CN114634412A - 一种高纯度龙胆酸及其应用 - Google Patents
一种高纯度龙胆酸及其应用 Download PDFInfo
- Publication number
- CN114634412A CN114634412A CN202210537742.2A CN202210537742A CN114634412A CN 114634412 A CN114634412 A CN 114634412A CN 202210537742 A CN202210537742 A CN 202210537742A CN 114634412 A CN114634412 A CN 114634412A
- Authority
- CN
- China
- Prior art keywords
- gentisic acid
- acid
- purity
- gentisic
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C65/00—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C65/01—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups
- C07C65/03—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups monocyclic and having all hydroxy or O-metal groups bound to the ring
- C07C65/05—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups monocyclic and having all hydroxy or O-metal groups bound to the ring o-Hydroxy carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/06—Free radical scavengers or antioxidants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/02—Preparation of carboxylic acids or their salts, halides or anhydrides from salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
- C07C51/412—Preparation of salts of carboxylic acids by conversion of the acids, their salts, esters or anhydrides with the same carboxylic acid part
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/47—Separation; Purification; Stabilisation; Use of additives by solid-liquid treatment; by chemisorption
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/48—Separation; Purification; Stabilisation; Use of additives by liquid-liquid treatment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/487—Separation; Purification; Stabilisation; Use of additives by treatment giving rise to chemical modification
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Toxicology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本申请公开了一种高纯度龙胆酸,其纯度≥99.50%,其包含:龙胆酸、水杨酸和杂质A,其中,所述杂质A的结构式为
Description
技术领域
本申请涉及医药化工领域。具体地,本申请涉及一种高纯度龙胆酸及其应用。
背景技术
龙胆酸,化学名称2,5-二羟基苯甲酸,也称为5-羟基水杨酸,是水杨酸经肾代谢之后的次要产物,临床上常用作抗病毒剂、抗菌剂及止痛剂(钠盐)。龙胆酸属氢醌类化合物,容易发生氧化反应,在制药工业上还用作抗氧化赋形剂。龙胆酸已在国外作为药用辅料。
现有技术中有采用苯甲酸为原料通过氯化、水解等步骤合成产物,但用该方法合成出来的主要产物有五种,产物难分离纯化,导致最终收率仅为5%左右,难实现工业化生产。
现行工艺中主要通过氢醌的科贝尔-施密特反应来制备龙胆酸,但该方法制得的产品含有大量的异构体2,4-二羟基苯甲酸和2,6-二羟基苯甲酸,不但产品纯度较低,而且色泽较深,为黄色粉末结晶,需要经过多次的重结晶纯化才能得到符合规格的白色龙胆酸产品,收率低,成本高。
专利CN100363322C公开了一种生产羟基苯甲酸化合物的方法,该方法包括从酚类化合物制备酚类化合物的碱金属盐,该碱金属盐与二氧化碳反应,其中,从酚类化合物制备酚类化合物的碱金属盐包括以下步骤:a)使碱金属醇盐与过量酚类化合物反应,所述酚类化合物过量于碱金属醇盐,形成酚类化合物碱金属盐;和b)在进行a)的同时,从反应中蒸馏出所形成的醇。该方法不采用非质子极性有机溶剂。
专利CN1684935A公开了一种羟基苯甲酸类的制备方法,是使本酚类与碱金属化合物进行脱水反应得到苯酚类的碱金属盐后,使该苯酚类的碱金属盐与二氧化碳反应,制备羟基苯甲酸类的方法中,碱金属化合物和相对于碱金属化合物过量的苯酚类在160℃或160℃以上的温度下进行脱水反应。该发明的方法不使用非质子性极性有机溶剂,工艺简易、成本低廉。
专利CN102766042A公开一种制备高纯度龙胆酸的方法,包括:将水杨酸与冰醋酸加入到反应容器中搅拌均匀;升温后滴加溴素,反应得到溴代水杨酸,减压蒸馏回收冰醋酸;在氯化亚铜存在的条件下,加入液碱,升温,反应得到羟基水杨酸;冷却至常温,过滤除去氯化亚铜,调节pH值至2;滤出羟基水杨酸的混合物并加入到水中,调节pH值至6.9~7.1,冷却结晶得到高纯度的龙胆酸钠盐;将得到的龙胆酸钠盐加入到水中,调节pH值至2,在活性碳与草酸存在的条件下,升温回流;过滤,滤液冷却结晶即得到龙胆酸。
上述现有技术各方法得到的龙胆酸产品纯度均较差,收率均较低。
发明内容
为了克服现有技术存在的缺陷,本申请在现有技术的基础上,对催化剂、碱进行筛选,对反应温度等进行优化研究,并针对产品纯度低,杂质含量较多情况下,开发出后续的成盐、游离、脱色结晶工艺,最终得到一种合格的高纯度龙胆酸。
本申请的具体技术方案如下:
1. 一种高纯度龙胆酸,其特征在于,其纯度≥99.50%,其包含:龙胆酸、水杨酸和杂质A,其中,所述杂质A具有式(I)所示的结构:
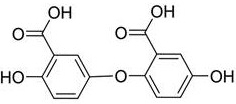
式(I);
优选地,所述高纯度龙胆酸中,所述水杨酸的质量百分比≤0.1%,所述杂质A的质量百分比≤0.1%;
优选地,所述高纯度龙胆酸还包含杂质B,其中,所述杂质B具有式(II)所示的结构:
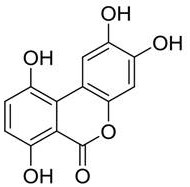
式(II);
优选地,所述高纯度龙胆酸中,所述杂质B的质量百分比≤0.1%。
2.根据项1所述的高纯度龙胆酸,其特征在于,其通过包括下述步骤的方法制备而成:
投料反应:将5-溴水杨酸加入无机碱溶液反应;
酸化:加入浓盐酸调节pH,析出固体,过滤;
萃取:向酸化步骤得到的滤液中加入萃取剂一,萃取,静置后出现絮状物;
过滤:过滤、分液,得到有机相一和水相,向水相中加入萃取剂二,萃取,再过滤、分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
析晶:浓缩有机相三,析出固体,加入有机溶剂一,得龙胆酸粗品;
成盐:将龙胆酸粗品加入有机溶剂二中,滴加含有有机碱的有机溶剂二的溶液,得龙胆酸有机碱盐湿品;
游离:将龙胆酸有机碱盐湿品加入水中,滴加浓盐酸调节pH,过滤,得龙胆酸精制品湿品;
脱色与结晶:将龙胆酸精制品湿品和脱色剂加入水中,过滤,得到龙胆酸晶体;
过滤与干燥:将龙胆酸晶体用水淋洗后再用有机溶剂一清洗,干燥,得高纯度龙胆酸。
3. 根据项2所述的高纯度龙胆酸,其特征在于,在投料反应步骤中,在铜催化剂存在的条件下,将5-溴水杨酸加入无机碱溶液,搅拌,升温使5-溴水杨酸与无机碱溶液反应,再降温,加入水;
优选地,所述无机碱溶液为氢氧化钠的水溶液或氢氧化钾的水溶液,优选为氢氧化钾的水溶液;
优选地,所述无机碱溶液的浓度为4~8mol/L,更优选为5~6mol/L;
优选地,所述无机碱与5-溴水杨酸的物质的量之比为4~7:1,更优选为5~6:1;
优选地,所述升温为升至90~114℃,更优选为升至105~114℃;
优选地,升温使5-溴水杨酸与无机碱溶液反应,HPLC中控5-溴水杨酸的含量小于1.5%,反应时间为20~48h,更优选为20~30h;
优选地,所述降温为降至20~30℃;
优选地,在铜催化剂和草酸存在的条件下,将5-溴水杨酸加入无机碱溶液;更优选地,所述草酸与5-溴水杨酸的物质的量之比为0.05~0.15:1;
优选地,所述铜催化剂为五水合硫酸铜或氧化亚铜,更优选为五水合硫酸铜;优选地,所述五水合硫酸铜与5-溴水杨酸的物质的量之比为0.05~0.15:1;优选地,所述氧化亚铜与5-溴水杨酸的物质的量之比为0.025~0.075:1。
优选地,所述水与5-溴水杨酸的物质的量之比为40~70:1,更优选为50~60:1。
4. 根据项2或3所述的高纯度龙胆酸,其特征在于,在酸化步骤中,调节pH至2.3~2.5;
优选地,在20~30℃条件下加入浓盐酸调节pH。
5. 根据项2~4中任一项所述的高纯度龙胆酸,其特征在于,在萃取步骤中,所述萃取剂一选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上;
优选地,所述萃取剂一为甲基叔丁基醚;
优选地,在萃取步骤中,向酸化步骤得到的滤液中加入萃取剂一和萃取助剂,所述萃取助剂优选为氯化钠;
优选地,所述甲基叔丁基醚与5-溴水杨酸的质量比为4~6:1,优选为5~5.5:1;
优选地,所述氯化钠与5-溴水杨酸的质量比为1.0~1.5:1;
优选地,在过滤步骤中,所述萃取剂二选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上;
优选地,所述萃取剂二与5-溴水杨酸的质量比为4~6:1,优选为5~5.5:1;
优选地,向漏斗中加入助滤剂,过滤、分液,得到有机相一和水相;更优选地,所述助滤剂为硅藻土。
6. 根据项2~5中任一项所述的高纯度龙胆酸,其特征在于,在析晶步骤中,浓缩有机相三,析出固体,加入有机溶剂一,搅拌,过滤并将滤饼干燥,得龙胆酸粗品;
优选地,所述有机溶剂一选自正庚烷、丙酮、乙酸乙酯、石油醚和氯仿中的一种或两种以上;
优选地,浓缩有机相三至析出大量固体;
优选地,在40~60℃条件下,更优选为在40~50℃条件下减压浓缩有机相三;
优选地,所述有机溶剂一与5-溴水杨酸的质量比为4~5.5:1,优选为4.5~5:1;
优选地,所述搅拌时间为1~2h;
优选地,在20~30℃条件下过滤;
优选地,所述滤饼在40~50℃条件下真空干燥2~4h,得龙胆酸粗品。
7. 根据项2~6中任一项所述的高纯度龙胆酸,其特征在于,在成盐步骤中,将龙胆酸粗品加入有机溶剂二中,搅拌,滴加含有有机碱的有机溶剂二的溶液,降温,过滤,得龙胆酸有机碱盐湿品;
优选地,在10~35℃条件下,滴加含有有机碱的有机溶剂二的溶液;
优选地,所述有机碱为二乙胺;
优选地,所述有机碱与5-溴水杨酸的物质的量之比为0.75~0.85:1;
优选地,所述有机溶剂二与龙胆酸粗品的质量比为8.6~10:1,优选为9~9.8:1;
优选地,所述有机溶剂二选自丙酮、异丙醇和四氢呋喃中的一种或两种以上;
优选地,所述降温为降至2~8℃。
8. 根据项2~7中任一项所述的高纯度龙胆酸,其特征在于,在游离步骤中,将龙胆酸有机碱盐湿品加入水中,搅拌,滴加浓盐酸调节pH,降温,过滤,得龙胆酸精制品湿品;
优选地,在20~30℃条件下滴加浓盐酸调节pH;
优选地,调节pH至1~2;
优选地,所述水与龙胆酸有机碱盐湿品的质量比为4~6:1;
优选地,所述降温为降至0~10℃。
9. 根据项2~8中任一项所述的高纯度龙胆酸,其特征在于,在脱色与结晶步骤中,将龙胆酸精制品湿品和脱色剂加入水中,搅拌,升温,过滤并将滤液降温,得到龙胆酸晶体;
优选地,将龙胆酸精制品湿品、脱色剂和草酸加入水中;
优选地,所述脱色剂选自活性炭、氧化铝和白土中的一种或两种以上;
优选地,升温后保温0.5~6h,优选为0.5~4h,更优选为0.5~1.5h;
优选地,所述升温为升至65~75℃;
优选地,所述活性炭与龙胆酸精制品湿品的质量比为0.05~0.1:1;
优选地,所述草酸与龙胆酸精制品湿品的质量比为0.01~0.03:1;
优选地,所述水与龙胆酸精制品湿品的质量比为4~6:1;
优选地,过滤并将滤液降温至0~10℃,得到龙胆酸晶体。
10. 根据项2~9中任一项所述的高纯度龙胆酸,其特征在于,在过滤与干燥步骤中,所述干燥条件为:40~55℃,真空干燥10~15h。
11. 根据项1~10中任一项所述的高纯度龙胆酸在制备抗病毒剂、抗菌剂、止痛剂或抗氧化赋形剂中的应用。
发明的效果
本申请的高纯度龙胆酸的纯度控制在99.5%以上,各种有机杂质均可控制在0.1%以下,可用于药用辅料,且本申请的高纯度龙胆酸通过特定的制备方法制得,高纯度龙胆酸的收率可达18~22%,适于大规模工业生产。
具体实施方式
以下对本申请的示范性实施方式做出说明,其中包括本申请实施方式的各种细节以助于理解,应当将它们认为仅仅是示范性的。因此,本领域普通技术人员应当认识到,可以对这里描述的实施方式做出各种改变和修改,而不会背离本申请的范围和精神。同样,为了清楚和简明,以下的描述中省略了对公知功能、结构和试剂的描述。
本申请提供一种高纯度龙胆酸,其特征在于,其纯度≥99.5%,其包含:龙胆酸、水杨酸和杂质A。其中,所述杂质A具有式(I)所示的结构:
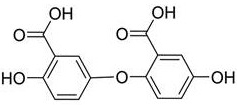
式(I)。
在一个具体实施方式中,所述高纯度龙胆酸中,所述水杨酸的质量百分比≤0.1%,例如可为0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.09%、0.1%等,所述杂质A的质量百分比≤0.1%,例如可为0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.09%、0.1%等。
在一个具体实施方式中,所述高纯度龙胆酸包含:龙胆酸、水杨酸、杂质A和杂质B,其中,所述杂质B具有式(II)所示的结构:
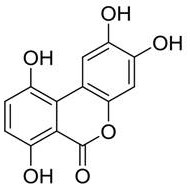
式(II)。
在一个具体实施方式中,所述高纯度龙胆酸中,所述杂质B的质量百分比≤0.1%,例如可为0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.09%、0.1%等。
在一个具体实施方式中,所述高纯度龙胆酸包含:龙胆酸、水杨酸和杂质A,不包含杂质B。
在一个具体实施方式中,所述高纯度龙胆酸包含杂质C(2,3-二羟基苯甲酸),所述高纯度龙胆酸中,所述2,3-二羟基苯甲酸的质量百分比≤0.1%,例如可为0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.09%、0.1%等。
本申请中的“龙胆酸”,其英文名为:Gentisic Acid,化学名为:2,5-二羟基苯甲酸,化学文摘(CAS)号为:490-79-9,其它名称为:DHB、5-羟基水杨酸,分子式为:C7H6O4,分子量为:154.12;其为白色至淡黄色粉末,在甲醇中易溶,在乙腈中溶解,在水中微溶,熔点为207~210℃。龙胆酸的用途为药用辅料,在制剂中用作自由基淬灭剂,降低放射性核素对前体药物辐射自分解效应。
在一个具体实施方式中,本申请的高纯度龙胆酸通过包括下述步骤的方法制备而成:
(1)投料反应:将5-溴水杨酸加入无机碱,催化剂溶液反应;
(2)酸化:加入浓盐酸调节pH,析出固体,过滤;
(3)萃取:向酸化步骤得到的滤液中加入萃取剂一,萃取,静置后出现絮状物;
(4)过滤:过滤、分液,得到有机相一和水相,向水相中加入萃取剂二,萃取,再过滤、分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
(5)析晶:浓缩有机相三,析出固体,加入有机溶剂一,得龙胆酸粗品;
(6)成盐:将龙胆酸粗品加入有机溶剂二中,滴加含有有机碱的有机溶剂二的溶液,得龙胆酸有机碱盐湿品;
(7)游离:将龙胆酸有机碱盐湿品加入水中,滴加浓盐酸调节pH,过滤,得龙胆酸精制品湿品;
(8)脱色与结晶:将龙胆酸精制品湿品和脱色剂加入结晶溶剂水中,过滤,得到龙胆酸精制品;
(9)过滤与干燥:将龙胆酸精制品用水淋洗后再用有机溶剂一清洗,干燥,得高纯度龙胆酸。
本申请中的“5-溴水杨酸”,其英文名为:5-Bromosalicylic acid,其它化学名称为:5-溴-2-羟基苯甲酸,化学文摘(CAS)号为:89-55-4,分子式为:C7H5BrO3,分子量为:217.02;按无水物计算,其含C7H5BrO3不少于98.0%;其为白色或类白色结晶性粉末。
在一个具体实施方式中,在投料反应步骤中,在铜催化剂存在的条件下,将5-溴水杨酸加入无机碱溶液,搅拌,升温使5-溴水杨酸与无机碱溶液反应,再降温,加入水。
在一个具体实施方式中,在投料反应步骤中,所述无机碱溶液为氢氧化钠的水溶液或氢氧化钾的水溶液,优选为氢氧化钾的水溶液;所述无机碱溶液的浓度为4~8mol/L,例如可为4mol/L、4.5mol/L、5mol/L、5.5mol/L、6mol/L、6.5mol/L、7mol/L、7.5mol/L、8mol/L等,无机碱溶液的浓度越低反应越慢,考虑成本及操作简便性优选为5~6mol/L的无机碱溶液。
在一个具体实施方式中,在投料反应步骤中,所述无机碱与5-溴水杨酸的物质的量之比,即摩尔量之比为4~7:1,例如可为4:1、4.5:1、5:1、5.3:1、5.5:1、6:1、6.5:1、7:1等,优选为5~6:1。
在一个具体实施方式中,在投料反应步骤中,所述升温为升至0~114℃,例如可为90℃、100℃、101℃、102℃、103℃、104℃、105℃、106℃、107℃、108℃、109℃、110℃、111℃、112℃、113℃、114℃等,优选为升至105~114℃。114℃为反应体系沸点,无法升至更高温度,温度控制在该范围内,反应时间短且原料转化完全。
在一个具体实施方式中,在投料反应步骤中,升温使5-溴水杨酸与无机碱溶液反应,HPLC中控5-溴水杨酸的含量小于1.5%,反应时间为20~48h,例如可为20h、22h、24h、26h、28h、30h、32h、34h、36h、38h、40h、42h、44h、46h、48h等,优选为20~30h。将反应时间控制在该范围内,反应完全且杂质少。
在一个具体实施方式中,在投料反应步骤中,HPLC中控5-溴水杨酸的含量的方法包括下述步骤:
5-溴水杨酸定位溶液:取5-溴水杨酸对照品适量,加溶剂使溶解并稀释制成每1mL中约含6μg的溶液,摇匀;
供试品溶液:取反应液适量,用溶剂稀释400倍(如取0.025mL稀释至10mL),摇匀,过滤,取滤液即得;
色谱条件:用十八烷基硅烷键合硅胶(Thermo Acclaim 120 ÅC18,4.6mm×250mm,5μm,或效能相当的色谱柱)为填充剂;以0.05%磷酸水溶液为流动相A,以乙腈为流动相B;捕集柱:Welch Ghost-Buster ColumnII 4.0×50mm或其他适宜的捕集柱;流速为每分钟1.0mL,进行线性梯度洗脱;柱温为40℃;检测波长为220nm;进样体积为10μL;
测定法:精密量取供试品溶液注入液相色谱仪,记录色谱图;
限度供试品溶液色谱图中,按面积归一化法计算,5-溴水杨酸峰面积不得大于总峰面积的1.5%。
在一个具体实施方式中,在投料反应步骤中,所述降温为降至20~30℃,例如可为20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃、30℃等。
在一个具体实施方式中,在投料反应步骤中,在铜催化剂和草酸存在的条件下,将5-溴水杨酸加入无机碱溶液。草酸作为抗氧化剂使用,易溶水除去。
在一个具体实施方式中,在投料反应步骤中,草酸与5-溴水杨酸的物质的量之比,即摩尔量之比为0.05~0.15:1,例如可为0.05:1、0.06:1、0.07:1、0.08:1、0.09:1、0.1:1、0.11:1、0.12:1、0.13:1、0.14:1、0.15:1等。草酸含量控制在上述范围内,产品收率高。
在一个具体实施方式中,在投料反应步骤中,所述铜催化剂为五水合硫酸铜或氧化亚铜,由于硫酸铜可溶于水,后处理方便,且可使反应更彻底,所以最优选为五水合硫酸铜。
在一个具体实施方式中,在投料反应步骤中,所述铜催化剂为五水合硫酸铜,所述五水合硫酸铜与5-溴水杨酸的物质的量之比,即摩尔量之比为0.05~0.15:1,例如可为0.05:1、0.06:1、0.07:1、0.08:1、0.09:1、0.1:1、0.11:1、0.12:1、0.13:1、0.14:1、0.15:1等。将五水合硫酸铜含量控制在上述范围内,反应时间更短,转化率更高,且五水合硫酸铜溶解完全。
在一个具体实施方式中,在投料反应步骤中,所述铜催化剂为氧化亚铜,所述氧化亚铜与5-溴水杨酸的物质的量之比,即摩尔量之比为0.025~0.075:1,例如可为0.025:1、0.03:1、0.035:1、0.04:1、0.045:1、0.05:1、0.055:1、0.06:1、0.065:1、0.07:1、0.075:1等。
在一个具体实施方式中,在投料反应步骤中,所述水与5-溴水杨酸的物质的量之比,即摩尔量之比为40~70:1,例如可为40:1、41.2:1、42:1、44:1、46:1、48:1、50:1、52:1、54:1、54.9:1、55:1、56:1、58:1、60:1、62:1、64:1、66:1、68:1、68.6:1、70:1等,优选为50~60:1。
在一个具体实施方式中,在酸化步骤中,调节pH至2.3~2.5,例如可为2.3、2.35、2.4、2.45、2.5等。将pH控制在该范围内,能够有效去除水杨酸,收率更高,且后续甲基叔丁基醚萃取的色素杂质最少,产品颜色较浅。
在一个具体实施方式中,在酸化步骤中,在20~30℃条件下加入浓盐酸调节pH,例如可在20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃、30℃等条件下加入浓盐酸调节pH。
在一个具体实施方式中,在萃取步骤中,所述萃取剂一选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上;优选为甲基叔丁基醚。
在一个具体实施方式中,在萃取步骤中,所述甲基叔丁基醚与5-溴水杨酸的质量比为4~6:1,例如可为4:1、4.1:1、4.2:1、4.3:1、4.4:1、4.44:1、4.5:1、4.6:1、4.7:1、4.8:1、4.9:1、5:1、5.1:1、5.18:1、5.2:1、5.3:1、5.4:1、5.5:1、5.6:1、5.7:1、5.8:1、5.92:1、6:1等,优选为5~5.5:1。
在一个具体实施方式中,在萃取步骤中,所述氯化钠与5-溴水杨酸的质量比为1.0~1.5:1,例如可为1.0:1、1.1:1、1.2:1、1.3:1、1.4:1、1.5:1等。
在一个具体实施方式中,在过滤步骤中,所述萃取剂二选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上。
在一个具体实施方式中,在过滤步骤中,所述萃取剂二与5-溴水杨酸的质量比为4~6:1,例如可为4:1、4.1:1、4.2:1、4.3:1、4.4:1、4.44:1、4.5:1、4.6:1、4.7:1、4.8:1、4.9:1、5:1、5.1:1、5.18:1、5.2:1、5.3:1、5.4:1、5.5:1、5.6:1、5.7:1、5.8:1、5.92:1、6:1等,优选为5~5.5:1。
在一个具体实施方式中,在过滤步骤中,向布氏漏斗中加入硅藻土,过滤、分液,得到有机相一和水相。
在一个具体实施方式中,在析晶步骤中,浓缩有机相三,析出固体,加入有机溶剂一,搅拌,过滤并将滤饼干燥,得龙胆酸粗品;所述有机溶剂一选自正庚烷、丙酮、乙酸乙酯、石油醚和二氯甲烷中的一种或两种以上。
在一个具体实施方式中,在析晶步骤中,在40~60℃条件下,例如可为40℃、42℃、44℃、46℃、48℃、50℃、52℃、54℃、56℃、58℃、60℃等条件下,优选为在40~50℃条件下,减压浓缩有机相三至析出大量固体。将温度控制在该范围内,产品纯度高,浓缩时间更短。
在一个具体实施方式中,在析晶步骤中,所述有机溶剂一与5-溴水杨酸的质量比为4~5.5:1,例如可为4:1、4.08:1、4.1:1、4.2:1、4.3:1、4.4:1、4.5:1、4.6:1、4.7:1、4.8:1、4.9:1、5:1、5.1:1、5.2:1、5.3:1、5.44:1、5.5:1等,优选为4.5~5:1。
在一个具体实施方式中,在析晶步骤中,所述搅拌时间为1~2h,例如可为1h、1.1h、1.2h、1.3h、1.4h、1.5h、1.6h、1.7h、1.8h、1.9h、2h等;在20~30℃条件下过滤,例如可为20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃、30℃等。
在一个具体实施方式中,在析晶步骤中,所述滤饼在40~50℃条件下真空干燥2~4h,得龙胆酸粗品,例如可在40℃、41℃、42℃、43℃、44℃、45℃、46℃、47℃、48℃、49℃、50℃条件下真空干燥2h、2.2h、2.4h、2.6h、2.8h、3h、3.2h、3.4h、3.6h、3.8h、4h等。
单独通过溶剂结晶法无法有效提纯,而龙胆酸具有羧酸结构,因此本申请开发成盐步骤和游离步骤。
在一个具体实施方式中,在成盐步骤中,将龙胆酸粗品加入有机溶剂二中,搅拌,滴加含有有机碱的有机溶剂二的溶液,降温,过滤,得龙胆酸有机碱成盐湿品。
在一个具体实施方式中,在成盐步骤中,在10~35℃条件下,例如可为10℃、12℃、14℃、15℃、18℃、20℃、22℃、25℃、27℃、30℃、32℃、34℃、35℃等条件下,滴加含有有机碱的有机溶剂二的溶液。将温度控制在上述范围内,后续降温析出晶体时不容易爆析,杂质较少。
在一个具体实施方式中,在成盐步骤中,所述有机碱为二乙胺,所述有机溶剂二选自丙酮、异丙醇和四氢呋喃中的一种或两种以上。
在一个具体实施方式中,在成盐步骤中,所述有机碱与5-溴水杨酸的物质的量之比,即摩尔量之比为0.75~0.85:1,例如可为0.75:1、0.76:1、0.77:1、0.78:1、0.79:1、0.8:1、0.81:1、0.82:1、0.83:1、0.84:1、0.85:1等,优选为0.75~0.81:1。控制有机碱含量在上述范围内,可使得成盐充分且杂质较少。
在一个具体实施方式中,在成盐步骤中,所述有机溶剂二与龙胆酸粗品的质量比为8.6~10:1,例如可为8.6:1、8.7:1、8.8:1、8.9:1、9:1、9.1:1、9.2:1、9.3:1、9.4:1、9.42:1、9.5:1、9.6:1、9.7:1、9.8:1、9.9:1、10:1等,优选为9~9.8:1。控制丙酮在上述范围内,可使得收率更高,且龙胆酸粗品溶解更完全。
在一个具体实施方式中,在成盐步骤中,所述降温为降至2~8℃,例如可为2℃、2.5℃、3℃、3.5℃、4℃、4.5℃、5℃、5.5℃、6℃、6.5℃、7℃、7.5℃、8℃等。温度控制在上述范围内,可获得更高的纯度和收率。
在一个具体实施方式中,在成盐步骤中,滴加含有有机碱的有机溶剂二的溶液完毕后,保温0.3~0.8h,再降温,例如可保温0.3h、0.4h、0.5h、0.6h、0.7h、0.8h等。
在一个具体实施方式中,在成盐步骤中,所述降温梯度为15~25℃/h,例如可为15℃/h、17℃/h、19℃/h、20℃/h、22℃/h、23℃/h、25℃/h等。
在一个具体实施方式中,在成盐步骤中,降温后保温0.3~0.8h,过滤,例如可保温0.3h、0.4h、0.5h、0.6h、0.7h、0.8h等。
在一个具体实施方式中,在游离步骤中,将龙胆酸有机碱盐湿品加入水中,搅拌,滴加浓盐酸调节pH,降温,过滤,得龙胆酸精制品湿品。
在一个具体实施方式中,在游离步骤中,在20~30℃条件下滴加浓盐酸调节pH,例如可在20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃、30℃等条件下滴加浓盐酸调节pH。
在一个具体实施方式中,在游离步骤中,调节pH至1~2,例如可为1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2等。pH控制在该范围内,可使龙胆酸游离更完全,产品收率更高。
在一个具体实施方式中,在游离步骤中,所述水与龙胆酸有机碱盐湿品的质量比为4~6:1,例如可为4:1、4.2:1、4.4:1、4.6:1、4.8:1、5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1等。将水控制在上述范围内,可使龙胆酸有机碱盐湿品溶解完全且产品收率更高。
在一个具体实施方式中,在游离步骤中,所述降温为降至0~10℃,例如可为0℃、1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃等,所述降温梯度为15~25℃/h,例如可为15℃/h、17℃/h、19℃/h、20℃/h、22℃/h、23℃/h、25℃/h等。
在一个具体实施方式中,在游离步骤中,降温后保温0.3~0.8h,过滤,例如可保温0.3h、0.4h、0.5h、0.6h、0.7h、0.8h等。
在一个具体实施方式中,在脱色与结晶步骤中,将龙胆酸精制品湿品和脱色剂加入水中,搅拌,升温,过滤并将滤液降温,得到龙胆酸晶体;所述脱色剂不限于活性炭、氧化铝和白土中的一种或一种以上。
在一个具体实施方式中,在脱色与结晶步骤中,将龙胆酸精制品湿品、脱色剂和草酸加入水中,草酸作为抗氧化剂。
在一个具体实施方式中,在脱色与结晶步骤中,升温后保温0.5~6h,例如可为0.5h、0.6h、0.7h、0.8h、0.9h、1h、1.2h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h等,优选为0.5~1.5h。将保温时间控制在该范围内,可使龙胆酸精制品湿品溶解完全,且进一步控制杂质B含量。
在一个具体实施方式中,在脱色与结晶步骤中,所述升温为升至65~75℃,例如可为65℃、66℃、67℃、68℃、69℃、70℃、71℃、72℃、73℃、74℃、75℃等。将温度控制在该范围内,可使龙胆酸精制品湿品溶解完全,且提高最终产品纯度。
在一个具体实施方式中,在脱色与结晶步骤中,所述活性炭与龙胆酸精制品湿品的质量比为0.05~0.1:1,例如可为0.05:1、0.06:1、0.07:1、0.08:1、0.09:1、0.1:1等。将活性炭与龙胆酸精制品湿品的质量比控制在该范围内,可提高色素去除效率。
在一个具体实施方式中,在脱色与结晶步骤中,所述草酸与龙胆酸精制品湿品的质量比为0.01~0.03:1,例如可为0.01:1、0.015:1、0.02:1、0.025:1、0.03:1等。
在一个具体实施方式中,在脱色与结晶步骤中,所述水与龙胆酸精制品湿品的质量比为4~6:1,例如可为4:1、4.2:1、4.4:1、4.6:1、4.8:1、5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1等。将水与龙胆酸精制品湿品的质量比控制在该范围内,可使龙胆酸精制品湿品,并进一步提高产品收率。
在一个具体实施方式中,在脱色与结晶步骤中,过滤并将滤液降温至0~10℃,例如可为0℃、1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃等,保温0.3~0.8h,例如可为0.3h、0.4h、0.5h、0.6h、0.7h、0.8h等,得到龙胆酸晶体。
在一个具体实施方式中,在过滤与干燥步骤中,所述干燥条件为:40~55℃下真空干燥10~15h,例如可在40℃、42℃、44℃、46℃、48℃、50℃、52℃、54℃、55℃等条件下真空干燥10h、11h、12h、13h、14h、15h等。
在一个具体实施方式中,本申请中的搅拌均在氮气保护下进行。
在一个具体实施方式中,所述水为纯化水。
本申请还提供一种上述任一项所述的高纯度龙胆酸在制备抗病毒剂、抗菌剂、止痛剂或抗氧化赋形剂中的应用。
实施例
本申请对试验中所用到的材料以及试验方法进行一般性和/或具体的描述,在下面的实施例中,所用材料或仪器未注明生产厂商者,均为可以通过市购获得的常规材料或仪器。
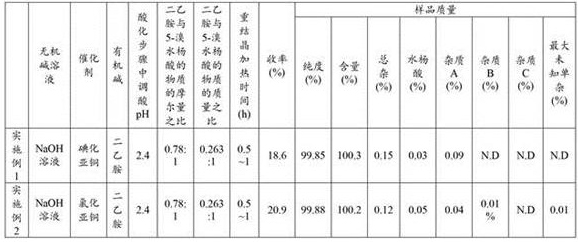
实施例1
投料反应:反应釜中加入5.0mol/L NaOH溶液,加入5-溴水杨酸50g,碘化亚铜0.5g,草酸0.5g;氮气保护下,开启搅拌;升温至100~120℃反应24h,取样中控HPLC(5-溴水杨酸)≤1.5%;降温至20~30℃,加入水;
酸化:控温20~30℃,加入浓盐酸调节反应液pH至(2.4),有固体析出,过滤;
萃取:滤液中加入氯化钠50g和甲基叔丁基醚250g,搅拌萃取,静置后出现絮状物;
过滤:布氏漏斗中加入硅藻土5g,过滤,分液,得到有机相一和水相;水相中加入甲基叔丁基醚125g,过滤,分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
析晶:40~50℃减压浓缩有机相三至析出大量固体,加入正庚烷360g;搅拌1~2h,室温20~30℃过滤;滤饼40~50℃真空干燥2~4h,得龙胆酸粗品16g;
成盐:反应釜中加入丙酮250g,加入龙胆酸粗品,氮气保护下,开启搅拌;控温10~35℃,滴加含二乙胺13.15g的丙酮溶液36g,滴加完毕,保温0.5h;关闭加热,降温(15~25℃/h)至2~8℃,保温0.5h,过滤,得到龙胆酸二乙胺盐湿品约25g;
游离:反应釜中加入水200g,加入龙胆酸二乙胺盐湿品;氮气保护下,开启搅拌,控温20~30℃,滴加浓盐酸调节pH;降温0~10℃,保温0.5h,过滤,得龙胆酸精制品湿品;
脱色与结晶:反应釜中加入水250g,加入龙胆酸精制品湿品,加入活性炭0.2g,加入草酸0.1g;氮气保护下,开启搅拌,升温至65~75℃,保温0.5~1h,趁热过滤;滤液降温0~10℃,保温0.5h;
过滤与干燥:用水淋洗,再用正庚烷22g清洗滤饼,滤饼在40~55℃下真空干燥12h,得高纯度龙胆酸11g。
实施例2
投料反应:反应釜中加入5.0mol/L NaOH溶液,加入5-溴水杨酸200g,氯化亚铜1.8g,草酸2g;氮气保护下,开启搅拌;升温至100~120℃反应24h,取样中控HPLC(5-溴水杨酸)≤1.5%;降温至室温(20~30℃),加入水;
酸化:控温20~30℃,加入浓盐酸调节反应液pH至2.4,有固体析出,过滤;
萃取:滤液中加入氯化钠200g和甲基叔丁基醚1000g,搅拌萃取,静置后出现絮状物;
过滤:布氏漏斗中加入硅藻土50g,过滤,分液,得到有机相一和水相;水相中加入甲基叔丁基醚500g,过滤,分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
析晶:40~50℃减压浓缩有机相三至析出大量固体,加入正庚烷500g;搅拌1~2h,室温20~30℃过滤;滤饼40~50℃真空干燥2~4h,得龙胆酸粗品68g;
成盐:反应釜中加入丙酮450g,加入龙胆酸粗品,氮气保护下,开启搅拌;控温10~35℃,滴加二乙胺52.60g的丙酮溶液150g,滴加完毕,保温0.5h;关闭加热,降温(15~25℃/h)至2~8℃,保温0.5h,过滤,得到龙胆酸二乙胺盐湿品96g;
游离:反应釜中加入水,加入龙胆酸二乙胺盐湿品;氮气保护下,开启搅拌,控温20~30℃,滴加浓盐酸调节pH;降温0~10℃,保温0.5h,过滤,得龙胆酸精制品湿品;
脱色与结晶:反应釜中加入水1000g,加入龙胆酸精制品湿品,加入活性炭1g,加入草酸0.5g;氮气保护下,开启搅拌,升温至65~75℃,保温0.5~1h,趁热过滤;滤液降温0~10℃,保温0.5h;
过滤与干燥:用水淋洗,再用正庚烷200g清洗滤饼,滤饼在40~55℃下真空干燥12.0h,得高纯度龙胆酸48g。
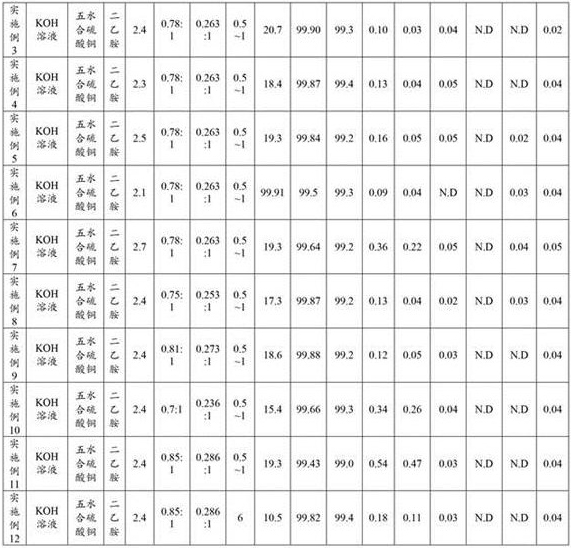
实施例3
投料反应:反应釜中加入5.0mol/L KOH溶液,加入5-溴水杨酸500g,五水合硫酸铜5.0g,草酸5g;氮气保护下,开启搅拌;升温至100~120℃反应24h,取样中控HPLC(5-溴水杨酸)≤1.5%;降温至室温(20~30℃),加入水;
酸化:控温20~30℃,加入浓盐酸调节反应液pH至2.4,有固体析出,过滤;
萃取:滤液中加入氯化钠500g和甲基叔丁基醚250g,搅拌萃取,静置后出现絮状物;
过滤:布氏漏斗中加入硅藻土50g,过滤,分液,得到有机相一和水相;水相中加入甲基叔丁基醚125g,过滤,分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
析晶:40~50℃减压浓缩有机相至析出大量固体,加入正庚烷250g;搅拌1~2h,室温20~30℃过滤;滤饼40~50℃真空干燥2~4h,得龙胆酸粗品160g;
成盐:反应釜中加入丙酮1200g,加入龙胆酸粗品,氮气保护下,开启搅拌;控温10~35℃,滴加二乙胺131.50g的丙酮溶液280g,滴加完毕,保温0.5h;关闭加热,降温(15~25℃/h)至2~8℃,保温0.5h,过滤,得到龙胆酸二乙胺盐湿品;
游离:反应釜中加入水2000g,加入龙胆酸二乙胺盐湿品;氮气保护下,开启搅拌,控温20~30℃,滴加浓盐酸调节pH;降温0~10℃,保温0.5h,过滤,得龙胆酸精制品湿品;
脱色与结晶:反应釜中加入水2000g,加入龙胆酸精制品湿品,加入活性炭3g,加入草酸1.5g;氮气保护下,开启搅拌,升温至65~75℃,保温0.5~1h,趁热过滤;滤液降温0~10℃,保温0.5h;
过滤与干燥:用水淋洗,再用正庚烷400g清洗滤饼,滤饼在40~55℃下真空干燥12h,得高纯度龙胆酸110g。
实施例4
本实施例与实施例3的不同之处在于,酸化步骤中,调节反应液pH至2.3。
实施例5
本实施例与实施例3的不同之处在于,酸化步骤中,调节反应液pH至2.5。
实施例6
本实施例与实施例3的不同之处在于,酸化步骤中,调节反应液pH至2.1。
实施例7
本实施例与实施例3的不同之处在于,酸化步骤中,调节反应液pH至2.7。
实施例8
本实施例与实施例3的不同之处在于,成盐步骤中,二乙胺与5-溴水杨酸的物质的量之比为0.75:1。
实施例9
本实施例与实施例3的不同之处在于,成盐步骤中,二乙胺与5-溴水杨酸的物质的量之比为0.81:1。
实施例10
本实施例与实施例3的不同之处在于,成盐步骤中,二乙胺与5-溴水杨酸的物质的量之比为0.7:1。
实施例11
本实施例与实施例3的不同之处在于,成盐步骤中,二乙胺与5-溴水杨酸的物质的量之比为0.85:1。
实施例12
本实施例与实施例3的不同之处在于,脱色与结晶步骤中,升温后保温6h,趁热过滤。
将各实施例的原料、试剂、反应条件及所得样品检测结果列于下表1中。
表1
以上所述,仅是本申请的较佳实施例而已,并非是对本申请作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本申请技术方案内容,依据本申请的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本申请技术方案的保护范围。
Claims (15)
1.一种高纯度龙胆酸,其特征在于,其纯度≥99.50%,其中包含:龙胆酸、水杨酸和杂质A,其中,所述杂质A具有式(I)所示的结构:

式(I)。
2.根据权利要求1所述的高纯度龙胆酸,其特征在于,所述水杨酸的质量百分比≤0.1%,所述杂质A的质量百分比≤0.1%。
3.根据权利要求1所述的高纯度龙胆酸,其特征在于,所述高纯度龙胆酸还包含杂质B,其中,所述杂质B具有式(II)所示的结构:
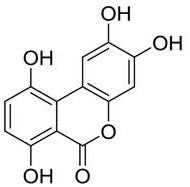
式(II)。
4.根据权利要求3所述的高纯度龙胆酸,其特征在于,所述高纯度龙胆酸中,所述杂质B的质量百分比≤0.1%。
5.根据权利要求1~4中任一项所述的高纯度龙胆酸,其特征在于,其通过包括下述步骤的方法制备而成:
投料反应:将5-溴水杨酸加入无机碱溶液反应;
酸化:加入浓盐酸调节pH,析出固体,过滤;
萃取:向酸化步骤得到的滤液中加入萃取剂一,萃取,静置后出现絮状物;
过滤:过滤、分液,得到有机相一和水相,向水相中加入萃取剂二,萃取,再过滤、分液,得到有机相二,将有机相二与有机相一合并得到有机相三;
析晶:浓缩有机相三,析出固体,加入有机溶剂一,得龙胆酸粗品;
成盐:将龙胆酸粗品加入有机溶剂二中,滴加含有有机碱的有机溶剂二的溶液,得龙胆酸有机碱盐湿品;
游离:将龙胆酸有机碱盐湿品加入水中,滴加浓盐酸调节pH,过滤,得龙胆酸精制品湿品;
脱色与结晶:将龙胆酸精制品湿品和脱色剂加入水中,过滤,得到龙胆酸晶体;
过滤与干燥:将龙胆酸晶体用水淋洗后再用有机溶剂一清洗,干燥,得高纯度龙胆酸。
6.根据权利要求5所述的高纯度龙胆酸,其特征在于,在酸化步骤中,调节pH至2.3~2.5。
7.根据权利要求5所述的高纯度龙胆酸,其特征在于,在萃取步骤中,所述萃取剂一选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上。
8.根据权利要求5所述的高纯度龙胆酸,其特征在于,在过滤步骤中,所述萃取剂二选自二氯甲烷、乙酸乙酯、乙酸异丙酯、甲苯和甲基叔丁基醚中的任意一种或两种以上。
9.根据权利要求5所述的高纯度龙胆酸,其特征在于,在析晶步骤中,所述有机溶剂一选自正庚烷、丙酮、乙酸乙酯、石油醚和氯仿中的一种或两种以上。
10.根据权利要求5所述的高纯度龙胆酸,其特征在于,所述有机碱与5-溴水杨酸的物质的量之比为0.75~0.85:1。
11.根据权利要求5所述的高纯度龙胆酸,其特征在于,所述有机碱为二乙胺。
12.根据权利要求5所述的高纯度龙胆酸,其特征在于,在成盐步骤中,所述有机溶剂二选自丙酮、异丙醇和四氢呋喃中的一种或两种以上。
13.根据权利要求5所述的高纯度龙胆酸,其特征在于,在脱色与结晶步骤中,将龙胆酸精制品湿品和脱色剂加入水中,搅拌,升温,过滤并将滤液降温,得到龙胆酸晶体。
14.根据权利要求13所述的高纯度龙胆酸,其特征在于,在脱色与结晶步骤中,升温后保温0.5~6h。
15.根据权利要求1~14中任一项所述的高纯度龙胆酸在制备抗病毒剂、抗菌剂、止痛剂或抗氧化赋形剂中的应用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210537742.2A CN114634412A (zh) | 2022-05-18 | 2022-05-18 | 一种高纯度龙胆酸及其应用 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210537742.2A CN114634412A (zh) | 2022-05-18 | 2022-05-18 | 一种高纯度龙胆酸及其应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN114634412A true CN114634412A (zh) | 2022-06-17 |
Family
ID=81953163
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210537742.2A Pending CN114634412A (zh) | 2022-05-18 | 2022-05-18 | 一种高纯度龙胆酸及其应用 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114634412A (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716308A (zh) * | 2022-05-18 | 2022-07-08 | 北京先通国际医药科技股份有限公司 | 一种高纯度龙胆酸的制备方法及其应用 |
| CN117024292A (zh) * | 2023-08-08 | 2023-11-10 | 深圳杉海创新技术有限公司 | 一种龙胆酸离子盐、其制备方法及日化产品或食品或药品 |
| CN119074969A (zh) * | 2024-11-06 | 2024-12-06 | 云南省药物研究所 | 一种高纯度可供注射用龙胆酸辅料的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2588679A (en) * | 1950-08-17 | 1952-03-11 | Dow Chemical Co | Purification of gentisic acid |
| JP2004189608A (ja) * | 2002-12-06 | 2004-07-08 | Sumikin Air Water Chemical Inc | 2,5−ジヒドロキシ安息香酸の精製方法 |
| CN102766042A (zh) * | 2012-07-04 | 2012-11-07 | 天长市禾益化学药品有限公司 | 一种制备高纯度龙胆酸的方法 |
-
2022
- 2022-05-18 CN CN202210537742.2A patent/CN114634412A/zh active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2588679A (en) * | 1950-08-17 | 1952-03-11 | Dow Chemical Co | Purification of gentisic acid |
| JP2004189608A (ja) * | 2002-12-06 | 2004-07-08 | Sumikin Air Water Chemical Inc | 2,5−ジヒドロキシ安息香酸の精製方法 |
| CN102766042A (zh) * | 2012-07-04 | 2012-11-07 | 天长市禾益化学药品有限公司 | 一种制备高纯度龙胆酸的方法 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716308A (zh) * | 2022-05-18 | 2022-07-08 | 北京先通国际医药科技股份有限公司 | 一种高纯度龙胆酸的制备方法及其应用 |
| CN117024292A (zh) * | 2023-08-08 | 2023-11-10 | 深圳杉海创新技术有限公司 | 一种龙胆酸离子盐、其制备方法及日化产品或食品或药品 |
| CN117024292B (zh) * | 2023-08-08 | 2024-09-24 | 深圳杉海创新技术有限公司 | 一种龙胆酸离子盐、其制备方法及日化产品或食品或药品 |
| CN119074969A (zh) * | 2024-11-06 | 2024-12-06 | 云南省药物研究所 | 一种高纯度可供注射用龙胆酸辅料的制备方法 |
| CN119074969B (zh) * | 2024-11-06 | 2025-04-01 | 云南省药物研究所 | 一种高纯度可供注射用龙胆酸辅料的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114634412A (zh) | 一种高纯度龙胆酸及其应用 | |
| CN108191749B (zh) | 一种氟啶虫酰胺及其中间体4-三氟甲基烟酸的制备方法 | |
| CN106256824B (zh) | 一种高纯度德拉沙星葡甲胺盐的制备方法 | |
| CN109336833B (zh) | 一种1,4,7,10-四氮杂环十二烷-1,4,7-三乙酸的制备方法 | |
| CN108623486A (zh) | 一种沙丁胺醇中间体ⅴ盐酸盐的制备方法 | |
| CN114716308A (zh) | 一种高纯度龙胆酸的制备方法及其应用 | |
| CN116283629B (zh) | 一种5-氨基-2-硝基苯甲酸的制备方法 | |
| CN101270124B (zh) | 一种提纯制备高纯度荧光素及荧光素盐的新方法 | |
| KR100235808B1 (ko) | 케토산의 제조방법 | |
| CN103420862B (zh) | 一种钆塞酸二钠中间体化合物的金属盐、其晶型及制备方法 | |
| CN108383745B (zh) | 一种醋氯芬酸的制备方法 | |
| CN102816060B (zh) | 高纯度克利贝特的制备方法 | |
| CN102010325A (zh) | 一种对羟基苯乙酸的合成方法 | |
| CN108440374B (zh) | 一种阿西美辛的制备方法 | |
| CN114591236A (zh) | 一种茚达特罗的改进制备方法 | |
| CA1053687A (en) | Purification of coumarin and alkylated derivatives of it | |
| CN113929649B (zh) | 一种科里内酯衍生物的制备方法 | |
| CN115611739B (zh) | 一种苯培酸中间体的制备方法及其中间体 | |
| CN112624936A (zh) | 一种托芬那酸的水相合成方法 | |
| CN114671836B (zh) | 一种胺碘酮杂质c的合成方法 | |
| CN114478295B (zh) | 一种泛影酸的合成方法 | |
| CN114573489B (zh) | 一种卡前列素的分离方法 | |
| KR100359503B1 (ko) | 방향족 프로피온산 유도체의 제조방법 | |
| CN120398705A (zh) | 一种丁卡因的高效制备方法 | |
| WO2025077873A1 (zh) | 一种普瑞巴林中间体的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |