data
- x' is the normalized data
- x min and x ma x are respectively the minimum and maximum values of the original value (abundance).
- the above equation is a linear transformation that preserves all abundance ratios of the original data after normalization.
- the clinical data were either discrete or continuous variables.
- continuous variables were transformed into discrete values through a discretization step. This process involves transforming a continuous-valued variable into a discrete variable by creating a set of contiguous intervals (or bins) that span the range of values of the variable. Grouping numerical features into interval-based groups is beneficial for classification and can significantly improve model performance.
- the number of samples in each phenotypic group was calculated and the final phenotype of the subject was determined by the phenotypic group with the largest number of samples.
- the phenotype thus determined was compared with the observed phenotype (affected by a pathology or a pathological state, not affected). Finally, a lollipop plot was generated to visualize this longitudinal follow-up analysis approach using the ggpubr package (v0.4.0).
- Example 1 Predictive diagnosis of preterm birth using a deep neural network trained with vaginal microbiota data.
- vaginal microbiota in relation to preterm birth using the English keywords: “vaginal microbiome”, “shotgun metagenomics” and “premature birth”: Feehily et al., 2020; Fett Stamm et al, 2019; Goltsman et al, 2018; Pace et al, 2021; Tortelli et al. 2021.
- TB term birth
- PTB preterm birth
- the performance of the optimized deep neural network was compared with three state-of-the-art classification algorithms: k-nearest neighbors (KNN), logistic regression (LR), and support vector machine (SVM). All these models were implemented in Python (version 3.9.10).
- the scikit-learn library (vO.24.2) was used. Each model was trained on the same data set, i.e., the 1290 samples.
- the best hyperparameters and configurations were identified using the grid-search cross-validation (GSCV) method of scikit-learn.
- the GSCV method identifies the best combination of hyperparameters during the 10-fold cross-validation process to achieve the optimal performance of the models.
- MetaPhlAn 3 (Beghini et al. 2021) uses clade-specific marker genes to identify the presence and relative abundance of microorganisms from metagenomic data. MetaPhlAn3 was used to process shotgun metagenomic data from the Fettweis cohort with default parameters and using the CHOCOPhlAnSGB database (Jan21 release). Species-level microbial diversity profiles were used as inputs for training a deep neural network. The performance of the resulting model was compared to a deep neural network model trained with diversity data obtained by pre-processing the same sequencing data with RiboTaxa. Results
- Example 2 Comparison of the performance of models trained on metabarcoding data versus direct metagenomics data for the predictive diagnosis of preterm birth.
- the Fettweis et al. study included 232 women whose vaginal samples were analyzed by both direct shotgun metagenomics and metabarcoding (sequencing of the 16S rDNA V3-V4 region).
- Raw shotgun metagenomics data (952 Gb) and metadata for the Fettweis et al. cohort were obtained after National Institute of Health data access approval. This dataset represented 173 women who delivered at term (667 vaginal samples, scored TB) and 55 women who delivered preterm (155 vaginal samples, scored PTB).
- Raw metabarcoding data (58 Gb) from 749 TB samples (173 women) and 205 PTB samples (55 women) were open access and were downloaded from HMP DACC (https://portal.hmpdacc.org).
- the reads were dereplicated to obtain unique sequences or ASVs (Amplicon Sequence Variants) with their abundance (number of reads corresponding to each unique sequence).
- the sample inference algorithm was then applied to correct the dereplicated sequences from the quality profiles of the raw sequences.
- the pairs of reads thus obtained were merged to obtain the complete amplicon sequences.
- Example 3 Predictive diagnosis of ECUN using a deep neural network trained with data from fecal microbiota.
- --max_read_length 301
- --insert_mean 120
- --insert_stddev 100.
- Model evaluation on external data [229] To further evaluate the performance of the optimized model, 50 fecal samples from 17 preterm infants including 7 who developed ECIIN, from the CORTECs cohort followed by the inventors, were analyzed. In addition, 40 infants from two published cohorts (Ward et al. 2023 and Schwartz et al. 2023) were also included to test the model performance.
- the constitution of the CORTECs cohort was approved by the ethics committee of CPP-Sud-Est VI (protocol code 2021/CE 26, the approval date is May 4, 2021).
- the CORTECs cohort aims to address prenatal and postnatal risk factors for ECU N. All prematurely born children hospitalized in the neonatal intensive care unit (NICU) of the Clermont-Ferrand University Hospital (France) were proposed to enter the cohort.
- Written informed consent was obtained from the families of study participants before enrollment. Infant stools were collected daily during their NICU stay, between May 2021 and June 2022. Stools were collected in a diaper using a sterile loop and then dispensed into eNAT buffer (Copan) before being briefly held at 4°C. Samples were stored at -80°C until DNA extraction.
- Cases of ECUN were identified by physicians based on systemic and abdominal findings and radiographic features. They were stratified according to disease severity according to Bell stages. Cases of ECUN were matched to a control preterm infant (two to one case) who did not develop ECUN. Case-control matching was based on gestational age at delivery, mode of delivery, sex, birth weight, and pre- and postnatal antibiotics. For each ECUN infant, available samples were selected within a 1-week window before the onset of ECUN and samples from corresponding control cases were matched according to the age of the ECUN subject.
- RNA capture probes were obtained by in vitro transcription. 500 ng of libraries were were mixed with 2.5 ⁇ g salmon sperm DNA and incubated with 500 ng biotinylated probes in hybridization buffer for 24 h at 65°C. Probe/target heteroduplexes were captured using 500 ⁇ g streptavidin-coated paramagnetic beads (Dynabeads M-280 Streptavidin, Invitrogen).
- Captured DNA fragments were eluted with 50 ⁇ L of 0.1 M NaOH and transferred to a sterile tube containing 70 ⁇ L of 1 M Tris-HCl buffer pH 7.5. Captured DNA was amplified by PCR with 25 cycles using primers complementary to Illumina adapters. To increase enrichment efficiency, a second capture cycle was performed. Captured DNA was then sequenced on the Illumina MiSeq 2 x 300 bp platform.
- [236]Raw sequencing data from the three cohorts were processed using the RiboTaxa pipeline and all input data were normalized or transformed as described previously. Species that were not present in the training samples were excluded as the model cannot account for them. For each sample, the relative abundance table of microorganisms at the species level concatenated with the subject's clinical data was used as input to the trained model. Each prediction was compared to the child's phenotype (control or ECIIN). SHAP plots were also generated. The final prediction of the children was also determined using longitudinal samples from the same infant using the same longitudinal follow-up analysis approach.
- Example 4 Predictive diagnosis of type 1 diabetes in children using deep neural network
- the primary objectives of the prospective study were to identify environmental and genetic factors triggering or protecting against the development of islet cell antibodies or type 1 diabetes (Rewers et al., 2018). For this, 7013 children from the general population were recruited, with a predetermined risk of type 1 diabetes of 3% and 788 children with first-degree relatives with type 1 diabetes and with a predetermined risk of type 1 diabetes of 10%. Medical visits took place quarterly until the age of 4 years, then every 6 months until the age of 15 years. Participants were followed by blood sampling every three months for measurements of autoantibodies directed against islet cells and detection of diabetes. Stool samples were collected longitudinally between 3 and 72 months of life to characterize the gut microbiota by metabarcoding and direct metagenomic sequencing.
- the IA+DT1 and T1D children were grouped into a single group of type 1 diabetic children for model training, subsequently designated as TD1.
- training was performed on the entire dataset (4707 samples from 144 control children and 110 T1D children) to produce a “no prior” model, and three subsets of the data were created based on the month in which T1D was diagnosed. For this, the child’s day of life at diagnosis was converted into months by dividing it by 30 days.
- T1D model “24-48 months” 2361 samples from 68 control children and 52 T1D children
- T1D model “48-72 months” (1101 samples from 23 control children and 20 T1D children)
- T1D model “24-72 months” 3193 samples from 83 control children and 66 T1D children.
- the models were designated by an interval of children’s ages at the time of T1D diagnosis and included in the model. These intervals cover at most a period of 2 to 6 years (24-72 months), corresponding to the period when the majority of T1D cases were diagnosed.
- Example 5 Predictive diagnosis of sepsis using deep neural network models
- Controlled, high-quality species-level relative abundance profiles and 5 clinical data were used to train a deep neural network to predict the risk of sepsis before the onset of infection leading to pathology.
- the deep neural network model was trained and then trained using 681 different features (637 microbial species and 44 clinical data groups). All species detected in all samples were retained, instead of applying a selection before training to preserve inter-individual variations in microbiota between infants. A total of 42,882 trainable parameters were tested and the optimal hyperparameter setting for the final model had 64 units (neurons) in the 1st hidden layer and a total of 3 hidden layers (Table 12). The model training was performed on: i86linux32, 4.0 GB RAM x 8 cores (32.8 GB total), without GPU and took 2 min.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
La présente demande concerne un procédé in vitro de diagnostic prédictif d'une pathologie à partir d'au moins un échantillon biologique prélevé chez un sujet et comprenant des microorganismes, le procédé comprenant l'identification et l'abondance relative desdits microorganismes présents dans l'échantillon Le diagnostic étant réalisé à l'aide d'un modèle d'intelligence artificielle pré-entrainé sur la base d'un jeu d'entrainement où le jeu de données labellisées comprend des profils de sujets d'entrainement, chaque profil de sujet d'entrainement comprenant l'identité et l'abondance relative de l'ensemble des microorganismes identifiés dans au moins un échantillon dudit sujet d'entrainement sans aucune présélection, où chaque profil de sujet d'entrainement est labellisé avec le phénotype du sujet d'entrainement dont il est issu.
Description
Description
Procédé de diagnostic prédictif d’une pathologie ou d’un état pathologique
[1]La présente demande concerne un procédé, notamment in vitro, de diagnostic ou de diagnostic prédictif d’une pathologie ou d’un état pathologique à partir d’un échantillon biologique prélevé chez un sujet. Selon un aspect particulier, l’invention concerne un procédé, notamment in vitro, de diagnostic prédictif d’une pathologie de l’appareil digestif ou d’une pathologie extra-digestive d’un sujet à partir de l’analyse du microbiote présent dans un échantillon biologique prélevé dans l’appareil digestif, et/ou hors de l’appareil digestif comme le vagin et/ou dans les selles d’un sujet. Encore plus particulièrement, l’invention concerne un procédé de diagnostic de l’entérocolite ulcéro-nécrosante (ECU N) du nouveau-né prématuré à partir d’un échantillon biologique prélevé dans ses selles. Selon un autre aspect particulier, la présente invention a pour objet le diagnostic prédictif d’un accouchement prématuré à partir d’un échantillon biologique prélevé dans le vagin chez une femme enceinte. Le présent procédé se situe donc dans le domaine du diagnostic, du diagnostic prédictif, notamment in vitro, et de la médecine personnalisée.
[2]L’accouchement prématuré est une cause majeure de la morbidité et de la mortalité des nouveau-nés. Une part des accouchements prématurés spontanés semble provenir d’une réaction d’inflammation suite à une infection du tractus génital. Cependant, une large part des accouchements prématurés reste sans cause identifiée, sans signes cliniques. Malgré différentes études relatives au microbiote vaginal et à la survenue d’un accouchement prématuré, il existe actuellement un besoin pour une méthode clinique fiable de prédiction de la survenue d’un accouchement prématuré. Actuellement, les cliniciens ne disposent malheureusement d’aucun outil fiable pour prédire le risque d’une naissance prématurée.
[3]Le brevet EP 3161167 décrit une méthode d’évaluation d’un risque d’accouchement prématuré fondée sur la détection, dans un échantillon vaginal ou cervical obtenu par écouvillonnage chez la femme enceinte, de la quantité des bactéries suivantes : Vimonas micra, Ureaplasma urealyticum ou Ureaplasma parvum, Atopobium vaginae, Peptoniphilus lacrimalis, Megasphaera cerevisiae et Parvibacter caecicola, par rapport à un niveau de référence. La quantification des bactéries est réalisée par amplification d’une petite région de l’ADN ribosomique (ADNr 16S) par réaction en chaîne de la polymérase (en anglais : Polymerase Chain Reaction ou PCR) quantitative (qPCR).
[4]Le brevet EP 2 972 308 B9 décrit un biomarqueur peptidique sérique ou plasmatique, produit par des cellules humaines, et non par le microbiote, dont la détection est utilisée dans une méthode d’évaluation d’un risque d’accouchement prématuré.
[5]La demande internationale WO 2020/227053 décrit un procédé de détermination du risque de naissance prématurée comprenant la détermination de l’abondance de Saccharibacteria TM7- H1 et optionnellement de BVAB1, Sneathia amnii et Prevotella dans un échantillon vaginal d’une femme enceinte, à partir de la séquence nucléotidique d’une petite portion de l’ADNr 16S des microorganismes.
[6]Ces exemples illustrent la possibilité d’une relation entre la nature du microbiote et l’état physiologique ou pathologique d’un sujet. Mais il est également connu que la complexité des microbiotes rend difficile la détermination des signatures microbiennes spécifiques et prédictives caractéristiques d’un état pathologique. Cette situation est rendue d’autant plus complexe du fait de très fortes variations interindividuelles. A ce jour, plusieurs techniques d’analyse du microbiote existent. Cependant, les approches actuelles ne permettent pas une caractérisation précise des microbiotes.
[7]Les gènes exprimant la petite sous-unité de l’ARN ribosomique (ARNr), c’est-à-dire les gènes appelés ADN ribosomique 16S « ADNr 16S » pour les microorganismes procaryotes, tels que notamment les bactéries et les archées, et « ADNr 18S » pour les eucaryotes, incluant notamment les levures, sont utilisés pour permettre la description de la structure du microbiote (Chakoory et al., 2022).
[8]Les publications de Park et al en 2021 et en 2022 décrivent un procédé de prédiction de la probabilité d’une naissance prématurée à partir de la détection d’un nombre restreint de microorganismes présents dans le microbiome vaginal.
[9]Dans la publication de Park et al. de 2021 , le procédé comprend la quantification simultanée par amplification qPCR de fragments spécifiques de petite taille de l’ADN de chacun des 10 microorganismes suivants : Lactobacillus crispatus, Lactobacillus iners, Weissella koreensis, Bacteroides fragilis, Prevotella bivia, Prevotella amnii, Prevotella salivae, Ureaplasma urealyticum, Ureaplasma parvum, Gardnerella vaginalis.
[10]Dans la publication de Park ét al, de 2022, sur la base d’une approche de séquençage d’une petite région V3-V4 du gène d’ADNr 16S et d’études bibliographiques, les prédictions de la probabilité d’une naissance prématurée sont établies sur la base de 10 bactéries (Lactobacillus crispatus, Lactobacillus fornicalis, Lactobacillus gasseri, Lactobacillus iners, Lactobacillus jensenii, Gardnerella vaginalis, Ureaplasma parvum, Atopobium vaginae, Prevotella timonensis et Peptoniphilus grossensis) ainsi que 7 bactéries supplémentaires sur la base de travaux antérieurs d’autres auteurs (Bifidobacterium breve, Dialister proprionicifaciens, Lactobacillus paracasei, Mobiluncus curtisii, Prevotella disiens, Staphylococcus aureus, Streptococcus anginosus). Mais l’exploitation conjointe de ces données n’a pas permis une utilisation clinique documentée selon l’état de l’art.
[11]Par conséquent, il existe une nécessité de développer un procédé permettant de prédire la probabilité d’un accouchement prématuré de façon plus fiable, prenant en compte la variabilité inter-individuelle et les espèces faiblement représentées. Les suivis des femmes enceintes permettraient d’identifier les femmes à risque et d’anticiper la prise en charge des nouveau- nés.
[12]Par ailleurs, l’entérocolite ulcéro-nécrosante (ECU N) est l'urgence gastro-intestinale potentiellement mortelle la plus courante rencontrée par les prématurés dans les unités de soins intensifs pour les nouveau-nés. Elle est définie en tant qu’inflammation ulcéreuse de la paroi intestinale. La pratique clinique actuelle pour diagnostiquer l’ECUN se fonde sur les résultats cliniques, radiologiques et hématologiques constituant les critères de Bell, selon une revue récente (D'Angelo et al., 2018). Les signes cliniques d'un début d’ECUN sont souvent très discrets et peuvent d'abord se manifester par une intolérance alimentaire et des symptômes non spécifiques (malaise, bradycardie) avant que les symptômes gastrointestinaux ne deviennent évidents. Ceux-ci incluent une augmentation des résidus gastriques, des selles sanglantes et une distension abdominale ; ceux-ci peuvent évoluer vers une hypotonie généralisée, une léthargie et une insuffisance cardio-respiratoire, qui peuvent également être présents lors d'autres affections néonatales, notamment la septicémie et les infections intestinales virales. Si la maladie n'est pas diagnostiquée et traitée à un stade précoce, elle peut entraîner une septicémie grave, une perforation intestinale, ainsi qu'une morbidité (nécrose digestive, insuffisance intestinale chronique) et une mortalité importantes (jusqu’à 40% pour les formes sévères).
[13]A ce jour, les cliniciens n’ont aucun outil diagnostic fiable de prédiction de l’ECUN. La physiopathologie de l’ECUN reste mal comprise et des méthodes efficaces pour sa détection précoce doivent encore être établies. Par conséquent, les efforts actuels pour comprendre et prédire l’ECUN se concentrent sur l’étude de ses facteurs de risque. La naissance prématurée représente le facteur de risque le plus important pour le développement de l’ECUN. Chez les nouveau-nés ayant un très faible poids à la naissance (<1 ,5 kg à la naissance), l'incidence de l’ECUN varie de 5 % à 13 %. De plus, l'administration prolongée d'antibiotiques au cours de la première semaine de vie et la substitution du lait maternel par du lait maternisé ou infantile sont fréquemment liées à l'apparition ultérieure de l’ECUN.
[14]La colonisation du microbiote intestinal a été largement considérée comme jouant un rôle dans le développement de l’ECUN chez les nouveau-nés prématurés, mais comme dans le cas de la probabilité d’une naissance prématurée évaluée à partir du microbiome vaginal, la complexité des microbiotes rend difficile la détermination des signatures microbiennes spécifiques et prédictives d’un état physiologique ou pathologique, ne permettant pas l’identification d’un seul agent pathogène opportuniste ou d’une communauté microbienne
pathogène comme cause de l’ECUN. Cet échec est principalement dû à l’établissement précoce et très dynamique du microbiote intestinal néonatal, influencé par de nombreux facteurs, notamment l’environnement, le sexe, l’âge gestationnel, le mode d’accouchement, le mode d’alimentation et les traitements antibiotiques.
[15]Par conséquent, il existe également un besoin très important de disposer d’un procédé fiable et reproductible de diagnostic prédictif des pathologies affectant les nouveau-nés, notamment les nouveau-nés prématurés. Un tel diagnostic prédictif permettrait d’identifier les nouveau- nés à risque et d’anticiper la prise en charge de pathologies susceptibles de gravement affecter leur vie.
[16]Pour donner un troisième exemple de la possibilité d’une relation entre la nature du microbiote et l’état physiologique ou pathologique d’un sujet, le diabète de type I (DT1) est une maladie auto-immune qui résulte de la destruction des cellules bêta du pancréas par les lymphocytes du patient. Cette destruction aboutit à l’incapacité pour le patient de sécréter l’insuline, ce qui conduit à l’impossibilité d’utiliser le glucose comme ressource énergétique, donc à une hyperglycémie en même temps qu’une carence énergétique intracellulaire. Le sucre en excès dans le sang est retrouvé dans les urines.
[17]Le DT1 affecte les enfants et les adultes jeunes. A court terme, il est responsable d’une dégradation importante de la qualité de vie puisque les sujets atteints doivent adapter en permanence leurs apports d’insuline (par voie sous-cutanée) à la glycémie, aux apports alimentaires et aux dépenses énergétiques. A moyen et long terme, l’hyperglycémie chronique entraîne des altérations multiviscérales, en particulier nerveuses et vasculaires.
[18]L’incidence du DT1 est en augmentation continue depuis au moins 1988. En France, elle est de 18 pour 100 000 chez les moins de 15 ans, sur la période 2013-2015, soit une prévalence de l'ordre de 1 ,3 pour 1 000. L'incidence du diabète du sujet jeune augmente de 3 à 4 % par an, en même temps que l’âge de début s’abaisse (Gale E 2002).
[19]L’activation immunitaire est multifactorielle et dépend en partie du système HLA, et d’événement infectieux postnatals. Il existe ainsi une agrégation familiale de cas, une association à d’autres maladies auto-immunes, et un lien possible avec certains agents viraux notamment les coxsackievirus du groupe B.
[20]Après la destruction des cellules bêta du pancréas le seul traitement repose sur l’insulinothérapie substitutive à vie. A ce jour le seul traitement curatif est la greffe de cellules bêta allogénique, qui est un traitement compliqué, nécessitant une immunosuppression prolongée, avec des résultats moyens.
[21 ]Le diagnostic du diabète de type I repose sur la mise en évidence d’une hyperglycémie, d’une glycosurie, et d’une activation du système immunitaire dirigée contre les cellules bêta, dont témoigne la présence d’anticorps anti GAD, anti Zn T8, et anti-insuline. Cette activation immunitaire précède la maladie de plusieurs mois, et une nouvelle stratégie émerge qui consiste à détecter des enfants à haut risque de développer un diabète de type I dans la fratrie d’un enfant déjà atteint, à lui proposer un traitement immunomodulateur. Ce repérage des enfants à haut risque repose à ce jour exclusivement sur la présence ou non d’auto-anticorps. Cependant tous les enfants qui ont des auto-anticorps ne développent pas un diabète de type I.
[22]En effet la mise en action d’une réponse immunitaire est tributaire d’un équilibre entre populations activatrices et inhibitrices de la réaction immunitaire, cet équilibre étant susceptible d’être largement influencé par des agents exogènes en particulier viraux et bactériens. Dans ce contexte l’hypothèse qu’une dysbiose digestive puisse entraîner une activation immunitaire est une piste prometteuse. Une cohorte internationale d’enfants à risque de diabète de type I (Vatanen T, Nature. 2018 Oct;562(7728):589-594) a permis d’étudier le microbiote digestif de ces enfants en comparaison avec celui d’enfants n’ayant pas développé la pathologie, sans pouvoir cependant identifier, avec les méthodes utilisées, de taxa microbiens caractéristiques de l’une ou l’autre des situations (pathologiques et saines). Par conséquent, il existe également un besoin très important de disposer d’un procédé fiable et reproductible de diagnostic prédictif du diabète de type I en se basant sur l’analyse du microbiote prélevé dans les selles d’enfants à risque. Un tel diagnostic prédictif permettrait d’identifier les enfants susceptibles de développer la maladie et d’anticiper la prise en charge de cette pathologie chronique affectant la qualité de vie et pouvant entrainer de graves séquelles voire le décès sans prise en charge adaptée.
[23]La possibilité d’identifier précocement des enfants à haut risque de développer une autoimmunité puis un diabète permettrait une révolution thérapeutique vers une médecine préventive personnalisée pour cette maladie extrêmement handicapante. En effet des traitements préventifs immunomodulateurs récents actuellement disponibles ont prouvé leur efficacité dans la prévention de la maladie diabétique chez des enfants à très haut risque. Cependant ces traitements ne sont pas dénués d’effets indésirables, et doivent être utilisés de manière ciblée.
[24]Pour donner un quatrième exemple de la possibilité d’une relation entre la nature du microbiote et l’état physiologique ou pathologique d’un sujet, le sepsis néonatal est une maladie due à la présence dans le sang d’un agent infectieux, le plus souvent de nature bactérienne. Cette situation est potentiellement gravissime par deux menaces : la défaillance hémodynamique due à la réaction inflammatoire disséminée (choc septique), et la dissémination bactérienne
dans des sites vitaux, notamment les méninges (méningite purulente). Elle nécessite donc un diagnostic et un traitement urgent, qui repose sur l’administration d’antibiotiques par voie intraveineuse. Ceux-ci ciblent dans un premier temps les germes les plus fréquemment impliqués (antibiothérapie probabiliste) ; dès la bactérie identifiée, l’antibiothérapie est adaptée afin de limiter le plus possible la sélection de souches résistantes aux antibiotiques.
[25]Le sepsis néonatal affecte environ 1 nouveau-né à terme sur 1000. Dans la situation d’une grossesse et d’une naissance normales, la prévention repose sur les antécédents de la mère et la détection du portage vaginal de streptocoque B. En cas de portage, une antibiothérapie est administrée à la mère pendant le travail, de telle sorte que le nouveau-né est protégé même en cas de transmission de streptocoque lors de la naissance.
[26]En revanche, en cas de prématurité, le sepsis néonatal est beaucoup plus fréquent, atteignant plus d’un enfant sur quatre. Cette fréquence accrue est due à la fragilité des enfants prématurés, à la présence de matériel invasif (cathéter, sondes) et à l’hospitalisation prolongée (germes hospitaliers, manipulations pluriquotidiennes par de nombreux soignants). Les germes responsables de sepsis sont le plus souvent retrouvés dans le tube digestif des enfants, et parfois sur la peau notamment en cas de cathéter à demeure.
[27]Le diagnostic du sepsis repose actuellement sur l’association de symptômes non-spécifiques (fièvre, malaises, tachycardie, vomissements etc.), de marqueurs sanguins de la réponse inflammatoire (polynucléose neutrophile, élévation de la CRP) et parfois de la mise en évidence d’une bactérie dans le sang (par hémoculture). Ce dernier examen doit être réalisé avant toute antibiothérapie (qui en masquerait le résultat), et nécessite un volume sanguin considérable (au moins 1 ml, soit 2% du volume sanguin total d’un prématuré de 500 grammes). L’identification d’un germe met en général 1 à 2 jours, et la caractérisation de sa sensibilité aux antibiotiques peut prendre jusqu’à une semaine.
[28]L’adaptation du traitement est donc tardive, exposant le nouveau-né à une antibiothérapie à spectre inutilement large (avec pour conséquence un déséquilibre du microbiote digestif et la sélection de souches résistantes).
[29]Par conséquent, il existe également un besoin très important de disposer d’un procédé fiable et reproductible de diagnostic prédictif du sepsis en se basant sur l’analyse du microbiote prélevé dans les selles d’enfants à risque. Un tel diagnostic prédictif permettrait d’identifier les enfants à risque et d’anticiper la prise en charge de cette pathologie chronique susceptible de gravement affecter leur vie.
[30]La prédiction du sepsis néonatal permettrait une surveillance accrue des nouveau-nés à risque, et autoriserait un traitement plus précoce en cas de symptômes. De plus, la caractérisation a
priori des germes probablement responsables, portés notamment dans le tube digestif du nouveau-né, permettrait de prescrire d’emblée un traitement plus adapté au profil de ces bactéries.
[31]ll est connu de l’art antérieur plusieurs méthodes d’analyse du microbiote dans un but diagnostic ou diagnostic prédictif d’une pathologie.
[32]Une première méthode dite de « métabarcoding » permet de déterminer des taxa présents dans un échantillon grâce à leur signature génétique, unique pour chaque taxa. L'idée est d'avoir un fragment d'ADN présent chez tous les taxa à analyser et qui constitue un marqueur génétique. Ce marqueur est un fragment d'ADN encadré par des régions très conservées et donc les plus « universelles » possibles, et qui, une fois séquencé, montre des variations de séquences génétiques entre taxa différents. Dans le cadre du microbiote, cette méthode comprend souvent l’amplification de fragments d’une taille comprise entre 300 à 470 paires de bases des régions V3 et/ou V4 du gène exprimant l’ARNr 16S. Cependant cette méthode présente plusieurs limites : des biais sont susceptibles d’être générés lors de l’étape d’amplification réalisée par PCR et peuvent altérer la vision de la diversité réelle du microbiote. En effet, il est connu que les amorces utilisées qui ne peuvent pas être « universelles » pour amplifier les séquences nucléotidiques vont favoriser l’amplification des séquences de certains microorganismes au détriment d’autres, résultant en une abondance possiblement erronée des microorganismes voire la non-détection de certains micro-organismes. En outre, la faible longueur des fragments d’ADN séquencés n’apporte qu’une faible résolution taxonomique, ne permettant pas de décrire les communautés microbiennes au niveau de l’espèce.
[33]Une autre méthode comprenant une étape de séquençage métagénomique direct (en anglais « shotgun ») suivie d’une étape d’assemblage pour générer des génomes complets (en anglais : Metagenome Assembled Genomes ou MAG) et d’une étape d’affiliation des MAGs conduit à une identification restreinte aux espèces dominantes.
[34]Une autre méthode comprend une étape de séquençage métagénomique direct suivie d’une affiliation des lectures brutes non assemblées d’une taille inférieure à 300 paires de bases d’une partie du gène exprimant l’ARNr 16S. L’affiliation de ces séquences de petite taille conduit à une faible résolution d’identification microbienne et à une surestimation de la diversité, notamment par détection de faux positifs.
[35] Il existe donc un besoin pour l’obtention de diagnostic et de diagnostic prédictif plus fins, fiables, reproductibles et relativement rapides à mettre en œuvre, de sorte à pouvoir être utilisable par les cliniciens dans leurs prises de décisions.
Description de l’invention
[36]Les inventeurs ont réussi à développer un unique procédé permettant de répondre aux différentes problématiques susmentionnées. Ce procédé comprend avantageusement l’emploi de l’ensemble des microorganismes identifiées dans le microbiote d’un sujet par un modèle d’intelligence artificielle pour établir un diagnostic ou un diagnostic prédictif d’une pathologie ou d’un état pathologique.
[37]La présente invention a ainsi pour premier objet un procédé, notamment in vitro, de diagnostic ou de diagnostic prédictif d’une pathologie ou d’un état pathologique chez un sujet, à partir d’au moins un échantillon biologique prélevé chez le sujet et contenant des microorganismes, ledit procédé comprenant les étapes suivantes : a) séquençage, à partir de l’acide nucléique isolé de l’échantillon du sujet, des séquences nucléotidiques correspondant à au moins une séquence d’intérêt sélectionnée dans le groupe consistant en : un fragment d’un gène exprimant l’ARN ribosomique (ARNr) 16S, un fragment d’un gène exprimant l’ARNr 18S, un fragment de l’ARNr 16S, un fragment de l’ARNr 18S, b) à partir du séquençage de l’étape a), détermination de l’identité et de l’abondance relative des microorganismes présents dans ledit échantillon sans aucune présélection, c) détermination du diagnostic prédictif de ladite pathologie ou de l’état pathologique par un modèle d’intelligence artificielle à partir au moins des abondances des identités obtenues à l’étape b), ledit modèle d’intelligence artificielle ayant préalablement été entraîné sur la base d’un jeu de données labellisées, où le jeu de données labellisées comprend des profils de sujets d’entrainement, chaque profil de sujet d’entrainement comprenant l’identité et l’abondance relative de l’ensemble des microorganismes identifiés dans au moins un échantillon dudit sujet d’entrainement, où chaque profil de sujet d’entrainement est labellisé avec le phénotype du sujet d’entrainement dont il est issu, et où des données de l’étape b) sont uniquement exclues les abondances des identités des microorganismes qui n’étaient pas présentes dans le jeu de données labellisées.
[38]Le label du phénotype attribué à chaque sujet d’entrainement dépend de la destinée du procédé selon l’invention et du type de données utilisées pour l’entrainement. Le jeu de données labellisées comprend au moins deux états différents pour les phénotypes et notamment des états antinomiques : un phénotype positif associé à un diagnostic/diagnostic
prédictif positif et un phénotype négatif associé à un diagnostic/diagnostic prédictif négatif. Ainsi, pour un diagnostic, le phénotype de sujet d’entrainement peut être classé « non atteint » ou « atteint » de la pathologie ou l’état pathologique ou encore « sain » et « malade », ces types de classement étant synonymes. Pour un diagnostic prédictif, le phénotype de sujet d’entrainement peut être classé en « ayant développé » ou « n’ayant pas développé » la pathologie ou l’état pathologique ou encore « avec apparition » ou « sans apparition » de la pathologie ou l’état pathologique, ces types de classement étant synonymes.
[39]L’invention présente l’avantage d’entrainer plus efficacement le modèle d’intelligence artificielle en utilisant l’identité de l’ensemble des microorganismes identifiés dans le jeu de données labellisées. L’absence d’étape de présélection d’identité de microorganismes dans le jeu de données labellisées d’entrainement du modèle d’intelligence artificielle permet de conserver toute la diversité et la variabilité individuelle des microbiotes et toutes les interactions microbiennes associées dans le cadre d’une pathologie ou d’un état pathologique déterminé.
[40]En outre, le procédé selon l’invention présente l’avantage de restreindre au minimum (voire de n’appliquer aucune restriction) l’exclusion des identités des microorganismes des données de l’étape b) transmises au modèle d’intelligence artificielle lors de l’étape c), permettant de conserver au maximum la diversité microbienne présente dans l’échantillon du sujet. En effet, la sélection des identités envoyées au modèle d’intelligence artificielle ne se fait aucunement sur la base d’une abondance relative trop faible dans l’échantillon du sujet ou du leur absence d’implication connue dans la pathologie ou l’état pathologique, mais seulement sur la base de leur présence dans le jeu de données d’entrainement. Ainsi, si le jeu de données est suffisamment grand et exhaustif, aucune identité de microorganismes n’est exclue des données transmises au modèle d’intelligence artificielle pour réaliser l’étape c).
[41 ]l I n’était pas évident qu’employer l’identité de l’ensemble des microorganismes sans sélection préalable lors de l’entrainement puisse donner des résultats pertinents. Cela est même contraire à ce qui était attendu. En effet, il est traditionnellement considéré que des données complexes de haute dimensionnalité, utilisées en entrée d’un modèle d’intelligence artificielle, peuvent contenir du bruit et des informations non pertinentes qui peuvent nuire à l’apprentissage et donc aux performances du modèle (Botteghi, N., Guo, M. & Brune, C. Deep kernel learning of dynamical models from high-dimensional noisy data. Sci Rep 12, 21530 (2022)). La recherche de signatures microbiennes pour le diagnostic et le diagnostic prédictif de pathologies et d’états pathologiques est particulièrement complexe du fait des très fortes variations interindividuelles du microbiote. Le microbiote de chaque individu est effectivement influencé par de nombreux facteurs relevant notamment du mode de vie, de l’alimentation et de l’environnement de ce dernier. C’est d’ailleurs pourquoi, bien qu’à ce jour, plusieurs
techniques d’analyse du microbiote existent, elles ne permettent pas une caractérisation précise entre microbiotes et pathologies, du risque de développer lesdites pathologies, ou de l’évolution de ces dernières. Ainsi, le résultat le plus probable aurait était l’obtention d’un grand nombre de diagnostic faux positifs ou faux négatifs.
[42]C’est pourquoi, alors que l’état de l’art montrait que la complexité des microbiotes rendait difficile la détermination des signatures microbiennes spécifiques et prédictives caractéristiques d’un état pathologique ou d’une pathologie, situation rendue d’autant plus complexe du fait de très fortes variations interindividuelles, tous ensemble ces aspects complexes transmis au modèle d’intelligence artificielle préalablement entrainé selon l’invention ont permis contre toute attente d’obtenir des résultats de diagnostic prédictif et de diagnostic d’une grande finesse, fiables, reproductibles et relativement rapides à mettre en œuvre. Le procédé de l’invention répond ainsi à un besoin clinique auparavant non satisfait et fournit une information simple et de qualité à un clinicien.
[43]ll s’agit donc ici d’une avancée majeure permettant de révéler des liens entre ces communautés de microorganismes et des pathologies et états pathologiques, que ces derniers soient déjà présents chez le sujet, qu’ils évoluent ou bien qu’ils se développent ou surviennent a posteriori. L’établissement de diagnostics prédictifs permettent avantageusement d’anticiper les prises en charge du sujet, voire d’effectuer des traitements préventifs.
[44]Le procédé de l’invention prend en compte comme identité de chaque microorganisme la classification par rang taxonomique, ce rang étant de préférence l’espèce du microorganisme. Aucune présélection n’est réalisée lors de l’identification, notamment sur la base de leur abondance relative et/ou de leur implication connue dans le diagnostic ou le diagnostic prédictif.
[45]Selon un mode de réalisation, les microorganismes du jeu de données labellisées ainsi que ceux de l’étape b) sont identifiés au niveau du même rang taxonomique. Ce rang est notamment choisi depuis le phylum jusqu’à l’espèce, et est de préférence l’espèce.
[46]Alternativement, lors de l’entrainement du modèle d’intelligence artificielle et lors de l’étape b), l’identité de chaque microorganisme correspond au rang taxonomique le plus confiant, qui peut être une espèce, un genre, une famille, un ordre, une classe ou un phylum. Ainsi dans ce cas, que ce soit pour le jeu de données labellisées ou l’identification de l’étape b), les identités des microorganismes n’auront pas toute le même rang. Cet aspect permet de manière avantageuse de conserver la maximum d’exhaustivité du jeu de données labellisées lors de l’entrainement du modèle d’intelligence. Dans le cas où il n’est pas possible d’attribuée une espèce à une séquence nucléotidique ou à un ensemble de séquences, il lui/leur sera attribuée le niveau taxonomique le plus confiant, qui pourra être un genre, une famille, un ordre, une
classe ou un phylum, (et potentiellement suivi du terme « non classé »), ainsi que son/leur abondance.
[47]Par « rang taxonomique le plus confiant », on entend le rang taxonomique le plus précis obtenable à partir de la séquence nucléotidique ou de l’ensemble de séquence nucléotidique utilisé pour identifier un microorganisme. L’obtention du rang le plus confiant dépend de différents facteurs, décrits en détail plus loin.
[48]La diversité des microbiotes donnée au modèle d’intelligence artificielle lors de son entrainement peut être assurée par l’emploi de données de sujets d’entrainement d’origines multinationales, notamment multi-continentales, notamment encore de l’ensemble des continents. Ainsi, les sujets d’entrainement sont répartis en différents groupes d’origine géographique. En particulier, la répartition des sujets dans les différents groupes est la plus représentative possible de la diversité géographique.
[49]Selon un mode de réalisation de l’invention, le jeu de données labellisées comprend au moins une donnée clinique déterminée, où chaque profil de sujet d’entrainement comprend une valeur pour la ou chaque donnée clinique déterminée, et où l’étape c) comprend la fourniture au modèle d’intelligence artificielle de la valeur correspondante du sujet pour la ou chaque donnée clinique déterminée.
[50]Selon un mode de réalisation de l’invention, le procédé selon l’invention présente ainsi l’avantage, à partir d’un simple prélèvement de microbiote vaginal pendant la grossesse, au 1er trimestre et/ou au 2ème trimestre et/ou 3ème trimestre, et de son séquençage, de prédire avec une forte certitude la survenue d’une naissance prématurée ou d’une naissance à terme.
[51]Notamment, le procédé de l’invention permet le diagnostic prédictif de la survenue d’un accouchement prématuré dont l’exactitude peut notamment atteindre 88 %. Un tel degré de fiabilité est non égalé parmi les procédés de diagnostic d’accouchement prématuré à ce jour.
[52]Selon un autre mode de réalisation, le procédé selon l’invention présente également l’avantage, à partir d’un simple prélèvement de microbiote dans les selles d’un sujet, et de son séquençage, de déterminer avec une forte certitude le développement d’une maladie de l’appareil digestif ou d’une maladie extra-digestive. Cette approche peut avantageusement être utilisée dans le cadre de médecine personnalisée pour évaluer la pertinence d’un suivi clinique plus précis et/ou le recours à un traitement thérapeutique.
[53]Ainsi, le procédé de l’invention permet une prédiction fiable de l’entérocolite ulcéro-nécrosante avec une exactitude pouvant notamment atteindre 94,9 %. Un tel degré de fiabilité est très utile pour identifier les nouveau-nés prématurés à risque, renforcer la surveillance et permettre
des réponses thérapeutiques rapides évitant d'éventuels problèmes de santé graves. A cet effet, le procédé de l’invention permet de diagnostiquer précocement et très efficacement l’ECUN et de distinguer tout aussi efficacement les nourrissons non affectés.
[54]Selon un mode de réalisation de l’invention, le procédé est destiné au diagnostic prédictif du diabète de type I chez un enfant. Le procédé selon l’invention, de manière similaire permet également de prédire de manière fiable la survenue d’un diabète de type I (DT1), avec une exactitude pouvant notamment atteindre 73,6 %. Le procédé de l’invention permet ainsi d’identifier précocement des enfants à haut risque de développer une auto-immunité puis un diabète permettrait une révolution thérapeutique vers une médecine préventive personnalisée pour éviter les conséquences handicapantes de la pathologie.
[55]Selon un mode de réalisation de l’invention, le procédé a pour but un diagnostic prédictif du sepsis néonatal chez un nourrisson. Le procédé selon l’invention permet encore de prédire de manière fiable la survenue de sepsis, avec une exactitude pouvant atteindre 92,3 %. Le procédé de l’invention permet ainsi d’identifier les nouveau-nés prématurés à risque, renforcer la surveillance et d’adapter le traitement au profil de ces bactéries impliquées dans la pathologie.
[56]L’invention a également pour objet un procédé d’entrainement d’un modèle d’intelligence artificielle destiné à obtenir un diagnostic ou un diagnostic prédictif, ledit procédé utilisant un jeu de données labellisées comprenant des profils de sujets d’entrainement, où chaque profil de sujet d’entrainement comprend l’identité et l’abondance relative de l’ensemble des microorganismes identifiés dans au moins un échantillon dudit sujet d’entrainement sans aucune présélection, et où chaque profil est labellisé avec le phénotype du sujet d’entrainement dont il est issu.
[57]Les caractéristiques décrites plus haut et plus bas en relation avec le jeu de données labellisés et de manière générale au modèle d’intelligence artificielle et à son entrainement s’appliquent mutatis mutandis au présent objet.
[58]Le procédé d’entrainement selon l’invention permet d’obtenir un modèle d’intelligence artificielle plus fiable et plus précis dans ces prédictions, pour les raisons susmentionnées.
[59]Ce procédé d’entrainement a notamment permis d’identifier des microorganismes qui seraient des acteurs clés de diverses pathologies, d’états pathologiques et d’absence de ces derniers. Des microorganismes peuvent ainsi être identifiés comme pouvant jouer le rôle de probiotiques ou pour le développement de nouveaux traitements, voire de nouveaux diagnostics et diagnostic prédictif. Dans ce cadre, grâce au procédé selon l’invention, les
inventeurs ont pu constater l’association de plusieurs espèces de microorganismes à la présence d’une pathologie donnée, d’une part, et constater l’association de plusieurs espèces de microorganismes à l’absence d’une pathologie donnée, d’autre part.
[60]Notamment, les inventeurs ont constaté que plusieurs espèces de Lactobacillus étaient associées à des cas non-ECU N, tandis que plusieurs autres espèces bactériennes telles que : Enterobacter non classées, Enterobacteriaceae non classées, Enterococcus faecalis, Klebsiella non classées, Haemophilus parainfluenzae, Enterococcus durans et Enterobacter cancerogenus étaient associées aux cas d’ECU N. Ces résultats suggèrent que le diagnostic du sujet est fonction à la fois des taxons dominants, sous-dominants voire rares, soulignant qu'aucune espèce individuelle ou groupe taxonomique d'espèces n'est exclusivement responsable d'un risque accru d’ECUN. Au lieu de cela, sans être tenus par aucune théorie, les inventeurs suggèrent probable que divers consortia microbiens puissent provoquer des cascades inflammatoires entraînant l’apparition de l’ECUN.
[61]Les données obtenues à l’aide du procédé d’entrainement permettent donc en outre de disposer d’une cartographie précise des microorganismes associés à la présence d’un état pouvant conduire à une pathologie ou un état pathologique, et des microorganismes associés à l’absence d’un état conduisant à une pathologie ou d’un état pathologique, d’autre part.
[62]Selon un aspect particulier, le procédé selon l’invention présente également l’avantage de ne pas augmenter le nombre d’examens obstétriques sur les femmes enceintes réalisés au cours de la grossesse, dans la mesure où l’échantillon vaginal peut être récupéré au cours d’un examen déjà programmé.
[63]Selon un autre aspect particulier, le procédé selon l’invention permet avantageusement de réaliser des interventions thérapeutiques précoces afin de prévenir le développement ou les pires complications d’une pathologie extra-digestive à partir de l’analyse du microbiome intestinal, ou fécal, d’un sujet.
[64]La présente invention a également pour objet un produit programme d’ordinateur comprenant des instructions exécutables, qui lorsqu’elles sont exécutées sur un ordinateur permettent la mise en œuvre de l’étape c) de détermination du diagnostic/diagnostic prédictif du procédé selon l’invention. Les caractéristiques précédemment et subséquemment décrites en relation avec le modèle d’intelligence artificielle s’appliquent mutatis mutandis au présent objet.
[65]Selon un mode de réalisation de l’invention, le produit programme d’ordinateur comprend des instructions permettant le diagnostic prédictif d’un accouchement prématuré chez un sujet.
[66]Selon un mode de réalisation, le produit programme d’ordinateur comprend des instructions permettant le diagnostic prédictif d’ECUN chez un sujet.
[67]Selon un mode de réalisation, le produit programme d’ordinateur comprend des instructions permettant le diagnostic prédictif du diabète de type I chez un sujet.
[68]Selon un mode de réalisation, le produit programme d’ordinateur comprend des instructions permettant le diagnostic prédictif du sepsis chez un sujet.
[69]L’invention a également pour objet l’utilisation d’un produit programme d’ordinateur selon l’invention pour le diagnostic/diagnostic prédictif d’une pathologie ou d’un état pathologique. Les caractéristiques précédemment et subséquemment décrites en relation avec le procédé de diagnostic/diagnostic prédictif selon l’invention s’appliquent mutatis mutandis au présent objet.
[70]L’invention a enfin pour objet la prise en charge ou le traitement d’un sujet dont le diagnostic ou le diagnostic positif à une pathologie ou à un état pathologique a été déterminé comme positif grâce au procédé de diagnostic/diagnostic prédictif de l’invention. Ledit traitement peut être un traitement curatif ou bien un traitement prophylactique en fonction de la situation. La prise en charge peut être une surveillance clinique renforcée, notamment dans le cadre du diagnostic prédictif d’un accouchement prématuré.
Description détaillée de l’invention
[71]La présente invention a ainsi pour premier objet un procédé, notamment in vitro, de diagnostic ou de diagnostic prédictif d’une pathologie ou d’un état pathologique chez un sujet, à partir d’au moins un échantillon biologique prélevé chez le sujet et contenant des microorganismes.
[72]Par « diagnostic », on entend dans l’invention la détermination de la présence ou de l’absence d’une pathologie ou d’un état pathologique chez un sujet. Un diagnostic positif est compris dans l’invention comme correspondant à la détermination de la présence de la pathologie ou de l’état pathologique chez le sujet. Un diagnostic négatif est compris comme correspondant à la détermination de l’absence de la pathologie ou de l’état pathologique chez le sujet.
[73]Par « diagnostic prédictif », on entend dans l’invention la détermination du risque de développer/de survenue/d’apparition une pathologie ou la survenue d’un état pathologique chez un sujet ne présentant aucun symptôme. Le diagnostic prédictif positif est compris dans la présente invention comme un fort risque d’apparition de la pathologie ou de l’état pathologique. A l’inverse, un diagnostic prédictif négatif est compris dans la présente invention comme un faible risque d’apparition de la pathologie ou de l’état pathologique.
[74]Un diagnostic/diagnostic prédictif positif peut être considéré comme déterminé lorsque la certitude associée est de plus de 50%, de préférence une certitude supérieure ou égale à 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % ou égale à 100 %. De même, un diagnostic/diagnostic prédictif négatif peut être considéré comme déterminé lors que la certitude associée est de plus de 50%, de préférence une certitude supérieure ou égale à 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % ou égale à 100 %.
[75]On entend par « pathologie » une maladie, un déséquilibre biologique ou un inconfort. La pathologie correspond notamment à une pathologie digestive, à une pathologie extra-digestive ou encore à une pathologie du nouveau-né, en particulier les entérocolites du type, plus particulièrement l’entérocolite ulcéro-nécrosante (ECIIN). Par « entérocolite ulcéro- nécrosante » on entend une maladie caractérisée par l’inflammation et la nécrose de la muqueuse intestinale. Encore plus particulièrement, parmi lesdites pathologies digestives on peut citer : les cancers digestifs, c’est-à-dire affectant au moins un des organes de l’appareil digestif, les maladies inflammatoires chroniques, telles que notamment la maladie de Crohn, la rectocolite hémorragique, le syndrome de l’intestin irritable et la maladie cœliaque.
[76]La pathologie est avantageusement soit une pathologie de l’organe où est prélevé l’échantillon biologique, ou bien une pathologie d’un autre organe de l’environnement où l’échantillon est prélevé.
[77]Par « pathologie extra-digestive », on entend un état ou une pathologie n’affectant pas directement un organe du système digestif mais dont l’une des conséquences est susceptible d’affecter directement ou indirectement le microbiote de l’appareil digestif et réciproquement. Parmi les états et pathologies extra-digestives, ou non-digestives, dont un diagnostic prédictif peut être réalisé par un procédé selon l’invention, on peut citer : le diabète, le sepsis, l’obésité, les maladies cardio-vasculaires, les maladies métaboliques, les maladies hépatiques, les maladies rénales, les maladies uro-génitales, les maladies pulmonaires, les maladies articulaires, les maladies musculaires, les maladies inflammatoires, l’asthme, les allergies, l’arthrite, les maladies neurodégénératives (Parkinson, Alzheimer...), les maladies psychiatriques, les maladies comportementales, tous types de cancers pour tous types d’organes.
[78]Par « état pathologique », on entend un état d'altération des fonctions, de la morphologie ou de la santé d'un organe ou organisme dont on connait ou non la cause, et qui se caractérise par la présence ou l'absence d'un ou plusieurs signes. Un état pathologique correspond notamment à un accouchement prématuré.
[79]Par « état ou pathologie du système digestif », on entend un état ou une pathologie affectant au moins un organe choisi parmi : la bouche, les glandes salivaires, le pharynx, l’œsophage, l’estomac, le pancréas, le foie, la vésicule biliaire, le canal cholédoque, l’intestin grêle et le gros intestin. Le gros intestin comprend le côlon ascendant, le colon transverse, le côlon sigmoïde et le rectum. Selon un aspect particulier du procédé de l’invention, ladite pathologie est une pathologie intestinale.
[80]On entend par « accouchement prématuré » un accouchement survenant avant le début de la 37ème semaine d’aménorrhée.
[81]Selon un aspect particulier du procédé de l’invention, ladite pathologie est une pathologie digestive d’un sujet choisi parmi : les enfants, les nourrissons (les enfants au-delà de leurs premier mois de vie et jusqu’à l’âge de 24 ou 30 mois) et les nouveau-nés (enfants de moins de 28 jours selon la définition de l’Organisation Mondiale de la Santé), lesdits nouveau-nés étant nés à terme, soit entre la 37ème semaine et la fin de la 40ème semaine d’aménorrhée, ou prématurés, c’est-à-dire nés avant la 37ème semaine d’aménorrhée.
[82]On entend par « sujet », un animal ou un être humain, l’animal étant notamment un mammifère. Selon un mode de réalisation particulier de l’invention, le stade de développement du sujet est choisi parmi : adulte (à partir de 18 ans), adolescent (12 - 17 ans), enfant (2 - 11 ans), nourrisson (28 jours - 23 mois), nouveau-né (0 - 27 jours) et nouveau-né prématuré (< 37 semaines d’aménorrhée). Selon un aspect particulier du procédé de l’invention, le sujet est une femme enceinte, un nouveau-né, un nourrisson ou un enfant humain.
[83]On entend par « échantillon biologique », tout échantillon du sujet contenant des microorganismes. En particulier, ledit échantillon biologique est choisi parmi : un prélèvement de l’appareil digestif, un prélèvement d’excrétions, en particulier un échantillon de selle du sujet, un prélèvement vaginal, un prélèvement cervical, un prélèvement cutané, et tout autre prélèvement biologique contenant des microorganismes.
[84]Le prélèvement de l’échantillon est en particulier réalisé de manière conventionnelle et bien connue par une personne spécialiste. Un échantillon biologique donné comprend une communauté de microorganismes désignée par le terme « microbiote ».
[85]Selon un mode de réalisation, l’échantillon peut correspondre au regroupement de plusieurs échantillons prélevés à des zones diverses d’une région de prélèvement chez le sujet, afin de tenter d’obtenir le maximum de diversité des microorganismes.
[86]On entend par « microorganisme », tout microorganisme unicellulaire ou multicellulaire tel que, mais sans limitation, les bactéries, les archées, les virus, les eucaryotes unicellulaires tels que les levures, etc.
[87]Parmi les microbiotes hébergés par un sujet humain, on peut distinguer le microbiote cutané, le microbiote mucosal, le microbiote pulmonaire, le microbiote bucco-dentaire, le microbiote vaginal, le microbiote urinaire, et les microbiotes de l’appareil digestif (microbiote buccal ou salivaire, microbiote de l’estomac, microbiote de l’intestin grêle, microbiote colique, microbiote anal). Le microbiote présent dans les selles, ou microbiote fécal correspond à l’ensemble des microorganismes retrouvés dans les selles faisant suite au transit dans le système digestif d’un sujet, pouvant être le reflet du microbiote intestinal au sens large avec une plus forte proximité avec le microbiote colique. Des microorganismes transitoires peuvent aussi être retrouvés dans ce microbiote. On entend par « microbiome » l’ensemble des génomes portant les gènes hébergés par les microorganismes constituant le microbiote. Le microbiome peut aussi être considéré comme étant l’ensemble des microorganismes y compris leurs génomes dans un environnement biologique particulier comme par exemple le côlon.
[88]Par « appareil digestif » on entend l’ensemble des organes des animaux pluricellulaires qui reçoit la nourriture, la digère pour en extraire des nutriments et excrète les déchets sous forme de matière fécale. Parmi les organes de l’appareil digestif humain, on peut citer : la bouche, les glandes salivaires, le pharynx, l’œsophage, l’estomac, le pancréas, le foie, la vésicule biliaire, le canal cholédoque, l’intestin grêle et le gros intestin. Le gros intestin comprend le côlon ascendant, le colon transverse, le côlon sigmoïde et le rectum. Par « excrétion » on entend les déchets inutilisables ou toxiques qui sont rejetés par le sujet comme l’urine, les matières fécales ou selles, ou des produits de sécrétion comme la bile ou la salive.
Etape a)
[89]L’étape a) correspond au séquençage de l’acide nucléique des microorganismes présents dans le ou les échantillons biologiques, le dit acide nucléique ayant été au préalable isolé de l’échantillon.
Extraction de l’acide nucléique depuis l’échantillon
[90]On entend par « acide nucléique » l’ensemble des molécules d’acides nucléiques présentes dans l’échantillon biologique, notamment l’acide désoxyribonucléique (ADN) et l’acide ribonucléique (ARN), parmi lesquels respectivement les gènes exprimant l’ARN ribosomique (ARNr) 16S et/ou ceux exprimant l’ARNr 18S, en particulier l’ARNr et encore plus particulièrement l’ARNr 16S et l’ARNr 18S.
[91 ]Par « gène exprimant l’ARNr 16S » on entend la séquence nucléotidique d’ADN comprenant la séquence nucléotidique codant l’ARNr 16S. Un gène exprimant un ARNr 16S est également appelé « ADNr 16S ».
[92]Par « gène exprimant l’ARNr 18S » on entend la séquence nucléotidique d’ADN comprenant la séquence nucléotidique d’ADN codant l’ARNr 18S. Un gène exprimant un ARNr 18S est également appelé « ADNr 18S ».
[93]Les gènes exprimant la petite sous-unité de l’ARNr, c’est-à-dire les gènes appelés « ADNr 16S » pour les microorganismes procaryotes, tels que notamment les bactéries et les archées, et « ADNr 18S » pour les eucaryotes, incluant notamment les levures, sont utilisés pour permettre la description de la structure du microbiote (Chakoory ét al., 2022).
[94]Afin d’isoler l’acide nucléique depuis l’échantillon, tout kit commercial d’extraction d’acides nucléiques peut être utilisé. Il est à noter que le rendement (quantité d’acides nucléiques) des kits ainsi que la qualité des acides nucléiques peut varier en fonction du type d’échantillon. Il est en général nécessaire de comparer l’efficacité des kits pour sélectionner le plus performant. L’extraction peut être réalisée manuellement ou à l’aide d’automate. Outre les kits commerciaux, il existe des procédés d’extraction pour lesquels les réactifs sont produits directement en laboratoire. Il existe également des standards de protocole d’extraction ayant pour but d’homogénéiser les procédures d’extractions des acides nucléiques dans le monde entier. En particulier, dans le cadre de l’ECUN, il peut notamment être utilisé le protocole H publié par l’IHMS (International Human Microbiome Standards) pour l’extraction d’ADN à partir des selles de nouveau-nés: (voir IHMS (human-microbiome.org)).
[95]Selon un mode de réalisation de l’invention, le procédé comprend l’isolement de l’acide nucléique issu d’une pluralité de microorganismes présents dans ledit échantillon biologique, en particulier de l’ensemble des microorganismes.
Séquençage de l’acide nucléique
[96]L’acide nucléique isolé est ensuite séquencé afin d’obtenir les séquences nucléotidiques correspondant à au moins une séquence d’intérêt choisie dans le groupe consistant en : un fragment d’un gène exprimant l’ARNr 16S, un fragment d’un gène exprimant l’ARNr 18S, un fragment de l’ARNr 16S et un fragment de l’ARNr 18S (ci-après nommées « séquences d’intérêt »). En effet, l’ADNr 16S, l’ADNr 18s, l’ARNr 16S et l’ARNr 18S sont très conservés chez tous les microorganismes, mais comprennent aussi des variations discriminantes entre taxa qui permet ainsi d’analyser les séquences appartenant aux microorganismes et par ailleurs de les distinguer. Ainsi, le but de l’étape de séquençage est de récupérer l’ensemble des séquences correspondants à au moins une séquence d’intérêt. Bien entendu par
« ensemble des séquences », il est entendu l’ensemble des séquences que la méthode de séquençage permet d’obtenir. Le point essentiel ici étant qu’il n’y a pas de discrimination de certaines séquences d’intérêt parmi celles trouvées dans l’échantillon, aucune présélection n’est effectuée. L’analyse utilise l’entièreté des données de séquençage.
[97]Selon un mode de réalisation préféré, il est obtenu les séquences nucléotidiques correspondant à au moins une séquence d’intérêt choisie dans le groupe consistant en : un fragment d’un gène exprimant l’ARNr 16S et un fragment d’un gène exprimant l’ARNr 18S.
[98]On entend par « séquençage » tout procédé connu destiné à déterminer la séquence nucléotidique d’un acide nucléique. Parmi ces procédés, le séquençage métagénomique direct dit « shotgun » est préféré, et est notamment décrit dans le document Quince C, et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017 Sep 12;35(9):833-844. Brièvement, ce type de séquençage comprend la fragmentation de l’acide nucléique isolé en fragments dont la taille varie en fonction de la plateforme de séquençage employée (typiquement de 200 à 550 pb en moyenne pour la plateforme Illumina® et de quelques dizaines de bases à > 100 000 pb pour la plateforme Nanopore®), qui sont subséquemment liés à des adaptateurs (spécifiques ici aussi à la plateforme employée) pour la préparation de la librairie de séquençage. Les librairies obtenues sont ensuite séquencées à l’aide d’une plateforme de séquençage haut débit (typiquement Illumina® ou Nanopore®). Les séquences obtenues sont ensuite filtrées de façon à retirer les séquences de mauvaise qualité et les séquences correspondant au génome du sujet, selon des principes bien établis dans le domaine technique. Les séquences filtrées sont ensuite organisées en vue de leur identification, comme vu plus loin en détail.
[99]L’utilisation des données de séquençage d’Illumina® issues d’approches de capture de gènes par hybridation est aussi privilégiée et notamment décrite dans le document Comtet-Marre, Sophie & Chakoory, Oshma & Peyret, Pierre, (2022), Targeted 16S rRNA Gene Capture by Hybridization and Bioinformatic Analysis. Brièvement l’acide nucléique isolé est fragmenté et lié à des adaptateurs de séquençage de manière similaire à la méthode « shotgun ». En parallèle, des sondes oligonucléotidiques, notamment biotinylées, complémentaires des séquences d’intérêt sont synthétisées puis hybridées avec les librairies de séquençage. Les complexes formés sont capturés, notamment à l’aide de billes magnétiques recouvertes de streptavidine, et amplifiés par PCR à l’aide d’amorces complémentaires aux adaptateurs. Les fragments capturés et amplifiés sont séquencés avec une plateforme de séquençage haut débit, puis filtrées, comme décrit précédemment. Les séquences filtrées sont ensuite organisées. Ainsi, dans ce cadre, selon un mode de réalisation particulier de l’invention, ledit procédé comprend une étape préliminaire d’isolement spécifique de l’acide nucléique issu d’une pluralité de microorganismes présent dans ledit échantillon biologique.
[100]Le séquençage peut également être du type « séquençage d’amplicons » ou « metabarcoding » notamment décrit dans le document Durazzi, F., Sala, C., Castellani, G. et al. Comparison between 16S rRNA and shotgun sequencing data for the taxonomie characterization of the gut microbiota. Sci Rep 11 , 3030 (2021). Néanmoins, ce type de séquençage est moins privilégié dans la mesure où il implique une amplification préliminaire par PCR de portions de l’ADNr 16S ou de l’ADNr 18S à l’aide d’amorces, notamment à l’aide d’amorces dites universelles qui peuvent conduire à une surreprésentation biaisée de certains microorganismes ou à l’exclusion de certains microorganismes. L’emploi d’amorces spécifiques de groupes taxonomiques microbiens peuvent également conduire à l’exclusion d’une partie des microorganismes présents dans l’échantillon analysé. Les séquences amplifiées sont liées à des adaptateurs spécifiques pour produire des banques de séquençage et séquencées à l’aide d’une plateforme de séquençage à haut débit, de manière similaire à ce qui est décrit au-dessus.
[101]Par « fragment » d’une séquence nucléotidique, il est entendu un fragment d’au moins 20% de la longueur de cette séquence. Par « un fragment d’au moins 20 % », on entend un fragment d’au moins 20 %, au moins 25 %, au moins 30 %, au moins 35 %, au moins 40 %, au moins 45 %, au moins 50 %, au moins 55 %, au moins 60 %, au moins 65 %, au moins 70 %, au moins 75 %, au moins 80 %, au moins 85 %, au moins 90 %, au moins 95 %, au moins 97 %, au moins 98 %, au moins 99 % ou 100 % de la séquence nucléotidique considérée.
[102]Le fragment d’ADNr 16S et/ou d’ARNr 16S séquencé des microorganismes appartient notamment aux procaryotes. Additionnellement, le fragment d’ADNr 18S et/ou d’ARNr 18S appartient également aux eucaryotes et micro-eucaryotes.
Etape b)
[103]Le but de l’étape b) est d’identifier l’ensemble des microorganismes présents dans l’échantillon à partir du séquençage de l’étape a) ainsi que leur abondance relative, et de fournir des données pertinentes d’entrées au modèle d’intelligence artificielle pour la détermination du diagnostic. Ici encore par « ensemble des microorganismes », il est entendu la totalité des microorganismes identifiables selon la méthode de séquençage employée. Le fait d’identifier l’ensemble des microorganismes présents dans l’échantillon et de fournir cet ensemble (dépourvu des identités absentes du jeu d’entrainement) au modèle d’intelligence artificielle permet de conserver le maximum de variabilité individuelle du sujet ainsi que les interactions microbiennes associées dans le cadre d’une pathologie ou d’un état pathologique déterminé et d’assurer un diagnostic/diagnostic prédictif personnalisé.
[104]A cet effet, selon un mode de réalisation de l’invention, le procédé comprend l’organisation des séquences séquencées pour reconstruire la séquence nucléotidique d’au moins une partie
de gène exprimant l’ARNr 16S et/ou de gène exprimant l’ARNr 18S. En particulier, l’étape b) comprend notamment en premier lieu une étape d’organisation des séquences obtenues à l’étape a) par leur alignement avec des séquences connues de microorganismes présents dans une base de données. Lesdites séquences connues comprennent au moins ladite séquence d’intérêt sélectionnée pour le plus grand nombre de microorganismes connus, afin de déterminer des correspondances directes ou de reconstruire des séquences de nouveaux microorganismes et/ou d’obtenir des séquences plus longues afin d'augmenter la fiabilité de l’identité des microorganismes présents dans l’échantillon biologique du sujet. Dans le cadre du séquençage metabarcoding, l’organisation se fait notamment par correspondance directe. Dans le cadre de la méthode « shotgun » ou de la capture de gènes par hybridation, l’organisation peut se faire par correspondance directe et/ou reconstruction.
[105]L’ensemble déterminé de microorganismes est notamment sélectionné parmi ceux disponibles dans des bases de données en ligne, notamment publiques. Parmi ces bases de données publiques, la base SILVA (https://arb-silva.de). Un autre exemple de bases de données est la base « Greengenes » (https://greengenes.secondgenome.com/). La personne du métier peut ainsi aisément déterminer si une séquence nucléotidique donnée est issue d’un microorganisme connu ou inconnu, ou du sujet humain ou animal.
[106]Ainsi, selon un mode de réalisation particulier, le procédé selon l’invention comprend une étape de reconstruction d’au moins une partie de la séquence du gène exprimant l’ARNr 16S et/ou de la séquence du gène exprimant l’ARNr 18S des microorganismes présents dans l’échantillon biologique. Bien entendu, la longueur reconstructible dépend de la longueur séquencée du fragment de la séquence d’intérêt et de l’effort de séquençage c’est-à-dire du nombre de lectures généré lors du séquençage (profondeur de séquençage).
[107]Plus particulièrement, dans un mode de réalisation particulier, lors de l’étape de reconstruction d’au moins une séquence nucléotidique, au moins 70 % de la longueur du gène exprimant l’ARNr 16S et/ou au moins 70 % de la longueur de l’ARNr 16S est reconstruite. Une augmentation de la taille de la partie reconstruite permet une meilleure finesse dans la détermination de l’identité du microorganisme, permettant d’aller jusqu’au rang taxonomique de l’espèce. La longueur d’un gène d’ADNr 16S étant d’environ 1500 paires de bases en moyenne, une séquence nucléotidique d’au moins 70 % de la longueur du gène comprend environ 1050 paires de bases, en moyenne.
[108]Selon un mode de réalisation de l’invention, il utilise l’ensemble des données métagénomiques du microbiote qui permettent ensuite la reconstruction de séquences d’intérêt complètes et une affiliation précise des microorganismes de la communauté microbienne au niveau du genre ou de l’espèce, voire l’identification de nouveaux microorganismes.
[109]L’étape d’organisation est notamment suivie d’une étape de classification par rangs taxonomiques des correspondances et/ou reconstructions permettant de déterminer l’identité des microorganismes présents dans l’échantillon biologique du sujet.
[110]L’identification peut notamment être complétée par des analyses phylogénétiques afin de situer les nouveaux microorganismes par rapport aux microorganismes connus les plus proches.
[111]Par « détermination de l’identité », on entend l’identification des microorganismes, en suivant une nomenclature, organisée en catégories hiérarchisées (classification par rangs taxonomiques), autrement dit en rangs taxonomiques, ces catégories consistent en l’appartenance au domaine du vivant (rang le moins précis) à la définition de l’espèce (rang le plus précis). Les rangs taxonomiques d’intérêt s’étendent depuis le phylum jusqu’à l’espèce. La classification taxonomique est réalisée par comparaison de chaque séquence d’intérêt reconstruite ou dont la correspondance est attribuée avec, des séquences d’ADNr 16S et/ou des séquences d’ADNr 18S contenues dans des bases de données. Parmi les bases de données publiques utilisables, on peut notamment citer à nouveau la base SILVA. Le rang taxonomique le plus confiant identifiable dépend de plusieurs paramètres dont le type de séquençage, les paramètres du séquençage, l’ensemble déterminé de microorganismes employé pour l’alignement (voir plus bas), etc. L’invention présente ainsi l’avantage de prendre en compte chaque identification déterminée. Il n’y a ainsi aucune présélection réalisée, permettant de préserver toute la diversité de l’échantillon du sujet. Cette exhaustivité participe à l’obtention d’un diagnostic/diagnostic prédictif de plus grande qualité qu’avec les méthodes de l’art antérieur. Selon un mode de réalisation, le même rang taxonomique parmi les rangs taxonomiques d’intérêt est conservé pour l’ensemble des séquences. Selon un mode de réalisation préféré, le rang taxonomique le plus précis parmi les rangs taxonomiques d’intérêt pour chaque séquence est déterminé. Ce second aspect permet une meilleure identification de la diversité microbienne de l’échantillon, et assure un diagnostic plus fiable.
[112]Par « détermination de l’abondance relative », on entend la détermination pour chacun des microorganismes considérés pour le procédé selon l’invention, de l’abondance du microorganisme rapportée à l’abondance totale des microorganismes considérés pour le procédé selon l’invention. La détermination de l’abondance dépend de la méthode de séquençage employée, et est bien connue de l’homme du métier.
Etape c)
[113]Lors de cette étape, un modèle d’intelligence artificielle préalablement entraîné sur la base d’un jeu de données labellisées détermine le diagnostic/diagnostic prédictif sur la base des
données obtenues à l’étape b). Le modèle d’intelligence artificielle peut également prendre en entrée en outre au moins une donnée clinique du sujet, comme il sera vu en détail plus loin.
[114]Le modèle d’intelligence artificielle présente ainsi une structure interne reflétant la relation entre d’une part (1) l’abondance relative des microorganismes au sein de l’échantillon, ainsi qu’optionnellement au moins une donnée clinique du sujet, et d’autre part (2) le diagnostic/diagnostic prédictif de la pathologie ou de l’état pathologique.
[115]Le modèle d’intelligence artificielle est un modèle d’apprentissage supervisé et correspond notamment à un modèle de classification, à un modèle d’apprentissage profond, à un réseau de neurones (en anglais « neural network » ou NN), à un réseau de neurones profonds (en anglais « deep neural network »), à un arbre de décision, à un modèle des K-plus proches voisins (en anglais « k-nearest neighbors » ou KNN), une forêt aléatoire (en anglais « random forest » ou RF), à une classification naïve bayésienne (en anglais « naive bayes » ou NB), à un algorithme « Boosting de gradient extrême» (en anglais Extreme gradient boosting ou XGBoost), à une régression logistique ou encore à une machine à vecteur de support (en anglais « support-vector machine » ou SVM). En particulier, le modèle d’intelligence artificielle est un réseau de neurones profonds avec une couche d’entrée composé de neurones équivalent au nombre de caractéristiques dans les données d’entrainement, suivi d’une ou plusieurs couches cachées et une couche de sortie qui donne le résultat du diagnostic/diagnostic prédictif.
Entrainement
[116]Par « préalablement entraîné » on entend un processus permettant au modèle d’intelligence artificielle d’apprendre à partir d’un jeu de données d’entrainement labellisées à associer de manière pondérée l’identité et l’abondance de microorganismes présents dans des échantillons de sujets, et optionnellement au moins une donnée clinique de ces sujets, au diagnostic/diagnostic prédictif correspondant.
[117]L’invention concerne ainsi également un procédé d’entrainement d’un modèle d’intelligence artificielle destiné à obtenir un diagnostic ou un diagnostic prédictif, ledit procédé utilisant un jeu de données labellisées.
[118]Le jeu de données labellisé ou jeu d’entrainement comprend des profils de sujets d’entrainement. Les sujets d’entrainements appartiennent à la même espèce que le sujet dont le ou les échantillons sont analysées dans le procédé de l’invention. De manière à renforcer l’entrainement, les sujets d’entrainements proviennent de manière avantageuse de divers nations, et notamment de divers continents. Une parité entre les types de sexe des sujets dans le jeu d’entrainement est également avantageux, en fonction bien entendu de la pathologie ou
de l’état pathologique considéré. Ces différents aspects permettent d’obtenir une meilleure représentativité des microbiotes de sujet . En effet, contrairement à l’art antérieur qui se focalise sur une restriction des microorganismes analysés, le principe de l’invention est de conserver toute la diversité du microbiote de chacun des sujets d’entrainement, pour que le modèle d’intelligence artificielle puisse déterminer l’ensemble des relations possibles, indépendamment de tout biais introduit par les connaissances à un instant déterminé. Contrairement à ce qui pouvait être attendu avec des données d’entrées aussi complexes, les résultats obtenus suite à l’entrainement donne une excellente justesse de prédiction de diagnostic/diagnostic prédictif de l’état physiologique ou pathologique pour lequel le modèle d’intelligence artificielle a été entraîné. Les inventeurs ont ainsi pu montrer que des microorganismes avec une abondance relative très faible, généralement exclus de l’entrainement pour cette raison, se sont avérés très pertinents pour déterminer le diagnostic prédictif de pathologies et états pathologiques. Ce que l’on pouvait considéré comme du bruit précédemment, est démontré ici comme point discriminant.
[119]Les sujets d’entrainement peuvent notamment être spécifiquement recrutés pour cet objectif, ou bien être issus d’une ou plusieurs bases de données, en particulier publiques, et plus particulièrement des bases de données de cohortes de sujets les plus exhaustives et diversifiées à disposition. Ces bases de données comprennent notamment des données de séquençage brutes issues d’un ou plusieurs échantillons de chaque sujet, et optionnellement au moins une donnée clinique de chaque sujet.
[120]Les sujets d’entrainements sont notamment dissociés en deux groupes, à savoir un groupe d’entrainement et un groupe de test. Le groupe d’entrainement permet de former le modèle d’intelligence artificielle, et le groupe de test permet de qualifier ses performances. Typiquement, le groupe d’entrainement représente 80% de l’ensemble des sujets d’entrainement, et le groupe de test 20%.
[121]Les profils de sujets d’entrainement comprennent chacun l’identité et l’abondance relative des microorganismes identifiés présents dans au moins un échantillon du sujet d’entrainement, ainsi qu’optionnellement au moins une donnée clinique du sujet d’entrainement. Les abondances relatives sont notamment obtenues par la mise en œuvre des étapes a) et b) décrites ci-dessus sur des échantillons de sujets, ou de l’unique étape b) sur des données de séquençage d’échantillons de sujets. Les identités des microorganismes (et donc leur abondance) peuvent être restreintes pour l’entrainement à un même rang taxonomique donné de sorte que l’ensemble des microorganismes sont identifiés au niveau du même rang, partant du phylum et jusqu’à l’espèce. Mais aucune présélection n’est réalisée sur les microorganismes identifiés, notamment sur la base de leur abondance relative et/ou de leur implication connue dans le diagnostic ou le diagnostic prédictif. Selon un mode de réalisation
préféré, aucune restriction quant au rang taxonomique n’est réalisée, et le rang taxonomique le plus confiant est conservé pour toutes les identités.
[122]Lorsque le procédé de l’invention est destiné au diagnostic d’un accouchement précoce de la femme enceinte, le ou les échantillons de chaque sujet d’entrainement sont notamment prélevés au cours du même trimestre, et typiquement au cours du 1er, 2ème ou 3ème trimestre, voire du même mois.
[123]La supervision de l’apprentissage est réalisée par la labellisation des profils de sujets d’entrainement avec leur phénotype. Les sujets sont classés en au moins deux phénotypes, et de préférence en deux phénotypes antinomiques. Dans le cadre d’un diagnostic, les phénotypes des sujets sont notamment atteint/non atteint de la pathologie/de l’état pathologique. Concernant le diagnostic prédictif, les phénotypes des sujets sont notamment avec apparition/sans apparition de la pathologie ou de l’état pathologique. De manière avantageuse, le jeu d’entrainement comprend un nombre équilibré de chaque phénotype, ou bien une plus grande proportion de phénotype positif.
[124]Les données des sujets d’entrainement sont notamment normalisées. Cette normalisation est en particulier du type min-max sur l’ensemble du jeu d’entrainement. Ce type de normalisation correspond à une transformation linéaire des caractéristiques dans une plage uniforme, tout en conservant tous les rapports de distance de la donnée d'origine. Cela est réalisé pour éviter que les valeurs numériques des caractéristiques (abondances des microorganismes) plus grandes ne surpassent celles des caractéristiques numériques plus petites, minimisant ainsi le biais dans la discrimination des états pathologiques. L'objectif principal est d'assurer la comparabilité des données à travers les échantillons microbiens ou les groupes d'échantillons, tels que ceux classés comme malades ou sains. En effet, la grande variabilité des tailles de des bases de données et de la profondeur de séquençage induit de fortes dépendances parmi les abondances des différents taxons. Ainsi, la normalisation des données garantit que toutes les caractéristiques (taxons) dans les données contribuent de manière égale au processus d'apprentissage, bien que toutes les caractéristiques ne soient pas également importantes pour la décision de classification.
[125]Lorsqu’au moins une donnée clinique est employée dans les données d’entrée en sus des données relatives aux microorganismes, elle est bien entendue pertinente vis-à-vis de la pathologie ou de l’état pathologique pour lequel le diagnostic/diagnostic prédictif est réalisé. Par « au moins une donnée clinique » on entend une, deux, trois, quatre, cinq, six, sept, huit, neuf, dix ou plus de dix données cliniques caractéristiques du sujet.
[126]Notamment, dans le cas d’une pathologie du nouveau-né, les données cliniques peuvent appartenir au sujet lui-même ou bien à sa mère. Dans ce cadre, peut être notamment utilisée
au moins une des données ci-dessous:
- l’âge réel du sujet auquel le prélèvement a été effectué, en nombre de jours de vie
- le poids à la naissance du sujet,
- l’âge gestationnel de l’enfant à la naissance,
- le mode de naissance (voie basse ou césarienne) du sujet,
- le genre du sujet (masculin, féminin),
- le dosage de composants ou marqueurs sanguins du sujet ou de la mère,
- le dosage de composants ou marqueurs fécaux du sujet ou de la mère,
- la présence d’au moins une autre pathologie chez le sujet ou bien la mère,
- l’administration d’un traitement médical au sujet ou bien à la mère,
- l’ethnie/la nationalité de la mère,
- l’alimentation de la mère et/ou du nouveau-né,
- le mode de vie de la mère (activité physique, consommation d’alcool, de tabac, de drogues, etc.).
[127]Par « ethnie », on entend un groupe de personnes que rapprochent un certain nombre de caractères. Dans un procédé selon l’invention, la caractéristique « ethnie » est notamment choisie dans le groupe constitué par : « Africain-Américain », « Américain-Indien », « Noir », « Blanc », « Caucasien », « Hispanique », « Asiatique », « Multi-ethnie ».
[128]Lorsque des données cliniques sont utilisées, ces dernières sont notamment encodées de la manière suivante : les données catégorielles (comme par exemple le genre et le mode de naissance dans le cas des nouveau-nés) sont converties en vecteurs en utilisant un « encodage 1 parmi n » (en anglais one-hot encoding), c'est-à-dire que tous les éléments du vecteur sont convertis en 0 sauf la variable catégorique qui est convertie en 1 . Les données à valeurs continues (âge réel, poids à la naissance et l’âge gestationnel dans le cas des nouveau-nés) sont transformées en une variable discrète en créant un ensemble d'intervalles contigus (« bin » en anglais) qui couvrent la plage des valeurs de la variable. La donnée clinique « jour de vie » est discrétisée en intervalles avec un pas croissant de 9 (de 0 à 99 jours) et 99 (100 à 499 jours). Un pas de temps de 1 pourrait aussi être considéré sur les 3 premières semaines de vie où apparait le plus fréquemment la pathologie. La donnée clinique « poids » est discrétisée en intervalles avec un pas croissant de 99 (de 500 à 2899 grammes). Le poids des enfants pourra aussi être suivi si nécessaire par intervalle de 9 tout au long des 3 premières semaines de vie jusqu’à l’apparition éventuelle de la pathologie. L'âge gestationnel à la naissance peut être converti en facteurs en raison du nombre limité de valeurs.
[129]Dans le cas du diagnostic d’un accouchement prématuré, ladite donnée clinique est notamment choisie parmi :
- la durée de gestation,
- l’âge de la femme enceinte,
- l’ethnie de la femme enceinte, et
- une combinaison de ces données cliniques.
La durée de la gestation peut notamment être exprimée en nombre de semaine de gestation ou désignée par la période à laquelle est réalisé le prélèvement de l’échantillon biologique.
[130]Cette période est notamment choisie parmi : le premier trimestre de grossesse, le deuxième trimestre de grossesse, le troisième trimestre de grossesse.
[131]L’âge de la femme enceinte, dans un procédé selon l’invention, peut être défini en nombre d’années ou par son appartenance à une tranche d’âge. Plus particulièrement, l’âge de la femme enceinte peut être attribué à l’un des deux groupes suivants : « inférieur à 35 ans » et « égal ou supérieur à 35 ans ».
[132]En amont de l’apprentissage, l’ensemble des microorganismes présents dans chaque profil de sujet d’entrainement est compilé, de sorte à déterminer le nombre d’entrées d’abondance d’identités de microorganismes du modèle d’intelligence artificielle. Selon un mode de réalisation, le modèle d’intelligence artificielle comprend au moins 500 entrées d’abondance d’identités de microorganismes, notamment au moins 600 entrées, en particulier au moins 700 entrées, notamment au moins 1000 entrées, particulièrement au moins 1300 entrées.
[133]Selon un mode de réalisation, le modèle d’intelligence artificielle comprend au moins 10 entrées de données cliniques déterminées, notamment au moins 20, particulièrement au moins 30, en particulier au moins 40.
[134]Selon un mode de réalisation de l’invention, le procédé étant destiné au diagnostic prédictif de l’accouchement précoce chez une femme enceinte, le modèle d’intelligence artificielle comprend au moins 600 entrées d’abondance d’identités de microorganismes et optionnellement au moins 10, notamment au moins 15, entrées de données cliniques déterminées.
[135]Selon un mode de réalisation de l’invention, le procédé étant destiné au diagnostic prédictif de l’ECUN, le modèle d’intelligence artificielle comprend au moins 1000, notamment au moins 1300, entrées d’abondance d’identités de microorganismes et optionnellement au moins 40, notamment au moins 45, entrées de données cliniques déterminées.
[136]Selon un mode de réalisation de l’invention, le procédé étant destiné au diagnostic prédictif de du diabète de type I, le modèle d’intelligence artificielle comprend au moins 1000, notamment au moins 1300, entrées d’abondance d’identités de microorganismes et optionnellement au moins 40 entrées de données cliniques déterminées.
[137]Selon un mode de réalisation de l’invention, le procédé étant destiné au diagnostic prédictif de du sepsis, le modèle d’intelligence artificielle comprend au moins 600, notamment au moins 1300, entrées d’abondance de microorganismes et optionnellement au moins 40 entrées de données cliniques déterminées.
Signatures de microorqanismes issues de l’entrainement
[138]Le procédé d’entrainement selon l’invention permet de mettre en évidence différentes signatures de microorganismes caractéristiques d’un diagnostic/diagnostic prédictif positif (ci- après « premières signatures ») ou négatif (ci-après « secondes signatures). Par « signature », on entend un ensemble d’identités de microorganismes. Ce procédé permet en outre la découverte de nouveaux microorganismes.
[139]Selon cet aspect de l’invention, une première signature de microorganismes associée à un diagnostic d’apparition et/ou de développement d’ECUN, notamment obtenue par un procédé selon l’invention, se caractérise notamment par la présence de microorganismes de l’espèce :
- Enterobacter non classées,
- Enterobacteriaceae non classées,
- Enterococcus faecalis,
- Klebsiella non classées,
- Haemophilus parainfluenzae,
- Enterococcus durans et
- Enterobacter cancerogenus.
[140]Ces microorganismes ont en effet été retrouvés, notamment en plus grande quantité, dans les échantillons biologiques statistiquement associés au diagnostic de présence d’ECUN (c’est-à- dire avec une probabilité de plus 50%).
[141]Une première signature associée à une probabilité élevée d’accouchement prématuré, notamment obtenue par un procédé selon l’invention, se caractérise notamment par la présence de microorganismes du genre :
- Anaerococcus,
- Peptoniphilus,
- Prevotella, en particulier Prevotella bivia,
- Gardnerella en particulier Gardnerella vaginalis,
- Sneathia en particulier S neat hi a amnii.
[142]En effet, ces microorganismes ont été découverts comme présents ou présents en plus grande quantité dans les échantillons biologiques statistiquement associés à une probabilité élevée d’accouchement prématuré (plus de 50%).
[143]Selon cet aspect, une seconde signature associée à une pluralité de microorganismes statistiquement associée à un diagnostic d’absence d’ECUN, notamment obtenue par un procédé selon l’invention, est caractérisée notamment par la présence de microorganismes de plusieurs espèces de Lactobacillus associées à des cas non-ECUN. En effet, ces microorganismes ont été découverts comme présents ou présents en plus grande quantité dans les échantillons biologiques statistiquement associés à une prédiction d’absence d’ECUN. La seconde signature associée à un diagnostic d’absence d’ECUN peut comprendre d’autres microorganismes, tels que : les genres Bifidobacterium, Bacteroides, les espèces Bifidobacterium longum, , Bacteroides fragilis, Lactobacillus casei.
[144]Une seconde signature associée à une probabilité élevée d’accouchement à terme (plus de 70%), notamment obtenue par un procédé selon l’invention, est caractérisé notamment par la présence de microorganismes de la famille Christensenellaceae et du genre :
- Bacteroides, ou
- Lactobacillus, en particulier Lactobacillus crispatus.
[145]En effet, ces microorganismes ont été découverts présents ou présents en plus grande quantité dans les échantillons biologiques statistiquement associés à une probabilité élevée d’accouchement à terme.
Diagnostic et Diagnostic prédictif
[146]Le diagnostic/diagnostic prédictif est déterminée à partir des identités et des abondances de microorganismes déterminés lors de l’étape b). De ces données obtenues à l’étape b) ne sont épurées celles des microorganismes absents du jeu d’entrainement. En ce sens, plus le jeu d’entrainement est conséquent, plus il y a de chances qu’il soit exhaustif, et qu’aucune épuration ne soit réalisée dans les données obtenues à l’étape b). Néanmoins, dans l’éventualité où un échantillon d’un sujet était découvert comme comprenant une identité de microorganisme qui n’était pas présente dans le jeu d’entrainement, il est possible a posteriori de ré-entrainer le modèle d’intelligence artificielle avec cette nouvelle entrée. Il est ainsi possible d’obtenir un enrichissement continu du modèle d’intelligence artificielle, et donc une justesse continuellement améliorée des prédictions.
[147]Les données conservées de l’étape b) suite à l’exclusion des microorganismes absents du jeu de données d’entrainement sont notamment normalisées. Cette normalisation est en particulier du type min-max sur la base du jeu d’entrainement.
[148]Le diagnostic/diagnostic obtenu lors de l’étape c) peut notamment être associé à une certitude/un indice de confiance, allant typiquement de 0 à 1 , reflétant la probabilité de correspondance. Ainsi, le modèle d’intelligence artificielle peut déterminer un diagnostic positif
d’une pathologie avec un indice de confiance de 0,8, indiquant qu’il y a 80% de chance que le microbiote analysé soit associé à cette pathologie. Dans le sens contraire, le modèle d’intelligence artificielle peut déterminer un diagnostic négatif avec un indice de confiance de 0,8, indiquant qu’il y a 80% de chance que le microbiote analysé ne soit associé à la pathologie et donc 20% de chance qu’il le soit.
Etape d)
[149]Le procédé selon l’invention peut comprendre une étape d) de compilation de plusieurs diagnostic/diagnostic prédictif pour une détermination finale du diagnostic/diagnostic prédictif.
[150]Selon un mode de réalisation particulier de l’invention, aux moins deux échantillons biologiques du sujet sont utilisés, en particulier au moins trois. Par « au moins deux échantillons biologiques » on entend deux, trois, quatre, cinq, six, sept, huit, neuf, dix ou plus de dix échantillons biologiques provenant du même sujet. Les échantillons peuvent être prélevés à un même moment, ou bien à des temporalités différentes.
[151]Selon un mode de réalisation de l’invention, lorsque plusieurs échantillons biologiques sont utilisés pour un même sujet, les étapes a) à c) sont réalisées sur chaque échantillon, de sorte que l’étape d) comprend la compilation du diagnostic/diagnostic prédictif obtenu à l’étape c) pour chaque échantillon et la détermination finale du diagnostic/diagnostic prédictif. Ainsi, le diagnostic/diagnostic prédictif peut être considéré comme positif/négatif si plus de 50 % du résultat des étape c) correspondent à cet état.
[152]Selon un mode de réalisation de l’invention, lorsque les échantillons sont prélevés à un même moment, l’étape d) permet de renforcer un premier diagnostic déterminé à la première étape c), afin notamment de pallier une potentielle sélection des microorganismes non souhaitée par le choix de la zone de prélèvement dans une région de prélèvement. Ainsi, les échantillons sont notamment prélevés dans des zones différentes d’une même région de prélèvement, afin de s’assurer de l’exhaustivité de la représentation des microorganismes dans la région du prélèvement du sujet.
[153]Selon un mode de réalisation de l’invention, lorsque les échantillons sont prélevés à des temporalités différentes, l’étape d) permet d’obtenir un suivi des modifications du microbiote du sujet et notamment le changement de son phénotype (de malade à sain suite à un traitement, ou bien de sain à malade), permettant à un clinicien de confirmer un effet curatif ou de prendre les mesures nécessaires à l’apparition d’une pathologie ou d’un état pathologique.
Légende des figures
[154]La présente invention est expliquée davantage par les figures et les exemples ci-après.
[155]La figure 1 montre une vue d’ensemble des étapes suivies pour un mode de réalisation du procédé de diagnostic d’une pathologie selon l’invention à partir de l’identification des microorganismes et de leur abondance dans l’échantillon d’un sujet, suivi par une étape de prédiction du diagnostic/diagnostic prédictif en utilisant le modèle de DNN entrainé et optimisé.
[156]La figure 2 illustre les étapes d’un exemple d’entraînement d’un modèle de réseau de neurones profonds selon l’invention et le réglage de ses hyperparamètres permettant l’optimisation de la prédiction du diagnostic/diagnostic prédictif.
[157]La figure 3 illustre les performances de prédiction obtenues par le modèle de réseau de neurones profonds en fonction des données d’entrée fournies. Les données d’entrée sont les données issues du séquençage métagénomique direct (cohorte Fettweis) traitées par RiboTaxa ou par MetaPhlAn3. MetaPhlAn3 utilise les lectures de haute qualité issues de séquençage métagénomique direct pour les comparer à une base de génomes références de microorganismes accessible à l’adresse : segatalab.cibio.unitn.it/data/Pasolli_et_al.html et déterminer la composition taxonomique du microbiote analysé (du domaine jusqu’à l’espèce) et les abondances relatives des microorganismes identifiés (fichier TSV).
[158]La figure 4 représente la performance de modèles de réseaux de neurones profonds entrainés sur des données issues de séquençage métagénomique direct et de metabarcoding au niveau du genre.
[159]La figure 5 représente la structure finale d’un modèle d’intelligence artificielle (modèle de réseau de neurones profonds entrainé) selon l’invention optimisé pour prédire l’ECUN.
[160]La figure 6 représente le taux de vrais positifs (en ordonnées) en fonction du taux de faux positifs (en abscisses) dans le cadre de la prédiction de la survenue de l’ECUN, où l’AUC est égale à 0,987.
[161] La figure 7 représente la précision (en ordonnées) en fonction de la sensibilité (en abscisses) dans le cadre de la prédiction de la survenue de l’ECUN, où l’AUC est égale à 0,992.
[162]La figure 8 représente les 20 caractéristiques d’entrée du modèle de réseau de neurones profonds entrainé contribuant le plus à la prédiction des phénotypes ECU N ou non-ECU N résumées par l'explicateur SHAP.
[163]La figure 9 illustre l’analyse du suivi longitudinal d’échantillons suite à la prédiction du modèle de réseau de neurones profonds entrainé dans le cadre de la prédiction de la survenue de l’ECUN. Le cercle non étiqueté à gauche représente le phénotype réel du nourrisson. Les échantillons des nourrissons sans pathologie sont indiqués en gris foncé et les échantillons des nourrissons ECU N en gris clair. Chaque cercle étiqueté représente un échantillon collecté chez chacun des nourrissons et les nombres à l’intérieur des cercles correspondent au jour du prélèvement (en jours de vie). La couleur de ces cercles représente le phénotype prédit par le réseau de neurones selon le même code couleur que les cercles non étiquetés. Le carré simple représente les échantillons qui ont été reclassés dans le groupe « contrôle » et le double carré représente les échantillons qui ont été reclassés dans le groupe « ECUN ».
[164]Les figures 10 et 11 représentent des exemples de graphiques SHAP illustrant les caractéristiques (micro-organismes) les plus importants qui influencent la prédiction vers le phénotype contrôle dans la cohorte de CORTECs. Pour chaque caractéristique, les valeurs négatives associées aux flèches correspondent aux valeurs SHAP associées à une contribution vers la prédiction du phénotype contrôle (f(x)=0). Le libellé à côté de chaque caractéristique (micro-organisme) représente son abondance dans l'échantillon.
[165]Les figures 12 et 13 représentent des exemples de graphiques SHAP illustrant les caractéristiques (micro-organismes) les plus importants qui influencent la prédiction vers l’ECUN dans la cohorte de CORTECs. Pour chaque caractéristique, les valeurs positives associées aux flèches correspondent aux valeurs SHAP associées à une contribution vers la prédiction du phénotype ECUN (f(x)=1). Le libellé à côté de chaque caractéristique (microorganisme) représente son abondance dans l'échantillon.
[166]La figure 14 représente les 20 caractéristiques d’entrée du modèle de réseau de neurones profonds entrainé contribuant le plus à la prédiction des phénotypes DT1 ou non-DT1 résumées par l'explicateur SHAP.
[167]La figure 15 représente l’approche d'analyse longitudinale des prédictions réalisées sur l’ensemble des échantillons d’enfants qui avaient au moins 3 échantillons dans l’ensemble test « sepsis ». Le phénotype final de l’enfant est déterminé par le groupe phénotypique ayant le plus grand nombre d'échantillons d’un même état.
[168]La figure 16 représente les 20 caractéristiques d’entrée du modèle de réseau de neurones profonds entrainé contribue le plus à la prédiction des phénotypes sepsis résumées par l'explicateur SHAP.
Exemples
Recueil des données d’entrainement
[169]Les inventeurs ont collecté des données brutes de séquençage de microbiotes et les données cliniques associées de cohortes de patients constituées dans le cadre d’études de différentes pathologies et états pathologiques : accouchement prématuré (AP), entérocolite ulcéro- nécrosante (ECUN), sepsis et diabète de type 1 (DT1).
[170]La première étape a consisté à sélectionner des publications scientifiques pertinentes ayant mis à disposition ces données. Une recherche par mots clés précis a été effectuée dans les bases de données de publications PubMed et Google Scholar. Les données de séquençage des microbiotes devaient avoir été obtenues par séquençage métagénomique direct, dit « shotgun ». Seules les études prospectives avec prélèvements avant le déclenchement de la pathologie ou de l'état pathologique, permettant un diagnostic prédictif, ont été retenues. De plus, l'inclusion de sujets contrôles était requise.
Traitement bioinformatique des données de séquençage métagénomique shotgun
[171]Les données de métagénomique « shotgun » ont été traitées avec le chainage bioinformatique RiboTaxa (Chakoory et al., 2022) afin obtenir les profils taxonomiques des microbiotes (identification des microorganismes à tous les rangs taxonomiques et abondances relatives associées). L’approche de RiboTaxa consiste en la reconstruction des séquences d'ADNr 16S et 18S à l'aide de bases de données de référence, ici, la base de données SILVA SSU 138.1 NR99 (Quast ét al., 2013), permettant ensuite une identification des microorganismes jusqu’au niveau de l’espèce. RiboTaxa effectue le contrôle qualité des lectures brutes, la reconstruction des séquences d’ADNr 16 et 18S, la détermination de leur abondance relative et de l’identité des microorganismes.
[172]Pour chaque échantillon, les lectures brutes ont été fournies comme entrée dans RiboTaxa. Les lectures ont été traitées pour supprimer les adaptateurs Illumina, les artefacts Illumina connus et pour couper les extrémités des lectures lorsque le score de qualité des bases se trouvaient en dessous de Q20. Les lectures résultantes contenant plus d'un « N », ou avec des scores de qualité inférieurs à 20 en moyenne sur la lecture, ou une longueur inférieure à 60 pb, ont été rejetées.
[173]Les lectures de haute qualité ont ensuite été assemblées en séquence d’ADNr 16S et 18S complètes à presque complètes à l’aide de deux assembleurs inclus dans RiboTaxa. MetaRib (Xue ét al., 2020) prend en entrée l’ensemble des lectures de haute qualité tandis qu’EMIRGE (Miller et al., 2011) utilise uniquement les lectures correspondant à de l’ADNr 16S et 18S
filtrées avec SortMeRNA (Kopylova et al., 2012). La double approche de reconstruction (EMIRGE et MetaRib) permet de maximiser la reconstruction des gènes exprimant l’ARNr 16S/18S et de décrire le plus précisément la structure des microbiotes. Bien que les deux assembleurs (EMIRGE et MetaRib) nécessitent une base de données de référence (ici SILVA, qui est la plus complète et de haute qualité), il est possible de reconstruire des séquences très distantes des séquences de référence, ce qui permet ainsi d’identifier de nouveaux microorganismes qui ne seraient pas identifiés par les autres approches (PCR quantitative, analyses classiques de données métagénomiques, amplification par PCR d’une portion du gène exprimant l’ARNr 16S puis séquençage).
[174]Pour la reconstruction du gène exprimant l’ARNr 16S/18S, les paramètres par défaut ont été utilisés, à l'exception des paramètres qui dépendent exclusivement de la longueur de séquençage des données d'entrée :
- le paramètre A « max_read_length » représente la taille de lecture la plus longue de l'ensemble de données d'entrée,
- le paramètre B « insert_mean » représente la taille moyenne des inserts des lectures pairées et
- le paramètre C « insert_stddev » représente l’écart-type de la distribution de taille des inserts des lectures pairées.
Les paramètres B et C ont été estimés à l'aide du script « mean_size.py », accessible à l’adresse : gist.github.com/timoast/af73c0e9fac00187ee49.
[175]Les séquences d’ADNr 16S et 18S reconstruites ont ensuite été regroupées avec un seuil d’identité de 97% puis classées à différents rangs taxonomiques, du domaine à l'espèce, en utilisant la base de données SILVA. Après avoir éliminé l’ADNr 18S humain considéré comme contaminant, les abondances relatives ont été calculées par RiboTaxa.
[176]Tous les tableaux de taxonomie obtenus ont été regroupés en un seul tableau contenant tous les profils au niveau du phylum, classe, ordre, famille, genre et de l’espèce à l'aide du script RiboTaxa_group_taxonomy.sh de RiboTaxa.
[177]Pour l’entrainement du modèle d’intelligence artificielle ci-dessous, tous les microorganismes identifiés dans tous les échantillons ont été conservés, au lieu d'appliquer une sélection avant l'entraînement, afin de conserver la diversité microbienne et les interactions microbiennes inter-individuelles.
Modèle d’intelligence artificielle
[178]Pour le diagnostic prédictif de chaque pathologies/états pathologiques présentés en exemples ci-dessous, un modèle de réseau de neurones profonds entièrement connectés, correspondant au précédemment décrit « produit programme d’ordinateur », a été implémenté et optimisé sur une même stratégie, en utilisant le langage de programmation Python et des librairies dédiées telles que scikit-learn, Tensorflow (https://tensorflow.org), Keras (https://github.com/keras-team/keras-tuner) et Adam (Kingma and Ba, 2017).
[179]L’architecture du réseau de neurones profonds se compose d’une couche d’entrée dont le nombre de neurones dépend du nombre de caractéristiques d’entrée (nombre de microorganismes identifiés et nombre et nature des données cliniques), des couches cachées dont leur nombre et le nombre de neurones correspondants sont déterminés lors de l’entrainement et l’optimisation du modèle, et une couche de sortie contenant 2 neurones, l’un pour une sortie « pathologie/état pathologique », l’autre pour une sortie « pas de pathologie/état pathologique ».
[180]Afin d’obtenir le modèle le plus performant possible, différentes fonctions mathématiques ont été sélectionnées et les valeurs des hyperparamètres du réseau de neurones profonds ont été optimisées en fonction des données d’entrainement obtenues pour chacune des pathologies.
[181] La fonction d'activation d’unité linéaire rectifiée (ReLLI) a été utilisée pour toutes les couches cachées. Les fonctions d'activation jouent un rôle important dans l’entraînement des réseaux de neurones en apportant la non-linéarité nécessaire au modèle pour apprendre des représentations complexes. La technique d'abandon des neurones sur chaque couche cachée a également été employée afin d'atténuer le surapprentissage du réseau de neurones, phénomène conduisant à une mauvaise généralisation du modèle et à des performances réduites sur de nouvelles données. L'abandon des neurones (ou dropout en anglais) est une méthode d'apprentissage qui implique la suppression aléatoire de neurones pendant l’entrainement du modèle, les nœuds supprimés étant exclus des étapes suivantes. La fonction d'activation de la couche de sortie utilise la fonction Softmax pour attribuer une valeur basée sur une probabilité comprise entre 0 et 1 à chaque classe (pathologie/état pathologique, pas de pathologie/état pathologique). Cette valeur permet au modèle de prendre une décision ‘risque de pathologie’ ou ‘pas de risque de pathologie’.
[182]Différentes valeurs d’autres hyperparamètres ont été testées. Le nombre d’epochs (nombre de fois où le jeu de données complet est propagé dans le réseau de neurones) a varié de 1 à 40. La perte d’entropie croisée entre la valeur cible et la valeur prédite a été optimisée au cours des epochs avec des taux d'apprentissage, allant de 0,0001 à 0,01. Le nombre de couches cachées a varié de 1 à 3 et le nombre de neurones dans la première couche cachée de 32 à
512 avec un pas croissant de 32. Pour faciliter la convergence du modèle, le nombre de neurones dans les couches cachées était défini à la moitié de celui de la couche précédente. Ces optimisations ont été implémentées à l'aide de Keras (https://github.com/keras- team/keras-tuner).
[183]Pour définir la meilleure combinaison d’hyperparamètres, l'ensemble des données d’entrainement a été divisé avec un rapport 8:2 pour obtenir 80% de données d'apprentissage et 20 % de données test. Une validation croisée K-Fold a été appliquée avec les données d’apprentissage (Figure 2). Celles-ci ont été divisées en K sous-ensembles de taille presque égale ; K-1 sous-ensembles étant utilisés pour l’entrainement du modèle et le sous-ensemble restant pour la validation du modèle produit. De cette manière K modèles ont été construits, avec à chaque fois une redistribution des K sous-ensembles et la définition de nouveaux hyperparamètres. La meilleure combinaison d’hyperparamètres pour chaque modèle a été sélectionnée en faisant la moyenne de la métrique de précision des K modèles. Le modèle optimisé a ensuite été entrainé en un modèle de classification final en utilisant l'ensemble des données d'apprentissage et testé sur les données test.
[184]Les performances du modèle de réseau de neurones profonds optimisé ont été estimées sur les données test (20% de l'ensemble des données) en comparant le phénotype prédit par le modèle et le phénotype observé chez le sujet. Par exemple, si le modèle classe correctement un échantillon provenant d’un sujet atteint d’une pathologie ou d’un état pathologique, il est considéré comme un vrai positif (TP pour True Positive), sinon il s'agit d'un faux négatif (FN pour False Negative). En revanche, si le modèle classe correctement un échantillon provenant d’un sujet non atteint d’une pathologie ou d’un état pathologique, il est considéré comme un vrai négatif (TN pour True Negative), sinon il s'agit d'un faux positif (FP pour False Positive). En raison du déséquilibre de classe (les échantillons provenant de sujets atteints par la pathologie ou l’état pathologique sont en général moins abondants dans les jeux de données), les performances du modèle ont été mesurées grâce à plusieurs métriques : l’exactitude (accuracy en anglais ; nombre total de prédictions justes sur le nombre total de sujets), la sensibilité (taux de sujets atteints de la pathologie correctement prédits par le modèle ou taux de vrais positifs), la spécificité (taux de sujets non atteints de la pathologie correctement prédits par le modèle ou taux de vrais négatifs), l'aire sous la courbe (AUC pour Area Under the Curve) de la caractéristique de fonctionnement du récepteur (ROC pour Receiver Operating Characteristic) /AUROC, et l’AUC de précision-sensibilité (PR-AUC pour Precision-Recall AUC).
[185]L’exactitude est calculée comme suit :
TP + TN
Exactitude =
TP + FP + TN + FN
[186]La sensibilité est calculée comme suit :
Sensibilité

[187]La spécificité est calculée comme suit : Spécificité

[188]Enfin, l’AUROC correspond à l’aire sous la courbe ROC qui montre la sensibilité (taux de vrais positifs) en fonction de la spécificité (taux de vrais négatifs). Le PR-AIIC mesure la sensibilité sur la précision (rapport des TP au nombre total de TP et FP). Les AUC ont été calculées à l'aide du package scikit-learn (Pedregosa et al., 2011) et tracées à l'aide de matplotlib (Hunter, 2007) (v3.1). Les intervalles de confiance (IC) à 95 % des AUC ont été estimés à l'aide de la méthode bootstrap (Efron and Tibshirani, 1994) avec 1 000 itérations. Les courbes ROC et le tracé de Sankey ont été générés respectivement à l'aide de matplotlib et de plotly (v5.15.0).
[189]Une approche SHAP (SHapley Additive exPlanations), a été exploitée pour expliquer le résultat de tout modèle d'apprentissage automatique. Les modèles peuvent être interprétés en calculant l'importance des données d’entrée liées aux performances de classification du modèle. L'importance des éléments d’entrée (métadonnées, microorganismes) a été calculée à l'aide de SHAP. La fonction DeepExplainer de SHAP est une méthode permettant de décomposer la sortie d'un réseau de neurones profonds (prédiction) en attribuant des valeurs de contributions à chaque donnée de l’entrée du réseau de neurones. Cette fonction permet de mettre en évidence les données d’entrée ayant le plus de poids dans la prédiction d’un phénotype.
Normalisation et vectorisation des données d’entrainement
[190]Les abondances relatives ont ensuite été normalisées pour éviter l'influence de taxons très abondants via la transformation ci-dessous, appelée normalisation min-max :
[191]
X — (x - Xmin) / (Xmax ~ Xmin)
[192]où : x est la donnée originale, x' est la donnée normalisée. xmin et xmax sont respectivement les valeurs minimale et maximale de la valeur d'origine (abondance). L'équation ci-dessus est une transformation linéaire qui conserve tous les ratios d'abondance des données d'origine après normalisation.
[193]Par ailleurs, une ou plusieurs données cliniques ont été utilisées en fonction de la pathologie ou de l’état pathologique pour lequel un diagnostic ou un diagnostic prédictif était réalisé.
[194]Les données cliniques étaient soit des variables discrètes ou continues. Pour mieux gérer les données, les variables continues ont été transformées en valeurs discrètes par une étape de discrétisation. Ce processus consiste à transformer une variable à valeur continue en une variable discrète en créant un ensemble d'intervalles (ou compartiments) contigus qui s'étendent sur la plage des valeurs de la variable. Le regroupement des caractéristiques numériques en groupes basés sur des intervalles est bénéfique pour la classification et peut améliorer considérablement les performances du modèle.
[195]L'étape suivante consistait à appliquer une technique d’encodage 1 parmi n (en anglais one- hot encoding) sur toutes les données discrètes à l'aide de LabelEncoder de la librairie scikit- learn (Pedregosa et al., 2011). Ainsi, les valeurs discrètes ont été vectorisées, c'est-à-dire que tous les éléments du vecteur ont été convertis en 0 sauf la variable catégorielle, qui a été convertie en 1.
[196]Pour chacun des jeux de données, un ensemble de données comprenant les données cliniques vectorisées et les abondances microbiennes normalisées ont servies de données d’entrée pour l’entrainement du modèle.
Analyse longitudinale des prédictions effectuée sur l’ensemble des échantillons d’un même sujet.
[197]Pour les jeux de données ECUN, sepsis et DT1 , des échantillons de selles ont été prélevés en série pour un même sujet permettant une analyse longitudinale des prédictions effectuées pour un même enfant. Cette approche permettait ici de mesurer la capacité d’un modèle à être performant dès le premier échantillon malgré la dynamique des microbiotes. Dans cette approche, un sujet était considéré comme correctement classé lorsque tous ses échantillons étaient correctement classés. Les sujets pour lesquels au moins un échantillon était mal classé étaient considérés comme mal classés. Les inventeurs ont aussi profité de l'échantillonnage longitudinal des sujets pour explorer l’évolution du microbiote au cours du temps et redéterminer le phénotype final de chaque sujet mal classé par le réseau de neurones profonds. Les sujets pour lesquels la prédiction du phénotype était inégal selon les échantillons et qui avaient au moins 3 échantillons dans l'ensemble de données test ont été identifiés. Le
nombre d'échantillons dans chaque groupe phénotypique a été calculé et le phénotype final du sujet était déterminé par le groupe phénotypique ayant le plus grand nombre d'échantillons. Le phénotype ainsi déterminé a été comparé avec le phénotype observé (atteint d’une pathologie ou d’un état pathologique, non atteint). Enfin, un tracé en sucette a été généré pour visualiser cette approche d’analyse du suivi longitudinal à l'aide du package ggpubr (v0.4.0).
Exemple 1 : Diagnostic prédictif de l’accouchement prématuré à l’aide d’un réseau de neurones profonds entrainé avec des données de microbiote vaginal.
Recueil des données du jeu d’entrainement
[198]Les inventeurs ont sélectionné cinq études s’étant intéressé au microbiote vaginal en lien avec l’accouchement prématuré grâce aux mots-clés anglais : « vaginal microbiome », « shotgun metagenomics » et « premature birth » : Feehily ét al., 2020 ; Fettweis ét al, 2019 ; Goltsman ét al, 2018 ; Pace ét al, 2021 ; Tortelli ét al. 2021.
[199]Les données brutes et les métadonnées associées ont été obtenues pour chaque cohorte sous les numéros d’accession listés dans le tableau 1 ou sur demande. ENA représente European National Archive, NIH représente National Institute of Health, SRA représente Sequence Read Archive.
Tableau 1

[200]Pour chaque cohorte, les métadonnées d'échantillons suivantes ont été retenues :
- phénotype de naissance à terme (TB pour Term Birth) ou de naissance prématurée (PTB pour preterm birth),
- moment de collecte de l'échantillon : 1er trimestre de grossesse soit de 1-13 semaines de
gestation, 2ème trimestre de grossesse soit de 14-26 semaines de gestation, 3ème trimestre de grossesse soit >= 27 semaines de gestation,
- âge des participantes (inférieur à 35 ans, supérieur ou égal à 35 ans),
- groupe ethnique (Africain-Américain, Américain-Indien, Asiatique, Noir, Caucasien, Hispanique, Multi-ethnie, Blanc) et
- l'identifiant (ID) de la participante.
[201]Un total de 1290 échantillons a été récupéré. Seuls les échantillons prélevés au cours de la grossesse ont été utilisés. Le tableau 2 représente les propriétés générales des études individuelles incluses pour l’entraînement du réseau de neurones profonds. Celles-ci présentent le nombre d’échantillons ou le nombre de participantes, TB représente une naissance à terme (en anglais Term Birth) et PTB une naissance prématurée (en anglais PreTerm Birth).
Tableau 2

Prétraitement des données
[202]Lors du prétraitement à l’aide de RiboTaxa, pour la reconstruction des gènes d’ADNr 16S/18S, les paramètres A, B et C décrits dans le tableau 3 suivant ont été utilisés.
Tableau 3
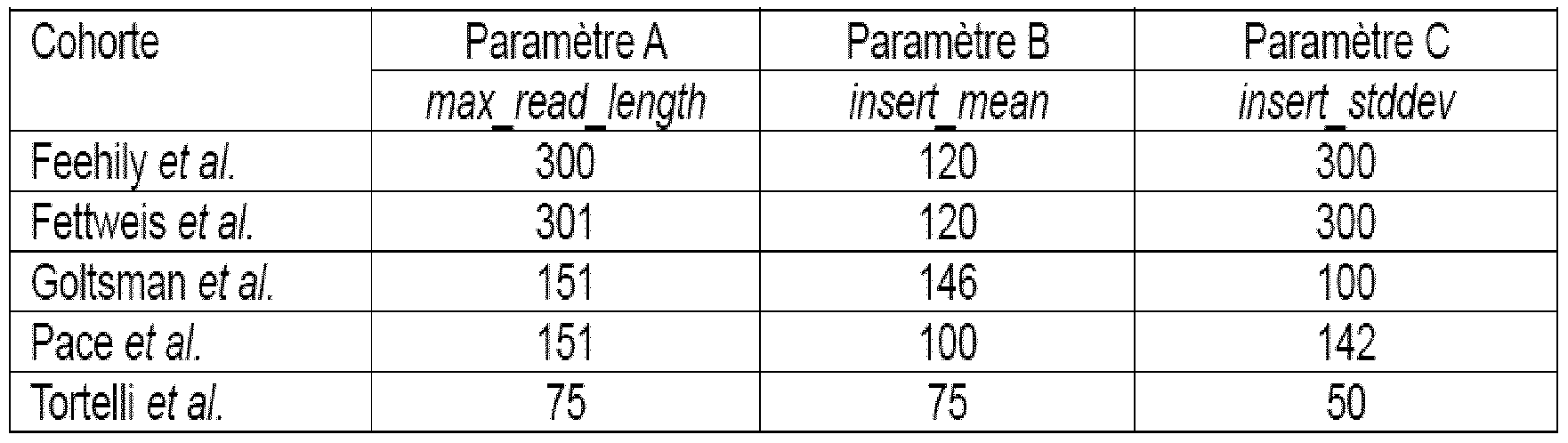
[204]Les profils taxonomiques au niveau de l'espèce ainsi que les données cliniques contenant les informations sur l'ethnicité, l'âge, le phénotype de la participante et le moment de la collecte d'échantillons ont été utilisés pour entrainer un réseau de neurones profonds.
Comparaison du réseau de neurones profonds optimisé avec d’autres modèles d’apprentissage
[205] Les performances du réseau de neurones profond optimisé ont été comparées à celles de trois algorithmes de classification de pointe : le modèle k-plus proches voisins (KNN), la régression logistique (LR) et la machine à vecteurs de support (SVM). Tous ces modèles ont été implémentés en Python (version 3.9.10). La librairie scikit-learn (vO.24.2) a été utilisée. Chaque modèle a été entraîné à partir des mêmes données, soit les 1290 échantillons. Les meilleurs hyperparamètres et configurations ont été identifiés en utilisant la méthode de validation croisée par recherche en grille (GSCV) de scikit-learn. La méthode GSCV identifie la meilleure combinaison d'hyperparamètres lors du processus de validation croisée à 10 plis (10-fold) pour obtenir les performances optimales des modèles.
[2061Comparaison du réseau de neurones profonds entraînés avec des données de diversité microbienne obtenues avec RiboTaxa et MetaPhlAn3
MetaPhlAn 3 (Beghini et al. 2021) utilise des gènes marqueurs spécifiques de clades permettant d’identifier la présence et l’abondance relative de microorganismes à partir de données métagénomiques. MetaPhlAn3 a été utilisé pour traiter les données de métagénomique « shotgun » de la cohorte Fettweis avec les paramètres par défaut et en utilisant la base de données CHOCOPhlAnSGB (version Jan21). Les profils de diversité microbienne au niveau de l’espèce ont été utilisés comme données d’entrée pour l’entrainement d’un réseau de neurones profonds. Les performances du modèle obtenu ont été comparées à un modèle de réseau de neurones profonds entraînés avec les données de diversité obtenues par pré-traitement des mêmes données de séquençage avec RiboTaxa.
Résultats
[207]Le traitement par le chainage bio-informatique RiboTaxa des données de séquençage métagénomique des cinq études a permis d’obtenir des séquences d’ADNr 16S ou 18S complètes à quasi-complètes avec une longueur minimum de 1045 bases. Une description précise du microbiote vaginal a ainsi été obtenue pour chaque échantillon, cette description comprend une identification au niveau de l’espèce et l’abondance relative de chaque espèce. L’approche de reconstruction des gènes exprimant l’ARNr 16S et/ou 18S permet de reconstruire des séquences très distantes des séquences de référence, ce qui permet ainsi d’identifier de nouveaux microorganismes qui ne seraient pas identifiés par les autres approches (PCR quantitative, analyses classiques de données métagénomiques, amplification par PCR d’une portion du gène exprimant l’ARNr 16S puis séquençage).
[208]Les données d’entrée composées des profils de microbiote vaginal associées aux quatre métadonnées (phénotype, ethnie, âge, moment de la collecte de l'échantillon) ont permis d’effectuer un apprentissage par réseaux de neurones profonds permettant de distinguer des accouchements à terme des accouchements prématurées. L’ensemble de données d’entrainement comprenaient 17 valeurs catégorielles (données cliniques vectorisées) et 636 valeurs numériques (abondances microbiennes normalisées).
[209]Le tableau 4 suivant rassemble les caractéristiques du réseau de neurones profonds obtenu.
Tableau 4

[210]L'évaluation du modèle final a été réalisée sur l'ensemble test composé de 239 échantillons n’ayant pas servi à la construction du modèle d’apprentissage d’intelligence artificielle. L’exactitude du diagnostic atteint 84,10%, tandis que la sensibilité et la spécificité atteignent 63,41% et 88,38% respectivement. Dans des essais répétés du réseau de neurones profonds, les inventeurs ont démontré un AUROC de 0,877 ± 0.11.
[211]Sur un même jeu de données d’entrée, les performances du réseau de neurones profonds (DNN) ont été supérieures comparées aux modèles de régression logistique (LR), des K-plus proches voisins (KNN) et d’une machine à vecteur de support SVM qui démontrent une exactitude similaire, tout de même de qualité (Tableau 5).
Tableau 5

[212]Les performances de prédiction du risque de survenue d’un accouchement prématuré ont été améliorées en focalisant l’entrainement d’un modèle sur les données provenant uniquement des échantillons prélevés au cours du deuxième trimestre. Le modèle a alors montré une sensibilité supérieure de 10% à 73,40% tout en conservant de très bonnes exactitude et spécificité à 82,58% et 85,61 % respectivement. Ce résultat montre que la sélection pertinente des données d’entrée est nécessaire pour obtenir les résultats les plus performants.
[213]La stratégie d’obtention des profils de diversité microbienne par reconstruction du gène exprimant l’ARNr 16S et/ou 18S a permis d’obtenir les meilleures performances comparée à l’utilisation d’autres gènes marqueurs (Figure 3).
Exemple 2 : Comparaison des performances de modèles entraînés sur des données de metabarcoding versus des données de métagénomique directe pour le diagnostic prédictif de l’accouchement prématuré.
Recueil des données pour le jeu d’entrainement
[214]L’étude de Fettweis et al. a porté sur 232 femmes dont les échantillons vaginaux ont été analysés à la fois par métagénomique directe dite « shotgun » et par metabarcoding (séquençage de la région V3-V4 de l’ADNr 16S). Les données brutes de métagénomique « shotgun » (952 Gb) et les métadonnées de la cohorte de Fettweis et al. ont été obtenues après l'approbation d’accès aux données par le National Institute of Health. Ce jeu de données représentait 173 femmes qui avaient accouché à terme (667 échantillons vaginaux, notés TB) et 55 femmes qui avaient accouché prématurément (155 échantillons vaginaux, notés PTB). Les données brutes de metabarcoding (58 Gb) appartenant à 749 échantillons TB (173 femmes) et 205 échantillons PTB (55 femmes) étaient en libre accès et ont été téléchargés depuis HMP DACC (https://portal.hmpdacc.org).
Prétraitement des données d’entrainement
[215]Pour les données issues de séquençage « shotgun », le chainage RiboTaxa a été utilisé. Pour la reconstruction du gène exprimant l'ARNr 16S et/ou 18S, les paramètres A, B et C ont été les suivants : --max_read_length = 301 , --insert_mean = 120, --insert_stddev = 300.
[216]Les données de séquençage metabarcoding ont été traitées avec DADA2 (package R 1.16). Une première étape de filtre contrôle qualité des lectures a été réalisée avec des paramètres standard : maxN=0, truncQ=2, rm.phix=TRUE et maxEE=2. Après apprentissage des taux d’erreur avec la fonction « learnErrors », les lectures ont été dérépliquées pour obtenir des séquences uniques ou ASVs (Amplicon Sequence Variants) assorties de leur abondance (nombre de lectures correspondant à chaque séquence unique). L'algorithme d'inférence d'échantillons a ensuite été appliqué afin de corriger les séquences dérépliquées à partir des profils de qualité des séquences brutes. Les paires de lectures ainsi obtenues ont été fusionnées pour obtenir les séquences d’amplicons complets. Finalement, les séquences chimériques ont été identifiées et éliminées et les ASVs restants ont été classés taxonomiquement avec la fonction « assignTaxonomy » et la base de données SILVA SSU 138.1 NR99 (Quast et al., 2013, https://benjjneb.github.io/dada2/training.html) ont été utlisées. Les abondances absolues d’ASVs au sein de chaque échantillon ont été converties en abondances relatives en utilisant la fonction « transform_sample_counts » du package R phyloseq (2.10).
[217]Etant donné que l’approche de séquençage metabarcoding se concentre sur l’analyse d’une portion de l’ADNr 16S, l’analyse taxonomique ne peut être réalisée au niveau de l’espèce. De ce fait, les identifications de microorganismes n’ont été réalisées qu’au niveau du genre et les deux approches de séquençage ont été comparées avec le rang taxonomique du genre.
[218]Les profils taxonomiques microbiens au niveau du genre, obtenus à partir des données de métagénomique shotgun et de metabarcoding, ainsi que les données cliniques (ethnicité, âge, phénotype et moment de la collecte des échantillons) ont été préalablement transformés comme décrit préalablement.
Entraînements de réseaux de neurones profonds
[219]Un réseau de neurones profonds a été implémenté et entraînés pour chacune des données de métagénomique directe et de metabarcoding, puis les performances des modèles produits ont été évalués avec l’ensemble de données test.
Résultats
[220]Le modèle entraîné sur les données issues du metabarcoding (au niveau du genre) a atteint une exactitude de 80,10 % (sur un total de 191 échantillons des données test), une spécificité de 86,84 % (sur 152 échantillons TB) et une sensibilité moindre de 53,84 % (sur 39 échantillons PTB) (Figure 4).
[221 ]En ce qui concerne les données issues de métagénomique directe, le modèle entrainé au rang taxonomique du genre a permis une amélioration de près de 10% de la sensibilité atteignant 63,33% (sur 33 échantillons PTB) pour une spécificité de 87,12% (sur 132 échantillons TB).
[222]Ces résultats illustrent que l’approche de reconstruction des gènes exprimant l’ADNr 16S et/ou 18S permet une meilleure identification des microorganismes grâce à des séquences longues d’ADNr et conduit ainsi à un modèle plus performant comparé au metabarcoding qui fournit des séquences courtes d’ADNr. Les biais liés à l’amplification PCR des amplicons, inhérents au metabarcoding, peuvent également impacter la représentativité de la diversité microbienne et dégrader les performances du modèle de classification.
Exemple 3 : Diagnostic prédictif de l’ECUN à l’aide d’un réseau de neurones profonds entrainé avec des données issues de microbiote fécal.
Recueil des données pour le jeu d’entrainement
[223]Les mots-clés (en anglais) suivants ont été utilisés pour identifier les études s’étant intéressé à l’ECUN chez le nouveau-né prématuré et ayant inclus des prélèvements de selles : « premature infants » ET (« stool microbiome » OU « intestinal microbiome») ET « shotgun metagenomics » ET « necrotizing enterocolitis ». A la fin du processus de sélection, deux études ont été retenues : Masi ét al. (2021) et Olm ét al. (2019).
[224]Les données brutes de séquençage métagénomique shotgun et les métadonnées de Masi et al. (2021) ont été téléchargées à partir de l’ENA dans le cadre du BioProject PRJEB39610 (n = 524 ; 974,51 Go). En plus de sa propre cohorte, Olm ét al. (2019) ont également utilisé des données de séquençage provenant de différents ensembles de données précédemment publiés. Toutes les données brutes et métadonnées utilisées dans la cohorte Olm (n=1038 au total) ont été téléchargées à partir de SRA sous les BioProjects : PRJNA294605 (n = 141 ; 596,53 Go), PRJNA417343 (n = 184 ; 152,21 Go) PRJNA396794 (n = 295 ; 1 ,35). Tb), PRJNA376566 (n = 358 ; 905,22 Go) et étude SRA SRP052967 (n = 60 ; 114,21 Go).
[225]Au total 1 305 échantillons contrôles (de 160 nourrissons) et 257 échantillons ECU N (de 48 nourrissons ayant développé l’ECUN) ont été utilisés pour l’entrainement d’un modèle. Aucun échantillon collecté après l'apparition de l’ECUN n’a été analysé. Cinq caractéristiques de données cliniques communes aux deux études ont été collectées : le phénotype (contrôle, ECUN), le mode de naissance (vaginal, césarienne), le genre (garçon, fille), l'âge gestationnel à la naissance (en semaines), le jour de vie (ou en anglais day of life, DOL, en jours) et le poids à la naissance du nouveau-né (en grammes) et l'identification du nourrisson. Seuls Masi et al. (2021) ont indiqué que les enfants de leur cohorte avaient reçu des probiotiques (Lactobacillus acidophilus, Bifidobacterium inf antis et B. bifidum).
[226]Les données cliniques des sujets sont présentées dans le tableau 6.
Tableau 6

Prétraitement des données d’entrainement
[227]Lors du prétraitement des données de séquençage à l’aide de RiboTaxa, pour la reconstruction du gène exprimant l'ARNr 16S et/ou 18S, les paramètres A, B et C ont été les suivants :
- cohorte Masi : --max_read_length = 151, --insert_mean = 144, --insert_stddev = 100 ;
- cohorte Olm : --max_read_length = 301, --insert_mean = 120, --insert_stddev = 100.
[228]Pour l’apprentissage du modèle, comme indiqué précédemment, les profils d’abondance des espèces microbiennes ont été normalisés et les données cliniques ont été discrétisées et vectorisées. Un ensemble de données comprenant 47 valeurs catégorielles et 1 282 valeurs numériques (abondances microbiennes normalisées) pour chacun des échantillons a été obtenu.
Evaluation du modèle sur des données externes
[229]Pour évaluer davantage les performances du modèle optimisé, 50 échantillons fécaux de 17 prématurés dont 7 ayant développé une ECIIN, issus de la cohorte CORTECs suivie par les inventeurs, ont été analysés. De plus, 40 nourrissons issus de deux cohortes publiées (Ward et al. 2023 et Schwartz et al. 2023) ont également été inclus pour tester les performances du modèle.
[230]La constitution de la cohorte CORTECs a été approuvée par le comité d'éthique du CPP-Sud- Est VI (code protocole 2021/CE 26, la date d'approbation est le 4 mai 2021). La cohorte CORTECs vise à traiter les facteurs de risque prénatals et postnatals d’ECU N. Tous les enfants nés prématurément hospitalisés dans l'unité de soins intensifs néonatals (USIN) du CHU de Clermont-Ferrand (France) ont été proposés pour entrer dans la cohorte. Un consentement éclairé écrit a été obtenu des familles des participants à l'étude avant l'inscription. Les selles des nourrissons ont été collectées quotidiennement pendant leur séjour à l'USIN, entre mai 2021 et juin 2022. Les selles ont été collectées dans une couche à l'aide d'une oese stérile, puis distribuées dans un tampon eNAT (Copan) avant d’être maintenues brièvement à 4 ° C. Les échantillons ont été conservés à -80 ° C jusqu'à l'extraction de l'ADN.
[231]Les cas d’ECUN ont été identifiés par les médecins sur la base de signes systémiques et abdominaux et de caractéristiques radiographiques. Ils ont été stratifiés selon la gravité de la maladie selon les stades de Bell. Les cas d’ECUN ont été appariés à un nouveau-né prématuré contrôle (deux pour un cas) qui n'a pas développé d’ECUN. L'appariement cas-contrôles était basé sur l'âge gestationnel à l'accouchement, le mode d'accouchement, le sexe, le poids à la naissance et les antibiotiques pré et postnatals. Pour chaque nourrisson ECUN, les échantillons disponibles ont été sélectionnés dans une fenêtre d'une semaine avant le début de l’ECUN et les échantillons des cas contrôles correspondants ont été appariés en fonction de l'âge du sujet ECUN.
[232]L'ADN génomique a été extrait à l'aide du protocole opératoire standard pour les échantillons fécaux (protocole H) recommandé par les normes internationales du microbiome humain (IHMS SOP 07 V1). La qualité de l'ADN a été évaluée à l'aide du fluoromètre Nanodrop 2000 (Thermo Scientific) et du système Agilent 4150 TapeStation avec des ScreenTape ADN génomique (Agilent). La quantité d'ADN a été évaluée à l'aide du fluoromètre Qubit 3 (Invitrogen) avec le kit de test Qubit dsDNA High Sensitivity (Invitrogen). Capture par hybridation du gène exprimant l'ARNr 16S et traitement des données de séquençage : les sondes de capture ont été conçues pour cibler le gène exprimant l’ARNr 16S (Gasc et al., 2016). Des librairies de séquençage ont été produites pour chaque échantillon à l'aide du kit de préparation de librairies Nextera XT. L'expérience de capture de gènes a été réalisée selon le protocole décrit par Ribière ét al. (2016) et Comtet-Marre et al. (2023). En bref, des sondes de capture ARN biotinylées ont été obtenues par transcription in vitro. 500 ng de librairies ont
été mélangés avec 2,5 pg d'ADN de sperme de saumon et incubés avec 500 ng de sondes biotinylées dans un tampon d'hybridation pendant 24 h à 65°C. Les hétéroduplex sonde/cible ont été capturés à l'aide de 500 pg de billes paramagnétiques recouvertes de streptavidine (Dynabeads M-280 Streptavidin, Invitrogen). Les billes ont été collectées à l'aide d'un support magnétique (Ambion), lavées une fois avec 500 pL de tampon 1 x SSC/0,1 % SDS, puis trois fois avec 500 pL de tampon 0,1 x SSC/0,1 % SDS préchauffé à 65°C. Les fragments d'ADN capturés ont été élués avec 50 pL de NaOH 0,1 M et transférés dans un tube stérile contenant 70 pL de tampon Tris-HCI 1 M pH 7,5. L'ADN capturé a été amplifié par PCR avec 25 cycles en utilisant des amorces complémentaires aux adaptateurs Illumina. Pour augmenter l’efficacité de l’enrichissement, un deuxième cycle de capture a été effectué. L'ADN capturé a ensuite été séquencé sur la plate-forme Illumina MiSeq 2 x 300 pb.
[233]Pour la cohorte de Ward étal. 2023, les nourrissons ont été recrutés dans deux unités de soins intensifs néonatals de niveau III (USIN) à Cincinnati (USA) et une USIN de niveau III à Birmingham (UK). Les cas d’ECUN rapportés étaient au stade II ou III de Bell. Un total de 115 données de séquençage métagénomique direct ont été utilisées, provenant de 3 nouveau-nés ECU N (9 échantillons) appariés à un total de 35 nouveau-nés prématurés contrôles (106 échantillons). Les échantillons de selles ont été collectés entre les jours 3 et 22 de vie. Les données brutes et les métadonnées ont été téléchargées depuis l'ENA (BioProject PRJNA63661).
[234]Schwartz et al. 2023 est une étude prospective américaine visant àétudier les facteurs associés à l'infection sanguine et au microbiome intestinal en unité de soins intensifs néonatals. Dans cette cohorte, deux nourrissons (8 échantillons) ont développé une entérocolite ulcéro-nécrosante (ECU N) et ont été sélectionnés. Les données brutes et les métadonnées ont été téléchargées depuis le dépôt NCBI (BioProject PRJNA884103).
[235]Pour les 3 cohortes, les données cliniques comprenaient les phénotypes (contrôle, NEC), le mode de naissance (vaginal, césarienne), le sexe (masculin, féminin), l'âge gestationnel (en semaines), le jour de vie (en jours) et le poids de naissance du nouveau-né (g) ainsi que l'identifiant de l'enfant (Tableau 7).
Tableau 7

[236]Les données brutes de séquençage issues des trois cohortes ont été traitées à l'aide du pipeline RiboTaxa et l’ensemble des données d’entrée ont été normalisées ou transformées comme décrit précédemment. Les espèces qui n'étaient pas présentes dans les échantillons utilisés pour l’apprentissage ont été exclues étant donné que le modèle ne peut pas les prendre en compte Pour chaque échantillon, le tableau d'abondances relatives des microorganismes au niveau de l’espèce concaténé avec les données cliniques du sujet a été utilisé comme entrée dans le modèle entraîné. Chaque prédiction a été comparée au phénotype de l’enfant (contrôle ou ECIIN). Des tracés SHAP ont également été générés. La prédiction finale des enfants a aussi été déterminée grâce aux échantillons longitudinaux provenant du même nourrisson en utilisant la même approche d’analyse de suivi longitudinal.
Résultats
[237]Toutes les données de séquençage ont été analysées avec le pipeline RiboTaxa (Chakoory et al., 2022), permettant la reconstruction de gènes d’ADNr 16S complets à presque complets pour fournir une description précise du microbiote intestinal jusqu'au niveau de l'espèce, comprenant l'identification des microorganismes dominants (>1 %), sous-dominants (<1%) et rares (<0, 1 %) permettant ainsi d’obtenir la meilleure représentativité du microbiote.
[238]ll est fréquemment mis en évidence que les bactéries des Enterobacteriaceae sont plus abondantes chez les enfants qui vont développer une ECIIN. L’analyse différentielle de la diversité des données de microbiote fécal destinées à l’entrainement montre également une abondance relatives moyennes d'Enterobacter non classées et d'entérobactéries non classées significativement plus élevées dans les échantillons ECIIN comparés aux échantillons de prématurés contrôles (p <0,05, Welch’s f-test). Malgré ces observations répétées dans les études, elles ne représentent toujours pas une signature microbienne fiable du risque d’ECUN car elles ne sont pas universellement retrouvées et que la notion de seuil d’abondance relative associé est difficile à déterminer.
[239]Pour pallier cette problématique, un réseau de neurones profonds a été développé et entrainé à l'aide de 1 402 caractéristiques (1 355 espèces microbiennes identifiées dans les selles et 47 données cliniques : 10 groupes d'âge gestationnel, 18 groupes de poids, 15 DOL, 2 modes de naissance et 2 groupes de sexe) (Figure 5). Le modèle final contenait 448 unités (neurones) dans la première couche cachée et un total de 3 couches cachées. L’entrainement du modèle a été réalisé en moins de 5 min sur un ordinateur i86linux32, 4,0 Go de RAM x 8 cœurs (32,8 Go au total)
[240]Le tableau 8 suivant rassemble les caractéristiques du réseau de neurones profonds obtenu.
[241]Tableau 8

[242]L'évaluation du modèle final a été réalisée sur l'ensemble test composé de 313 échantillons (provenant de 140 nourrissons). Le modèle a présenté une excellente exactitude de 94,9 %, une spécificité de 95,8 % (249 sur 260 échantillons contrôles) et une très bonne sensibilité de 90,6 % (48 sur 53 échantillons ECUN). Dans des essais répétés du réseau de neurones profonds, les inventeurs ont démontré un AUROC de 0,987 ± 0,01 (Figure 6), suggérant un bon équilibre entre sensibilité et spécificité et une valeur PR-AUC de 0,992±0,002 (Figure 7). De manière intéressante, Olm et al. ont appliqué une classification améliorée par gradient pour distinguer les nourrissons ECUN des contrôles à l'aide de données taxonomiques et ont obtenu seulement une précision de 64 % (Olm et al., 2019).
[243]Chez 92,8% des nourrissons ECUN (26 sur 28) et 90,1 % des enfants contrôles (101 sur 112) le diagnostic prédictif a été correct pour l’ensemble des échantillons issus d’un même enfant,
démontrant la robustesse du diagnostic malgré la colonisation dynamique du microbiote intestinal des nouveau-nés.
[244]Du fait de la gravité des conséquences de la survenue de l’ECUN chez les nouveau-nés prématurés, les inventeurs ont cherché à améliorer les performances du modèle en utilisant une stratégie de vote majoritaire, déterminant un diagnostic prédictif à partir du phénotype majoritaire prédit pour les différents échantillons d’un même enfant, lorsqu’ils étaient disponibles.
[245] Dans cette étude, seulement 16 échantillons (provenant de 16 nourrissons) sur 313 échantillons testés ont été mal classés par le réseau de neurones profonds. Parmi les 16 échantillons mal classés, 6 appartenaient à 6 nourrissons (2 contrôles et 4 ECIIN) pour lesquels plus de trois échantillons en série étaient présents dans l’ensemble de données test. Ainsi, 22 échantillons longitudinaux appartenant aux 6 nourrissons ont été considérés. Cette approche a permis de déterminer le bon phénotype de chaque enfant.
[246]L’approche SHAP implémentée dans le réseau de neurones profonds permet l’identification des espèces clés contribuant à la prédiction du modèle, pouvant s’apparenter à des signatures microbiennes complexes de la pathologie ou de l’état sain. Les 20 caractéristiques les plus importantes contribuant à la prédiction du modèle sont présentées dans la figure 8.
[247]Les quatre contributeurs les plus importants étaient des espèces de Lactobacillus spp. et leurs valeurs SHAP élevées étaient associées à des échantillons d’enfants contrôles. L’approche de caractérisation des microbiotes basées sur l’ADNr 16S montre ici toute sa puissance avec l’identification par RiboTaxa de deux bactéries non classées, qui ne pourraient pas être révélées avec d’autres approches d’analyse de données de séquençage métagénomique. Ces bactéries, bacterium_129 et bacterium_ARbO3, ont contribué à la prédiction du phénotype contrôle. L'analyse phylogénétique a révélé que la bactérie_129 était potentiellement une nouvelle espèce de Lactobacillus partageant une identité de 96,32 % avec la souche Dwan5 de L. casei, tandis que la bactérie_ARbO3 partageait une identité de 99,71 % avec la souche ADY07 de Bacillus cereus.
[248]En revanche, les espèces affiliées Enterobacter non classé, Syntrophomonas non cultivé, Streptomyces vanillaeus, Enterobacteriaceae non classé et Enterococcus faecalis ont le plus contribué à la classification ECUN.
[249] Il est intéressant de noter que des espèces peu abondantes telles que Ruminococcus sp., Staphylococcus non cultivé, Streptococcus parasanguinis et Proteus spp. ont également été observées comme contribuant à la classification ECUN, tandis que les Bifidobacterium non classées étaient associés à des échantillons d’enfants prématurés contrôles.
[250]Les performances prédictives du modèle proviennent principalement de données de microbiome. L'exclusion de caractéristiques cliniques lors de l’apprentissage a abouti à des valeurs de performance (AUROC = 0,931 , PR-AIIC = 0,956) qui n'étaient pas significativement différentes de celles du modèle incluant les données cliniques (p > 0,05, test U de Mann- Whitney entre ROC et courbes de précision-spécificité avec et sans métadonnées).
[251]Pour évaluer les performances du modèle sur des données extérieures à celles utilisées pour l’entrainement du modèle, les inventeurs ont utilisé les données de 3 cohortes (France, USA, Angleterre).
[252]Dans la cohorte CORTECs, les espèces de la famille des Enterobacteriaceae (Klebsiella non classées, Escherichia-Shigella non classées et Enterobacter spp.) qui sont couramment associées dans la pathogenèse de l’ECUN étaient présents dans les deux groupes et variaient en abondance relative d'un nourrisson à l'autre. Sur l'ensemble des échantillons de la cohorte, le réseau de neurones profonds optimisé suivi par l’approche d’analyse longitudinale des prédictions effectuée sur l’ensemble des échantillons des enfants illustrée par la figure 9, a démontré une sensibilité de 100 % (7 nourrissons ECUN stade 1a, ce qui correspond à 21 échantillons) et une spécificité de 80 % (8 contrôles sur 10, ce qui correspond à 23 échantillons).
[253]De même, sur la cohorte de Ward, une sensibilité de 100% (3 nourrissons atteints d’ECUN représentant 9 échantillons) et une spécificité de 86% (30 nourrissons témoins sur 35, ce qui correspond à 90 échantillons) ont été atteintes. Sur la cohorte de Schwartz, une sensibilité de 100% (2 nourrissons atteints de NEC, ce qui correspond à 8 échantillons) a été obtenue.
[254]En synthèse, la prédiction du phénotype d’échantillons provenant des 3 cohortes externes à l’entrainement a abouti à une sensibilité de 100% et une spécificité de 84,4%. Ainsi, les inventeurs ont réalisé un modèle très performant, capable de classer efficacement des échantillons provenant de différentes zones et pratiques de l'USIN malgré l'hétérogénéité du microbiome entre les cohortes.
[255]Parmi les caractéristiques contribuant aux différentes prédictions (figures 10 et 11), la prédiction des échantillons contrôle était principalement liée à la présence d'une abondance plus élevée de Lactobacillus spp. dont L. rhamnosus, L. casei et Lactobacillus sp. En revanche, une prédiction ECUN était liée à une abondance plus élevée d Enterococcus faecalis, Veillonella ratti, Klebsiella non classées, Enterococcus durans, Enterobacter cancerogenus, Clostridium neonatale ou C. perfringens. Des espèces peu abondantes telles que Staphylococcus non cultivés, Haemophilus parainfluenzae et Staphylococcus epidermidis ont contribué à la prédiction de l’ECUN dans certains échantillons, mettant en évidence une tendance à la co-variation entre espèces dominantes et rares suggérant l’existence d’un
réseau complexe d’interactions écologiques entre ces espèces. De manière intéressante, il a été observé qu’en fonction des enfants les profils de contribution des microorganismes variaient, démontrant tout l’intérêt de considérer le maximum de microorganismes pour l’entrainement du modèle afin d’effectivement prendre en compte toute la variabilité interindividuelle des microbiotes. Il existe donc plusieurs signatures microbiennes pour une même pathologie renforçant l’intérêt de ne pas sélectionner un nombre restreint de microorganismes pour l’entrainement des modèles de diagnostic prédictif.
[256]Les données cliniques contribuaient également à la classification des échantillons dans l’un des deux phénotypes. Un poids à la naissance <800 g et un âge gestationnel <30 semaines étaient les deux facteurs souvent associés à l’ECUN, tandis qu'un accouchement par voie basse et un âge gestationnel >31 semaines étaient associées aux échantillons des nourrissons non-ECUN.
Exemple 4 : Diagnostic prédictif du diabète de type 1 chez l’enfant à l’aide du réseau de neurones profonds
Recueil des données pour le jeu d’entrainement
[257]Les mots-clés (en anglais) suivants ont été utilisés pour identifier les études s’étant intéressé au diabète de type 1 chez l’enfant et ayant réalisé des prélèvements de selles avant l’identification de la pathologie « infants » ET (« stool microbiome » OU
« intestinal microbiome») ET « shotgun metagenomics » ET « Type 1 diabetes ». Cette recherche a abouti à l’identification d’une étude internationale « The Environmental Determinants of Diabetes in the Young (TEDDY) » réalisée aux États-Unis (Colorado, Floride, Washington) et en Europe (Finlande, Allemagne, Suède) (TEDDY Study Group, 2008).
[258]Les objectifs principaux de l'étude prospective visaient à identifier les facteurs environnementaux et génétiques déclenchant ou protégeant du développement d'anticorps anti-îlots de Langherans ou de diabète de type 1 (Rewers ét al., 2018). Pour cela, 7013 enfants de la population générale ont été recrutés, présentant un risque prédéterminé de diabète de type 1 de 3 % et 788 enfants ayant des parents au premier degré atteints de diabète de type 1 et présentant un risque prédéterminé de diabète de type 1 de 10 %. Les visites médicales ont eu lieu trimestriellement jusqu'à l'âge de 4 ans, puis tous les 6 mois jusqu'à l'âge de 15 ans. Les participants ont été suivis par prélèvement sanguin tous les trois mois pour des mesures d'auto-anticorps dirigés contre les cellules des îlots de Langerhans et de détection du diabète. Des échantillons de selles ont été collectés longitudinalement entre 3 et 72 mois de vie pour caractériser le microbiote intestinal par metabarcoding et par séquençage métagénomique direct. Chaque enfant atteint de diabète a été apparié à un ou deux contrôles.
[259]Dans cet exemple, seules les données de séquençage métagénomique direct ont été utilisées et les données des enfants présentant des auto-anticorps sans diabète de type 1 ont été exclues. Les données des témoins de ces enfants ont également été exclues. Ainsi les inventeurs ont utilisé un total de 6 955 données métagénomiques correspondant respectivement à 1 975 échantillons IA+DT1 (provenant de 91 enfants IA+DT1 dont le test était positif pour un ou plusieurs auto-anticorps et qui ont été diagnostiqués du diabète de type 1), 273 échantillons DT1 (provenant de 19 enfants DT1 dont le test était négatif pour un ou plusieurs auto-anticorps mais qui ont été diagnostiqués du diabète de type 1) et 4 707 échantillons témoins (provenant de 468 enfants contrôles des enfant DT1 et IA+DT1). Cinq données cliniques ont été agrégées : le phénotype (contrôle, IA+DT1 , DT1), le sexe (garçon, fille), le mois de vie au moment du prélèvement (en mois), l'identification de l’enfant et le jour de vie de l’enfant au moment où le DT1 a été diagnostiqué (en jours). L’information d’appariement des enfants a également été enregistrées. Les données brutes (4,96 Tb) et les métadonnées ont été reçues après l'approbation d’accès aux données par le National Institute of Health.
[260]Les données cliniques des sujets sont présentées dans le tableau 9
Tableau 9
 Prétraitement des données d’entrainement
Prétraitement des données d’entrainement
[261]Lors du prétraitement à l’aide de RiboTaxa, pour la reconstruction du gène d’ADNr 16S/18S, les paramètres A, B et C ont été les suivants : --max_read_length = 102, --insert_mean = 200, --insert_stddev = 100.
[262]Les enfants IA+DT 1 et DT1 ont été regroupés en un seul groupe d'enfants diabétiques de type 1 pour l'entraînement du modèle, désigné comme TD1 par la suite. Ainsi, l’entraînement a été réalisé sur l’ensemble des données (4707 échantillons provenant de 144 enfants contrôles et 110 enfants DT1) pour produire un modèle « sans a priori », et trois sous-ensembles de données ont été créés en fonction du mois où le DT1 a été diagnostiqué. Pour cela, le jour de vie de l’enfant au moment du diagnostic a été converti en mois en le divisant par 30 jours. Les groupes ont ensuite été établi comme suit : modèle DT1 « 24-48 mois » (2361 échantillons provenant de 68 enfants contrôles et 52 enfants DT1), modèle DT1 « 48-72 mois » (1101 échantillons provenant de 23 enfants contrôles et 20 enfants DT1) et modèle DT1 « 24-72 mois » (3193 échantillons provenant de 83 enfants contrôles et 66 enfants DT1). Pour chaque groupe, seuls les enfants contrôles appariés avec les enfants DT1 inclus ont été conservés. Seuls les échantillons prélevés avant le diagnostic du TD1 ont été conservés. Les modèles ont été désignés par un intervalle d’âges des enfants au moment du diagnostic du DT1 et inclus dans le modèle. Ces intervalles couvrent au maximum une période de 2 à 6 ans (24-72 mois), correspondant à la période où la majorité des cas de DT1 ont été diagnostiqués.
Résultats
[263]Les profils d'abondances relatives contrôlés et de haute qualité au niveau des espèces ainsi que trois données cliniques (phénotype, sexe, mois de vie au moment du prélèvement) ont été utilisés pour entraîner 4 réseaux de neurones profonds pour le diagnostic prédictif du risque de DT1.
[264]Chaque modèle avait un nombre de caractéristiques d’entrée différent : modèle DT 1 « sans a priori » (1476 espèces microbiennes, 71 groupes de données cliniques : 69 groupes de prélèvements et 2 groupes de sexe), modèle DT 1 « 24-48 mois » (1305 espèces microbiennes et 42 groupes de données cliniques : 40 groupes de prélèvement et 2 groupes de sexe), modèle DT 1 « 48-72 mois » (1014 espèces microbiennes et 17 groupes de données cliniques : 15 groupes de prélèvement et 2 groupes de sexe) et modèle DT1 « 24-72 mois » (1354 espèces microbiennes et 59 groupes de données cliniques : 57 groupes de prélèvement et 2 groupes de sexe). Pour chaque modèle, toutes les espèces détectées dans tous les échantillons ont été conservés.
[265]Les hyperparamètres ont variés pour chacun des modèles (Tableau 10). L’apprentissage des modèles a été réalisée sur : i86linux32, 4,0 Go de RAM x 8 cœurs (32,8 Go au total), sans GPU, et s'est déroulée en 2 min au maximum.
[266]Le tableau 10 suivant rassemble les principaux hyperparamètres optimaux des modèles de réseaux de neurones profond ainsi obtenus.
Tableau 10

[267]Les performances des modèles ciblés sur une fenêtre « âge de déclenchement du DT 1 » ont été globalement les meilleures en comparaison de celles du modèle « sans a priori » prenant l’ensemble des données (Tableau 11), avec notamment une sensibilité allant de 70,8% à 76,5% pour ces modèles contre 63% pour le modèle « sans a priori ». Ce résultat illustre une nouvelle fois l’importance de sélectionner les données de manière pertinente.
Tableau 11

[268]L'échantillonnage en série des enfants réalisé sur la cohorte TEDDY a été utilisé pour appliquer une approche d'analyse longitudinale des prédictions effectuées sur l’ensemble des échantillons de chaque enfant comme décrit précédemment. Cette approche a permis d’obtenir pour les modèles ciblés sur les tranches d’âges, l’identification correcte de 63,2% à
81 ,3% des enfants ayant développé plus tard le TD1 et de 68,1 % à 71 % des enfants non atteints de diabète de type 1 .
[269]La prédiction de la pathologie de diabète de type 1 est pondérée par un ensemble de microorganismes comme illustré dans la figure 14.
Exemple 5 : Diagnostic prédictif de sepsis à l’aide de modèles de réseaux de neurones profonds
Recueil des données pour le jeu d’entrainement
[270]Les mots-clés (en anglais) suivants ont été utilisés pour identifier les études s’étant intéressé au sepsis chez les nouveau-nés : («newborns » OU « premature infants ») ET (« stool microbiome » OU « intestinal microbiome») ET « shotgun metagenomics » ET (« sepsis » OU « bloodstream infection »). Les inventeurs ont également sélectionné des études incluant des informations cliniques telles que : mode de naissance (voie basse ou césarienne), genre (masculin-féminin), âge gestationnel (en semaines), âge réel (en jours de vie) et poids à la naissance (en grammes). Finalement deux études ont été retenues.
[271]Les données brutes de séquençage métagénomique et les métadonnées de Heston et al., 2023 ont été téléchargées à partir de Sequence Read Archive (SRA) dans le cadre du BioProject PRJNA947616 (n = 622 ; 1 ,17 Tb). Les données brutes de la cohorte de Schwartz et al., 2023 ont été téléchargées à partir de SRA sous le BioProject PRJNA884103 (n = 195, 234,7 Go) et les métadonnées ont été reçues des auteurs de l'étude.
[272]Un total de 418 et 167 données métagénomiques ont été extraites respectivement de Heston et al., 2023 et Schwartz et al., 2023. Les enfants qui ont développé d’autres pathologies comme l’entérocolite ulcéro-nécrosante ainsi que les enfants nés à terme (>37 semaines d’aménorrhée) ont été exclus. De plus, aucun échantillon collecté après l'apparition de sepsis n’a été analysé. Cinq caractéristiques de métadonnées cliniques ont été collectées et rapportées dans les deux études, telles que les phénotypes (contrôle, sepsis), le mode de naissance (vaginal, césarienne), le genre (garçon, fille), l'âge gestationnel à la naissance (en semaines), le jour de vie (ou DOL pour day of life en anglais, en jours), le poids à la naissance du nouveau-né (en grammes) et l'identification du nourrisson.
Prétraitement des données d’entrainement
[273]Lors du prétraitement à l’aide de RiboTaxa, pour la reconstruction du gène exprimant l'ARNr 16S/18S, les paramètres A, B et C ont été les suivants :
- cohorte Heston : --max_read_length = 152, --insert_mean = 144, --insert_stddev = 124;
- cohorte Schwartz : --max_read_length = 302, --insert_mean = 268, --insert_stddev = 144
[274]Les données de diversité microbienne ont été normalisées et les données cliniques ont été discrétisées et vectorisées pour obtenir 44 valeurs catégorielles (9 groupes d'âge gestationnel, 16 groupes de poids, 15 groupes de DOL, 2 modes de naissance et 2 groupes de sexe) et 637 valeurs numériques (abondances microbiennes).
Résultats
[275]Des données de séquençage métagénomique direct (« shotgun ») ont été utilisées pour décrire le microbiote à haute résolution (au niveau de l’espèce). 585 données d’échantillons de selles métagénomiques (486 provenant de 87 nouveau-nés prématurés et 99 provenant de 29 nourrissons prématurés ayant développé ultérieurement un sepsis) ont été analysés à l'aide de RiboTaxa (Chakoory et al., 2022), permettant l'identification d'un total de 637 espèces uniques. Cette uniformité permet à un modèle unique de prendre en charge les données de divers protocoles d’étude. Les profils d'abondances relatives contrôlés et de haute qualité au niveau de l’espèce ainsi que 5 données cliniques (âge gestationnel, poids à la naissance, jour de vie au moment du prélèvement, mode de naissance, et sexe de l’enfant) ont été utilisés pour entraîner un réseau de neurones profonds permettant de prédire le risque de sepsis avant l’apparition de l’infection conduisant à la pathologie.
[276]Le modèle de réseau de neurones profonds a été formé puis entrainé à l'aide de 681 caractéristiques différentes (637 espèces microbiennes et 44 groupes de données cliniques). Toutes les espèces détectées dans tous les échantillons ont été conservées, au lieu d'appliquer une sélection avant l'entraînement pour conserver les variations interindividuelles de microbiotes entre les nourrissons. Un total de 42 882 paramètres entraînables a été testé et le réglage optimal des hyperparamètres pour le modèle final avait 64 unités (neurones) dans la 1ère couche cachée et un total de 3 couches cachées (Tableau 12). L’apprentissage du modèle a été réalisée sur : i86linux32, 4,0 Go de RAM x 8 cœurs (32,8 Go au total), sans GPU et s'est déroulée en 2 min.
[277]Le tableau 12 suivant résume les caractéristiques principales du modèle de réseau de neurones profonds obtenu.
Tableau 12

[278]L'évaluation du réseau de neurones profonds entraîné a été réalisée sur l'ensemble de données test composé de 117 échantillons (provenant de 60 enfants contrôles et 14 enfants atteints de sepsis). Le modèle a démontré une exactitude de 92,3 %, une sensibilité de 72,2% et une spécificité de 96,0 %. Dans des essais répétés, les inventeurs ont démontré un AUROC de 0,941 ± 0,013 et une valeur PR-ALIC de 0,942± 0,011 suggérant un bon équilibre entre sensibilité et spécificité.
[279]Parmi les enfants qui ont développé par la suite un sepsis, 72,2% ont présenté un risque de sepsis à tous leurs tests. À l'inverse, 96% des enfants contrôles n'ont montré aucun risque de sepsis à aucun de leurs tests. Le modèle apporte donc un excellent diagnostic prédictif permettant d’identifier dès le premier échantillon prélevé le risque de survenue de sepsis.
[280]Lorsque cela a été possible, les enfants qui avaient des échantillons prédits avec le mauvais phénotype, une analyse longitudinale des prédictions effectuées sur l’ensemble de leurs échantillons a été réalisée sur le principe décrit précédemment. Deux nouveau-nés contrôles avaient chacun 3 échantillons permettant d’effectuer cette analyse et la majorité de leurs échantillons ont été prédits avec le phénotype correct (Figure 15), permettant ainsi d’identifier correctement 96,7 % des enfants n’ayant pas développé le sepsis.
[281 ]La décomposition des contributions des différentes données d’entrée a montré que l’apport des données cliniques était notable. Elles représentaient 11 des 20 caractéristiques les plus importantes contribuant à la validation du modèle (Figure 16). Les caractéristiques d’âge gestationnel de 25 et 28 semaines d’aménorrhée et de poids 500-599 grammes ont été associées à la prédiction « sepsis » tandis que les âges gestationnels de 29 et 30 semaines d’aménorrhée étaient associés à la prédiction « contrôle », témoignant de la fragilité observée des enfants les plus prématurés. Cette liste comprenait également des microorganismes tels que les espèces de Bifidobacterium associées à la prédiction du groupe contrôle et souvent corrélé avec l'alimentation, en particulier avec l'allaitement maternel, tandis que les espèces Streptococcus et Staphylococcus étaient associées à la prédiction du groupe sepsis.
[282]Ainsi pour évaluer l’importance des jeux de données cliniques dans la prédiction de sepsis chez les nourrissons, les inventeurs ont exclus les données de diversité microbienne et le nouveau modèle a été entrainé uniquement sur les cinq données cliniques qui représentait 44 caractéristiques d’entrée pour le réseau de neurones profonds. Le modèle nouvellement développé a montré une baisse au niveau de la sensibilité, avec 61 ,1 % (contre 72,2 %) mais toujours avec une très bonne spécificité de 97,0 %. Ce résultat confirme le poids important des données cliniques dans le diagnostic prédictif du sepsis et l’apport nécessaire des données de microbiote fécal pour obtenir une plus grande sensibilité.
[283]La performance du modèle de réseau de neurones profond entrainé avec les jeux de données « microbiote combiné avec les données cliniques » a aussi été testée en traitant les données de métagénomique à différents niveaux taxonomiques (Phylum, Classe, Ordre, Famille, Genre). Les modèles ont été évalués avec le même ensemble de données test de 117 échantillons d’enfants contrôles et de 18 échantillons d’enfants ayant développé un sepsis.
[284]Le tableau 13 résume les performances des différents modèles élaborés.
Tableau 13

[285]Ces résultats démontrent que les modèles de réseau de neurones profonds entrainés sur des données de diversité microbienne retenues au rang taxonomique de l’ordre et de la classe sont plus performants qu’au rang taxonomique de l’espèce. Néanmoins les classes/ordres associés au groupe sepsis restent très large et ne permettent pas une identification précise des espèces potentiellement liées à un risque de sepsis. En revanche, le modèle entraîné sur les espèces est légèrement moins performant mais permet de remonter une liste de microorganismes impliqués dans la pathologie, ce qui pourraient permettre aux cliniciens d'adapter le traitement en fonction des microorganismes identifiés chez les nourrissons.
Claims
1 . Procédé in vitro de diagnostic prédictif d’une pathologie ou d’un état pathologique chez un sujet, à partir d’au moins un échantillon biologique prélevé chez le sujet et contenant des microorganismes, ledit procédé comprenant les étapes suivantes : a) séquençage, à partir de l’acide nucléique isolé dudit au moins un échantillon biologique, des séquences nucléotidiques correspondant à au moins une séquence d’intérêt sélectionnée dans le groupe consistant en : un fragment d’un gène exprimant l’ARN ribosomique (ARNr) 16S, un fragment d’un gène exprimant l’ARNr 18S, un fragment de l’ARNr 16S, un fragment de l’ARNr 18S, b) à partir du séquençage de l’étape a), détermination de l’identité et de l’abondance relative des microorganismes présents dans ledit échantillon, c) détermination du diagnostic prédictif de ladite pathologie ou de l’état pathologique par un modèle d’intelligence artificielle à partir au moins des abondances des identités obtenues à l’étape b), ledit modèle d’intelligence artificielle ayant préalablement été entraîné sur la base d’un jeu de données labellisées, où le jeu de données labellisées comprend des profils de sujets d’entrainement, chaque profil de sujet d’entrainement comprenant l’identité et l’abondance relative de l’ensemble des microorganismes identifiés dans au moins un échantillon dudit sujet d’entrainement sans aucune présélection, où chaque profil de sujet d’entrainement est labellisé avec le phénotype du sujet d’entrainement dont il est issu, ledit phénotype de sujet d’entrainement étant classé sans apparition ou avec apparition de la pathologie ou de l’état pathologique, et où des données de l’étape b) sont uniquement exclues les abondances des identités des microorganismes qui n’étaient pas présentes dans le jeu de données labellisées.
2. Procédé selon la revendication 1 , dans lequel lors de l’entrainement du modèle d’intelligence artificielle et lors de l’étape b), l’identité de chaque microorganisme correspond au rang taxonomique le plus confiant.
3. Procédé selon la revendication 1 ou 2, dans lequel les sujets d’entrainement ont des origines multinationales.
4. Procédé selon la revendication 1 ou 2, dans lequel le jeu de données labellisées comprend au moins une donnée clinique déterminée, où chaque profil de sujet d’entrainement
comprend une valeur pour la ou chaque donnée clinique déterminée, et où l’étape c) comprend la fourniture au modèle d’intelligence artificielle de la valeur correspondante du sujet pour la ou chaque donnée clinique déterminée.
5. Procédé selon l’une des revendications 1 à 3, dans lequel au moins deux échantillons biologiques sont utilisés, où les étapes a) à c) sont réalisées sur chaque échantillon et où le procédé comprend une étape d) de compilation du diagnostic prédictif obtenu pour chaque échantillon et de détermination finale du diagnostic prédictif.
6. Procédé selon l’une quelconque des revendications 1 à 4, dans lequel l’étape b) comprend l’organisation des séquences nucléotidiques obtenues à l’étape a) pour reconstruire la séquence d’au moins 70 % de la longueur de ladite au moins une séquence d’intérêt sélectionnée.
7. Procédé selon l’une quelconque des revendications 1 à 5, étant destiné au diagnostic prédictif d’un accouchement précoce chez une femme enceinte.
8. Procédé selon les revendications 3 et 6, dans lequel ladite au moins une catégorie de donnée clinique déterminée est sélectionnée dans le groupe consistant en : l’âge, l’ethnie, le trimestre de la grossesse et une combinaison de celles-ci.
9. Procédé selon l’une quelconque des revendications 1 à 5, étant destinée au diagnostic prédictif de l’entérocolite ulcéro-nécrosante chez un nourrisson.
10. Procédé selon les revendications 3 et 8, dans lequel ladite au moins une catégorie de donnée clinique déterminée est sélectionnée dans le groupe consistant en : l’âge en nombre de jours depuis la naissance, le poids à la naissance, l’âge gestationnel, le mode de naissance, le genre, l’alimentation de la mère du nourrisson, le résultat du dosage de composants ou marqueurs sanguins, l’administration d’un traitement médical, la présence d’au moins une autre pathologie et une combinaison de celles-ci.
11 . Procédé selon l’une quelconque des revendications 1 à 5, étant destinée au diagnostic prédictif du diabète de type I chez un enfant.
12. Procédé selon l’une quelconque des revendications 1 à 5, étant destinée au diagnostic prédictif du sepsis néonatal chez un nourrisson.
13. Produit programme d’ordinateur comprenant des instructions exécutables, qui lorsqu’elles sont exécutées sur un ordinateur permettent la mise en œuvre de l’étape c) du procédé selon l’une quelconque des revendications 1 à 12.
14. Procédé d’entrainement d’un modèle d’intelligence artificielle destiné à obtenir un diagnostic prédictif, ledit procédé utilisant un jeu de données labellisées comprenant des profils de sujets d’entrainement, où chaque profil de sujet d’entrainement comprend l’identité et l’abondance relative de l’ensemble des microorganismes identifiés dans au moins un échantillon dudit sujet d’entrainement sans aucune présélection, et où chaque profil de sujet d’entrainement est labellisé avec le phénotype du sujet d’entrainement dont il est issu, ledit phénotype de sujet d’entrainement étant classé en ayant développé ou n’ayant pas développé la pathologie ou l’état pathologique.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR2308145A FR3151603A1 (fr) | 2023-07-27 | 2023-07-27 | Procédé de détection d’une pathologie |
| FRFR2308145 | 2023-07-27 | ||
| FRFR2313206 | 2023-11-28 | ||
| FR2313206A FR3155837A1 (fr) | 2023-11-28 | 2023-11-28 | Procédé de prédiction d’un état ou d’une pathologie |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025022019A1 true WO2025022019A1 (fr) | 2025-01-30 |
Family
ID=91961642
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/071489 Pending WO2025022019A1 (fr) | 2023-07-27 | 2024-07-29 | Procédé de diagnostic prédictif d'une pathologie ou d'un état pathologique |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025022019A1 (fr) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3161167A1 (fr) | 2014-06-30 | 2017-05-03 | The Chinese University Of Hong Kong | Détection de taxons bactériens permettant de prédire des issues défavorables de la grossesse |
| US20170159108A1 (en) * | 2014-05-06 | 2017-06-08 | Is-Diagnostics Ltd. | Microbial population analysis |
| WO2020227053A1 (fr) | 2019-05-03 | 2020-11-12 | Virginia Commonwealth University | Marqueurs du microbiome vaginal pour la prédiction et la prévention de la naissance prématurée et d'autres issues de grossesse défavorables |
| EP2972308B9 (fr) | 2013-03-15 | 2021-01-20 | Sera Prognostics, Inc. | Biomarqueurs et procédés de prédiction d'une naissance prématurée |
| US20210381054A1 (en) * | 2018-10-31 | 2021-12-09 | Coyote Diagnostics Lab (Beijing) Co., Ltd. | Methods, systems and kits for predicting premature birth condition |
| US20220081708A1 (en) * | 2019-01-03 | 2022-03-17 | Evolve Biosystems, Inc. | Diagnosis and treatment of dysbiosis-associated with nec |
-
2024
- 2024-07-29 WO PCT/EP2024/071489 patent/WO2025022019A1/fr active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2972308B9 (fr) | 2013-03-15 | 2021-01-20 | Sera Prognostics, Inc. | Biomarqueurs et procédés de prédiction d'une naissance prématurée |
| US20170159108A1 (en) * | 2014-05-06 | 2017-06-08 | Is-Diagnostics Ltd. | Microbial population analysis |
| EP3161167A1 (fr) | 2014-06-30 | 2017-05-03 | The Chinese University Of Hong Kong | Détection de taxons bactériens permettant de prédire des issues défavorables de la grossesse |
| US20210381054A1 (en) * | 2018-10-31 | 2021-12-09 | Coyote Diagnostics Lab (Beijing) Co., Ltd. | Methods, systems and kits for predicting premature birth condition |
| US20220081708A1 (en) * | 2019-01-03 | 2022-03-17 | Evolve Biosystems, Inc. | Diagnosis and treatment of dysbiosis-associated with nec |
| WO2020227053A1 (fr) | 2019-05-03 | 2020-11-12 | Virginia Commonwealth University | Marqueurs du microbiome vaginal pour la prédiction et la prévention de la naissance prématurée et d'autres issues de grossesse défavorables |
Non-Patent Citations (10)
| Title |
|---|
| CHAKOORY OSHMA ET AL: "RiboTaxa: combined approaches for rRNA genes taxonomic resolution down to the species level from metagenomics data revealing novelties", NAR GENOMICS AND BIOINFORMATICS, vol. 4, no. 3, 9 July 2022 (2022-07-09), XP093175186, ISSN: 2631-9268, DOI: 10.1093/nargab/lqac070 * |
| COMTET-MARRE, SOPHIECHAKOORYOSHMAPEYRET, PIERRE, TARGETED 16S RRNA GENE CAPTURE BY HYBRIDIZATION AND BIOINFORMATIC ANALYSIS, 2022 |
| DURAZZI, F.SALA, C.CASTELLANI, G. ET AL.: "Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota", SCI REP, vol. 11, 2021, pages 3030 |
| KOSTIC ALEKSANDAR D ET AL: "The Dynamics of the Human Infant Gut Microbiome in Development and in Progression toward Type 1 Diabetes", CELL HOST & MICROBE, ELSEVIER, NL, vol. 17, no. 2, 5 February 2015 (2015-02-05), pages 260 - 273, XP029139280, ISSN: 1931-3128, DOI: 10.1016/J.CHOM.2015.01.001 * |
| MARÍA CERNADA ET AL: "Sepsis in preterm infants causes alterations in mucosal gene expression and microbiota profiles compared to non-septic twins", SCIENTIFIC REPORTS, vol. 6, no. 1, 1 May 2016 (2016-05-01), XP055610070, DOI: 10.1038/srep25497 * |
| PARK SUNWHA ET AL: "Predicting preterm birth through vaginal microbiota, cervical length, and WBC using a machine learning model", FRONTIERS IN MICROBIOLOGY, vol. 13, 2 August 2022 (2022-08-02), Lausanne, XP093175085, ISSN: 1664-302X, DOI: 10.3389/fmicb.2022.912853 * |
| PRISCILA T DOBBLER ET AL: "Low Microbial Diversity and Abnormal Microbial Succession Is Associated with Necrotizing Enterocolitis in Preterm Infants", FRONTIERS IN MICROBIOLOGY, vol. 8, 1 November 2017 (2017-11-01), pages 1 - 12, XP055723441, DOI: 10.3389/fmicb.2017.02243 * |
| QUINCE C ET AL.: "Shotgun metagenomics, from sampling to analysis", NAT BIOTECHNOL., vol. 35, no. 9, 12 September 2017 (2017-09-12), pages 833 - 844, XP055831282, DOI: 10.1038/nbt.3935 |
| VATANEN T, NATURE., vol. 562, no. 7728, October 2018 (2018-10-01), pages 589 - 594 |
| WANI ATIF KHURSHID ET AL: "Metagenomics and artificial intelligence in the context of human health", INFECTION , GENETICS AND EVOLUTION, ELSEVIER, AMSTERDAM, NL, vol. 100, 10 March 2022 (2022-03-10), XP087017703, ISSN: 1567-1348, [retrieved on 20220310], DOI: 10.1016/J.MEEGID.2022.105267 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Baud et al. | Microbial diversity in the vaginal microbiota and its link to pregnancy outcomes | |
| Zmora et al. | Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features | |
| Pulikkan et al. | Gut microbial dysbiosis in Indian children with autism spectrum disorders | |
| Brooks et al. | Gut microbiota diversity across ethnicities in the United States | |
| Papa et al. | Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease | |
| Chiu et al. | Systematic analysis of the association between gut flora and obesity through high‐throughput sequencing and bioinformatics approaches | |
| Patrone et al. | Gut microbiota profile in systemic sclerosis patients with and without clinical evidence of gastrointestinal involvement | |
| Hansen et al. | Microbiota ofDe-NovoPediatric IBD: IncreasedFaecalibacterium Prausnitziiand Reduced Bacterial Diversity in Crohn's But Not in Ulcerative Colitis | |
| Suchodolski et al. | 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease | |
| Biagi et al. | Gut microbiome in Down syndrome | |
| Zhu et al. | Nasopharyngeal airway dual-transcriptome of infants with severe bronchiolitis and risk of childhood asthma: A multicenter prospective study | |
| Tang et al. | Prospective study reveals a microbiome signature that predicts the occurrence of post-operative enterocolitis in Hirschsprung disease (HSCR) patients | |
| CN105473738B (zh) | 结直肠癌生物标志物 | |
| EP3347496A1 (fr) | Méthode et système de diagnostics dérivés du microbiome et traitements thérapeutiques pour la santé bucco-dentaire | |
| CN108348167B (zh) | 用于脑-颅面健康相关病症的源自微生物群系的诊断及治疗方法和系统 | |
| US12596121B2 (en) | Companion diagnostic method for use in the treatment of irritable bowel syndrome with dietary interventions or faecal microbiota transplant | |
| Ding et al. | A metagenomic study of the gut microbiome in PTB’S disease | |
| US20190100789A1 (en) | Method and system for microbiome-derived diagnostics and therapeutics infectious disease and other health conditions associated with antibiotic usage | |
| Chamorro et al. | Landscapes and bacterial signatures of mucosa-associated intestinal microbiota in Chilean and Spanish patients with inflammatory bowel disease | |
| Escribano et al. | Influence of a Serratia marcescens outbreak on the gut microbiota establishment process in low-weight preterm neonates | |
| Ferrocino et al. | Mycobiota composition and changes across pregnancy in patients with gestational diabetes mellitus (GDM) | |
| US20250210197A1 (en) | Machine learning for differentiating among multiple diseases | |
| US20250285756A1 (en) | Two competing guilds as core microbiome signature for human diseases | |
| Liu et al. | Individualized network analysis reveals a link between the gut microbiome, diet intervention and Gestational Diabetes Mellitus | |
| WO2025022019A1 (fr) | Procédé de diagnostic prédictif d'une pathologie ou d'un état pathologique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24745976 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024745976 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |