A presente invenção refere-se a um método para síntese assi- métrica de (1S, 2R)-Milnaciprano bem como a um intermediário clorado na forma (1S, 2R) enantiomérica principal.
Milnaciprano é um antidepressivo inibidor da recaptação de se- rotonina-noradrenalina recomendado no tratamento de depressão (FR 2 508 035).
Muitas sínteses do composto racêmico foram descritas na litera- tura (EP 0 377 381; EP 0 200 638; EP 1 757 597; EP 1 767 522; EP 1 845 084; EP 1 770 084; Shuto S. e outro,J. Med. Chem. 1995, 38, 2964-2968).
Além disso, foi recentemente demonstrado que o enantiômero (1S, 2R)-milnaciprano é mais ativo do que a mistura racêmica (Viazzo P. e outro,Tetrahedron Lett. 1996, 37, 26, 4519-4522).
Um primeiro método para obter este enantiômero em forma enri- quecida foi a separação ou resolução de enantiômeros da mistura racêmica (Bonnaud B. e outro, J. Chromatogr. 1985, 318, 398-403). Entretanto, tal mé- todo não é industrialmente eficaz quanto aos custos visto que existe uma perda de pelo menos metade do produto. As sínteses enantiosseletivas fo- ram aquelas desenvolvidas para preparar milnaciprano enantiomericamente enriquecido (Doyle M. P. and Hu W. Adv. Synth. Catai. 2001, 343, 299-302; Roggen H. e outro, Bloorg. Med. Chem. 2007, 17, 2834-2837; Shuto S. e outro, Tetrahedron Lett. 1996, 37, 641-644; Wang X. -Q. e outro, Chinese journal of Pharmaceuticals 2004, 35, 259-260; WO 2005/118 564).
Entretanto, a maioria destas sínteses usa azida de sódio como um reagente, que pode dificilmente ser contemplada industrialmente por causa de sua toxicidade e de sua instabilidade que pode levar a uma explo- são. Entretanto existe ainda uma necessidade significante de novos métodos para sintetizar (1S, 2R)-milnaciprano que são mais seguros, mais econômi- cos e mais eficientes.
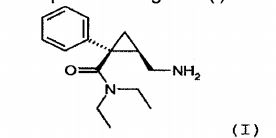
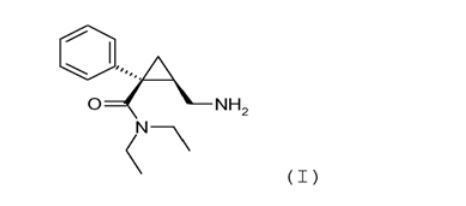
Desse modo, mais particularmente o objetivo da invenção é um método para a síntese de um sal de adição de ácido farmaceuticamente a- ceitável de (1S, 2R)-milnaciprano da seguinte (I):
compreendendo as seguintes etapas sucessivas: (a) reação de fenilacetonitrila e de (R)-epicloridrina na presença de uma base contendo um metal alcalino, seguido por um tratamento básico, e em seguida por um tratamento ácido para obter a lactona da seguinte fórmula (II):
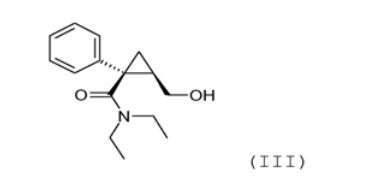
(b) reação de a lactona (II) obtida na etapa anterior (a) com MNEt2, em que M representa um metal alcalino, ou com NHEt2 na presença de um complexo de ácido-amina de Lewis em que a amina é selecionada de dietilamina, trietilamina, di-isopropiletilamina, N, N-dietilanilina, N,N- dimetilbenzilamina, N-metilpiperidina, N-metilmorfolina, N,N'- dimetilpiperazina e tetramina de hexametileno, para obter o álcool de amida da seguinte fórmula (III):
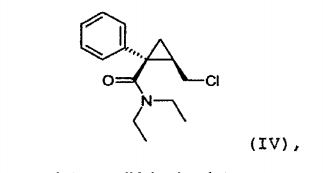
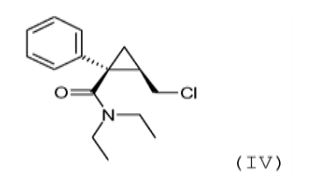
(c) reação do álcool de amida de fórmula (III) obtido na etapa an- terior (b) com cloreto de tionila para obter a amida clorada da seguinte fórmula (IV): (IV) (d) reação da amida clorada de fórmula (IV)
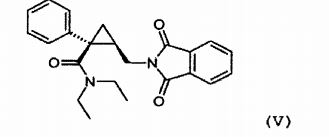
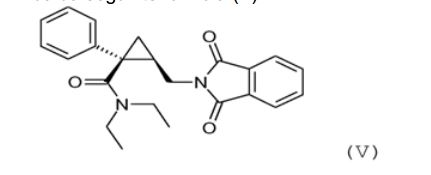
obtida na etapa an- terior (c) com um sal de ftalimida, tal como o sal de potássio, para obter o derivado de ftalimida da seguinte fórmula (V): (e) hidrólise do grupo de ftalimida do derivado de ftalimida de fórmula (V)
obtida na etapa anterior (d) para obter (1S, 2R)-milnaciprano, e (f) salificação de (1S, 2R)-milnaciprano obtido na etapa anterior (e) em um sistema adequado de solventes, na presença de um ácido farma- ceuticamente aceitável.
Na presente invenção, « farmaceuticamente aceitável » descre- ve o que é útil na presença de uma composição farmacêutica que é geral- mente segura, não tóxica e nem biologicamente nem de outra maneira inde- sejável, e que é aceitável para uso veterinário bem como para uso farmacêu- tico humano.
Um « sal de adição de ácido farmaceuticamente aceitável » de um composto é entendido indicar, na presente invenção, sais que são far- maceuticamente aceitáveis, como definido aqui, que têm a atividade farma- cológica desejada do composto origem e que são obtidos pela adição de um ácido farmaceuticamente aceitável sobre o composto.
Por « ácido farmaceuticamente aceitável » notavelmente enten- de-se ácidos inorgânicos tais como ácido clorídrico, ácido hidrobrômico, áci- do sulfúrico, ácido nítrico, ácido fosfórico, e similares, ou ácidos orgânicos tais como ácido acético, ácidos benzenossulfônico, ácido benzoico, ácido canfor-sulfônico, ácido cítrico, ácido etanossulfônico, ácido fumárico, ácido gluco-heptônico, ácido glucônico, ácido glutâmico, ácido glicólico, ácido hi- droxinaftoico, ácido 2-hidroxietanossulfônico, ácido láctico, ácido maleico, ácido málico, ácido mandélico, ácido metanossulfônico, ácido mucônico, áci- do 2-naftalenossulfônico, ácido propiônico, ácido salicílico, ácido sucínico, ácido dibenzoil-L-tartárico, ácido tartárico, ácido p-toluenossulfônico, ácido trimetilacético, ácido trifluoroacético, e similares. Preferivelmente, este é ácido clorídrico.
Etapa (a):
Esta etapa corresponde à seguinte sequência de reação: (2)
Por « base contendo um metal alcalino » entende-se, no sentido da presente invenção, uma base de fórmula RM, em que: - M representa um metal alcalino, e, em particular, sódio (Na), potássio (K) ou lítio (Li), e - R representa um átomo de hidrogênio, uma alquila (tal como butila ou hexila), alcóxi (tal como terciobutilóxi) ou grupo NR12, com R1 re- presentando um átomo de hidrogênio, uma alquila (tal como isopropila) ou grupo Si (CH3)3.
Por « alquila » entende-se, no sentido da presente invenção, uma cadeia de hidrocarboneto linear ou ramificada, saturada, compreenden- do de 1 a 6 átomos de carbono. Em particular, esta será um grupo butila, hexila ou isopropila.
Por « alcóxi » entende-se no sentido da presente invenção, um grupo alquila como definido acima, ligado ao resto da molécula por meio de um átomo de oxigênio. Em particular, este será um grupo terciobutilóxi.
A base contendo um metal alcalino em particular será seleciona- da a partir de NaH, NaNH2, hexametildisilazano de potássio ou lítio (KHMDS ou LiHMDS), butil lítio, hexil lítio, terciobutilado de sódio ou potássio ou di- isopropilamida de lítio (LDA). Vantajosamente, esta será NaH ou NaNH2, e preferivelmente esta será NaNH2.
Com o tratamento básico subsequente, é possível hidrolisar a função de nitrila do composto (3) em um ácido carboxílico para obter o com- posto (4). Um hidróxido de metal alcalino é parcialmente adequado para este tratamento, tal como NaOH ou KOH, e, em particular, NaOH.
Além disso, com o tratamento de ácido é possível ciclizar o deri- vado de ácido hidroxila (4) na lactona (II). Um ácido particularmente adequa- do para este tratamento é ácido clorídrico, notavelmente em uma solução aquosa, por exemplo, em 25 %.
As etapas (al), (a2) e (a3) vantajosamente serão realizadas em um mesmo reator, sem afastamento dos produtos intermediários (3) e (4) (um método descrito como um método de um pote). Sob estas condições, um mesmo e único solvente vantajosamente será usado para estas 3 eta- pas, e preferivelmente este será tolueno, a base e o ácido das etapas (a2) e (a3) sendo, entretanto, vantajosamente introduzidas na forma de uma solu- ção aquosa.
Etapa (b):
Por « metal alcalino » é mais particularmente entendido sódio, potássio e lítio.
MNEt2 pode notavelmente ser obtido por reação de NHEt2 com um alcóxido de metal alcalino. MNEt2 em seguida será vantajosamente for- mado in situ, isto é, pela adição de dois reagentes, NHEt2 e alcóxido de me- tal alcalino, no meio de reação contendo a lactona.
Por « alcóxido de metal alcalino », entende-se no sentido da presente invenção um composto de fórmula Alq-O-M, em que M representa um metal alcalino como definido acima e Alq representa uma cadeia de hi- drocarboneto linear ou ramificada, saturada, incluindo de 1 a 6, preferivel- mente de 1 a 4 átomos de carbono. Esta será em particular MeONa, MeOK, EtONa ou também EtOK.
Quando M = Li, LiNEt2 pode ser formado pela adição de um de- rivado de lítio, tal como butil lítio, em NHEt2. Neste caso, LiNEt2 preferivel- mente será preparado de ante mão, antes de ser introduzido no meio de re- ação contendo a lactona.
Por « derivado de lítio », notavelmente entende-se, no sentido da presente invenção, um derivado de fórmula Alq' Li com Alq' representan- do uma cadeia de hidrocarboneto linear ou ramificada, saturada, incluindo de 1 a 6, preferivelmente de 1 a 4 átomos de carbono. Este em particular é butil lítio.
Por « ácido de Lewis », entende-se, no sentido da presente in- venção, uma entidade química capaz de aceitar um dupleto de elétron e, portanto, capaz de formar um complexo com o átomo de oxigênio da carbo- nila C=O de uma lactona (II). Com isto, a carbonila de uma lactona pode ser ativada e, portanto, a adição do composto nucleofílico (NHEt2) sobre a última pode ser promovida. Em particular, o ácido de Lewis pode ser AICI3.
Preferivelmente, esta etapa será realizada na presença de dieti- lamina e um complexo AICl3-NHEt2-
Esta etapa pode notavelmente ser realizada em tolueno como um solvente, incluindo no caso do uso de NHEt2 na presença de um ácido de Lewis, ao mesmo tempo em que uma reação de acilação de Friedel- Crafts poderia ter sido esperada entre a lactona e o tolueno na presença de um ácido de Lewis tal como AICI3.
Preferivelmente, esta etapa será realizada na presença de NHEt2 e AICI3 como um ácido de Lewis.
Etapa (c):
Durante esta etapa de cloração, o ácido clorídrico é formado. É importante promover este composto antes da etapa seguinte. Usando-se um solvente tal como tolueno, sua remoção pode ser facilitada por concentração do meio de reação.
De fato, com tolueno, é possível remover ácido clorídrico por co- evaporação mais facilmente do que com um solvente tal como cloreto de metileno, por causa do seu ponto de fusão superior.
Etapa (d):
Esta etapa será vantajosamente conduzida com o sal de potás- sio de ftalimida. A reação pode vantajosamente ser conduzida em tolueno como um solvente.
Etapa (e):
Esta etapa de hidrólise do derivado de ftalimida em uma amina primária é vantajosamente realizada por reação com hidrazina, uma alquila- mina tal como metilamina, ou uma hidroxialquilamina tal como etanolamina.
Por « alquilamina », entende-se, no sentido da presente inven- ção, uma amina de fórmula Alq' 'NH2 com Alq" representando uma cadeia de hidrocarboneto linear ou ramificada, saturada, incluindo de 1 a 6, preferi- velmente de 1 a 4 átomos de carbono. Em particular, esta é metilamina.
Por « hidroxialquilamina », entende-se no sentido da presente invenção, uma hidroxil-amina de fórmula HO-R2-NH2 com R2 representando uma cadeia de hidrocarboneto linear ou ramificada, saturada, incluindo de 1 a 6, preferivelmente de 1 a 4 átomos de carbono. Em particular esta é etano- lamina.
Preferivelmente, esta etapa será realizada na presença de eta- nolamina.
Esta etapa pode vantajosamente ser realizada em um solvente tal como tolueno. Entretanto, hidrazina, alquilamina ou hidroxialquilamina pode ser adicionada na forma de uma solução aquosa.
Etapa (f):
Com esta etapa, é possível salificar (1S, 2R)-milnaciprano obtido na etapa anterior (e) e ao mesmo tempo purificar o sal de adição de ácido de (1S, 2R)-milnaciprano por cristalização e em seguida filtragem.
Preferivelmente, esta etapa será realizada na presença de ácido clorídrico para formar cloridrato de (1S, 2R)-milnaciprano.
Vantajosamente, o sistema de solventes usados para a salifica- ção compreenderá tolueno, e preferivelmente será uma mistura de tolueno, acetato de isopropila e isopropanol.
Preferivelmente, esta mistura terá a seguinte composição, relati- vamente ao volume total dos solventes: - 0% a 50%, vantajosamente de 30% a 40%, por volume de tolu- eno, - 40% a 90%, vantajosamente de 50% a 80%, por volume de a- cetato de isopropila, e - 5% a 25%, vantajosamente de 10% a 20%, por volume de iso- propanol.
Em particular, as etapas de (a) a (e) vantajosamente serão reali- zadas em um meio de reação compreendendo um mesmo e único solvente tal como tolueno.
De fato, usando-se um mesmo e único solvente na maior parte das etapas (exceto para a última etapa de salificação), é possível simplificar o procedimento para preparar o composto e reduzir o custo do mesmo na medida em que o solvente não tenha que ser mudado em cada etapa. Sob estas condições, não é, portanto, necessário isolar os intermediários de rea- ção mesmo se as etapas de extração puderem ser realizadas para remover algumas impurezas que podem ser importunas para a própria progressão das etapas seguintes.
Os inventores, desse modo, descobriram que o todo da sequên- cia de reação pode inesperadamente ser realizado com um mesmo e único solvente para as etapas de (a) a (e), e preferivelmente com tolueno.
Sob estas condições, será vantajoso não isolar qualquer dos produtos intermediários obtidos nas etapas de (a) a (d), e preferivelmente de (a) a (e), do meio de reação. Desse modo, entende-se que os produtos in- termediários obtidos sempre serão em solução no meio de reação, preferi- velmente em tolueno, e nunca serão isolados na forma seca ou quase seca. As etapas de concentração do meio de reação podem, entretanto, ser reali- zadas, em particular, após as etapas de extração, porém será vantajoso não evaporar a seco o meio de reação notavelmente por razões de custo e con- veniência. Isto tem a vantagem adicional de evitar perdas de produto adicio- nais durante as etapas de purificação intermediárias.
Desse modo, com tal método, é possível obter (1S, 2R)- milnaciprano com um excesso enantiomérico (ee) de pelo menos 95%, e preferivelmente de pelo menos 98%, e vantajosamente com uma produção maior do que 40%, preferivelmente maior do que 45%, relativamente à (R)- epicloridrina usada como produto de partida.
O objeto da presente invenção é também o composto da seguin- te fórmula (IV), na forma (1S, 2R) enantiomérica: (IV),
em particular, como um intermediário de síntese.
Este composto é vantajosamente obtido com um excesso anan- tiomérico maior do que 90%, preferivelmente maior do que 95%, e ainda pre-ferivelmente maior do que 98%.
A presente invenção será melhor entendida à luz dos exemplos não limitantes que seguem.
EXEMPLOS:
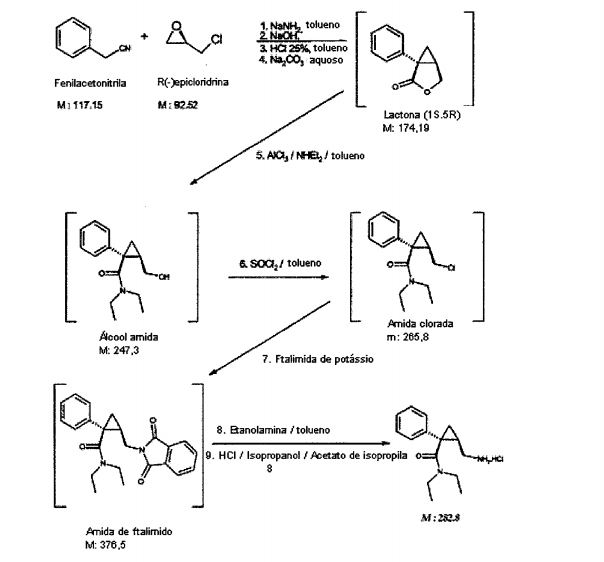
Cloridrato de (1S, 2R)-milnaciprano, em uma base de 41 kg de produto acabado, é sintetizado de acordo com o esquema seguinte e proce- 10 dimento operante:
28 kg de amida de sódio (682 moles) são suspensos em 400 L de tolueno e em seguida, sob agitação intensa, 85,5 kg de fenilacetonitrila (729,5 moles) diluídos em 10 L de tolueno são despejado em uma temperatura compreendendo entre 0°C e 5°C. O meio de reação é agitado durante pelo menos 1 hora a 10°C. 27 kg de epicloridrina quiral (292 moles) em solução em 20 L de tolueno são adicionados ao mesmo tempo em que mantendo a temperatura a 10°C. No final do despejo, o meio é agitado durante pelo menos 2 horas. A hidrólise é realizada despejando-se o meio de reação sobre uma solução aquosa de 240 L ao mesmo tempo em que mantendo a temperatura entre 5°C e 40°C. Após concentração da solução obtida, 115 kg de 30% de soda são adicionados e o meio é aquecido para 95 C para permitir a hidrólise das funções de nitrila. O meio é lavado duas vezes com 190 L de tolueno. As fases de tolueno são removidas e a fase aquosa é recuperada após adição de 270 L de tolueno e acidificado por uma solução de ácido clorídrico a 25% até um pH compreendido entre 1 e 2. O meio é em seguida aquecido para 60°C durante pelo menos 3 horas. Após decantação, a fase de tolueno contendo a lactona é lavada com 140 L de água neutralizada por uma solução de carbonato de sódio a 10% até um pH compreendido entre 8 e 9 e em seguida novamente lavada com 140 L de água. A fase de tolueno obtida é concentrada até um volume de 120 L contendo 38 kg de lactona (218 moles).
Etapa 5:
34 kg de cloreto de alumínio (255 moles) são suspensos em 240 L de tolueno e em seguida 38,3 kg de dietilamina (523,5 moles) são adicionados ao mesmo tempo em que mantendo a temperatura entre 15°C e 30°C. O concentrado de lactona (38 kg) obtido anteriormente é despejado sobre o meio mantido a 25°C. O meio de reação é agitado durante pelo menos 1 hora e 30 minutos. A formação de um precipitado é observada.
Este meio de reação é hidrolisado com 345 L de água e em seguida filtrado após adição de um adjuvante de filtragem.
Após decantação, a fase orgânica é lavada duas vezes com 235
L e 175 L de água e em seguida concentrada até um concentrado de álcool de amida de 110 L ser obtido.
Etapa 6:
24,7 kg de cloreto de tionila (207 moles) são despejados sobre o concentrado dentro de 1 hora a 25°C sob agitação intensa. O meio de rea- ção é concentrado a vácuo limitando-se a temperatura a 50°C. Esta opera- ção de concentração é repetida duas vezes após adicionar duas vezes 62 L de tolueno, para obter um concentrado de amida clorada.
Etapa 7:
O concentrado de amida clorada obtido na etapa anterior é des- pejado sobre uma suspensão de ftalimida de potássio (51,9 kg de ftalimida de potássio (280 moles) em 155 L de tolueno), e o meio é aquecido para 85°C durante pelo menos 3 horas. O meio de reação é resfriado para 45°C, lavado duas vezes com 130L de água. Após decantação, a fase de tolueno obtida contém cerca de 74 kg de ftalimido-amida (196,5 moles).
Etapa 8:
92,4 kg de etanolamina (1513 moles) são introduzidos na solu- ção de tolueno de ftalimido-amida sob agitação intensa; o meio é aquecido para 82,5°C durante 2 horas. Após resfriamento e adição de 247 L de tolue- no, o meio de reação é lavado com 225 L de solução de NaCI a 20% de sali- na aquosa. Após 2 contraextrações da fase aquosa com 52 L de tolueno, as fases de tolueno são agrupadas e lavadas duas vezes com 225 L de solução de NaCI a 20% de salina. Após decantação, 185 L de água são adicionados sobre a fase de tolueno e o meio é acidificado para um pH compreendido entre 2 e 3 com 25% de ácido clorídrico. Após decantação, a fase orgânica do ácido é novamente extraída com 74 L de água. A fase orgânica é em se- guida removida. As fases aquosas de ácido agrupadas são extraídas duas vezes com 370 e 150 L de tolueno após retornar para um pH básico com- preendido entre 12 e 13 com uma solução de soda a 20% aquosa. As fases orgânicas agrupadas são lavadas com 80 L de água e em seguida concen- tradas.
Etapa 9:
Sobre o concentrado de tolueno, são adicionados 283 L de acetato de isopropila e 48,4 L de isopropanol. Uma solução de ácido clorídrico a 5N em isopropanol é despejada sobre esta solução orgânica, até um pH compreendido entre 3 e 4 (cerca de 30 L de solução) em uma temperatura 5 de 30°C. Durante a introdução da solução de ácido, os precipitados de clori- drato, o meio é resfriado para 10°C e mantido durante pelo menos 2 horas nesta temperatura. A suspensão é filtrada, lavada 3 vezes com 56 L de acetato de isopropila. O produto obtido é secado a vácuo a 70°C. 41 kg de clori- drato de (1S, 2R)-milnaciprano (145 moles) são obtidos, isto é, uma produ- 10 ção de 9,6% em relação à epicloridrina quiral.