BRPI0717156B1 - inibidores do co-transportador 2 da glicose de sódio e composição farmacêutica que os compreende - Google Patents
inibidores do co-transportador 2 da glicose de sódio e composição farmacêutica que os compreende Download PDFInfo
- Publication number
- BRPI0717156B1 BRPI0717156B1 BRPI0717156-0A BRPI0717156A BRPI0717156B1 BR PI0717156 B1 BRPI0717156 B1 BR PI0717156B1 BR PI0717156 A BRPI0717156 A BR PI0717156A BR PI0717156 B1 BRPI0717156 B1 BR PI0717156B1
- Authority
- BR
- Brazil
- Prior art keywords
- mmol
- foi
- alkyl
- chloro
- phenyl
- Prior art date
Links
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 13
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 title description 10
- 150000001875 compounds Chemical class 0.000 claims abstract description 153
- 125000003118 aryl group Chemical group 0.000 claims description 59
- 229910052739 hydrogen Inorganic materials 0.000 claims description 58
- 239000001257 hydrogen Substances 0.000 claims description 58
- 125000000217 alkyl group Chemical group 0.000 claims description 57
- 125000000623 heterocyclic group Chemical group 0.000 claims description 36
- 150000002431 hydrogen Chemical class 0.000 claims description 34
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 21
- 150000003839 salts Chemical class 0.000 claims description 14
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 150000002367 halogens Chemical class 0.000 claims description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 9
- 239000011737 fluorine Substances 0.000 claims description 9
- 229910052731 fluorine Inorganic materials 0.000 claims description 9
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 5
- QKDRXGFQVGOQKS-CRSSMBPESA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methylsulfanyloxane-3,4,5-triol Chemical group C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](SC)O2)O)=CC=C1Cl QKDRXGFQVGOQKS-CRSSMBPESA-N 0.000 claims description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 abstract description 21
- 239000008103 glucose Substances 0.000 abstract description 21
- 239000011734 sodium Substances 0.000 abstract description 14
- 238000011282 treatment Methods 0.000 abstract description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 abstract description 6
- 206010012601 diabetes mellitus Diseases 0.000 abstract description 6
- 229910052708 sodium Inorganic materials 0.000 abstract description 6
- 208000037765 diseases and disorders Diseases 0.000 abstract description 3
- 239000003112 inhibitor Substances 0.000 abstract description 3
- 208000008589 Obesity Diseases 0.000 abstract description 2
- 235000020824 obesity Nutrition 0.000 abstract description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 179
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 145
- -1 Oxo-D-Xylopyranosyl-Substituted Phenyl Chemical class 0.000 description 79
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 76
- 244000257039 Duranta repens Species 0.000 description 66
- 235000019439 ethyl acetate Nutrition 0.000 description 66
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 54
- 239000000243 solution Substances 0.000 description 51
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 48
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 46
- 238000006243 chemical reaction Methods 0.000 description 46
- 238000002360 preparation method Methods 0.000 description 44
- 239000000203 mixture Substances 0.000 description 40
- 238000000034 method Methods 0.000 description 36
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 35
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 35
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 32
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 31
- 238000000524 positive electrospray ionisation mass spectrometry Methods 0.000 description 31
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 28
- 241001459693 Dipterocarpus zeylanicus Species 0.000 description 26
- 239000012267 brine Substances 0.000 description 26
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 26
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 25
- 230000015572 biosynthetic process Effects 0.000 description 25
- 239000007787 solid Substances 0.000 description 25
- 238000003786 synthesis reaction Methods 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- 201000010099 disease Diseases 0.000 description 23
- 238000003818 flash chromatography Methods 0.000 description 23
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 22
- 210000004027 cell Anatomy 0.000 description 20
- 239000000499 gel Substances 0.000 description 20
- 239000000741 silica gel Substances 0.000 description 20
- 229910002027 silica gel Inorganic materials 0.000 description 20
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 19
- 125000000547 substituted alkyl group Chemical group 0.000 description 19
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 229910004298 SiO 2 Inorganic materials 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 17
- 238000004587 chromatography analysis Methods 0.000 description 16
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 16
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000007983 Tris buffer Substances 0.000 description 15
- 238000000746 purification Methods 0.000 description 14
- 150000002148 esters Chemical class 0.000 description 13
- 208000035475 disorder Diseases 0.000 description 12
- 239000002552 dosage form Substances 0.000 description 12
- 238000000605 extraction Methods 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 12
- 239000011541 reaction mixture Substances 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 239000000377 silicon dioxide Substances 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- 125000003107 substituted aryl group Chemical group 0.000 description 11
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 10
- 238000010521 absorption reaction Methods 0.000 description 10
- 229910052681 coesite Inorganic materials 0.000 description 10
- 229910052906 cristobalite Inorganic materials 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 229910052682 stishovite Inorganic materials 0.000 description 10
- 229910052905 tridymite Inorganic materials 0.000 description 10
- 239000000872 buffer Substances 0.000 description 9
- 210000002700 urine Anatomy 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Natural products CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 8
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical group CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 8
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 240000008042 Zea mays Species 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 235000019341 magnesium sulphate Nutrition 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 229950010131 puromycin Drugs 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 5
- 125000003545 alkoxy group Chemical class 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000002953 preparative HPLC Methods 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 239000011701 zinc Substances 0.000 description 5
- FRDUXQSWUHUIGB-LXHROKJGSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-ethylsulfonyloxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](O2)S(=O)(=O)CC)O)=CC=C1Cl FRDUXQSWUHUIGB-LXHROKJGSA-N 0.000 description 4
- XEEROTCYZVZEEI-MJCUULBUSA-N (2s,3r,4r,5s,6s)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methoxyoxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](OC)O2)O)=CC=C1Cl XEEROTCYZVZEEI-MJCUULBUSA-N 0.000 description 4
- 239000005695 Ammonium acetate Substances 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- 239000007832 Na2SO4 Substances 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 102000003673 Symporters Human genes 0.000 description 4
- 108090000088 Symporters Proteins 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- 235000019257 ammonium acetate Nutrition 0.000 description 4
- 229940043376 ammonium acetate Drugs 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 150000001540 azides Chemical class 0.000 description 4
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 4
- 229940098773 bovine serum albumin Drugs 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 125000003396 thiol group Chemical class [H]S* 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- UTJMJMFVPBQKQV-MHMIHQHRSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-propan-2-yloxyoxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](OC(C)C)O2)O)=CC=C1Cl UTJMJMFVPBQKQV-MHMIHQHRSA-N 0.000 description 3
- UTJMJMFVPBQKQV-VROINQGHSA-N (2s,3r,4r,5s,6s)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-propan-2-yloxyoxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](OC(C)C)O2)O)=CC=C1Cl UTJMJMFVPBQKQV-VROINQGHSA-N 0.000 description 3
- PNYGUECZGOHYPH-TWGBQZSLSA-N (2s,3s,4r,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-(hydroxymethyl)-1-methylpiperidine-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@H]2N([C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)C)=CC=C1Cl PNYGUECZGOHYPH-TWGBQZSLSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 241001282138 Antigonia capros Species 0.000 description 3
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 3
- 240000000233 Melia azedarach Species 0.000 description 3
- 108091006277 SLC5A1 Proteins 0.000 description 3
- 102000058090 Sodium-Glucose Transporter 1 Human genes 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- MJOQJPYNENPSSS-XQHKEYJVSA-N [(3r,4s,5r,6s)-4,5,6-triacetyloxyoxan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1CO[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O MJOQJPYNENPSSS-XQHKEYJVSA-N 0.000 description 3
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 3
- 239000012346 acetyl chloride Substances 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000029142 excretion Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 235000012631 food intake Nutrition 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 229930182470 glycoside Natural products 0.000 description 3
- 150000002338 glycosides Chemical class 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 235000009200 high fat diet Nutrition 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000008297 liquid dosage form Substances 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 125000001997 phenyl group Chemical class [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 3
- JFVIJDSBSHXAJV-VIYGXFRKSA-N (2s,3r,4r,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-fluoro-6-methoxyoxane-3,4-diol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@@H](F)[C@H](OC)O2)O)=CC=C1Cl JFVIJDSBSHXAJV-VIYGXFRKSA-N 0.000 description 2
- SRARBFGCMPBDLS-YRXWBPOGSA-N (2s,3r,4r,5s)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-hydroxyiminooxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)C(=NO)O2)O)=CC=C1Cl SRARBFGCMPBDLS-YRXWBPOGSA-N 0.000 description 2
- XEEROTCYZVZEEI-AUGMSIGLSA-N (2s,3r,4r,5s)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methoxyoxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)C(OC)O2)O)=CC=C1Cl XEEROTCYZVZEEI-AUGMSIGLSA-N 0.000 description 2
- YSQLKZLRLCWBQK-OKLPXFMBSA-N (2s,3r,4r,5s)-2-[4-chloro-3-[(4-hydroxyphenyl)methyl]phenyl]-6-methoxyoxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)C(OC)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O)=CC=2)=C1 YSQLKZLRLCWBQK-OKLPXFMBSA-N 0.000 description 2
- BTTSHPDHZXNGNH-LXHROKJGSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-ethylsulfanyloxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](SCC)O2)O)=CC=C1Cl BTTSHPDHZXNGNH-LXHROKJGSA-N 0.000 description 2
- UQCUYCVVIWVRHT-ASTSIXKLSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-ethylsulfinyloxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](O2)S(=O)CC)O)=CC=C1Cl UQCUYCVVIWVRHT-ASTSIXKLSA-N 0.000 description 2
- SJQNCNOQTGFQMR-CRSSMBPESA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methoxythiane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](OC)S2)O)=CC=C1Cl SJQNCNOQTGFQMR-CRSSMBPESA-N 0.000 description 2
- BWAFPXYSRXDICQ-WCIQWLHISA-N (2s,3r,4s,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]oxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)CO2)O)=CC=C1Cl BWAFPXYSRXDICQ-WCIQWLHISA-N 0.000 description 2
- AGHNXKZBDRQJSE-TWGBQZSLSA-N (2s,3s,4r,5r,6r)-2-[3-[(4-ethoxyphenyl)methyl]phenyl]-6-(hydroxymethyl)-1-methylpiperidine-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC=CC([C@@H]2N([C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)C)=C1 AGHNXKZBDRQJSE-TWGBQZSLSA-N 0.000 description 2
- BDWPSWXDSWVNLZ-XMLOSSLDSA-N (2s,3s,4r,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-methyl-3,4,5-tris[(e)-prop-1-enoxy]-6-[[(e)-prop-1-enoxy]methyl]piperidine Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@H]2N([C@H](CO\C=C\C)[C@@H](O\C=C\C)[C@H](O\C=C\C)[C@H]2O\C=C\C)C)=CC=C1Cl BDWPSWXDSWVNLZ-XMLOSSLDSA-N 0.000 description 2
- HMNGIDFDTPKVKB-TXVWBRJLSA-N (2s,3s,4r,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-(hydroxymethyl)piperidine-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)N2)O)=CC=C1Cl HMNGIDFDTPKVKB-TXVWBRJLSA-N 0.000 description 2
- BKUPFVJDKGLHSR-ANULTFPQSA-N (2s,3s,4s,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-methylpiperidine-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@H]2N(C[C@@H](O)[C@H](O)[C@H]2O)C)=CC=C1Cl BKUPFVJDKGLHSR-ANULTFPQSA-N 0.000 description 2
- BQSGPTNJDRCQCQ-FYQPLNBISA-N (2s,3s,4s,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4,5-trihydroxypiperidine-1-carboxylic acid Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@H]2N(C[C@@H](O)[C@H](O)[C@H]2O)C(O)=O)=CC=C1Cl BQSGPTNJDRCQCQ-FYQPLNBISA-N 0.000 description 2
- DBMXIMGIUITTBC-ZQROTMPRSA-N (3r,4s,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-ol Chemical compound C1=CC(OCC)=CC=C1CC1=CC(C2(O)[C@@H]([C@@H](OCC=3C=CC=CC=3)[C@H](OCC=3C=CC=CC=3)[C@@H](COCC=3C=CC=CC=3)O2)OCC=2C=CC=CC=2)=CC=C1Cl DBMXIMGIUITTBC-ZQROTMPRSA-N 0.000 description 2
- CVLDCIUEZOEBBY-JPEWJUICSA-N (3s,4r,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-methyl-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)piperidine Chemical compound C1=CC(OCC)=CC=C1CC1=CC(C2N(C(COCC=3C=CC=CC=3)[C@@H](OCC=3C=CC=CC=3)[C@H](OCC=3C=CC=CC=3)[C@H]2OCC=2C=CC=CC=2)C)=CC=C1Cl CVLDCIUEZOEBBY-JPEWJUICSA-N 0.000 description 2
- YZWOETIOLJSKIN-WYGKVCCSSA-N (3s,4r,5r,6s)-6-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]oxane-2,3,4,5-tetrol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)C(O)O2)O)=CC=C1Cl YZWOETIOLJSKIN-WYGKVCCSSA-N 0.000 description 2
- VRTZOIPOBBAYPL-UHFFFAOYSA-N 1,3-dioxole-4-carbaldehyde Chemical compound O=CC1=COCO1 VRTZOIPOBBAYPL-UHFFFAOYSA-N 0.000 description 2
- VPSAUIRUSBYTSQ-MLNNCEHLSA-N 1-[(2s,3s,4s,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4,5-trihydroxypiperidin-1-yl]ethanone Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@H]2N(C[C@@H](O)[C@H](O)[C@H]2O)C(C)=O)=CC=C1Cl VPSAUIRUSBYTSQ-MLNNCEHLSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical compound OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 description 2
- ZUNCHZBITMUSRD-UHFFFAOYSA-N 4-bromo-1-chloro-2-[(4-ethoxyphenyl)methyl]benzene Chemical compound C1=CC(OCC)=CC=C1CC1=CC(Br)=CC=C1Cl ZUNCHZBITMUSRD-UHFFFAOYSA-N 0.000 description 2
- WSUBHQBKDTYHQE-FYQPLNBISA-N C1=CC(OCC)=CC=C1CC1=CC([C@H](O)[C@H](O)[C@@H](O)[C@H](O)C=NO)=CC=C1Cl Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H](O)[C@H](O)[C@@H](O)[C@H](O)C=NO)=CC=C1Cl WSUBHQBKDTYHQE-FYQPLNBISA-N 0.000 description 2
- 0 C[C@](C(CC(C1)O)O)(c(cc2Cc(cc3)ccc3OC*)ccc2Cl)N1C(NCC=C)=O Chemical compound C[C@](C(CC(C1)O)O)(c(cc2Cc(cc3)ccc3OC*)ccc2Cl)N1C(NCC=C)=O 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 101000716688 Homo sapiens Sodium/glucose cotransporter 1 Proteins 0.000 description 2
- 101000716682 Homo sapiens Sodium/glucose cotransporter 2 Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical compound ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- AZAQSUOEPAQLFO-KMDXXIMOSA-N [(2s,3r,4s)-3-acetyloxy-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4-dihydro-2h-pyran-4-yl] acetate Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](OC(C)=O)C=CO2)OC(C)=O)=CC=C1Cl AZAQSUOEPAQLFO-KMDXXIMOSA-N 0.000 description 2
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- UIJGNTRUPZPVNG-UHFFFAOYSA-N benzenecarbothioic s-acid Chemical compound SC(=O)C1=CC=CC=C1 UIJGNTRUPZPVNG-UHFFFAOYSA-N 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- ABBZJHFBQXYTLU-UHFFFAOYSA-N but-3-enamide Chemical compound NC(=O)CC=C ABBZJHFBQXYTLU-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000005587 carbonate group Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 229940092727 claro Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 230000002844 continuous effect Effects 0.000 description 2
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 229960003834 dapagliflozin Drugs 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 125000004404 heteroalkyl group Chemical group 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 2
- 102000052194 human SLC5A1 Human genes 0.000 description 2
- 102000052543 human SLC5A2 Human genes 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical group C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- CXHHBNMLPJOKQD-UHFFFAOYSA-M methyl carbonate Chemical compound COC([O-])=O CXHHBNMLPJOKQD-UHFFFAOYSA-M 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- FTEIFTIWXGDVGS-VAFBSOEGSA-N n-[(2s,3s,4r,5r,6s)-6-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4,5-trihydroxyoxan-2-yl]-n-propylacetamide Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](N(CCC)C(C)=O)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC)=CC=2)=C1 FTEIFTIWXGDVGS-VAFBSOEGSA-N 0.000 description 2
- 125000003835 nucleoside group Chemical group 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- 101150002764 purA gene Proteins 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 239000012363 selectfluor Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- QJDUDPQVDAASMV-UHFFFAOYSA-M sodium;ethanethiolate Chemical compound [Na+].CC[S-] QJDUDPQVDAASMV-UHFFFAOYSA-M 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical group C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- YKXCWZVUWWQSAV-BTVCFUMJSA-N (2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O YKXCWZVUWWQSAV-BTVCFUMJSA-N 0.000 description 1
- JFVIJDSBSHXAJV-DQKSBWHRSA-N (2s,3r,4r,5r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-fluoro-6-methoxyoxane-3,4-diol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@@H](F)C(OC)O2)O)=CC=C1Cl JFVIJDSBSHXAJV-DQKSBWHRSA-N 0.000 description 1
- XEEROTCYZVZEEI-CRSSMBPESA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methoxyoxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](OC)O2)O)=CC=C1Cl XEEROTCYZVZEEI-CRSSMBPESA-N 0.000 description 1
- LITRERKYGZPHDC-CRSSMBPESA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methylsulfonyloxane-3,4,5-triol Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@H](O2)S(C)(=O)=O)O)=CC=C1Cl LITRERKYGZPHDC-CRSSMBPESA-N 0.000 description 1
- IAPVMXRKLOSMDT-ODSPINPYSA-N (2s,3s,4r,5r,6r)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-3,4,5-tris[(e)-prop-1-enoxy]-6-[[(e)-prop-1-enoxy]methyl]piperidine Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O\C=C\C)[C@H](O\C=C\C)[C@@H](CO\C=C\C)N2)O\C=C\C)=CC=C1Cl IAPVMXRKLOSMDT-ODSPINPYSA-N 0.000 description 1
- FAGHSJFNVFZCES-OWSLCNJRSA-N (2s,3s,4s,5r)-2-[4-chloro-3-[(4-hydroxyphenyl)methyl]phenyl]piperidine-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)CN[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O)=CC=2)=C1 FAGHSJFNVFZCES-OWSLCNJRSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- 125000006039 1-hexenyl group Chemical group 0.000 description 1
- 125000006023 1-pentenyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical group C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000006069 2,3-dimethyl-2-butenyl group Chemical group 0.000 description 1
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- 125000006027 3-methyl-1-butenyl group Chemical group 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- ANXBXSSIJIPYCC-ZYGPMETQSA-N C(C)(=O)O[C@@H]1[C@@H](C(O[C@H]([C@H]1OC(C)=O)C1=CC(=C(C=C1)Cl)CC1=CC=C(C=C1)OCC)O)CC(=O)O Chemical compound C(C)(=O)O[C@@H]1[C@@H](C(O[C@H]([C@H]1OC(C)=O)C1=CC(=C(C=C1)Cl)CC1=CC=C(C=C1)OCC)O)CC(=O)O ANXBXSSIJIPYCC-ZYGPMETQSA-N 0.000 description 1
- YTVZZMCTJLRAQM-UHFFFAOYSA-N CCOC1=CC=C(C=C1)CC2=C(C=CC(=C2)C(=O)C(CCC(=O)C)OCC3=CC=CC=C3)Cl Chemical compound CCOC1=CC=C(C=C1)CC2=C(C=CC(=C2)C(=O)C(CCC(=O)C)OCC3=CC=CC=C3)Cl YTVZZMCTJLRAQM-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 239000004128 Copper(II) sulphate Substances 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical class CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 229920002274 Nalgene Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229910019020 PtO2 Inorganic materials 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 229910018557 Si O Inorganic materials 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 241000282539 Theropithecus gelada Species 0.000 description 1
- YPWFISCTZQNZAU-UHFFFAOYSA-N Thiane Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- HXBPYFMVGFDZFT-UHFFFAOYSA-N allyl isocyanate Chemical compound C=CCN=C=O HXBPYFMVGFDZFT-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 239000004305 biphenyl Chemical group 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229940045348 brown mixture Drugs 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 229940106681 chloroacetic acid Drugs 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000011872 intimate mixture Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005468 isobutylenyl group Chemical group 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 239000006201 parenteral dosage form Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- IOUVKUPGCMBWBT-QNDFHXLGSA-N phlorizin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 IOUVKUPGCMBWBT-QNDFHXLGSA-N 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Inorganic materials [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- ILJSQTXMGCGYMG-UHFFFAOYSA-N triacetic acid Chemical compound CC(=O)CC(=O)CC(O)=O ILJSQTXMGCGYMG-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000007762 w/o emulsion Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D291/00—Heterocyclic compounds containing rings having nitrogen, oxygen and sulfur atoms as the only ring hetero atoms
- C07D291/02—Heterocyclic compounds containing rings having nitrogen, oxygen and sulfur atoms as the only ring hetero atoms not condensed with other rings
- C07D291/06—Six-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7032—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a polyol, i.e. compounds having two or more free or esterified hydroxy groups, including the hydroxy group involved in the glycosidic linkage, e.g. monoglucosyldiacylglycerides, lactobionic acid, gangliosides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D335/00—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom
- C07D335/02—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/14—Acyclic radicals, not substituted by cyclic structures attached to a sulfur, selenium or tellurium atom of a saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Crystallography & Structural Chemistry (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Hydrogenated Pyridines (AREA)
- Saccharide Compounds (AREA)
- Pyrane Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

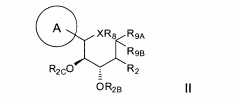

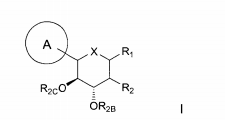
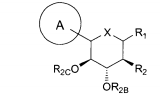
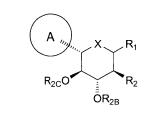
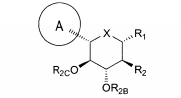


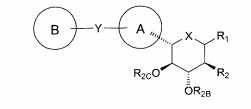

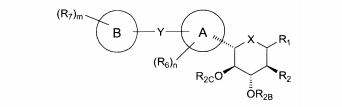



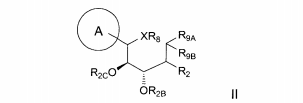





dores de SGLT2 é descrita, por exemplo, nos pedidos de patente U.S. N°s 10/540.519; 10/734.573; 11/247.216; 11/247.356; e nos pedidos de patente internacionais N°s WO 03/020737; WO 2004/058790; WO 2004/080990; WO 2004/089967; WO 2005/011592; WO 2005/012242; WO 2005/012243; WO 2005/012318; WO 2005/021566; e WO 2005/085265.


6.1. Exemplo 1: Síntese de (2S.3R.4R.5S)-2-[4-Cloro-3-(4-etóxi-benzil)-fenin- 6-metóxi-tetraidro-piran-3,4,5-triol

- A. Preparação de [(3aS,5S,6R,6aS)-6-(terc-butil-dimetil-silaniló-xi)-2.2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-5-il]-metanol. Este composto foi sintetizado usando os procedimentos conhecidos na técnica. Ver, por exemplo, Nucleosides Nucleotides. 20:649-652 (2001) e suas referências.
- B. Preparação de (3aS,5R.6R,6aS)-6-(terc-butil-dimetil-silaniló-xi)-2,2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-5-carbaldeído. A uma solução de cloreto de oxalila (0,76 ml, 8,7 mmols) em CH2CI2 (55 ml) sob N2 em -78°C foi adicionado por gotejamento uma solução de DMSO (0,84 ml, 11,8 mmols) em CH2CI2 (5 ml). Após 15 minutos, o álcool da etapa A (2,40 g, 7,9 mmols) em CH2CI2 (20 ml) foi adicionado por gotejamento. Após 15 minutos, NEt3 foi adicionado lentamente. A reação foi deixada aquecer lentamente para a temperatura ambiente durante 105 minutos, depois extinta com H2O, diluída com Et2O, e lavada com H2O, NaHCO3sat aq., e salmoura. As fases orgânicas combinadas foram extraídas outra vez com Et2O, que foi lavado pela mesma sequência. As fases orgânicas combinadas foram secadas por MgSO4, filtradas e concentradas sob vácuo para fornecer (3aS,5R,6R,6aS)-6-(terc-butil-dimetil-silanilóxi)-2,2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-5-carbaldeído (2,4 g, cerca de 64% pura por RMN). O produto foi realizado sem mais purificação.
- C. Preparação de 4-bromo-1-cloro-2-(4-etóxi-benzil)-benzeno. Este composto foi preparado como descrito no pedido de patente U.S. N° 10/745.075 de Deshpande et al., filed December 23, 2003.
- D. Preparação de (S)-[(3aS,5S,6R,6aS)-6-(terc-butil-dimetil-silanilóxi)-2,2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-5-il]-[4-cloro-3-(4-etóxi-benzil)-fenil]-metanol. A uma solução de 4-bromo-1-cloro-2-(4-etóxi-benzil)-benzeno da etapa C (3,6 g, 11,1 mmols) em THF (60 ml) sob N2 em -78°C foi adicionado por gotejamento BuLi (2,5 M em hexanos, 4,4 ml, 11,1 mmols). Após 30 minutos, o aldeído da etapa B (2,4 g, 64% pura, 5,1 mmols) em THF (20 ml) foi adicionado por gotejamento, e a reação foi agitada durante 30 min em -78°C, deixada aquecer para a temperatura ambiente e agitada durante 60 minutos, extinta com NH4CI sat. aq., diluída com Et2O, e lavada com H2O e salmoura. As lavagens aquosas combinadas foram extraídas outra vez com Et2O, que foi lavado pela mesma sequência. Os extratos orgânicos combinados foram secados por MgSO4, filtradas e concentradas sob vácuo. O resíduo foi purificado por cromatografia instantânea (120g Si-O2, 0-20% EtOAc: Hexanos, 75 minutos, 85 ml/min) para fornecer (S)-[(3aS,5S,6R,6aS)-6-(terc-butil-dimetil-silanilóxi)-2,2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-5-il]-[4-cloro-3-(4-etóxi-benzil)-fenil]-metanol puro (0,84 g, 1,5 mmol, 30%) acrescido do epímero C5 (0,83g) e algumas frações misturadas (0,51 g).
- E. Preparação de (2S,3R,4R,5S)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-metóxi-tetraidro-piran-3,4,5-triol. Uma solução de 0,35 M HCI em MeOH foi preparada mediante a adição de AcCI (0,25 ml, 3,5 mmols) em MeOH (10 ml) e agitação durante 15 minutos O álcool da etapa D (0,84g, 1,5 mmol) foi tratado com esta solução durante 16 horas em temperatura ambiente e 2 horas a 80°C em um frasco lacrado. A reação foi esfriada para a temperatura ambiente, extinta com K2CO3 até que básica, diluída com CH2CI2, filtrada e concentrada sob vácuo. O produto foi purificado por cromatografia instantânea (40 g SiO2, 0 a 10% MeOH: CH2CI2, 60 minutos, 35 ml/min), colocada em suspensão em H2O, e liofilizada para fornecer (2S,3R,4R,5S)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-metóxi-tetraidro-piran-3,4,5-triol (0,46 g, 1,1 mmol, 75%) como um sólido branco. RMN revelou uma relação DE 1,2:1 de α e β anômeros.
6.2. Exemplo 2: Síntese de (3S,4R,5R,6S)-6-[4-Cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-2,3,4,5-tetraol
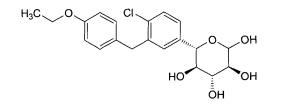
1H RMN (400 MHz, metanol-d4) δ ppm 7,34 (dd, J = 8,08, 4,04 Hz, 1 H), 7,22 - 7,30 (m, 2 H), 7,09 (d, J = 8,34 Hz, 2 H), 6,80 (d, J = 8,08 Hz, 2 H), 5,16 (d, J = 3,79 Hz, 1 H α), 4,65 (d, J = 9,60 Hz, 1 H α ou β), 4,59 (d, J = 7,58 Hz, 1 H α ou β), 4,14 (d, J = 9,60 Hz, 1 H α ou β), 3,96 - 4,07 (m, 4 H), 3,76 (t, J = 9,35 Hz, 1 H α ou β), 3,50 (dd, J = 9,60, 3,79 Hz, 1 H α ou β), 3,43 (t, J = 9,09 Hz, 1 H α ou β), 3,23 - 3,29 (m, 1,5 H), 1,36 (t, J = 7,07 Hz, 3 H). MS (ES+) [M + NH4]+ = 412.
6.3. Exemplo 3: Síntese de (2S,3R.4R,5S)-2-[4-Cloro-3-(4-etóxi-benzil)-fenil]- 6-etóxi-tetraidro-piran-3,4,5-triol
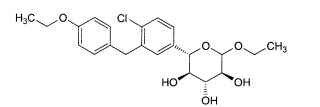
1H RMN (400 MHz, Clorofórmio-d) δ ppm: 7,28 - 7,32 (m, 1 H), 7,14 (m, 2 H), 7,02 (d, J = 8,84 Hz, 2 H), 6,72 - 6,76 (m, 2 H), 4,88 (d, J =4.04 Hz, 1 H α), 4,37 (d, J = 9,60 Hz, 1 H α), 4,33 (d, J = 7,83 Hz, 1 H β), 4,06 (d, J = 9,35 Hz, 1 H β), 3,89 - 4,02 (m, 4 H), 3,36 - 3,87 (m, 5 H), 2,62 (s, 1 H β), 2,54 (s, 1 H α), 2,41 (d, J = 1,52 Hz, 1 H β), 2,02 (d, J = 10,36 Hz, 1 H α), 1,92 (d, J = 2,53 Hz, 1 H), 1,32 (t, J = 6,95 Hz, 3 H), 1,13 - 1,19 (m, 3 H). MS (ES+) [M + NH4]+ = 440.
6.4 Exemplo 4: Síntese de (2S,3R,4R,5S,6S)-2-[4-Cloro-3-(4-etóxi-benzil)- fenil]-6-isopropóxi-tetraidro-piran-3,4,5-triol e (2S,3R,4R,5S,6R)-2-[4-Cloro-3- (4-etóxi-benzil)-fenil]-6-isopropóxi-tetraidro-piran-3,4,5-triol

6.5. Exemplo 5: Síntese de (2S,3R.4R,5S,6R)-2-[4-Cloro-3-(4-etóxi-benzil)- fenill-6-metóxi-tetraidro-piran-3,4,5-triol e (2S,3R,4R,5S,6S)-2-r4-Cloro-3-(4- etóxi-benzil)-fenil]-6-metóxi-tetraidro-piran-3,4,5-triol
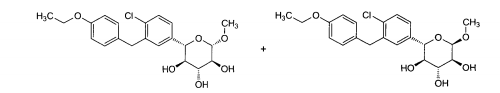
- A. Preparação de éster (3S,4R,5S,6S)-2,4,5-triacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-3-il de ácido acético. O álcool do E-xemplo 1, etapa D (6,80 g, 12,4 mmols) foi tratado com 3:2 AcOH/H2O (62 ml) em 100°C durante 22 horas. A reação foi concentrada sob vácuo, em evaporador rotatório 3 vezes com tolueno, e colocada sob vácuo elevado. O resíduo foi tratado com anidrido acético (9,4 ml, 99,2 mmols) em piridina (25 ml) durante 16 horas. A reação foi extinta com H2O, agitada 1 hora, diluída com Et2O, lavada com 1 M NaHSO4aq., H2O, NaHCO3 sat. aq., e salmoura (com extração de retorno), secada por MgSO4, filtrada e concentrada sob vácuo. O resíduo foi purificado por cromatografia instantânea (120 g SiO2, 0 a 50% EtOAc/Hex) para fornecer éster (3S,4R,5S,6S)-2,4,5-triacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-3-il de ácido acético (6,10 g, 10,9 mmols, 87%).
- B. Preparação de éster (2S,3S,4R,5S,6S)-4,5-diacetóxi-2-bromo-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-3-ílico de ácido acético. O tetra-acetato de etapa A (8,08 g, 14,4 mmols) foi tratado com 33% HBr em AcOH (30 ml) durante 1 hora. A reação foi diluída com CH2CI2 (60 ml), agitada durante 30 minutos, diluída com mais DCM, lavada 3x com H2O gelado e com NaHCO3 sat. aq. (com extração de retorno), secada por MgSO4, filtrada, e concentrada sob vácuo para fornecer éster (2S,3S,4R,5S,6S)-4,5-diacetóxi-2-bromo-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-3-ílico de ácido acético.
- C. Preparação de éster (2S,3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-2-metóxi-tetraidro-piran-3-ílico de ácido acético. O brometo bruto da etapa B (8,4 g, 14,4 mmols) e ZnO (1,2 g, 14,4 mmols) foram dissolvidos em MeOH (144 ml) e aquecidos em 70°C durante 1 hora. A reação foi esfriada para a temperatura ambiente, filtrada através de celita com EtOAc, e concentrada sob vácuo. O resíduo foi recristalizado a partir de MeOH em duas bateladas para fornecer éster (2S,3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-2-metóxi-tetraidro-piran-3-ílico de ácido acético (5,98 g, 11,2 mmols, 78%) como o β-anômero puro.
- D. Preparação de (2S,3R,4R,5S,6S)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-metóxi-tetraidro-piran-3,4,5-triol. Triacetato recristalizado da etapa C (5,98 g, 11,2 mmols) foi tratado com K2CO3 (7,7 g, 56 mmols) em MeOH (112 ml) com agitação vigorosa durante 1 hora. A reação foi filtrada através de celita e concentrada sob vácuo. O resíduo foi dissolvido em DCM, lavada com H2O e salmoura, secada por MgSO4, filtrada e concentrada sob vácuo. O resíduo foi passado através de um tampão de sílica-gel com 5% Me-OH:CH2CI2, concentrado sob vácuo, colocado em suspensão em H2O, e liofi-lizado para fornecer (2S,3R,4R,5S,6S)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-metóxi-tetraidro-piran-3,4,5-triol (4,37 g, 10,7 mmols, 96%) como um sólido branco.
6.6 Exemplo 6: Síntese de N-{(2S,3S,4R,5R,6S)-6-[4-Cloro-3-(4-etóxi- benzil)-fenil]-3,4,5-triidróxi-tetraidro-piran-2-il}-N-propil-acetamida
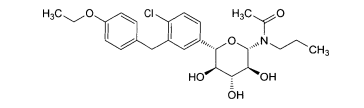
1H RMN (400 MHz, MeOD) δ ppm 7,29 - 7,40 (m, 1 H), 7,16 - 7,26 (m, 2 H), 7,08 (d, J = 8,6 Hz, 2 H), 6,81 (d, J = 8,6 Hz, 2 H), 5,59 (d, J =8,6 Hz, 0,33 H), 4,98 (d, J = 11,9 Hz, 0,67 H), 4,25 (d, J = 9,3 Hz, 0,67 H), 4,17 (d, J = 9,9 Hz, 0,33 H), 3,92 - 4,06 (m, 4 H), 3,46 - 3,64 (m, 3 H), 3,06 - 3,28 (m, 2 H), 2,16 (s, 3 H), 1,49 - 1,68 (m, 2 H), 1,36 (t, J = 6,9 Hz, 3 H), 0,93 (t, J = 7,5 Hz, 1 H), 0,87 (t, J = 7,5 Hz, 2 H). MS (ES+) [M + H]+ = 478. 6.7 Exemplo 7: Síntese de (2R,3S,4S,5S)-5-[4-Cloro-3-(4-etóxi-benzil)-fenil]-2,3,4,5-tetraidróxi-pentanal oxima
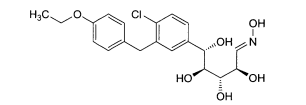
Isômero principal 1H RMN (400 MHz, MeOD) δ ppm 7,31 - 7,36 (m, 2 H), 7,23 - 7,30 (m, 2 H), 7,10 (d, J = 8,8 Hz, 2 H), 6,80 (d, J = 8,6 Hz, 2 H), 4,63 (d, J = 8,1 Hz, 1 H), 4,28 (t, J = 6,8 Hz, 1 H), 3,96 - 4,03 (m, 4 H), 3,90 - 3,94 (m, 1 H), 3,59 (dd, J = 8,0, 1,6 Hz, 1 H), 1,36 (t, J = 6,9 Hz, 3 H); MS (ES+) [M + H]+ = 410.
6.8. Exemplo 8: Síntese de (3S,4R,5R,6S)-6-[4-Cloro-3-(4-etóxi-benzil)-fenil]- 3,4,5-triidróxi-tetraidro-piran-2-ona oxima

- A. Preparação de éster (3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3- (4-etóxi-benzil)-fenil]-2-hidróxi-tetraidro-piran-3-ílico de ácido acético. O tetra- acetato do Exemplo 5, etapa A (200 mg, 0,36 mmol) foi tratado com benzi- lamina (39 μl, 0,36 mmol) em DMF (1,8 ml) durante 2 horas. A reação diluída com Et2O, lavada com 1 M NaHSO4aq., H2O, NaHCO3 sat. aq., e salmoura, secada por MgSO4, filtrada e concentrada sob vácuo. O resíduo foi purifica-do por cromatografia instantânea (12 g SiO2, 0 - 50% EtOAc:Hex.) para fornecer éster (3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-2- hidróxi-tetraidro-piran-3-ílico de ácido acético (142 mg, 0,27 mmol, 77%) como uma relação 3:1 de anômeros.
- B. Preparação de éster (3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3- (4-etóxi-benzil)-fenil]-2-[(Z)-hidroxiiminol-tetraidro-piran-3-ílico de ácido acético. O composto da etapa A (142 mg, 0,27 mmol) e cloridrato de hidroxila- mina (57 mg, 0,82 mmol) foram dissolvidos em piridina (1,4 ml). A reação foi agitada durante 6 horas, diluída com EtOAc, lavada com 1 M NaHSO4 aq., H2O, NaHCO3 sat. aq., e salmoura (com extração de retorno), secada por Na2SO4, filtrada e concentradas sob vácuo. O resíduo foi dissolvido em CH2CI2, esfriado para -78°C, e tratado com DBU (49 μL, 0,33 mmol) seguido por N-clorossuccinimida (44 mg, 0,33 mmol). A reação foi agitada durante 20 minutos em -78°C, depois deixada aquecer para a temperatura ambiente durante 15 minutos. A reação foi diluída com EtOAc, lavada com H2O e salmoura (com extração de retorno), secada por MgSO4, filtrada e concentradas sob vácuo. O resíduo foi purificado por cromatografia instantânea (12 g SiO2, 0 - 50% EtOAc:Hex.) para fornecer éster (3S,4R,5S,6S)-4,5-diacetóxi- 6-[4-cloro-3-(4-etóxi-benzil)-fenil]-2-[(Z)-hidroxiimino]-tetraidro-piran-3-ílico de ácido acético (97 mg, 0,18 mmol, 67%).
- C. Preparação de (3S,4R,5R,6S)-6-[4-cloro-3-(4-etóxi-benzil)- fenill-3,4,5-triidróxi-tetraidro-piran-2-ona oxima. O composto da etapa B (97 mg, 0,18 mmol) foi tratado com 7,0 M NH3 em MeOH (1,8 ml) durante 1 hora. A reação foi concentrada sob vácuo, e o resíduo foi purificado por croma- tografia instantânea (12 g SiO2, 0 - 12% MeOH:CH2CI2), colocado em suspensão em H2O, e liofilizado para fornecer (3S,4R,5R,6S)-6-[4-cloro-3-(4- etóxi-benzil)-fenil]-3,4,5-triidróxi-tetraidro-piran-2-ona oxima (57 mg, 0,14 mmol, 77%) como um sólido branco.
6.9. Exemplo 9: Síntese de (2S,3R,4R,5R)-2-[4-Cloro-3-(4-etóxi-benzil)-fenil]- 5-flúor-6-metóxi-tetraidro-piran-3,4-diol
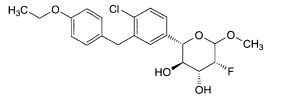
- A. Preparação de diacetato de (2S,3R,4S)-2-(4-cloro-3-(4- etoxibenzil)fenil)-3,4-diidro-2H-piran-3,4-di-ila. Em um frasco carregado com 282 mg de tetra-acetato do Exemplo 5, etapa A (0,5 mmol), 1,25 ml de HBr (33% em HOAc) foi adicionado. A reação foi agitada durante uma hora, diluída com 50 ml diclorometano e extinta mediante despejo em água gelada. A camada orgânica foi separada e lavada com NaHCO3 saturado aquoso e salmoura. Após secagem por sulfato de magnésio, os solventes foram concentrados in vacuo. O resíduo bruto foi absorvido em 0,5 ml de diclorometano e adicionado a uma suspensão de sulfato de cobre(ll) (20 mg, 0,125 mmol), pó de Zn (82 mg, 1,25 mmol), e acetato de sódio (984 mg, 12 mmols) em 2,5 ml de ácido acético/água (3:2 v:v). Esta mistura foi deixada agitar em temperatura ambiente durante 4 h, após o que a reação foi carregada novamente com 20 mg de sulfato de cobre(ll) e 82 mg de pó de Zn e agitada durante mais 18 h. A mistura foi extinta com água, extraída com acetato de eti- la. A camada orgânica foi secada por sulfato de magnésio e removida in vacuo. A cromatografia instantânea forneceu diacetato de (2S,3R,4S)-2-(4- cloro-3-(4-etoxibenzil)fenil)-3,4-diidro-2H-piran-3,4-di-ila (32 mg, 16% de rendimento).
- B. Preparação de (2S,3R,4R,5R,6R)-2-(4-cloro-3-(4-etóxi- benzil)-fenil)-5-flúor-6-metóxi-tetraidro-2H-piran-3,4-diol. Selectfluor® (45 mg, 0,128 mmol) foi adicionado a uma solução de composto da etapa A (38 mg, 0,0853 mmol) em 0,4 ml de acetonitrila:metanol (1:1 v:v). A reação foi agitada em temperatura ambiente e monitorada por conclusão por LCMS. A reação foi extinta com 2 ml de NH4CI saturado aquoso e extraída com éter dietí- lico (2x5 ml). Os orgânicos extraídos foram secados por sulfato de sódio e concentrados in vacuo. A cromatografia instantânea (5 a 10% acetato de etila/hexanos) forneceu o produto fluorado. Carbonato de potássio (5 mg) foi depois adicionado a uma solução deste produto isolado em 0,5 ml de metanol. A reação foi agitada em temperatura ambiente durante 2 h, após o que foi extinta com 2 ml de água e extraída com acetato de etila (2x4 ml). A camada orgânica foi filtrada por um tampão de sílica e concentrada para fornecer 6,3 mg de (2S,3R,4R,5R,6R)-2-(4-cloro-3-(4-etóxi-benzil)-fenil)-5-flúor- 6-metóxi-tetraidro-2H-piran-3,4-diol como um óleo transparente.
6.10. Exemplo 10: Síntese de (2S,3R,4R,5S)-2-[4-Cloro-3-(4-hidróxi-benzil)- fenil]-6-metóxi-tetraidro-piran-3,4,5-triol

- A. Preparação de [4-(5-bromo-2-cloro-benzil)-fenóxfl-terc-butil- dimetil-silano. Este composto foi preparado como descrito na publicação do pedido de patente U.S. N° 2006/0251728 de Himmelsbach et al., publicado em 09 de novembro de 2006.
- B. (S)-{3-[4-(terc-butil-dimetil-silanilóxi)-benzil]-4-cloro-fenil}- [(3aS,5S,6R,6aS)-6-(terc-butil-dimetil-silanilóxi)-2,2-dimetil-tetraidro-furo[2,3- d][1,3]dioxol-5-il]-metanol. Uma solução de 0,85 g (2,07 mmols) do composto da etapa A em 4,14 ml de éter dietílico foi esfriada para -78°C sob uma atmosfera inerte. A isto foi adicionado 2,66 ml de terc-butillítio (1,55 M em he- xanos, 4,14 mmols) através de seringa durante 5 minutos. A reação foi agitada em -78°C durante 30 minutos. Uma solução de 0,5 g (1,65 mmol) de composto do Exemplo 1, etapa B em 1,65 ml de éter dietílico foi adicionada. Esta mistura de reação foi agitada em -78°C durante 30 minutos seguido por 1,5 h a 0°C. A reação bruta foi filtrada por um tampão de sílica-gel com éter dietílico em excesso, que foi subsequentemente removido in vacuo. O produto obtido é aproximadamente uma relação 1,2:1 de diastereômeros no álcool secundário recentemente formado. Os diastereômeros foram facilmente separados por cromatografia em sílica-gel (gradiente 4 a 8% acetato de eti- la/hexanos). Rendimento: 40% (diastereômero desejado), 58% (diastereô- mero indesejado).
- C. Preparação de (2S,3R,4R,5S)-2-[4-cloro-3-(4-hidróxi-benzil)- fenil]-6-metóxi-tetraidro-piran-3,4,5-triol. Cloreto de acetila (0,17 ml) foi adicionado a 7 ml de metanol e agitado durante 15 minutos em temperatura ambiente. Esta solução foi transferida para um frasco carregado com 0,446 g de composto da etapa B, que foi depois lacrado e aquecido para 80°C durante 1 h. A reação foi esfriada para a temperatura ambiente e extinta com 50 ml de bicarbonato de sódio aquoso saturado. Esta camada aquosa foi extraída com acetato de etila (3 x 50 ml). As camadas orgânicas combinadas foram lavadas com salmoura, secadas por sulfato de magnésio, e o solvente foi removido in vacuo. O resíduo foi purificado por cromatografia em sílica- gel (gradiente 0 a 20% metanol/diclorometano) para fornecer aproximadamente uma mistura 1:1 de anômeros α:β. Rendimento: 65%.
6.11 Exemplo 11: Síntese de (2S,3R,4S,5R)-2-[4-Cloro-3-(4-etóxi-benzil)- fenil]-tetraidro-piran-3,4,5-triol
1H RMN (400 MHz, Acetona) δ ppm 7,35 - 7,41 (m, 2 H), 7,30 (dd, J = 8,34, 2,02 Hz, 1 H), 7,16 (d, J = 7,58 Hz, 2 H), 6,83 (d, J = 8,59 Hz, 2 H), 4,93 - 5,01 (m, 1 H), 4,74 (d, J = 3,79 Hz, 0,5 Hα), 4,42 (d, J = 9,60 Hz, 0,5 Hα), 4,33 (d, J = 7,58 Hz, 0,5 Hβ), 4,20 (d, J = 9,60 Hz, 0,5 Hβ), 4,05 (t, J = 2,53 Hz, 2 H), 4,05 (d, J = 5,31 Hz, 2 H), 3,93 (dd, J = 10,11, 4,80 Hz, 1 H), 3,75 - 3,89 (m, 2 H), 3,72 (t, J = 9,09 Hz, 1 H), 3,50 (t, J = 9,09 Hz, 1 H), 3,41 (s, 1,5 Hβ), 3,35 (s, 1,5 Hα), 3,29 - 3,34 (m, 3 H), 2,16 - 2,27 (m, 1 H), 1,97 - 2,04 (m, 1 H). MS (ES+) [M + NH4]+ = 468.
6.12. Exemplo 12: Síntese de (2S,3S,4S,5R)-2-[4-Cloro-3-(4-hidróxi-benzil)- fenil]-piperidina-3,4,5-triol
- A. Preparação de ((3aS,5S,6R,6aS)-5-{azido-[(S)-4-cloro-3-(4- etóxi-benzil)-fenil]-metil}-2,2-dimetil-tetraidro-furo[2,3-d][1,3]dioxol-6-ilóxi)- terc-butil-dimetil-silano. A uma solução do epímero C5 de álcool do Exemplo 1, etapa D (682 mg, 1,24 mmol) e PPh3 (489 mg, 1,87 mmol) em THF (6,2 ml) foi adicionado DIAD (366 μl- 1.87 mmol) seguido por difenil fosforil azido (DPPA, 323 μl, 1,49 mmol). A reação foi agitada durante 1,5 hora, extinta com NH4CI sat. aq., diluída com Et2O, lavada com H2O e salmoura (com extração de retorno), secada por MgSO4, e concentrada sob vácuo. O resíduo foi purificado por cromatografia instantânea (40 g SiO2, 0 - 8% EtOAc:Hex.) para fornecer ((3aS,5S,6R,6aS)-5-{azido-[(S)-4-cloro-3-(4-etóxi-benzil)-fenil]- metil}-2,2-dimetil-tetraidro-furo[2,3-d][1,3]díoxol-6-ilóxi)-terc-butil-dimetil- silano (636 mg, 1,11 mmol, 89%) como um óleo amarelo.
- B. Preparação de (2R.3S,4S,5S)-5-{azido-[(S)-4-cloro-3-(4-etóxi- benzil)-fenill-metil)-tetraidro-furan-2,3,4-triol. Cloreto de acetila (0,175 ml, 2,45 mmols) foi adicionado a MeOH (7 ml). A solução foi agitada 15 minutos, depois adicionada ao azido da etapa A (392 mg, 0,68 mmol). A reação foi agitada durante 16 horas, depois concentrada sob vácuo, em evaporador rotatório 2 vezes com MeOH, e colocada no vácuo elevado para fornecer um sólido branco. O sólido foi tratado com 1:1 AcOH:H2O (7 ml) em 100°C durante 2,5 horas. A reação foi concentrada sob vácuo, em evaporador rotatório 2 vezes com tolueno, e colocada no vácuo elevado. O resíduo foi purificado por cromatografia instantânea (40 g SiO2, 0 - 6% MeOH:CH2CI2) para fornecer (2R,3S,4S,5S)-5-{azido-[(S)-4-cloro-3-(4-etóxi-benzil)-fenil]-metil}- tetraidro-furan-2,3,4-triol (223 mg, 0,53 mmol, 78%) como um mistura de a- nômeros.
- C. Preparação de (2S,3S,4S,5R)-2-(4-cloro-3-(4-etóxi-benzil)- fenil]-piperidina-3,4,5-triol. O composto da etapa B (216 mg, 0,52 mmol) foi hidrogenado sob pressão atmosférica H2 por PtO2 (6 mg, 0,026 mmol) em MeOH (5 ml) com AcOH (0,25 ml) durante 6 horas. A reação foi filtrada, concentrada sob vácuo, diluída com EtOAc, lavada com 10% K2CO3 aq. e salmoura, secada por Na2SO4, filtrada e concentrada sob vácuo. Uma parte do material (cerca de 55 mg) foi purificado HPLC prep (coluna Sunfire C18 30 x 100 mm, 20 - 70% MeCN:H2O (10 mM NH4OAc), 15 minutos, 45 ml/min) e liofilizada para fornecer (2S,3S,4S,5R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]- piperidina-3,4,5-triol (27 mg, 0,071 mmol) como um sólido branco.
6.13. Exemplo 13: Síntese de (2S,3R,4R,5S,6R)-2-[4-Cloro-3-(4-etóxi- benzil)-fenil]-6-etanossulfinil-tetraidro-piran-3,4,5-triol e (2S,3R,4R,5S,6R)-2- [4-Cloro-3-(4-etóxi-benzil)-fenil]-6-etanossulfonil-tetraidro-piran-3,4,5-triol

- A. Preparação de (2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-6-etilsulfanil-tetraidro-piran-3,4,5-triol. A uma solução de brometo do Exemplo 5, etapa B (291 mg, 0,50 mmol) em EtOH (5 ml) a 0°C foi adicionado NaSEt (84 mg, 1,0 mmol). A reação foi agitada 30 minutos, depois diluída com EtOAc, lavada com NaOH aq. diluído e com salmoura (com extração de retorno), secada por Na2SO4, filtrada e concentrada sob vácuo. O resíduo foi purificado por cromatografia instantânea (40 g SiO2, 0 a 7% MeOH:CH2CI2), colocado em suspensão em H2O, e liofilizado para fornecer (2S,3R,4R, 5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-etilsulfanil-tetraidro-piran-3,4,5- triol (126 mg, 0,29 mmol, 58%) como um pó branco.
- B. Preparação de (2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-6-etanossulfinil-tetraidro-piran-3,4,5-triol e (2S,3R,4R,5S,6R)-2-[4- Cloro-3-(4-etóxi-benzil)-fenil]-6-etanossulfonil-tetraidro-piran-3,4,5-triol. A uma solução de composto da etapa A (10 mg, 0,023 mmol) em AcOH (0,5 ml) foi adicionado H2O2 (35% em peso de solução em H2O, 3 mg, 0,092 mmol, 9 μl). A mistura foi agitada em temperatura ambiente durante 2 horas antes de ser concentrada sob vácuo. A purificação da mistura por cromato- grafia em sílica-gel (5% MeOH/CH2CI2) proporcionou (2S,3R,4R,5S,6R)-2-[4- cloro-3-(4-etóxi-benzil)-fenil]-6-etanossulfinil-tetraidro-piran-3,4,5-triol (como uma mistura de diastereômeros em enxofre) (2 mg, 19%) e (2S,3R,4R, 5S,6R)-2-[4-Cloro-3-(4-etóxi-benzil)-fenil]-6-etanossulfonil-tetraidro-piran- 3,4,5-triol (5mg, 46%) ambos como sólido brancos.
(2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-etanossul- fonil-tetraidro-piran-3,4,5-triol: 1H RMN (400 MHz, metanol) δ ppm 7,28 (m, 1 H), 7,16 (m, 2 H), 6,99 (d, J = 8,6 Hz, 2 H), 6,71 (d, J = 8,6 Hz, 2 H), 4,46 (d, J = 9,6 Hz, 1 H), 4,19 (d, J = 9,4 Hz, 1 H), 3,90 (m, 4 H), 3,81 (t, J = 9,3 Hz, 1 H), 3,46 (t, J = 9,1 Hz, 1 H), 3,24 (t, J = 9,1 Hz, 1 H), 2,98 (m, 2 H), 1,26 (t, J = 6,8 Hz, 3 H), 1,18 (t, J = 7,6 Hz, 3 H); MS (ES+) [M + NH4]+ = 488.6.14. Exemplo 14: Síntese de Éster (2R,3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3-(4 etóxi-benzil)-fenill-2-metilsulfanil-tetraidro-piran-3-il de ácido acéti- co
- A. Preparação de (2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-6-metilsulfanil-tetraidro-piran-3,4,5-triol. A uma solução de brometo do Exemplo 5, etapa B (347 mg, 0,60 mmol) em EtOH (6 ml) a 0°C foi adicionado NaSMe (70 mg, 0,72 mmol). A reação foi agitada 30 minutos, depois diluída com EtOAc, lavada com NaOH aq. diluído e com salmoura (com extração de retorno), secada por Na2SO4, filtrada e concentrada sob vácuo. O resíduo foi purificado por cromatografia instantânea (40 g SiO2, 0-7% Me- OH:CH2CI2), colocada em suspensão em H2O, e liofilizada para fornecer (2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-metilsulfanil-tetraidro- piran-3,4,5-triol (212 mg, 0,43 mmol, 72%) como um pó branco.
- B. Preparação de éster (2R,3S,4R,5S,6S)-4,5-diacetóxi-6-[4- cloro-3-(4-etóxi-benzil)-fenil]-2-metilsulfanil-tetraidro-piran-3-ílico de ácido acético. Triol da etapa A (45 mg, 0,11 mmol) foi tratado com anidrido acético (60 □I, 0,64 mmol) em piridina (0,5 ml) durante 16 horas. A reação foi diluída com Et2O, lavada com 1 M NaHSO4aq., H2O, NaHCO3 sat. aq., e salmoura (com extração de retorno), secada por MgSO4, filtrada, e concentrada sob vácuo. O resíduo foi purificado por cromatografia instantânea (4 g SiO2, 0 - 25% EtOAc/Hex), colocada em suspensão em H2O, e liofilizada em éster (2R,3S,4R,5S,6S)-4,5-diacetóxi-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-2- metilsulfanil-tetraidro-piran-3-ílico de ácido acético (46 mg, 0,087 mmol, 79%) como um sólido branco.
Exemplo 15: Síntese de (2S,3R,4R,5S,6R)-2-(4-Cloro-3-(4-etóxi-benzil)- fenil]-6-metanossulfonil-tetraidro-piran-3,4,5-triol

1H RMN (400 MHz, metanol) δ ppm 7,28 (m, 1 H), 7,27 (m, 2 H), 7,10 (d, J = 8,4 Hz, 2 H), 6,81 (d, J = 8,4 Hz, 2 H), 4,53 (d, J = 9,6 Hz, 1 H), 4,30 (d, J = 9,6 Hz, 1 H), 4,00 (m, 4 H), 3,88 (t, J = 9,1 Hz, 1 H), 3,55 (t, J = 9,1 Hz, 1 H), 3,35 (t, J = 9,1 Hz, 1 H), 2,92 (s, 3 H), 1,36 (t, J = 6,8 Hz, 3 H); MS (ES+) [M + NH4]+ = 474.
6.16. Exemplo 16: Síntese de 1-((2S,3S,4S,5R)-2-[4-Cloro-3-(4-etóxi-benzil)- fenil]-3,4,5-triidróxi-piperidin-1-il}-etanona
1H RMN (400 MHz, MeOD) δ ppm 7,33 (d, J = 8,34 Hz, 1 H), 7,18 (dd, J = 8,46, 2,15 Hz, 1 H), 7,11 (d, J = 1,77 Hz, 1 H), 7,07 (d, J = 8,84 Hz, 2 H), 6,81 (d, J = 8,84 Hz, 2 H), 3,96 - 4,03 (m, 4 H), 3,83 - 3,89 (m, 1 H), 3,73 - 3,77 (m, 1 H), 3,55 - 3,59 (m, 1 H), 2,09 (br. s., 3 H), 1,36 (t, J = 6,95 Hz, 3 H); MS (ES+) [M + H]+ = 420.
6.17. Exemplo 17: Síntese de éster metílico de ácido (2S,3S,4S,5R)-2-[4- Cloro-3-(4-etóxi-benzil)-fenin-3,4,5-triidróxi-piperidina-1-carboxílico
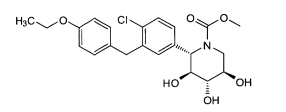
1H RMN (400 MHz, MeOD) δ ppm 7,32 (d, J = 8,34 Hz, 1 H), 7,15 (dd, J = 8,34, 2,02 Hz, 1 H), 7,10 (d, J = 2,27 Hz, 1 H), 7,04 - 7,09 (m, 2 H), 6,81 (d, J = 8,59 Hz, 2 H), 4,80 (d, J = 6,06 Hz, 1 H), 4,00 (q, J = 7,07 Hz, 5 H), 3,81 - 3,86 (m, 1 H), 3,70 - 3,73 (m, 1 H), 3,60 (s, 3 H), 3,54 - 3,59 (m, 1 H), 3,46 (dd, J = 14,40, 3,28 Hz, 1 H), 1,36 (t, J = 6,95 Hz, 3 H); MS (ES+) [M + H]+ = 436.
6.18. Exemplo 18: Síntese de alil amida de ácido (2S,3S,4S.5R)-2-[4-cloro-3- (4-etóxi-benzil)-fenin-3,4,5-triidróxi-piperidina-1-carboxílico

1H RMN (400 MHz, MeOD) δ ppm 7,32 (d, J = 8,1 Hz, 1 H), 7,16 - 7,20 (m, 2 H), 7,07 (d, J = 8,8 Hz, 2 H), 6,80 (d, J = 8,8 Hz, 2 H), 5,68 - 5,79 (m, J = 17,2, 10,2, 5,3, 5,2 Hz, 1 H), 4,92 - 5,00 (m, 2 H), 4,77 (d, J = 6,3 Hz, 1 H), 3,94 - 4,05 (m, 4 H), 3,86 (dd, J = 14,0, 3,4 Hz, 1 H), 3,69 - 3,81 (m, 3 H), 3,59 - 3,68 (m, 1 H), 3,56 (dd, J = 7,3, 5,1 Hz, 1 H), 3,47 (dd, J = 13,9, 3,5 Hz, 1 H), 1,36 (t, J = 6,9 Hz, 3 H); MS (ES+) [M + H]+ = 461.
6.19. Exemplo 19: Síntese de (2S,3S,4S,5R)-2-[4-Cloro-3-(4-etóxi-benzil)- fenil]-1-metil-piperidina-3,4,5-triol

1H RMN (400 MHz, MeOD) δ ppm 7,35 (d, J = 8,1 Hz, 1 H), 7,20 - 7,24 (m, 1 H), 7,17 (dd, J = 8,2, 1,9 Hz, 1 H), 7,09 (d, J = 8,6 Hz, 2 H), 6,80 (d, J = 8,6 Hz, 2 H), 4,03 (s, 2 H), 3,99 (q, J = 7,1 Hz, 2 H), 3,64 (ddd, J = 10,5, 9,2, 4,8 Hz, 1 H), 3,33 - 3,37 (m, 1 H), 3,21 (t, J = 9,0 Hz, 1 H), 3,03 (dd, J = 11,1, 4,8 Hz, 1 H), 2,74 (d, J = 9,3 Hz, 1 H), 2,15 (t, J = 10,9 Hz, 1 H), 1,95 (s, 3 H), 1,36 (t, J = 6,9 Hz, 3 H); MS (ES+) [M + H]+ = 392.
6.20. Exemplo 20: Síntese de (2S,3S,4R,5R,6R)-2-[3-(4-Etóxi-benzil)-fenil]-6- hidroximetil-1-metil-piperidina-3,4,5-triol

- A. Preparação de (3R,4S,5R,6R)-3,4,5-tris(benzilóxi)-6-(ben- ziloximetil) tetraidro-2H-piron-2-ona. Tetra-O-benzil-D-glicopiranose (2,07 g, 3,8 mmols) foi dissolvido em DMSO (10,1 ml). A esta mistura foi adicionado anidrido acético (7,0 ml) e agitada em temperatura ambiente durante a noite. À mistura de reação gelo foi adicionado e agitada durante 1 h. A mistura foi extraída com éter (3 x 20 ml). O extrato foi lavado com água (2x10 ml), bicarbonato de sódio aquoso (2 x 10 ml), salmoura, secado (sulfato de sódio) e concentrado sob vacuo. A cromatografia de coluna em sílica-gel instantânea com 0 - 25% etilacetato/Hexano resultou em 1,712 g de (3R,4S,5R,6R)- 3,4,5-tris(benzilóxi)-6-(benziloximetil) tetraidro-2H-piron-2-ona (83%).
- B. Preparação de (3R,4S,5R,6R)-3,4,5-tris(benzilóxi)-6-(benzi- loximetil)-2-(4-cloro-3-(4-etoxibenzil)fenil)tetraidro-2H-piran-2-ol. n-Butil lítio (2,5N em hexano) (1,263 ml, 3,16 mmols) foi adicionado por gotejamento a uma solução de composto do Exemplo 1, etapa C (1,028 g, 3,16 mmols) em THF anidro (15 ml) em -78 °C. Após agitação durante 30 min em -78°C, uma solução do composto da etapa A (1,7 g, 3,16 mmols) em THF anidro (10 ml) foi adicionada por gotejamento e agitada durante 1 h enquanto se deixa a- quecer para a temperatura ambiente. Cloreto de amónio aquoso (10 ml) foi adicionado à mistura de reação, THF removido sob vácuo, e a camada a- quosa extraída com acetato de etila (2 x 20 ml). As fases orgânicas combinadas lavadas com salmoura, secadas (sulfato de sódio) e concentradas sob vácuo. A mistura bruta purificada por cromatografia de coluna em sílica-gel instantânea com 0 - 20% acetato de etila/Hexano para fornecer 712 mg de (3R,4S,5R,6R)-3,4,5-tris(benzilóxi)-6-(benziloximetil)-2-(4-cloro-3-(4- etoxibenzil)fenil)tetraidro-2H-piran-2-ol (29%). M + H2O = 802,1.
- C. Preparação de (2R,3R,4S)-2,3,4,6-tetraguis(benzilóxi)-1-(4- cloro-3-(4-etoxibenzil)fenil)hexano-1,5-diona. A uma solução agitada de reagente Dess-Martin (500 mg, excesso) em CH2CI2 (10 ml) foi adicionado o composto da etapa B (500 mg, 0,6 mmol)) em diclorometano anídrico(10 ml) e agitada durante a noite. A mistura de reação extinta com 1N hidróxido de sódio (3 ml), extraída com diclorometano (2 x10 ml), as frações orgânicas combinadas foram lavadas com salmoura, secadas por sulfato de sódio, concentradas sob pressão reduzida para obter 487 mg de produto bruto. (M + H2O = 800,1).
- D. Preparação de (3R,4R,5S)-3,4,5-tris(benzilóxi)-2-(benziloxi- metil)-6-(4-cloro-3-(4-etoxibenzi)fenil)piperidina. Uma solução do composto da etapa C (400 mg, 0,5 mmol), 7N amônia em MeOH (1,0 ml) e peneiras moleculares 4 A recentemente ativadas (250 mg) em diclorometano (20 ml) foram submetidas a refluxo durante a noite. A mistura de reação foi esfriada para a temperatura ambiente, depois cianoboroidreto de sódio (160 mg, 2,55 mmols) foi adicionado e submetido a refluxo durante 2 h adicionais. A mistura de reação foi filtrada, diluída com diclorometano (20 ml), lavada com água, salmoura, secada (sulfato de sódio), e concentrada sob pressão reduzida. A cromatografia em sílica-gel (gradiente 50 a 100% acetonitrila contendo 0,1% acetato de amônio/água) forneceu (3R,4R,5S)-3,4,5-tris(benzilóxi)-2-(benzi- loximetil)-6-(4-cloro-3-(4-etoxibenzil)fenil)piperidina (136 mg, 34%).
- E. Preparação de (3R,4R,5S)-3,4,5-tris(benzilóxi)-2-(benziloxi- metil)-6-(4-cloro-3-(4-etoxibenzil)fenil)-1-metilpiperidina. Composto da etapa D (50 mg, 0,065 mmol) foi dissolvido em acetonitrila (1 ml) e tratado com carbonato de potássio (18 mg, 0,13 mmol) durante 30 minutos A esta mistura iodometano (20 uL, 0,32 mmol) foi adicionado e agitado durante a noite. A mistura de reação foi diluída com acetato de etila (10 ml), lavada com água, salmoura, secada (sulfato de sódio), e concentrada sob vácuo. A cromatografia em sílica-gel (gradiente 50 a 100% acetonitrila contendo 0,1% acetato de amônio/água) forneceu (3R,4R,5S)-3,4,5-tris(benzilóxi)-2-(benziloximetil)- 6-(4-cloro-3-(4-etoxibenzil) fenil)-1-metilpiperidina (29 mg, 56%). MH+ 782,1.
- F. Preparação de (2S,3S,4R,5R,6R)-2-[3-(4-etóxi-benzil)-fenil1-6- hidroximetil-1-metil-piperidina-3,4,5-triol. O composto da etapa E (50 mg) em metanol e ácido acético (25 uL) foi tratado 5% líquido Pd-C (10 mg) sob atmosfera de H2 durante 4 h. A mistura de reação foi filtrada através de um tampão de celite e concentrada. A cromatografia em sílica-gel (gradiente 10 a 100% acetonitrila contendo 0,1% acetato de amônio/água) forneceu (2S,3S,4R,5R,6R)-2-[3-(4-etóxi-benzil)-fenil]-6-hidroximetil-1-metil-piperidina- 3,4,5-triol (6 mg, 70%).
6.21. Exemplo 21: Síntese de (2S,3R,4R,5S,6R)-2-(4-cloro-3-(4-etoxibenzil) fenil)-6-metoxitetraidro-2H-tiopiran-3,4,5-triol

- A. Preparação de benzotioato de (S-(1S)-((3aS,6S,6aS)-6-(terc- butildimetilsililóxi)-2,2-dimetiltetraidrofuro[2,3-d][1,3]dioxol-5-il)(4-cloro-3-(4- etoxibenzil)fenil) metila. Dietilazodicarboxilato (150 μL, 0,914 mmol) foi adicionado a uma solução de trifenilfosfina (240 mg, 0,914 mmol) em 1,0 ml de THF em temperatura ambiente. Após uma hora, o epímero C5 do Exemplo 1, etapa D (167 mg, 0,305 mmol) foi adicionado em 0,5 ml de THF através de seringa e foi seguido pela adição de ácido tiobenzóico (110 μL, 0,914 mmol) através de seringa. Esta solução laranja foi agitada durante 22 horas em temperatura ambiente. Após remoção dos solventes in vacuo, o resíduo foi purificado por cromatografia instantânea (gradiente 0 a 10% acetato de etila/hexanos) para fornecer o composto do título como um óleo amarelo claro (104 mg, 50% rendimento). MS (ES+) [M + NH4]+ = 566.
- B. Preparação de (2S,3R,4R,5S,6R)-2-(4-cloro-3-(4-etoxibenzil) fenil)-6-metoxitetraidro-2H-tiopiran-3,4,5-triol. Metóxido de sódio (0,3 ml de uma 4,3M solução em metanol) foi adicionado a uma solução do composto da etapa A (104 mg, 0,152 mmol) em 6 ml de metanol. Após 30 minutos, a reação foi diluída com 20 ml de acetato de etila e lavada com água e salmoura (20 ml cada). A camada orgânica foi secada com sulfato de magnésio, filtrada e os solventes removidos in vacuo. O resíduo foi purificado rapidamente por cromatografia instantânea (5% acetato de etila/hexanos) e o produto foi levado adiante diretamente para impedir a formação de dissulfe- to.
1H RMN (400 MHz, acetona-d6) δ ppm 7,33 (m, 2 H), 7,25 (dd, J = 2,27, 8,34 Hz, 1 H), 7,13 (d, J = 8,59 Hz, 2 H), 6,82 (d, J = 8,59 Hz, 2 H), 4,48 (d, J = 3,03 Hz, 1 H), 4,02 (s, 2 H), 3,99 (q, J = 7,07 Hz, 2 H), 3,91 (d, J = 10,36 Hz, 1 H), 3,80 - 3,85 (m, 2 H), 3,68 (dd, J = 8,37, 9,35 Hz, 1 H), 3,42 (s, 3 H), 1,33 (t, J = 7,07 Hz, 3 H). MS (ES+) [M + NH4]+ = 424.
6.22. Exemplo 22: Síntese de (2S,3S,4R,5R,6R)-2-[4-Cloro-3-(4-etóxi- benzil)-fenil]-6-hidroximetil-piperidina-3,4,5-triol

- A. Preparação de (2R,3R,4S,5R,6S)-3,4,5-tris-alilóxi-2-aliloxi- metil-6-metóxi-tetraidro-pirano. A uma solução de α-D-metilglicosídeo (3 g, 15,45 mmols) em DMF (50 ml) foi adicionado NaH (60% dispersão em óleo mineral, 3,34 g, 0,14 mol). Durante esta adição, uma suspensão espessa se forma e uma quantidade adicional de DMF (15 ml) foi adicionada para recobrar em solução. Após agitação em temperatura ambiente durante 30 minutos, a mistura foi esfriada para 0°C e brometo de alila (17 g, 0,14 mol, 12 ml) foi adicionado lentamente. A mistura foi depois deixada aquecer para a temperatura ambiente e agitada durante 18 horas. MeOH foi cuidadosamente adicionado à mistura marrom clara para extinguir o NaH em excesso e depois a mistura foi concentrada. O resíduo foi diluído com CH2CI2 e lavado com H2O, secado (MgSO4) e concentrado para proporcionar um óleo amarelo. A purificação por cromatografia em sílica-gel (20% EtOAc/hexanos) proporcionou (2R,3R,4S,5R,6S)-3,4,5-tris-alilóxi-2-aliloximetil-6-metóxi-tetraidro- pirano (4,06 g, 11,47 mmols, 74%) como um óleo incolor. TLC: Rf = 0,20, 20% EtOAc/hexanos.
- B. Preparação de (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6-aliloximetil- tetraidro-piran-2-ol. Uma solução de composto da etapa A (10g, 0,028 mol) em AcOH (400 ml) foi aquecida para 90°C. TfOH (2 N solução em H2O, 16,69 g, 0,112 mol, 56 ml) foi adicionado e a mistura agitada em 90 °C durante 75 minutos. A solução foi esfriada e diluída com CH2CI2, lavada com H2O (x3), NaHCO3 sat., secada (MgSO4) e concentrada para fornecer um sólido amarelo. A purificação por cromatografia em sílica-gel (20% - 40% EtOAc/hexanos) proporcionou (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6-aliloximetil- tetraidro-piran-2-ol como um mistura de anômeros (5,85 g, 17,2 mmols, 61%) como um sólido branco. TLC: Rf = 0,40, 40% EtOAc/hexanos.
- C. Preparação de (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6-aliloximetil- tetraidro-piran-2-ona. Cloreto de oxalila (2,75 g, 21,7 mmols, 1,89 ml) foi dissolvido em CH2CI2 (90 ml) e a mistura esfriada para -78 °C. DMSO (3,39 g, 43,4 mmols, 3,08 ml) foi adicionado como uma solução em CH2CI2 (60 ml). A mistura foi agitada em -78°C durante 15 minutos e depois o composto da etapa B (6,70 g, 19,7 mmols) foi adicionado como uma solução em CH2CI2 (150 ml). A mistura de reação foi agitada durante mais 15 minutos em -78°C e Et3N (9,97 g, 98,5 mmols, 13,7 ml) adicionado. A mistura foi agitada em 78°C durante mais 5 minutos e depois deixada aquecer para a temperatura ambiente durante 30 minutos. A reação foi extinta com H2O e a camada orgânica separada, lavada duas vezes com H2O, secada e concentrada para fornecer um óleo amarelo-pálido. A purificação por cromatografia em sílica- gel (15% EtOAc/hexanos) proporcionou (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6- aliloximetil-tetraidro-piran-2-ona (2,49 g, 7,37 mmols, 37%) como um óleo incolor. TLC: Rf = 0,40, 20% EtOAc/hexanos.
- D. Preparação de (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6-aliloximetil-2- [4-cloro-3-(4-etóxi-benzil)-fenil]-tetraidro-piran-2-ol. O composto do Exemplo 1, Etapa C (2,37 g, 7,31 mmols) foi dissolvido em THF (25 ml) e esfriado para -78°C. n-BuLi (2,5 N solução em hexanos, 0,47 g, 7,31 mmols, 2,92 ml) foi adicionado por gotejamento e a solução agitada durante 15 minutos. O composto da etapa C (2,47 g, 7,31 mmols) foi adicionado como uma solução em THF (25 ml) e a mistura de reação agitada em -78°C durante mais 15 minutos antes de ser deixada aquecer para a temperatura ambiente durante 30 minutos. A reação foi extinta com NH4CI sat. e a camada orgânica separada. A camada aquosa foi extraída outra vez com Et2O e os orgânicos combinados secados e concentrados para fornecer um óleo amarelo. A purificação por cromatografia em sílica-gel (10% - 20% EtOAc/hexanos) proporcionou (3R,4S,5R,6R)-3,4,5-tris-alilóxi-6-aliloximetil-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-tetraidro-piran-2-ol (0,95 g, 1,63 mmol, 22%) como um óleo incolor. MS (ES+) [M + NH4]+ = 602.
- E. Preparação de (2R,3R,4S)-2,3,4,6-tetraguis-alilóxi-1-[4-cloro- 3-(4-etóxi-benzil)-fenill-hexano-1,5-diona. A uma solução do composto da etapa D (0,93 g, 1,59 mmol) em CH2CI2 (25 ml) foi adicionado periodinano Dess-Martin (0,68 g, 1,59 mmol). A mistura foi agitada em temperatura ambiente durante 1 hora e depois uma segunda porção de periodinano Dess- Martin (1 eqiv.) foi adicionada. A agitação continuou durante mais uma hora e depois a reação foi extinta com 1N NaOH (~4 ml). H2O foi adicionado e a camada orgânica separada. A camada aquosa foi extraída outra vez com CH2CI2, secada e concentrada para fornecer um sólido ceroso amarelo. A purificação por cromatografia em sílica-gel (15% - 20% EtOAc/hexanos) pro-porcionou (2R,3R,4S)-2,3,4,6-tetraquis-alilóxi-1-[4-cloro-3-(4-etóxi-benzil)- fenil]-hexano-1,5-diona (0,60 g, 1,03 mmol, 65%) como um sólido branco. MS (ES+) [M + NH4]+ = 600.
- F. Preparação de (2R,3R,4R,5S,6S)-3,4,5-tris-alilóxi-2-aliloxime- til-6-r4-cloro-3-(4-etóxi-benzil)-fenil]-piperidina. A uma solução do composto da etapa E (0,60 g, 1,03 mmol) em MeOH (12 ml) foi adicionado 4 A MS seguido por formiato de amônio (0,13 g, 2,06 mmols). NaBH3CN (0,14 g, 2,3 mmols) foi depois adicionado de uma vez e a mistura agitada em temperatura ambiente durante 1 hora 30 minutos. A mistura de reação foi depois filtrada e concentrada. A purificação por cromatografia em sílica-gel (10% - 20% EtOAc/hexanos) proporcionou (2R,3R,4R,5S,6S)-3,4,5-tris-alilóxi-2-aliloxi- metil-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-piperidina (155 mg, 0,27 mmol, 27%). MS (ES+) [M + H]+ = 568.
- G. Preparação de (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-ben- zil)-fenin-3,4,5-tris-[((E)-propenil)óxi]-6-[((E)-propenil)oximetill-piperidina. lr(COD)[PCH3Ph2]PF6 (8 mg, 30% em mol) em THF (0,3 ml) foi agitado sob uma atmosfera de H2 até que a cor mudou de vermelho para amarelo-pálido (~5 minutos). O composto da etapa F (19 mg, 0,033 mol) em THF (0,5 ml) foi depois adicionado e a mistura agitada em temperatura ambiente durante 45 minutos e depois concentrada. A purificação por cromatografia em sílica-gel (20% EtOAc/hexanos) proporcionou (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi- benzil)-fenil]-3,4,5-tris-[((E)-propenil)óxi]-6-[((E)-propenil)oximetil]-piperidina (15 mg, 0,026 mmol, 80%) como um óleo incolor. MS (ES+) [M + H]+ = 568.
- H. Preparação de (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenin-6-hidroximetil-piperidina-3,4,5-triol. O composto da etapa G (15 mg, 0,026 mmol) foi dissolvido em uma solução de THF/AcOH/1NHCI (0,2 ml:0,3 ml:0,15 ml) e aquecido para 70°C durante 30 minutos. A mistura foi concentrada para fornecer um óleo amarelo claro. A purificação por HPLC preparativa (Sunfire C18, 30 x 100 mm, 5 μm, 10% - 100% B durante 15 minutos) proporcionou (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6-hidroxi- metil-piperidina-3,4,5-triol (5 mg, 0,012 mmol, 46%) como um sólido branco. MS (ES+) [M + H]+ = 408.
6.23. Exemplo 23: Síntese de (2S,3S,4R,5R,6R)-2-[4-Cloro-3-(4-etóxi- benzil)-fenil]-6-hidroximetil-1-metil-piperidina-3,4,5-triol
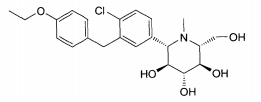
- A. Preparação de (2R,3R,4R,5S,6S)-3,4,5-tris-alilóxi-2-aliloxi- metil-6-[4-cloro-3-(4-etóxi-benzil)-fenil]-1-metil-piperidina. A uma solução do composto do Exemplo 22, Etapa F (135 mg, 0,24 mmol) em MeCN foi adicionado K2CO3 (164 mg, 1,19 mmol). A mistura foi agitada durante 30 minutos e depois Mel (676 mg, 4,76 mmols) foi adicionado. A agitação continuou em temperatura ambiente durante 8 horas, depois a mistura foi filtrada e concentrada. A purificação por cromatografia em sílica-gel (10% EtO- Ac/hexanos) proporcionou (2R,3R,4R,5S,6S)-3,4,5-tris-alilóxi-2-aliloximetil-6- [4-cloro-3-(4-etóxi-benzil)-fenil]-1-metil-piperidina (90 mg, 0,15 mmol, 65%) como um óleo incolor. MS (ES+) [M + H]+ = 582.
- B. Preparação de (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-1-metil-3,4,5-tris-[((E)-propenil)óxi]-6-[((E)-propenil)oximetil]-piperidina. lr(COD)[PCH3Ph2]PF6 (27 mg, 30% em mol) em THF (1 ml) foi agitado sob uma atmosfera de H2 até que a cor mudou de vermelho para amarelo-pálido (~5 minutos). O composto da etapa A (62 mg, 0,11 mol) em THF (1,5 ml) foi depois adicionado e a mistura agitada em temperatura ambiente durante 45 minutos e depois concentrada. A purificação por cromatografia em sílica-gel (20% EtOAc/hexanos) proporcionou (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi- benzil)-fenil]-1-metil-3,4,5-tris-[((E)-propenil)óxi]-6-[((E)-propenil)oximetil]- piperidina (62 mg, 0,11 mmol, 100%) como um óleo incolor. MS (ES+) [M + H]+ = 582.
- C. Preparação de (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-benzil)- fenil]-6-hidroximetil-1-metil-piperidina-3,4,5-triol. O composto da etapa B (54 mg, 0,093 mmol) foi dissolvido em uma solução de THF/AcOH/1 NHCI (0,5 ml:0,6 ml:0,30 ml) e aquecido para 70°C durante 30 minutos. A mistura foi 5 concentrada para fornecer um óleo amarelo-pálido. A purificação por HPLC preparativa (Sunfire C18, 30 x 100 mm, 5 μm, 10% - 100% B durante 15 minutos) proporcionou (2S,3S,4R,5R,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6- hidroximetil-1-metil-piperidina-3,4,5-triol (22 mg, 0,052 mmol, 56%) como um sólido branco. MS (ES+) [M + H]+ = 422.




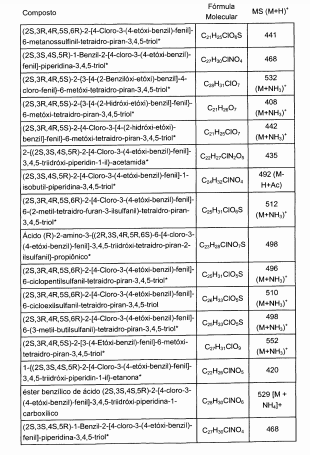

y = A + ((B-A)/(1+((C/x)^D)))
em que A é o valor y mínimo; B é o valor y máximo; C é a IC50; e D é a inclinação. O cálculo da IC50 é executado usando o software XLFit4 (ID Business Solutions Inc., Bridgewater, NJ 08807) para Microsoft Excel (a equação acima é model 205 deste software).
Claims (19)
- Composto, caracterizado pelo fato de que apresenta a fórmula:ou um sal farmaceuticamente aceitável deste, em que:
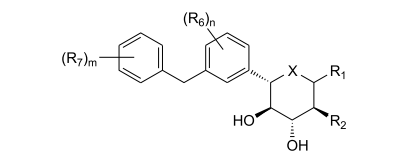
X é O, S ou NR3;
quando X for O, R1 é OR1A, SR1A, SOR1A, SO2R1A ou N(R1A)2; quando X for S, R1 é hidrogênio, OR1A, SR1A, SOR1A, ou SO2R1A;
quando X for NR3, R1 é OR1A, SR1A, SOR1A, SO2R1A, ou R1A; cada R1A é independentemente hidrogênio, alquila, arila ou heterociclo;
R2 é flúor ou OR2A;
cada um de R2A, R2B, e R2C é independentemente hidrogênio, alquila, C(O)alquila, C(O)arila ou arila;
R3 é hidrogênio, C(O)R3A, CO2R3A, CON(R3b)2, alquila, arila ou heterociclo;
cada R3A é independentemente alquila ou arila;
cada R3B é independentemente hidrogênio ou alquila ou arila;
cada R6 é independentemente hidrogênio, hidróxi, halogênio, amino, ciano, nitro, C≡CR6A, OR6A, SR6A, SOR6A, SO2R6A, C(O)R6A, CO2R6A, CO2H, CON(R6A)(R6A), CONH(R6A), CONH2, NHC(O)R6A, NHSO2R6A, alquila, arila, ou heterociclo;
cada R6a é independentemente alquila, arila ou heterociclo;
cada R7 é independentemente hidrogênio, hidróxi, halogênio, amino, ciano, nitro, C≡CR 7 a, OR7A, SR7A, SOR7A, SO2R7A, C(O)R 7 A, CO2R7A, CO2H, CON(R 7 A)(R 7 A), CONH(R 7 A), CONH2, NHC(O)R 7 A, NHSO2R7A, alquila, arila ou heterociclo;
cada R7A independentemente alquila, arila ou heterociclo;
m é 1 a 3;
n é 1 a 3;
"alquila" significa um hidrocarboneto de cadeia reta, ramificada e/ou cíclica tendo de 1 a 20 átomo de carbono; e
"arila" significa um anel aromático ou um sistema de anel aromático ou parcialmente aromático composto de átomos de carbono e hidrogênio. - Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R1 é OR1A.
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R1 é SR1A
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R1 é SOR1A.
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R1 é SO2R1A
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R1 é hidrogênio ou R1A
- Composto de acordo com qualquer uma das reivindicações 2 a 6, caracterizado pelo fato de que R1A ou pelo menos um R1A é hidrogênio ou alquila tendo de 1 a 4 átomos de carbono.
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R6 é hidrogênio, hidróxi, halogênio, OR6A ou alquila tendo de 1 a 4 átomos de carbono.
- Composto de acordo com a reivindicação 3, caracterizado pelo fato de que R7 é hidrogênio, C≡CR7A, OR7A ou alquila tendo de 1 a 4 átomos de carbono.
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que é da fórmula:
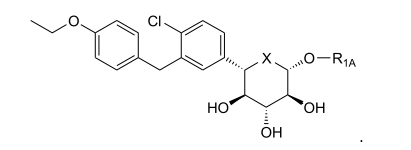
- Composto de acordo com a reivindicação 2, caracterizado pelo fato de que é da fórmula:

- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que é da fórmula:

- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que é da fórmula:

- Composto de acordo com a reivindicação 3, caracterizado pelo fato de que é da fórmula:ou da fórmula:


- Composto de acordo com qualquer uma das reivindicações 10 a 13, caracterizado pelo fato de que X é O.
- Composto de acordo com qualquer uma das reivindicações 10 a 15, caracterizado pelo fato de que R1Aé hidrogênio, metila ou etila.
- Composto de acordo com a reivindicação 1, caracterizado pelo fato de que é (2S,3R,4R,5S,6R)-2-[4-cloro-3-(4-etóxi-benzil)-fenil]-6- metilsulfanil-tetrahidro-piran-3,4,5-triol ou um sal farmaceuticamente aceitável do mesmo.
- Composição farmacêutica, caracterizada pelo fato de que compreende um composto como definido em qualquer uma das reivindicações 1 a 17, e um diluente ou excipiente farmaceuticamente aceitável.
- Composição farmacêutica de acordo com a reivindicação 18, caracterizada pelo fato de que compreende (2S,3R,4R,5S,6R)-2-[4-cloro-3- (4-etóxi-benzil)-fenil]-6-metilsulfanil-tetrahidro-piran-3,4,5-triol ou um sal farmaceuticamente aceitável do mesmo, e um diluente ou excipiente farmaceuticamente aceitável.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US84815606P | 2006-09-29 | 2006-09-29 | |
| US60/848,156 | 2006-09-29 | ||
| US90571407P | 2007-03-08 | 2007-03-08 | |
| US60/905,714 | 2007-03-08 | ||
| US94878007P | 2007-07-10 | 2007-07-10 | |
| US60/948,780 | 2007-07-10 | ||
| PCT/US2007/079654 WO2008042688A2 (en) | 2006-09-29 | 2007-09-27 | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0717156A2 BRPI0717156A2 (pt) | 2013-10-15 |
| BRPI0717156B1 true BRPI0717156B1 (pt) | 2020-06-30 |
| BRPI0717156B8 BRPI0717156B8 (pt) | 2021-05-25 |
Family
ID=38947681
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0717156A BRPI0717156B8 (pt) | 2006-09-29 | 2007-09-27 | inibidores do co-transportador 2 da glicose de sódio e composição farmacêutica que os compreende |
Country Status (24)
| Country | Link |
|---|---|
| US (5) | US7781577B2 (pt) |
| EP (2) | EP2308841B1 (pt) |
| JP (3) | JP5283625B2 (pt) |
| KR (2) | KR101492277B1 (pt) |
| AR (1) | AR063047A1 (pt) |
| AT (1) | ATE496888T1 (pt) |
| AU (1) | AU2007304971B2 (pt) |
| BR (1) | BRPI0717156B8 (pt) |
| CA (1) | CA2664688C (pt) |
| DE (1) | DE602007012292D1 (pt) |
| DK (2) | DK2089361T5 (pt) |
| EA (1) | EA016511B1 (pt) |
| ES (1) | ES2477216T3 (pt) |
| FR (1) | FR19C1053I2 (pt) |
| HU (1) | HUS1900038I1 (pt) |
| IL (1) | IL197836A (pt) |
| MX (1) | MX2009003305A (pt) |
| NL (1) | NL301003I2 (pt) |
| NO (1) | NO345139B1 (pt) |
| NZ (1) | NZ575811A (pt) |
| PL (2) | PL2308841T3 (pt) |
| PT (2) | PT2089361E (pt) |
| TW (1) | TWI499414B (pt) |
| WO (1) | WO2008042688A2 (pt) |
Families Citing this family (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI499414B (zh) * | 2006-09-29 | 2015-09-11 | Lexicon Pharmaceuticals Inc | 鈉與葡萄糖第2型共同運輸體(co-transporter 2)的抑制物與其應用方法 |
| WO2008109591A1 (en) * | 2007-03-08 | 2008-09-12 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| AU2008279424B2 (en) * | 2007-07-26 | 2013-06-13 | Lexicon Pharmaceuticals, Inc. | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
| EP2025674A1 (de) | 2007-08-15 | 2009-02-18 | sanofi-aventis | Substituierte Tetrahydronaphthaline, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| AU2009270936B2 (en) | 2008-07-15 | 2014-12-18 | Theracos, Inc. | Deuterated benzylbenzene derivatives and methods of use |
| TWI472521B (zh) * | 2008-07-17 | 2015-02-11 | Lexicon Pharmaceuticals Inc | (2s,3r,4r,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(甲硫)四氫-2h-哌喃-3,4,5-三醇的固體形態與其使用方法 |
| EA021983B1 (ru) | 2009-11-02 | 2015-10-30 | Пфайзер Инк. | Производные диоксабицикло[3.2.1]октан-2,3,4-триола |
| WO2011070592A2 (en) | 2009-12-09 | 2011-06-16 | Panacea Biotec Ltd. | Novel sugar derivatives |
| TWI562775B (en) * | 2010-03-02 | 2016-12-21 | Lexicon Pharmaceuticals Inc | Methods of using inhibitors of sodium-glucose cotransporters 1 and 2 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| US8530413B2 (en) | 2010-06-21 | 2013-09-10 | Sanofi | Heterocyclically substituted methoxyphenyl derivatives with an oxo group, processes for preparation thereof and use thereof as medicaments |
| TW201215387A (en) | 2010-07-05 | 2012-04-16 | Sanofi Aventis | Spirocyclically substituted 1,3-propane dioxide derivatives, processes for preparation thereof and use thereof as a medicament |
| TW201221505A (en) | 2010-07-05 | 2012-06-01 | Sanofi Sa | Aryloxyalkylene-substituted hydroxyphenylhexynoic acids, process for preparation thereof and use thereof as a medicament |
| TW201215388A (en) | 2010-07-05 | 2012-04-16 | Sanofi Sa | (2-aryloxyacetylamino)phenylpropionic acid derivatives, processes for preparation thereof and use thereof as medicaments |
| WO2012025857A1 (en) * | 2010-08-23 | 2012-03-01 | Hetero Research Foundation | Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors |
| CN102453026A (zh) | 2010-10-27 | 2012-05-16 | 上海艾力斯医药科技有限公司 | C-芳基葡糖苷衍生物、制备方法及其应用 |
| TWI631963B (zh) * | 2011-01-05 | 2018-08-11 | 雷西肯製藥股份有限公司 | 包含鈉-葡萄糖共同輸送體1與2之抑制劑的組合物與應用方法 |
| WO2012165914A2 (en) | 2011-06-01 | 2012-12-06 | Green Cross Corporation | Novel diphenylmethane derivatives as sglt2 inhibitors |
| BR112013031032A2 (pt) | 2011-06-03 | 2016-11-29 | Boehringer Ingelheim Int | inibidores de sglt-2 para o tratamento de distúrbios metabólicos em pacientes tratados com agentes neurolépticos |
| WO2012172566A2 (en) * | 2011-06-13 | 2012-12-20 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9018249B2 (en) | 2011-09-13 | 2015-04-28 | Panacea Biotec Limited | SGLT inhibitors |
| EP2760862B1 (en) | 2011-09-27 | 2015-10-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| PL3489226T3 (pl) * | 2012-11-20 | 2021-08-02 | Lexicon Pharmaceuticals, Inc. | Inhibitory kotransportera glukozowo-sodowego 1 |
| EP2952206B1 (en) | 2013-02-04 | 2018-10-24 | Taisho Pharmaceutical Co., Ltd. | Prophylactic or therapeutic drug for constipation |
| CN103073606B (zh) * | 2013-02-05 | 2016-05-18 | 中国医药研究开发中心有限公司 | 5’-s-(4,4’-二甲氧基三苯甲基)-2’-脱氧肌苷的合成与制备方法 |
| DK2981269T3 (da) * | 2013-04-04 | 2023-10-23 | Boehringer Ingelheim Vetmedica Gmbh | Behandling af stofskifteforstyrrelser hos hestedyr |
| MX376097B (es) | 2013-12-17 | 2025-03-07 | Boehringer Ingelheim Vetmedica Gmbh | Inhibidores de sglt2 para usarse en el tratamiento o prevención de trastornos metabólicos en animales felinos. |
| CA2932674C (en) | 2014-01-23 | 2023-01-24 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in canine animals |
| FI3721882T3 (fi) | 2014-04-01 | 2024-09-24 | Boehringer Ingelheim Vetmedica Gmbh | Aineenvaihduntahäiriöiden hoito hevoseläimissä |
| EP4403230A3 (en) | 2014-09-25 | 2025-01-08 | Boehringer Ingelheim Vetmedica GmbH | Combination treatment of sglt2 inhibitors and dopamine agonists for preventing metabolic disorders in equine animals |
| AU2016310535B2 (en) | 2015-08-27 | 2021-08-19 | Boehringer Ingelheim Vetmedica Gmbh | Liquid pharmaceutical compositions comprising SGLT-2 inhibitors |
| JP6727419B2 (ja) * | 2016-05-25 | 2020-07-22 | クリスタル ファーマシューティカル(スーチョウ)カンパニー,リミテッド | ナトリウム−グルコース共輸送体阻害薬の新規な結晶形及びその製造方法並びに用途 |
| BR112019005930A2 (pt) | 2016-10-19 | 2019-06-11 | Boehringer Ingelheim Int | combinações compreendendo um inibidor de ssao/vap-1 e um inibidor de sglt2, e usos das mesmas |
| JP6917527B2 (ja) | 2018-01-05 | 2021-08-11 | シャンドン ダンホン ファーマスーティカル カンパニー リミテッドShandong Danhong Pharmaceutical Co., Ltd. | Sglt阻害剤及びその応用 |
| WO2019166958A1 (en) * | 2018-02-28 | 2019-09-06 | Mylan Laboratories Ltd | Process for the preparation of sotagliflozin |
| US20210113561A1 (en) | 2018-04-17 | 2021-04-22 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition, methods for treating and uses thereof |
| US20210238170A1 (en) * | 2018-05-09 | 2021-08-05 | Janssen Pharmaceutica Nv | 5,5-Difluoro- and 5-Fluoro-5-Methyl-C-Glycoside Derivatives Useful As Dual SGLT1 / SGLT2 Modulators |
| WO2020039394A1 (en) | 2018-08-24 | 2020-02-27 | Novartis Ag | New drug combinations |
| US12213970B2 (en) | 2018-10-29 | 2025-02-04 | Boehringer Ingelheim International Gmbh | Pyridinyl sulfonamide derivatives, pharmaceutical compositions and uses thereof |
| DK3873600T5 (da) | 2018-10-29 | 2024-03-18 | Boehringer Ingelheim Int | Pyridinylsulfonamidderivater, farmaceutiske sammensætninger og anvendelser deraf |
| AU2020231322A1 (en) * | 2019-03-01 | 2021-09-02 | Lexicon Pharmaceuticals, Inc. | Use of sotagliflozin for the treatment of patients with type 1 diabetes mellitus |
| KR102951944B1 (ko) | 2019-07-05 | 2026-04-10 | 산동 단홍 파머수티컬 컴퍼니., 리미티드. | SGLTs 억제제의 결정형 및 그 응용 |
| CN114585358B (zh) | 2019-07-26 | 2023-11-10 | 东宝紫星(杭州)生物医药有限公司 | 一种SGLTs/DPP4抑制剂及其应用 |
| US12421269B2 (en) | 2019-07-26 | 2025-09-23 | Cgenetech (Suzhou, China) Co., Ltd. | SGLT2/DPP4 inhibitor and application thereof |
| EP3771480A1 (en) * | 2019-08-01 | 2021-02-03 | Lexicon Pharmaceuticals, Inc. | Continuous process for preparing the crystalline form ii of sotagliflozin |
| EP4064854A1 (en) | 2019-11-28 | 2022-10-05 | Boehringer Ingelheim Vetmedica GmbH | Use of sglt-2 inhibitors in the drying-off of non-human mammals |
| JP7423800B2 (ja) | 2020-02-17 | 2024-01-29 | ベーリンガー インゲルハイム フェトメディカ ゲーエムベーハー | ネコにおける心臓疾患の予防および/または治療のためのsglt-2阻害剤の使用 |
| CN111763197A (zh) * | 2020-07-08 | 2020-10-13 | 青岛大学 | 一种新型手性吲哚类化合物的合成方法 |
| EP4249479A4 (en) * | 2020-11-19 | 2024-12-18 | Beijing Increase Innovative Drug Co., Ltd. | GLUCOSIDE DERIVATIVE, ITS PREPARATION METHOD AND ITS APPLICATION |
| US20240269105A1 (en) | 2021-07-28 | 2024-08-15 | Boehringer Ingelheim Vetmedica Gmbh | Use of sglt-2 inhibitors for the prevention and/or treatment of hypertension in non-human mammals |
| CN117715639A (zh) | 2021-07-28 | 2024-03-15 | 勃林格殷格翰动物保健有限公司 | Sglt-2抑制剂用于预防和/或治疗非人哺乳动物中的肾脏病的用途 |
| MX2024001184A (es) | 2021-07-28 | 2024-02-27 | Boehringer Ingelheim Vetmedica Gmbh | Uso de inhibidores de sglt-2 para la prevencion y/o tratamiento de cardiopatias en mamiferos no humanos, que excluye felinos, en particular, caninos. |
| WO2023227492A1 (en) | 2022-05-25 | 2023-11-30 | Boehringer Ingelheim Vetmedica Gmbh | Aqueous pharmaceutical compositions comprising sglt-2 inhibitors |
| JP2026508912A (ja) | 2023-03-06 | 2026-03-13 | ベーリンガー インゲルハイム フェトメディカ ゲーエムベーハー | 液体医薬組成物、特に1種又は複数のsglt-2阻害剤を含む液体医薬組成物の送達のためのシステム |
| TW202508593A (zh) | 2023-05-24 | 2025-03-01 | 德商百靈佳殷格翰維美迪加股份有限公司 | 包含一或多種sglt-2抑制劑及匹莫苯坦(pimobendan)及/或替米沙坦(telmisartan)之非人類哺乳動物心臟病之組合治療及/或預防 |
| CN121175053A (zh) | 2023-05-24 | 2025-12-19 | 勃林格殷格翰动物保健有限公司 | 包含一或多种sglt-2抑制剂及替米沙坦的非人类哺乳动物中肾脏疾病和/或高血压的组合治疗和/或预防 |
| US20250108032A1 (en) | 2023-09-28 | 2025-04-03 | Lexicon Pharmaceuticals, Inc. | Methods of treating type 1 diabetes and kidney disease |
| US20250114323A1 (en) | 2023-10-06 | 2025-04-10 | Lexicon Pharmaceuticals, Inc. | Methods of reducing the risk of cardiovascular events in patients with left ventricular hypertrophy |
| WO2025160910A1 (en) * | 2024-02-01 | 2025-08-07 | New Wish Biotechnology Wuxi Co., Ltd. | Indazole compound and pharmaceutical composition, preparation method and use thereof |
Family Cites Families (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3870699A (en) * | 1973-03-06 | 1975-03-11 | Upjohn Co | Lincomycin analogs |
| CN1020944C (zh) | 1990-01-30 | 1993-05-26 | 阿图尔-费希尔股份公司费希尔厂 | 紧固件 |
| WO1995016049A1 (en) | 1993-12-08 | 1995-06-15 | The Scripps Research Institute | Catalytic chemo-enzymatic asymmetric synthesis of carbohydrates |
| US6214331B1 (en) * | 1995-06-06 | 2001-04-10 | C. R. Bard, Inc. | Process for the preparation of aqueous dispersions of particles of water-soluble polymers and the particles obtained |
| JP2885191B2 (ja) | 1996-07-09 | 1999-04-19 | 日本電気株式会社 | 液晶プロジェクタ |
| JP3928197B2 (ja) | 1997-01-23 | 2007-06-13 | 住友化学株式会社 | アリール置換芳香族類の製造方法 |
| US6664399B1 (en) | 1999-09-02 | 2003-12-16 | E. I. Du Pont De Nemours & Company | Triazole linked carbohydrates |
| PH12000002657B1 (en) | 1999-10-12 | 2006-02-21 | Bristol Myers Squibb Co | C-aryl glucoside SGLT2 inhibitors |
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| ES2254376T3 (es) | 2000-03-17 | 2006-06-16 | Kissei Pharmaceutical Co., Ltd. | Derivados glucopiranosiloxibencilbenceno, preparaciones medicinales que los contienen e intermediarios para la preparacion de los indicados derivados. |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US6653290B2 (en) | 2000-10-06 | 2003-11-25 | Bristol-Myers Squibb Company | Tumor proliferation inhibitors |
| AU2002223127A1 (en) | 2000-11-30 | 2002-06-11 | Kissei Pharmaceutical Co., Ltd. Intellectual Property | Glucopyranosyloxybenzyl benzene derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| PL209375B1 (pl) | 2000-12-28 | 2011-08-31 | Kissei Pharmaceutical | Pochodne glukopiranozyloksypirazolu, kompozycja farmaceutyczna zawierająca takie pochodne i zastosowanie tych pochodnych do wytwarzania kompozycji farmaceutycznej |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2002083066A2 (en) | 2001-04-11 | 2002-10-24 | Bristol-Myers Squibb Company | Amino acid complexes of c-aryl glucosides for treatment of diabetes and method |
| FR2826653B1 (fr) | 2001-06-29 | 2005-10-14 | Servier Lab | Nouveaux derives de pyrido-pyrido-pyrrolo[3,2-g]pyrrolo [3,4-e]-indole et pyrido-pyrrolo[2,3-a]pyrrolo[3,4-c] carbazole, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| WO2003011880A1 (fr) | 2001-07-31 | 2003-02-13 | Kissei Pharmaceutical Co., Ltd. | Derive de glucopyranosyloxybenzylbenzene, composition medicinale contenant ce derive, usage medicinal de cette composition et produit intermediaire pour produire cette composition |
| PE20030329A1 (es) | 2001-08-08 | 2003-05-12 | Takeda Chemical Industries Ltd | Compuesto biciclico con actividad antagonista de ccr5 y produccion del mismo |
| US20030087843A1 (en) | 2001-09-05 | 2003-05-08 | Washburn William N. | O-pyrazole glucoside SGLT2 inhibitors and method of use |
| EP1432721A4 (en) | 2001-09-13 | 2008-02-20 | Bristol Myers Squibb Co | PROCESS FOR THE PREPARATION OF REBECCAMYCIN AND ANALOGUE THEREOF |
| FR2831169B1 (fr) | 2001-10-22 | 2003-12-12 | Servier Lab | Nouveaux derives d'hydroxyalkyle indolocarbazole, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| US7521568B2 (en) | 2001-12-14 | 2009-04-21 | Purdue Research Foundation | Enantiopure 4-deoxypentenosides, dihydropyrans and tetrahydropyrans and syntheses thereof |
| US6927294B1 (en) * | 2002-03-08 | 2005-08-09 | University Of Southern California | Nitrogen-containing heterocycles |
| DE10213228A1 (de) | 2002-03-25 | 2003-10-16 | Bayer Ag | Cyclopenten-Derivate |
| US6562791B1 (en) | 2002-03-29 | 2003-05-13 | Council Of Scientific And Industrial Research | Glucopyranoside, process for isolation thereof, pharmaceutical composition containing same and use thereof |
| RU2322449C2 (ru) * | 2002-08-09 | 2008-04-20 | Тайсо Фармасьютикал Ко., Лтд. | ПРОИЗВОДНЫЕ АРИЛ 5-ТИО-β-D-ГЛЮКОПИРАНОЗИДА И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА ПРИ ДИАБЕТЕ, СОДЕРЖАЩИЕ ИХ |
| AU2002951247A0 (en) | 2002-09-06 | 2002-09-19 | Alchemia Limited | Compounds that interact with kinases |
| FR2845995A1 (fr) | 2002-10-16 | 2004-04-23 | Servier Lab | Nouveaux derives de pyrrolo[3,4-c]carbazole et de pyrido[2,3-b]pyrrolo[3,4-e]indole, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| FR2845996A1 (fr) | 2002-10-16 | 2004-04-23 | Servier Lab | Nouveaux derives de[3,4-a:3,4-c]carbazole, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| KR20050089156A (ko) | 2002-12-04 | 2005-09-07 | 깃세이 야쿠힌 고교 가부시키가이샤 | 고혈당증에 기인하는 질환의 예방 또는 치료제 |
| DE10258008B4 (de) | 2002-12-12 | 2006-02-02 | Sanofi-Aventis Deutschland Gmbh | Heterocyclische Fluorglycosidderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zur Herstellung dieser Arzneimittel |
| DE10258007B4 (de) | 2002-12-12 | 2006-02-09 | Sanofi-Aventis Deutschland Gmbh | Aromatische Fluorglycosidderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zur Herstellung dieser Arzneimittel |
| AU2003289440A1 (en) | 2002-12-25 | 2004-07-22 | Kissei Pharmaceutical Co., Ltd. | Nitrogen-containing heterocycic derivatives, medicinal compositions containing the same and medicinal use thereof |
| BR0317929A (pt) | 2003-01-03 | 2006-04-11 | Bristol Myers Squibb Co | métodos de produzir inibidores de sglt2 de glicosìdeo de c-arila |
| ES2567571T3 (es) | 2003-03-14 | 2016-04-25 | Astellas Pharma Inc. | Derivados de C-glucósido y sales de los mismos |
| WO2004089966A1 (ja) | 2003-04-01 | 2004-10-21 | Taisho Pharmaceutical Co., Ltd. | 選択的なヘテロアリール 5-チオ-β-D-アルドヘキソピラノシドの製造法 |
| CN1761676A (zh) | 2003-04-01 | 2006-04-19 | 大正制药株式会社 | 杂芳基5-硫代-β-D-吡喃葡糖苷衍生物及含有该衍生物的糖尿病治疗药 |
| UA86042C2 (en) | 2003-08-01 | 2009-03-25 | Янссен Фармацевтика Н.В. | Substituted indazole-o-glucosides |
| RS20060320A (sr) | 2003-08-01 | 2008-08-07 | Janssen Pharmaceutica N.V., | Supstituisani indazol-o-glukozidi |
| EA010422B1 (ru) | 2003-08-01 | 2008-08-29 | Янссен Фармацевтика Н.В. | Замещённые индол-о-глюкозиды |
| TW200524951A (en) | 2003-08-01 | 2005-08-01 | Janssen Pharmaceutica Nv | Substituted benzimidazole-, benztriazole-, and benzimidazolone-O-glucosides |
| EP1679965A4 (en) | 2003-08-01 | 2009-05-27 | Janssen Pharmaceutica Nv | SUBSTITUTED ANELLIERED HETEROCYCLIC C-GLYCOSIDES |
| EP1660509B1 (de) | 2003-08-26 | 2009-02-04 | Boehringer Ingelheim International GmbH | Glucopyranosyloxy-pyrazole, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| US7375090B2 (en) | 2003-08-26 | 2008-05-20 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy-pyrazoles, pharmaceutical compositions containing these compounds, the use thereof and processed for the preparation thereof |
| JP2007508314A (ja) | 2003-10-08 | 2007-04-05 | プレジデント・アンド・フェロウズ・オブ・ハーバード・カレッジ | ピロバレロン類縁体及びそれらの治療的使用 |
| US7371732B2 (en) | 2003-12-22 | 2008-05-13 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy-substituted aromatic compounds, medicaments containing such compounds, their use and process for their manufacture |
| DE102004017793A1 (de) | 2004-02-21 | 2005-09-08 | Biofrontera Discovery Gmbh | Trioxacarcine und deren Verwendung |
| EP1718724B1 (en) | 2004-02-24 | 2008-08-27 | Novozymes A/S | Enzyme stabilization in liquid detergents |
| EP1724277A4 (en) | 2004-03-04 | 2012-05-02 | Kissei Pharmaceutical | ACCOLE HETEROCYCLIC DERIVATIVE, DRUG-CONTAINING COMPOSITION THEREOF, AND THE USE THEREOF |
| ATE557013T1 (de) | 2004-03-16 | 2012-05-15 | Boehringer Ingelheim Int | Glucopyranosyl-substituierte benzol-derivate, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| WO2006002233A2 (en) | 2004-06-22 | 2006-01-05 | Wall, Michael, A. | Alvaradoins e-n, new antitumor and cytotoxic anthracenone g-glycosides |
| US7393836B2 (en) * | 2004-07-06 | 2008-07-01 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| EP1813608B8 (en) | 2004-07-08 | 2009-09-09 | Basf Se | Preparation of alpha-hydroxy ketones |
| EP1783122A4 (en) | 2004-07-08 | 2008-12-10 | Astellas Pharma Inc | PROCESS FOR PREPARING AZULATE DERIVATIVES AND INTERMEDIATE PRODUCTS FOR THEIR SYNTHESIS |
| DE102004034690A1 (de) | 2004-07-17 | 2006-02-02 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Methyliden-D-xylopyranosyl-und Oxo-D-xylopyranosyl-substituierte Phenyle, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| EP1782828B1 (en) | 2004-07-21 | 2016-06-29 | Kissei Pharmaceutical Co., Ltd. | Progression inhibitor for disease attributed to abnormal accumulation of liver fat |
| TW200606129A (en) | 2004-07-26 | 2006-02-16 | Chugai Pharmaceutical Co Ltd | Novel cyclohexane derivative, its prodrug, its salt and diabetic therapeutic agent containing the same |
| EP1773800A1 (de) | 2004-07-27 | 2007-04-18 | Boehringer Ingelheim International GmbH | D-glucopyranosyl-phenyl-substituierte cyclen, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| DE102004046583A1 (de) | 2004-09-23 | 2006-03-30 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | D-Xylopyranosyl-phenyl-substituierte Cyclen, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| WO2006018150A1 (de) * | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituierte cyclen, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| DE102004039096A1 (de) | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | D-Xylopyranosyl-phenyl-substituierte Cyclen, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| DE102004048388A1 (de) | 2004-10-01 | 2006-04-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | D-Pyranosyl-substituierte Phenyle, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| WO2006037335A2 (en) | 2004-10-06 | 2006-04-13 | Recepticon Aps | Use of compounds for the prevention of drug-induced cell toxicity |
| US7622451B2 (en) | 2004-11-03 | 2009-11-24 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| WO2006050501A2 (en) | 2004-11-03 | 2006-05-11 | University Of Kansas | Novobiocin analogues as anticancer agents |
| TW200637839A (en) | 2005-01-07 | 2006-11-01 | Taisho Pharmaceutical Co Ltd | 1-thio-d-glucitol derivatives |
| ATE445608T1 (de) | 2005-02-23 | 2009-10-15 | Boehringer Ingelheim Int | Glucopyranosylsubstituierte ((hetero)arylethynyl- benzyl)-benzenderivative und deren verwendung als inhibitoren des natriumabhängigen glucose- cotransporters typ 2 (sglt2) |
| CN101160315A (zh) | 2005-03-25 | 2008-04-09 | 印斯拜尔药品股份有限公司 | 细胞骨架的活性化合物,组合物及用途 |
| US20060240496A1 (en) | 2005-04-21 | 2006-10-26 | Lakshmi Anne | Immunogens, derivatives and immunoassay for ethyl glucuronide |
| US7723309B2 (en) | 2005-05-03 | 2010-05-25 | Boehringer Ingelheim International Gmbh | Crystalline forms of 1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments |
| CA2620566A1 (en) | 2005-08-30 | 2007-03-08 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US8253752B2 (en) | 2006-07-20 | 2012-08-28 | Qualcomm Incorporated | Method and apparatus for encoder assisted pre-processing |
| CL2007002166A1 (es) | 2006-07-24 | 2008-01-25 | Tetralogic Pharm Corp | Compuestos derivados de heterociclos de nitrogeno, antagonistas de los inhibidores de las proteinas de la apoptosis; sus composiciones farmaceuticas; y uso de dichos compuestos para el tratamiento del cancer. |
| TWI499414B (zh) * | 2006-09-29 | 2015-09-11 | Lexicon Pharmaceuticals Inc | 鈉與葡萄糖第2型共同運輸體(co-transporter 2)的抑制物與其應用方法 |
| US20080107744A1 (en) * | 2006-11-06 | 2008-05-08 | Jack Fa-De Chu | Injectable hollow tissue filler |
| WO2008109591A1 (en) * | 2007-03-08 | 2008-09-12 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| AU2008279424B2 (en) * | 2007-07-26 | 2013-06-13 | Lexicon Pharmaceuticals, Inc. | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
| US8015005B2 (en) | 2008-02-15 | 2011-09-06 | Motorola Mobility, Inc. | Method and apparatus for voice searching for stored content using uniterm discovery |
| TWI472521B (zh) * | 2008-07-17 | 2015-02-11 | Lexicon Pharmaceuticals Inc | (2s,3r,4r,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(甲硫)四氫-2h-哌喃-3,4,5-三醇的固體形態與其使用方法 |
| PL3489226T3 (pl) * | 2012-11-20 | 2021-08-02 | Lexicon Pharmaceuticals, Inc. | Inhibitory kotransportera glukozowo-sodowego 1 |
-
2007
- 2007-09-19 TW TW096134994A patent/TWI499414B/zh active
- 2007-09-27 AT AT07843301T patent/ATE496888T1/de active
- 2007-09-27 DK DK07843301.8T patent/DK2089361T5/da active
- 2007-09-27 NZ NZ575811A patent/NZ575811A/en unknown
- 2007-09-27 PL PL10194063T patent/PL2308841T3/pl unknown
- 2007-09-27 MX MX2009003305A patent/MX2009003305A/es active IP Right Grant
- 2007-09-27 BR BRPI0717156A patent/BRPI0717156B8/pt active IP Right Grant
- 2007-09-27 PT PT07843301T patent/PT2089361E/pt unknown
- 2007-09-27 US US11/862,690 patent/US7781577B2/en active Active
- 2007-09-27 PT PT101940633T patent/PT2308841E/pt unknown
- 2007-09-27 CA CA2664688A patent/CA2664688C/en active Active
- 2007-09-27 DE DE602007012292T patent/DE602007012292D1/de active Active
- 2007-09-27 WO PCT/US2007/079654 patent/WO2008042688A2/en not_active Ceased
- 2007-09-27 PL PL07843301T patent/PL2089361T4/pl unknown
- 2007-09-27 DK DK10194063.3T patent/DK2308841T3/da active
- 2007-09-27 AU AU2007304971A patent/AU2007304971B2/en active Active
- 2007-09-27 KR KR1020097008685A patent/KR101492277B1/ko active Active
- 2007-09-27 EP EP10194063.3A patent/EP2308841B1/en active Active
- 2007-09-27 EA EA200970337A patent/EA016511B1/ru not_active IP Right Cessation
- 2007-09-27 EP EP07843301A patent/EP2089361B1/en active Active
- 2007-09-27 ES ES10194063.3T patent/ES2477216T3/es active Active
- 2007-09-27 KR KR1020147033635A patent/KR20150002889A/ko not_active Withdrawn
- 2007-09-27 JP JP2009530593A patent/JP5283625B2/ja active Active
- 2007-09-28 AR ARP070104306A patent/AR063047A1/es active IP Right Grant
-
2009
- 2009-03-26 IL IL197836A patent/IL197836A/en active IP Right Grant
- 2009-04-28 NO NO20091700A patent/NO345139B1/no unknown
-
2010
- 2010-08-18 US US12/858,666 patent/US8476413B2/en active Active
-
2012
- 2012-11-20 JP JP2012254023A patent/JP5701845B2/ja active Active
-
2013
- 2013-06-25 US US13/925,981 patent/US9365602B2/en active Active
-
2015
- 2015-02-17 JP JP2015028510A patent/JP5889453B2/ja active Active
-
2016
- 2016-05-16 US US15/155,780 patent/US20170087172A1/en not_active Abandoned
-
2018
- 2018-03-27 US US15/937,496 patent/US20190099436A1/en not_active Abandoned
-
2019
- 2019-08-15 HU HUS1900038C patent/HUS1900038I1/hu unknown
- 2019-08-30 NL NL301003C patent/NL301003I2/nl unknown
- 2019-09-19 FR FR19C1053C patent/FR19C1053I2/fr active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0717156B1 (pt) | inibidores do co-transportador 2 da glicose de sódio e composição farmacêutica que os compreende | |
| US7846945B2 (en) | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use | |
| RU2667060C2 (ru) | Производные маннозы для лечения бактериальных инфекций | |
| CN103254119B (zh) | 钠-葡萄糖协同转运蛋白2的抑制剂及其用法 | |
| RU2678327C2 (ru) | Производные маннозы для лечения бактериальных инфекций | |
| ES2362684T3 (es) | Inhibidores del cotransportador 2 de sodio-glucosa y métodos para su utilización. | |
| HK1124863B (en) | Inhibitors of sodium glucose co-transporter 2 and methods of their use | |
| HK1183020B (en) | Inhibitors of sodium glucose co-transporter 2 and methods of their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B06T | Formal requirements before examination [chapter 6.20 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 30/06/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 27/09/2007 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |