CN101007795B - Synthetic method of paclitaxel and docetaxel - Google Patents
Synthetic method of paclitaxel and docetaxel Download PDFInfo
- Publication number
- CN101007795B CN101007795B CN2007100669400A CN200710066940A CN101007795B CN 101007795 B CN101007795 B CN 101007795B CN 2007100669400 A CN2007100669400 A CN 2007100669400A CN 200710066940 A CN200710066940 A CN 200710066940A CN 101007795 B CN101007795 B CN 101007795B
- Authority
- CN
- China
- Prior art keywords
- compound
- solvent
- docetaxel
- reaction
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Epoxy Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明提供紫杉醇及多烯紫杉醇的合成方法,是以10-DAB III为中间体,先选择性地保护C-7羟基,然后对C-10羟基进行酰化或保护,接着用保护的侧链酸与C-13羟基进行偶联,最后水解脱除保护基即得目标化合物紫杉醇或多烯紫杉醇。本发明方法克服目前几种保护基存在的缺点,开发出一种新的保护基——咪唑基硫羰基;为了避免C-2’差向异构化的形成及剧烈的反应条件,合成了2-单取代-1,3-噁唑烷型侧链酸;设计合理,所用试剂价格低廉、反应步骤少以及反应条件温和。本发明方法为紫杉醇的工业化生产提供了有利条件。同时为多烯紫杉醇的合成提供了一种新途径。The invention provides a synthetic method of paclitaxel and docetaxel, which uses 10-DAB III as an intermediate, first selectively protects the C-7 hydroxyl group, then acylates or protects the C-10 hydroxyl group, and then uses the protected side chain The acid is coupled with the C-13 hydroxyl group, and finally the protecting group is removed by hydrolysis to obtain the target compound paclitaxel or docetaxel. The inventive method overcomes the shortcoming that several protecting groups exist at present, develops a kind of new protecting group---imidazolylthiocarbonyl; In order to avoid the formation of C-2' epimerization and severe reaction conditions, synthesized 2 -Monosubstituted-1,3-oxazolidine type side chain acid; reasonable design, low price of reagents, few reaction steps and mild reaction conditions. The method of the invention provides favorable conditions for the industrial production of paclitaxel. At the same time, it provides a new way for the synthesis of docetaxel.
Description
技术领域technical field
本发明属化合物合成,主要涉及抗癌药物紫杉醇及多烯紫杉醇的合成方法。The invention belongs to compound synthesis, and mainly relates to the synthesis method of anticancer drugs paclitaxel and docetaxel.
背景技术Background technique
紫杉醇(Paclitaxel,商品名Taxol,Formula Ia)是从红豆杉属(Taxus)植物中分离出的一种紫杉烷二萜类化合物。其结构新颖、抗癌机理独特、抗癌效果显著、抗癌谱广,被认为是迄今所发现最好的抗癌药物之一。继美国之后,目前紫杉醇作为一线抗癌药物已在40多个国家被批准上市。然而,紫杉醇供应匮乏大大限制了它在临床上的应用。目前,药用紫杉醇的主要来源是从天然红豆杉属植物的树皮中提取分离。但由于该种属植物数量少,生长慢,含量低,且提取难度颇大,从长远考虑根本无法满足日益增长的临床需求。Paclitaxel (trade name Taxol, Formula Ia) is a taxane diterpenoid isolated from Taxus plants. Its novel structure, unique anti-cancer mechanism, remarkable anti-cancer effect and broad anti-cancer spectrum are considered to be one of the best anti-cancer drugs discovered so far. After the United States, paclitaxel as a first-line anticancer drug has been approved for marketing in more than 40 countries. However, the lack of paclitaxel supply greatly limits its clinical application. At present, the main source of medicinal paclitaxel is to extract and isolate from the bark of the natural Taxus genus. However, due to the small number of plants in this genus, slow growth, low content, and rather difficult extraction, it is impossible to meet the growing clinical needs in the long run.
多烯紫杉醇(Docetaxel,商品名Taxotere,Formula Ib)是抗癌新药紫杉醇经化学修饰的衍生物,其抗肿瘤效果和毒副作用明显优于紫杉醇。临床上多烯紫杉醇作为化疗药物已开始用于乳腺癌、肺癌、胃癌和食管癌等的治疗,具有较好效果,且副作用较少。然而,多烯紫杉醇的供应远远不能满足市场的需求。Docetaxel (Docetaxel, trade name Taxotere, Formula Ib) is a chemically modified derivative of the new anticancer drug paclitaxel, and its antitumor effect and side effects are significantly better than paclitaxel. Clinically, docetaxel has been used as a chemotherapeutic drug in the treatment of breast cancer, lung cancer, gastric cancer and esophageal cancer, etc., with good effect and less side effects. However, the supply of docetaxel is far from meeting the demand of the market.
近年来,紫杉醇化学全合成获得成功,但合成路线复杂,成本过高,故而仅具有研究意义,尚无商业价值。相比较而言,紫杉醇类化学半合成是具有实用价值的一种制备方法,因而显得尤为重要。In recent years, the chemical total synthesis of paclitaxel has been successful, but the synthetic route is complicated and the cost is too high, so it is only of research significance and has no commercial value. In comparison, the chemical semi-synthesis of paclitaxel is a preparation method with practical value, so it is particularly important.
半合成研究一般都是以与紫杉醇大环结构类似的10—去乙酰基巴卡亭(10-deacetylbaccatin III,10-DAB III)为中间体,通过与紫杉醇或多烯紫杉醇的侧链对接而制得紫杉醇或多烯紫杉醇。10-DAB III可从来源丰富的红豆杉属植物的针叶中提取而得,且得率高,从而为紫杉醇和多烯紫杉醇的半合成提供了充足的保障。Semi-synthetic studies generally use 10-deacetylbaccatin (10-deacetylbaccatin III, 10-DAB III), which is similar in structure to the macrocycle of paclitaxel, as an intermediate, and is prepared by docking with the side chain of paclitaxel or docetaxel. Get paclitaxel or docetaxel. 10-DAB III can be extracted from the needles of the genus Taxus with a high yield, which provides sufficient guarantee for the semi-synthesis of paclitaxel and docetaxel.


一般来讲,以10-DAB III为前体物来半合成紫杉醇及多烯紫杉醇的路线包括以下几步:Generally speaking, the semi-synthetic route of paclitaxel and docetaxel using 10-DAB III as a precursor includes the following steps:
(一)在C-7和C-10位羟基选择性地保护(1) Selective protection of hydroxyl groups at C-7 and C-10
在10-DAB III(Formula II)分子结构中存在四个羟基,分别位于C-1、7、10和13位。这些羟基的反应活性不同,反应活性顺序由快到慢是C-7>C-10>C-13>C-1。此外,C-7羟基在碱性条件下极易差向异构化。所以,C-7羟基的保护基必须要求在温和的反应条件下可选择性地上保护,又可选择性地脱保护。在紫杉醇的半合成研究中,已报道用作C-7羟基保护基的基团有四种:三乙基硅基(TES)、三氯乙氧羰酰基(Troc)、三氯乙酰基和氯乙酰基。其中,三乙基硅基是最常用的保护基,然而在C-7羟基保护的反应中,其最佳反应条件是需要大大过量(20倍等摩尔量)昂贵的三乙基硅基;采用三氯乙氧羰酰基作为C-7羟基保护基,由于该基团对C-7和C-10两羟基的反应几乎没有选择性,故而在合成得到紫杉醇的过程中需要多几步反应,另外在对C-7羟基进行保护与脱保护时均需要剧烈的反应条件(TetrahedronLetters,1992,pp5185-5188);专利US6130336采用间歇加入三氯乙酰基保护基的方法对C-7羟基进行选择性保护;至于氯乙酰基,由于其较低的反应活性选择性,所以在反应中需要其严格的量化条件。There are four hydroxyl groups in the molecular structure of 10-DAB III (Formula II), which are located at C-1, 7, 10 and 13 respectively. The reactivity of these hydroxyl groups is different, and the order of reactivity from fast to slow is C-7>C-10>C-13>C-1. In addition, the C-7 hydroxyl group is extremely prone to epimerization under basic conditions. Therefore, the protecting group of the C-7 hydroxyl group must be selectively protected and deprotected under mild reaction conditions. In the semi-synthetic study of paclitaxel, four groups have been reported as C-7 hydroxyl protecting groups: triethylsilyl (TES), trichloroethoxycarbonyl (Troc), trichloroacetyl and chlorine Acetyl. Wherein, triethylsilyl is the most commonly used protecting group, yet in the reaction of C-7 hydroxyl protection, its optimum reaction condition is to need the expensive triethylsilyl of large excess (20 times equimolar quantity); Trichloroethoxycarbonyl is used as C-7 hydroxyl protecting group, because this group has almost no selectivity to the reaction of C-7 and C-10 two hydroxyl groups, so it needs several more steps of reaction in the process of obtaining paclitaxel in synthesis, in addition When protecting and deprotecting the C-7 hydroxyl group, severe reaction conditions are required (Tetrahedron Letters, 1992, pp5185-5188); the patent US6130336 adopts the method of adding trichloroacetyl protecting group intermittently to selectively protect the C-7 hydroxyl group ; As for the chloroacetyl group, due to its low reactivity selectivity, strict quantification conditions are required in the reaction.
在C-7羟基保护之后,对C-10羟基进行酰化作用得到7-位羟基保护的巴卡亭III;或在C-7羟基保护之后,采用氯乙酰氯等试剂对C-10羟基进行保护,可得到7,10-位羟基保护的10-去乙酰巴卡亭III。After the C-7 hydroxyl group is protected, the C-10 hydroxyl group is acylated to obtain the 7-hydroxyl protected baccatin III; Protection can give 10-deacetylbaccatin III with 7,10-hydroxyl protection.
(二)保护的苯基异丝氨酸侧链(C13侧链)与C-13羟基选择性地进行酯化对接(2) Selective docking of the protected phenylisoserine side chain (C13 side chain) with the C-13 hydroxyl group
由于巴卡亭分子中的C-13羟基是位于半球形紫杉烷骨架的凹面区域,因而被遮挡的C-13羟基很难与侧链酸进行酯化作用,所以此步反应是合成过程中的关键。为了提高酯化产率及降低副产物的形成,苯基异丝氨酸侧链部分通常要进行适当的保护,常用保护的C-13侧链有三大类:直线型、四元环β—内酰胺型和五元环噁唑烷型。Since the C-13 hydroxyl group in the baccatin molecule is located in the concave area of the hemispherical taxane skeleton, it is difficult for the blocked C-13 hydroxyl group to undergo esterification with the side chain acid, so this step is a step in the synthesis process. key. In order to improve the yield of esterification and reduce the formation of by-products, the side chain of phenylisoserine is usually properly protected. There are three types of C-13 side chains commonly used for protection: linear type, four-membered ring β-lactam type and five-membered ring oxazolidine type.
(三)侧链及巴卡亭两部分的脱保护(3) Deprotection of the side chain and the two parts of baccatin
在脱保护过程中,常常需要将侧链及巴卡亭两部分分步进行,同时也会脱掉侧链N-保护基。很明显,如果能在一步反应中同时在侧链及巴卡亭两部分脱保护,且能保留N-保护基,这样对合成紫杉醇及多烯紫杉醇将会大大简化合成工艺。In the deprotection process, it is often necessary to carry out the side chain and baccatin step by step, and at the same time, the N-protecting group of the side chain will also be removed. Obviously, if the side chain and bacatine can be simultaneously deprotected in one step reaction, and the N-protecting group can be retained, the synthesis process of paclitaxel and docetaxel will be greatly simplified.
发明内容Contents of the invention
本发明目的是为半合成紫杉醇及多烯紫杉醇提供一种新的合成方法,是以10-DABIII为中间体,先选择性地保护C-7羟基,然后对C-10羟基进行酰化或保护,接着用保护的侧链酸与C-13羟基进行偶联,最后水解脱除保护基即得目标化合物紫杉醇或多烯紫杉醇。The purpose of the present invention is to provide a new synthetic method for semi-synthetic paclitaxel and docetaxel, which uses 10-DABIII as an intermediate to selectively protect the C-7 hydroxyl group, and then acylate or protect the C-10 hydroxyl group. , and then use the protected side chain acid to couple with the C-13 hydroxyl group, and finally hydrolyze and remove the protecting group to obtain the target compound paclitaxel or docetaxel.
本发明所提供的半合成紫杉醇及多烯紫杉醇的合成方法具体通过以下步骤实现:The synthetic method of semi-synthetic paclitaxel and docetaxel provided by the present invention is specifically realized through the following steps:
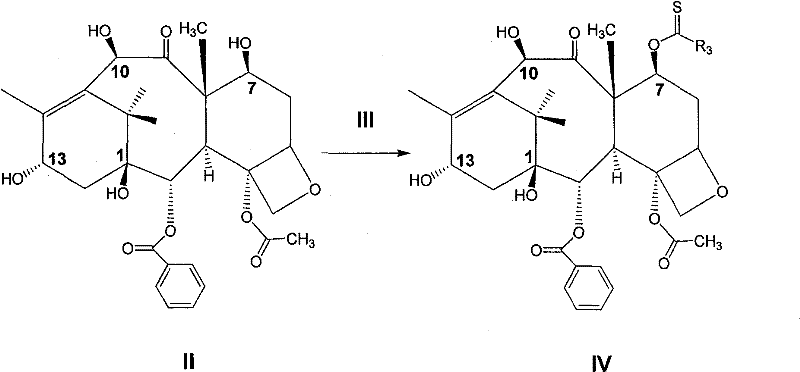
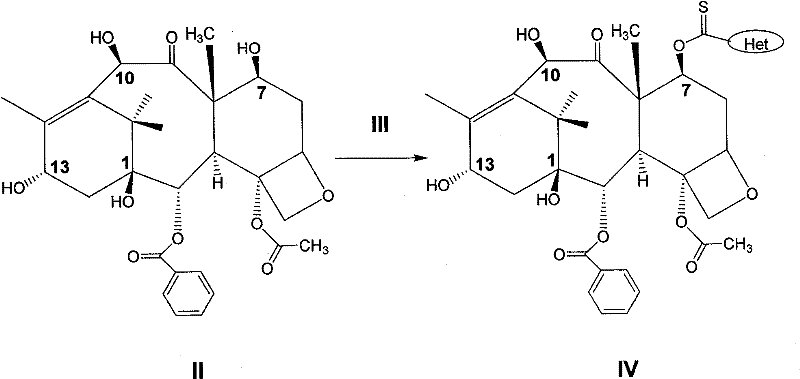
(1)10—去乙酰基巴卡亭(10-DAB III,化合物II)与化合物(III)在溶剂中进行反应,得到化合物(IV)。(1) 10-deacetylbaccatin (10-DAB III, compound II) reacts with compound (III) in a solvent to obtain compound (IV).


或or

其中R3-代表脂肪基、芳香基;X-代表卤素基、羟基;
上述为制备化合物(IV)的反应式,在该步反应中所用到的化合物(III)可以是酰卤化合物,也可以是杂环化合物,通常采用芳香酰卤或芳香杂环化合物,如硫代羰基二咪唑。反应的温度可以在-78~100℃之间,通常是在室温条件下进行,产率65~85%。所用到的溶剂可以是醚类溶剂,如1,4-二氧六环、二甲氧基乙烷、四氢呋喃等;也可以是芳香溶剂,如苯、甲苯、二甲苯等;还可以是氯代溶剂,如二氯甲烷、二氯乙烷、氯仿等,通常采用氯仿。所得产物(IV)的纯化可以通过重结晶的方法,如乙酸乙酯一正己烷;也可以进行快速硅胶柱层析。The above is the reaction formula for preparing compound (IV). The compound (III) used in this step reaction can be an acyl halide compound or a heterocyclic compound, usually an aromatic acyl halide or an aromatic heterocyclic compound, such as thio Carbonyldiimidazole. The reaction temperature can be between -78-100°C, usually at room temperature, and the yield is 65-85%. The solvent used can be an ether solvent, such as 1,4-dioxane, dimethoxyethane, tetrahydrofuran, etc.; it can also be an aromatic solvent, such as benzene, toluene, xylene, etc.; it can also be chlorinated Solvents, such as dichloromethane, dichloroethane, chloroform, etc., usually use chloroform. The resulting product (IV) can be purified by recrystallization, such as ethyl acetate-n-hexane; flash silica gel column chromatography can also be performed.
(2)化合物(IV)与乙酰化试剂在溶剂中进行反应,得到化合物(Va);化合物(IV)与氯乙酰氯等试剂在溶剂中进行反应,对C-10羟基进行保护得到化合物(Vb)。(2) Compound (IV) reacts with an acetylating reagent in a solvent to obtain compound (Va); compound (IV) reacts with reagents such as chloroacetyl chloride in a solvent to protect the C-10 hydroxyl group to obtain compound (Vb ).

此处R2-代表乙酰基、氯乙酰基、三氯乙氧羰酰基、苄氧羰酰基、三氯乙酰基;R3-代表脂肪基、芳香基;
上述为制备化合物(V)的反应式,在该步反应中所用到的乙酰化试剂可以是乙酰氯、乙酸酐、冰乙酸等,通常采用乙酰氯或乙酸酐;对C-10羟基保护所用到的试剂可以是氯乙酰氯、三氯乙氧羰酰氯、苄氧羰酰氯、三氯乙酰氯。使用的捕酸剂是一些弱碱,如吡啶、三乙胺、哌啶等。反应的温度可以在-78~100℃之间,通常在0~30℃之间。使用的溶剂可以是醚类溶剂,如四氢呋喃等;也可以是氯代溶剂,如二氯甲烷、氯仿等,还可以是吡啶,通常采用的是吡啶。重结晶和快速柱层析均是可采用的纯化方法。The above is the reaction formula for preparing compound (V). The acetylating reagent used in this step reaction can be acetyl chloride, acetic anhydride, glacial acetic acid, etc., usually using acetyl chloride or acetic anhydride; used for C-10 hydroxyl protection. The reagent can be chloroacetyl chloride, trichloroethoxycarbonyl chloride, benzyloxycarbonyl chloride, trichloroacetyl chloride. The acid scavenger used is some weak bases, such as pyridine, triethylamine, piperidine, etc. The reaction temperature can be between -78°C and 100°C, usually between 0°C and 30°C. The solvent used can be an ether solvent, such as tetrahydrofuran, etc.; it can also be a chlorinated solvent, such as methylene chloride, chloroform, etc., or it can be pyridine, which is usually pyridine. Both recrystallization and flash column chromatography are available purification methods.
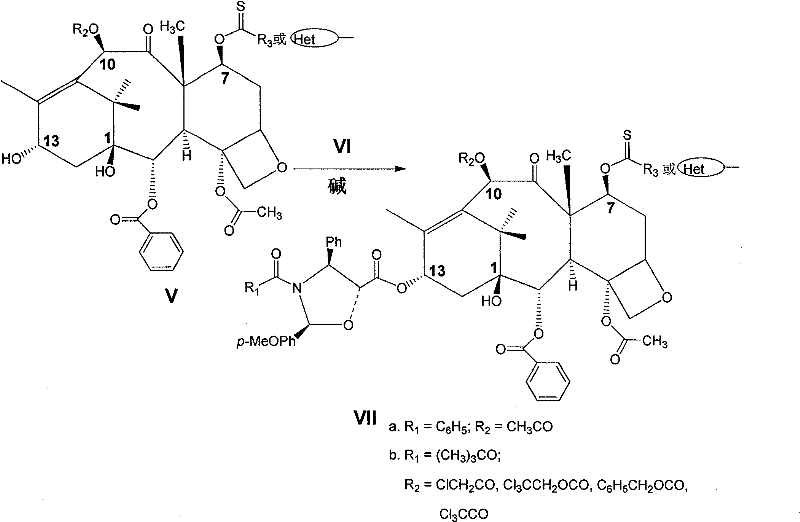
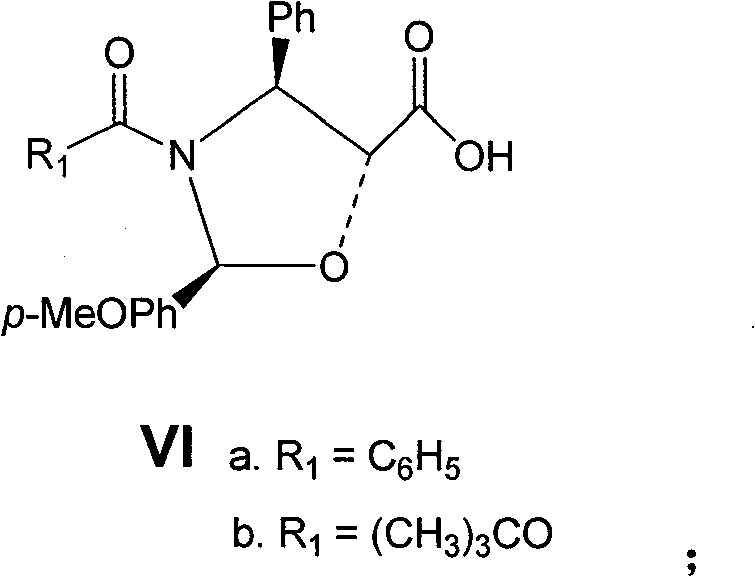
(3)化合物(Va)中的C-13羟基与保护的侧链酸(VIa)在碱存在下在溶剂中进行酯化偶联,得到化合物(VIIa);化合物(Vb)中的C-13羟基与保护的侧链酸(VIb)在碱存在下在溶剂中进行酯化偶联,得到化合物(VIIb)。(3) The C-13 hydroxyl in compound (Va) and the protected side chain acid (VIa) carry out esterification coupling in a solvent in the presence of a base to obtain compound (VIIa); C-13 in compound (Vb) The hydroxyl group and the protected side chain acid (VIb) are esterified and coupled in a solvent in the presence of a base to obtain compound (VIIb).


此处R1-代表苯基、叔丁氧基;R2-代表乙酰基、氯乙酰基、三氯乙氧羰酰基、苄氧羰酰基、三氯乙酰基;R3-代表脂肪基、芳香基:
上述为制备化合物(VII)的反应式,在该步反应中所用到的碱包括两部分:一部分是缩合剂羰二酰亚胺类,如二环己基羰二酰亚胺(DCC)、二异丙基羰二酰亚胺(DIC)等;另一部分是激活剂,如4-二甲氨基吡啶等。可选用的溶剂可以是醚性溶剂,如1,4-二氧六环、二甲氧基乙烷、四氢呋喃等;可以是芳香溶剂,如苯、甲苯、二甲苯等;可以是氯代溶剂,如二氯甲烷、二氯乙烷、氯仿等;也可以是酰胺溶剂,如二甲基甲酰胺、二甲基乙酰胺等;还可以是它们的组合,如苯—四氢呋喃、甲苯—二氯甲烷等。该步反应可以温控在-78~100℃之间,通常在0~30℃之间。所得产物(VII)的粗品可采用快速柱层析进行纯化,也可不经纯化直接进入下一步反应。Above-mentioned is the reaction formula of preparing compound (VII), and the alkali used in this step reaction comprises two parts: a part is condensing agent carbonyl imides, as dicyclohexyl carbonyl imide (DCC), diiso Propyl carbonyl imide (DIC), etc.; the other part is an activator, such as 4-dimethylaminopyridine, etc. The optional solvent can be an ether solvent, such as 1,4-dioxane, dimethoxyethane, tetrahydrofuran, etc.; it can be an aromatic solvent, such as benzene, toluene, xylene, etc.; it can be a chlorinated solvent, Such as dichloromethane, dichloroethane, chloroform, etc.; it can also be an amide solvent, such as dimethylformamide, dimethylacetamide, etc.; it can also be a combination of them, such as benzene-tetrahydrofuran, toluene-dichloromethane wait. The reaction temperature of this step can be controlled between -78°C and 100°C, usually between 0°C and 30°C. The crude product of the obtained product (VII) can be purified by flash column chromatography, or directly enter the next reaction without purification.
(4)化合物(VIIa)在酸性条件下进行一步反应脱除C-7保护基及噁唑环保护基,得到目标产物紫杉醇(Ia);化合物(VIIb)在酸性条件下进行一步反应脱除C-7保护基、C-10保护基及噁唑环保护基,得到目标产物多烯紫杉醇(Ib)。(4) Compound (VIIa) carries out a one-step reaction under acidic conditions to remove the C-7 protecting group and the oxazole ring protecting group to obtain the target product paclitaxel (Ia); compound (VIIb) carries out a one-step reaction under acidic conditions to remove C -7 protecting group, C-10 protecting group and oxazole ring protecting group, obtain target product docetaxel (Ib).

此处R1-代表苯基、叔丁氧基;R2-代表乙酰基、氯乙酰基、三氯乙氧羰酰基、苄氧羰酰基、三氯乙酰基;R3-代表脂肪基、芳香基;
上述为制备目标化合物紫杉醇及多烯紫杉醇的反应式,在该步反应中所用到的弱酸可以是有机酸,如甲酸、乙酸、三氟乙酸、对甲苯磺酸等;也可以是无机酸的水溶液,如稀盐酸、稀硫酸、磷酸等;还可以是它们的组合,如对甲苯磺酸-稀盐酸、三氟乙酸-乙酸等。反应的温度可以在-78~100℃之间,通常是室温条件。可选用的溶剂可以是水、四氢呋喃、二氧六环、乙腈、甲醇、乙醇等。所得粗品经过柱层析可得纯产物。The above is the reaction formula for preparing the target compound paclitaxel and docetaxel. The weak acid used in this step reaction can be an organic acid, such as formic acid, acetic acid, trifluoroacetic acid, p-toluenesulfonic acid, etc.; it can also be an aqueous solution of an inorganic acid , such as dilute hydrochloric acid, dilute sulfuric acid, phosphoric acid, etc.; it can also be a combination of them, such as p-toluenesulfonic acid-dilute hydrochloric acid, trifluoroacetic acid-acetic acid, etc. The reaction temperature can be between -78°C and 100°C, usually at room temperature. The optional solvent can be water, tetrahydrofuran, dioxane, acetonitrile, methanol, ethanol, etc. The resulting crude product can be purified by column chromatography.
本发明提供的紫杉醇合成新方法具有以下特点,(1)克服目前几种保护基存在的缺点,开发出一种新的保护基——咪唑基硫羰基。(2)为了避免C-2’差向异构化的形成及剧烈的反应条件,合成了2-单取代-1,3-噁唑烷型侧链酸。(3)设计合理,所用试剂价格低廉、反应步骤少以及反应条件温和等。本发明方法为紫杉醇的工业化生产提供了有利条件。同时,本发明为多烯紫杉醇的合成提供了一种新途径。The new method for synthesizing paclitaxel provided by the invention has the following characteristics: (1) Overcome the shortcomings of several protecting groups at present, and develop a new protecting group——imidazolylthiocarbonyl. (2) In order to avoid the formation of C-2' epimerization and severe reaction conditions, a 2-monosubstituted-1,3-oxazolidine type side chain acid was synthesized. (3) The design is reasonable, the reagents used are cheap, the reaction steps are few, and the reaction conditions are mild. The method of the invention provides favorable conditions for the industrial production of paclitaxel. At the same time, the invention provides a new way for the synthesis of docetaxel.
具体实施方式Detailed ways
下面将通过实施例对本发明作进一步的说明。The present invention will be further described below through embodiment.
实施例1:7-(咪唑基)硫代羰氧基-10-去乙酰巴卡亭-III(IV)的制备Example 1: Preparation of 7-(imidazolyl)thiocarbonyloxy-10-deacetylbaccatin-III(IV)
将1.25g(2.3mmol)10-去乙酰巴卡亭-III溶入110mL无水氯仿中,室温搅拌下加入0.82g(4.6mmol)1,1’-硫代羰基二咪唑,继续搅拌,体系呈亮黄透明,TLC监测反应进程。反应结束后,氯仿稀释,依次采用水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经乙酸乙酯一正己烷重结晶纯化,得白色固体化合物IV1.24g(72.6%)。m.p.145-148℃;IR(KBr)v:3445,1717,1245cm-1;1HNMR(400MHz,DMSO-d6)δ:8.31(s,1H),8.05(d,J=7.2Hz,2H),7.69(m,1H),7.67(s,1H),7.58(t,J1=8.0Hz,J2=7.2Hz,2H),7.09(s,1H),6.11(t,J1=9.2Hz,J2=8.4Hz,1H),5.51(d,J=6.8Hz,1H),5.29(d,J=4.4Hz,1H),5.24(s,1H),5.18(s,1H),5.02(d,J=9.2Hz,1H),4.63(s,1H),4.46(s,1H),4.14(s,2H),4.04(d,J=6.8Hz,1H),2.73(m,1H),2.25(s,3H),2.21(m,2H),1.94(s,3H),1.90(m,1H),1.85(s,3H),0.96(s,3H),0.94(s,3H);13CNMR(100MHz,DMSO-d6)δ:209.23,182.74,169.99,165.29,141.88,136.91,134.40,133.28,130.76,130.11,129.53(2C),128.69(2C),118.56,82.70,81.86,79.45,76.93,75.39,74.69,74.39,69.95,55.38,46.16,42.27,39.09,31.21,26.57,22.14,20.07,14.79,11.22;LRMS calcd for C33H38N2O10S[M]+654.2,found654.4.Dissolve 1.25g (2.3mmol) of 10-deacetylbaccatin-III into 110mL of anhydrous chloroform, add 0.82g (4.6mmol) of 1,1'-thiocarbonyldiimidazole under stirring at room temperature, and continue stirring, the system is Bright yellow and transparent, TLC monitors the reaction process. After the reaction, it was diluted with chloroform, and washed with water and saturated sodium chloride solution successively. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by recrystallization from ethyl acetate-n-hexane to obtain 1.24 g (72.6%) of compound IV as a white solid. mp145-148°C; IR(KBr)v: 3445, 1717, 1245cm -1 ; 1 HNMR (400MHz, DMSO-d 6 ) δ: 8.31(s, 1H), 8.05(d, J=7.2Hz, 2H), 7.69(m, 1H), 7.67(s, 1H), 7.58(t, J 1 =8.0Hz, J2=7.2Hz, 2H), 7.09(s, 1H), 6.11(t, J 1 =9.2Hz, J 2 =8.4Hz, 1H), 5.51(d, J=6.8Hz, 1H), 5.29(d, J=4.4Hz, 1H), 5.24(s, 1H), 5.18(s, 1H), 5.02(d, J=9.2Hz, 1H), 4.63(s, 1H), 4.46(s, 1H), 4.14(s, 2H), 4.04(d, J=6.8Hz, 1H), 2.73(m, 1H), 2.25( s, 3H), 2.21(m, 2H), 1.94(s, 3H), 1.90(m, 1H), 1.85(s, 3H), 0.96(s, 3H), 0.94(s, 3H); 13 CNMR( 100MHz, DMSO-d6)δ: 209.23, 182.74, 169.99, 165.29, 141.88, 136.91, 134.40, 133.28, 130.76, 130.11, 129.53(2C), 128.69(2C), 118.56, 82.70, 81.856, 76 74.69, 74.39, 69.95, 55.38, 46.16, 42.27, 39.09, 31.21, 26.57, 22.14, 20.07, 14.79, 11.22; LRMS calcd for C 33 H 38 N 2 O 10 S[M] + 654.2, found 654.4.
实施例2:7-(咪唑基)硫代羰氧基-巴卡亭-III(V)的制备Example 2: Preparation of 7-(imidazolyl)thiocarbonyloxy-baccatin-III(V)
将1.11g(1.7mmol)化合物IV溶入15mL无水吡啶中,通氮气,搅拌,滴加3.3ml(34mmol)乙酸酐,室温下继续搅拌24h。加入等体积的乙酸乙酯和水,搅拌,静置分层,水相用乙酸乙酯提取两次,合并有机相,用饱和的硫酸铜溶液洗涤至完全除去吡啶,再依次用水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为3∶4),得白色固体化合物V0.97g(71.7%)。m.p.241-244℃;IR(KBr)v:3466,1722,1236cm-1;1HNMR(400MHz,DMSO-d6)δ:8.15(s,1H),8.04(d,J=7.2Hz,2H),7.69(t,J1=7.2Hz,,J2=8.0Hz,1H),7.59(t,J=7.6Hz,2H),7.54(s,1H),7.03(s,1H),6.16(s,1H),6.12(m,1H),5.49(d,J=7.2Hz,1H),5.44(d,J=4.4Hz,1H),5.05(d,J=5.2Hz,1H),4.65(s,2H),4.13(s,2H),3.95(d,J=7.2Hz,1H),2.86(m,1H),2.25(s,3H),2.19(m,2H),1.95(s,6H),1.86(m,1H),1.84(s,3H),0.98(s,6H);13CNMR(100MHz,DMSO-d6):202.69,183.07,170.12,168.47,165.25,146.25,136.85,133.39,130.36,130.06,129.99,129.59(2C),128.76(2C),118.47,82.69,81.35,79.46,76.76,75.51,75.25,74.01,66.00,55.51,46.66,42.44,39.10,31.09,26.54,22.18,20.56,20.33,15.27,11.22;LRMS calcdfor C35H40N2O11S[M]+696.2,found 696.5.1.11g (1.7mmol) of compound IV was dissolved in 15mL of anhydrous pyridine, nitrogen was blown, stirred, 3.3ml (34mmol) of acetic anhydride was added dropwise, and stirring was continued at room temperature for 24h. Add an equal volume of ethyl acetate and water, stir, let stand to separate layers, extract the aqueous phase twice with ethyl acetate, combine the organic phases, wash with saturated copper sulfate solution until pyridine is completely removed, and then successively water and saturated chlorine NaCl solution for washing. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 3:4) to obtain 0.97 g (71.7%) of compound V as a white solid. mp241-244°C; IR(KBr)v: 3466, 1722, 1236cm -1 ; 1 HNMR (400MHz, DMSO-d 6 ) δ: 8.15(s, 1H), 8.04(d, J=7.2Hz, 2H), 7.69(t, J1=7.2Hz, J2 =8.0Hz, 1H), 7.59(t, J=7.6Hz, 2H), 7.54(s, 1H), 7.03(s, 1H), 6.16(s, 1H ), 6.12(m, 1H), 5.49(d, J=7.2Hz, 1H), 5.44(d, J=4.4Hz, 1H), 5.05(d, J=5.2Hz, 1H), 4.65(s, 2H ), 4.13(s, 2H), 3.95(d, J=7.2Hz, 1H), 2.86(m, 1H), 2.25(s, 3H), 2.19(m, 2H), 1.95(s, 6H), 1.86 (m, 1H), 1.84(s, 3H), 0.98(s, 6H); 13 CNMR (100MHz, DMSO-d 6 ): 202.69, 183.07, 170.12, 168.47, 165.25, 146.25, 136.85, 133.39, 130.36, 130.06 , 129.99, 129.59(2C), 128.76(2C), 118.47, 82.69, 81.35, 79.46, 76.76, 75.51, 75.25, 74.01, 66.00, 55.51, 46.66, 42.44, 39.10, 31.09, 25.34, 20.25 , 11.22; LRMS calcdfor C 35 H 40 N 2 O 11 S[M] + 696.2, found 696.5.
实施例3:13-(2’-苯基-4’-对甲氧基苯基N-苯甲酰基-噁唑烷-1-羰氧基)-7-(咪唑基)硫代羰氧基-巴卡亭-III(VIIa)的制备Example 3: 13-(2'-phenyl-4'-p-methoxyphenyl N-benzoyl-oxazolidine-1-carbonyloxy)-7-(imidazolyl)thiocarbonyloxy - Preparation of baccatin-III (VIIa)
将0.77g(1.1mmol)化合物V溶入120mL无水甲苯和二氯甲烷(体积比为2∶1)中,搅拌下加入1.33g(3.3mmol)化合物VIa,40.3mg(0.33mmol)N,N’-二甲氨基吡啶(DMAP),搅拌10min后,加入340mg(1.65mmol)二环己基羰二酰亚胺(DCC),稍后体系变浑浊。室温条件下继续搅拌,TLC检测反应进程。反应完毕后,将浑浊物虑除,乙酸乙酯稀释,依次用饱和的氯化铵溶液、碳酸氢钠溶液和氯化钠溶液洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为1∶2),得白色固体化合物VIIa 1.02g(75%)。m.p.157-161℃;IR(KBr)v:3428,1726,1653,1245 cm-1;1HNMR(400MHz,DMSO-d6)δ:8.18(s,1H),8.02(d,J=7.2Hz,2H),7.69(t,J1=7.6Hz,J2=7.2Hz,1H),7.59(t,J=7.6Hz,2H),7.56(s,1H),7.44-7.26(m,12H),7.04(s,1H),6.96(d,J=8.0Hz,2H),6.63(s,1H),6.20(s,1H),6.17-6.11(m,2H),5.58(d,J=6.8Hz,1H),5.51(s,1H),5.16(s,1H),5.09(s,1H),5.03(d,J=9.2Hz,1H),4.12(s,2H),3.95(d,J=7.2Hz,1H),3.78(s,3H),2.84(m,1H),2.38(m,1H),2.25(m,1H),2.14(s,3H),1.99(s,3H),1.96(s,3H),1.87(s,3H),1.86(m,1H),1.16(s,3H),1.03(s,3H);13CNMR(100MHz,DMSO-d6)δ:202.03,183.02,170.39,169.35,169.32,168.39,165.13,159.59(2C),156.58,140.14,139.08,136.86,135.79,133.53,132.62,130.39,129.68,129.58(2C),129.41,128.74,128.52(2C),128.39(2C),128.22(2C),127.91,126.84(2C),126.49(2C),118.46,113.63(2C),90.14,82.60,82.11,80.99,79.65,76.66,75.23,74.65,73.84,70.93,66.09,55.54,55.16,47.48,43.01,35.15,31.02,25.30,21.60,21.10,20.23,14.18,11.21;LRMS calcd for C59H59N3O15S[M]+1081.3,found1081.0.Dissolve 0.77g (1.1mmol) of compound V into 120mL of anhydrous toluene and dichloromethane (volume ratio is 2:1), add 1.33g (3.3mmol) of compound VIa, 40.3mg (0.33mmol) N, N '-Dimethylaminopyridine (DMAP), after stirring for 10 min, 340 mg (1.65 mmol) of dicyclohexylcarbonyl imide (DCC) was added, and the system became turbid after a while. Stirring was continued at room temperature, and the reaction progress was detected by TLC. After the reaction was completed, the turbidity was filtered off, diluted with ethyl acetate, and washed with saturated ammonium chloride solution, sodium bicarbonate solution and sodium chloride solution in sequence. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 1:2) to obtain 1.02 g (75%) of compound VIIa as a white solid. mp157-161°C; IR(KBr)v: 3428, 1726, 1653, 1245 cm -1 ; 1 HNMR (400MHz, DMSO-d 6 ) δ: 8.18(s, 1H), 8.02(d, J=7.2Hz, 2H), 7.69(t, J 1 =7.6Hz, J 2 =7.2Hz, 1H), 7.59(t, J=7.6Hz, 2H), 7.56(s, 1H), 7.44-7.26(m, 12H), 7.04(s, 1H), 6.96(d, J=8.0Hz, 2H), 6.63(s, 1H), 6.20(s, 1H), 6.17-6.11(m, 2H), 5.58(d, J=6.8Hz , 1H), 5.51(s, 1H), 5.16(s, 1H), 5.09(s, 1H), 5.03(d, J=9.2Hz, 1H), 4.12(s, 2H), 3.95(d, J= 7.2Hz, 1H), 3.78(s, 3H), 2.84(m, 1H), 2.38(m, 1H), 2.25(m, 1H), 2.14(s, 3H), 1.99(s, 3H), 1.96( s, 3H), 1.87 (s, 3H), 1.86 (m, 1H), 1.16 (s, 3H), 1.03 (s, 3H); 13 CNMR (100MHz, DMSO-d 6 ) δ: 202.03, 183.02, 170.39 , 169.35, 169.32, 168.39, 165.13, 159.59 (2c), 156.58, 140.14, 139.08, 136.86, 135.79, 133.53, 132.62, 130.39, 129.58 (2C), 129.74, 128.52 (2C), 128.74, 128.52 (2C), 128.5.52 (28.74, 128.52 (28.74, 128.52 (28.74). , 128.22(2C), 127.91, 126.84(2C), 126.49(2C), 118.46, 113.63(2C), 90.14, 82.60, 82.11, 80.99, 79.65, 76.66, 75.23, 74.65, 73.84, 70.93, 65.56, 5 , 47.48, 43.01, 35.15, 31.02, 25.30, 21.60, 21.10, 20.23, 14.18, 11.21; LRMS calcd for C 59 H 59 N 3 O 15 S[M] + 1081.3, found 1081.0.
实施例4:紫杉醇(Ia)的制备Embodiment 4: the preparation of paclitaxel (Ia)
将0.82g(0.76mmol)化合物VIIa溶入30mL甲醇中,室温搅拌下加入145mg(0.84mmol)对甲苯磺酸,反应3h后,再加入10mL 0.1N的盐酸水溶液,TLC检测反应进程。反应结束后,乙酸乙酯稀释,再依次用饱和的碳酸氢钠溶液、水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压浓缩,粗品经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为1∶1),得白色固体化合物Ia 0.56g(67%)。m.p.206-209℃;1HNMR(400MHz,DMSO-d6)δ:8.89(d,J=8.8Hz,1H),7.99(d,J=9.2Hz,1H),7.97(d,J=8.0Hz,2H),7.88(d,J=8.4Hz,2H),7.73-7.36(m,9H),7.22(d,J=8.4Hz,1H),6.29(s,1H),6.17(d,J=7.6Hz,1H),5.89(t,J1=8.8Hz,J2=9.2Hz,1H),5.42-5.40(m,2H),4.92-4.90(m,2H),4.70(s,1H),4.58(t,J1=7.6Hz,J2=8.4Hz,1H),4.41(d,J=4.0Hz,2H),4.11-4.09(m,1H),3.61(d,J=7.6Hz,1H),2.32(m,1H),2.22(s,3H),2.12(m,1H),2.11(s,3H),1.88(m,2H),1.79(s,3H),1.50(s,3H),1.07(s,3H),1.05(s,3H);LRMS calcd for C47H51NO14[M]+853.3,found 853.0.0.82g (0.76mmol) of compound VIIa was dissolved in 30mL of methanol, and 145mg (0.84mmol) of p-toluenesulfonic acid was added under stirring at room temperature. After reacting for 3 hours, 10mL of 0.1N hydrochloric acid aqueous solution was added, and the reaction progress was detected by TLC. After the reaction was completed, it was diluted with ethyl acetate, and then washed with saturated sodium bicarbonate solution, water and saturated sodium chloride solution in sequence. Dry over anhydrous sodium sulfate, filter, and concentrate under reduced pressure. The crude product is purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 1:1) to obtain 0.56 g (67%) of white solid compound Ia . mp206-209°C; 1 HNMR (400MHz, DMSO-d 6 ) δ: 8.89(d, J=8.8Hz, 1H), 7.99(d, J=9.2Hz, 1H), 7.97(d, J=8.0Hz, 2H), 7.88(d, J=8.4Hz, 2H), 7.73-7.36(m, 9H), 7.22(d, J=8.4Hz, 1H), 6.29(s, 1H), 6.17(d, J=7.6 Hz, 1H), 5.89(t, J 1 =8.8Hz, J 2 =9.2Hz, 1H), 5.42-5.40(m, 2H), 4.92-4.90(m, 2H), 4.70(s, 1H), 4.58 (t, J1 = 7.6Hz, J2 = 8.4Hz, 1H), 4.41 (d, J = 4.0Hz, 2H), 4.11-4.09 (m, 1H), 3.61 (d, J = 7.6Hz, 1H) , 2.32(m, 1H), 2.22(s, 3H), 2.12(m, 1H), 2.11(s, 3H), 1.88(m, 2H), 1.79(s, 3H), 1.50(s, 3H), 1.07(s, 3H), 1.05(s, 3H); LRMS calcd for C 47 H 51 NO 14 [M] + 853.3, found 853.0.
实施例5:7-(苯基)硫代羰氧基-10-去乙酰巴卡亭-III(IV)的制备Example 5: Preparation of 7-(phenyl)thiocarbonyloxy-10-desacetylbaccatin-III(IV)
将1.25g(2.3mmol)10-去乙酰巴卡亭-III溶入110mL无水氯仿中,室温搅拌下加入0.64g(4.6mmol)硫代苯甲酸,继续搅拌,TLC监测反应进程。反应结束后,氯仿稀释,依次采用水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经乙酸乙酯—正己烷重结晶纯化,得化合物IV。Dissolve 1.25g (2.3mmol) of 10-deacetylbaccatin-III into 110mL of anhydrous chloroform, add 0.64g (4.6mmol) of thiobenzoic acid under stirring at room temperature, continue stirring, and monitor the reaction progress by TLC. After the reaction, it was diluted with chloroform, and washed with water and saturated sodium chloride solution successively. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by ethyl acetate-n-hexane recrystallization to obtain compound IV.
实施例6:7-(苯基)硫代羰氧基-10-三氯乙酰基-10-去乙酰巴卡亭-III(V)的制备Example 6: Preparation of 7-(phenyl)thiocarbonyloxy-10-trichloroacetyl-10-desacetylbaccatin-III(V)
将1.13g(1.7mmol)化合物IV溶入15mL无水吡啶中,通氮气,搅拌,滴加6.18g(34mmol)三氯乙酰氯,室温下继续搅拌24h。加入等体积的乙酸乙酯和水,搅拌,静置分层,水相用乙酸乙酯提取两次,合并有机相,用饱和的硫酸铜溶液洗涤至完全除去吡啶,再依次用水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为3∶4),得化合物V。Dissolve 1.13g (1.7mmol) of compound IV in 15mL of anhydrous pyridine, blow nitrogen gas, stir, add dropwise 6.18g (34mmol) of trichloroacetyl chloride, and continue stirring at room temperature for 24h. Add an equal volume of ethyl acetate and water, stir, let stand to separate layers, extract the aqueous phase twice with ethyl acetate, combine the organic phases, wash with saturated copper sulfate solution until pyridine is completely removed, and then successively water and saturated chlorine NaCl solution for washing. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 3:4) to obtain compound V.
实施例7:13-(2’-苯基-4’-对甲氧基苯基-N-叔丁氧羰基-噁唑烷-1-羰氧基)-7-(苯基)硫代羰氧基-10-三氯乙酰基-10-去乙酰巴卡亭-III(VIIb)的制备Example 7: 13-(2'-phenyl-4'-p-methoxyphenyl-N-tert-butoxycarbonyl-oxazolidine-1-carbonyloxy)-7-(phenyl)thiocarbonyl Preparation of Oxy-10-trichloroacetyl-10-desacetylbaccatin-III(VIIb)
将0.89g(1.1mmol)化合物V溶入120mL无水甲苯和二氯甲烷(体积比为2∶1)中,搅拌下加入1.32g(3.3mmol)化合物VIb,40.3mg(0.33mmol)N’N’-二甲氨基吡啶(DMAP),搅拌10min后,加入340mg(1.65mmol)二环己基羰二酰亚胺(DCC),稍后体系变浑浊。室温条件下继续搅拌,TLC检测反应进程。反应完毕后,将浑浊物虑除,乙酸乙酯稀释,依次用饱和的氯化铵溶液、碳酸氢钠溶液和氯化钠溶液洗涤。无水硫酸钠干燥,过虑,减压除去溶剂。浓缩物经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为1∶2),得化合物VIIb。Dissolve 0.89g (1.1mmol) of compound V into 120mL of anhydrous toluene and dichloromethane (2:1 by volume), and add 1.32g (3.3mmol) of compound VIb under stirring, 40.3mg (0.33mmol) N'N '-Dimethylaminopyridine (DMAP), after stirring for 10 min, 340 mg (1.65 mmol) of dicyclohexylcarbonyl imide (DCC) was added, and the system became turbid after a while. Stirring was continued at room temperature, and the reaction progress was detected by TLC. After the reaction was completed, the turbidity was filtered off, diluted with ethyl acetate, and washed with saturated ammonium chloride solution, sodium bicarbonate solution and sodium chloride solution in sequence. Dry over anhydrous sodium sulfate, filter, and remove the solvent under reduced pressure. The concentrate was purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 1:2) to obtain compound VIIb.
实施例8:多烯紫杉醇(Ib)的制备Embodiment 8: the preparation of docetaxel (Ib)
将0.91g(0.76mmol)化合物VIIb溶入30mL甲醇中,室温搅拌下加入145mg(0.84mmol)对甲苯磺酸,反应3h后,再加入10mL 0.1N的盐酸水溶液,TLC检测反应进程。反应结束后,乙酸乙酯稀释,再依次用饱和的碳酸氢钠溶液、水和饱和的氯化钠溶液进行洗涤。无水硫酸钠干燥,过虑,减压浓缩,粗品经快速柱色谱纯化(洗脱剂为乙酸乙酯—环己烷,体积比为1∶1),得化合物Ib。0.91g (0.76mmol) of compound VIIb was dissolved in 30mL of methanol, and 145mg (0.84mmol) of p-toluenesulfonic acid was added under stirring at room temperature. After reacting for 3h, 10mL of 0.1N hydrochloric acid aqueous solution was added, and the reaction progress was detected by TLC. After the reaction was completed, it was diluted with ethyl acetate, and then washed with saturated sodium bicarbonate solution, water and saturated sodium chloride solution in sequence. Dry over anhydrous sodium sulfate, filter, and concentrate under reduced pressure. The crude product is purified by flash column chromatography (eluent: ethyl acetate-cyclohexane, volume ratio: 1:1) to obtain compound Ib.
Claims (6)





Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007100669400A CN101007795B (en) | 2007-01-26 | 2007-01-26 | Synthetic method of paclitaxel and docetaxel |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007100669400A CN101007795B (en) | 2007-01-26 | 2007-01-26 | Synthetic method of paclitaxel and docetaxel |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101007795A CN101007795A (en) | 2007-08-01 |
| CN101007795B true CN101007795B (en) | 2010-08-18 |
Family
ID=38696492
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007100669400A Expired - Fee Related CN101007795B (en) | 2007-01-26 | 2007-01-26 | Synthetic method of paclitaxel and docetaxel |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101007795B (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011134067A1 (en) * | 2010-04-29 | 2011-11-03 | 6570763 Canada Inc. | Novel amino acid molecule and uses thereof |
| CN107056767B (en) * | 2015-12-04 | 2022-07-15 | 江苏恩华络康药物研发有限公司 | Process and intermediates for the preparation of water-soluble taxane derivatives |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1158126A (en) * | 1995-06-20 | 1997-08-27 | 法玛西雅厄普约翰公司 | Method for preparing paclitaxel and derivatives thereof |
| CN1670018A (en) * | 2004-03-18 | 2005-09-21 | 中国科学院大连化学物理研究所 | A kind of preparation method of paclitaxel |
-
2007
- 2007-01-26 CN CN2007100669400A patent/CN101007795B/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1158126A (en) * | 1995-06-20 | 1997-08-27 | 法玛西雅厄普约翰公司 | Method for preparing paclitaxel and derivatives thereof |
| CN1670018A (en) * | 2004-03-18 | 2005-09-21 | 中国科学院大连化学物理研究所 | A kind of preparation method of paclitaxel |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101007795A (en) | 2007-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU678423B2 (en) | 2-debenzoyl-2-acyl taxol derivatives and methods for making same | |
| US20190142784A1 (en) | 9,10-Alpha,Alpha-OH-Taxane Analogs and Methods for Production Thereof | |
| US5821363A (en) | Antineoplastic use and pharmaceutical compositions containing them | |
| US6500966B1 (en) | Process for the preparation of taxanes from 10-deacetylbaccatin III | |
| JP2002505326A (en) | Synthesis of paclitaxel from baccatin III by protection of 7-hydroxyl using strong base and electrophile | |
| CN101007795B (en) | Synthetic method of paclitaxel and docetaxel | |
| CN103012328B (en) | Method for preparing second-generation taxol anticancer drug Cabazitaxel | |
| US6891050B2 (en) | Process for the preparation of taxanes such as paclitaxel, docetaxel and structurally similar analogs | |
| RS52438B (en) | PREPARATION OF 9-DIHYDRO-13-ACETYLBACCATINE TAXAN III | |
| HU229373B1 (en) | A process for the preparation of paclitaxel | |
| AU2002234535A1 (en) | A process for the preparation of paclitaxel | |
| CN103012330A (en) | Preparation method of taxol anticancer drugs XRP6258 | |
| CN106632297A (en) | Docetaxel side chain 2'-derived novel taxanes antitumor compound as well as synthesis method and application thereof | |
| CN101448839B (en) | A process for the preparation of a taxane derivative | |
| CN101128448A (en) | Method for preparing paclitaxel | |
| CN102757410A (en) | Simple and efficient method for preparing taxol analog Larotaxel | |
| CN104610247A (en) | Semi-synthetic taxane derivative as well as preparation method and application thereof | |
| JP5870197B2 (en) | Method for producing taxane derivative | |
| EP0747372A1 (en) | Taxane derivatives from 14-beta-hydroxy-10 deacetybaccatin III |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20100818 Termination date: 20110126 |