REIVINDICAÇÃO DE PRIORIDADE PARA PEDIDOS RELACIONADOS Este pedido reivindica a prioridade de pedido provisório US número de série 60/841.097, depositado em 30 de Agosto de 2006, que é incorporado aqui por referência em sua totalidade.
CAMPO DA INVENÇÃO
O campo da invenção refere-se a novas formas de sal sólido de alguns inibidores seletivos de dipeptidil peptidase IV. A invenção refere-se a sais tartarato e citrato de compostos de ácido pirrolidinilaminoacetil pirrolidina borônico, inibidores potentes e seletivos de DPP-IV. Estes sais novos, que são quimicamente e fisicamente estáveis durante pelo menos um período de 12 meses, são sólidos substancialmente não deliquescentes e são apropriados para formulação como medicamentos.
FUNDAMENTOS DA INVENÇÃO
Dipeptidil peptidase-IV (DPP-IV) é uma serina protease que pertence a um grupo de amino-dipeptidases que inclui DPP-VII, DPP-VIIL DPP-IX, e proteína de ativação de fíbroblastos (FAP). DPP-IV, que catalisa a liberação de um dipeptídeo N-terminal de proteínas, desempenha, como se acredita, um papel no controle de metabolismo de glicose. Administração in vivo de inibidores sintéticos de DPP-IV previne degradação N-terminal do peptídeo-1 de tipo glucagona do hormônio insulinotrópico (GLP-I) e peptídeo inibitório gástrico (GIP) resultando em aumentada secreção de insulina e tolerância a glicose melhorada. Ácidos aminoacetil pirrolidina borônicos substituídos têm sido desenvolvidos como inibidores para o tratamento de pacientes com controle glicêmico prejudicado tais como Diabetes Melitus e condições relacionadas. Consequentemente, é desejável formular tais inibidores como materiais farmacêuticos sólidos convenientes para uso. Bases livres e sais de muitos compostos farmacêuticos são tais formulações farmacêuticas típicas. Entretanto, as formas de base livre de alguns ácidos aminoacetil pirrolidina borônicos não são sólidos estáveis, prontamente absorvendo umidade atmosférica. Além disso, alguns sais destas bases livres são igualmente sólidos não estáveis.
Pedido US número de série. 10/514.575, depositado em 15 de Novembro de 2004, intitulado "Heterocyclic Boronic Acid Compounds,"e publicado em 15 de Março de 2007 como US 2007/0060547, descreve uma classe grande de composto de ácido pirrolidina borônico que inibe dipeptidil peptidase-IV. Além disso, Pedido provisório US número de série 11/381.085, depositado em Io de Maio de 2006, intitulado "Pyrrolidine Compounds and Methods for Selective Inhibition of Dipeptidyl Peptidase-IV," e publicado em 23 de Novembro de 2006 como US 2006/0264400, descreve um classe mais específica de compostos de ácido pirrolidinilaminoacetil pirrolidina borônico que seletivamente inibem DPP-IV. Uma forma de realização desta invenção mostra uma promessa particular como um medicamento. Entretanto, embora biologicamente efetivo como uma base livre, composto de base livre não é fisicamente e quimicamente estável como um sólido em armazenamento.
Assim, existe uma necessidade para uma forma de sal quimicamente estável do composto para uso em formulações farmacêuticas para tratamento de más condições em que inibição seletiva de DPP-IV é indicado em forma medicinal.
SUMÁRIO DA INVENÇÃO
A presente invenção refere-se a formas de sal orgânico fisicamente e quimicamente estáveis de ácidos pirrolidinilaminoacetil- pirrolidina-2-borônico inibidores de DPP-IV, compostos de fórmula (1) como definido aqui. Os sais citrato e tartarato de compostos de fórmula (1) são sólidos substancialmente não deliquescentes e podem ser armazenados sob condições ambientes durante pelo menos 12 meses sem decomposições. As formas sólidas destes compostos são apropriadas para processamento, formulação, armazenamento, e administração dos inibidores de DPP-IV, que 5 podem ser úteis como medicamentos ingeridos oralmente para o tratamento de diabetes. Métodos de preparar as formas de sal, métodos de usar as formas de sal, composições farmacêuticas incluindo as formas de sal, e combinações farmacêuticas incluindo as formas de sal, também são providos.
Uma forma de realização de acordo com a invenção provê um 10 sal citrato ou tartarato de um composto pirrolidina de fórmula (I):
um isômero cíclico do mesmo ou qualquer hidrato ou solvato do mesmo; em que R2 e R3 independentemente são -OH, -CT M+ em que M+ é um cátion, uma hidróxila contendo um grupo protetor de ácido borônico, ou um grupo capaz de ser hidrolisado para um grupo hidroxila em uma solução 15 aquosa em pH fisiológico ou em fluidos biológicos; ou R e R juntos com o átomo de boro ao qual eles são fixados formam um grupo cíclico capaz de ser hidrolisado em um grupo de ácido borônico; as linhas onduladas em carbonos assimétricos Ca e Cb independentemente indicam para cada carbono assimétrico uma configuração R, uma configuração S, ou uma mistura de 20 ambas configurações, tal que todos os estereoisômeros e todas as misturas estereoméricas estão incluídas; e a porção tartarato tem qualquer configuração estereomérica ou mistura da mesma.
Em uma forma de realização da invenção, a porção tartarato pode ser um L-tartarato, a D-tartarato, um meso-tartarato, ou qualquer 25 combinação dos mesmos. Em outra forma de realização, a porção tartarato é um L-tartarato.
Em uma forma de realização, os grupos R e R podem ser ambos grupos hidroxila, tal que o átomo de boro está contido em um grupo de ácido borônico ligado à pirrolidina posição 2, deste modo, uma porção boro- 5 prolina. Em outra forma de realização, R2 e R3 de fórmula (I) podem ambos ser grupos hidroxila e o sal tartarato ser um sal L-tartarato.
Em uma forma de realização, o sal reivindicado pode ser qualquer hidrato ou solvato do mesmo.
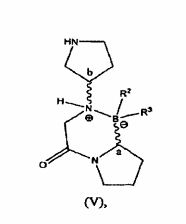
Outra forma de realização é dirigida a isômero cíclico de fórmula (V):
em que Ca e Cb cada independentemente tem uma configuração R, uma configuração S, ou uma mistura de ambas configurações tal que todos os estereoisômeros e todas as misturas estereoméricas estão incluídos.
Outra forma de realização é dirigida a um sal L-tartarato de um 15 composto de fórmula (V). Ainda outra forma de realização é dirigida a um sal L- tartarato de um composto de fórmula (V) em que R e R são ambos grupos hidroxila.
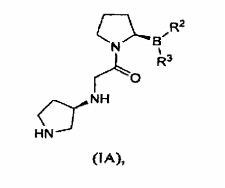
Outra forma de realização da invenção é dirigida a um sal citrato ou tartarato de um composto de fórmula (IA):
em que a pureza estereomérica é pelo menos cerca de 55%, ou pelo menos cerca de 90%, ou pelo menos cerca de 98%. Uma forma de realização provê um sal em que R2 e R3 são ambos grupos hidroxila. Outra forma de realização provê um sal L-tartarato do composto de fórmula (IA). Outra forma de realização provê um sal L-tartarato do composto de fórmula (IA) em que R e R são ambos grupos hidroxila.
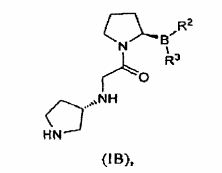
Outra forma de realização da invenção é dirigida a um sal citrato ou tartarato de um composto de (IB):
em que a pureza estereomérica é pelo menos cerca de 55%, ou pelo menos cerca de 90%, ou pelo menos cerca de 98%. Uma forma de realização provê um sal em que R2 e R3 são ambos grupos hidroxila. Outra forma de realização provê um sal L-tartarato do composto de fórmula (IB). Outra forma de realização provê um sal L-tartarato do composto de fórmula (IB) em que R e R são ambos grupos hidroxila.
Outra forma de realização da invenção provê formas sólidas substancialmente não deliquescentes dos sais citrato e tartarato de compostos de qualquer uma das fórmulas (I), (IA), (IB), ou (V), ou formas cíclicas, hidratos, ou solvatos dos mesmos.
Uma forma de realização da invenção também é dirigida a uma composição farmacêutica contendo um sal citrato ou tartarato da invenção e um carreador farmaceuticamente aceitável. Uma composição farmacêutica pode ser formulada para ser dosada por via administrativa incluindo mas não limitado a injeção parenteral, oral, bucal, retal e outros.
Uma forma de realização da invenção é dirigida a um método de tratamento de uma má condição que pode ser regulada ou normalizada através de inibição de DPP-IV. O método envolve administração de uma quantidade efetiva de sal citrato ou tartarato da invenção para mamíferos, tais como humanos, para afetar uma má condição que pode ser regulada ou normalizada através de inibição de DPP-IV.
Uma forma de realização da invenção é dirigida a uma combinação farmacêutica de sal citrato ou tartarato da invenção e um ou mais outros medicamentos que aumenta a secreção de insulina, aumenta a sensibilidade à insulina, reduz a absorção de açúcar do trato gastrointestinal, melhora o efeito de proteínas ou peptídeos endógenos que afetam o controle glicêmico, provê uma substituição para proteínas ou peptídeos endógenos que afetam controle glicêmico, ou qualquer combinação dos mesmos. A combinação farmacêutica pode ser formulada de acordo com a invenção como uma composição farmacêutica.
A invenção também é dirigida a um processo para preparar um sal citrato ou tartarato da invenção, um método para preparar uma composição farmacêutica da invenção, e o uso de sal citrato ou tartarato da invenção em um método para a preparação de um medicamento para tratar uma má condição que pode ser regulada ou normalizada através de inibição de DPP- IV.
DESCRIÇÃO DOS DESENHOS
FIG. 1 é um diagrama esquemático ilustrando a formação do sal L-tartarato de ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]-acetil}- pirrolidina-2-borônico.
FIG. 2 é um gráfico ilustrando a absorção de umidade do sal L-tartarato de ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]-acetil}-pirrolidina- 2-borônico com aumento de umidade relativa.
FIG. 3 é um cromatograma obtido de análise de cromatografia de líquido de alto desempenho (HPLC) de L-tartarato.
FIG. 4 é um cromatograma obtido de análise HPLC do composto pirrolidina.
FIG. 5 é um cromatograma obtido de análise HPLC do sal L- tartarato do composto pirrolidina.
FIG. 6 é um ampliação dos picos correspondendo ao sal L- tartarato do composto pirrolidina.
FIG. 7 é um cromatograma obtido de análise HPLC do produto secado por pulverização, mostrando que o sal L-tartarato da invenção foi isolado por secagem por pulverização sem degradação de produto.
FIG. 8 é um gráfico ilustrando a absorção de umidade típica do sal tartarato da invenção durante cinco dias a 45%, 58%, 63% e 76% de umidade relativa.
FIG. 9 é um cromatograma obtido de análise HPLC do sal L- tartarato da invenção antes da incubação de 2 meses em uma câmara de umidade relativa de 45%.
FIG. 10 é um cromatograma obtido de análise HPLC do sal L- tartarato da invenção após incubação de 2 meses em uma câmara de umidade relativa de 45%.
DESCRIÇÃO DETALHADA DA INVENÇÃO
Definições
Salvo especificado em contrário, todos os termos técnicos e científicos usados aqui têm o mesmo significado como comumente entendido pelo versado na técnica à qual esta invenção pertence. Embora métodos e materiais similares ou equivalente àqueles descritos aqui possam ser usados para praticar a invenção, métodos e materiais apropriados estão descritos abaixo. Todas publicações, pedidos de patente, patentes e outras referências citadas aqui são incorporadas por referência em sua totalidade. O presente relatório provê definições selecionadas de alguns termos, e estas definições são preferidas com relação a outras definições no caso de existirem discrepâncias. Além disso, os materiais, métodos, e exemplos são ilustrativos apenas e não se destinam a ser limitantes.
Outras aspectos e vantagens da invenção estarão evidentes da seguinte descrição detalhada e das reivindicações.
O termo "configuração absoluta" em conexão com um carbono assimétrico é determinado considerando o formato tetraédrico das ligações de carbono assimétricas e atribuindo uma prioridade de 1 a 4 para cada uma dos grupos ligados ao carbono assimétrico com o grupo tendo o número atômico mais elevado tendo a primeira prioridade. Se o tetraedro é visto de um lado remoto do grupo 4, uma configuração absoluta R é atribuída quando os grupos 1-3 estão em uma arranjo no sentido horário, e uma configuração absoluta S é atribuída quando os grupos 1-3 estão em um arranjo no sentido anti-horário.
O termo "e/ou." como usado aqui, significa um membro do grupo nomeado ou qualquer combinação dos mesmos. O termo "ou" é um "ou" inclusivo salvo especificado em contrário.
O termo "condições ambientes" inclui temperaturas variando de cerca de 15°C a cerca de 45°C5 por exemplo 20°C, 25°C, 30°C, 35°C, e umidade relativa variando de cerca de 20% a cerca de 65%, por exemplo 25%, 30%, 35%, 40%, 45%, 50%, 55%, e 60%.
O termo "carbono assimétrico" significa um átomo de carbono covalentemente ligado a quatro grupos diferentes.
O termo "degeneração de célula beta" se destina a incluir perda de função de célula beta, disfunção de célula beta, e morte de células beta, tais como necrose ou apoptose de células beta.
O termo "Diabetes Melitus e condições relacionadas"refere-se a diabetes tipo 1 diabetes, diabetes tipo 2, diabetes gestacional, MODY, tolerância a glicose diminuída, abstinência de glicose diminuída, hiperglicemia, metabolismo de glicose diminuído, resistência à insulina, obesidade, complicações diabéticas, e outros.
O termo "complicações diabéticas"refere-se a condições, doenças e enfermidades associadas com diabetes incluindo retinopatias, neuropatias, neffopatias, cardiomiopatias, dermopatias, ateroesclerose, doença de artéria coronária e outras complicações conhecidas de diabetes.
O termo "diastereômero" significa um membro de um grupo de dois ou mais estereoisômeros tendo pelo menos dois carbonos assimétricos tal que estes estereoisômeros não são imagens de espelho uns dos outros.
Os termos "DPP-VII, DPP-VIII, DPP-IX e FAP" significam respectivamente amino dipeptidil peptidase VII, VIII, IX e proteína de ativação de fibroblasto. As enzimas DPP decompõem porções de dipeptídeo no N-término de seus substratos de proteína ou oligopeptídeo. Em particular, o termo "DPP-IV"denota dipeptidil peptidase IV (EC 3.4.14.5; DPP-IV), também conhecida como "CD-26."DPP-IV preferencialmente decompõe um dipeptídeo do N-término de uma cadeia de polipeptídeo contendo um resíduo de prolina ou alanina na penúltima posição.
O termo "enantiômero" significa um membro de um par de estereoisômeros tendo a mesma estrutura molecular e pelo menos um carbono assimétrico tal que os estereoisômeros do par são as imagens de espelho uns dos outros. Se o enantiômero contém dois ou mais carbonos assimétricos, o par enantiomérico terá assimetria oposta em cada carbono assimétrico.
O termo "base livre"refere-se ao composto de ácido pirrolidinilamino-acetil pirrolidina borônico de fórmula I ou V, em que os grupos amina não são protonados.
O termo "grupo que pode ser hidrolisado para uma hidroxila" como usado aqui refere-se a um grupo éster formado a partir da combinação de um álcool alifático ou aromático ou diol e um ácido borônico.
O termo "inibidor"(e seu verbo e gerúndio correspondentes) significa um composto, incluindo um sal do mesmo, que reversivelmente, irreversivelmente, ou temporariamente interage com uma enzima para reduzir, modificar, retardar ou bloquear sua atividade enzimática no seu substrato normal. A interação pode ocorrer dentro ou no sítio enzimático ou em um sítio alostérico associado com a enzima.
O termo "grupo N-protetor" ou "N-protegido" como usado aqui refere-se àqueles grupos pretendidos para proteger o N-término de um aminoácido ou peptídeo ou para proteger um grupo amino contra reações indesejáveis durante procedimentos sintéticos. Grupos N-protetores comumente usados estão descritos em T.W. Greene, P. G. Wuts, "Protective Groups In Organic Synthesis, 3aed." (John Wiley & Sons, Nova Iorque (1999)), que é aqui incorporado por referência. Grupos N-protetores incluem grupos acila tais como formila, acetila, propionila, pivaloila, t-butilacetila, 2- cloroacetila, 2-bromoacetila, trifluoroacetila, tricloroacetila, ftalila, o- nitrofenoxiacetila, a-clorobutirila, benzoíla, 4-clorobenzoila, 4- bromobenzoila, 4-nitrobenzoila, e outros; grupos sulfonila tais como benzenossulfonila, p-toluenossulfonila e outros; grupos formando carbamato tais como benziloxicarbonila, p-clorobenziloxicarbonila, p- metoxibenziloxicarbonila, p-nitrobenziloxicarbonila, 2- nitrobenziloxicarbonila, p-bromobenziloxicarbonila, 3,4- dimetoxibenziloxicarbonila, 3,5-dimetoxibenziloxicarbonila, 2.4- dimetoxibenziloxicarbonila, 4-metoxibenziloxicarbonila, 2-nitro-4,5- dimetoxibenziloxicarbonila, 3,4,5-trimetoxibenziloxicarbonila, 1 -(p- bifenilil)-! -metiletoxicarbonila, α,α-dimetil-3,5-dimetoxibenziloxicarbonila, benzidriloxicarbonila, t-butiloxicarbonila, diisopropilmetoxicarbonila, isopropiloxicarbonila, etoxicarbonila, metoxicarbonila, aliloxicarbonila, 2,2,2,-tricloroetoxicarbonila, fenoxicarbonila, 4-nitrofenoxicarbonila, fluorenil-9-metoxicarbonila, ciclopentiloxicarbonila, adamantiloxicarbonila, ciclohexiloxicarbonila, feniltiocarbonil e outros; grupos alquila tais como benzila, trifenilmetila, benziloximetila e outros; e grupos silila tais como trimetilsilila e outros. Grupos N-protetores preferidos são formila, acetila, benzoila, pivaloila, t-butilacetila, fenilsulfonila, benzila, 9- fluorenilmetiloxicarbonila (Fmoc), t-butiloxicarbonila (Boc) e benziloxicarbonila (Cbz).
O termo "opticamente ativo" significa um composto orgânico contendo pelo menos um carbono assimétrico tal que uma solução do composto orgânico irá girar luz polarizada plana.
O termo "mistura opticamente ativa" significa uma mistura de compostos opticamente ativos em solução que irá girar luz polarizada plana. A mistura opticamente ativa pode ser uma mistura de diastereômeros ou uma mistura desigual de enantiômeros.
O termo "pró-droga" significa um composto farmaceuticamente aceitável que converterá para o ingrediente ativo ou um metabólito ativo do mesmo em administração da pró-droga a um organismo vivo, preferivelmente um mamífero, mais preferivelmente um humano. A conversão pode ocorrer por ação enzimática, hidrólise química, oxidação, redução ou qualquer outro processo fisiológico in vivo para reação química ou bioquímica.
O termo "mistura racêmica" significa um par enantiomérico de proporções iguais tal que eles cancelam cada rotação do outro de luz polarizada plana.
Como usada aqui, a frase "sal da invenção" refere-se a um sal citrato bem como um sal tartarato do composto de ácido pirrolidinilaminoacetil pirrolidina borônico de fórmula I ou V. Como usado aqui, um termo singular inclui o plural, e do mesmo modo, um termo plural inclui o singular. Deste modo, a frase "um sal da invenção" refere-se a um espécie estereoisomérica simples, as várias espécies estereoisoméricas, e misturas das mesmas. Exemplos não limitantes de um sal da invenção incluem (a) o sal L-, D-, ou meso-tartarato de ácido (2R)-I-{7-[(9R)- pirrolidina-9-ilamino]-acetil}-pirrolidina-2-borônico e misturas dos mesmos e (b) o sal L-, D-, ou meso-tartarato de ácido (2R)-1 -{7-[(9S)-pirrolidina-9- ilamino]-acetil}-pirrolidina-2-borônico e misturas dos mesmos. Um sal da invenção também inclui o sal citrato de (a) ácido (2R)-l-{7-[(9R)-pirrolidina- 9-ilamino]-acetil}-pirrolidina-2-borônico; (b) ácido (2R)-l-{7-[(9S)- pirrolidina-9-ilamino]-acetil}-pirrolidina-2-borônico; e (c) qualquer combinação dos mesmos.
O termo "higroscópico" como o termo é usado aqui refere-se a uma propriedade de uma substância sólida de vapor d' água absorvido espontaneamente de ar úmido ou gás que está em contato com a substância. Um substância sólida "deliquesce"ou é "deliquescente" quando é higroscópica e, em absorção de vapor de água suficiente, dissolve na água que ela absorveu.
O termo "estereoisômero" significa uma das configurações absolutas de uma molécula orgânica simples tendo pelo menos um carbono assimétrico. Incluídos dentro da definição de um estereoisômero estão enantiômeros e diastereômeros. Um estereoisômero tem uma configuração absoluta cerca de cada dos carbonos assimétricos da molécula orgânica. Uma molécula orgânica com um carbono assimétrico apresenta dois estereoisômeros. Um molécula orgânica com dois carbonos assimétricos apresenta quatro estereoisômeros. Uma molécula orgânica com três carbonos assimétricos apresenta oito estereoisômeros. A projeção de luz polarizada plana através de uma solução contendo um estereoisômero causará rotação do plano polarizado.
O termo "mistura estereomérica" significa uma mistura de dois ou mais estereoisômeros e inclui enantiômeros, diastereômeros e combinações dos mesmos. A mistura estereomérica pode ou não pode ser opticamente ativa.
O termo "pureza estereomérica" em uma porcentagem dada significa que o estereoisômero designado predomina nesta porcentagem dada em uma mistura de estereoisômeros.
Salvo especifícamente indicado, as definições de termos para grupos químicos, grupos funcionais, porções e reações químicas descritas aqui seguem as definições providas em tais livros de texto e estudos de química orgânica como "Basic Principles of Organic Chemistry", Roberts e Caserio, W. A. Benjamin & Co. Nova Iorque, N.Y, 1965; "Advanced Organic Chemistry", 4a’ed., Jerry March, Wiley Interscience, Nova Iorque, N.Y. 1992; T. W. Greene, P. G. Wuts, "Protective Groups In Organic Synthesis, 3aEd." (John Wiley & Sons, Nova Iorque (1999), e Hawley's Condensed Chemical Dictionary, Ila. Ed., Sax e Lewis, Van Nostrand, Reinhold, Nova Iorque, N.Y., 1987. Ademais, as definições para termos estereoquímicos são baseadas em "Stereochemistry of Carbon Compounds", Ernest Eliel, McGraw-Hill publisher, Nova Iorque, N.Y. 1962. As descrições destes livros de texto estão incorporadas aqui por referência.
DESCRIÇÃO DETALHADA
Os sais citrato ou tartarato de um composto de fórmula (I), incluindo isômero cíclicos, hidratos e solvatos dos mesmos, referidos aqui como "sais da invenção," têm propriedades benéficas que os tomam úteis, inter alia, na preparação de formulações farmacêuticas para tratamento de diabetes. Estas propriedades incluem, por exemplo, bioatividade para inibição in vivo de DPP-IV, bom rendimento em preparação, boa solubilidade e taxa de dissolução em água ou fluidos corporais, e estabilidade física e química sob condições ambientes durante períodos prolongados de tempo que permitem processamento, formulação, armazenamento, e administração simples e fácil dos sais da invenção.
A presente invenção envolve a descoberta inesperada de que um sal citrato ou tartarato de um composto de ácido pirrolidinilaminoacetil pirrolidina borônico, por exemplo, um sal citrato ou tartarato de ácido l-{7- pirrolidina-9-ilamino]-acetil}-pirrolidina-2-borônico, um composto de fórmula (I) que é um inibidor de dipeptidil peptidase IV seletivo, tem boa estabilidade física como um sólido, favorável para processamento, formulação, armazenamento, e administração. As base livres dos compostos de fórmula (I), tais como os compostos de fórmulas (IA) e (IB), e uma variedade de outras formas de sal, foram verificadas como tendo estabilidade física fraca sob condições ambientes. A base livre, e sais da base livre com uma variedade de ácidos orgânicos, foram verificados como sendo ou óleos ou sólidos altamente deliquescentes.
Referindo-se à Tabela 1, abaixo, é evidente que a forma de base livre e vários outros sais do composto de fórmula (I), em que R2 e R3 são ambos hidroxila, quer não formam prontamente um sólido, ou se sólidos forem formados, os sólidos deliquescem em condições ambiente, e deste modo não poderiam ser usados para o desenvolvimento de uma forma de dosagem sólida. O sal citrato e tartarato da invenção prontamente formou um sólido. Em adição, o sólido não deliquesceu e permaneceu processável sob condições ambientes. Em temperatura ambiente e menos do que 70% de umidade, por exemplo a 63% de umidade relativa, embora um sal da invenção possa inicialmente absorver alguma água, os patamares de absorção de água e o sal retém sua forma de pó sólido em equilíbrio, o que permite o desenvolvimento de formas de dosagem sólidas. Deste modo, os sais citrato e tartarato da invenção têm estabilidade física desejável em que embora eles absorvam água atmosférica limitada, eles permanecem sólidos fisicamente e quimicamente estáveis. Por esta razão eles são particularmente bem adaptados para a preparação de comprimidos e cápsulas, bem como outras formulações farmacêuticas sólidas. Estes sais da invenção também têm boa processabilidade em que eles não são pegajosos. Consequentemente, eles podem ser medidos e divididos em alíquotas em quantidades precisas e reproduzíveis. Deste modo, os sais da invenção têm propriedades que facilitam a preparação de várias formulações farmacêuticas. E, como é mostrado na Tabela 5, e discutido abaixo, sais da invenção podem ser armazenados sob condições ambientes na presença de umidade atmosférica por períodos de tempo prolongados, pelo menos 12 meses, sem qualquer decomposição significante ocorrendo.
Uma forma de realização da invenção provê sal citrato ou tartarato de um composto de ácido pirrolidinilaminoacetil pirrolidina borônico de fórmula (I), um sal da invenção. O sal da invenção pode ser qualquer combinação estereoisomérica do ácido e base livre. Como usada aqui, o termo "ácido"refere-se a citrato ou tartarato, enquanto o termo "base livre" ou "composto pirrolidina"refere-se ao ácido pirrolidinilaminoacetil pirrolidina borônico. Citrato não é quiral, mas tartarato é, e tartarato inclui, por exemplo, L- tartarato, D-tartarato, meso-tartarato, bem como uma estereomistura tais como um mistura racêmica, uma mistura diaestereoisomérica, uma mistura de um par enantiomérico e um diastereômero ou uma mistura opticamente ativa de pelo menos dois estereoisômeros. O composto pirrolidina pode ser uma forma linear ou cíclica tendo a estrutura de fórmula (I) ou (V) respectivamente. O composto pirrolidina preferido pode ser ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]- acetil} -pirrolidina-2-borônico ou ácido (2R)-l-{7-[(9S)-pirrolidina-9-ilamino]- acetil}-pirrolidina-2-borônico. Deste modo, um sal da invenção preferido inclui o sal citrato ou tartarato de ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]-acetil}- pirrolidina-2-borônico ou ácido (2R)-l-{7-[(9S)-pirrolidina-9-ilamino]-acetil}- pirrolidina-2-borônico. Mais especifícamente, o sal da invenção preferido é o sal L-tartarato de ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]-acetil}-pirrolidina-2- borônico, que é, um sal L-tartarato de estrutura (IA):

em que R2 e R3são ambos grupos hidroxila.
Um sal da invenção demonstra propriedades físicas melhoradas que possibilitam o pronto isolamento de sólidos e formulação subsequente em uma forma de dosagem sólida. Em contraste, as bases livres correspondentes foram encontradas para deliquescer sob condições ambientes. Também foi verificado que muitos dos sais avaliados (ver Tabela 1) quer não prontamente formaram um sólido, ou, se um sólido foi formado, deliquesceram ou tomaram-se um material pastoso, intratável em condições ambiente.
Surpreendentemente, dois sais ácidos orgânicos, os sais tartarato e citrato de base livre, foram verificados como não mostrando a mesma instabilidade física e química como outros sais. Os sais da invenção, por exemplo o sal mono de ácido (2R,3R)-tartárico (ácido L-tartárico) preferido de ácido l-{7-[(9R)-pirrolidina-9-ilamino]-acetil}-pirrolidina-2- borônico, prontamente formou sólidos e não foram verificados como deliquescendo sob condições ambientes. Sais da invenção exibiram estabilidade física melhorada, tais como estabilidade física sob condições de temperatura elevada e umidade.
Referindo-se à Tabela 5, abaixo, pode ser visto que amostras do sal L-tartarato de ácido (2R)-l-{7-[(9R)-pirrolidina-9-ilamino]-acetil)- pirrolidina-2-borônico poderíam ser armazenadas por períodos prolongados de umidade relativa razoavelmente elevada (UR); por exemplo armazenamento por 6 meses a 75% de UR, e armazenamento por 12 meses a 60% de UR, ambos a 25°C, foram observados não causar nenhuma decomposição detectável dentro do erro estatístico.
Uma forma hidratada do sal tartarato ou citrato da invenção pode ser usada em preparação de formas de dosagem. Para preparar estas formas, o sal da invenção é deixado se equilibrar sob condições ambientes para prover um hidrato. A quantidade de água absorvida no hidrato pode ser determinada e auxílio apropriado é dado para o peso da água em qualquer preparação subsequente, possibilitando medição exata da dosagem desejada do sal. As condições atmosféricas sob as quais as formas de dosagem farmacêuticas são preparadas são apropriadamente controladas para assegurar quantidades precisas e reproduzíveis do composto ativo que são medidas durante o processo de fabricação. Uma vez que as formas de dosagem estão preparadas, exposição à variação de umidade não afetará a liberação da dose do sal desejada mesmo que o peso total real da forma de dosagem sólida individual possa variar devido à absorção subsequente ou dessorção de água. Por exemplo, Exemplo 5 e Tabela 3 abaixo ilustram administração de preparação de formulação exata como função de absorção de água.
A invenção inclui o sal citrato e tartarato de todos os estereoisômeros de um composto pirrolidina de fórmula (I), incluindo enantiômeros, diastereômeros, bem como os racematos e misturas estereoisoméricas. As misturas podem ou não podem ser opticamente ativas. Em algumas formas de realização, o sal da invenção pode ter uma pureza óptica de pelo menos cerca de 55%, preferivelmente 80%, mais preferivelmente 90%, o mais preferível 98%. Em outras formas de realização, o sal é um enantiômero opticamente enriquecido de tartarato e/ou o composto pirrolidina. Em ainda outras formas de realização, o sal é uma mistura de estereoisômeros incluindo mas não limitado a mistura desiguais de enantiômeros e/ou misturas de diastereômeros do composto de tartarato e/ou pirrolidina.
Métodos de tratamento
Uma forma de realização da invenção provê um método de inibição de dipeptidil peptidase-IV compreendendo contactar a enzima, dipeptidil peptidase-IV, com um sal da invenção em qualquer das suas formas como descrito acima. O contato pode ser efetuado in vitro tais como através de um teste diagnóstico ou um teste de triagem, ou in vivo através de uma via administrativa apropriada como discutido abaixo.
Os métodos in vivo de acordo com a invenção envolvem um sal da invenção em seu papel como um inibidor seletivo de DPP-IV. Por exemplo, a invenção provê um método de tratamento de um mamífero (tais como um humano) sofrendo de uma má condição que pode ser regulada ou normalizada através de inibição de DPP-IV. Os métodos da invenção são efetuados administrando ao mamífero (por exemplo, um humano) uma quantidade efetiva de um sal da invenção para tratar, controlar, melhorar ou prevenir a má condição. Tratamento é efetuado através de inibição de DPP- IV. Administração é tipicamente efetuada através do uso de uma composição farmacêutica contendo um sal da invenção. Para uso in vivo como um inibidor de DPP-IV, um sal da invenção pode ser formulado em qualquer maneira como descrito aqui.
Más condições que podem ser tratadas usando um sal da invenção são aquelas que podem ser reguladas ou normalizadas por inibição de DPP-IV. Estas más condições são conhecidas como sendo o resultado, pelo menos em parte, da presença ou ausência reduzida, ou atividade alterada, de peptídeos regulados pela enzima DPP-IV, especialmente no contexto do seu papel fisiológico em controle glicêmico. Deste modo, estas más condições incluem aquelas caracterizadas por controle glicêmico prejudicado tais como Diabetes Melitus e condições relacionadas. Por exemplo, a má condição pode ser diabetes tipo 1, diabetes tipo 2, diabetes gestacional. MODY, tolerância a glicose enfraquecida, abstinência de glicose diminuída, hiperglicemia, metabolismo de glicose diminuído, tolerância a glicose enfraquecida (IGT) e sua progressão para a diabetes Tipo II, hiperinsulinemia, obesidade, degeneração de célula beta (em particular apoptose de células beta), a progressão de diabetes tipo II não requerendo insulina para diabetes tipo II requerendo insulina; perda do número e/ou do tamanho de células beta em um indivíduo mamífero, e complicações diabéticas tais como retinopatia, neuropatia, cardiomiopatia, dermopatia, infecção relacionada a diabetes, ateroesclerose, doença de artéria coronária, derrame e más condições similares.
Em outras formas de realização de método de tratamento de acordo com a invenção, resistência à insulina é um componente de má condição que pode ser regulada ou normalizada por inibição de DPP-IV. Por exemplo, as más condições podem incluir abstinência de glicose diminuída, tolerância a glicose enfraquecida, síndrome de ovário policístico e outros. Em ainda outras formas de realização, a má condição que pode ser regulada ou normalizada por inibição de DPP-IV envolve uma diminuição de neogenese de ilhotas, sobrevivência de células β, ou biossíntese de insulina.
Um sal da invenção pode ser administrado a um mamífero, especialmente um humano em necessidade de tal tratamento, prevenção, eliminação, alívio ou melhora das várias más condições mencionadas acima. Tais mamíferos incluem, sem limitação, animais domésticos tais como animais domésticos e animais de criação, bem como animais não domésticos tais como animais selvagens.
A quantidade de sal a ser administrada ao paciente pode ser qualquer quantidade apropriada para inibir DPP-IV que resulta em tratamento da condição e um efeito benéfico para o paciente. A quantidade do sal a ser administrada pode ser uma dose efetiva ou uma fração apropriada do mesmo. Tais quantidades dependerão dos parâmetros de paciente individuais incluindo idade, condição física, tamanho, peso, a condição sendo tratada, a severidade da condição, e qualquer tratamento concorrente. Fatores que determinam dosagens apropriadas são bem conhecidos para os versados na técnica, e pode ser dirigidos através da experimentação de rotina, usando habilidade e treinamento do médico clínico. Por exemplo, a determinação das propriedades farmacocinéticas e farmacodinâmicas pode ser feita usando análises químicas e biológicas padrões e através do uso de técnicas de modelagem matemática conhecidas nas técnicas farmacológicas. A utilidade terapêutica e regime de dosagem podem ser extrapolados dos resultados de tais técnicas e através do uso de modelos farmacocinéticos e/ou farmacodinâmicos apropriados.
A dose administrada de um composto pirrolidina da invenção pode ser ajustada de acordo com a idade, peso e condição do paciente, bem como a via de administração, forma e regime de dosagem e o resultado desejado. A escolha de dosagem final, via e formulação farmacêutica podem ser determinadas pelo médico atendendo ao paciente, cuja sabedoria e discernimento irão guiar o processo. Entretanto, um paciente pode insistir em uma dose menor ou dose mais tolerável por razões médicas, razões psicológicas ou virtualmente quaisquer outras razões.
Preferivelmente, o sal da invenção pode ser administrado em uma dose a partir de 0,1 a 30 mg do sal per kg de peso do mamífero, mais preferivelmente 2 a 15 mg/kg de peso do mamífero. A faixa de dose para humanos adultos é geralmente de cerca de 0,5 a cerca de 2.400 mg/dia, preferivelmente cerca de 10 mg a cerca de 1.050 mg/dia, e mais preferivelmente cerca de 50 mg a cerca de 750 mg/dia. Ele pode ser administrado em uma dose simples ou na forma de doses múltiplas dadas 4 vezes por dia. Estas doses são baseadas no peso da base livre sozinha. Correção para contribuição de peso pelo componente de tartarato ou citrato e a água de hidratação seria feito para chegar ao peso real da forma hidratada do sal a ser administrado. Em alguns casos, pode ser útil começar com uma dosagem maior e quando a condição estiver sobre controle reduzir a dosagem. Deste modo, pode ser vantajoso administrar uma dose inicial de cerca de 70 mg a cerca de 2.400 mg o primeiro dia então uma dose menor de cerca de 20 a cerca de 1.200 mg nos dias subsequentes. Em outros casos, pode ser útil iniciar a terapia em uma dose menor e aumentar a dosagem se necessário. A dosagem exata dependerá do modo de administração, de terapia desejada, forma em que administrado, o indivíduo a ser tratado e o peso corporal do indivíduo a ser tratado, e a preferência e experiência do médico ou veterinário tratando do caso. Dosagem é descrita em termos da base livre e é ajustada consequentemente para o sal citrato ou tartarato.
O uso de um sal da invenção também inclui a fabricação de um remédio e um método de tratamento usando tal remédio na forma de uma combinação farmacêutica e/ou uma composição farmacêutica.
Combinações farmacêuticas e seu uso em tratamento
Um sal da invenção pode ser combinado com um segundo medicamento para formar uma combinação farmacêutica da invenção. O segundo medicamento é um agente conhecido para tratar, controlar, ou prevenir uma má condição que pode ser regulada ou normalizada através de inibição de DPP-IV. As más condições tratadas por tais combinações são aquelas que podem ser reguladas ou normalizadas através de inibição de DPP- IV.
O segundo medicamento pode também incluir uma quantidade terapeuticamente efetiva de um medicamento conhecido como agente antidiabético incluindo mas não limitado a um agente que aumenta secreção de insulina, um agente que aumenta sensibilidade à insulina, um agente que reduz a absorção de açúcar do trato gastrointestinal, um agente que melhora o efeito de proteínas ou peptídeos endógenos que desempenham um papel em controle glicêmico, ou um agente que atua na terapia de substituição para proteínas ou peptídeos endógenos que têm um papel conhecido no controle glicêmico. Tais agentes incluem mas não estão limitados a gliburida (por exemplo Micronase e Diabeta), glipizida (por exemplo Glucotrol), nateglinida (por exemplo Starlix), repaglinida (por exemplo Prandin), metformina (por exemplo Glucophage), rosiglitazona (por exemplo Avandia), acarbose (por exemplo Precose), miglitol (por exemplo Glyset), exenatida (por exemplo Byetta), e insulina (por exemplo Humulin e Novolin). Agentes exemplares adicionais incluem mas não estão limitados a biguanidas, clorpropamida, a glucagon como peptídeo-1 (GLP-I) ou miméticos dos mesmos tais como LY315902 ou LY307161, glimepirida, meglitinida, fenformina, pioglitazona, sulfonil uréias, troglitazona, G1 -262570, isaglitazona, JTT-501, NN-2344, L895645, YM-440, R-l 19702, AJ9677, KAD1 129, APR-HO39242, GW- 409544, KRP297, AC2993, Exendin-4, e NN221 1 . As estruturas químicas, nomes comuns e estudos farmacológicos dos compostos precedentes designados por letras e números estão prontamente disponíveis na internet, por exemplo, com entrada da designação de letra/número como um termo de busca no sítio da rede de busca GOOGLE.
Um sal da invenção pode ser usado em combinação com um ou mais segundos medicamentos úteis como agentes antidiabéticos (empregados para tratar diabetes e doenças relacionadas). O segundo medicamento pode ser administrado oralmente na mesma dosagem com o sal da invenção, ou em uma forma de dosagem oral separada. O sal da invenção e o segundo medicamento podem também ser administrados, por exemplo por injeção, separadamente, simultaneamente ou como uma mistura.
Combinação farmacêutica da invenção pode ser formulada como uma composição farmacêutica de um carreador farmaceuticamente aceitável juntamente com um sal da invenção e uma ou mais segundos medicamentos.
Na combinação farmacêutica da invenção, o sal da invenção está tipicamente presente em uma relação em peso para o segundo medicamento a partir de cerca de 0,01:1 a cerca de 200:1, dependendo da identidade do segundo medicamento.
O uso de um sal da invenção em combinação com um ou outros agentes antidiabéticos pode produzir resultados antihiperglicêmicos maiores que possível com cada um desses agentes antidiabéticos sozinhos. O uso de um sal da invenção em combinação com um ou mais outros agentes antidiabéticos pode também produzir um efeito sinergístico em que o resultado antihiperglicêmico pode ser maior que os efeitos aditivos antihiperglicêmicos combinados produzidos por estes agentes antidiabéticos.
A quantidade efetiva de um segundo medicamento formulado como um componente da combinação farmacêutica da invenção seguirá as recomendações do fabricante do segundo medicamento, o julgamento do médico assistente e será guiado pelos protocolos e fatores administrativos para quantidades e dosagem como indicado em PHYSICIAN'S DESK REFERENCE (PDR).
A dose administrada de um sal da invenção dentro da combinação farmacêutica será cuidadosamente ajustada de acordo com a idade, peso e condição do paciente, bem como a via de administração, forma e regime de dosagem e o resultado desejado. A última escolha de dosagem, via e formulação farmacêutica será determinada pelo médico atendendo ao paciente, cuja sabedoria e discernimento irão guiar este processo.
As composições descritas acima podem ser administradas nas formas de dosagem como descrito acima em doses simples divididas de uma a quatro vezes diariamente. Pode ser aconselhável iniciar um paciente em uma combinação de dose baixa e trabalhando gradualmente para uma combinação de dose elevada.
Composições farmacêuticas da invenção
A invenção inclui uma composição farmacêutica contendo um sal da invenção, com ou sem outro medicamento como descrito acima, em associação com um carreador farmacêutico. Uma composição farmacêutica pode ser formulada com uma ou mais carreadores, tais como carreadores sólidos ou líquidos convencionais ou diluentes e aditivos farmacêuticos de um tipo apropriado para o modo de administração desejado. Um sal da invenção em uma composição farmacêutica pode ser administrado a espécies mamíferas, especialmente humanos, por uma via oral, bucal, retal, pulmonar ou similar, por exemplo, na forma de comprimidos, cápsulas, grânulos ou pós. Pode ser administrado por uma via parenteral na forma de preparações injetáveis.Pode ser administrado por uma via transdérmica quer por um sinal de liberação para distribuição transdérmica ou por eletrotransporte usando um dispositivo de distribuição apropriado.
Uma composição farmacêutica contendo um sal da invenção pode ser preparada por técnicas convencionais, por exemplo como descrito em Remington: The Science and Practice of Pharmacy, 19aEd., 1995. A composição pode aparecer em uma forma convencional, por exemplo, cápsula, comprimido, aerossol, solução, suspensão ou em uma forma apropriada para aplicação tópica.
Uma composição farmacêutica típica inclui um sal da invenção formulado com um carreador farmaceuticamente aceitável que pode ser um excipiente ou a diluente, ou pode ser encerrado dentro de um carreador que pode estar na forma de um líquido, cápsula, sachê, comprimido, papel ou outro recipiente. Ao fazer a composição, técnicas convencionais para a preparação de composições farmacêuticas pode ser usadas.
Por exemplo, um sal da invenção pode ser misturado com um carreador, ou diluído por um carreador, ou fechado dentro de um carreador, que pode estar na forma de uma ampola, cápsula, comprimido, sachê, papel, ou outro recipiente. Quando o carreador serve como um diluente, pode ser sólido, semi-sólido, ou material líquido que atua como um carreador, excipiente, ou meio de cultura para o composto ativo. O sal pode ser absorvido em um recipiente sólido granular por exemplo em um sachê. Alguns exemplos de carreadores apropriados incluem, sem limitação,água, soluções de sal, álcoois, polietileno glicóis, óleo de casto poliidroxietoxilado, óleo de amendoim, óleo de oliva, gelatina, lactose, terra alba, sacarose, dextrina, magnésio carbonato, açúcar, ciclodextrina, amilose, estearato de magnésio, talco, gelatina, ágar, pectina, acácia, ácido esteárico ou éteres de celulose de alquila inferior, ácido silícico, ácidos graxos, aminas de ácido graxo, monoglicerídeos de ácido graxo e diglicerídeos, ésteres de ácido graxo de pentaeritritol, polioxietileno, hidroximetilcelulose e polivinilpirrolidona. Similarmente, o carreador ou diluente pode incluir qualquer material de liberação prolongada, conhecido na técnica, tais como monoestearato de glicerila ou distearato de glicerila, sozinho ou misturado com cera.
Uma formulação pode ser misturada com agentes auxiliares que não reagem nocivamente com o composto pirrolidina. Tais aditivos podem incluir, sem limitação, agentes umectantes, agentes emulsificantes e de suspensão, sal para influenciar pressão osmótica, tampões e/ou agentes de conservação de substâncias colorantes, agentes adoçantes ou agentes aromatizantes. Uma composição farmacêutica pode também ser esterilizada se desejado.
A via de administração pode ser qualquer via que efetivamente transporte o sal da invenção para o sítio apropriado ou desejado de ação, tais como oral, nasal, pulmonar, bucal, retal, subdérmica, intradérmica, transdérmica ou depósito, subcutâneo, intravenoso, intra-uretral, intramuscular, intranasal, solução oftálmica ou um linimento, a via oral sendo preferida.
Se o carreador for usado para administração oral, a preparação pode ser comprimida, colocada em uma cápsula de gelatina dura em forma de pó ou grânulo, ou pode estar na forma de um comprimido ou pastilha expectorante. Se um carreador líquido for usado, a preparação pode estar na forma de um xarope, emulsão, cápsula de gelatina macia ou líquido injetável estéril tais como uma suspensão ou solução líquida aquosa ou não aquosa.
Formas de dosagem injetáveis geralmente incluem suspensões aquosas ou suspensões de óleo que podem ser preparadas usando um dispersante apropriado ou agentes umectantes e um agente de suspensão. Formas injetáveis podem estar em fase de solução ou na forma de a suspensão, que é preparada com um solvente ou diluente. Solventes aceitáveis ou carreadores incluem água esterilizada, solução de Ringer, ou uma solução salina aquosa isotônica. Altemativamente, óleos estéreis podem ser empregados como solventes ou agentes de suspensão. Preferivelmente, o óleo ou ácido graxo não é volátil, incluindo óleos naturais ou sintéticos, ácidos graxos, mono-, di- ou tri-glicerídeos.
Para injeção, uma composição farmacêutica pode ser um pó apropriado para reconstituição com uma solução apropriada como descrito acima. Exemplos desses incluem, mas não estão limitados a, pós secos por congelamento, secos por rotação ou secos por pulverização, pós amorfos, grânulos, precipitados, ou particulados. Para injeção, uma composição farmacêutica pode opcionalmente conter estabilizadores, modificadores de pH, tensoativos, modificadores de biodisponibilidade e combinações dos mesmos. Um sal da invenção pode ser formulado para administração parenteral por injeção tais como por injeção de bolo ou infusão contínua. Uma forma de dosagem de unidade pode ser em ampolas ou em recipientes multi- doses.
Uma composição farmacêutica da invenção pode ser projetada para prover liberação rápida, prolongada, ou atrasada do ingrediente ativo após administração ao paciente empregando procedimentos bem conhecidos na técnica. Deste modo, a composição farmacêutica pode também ser formulada para liberação controlada ou para liberação lenta.
Uma composição farmacêutica da invenção pode incluir, por exemplo, micelas ou lipossomas, ou alguma outra forma encapsula, ou pode ser administrada em uma forma de liberação prolongada, ou uma forma revestida entérica para prover uma armazenamento prolongado e/ou efeito de distribuição. Portanto, a composição farmacêutica pode ser comprimida em grânulos ou cilindros e implantada intramuscularmente ou subcutaneamente como injeções de depósitos ou como implantes tais como stents. Tais implantes podem empregar materiais inertes conhecidos tais como silicones e polímeros biodegradáveis, por exemplo, polilactídeo-poliglicolídeo. Exemplos outros polímeros biodegradáveis incluem poli (ortoésteres) e poli (anidridos).
Um sal da invenção pode ser formulado como um implante de liberação sustentado ou material implantável apropriado para administração contínua sobre um período de tempo significante. Implantes de liberação típicos sustentados são formados de polímeros e de polímeros biodegradáveis farmaceuticamente aceitáveis tais como polímeros e copolímeros de ácido lático, lactídeo, ácido glicólico, glicolídeo, ácido capróico e caprolactona. A dose e quantidade de um sal da invenção dentro do implante será calculada para entregar o nível de sangue de dose simples desejado do sal.
Para administração nasal, uma composição farmacêutica pode conter um sal da invenção dissolvido ou suspendido em um carreador líquida, em particular um carreador aquoso, para aplicação de aerossol. O carreador pode conter aditivos tais como agentes solubilizantes, por exemplo, propileno glicol, tensoativos, melhoradores de absorção tais como lecitina (fosfatidilcolina) ou ciclodextrina, ou conservantes tais como parabenos.
Para aplicação parenteral, particularmente apropriados são soluções ou suspensões injetáveis, preferivelmente soluções aquosas com um sal da invenção dissolvido em óleo de rícino poliidroxilado.
Comprimidos, drágeas, ou cápsulas tendo talco e/ou um carreador de carboidrato ou aglutinante ou afins são particularmente apropriados para aplicação oral. Carreadores preferidos para comprimidos, drágeas, ou cápsulas incluem lactose, amido de milho, e/ou amido de batata. Um xarope ou elixir pode ser usado em casos onde um carreador adoçado pode ser empregado.
Um comprimido típico pode ser preparado por técnicas de formação de comprimidos convencionais como segue.
Um comprimido típico tendo um equivalente de 400 mg de força de base livre pode ser preparado formulando 650 mg do sal L-tartarato, 215 mg de celulose microcristalina, 50 mg de fosfato de cálcio dibásico, 20 mg de copolividona., 50 mg de crospovidona. 10 mg de dióxido de silicone coloidal, 5 mg de estearato de magnésio e 3% de opadry AMB sólido. Outra composição de comprimido típica tendo 50 mg de força de base livre pode ser formulada combinando 80 mg do sal L-tartarato, 815 mg de celulose microcristalina, 50 mg de fosfato de cálcio dibásico, 20 mg de copolividona, 20 mg de crospovidona, 10 mg de dióxido de silicone coloidal, 5 mg de estearato de magnésio e 3 % opadry AMB sólido. O sal pode ser submetido a moagem e peneiramento através de uma tela de malha 20. O sal moído e telado pode ser misturado com celulose microcristalina, copolividona, crospovidona e dióxido de silicone coloidal em um misturador em formato em V apropriado para um tempo e rpm apropriados. A composição resultante pode ser misturada com o estearato de magnésio lubrificante. Comprimidos podem ser comprimidos usando a mistura lubrificada em um peso de comprimido teórico de 1000 mg. Parte dos comprimidos prensados podem ser revestidos usando Opadry AMB. Revestimento pode ser continuado até um ganho de peso de sólido apropriado ser realizado.
Uma outra cápsula típica para administração oral contém um sal da invenção (200 mg de base livre equivalente), lactose (75 mg) e estearato de magnésio (15 mg). A mistura é passada através de uma tela de malha 60 e embalada em uma cápsula de gelatina No. 1.
Uma preparação injetável típica é produzida colocando assepticamente um sal da invenção (em 200 mg de base livre equivalente) em um frasco, secando por congelamento assepticamente e vedando. Para uso, os conteúdos do frasco são misturados com 2 mL de salina fisiológica, para produzir uma preparação injetável.
Um sal da invenção pode ser distribuído em forma de dosagem de unidade de cerca de 0,5 a cerca de 2.000 mg de ingrediente ativo junto com um carreador farmaceuticamente aceitável por dosagem de unidade. Usualmente, uma forma de dosagem apropriado para administração oral, nasal, pulmonar ou transdérmica inclui de cerca de 0,5 mg a cerca de 2.000 mg, preferivelmente de cerca de 10 mg a cerca de 1.000 mg por dia, mais preferivelmente de cerca de 50 mg a cerca de 750 mg, de um sal (como medido pela base livre equivalente) misturado com um carreador farmaceuticamente aceitável ou diluente.
As propriedades de liberação da droga de comprimidos feitos de acordo com a invenção indicaram distribuição apropriada do composto pirrolidina. Amostras dos comprimidos foram testadas para fluxo, dureza, desintegração e liberação de droga. Em particular, fluxo apropriado foi estimado por índice de Carr e uma observação visual em misturas lubrificadas. Comprimidos prensados tiveram dureza aceitável de cerca de 12kP. Comprimidos também tiveram tempo de desintegração aceitável (menos do que 15 minutos). Dados sem comparação controlada mostram liberação in vitro apropriada (mais do que 90 % em 30 minutos).
Quando preparando uma formulação farmacêutica contendo um sal da invenção, a propriedade física do sal deve ser levada em conta.Por exemplo, o sal L-tartarato permaneceu um sólido processável por vários dias em temperatura ambiente uma umidade relativa de 75%. Também melhorou a estabilidade química relativa à base livre a 25°C/60% UR, 40°C/75% UR e 60°C/ umidade de ambiente, e permaneceu um sólido processável a 63% de umidade relativa e temperatura ambiente por dois meses. Deste modo, a temperatura apropriada e umidade relativa devem ser selecionadas e mantidas durante a preparação de formulações farmacêuticas contendo um sal da invenção. Se temperatura ambiente for selecionada, por exemplo, um sal da invenção pode ser mantido a umidade moderada, que está abaixo de 65% umidade relativa, ou preferivelmente, abaixo de 60 % da umidade relativa.
Em adição, embora um sal da invenção seja fisicamente estável em condições ambientes, sua higroscopicidade deve ser levada em contra. Deste modo, quando preparando uma formulação farmacêutica contendo um sal da invenção, formas higroscopicamente estáveis do sal devem ser usadas. Um sal da invenção é higroscopicamente usado quando seu peso é estável sob uma temperatura apropriada e umidade relativa. Deste modo, anterior a medição, um sal da invenção deve ser equilibrado para a temperatura apropriada e umidade.
Além disso, quando pesando ou de outra maneira medindo um sal da invenção na preparação de composições farmacêuticas, a quantidade de água absorvida deve ser levada em conta. Por exemplo, para conseguir uma dose efetiva apropriada, o sal da invenção, equilibrado para a temperatura selecionada e umidade, pode ser preparado e os pesos de água de hidratação medidos. A medição de água e correlação de peso consequente irão dar a quantidade real de sal para uma dose efetiva selecionada.
Se um desintegrante for incluído na formulação de um comprimido ou cápsula, então a formulação deve ser projetada e preparada de tal forma a evitar colapso de modo precoce e desintegração do comprimido ou cápsula resultando de reação do desintegrante com água absorvida pelo sal.
Uma combinação farmacêutica da invenção pode ser formulada como uma composição farmacêutica empregando todas as formas de realização, carreadores, projetos de vias, e outros aspectos acima descritos para formulação de uma composição farmacêutica de um sal sozinho.
A invenção também abrange pró-drogas de um sal da invenção que em administração sofre conversão química por processos metabólicos antes de tomar-se uma substância farmacológicas ativa. Em geral, tais pró- drogas serão derivados funcionais de um sal da invenção que são prontamente convertíveis in vivo em um sal da invenção. Procedimentos convencionais para a seleção e preparação de derivados de pró-drogas apropriados são descritos, por exemplo, em "Design de Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
Deste modo, outro aspecto da invenção provê uma composição farmacêutica de um sal da invenção, sozinho ou em combinação com outro tipo de agente antidiabético de tipo e/ou outro tipo de agente terapêutico.
Formas de realização adicionais da invenção são representadas por: (1) uma composição farmacêutica incluindo um sal da invenção, como descrito acima, junto com pelo menos um carreador farmaceuticamente aceitável ou diluente; (2) métodos de fazer uma composição farmacêutica de um sal da invenção em que o carreador farmaceuticamente aceitável ou diluente é apropriado para administração oral; (3) métodos de fazer uma composição farmacêutica de um sal da invenção apropriado para administração oral ainda incluindo a etapa de formular a composição em um comprimido ou cápsula; (4) métodos de fazer uma composição farmacêutica de um sal da invenção em que o carreador farmaceuticamente aceitável ou diluente é apropriado para administração parenteral; (5) métodos de fazer uma composição farmacêutica de um sal da invenção apropriada para administração parenteral ainda incluindo a etapa de liofilizar a composição para formar uma preparação liofilizada.
Atividade inibitória de DPP-IV de um sal da invenção pode ser determinada pelo uso de um sistema de análise in vitro. Constantes de inibição (valores Kj ou IC50) para os inibidores de DPP-IV da invenção podem ser determinados pelo método descrito abaixo.
Métodos de preparar um sal da invenção
Uma forma de realização da invenção também provê um processo para preparar um sal da invenção. Preparação do composto pirrolidina de base livre é provido em pedido de patente US número de série 60/704.380, depositado em Io de Agosto de 2005, e em Pedido de patente
U.S. No. de série 10/514.575, depositado em 15 de Novembro de 2004. Um esquema sintético geral para preparação de um sal tartarato da invenção é mostrado abaixo como Esquema 1
O sal da invenção pode ser obtido como o produto direto de 5 síntese de composto como ilustrado no esquema sintético acima (ver etapa 8) e descrito em detalhes no Exemplo 4 abaixo. Na alternativa, a base livre pode ser dissolvida em um solvente apropriado, e então o sal pode ser isolado evaporando o solvente. Um solvente apropriado pode ser, por exemplo, um solvente orgânico hidroxílico ou um solvente orgânico aprótico polar, ou água. O solvente pode ser evaporado de numerosas formas incluindo, sem limitação, por secagem por pulverização ou secagem por congelamento, ou de outra forma separando o sal e solvente, por exemplo por precipitação. Deste modo, um exemplo de um método para preparar um sal da invenção inclui, sem limitação, dissolver a base livre em um alcanol de 1 a 6 carbonos que contém uma quantidade apropriada de um ácido apropriado, por exemplo uma quantidade estequiométrica de citrato ou tartarato, e então o sal pode ser isolado evaporando o solvente. Altemativamente, o ácido pode ser adicionado a uma solução de base livre, e então um sal é isolado por remoção do solvente. Em adição, os processos para formar um sal farmaceuticamente aceitável de um composto de amina tais como um composto pirrolidina da invenção são bem conhecidos na técnica. Ver, por exemplo, "The Practice de Medicinal Chemistry, 2a. edição", por Camille G. Wermuth, Academic Press, Nova Iorque, N. Y., 1996.
A invenção será ainda descrita nos seguintes exemplos, que não limitam o escopo da invenção descrita nas reivindicações.
Exemplos
A preparação e atividade biológica da base livre são descritas em Pedido de Patente U.S número de série 11/381.085, depositado em Io de Maio de 2006, incorporada aqui por referência.
Exemplo 1 —Preparação de sal
O seguinte procedimento foi usado para preparar ou em tentativas para preparar vários sais do composto pirrolidina de fórmula I (ver Figura 1). Para um frasco de 1L sob uma manta de nitrogênio, 50 mmol do composto de base livre foram adicionados, e então 250 mL de álcool isopropílico (IPA) foram adicionados. A mistura foi agitada por 20-30 minutos para dissolver o sólido completamente. Para um segundo frasco de 500 mL sob uma manta de nitrogênio, 50 mmol do ácido desejado (ácido tartárico ou cítrico) foram adicionados, e então 250 mL de IPA foram adicionados. A mistura foi aquecida suavemente a 30°C e agitada por 10-20 minutos até o sólido dissolver. A solução de ácido foi adicionada rapidamente para solução de base livre, e a mistura foi agitada a temperatura ambiente por 0,5 horas antes de filtração. Qualquer bolo resultante foi secado a ar por 0,5 horas e então lavado com 150 mL de éter butil metílico terciário (MTBE), secado a ar durante 1-2 horas, e então secado em um forno a vácuo a 40-50°C 5 durante um período durante a noite.
Tentativas foram feitas para criar o acetato, adipato, sulfonato de 10-cânfora, citrato, decanoato, hidrobromato, L-ascorbato, L-glutamato, L- lactato, L-e R-tartarato, sulfonato de 2-naftaleno, palmitato, e sal succinato do composto de pirrolidina. Os resultados são resumidos na tabela 1 abaixo.
Somente os sais citrato e L-, D-, e DL-tartarato puder deram sólidos; liquefeito na filtração am ser isolados. Os outros sais ou não deram sólidos ou deram sólidos que foram liquefeitos na filtração. Todos os sais eram amorfos por calorimetria de varredura diferencial (DSC). Os sais citrato e D-tartarato não foram estáveis quando submetidos a 75% de umidade relativa (UR) mas foram estáveis a 63% de UR. Em contraste, o sal L- tartarato foi estável quando submetido a 75% de UR. Para o sal L-tartarato, um total de 18,12 g de um sólido creme colorido foi obtido, 92,6% de teoria.
O sal L-tartarato de ácido (2R)-l-{7-[(9R)-pirrolidin-9- ilamino]-acetil}-pirrolidina-2-borônico tem a fórmula molecular CioH2oBN3θ3-C4H4θ6e um peso molecular de 391,25 g/mole. Ele é um sólido amorfo de tom branco a marrom pálido que é livremente solúvel em água e álcoois (> 2000 mg/mL). Ele é moderadamente higroscópico e permanece um sólido processável em condições ambientes. A sorção de água do sal tartarato é ilustrada na figura 2.
Exemplo 2 -Procedimento de secagem por pulverização
Para a secagem por pulverização, um secador por pulverização em escala laboratorial SD011 (BUCHI, modelo B-290 Advanced), equipado com um bocal de dois fluidos foi usado. A tampa do bocal e o e diâmetro foram 1,5 e 0,7 mm, respectivamente. Um ciclone de desempenho elevado foi usado para coletar o produto secado.
O fluxo de nitrogênio de secagem foi controlado pela velocidade do ventilador (F_secagem). Nestes testes, o ventilador circulou nitrogênio a 100% de capacidade (taxa de fluxo a aproximadamente 35 kg/h). O fluxo de nitrogênio atomizante (átomo F), controlado por uma válvula de agulha, foi fixado a 357 L/h (30 mm de altura no rotâmetro). Antes de iniciar cada teste, o secador foi estabilizado com água deionizada. O fluxo de solução (F-alimentação) foi fixado a 9 mL/min (30% da velocidade da bomba de alimentação peristáltica). A temperatura de entrada (T_ entrada ) foi controlada por um aquecedor elétrico (HX) e foi ajustada a fim de obter a marcação da temperatura de saída (T saída) (100°C). Os parâmetros operacionais foram definidos com base em um teste usado previamente realizado em SD011 para 17DC01 (MAR-001-026).
Produtos coletados do ciclone e do fundo da câmera de secagem foram combinados.
Exemplo 3 -Preparação do sal L-tartarato por secagem por pulverização
Uma solução de 1,00 g do composto de base livre e 4,06 g de água deionizada, e uma solução de 0,62 g de ácido L-tartárico e 2,44 g de água deionizada, foram agitadas durante 15 minutos a 20-25°C. Então a solução de ácido L-tartárico foi adicionada na solução de composto de base livre e a mistura foi agitada durante 15 minutos a 20-25°C. Antes da secagem por pulverização, a mistura de reação tem um teor sólido de aproximadamente 25% (p/p). Este procedimento foi escalado até converter 500 g da base livre para o sal L-tartarato em três bateladas.
Resultados para os produtos secos por pulverização das três reações diferentes são resumidos na tabela 2.
* Produto secado por pulverização pode ser coletado no ciclone (maioria) ou no fundo da câmera de secagem (produtos sensíveis a temperatura podem ter menor pureza na câmara devido à exposição em maior temperatura durante períodos mais longos de tempo). Nesta batelada, a quantidade coletada na câmara foi tão pequena que não foi feita análise.
Além disso, a solução antes da pulverização e o produto secado por pulverização foram analisados para pureza por HPLC usando o seguinte método. Uma coluna de Waters Symmetry C18 (3,0 x 150 mm, 3,5 um) foi usada. A fase móvel A consistiu de 25 mM de ácido octanossulfônico de sódio, 0,1% de TFA em 90:10 água:metanol. A fase móvel B consistiu de 25 mM de ácido octanossulfônico de sódio, 0,1% de TFA em 25:75 água:metanol. Temperatura de coluna foi de 60°C, e a detecção de comprimento de onda foi de 210 nm. O volume de injeção foi de 15 pL; taxa de fluxo foi de 1,0 mL/minuto durante um tempo de ciclo de 70 minutos. O gradiente foi como a seguir:
Nota: Ciclo da coluna a 0,1 mL/min até coluna ser equilibrada a 60°C.
Os cromatogramas resultantes mostrados nas figuras 3 a 6 correspondem aos para tartarato, o composto pirrolidina e o sal L-tartarato, respectivamente.
O produto seco por pulverização das três bateladas de reação foram combinados e o cromatograma obtido por análise HPLC do produto seco por pulverização combinado é ilustrado na figura 7.
Em resumo, o sal L-tartarato de ácido (2R)-l-{7-[(9R)- pirrolidin-9-ilamino]-acetil}-pirrolidina-2-borônico isolado por secagem por pulverização tem uma degradação mínima do produto ou nenhuma degradação.
Exemplo 4 -Preparação de um sal tartarato da invenção como um produto direto de síntese de composto
Um sal tartarato da invenção foi preparado a partir da base livre protegida como a seguir. Um reator foi carregado 1,0 kg da base livre protegida, 0,40 kg do ácido tartárico, e 2,0 kg (2L) de água purificada. A mistura foi agitada durante não menos do que 1 hora, enquanto o teor do reator foi mantido abaixo de 30°C como a reação inicial foi algumas vezes exotérmica. Então 0,33 kg de ácido fenil-borônico e 3,7 kg de éter terc-butil metílico (5 L) foram adicionados, e a mistura foi agitada durante não menos do que 2 horas a 15°C a 25°C. Uma amostra foi coletada e analisada por HPLC. Se a mistura de reação tem mais do que 0,5% base livre, foi agitada durante outra 1-3 hora e amostrada novamente Se a mistura tem 0,5% ou menos da base livre, então a agitação foi parada e as camadas deixadas separar durante não menos do que 15 minutos. A fase orgânica foi descartada, e a fase aquosa de fundo usada na próxima etapa.
A fase aquosa foi extraída com 4,3 kg de 2-metil-tetra- hidrofurano (5 L) por agitação durante não menos do que 10 minutos, e então aquoso e camadas orgânicas foram deixadas para separar durante não menos do que 15 minutos. A fase orgânica foi novamente descartada, e a fase aquosa submetida a dois mais ciclos de extração com 4,3 kg de 2-metil-tetra- hidrofurano (5 L) como descrito.
A fase aquosa resultante foi reagida com 3,7 kg de éter terc- butil metílico (5 L), e a mistura agitada durante não menos do que 10 minutos. Então as camadas foram deixadas separar durante não menos do que 15 minutos. Novamente, a fase orgânica foi descartada, e a fase aquosa de fundo coletada. Uma amostra foi coletada para análise HPLC.
A fase aquosa resultante foi filtrada com acabamento e uma amostra analisada por HPLC. Solventes residuais foram removidos por aplicação de um vácuo a -0,8 a -0,9 bar durante 2 horas a 35°C a 50°C. Uma amostra da solução resultante foi coletada para análise HPLC.
Uma parte da solução foi transferida para bandejas do secador por congelamento e congeladas. As bandejas congeladas foram então removidas e colocadas no congelador de produto e mantidas ai. O resto da solução do produto foi carregado em bandejas do secador de congelamento e submetidas à secagem por congelamento. Então as bandejas previamente congeladas foram submetidas a secagem por congelamento.
Exemplo 5 -Propriedades do sal tartarato
Estabilidade física em câmaras de umidade. O sal tartarato foi preparado em uma escala de 50 mmol para dar 18,12 g de um sólido creme colorido. Quatro amostras de 1,0 g do sal foram colocados em quatro câmaras de umidade separadas a 45%, 58%, 63%, e 76% de umidade relativa em temperatura ambiente (20,00°C a 22,22°C). As amostras foram pesadas de hora em hora primeiro, e então em intervalos diários. Resultados são ilustrados na figura 8.
Após cinco dias, as amostras ganharam entre 9% a 13% água a UR de 45% a 63%, com a maior parte do ganho ocorrendo nos primeiros três dias. A taxa de absorção de água foi proporcional na umidade relativa, com pequena diferença entre 58% de UR e 63% de UR. Assim, o sal tartarato foi fisicamente estável (isto é, permaneceu um sólido escoável) em temperatura ambiente e UR até 63%.
Uma amostra do sal tartarato foi armazenada em um recipiente aberto em temperatura ambiente e 45% de UR durante dois meses. O material manteve boas propriedades de fluxo após dois meses.
Estabilidade química. Estabilidade química foi determinada usando HPLC. O sal L-tartarato foi continuamente mantido em câmara a 45% de UR durante dois meses. Figuras 9 e 10 são os cromatogramas de HPLC para sal anterior a e após dois meses a 45% de UR. Pouca ou nenhuma mudança na pureza foi observada.
A estabilidade química do sal tartarato e a base livre também foi determinada após um e três meses a 25°C, 40°C e 60°C. Resultados são resumidos na seguinte tabela.
Análise da atividade biológica e seletividade da base livre é descrita no pedido de patente US número de série 11/381.085, depositado em 1 de maio de 2006 incorporado aqui por referência. A atividade biológica e seletividade do sal tartarato foram comparadas com as da base livre usando o mesmo método como descrito no pedido 10/381.085. Resultados indicam que a base livre e o sal L-tartarato tem potências comparáveis e seletividade contra os vários DPP testados. Especifícamente, como a base livre, o sal tartarato mostrou seletividade excelente para DPP-IV relativo a DPP-VIII.
Exemplo 7 -Formação de comprimidos
Comprimidos tendo 400 mg e 50 mg de um sal da invenção foram preparados como a seguir. O sal foi moído e peneirado através de uma tela de malha 20 e peneirado através de uma tela de malha 20 e então misturado com celulose microcristalina, copolividona, crospovidona e dióxido de silício coloidal em um liquidificador tipo de formato em V apropriado durante 10 minutos a 25 rpm. A composição resultante foi misturada com estearato de magnésio lubrificante durante 2 minutos a 25 rpm. Comprimidos foram comprimidos usando a mistura lubrificada em um peso de comprimido teórico de 1000 mg usando uma ferramenta de comprimido conformado em cápsula modificado de 1,99 cm x 0,94 cm. Partes dos comprimidos prensados foram revestidas usando Opadry AMB. O revestimento foi continuado até aproximadamente um ganho em peso de 3% em sólidos ser obtido. As composições dos comprimidos são as seguintes.
Tabela 5
Estudo de estabilidade 12 meses do sal tartarato
Tabela 5 mostra os resultados de um estudo de estabilidade de 12 meses em várias temperaturas e umidades relativas para o sal L-tartarato do composto de fórmula (IA) em que R e R são ambos OH. Como pode ser visto, substancialmente nenhuma mudança na pureza química foi observada 5 sob qualquer uma das condições testadas, e a aparência física não muda daquela de um sólido de branco indefinido exceto no ponto de tempo de 12 meses a 25°C, um leve tom amarelado foi observado.
Apesar da invenção ter sido descrita em conjunto com a descrição detalhada, a descrição acima se destina a ilustrar e não limitar o 10 escopo da invenção, que é definido pelo escopo das reivindicações anexas.
Outros aspectos, vantagens e modificações estão dentro do escopo das seguintes reivindicações.