BRPI0706992B1 - Composto de fórmula (i) e formulação farmacêutica - Google Patents
Composto de fórmula (i) e formulação farmacêutica Download PDFInfo
- Publication number
- BRPI0706992B1 BRPI0706992B1 BRPI0706992-8A BRPI0706992A BRPI0706992B1 BR PI0706992 B1 BRPI0706992 B1 BR PI0706992B1 BR PI0706992 A BRPI0706992 A BR PI0706992A BR PI0706992 B1 BRPI0706992 B1 BR PI0706992B1
- Authority
- BR
- Brazil
- Prior art keywords
- ring
- pyrone
- hydrogen
- indole
- methoxy
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 87
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 11
- 150000003839 salts Chemical class 0.000 claims abstract description 26
- 125000003118 aryl group Chemical group 0.000 claims description 36
- -1 nitro, amino, cyano, cyanamido, guanidino, amidino Chemical group 0.000 claims description 29
- 239000001257 hydrogen Substances 0.000 claims description 26
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 25
- 125000001041 indolyl group Chemical group 0.000 claims description 25
- ZPSJGADGUYYRKE-UHFFFAOYSA-N 2H-pyran-2-one Chemical group O=C1C=CC=CO1 ZPSJGADGUYYRKE-UHFFFAOYSA-N 0.000 claims description 23
- 229910052760 oxygen Inorganic materials 0.000 claims description 19
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 18
- 239000001301 oxygen Substances 0.000 claims description 18
- 125000000217 alkyl group Chemical group 0.000 claims description 16
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 16
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 15
- 229910052717 sulfur Inorganic materials 0.000 claims description 15
- 239000011593 sulfur Substances 0.000 claims description 15
- CVQUWLDCFXOXEN-UHFFFAOYSA-N Pyran-4-one Chemical class O=C1C=COC=C1 CVQUWLDCFXOXEN-UHFFFAOYSA-N 0.000 claims description 12
- 125000005842 heteroatom Chemical group 0.000 claims description 12
- 229910052757 nitrogen Inorganic materials 0.000 claims description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 12
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 12
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 11
- 239000002904 solvent Substances 0.000 claims description 10
- 125000003342 alkenyl group Chemical group 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 9
- 239000002671 adjuvant Substances 0.000 claims description 7
- 125000000304 alkynyl group Chemical group 0.000 claims description 7
- 125000005843 halogen group Chemical group 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 239000003755 preservative agent Substances 0.000 claims description 7
- 125000004423 acyloxy group Chemical group 0.000 claims description 6
- 239000003995 emulsifying agent Substances 0.000 claims description 6
- 125000001072 heteroaryl group Chemical group 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 125000005504 styryl group Chemical group 0.000 claims description 6
- 150000003573 thiols Chemical class 0.000 claims description 6
- 125000003545 alkoxy group Chemical group 0.000 claims description 5
- 125000005018 aryl alkenyl group Chemical group 0.000 claims description 5
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 5
- 125000004122 cyclic group Chemical group 0.000 claims description 5
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 claims description 4
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 4
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 3
- 150000001356 alkyl thiols Chemical class 0.000 claims description 3
- 229910052786 argon Inorganic materials 0.000 claims description 3
- 125000005015 aryl alkynyl group Chemical group 0.000 claims description 3
- 125000001821 azanediyl group Chemical group [H]N(*)* 0.000 claims description 3
- 229910052799 carbon Inorganic materials 0.000 claims description 3
- 125000001424 substituent group Chemical group 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 34
- 238000011282 treatment Methods 0.000 abstract description 32
- 239000003814 drug Substances 0.000 abstract description 17
- 201000010099 disease Diseases 0.000 abstract description 13
- 229940079593 drug Drugs 0.000 abstract description 12
- 230000002265 prevention Effects 0.000 abstract description 12
- 229940054051 antipsychotic indole derivative Drugs 0.000 abstract description 3
- 238000004519 manufacturing process Methods 0.000 abstract description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 54
- DRLFMBDRBRZALE-UHFFFAOYSA-N melatonin Chemical compound COC1=CC=C2NC=C(CCNC(C)=O)C2=C1 DRLFMBDRBRZALE-UHFFFAOYSA-N 0.000 description 45
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 45
- 229960003987 melatonin Drugs 0.000 description 44
- YJPIGAIKUZMOQA-UHFFFAOYSA-N Melatonin Natural products COC1=CC=C2N(C(C)=O)C=C(CCN)C2=C1 YJPIGAIKUZMOQA-UHFFFAOYSA-N 0.000 description 41
- 239000000203 mixture Substances 0.000 description 39
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 34
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 32
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 23
- 230000000694 effects Effects 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- 208000035475 disorder Diseases 0.000 description 21
- 230000007958 sleep Effects 0.000 description 21
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 19
- 206010022489 Insulin Resistance Diseases 0.000 description 18
- 238000012360 testing method Methods 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 17
- 241000699670 Mus sp. Species 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- 239000007787 solid Substances 0.000 description 15
- 238000003786 synthesis reaction Methods 0.000 description 15
- 229910001868 water Inorganic materials 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 102000004877 Insulin Human genes 0.000 description 14
- 108090001061 Insulin Proteins 0.000 description 14
- 229940049706 benzodiazepine Drugs 0.000 description 14
- 229940125396 insulin Drugs 0.000 description 14
- 208000019116 sleep disease Diseases 0.000 description 14
- 239000011734 sodium Substances 0.000 description 14
- 210000003169 central nervous system Anatomy 0.000 description 13
- JTEJPPKMYBDEMY-UHFFFAOYSA-N 5-methoxytryptamine Chemical compound COC1=CC=C2NC=C(CCN)C2=C1 JTEJPPKMYBDEMY-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 238000009472 formulation Methods 0.000 description 12
- 241001465754 Metazoa Species 0.000 description 11
- 238000009739 binding Methods 0.000 description 11
- 210000004027 cell Anatomy 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 230000037396 body weight Effects 0.000 description 10
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 10
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 10
- 238000003760 magnetic stirring Methods 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000012300 argon atmosphere Substances 0.000 description 9
- 230000027455 binding Effects 0.000 description 9
- 230000033228 biological regulation Effects 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 238000000034 method Methods 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 206010020772 Hypertension Diseases 0.000 description 8
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- 210000004556 brain Anatomy 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 235000021588 free fatty acids Nutrition 0.000 description 8
- 208000011580 syndromic disease Diseases 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 102000027484 GABAA receptors Human genes 0.000 description 7
- 108091008681 GABAA receptors Proteins 0.000 description 7
- 238000010268 HPLC based assay Methods 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 206010028980 Neoplasm Diseases 0.000 description 7
- 208000008589 Obesity Diseases 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 230000027288 circadian rhythm Effects 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 230000000147 hypnotic effect Effects 0.000 description 7
- 235000020824 obesity Nutrition 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 7
- 238000009987 spinning Methods 0.000 description 7
- 102000040125 5-hydroxytryptamine receptor family Human genes 0.000 description 6
- 108091032151 5-hydroxytryptamine receptor family Proteins 0.000 description 6
- 229940097276 5-methoxytryptamine Drugs 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 239000013543 active substance Substances 0.000 description 6
- 210000001789 adipocyte Anatomy 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 150000001557 benzodiazepines Chemical class 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 230000002354 daily effect Effects 0.000 description 6
- 206010012601 diabetes mellitus Diseases 0.000 description 6
- 230000002124 endocrine Effects 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 230000004770 neurodegeneration Effects 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 208000020016 psychiatric disease Diseases 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 238000002390 rotary evaporation Methods 0.000 description 6
- 208000012672 seasonal affective disease Diseases 0.000 description 6
- 230000001624 sedative effect Effects 0.000 description 6
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 6
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 5
- 229940116892 5 Hydroxytryptamine 2B receptor antagonist Drugs 0.000 description 5
- 102100024956 5-hydroxytryptamine receptor 2B Human genes 0.000 description 5
- 101710138092 5-hydroxytryptamine receptor 2B Proteins 0.000 description 5
- 208000019901 Anxiety disease Diseases 0.000 description 5
- 208000019888 Circadian rhythm sleep disease Diseases 0.000 description 5
- 102000005915 GABA Receptors Human genes 0.000 description 5
- 108010005551 GABA Receptors Proteins 0.000 description 5
- 102000016267 Leptin Human genes 0.000 description 5
- 108010092277 Leptin Proteins 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 206010036618 Premenstrual syndrome Diseases 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 5
- 230000032683 aging Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 206010015037 epilepsy Diseases 0.000 description 5
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 5
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 5
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 5
- 229940039781 leptin Drugs 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- SQMCFUSVGSBKFK-UHFFFAOYSA-N sodium;5-(cyclohexen-1-yl)-1,5-dimethyl-1,3-diazinane-2,4,6-trione Chemical compound [Na+].O=C1N(C)C(=O)NC(=O)C1(C)C1=CCCCC1 SQMCFUSVGSBKFK-UHFFFAOYSA-N 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 208000006561 Cluster Headache Diseases 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 102000001419 Melatonin receptor Human genes 0.000 description 4
- 108050009605 Melatonin receptor Proteins 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- 208000019695 Migraine disease Diseases 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- 239000007832 Na2SO4 Substances 0.000 description 4
- 208000002193 Pain Diseases 0.000 description 4
- 206010062519 Poor quality sleep Diseases 0.000 description 4
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 210000000577 adipose tissue Anatomy 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 4
- 230000036506 anxiety Effects 0.000 description 4
- 230000003542 behavioural effect Effects 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 230000002060 circadian Effects 0.000 description 4
- 208000018912 cluster headache syndrome Diseases 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 208000002173 dizziness Diseases 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000013265 extended release Methods 0.000 description 4
- 230000004190 glucose uptake Effects 0.000 description 4
- 201000001421 hyperglycemia Diseases 0.000 description 4
- 230000001771 impaired effect Effects 0.000 description 4
- 206010022437 insomnia Diseases 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 230000033001 locomotion Effects 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 4
- 150000004682 monohydrates Chemical class 0.000 description 4
- 208000015122 neurodegenerative disease Diseases 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 239000000932 sedative agent Substances 0.000 description 4
- 229940076279 serotonin Drugs 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 230000004584 weight gain Effects 0.000 description 4
- 235000019786 weight gain Nutrition 0.000 description 4
- 102100024959 5-hydroxytryptamine receptor 2C Human genes 0.000 description 3
- 101710138093 5-hydroxytryptamine receptor 2C Proteins 0.000 description 3
- 208000030507 AIDS Diseases 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 208000024827 Alzheimer disease Diseases 0.000 description 3
- 206010053567 Coagulopathies Diseases 0.000 description 3
- ORGPJDKNYMVLFL-UHFFFAOYSA-N Coumalic acid Chemical compound OC(=O)C=1C=CC(=O)OC=1 ORGPJDKNYMVLFL-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 208000010412 Glaucoma Diseases 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 208000002033 Myoclonus Diseases 0.000 description 3
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 229960003529 diazepam Drugs 0.000 description 3
- 230000035558 fertility Effects 0.000 description 3
- 208000031424 hyperprolactinemia Diseases 0.000 description 3
- 239000003326 hypnotic agent Substances 0.000 description 3
- 208000026278 immune system disease Diseases 0.000 description 3
- 239000003701 inert diluent Substances 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 206010027599 migraine Diseases 0.000 description 3
- 230000037023 motor activity Effects 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 239000002858 neurotransmitter agent Substances 0.000 description 3
- 230000036407 pain Effects 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 208000006155 precocious puberty Diseases 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 201000000980 schizophrenia Diseases 0.000 description 3
- 230000001932 seasonal effect Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 230000004622 sleep time Effects 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- GBBSUAFBMRNDJC-MRXNPFEDSA-N (5R)-zopiclone Chemical compound C1CN(C)CCN1C(=O)O[C@@H]1C2=NC=CN=C2C(=O)N1C1=CC=C(Cl)C=N1 GBBSUAFBMRNDJC-MRXNPFEDSA-N 0.000 description 2
- RLWWKLWEBQOOAB-UHFFFAOYSA-N 2-(hydroxymethyl)-5-methoxypyran-4-one Chemical compound COC1=COC(CO)=CC1=O RLWWKLWEBQOOAB-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- XPCTZQVDEJYUGT-UHFFFAOYSA-N 3-hydroxy-2-methyl-4-pyrone Chemical compound CC=1OC=CC(=O)C=1O XPCTZQVDEJYUGT-UHFFFAOYSA-N 0.000 description 2
- UUUICOMMFFTCHS-UHFFFAOYSA-N 5-hydroxy-4-oxopyran-2-carboxylic acid Chemical compound OC(=O)C1=CC(=O)C(O)=CO1 UUUICOMMFFTCHS-UHFFFAOYSA-N 0.000 description 2
- 102100036321 5-hydroxytryptamine receptor 2A Human genes 0.000 description 2
- 101710138091 5-hydroxytryptamine receptor 2A Proteins 0.000 description 2
- 102100040385 5-hydroxytryptamine receptor 4 Human genes 0.000 description 2
- 101710150225 5-hydroxytryptamine receptor 4 Proteins 0.000 description 2
- JQYYVBCIIMRLDL-UHFFFAOYSA-N 5-methoxy-4-oxopyran-2-carboxylic acid Chemical compound COC1=COC(C(O)=O)=CC1=O JQYYVBCIIMRLDL-UHFFFAOYSA-N 0.000 description 2
- AUZCNXBFVCKKHV-UHFFFAOYSA-N 6-oxopyran-2-carboxylic acid Chemical compound OC(=O)C1=CC=CC(=O)O1 AUZCNXBFVCKKHV-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- 102100033367 Appetite-regulating hormone Human genes 0.000 description 2
- 206010003591 Ataxia Diseases 0.000 description 2
- 208000020706 Autistic disease Diseases 0.000 description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 2
- 241000167854 Bourreria succulenta Species 0.000 description 2
- 206010006550 Bulimia nervosa Diseases 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- PBAYDYUZOSNJGU-UHFFFAOYSA-N Chelidonic acid Chemical compound OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000000094 Chronic Pain Diseases 0.000 description 2
- 208000017164 Chronobiology disease Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- 201000010374 Down Syndrome Diseases 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 2
- 208000001640 Fibromyalgia Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 101800001586 Ghrelin Proteins 0.000 description 2
- 208000002705 Glucose Intolerance Diseases 0.000 description 2
- 206010056438 Growth hormone deficiency Diseases 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 208000023105 Huntington disease Diseases 0.000 description 2
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 2
- CBIAWPMZSFFRGN-UHFFFAOYSA-N Indiplon Chemical compound CC(=O)N(C)C1=CC=CC(C=2N3N=CC(=C3N=CC=2)C(=O)C=2SC=CC=2)=C1 CBIAWPMZSFFRGN-UHFFFAOYSA-N 0.000 description 2
- 102000003746 Insulin Receptor Human genes 0.000 description 2
- 108010001127 Insulin Receptor Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 208000028017 Psychotic disease Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 206010038910 Retinitis Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 206010039897 Sedation Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 208000010513 Stupor Diseases 0.000 description 2
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 2
- 208000034972 Sudden Infant Death Diseases 0.000 description 2
- 206010042440 Sudden infant death syndrome Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000012317 TBTU Substances 0.000 description 2
- 206010047700 Vomiting Diseases 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000005024 alkenyl aryl group Chemical group 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- VIROVYVQCGLCII-UHFFFAOYSA-N amobarbital Chemical compound CC(C)CCC1(CC)C(=O)NC(=O)NC1=O VIROVYVQCGLCII-UHFFFAOYSA-N 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 230000036592 analgesia Effects 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940125708 antidiabetic agent Drugs 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 239000000164 antipsychotic agent Substances 0.000 description 2
- 229940005529 antipsychotics Drugs 0.000 description 2
- 229940125717 barbiturate Drugs 0.000 description 2
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N benzopyrrole Natural products C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000023555 blood coagulation Effects 0.000 description 2
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- 235000019693 cherries Nutrition 0.000 description 2
- 230000019771 cognition Effects 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 201000001098 delayed sleep phase syndrome Diseases 0.000 description 2
- 208000033921 delayed sleep phase type circadian rhythm sleep disease Diseases 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 230000001804 emulsifying effect Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 229940093471 ethyl oleate Drugs 0.000 description 2
- 230000002461 excitatory amino acid Effects 0.000 description 2
- 239000003257 excitatory amino acid Substances 0.000 description 2
- 208000030533 eye disease Diseases 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 230000003371 gabaergic effect Effects 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 2
- 230000009229 glucose formation Effects 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 2
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 2
- 201000008980 hyperinsulinism Diseases 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 229950003867 indiplon Drugs 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 150000002475 indoles Chemical class 0.000 description 2
- 208000000509 infertility Diseases 0.000 description 2
- 230000036512 infertility Effects 0.000 description 2
- 231100000535 infertility Toxicity 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 210000004153 islets of langerhan Anatomy 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002974 melatonin derivative Substances 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000036651 mood Effects 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 210000000663 muscle cell Anatomy 0.000 description 2
- 230000001613 neoplastic effect Effects 0.000 description 2
- 230000000955 neuroendocrine Effects 0.000 description 2
- 201000001119 neuropathy Diseases 0.000 description 2
- 230000007823 neuropathy Effects 0.000 description 2
- 150000005830 nonesterified fatty acids Chemical class 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 229940127240 opiate Drugs 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 208000033808 peripheral neuropathy Diseases 0.000 description 2
- 230000010363 phase shift Effects 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000036280 sedation Effects 0.000 description 2
- 230000009758 senescence Effects 0.000 description 2
- 239000000952 serotonin receptor agonist Substances 0.000 description 2
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 201000002859 sleep apnea Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000008733 trauma Effects 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 229960004010 zaleplon Drugs 0.000 description 2
- HUNXMJYCHXQEGX-UHFFFAOYSA-N zaleplon Chemical compound CCN(C(C)=O)C1=CC=CC(C=2N3N=CC(=C3N=CC=2)C#N)=C1 HUNXMJYCHXQEGX-UHFFFAOYSA-N 0.000 description 2
- 229960001475 zolpidem Drugs 0.000 description 2
- ZAFYATHCZYHLPB-UHFFFAOYSA-N zolpidem Chemical compound N1=C2C=CC(C)=CN2C(CC(=O)N(C)C)=C1C1=CC=C(C)C=C1 ZAFYATHCZYHLPB-UHFFFAOYSA-N 0.000 description 2
- 229960000820 zopiclone Drugs 0.000 description 2
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 1
- VRYALKFFQXWPIH-PBXRRBTRSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)CC=O VRYALKFFQXWPIH-PBXRRBTRSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- FJIKWRGCXUCUIG-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1-methyl-3h-1,4-benzodiazepin-2-one Chemical compound O=C([C@H](O)N=1)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1Cl FJIKWRGCXUCUIG-HNNXBMFYSA-N 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- ZEHYJZXQEQOSON-AATRIKPKSA-N (e)-1-chloro-3-ethylpent-1-en-4-yn-3-ol Chemical compound CCC(O)(C#C)\C=C\Cl ZEHYJZXQEQOSON-AATRIKPKSA-N 0.000 description 1
- QECAKYKTTYQVKX-RMKNXTFCSA-N (e)-3-(2,5-dihydropyrrol-1-yl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one Chemical compound COC1=C(OC)C(OC)=CC(C(=O)\C=C\N2CC=CC2)=C1 QECAKYKTTYQVKX-RMKNXTFCSA-N 0.000 description 1
- UNFQKKSADLVQJE-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-(3-methyl-5-oxo-4h-imidazol-2-yl)urea;hydrate Chemical compound O.CN1CC(=O)N=C1NC(=O)NC1=CC=CC(Cl)=C1 UNFQKKSADLVQJE-UHFFFAOYSA-N 0.000 description 1
- WYCMFCPHWDZHMR-UHFFFAOYSA-N 1-(4,7-dimethyl-6,6a,8,9,10,10a-hexahydroindolo[4,3-fg]quinoline-9-yl)-n,n-dimethylmethanesulfonamide Chemical compound C1=CC(C2CC(CN(C)C2C2)CS(=O)(=O)N(C)C)=C3C2=CN(C)C3=C1 WYCMFCPHWDZHMR-UHFFFAOYSA-N 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- KYWMWUUMCDZISK-UHFFFAOYSA-N 2,2,5,5-tetrakis(trifluoromethyl)-1h-imidazol-4-amine Chemical compound NC1=NC(C(F)(F)F)(C(F)(F)F)NC1(C(F)(F)F)C(F)(F)F KYWMWUUMCDZISK-UHFFFAOYSA-N 0.000 description 1
- KGLZWCCDTHHPLO-UHFFFAOYSA-N 2,3-dihydropyridine-3-carboxamide Chemical compound NC(=O)C1CN=CC=C1 KGLZWCCDTHHPLO-UHFFFAOYSA-N 0.000 description 1
- TXVAYRSEKRMEIF-UHFFFAOYSA-N 2-(5-methoxy-1h-indol-3-yl)ethylazanium;chloride Chemical compound Cl.COC1=CC=C2NC=C(CCN)C2=C1 TXVAYRSEKRMEIF-UHFFFAOYSA-N 0.000 description 1
- FXZJKVODWNYPKK-UHFFFAOYSA-N 3-[3-[4-(3-chlorophenyl)piperazin-1-yl]propyl]-1h-quinazoline-2,4-dione Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(C4=CC=CC=C4NC3=O)=O)CC2)=C1 FXZJKVODWNYPKK-UHFFFAOYSA-N 0.000 description 1
- MVQVNTPHUGQQHK-UHFFFAOYSA-N 3-pyridinemethanol Chemical compound OCC1=CC=CN=C1 MVQVNTPHUGQQHK-UHFFFAOYSA-N 0.000 description 1
- PCTRYMLLRKWXGF-UHFFFAOYSA-N 4-(butylamino)-1-ethyl-6-methyl-5-pyrazolo[3,4-b]pyridinecarboxylic acid ethyl ester Chemical compound CCCCNC1=C(C(=O)OCC)C(C)=NC2=C1C=NN2CC PCTRYMLLRKWXGF-UHFFFAOYSA-N 0.000 description 1
- QXIUMMLTJVHILT-UHFFFAOYSA-N 4-[3-(tert-butylamino)-2-hydroxypropoxy]-1H-indole-2-carbonitrile Chemical compound CC(C)(C)NCC(O)COC1=CC=CC2=C1C=C(C#N)N2 QXIUMMLTJVHILT-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- UAHFGYDRQSXQEB-PWPYQVNISA-N 4-nle-α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 UAHFGYDRQSXQEB-PWPYQVNISA-N 0.000 description 1
- UFMDRPPBUAPWBK-UHFFFAOYSA-N 4-oxopyran-2-carboxylic acid Chemical compound OC(=O)C1=CC(=O)C=CO1 UFMDRPPBUAPWBK-UHFFFAOYSA-N 0.000 description 1
- 108091005436 5-HT7 receptors Proteins 0.000 description 1
- NFFXEUUOMTXWCX-UHFFFAOYSA-N 5-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]-2-methoxy-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound C1=C(C(=O)NCC=2C=CC(=CC=2)C(F)(F)F)C(OC)=CC=C1CC1SC(=O)NC1=O NFFXEUUOMTXWCX-UHFFFAOYSA-N 0.000 description 1
- 102100022738 5-hydroxytryptamine receptor 1A Human genes 0.000 description 1
- 101710138638 5-hydroxytryptamine receptor 1A Proteins 0.000 description 1
- CDAZYPQZWWYWDX-UHFFFAOYSA-N 7-chloro-n-(cyclopropylmethyl)-4-hydroxy-5-phenyl-3h-1,4-benzodiazepin-2-imine Chemical compound N1=C2C=CC(Cl)=CC2=C(C=2C=CC=CC=2)N(O)CC1=NCC1CC1 CDAZYPQZWWYWDX-UHFFFAOYSA-N 0.000 description 1
- ASXGJMSKWNBENU-UHFFFAOYSA-N 8-OH-DPAT Chemical compound C1=CC(O)=C2CC(N(CCC)CCC)CCC2=C1 ASXGJMSKWNBENU-UHFFFAOYSA-N 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- FDQGNLOWMMVRQL-UHFFFAOYSA-N Allobarbital Chemical compound C=CCC1(CC=C)C(=O)NC(=O)NC1=O FDQGNLOWMMVRQL-UHFFFAOYSA-N 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 208000006096 Attention Deficit Disorder with Hyperactivity Diseases 0.000 description 1
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 description 1
- 206010004385 Benign neoplasm of prostate Diseases 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- UMSGKTJDUHERQW-UHFFFAOYSA-N Brotizolam Chemical compound C1=2C=C(Br)SC=2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl UMSGKTJDUHERQW-UHFFFAOYSA-N 0.000 description 1
- QORQZMBCPRBCAB-UHFFFAOYSA-M Butabarbital sodium Chemical compound [Na+].CCC(C)C1(CC)C(=O)NC([O-])=NC1=O QORQZMBCPRBCAB-UHFFFAOYSA-M 0.000 description 1
- 102000000584 Calmodulin Human genes 0.000 description 1
- 108010041952 Calmodulin Proteins 0.000 description 1
- 206010007270 Carcinoid syndrome Diseases 0.000 description 1
- 201000005947 Carney Complex Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 1
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 206010010219 Compulsions Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 208000032538 Depersonalisation Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 description 1
- 208000007590 Disorders of Excessive Somnolence Diseases 0.000 description 1
- 206010013754 Drug withdrawal syndrome Diseases 0.000 description 1
- 206010014486 Elevated triglycerides Diseases 0.000 description 1
- 208000027534 Emotional disease Diseases 0.000 description 1
- 208000008967 Enuresis Diseases 0.000 description 1
- ZFDIRQKJPRINOQ-HWKANZROSA-N Ethyl crotonate Chemical compound CCOC(=O)\C=C\C ZFDIRQKJPRINOQ-HWKANZROSA-N 0.000 description 1
- 229940123457 Free radical scavenger Drugs 0.000 description 1
- 230000009508 GABAergic inhibition Effects 0.000 description 1
- ZXRVKCBLGJOCEE-UHFFFAOYSA-N Gaboxadol Chemical compound C1NCCC2=C1ONC2=O ZXRVKCBLGJOCEE-UHFFFAOYSA-N 0.000 description 1
- 102000058061 Glucose Transporter Type 4 Human genes 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- JMBQKKAJIKAWKF-UHFFFAOYSA-N Glutethimide Chemical compound C=1C=CC=CC=1C1(CC)CCC(=O)NC1=O JMBQKKAJIKAWKF-UHFFFAOYSA-N 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 102000018997 Growth Hormone Human genes 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- WYCLKVQLVUQKNZ-UHFFFAOYSA-N Halazepam Chemical compound N=1CC(=O)N(CC(F)(F)F)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 WYCLKVQLVUQKNZ-UHFFFAOYSA-N 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 208000031773 Insulin resistance syndrome Diseases 0.000 description 1
- 206010022491 Insulin resistant diabetes Diseases 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- PWWVAXIEGOYWEE-UHFFFAOYSA-N Isophenergan Chemical compound C1=CC=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 PWWVAXIEGOYWEE-UHFFFAOYSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- HYMLWHLQFGRFIY-UHFFFAOYSA-N Maltol Natural products CC1OC=CC(=O)C1=O HYMLWHLQFGRFIY-UHFFFAOYSA-N 0.000 description 1
- 206010026749 Mania Diseases 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- NPPQSCRMBWNHMW-UHFFFAOYSA-N Meprobamate Chemical compound NC(=O)OCC(C)(CCC)COC(N)=O NPPQSCRMBWNHMW-UHFFFAOYSA-N 0.000 description 1
- JEYCTXHKTXCGPB-UHFFFAOYSA-N Methaqualone Chemical compound CC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1C JEYCTXHKTXCGPB-UHFFFAOYSA-N 0.000 description 1
- UEQUQVLFIPOEMF-UHFFFAOYSA-N Mianserin Chemical compound C1C2=CC=CC=C2N2CCN(C)CC2C2=CC=CC=C21 UEQUQVLFIPOEMF-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 206010027603 Migraine headaches Diseases 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000000112 Myalgia Diseases 0.000 description 1
- LUINDDOUWHRIPW-UHFFFAOYSA-N N-[2-(6-chloro-5-methoxy-1H-indol-3-yl)ethyl]acetamide Chemical compound C1=C(Cl)C(OC)=CC2=C1NC=C2CCNC(C)=O LUINDDOUWHRIPW-UHFFFAOYSA-N 0.000 description 1
- YLXDSYKOBKBWJQ-LBPRGKRZSA-N N-[2-[(8S)-2,6,7,8-tetrahydro-1H-cyclopenta[e]benzofuran-8-yl]ethyl]propanamide Chemical compound C1=C2OCCC2=C2[C@H](CCNC(=O)CC)CCC2=C1 YLXDSYKOBKBWJQ-LBPRGKRZSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 241001181114 Neta Species 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- 208000000224 Night Terrors Diseases 0.000 description 1
- 229940121985 Non-benzodiazepine hypnotic Drugs 0.000 description 1
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 description 1
- 206010057342 Onychophagia Diseases 0.000 description 1
- 102000003840 Opioid Receptors Human genes 0.000 description 1
- 108090000137 Opioid Receptors Proteins 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 102000023984 PPAR alpha Human genes 0.000 description 1
- 102000000536 PPAR gamma Human genes 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 206010033888 Paraphilia Diseases 0.000 description 1
- 235000011925 Passiflora alata Nutrition 0.000 description 1
- 235000000370 Passiflora edulis Nutrition 0.000 description 1
- 235000011922 Passiflora incarnata Nutrition 0.000 description 1
- 240000002690 Passiflora mixta Species 0.000 description 1
- 235000013750 Passiflora mixta Nutrition 0.000 description 1
- 235000013731 Passiflora van volxemii Nutrition 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 1
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 1
- RMUCZJUITONUFY-UHFFFAOYSA-N Phenelzine Chemical compound NNCCC1=CC=CC=C1 RMUCZJUITONUFY-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010070606 Post stroke depression Diseases 0.000 description 1
- 208000021129 Postpartum disease Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- MWQCHHACWWAQLJ-UHFFFAOYSA-N Prazepam Chemical compound O=C1CN=C(C=2C=CC=CC=2)C2=CC(Cl)=CC=C2N1CC1CC1 MWQCHHACWWAQLJ-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- IKMPWMZBZSAONZ-UHFFFAOYSA-N Quazepam Chemical compound FC1=CC=CC=C1C1=NCC(=S)N(CC(F)(F)F)C2=CC=C(Cl)C=C12 IKMPWMZBZSAONZ-UHFFFAOYSA-N 0.000 description 1
- 208000012322 Raynaud phenomenon Diseases 0.000 description 1
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 108091006300 SLC2A4 Proteins 0.000 description 1
- ZRIHAIZYIMGOAB-UHFFFAOYSA-N Secbutobarbitone Natural products CCC(C)C1(CC)C(=O)NC(=O)NC1=O ZRIHAIZYIMGOAB-UHFFFAOYSA-N 0.000 description 1
- 201000001880 Sexual dysfunction Diseases 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 206010040991 Sleep disorders and disturbances Diseases 0.000 description 1
- 206010041010 Sleep terror Diseases 0.000 description 1
- 206010041250 Social phobia Diseases 0.000 description 1
- 208000027520 Somatoform disease Diseases 0.000 description 1
- 206010041347 Somnambulism Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- SEQDDYPDSLOBDC-UHFFFAOYSA-N Temazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 SEQDDYPDSLOBDC-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- KLBQZWRITKRQQV-UHFFFAOYSA-N Thioridazine Chemical compound C12=CC(SC)=CC=C2SC2=CC=CC=C2N1CCC1CCCCN1C KLBQZWRITKRQQV-UHFFFAOYSA-N 0.000 description 1
- 208000016620 Tourette disease Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 1
- 206010046543 Urinary incontinence Diseases 0.000 description 1
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- 208000021017 Weight Gain Diseases 0.000 description 1
- GDSCFOSHSOWNDL-UHFFFAOYSA-N Zolasepam Chemical compound N=1CC(=O)N(C)C(N(N=C2C)C)=C2C=1C1=CC=CC=C1F GDSCFOSHSOWNDL-UHFFFAOYSA-N 0.000 description 1
- MOZPSIXKYJUTKI-RLXJOQACSA-N [1-[2-(methanesulfonamido)ethyl]piperidin-4-yl]methyl 1-(tritritiomethyl)indole-3-carboxylate Chemical compound C12=CC=CC=C2N(C([3H])([3H])[3H])C=C1C(=O)OCC1CCN(CCNS(C)(=O)=O)CC1 MOZPSIXKYJUTKI-RLXJOQACSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 229960003148 adinazolam Drugs 0.000 description 1
- GJSLOMWRLALDCT-UHFFFAOYSA-N adinazolam Chemical compound C12=CC(Cl)=CC=C2N2C(CN(C)C)=NN=C2CN=C1C1=CC=CC=C1 GJSLOMWRLALDCT-UHFFFAOYSA-N 0.000 description 1
- 239000000808 adrenergic beta-agonist Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 201000007930 alcohol dependence Diseases 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 230000008856 allosteric binding Effects 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- PMMURAAUARKVCB-UHFFFAOYSA-N alpha-D-ara-dHexp Natural products OCC1OC(O)CC(O)C1O PMMURAAUARKVCB-UHFFFAOYSA-N 0.000 description 1
- 229960004538 alprazolam Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960000836 amitriptyline Drugs 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 229960001301 amobarbital Drugs 0.000 description 1
- 229960002519 amoxapine Drugs 0.000 description 1
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 description 1
- 230000001195 anabolic effect Effects 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 239000000883 anti-obesity agent Substances 0.000 description 1
- 239000003529 anticholesteremic agent Substances 0.000 description 1
- 229940127226 anticholesterol agent Drugs 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- 239000003420 antiserotonin agent Substances 0.000 description 1
- 230000000338 anxiogenic effect Effects 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 description 1
- 210000003403 autonomic nervous system Anatomy 0.000 description 1
- CREXVNNSNOKDHW-UHFFFAOYSA-N azaniumylideneazanide Chemical group N[N] CREXVNNSNOKDHW-UHFFFAOYSA-N 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- AIZFEOPQVZBNGH-UHFFFAOYSA-N bentazepam Chemical compound C1=2C=3CCCCC=3SC=2NC(=O)CN=C1C1=CC=CC=C1 AIZFEOPQVZBNGH-UHFFFAOYSA-N 0.000 description 1
- 229950001957 bentazepam Drugs 0.000 description 1
- 229960001303 benzoctamine Drugs 0.000 description 1
- GNRXCIONJWKSEA-UHFFFAOYSA-N benzoctamine Chemical compound C12=CC=CC=C2C2(CNC)C3=CC=CC=C3C1CC2 GNRXCIONJWKSEA-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- IEPBPSSCIZTJIF-UHFFFAOYSA-N bis(2,2,2-trichloroethyl) carbonate Chemical compound ClC(Cl)(Cl)COC(=O)OCC(Cl)(Cl)Cl IEPBPSSCIZTJIF-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 208000022266 body dysmorphic disease Diseases 0.000 description 1
- 208000030963 borderline personality disease Diseases 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 229960003051 brotizolam Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960001058 bupropion Drugs 0.000 description 1
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 1
- 229940015694 butabarbital Drugs 0.000 description 1
- UZVHFVZFNXBMQJ-UHFFFAOYSA-N butalbital Chemical compound CC(C)CC1(CC=C)C(=O)NC(=O)NC1=O UZVHFVZFNXBMQJ-UHFFFAOYSA-N 0.000 description 1
- 229960002546 butalbital Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- HLSLSXBFTXUKCY-UHFFFAOYSA-N capuride Chemical compound CCC(C)C(CC)C(=O)NC(N)=O HLSLSXBFTXUKCY-UHFFFAOYSA-N 0.000 description 1
- 229950003152 capuride Drugs 0.000 description 1
- ITMSAWKLJVGBIT-UHFFFAOYSA-N carbocloral Chemical compound CCOC(=O)NC(O)C(Cl)(Cl)Cl ITMSAWKLJVGBIT-UHFFFAOYSA-N 0.000 description 1
- 229950003854 carbocloral Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000003280 chronobiological effect Effects 0.000 description 1
- 230000008632 circadian clock Effects 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 229960004606 clomipramine Drugs 0.000 description 1
- 229950000551 cloperidone Drugs 0.000 description 1
- 229960004362 clorazepate Drugs 0.000 description 1
- XDDJGVMJFWAHJX-UHFFFAOYSA-N clorazepic acid Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)O)N=C1C1=CC=CC=C1 XDDJGVMJFWAHJX-UHFFFAOYSA-N 0.000 description 1
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 description 1
- 229960004170 clozapine Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000011970 concomitant therapy Methods 0.000 description 1
- 230000002844 continuous effect Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000036757 core body temperature Effects 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 1
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- UPMOVJBGNREKJV-CQOQZXRMSA-N dexclamol Chemical compound C12=CC=CC=C2CCC2=CC=CC3=C2[C@@H]1CN1CC[C@@](C(C)C)(O)C[C@@H]13 UPMOVJBGNREKJV-CQOQZXRMSA-N 0.000 description 1
- 229950005215 dexclamol Drugs 0.000 description 1
- 125000004985 dialkyl amino alkyl group Chemical group 0.000 description 1
- ATKXDQOHNICLQW-UHFFFAOYSA-N dichloralphenazone Chemical compound OC(O)C(Cl)(Cl)Cl.OC(O)C(Cl)(Cl)Cl.CN1C(C)=CC(=O)N1C1=CC=CC=C1 ATKXDQOHNICLQW-UHFFFAOYSA-N 0.000 description 1
- 229960005422 dichloralphenazone Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 1
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical compound C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 229960000520 diphenhydramine Drugs 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229960005426 doxepin Drugs 0.000 description 1
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 208000024732 dysthymic disease Diseases 0.000 description 1
- 230000007937 eating Effects 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 201000010063 epididymitis Diseases 0.000 description 1
- CDCHDCWJMGXXRH-UHFFFAOYSA-N estazolam Chemical compound C=1C(Cl)=CC=C(N2C=NN=C2CN=2)C=1C=2C1=CC=CC=C1 CDCHDCWJMGXXRH-UHFFFAOYSA-N 0.000 description 1
- 229960002336 estazolam Drugs 0.000 description 1
- GBBSUAFBMRNDJC-INIZCTEOSA-N eszopiclone Chemical compound C1CN(C)CCN1C(=O)O[C@H]1C2=NC=CN=C2C(=O)N1C1=CC=C(Cl)C=N1 GBBSUAFBMRNDJC-INIZCTEOSA-N 0.000 description 1
- 229960001578 eszopiclone Drugs 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 1
- 229960001690 etomidate Drugs 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000004880 explosion Methods 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 229950002489 fenobam Drugs 0.000 description 1
- 229960002297 fenofibrate Drugs 0.000 description 1
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 1
- 229960002200 flunitrazepam Drugs 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 229960003528 flurazepam Drugs 0.000 description 1
- SAADBVWGJQAEFS-UHFFFAOYSA-N flurazepam Chemical compound N=1CC(=O)N(CCN(CC)CC)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F SAADBVWGJQAEFS-UHFFFAOYSA-N 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229950004346 gaboxadol Drugs 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 230000004110 gluconeogenesis Effects 0.000 description 1
- 230000006377 glucose transport Effects 0.000 description 1
- 229960002972 glutethimide Drugs 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 230000004116 glycogenolysis Effects 0.000 description 1
- 210000002149 gonad Anatomy 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 229960002158 halazepam Drugs 0.000 description 1
- 230000008821 health effect Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- ZQDWXGKKHFNSQK-UHFFFAOYSA-N hydroxyzine Chemical compound C1CN(CCOCCO)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZQDWXGKKHFNSQK-UHFFFAOYSA-N 0.000 description 1
- 229960000930 hydroxyzine Drugs 0.000 description 1
- 208000013403 hyperactivity Diseases 0.000 description 1
- 206010020765 hypersomnia Diseases 0.000 description 1
- 239000005554 hypnotics and sedatives Substances 0.000 description 1
- 230000002631 hypothermal effect Effects 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000005032 impulse control Effects 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 208000035231 inattentive type attention deficit hyperactivity disease Diseases 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- FPCCSQOGAWCVBH-UHFFFAOYSA-N ketanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=O)=O)CC1 FPCCSQOGAWCVBH-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- BEJNERDRQOWKJM-UHFFFAOYSA-N kojic acid Chemical compound OCC1=CC(=O)C(O)=CO1 BEJNERDRQOWKJM-UHFFFAOYSA-N 0.000 description 1
- 229960004705 kojic acid Drugs 0.000 description 1
- WZNJWVWKTVETCG-UHFFFAOYSA-N kojic acid Natural products OC(=O)C(N)CN1C=CC(=O)C(O)=C1 WZNJWVWKTVETCG-UHFFFAOYSA-N 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000013016 learning Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 230000004130 lipolysis Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960001078 lithium Drugs 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 229960004033 lormetazepam Drugs 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 208000024714 major depressive disease Diseases 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940043353 maltol Drugs 0.000 description 1
- 210000005171 mammalian brain Anatomy 0.000 description 1
- 229960004090 maprotiline Drugs 0.000 description 1
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 description 1
- SFITWQDBYUMAPS-UHFFFAOYSA-N mecloqualone Chemical compound CC1=NC2=CC=CC=C2C(=O)N1C1=CC=CC=C1Cl SFITWQDBYUMAPS-UHFFFAOYSA-N 0.000 description 1
- 229950007403 mecloqualone Drugs 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 230000001193 melatoninergic effect Effects 0.000 description 1
- 230000012241 membrane hyperpolarization Effects 0.000 description 1
- 230000010291 membrane polarization Effects 0.000 description 1
- 230000015654 memory Effects 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- ALARQZQTBTVLJV-UHFFFAOYSA-N mephobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)N(C)C1=O ALARQZQTBTVLJV-UHFFFAOYSA-N 0.000 description 1
- 229960004815 meprobamate Drugs 0.000 description 1
- WZHJKEUHNJHDLS-QTGUNEKASA-N metergoline Chemical compound C([C@H]1CN([C@H]2[C@@H](C=3C=CC=C4N(C)C=C(C=34)C2)C1)C)NC(=O)OCC1=CC=CC=C1 WZHJKEUHNJHDLS-QTGUNEKASA-N 0.000 description 1
- 229960004650 metergoline Drugs 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 229960002803 methaqualone Drugs 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- 229960001703 methylphenobarbital Drugs 0.000 description 1
- 229960003955 mianserin Drugs 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229950010642 midaflur Drugs 0.000 description 1
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 1
- 229960003793 midazolam Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- VDCLSGXZVUDARN-UHFFFAOYSA-N molecular bromine;pyridine;hydrobromide Chemical compound Br.BrBr.C1=CC=NC=C1 VDCLSGXZVUDARN-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 208000013465 muscle pain Diseases 0.000 description 1
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 1
- 201000003631 narcolepsy Diseases 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 1
- 229960001800 nefazodone Drugs 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000008587 neuronal excitability Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960004738 nicotinyl alcohol Drugs 0.000 description 1
- 229960001454 nitrazepam Drugs 0.000 description 1
- KJONHKAYOJNZEC-UHFFFAOYSA-N nitrazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1 KJONHKAYOJNZEC-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 229960001158 nortriptyline Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229960005343 ondansetron Drugs 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000016087 ovulation Effects 0.000 description 1
- 208000027753 pain disease Diseases 0.000 description 1
- 230000008533 pain sensitivity Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 208000023515 periodic limb movement disease Diseases 0.000 description 1
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229960000964 phenelzine Drugs 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 238000001126 phototherapy Methods 0.000 description 1
- 125000005544 phthalimido group Chemical group 0.000 description 1
- 208000024335 physical disease Diseases 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 208000028173 post-traumatic stress disease Diseases 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 229960004856 prazepam Drugs 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 206010036596 premature ejaculation Diseases 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- 230000000216 proconvulsive effect Effects 0.000 description 1
- 229960003910 promethazine Drugs 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 229960004134 propofol Drugs 0.000 description 1
- OLBCVFGFOZPWHH-UHFFFAOYSA-N propofol Chemical compound CC(C)C1=CC=CC(C(C)C)=C1O OLBCVFGFOZPWHH-UHFFFAOYSA-N 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 229960002601 protriptyline Drugs 0.000 description 1
- BWPIARFWQZKAIA-UHFFFAOYSA-N protriptyline Chemical compound C1=CC2=CC=CC=C2C(CCCNC)C2=CC=CC=C21 BWPIARFWQZKAIA-UHFFFAOYSA-N 0.000 description 1
- 230000008141 pubertal development Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- JQRYUMGHOUYJFW-UHFFFAOYSA-N pyridine;trihydrobromide Chemical compound [Br-].[Br-].[Br-].C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1 JQRYUMGHOUYJFW-UHFFFAOYSA-N 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 229960001964 quazepam Drugs 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229960001150 ramelteon Drugs 0.000 description 1
- 230000004461 rapid eye movement Effects 0.000 description 1
- 230000036385 rapid eye movement (rem) sleep Effects 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229950004797 reclazepam Drugs 0.000 description 1
- MQGIGGJUPITZSE-UHFFFAOYSA-N reclazepam Chemical compound C12=CC(Cl)=CC=C2N(C=2OCC(=O)N=2)CCN=C1C1=CC=CC=C1Cl MQGIGGJUPITZSE-UHFFFAOYSA-N 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 230000003893 regulation of appetite Effects 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 230000003938 response to stress Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 229950004692 roletamide Drugs 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229960002060 secobarbital Drugs 0.000 description 1
- KQPKPCNLIDLUMF-UHFFFAOYSA-N secobarbital Chemical compound CCCC(C)C1(CC=C)C(=O)NC(=O)NC1=O KQPKPCNLIDLUMF-UHFFFAOYSA-N 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 230000000276 sedentary effect Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000003352 sequestering agent Substances 0.000 description 1
- 230000000862 serotonergic effect Effects 0.000 description 1
- 239000003215 serotonin 5-HT2 receptor antagonist Substances 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 231100000872 sexual dysfunction Toxicity 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 230000036299 sexual function Effects 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 229910001923 silver oxide Inorganic materials 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 208000022925 sleep disturbance Diseases 0.000 description 1
- 230000003860 sleep quality Effects 0.000 description 1
- 208000020685 sleep-wake disease Diseases 0.000 description 1
- 230000037322 slow-wave sleep Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- AEQFSUDEHCCHBT-UHFFFAOYSA-M sodium valproate Chemical compound [Na+].CCCC(C([O-])=O)CCC AEQFSUDEHCCHBT-UHFFFAOYSA-M 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- WZAIVXXKOAWTGQ-UHFFFAOYSA-N spiro[2,3-dihydronaphthalene-4,3'-piperidine]-1,2',6'-trione Chemical compound O=C1NC(=O)CCC11C2=CC=CC=C2C(=O)CC1 WZAIVXXKOAWTGQ-UHFFFAOYSA-N 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 229950003877 suproclone Drugs 0.000 description 1
- IBAUKGNDWVSETP-UHFFFAOYSA-N suproclone Chemical compound C1CN(C(=O)CC)CCN1C(=O)OC1C(SCCS2)=C2C(=O)N1C1=CC=C(C=CC(Cl)=N2)C2=N1 IBAUKGNDWVSETP-UHFFFAOYSA-N 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 210000000225 synapse Anatomy 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960003188 temazepam Drugs 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 229960002784 thioridazine Drugs 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 229950002859 tracazolate Drugs 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- ZFDIRQKJPRINOQ-UHFFFAOYSA-N transbutenic acid ethyl ester Natural products CCOC(=O)C=CC ZFDIRQKJPRINOQ-UHFFFAOYSA-N 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 229960003991 trazodone Drugs 0.000 description 1
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- HFFLGKNGCAIQMO-UHFFFAOYSA-N trichloroacetaldehyde Chemical compound ClC(Cl)(Cl)C=O HFFLGKNGCAIQMO-UHFFFAOYSA-N 0.000 description 1
- 208000002271 trichotillomania Diseases 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 229950004526 uldazepam Drugs 0.000 description 1
- DTMPGSXFUXZBDK-UHFFFAOYSA-N uldazepam Chemical compound C12=CC(Cl)=CC=C2N=C(NOCC=C)CN=C1C1=CC=CC=C1Cl DTMPGSXFUXZBDK-UHFFFAOYSA-N 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229940102566 valproate Drugs 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 229960003726 vasopressin Drugs 0.000 description 1
- 229960004688 venlafaxine Drugs 0.000 description 1
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 description 1
- 230000002618 waking effect Effects 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- 229960001366 zolazepam Drugs 0.000 description 1
- 230000003820 β-cell dysfunction Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Addiction (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Hospice & Palliative Care (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Psychology (AREA)
- Immunology (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
Abstract
composto, formulação farmacêutica, e uso de um composto, sal ou estereoisômero a presente invenção refere-se a novos derivados de pirona-indol, às formulações farmacêuticas que contêm os mesmos e à utilização dos compostos na manufatura de medicamentos para o tratamento ou a prevenção de várias doenças.
Description
[001] Foi verificado que as alfa e gama-pironas são classes de compostos que são ligados a diversas características comportamentais e farmacológicas que incluem os efeitos sedativos, antiansiolíticos, neuroprotetores e antioxidantes. Especificamente, um derivado de gama-pirona chamado maltol foi isolado da flor do maracujá, e foi verificado que causa a sedação do sistema nervoso central (SNC) e uma redução na agitação induzida por cafeína e na motilidade espontânea em animais; estes efeitos são mediados através da ativação dos receptores do ácido gama-aminobutírico (GABA) (Soulimani et al., J. Ethnopharmacology 57: 11, 1997; Dhawan et al., J. Ethnopharmacology 78: 165-70, 2001). Foi mostrado que outros elementos desta família, os ácidos gama-pironas comênico, inecônico e quelidônico exercem efeitos sedativos através da interação com os receptores opióides (Pedido de Patente Norte-americano N° 2003/0181516).
[002] A superfamília do receptor de GABAa representa uma das classes de receptores através da qual o neurotransmissor inibitório principal, GABA, age.Extensamente, embora desigualmente distribuídos através do cérebro de mamífero, estes receptores e, em particular, um complexo de proteínas chamado de receptor de GABAa, causa alterações na condutância de cloreto e na polarização da membrana (Mehta e Ticku, Brain Res. Brain Rev. 29:196-217, 1999).
[003] As drogas de benzodiazepina exercem as suas ações hipnótica, analgésica e antiansiolítica ao interagirem com os sítios de ligação de benzodiazepina no receptor de GABAa. Além do sítio de ligação à benzodiazepina, o receptor de GABAa contém diversos sítios de interação distintos com outras classes de drogas que modulam as atividades GABAérgicas, que incluem agentes hipnóticos que não de benzodiazepina (por exemplo, zolpidem, zaleplon, indiplon, zopiclone) (Sanger, CNS Drugs 18 (Suppl. 1): 9-15, 2004), esteróides, pictrotoxina e barbituratos. Os sítios de ligação de benzodiazepina e que não de benzodiazepina no complexo do receptor de GABAa não são sobrepostos ao GABA ou a quaisquer outros sítios de ligação de drogas (consultar, por exemplo, Cooper, et al., The Biochemical Basis of Neuropharmacology, 6a ed., pp. 145-148, Oxford University Press, New York, 1991). Os estudos eletrofisiológicos indicam que a ação principal das benzodiazepinas e que não de benzodiazepinas é a intensificação da inibição GABAérgica de excitabilidade neuronal. Isto se dá devido à potenciação do influxo de cloreto induzido por GABA nas células e subsequentemente a hiperpolarização da membrana. A modulação alossetérica clinicamente importante dos receptores de GABA por benzodiazepinas e por não- benzodiazepinas foi uma área de descoberta farmacológica intensa nos últimos anos. Os agonistas que agem no sítio da benzodiazepina são conhecidos por exibir efeitos antiansiolíticos, sedativos e hipnóticos, sendo que os compostos que agem como agonistas inversos neste sítio provocam efeitos ansiogênicos, intensificadores da cognição e pró- convulsivantes (Dawson et al., CNS Spectr. 10:21-7, 2005).
[004] Os principais distúrbios para os quais os receptores GABAa representam alvos terapêuticos importantes incluem distúrbios de ansiedade, distúrbios cognitivos, epilepsias, distúrbios do humor, esquizofrenia, dor e distúrbios do sono. Os moduladores do receptor de GABA são conhecidos por desempenhar um papel importante no sono, e os moduladores alostéricos positivos dos receptores de GABAa são extensamente utilizados para promover e manter o sono em uma variedade de distúrbios primários e secundários do sono (Sanger, CNS Drugs, 18 (Suppl. 1): 9-15, 2004).
[005] Embora as benzodiazepinas tenham um longo histórico de utilização farmacêutica como antiansiolíticos, estes compostos frequentemente exibem uma série de efeitos colaterais não desejados. Estes podem incluir a dificuldade cognitiva, sedação, ataxia, potenciação de efeitos do etanol, risco de quedas aumentado e uma tendência à tolerância e à dependência de drogas. Um aspecto importante destas atividades é o efeito diário residual que tem por resultado a dificuldade de vigilância durante o dia. Portanto, novos moduladores do receptor de GABA com efeitos colaterais menos refratários são procurados.
[006] Os compostos de indol, especificamente aqueles relacionados à serotonina (5-hidróxitriptamina; 5-HT) e à melatonina (N-aceti1-5-metóxi-triptamina) têm efeitos profundos no SNC e afetam, desse modo, o sono, a vigília, o apetite e o humor. Há uma série extensa de áreas clinicamente relevantes em que a participação do sistema da melatonina foi demonstrada (Bubenik et al. Biol. Signal s Recept. 7: 195-219, 1998). Estas incluem a regulação da temperatura corpórea central (Strassman et al., J. Appl. Physiol. 71: 2178-2182, 1991; Krauchi et al. J. Appl, Physiol. 83: 134-9, 1997), respostas imunológicas, (Maestroni e Conti, J. Neuroimmun. 28:167-176 1990; Fraschini et al., Acta Oncol. 29:775-776, 1990; Guerrero e Rei ter, Endocr. Res. 18:91-113, 1992), o desenvolvimento puberal, a ovulação, a reprodução sazonal, a gordura retroperitoneal e epididimal, bem como os níveis de insulina do plasma, leptina, hormônio do crescimento e grelina (Rasmussen et al., Endocrinology 140:1009-12, 1999; Cramer et al., Arzeneim-Forsch 26:1076-1078, 1976; Wright et al., Clin. Endocrinol. 24: 375-382, 1986, Paccotti et al, Chronobiology 15: 27 9-288, 1988; Valcavi et al., Clin. Endocrinol. 39: 139-199, 1993; Mustonen et al. Endocrine 16:43-6, 2001), ritmos do cortisol, pressão ocular (Sampes et al. Curr. Eye Res. 7:649-653, 1988; Rhode et al., Ophthalmic Res. 25: 10-15, 1993), pressão sanguínea (Scheer et al., Hypertension 43-192-7, 2004), metabolismo da glicose, grelina, leptina e massa de gordura corpórea, vasopressina e excreção de urina (Song et al., FASEB J. 11:93-100, 1997; Yasin et al. Brain Res. Bull. 39:1-5, 1997). Em alguns casos, os distúrbios psiquiátricos podem ter etiologias cronobiológicas subjacentes (por exemplo, distúrbio afetivo sazonal) e são candidatos definitivos para a terapia de melatonina (Miller, Altern. Med. Rev. 10:5-13, 2005). A melatonina também age como um removedor de radicais livres e um antioxidante (Pooggeler et al., J. Pineal Res. 14: 151-168, 1993).
[007] Há uma forte evidência que a melatonina regula especificamente o sono e a vigília nos seres humanos. A melatonina foi administrada para re-sincronizar os ritmos circadianos que estão fora da fase com o ciclo fotoperiódico local. Por exemplo, os distúrbios do sono/vigília causados pelo cruzamento rápido de zonas de tempo (tontura de fuso horário), pacientes com a síndrome da fase do sono atrasada (DSPS), turno de trabalho e cegueira total, podem ser tratados com a melatonina ou com análogos de melatonina (consultar as Patentes Norte- americanas N°. 4.600.723 e 4 .666.086 de Short et al. e 5.242.941 de Lewy et al.). Além disso, a melatonina tem propriedades sedativas hipnóticas diretas em indivíduos humanos normais e insones (por exemplo, Luboshizslcy et al. Sleep Med. Rev. 2:191-202, 1998; Patente Norte- americana N°. 5.403. 851 de D'Orlando et al.). Foi demonstrado que os distúrbios do sono em pessoas idosas respondem ao tratamento com melatonina (Garfinkel et al., Lancet 346:541-543, 1995; Pandi-Perumal et al., Exp. Gerontol. 40:911-25, 2005; Patente Norte-americana N° 5.498.423 de Zisapel). A melatonina e seus análogos reduzem a latência ao início do sono nos pacientes com insânia (Roth et l., Sleep, 28:303-7, 2005, Zhdanova et al. CI in. Pharmacol. Ther. 57:552-8, 1995) ou a depressão (Pape et al., Neuropsychopharmacology 28:694-703, 2003) e intensificam particularmente o valor reparador do sono em pacientes com insânia, tendo por resultado a vigília intensificada durante o dia (Zisapel, Pedido de Patente PCT WO N° 03/015690).
[008] Há um amplo espectro de respostas sintomáticas aos tratamentos com melatonina em distúrbios diferentes. Estes incluem ansiedade (Loiseu Eur. Neuropsychopharmacol. 2005), crises convulsivas (Munoz-Hoyos et al., J. Chi Id. Neurol. 13:501-9, 1998), dor (Ray et al. 15 J. Indian Med. Sci. 58:122-30, 2004) cefaléia em salvas e enxaqueca (Peres, Cephalalgia 25:403-11, 2005), depressão, mania e esquizofrenia (consultar Dobocovich “Antidepressant Agents”, Patente Norte-americana N°. 5.093.352; Shamir et al., J. Clin. Psychopharmacol. 20: 691-4, 2000) glaucoma, envelhecimento, estresse (Armstrong e Redman, Med. Hypotheses 34:300-309, 1991; Reiter, Bioassays 14:169-175, 1992), hipertensão (Scheer et al., Hypertension 43:192-7 2004, Zisapel, Pedido de Patente Norte-americano N° 10/169.467), síndrome da abstinência de drogas (Zisapel, Patente Norte- americana N°. 6.469.044), osteoporose (Cardinali et al., J. Pineal Res. 34:81-7, 2003), vários cânceres (Gonzalez et al., Melanoma Res. 1:237-243, 1991; Lissoni et al., Eur. J. Cancer 29A:185-189, 1993; Blask et al., Endocrine 27: 179-88 2005; Patente Norte-americana N° 5.196.435 de Clemens et al. e 5.272.141 de Fraschini et al.) ,tumores benignos e doenças proliferativas tais como a Hiperplasia Prostática Benigna (BPH) (Patente Norte-americana N O. 5.750. 557 e Patente Européia N° 0565296B de Zisapel), psoríase, contracepção e fertilidade, puberdade precoce, síndrome pré-menstrual e hiperprolactinemia (Pevre et. al., J. Clin. Endocrinol. Metab. 47:1383-1388, 1978: Purry et al., Ain. J. Psychiatry 144:762766, 1987: Waldhauser et al., Clin, Endocrinol. Metab. 73:793796, 1991; Bispink et al., J. Pineal Res. 8:97-106, 1990; Cagnacci et. al., J. Clin Endocriniool. Metab. 73:783-796, 1991; Voordouw et. al., J. Clin Endocriniool. Metab. 74:10-108, 1992; vide as Patentes Norte Americanas N° 4.855.305 e 4.945.103 de Cohen et al., e 5.272.141 de Fraschini et al.).
[009] A melatonina é benéfica para o tratamento e a prevenação de distúrbios neurodegenerativos (Skene et al., brain Ver. 528:170-174, 1990; feng et al., J Pineal Res. 37:129-36, 2004), acidente vascular cerebral isquêmico (Cho et. al., Brain Research 755:335-338, 1997; Reiter et. al., Exp. Bio Med.230:104-17, 2005), doença de Alzheimer (Pappola et. al., J. Neurosci. 17:11683-90, 1997; Feng e Zhang, Free Radic. Biol. Med. 37:1790801, 2004) e síndrome da morte infantil repentina (SIDS) Patente Norte Americana N° 5.500.225 de Laudon et. al.).
[010] Três subtipos de receptores de melatonina foram identificados até agora: MT-1, MT-2 e reductase de ribosídeo-quinona de diidronicotinamida 2 que é denominado algumas vezes como receptores de melatonina MT-3 ou ML2 (Dubocovich et. al., 1-UPHAR media, Londres, Reino Unido, 18793, 1998; Maillet et al., FEBS Lett. 3: 578-116-20, 2004). O MT-I fica localizado no SNC e em órgãos periféricos tais como os rins e o trato urogenital, sendo que o MT-2 fica localizado principalmente no sistema nervoso central. Não há nenhuma atividade fisiológica atribuída (ML2) aos sítios MT-3. Além disso, a melatonina interage com as proteínas intracelulares tais como a calmodulina (Anton-Tay et al., J. Pineal Res. 24:35-42, 1998) e proteínas associadas à tubulina (Cardinale et al., J. Pineal Res. 23: 32-9, 1997). Os padrões de retenção da melatonina radioativa inj etados em camundongos demonstram a acumulação de melatonina no cérebro, na pituitária, nos pulmões, no coração, nas gônadas e nos órgãos sexuais 5 acessórios (Withyachumnarnkul et al. Life sci. 12:1575-65, 1986).
[011] É evidente que existe uma ampla faixa de utilizações terapêuticas para a melatonina e seus análogos. Consequentemente, é de interesse contínuo a identificação de 10 novos compostos que interagem com o sistema melatoninérgico como agentes terapêuticos potenciais (ZIotos, Arch. Pharm. Chem. Life Sci. 338:229-247, 2005). Estes compostos podem propiciar uma duração mais longa, localização seletiva e uma eficácia maior em relação àqueles de melatonina.
[012] A serotonina (5-HT) é conhecida agora como moduladora de numerosos sistemas fisiológicos e comportamentais de modo que explicam muitas drogas à base de 5-HT utilizadas como tratamentos em condições clínicas muito diferentes. Há terapêuticas extensivas direcionadas para aumentar ou diminuir a função de 5-HT em sítios selecionados, em condições clínicas extensamente diferentes. As sondas de turnover de 5-HT no SNC e no tecido periférico demonstraram que as alterações no metabolismo de 5-HT estão associadas com uma ampla série de condições clínicas e foi demonstrado que muitas drogas, tais como os antidepressivos, antipsicóticos e antiansiolíticos alteram a função de 5-HT em diversos distúrbios. O desenvolvimento e a utilização clínica difundida dos inibidores seletivos da recaptação de 5-HT (SSRI) e a delineação pré-clínica dos subtipos múltiplos do receptor de 5-HT e de seus acoplamentos a sistemas do mensageiro intracelular e ao desenvolvimento de drogas que agem seletivamente nestes sistemas, catalisaram uma explosão de novas informações sobre pesquisa neste campo. Está agora claro que os sistemas de 5-HT são extremamente diversos e que estão envolvidos em uma pluralidade de processos fisiológicos e comportamentais. Por outro lado, o desenvolvimento de agonistas e antagonistas específicos do receptor de 5-HT conduziram a intervenções terapêuticas alvejadas mais específicas, tais como a utilização do agonista de 5-HT, a sumatriptina, na enxaqueca e na cefaleia em salvas e do antagonista de 5-HT3, o ondansetron, no controle da náusea e da emese.
[013] Há um número extensivo de áreas clinicamente relevantes em que a participação do sistema de 5- HT foi demonstrada. Estes incluem a regulação do humor, do medo e da ansiedade, aprendizagem e memória, controle cognitivo e regulação de apetite e da alimentação, do sono, da função sexual, o controle dos impulsos, a regulação comportamental desenvolvimental, o envelhecimento e a neurodegeneração, a motivação e a recompensa, a sensibilidade à dor, a emese, o mioclônus, a regulação neuroendócrina, a regulação do ritmo circadiano, a resposta ao estresse e a síndrome carcinóide.
[014] Há um amplo espectro de respostas sintomáticas aos tratamentos seletivos do inibidor de recaptação de serotonina (SSRI) em distúrbios diferentes. A disponibilidade aumentada de uma série de SSRI para a utilização clínica conduziu às experimentações de tratamento em uma ampla variedade de condições clínicas diferentes. Os estudos controlados com placebo demonstraram resultados positivos no tratamento com o SSRI em: depressão, transtorno obsessivo- compulsivo (TOC) síndrome do pânico, síndrome pré- menstrual, bulimia nervosa, distúrbio autístico, neuropatia diabética e obesidade diabética. O amplo espectro de condições clínicas diferentes que foram relatadas para demonstrar uma resposta sintomática após o tratamento com SSRI inclui a depressão profunda, depressão secundária à condição médica, depressão pós-derrame, distimia, distúrbio afetivo sazonal, TOC, síndrome do pânico, fobia social, distúrbio da personalidade limítrofe, síndrome da despersonalização, síndrome corporal dismórfica, síndrome pré-menstrual, distúrbios do pós-parto, 5 bulimia nervosa, distúrbio do estres se pós - traumático, distúrbio autístico, déficit de atenção, distúrbio de hiperatividade, síndrome de Tourette, tricotilomania, onicofagia, síndrome de Prader-Wi11i, parafilias e compulsões sexuais, ejaculação precoce, profilaxia da enxaqueca, neuropatia diabética, síndromes de dor, obesidade, ganho de peso em fumantes, alcoolismo, risco emocional após lesão no cérebro, paralisia do sono, ciúme patológico, esquizofrenia crônica, comportamento autodestrutivo, artrite, fenômeno de Raynaud, fibromialgia, síndrome da fadiga crônica, síndrome do intestino irritável, síncope da inclinação à direita, mioclônus intencional e regulação neuroendócrina.
[015] Os dados pré-clínicos em 5-HTindicam que os sistemas de 5-HT são predominantemente modulatórios e que a maioria dos efeitos da 5-HT interagem com o status crescente de outros sistemas de neurotransmissor envolvidos. A neuroanatomia do sistema de 5-HTsugere que até ou mais da 5- HT liberada pode não estar nas sinapses. Desse modo, não se esperaria que os efeitos da 5-HTestivessem localizados ou que demonstrassem as propriedades associadas com os sistemas que mais diretamente mediam a neurotransmissão. A natureza modulatória dos sistemas de 5-HT pode ser vista em nível clínico através das interações com outros sistemas de neurotransmissores. Em animais que se comportam, a atividade dos neurônios serotonérgicos do cérebro está intimamente ligada ao ciclo de dormir-acordar-levantar: a taxa mais elevada de ativação ao acordar ou levantar ativo, nível intermediário de descarga durante estados em repouso e sono de ondas lentas, e o silêncio virtual durante o sono do movimento rápido dos olhos. Alguns compostos de SSRI são associados com a perda de peso refrataria ou o ganho excessivo de peso, a insânia e a disfunção sexual.
[016] A participação difundida dos sistemas de 5-HT na modulação de funções fisiológicas de um grande número de sistemas biológicos diferentes e importantes, acoplados com o progresso rápido de abordagem biológica molecular no descobrimento de novos subtipos de receptor de 5-HT deve promover a atividade de pesquisa aumentada direcionada ao desenvolvimento de moduladores de 5-HT clinicamente aplicáveis que podem ser dotados com outras propriedades farmacológicas a fim de otimizar os parâmetros de utilização de drogas para o efeito clínico.
[017] Os novos compostos relacionados à melatonina ou à serotonina e às pironas, mas com perfis farmacológicos ou farmacocinéticos diferentes destas moléculas, são provavelmente novos produtos farmacêuticos importantes. Por exemplo, vide a Patente Norte-americana N O 5.403.851, que descreve a utilização de triptaminas substituídas, fenilalquilaiminas e compostos relacionados, a fim de tratar uma série de indicações farmacêuticas que incluem distúrbios do sono, indicações endócrinas, distúrbios do sistema imunológico, etc. O Pedido de Patente PCT WO N O. 87/00432 descreve as composições para o tratamento ou a prevenção de a psoríase que contêm melatonina ou compostos relacionados. A Patente Norte-americana 5.122.535 descreve a produção de melatonina e de análogos desta para várias finalidades terapêuticas, que incluem a administração de melatonina em combinação com uma azidotimidina para o tratamento da AIDS. Os análogos de melatonina à base das propriedades bioisostéricas do anel naftalênico e do anel de indol foram descritos em J. Med. Chem. 1992, 35: 1484-1485, Pedido de Patente Europeu 662471 A2 950712 de Depreux et al., WO 9529173 AI 951102 de Ladlow et ai., Patentes Norte- americanas N° 5.151.446 de Horn et al., 5.194.614 de Adrieux et al. e 5.276.051 de Lesieur et al. A melatonina e seus análogos podem potencializar os efeitos de moduladores do receptor de GABA (Zisapel, Pedido de Patente Norte- americano N° US20055175692, Zisapel, Patente Norte - americana N°. 469. 044).
[018] O diabetes resistente à insulina e o diabetes não dependente da insulina são predominantes em até da população, dependendo da idade e da natureza do subconjunto. Nos Estados Unidos somente, 16 milhões de pessoas têm o diabetes tipo 2 e 13 milhões tem a tolerância a glicose prejudicada. Realmente, o diabetes tipo 2 atingiu proporções epidêmicas em todo o mundo. Por volta de 2025, estima-se que 300 milhões de pessoas terão diabetes, sendo que a maioria irá habitar a China, a índia e os Estados Unidos. Por causa de uma população obesa, envelhecendo e cada vez mais sedentária com dietas insalubres e que se alteram, a resistência à insulina também está aumentando de modo alarmante (já é duas a três vezes mais predominante do que o diabetes tipo 2).
[019] A resistência à insulina ocorre geralmente cedo no desenvolvimento do diabetes tipo 2. Um equilíbrio alterado no sistema nervoso autônomo em determinados trajetos endócrinos e inflamatórios poderia contribuir para o desenvolvimento de resistência à insulina. No diabetes, a hiperglicemia agrava adicionalmente a resistência à insulina bem como a disfunção das células beta, mas os mecanismos que causam este fenômeno, isto é, a glicotoxicidade, não são inteiramente compreendidos. A resistência à insulina pode ser demonstrada em parentes saudáveis de primeiro grau de pacientes que sofrem de diabetes tipo 2, parentes estes que também têm um risco elevado de desenvolver o diabetes tipo 2.
[020] A hiperglicemia de jejum do diabetes tipo 2 existe na presença de hiperinsulinemia; isto reflete a presença de resistência à insulina no fígado com glicogenólise e gliconeogênese resultantes. Além da supressão de insulina prejudicada da produção de glicose hepática, uma diminuição da captação de glicose mediada pela insulina pelas células musculares contribui (em aproximadamente 50%) à hiperglicemia resultante.
[021] A tolerância à glicose declina com a idade por causa de: 1) resistência à insulina aumentada do receptor de células; 2) distúrbios do pós-receptor intracelular e 3) sensibilidade diminuída à insulina e à glicose das células beta de ilhotas pancreáticas. A resistência à insulina, com hiperinsulinemia e/ou hiperglicemia secundárias, contribui para muitos distúrbios associados com o envelhecimento, isto é, a hipertensão, a obesidade, a aterosclerose, anomalias de lipídeos, coagulopatias e perturbações metabólicas crônicas que incluem o diabetes tipo 2. A insulina é um dos hormônios anabólicos mais importantes no corpo e é essencial para o controle do metabolismo de carboidratos, lipídeos e proteínas. A insulina é secretada das células beta no pâncreas endócrino. Ela age ao se ligar ao receptor de insulina de transmembrana nas células alvo, e este ativa o domínio de cinase de tirosina na parte intracelular do receptor, conduzindo à fosforilação de substratos do receptor de insulina (IRS). Isto inicia uma cascata de reações de sinalização na célula, conduzindo a efeitos metabólicos. Os tecidos alvo principais de ação metabólica da insulina são os músculos, o fígado e o tecido adiposo. A insulina estimula a captação de glicose em tecidos sensíveis à insulina, principalmente no músculo esqueletal, e inibe a produção de glicose no fígado e promove a armazenagem de glicogênio no fígado e no músculo esqueletal. Ela promove a distribuição de ácidos graxos não-esterificados (NEFA) ao tecido adiposo, onde eles são armazenados como triglicerídeos, e a lipólise nas células de gordura é inibida. Em geral, a síntese de proteína total é aumentada.
[022] Pesquisas recentes sugerem que há uma expressão elevada do fator- α de necrose de tumor de citocina (TNF-α) nos adipócitos de indivíduos obesos e que este TNF- α é um contribuinte principal para a resistência à insulina e ao subsequente diabetes tipo 2 da obesidade. O TNF- α é um regulador importante dos processos de apoptose e, modula, desse modo, o volume do tumor, de tecidos adiposos e musculares. Ele é produzido não somente pelas células imunocompetentes, mas também pelos adipócitos e pelas células musculares. Esta citocina é ativada nos tumores e na obesidade, entre outras condições. Ao agir na fosforilação de IRS-I e na 3-cinase de fosfatidilinositol (PI-3), ao modificar a resistência através da regulação da síntese do transportador de glicose responsivo à insulina GLUT4 e através da interferência com a sinalização de insulina (talvez através da leptina) o TNF- α promove a resistência à insulina e a anorexia.
[023] Independentemente da causa, a resistência insulina é associada com os efeitos difundidos e adversos saúde. Isto é verdadeiro até mesmo quando a tolerância à glicose é prejudicada apenas ligeiramente, mas não ainda na escala diabética aberta. São marcantes entre os efeitos adversos a predisposição à doença vascular que afeta grandes vasos sanguíneos e uma associação com a hipertensão e a dislipidemia (triglicerídeos elevados e HDL diminuído). De fato, esta combinação de 1) intolerância à glicose, 2) resistência à insulina, 3) hipertensão e 4) dislipidemia é suficientemente comum para ter adquirido o nome de Síndrome X, síndrome de resistência à insulina ou síndrome de Reaven. Clinicamente, define centenas de milhões de pessoas em todo o mundo.
[024] Em vista da discussão acima, os derivados de pirona-indol devem ser de utilização terapêutica para uma variedade de doenças e condições, particularmente aquelas associadas com a melatonina, a 5-HT, a insulina e a desregulação GABAérgica. A presente invenção refere-se à necessidade de compostos terapeuticamente mais avançados do que aqueles visados para modular uma destas classes sozinha. Tal agente que age como MT-1 e MT-2 ou agonistas/antagonistas do receptor de serotonina com propriedades de modulação do receptor de GABA adicionais pode prover novas drogas com, mas sem ficar a ela limitada, a eficácia sedativa com benefícios clínicos adicionais, tal como a melhoria do sono com efeitos benéficos na vigília durante o dia. Devido a este modo de ação exclusivo, estes agentes não irão exibir efeitos colaterais típicos relacionados às benzodiazepinas, tais como a tolerância e os sintomas da interrupção de drogas.
[025] Além disso, a presente invenção refere-se à necessidade de novos derivados melatoninérgicos que afetam a resistência à insulina e o diabetes tipo II.
[026] Supõe-se que todo o conteúdo das patentes, dos pedidos de patente e dos artigos de literatura citados acima são incorporados na presente invenção a título de referência.
[027] A invenção refere- se aos compostos que têm a fórmula (I): Ar-B-Ar' (I) em que: B representa: _ X-Y-Z-, em que X representa - (CH2n (em que n é 0-6) em que a porção alquila é linear ou ramificada, Y representa oxigênio, enxofre, >NH ou está ausente, Z representa >C = O, >O, >COO ou está ausente, em que pelo menos um de X, Y e Z deve estar presente; Ar representa um sistema de anel de núcleo de indol:
[028] Ar' representa um sistema de anel do núcleo de alfa, beta ou gama-pirona: em que cada um de R1-4 substitui o anel Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1’-2’ substitui o anel Ar' em qualquer posição disponível e em que cada um de R1-4 representa independentemente hidrogênio, oxigênio, halo, halo C1-5 alquila, arila, acila, um grupo heterocíclico Cl-7 que contêm um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; alquila C1-5, alquenila C2-5, arilalquinila C2-5, alquilarila Cl -5, alquenilarila C2-5, alquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamina Cl-5, alquilamido Cl-5, hidróxi, tiol, acilóxi, azido, alcóxi Cl-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, aralalquinila ou estirila podem ser opcionalmente substituídos por anel por um a quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, halo-alquila C1-5, arila, um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; um grupo heteroarila que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila Cl-5, alquenila C2-5, alquinila C25, alquilarila Cl-5, arilalquenila C1-5, arilalquinila C2-5 alquinila, hidróxi alquila C1-5 nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, azido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e R3 ou R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e R2, incluindo a posição N e Ar' pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1 ou por R2; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
em que cada um de R1-4 substitui o anel Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1’-2’ substitui o anel Ar' em qualquer posição disponível e em que cada um de R1-4 representa independentemente hidrogênio, oxigênio, halo, halo C1-5 alquila, arila, acila, um grupo heterocíclico Cl-7 que contêm um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; alquila C1-5, alquenila C2-5, arilalquinila C2-5, alquilarila Cl -5, alquenilarila C2-5, alquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamina Cl-5, alquilamido Cl-5, hidróxi, tiol, acilóxi, azido, alcóxi Cl-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, aralalquinila ou estirila podem ser opcionalmente substituídos por anel por um a quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, halo-alquila C1-5, arila, um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; um grupo heteroarila que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila Cl-5, alquenila C2-5, alquinila C25, alquilarila Cl-5, arilalquenila C1-5, arilalquinila C2-5 alquinila, hidróxi alquila C1-5 nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, azido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e R3 ou R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e R2, incluindo a posição N e Ar' pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1 ou por R2; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
[029] Tal como utilizado na presente invenção, "arila" representa fenila ou naftila.
[030] Sem detrimento da generalidade dos compostos da presente invenção, um subgrupo de compostos presentemente preferidos é definido de modo que na fórmula (I), X é (CH2)2-, Y é >NH ou >0, Z é >C = O, Ar é um indol que contém uma ligação, R3, a X na posição 3 do anel de indol, RI é metóxi na posição 5 do anel de indol, sendo que cada um de R2 e R4 é hidrogênio e (a) Ar' é uma gama-pirona ligada a Z na posição 2 do anel de pirona, R1’, é hidrogênio ou um grupo hidróxi na posição 5 do anel de pirona, e R2 é hidrogênio ou um grupo carbóxi na posição 6 do anel de gama-pirona ou (b) Ar' é um anel de alfa-pirona ligado a Z na posição 5 do anel de pirona, cada um de RI, e RT é hidrogênio nas posições 3, 4 ou 6 do anel de pirona; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
[031] A presente invenção também inclui em seu âmbito as composições farmacêuticas que contêm como substância ativa uma quantidade terapeuticamente eficaz de um composto da fórmula (I) ou um sal farmaceuticamente aceitável deste bem como qualquer estereoisômero, englobado pela fórmula (I), em associação com um ou mais diluentes, conservantes, solubilizantes, emulsificantes, adjuvantes, excipientes ou veículos farmaceuticamente aceitáveis utilizados convencionalmente nas formulações farmacêuticas e veterinárias. As presentes formulações farmacêuticas podem ser adotadas para a administração a seres humanos e/ou a animais.
[032] Os compostos da fórmula (I) são úteis para o tratamento e/ou a prevenção e/ou a minimização da resistência à insulina e ao diabetes tipo II, a perda neuronal associada com o acidente vascular cerebral, a isquemia, o trauma do sistema nervoso central (SNC), distúrbios do SNC incluindo doenças neurodegenerativas (tais como o mal de Alzheimer, esclerose lateral amiotrófica (ELA), o mal de Huntington, o mal de Parkinson e a síndrome de Down); para o tratamento ou a prevenção das consequências adversas da superestimulação dos aminoácidos excitatórios; para o tratamento ou a prevenção de distúrbios psiquiátricos, epilepsia e outros distúrbios convulsivos, ansiedade, distúrbios do sono incluindo a insônia, doenças psiquiátricas (por exemplo, depressão, psicose) dor crônica (analgesia) glaucoma, retinite por citomegalovírus (CMV) e incontinência urinária e 30 indução à anestesia, bem como intensificação da cognição e impedimento e tratamento de sintomas da tolerância e da abstinência de opiatos.
[033] Por meio de uma elaboração ou de uma explanação adicional, as condições que são contempladas presentemente podem ser receptivas ao tratamento pela administração dos presentes compostos, tais condições incluem impotência; distúrbios cardiovasculares (incluindo a hipertensão); distúrbios de coagulação sanguínea; distúrbios inflamatórios; neuropatia; distúrbios com base cronobiológica (por exemplo, tontura de fuso horário); distúrbios do sono circadianos (tais como a síndrome do sono atrasado, os problemas com turnos de trabalho e distúrbios relacionados às estações, por exemplo, o distúrbio afetivo sazonal (SAD)); indicações endócrinas (por exemplo, contracepção e infertilidade, puberdade precoce, síndrome pré-menstrual, hiperprolactinemia e deficiência do hormônio do crescimento) doenças neoplásticas (incluindo câncer outras doenças proliferativas (benignas e crescimento de tumor da próstata)' distúrbios do sistema imunológico incluindo a AIDS; condições associadas com a senescência; doenças oftalmológicas; cefaléia em salvas; enxaqueca; proteção da pele; distúrbios da estabilização do diabetes e de ganho de peso (leptina, obesidade); para prover a proteção da pele e como um auxílio à criação de animais (por exemplo, regulação da fertilidade, puberdade e cor da pelagem).
[034] A presente invenção refere-se aos compostos que têm a fórmula (I): Ar-B-Ar' (I) em que: -B-representa: X-Y-Z em que X representa -(CH2)2- (em que n é 0-6) em que a porção alquila é linear ou ramificada, Y representa oxigênio, enxofre, >NH ou está ausente; Z representa >C = 0 ou > 0 ou >COO ou está ausente; em que pelo menos um de X, Y e Z deve estar presente; o sistema de anel de Ar representa um núcleo de indol:
[035] Ar' representa um sistema de anel do núcleo de alfa, beta ou gama-pirona: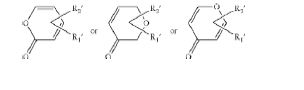 em que cada um de R1-4 substitui os sistemas de anel de Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1-2 substitui o sistema de anel de Ar' em qualquer posição disponível e em que cada um de R1-4 e R1’-2’ representa independentemente hidrogênio, oxigênio, halo, halo-C1-5 alquila, arila, acila, um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila C1-5, alquenila C2-5, alquinila, alquilarila Cl-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamina Cl-5, alquilamido Cl-5, hidróxi, tiol, acilóxi, azido, alcóxi Cl-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, arilalquinila ou estirila podem ser opcionalmente substituídos em anel por um a quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, haloalquila Cl-5, arila, um grupo heterocíclico C5-7 que contém de um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila que contém de um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila Cl-5, alquenila C2-5, alquinila C25, alquenilarila C2-5, arilalquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, az ido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e R3 ou R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e por R2 incluindo a posição N e Ar' pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1’ ou R2’; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
em que cada um de R1-4 substitui os sistemas de anel de Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1-2 substitui o sistema de anel de Ar' em qualquer posição disponível e em que cada um de R1-4 e R1’-2’ representa independentemente hidrogênio, oxigênio, halo, halo-C1-5 alquila, arila, acila, um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila C1-5, alquenila C2-5, alquinila, alquilarila Cl-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamina Cl-5, alquilamido Cl-5, hidróxi, tiol, acilóxi, azido, alcóxi Cl-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, arilalquinila ou estirila podem ser opcionalmente substituídos em anel por um a quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, haloalquila Cl-5, arila, um grupo heterocíclico C5-7 que contém de um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila que contém de um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila Cl-5, alquenila C2-5, alquinila C25, alquenilarila C2-5, arilalquinila C2-5, hidróxi alquila Cl-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, az ido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e R3 ou R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e por R2 incluindo a posição N e Ar' pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1’ ou R2’; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
[036] Tal como utilizado na presente invenção, "arila" representa fenila ou naftila.
[037] Também Tal como utilizado na presente invenção, a referência a “um” composto, sal ou estereoisômero da fórmula (I) se presta a englobar “um ou mais” tais compostos, sais ou estereoisômeros. Além disso, a referência a um "composto" da fórmula (I) tal como na discussão abaixo de formulações farmacêuticas, também se presta a incluir um sal ou um estereoisômero do composto.
[038] Em uma realização preferida, X é -(CH2)n-, em que n é qualquer um de O a 6, e preferivelmente qualquer um de 1 a 6, Y é ou e Z é >CO.
[039] Sem detrimento da generalidade dos compostos da presente invenção, em uma realização preferida dos compostos definida pela fórmula (I), X é -(CH2)2-, Y é >NH ou >O, Z é >C=O, Ar é um indol que contém uma ligação, R3 a X na posição 3 do anel de indol, R1 é metóxi na posição 5 do anel de indol, sendo que cada um de R2 e R4 é hidrogênio, Ar’ é uma gama-pirona ligada a Z na posição 2 do anel de pirona, RI, é hidrogênio ou um grupo hidróxi na posição 5 do anel de pirona e R2, é hidrogênio ou um grupo carbóxi na posição 6 do anel de gama-pirona; ou um sal ou um estereoisômero farmaceuticamente aceitável destes. Em uma segunda realização preferida, Ar é tal como definido acima e Ar' é um anel de alfa-pirona ligado a Z na posição 5 do anel de alfa-pirona e RI, e RT são hidrogênios; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
[040] A presente invenção também inclui dentro de seu âmbito a preparação das composições que contêm um composto da fórmula (1) as composições são úteis como medicamentos. As composições farmacêuticas contêm como substância ativa uma quantidade terapeuticamente eficaz de um composto da fórmula (I) ou um sal farmaceuticamente aceitável deste bem como qualquer estereoisômero abrangido pela fórmula (I) em associação com um ou mais diluentes, conservantes, solubilizantes, emulsificantes, adjuvantes, excipientes e veículos farmaceuticamente aceitáveis utilizados convencionalmente nas formulações farmacêuticas e veterinárias. As presentes formulações farmacêuticas podem ser adaptadas para a administração a seres humanos e/ou a animais.
[041] Formulação farmacêutica, de acordo com a invenção, é caracterizada preferivelmente por pelo menos uma das seguintes características: (i) é adaptada para ser administrada pelas vias de administração oral, parenteral (por exemplo, injeção intramuscular, intraperitoneal, intravenosa ou subcutânea ou implante), nasal, vaginal, retal, sublingual ou tópica e pode ser formulada nas formas de dosagem apropriadas para cada via de administração; (ii) está na forma de dosagem unitária, sendo que cada dosagem unitária compreende uma quantidade de pelo menos um composto da fórmula (I) que está na faixa de aproximadamente 2,5 μg a 25 mg/kg de peso corpóreo; (iii) é uma formulação de liberação prolongada, em que pelo 5 menos um composto da fórmula (I) é liberado a uma taxa controlada predeterminada.
[042] As formulações adicionais podem ser caracterizadas pelo fato de que podem ser administradas sozinhas ou em combinação com ou conjuntamente com outros compostos que são conhecidos no estado da técnica como sendo úteis para a prevenção e o tratamento de distúrbios do sistema nervoso central (SNC) e de distúrbios metabólicos, incluindo, mas sem ficar limitados, doenças neurodegenerativas, distúrbios do sono, resistência à insulina e diabetes tipo II.
[043] Os sais farmaceuticamente aceitáveis apropriados dos compostos da fórmula (1) de utilização na presente invenção incluem os sais que podem, por exemplo, ser formados ao misturar uma solução do composto com uma solução de um ácido não- tóxico farmaceuticamente aceitável, tais como o ácido clorídrico, o ácido fumárico, o ácido maléico, o ácido succínico, o ácido acético, o ácido cítrico, o ácido tartárico, o ácido carbônico, o ácido fosfórico ou o ácido sulfúrico. Os grupos sais de amina também podem compreender os sais de amônio quaternário em que o átomo de amino nitrogênio contém um grupo alquila, alquenila, alquinila ou aralquila. Onde o composto contém um grupo ácido, por exemplo, um grupo ácido carboxílico, a presente invenção também contempla os sais farmaceuticamente aceitáveis dos mesmos, preferivelmente os sais atóxicos destes, tais como os sais de sódio, de potássio e de cálcio dos mesmos.
[044] Os compostos da fórmula (I) podem ser administrados aos mamíferos para o tratamento e/ou a prevenção da resistência à insulina e o diabetes tipo II; a perda neuronal associada com acidente vascular cerebral; isquemia; trauma do sistema nervoso central (SNC); distúrbios do SNC que incluem doenças neurodegenerativas (tais como o mal de Alzheimer, esclerose lateral amiotrófica (ELA), mal de Huntington, mal de Parkinson e síndrome de Down); as consequências adversas da superestimulação dos aminoácidos excitatórios; doenças psiquiátricas; epilepsia e outros distúrbios convulsivos; ansiedade; distúrbios do sono incluindo a insônia; doenças psiquiátricas (por exemplo, depressão, psicose); dor crônica (analgesia); glaucoma; retinite por citomegalovírus (CMV); incontinência urinária; e sintomas da tolerância e da abstinência de opiatos. Os compostos também podem ser administrados para induzir a anestesia, bem como para intensificar a cognição.
[045] Além disso, os compostos da invenção podem ser administrados a um mamífero para o tratamento e/ou a prevenção da impotência ; distúrbios cardiovasculares(incluindo a hipertensão, distúrbios coagulação sanguínea) distúrbios inflamatórios; neuropatia; distúrbios com base cronobiológica (por exemplo, tontura de fuso horário); distúrbios do sono circadianos (tais como a síndrome do sono atrasado, os problemas com turnos de trabalho e distúrbios relacionados às estações do ano (por exemplo, distúrbio afetivo sazonal (SAD)); indicações endócrinas (por exemplo, contracepção, infertilidade, puberdade precoce, síndrome pré-menstrual, hiperprolactinemia e deficiência do hormônio do crescimento); doenças neoplásticas (incluindo e câncer outras doenças proliferativas (benignas crescimento de tumor da próstata); distúrbios do sistema imunológico incluindo a AIDS; as condições associadas com a senescência; doenças oftalmológicas; cefaleia em salvas; enxaqueca; distúrbios do ganho do peso (leptina, obesidade); para prover a proteção à pele e como um auxílio à criação de animais, por exemplo, regulação da fertilidade, da puberdade e da cor da pelagem.
[046] Tal como utilizado na presente invenção, “tratar” significa aliviar ou curar uma doença, um distúrbio ou uma condição ou facilitar pelo menos um sintoma da doença, do distúrbio ou da condição.
[047] Em realizações preferidas, a doença ou o distúrbio é sofrido por seres humanos e os compostos da invenção são 10 administrados aos seres humanos.
[048] Os compostos da invenção podem ser administrados sozinhos ou em combinação com outros agentes conhecidos como benéficos no tratamento da doença, do distúrbio ou da condição a ser tratada. Tal como utilizado na presente invenção, “em combinação” significa que o composto da fórmula (I) e outro agente pode ser co-administrado, em terapia concomitante ou em uma combinação física fixa ou pode ser administrado em períodos separados, mas para complementar um ao outro.
[049] Em uma realização preferida, os compostos da fórmula (1) podem ser administrados para alterar os ritmos circadianos ou para melhorar a qualidade do sono ou para o tratamento ou a prevenção de distúrbios do sono ou distúrbios do sono em um mamífero, especialmente um ser humano. Além disso, os compostos da fórmula (I) podem ser administrados para aumentar a eficiência do sono e para aumentar a manutenção do sono. Os distúrbios e as perturbações do sono que podem ser tratados ou impedidos através da administração dos compostos da fórmula (I) incluem os problemas do sono associados com a insânia, hipersonia, apneia do sono, narcolepsia, mioclônus noturno, interrupções do sono REM, tontura de fuso horário, distúrbios do sono relacionados a turnos de trabalho, dissonias, terror noturno, insônias associadas com a depressão ou com os distúrbios emocionais e sonambulismo e enurese, bem como os distúrbios do sono que acompanham o envelhecimento, as condições associadas com a ritmicidade circadiana, os distúrbios mentais e físicos associados com viagens através dos fusos-horários e com programações de turnos de trabalho rotatórias ou síndrome tal como a fibromialgia que é manifestada pelo sono não-reparador e por dor muscular ou a apneia do sono que é associada com os distúrbios respiratórios durante o sono.
[050] No tratamento ou na prevenção das condições antecedentes, definidas amplamente como distúrbios do ritmo circadiano ou distúrbios do sono, o composto da fórmula (I) pode ser administrado sozinho ou em combinação com outros compostos conhecidos no estado da técnica como sendo úteis para intensificar a qualidade do sono e para a prevenção e o tratamento de distúrbios e perturbações do sono, incluindo, por exemplo, sedativos, hipnóticos, antiansiolíticos, antipsicóticos, agentes antiansiedade, tranquilizantes leves, agonistas antagonistas de me latonina, melatonina, benzodiazepinas, barbituratos, antagonistas de 5-HT-2 e outros ainda, como: adinazolam, alobarbital, alonimida, alprazolam, amitriptilina, amobarbi tal, amoxapina, bentazepam, benzoctamina, brotizolam, bupropion, buspriona, butabarbital, butalbital capurida, carbocloral, betaína cloral, hidrato de cloral, clordiazepóxido, clomipramina, cloperidona, clorazepato, cloretato, clozapina, ciprazepam, de sipramina, dexclamol, diazepam, dicloralfenazona, divalproex, difenidramina, doxepin, estazolam, eszopiclona, etclorvinol, etomidato, fenobam, flunitrazepam, flurazepam, fluvoxaimina, fluoxetina, forazepam, gaboxadol, glutetimida, . halazepam, hidroxizine, imiprainine, indiplon, lítio, lorazepam, lormetazepam, maprotiline, mecloqualona, melatonina, mefobarbital, meprobamato, metaqualona, midaflur, midazolam, nefazodona, nisobainato, nitrazepam, nortriptilina, oxazepam, paraldeído, paroxetina, pentobarbital, perlapine, perfenazina, fenelzina, fenobarbital, prazepam, prometazine, propofol, protriptilina, quazepam, ramelteon, reclazepam, roletamida, secobarbital, sertralina, suproclona, temazepam, tioridazine, tracazolato, tranilcipromaina, trazodona, triazolam, trepipain, tricetamida, triclofos, trifluoperazina, trimetozina, trimipramina, uldazepam, valproato, venlafaxina, zaleplon, zolazepam, zolpidem, zopiclona e os sais e combinações dos mesmos, e outros ainda.
[051] As combinações de um ou de mais destes agentes terapêuticos conhecidos com um composto da fórmula (I) irão conferir efeitos adicionais, complementares e frequentemente sinergísticos para intensificar as propriedades desejáveis do agente terapêutico conhecido.
[052] O composto da fórmula (I) sozinho ou em combinação com um dos agentes terapêuticos conhecidos acima mencionados pode ser administrado adicionalmente em combinação a métodos de tratamento físicos, tais como a terapia de luz (tal como descrito nas Patentes Norte-americanas N° 5.447.527 e 5.562.719, sendo que ambas são incorporadas na presente invenção a título de referência).
[053] Em uma outra realização, os compostos da fórmula (I) podem ser administrados em combinação comum agente antidiabético, tal como a insulina, sulfoniluréias, biguanidas (tais como a metformina) inibidores de alfaglicosidase (tais como a acarbose) agonistas do gama receptor ativados pelo proliferador de peroxissomo (PPARgama) tais como tiazolidinedionas, que incluem a pioglitazona e rosiglitazona, agentes para a diminuição do colesterol tais como inibidores da reductase de HMG-CoA (lovastatina, sinvastatina, pravastatina, fluvastatina, atoivastatina, rivastatina, itavastatina e outras estatinas), sequestrantes (derivados de colestiramina, colestipol e dialquilaminoalquila de um dextrano reticulado) álcool nicotinílico, ácido nicotínico ou um sal deste, os agonistas de PPARalbfa (genfibrozil, clofibrato, fenofibrato e bezafibrato), probucol, agonistas de PPARalfa/gama, tais como KRP-297, agentes antiobesidade, tais como fenfluramina, dexfenflurainina, fentiramina, subitramina, orlistat, inibidores do neuropeptídeo Y5, agonistas do receptor beta adrenérgico, inibidores da dipeptidil peptidase- 4 e inibidores de PTP-IB.
[054] Quando um composto da fórmula (I) é administrado em combinação com um outro agente terapêutico, tal como um agente antidiabético ou um agente para tratar um distúrbio do sono ou distúrbio do ritmo circadiano, o composto da fórmula (I) e o agente terapêutico conhecido podem ser administrados independentemente em uma dosagem diária que varia de um centésimo a uma vez os níveis de dosagem que são eficazes quando os compostos são administrados sozinhos.
[055] Os compostos da fórmula (I) podem ser formulados na composição farmacêutica apropriada para as vias de administração oral parenteral (por exemplo, injeção intramuscular, intraperitoneal, intravenosa ou subcutânea, ou implante) nasal, vaginal, retal, sublingual ou tópica. As composições podem compreender um ou mais diluentes, conservantes, solubilizantes, emulsificantes, adjuvantes, excipientes e/ou veículos farmaceuticamente aceitáveis.
[056] As formas de dosagem sólidas para a administração oral incluem cápsulas, comprimidos, pílulas, pós e grânulos. Em tais formas de dosagem sólidas, o composto ativo é misturado com pelo menos um veículo farmaceuticamente aceitável inerte tal como a sacarose, a lactose ou o amido. Tais formas de dosagem também podem compreender, como é a prática normal, substâncias adicionais com exceção dos diluentes inertes, por exemplo, agentes lubrificantes tais como o estearato de magnésio. Os exemplos de adjuvantes que podem ser incorporados em comprimidos, cápsulas e outros ainda são os seguintes: um aglutinante tal como a goma de tragacanto, acácia, amido de milho ou gelatina; um excipiente tal como a celulose microcristalina; um agente desintegrante tal como o amido de milho, amido pré-gelatinizado, ácido algínico, e outros ainda; um lubrificante tal como o estearato de magnésio; um agente adoçante tal como a sacarose, a lactose ou a sacarina; um agente flavorizante tal como hortelã-pimenta, óleo de gualtéria ou cereja. No caso de cápsulas, comprimidos e pílulas, as formas de dosagem também podem compreender agentes de tamponamento. Quando a forma de dosagem unitária é uma cápsula, pode conter, além dos materiais do tipo acima, um portador líquido tal como um óleo graxo. Vários outros materiais podem estar presentes como revestimento ou então modificam a forma física da dosagem , unitária. Os comprimidos e as pílulas também podem ser preparados com revestimentos entéricos e os comprimidos podem ser revestidos com goma-laca, açúcar ou ambos.
[057] As formas de dosagem líquidas para a administração oral incluem emulsões, soluções, suspensões, xaropes e elixires farmaceuticamente aceitáveis que contêm os diluentes inertes utilizados geralmente no estado da técnica, como a água. Além de tais diluentes inertes, as composições também podem incluir agentes adjuvantes, tais como os agentes de umidificação, emulsificação e suspensão e agentes adoçantes, flavorizantes e perfumantes. Um xarope ou um elixir pode conter o composto ativo, sacarose como um agente adoçante, metil e propil parabenos como conservantes, um corante e um flavorizante tal como o sabor de cereja ou de laranja.
[058] Os preparados de acordo com a presente invenção para a administração parenteral incluem soluções, suspensões ou emulsões aquosas ou não-aquosas estéreis. As composições estéreis para a injeção podem ser formuladas de acordo com a prática farmacêutica convencional ao dissolver ou suspender a substância ativa em um veículo tal como a água para injeção, um óleo vegetal natural como o óleo de gergelim, óleo de coco, óleo de amendoim, óleo de semente de algodão, etc., ou um veículo gorduroso sintético tal como o oleato de etila ou outros ainda. Os tampões, conservantes, antioxidantes e outros ainda podem ser incorporados conforme necessário. os exemplos de solventes ou de veículos não-aquosos incluem o propileno glicol, o polietileno glicol, óleos vegetais, tais como o azeite de oliva e o óleo de milho, gelatina e ésteres orgânicos injetáveis, tal como o oleato de etila. Tais formas de dosagem também podem conter adjuvantes tais como agentes de conservação, umidificação, emulsificação e dispersão. Eles podem ser esterilizados, por exemplo, pela filtração através de um filtro de retenção de bactérias, ao incorporar os agentes esterilizantes nas composições, ao irradiar as composições, ou ao aquecer as composições. Eles também podem ser manufaturados na forma de composições sólidas estéreis que podem ser dissolvidas em água estéril ou em algum outro meio injetável estéril imediatamente antes do uso.
[059] As composições para a administração retal ou vaginal são preferivelmente supositórios que podem conter, além da substância ativa, excipientes tais como a manteiga de cacau ou uma cera para supositório. As composições para a administração nasal ou sublingual também são preparadas com os excipientes padrão bem conhecidos no estado da técnica.
[060] A dosagem do agente ativo nas composições da presente invenção pode variar, contanto que uma quantidade terapêutica seja administrada. O agente ativo é administrado desejavelmente a um paciente (humano ou animal) com necessidade de tal tratamento nas dosagens que irão propiciar a eficácia farmacêutica mais favorável. A dosagem selecionada depende da natureza e da gravidade da doença ou do distúrbio a ser tratado, do efeito terapêutico desejado, da via de administração e da duração do tratamento. Uma quantidade de dosagem também pode variar dependendo do peso do paciente e de outros fatores. Por exemplo, o efeito de um composto da fórmula (I) que induz um deslocamento de fase em um marcapasso circadiano central pode ser dependente do ambiente e da época circadiana da administração. O mesmo composto pode induzir um avanço de fase, um atraso de fase ou tem um efeito menor em um ritmo circadiano particular, dependendo do tempo de administração circadiana. A dose irá variar de paciente para paciente, dependendo da natureza e da gravidade da doença, do peso do paciente, das dietas especiais que estiverem sendo seguidas pelo paciente, de medicação simultânea, da biodisponibilidade do composto quando da administração e de outros fatores que um técnico no assunto irá reconhecer.
[061] No tratamento de uma condição de acordo com a presente invenção, um nível diário de dosagem apropriado será geralmente de aproximadamente 2,5 μg a 25 mg/kg do peso corpóreo do paciente. A quantidade de dosagem diária pode ser administrada em uma dose única ou em doses múltiplas por dia. Preferivelmente, o nível de dosagem será de aproximadamente 2,5 μg de aproximadamente 20 mg/kg de peso corpóreo do paciente; com mais preferência, de aproximadamente 2,5 μg a aproximadamente 10 mg/kg de peso corpóreo do paciente. Por exemplo, para obter um efeito de deslocamento de fase do ritmo circadiano, a restauração do relógio circadiano interno, encurtando o tempo de retomada de ritmos circadianos, aliviando um distúrbio do ritmo circadiano ou intensificando a qualidade do sono, um nível de dosagem apropriado varia de aproximadamente 2,5 μg a aproximadamente 25 mg/kg do peso corpóreo do paciente, preferivelmente de aproximadamente 2,5 μg a aproximadamente 20 mg/kg do peso corpóreo do paciente, e especialmente de aproximadamente 2,5 μg a 10 mg/kg do peso corpóreo do paciente. Em mamíferos maiores, por exemplo, seres humanos, uma dose diária indicada típica para a administração oral é de aproximadamente 0,2 a aproximadamente 1000 mg. Preferivelmente, a dosagem oral diária fica compreendida na faixa de aproximadamente 0,5 a aproximadamente 50 mg, e com mais preferência na faixa de aproximadamente 2,5 a aproximadamente 20 mg. Ao utilizar uma formulação injetável ou tópica, um nível de dosagem preferido varia de aproximadamente 2,5 μg a 5 mg/kg do peso corpóreo do paciente e especialmente de aproximadamente 2,5 μg a 1 mg/kg do peso corpóreo do paciente. Em mamíferos maiores, por exemplo, os seres humanos, uma dose indicada típica é de aproximadamente 100 μg a 100 mg i.v. Um composto pode ser administrado em um regime de uma vez a diversas vezes por dia, por exemplo, uma a quatro vezes por dia, preferivelmente uma vez ou duas vezes por dia.
[062] As formulações da presente invenção podem estar na forma de liberação imediata, ou, onde apropriado, como formulações sólidas para a administração oral, podem estar em formas de liberação prolongada. As formulações de liberação prolongada incluem formulações de liberação retardada, sustentada, pulsada ou controlada. As formulações de liberação prolongada apropriadas úteis para as finalidades da presente invenção incluem os tipos de formulação descritos nas Patentes 6.106.864; 7.053.122 e 7.118.762, incorporadas na presente invenção a título de referência. Os detalhes de outros tipos de tecnologias de liberação apropriadas, tais como dispersões de energia elevada e partículas osmóticas e revestidas podem ser encontrados, por exemplo, em Verma, R. e S. Garg, Pharmaceutical Technology On -Linet 25(2) 1-14 (2001) também incorporado na presente invenção a título de referência.
[063] O período de tempo em que uma formulação de liberação prolongada libera o composto varia com base na indicação e nos níveis terapêuticos alvo. Para a insônia, por exemplo, é desejável limitar os efeitos farmacológicos do composto administrado ao período noturno, por exemplo, aproximadamente oito horas. Para o tratamento antidiabetes, é desejável que o composto tenha efeito continuamente, por exemplo, uma eficácia de doze horas com administração da formulação duas vezes ao dia, de manhã e à noite.
[064] A invenção será ilustrada pelos seguintes exemplos. Deve ficar compreendido que os seguintes exemplos são ilustrativos somente e não se prestam a limitar o âmbito da presente invenção de maneira alguma. Exemplo 1 N-[2-(5-metóxi-indol-3-il)-etil]-comenamida Esquema de Reação para a síntese da síntese de N-[2- (5-metóxi-indol-3-il)-etil]-comenamida
Esquema de Reação para a síntese da síntese de N-[2- (5-metóxi-indol-3-il)-etil]-comenamida
[065] Sob uma atmosfera de argônio, um frasco com três gargalos de fundo arredondado de 100 ml foi carregado com ácido comênico (560 mg, 1 equivalente) e 5-metóxi- triptamina (750 mg, 1,1 equivalente) dissolvido em DMF (20 ml) e colocado a 0°C por meio de um banho de gelo. HOBt (1- monoidrato de hidróxibenxotriazol, 535 mg, 1,1 equivalente), EDC (cloridreto de 1-(3 -dimetilaminopropil)-3 -etil carbodiimida, 760 mg, 1,1 equivalente) e trietilamina (1,25 ml, 2,5 equivalentes) foram então adicionados com agitação magnética. A mistura foi agitada por mais quinze minutos a 0°C e colocada para reagir subsequentemente por 48 horas à temperatura ambiente. Água (25 ml) foi então adicionada e a mistura foi extraída completamente com diclrometano (6x30 ml). As fases orgânicas combinadas foram secadas sobre Na2SO4 e o solvente foi removido por evaporação giratória. O componente bruto foi submetido à cromatografia sobre uma coluna em gel de sílica ao eluir com diclorometano/metanol 95/5. O produto foi recuperado como um óleo grosso, o qual foi separado três vezes com éter dietílico para se obter um sólido marrom (180 mg, rendimento de 15%).
[066] Dados experimentais para N-[2-(5-metóxi- indol-3-il)-etil] comenamida MS (ESI POS): 329 (M + H), 351 (M + Na), 392 (M + Na + CH3CN) Ensaio de HPLC: 97% 1H NMR (CDCl3, 400 MHz) δ 3,06 (t,j = 6,7 Hz, 2H, CH2CH2NH), 3,76-3,79 (m, 2H, CH2CH2NH), 3, 84 (s, 3H OCH3), 6,32 (br s, 1H, OH), 6, 76 (br s, 1H, CH2CH2NH), 6,9 (dd, J1 = 2,3 Hz, J2 = 8,8 Hz, 1H, H aromático) 7, 04 (d, j = 2,3 Hz, 1H, H, aromático) 7, 06 (d, 2, 3 Hz, 1H, H aromático) 7, 27 (s, 1H, CH), 7,29 (d, J = 8,8 Hz, 1H, H aromático) 7,73 (s, 1H, CH, 7,96 (br s, 1H, NH). Exemplo 2 Éster O-[2-(5-metóxi-indol-3-il)-etil]-comênico Esquema de Reação para síntese do éster O-[2-(5- metóxi-indol-3-il)-etil]-comênico
Esquema de Reação para síntese do éster O-[2-(5- metóxi-indol-3-il)-etil]-comênico
[067] Sob uma atmosfera de argônio, um frasco de três gargalos de fundo arredondado de 100 ml foi carregado com ácido comênico (300 mg, 1 equivalente) e 5-metóxi triptofol (365 mg, 1 equivalente) dissolvido em CH2C1/DMF (10/5 ml, respectivamente). DDC (diciclohexil carbodiimida, 435 mg, 1,1 equivalente) e DMAP (4 -dimetilaminopiridina, 45 mg, 0,2 equivalente) foram então adicionados com agitação magnética. Depois que a mistura foi agitada por dezesseis horas à temperatura ambiente, o precipitado branco que se formou foi descartado por meio de filtração através de um funil Buchner. Do material filtrado claro, o solvente foi removido por evaporação giratória. O produto bruto foi então foi submetido à cromatografia sobre uma coluna em gel de sílica ao eluir com 250 ml de CH2C1 seguido por diclorometano/metanol 97/3. As frações que contêm o produto foram combinadas e concentradas e o sólido resultante foi recristalizado a partir do cicloexano/acetato de etila. O éster O-[2-(5-metóxi-indol-3- il)-etil]-comênico puro foi obtido como um sólido amarelo claro (250 mg, rendimento de 40%).
[068] Dados experimentais para o éster 0- [2- (5-metóxi-indol-3-il) - etil] –comênico MS (ESI POS): 330 (M + H), 352 (M + Na), 393 (M + Na + CH3CN) Ensaio de HPLC: 97% 1H NMR (CDCl3, 400 MHz) δ 3,18-3,22 (m, 2H, CH2CH20), 3,87 (s, 3H, OCH3), 4,60-4,64 (m, 2H, CH2CH20), 6,40 (br s, 1H, OH), 6,88 (dd, J, = 2,2 Hz, J2= 8,8 Hz, 1H, H aromático), 7,067,08 (m, 2H, H aromático + CH) , 7,22 (s, 1H, H aromático), 7,25-7,28 (m, 1H, H aromático), 7,96-8,0 (s + br s, 2H, NH+ CH). Exemplo 3 N-[2-5-metóxi-indol-3-il)-etil]-quelidonamida Esquema de Reação para a síntese de N-[2-5-metóxi- indol-3-il)-etil]-quelidonamida
Esquema de Reação para a síntese de N-[2-5-metóxi- indol-3-il)-etil]-quelidonamida i) DMF, HOBt 1,1 equivalentes, EDC 1,1 equivalentes, Net3 2,5 equivalentes, temperatura ambiente, 24 horas.
i) DMF, HOBt 1,1 equivalentes, EDC 1,1 equivalentes, Net3 2,5 equivalentes, temperatura ambiente, 24 horas.
[069] Em um frasco de quatro gargalos de fundo arredondado de 100 ml mantido sob uma atmosfera de argônio, 5- metóxi-triptamina (350 mg, 1,1 equivalente) foi dissolvida em 10 ml de DMF. Sob agitação magnética, ácido quelidônico (310 mg, 1,1 equivalente) foi adicionado. A solução resultante foi colocada a 0°C por meio de um banho de gelo e HOBt (1-monoidrato de hidróxi benxotriazol, 250 mg, 1,1 equivalente), EDC (cloridreto de 1-(3-dimetilaminopropil)-3- etil carbodiimida, 350 mg, 1,1 equivalente) e trietilamina (0,6 ml, 2,5 equivalentes) foram então adicionados sob agitação magnética. A mistura permaneceu sob agitação por mais quinze minutos a 0°C e foi colocada subsequentemente para reagir por 48 horas à temperatura ambiente. 0 curso da reação foi seguido por HPLC- MS. Os materiais precipitados foram removidos por meio de filtração. Água (100 ml) foi adicionada ao material filtrado e a mistura foi extraída com diclorometano (3 x 50 ml). As fases orgânicas combinadas foram secadas sobre Na2SO4 e o solvente foi removido por evaporação giratória. 0 produto bruto foi então foi submetido à cromatografia sobre uma coluna em gel de sílica pela eluição inicial com diclorometano/etanol 8/2. Após a eluição de um produto lateral, a polaridade do eluente foi aumentada (diclorometano/etanol l/l) e o produto foi recuperado como um sólido amarelo pálido (70 mg, rendimento de 11%) .
[070] Dados experimentais para N- [2- (5-metóxi- indol-3-il) -etil] - que1idonamida MS (ESI POS): 357 (M + H), 374 (M + Na) , 398 (M + H + CH3CN) Ensaio de HPLC: 97% 1H NMR (DMSO-de 400 MHz) δ 2,91(t,J = 7,5 Hz, 2H, CH2CH2NH), 3,50-3,55 (m, 2H, CH2CH2NH), 3,76 (s, 3H, OCH3), 6,64-6,71 (m, 3H), 7,07 (d, J = 2,6 Hz, 1H), 7,13 (d, J = 2,1 Hz, 1H), 7,20 (d, J = 8,8 Hz, 1H), 8,29 (s, 1H, H), 8,92 (br t, J = 5,8 Hz, 1H, CH2CH2NH), 10,62 (br s, 1H, COOH). Exemplo 4 N-[2-(5-metóxiindol-3-il)-etil]-cumalilamida Procedimento geral para a síntese de N-[2-(5-metóxiindol-3-il)-etil]-cumalilamida
Procedimento geral para a síntese de N-[2-(5-metóxiindol-3-il)-etil]-cumalilamida
[071] Sob uma atmosfera de argônio, um frasco de três gargalos de fundo arredondado de 100 ml foi carregado com ácido cumálico (600 mg, 1 equivalente) e 5-metóxi-triptamina (900 mg, 1,1 equivalente), dissolvido em DMF (25 ml) e trazido a 0°C por meio de um banho de gelo. HOBt (1-monoidrato de (cloridreto de 1- (3-dimetilaminopropil)-3-etil carbodiimida 900 mg, 1,1 equivalente) e trietilamina (1,5 ml, 2,5 equivalentes) foram então adicionados sob agitação magnética. A mistura foi agitada por mais quinze minutos a 0°C e colocada para reagir subsequentemente por 48 horas à temperatura ambiente. O curso da reação foi seguido por HPLC-MS. Água (40 ml) foi então adicionada e a mistura foi extraída completamente com diclorometano (6 x 30 ml). As fases orgânicas combinadas foram secadas sobre Na2S04 e o solvente foi removido por evaporação giratória. O produto bruto foi então submetido à cromatografia sobre uma coluna em gel de sílica ao eluir com diclorometano/metanol 9515 e o produto foi recuperado (130 mg, rendimento de 9,5%).
[072] Dados experimentais para N-[2-(5- metóxiindol-3-il)-etil]-cumalilamida MS (ESI POS): 313 (M + H), 335 (M + Na), 376 (M + Na + CH3CN) Ensaio de HPLC: 95% 1H NMR (CDCl3, 400 MHz) δ 3,09 (t, J = 6,1 Hz, 2H, CH2CH2NH), 3,70-3,74 (m, 2H, CH2CH2NH), 3,87 (s, 3H, OCH), 5,58 (d, J = 8,8 Hz, 1H, CH), 6,88-7,04 (m, 5H, 4 H aromático + 1 CH), 7,29 (d, J = 8,8 Hz, 1H, CH), 8,03 (br s, 1H, NH), 9,65 (br s, 1H, CH2CH2NH) . Exemplo 5 N-[2-(2-bromo-5-tnetóxi-indol-3-il)-etil]- cumalilamida
[073] Esquema de Reação para a síntese de N-[2- (5-metóxi-indol-3-il)-etil]-cumalilamida a) anidrido ftálico, TEA, tolueno, refluxo, até a manhã seguinte b) tribrometo de piridínio, THF/clorofórmio, - 10°C, 30 min. c) metilamina, EtOH, temperatura ambiente, 3 horas, d) ácido cumálico, NMM, TBTU, DMF, temperatura ambiente, 5 horas a. 5-metóxi-triptamina e anidreto ftálico foram refluxados em tolueno por dezesseis horas. A concentração da reação sob pressão reduzida resultou no produto bruto que foi utilizado na etapa seguinte sem purificação adicional. b. A ftaloiltriptamina bruta foi dissolvida em THF:CHC13(1:1) e a solução resultante foi resfriada até -10°C e então tratada com perbrometo de brometo de piridínio. A reação foi verificada por TLC e foi colocada para aquecer até a temperatura ambiente; CH2Cl2 foi adicionado. A solução foi lavada com Na2SO4 aquoso saturado e as camadas aquosas foram extraídas com CH2Cl2. As camadas orgânicas combinadas foram secadas (MgSO4), filtradas, concentradas sob pressão reduzida, e o produto bruto foi utilizado na etapa seguinte sem purificação adicional. c. O grupo ftalimido foi removido pelo tratamento com metilamina aquosa em etanol à temperatura ambiente. d. N-metilmorfolina foi adicionada a uma solução de ácido cumálico em dimetil formamida seguida por tetrafluoroborato de 2-(1H-benzotriazol-1-il) -1,1,3,3- tetrametil urônio (TBTU) sob uma atmosfera de nitrogénio. Depois que a mistura de reação foi agitada por vinte minutos à temperatura ambiente, 5-metóxi-triptamina foi adicionada lentamente e a mistura foi agitada por cinco horas. A DMF da mistura de reação foi removida sob vácuo elevado. O produto sólido foi dissolvido em CH2Cl2 e a fração orgânica resultante foi lavada com HCl 0,2N, NaHC03, 0,2N e água, e então secada (MgSO4), filtrada e concentrada sob pressão reduzida. O produto resultante foi purificado por meio de cromatografia de coluna.
a) anidrido ftálico, TEA, tolueno, refluxo, até a manhã seguinte b) tribrometo de piridínio, THF/clorofórmio, - 10°C, 30 min. c) metilamina, EtOH, temperatura ambiente, 3 horas, d) ácido cumálico, NMM, TBTU, DMF, temperatura ambiente, 5 horas a. 5-metóxi-triptamina e anidreto ftálico foram refluxados em tolueno por dezesseis horas. A concentração da reação sob pressão reduzida resultou no produto bruto que foi utilizado na etapa seguinte sem purificação adicional. b. A ftaloiltriptamina bruta foi dissolvida em THF:CHC13(1:1) e a solução resultante foi resfriada até -10°C e então tratada com perbrometo de brometo de piridínio. A reação foi verificada por TLC e foi colocada para aquecer até a temperatura ambiente; CH2Cl2 foi adicionado. A solução foi lavada com Na2SO4 aquoso saturado e as camadas aquosas foram extraídas com CH2Cl2. As camadas orgânicas combinadas foram secadas (MgSO4), filtradas, concentradas sob pressão reduzida, e o produto bruto foi utilizado na etapa seguinte sem purificação adicional. c. O grupo ftalimido foi removido pelo tratamento com metilamina aquosa em etanol à temperatura ambiente. d. N-metilmorfolina foi adicionada a uma solução de ácido cumálico em dimetil formamida seguida por tetrafluoroborato de 2-(1H-benzotriazol-1-il) -1,1,3,3- tetrametil urônio (TBTU) sob uma atmosfera de nitrogénio. Depois que a mistura de reação foi agitada por vinte minutos à temperatura ambiente, 5-metóxi-triptamina foi adicionada lentamente e a mistura foi agitada por cinco horas. A DMF da mistura de reação foi removida sob vácuo elevado. O produto sólido foi dissolvido em CH2Cl2 e a fração orgânica resultante foi lavada com HCl 0,2N, NaHC03, 0,2N e água, e então secada (MgSO4), filtrada e concentrada sob pressão reduzida. O produto resultante foi purificado por meio de cromatografia de coluna.
[074] Dados experimentais para N-[2-(2-bromo-5-metóxi-indol-3-il) - etil]-cumalilamida 1H NMR(CDCl3, 300 MHz) δ 10,00 MR (s, 1H, NH) , 8,00 (s, 1H, COOCH aromático), 7,06 (t, 1H, J = 9 Hz, CONH) , 6,786,67 (m, 4H, H aromático), 5,41 (d, 1H, J = 9,6 Hz, COCH aromático), 3,67 (s, 3H, OCH), 3,52 (q, 2H, J = 6,24 Hz), 2,87 (t, 2H, J = 6,3 Hz) Exemplo 6 N-[2-(5-metóxi-indol-3-il)-etil]-comanilamida
[075] Esquema de Reação para a síntese de N- [2- (5-metóxi-indol-3-il)-etil]-comanilamida i. DMF, HOBt 1,1 equivalente, EDC 1,1 equivalentes, Net3 2,5 equivalentes, temperatura ambiente, 6 horas.
i. DMF, HOBt 1,1 equivalente, EDC 1,1 equivalentes, Net3 2,5 equivalentes, temperatura ambiente, 6 horas.
[076] Sob uma atmosfera de argônio, um frasco com três gargalos de fundo arredondado de 100 ml foi carregado com ácido comânico (500 mg, 1 equivalente) e 5-metóxi- triptamina (760 mg, 1,1 equivalente), dissolvido em DMF (25 ml) e colocado a 0°C por meio de um banho de gelo. HOBt (1- monoidrato de hidróxi benxotriazol, 530 mg, 1,1 equivalente), EDC (cloridreto de 1-(3-dimetilininopropil)-3-etil carbodiimida, 750 mg, 1,1 equivalente) e trietilamina (1,25 ml, 2,5 equivalentes) foram então adicionados sob agitação magnética. A mistura foi agitada por mais quinze minutos a 0°C e colocada para reagir subsequentemente por seis horas à temperatura ambiente. O curso da reação foi seguido por HPLC- MS. Água (50 ml) foi então adicionada e a mistura foi extraída com diclorometano (3 x 50 ml). As fases orgânicas combinadas foram secadas sobre Na2SO4 o solvente foi removido por evaporação giratória. O produto bruto foi então submetido à cromatografia sobre uma coluna em gel de sílica ao eluir com diclorometano/metanol 95/5. O produto foi recuperado como um sólido amarelo brilhante (235 mg, rendimento de 21%).
[077] Dados experimentais para N-[2-(5-metóxi- indol-3-il)-etil]-comanilamida MS (ESI POS): 313 (M + H), 330 (M + H20), 335 (M + Na), 376 (M + Na + CH3CN) Ensaio de HPLC: 98% 1H NMR (DMSO-de, 400 MHz) δ 2,88-2,92 (m, 2H, CH2CH2NH), 3,48- 3,53 (m, 2H, CH2CH2NH), 3,75 (s, 3H, OCH) , 6,42 (dd, J1 = 2,3 Hz, J2 5,9 Hz, 1H, CH = CH), 6,71 (dd, J1= 2,1 Hz, J2= 8,8 Hz, 1H, H aromático), 6,78 (d, J = 2,3 Hz, 1H, H aromático), 7,04 (d, J = 2,3 Hz, 1H, CH), 7,13 (d, J = 2,1 Hz, 1H, H aromático), 7, 22 (d, J = 8,8 Hz, 1H, H aromático), 8,21 (d, J = 5,9 Hz, 1H, CH = CH-CO) , 9,04 (br t, J = 5,8 Hz, 1H, CH2CH2NH), 10,65 (br s, 1H, NH). Exemplo 7 N-[2-(5-metóxi-indol-3-il)-etil]-2-metóxi- comenamida
[078] Esquema de Reação para a síntese de N-[2- (5-metóxi-indol-3-il)-etil]-2-metóxi-comenamida i) CH3I 2,2 equivalentes, CH3ONa 1,1 equivalente, CH3OH, temperatura ambiente, 72 horas. ii) MnO2 16 equivalentes, CH3OH, refluxo, 1,5 hora; Ag20 1 equivalente, H2O, NaOH 1N, temperatura ambiente, 1 hora. iii) HOBt 1,1 equivalente, EDC 1,1 equivalente, Net3 5 equivalentes, temperatura ambiente, 16 horas.
i) CH3I 2,2 equivalentes, CH3ONa 1,1 equivalente, CH3OH, temperatura ambiente, 72 horas. ii) MnO2 16 equivalentes, CH3OH, refluxo, 1,5 hora; Ag20 1 equivalente, H2O, NaOH 1N, temperatura ambiente, 1 hora. iii) HOBt 1,1 equivalente, EDC 1,1 equivalente, Net3 5 equivalentes, temperatura ambiente, 16 horas.
[079] Procedimento geral para a síntese de N-[2- (5-metóxi-indol-3- il)-etil]- 2-metóxi-comenamida
[080] Em um frasco de quatro gargalos de fundo arredondado de 250 ml mantido sob uma atmosfera de argônio, 3,2 g de ácido kójico (1 equivalente) foram dissolvidos em 80 ml de metanol. Metóxido de sódio em solução metanólica (4,6 ml, 1,1 equivalente; Fluka, 5,4 M) foi então adicionado sob agitação magnética em uma porção. Após quinze minutos, uma solução de 2,95 ml (1,1 equivalente) de iodeto de metila em 10 ml de CH3OH foi adicionada por gotejamento e a solução resultante foi colocada para reagir à temperatura ambiente. O curso da reação foi seguido por TLC (diclorometano/metanol 9/1 como o eluente). Após sete horas, a conversão era de aproximadamente 50%; assim sendo, outro 1,1 equivalente de CH3I (2,95 ml em 10 ml de CH3OH) foi adicionado. A mistura de reação foi então reagida sob agitação à temperatura ambiente por mais 65 horas, depois do que água (400 ml) foi adicionada. A solução foi concentrada até um volume residual de aproximadamente 2530 ml e foi deixada em 4°C por catorze horas. O precipitado resultante foi coletado por meio de filtração, lavado com éter dietílico e secado sob o vácuo a 50°C. 2-hidroximetil-5- metóxi-4-piranona foi recuperada como um sólido cristalino amarelo (2,2 g, rendimento de 63%).
[081] Em um frasco de fundo arredondado de 250 ml, 2- hidroximetil-5-metóxi-4-piranona (2,2 g, 1 equivalente) foi dissolvida em 85 ml de metanol e 19,6 g de dióxido de manganês ativo foram adicionados (16 equivalentes) . A mistura de reação foi aquecida sob o refluxo por uma hora e meia, e a seguir resfriada até a temperatura ambiente. A parte insolúvel foi filtrada e a solução do material filtrado restante foi concentrada até aproximadamente um terço do volume inicial. A isto, 30 ml de água, 10 ml de NaOH 1N e 3,3g de óxido de prata (1 equivalente) foram adicionados. A mistura resultante foi reagida por uma hora à temperatura ambiente e então filtrada sobre um chumaço de Celite para eliminar os sais. O material filtrado foi concentrado sob pressão reduzida para remover o metanol do mesmo e foi então lavado com diclorometano. HCl 2N (12 ml) foi adicionado subsequentemente à fase solúvel em água para formar um precipitado, o qual foi coletado por meio de filtração, lavado com éter dietílico e secado sob vácuo a 50 °C. O ácido 5-metóxi-4-oxo-4H-piran-2-carboxílico foi obtido como um sólido branco (1,2 g, rendimento de 50%).
[082] Sob uma atmosfera de argônio, um frasco de três gargalos de fundo arredondado de 100 ml foi carregado com ácido 5- metóxi-4-oxo-4H-piran-2-carboxílico (340 mg, 1 equivalente) e cloridreto de 5-metóxi-triptamina (500 mg, 1,1 equivalente), dissolvidos em DMF (15 ml) e colocados a 0°C por meio de um banho de gelo. HOBt (1-monoidrato de hidróxi benxotriazol, 300 mg, 1,1 equivalente), EDC (cloridreto de l- (3- dimetilaminopropil)-3-etil carbodiimida, 425 mg, 1,1 equivalente) e trietilamina (0,98 ml, 3,5 equivalentes) foram então adicionados sob agitação magnética. A mistura foi agitada por mais quinze minutos a 0°C e colocada para reagir subsequentemente por dezesseis horas à temperatura ambiente. O curso da reação foi seguido por HPLC-MS. Água (25 ml) foi então adicionada e a mistura foi extraída com diclorometano (2 x 30 ml) . Após um tempo, uma suspensão apareceu nas fases orgânicas combinadas. O sólido assim formado foi então coletado por meio de filtração, lavado com diclorometano e secado a 50°C. O produto foi recuperado como um sólido branco (210 mg). Do material filtrado, o solvente foi removido por evaporação giratória. O resíduo sólido obtido foi triturado com diclorometano/éter de petróleo e colocado em repouso à temperatura ambiente por 24 horas. A mistura foi então filtrada para prover N-[2-(5-metóxi-indol-3-il)-etil]-2-metóxi-comenamida adicional (70 mg, rendimento de 42%).
[083] Dados experimentais para N- [2- (5-metóxi- indol-3-il) -etil] -2- metóxi - comenamida MS (ESI POS): 343 (M + H), 365 (M + Na), 406 (M + Na + CH3CN) Ensaio de HPLC: 98% 1HNMR (DMSO-de, 400 MHz) δ 2,87-2,91 NMR (m, 2H, CH2CH2NH), 3. 47-3,52 (m, 2H, CH2CH2NH), 3,70 (s, 3H, OCH), 3,74 (s, 3H, OCH), 6,70 (dd, J1 = 2,2 Hz, J= 8,8 Hz, 1H, H aromático), 6,83 (s, 1H, CH), 7,03 (d, J = 2,8 Hz, 1H, H aromático), 7,12 (d, J = 2,2 Hz, 1H, H aromático), 7,21 (d, J = 8,8 Hz, 1H, H aromático), 8,12 (s, 1H, CH), 9,02 (br t, J = 5,7 Hz, CH7CH2NH), 10,64 (br s, 1H, NH). Exemplo 8 N-[2-(5-metóxi-indol-3-il)-etil]-2-pirona-6- carboxamida
[084] Esquema de Reação para a síntese de N-[2-(5-metóxi-indol-3- il)-etil]-2-pirona-6-carboxamida i KOEt 0,998 equivalente, tolueno, temperatura ambiente, 18 horas; H2O/HCL 37%, temperatura ambiente, 30 minutos. ii HC1 37%, 100°C, 6 horas. iii DME, HOBt 1,1 equivalente, EDC 1,1 equivalente, Py 2,2 equivalentes, Neta 1,4 equivalente, temperatura ambiente, 3 horas.
i KOEt 0,998 equivalente, tolueno, temperatura ambiente, 18 horas; H2O/HCL 37%, temperatura ambiente, 30 minutos. ii HC1 37%, 100°C, 6 horas. iii DME, HOBt 1,1 equivalente, EDC 1,1 equivalente, Py 2,2 equivalentes, Neta 1,4 equivalente, temperatura ambiente, 3 horas.
[085] Procedimento geral para a síntese de N-[2- (5-metóxi-indol-3-il)-etil]-2-pirona-6-carboxamida
[086] Etapas 1 e 2 - em um frasco de quarto gargalos de fundo arredondado de 100 ml mantido sob uma atmosfera de argônio, 5,0 g de oxalato de dietila (1 equivalente) foram dissolvidos em 35 ml de tolueno seco. Etóxido de potássio (2,9 g, 0,998 equivalente) foi então adicionado sob agitação magnética em pequenas porções. A temperatura interna atingiu 40 °C e a suspensão inicial se transformou lentamente em uma solução alaranjada. Após duas horas, a solução foi colocada a 0°C por meio de um banho de gelo e o crotonato de etila (4,3 ml, 1 equivalente) foi adicionado por gotejamento durante um período de dez minutos. Após quinze minutos do final da adição, foi observada a formação de um precipitado amarelo do sal de potássio de 2,4- hexadieno-5-hidróxi-l,6-dioato. A suspensão foi colocada para reagir até a manhã seguinte à temperatura ambiente. A mistura de reação foi filtrada subsequentemente e o precipitado amarelo obtido foi lavado com cicloexano e éter dietílico e secado sob o vácuo a 50 °C para resultar em 4,9 g de um sólido amarelo. Este último foi dissolvido então em 70 ml de água, à qual 5 ml de HCl a 37% foram adicionados. Após alguns minutos, um precipitado amarelo foi formado. A suspensão foi agitada à temperatura ambiente por mais trinta 30 minutos e foi então armazenada a 4°C até a manhã seguinte. O intermediário de 2,4- hexadieno-5-hidróxi-1,6-dioato de dietila foi coletado por meio de filtração e lavado com água.
[087] O éster bruto obtido dessa maneira foi aquecido a 100 °C com 6 ml de ácido clorídrico concentrado. A suspensão inicial se transformou em uma solução quando a temperatura alcançou 60 °C. Após uma hora, um sólido amarelo começou a se formar. Após seis horas, a suspensão foi resfriada e o ácido de pirona foi filtrado. O volume do material filtrado foi reduzido por meio de evaporação; o líquido mãe residual foi resfriado e éter dietílico foi adicionado a fim de precipitar uma quantidade adicional de ácido, o qual foi então recuperado por meio de filtração.
[088] O ácido 2-pirona-6-carboxílico foi obtido juntamente com um sólido amarelo pálido (1,5 g, rendimento de 31%).
[089] Etapa 3 - Em um frasco de três gargalos de fundo arredondado de 100 ml mantido sob uma atmosfera de argônio, cloridreto 5-metóxi-triptamina (430 mg, 1,1 equivalente) foi suspenso em 1,2-dimetoxietano (DME, 15 ml). Piridina foi adicionada (0,34 ml, 2,2 equivalentes) e a suspensão foi agitada à temperatura ambiente por trinta minutos. O ácido 2- pirona-6-carboxílico (250 mg, 1 equivalente) foi então adicionado e a temperatura interna foi colocada a 0°C por meio de um banho de gelo. HOBt (1-monoidrato de hidróxi benxotriazol, 260 mg, 1,1 equivalente), EDC (cloridreto de 1- (3-dimetilaminopropil)-3-etil carbodiimida, 370 mg, 1,1 equivalente) e trietilamina (0,34 ml, 1,4 equivalente) foram então adicionados sob agitação magnética. A mistura foi agitada por mais quinze minutos a 0°C e colocada para reagir subsequentemente por três horas à temperatura ambiente. O curso da reação foi seguido por HPLC-MS. A solução obtida foi concentrada sob vácuo e o resíduo bruto foi purificado por cromatografia de coluna, ao eluir com diclorometano/metanol 98/2. N-[2-(5-metóxi-indol-3-il)-etil]- 2-pirona-6-carboxamida foi recuperado como um sólido amarelo (400 mg, rendimento de 72%). Dados experimentais para N-[2-(5-metóxi-indol-3-il) -etil]-2-pirona-6-carboxamida MS (ESI POS) : 313 (M + H), 330 (M + H2O), 376 (M + Na + CH3CN) Ensaio de HPLC: 97% 1H NMR (DMSO-de, 400 MHz) δ 2,87-2,91 (m, 2H, CH2CH7NH) , 3,47- 3,52 (m, 2H, CH2CH2NH), 3,75 (s, 3H, OCH), 6,55 (d, J = 9,4 Hz, 1 H, CH), 6,70 (dd, Ji= 2,9 Hz, J2= 8,8 Hz, 1H, H aromático), 7,02 (Br d, J = 6,6 Hz, 1H, CH), 7,06 (d, J = 2,1 Hz, 1H, H aromático), 7,13 (d, J = 2,2 Hz, 1H, H aromático), 7,22 (d, J = 8,8 Hz, 1H, H aromático), 7,67 (dd, J, = 6,6 Hz, J2 = 9,4 Hz, 1H, CH), 8,87 (br t, J = 5,8 Hz, 1H, CH2CH2NH), 10,65 (br s, 1H, NH).
[090] Potenciação do Tempo de Sono em Camundongos com Hexobarbital-Na Camundongos CD1 foram divididos aleatoriamente em grupos de sete camundongos cada um. Os camundongos em cada grupo receberam intraperitonealmente uma dose de um dos seguintes: 100 mg/kg de uma das substâncias de teste de éster O-[2-(5-metóxi- indol-3-il)-etil]-comênico, N-[2-(5-metóxi-indol-3-i1)-etil]-comenamida, N-[2-(5-1-3 metóxi-indol-3-il)- etil]- cumalilamida, N-[2-(5-metóxi-indol-3-il)-etil]quelidonamida, N-[2-(5-metóxi-indol-3-il)-etil]-comanilamida ou N-[2-(5-metóxi-indol-3-il)-etil]-2-metóxi- comenamida em solução salina (0,1 ml/10 g do peso corpóreo) ou solução salina sozinha. Quinze minutos depois, os camundongos receberam uma dose de 50 mg/kg de hexobarbital-Na intravenosamente. O tempo de sono foi medido em cada animal como o tempo de perda à recuperação do reflexo de ficar em pé.
[091] Conforme mostrado na Tabela 1 abaixo, 100 mg/kg i.p de éster O-[2-(5-metóxi-indol-3-il)-etil]-comênico, N[2-(5-metóxi-indol-3-i1)-etil]-cumalilamida e N-[2-(5- metóxiindol-3-il)-etil]-comenamida aumentaram significativamente o tempo de narcose com hexobarbital-Na e N- [2-(5-metóxi-indol-3-il)-etil]-quelidonamida e N-[2-(5-metóxi-indol-3-il)-etil]- comanilamida aumentaram moderadamente o tempo de narcose com hexobarbital-Na. Os resultados demonstram a potência hipnótica dos compostos através de um mecanismo de ligação alostérica positivo de GABAa. Tabela 1. Efeitos de 100 mg/kg dos compostos de teste no tempo de sono induzido por hexobarbital-Na em camundongos
[092] Ligação da 125I-melatonina nas Membranas de Células de CHO-K1
[093] Alíquotas das membranas suspensas de células recombinantes humanas CHO-K1 (ovário de hamster chinês) que expressam estavelmente os receptores de melatonina-1 ou de melatonina-2 (MT-1 ou MT-2) humanos ou de cérebro de hamster (MT-3) foram incubadas a 250C com 0,05 nM de 125I-melatonina em tampão (25 mM de HEPES, pH 7,4, 5 mM de MgCl2, 1 mM de CaCl2, 0,5% de BSA) ou com 0,1 nM, para MT-3 sozinho ou na presença de 1 nM, 10 nM, 0,1 μM, 1 μM e 10 μM das substâncias de teste N-[2-(5-metóxi-indol-3-i1)-etil]- comenamida, N-[2(5-metóxi- indol-3-il)-etil]-comanilamida, N- [2-(5-metóxiindol-3-il)-etil]-cumalilamida, N-[2-(5-metóxi- ind01-3-i1)etil]-2-metóxi-comenamida, N-[2-(5-metóxi-indol-3- il)-etil]-quelidonamida, éster O-[2-(5-metóxi-ind01-3-i1)- etil]-comênico, N-[2-(5-metóxi-indol-3-il)-etil]-2-pirona- 6carboxamida e N-[2-(2-bromo-5-metóxi-indol-3-il)-etil]- cumalilamida por três horas para MT-1, quatro horas para MT-2 e trinta minutos para MT-3. A reação de ligação foi completada e as membranas foram lavadas com 4 ml de tampão HEPES gelado por meio de filtração a vácuo. As membranas foram então coletadas, e os filtros contendo a 125I-melatonina ligada foram testados quanto à quantidade de radioatividade em um econtador. A ligação não-específica foi avaliada utilizando uma reação com 1 μM de 6-cloromelatonina (MT-1 e MT-2) ou 30 μM de melatonina (MT-3).
[094] Os resultados, mostrados nas Tabelas 2 e 3, demonstram a competição dos compostos na ligação específica de 125I-melatonina aos receptores de MT-1, MT-2 e MT-3. Foi mostrado que N-[2-(5-metóxi-indol-3-il)-etil]-comenamida e N- [2-(5-metóxi-indol-3-il)-etil]-comanilamida se ligam com uma afinidade elevada aos três subtipos de receptor de melatonina, sendo que foi mostrado que o restante dos compostos se liga com uma afinidade pelo menos moderada aos receptores de melatonina. Tabela 2. Efeitos dos compostos de teste na ligação aos receptores de MT-1 ou MT-2 Tabela 3
Tabela 3
[095] Subtipos de Receptor de Serotonina que se Ligam às Membranas das Células CHO-K1
[096] Alíquotas das membranas suspensas das células CHO- Kl recombinantes humanas que expressam estavelmente os receptores de 5-HTIA, 5-HT2A, 5-HTIB, 5-HT2B, 5- HT2C, 5-HT4, 5- HTG ou 5-HT7 humanos foram pré-incubadas a 25°C com 1,5 nM de [3H] 8-OH-DPAT (5-HTIA), 1,5 nM de [3H] Ketanserin (5-HTA2), 0,01 nM de [125I] Cyanopindolol (5-HTIB), 1 nM de [3H] Mesulergine (5-HT2C) , 0,7 nM de [3H] GR—113808 (5-HT4) ou a 37°C, 1,2 nM de [3H] LSD (5-HT2B, 5-HTG 5-HT7) em tampão (50 mM de Tris-HCl, pH 7,7) sozinho ou na presença de 1 nM, 10 nM, 0,1 μM, 1 μM e 10 μM de N-[2-(5-metóxi-indol-3-il)-etil]- comenamida, N-[2-(5-metóxi-indol-3-il)-etil]-cumalilamida, N— [2—(5-metóxi-indol-3-il)-etil]-comanilamida, N—[2-(5—metóxi— indol-3-il)-etil]-2-metóxi-comenamida ou N-[2-(5-metóxi- indol-3-il)-etil]-quelidonamida por sessenta minutos. A reação de ligação foi terminada e lavada com 4 ml de tampão gelado de 50 mM de Tris-HCl por filtração a vácuo. As membranas foram então coletadas, e os filtros contendo os ligandos ligados foram testados quanto à quantidade de radioatividade em um b— contador. A ligação não—específica foi avaliada utilizando uma reação com 10 μM de Metergoline (5-HT1a), 1 μM de Mianserin (5-HT2A e 5-HT2C) ou 10 μM de serotonina (5-HTIB, 5-HT2B, 5-HT4, 5-HT6 5-HT7).
[097] Os resultados, mostrados na Tabela 4 abaixo, demonstram a competição dos compostos na ligação de receptores de 5-HT específicos. Foi mostrado que N-[2-(5- metóxi-indol-3-il)-etil]-comenamida se liga com uma afinidade moderada aos receptores de 5-HTIA, 5-HT2B 5-HT7, N-[2-(5-metóxi- indol-3-il)-etil]-cumalilamida se liga com uma afinidade moderada aos receptores de 5-HT2B 5-HT7, N-[2-(5-metóxi-indol- 3-il)—etil] -comanilamida se liga com uma afinidade moderada ao receptor de 5-HTIB, N-[2-(5-metóxi-indol-3-il)-etil]-2- metóxi-comenamida se liga com uma afinidade moderada aos receptores de 5-HT2B 5-HT7 e N—[2—(5-metóxi-indol-3-il)-etil]- quelidonamida se liga com uma afinidade moderada aos receptores de 5-HTIA e 5-HTIB. Tabela 4. Efeitos dos compostos de teste na ligação aos receptores de 5-HT

[098] Os compostos hipnóticos causam uma depressão da atividade locomotora, capacidade de elevação sobre as patas traseiras reduzida, hipotermia e ataxia avaliada em uma barra 5 giratória em camundongos (Crabbe et al. Psychopharmacology, 161; 408-416, 2002).
[099] Os camundongos ficaram sem comer por dezesseis horas antes do tratamento. Os camundongos CDI machos, pesando 25-30g, foram tratados intraperitonealmente com melatonina, N- [2-(5-metóxi-indol-3-il)-etil]-comanilamida ou N-[2- (5-metóxi-indol-3-il)-etil]-comenamida em uma dose de 100 mg/kg.
[0100] Os movimentos horizontal (isto é, a locomoção) e vertical (isto é, elevação sobre as patas traseiras) foram medidos por cinco minutos duas vezes, trinta e sessenta minutos após o tratamento. Oito camundongos/grupo foram utilizados. O medidor da atividade do canal 4 é uma armação em formato de quadrado que contém gaiolas acrílicas permeáveis infravermelhas transparentes. As armações apresentam dois pares de tiras de feixe de luz para medir os movimentos horizontais, e dois pares para medir a elevação sobre as patas traseiras. Cada tira é equipada com dezesseis sensores infravermelhos.
[0101] No ensaio de motímetro, a melatonina, N- [2-(5-metóxi-indoI-3-i1)-etil]-comanilamida e N-[2-(5-metóxi- indol-3-il)-etil]-comenamida na dose intraperitoneal de 100 mg/kg não alteraram significativamente a atividade motora e a elevação sobre as patas traseiras medidas entre 30 e 35 minutos e 60 e 65 minutos após o tratamento (Tabela 5).
[0102] N-[2-(5-metóxi-indol-3-il)-etil]- cumalilamida na dose intraperitoneal de 100 mg/kg diminuiu significativamente a atividade motora e de elevação sobre as patas traseiras nos dois intervalos de tempo mencionados acima. Estes resultados 10 demonstram os efeitos hipnótico e sedativo de N-[2-(5-metóxiindol-3-il)-etil]-cumalilamida.
[0103] As incidências de animais correndo mais de 120 minutos na roda giratória foram anotadas e as significâncias foram calculadas pelo teste X2 não-paramétrico. Oito camundongos/grupo foram utilizados.
[0104] O aparelho de roda giratória é dividido em cinco zonas de teste, de modo que até cinco camundongos podem ser testados ao mesmo tempo. A roda foi especialmente usinada para prover uma alça apropriada para o animal. O diâmetro da roda é de 3,5 cm. A velocidade de rotação é de 15 rpm. Quando o animal cai da roda giratória, ele pressiona um botão para gravar automaticamente o tempo gasto na roda. No dia antes da experiência, os camundongos foram treinados a correr na roda giratória com 15 rpm. Diazepam foi administrado oralmente sessenta minutos antes do ensaio da roda giratória, e as substâncias de teste foram administradas intraperitonealmente quinze minutos antes do teste.
[0105] No teste da roda giratória, a dose de N- [2-(5-metóxi- indol-3-il)-etil]-comanilamida e de N-[2-(5- metóxiindol-3-il) -etil]-comenamida prejudicou de maneira dependente o desempenho dos camundongos na velocidade de rotação de 15 rpm (Tabela 6).
[0106] Diazepan na dose oral de 1,5 mg/kg potencializou significativamente o desempenho da roda giratória, prejudicando os efeitos de ambos os compostos em todas as três doses aplicadas. Estes resultados demonstram os efeitos hipnóticos sinergísticos de N-[2-(5-metóxi-indol-3- il)-etil] comanilamida e N-[2-(5-metóxi-indol-3-il)-etil]- comenamida administrados com diazepam do agente hipnóticode benzodiazepina.Tabela 5: Efeitos da melatonina e de N-[2-(5-metóxi- indol-3-il)-etil]-cumalilamida na atividade motora em camundongos (movimento horizontal) Tabela 6: Efeitos da interação de N-[2-(5-metóxi- indol-3-il)-etil]-comanilamida e de N-[2-(5-metóxi-indol-3- il)-etil] comenamida com diazepam no ensaio de roda giratória em camundongos
Tabela 6: Efeitos da interação de N-[2-(5-metóxi- indol-3-il)-etil]-comanilamida e de N-[2-(5-metóxi-indol-3- il)-etil] comenamida com diazepam no ensaio de roda giratória em camundongos
[108] Adipócitos ficaram sem glicose por uma hora em tampão de HEPES-sal contendo 2% de BSA livre de AGL. Os AGL (ácidos graxos livres) foram então adicionados às células nas concentrações indicadas (300 μΜ) pelos tempos indicados (três horas). Dez minutos antes do final do tratamento com AGL, as células foram estimuladas com insulin (20 nM)/melatonina (10 nM) /compostos de teste (10 nM) a 37°C. 2-[3H]-deoxi-d-glicose em 1 μCi/mL e 0,1 mM de 2-deoxiglicose não-etiquetada em tampão de KRP-HEPES foram adicionados e as células foram incubadas por dez minutos à temperatura ambiente. A captação de glicose não-especifica foi medida por incubações paralelas na presença de 10 μM de citocalasina B, bloqueando a captação de glicose mediada pelo transportador, e foi subtraída da captação total em cada ensaio. As células foram então lavadas três vezes com solução salina tamponada com fosfato gelado (STF) e solubilizadas em NaOH 1M por vinte minutos. A amostra foi então contada utilizando um contador de cintilação. A captação de 2- [3H]-deoxi-d-glicose foi testada em triplicatas para cada condição em pelo menos três experiências independentes. A captação de 2-[3H]-deoxi-d- glicose (contagens por minuto-cpm) é apresentada como média + SE de triplicatas em uma experiência representativa ou resultados de três experiências independentes. O teste ANOVA foi utilizado com significância de P < 0,05 (Tabela 7).
 (a:P < 0,05 versus grupo C, b:P < 0, 05 versus grupo D, c:P < 0,05 versus grupo da melatonina, d:P < 0,05 versus todos os outros grupos, ANOVA).
(a:P < 0,05 versus grupo C, b:P < 0, 05 versus grupo D, c:P < 0,05 versus grupo da melatonina, d:P < 0,05 versus todos os outros grupos, ANOVA).
[109] Adipócitos 3T3-L1 foram utilizados como um modelo in vitro para avaliar o efeito celular de derivados de pirona- indol e de melatonina na resistência à insulina iniciada pelo tratamento com AGL elevados. Nos adipócitos 3T3- L1, o tratamento com AGL prejudicou a sinalização de insulina os derivados de melatonina/pirona-indol aprimoraram o transporte da glicose. Portanto, a melatonina e os derivados de pirona- indol poderiam incrementar a resistência à insulina iniciada por AGL.
Claims (12)
1. COMPOSTO, caracterizado por compreender a fórmula: Ar - B-Ar' (I) em que: -B- representa: X-Y-Z, em que X representa -(CH2)n (em que n está entre 0 a 6) em que a porção alquila é linear ou ramificada; Y representa oxigênio, enxofre, >NH ou está ausente; Z representa >C = O, >O, >COO ou está ausente; em que pelo menos um dentre X, Y e Z deve estar presente; Ar representa um sistema de anel de núcleo de indol: Ar' representa um sistema de anel do núcleo de alfa, beta ou gama-pirona:
Ar' representa um sistema de anel do núcleo de alfa, beta ou gama-pirona: em que cada um de R1-4 substitui o sistema de anel de Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1’-R2’, substitui o sistema de anel de Ar' em qualquer posição disponível e em que cada um de R1-R4 e de R1’- R2’,representa independentemente hidrogênio, oxigênio, halo, halo-C1-5 alquila, , um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre, alquila C1-5, alquenila C2-5, alquinila C2-5, arilalquila Cl-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila C1-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamida C1-5, alquilamido C1-5, hidróxi, tiol, acilóxi, azido, alcóxi C1-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, aralalquinila ou estirila podem, opcionalmente, substituídos em anel por um ou quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, halo- C1-5 alquila, um grupo heterocíclico C1-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila C1-5, alquenila C2-5, alquinila C2-5, alquilarila C1-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila C15, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, azido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e tanto R3 quanto R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e R2, incluindo a posição N, e Ar’ pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1’ e R2’; ou um sal ou um estereoisômero farmaceuticamente aceitável destes; e em que o composto não é
em que cada um de R1-4 substitui o sistema de anel de Ar em qualquer posição disponível (incluindo a posição N) e cada um de R1’-R2’, substitui o sistema de anel de Ar' em qualquer posição disponível e em que cada um de R1-R4 e de R1’- R2’,representa independentemente hidrogênio, oxigênio, halo, halo-C1-5 alquila, , um grupo heterocíclico C5-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre; um grupo heteroarila C6-8 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio ou enxofre, alquila C1-5, alquenila C2-5, alquinila C2-5, arilalquila Cl-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila C1-5, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, alquilamida C1-5, alquilamido C1-5, hidróxi, tiol, acilóxi, azido, alcóxi C1-5, carbóxi, carbonilamido ou estirila; em que os ditos grupos arilalquila, arilalquenila, aralalquinila ou estirila podem, opcionalmente, substituídos em anel por um ou quatro substituintes selecionados independentemente do grupo que consiste em hidrogênio, halo, halo- C1-5 alquila, um grupo heterocíclico C1-7 que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; um grupo heteroarila que contém um a três heteroátomos selecionados independentemente entre nitrogênio, oxigênio e enxofre; alquila C1-5, alquenila C2-5, alquinila C2-5, alquilarila C1-5, arilalquenila C2-5, arilalquinila C2-5, hidróxi alquila C15, nitro, amino, ciano, cianamido, guanidino, amidino, acilamido, hidróxi, tiol, acilóxi, azido, alcóxi, carbóxi, carbonilamido, S-alquila ou alquiltiol; e tanto R3 quanto R4 pode incluir ou representar adicionalmente uma ligação a B; em que Ar pode ser ligado a B em qualquer posição no anel de Ar não substituído por R1 e R2, incluindo a posição N, e Ar’ pode ser ligado a B em qualquer carbono no anel de Ar' não substituído por R1’ e R2’; ou um sal ou um estereoisômero farmaceuticamente aceitável destes; e em que o composto não é com a condição de exclusão do composto, quando X - (CH2)2-, Y -NH2-, Z estar ausente e Ar' ser uma gama-pirona substituída.
com a condição de exclusão do composto, quando X - (CH2)2-, Y -NH2-, Z estar ausente e Ar' ser uma gama-pirona substituída.
2. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)n, em que n está entre 0 e 6, Y ser >NH ou >O e Z é >CO.
3. COMPOSTO, de acordo com a reivindicação 2, caracterizado por Ar' ser um sistema do anel de alfa-pirona.
4. COMPOSTO, de acordo com a reivindicação 2, caracterizado por Ar' ser um sistema do anel de beta-pirona.
5. COMPOSTO, de acordo com a reivindicação 2, caracterizado por Ar’ ser um sistema do anel de gama-pirona.
6. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)2-, Y ser >NH ou >O e Z é >CO, Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 ser um grupo metóxi na posição 5 do grupo do anel de indol e cada um de R2 e R4 ser hidrogênio; Ar’ é um anel de gama-pirona ligado a Z na posição 2 do anel de pirona; R1’ ser hidrogênio ou um grupo hidróxi na posição 5 do grupo do anel de pirona; e R2’ ser hidrogênio ou um grupo carbóxi na posição 6 do anel de gama-pirona; ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
7. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)2-, Y ser >NH ou >O e Z ser >CO; Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 ser um grupo metóxi na posição 5 do anel de indol e cada um de R2 e R4 ser hidrogênio; Ar’ ser um anel de alfa-pirona substituído por Z na posição 5 do anel de pirona; e cada um de R1’ e R2 ser hidrogênio ou um sal ou um estereoisômero farmaceuticamente aceitável destes.
8. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)2-, Y ser >NH; e Z ser >CO; Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 ser um grupo metóxi na posição 5 do anel de indol e cada um de R2 eR4 ser hidrogênio; Ar’ ser um anel de gama-pirona substituído por Z na posição 2 do anel de pirona; R1’ ser um grupo hidróxi na posição 5 do anel de pirona; e R2’ ser hidrogênio; ou um sal ou um estereoisômero destes.
9. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)2 -, Y é >O e Z é >CO; Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 ser um grupo metóxi na posição 5 do anel de indol e cada um de R2 e R4 ser hidrogênio; Ar' ser um anel de gama-pirona substituído por Z na posição 2 do anel de pirona; R1’ ser um grupo hidróxi na posição 5 do anel de pirona; e R2’ ser hidrogênio; ou um sal ou um estereoisômero destes.
10. COMPOSTO, de acordo com a reivindicação 1, caracterizado por: X ser -(CH2)2-, Y ser >NH e Z ser >CO; Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 ser um grupo metóxi na posição 5 do anel de indol e cada um de R2 e R4 ser hidrogênio; Ar' ser um anel de gama-pirona substituído por Z na posição 2 do anel de pirona; cada um de R1’ e R2’ ser hidrogênio; ou um sal ou um estereoisômero destes.
11. COMPOSTO, de acordo com a reivindicação 1, caracterizado por X ser -(CH2)2-, Y ser >NH e Z ser >CO; Ar ser um anel de indol; R3 ser uma ligação a X na posição 3 do anel de indol; R1 é um grupo metóxi na posição 5 do anel de indol e cada um de R2 e R4 ser hidrogênio; Ar' é um anel de alfa-pirona substituído por Z na posição 5 do anel de pirona; e R1’ e R2’ ser hidrogênio; ou um sal ou um estereoisômero destes.
12. FORMULAÇÃO FARMACÊUTICA, caracterizada por compreender uma quantidade terapeuticamente eficaz de um composto, de um sal ou de um estereoisômero, conforme definido nas reivindicações 1 a 11, em combinação com um ou mais diluentes, conservantes, solubilizantes, emulsificantes, adjuvantes, excipientes ou veículos farmaceuticamente aceitáveis.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77332206P | 2006-02-15 | 2006-02-15 | |
| US60/773,322 | 2006-02-15 | ||
| PCT/IB2007/000330 WO2007093880A2 (en) | 2006-02-15 | 2007-02-13 | Novel pyrone-indole derivatives and process for their preparation |
Publications (4)
| Publication Number | Publication Date |
|---|---|
| BRPI0706992A2 BRPI0706992A2 (pt) | 2011-04-12 |
| BRPI0706992B1 true BRPI0706992B1 (pt) | 2021-05-04 |
| BRPI0706992C1 BRPI0706992C1 (pt) | 2021-05-18 |
| BRPI0706992C8 BRPI0706992C8 (pt) | 2021-05-25 |
Family
ID=38283075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0706992A BRPI0706992C8 (pt) | 2006-02-15 | 2007-02-13 | composto de fórmula (i) e formulação farmacêutica |
Country Status (21)
| Country | Link |
|---|---|
| US (3) | US7635710B2 (pt) |
| EP (1) | EP1991541B1 (pt) |
| JP (1) | JP5248332B2 (pt) |
| KR (1) | KR101207736B1 (pt) |
| CN (1) | CN101374833B (pt) |
| AU (1) | AU2007216226B2 (pt) |
| BR (1) | BRPI0706992C8 (pt) |
| CA (1) | CA2642465C (pt) |
| DK (1) | DK1991541T3 (pt) |
| EA (1) | EA015605B1 (pt) |
| ES (1) | ES2428873T3 (pt) |
| IL (1) | IL193236A (pt) |
| NO (1) | NO339826B1 (pt) |
| NZ (1) | NZ569797A (pt) |
| PL (1) | PL1991541T3 (pt) |
| PT (1) | PT1991541E (pt) |
| SI (1) | SI1991541T1 (pt) |
| TW (1) | TWI501960B (pt) |
| UA (1) | UA104988C2 (pt) |
| WO (1) | WO2007093880A2 (pt) |
| ZA (1) | ZA200806799B (pt) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6262118B1 (en) * | 1999-06-04 | 2001-07-17 | Metabolex, Inc. | Use of (-) (3-trihalomethylphenoxy) (4-halophenyl) acetic acid derivatives for treatment of insulin resistance, type 2 diabetes and hyperlipidemia |
| US7576131B2 (en) * | 1999-06-04 | 2009-08-18 | Metabolex, Inc. | Use of (-) (3-trihalomethylphenoxy) (4-halophenyl) acetic acid derivatives for treatment of insulin resistance, type 2 diabetes, hyperlipidemia and hyperuricemia |
| CN103319467B (zh) * | 2013-06-15 | 2015-10-14 | 湖南科技大学 | 一种4-[5-羟基-4-吡喃酮-2-基亚甲氨基]-3-巯基-1,2,4-三唑化合物及用途 |
| ES2694049T3 (es) | 2014-01-14 | 2018-12-17 | Astellas Pharma Inc. | Compuesto de indol |
| RU2561045C1 (ru) * | 2014-07-15 | 2015-08-20 | Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования "Кубанский государственный университет" (ФГБОУ ВПО "КубГУ") | Нейропротекторное фармакологическое средство |
| WO2016039750A1 (en) | 2014-09-11 | 2016-03-17 | Halliburton Energy Services, Inc. | Cyanamide-based carbon dioxide and/or hydrogen sulfide scavengers and methods of use in subterranean operations |
| HUE059559T2 (hu) | 2015-02-25 | 2022-12-28 | Univ California | 5HT agonisták az epilepsziás rendellenességek kezelésére |
| CN109843286A (zh) * | 2016-08-23 | 2019-06-04 | 纽里姆药物有限公司 | 治疗瘙痒和/或搔痒的方法 |
| JP2020529440A (ja) | 2017-08-04 | 2020-10-08 | オービッド・セラピューティクス・インコーポレイテッドOvid Therapeutics Inc. | 糖尿病および関連する状態の処置におけるガボキサドールの使用 |
| EP3749697A4 (en) | 2018-02-05 | 2021-11-03 | Bio-Rad Laboratories, Inc. | CHROMATOGRAPHY RESIN WITH LIGAND MIXED MODE ANIONIC / HYDROPHOBIC EXCHANGE |
| WO2020061410A1 (en) | 2018-09-20 | 2020-03-26 | Ovid Therapeutics Inc. | Use of gaboxadol for the treatment of tourette syndrome, tics and stuttering |
| WO2020131856A1 (en) | 2018-12-17 | 2020-06-25 | Ovid Therapeutics Inc. | Use of gaboxadol for the treatment of non-24 hour sleep-wake disorder |
| US12478610B2 (en) | 2020-05-11 | 2025-11-25 | Orit MALKA | Compositions of tryptophol derivatives and 4-ethyl-phenol derivatives, and methods of using same |
| WO2022238905A1 (en) | 2021-05-11 | 2022-11-17 | Neurim Pharmaceuticals (1991) Ltd. | Method for diagnosing and treating subjects having single nucleotide polymorphisms in chromosome 2, 2:107,510,000-107,540,000 locus |
| IL321429A (en) | 2023-01-13 | 2025-08-01 | Neurim Pharmaceuticals 1991 Ltd | Piromelatine for treating parasomnias associated with loss of rem sleep atonia |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA222247A (en) | 1922-08-15 | Hubert Rogers Reginald | Pocket container for confections | |
| IT1196849B (it) | 1986-02-16 | 1988-11-25 | Rotta Research Lab | Nuovi derivati degli acidi 5 pentilammino 5 oxo pentaoico e 4 pentilammino 4 oxo butanoico ad attivita antagonista della colecistochinina e procedimento per la loro preparazione |
| AU8876591A (en) | 1990-11-02 | 1992-05-26 | Upjohn Company, The | Indole-3-methanamines useful as anti-diabetic, anti-obesity and anti-atherosclerotic agents |
| US5840751A (en) * | 1993-11-19 | 1998-11-24 | Warner-Lambert Company | 5,6-dihydropyrone derivatives as protease inhibitors and antiviral agents |
| EP0841063A1 (en) * | 1996-03-27 | 1998-05-13 | Toray Industries, Inc. | Ketone derivatives and medicinal use thereof |
| IL130169A (en) | 1999-05-27 | 2006-08-20 | Neurim Pharma 1991 | Indole derivatives, and pharmaceutical preparations, skin protection preparations, and cosmetics containing them |
| EP1324993A2 (en) * | 2000-04-18 | 2003-07-09 | Cytovia, Inc. | Substituted 1,4-thiazepine and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| PE20060303A1 (es) | 2004-06-23 | 2006-05-19 | Wyeth Corp | Metabolitos de indolilalquilamina como ligandos de 5-hidroxitriptamina-6 |
| US7834050B2 (en) * | 2006-03-29 | 2010-11-16 | Duke University | Small molecule insulin mimetics absent quinones |
-
2007
- 2007-02-12 US US11/705,030 patent/US7635710B2/en active Active
- 2007-02-13 ES ES07733884T patent/ES2428873T3/es active Active
- 2007-02-13 DK DK07733884.6T patent/DK1991541T3/da active
- 2007-02-13 PL PL07733884T patent/PL1991541T3/pl unknown
- 2007-02-13 UA UAA200808838A patent/UA104988C2/uk unknown
- 2007-02-13 SI SI200731333T patent/SI1991541T1/sl unknown
- 2007-02-13 NZ NZ569797A patent/NZ569797A/en unknown
- 2007-02-13 BR BRPI0706992A patent/BRPI0706992C8/pt active IP Right Grant
- 2007-02-13 JP JP2008554868A patent/JP5248332B2/ja active Active
- 2007-02-13 EP EP07733884.6A patent/EP1991541B1/en active Active
- 2007-02-13 EA EA200801580A patent/EA015605B1/ru unknown
- 2007-02-13 WO PCT/IB2007/000330 patent/WO2007093880A2/en not_active Ceased
- 2007-02-13 CA CA2642465A patent/CA2642465C/en active Active
- 2007-02-13 AU AU2007216226A patent/AU2007216226B2/en active Active
- 2007-02-13 KR KR1020087022304A patent/KR101207736B1/ko active Active
- 2007-02-13 PT PT77338846T patent/PT1991541E/pt unknown
- 2007-02-13 CN CN2007800039654A patent/CN101374833B/zh active Active
- 2007-02-26 TW TW096106545A patent/TWI501960B/zh active
-
2008
- 2008-08-04 IL IL193236A patent/IL193236A/en active IP Right Grant
- 2008-08-06 ZA ZA200806799A patent/ZA200806799B/xx unknown
- 2008-09-12 NO NO20083905A patent/NO339826B1/no unknown
-
2009
- 2009-11-04 US US12/612,001 patent/US8242163B2/en active Active
-
2012
- 2012-07-13 US US13/549,181 patent/US8569355B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0706992B1 (pt) | Composto de fórmula (i) e formulação farmacêutica | |
| TWI374141B (en) | Spiro-oxindole compounds and their uses as therapeutic agents | |
| CN110172051B (zh) | IRE-1α抑制剂 | |
| US9101613B2 (en) | Methods for treating neurological disease | |
| ES2376161T3 (es) | Indoles sustituidos y su utilización como antagonistas de integrinas. | |
| HU219715B (hu) | Kondenzált pirrol-karboxanilid-származékok | |
| JP2001512727A (ja) | 5ht−1受容体のリガンドとしてのニ環式化合物 | |
| JP2009516712A (ja) | 依存性イオンチャネルを調節するための組成物および方法 | |
| EP1189583B1 (en) | Indole derivatives | |
| WO2010101128A1 (ja) | アミド化合物 | |
| HK1126760B (en) | Novel pyrone-indole derivatives and process for their preparation | |
| JPWO2002042297A1 (ja) | ピペリジン化合物およびその医薬用途 | |
| AU6967400A (en) | Methods of administering APO B-secretion/MTP inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 13/02/2007, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME MEDIDA CAUTELAR DE 07/04/2021 - ADI 5.529/DF |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: REF. RPI 2626 DE 04/05/2021 QUANTO AO ENDERECO. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 13/02/2007 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |