CN115190813A - 整合应激反应途径的调节剂 - Google Patents
整合应激反应途径的调节剂 Download PDFInfo
- Publication number
- CN115190813A CN115190813A CN202180020265.6A CN202180020265A CN115190813A CN 115190813 A CN115190813 A CN 115190813A CN 202180020265 A CN202180020265 A CN 202180020265A CN 115190813 A CN115190813 A CN 115190813A
- Authority
- CN
- China
- Prior art keywords
- compound
- optionally substituted
- pharmaceutically acceptable
- hydrate
- tautomer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4192—1,2,3-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Electrochromic Elements, Electrophoresis, Or Variable Reflection Or Absorption Elements (AREA)
- Amplifiers (AREA)
Abstract
本发明涉及式(I)的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体, 其中R1、R2、R2a、R3、Ra1、Ra2、Ra3、Ra4、Ra5、Ra6、A1和A2具有如在说明书和权利要求书中指出的含义。本发明进一步涉及包含所述化合物的药物组合物,它们作为药物以及在用于治疗和预防一种或多种与整合应激反应有关的疾病或障碍的方法中的用途。
其中R1、R2、R2a、R3、Ra1、Ra2、Ra3、Ra4、Ra5、Ra6、A1和A2具有如在说明书和权利要求书中指出的含义。本发明进一步涉及包含所述化合物的药物组合物,它们作为药物以及在用于治疗和预防一种或多种与整合应激反应有关的疾病或障碍的方法中的用途。
Description
本发明涉及式(I)的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体

其中R1、R2、R2a、R3、Ra1、Ra2、Ra3、Ra4、Ra5、Ra6、A1和A2具有如在说明书和权利要求书中指出的含义。本发明进一步涉及包含所述化合物的药物组合物、它们作为药物以及在用于治疗和预防一种或多种与整合应激反应有关的疾病或障碍的方法中的用途。
整合应激反应(ISR)是所有真核生物共有的细胞应激反应(1)。ISR信号传递的调节异常具有重要的病理学后果,尤其是与炎症、病毒感染、糖尿病、癌症和神经变性疾病有关的后果。
ISR是不同类型的细胞应激的共同特征,导致在丝氨酸51上的真核翻译起始因子2(eIF2α)的α亚基的磷酸化,从而抑制正常蛋白合成和应激反应基因的表达(2)。在哺乳动物细胞中,所述磷酸化由四个eIF2α激酶的家族进行,即:PKR-样ER激酶(PERK)、双链RNA依赖性蛋白激酶(PKR)、亚铁血红素调节的eIF2α激酶(HRI)和一般性调控阻遏蛋白激酶2(GCN2),各自响应于不同的环境和生理应激(3)。
eIF2α与eIF2β和eIF2γ一起形成eIF2复合物,这是正常mRNA翻译起始的关键参与者(4)。eIF2复合物结合GTP和Met-tRNAi,从而形成三元复合物(eIF2-GTP-Met-tRNAi),其被核糖体募集用于翻译起始(5, 6)。
eIF2B是由5种亚基(α、β、γ、δ、ε)组成的异十聚体复合物,所述亚基一式两份地形成GEF活性十聚体(7)。
响应于ISR活化,磷酸化的eIF2α抑制eIF2B介导的GDP与GTP的交换,导致三元复合物形成减少,并因此抑制以核糖体与5'AUG起始密码子结合为特征的正常mRNA的翻译(8)。在三元复合物丰度降低的这些条件下,包括编码转录因子ATF4的mRNA在内的几种特定mRNA的翻译通过涉及上游ORF (uORF)的翻译改变的机制被活化(7, 9, 10)。这些mRNA通常包含一个或多个uORF,所述uORF通常在未受应激的细胞中起作用以限制核糖体向主要编码ORF的流动。例如,在正常条件下,在ATF的5' UTR中的uORF占据核糖体并阻止ATF4的编码序列的翻译。但是,在应激条件下,即在三元复合物形成减少的条件下,核糖体扫描经过这些上游ORF并在ATF4编码ORF处开始翻译的可能性增加。ATF4和其它以这种方式表达的应激反应因子随后控制着一系列其它应激反应基因的表达。急性期在于旨在恢复稳态的蛋白的表达,而慢性期导致促凋亡因子的表达(1, 11, 12, 13)。
ISR信号传递的标志物的上调已在多种病症中得到证实,其中包括癌症和神经变性疾病。在癌症中,ER应激调节的翻译增加对低氧条件的耐受性并促进肿瘤生长(14, 15,16),并且已经证明通过基因靶向对PERK的删除可以减慢从转化的PERK−/−小鼠胚胎成纤维细胞衍生出的肿瘤的生长(14, 17)。此外,最近的一份报告已经提供了如下的概念证据:使用在小鼠中的源自患者的异种移植模型,eIF2B的活化剂有效治疗一种侵袭性转移性前列腺癌(28)。总之,细胞保护性ISR信号传递的预防可能代表一种有效的抗增殖策略,其用于治疗至少某些形式的癌症。
此外,ISR信号传递的调节可以证明在保持突触功能和减少神经元衰退中是有效的,在特征在于错误折叠的蛋白和未折叠蛋白反应(UPR)的激活的神经变性疾病(诸如肌萎缩性侧索硬化(ALS)、额颞叶痴呆(FTD)、阿尔茨海默氏病(AD)、帕金森病(PD)和JakobCreutzfeld (朊病毒)疾病)中也是如此(18, 19, 20)。对于朊病毒病(存在的神经变性疾病的一个例子),其中已经表明,ISR信号传递的药理学以及遗传抑制可以使蛋白翻译水平正常化、挽救突触功能并预防神经元损失(21)。具体地,通过控制磷酸化eIF2α水平的磷酸酶的过表达来降低磷酸化eIF2α的水平会增加感染了朊病毒的小鼠的存活率,而持续的eIF2α磷酸化降低存活率(22)。
此外,控制蛋白表达水平对于适当脑功能的重要性的直接证据以影响eIF2和eIF2B的功能的罕见遗传性疾病的形式存在。破坏eIF2的复杂完整性且因此导致降低的正常蛋白表达水平的eIF2γ中的突变与智力障碍综合征(ID)相关(23)。在eIF2B的亚基中的部分功能丧失突变已被证实是罕见的脑白质营养不良白质消融性疾病(VWMD)的原因(24,25)。具体地,与ISRIB相关的小分子在VWMD小鼠模型中对eIF2B部分功能丧失的稳定化已被证实会减少ISR标记物并改善功能以及病理学终点(26, 27)。
eIF2α途径的调节剂描述于WO 2014/144952 A2中。WO 2017/193030 A1、WO 2017/193034 A1、WO 2017/193041 A1和WO 2017/193063 A1描述了整合应激途径的调节剂。WO2017/212423 A1、WO 2017/212425 A1、WO 2018/225093 A1、WO 2019/008506 A1和WO2019/008507 A1描述了ATF4途径的抑制剂。WO 2019/032743 A1、WO 2019/046779 A1、WO2020/167994 A1、WO 2020/168011 A1和WO 2020/181247 A1涉及真核起始因子2B调节剂。在WO 2020/77217 A1中,描述了可用于调节整合应激反应(ISR)和用于治疗相关的疾病、障碍和病症的化合物、组合物和方法。
描述整合应激途径的调节剂的其它文献是WO 2019/090069 A1、WO 2019/090074A1、WO 2019/090076 A1、WO 2019/090078 A1、WO 2019/090081 A1、WO 2019/090082 A1、WO2019/090085 A1、WO 2019/090088 A1、WO 2019/090090 A1、WO 2020/223536 A1、WO 2020/223538 A1、WO 2020/252207 A1、WO 2020/216764 A1、WO 2020/216766 A1、国际专利申请PCT/EP2021/051697、欧洲专利申请20203312.2、20203311.4和20203309.8。真核起始因子的调节剂描述于WO 2019/183589 A1中。WO 2019/118785 A2、WO 2019/236710 A1、WO2020/176428 A1和WO 2020/252205 A1描述了整合应激反应途径的抑制剂。作为ATF4抑制剂的杂芳基衍生物描述于WO 2019/193540 A1中。作为ATF4抑制剂的二环芳族环衍生物描述于WO 2019/193541 A1中。WO 2020/031107 A1和WO 2020/012339 A1描述了ATF4途径的抑制剂。
但是,仍然需要可用作整合应激反应途径的调节剂的具有良好药代动力学性能的新化合物。
因而,本发明的一个目的是提供一类新的作为整合应激反应途径的调节剂的化合物,其可以有效治疗整合应激反应途径相关的疾病并且其可以显示出改善的药学相关性能,包括活性、溶解度、选择性、ADMET性能和/或减少的副作用。
因此,本发明提供式(I)的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体

其中
Ra1、Ra2、Ra3、Ra4、Ra5、Ra6独立地选自H、卤素、C1-4烷基和A2a,其中C1-4烷基任选地被一个或多个取代基取代,所述取代基选自卤素、OH和O-C1-3烷基,其中所述取代基相同或不同,且
前提条件是,Ra1、Ra2、Ra3、Ra4、Ra5、Ra6中的仅一个是A2a;
A1是C5亚环烷基、C5亚环烯基或含有氮环原子的5元亚杂环基,前提条件是,用星号标记的环A1的环原子是碳原子,其中A1任选地被一个或多个相同或不同的R4取代;
每个R4独立地是氧代(=O),其中所述环是至少部分饱和的、硫代(=S),其中所述环是至少部分饱和的、卤素、CN、OR5或C1-6烷基,其中C1-6烷基任选地被一个或多个相同或不同的卤素取代;
R5是H或C1-6烷基,其中C1-6烷基任选地被一个或多个相同或不同的卤素取代;
A2是R6a或A2a;
R6a是OR6a1、SR6a1、N(R6a1R6a2)、C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个取代基取代,所述取代基选自卤素、OR6a3、CN和A2a,其中所述取代基相同或不同;
R6a1、R6a2独立地选自H、C1-6烷基、C2-6烯基、C2-6炔基和A2a,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个取代基取代,所述取代基选自卤素、CN、OR6a3、A2a和OA2a,其中所述取代基相同或不同;
R6a3是H或C1-4烷基,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;
A2a是苯基、C3-7环烷基、C4-12二环烷基或3-7元杂环基,其中A2a任选地被一个或多个相同或不同的R6取代;
每个R6独立地是R6b、OH、OR6b、卤素或CN,其中R6b是环丙基、C1-6烷基、C2-6烯基或C2-6炔基,其中R6b任选地被一个或多个相同或不同的卤素取代;或
两个R6连接以与它们所连接的原子一起形成环A2b;
A2b是苯基、C3-7环烷基或3-7元杂环基,其中A2b任选地被一个或多个相同或不同的R7取代;
每个R7独立地是C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代;
R1是H或C1-4烷基,优选H,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;
R2是H、F或C1-4烷基,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;且
R3是A3、C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的R8取代;或
R2和R3连接以与它们所连接的氧原子和碳原子一起形成环A3a,其中A3a是7-12元杂二环基,其中7-12元杂二环基任选地被一个或多个相同或不同的R10取代;
R2a是H或F,优选H;
每个R8独立地是卤素、CN、C(O)OR9、OR9、C(O)R9、C(O)N(R9R9a)、S(O)2N(R9R9a)、S(O)N(R9R9a)、S(O)2R9、S(O)R9、N(R9)S(O)2N(R9aR9b)、SR9、N(R9R9a)、NO2、OC(O)R9、N(R9)C(O)R9a、N(R9)SO2R9a、N(R9)S(O)R9a、N(R9)C(O)N(R9aR9b)、N(R9)C(O)OR9a、OC(O)N(R9R9a)或A3;
R9、R9a、R9b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素、或一个OH、或一个OC1-4烷基、或一个A3取代;
每个A3独立地是苯基、萘基、C3-7环烷基、3-7元杂环基或7-12元杂二环基,其中A3任选地被一个或多个相同或不同的R10取代;
每个R10独立地是卤素、CN、C(O)OR11、OR11、C(O)R11、C(O)N(R11R11a)、S(O)2N(R11R11a)、S(O)N(R11R11a)、S(O)2R11、S(O)R11、N(R11)S(O)2N(R11aR11b)、SR11、N(R11R11a)、NO2、OC(O)R11、N(R11)C(O)R11a、N(R11)S(O)2R11a、N(R11)S(O)R11a、N(R11)C(O)OR11a、N(R11)C(O)N(R11aR11b)、OC(O)N(R11R11a)、氧代(=O),其中所述环是至少部分饱和的、C1-6烷基,C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的R12取代;
R11、R11a、R11b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代;
每个R12独立地是卤素、CN、C(O)OR13、OR13、C(O)R13、C(O)N(R13R13a)、S(O)2N(R13R13a)、S(O)N(R13R13a)、S(O)2R13、S(O)R13、N(R13)S(O)2N(R13aR13b)、SR13、N(R13R13a)、NO2、OC(O)R13、N(R13)C(O)R13a、N(R13)SO2R13a、N(R13)S(O)R13a、N(R13)C(O)N(R13aR13b)、N(R13)C(O)OR13a或OC(O)N(R13R13a);
R13、R13a、R13b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代。
本发明的化合物代表1,3-二氧杂环己烷衍生物,它们可以根据本文或别处(例如在WO 98/56788 A2中)描述的途径和方法来制备。
在变量或取代基可以选自一组不同变体并且这样的变量或取代基出现超过一次的情况下,各个变体可以相同或不同。
在本发明的含义内,如下使用术语:
术语“任选地被取代的”是指未被取代的或被取代的。通常,但不限于,“一个或多个取代基”是指一个、两个或三个取代基,优选一个或两个取代基,且更优选一个取代基。通常,这些取代基可以相同或不同。
“烷基”是指直链或支链烃链。烷基碳的每个氢可以被进一步指定的取代基替代。
“烯基”是指含有至少一个碳-碳双键的直链或支链烃链。烯基碳的每个氢可以被进一步指定的取代基替代。
“炔基”是指含有至少一个碳-碳三键的直链或支链烃链。炔基碳的每个氢可以被进一步指定的取代基替代。
“C1-4烷基”是指具有1-4个碳原子的烷基链,如果存在于分子的末端,例如:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基,或者,当分子的两个部分通过烷基基团连接时,例如-CH2-、-CH2-CH2-、-CH(CH3)-、-CH2-CH2-CH2-、-CH(C2H5)-、-C(CH3)2-。C1-4烷基碳的每个氢可以被进一步指定的取代基替代。
“C1-6烷基”是指具有1-6个碳原子的烷基链,如果存在于分子的末端,例如:C1-4烷基、甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基,或者,当分子的两个部分通过烷基基团连接时,例如-CH2-、-CH2-CH2-、-CH(CH3)-、-CH2-CH2-CH2-、-CH(C2H5)-、-C(CH3)2-。C1-6烷基碳的每个氢可以被进一步指定的取代基替代。
“C2-6烯基”是指具有2-6个碳原子的烯基链,如果存在于分子的末端,例如:-CH=CH2、-CH=CH-CH3、-CH2-CH=CH2、-CH=CH-CH2-CH3、-CH=CH-CH=CH2,或者,当分子的两个部分通过烯基基团连接时,例如-CH=CH-。C2-6烯基碳的每个氢可以被进一步指定的取代基替代。
“C2-6炔基”是指具有2-6个碳原子的炔基链,如果存在于分子的末端,例如:-C≡CH、-CH2-C≡CH、CH2-CH2-C≡CH、CH2-C≡C-CH3,或者,当分子的两个部分通过炔基基团连接时,例如-C≡C-。C2-6炔基碳的每个氢可以被进一步指定的取代基替代。
“C3-7环烷基”或“C3-7环烷基环”是指具有3-7个碳原子的环烷基链,例如环丙基、环丁基、环戊基、环己基、环己烯基、环庚基。优选地,环烷基是指环丙基、环丁基、环戊基、环己基或环庚基。环烷基碳的每个氢可以被本文进一步指定的取代基替代。相应地定义术语“C3-5环烷基”或“C3-5环烷基环”。
“C5亚环烷基”是指具有5个碳原子的二价环烷基,即二价环戊基环。
“C5亚环烯基”是指二价亚环烯基,即二价环戊烯或环戊二烯。
“C4-12二环烷基”或“C4-12二环烷基环”是指具有4-12个碳原子的二环稠合的、桥连的或螺烷基链,例如六氢茚满、八氢并环戊二烯、双环[2.2.1]庚烷或螺(3.2)己烷。二环烷基碳的每个氢可以被本文进一步指定的取代基替代。
“卤素”是指氟、氯、溴或碘。通常优选地卤素是氟或氯。
“3-7元杂环基”或“3-7元杂环”是指具有3、4、5、6或7个环原子的环,其可以含有至多最大数目的双键(芳族环或完全饱和、部分饱和或不饱和的非芳族环),其中至少一个环原子直至4个环原子被选自硫(包括-S(O)-、-S(O)2-)、氧和氮(包括=N(O)-)的杂原子替代,且其中所述环经由碳或氮原子连接至分子的其余部分。3-7元杂环的例子是氮杂环丙烷、氮杂环丁烷、氧杂环丁烷、硫杂环丁烷、呋喃、噻吩、吡咯、吡咯啉、咪唑、咪唑啉、吡唑、吡唑啉、噁唑、噁唑啉、异噁唑、异噁唑啉、噻唑、噻唑啉、异噻唑、异噻唑啉、噻二唑、噻二唑啉、四氢呋喃、四氢噻吩、吡咯烷、咪唑烷、吡唑烷、噁唑烷、异噁唑烷、噻唑烷、异噻唑烷、噻二唑烷、环丁砜、吡喃、二氢吡喃、四氢吡喃、咪唑烷、吡啶、哒嗪、吡嗪、嘧啶、哌嗪、哌啶、吗啉、四唑、三唑、三唑烷、四唑烷、二氮杂环庚烷、氮杂环庚三烯或高哌嗪。术语“5-6元杂环基”或“5-6元杂环”相应地定义且包括5-6元芳族杂环基或杂环。术语“5元杂环基”或“5元杂环”相应地定义且包括5元芳族杂环基或杂环。
术语“含有氮环原子的5元亚杂环基”是指二价5元杂环,其中五个环原子中的至少一个是氮原子并且其中所述环经由碳或氮原子连接至分子的其余部分。
“饱和的4-7元杂环基”或“饱和的4-7元杂环”是指完全饱和的“4-7元杂环基”或“4-7元杂环”。
“4-7元至少部分饱和的杂环基”或“4-7元至少部分饱和的杂环”是指至少部分饱和的“4-7元杂环基”或“4-7元杂环”。
“5-6元芳族杂环基”或“5-6元芳族杂环”是指衍生自环戊二烯基或苯的杂环,其中至少一个碳原子被选自硫(包括-S(O)-、-S(O)2-)、氧和氮(包括=N(O)-)的杂原子替代。这样的杂环的例子是呋喃、噻吩、吡咯、咪唑、吡唑、噁唑、异噁唑、噻唑、异噻唑、噻二唑、三唑、四唑、吡啶、嘧啶、哒嗪、吡嗪、三嗪。
“5元芳族杂环基”或“5元芳族杂环”是指衍生自环戊二烯基的杂环,其中至少一个碳原子被选自硫(包括-S(O)-、-S(O)2-)、氧和氮(包括=N(O)-)的杂原子替代。这样的杂环的例子是呋喃、噻吩、吡咯、咪唑、吡唑、噁唑、异噁唑、噻唑、异噻唑、噻二唑、三唑、四唑。
“7-12元杂二环基”或“7-12元杂二环”是指具有7-12个环原子的两个环的杂环系统,其中两个环共享至少一个环原子,并且其可以含有至多最大数目的双键(芳族环或完全饱和、部分饱和或不饱和的非芳族环),其中至少一个环原子直到6个环原子被选自硫(包括-S(O)-、-S(O)2-)、氧和氮(包括=N(O)-)的杂原子替代,且其中所述环经由碳或氮原子连接至分子的其余部分。7-12元杂二环的例子是吲哚、吲哚啉、苯并呋喃、苯并噻吩、苯并噁唑、苯并异噁唑、苯并噻唑、苯并异噻唑、苯并咪唑、苯并咪唑啉、喹啉、喹唑啉、二氢喹唑啉、喹啉、二氢喹啉、四氢喹啉、十氢喹啉、异喹啉、十氢异喹啉、四氢异喹啉、二氢异喹啉、苯并氮杂环庚三烯、嘌呤或蝶啶。术语7-12元杂二环还包括两个环的螺结构如6-氧杂-2-氮杂螺[3,4]辛烷、2-氧杂-6-氮杂螺[3.3]庚烷-6-基或2,6-二氮杂螺[3.3]庚烷-6-基,或桥连杂环如8-氮杂-双环[3.2.1]辛烷或2,5-二氮杂双环[2.2.2]辛烷-2-基或3,8-二氮杂双环[3.2.1]辛烷。
“饱和的7-12元杂二环基”或“饱和的7-12元杂二环”是指完全饱和的7-12元杂二环基或7-12元杂二环。
“7-12元至少部分饱和的杂二环基”或“7-12元至少部分饱和的杂二环”是指至少部分饱和的“7-12元杂二环基”或“7-12元杂二环”。
“9-11元芳族杂二环基”或“9-11元芳族杂二环”是指两个环的杂环系统,其中至少一个环是芳族的,并且其中所述杂环系统具有9-11个环原子,其中两个环共享两个环原子,并且其可以含有至多最大数目的双键(完全或部分芳族的),其中至少一个环原子直到6个环原子被选自硫(包括-S(O)-、-S(O)2-)、氧和氮(包括=N(O)-)的杂原子替代,且其中所述环经由碳或氮原子连接至分子的其余部分。9-11元芳族杂二环的例子是吲哚、吲哚啉、苯并呋喃、苯并噻吩、苯并噁唑、苯并异噁唑、苯并噻唑、苯并异噻唑、苯并咪唑、苯并咪唑啉、喹啉、喹唑啉、二氢喹唑啉、二氢喹啉、四氢喹啉、异喹啉、四氢异喹啉、二氢异喹啉、苯并氮杂环庚三烯、嘌呤或蝶啶。术语“9-10元芳族杂二环基”或“9-10元芳族杂二环”相应地定义。
优选的式(I)的化合物是其中所含的一个或多个残基具有下面给出的含义的那些化合物,其中优选的取代基定义的所有组合都是本发明的主题。关于所有优选的式(I)的化合物,本发明还包括所有互变异构和立体异构形式及其所有比率的混合物,以及它们的药学上可接受的盐。
在本发明的优选实施方案中,下面提到的取代基独立地具有下列含义。因此,这些取代基中的一个或多个可以具有下面给出的优选的或更优选的含义。
优选地,Ra1、Ra2、Ra3、Ra4、Ra5、Ra6是H。
优选地,A1是含有氮环原子的5元亚杂环基,且其中A1任选地被一个或多个相同或不同的R4取代。
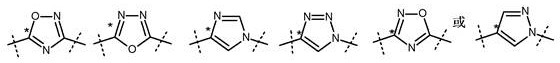
更优选地,A1是含有氮环原子的5元亚杂环基,其选自以下二价杂环:噁二唑、咪唑、咪唑烷、吡唑和三唑,甚至更优选三唑或噁二唑,甚至更优选噁二唑。A1任选地被一个或多个相同或不同的R4取代。
优选地,A1是未被取代的或被一个或两个相同或不同的R4取代,更优选地,A1是未被取代的。
优选地,R4独立地是氧代(=O),其中所述环是至少部分饱和的、卤素、CN、OR5或C1-6烷基,其中C1-6烷基任选地被一个或多个相同或不同的卤素取代。
优选地,R4是氧代,其中所述环是至少部分饱和的。
优选地,A1是

更优选地,A1是

甚至更优选地,A1是

在一个实施方案中,A2是R6a。在另一个实施方案中,A2是A2a。
优选地,R6a是C1-6烷基,其任选地被一个或多个卤素和/或一个A2a和/或一个OR6a3取代。更优选地,R6a是C1-6烷基,其任选地被一个或多个卤素和/或一个OR6a3取代。
优选地,R6a是OR6a1。
R6a1优选地是A2a或C1-6烷基,其中C1-6烷基任选地被一个或多个卤素和/或一个A2a和/或一个OR6a3取代。更优选地,R6a1是C1-6烷基,其任选地被一个或多个F和/或一个OR6a3取代。
优选地,R6a2是H。
优选地,R6a是OC1-4烷基;OC1-4烷基-OC1-4烷基,其中每个C1-4烷基任选地被1-3个F取代;或OCH2A2a。甚至更优选地,R6a是O(CH2)3CF3或O(CH2)2OCF3。
优选地,A2a是苯基;C3-7环烷基;或3-7元杂环基,其中A2a任选地被一个或多个相同或不同的R6取代。
优选地,A2a是苯基或5-6元芳族杂环基, 其中A2a任选地被一个或多个相同或不同的R6取代。优选的5-6元芳族杂环基优选地是吡啶基、吡嗪基、哒嗪基、吡唑基或1,2,4-噁二唑基。
优选地,A2a是C3-7环烷基,更优选环丁基,其中A2a任选地被一个或多个相同或不同的R6取代。
优选地,A2a是氮杂环丁基。
优选地,A2a被一个或两个相同或不同的R6取代。
优选地,R6独立地是F、Cl、CF3、OCH3、OCF3、CH3、CH2CH3或环丙基,优选F、Cl、CF3、OCH3、CH3、CH2CH3或环丙基。
甚至更优选地,A2是4-氯苯基、3-三氟甲氧基环丁基、3-三氟甲氧基-氮杂环丁烷-1-基、4,4,4-三氟丁基-1-氧基或2-三氟甲氧基乙氧基。
优选地,R2是H。
优选地,R3是A3。
优选地,A3是苯基、吡啶基、吡嗪基、嘧啶基(pyrimidazyl)、环丙基、环丁基或环己基,更优选苯基或吡啶基,且其中A3任选地被一个或多个相同或不同的R10取代。
优选地,A3被一个或两个相同或不同的R10取代。
优选地,R2和R3与它们所连接的氧和碳原子连接在一起以形成二氢苯并吡喃环,其中所述环任选地被一个或多个相同或不同的R10取代,优选地,所述环被一个或两个R10取代。
优选地,R10是CHF2。
优选地,R10独立地是F、Cl、Br、CF3、OCF3、CH=O、CH2OH或CH3;优选F、Cl、CF3、CH=O、CH2OH或CH3。甚至更优选F或Cl。
甚至更优选地,R3是3-氯-4-二氟甲基苯基或3,4-二氯苯基。
甚至更优选地,R3是4-氯-3-氟苯基或2-氯-3-氟吡啶-5-基,甚至更优选4-氯-3-氟苯基。
其中一些或全部上述基团具有优选的或更优选的含义的式(I)的化合物也是本发明的一个目的。
本发明的优选的具体化合物选自:
2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[顺式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4,4,4-三氟丁氧基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-[(6-氯-5-氟-3-吡啶基)氧基]-N-[反式-2-[5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-[3-氯-4-(二氟甲基)苯氧基]-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;或
2-(3,4-二氯苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺。
在可以发生式(I)的化合物的互变异构(如例如,酮-烯醇互变异构)的情况下,分别包括以及作为任何比率的混合物包括各种形式,如例如酮和烯醇形式。这同样适用于立体异构体,如例如对映异构体、顺/反异构体、构象异构体等。
尤其是,当在根据式(I)的化合物中给出对映异构或非对映异构形式时,单独每种纯形式和至少两种纯形式的任何比率的任何混合物都被式(I)包含并且是本发明的主题。
一种优选的化合物是具有如在式(Ia)中所示的相对构型的式(I)的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体

同位素标记的式(I)的化合物也在本发明范围内。用于同位素标记的方法是本领域已知的。优选的同位素是元素H、C、N、O和S的同位素。式(I)的化合物的溶剂化物和水合物也在本发明的范围内。
如果需要的话,可以通过本领域众所周知的方法分离异构体,例如,通过液相色谱法。通过使用例如手性固定相,这同样适用于对映异构体。另外,可以如下分离对映异构体:将它们转化为非对映异构体,即,与对映异构纯的辅助化合物偶联,随后分离所得的非对映异构体,并裂解辅助残基。可替换地,使用光学纯的起始材料、试剂和/或催化剂,可以由立体选择性合成获得式(I)的化合物的任何对映异构体。
在根据式(I)的化合物含有一个或多个酸性或碱性基团的情况下,本发明也包含它们的相应的药学上或毒理学上可接受的盐,尤其是它们的药学上可利用的盐。因此,根据本发明,可以例如作为碱金属盐、碱土金属盐或作为铵盐使用含有酸性基团的式(I)的化合物。这样的盐的更确切的例子包括钠盐、钾盐、钙盐、镁盐或与氨或有机胺例如乙胺、乙醇胺、三乙醇胺或氨基酸的盐。可以存在含有一个或多个碱性基团(即,可以被质子化的基团)的式(I)的化合物,并且其根据本发明可以以它们与无机或有机酸形成的加成盐的形式使用。合适的酸的例子包括:盐酸、氢溴酸、磷酸、硫酸、硝酸、甲磺酸、对甲苯磺酸、萘二磺酸、草酸、乙酸、酒石酸、乳酸、水杨酸、苯甲酸、甲酸、丙酸、新戊酸、二乙基乙酸、丙二酸、琥珀酸、庚二酸、富马酸、马来酸、苹果酸、氨基磺酸、苯基丙酸、葡糖酸、抗坏血酸、异烟酸、柠檬酸、己二酸,以及本领域技术人员已知的其它酸。如果式(I)的化合物在分子中同时含有酸性和碱性基团,则除了提到的盐形式之外,本发明还包括内盐或内铵盐(两性离子)。通过本领域技术人员已知的常规方法,如例如,通过在溶剂或分散剂中使这些与有机或无机酸或碱接触,或通过与其它盐的阴离子交换或阳离子交换,可以获得根据式(I)的各种盐。本发明还包括式(I)的化合物的所有盐,所述盐由于低生理学相容性,不直接适用于药物,但其可用作例如化学反应的中间体或用于制备药学上可接受的盐。
如下所示,本发明的化合物据信适用于调节整合应激反应途径。
整合应激反应(ISR)是所有真核生物共有的细胞应激反应(1)。ISR信号传递的调节异常具有重要的病理学后果,尤其是与炎症、病毒感染、糖尿病、癌症和神经变性疾病有关的后果。
ISR是不同类型的细胞应激的共同特征,导致在丝氨酸51上的真核翻译起始因子2(eIF2α)的α亚基的磷酸化,从而抑制正常蛋白合成和应激反应基因的表达(2)。在哺乳动物细胞中,所述磷酸化由四个eIF2α激酶的家族进行,即:PKR-样ER激酶(PERK)、双链RNA依赖性蛋白激酶(PKR)、亚铁血红素调节的eIF2α激酶(HRI)和一般性调控阻遏蛋白激酶2(GCN2),各自响应于不同的环境和生理应激(3)。
eIF2α与eIF2β和eIF2γ一起形成eIF2复合物,这是正常mRNA翻译起始的关键参与者(4)。eIF2复合物结合GTP和Met-tRNAi,从而形成三元复合物(eIF2-GTP-Met-tRNAi),其被核糖体募集用于翻译起始(5, 6)。
eIF2B是由5种亚基(α、β、γ、δ、ε)组成的异十聚体复合物,所述亚基一式两份地形成GEF活性十聚体(7)。
响应于ISR活化,磷酸化的eIF2α抑制eIF2B介导的GDP与GTP的交换,导致三元复合物形成减少,并因此抑制以核糖体与5'AUG起始密码子结合为特征的正常mRNA的翻译(8)。在三元复合物丰度降低的这些条件下,包括编码转录因子ATF4的mRNA在内的几种特定mRNA的翻译通过涉及上游ORF (uORF)的翻译改变的机制被活化(7, 9, 10)。这些mRNA通常包含一个或多个uORF,所述uORF通常在未受应激的细胞中起作用以限制核糖体向主要编码ORF的流动。例如,在正常条件下,在ATF的5' UTR中的uORF占据核糖体并阻止ATF4的编码序列的翻译。但是,在应激条件下,即在三元复合物形成减少的条件下,核糖体扫描经过这些上游ORF并在ATF4编码ORF处开始翻译的可能性增加。ATF4和其它以这种方式表达的应激反应因子随后控制着一系列其它应激反应基因的表达。急性期在于旨在恢复稳态的蛋白的表达,而慢性期导致促凋亡因子的表达(1, 11, 12, 13)。
ISR信号传递的标志物的上调已在多种病症中得到证实,其中包括癌症和神经变性疾病。在癌症中,ER应激调节的翻译增加对低氧条件的耐受性并促进肿瘤生长(14, 15,16),并且已经证明通过基因靶向对PERK的删除可以减慢从转化的PERK−/−小鼠胚胎成纤维细胞衍生出的肿瘤的生长(14, 17)。此外,最近的一份报告已经提供了如下的概念证据:使用在小鼠中的源自患者的异种移植模型,eIF2B的活化剂有效治疗一种侵袭性转移性前列腺癌(28)。总之,细胞保护性ISR信号传递的预防可能代表一种有效的抗增殖策略,其用于治疗至少某些形式的癌症。
此外,ISR信号传递的调节可以证明在保持突触功能和减少神经元衰退中是有效的,在特征在于错误折叠的蛋白和未折叠蛋白反应(UPR)的激活的神经变性疾病(诸如肌萎缩性侧索硬化(ALS)、额颞叶痴呆(FTD)、阿尔茨海默氏病(AD)、帕金森病(PD)和JakobCreutzfeld (朊病毒)疾病)中也是如此(18, 19, 20)。对于朊病毒病(存在的神经变性疾病的一个例子),其中已经表明,ISR信号传递的药理学以及遗传抑制可以使蛋白翻译水平正常化、挽救突触功能并预防神经元损失(21)。具体地,通过控制磷酸化eIF2α水平的磷酸酶的过表达来降低磷酸化eIF2α的水平会增加感染了朊病毒的小鼠的存活率,而持续的eIF2α磷酸化降低存活率(22)。
此外,控制蛋白表达水平对于适当脑功能的重要性的直接证据以影响eIF2和eIF2B的功能的罕见遗传性疾病的形式存在。破坏eIF2的复杂完整性且因此导致降低的正常蛋白表达水平的eIF2γ中的突变与智力障碍综合征(ID)相关(23)。在eIF2B的亚基中的部分功能丧失突变已被证实是罕见的脑白质营养不良白质消融性疾病(VWMD)的原因(24,25)。具体地,与ISRIB相关的小分子在VWMD小鼠模型中对eIF2B部分功能丧失的稳定化已被证实会减少ISR标记物并改善功能以及病理学终点(26, 27)。
本发明提供了要用于治疗本文提及的疾病或障碍的呈游离或药学上可接受的盐形式或呈溶剂化物、水合物、互变异构体或立体异构体形式的本发明的化合物。
因此,本发明的一个方面是用作上述药物的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体。
描述的治疗方法可以应用于哺乳动物诸如狗、猫、牛、马、兔、猴和人类。优选地,所述哺乳动物患者是人类患者。
因此,本发明提供了要用于治疗或预防一种或多种与整合应激反应有关的疾病或障碍的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物。
本发明的另一个方面是用于治疗或预防一种或多种与整合应激反应有关的障碍或疾病的方法的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物。
本发明的另一个方面是本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物用于制备药物的用途,所述药物用于治疗或预防一种或多种与整合应激反应有关的障碍或疾病。
本发明的再另一个方面是一种用于治疗、控制、延迟或预防需要治疗的哺乳动物患者中的一种或多种与整合应激反应有关的疾病或障碍的方法,其中所述方法包括给所述患者施用治疗有效量的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物。
本发明提供要用于治疗或预防一种或多种下述疾病或障碍的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体。
本发明的另一个方面是用于治疗或预防一种或多种下述障碍或疾病的方法的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物。
本发明的另一个方面是本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物用于制备药物的用途,所述药物用于治疗或预防一种或多种下述障碍或疾病。
本发明的再另一个方面是一种用于治疗、控制、延迟或预防需要治疗的哺乳动物患者中的一种或多种下述疾病或障碍的方法,其中所述方法包括给所述患者施用治疗有效量的本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或本发明的药物组合物。
疾病或障碍包括、但不限于脑白质营养不良,智力障碍综合征,神经变性疾病和障碍,肿瘤性疾病,感染性疾病,炎性疾病,肌肉骨骼疾病,代谢疾病,眼疾病以及选自器官纤维化、慢性和急性肝病、慢性和急性肺病、慢性和急性肾病、心肌梗塞、心血管疾病、心律失常、动脉粥样硬化、脊髓损伤、缺血性中风和神经性疼痛的疾病。
脑白质营养不良
脑白质营养不良的例子包括、但不限于白质消融性疾病(VWMD)和伴有CNS髓鞘形成不足的儿童共济失调(例如与eIF2或包括eIF2的信号转导或信号传递途径中的组分的功能受损相关)。
智力障碍综合征
智力障碍尤其是指这样的病症:其中人在智力功能如交流、照顾自己方面具有某些限制,和/或具有受损的社交技能。智力障碍综合征包括、但不限于与eIF2或包括eIF2的信号转导或信号传递途径中的组分的功能受损相关的智力障碍病症。
神经变性疾病/障碍
神经变性疾病和障碍的例子包括、但不限于亚历山大氏病、阿耳珀病、阿尔茨海默氏病、肌萎缩性侧索硬化、共济失调毛细血管扩张症、巴藤病(也被称作Spielmeyer-Vogt-Sjogren- Batten病)、牛海绵状脑病(BSE)、卡纳万病、科凯恩综合征、皮质基底变性、克雅病、额颞叶痴呆、Gerstmann-Straussler-Scheinker综合征、亨廷顿病、HIV相关的痴呆、肯尼迪病、克拉伯病、库鲁病、路易体痴呆、Machado-Joseph病(3型脊髓小脑性共济失调)、多发性硬化、多系统萎缩、发作性睡病、神经疏螺旋体病、帕金森病、佩梅病、皮克病、原发性侧索硬化、朊病毒疾病、进行性核上性麻痹、雷弗素姆氏病、山德霍夫氏病、谢耳德氏病、恶性贫血继发的亚急性脊髓混合变性、精神分裂症、脊髓小脑性共济失调(具有不同特征的多种类型)、脊髓性肌萎缩、Steele-Richardson-Olszewski病、脊髓痨和Tau病变。
神经变性疾病或障碍尤其选自阿尔茨海默氏病、帕金森病和肌萎缩性侧索硬化。
肿瘤性疾病
可以将肿瘤性疾病最广义地理解为起因于失控细胞生长的任何组织。在许多情况下,肿瘤至少导致庞大的组织块,其任选通过血管受到神经支配。它可以包含或不包含一个或多个转移/转移灶的形成。本发明的肿瘤性疾病可以是通过疾病和有关健康问题的国际统计分类第10次修订(International Statistical Classification of Diseases andRelated Health Problems 10th Revision)(ICD-10)类别C00-D48分类的任何肿瘤。
示例性地,根据本发明的肿瘤性疾病可以是一种或多种恶性肿瘤(肿瘤) (ICD-10类别C00-C97)的存在,可以是一种或多种原位肿瘤(ICD-10类别D00-D09)的存在,可以是一种或多种良性肿瘤(ICD-10类别D10-D36)的存在,或可以是一种或多种不确定或未知行为的肿瘤(ICD-10类别D37-D48)的存在。优选地,根据本发明的肿瘤性疾病是指一种或多种恶性肿瘤的存在,即,是恶性肿瘤形成(ICD-10类别C00-C97)。
在一个更优选的实施方案中,所述肿瘤性疾病是癌症。
可以将癌症最广义地理解为任何恶性肿瘤性疾病,即,一种或多种恶性肿瘤在患者中的存在。癌症可以是实体或血液学恶性肿瘤。本文涵盖但不限于白血病、淋巴瘤、癌和肉瘤。
在本文中尤其包括特征在于上调的ISR标志物的肿瘤性疾病,诸如癌症。
示例性的癌症包括、但不限于甲状腺癌、内分泌系统癌、胰腺癌、脑癌(例如多形性胶质母细胞瘤、神经胶质瘤)、乳腺癌(例如ER阳性的、ER阴性的、化学疗法抗性的、赫赛汀抗性的、HER2阳性的、多柔比星抗性的、他莫昔芬抗性的、导管癌、小叶癌、原发性的、转移性的)、子宫颈癌、卵巢癌、子宫癌、结肠癌、头颈癌、肝癌(例如肝细胞癌)、肾癌、肺癌(例如非小细胞肺癌、鳞状细胞肺癌、腺癌、大细胞肺癌、小细胞肺癌、类癌、肉瘤)、结肠癌、食管癌、胃癌、膀胱癌、骨癌、胃癌、前列腺癌和皮肤癌(例如黑素瘤)。
其它例子包括、但不限于骨髓瘤、白血病、间皮瘤和肉瘤。
另外的例子包括、但不限于髓母细胞瘤、霍奇金病、非霍奇金淋巴瘤、多发性骨髓瘤、神经母细胞瘤、神经胶质瘤、多形性胶质母细胞瘤、横纹肌肉瘤、原发性血小板增多、原发性巨球蛋白血症、原发性脑肿瘤、恶性胰腺胰岛瘤、恶性类癌、膀胱癌、恶化前的皮肤病变、睾丸癌、淋巴瘤、生殖尿道癌、恶性高钙血症、子宫内膜癌、肾上腺皮质癌、内分泌或外分泌胰腺肿瘤、髓样甲状腺癌、甲状腺髓样癌、黑素瘤、结肠直肠癌、乳头状甲状腺癌、肝细胞癌、佩吉特乳头病、叶状瘤、小叶癌、导管癌、胰腺星形细胞癌和肝星形细胞癌。
示例性的白血病包括、但不限于急性非淋巴细胞性白血病、慢性淋巴细胞性白血病、急性粒细胞性白血病、慢性粒细胞性白血病、急性早幼粒细胞性白血病、成年T-细胞白血病、白细胞不增多性白血病、白细胞增多性白血病、嗜碱细胞性白血病、胚细胞白血病、牛白血病、慢性粒细胞性白血病、皮肤白血病、胚细胞性白血病、嗜酸性粒细胞性白血病、Gross氏白血病、毛细胞白血病、成血细胞性白血病(hemoblastic leukemia)、成血细胞性白血病(hemocytoblastic leukemia)、组织细胞性白血病、干细胞白血病、急性单核细胞性白血病、白细胞减少性白血病、淋巴性白血病、成淋巴细胞性白血病、淋巴细胞性白血病、淋巴原性白血病、淋巴样白血病、淋巴肉瘤细胞白血病、肥大细胞白血病、巨核细胞白血病、微型成髓细胞白血病(micromyeloblastic leukemia)、单核细胞性白血病、成髓细胞性白血病、髓细胞性白血病、骨髓粒细胞性白血病、髓单核细胞性白血病、Naegeli白血病、浆细胞白血病、多发性骨髓瘤、浆细胞性白血病、早幼粒细胞性白血病、Rieder细胞白血病、希林氏白血病、干细胞白血病、亚白血性白血病和未分化细胞性白血病。
示例性的肉瘤包括、但不限于软骨肉瘤、纤维肉瘤、淋巴肉瘤、黑素肉瘤、粘液肉瘤、骨肉瘤、艾伯内西氏肉瘤、脂肪肉瘤、脂肉瘤、软组织腺泡状肉瘤、成釉细胞肉瘤、葡萄状肉瘤、绿色瘤肉瘤、绒毛膜癌、胚胎性肉瘤、肾母细胞瘤肉瘤、子宫内膜肉瘤、间质肉瘤、尤因氏肉瘤、筋膜肉瘤、成纤维细胞性肉瘤、巨细胞肉瘤、粒细胞肉瘤、霍奇金肉瘤、特发性多发性色素性出血性肉瘤、B细胞免疫母细胞肉瘤、淋巴瘤、T细胞免疫母细胞肉瘤、詹森肉瘤、卡波西氏肉瘤、库普弗细胞肉瘤、血管肉瘤、白血病性肉瘤、恶性间叶瘤肉瘤、骨膜外肉瘤、网状细胞肉瘤、劳斯肉瘤、浆液囊性肉瘤、滑膜肉瘤和毛细血管扩张性肉瘤。
示例性的黑素瘤包括、但不限于肢端雀斑痣样黑素瘤、无黑色素性黑素瘤、良性幼年型黑素瘤、克劳德曼氏黑素瘤(Cloudman's melanoma)、S91黑素瘤、哈-帕二氏黑素瘤、幼年型黑素瘤、恶性雀斑样黑素瘤、恶性黑素瘤、结节性黑色素瘤、甲下黑素瘤和浅表扩散性黑素瘤。
示例性的癌包括、但不限于甲状腺髓样癌、家族性甲状腺髓样癌、腺泡癌、腺泡状癌、囊性腺样癌、腺样囊性癌、腺癌、肾上腺皮质癌、肺泡癌、肺泡细胞癌、基底细胞癌、基底上皮细胞癌、基底细胞样癌、基底鳞状细胞癌、细支气管肺泡癌、细支气管癌、支气管癌、脑样癌、胆管细胞癌、绒毛膜癌、胶体癌、粉刺状癌、子宫体癌、筛状癌、铠甲状癌、皮肤癌、柱状癌、柱状细胞癌、管癌、导管癌、硬癌、胚胎性癌、脑样癌、表皮样癌、腺样上皮癌、外生性癌、溃疡性癌、纤维癌、胶样癌、胶状癌、巨细胞癌(giant cell carcinoma)、巨细胞癌(carcinoma gigantocellulare)、腺癌、粒层细胞癌、毛基质癌、血样癌、肝细胞癌、Hurthle细胞癌、胶样癌(hyaline carcinoma)、肾上腺样癌、婴儿胚胎癌、原位癌、表皮内癌、上皮内癌、Krompecher癌、Kulchitzky细胞癌、大细胞癌、豆状癌(lenticular carcinoma)、豆状癌(carcinoma lenticulare)、脂肪瘤样癌、小叶癌、淋巴上皮癌、髓样癌(carcinomamedullare)、髓样癌(medullary carcinoma)、黑色素癌、软癌、粘液癌(mucinouscarcinoma)、粘液癌(carcinoma muciparum)、粘液细胞癌、粘液表皮样癌、粘液癌(carcinoma mucosum)、粘液癌(mucous carcinoma)、粘液瘤样癌(carcinomamyxomatodes)、鼻咽癌、燕麦细胞癌、骨化癌、骨样癌、乳头状癌、门脉周癌、浸润前癌、刺细胞癌、脑样癌(pultaceous carcinoma)、肾细胞癌、储备细胞癌、肉瘤样癌、施奈德癌、硬癌、阴囊癌、印戒细胞癌、单纯癌、小细胞癌、马铃薯状癌、球状细胞癌、梭形细胞癌、髓样癌、鳞状癌、鳞状细胞癌、绳捆癌、血管扩张性癌(carcinoma telangiectaticum)、血管扩张性癌(carcinoma telangiectodes)、移行细胞癌、结节性皮癌(carcinoma tuberosum)、管状癌、结节性皮癌(tuberous carcinoma)、疣状癌和绒毛状癌。
感染性疾病
例子包括、但不限于由病毒引起的感染(诸如由以下病毒引起的感染:HIV-1: 人免疫缺陷病毒1型;IAV: 甲型流感病毒;HCV: 丙型肝炎病毒;DENV: 登革热病毒;ASFV: 非洲猪瘟病毒;EBV: 爱泼斯坦-巴尔病毒;HSV1: 单纯疱疹病毒1;CHIKV: 基孔肯亚病毒;HCMV: 人巨细胞病毒;SARS-CoV: 重度急性呼吸综合征冠状病毒;SARS-CoV-2: 重度急性呼吸综合征冠状病毒2)和由细菌引起的感染(诸如由军团菌属、布鲁杆菌属、芯卡体属(Simkania)、衣原体属、螺杆菌属和弯曲杆菌属引起的感染)。
炎性疾病
炎性疾病的例子包括、但不限于手术后认知功能障碍(外科手术后认知功能下降)、创伤性脑损伤、关节炎、类风湿性关节炎、银屑病关节炎、青少年特发性关节炎、多发性硬化、系统性红斑狼疮(SLE)、重症肌无力、青少年型糖尿病、1型糖尿病、格-巴二氏综合征、桥本脑炎、桥本甲状腺炎、强直性脊柱炎、银屑病、舍格伦综合征、血管炎、肾小球肾炎、自身免疫甲状腺炎、贝切特氏病、克罗恩氏病、溃疡性结肠炎、大疱性类天疱疮、结节病、鱼鳞病、格雷夫斯眼病、炎性肠病、阿狄森氏病、白癫风、哮喘、变应性哮喘、寻常痤疮、乳糜泻、慢性前列腺炎、炎性肠病、盆腔炎性疾病、再灌注损伤、结节病、移植排斥、间质性膀胱炎、动脉粥样硬化和特应性皮炎。
肌肉骨骼疾病
肌肉骨骼疾病的例子包括、但不限于肌营养不良、多发性硬化、Freidrich氏共济失调、肌萎缩障碍(例如,肌肉萎缩、少肌症、恶病质)、包涵体肌病、进行性肌萎缩、运动神经元病、腕管综合征、上髁炎、腱炎、背痛、肌肉痛、肌肉酸痛、重复性劳损障碍和麻痹。
代谢疾病
代谢疾病的例子包括、但不限于糖尿病(尤其是II型糖尿病)、非酒精性脂肪性肝炎(NASH)、非酒精性脂肪肝病(NAFLD)、Niemann-Pick病、肝纤维化、肥胖、心脏病、动脉粥样硬化、关节炎、胱氨酸病、苯丙酮尿症、增殖性视网膜病变和Kearns-Sayre病。
眼疾病
眼疾病的例子包括、但不限于任何闭塞性或炎症性视网膜血管疾病的水肿或新生血管形成,诸如虹膜红变、新生血管性青光眼、翼状胬肉、血管化青光眼滤泡、结膜乳头状瘤;脉络膜新生血管形成,诸如新生血管年龄相关性黄斑变性(AMD)、近视、前葡萄膜炎(prior uveitis)、创伤或特发性;黄斑水肿,诸如手术后黄斑水肿、继发于葡萄膜炎(包括视网膜和/或脉络膜炎症)的黄斑水肿、继发于糖尿病的黄斑水肿、和继发于视网膜血管闭塞性疾病(即视网膜分支和中央静脉闭塞)的黄斑水肿;由糖尿病引起的视网膜新生血管形成,诸如视网膜静脉闭塞、葡萄膜炎、颈动脉疾病引起的眼缺血综合征、眼或视网膜动脉阻塞、镰状红细胞性视网膜病变、其它缺血性或闭塞性新生血管视网膜病变、早产儿视网膜病变或伊耳斯氏病;和遗传性障碍,诸如VonHippel-Lindau综合征。
其它疾病
其它疾病包括、但不限于器官纤维化(诸如肝纤维化、肺纤维化或肾纤维化)、慢性和急性肝病(诸如脂肪肝病或肝皮脂腺病)、慢性和急性肺病、慢性和急性肾病、心肌梗塞、心血管疾病、心律失常、动脉粥样硬化、脊髓损伤、缺血性中风和神经性疼痛。
本发明的再另一个方面是一种药物组合物,其包含至少一种本发明的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体以及药学上可接受的载体,任选地与一种或多种其它生物活性化合物或药物组合物组合。
优选地,所述一种或多种生物活性化合物是除式(I)的化合物之外的整合应激反应途径的调节剂。
“药物组合物”是指一种或多种活性成分和构成载体的一种或多种惰性成分,以及由任何两种或更多种成分组合、络合或聚集而直接或间接形成的任何产品,或者由一种或多种成分分解而直接或间接形成的任何产品,或者由一种或多种成分的其它类型的反应或相互作用而直接或间接形成的任何产品。因此,本发明的药物组合物包括通过混合本发明的化合物和药学上可接受的载体而制成的任何组合物。
本发明的药物组合物可以包含一种或多种另外的化合物作为活性成分,如在组合物中的式(I)的化合物的混合物或整合应激反应途径的其它调节剂。
所述活性成分可以包含在一种或多种不同的药物组合物(药物组合物的组合)中。
术语“药学上可接受的盐”是指由药学上可接受的无毒的碱或酸(包括无机碱或酸和有机碱或酸)制备的盐。
所述组合物包括适用于口服、直肠、局部、胃肠外(包括皮下、肌肉内和静脉内)、眼(眼部)、肺(鼻或含服吸入)或鼻施用的组合物,尽管在任何给定情况下最合适的途径取决于所治疗的病症的性质和严重程度以及活性成分的性质。它们可以方便地存在于单位剂型中,并且可以通过药学领域众所周知的任何方法制备。
在实际应用中,式(I)的化合物可以作为活性成分与药物载体根据常规药物混配技术合并为紧密混合物。载体可以采用多种形式,取决于预期施用的制剂的形式,例如,口服或胃肠外(包括静脉内)。在制备口服剂型组合物时,在口服液体制剂(例如,混悬液、酏剂和溶液)的情况下,可以使用任何常用的药物介质,诸如水、二醇类、油类、醇类、调味剂、防腐剂、着色剂等;或在口服固体制剂(诸如粉剂、硬和软胶囊剂和片剂)的情况下,可以使用载体诸如淀粉、糖、微晶纤维素、稀释剂、造粒剂、润滑剂、粘合剂、崩解剂等,其中固体口服制剂比液体制剂优选。
由于片剂和胶囊剂容易施用,片剂和胶囊剂代表最有利的口服剂量单位形式,在这样的情况下,显而易见地使用固体药用载体。如果需要的话,可以通过标准的水性或非水性技术包衣片剂。这样的组合物和制剂应该含有至少0.1%的活性化合物。活性化合物在这些组合物中的百分比当然可以改变,并且可以方便地为所述单位的重量的大约2%至大约60%。活性化合物在这种治疗上有用的组合物中的量是可获得有效剂量的量。也可以鼻内施用活性化合物,例如作为液体滴剂或喷雾剂。
片剂、丸剂、胶囊剂等还可以含有:粘合剂诸如黄蓍胶、阿拉伯胶、玉米淀粉或明胶;赋形剂诸如磷酸二钙;崩解剂诸如玉米淀粉、马铃薯淀粉、海藻酸;润滑剂诸如硬脂酸镁;和甜味剂诸如蔗糖、乳糖或糖精。当剂量单位形式是胶囊时,除了上述类型的物质之外,它还可以含有液体载体诸如脂肪油。
各种其它物质可以作为包衣存在,或用于改变剂量单位的物理形式。例如,可以用虫胶、糖或两者包衣片剂。除了活性成分之外,糖浆剂或酏剂还可以含有:作为甜味剂的蔗糖,作为防腐剂的对羟苯甲酸甲酯和对羟苯甲酸丙酯,染料和调味剂例如樱桃或橙调味剂。
也可以胃肠外地施用式(I)的化合物。可以在与表面活性剂诸如羟丙基纤维素适当混合的水中制备这些活性化合物的溶液或混悬液。还可以在甘油、液体聚乙二醇和其在油中的混合物中制备分散体。在贮存和使用的一般条件下,这些制品含有防腐剂以阻止微生物的生长。
适合于注射使用的药物形式包括无菌水溶液或分散体,和用于即时制备无菌注射溶液或分散体的无菌粉末。在所有情况下, 该形式应该是无菌的,并且应该以易于注射的程度流动。它应当在制造和储存条件下是稳定的,并且应当针对微生物诸如细菌和真菌的污染作用进行保存。所述载体可以是溶剂或分散介质,其含有例如水、乙醇、多元醇(例如,甘油、丙二醇和液体聚乙二醇)、其合适混合物和植物油。
可以采用任何合适的施用途径以给哺乳动物、特别是人提供有效剂量的本发明的化合物。可以使用例如口服、直肠、局部、胃肠外、眼、肺、鼻途径等。剂型包括片剂、糖锭、分散体、混悬液、溶液、胶囊剂、乳膏剂、软膏剂、气雾剂等。优选地口服施用式(I)的化合物。
采用的活性成分的有效剂量可以随采用的特定化合物、施用模式、要治疗的病症和要治疗的病症的严重程度而变化。这样的剂量可以由本领域技术人员容易地确定。
用于合成本发明的优选实施方案的起始材料可以从商业可得来源诸如Array、Sigma Aldrich、Acros、Fisher、Fluka、ABCR购买,或者可以由本领域技术人员使用已知方法合成。
一般而言,几种方法可用于制备本发明的化合物。在某些情况下,可以组合各种策略。可以使用依序或会聚途径。下面描述了示例性合成途径。
实施例
I 化学合成
实验程序:
使用下述缩写和缩略语:
aq 水性的
ACN 乙腈
BF3·OEt2 三氟化硼乙醚络合物
盐水 NaCl在水中的饱和溶液
Burgess试剂 N-三乙铵基磺酰基氨基甲酸甲酯
CDI 二-1H-咪唑-1-基甲酮
CSA 樟脑磺酸
CV 柱体积
δ 以百万份数为单位的化学位移
DAST N,N-二乙基氨基三氟化硫
DCM 二氯甲烷
DMSO 二甲基亚砜
DMSO-d 6 氘代二甲基亚砜
DIPEA 二异丙基乙胺
DMF 二甲基甲酰胺
EDCI 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐
ELS 蒸发光散射
ESI+ 正电离模式
ESI- 负电离模式
EtOAc 乙酸乙酯
EtOH 乙醇
FCC 快速柱色谱法
g 克
HATU 1-[双(二甲基氨基)亚甲基]-1H-[1,2,3]三唑并[4,5-b]吡啶-1-鎓3-氧化物六氟磷酸盐
HCl 盐酸
HOAt 1-羟基-7-氮杂苯并三唑
HPLC 高效液相色谱法
h 小时
H2 氢气氛
J NMR耦合常数
LC 液相色谱法
LCMS 液相色谱法质谱法
MgSO4 硫酸镁
M 摩尔的
mg 毫克
MHz 兆赫
mL 毫升
min 分钟
mm 毫米
mM 毫摩尔的
mol 摩尔
nm 纳米
MW 微波
MTBE 甲基叔丁基醚
M/Z 质荷比
N2 氮气氛
Na2SO4 硫酸钠
NaBH4 硼氢化钠
NaClO2 亚氯酸钠
NaHCO3 碳酸氢钠
NH4Cl 氯化铵
NaH2PO4 磷酸二氢钠
NMM 4-甲基吗啉
NMR 核磁共振
NaOCl 次氯酸钠
PDA 光度二极管阵列
Pd/C 碳载钯
prep. 制备型
PTSA 对甲苯磺酸一水合物
r.t. 室温
RT 保留时间
satd 饱和的
sec 秒
TEMPO (2,2,6,6-四甲基哌啶-1-基)氧自由基(oxidanyl)
THF 四氢呋喃
Tol 甲苯
TsCl 4-甲苯磺酰氯
uPLC 超高效液相色谱法
UV 紫外
µL 微升。
分析型LCMS条件如下:
系统1 (S1): 酸性IPC方法(MS18和MS19)
使用Kinetex Core shell C18柱(2.1 mm×50 mm, 5 μm;温度: 40℃)和5-100%B的梯度(A= 0.1% 甲酸/H2O;B= 0.1% 甲酸/ACN)历时1.2 min、然后在100% B保持0.1min,在Shimadzu LCMS系统上进行分析型(MET/CR/1410) HPLC-MS。然后历时0.01 min施加100-5% B的第二梯度,注射体积为3 μL,流速为1.2 mL/min。使用SPD-M20A光电二极管阵列检测器(波谱范围: 200-400 nm),在215 nm记录UV波谱。使用2010EV检测器得到质谱图。使用Shimadzu LCMS-Solutions和PsiPort软件整合和报告数据。
系统2 (S2): 酸性IPC方法(MSQ2和MSQ4):
使用Waters UPLC® BEHTM C18柱(2.1 mm×50 mm, 1.7 μm;温度40℃)和5-100%B的梯度(A= 0.1% 甲酸/H2O: B= 0.1% 甲酸/ACN)历时1.1 min、然后在100% B保持0.25min,在Waters Acquity uPLC系统上进行分析型(MET/uPLC/1704) uHPLC-MS。然后历时0.05 min施加100-5% B的第二梯度并保持0.1 min,注射体积为1 μL,流速为0.9 mL/min。在具有200-400 nm的波谱范围的Waters Acquity PDA上在215 nm记录UV波谱。使用WatersQDa得到质谱图。使用Waters MassLynx和OpenLynx软件整合和报告数据。
系统3 (S3): 碱性IPC方法(MS16):
使用Waters UPLC® BEHTM C18柱(2.1 mm×30 mm, 1.7 μm;温度40℃)和5-100%B的梯度(A: 2 mM碳酸氢铵,缓冲至pH 10, B: ACN)历时0.75 min、然后在100% B保持0.1min,在Waters Acquity uPLC系统上进行分析型(MET/CR/1602) uHPLC-MS。然后历时0.05min施加100-5% B的第二梯度并保持0.1 min,注射体积为1 μL,流速为1 mL/min。在具有200-400 nm的波谱范围的Waters Acquity PDA上在215 nm记录UV波谱。使用WatersQuattro Premier XE得到质谱图。使用Waters MassLynx和OpenLynx软件整合和报告数据。
系统4 (S4): 酸性最终方法(MSQ1和MSQ2):
使用Phenomenex Kinetex-XB C18柱(2.1 mm×100 mm, 1.7 μM;温度: 40℃)和5-100% B的梯度(A = 0.1% 甲酸/H2O;B = 0.1% 甲酸/ACN)历时5.3 min、然后在100% B保持0.5 min,在Waters Acquity uPLC系统上进行分析型(MET/uPLC/AB101) uHPLC-MS。然后历时0.02 min施加100-5% B的第二梯度并保持1.18 min,注射体积为1 μL,流速为0.6 mL/min。使用Waters Acquity PDA检测器(波谱范围: 200-400 nm)在215 nm记录UV波谱。使用Waters SQD (MSQ1)或Waters Acquity QDA (MSQ2)得到质谱图。使用Waters MassLynx和OpenLynx软件整合和报告数据。
系统5 (S5): 酸性最终方法(MS18, MS19)
使用Waters Atlantis dC18柱(2.1 mm×100 mm, 3 μm;温度: 40℃)和5-100% B的梯度(A= 0.1% 甲酸/H2O;B= 0.1% 甲酸/ACN)历时5 min、然后在100% B保持0.4 min,在Shimadzu LCMS系统上进行分析型(MET/CR/1416) HPLC-MS。然后历时0.02 min施加100-5%B的第二梯度并保持1.58 min,注射体积为3 μL,流速为0.6 mL/min。使用SPD-M20A光电二极管阵列检测器(波谱范围: 200-400 nm)在215 nm记录UV波谱。使用2010EV检测器得到质谱图。使用Shimadzu LCMS-Solutions和PsiPort软件整合和报告数据。
系统6 (S6): 碱性最终方法(MS16)
使用Waters UPLC® BEHTM C18柱(2.1 mm×100 mm, 1.7 μm柱;温度: 40℃)和5-100%的梯度(A= 2 mM碳酸氢铵,缓冲至pH 10;B = ACN)历时5.3 min、然后在100% B保持0.5 min,在Waters Acquity uPLC系统上进行分析型(MET/uHPLC/AB105) uPLC-MS。然后历时0.02 min施加100-5% B的第二梯度并保持1.18 min,注射体积为1 μL且流速为0.6 mL/min。使用Waters Acquity光电二极管阵列检测器(波谱范围: 200-400 nm)在215 nm记录UV波谱。使用Waters Quattro Premier XE质量检测器得到质谱图。使用Waters MassLynx和OpenLynx软件整合和报告数据。
系统7 (S7): 碱性最终方法(MS10)
使用Phenomenex Gemini NX C18柱(2.0 mm×100 mm, 3 μm;温度: 40℃)和5-100% B的梯度(A= 2 mM碳酸氢铵,缓冲至pH 10;B = ACN)历时5.5 min、然后在100% B保持0.4 min,在Agilent G1312A系统上进行分析型(MET/CR/1603) (M6) HPLC-MS。然后历时0.02 min施加100-5% B的第二梯度并保持1.08 min,注射体积为3 μL且流速为0.6 mL/min。使用Waters 2996光电二极管阵列检测器(波谱范围: 200-400 nm)在215 nm记录UV波谱。当报告时,使用Water 2420检测器收集ELS数据。使用Waters ZQ质量检测器得到质谱图。使用Waters MassLynx和OpenLynx软件整合和报告数据。
纯化方法如下:
方法1: 酸性早期方法
使用Waters Sunfire C18柱(30 mm×100 mm, 10 μM;温度:室温)和10-95% B的梯度(A= 0.1% 甲酸/H2O;B= 0.1% 甲酸/ACN)历时14.44 min、然后在95% B保持2.11 min,在Gilson LC系统上进行纯化(P1) LC。然后历时0.2 min施加95-10% B的第二梯度,注射体积为1500 μL,流速为40 mL/min。使用Gilson检测器在215 nm记录UV波谱。
方法2: 酸性标准方法
使用Waters Sunfire C18柱(30 mm×10 mm, 10 μM;温度:室温)和30-95% B的梯度(A= 0.1% 甲酸/水;B= 0.1% 甲酸/ACN)历时11.00 min、然后在95% B保持2.10 min,在Gilson LC系统上进行纯化(P2) LC。然后历时0.2 min施加95-30% B的第二梯度,注射体积为1500 μL,流速为40 mL/min。使用Gilson检测器在215 nm记录UV波谱。
方法3: 碱性早期方法
使用Waters X-Bridge C18柱(30 mm×100 mm, 10 μM;温度:室温)和10-95% B的梯度(A= 0.2%氢氧化铵/H2O;B= 0.2%氢氧化铵/ACN)历时14.44 min、然后在95% B保持2.11 min,在Gilson LC系统上进行纯化(P3) LC。然后历时0.2 min施加95-10% B的第二梯度,注射体积为1500 μL,流速为40 mL/min。使用Gilson检测器在215 nm记录UV波谱。
方法4: 碱性标准方法
使用Waters X-Bridge C18柱(30 mm×10 mm, 10 μM;温度:室温)和30-95% B的梯度(A= 0.2%氢氧化铵/水;B= 0.2%氢氧化铵/ACN)历时11.00 min、然后在95% B保持2.10min,在Gilson LC系统上进行纯化(P4) LC。然后历时0.21 min施加95-30% B的第二梯度,注射体积为1500 μL,流速为40 mL/min。使用Gilson检测器在215 nm记录UV波谱。
方法5:使用酸性pH、标准洗脱方法的反相色谱法
使用适当的SNAP C18柱和10% B的梯度(A= 0.1% 甲酸/H2O;B= 0.1% 甲酸/ACN)经历1.7 CV、然后10-100% B经历19.5 CV和100% B经历2 CV,在Biotage Isolera系统上进行在反相硅胶上的FCC纯化(酸性pH,标准洗脱方法)。
方法6:使用中性pH、标准洗脱方法的反相色谱法
使用适当的SNAP C18柱和10% B的梯度(A= H2O;B= ACN)经历1.7 CV、然后10-100% B经历19.5 CV和100% B经历2 CV,在Biotage Isolera系统上进行在反相硅胶上的FCC纯化(中性pH,标准洗脱方法)。
NMR条件
除非另有说明,否则分别在Bruker Avance III HD 500 MHz、Bruker Avance IIIHD 400 MHz波谱仪或Bruker Avance III HD 250 MHz波谱仪上在500 MHz、400 MHz或250MHz记录1H NMR谱。化学位移δ以百万份数(ppm)为单位引用,并参考残余溶剂峰。使用以下缩写表示多重性和一般指定:s(单峰)、d(双峰)、t(三重峰)、q(四重峰)、dd(双二重峰)、ddd(双二重峰的二重峰)、dt(双三重峰)、dq(双四重峰)、hep(七重峰)、m(多重峰)、pent(五重峰)、td(三二重峰)、qd(四二重峰)、app.(明显)和br.(宽峰)。将耦合常数J引用到最接近的0.1 Hz。
一般合成
已经以> 95%的纯度合成了所有化合物,除非另外指出。
路线1的方案

中间体1 (步骤1.a): 2-[2-羟基-1-(羟基甲基)乙基]异吲哚啉-1,3-二酮
将异苯并呋喃-1,3-二酮(854 mg, 5.8 mmol)和2-氨基丙烷-1,3-二醇(521 mg,5.7 mmol)在甲苯(20 mL)中的溶液在110℃加热24 h。将反应混合物冷却至62℃并用MTBE(20 mL)处理,导致白色粉末的沉淀。将悬浮液搅拌1 h,然后热过滤以收集沉淀物。将沉淀物用温MTBE (20 mL)洗涤并在真空中干燥以提供为白色粉末的标题化合物(786 mg, 3.45mmol, 60%收率);1H NMR (500 MHz, DMSO-d 6) δ 7.87 - 7.81 (m, 4H), 4.89 - 4.84(m, 2H), 4.27 - 4.20 (m, 1H), 3.83 - 3.76 (m, 2H), 3.69 - 3.62 (m, 2H);M/Z:222 [M+H]+, ESI+, RT = 0.73 (S1)。
路线2的方案
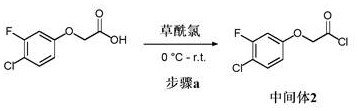
中间体2 (步骤2.a): 2-(4-氯-3-氟苯氧基)乙酰氯
在0℃向2-(4-氯-3-氟苯氧基)乙酸(5.16 g, 22.7 mmol)在DCM (45 mL)中的溶液中加入草酰氯(10 mL, 0.12 mol),随后加入DMF (81 μL, 1.11 mmol),并将混合物在室温搅拌17 h。将反应混合物在真空中浓缩以提供为橙色油的标题化合物(90% 纯度, 5.30g, 21.4 mmol, 94%收率);1H NMR (400 MHz, 氯仿-d) δ 7.31 (t, J = 8.6 Hz, 1H),6.75 (dt, J = 10.2, 2.9 Hz, 1H), 6.66 (ddd, J = 8.9, 2.9, 1.2 Hz, 1H), 4.96(s, 2H)。
使用对应的起始物料,根据通过中间体2举例说明的一般路线2合成表1中的中间体。
表1

路线3的方案
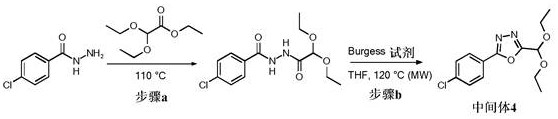
步骤3.a: 4-氯-N'-(2,2-二乙氧基乙酰基)苯甲酰肼
将4-氯苯甲酰肼(250 mg, 1.47 mmol)在2,2-二乙氧基乙酸乙酯(2.7 mL, 14.7mmol)中的悬浮液在110℃搅拌16 h。将反应混合物冷却至室温,用MeOH稀释,并将沉淀物通过过滤除去。将滤液在真空中浓缩并通过硅胶上的色谱法(0-100% EtOAc/庚烷)纯化以提供为淡黄色油的标题化合物(80% 纯度, 135 mg, 0.36 mmol, 25%收率);1H NMR (400MHz, 氯仿-d) δ 9.24 (d, J = 5.3 Hz, 1H), 8.93 (d, J = 4.8 Hz, 1H), 7.81 -7.76 (m, 2H), 7.48 - 7.41 (m, 2H) 3.82 - 3.64 (m, 4H), 1.32 - 1.24 (m, 6H);M/ Z: 299, 301, [M-H]-,ESI-,RT = 0.98 (S1)。
中间体4 (步骤3.b): 2-(4-氯苯基)-5-(二乙氧基甲基)-1,3,4-噁二唑
将4-氯-N'-(2,2-二乙氧基乙酰基)苯甲酰肼(135 mg, 0.45 mmol)和甲氧基羰基-(三乙基铵基)磺酰基亚胺盐(azanide) (0.43 g, 1.80 mmol)在无水THF (4.4 mL)中的悬浮液在微波辐射下在120℃搅拌10 min。将反应混合物在EtOAc和饱和NaHCO3水溶液之间分配,并将有机层分离,用盐水洗涤,经MgSO4干燥,并在真空中浓缩。将残余物通过硅胶上的色谱法(0-100% EtOAc/庚烷)纯化以提供为灰白色固体的标题化合物(92% 纯度, 68mg, 0.22 mmol, 49%收率);1H NMR (400 MHz, 氯仿-d) δ 8.10 - 8.03 (m, 2H), 7.54- 7.49 (m, 2H), 5.79 (s, 1H), 3.90 - 3.70 (m, 4H), 1.32 (t, J = 7.1, 6H);M/Z:283, 285 [M+H]+, ESI+, RT = 1.21 (S1)。
路线4的方案

步骤4.a: 2-[反式-2-[(苄氧基)甲基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮
将苄氧基乙醛(0.25 mL, 1.77 mmol)、PTSA (28 mg, 0.15 mmol)和2-[2-羟基-1-(羟基甲基)乙基]异吲哚啉-1,3-二酮(336 mg, 1.47 mmol, 中间体1)在甲苯(30 mL)中的溶液在Dean-Stark条件下回流加热18 h。将反应混合物冷却至室温并依次用饱和NaHCO3水溶液(2×10 mL)和盐水(10 mL)洗涤。将有机层经Na2SO4干燥,在真空中浓缩,并通过硅胶上的色谱法(0-35% EtOAc/庚烷)纯化以提供为无色油的标题化合物(303 mg, 0.82 mmol,55%收率);1H NMR (400 MHz, 氯仿-d) δ 7.90 - 7.82 (m, 2H), 7.79 - 7.71 (m, 2H),7.41 - 7.34 (m, 4H), 7.34 - 7.29 (m, 1H), 4.91 (t, J = 4.5 Hz, 1H), 4.74 -4.66 (m, 1H), 4.64 (s, 2H), 4.52 - 4.43 (m, 2H), 4.09 (dd, J = 10.8, 4.9 Hz,2H), 3.60 (d, J = 4.5 Hz, 2H)。
中间体5 (步骤4.b): 2-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮
将2-[反式-2-[(苄氧基)甲基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(393 mg, 1.06 mmol)和Pd/C (10%, 112 mg, 0.11 mmol)在EtOH (10 mL)和EtOAc (6 mL)中的悬浮液在H2下搅拌5 h。将反应混合物用N2吹扫,温热至接近回流,然后通过硅藻土垫过滤。将滤液在真空中浓缩以提供为白色固体的标题化合物(94% 纯度, 260mg, 0.93 mmol, 88%收率);1H NMR (400 MHz, 氯仿-d) δ 7.90 - 7.80 (m, 2H), 7.79- 7.66 (m, 2H), 4.78 (t, J = 4.3 Hz, 1H), 4.69 - 4.58 (m, 1H), 4.47 (dd, J =10.8 Hz, 2H), 4.07 (dd, J = 10.7, 4.8 Hz, 2H), 3.68 (d, J = 4.2 Hz, 2H), 1.92(s, 1H)。
路线5的方案

中间体6 (步骤5.a): 反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-1,3-二氧杂环己烷-2-甲酸
将TEMPO (0.13 g, 0.80 mmol)加入2-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(89% 纯度, 2.37 g, 8.01 mmol, 中间体5)在ACN(66 mL)和0.67 M的NaH2PO4水溶液(66 mL)中的溶液中。将反应混合物温热至35℃。加入NaClO2 (80%, 1.83 g, 16.0 mmol)在H2O (15 mL)中的溶液,随后加入NaOCl (5.0%, 0.5mL, 0.41 mmol)。将反应混合物在35℃搅拌20 h。加入另外的TEMPO (0.13 g, 0.80mmol)、NaClO2 (80%, 1.83 g, 16.0 mmol)和NaOCl (5.0%, 0.5 mL, 0.41 mmol),并将反应混合物在35℃搅拌24 h。加入另外的TEMPO (0.13 g, 0.80 mmol)、NaClO2 (80%, 1.83g, 16.0 mmol)和NaOCl (5.0%, 0.5 mL, 0.41 mmol),并将混合物在35℃搅拌24 h。将反应混合物在真空中浓缩。将水性残余物使用饱和NaHCO3水溶液碱化至pH 9,并用EtOAc (2×20 mL)洗涤。将水层冷却至0℃并通过缓慢加入1 M的HCl水溶液酸化至pH 2。将水层用EtOAc (3×20 mL)再萃取。将合并的有机萃取物用H2O (50 mL)洗涤,经Na2SO4干燥并在真空中浓缩以提供为灰白色粉末的标题化合物(2.10 g, 7.50 mmol, 94%收率);1H NMR(400 MHz, 甲醇-d) δ 7.90 - 7.86 (m, 2H), 7.85 - 7.81 (m, 2H), 5.11 (s, 1H),4.63 - 4.56 (m, 1H), 4.56 - 4.49 (m, 2H), 4.20 - 4.14 (m, 2H)。
路线6的方案

中间体7 (步骤6.a): [反式-5-氨基-1,3-二氧杂环己烷-2-基]甲醇
将2-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(93% 纯度, 904 mg, 3.19 mmol, 中间体5)和水合肼(0.6 mL, 12.8 mmol)在EtOH(49 mL)中的悬浮液在50℃搅拌2 h。将反应混合物冷却至室温并在真空中浓缩。将残余物悬浮于MeOH (20 mL)中,导致白色固体的沉淀。将固体在真空下过滤并将滤液在真空中浓缩。将残余物溶解在MeOH中并加载到SCX筒(2 g)上。将产物用MeOH洗涤,然后用7 M的NH3在MeOH中的溶液洗脱。将洗脱液收集并在真空中浓缩以提供为粘稠黄色油的标题化合物(299mg, 2.25 mmol, 70%收率);1H NMR (500 MHz, DMSO-d 6) δ 4.76 (s, 1H), 4.34 (t, J =4.7 Hz, 1H), 3.98 - 3.86 (m, 2H), 3.41 - 3.19 (m, 2H), 3.19 - 3.07 (m, 2H),2.75 (tt, J = 10.2, 5.0 Hz, 1H)。
步骤6.b: 2-(4-氯-3-氟苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺
将2-(4-氯-3-氟苯氧基)乙酸(459 mg, 2.3 mmol)和DIPEA (1.2 mL, 6.7 mmol)在无水DMF (10 mL)中的溶液用HATU (807 mg, 2.1 mmol)处理。将反应混合物在室温搅拌10 min。加入[反式-5-氨基-1,3-二氧杂环己烷-2-基]甲醇(299 mg, 2.25 mmol, 中间体7)并将反应混合物在室温搅拌19 h。将反应混合物用H2O (30 mL)稀释,然后用EtOAc (30mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,经无水Na2SO4干燥并在真空中浓缩。通过硅胶上的色谱法(0-100% EtOAc/庚烷)纯化,提供为灰白色固体的标题化合物(393 mg,1.20 mmol, 54%收率);1H NMR (400 MHz, 氯仿-d) δ 7.35 (t, J = 8.6 Hz, 1H), 6.76(dd, J = 10.2, 2.9 Hz, 1H), 6.71 - 6.63 (m, 1H), 6.12 (d, J = 8.0 Hz, 1H),4.61 (t, J = 4.4 Hz, 1H), 4.45 (s, 2H), 4.42 - 4.31 (m, 1H), 4.28 (dd, J =11.2, 4.8 Hz, 2H), 3.67 (dd, J = 6.6, 4.4 Hz, 2H), 3.51 (t, J = 10.7 Hz, 2H),2.81 (s, 4H), 1.80 (t, J = 6.6 Hz, 1H);M/Z: 320, 322 [M+H]+, ESI+, RT = 0.74(S2)。
中间体8 (步骤6.c): 反式-5-[2-(4-氯-3-氟苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-甲酸
在室温将NaOCl (5%, 0.23 mL, 0.18 mmol)、NaClO2 (80%, 843 mg, 7.38mmol)和TEMPO (58 mg, 0.369 mmol)加入2-(4-氯-3-氟苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺(393 mg, 1.23 mmol)在ACN (10 mL)和NaH2PO4 (0.67 M,10 mL, 6.8 mmol)中的溶液中。将反应混合物在35℃加热22 h。将反应混合物冷却至室温并用EtOAc (30 mL)和H2O (30 mL)稀释。将水层使用2 M的HCl水溶液酸化至pH 2,并用EtOAc (3×20 mL)萃取。将合并的有机层用盐水(30 mL)洗涤,经Na2SO4干燥并在真空中浓缩以提供为灰白色固体的标题化合物(88% 纯度, 274 mg, 0.723 mmol, 59%收率);1HNMR (500 MHz, DMSO-d 6) δ 8.12 (d, J = 7.8 Hz, 1H), 7.49 (t, J = 8.9 Hz, 1H),7.07 (dd, J = 11.4, 2.8 Hz, 1H), 6.85 (ddd, J = 9.0, 2.8, 1.2 Hz, 1H), 4.91(s, 1H), 4.54 (s, 2H), 4.12 - 3.91 (m, 3H), 3.66 - 3.53 (m, 2H);M/Z: 334, 336[M+H]+, ESI+, RT = 0.76 (S2)。
使用对应的起始物料,根据通过中间体8举例说明的一般路线6合成表2中的中间体。
表2

路线7的方案

步骤7.a: 顺式-3-羟基环丁烷-1-甲酸苄酯
在N2下在-40℃将NaBH4 (0.51 g, 13.5 mmol)加入3-氧代环丁烷甲酸苄酯(2.76g, 13.5 mmol)在MeOH (25 mL)中的溶液中并搅拌1 h。将反应混合物温热至0℃并用饱和NH4Cl水溶液(10 mL)淬灭。将反应混合物温热至室温并在真空中浓缩。将残余物溶解在H2O(50 mL)中,并用EtOAc (2×50 mL)萃取。将合并的有机萃取物用盐水(100 mL)洗涤,经Na2SO4干燥并在真空中浓缩以提供为无色油的标题化合物(2.73 g, 13.2 mmol, 98%收率);1H NMR (400 MHz, 氯仿-d) δ 7.41 - 7.29 (m, 5H), 5.14 (s, 2H), 4.27 - 4.15(m, 1H), 2.74 - 2.53 (m, 3H), 2.28 - 2.12 (m, 2H), 1.86 (d, J = 7.6 Hz, 1H)。
步骤7.b: 顺式-3-(三氟甲氧基)环丁烷-1-甲酸苄酯
在箔覆盖的烧瓶中在N2下在室温将2-氟吡啶(3.5 mL, 40.4 mmol)和三氟甲基三甲基硅烷(6.0 mL, 40.4 mmol)接连地逐滴加入顺式-3-羟基环丁烷-1-甲酸苄酯(2.78 g,13.5 mmol)、三氟甲磺酸银(10.43 g, 40.4 mmol)、Selectfluor (7.16 g, 20.2 mmol)和氟化钾(3.13 g, 53.9 mmol)在EtOAc (120 mL)中的溶液中。将反应混合物在室温搅拌20h,然后通过硅藻土过滤,用EtOAc (50 mL)洗涤。将滤液在真空中浓缩并通过硅胶上的色谱法(5-30% EtOAc/庚烷)纯化以提供为无色油的标题化合物(1.69 g, 6.16 mmol, 46%收率);1H NMR (400 MHz, 氯仿-d) δ 7.44 - 7.29 (m, 5H), 5.15 (s, 2H), 4.58 (p, J= 7.6 Hz, 1H), 2.83 - 2.73 (m, 1H), 2.65 (dtd, J = 10.0, 7.3, 2.6 Hz, 2H),2.60 - 2.47 (m, 2H)。
中间体10 (步骤7.c): 顺式-3-(三氟甲氧基)环丁烷甲酸
将顺式-3-(三氟甲氧基)环丁烷-1-甲酸苄酯(1.70 g, 6.20 mmol)和Pd/C (10%,0.66 g, 0.31 mmol)在EtOH (50 mL)中的悬浮液在H2下在室温搅拌18 h。将反应混合物用N2吹扫,然后通过硅藻土过滤,用EtOH (50 mL)洗涤。将滤液在真空中浓缩以提供为淡黄色油的标题化合物(1.06 g, 5.64 mmol, 91%收率);1H NMR (400 MHz, 氯仿-d) δ 9.11(s, 1H), 4.60 (p, J = 7.4 Hz, 1H), 2.84 - 2.62 (m, 3H), 2.55 (q, J = 10.2,9.5 Hz, 2H)。如通过1H NMR所观察到,含有10%的反式-异构体。
路线8的方案

步骤8.a: 咪唑-1-甲酸4,4,4-三氟丁酯
在0℃在N2下将4,4,4-三氟丁烷-1-醇(3.0 g, 23.4 mmol)在DCM (50 mL)中的溶液加入到CDI (5.70 g, 35.1 mmol)在THF (100 mL)中的溶液中并搅拌1 h。将反应混合物温热至室温并搅拌19 h。将反应混合物在真空中浓缩并通过硅胶上的色谱法(5-50%EtOAc/庚烷)纯化以提供为无色油的标题化合物(4.92 g, 22.1 mmol, 95%收率);1H NMR(400 MHz, 氯仿-d) δ 8.14 (s, 1H), 7.42 (t, J = 1.4 Hz, 1H), 7.14 - 7.04 (m,1H), 4.50 (t, J = 6.4 Hz, 2H), 2.37 - 2.19 (m, 2H), 2.11 (dq, J = 10.3, 6.6Hz, 2H);19F{1H} NMR (376 MHz, 氯仿-d) δ -66.31 (3F, s);M/Z: 223 [M+H]+, ESI+,RT = 0.73 (S2)。
中间体11 (步骤8.b): N-氨基氨基甲酸4,4,4-三氟丁酯
将咪唑-1-甲酸4,4,4-三氟丁酯(4.92 g, 22.1 mmol)和水合肼(4.4 mL, 88.6mmol)在DCM (70 mL)中的溶液在室温搅拌1.5 h。加入异丙醇(15 mL),并将有机层用H2O(100 mL)、饱和NaHCO3水溶液(100 mL)、盐水(100 mL)洗涤,经Na2SO4干燥,并在真空中浓缩以提供为无色油的标题化合物(2.54 g, 13.7 mmol, 62%收率);1H NMR (400 MHz, DMSO-d 6) δ 8.15 (s, 1H), 4.15 - 3.89 (m, 4H), 2.40 - 2.15 (m, 2H), 1.84 - 1.67 (m,2H);19F{1H} NMR (376 MHz, 氯仿-d) δ -64.89 (3F, s)。
使用路线8制备下述中间体。
中间体12: N-氨基氨基甲酸2-(三氟甲氧基)乙酯

1H NMR (400 MHz, DMSO-d 6) δ 8.30 (s, 1H), 4.23 (s, 4H), 4.07 (s, 2H);19F{1H} NMR (376 MHz, DMSO-d 6 ) δ -58.99 (3F, s)。
中间体13: 3-(三氟甲氧基)氮杂环丁烷-1-碳酰肼

1H NMR (400 MHz, DMSO-d 6) δ 9.92 (s, 2H), 9.45 (s, 1H), 5.22 (tt, J =6.9, 4.0 Hz, 1H), 4.33 (dd, J = 10.1, 6.7 Hz, 2H), 4.01 (dd, J = 10.1, 3.8Hz, 2H);19F{1H} NMR (376 MHz, 氯仿-d) δ -58.43 (3F, s)。
路线9的方案
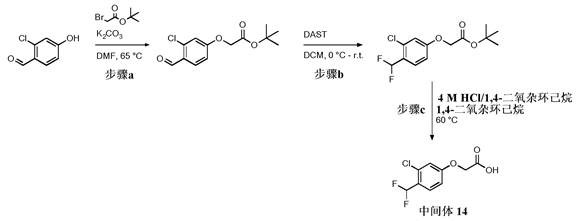
步骤9.a: 2-(3-氯-4-甲酰基苯氧基)乙酸叔丁酯
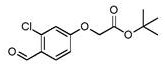
向2-氯-4-羟基苯甲醛(1.50 g, 9.58 mmol)在无水DMF (15 mL)中的溶液中加入溴乙酸叔丁酯(1.6 mL, 10.5 mmol),随后加入K2CO3 (2.65 g, 19.2 mmol),并将混合物在65℃搅拌2 h。将反应混合物冷却至室温,然后倒在水(100 mL)上。将得到的溶液用EtOAc(2×70 mL)萃取,并将合并的有机萃取物用盐水(100 mL)洗涤,经MgSO4干燥,并在真空中浓缩,以定量收率提供为灰白色固体的标题化合物(2.76 g, 9.89 mmol);1H NMR (400MHz, 氯仿-d) δ 10.34 (s, 1H), 7.90 (d, J = 8.7 Hz, 1H), 6.93 (d, J = 2.4 Hz,1H), 6.88 (dd, J = 8.7, 2.2 Hz, 1H), 4.58 (s, 2H), 1.49 (s, 9H);M/Z: 271, 273[M+H]+, ESI+, RT = 0.98 (S2)。
步骤9.b: 2-[3-氯-4-(二氟甲基)苯氧基]乙酸叔丁酯

在0℃向2-(3-氯-4-甲酰基苯氧基)乙酸叔丁酯(2.76 g, 9.89 mmol)在无水DCM(20 mL)中的溶液中逐滴加入DAST (2.6 mL, 19.8 mmol),并将混合物在室温搅拌3 h。缓慢地加入饱和NaHCO3水溶液(50 mL),并在室温搅拌1 h。将溶液用DCM (2×50 mL)萃取,并将合并的有机萃取物用饱和NaHCO3水溶液洗涤,经MgSO4干燥,并在真空中浓缩。将残余物通过硅胶上的色谱法(5-40% EtOAc/庚烷)纯化以提供为黄色油的标题化合物(2.02 g, 6.62mmol, 67%收率);1H NMR (400 MHz, 氯仿-d) δ 7.58 (d, J = 8.6 Hz, 1H), 7.05 -6.73 (m, 3H), 4.53 (s, 2H), 1.49 (s, 9H);M/Z: 291, 293 [M-H]-、ESI-、RT = 1.06(S2)。
中间体14 (步骤9.c): 2-[3-氯-4-(二氟甲基)苯氧基]乙酸

向2-[3-氯-4-(二氟甲基)苯氧基]乙酸叔丁酯(2.02 g, 6.62 mmol)在1,4-二氧杂环己烷(5 mL)中的溶液中加入4 M HCl/1,4-二氧杂环己烷(17 mL, 66.2 mmol),并将混合物在60℃搅拌4 h。将反应混合物冷却至室温并在真空中浓缩。加入H2O并将得到的沉淀物在真空下过滤,用H2O洗涤。然后将固体溶解在MeCN中,在真空下过滤并将滤液在真空中浓缩以提供为米色固体的标题化合物(1.51 g, 6.26 mmol, 95%收率);1H NMR (400 MHz,氯仿-d) δ 7.61 (d, J = 8.7 Hz, 1H), 7.05 - 6.74 (m, 3H), 4.73 (s, 2H);19F NMR(376 MHz, 氯仿-d) δ -113.69;M/Z: 235, 237 [M-H]-、ESI-、RT = 0.74 (S2)。
路线10的方案

步骤10.a: 2-(3,4-二氯苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺和2-(3,4-二氯苯氧基)乙酸[反式-5-[2-(3,4-二氯苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-基]甲酯
将[反式-5-氨基-1,3-二氧杂环己烷-2-基]甲醇(495 mg, 3.72 mmol, 中间体7)和DIPEA (3.9 mL, 22.3 mmol)在DCM (10 mL)中的溶液冷却至0℃并用2-(3,4-二氯苯氧基)乙酰氯(1.87 g, 7.81 mmol, 中间体3)在DCM (15 mL)中的溶液逐滴处理。将混合物温热至室温并搅拌3 h。将反应混合物冷却至0℃并用H2O (10 mL)缓慢地淬灭。将有机层分离,并用饱和NaHCO3水溶液(20 mL)和盐水(20 mL)洗涤,经Na2SO4干燥,并在真空中浓缩。将残余物通过硅胶上的色谱法(20-100% EtOAc/庚烷)纯化以提供为灰白色固体的标题化合物2-(3,4-二氯苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺(405mg, 1.14 mmol, 31%收率)和2-(3,4-二氯苯氧基)乙酸[反式-5-[2-(3,4-二氯苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-基]甲酯(92% 纯度, 800 mg, 1.37 mmol, 37%收率)。
2-(3,4-二氯苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺

(405 mg, 1.14 mmol, 31%收率);1H NMR (400 MHz, 氯仿-d) δ 7.39 (d, J =8.9 Hz, 1H), 7.04 (d, J = 2.9 Hz, 1H), 6.78 (dd, J = 8.9, 2.9 Hz, 1H), 6.11(d, J = 8.1 Hz, 1H), 4.60 (t, J = 4.4 Hz, 1H), 4.44 (s, 2H), 4.40 - 4.30 (m,1H), 4.30 - 4.23 (m, 2H), 3.66 (dd, J = 6.6, 4.4 Hz, 2H), 3.54 - 3.46 (m,2H), 1.81 (t, J = 6.6 Hz, 1H)。
2-(3,4-二氯苯氧基)乙酸[反式-5-[2-(3,4-二氯苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-基]甲酯

(92% 纯度, 800 mg, 1.37 mmol, 37%收率);1H NMR (500 MHz, 氯仿-d) δ7.36 (m, 2H), 7.02 (m, 2H), 6.81 - 6.75 (m, 2H), 6.14 (d,J = 8.1 Hz, 1H),4.71 (t,J = 4.4 Hz, 1H), 4.66 (s, 2H), 4.44 (s, 2H), 4.38 - 4.30 (m, 1H),4.30 - 4.21 (m, 4H), 3.52 - 3.44 (m, 2H)。
步骤10.b: 2-(3,4-二氯苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺

将2-(3,4-二氯苯氧基)乙酸[反式-5-[2-(3,4-二氯苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-基]甲酯(92% 纯度, 800 mg, 1.37 mmol)在H2O (2.8 mL)、THF (2.8 mL)和MeOH (2.8 mL)中的溶液用LiOH (43 mg, 1.78 mmol)处理并在室温搅拌1 h。将有机溶剂在真空中除去以提供水性残余物,将其用H2O (10 mL)稀释,并用EtOAc (2×10 mL)萃取。将合并的有机萃取物用盐水(20 mL)洗涤,经Na2SO4干燥,并在真空中浓缩以提供为灰白色固体的标题化合物(385 mg, 1.09 mmol, 73%收率);1H NMR (400 MHz, 氯仿-d) δ 7.39(d, J = 8.9 Hz, 1H), 7.05 (d, J = 2.9 Hz, 1H), 6.78 (dd, J = 8.9, 2.9 Hz,1H), 6.11 (d, J = 8.2 Hz, 1H), 4.60 (t, J = 4.4 Hz, 1H), 4.44 (s, 2H), 4.40 -4.31 (m, 1H), 4.30 - 4.20 (m, 2H), 3.66 (d, J = 4.4 Hz, 2H), 3.55 - 3.43 (m,2H), 1.80 (s, 1H)。
中间体15 (步骤10.c): 反式-5-[2-(3,4-二氯苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-甲酸

在室温将NaOCl (5.0%, 0.44 mL, 0.353 mmol)、NaClO2 (80%, 1.61 g, 14.1mmol)和TEMPO (111 mg, 0.705 mmol)加入2-(3,4-二氯苯氧基)-N-[反式-2-(羟基甲基)-1,3-二氧杂环己烷-5-基]乙酰胺(790 mg, 2.35 mmol)在ACN (38 mL)和0.67 M NaH2PO4(19 mL, 12.9 mmol)中的溶液中,并将混合物在35℃搅拌48 h。将反应混合物在室温在真空中浓缩以除去有机溶剂,并将得到的水溶液使用饱和NaHCO3水溶液碱化至pH 8/9。将水溶液用EtOAc (2×30 mL)洗涤并抛弃有机萃取物。然后将水溶液冷却至0℃并通过缓慢加入1 M的HCl水溶液酸化至pH 2/3。然后将得到的溶液用EtOAc (2×50 mL)萃取,并将合并的有机萃取物用H2O (50 mL)洗涤,经Na2SO4干燥,并在真空中浓缩以提供为泡沫状灰白色固体的标题化合物(90% 纯度, 760 mg, 1.95 mmol, 83%收率);1H NMR (400 MHz, DMSO-d 6) δ 8.11 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.9 Hz, 1H), 7.25 (d, J = 2.9Hz, 1H), 6.98 (dd, J = 8.9, 2.9 Hz, 1H), 4.91 (s, 1H), 4.55 (s, 2H), 4.08 -3.95 (m, 3H), 3.59 (m, 2H);M/Z: 348, 350, 352 [M-H]-、ESI-、RT = 2.61 (S4)。
路线11的方案

步骤11.a: 2-[反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]异吲哚啉-1,3-二酮
将BF3·OEt2 (0.12 mL, 0.97 mmol)在无水ACN (3 mL)中的溶液在80℃搅拌。逐滴加入2-[2-羟基-1-(羟基甲基)乙基]异吲哚啉-1,3-二酮(100 mg, 0.44 mmol, 中间体1)和2-(4-氯苯基)-5-(二乙氧基甲基)-1,3,4-噁二唑(94 μL, 0.44 mmol, 中间体4)在ACN (2 mL)中的单独溶液。将反应混合物搅拌1 h,然后加入另外的BF3·OEt2 (0.12 mL,0.97 mmol),并将反应混合物在80℃加热另外1 h。将反应冷却至室温,用饱和NaHCO3水溶液(0.1 mL)淬灭并在真空中浓缩。将残余物溶解在EtOAc (10 mL)中,并依次用饱和NaHCO3水溶液(10 mL)和盐水(10 mL)洗涤。将有机层经无水Na2SO4干燥,在真空中浓缩并通过硅胶上的色谱法(10-100% EtOAc/庚烷)纯化以提供为灰白色固体的标题化合物(70% 纯度, 33mg, 0.06 mmol, 13%收率),为非对映异构体的混合物;1H NMR (400 MHz, 氯仿-d) δ8.12 - 8.08 (m, 1H), 8.08 - 8.04 (m, 2H), 7.89 - 7.82 (m, 4H), 7.79 - 7.71(m, 4H), 7.54 - 7.49 (m, 3H), 6.04 (s, 1H), 4.90 - 4.80 (m, 2H), 4.73 - 4.65(m, 3H), 4.57 - 4.49 (m, 1H), 4.35 - 4.24 (m, 3H), 4.19 (dd, J = 11.0, 5.0Hz, 1H), 4.12 (q, J = 7.1 Hz, 3H), 2.04 (s, 4H), 1.73-1.59 (m, 4H);M/Z: 412,414 [M+H]+, ESI+, RT = 3.52, 3.67 (S4)。
步骤11.b: 反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-胺
将水合肼(0.02 mL, 0.14 mmol)和2-[反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]异吲哚啉-1,3-二酮(33 mg, 0.06 mmol)在EtOH (0.7 mL)中的悬浮液在室温搅拌2 h。将反应混合物在40℃加热25 h,然后在50℃加热另外6 h。将反应混合物冷却至室温,通过硅藻土垫过滤并在真空中浓缩以提供为白色固体的标题化合物(50% 纯度, 17 mg, 0.03 mmol, 54%收率);1H NMR (400 MHz, 氯仿-d) δ 8.04 (m,2H), 7.49 (m, 2H), 5.80 (s, 1H), 4.35 (dd, J = 11.4, 4.6 Hz, 2H), 4.09 (d, J=12.0 Hz,1H), 3.51 (t, J = 10.8 Hz, 2H), 3.36 - 3.25 (m, 2H);M/Z: 282 [M+H]+,ESI+, RT = 0.82 (S1)。
实施例1 (步骤11.c): 2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺
将反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-胺(50%,17 mg, 0.03 mmol)在DCM (0.5 mL)中的溶液用DIPEA (0.02 mL, 0.09 mmol)处理并冷却至0℃。缓慢地加入2-(4-氯-3-氟苯氧基)乙酰氯(7.4 mg, 0.03 mmol, 中间体2)在DCM(0.5 mL)中的溶液。将反应混合物温热至室温并搅拌1 h。加入2-(4-氯-3-氟苯氧基)乙酰氯(3.0 mg, 0.01 mmol, 中间体2),并将反应在室温搅拌1 h。将反应混合物冷却至0℃,用H2O淬灭并在真空中浓缩。将残余物通过制备型HPLC (方法4)纯化以提供为灰白色粉末的标题化合物(5.0 mg, 0.01 mmol, 35%收率);1H NMR (400 MHz, 氯仿-d) δ 8.07 - 8.01(m, 2H), 7.54 - 7.48 (m, 2H), 7.36 (t, J = 8.6 Hz, 1H), 6.84 - 6.76 (m, 2H),6.72 (ddd, J = 8.9, 2.9, 1.3 Hz, 1H), 6.03 (s, 1H), 4.53 - 4.44 (m, 4H), 4.39- 4.32 (m, 1H), 3.86 (dd, J = 11.6, 6.6 Hz, 2H);M/Z: 468, 470, 472 [M+H]+,ESI+, RT = 3.65 (S4)。
路线12的方案

步骤12.a: 2-[反式-2-[({[(苄氧基)羰基]氨基}氨基)羰基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮
在0℃在N2下将反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-1,3-二氧杂环己烷-2-甲酸(2.07 g, 7.39 mmol, 中间体6)在无水THF (49 mL)中的溶液通过依次加入氯甲酸异丁酯(0.9 mL, 7.02 mmol)和NMM (0.8 mL, 7.39 mmol)进行处理。加入N-氨基氨基甲酸苄酯(1.17 g, 7.02 mmol)并将反应在0℃搅拌30 min,然后在室温搅拌1 h。将反应冷却至0℃并用H2O (1 mL)淬灭。将得到的混合物在真空中浓缩并将残余物在EtOAc (50mL)和H2O (50 mL)之间分配。分离各层并将水层用EtOAc (20 mL)再萃取。将合并的有机萃取物用饱和NaHCO3水溶液(80 mL)、盐水(80 mL)洗涤,经无水Na2SO4干燥,并在真空中浓缩以提供为灰白色粉末的标题化合物(86%, 2.86 g, 5.78 mmol, 78%收率);1H NMR (400MHz, DMSO-d 6) δ 10.16 - 9.91 (m, 1H), 9.27 (s, 1H), 7.93 - 7.82 (m, 4H), 7.44- 7.27 (m, 5H), 5.09 (s, 2H), 5.03 (s, 1H), 4.44 - 4.28 (m, 3H), 4.22 - 4.12(m, 2H)。
步骤12.b: 反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-1,3-二氧杂环己烷-2-碳酰肼
将2-[反式-2-[({[(苄氧基)羰基]氨基}氨基)羰基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(86% 纯度, 2.86 g, 5.78 mmol)和Pd/C (10%, 0.62 g,0.578 mmol)在EtOH (160 mL)和EtOAc (50 mL)中的悬浮液在H2下搅拌3 h。加入另外的Pd/C (10%, 0.62 g, 0.578 mmol)并将反应混合物在H2下搅拌另外4 h。将反应容器用N2冲洗。将反应混合物加热至接近回流并通过硅藻土垫过滤,用热EtOH洗涤。将反应混合物在真空中浓缩并通过硅胶上的色谱法(0-7% MeOH/DCM)纯化以提供为淡黄色粉末的标题化合物(90% 纯度, 290 mg, 0.90 mmol, 15%收率);1H NMR (500 MHz, DMSO-d 6) δ 9.32 (s,1H), 7.93 - 7.79 (m, 4H), 4.93 (s, 1H), 4.41 - 4.25 (m, 5H), 4.17 - 4.08 (m,2H)。
步骤12.c: 反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-N'-[顺式-3-(三氟甲氧基)环丁烷羰基]-1,3-二氧杂环己烷-2-碳酰肼
在0℃将顺式-3-(三氟甲氧基)环丁烷甲酸(170 mg, 0.90 mmol, 中间体10)在无水THF (7.5 mL)中的溶液通过依次加入氯甲酸异丁酯(0.11 mL, 0.85 mmol)和NMM (0.1mL, 0.90 mmol)进行处理。将混合物搅拌15 min,然后加入反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-1,3-二氧杂环己烷-2-碳酰肼(90% 纯度, 290 mg, 0.90 mmol)在无水THF (5 mL)中的悬浮液。将反应温热至室温并搅拌3 h。将反应混合物冷却至0℃并通过缓慢加入H2O (5 mL)淬灭。将反应混合物在真空中浓缩,并将得到的残余物在H2O和EtOAc之间分配。将水层用EtOAc再萃取。将合并的有机萃取物用饱和NaHCO3水溶液和盐水洗涤,然后在真空中浓缩以提供为灰白色粉末的标题化合物(90% 纯度, 382 mg, 0.75 mmol, 84%收率);1H NMR (500 MHz, DMSO-d 6) δ 10.04 (s, 1H), 9.96 (s, 1H), 7.88 (s, 4H),5.04 (s, 1H), 4.89 - 4.71 (m, 1H), 4.46 - 4.26 (m, 3H), 4.26 - 4.10 (m, 2H),2.77 - 2.63 (m, 2H), 2.36 - 2.18 (m, 4H)。
步骤12.d: 2-[反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮
将反式-5-(1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-N'-[顺式-3-(三氟甲氧基)环丁烷羰基]-1,3-二氧杂环己烷-2-碳酰肼(85%, 365 mg, 0.68 mmol)和Burgess试剂(647 mg, 2.71 mmol)在无水THF (11 mL)中的悬浮液在微波瓶中在120℃照射3 min。将反应混合物在真空中浓缩并通过制备型HPLC (方法6)纯化以提供为灰白色固体的标题化合物(190 mg, 0.41 mmol, 61%收率);1H NMR (400 MHz, DMSO-d 6) δ 7.98 - 7.73 (m,4H), 6.09 (s, 1H), 4.97 - 4.87 (m, 1H), 4.55 - 4.39 (m, 3H), 4.29 - 4.20 (m,2H), 3.56 - 3.46 (m, 1H), 2.92 - 2.83 (m, 2H), 2.57 - 2.51 (m, 2H)。
中间体16 (步骤12.e): 反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-胺
将2-[反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(90% 纯度, 330 mg, 0.68 mmol)和水合肼(0.13 mL, 2.70 mmol)在EtOH (7.8 mL)中的溶液在40℃加热6 h,然后冷却至室温并搅拌18 h。将得到的沉淀物在真空下过滤,用EtOH洗涤。将合并的滤液收集并在真空中浓缩以提供为灰白色固体的标题化合物(77% 纯度, 250 mg, 0.62 mmol, 92%收率);1H NMR(400 MHz, DMSO-d 6) δ 5.78 (s, 1H), 4.97 - 4.83 (m, 1H), 4.14 - 4.02 (m, 2H),3.52 - 3.39 (m, 5H), 2.95 (tt, J = 10.2, 5.0 Hz, 1H), 2.90 - 2.80 (m, 2H),2.55 - 2.51 (m, 2H)。
实施例2 (步骤12.f): 2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺
将反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-胺(77% 纯度, 255 mg, 0.64 mmol, 中间体16)和DIPEA (0.33 mL, 1.90mmol)在DCM (8 mL)中的溶液冷却至0℃,并用2-(4-氯-3-氟苯氧基)乙酰氯(90% 纯度,173 mg, 0.70 mmol, 中间体2)处理。将反应温热至室温并搅拌1 h。将反应冷却至0℃并用H2O (0.1 mL)淬灭,然后将反应混合物在真空中浓缩。将残余物通过制备型HPLC (方法6)纯化,并将得到的固体溶解在ACN (10 mL)和H2O (10 mL)中。将混浊溶液加热以溶解物质,然后将其冷却4天。将得到的固体收集并通过制备型HPLC (方法3)纯化以提供为白色固体的标题化合物(62 mg, 0.13 mmol, 20%收率);1H NMR (500 MHz, 氯仿-d) δ 7.35 (t, J= 8.6 Hz, 1H), 6.79 (dd, J = 10.2, 2.9 Hz, 1H), 6.75 (d, J = 8.4 Hz, 1H),6.71 (ddd, J = 8.9, 2.9, 1.2 Hz, 1H), 5.93 (s, 1H), 4.72 (p, J = 7.6 Hz, 1H),4.49 (s, 2H), 4.43 (dd, J = 11.6, 3.8 Hz, 2H), 4.35 (dtt, J = 10.8, 7.4, 3.8Hz, 1H), 3.82 (dd, J = 11.6, 7.0 Hz, 2H), 3.38 (tt, J = 10.1, 7.8 Hz, 1H),2.94 - 2.86 (m, 2H), 2.77 - 2.68 (m, 2H);M/Z: 496, 498 [M+H]+, ESI+, RT = 3.50(S4)。
路线13的方案

步骤13.a: 2-(4-氯-3-氟苯氧基)-N-[2-羟基-1-(羟基甲基)乙基]-乙酰胺
将2-(4-氯-3-氟苯氧基)乙酸(200 mg, 0.98 mmol)、2-氨基丙烷-1,3-二醇(107mg, 1.17 mmol)和HOAt (160 mg, 1.18 mmol)溶解在无水DMF (5 mL)中。然后在0℃加入分成小份的EDCI (225 mg, 1.17 mmol)。将反应混合物温热至室温并搅拌17 h。将反应混合物用H2O (20 mL)淬灭,并用EtOAc (2×30 mL)萃取。将合并的有机萃取物用盐水(20mL)洗涤,经MgSO4干燥并在真空中浓缩。通过硅胶上的色谱法(0-100% EtOAc/庚烷)纯化,以定量收率提供为白色粉末的标题化合物(278 mg, 1.0 mmol);1H NMR (500 MHz, DMSO-d 6) δ 7.67 (d, J = 8.3 Hz, 1H), 7.49 (t, J = 8.9 Hz, 1H), 7.08 (dd, J = 11.4,2.9 Hz, 1H), 6.85 (ddd, J = 9.0, 2.9, 1.1 Hz, 1H), 4.74 - 4.63 (m, 2H), 4.54(s, 2H), 3.83 - 3.74 (m, 1H), 3.46 - 3.40 (m, 4H);M/Z: 278, 280 [M+H]+, ESI+,RT = 0.88 (S1)。
实施例3 (步骤13.b): 2-(4-氯-3-氟苯氧基)-N-[顺式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺
将1-(4-氯苯基)-1H-1,2,3-三唑-4-甲醛(50 mg, 0.241 mmol)、CSA (6 mg,0.03 mmol)和2-(4-氯-3-氟苯氧基)-N-[2-羟基-1-(羟基甲基)乙基]乙酰胺(67 mg, 0.24mmol)在无水甲苯(2 mL)中的悬浮液在110℃加热18 h。加入另外的2-(4-氯-3-氟苯氧基)-N-[2-羟基-1-(羟基甲基)乙基]乙酰胺(67 mg, 0.24 mmol)和CSA (6 mg, 0.03 mmol),并将反应混合物在110℃加热5 h。将反应混合物冷却至室温并加入盐水。将水层用EtOAc (2×50 mL)萃取,经MgSO4干燥,并在真空中浓缩。通过硅胶上的色谱法(0-100% EtOAc/庚烷)和随后的制备型HPLC (方法3)纯化,提供为白色粉末的标题化合物(10 mg, 0.02 mmol,8.6%收率),为顺式和反式异构体的混合物(93:7);1H NMR (500 MHz, 氯仿-d) δ 7.93(s, 1H), 7.70 - 7.65 (m, 2H), 7.55 - 7.49 (m, 2H), 7.40 - 7.34 (m, 1H), 7.30(t, J = 8.6 Hz, 1H), 6.78 (dd, J = 10.3, 2.9 Hz, 1H), 6.69 (dd, J = 8.9, 1.6Hz, 1H), 5.91 (s, 1H), 4.53 (s, 2H), 4.28 - 4.15 (m, 4H), 4.11 (d, J = 8.3Hz, 1H);M/Z: 467, 469 [M+H]+, ESI+, RT = 3.65 (S6)。
路线14的方案

步骤14.a: 2-[反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮
将1-(4-氯苯基)-1H-1,2,3-三唑-4-甲醛(270 mg, 1.30 mmol)、CSA (32 mg,0.14 mmol)和2-[2-羟基-1-(羟基甲基)乙基]异吲哚啉-1,3-二酮, 中间体1 (288 mg,1.30 mmol)在无水甲苯(11 mL)中的悬浮液在110℃加热1 h。将反应混合物冷却至室温并用冷DCM稀释。将得到的沉淀物在真空下过滤以提供为无色固体的标题化合物(305 mg,0.74 mmol, 57%收率);1H NMR (400 MHz, 氯仿-d) δ 8.11 (s, 1H), 7.89 (dd, J =5.5, 3.1 Hz, 2H), 7.79 (dd, J = 5.5, 3.1 Hz, 2H), 7.75 - 7.68 (m, 2H), 7.57 -7.50 (m, 2H), 6.02 (s, 1H), 4.86 - 4.67 (m, 3H), 428 - 4.19 (m, 2H);M/Z: 411,413 [M+H]+, ESI+, RT = 1.29 (S1)。
步骤14.b: 反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-胺
将水合肼(0.46 mL, 3.71 mmol)和2-[反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]-2,3-二氢-1H-异吲哚-1,3-二酮(305 mg, 0.74 mmol)在EtOH (7.4 mL)中的悬浮液在80℃加热1 h。将反应混合物冷却至室温并将得到的沉淀物在真空下过滤。然后将滤液在真空中浓缩以提供为无色固体的标题化合物(200 mg, 0.69mmol, 93%收率);1H NMR (400 MHz, DMSO-d 6) δ 8.88 (s, 1H), 8.06 - 7.89 (m, 2H),7.72 - 7.58 (m, 2H), 5.66 (s, 1H), 4.09 (dd, J = 11.3, 5.0 Hz, 2H), 3.52 -3.39 (m, 2H), 2.94 (tt, J = 10.3, 5.0 Hz, 1H);M/Z: 281, 283, 285 [M+H]+, ESI+,RT = 0.66 (S1)。
实施例4 (步骤14.c): 2-(4-氯-3-氟苯氧基)-N-[反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺
在0℃将2-(4-氯-3-氟苯氧基)乙酰氯(0.05 mL, 0.77 mmol, 中间体2)在DCM (1mL)中的溶液加入反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-胺(72 mg, 0.26 mmol)和Et3N (0.21 mL, 1.54 mmol)在DCM (1.2 mL)中的溶液中。将反应混合物温热至室温并搅拌15 min。将反应混合物用饱和NaHCO3水溶液稀释,通过过滤分离不溶物。将有机层收集,经Na2SO4干燥并在真空中浓缩。将残余物质(与通过抽滤分离的固体一起)与ACN一起研磨以提供为灰白色固体的标题化合物(87 mg, 0.18 mmol, 70%收率);1H NMR (400 MHz, DMSO-d 6) δ 8.94 (s, 1H), 8.17 (d, J = 7.6 Hz, 1H), 7.99 (d, J= 8.9 Hz, 2H), 7.67 (d, J = 8.9 Hz, 2H), 7.52 (t, J = 8.9 Hz, 1H), 7.10 (dd,J = 11.3, 2.8 Hz, 1H), 6.88 (dd, J = 8.9, 1.8, 1H), 5.80 (s, 1H), 4.57 (s,2H), 4.23 - 4.08 (m, 3H), 3.85 - 3.68 (m, 2H);M/Z: 511, 513, 515 [M+H]+, ESI+,RT = 3.58 (S4)。
路线15的方案

步骤15.a: 2-(4-氯-3-氟苯氧基)-N-[反式-2-{N'-[(4,4,4-三氟丁氧基)羰基]肼羰基}-1,3-二氧杂环己烷-5-基]乙酰胺
在0℃在N2下将NMM (80 μL, 0.7 mmol)和氯甲酸异丁酯(90 μL, 0.7 mmol)加入反式-5-[2-(4-氯-3-氟苯氧基)乙酰胺基]-1,3-二氧杂环己烷-2-甲酸(88%, 274 mg, 0.7mmol, 中间体8)在无水THF (6.5 mL)中的溶液中,并搅拌15 min。加入N-氨基氨基甲酸4,4,4-三氟丁酯(135 mg, 0.7 mmol, 中间体11),并将反应混合物温热至室温并搅拌17 h。将反应混合物冷却至0℃并加入另外的NMM (40 μL, 0.35 mmol)和氯甲酸异丁酯(45 μL,0.35 mmol)。将反应混合物温热至室温并搅拌1 h。将反应混合物用EtOAc (30 mL)稀释,并用H2O (30 mL)、饱和NaHCO3水溶液(30 mL)、盐水(30 mL)洗涤,经无水Na2SO4干燥并在真空中浓缩。将得到的固体与ACN一起研磨以提供为白色固体的标题化合物(222 mg, 0.442mmol, 61%收率);1H NMR (400 MHz, DMSO-d 6) δ 9.92 (s, 1H), 9.25 - 8.65 (m, 1H),8.10 (d, J = 7.6 Hz, 1H), 7.50 (t, J = 8.9 Hz, 1H), 7.08 (dd, J = 11.3, 2.8Hz, 1H), 6.85 (ddd, J = 9.0, 2.8, 1.1 Hz, 1H), 4.89 (s, 1H), 4.54 (s, 2H),4.14 - 3.96 (m, 5H), 3.67 - 3.51 (m, 2H), 2.39 - 2.19 (m, 2H), 1.87 - 1.61(m, 2H);19F{1H} NMR (376 MHz, 氯仿-d) δ -64.91- -65.02 (m, 3F), -113.96 (s,1F);M/Z: 502, 504 [M+H]+, ESI+, RT = 0.98 (S2)。
实施例5 (步骤15.b): 2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4,4,4-三氟丁氧基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺
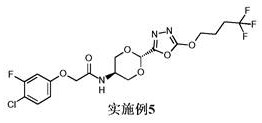
在室温向2-(4-氯-3-氟苯氧基)-N-[反式-2-{N'-[(4,4,4-三氟丁氧基)羰基]肼羰基}-1,3-二氧杂环己烷-5-基]乙酰胺(162 mg, 0.32 mmol)在ACN (5 mL)中的悬浮液中加入K2CO3 (223 mg, 1.6 mmol)和TsCl (154 mg, 0.81 mmol)。将得到的混合物加热至80℃并搅拌1.5 h。将反应混合物冷却至室温并用DCM (30 mL)稀释,用H2O (30 mL)、饱和NaHCO3水溶液(30 mL)、盐水(30 mL)洗涤,经无水Na2SO4干燥并在真空中浓缩。通过制备型HPLC (方法3)纯化,提供为粘稠的无色胶质的标题化合物(65 mg, 0.13 mmol, 41%收率);1H NMR (400 MHz, DMSO-d 6) δ 8.19 (d, J = 7.6 Hz, 1H), 7.50 (t, J = 8.9 Hz,1H), 7.08 (dd, J = 11.3, 2.8 Hz, 1H), 6.85 (dd, J = 8.9, 1.8 Hz, 1H), 5.82(s, 1H), 4.62 - 4.45 (m, 4H), 4.20 - 3.99 (m, 3H), 3.82 - 3.67 (m, 2H), 2.10- 1.92 (m, 2H);19F{1H} NMR (376 MHz, 氯仿-d) δ -64.89 (3F, s), -113.95 (1F,s);M/Z: 484, 486 [M+H]+, ESI+, RT = 3.42 (S4)。
使用路线15制备下述实施例。
实施例6: 2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺
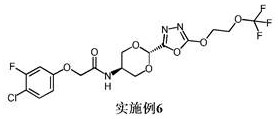
使用中间体8和中间体12;1H NMR (400 MHz, DMSO-d 6) δ 8.22 (d, J = 7.7 Hz,1H), 7.51 (t, J = 8.9 Hz, 1H), 7.08 (dd, J = 11.4, 2.8 Hz, 1H), 6.85 (ddd, J= 8.9, 2.8, 1.1 Hz, 1H), 5.84 (s, 1H), 4.79 - 4.66 (m, 2H), 4.56 (s, 2H),4.50 - 4.42 (m, 2H), 4.22 - 4.01 (m, 3H), 3.75 (t, J = 10.1 Hz, 2H);M/Z: 486,488 [M+H]+, ESI+, RT = 3.35 (S4)。
实施例7: 2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺
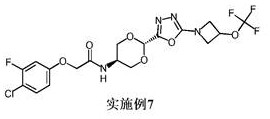
使用中间体8和中间体13;1H NMR (400 MHz, DMSO-d 6) δ 8.18 (d, J = 7.6 Hz,1H), 7.50 (t, J = 8.9 Hz, 1H), 7.08 (dd, J = 11.3, 2.8 Hz, 1H), 6.85 (ddd, J= 9.0, 2.8, 1.2 Hz, 1H), 5.78 (s, 1H), 5.32 (tt, J = 7.0, 4.1 Hz, 1H), 4.59 -4.43 (m, 4H), 4.26 (dd, J = 9.7, 4.0 Hz, 2H), 4.18 - 4.01 (m, 3H), 3.74 (t, J= 9.6 Hz, 2H);M/Z: 497, 499 [M+H]+, ESI+, RT = 3.26 (S4)。
实施例8: 2-[3-氯-4-(二氟甲基)苯氧基]-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺
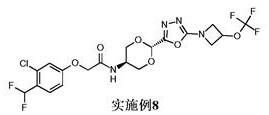
使用中间体9和中间体13;1H NMR (400 MHz, DMSO-d 6) δ 8.21 (d, J = 7.5 Hz,1H), 7.63 (d, J = 8.8 Hz, 1H), 7.28 - 6.97 (m, 3H), 5.79 (s, 1H), 5.32 (tt, J= 7.0, 4.1 Hz, 1H), 4.62 (s, 2H), 4.51 (dd, J = 9.6, 6.8 Hz, 2H), 4.26 (dd, J= 9.6, 4.0 Hz, 2H), 4.18 - 4.01 (m, 3H), 3.81 - 3.67 (m, 2H);19F NMR (376 MHz,DMSO-d 6) δ -58.47, -112.49 (d, J = 54.6 Hz);M/Z: 529, 531 [M+H]+, ESI+, RT =3.38 (S4)。
实施例9: 2-(3,4-二氯苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺

使用中间体15和中间体12: 1H NMR (500 MHz, 氯仿-d) δ 7.40 (d, J = 8.9Hz, 1H), 7.08 (d, J = 2.9 Hz, 1H), 6.85 - 6.79 (m, 2H), 5.83 (s, 1H), 4.77 -4.71 (m, 2H), 4.49 (s, 2H), 4.41 (dd, J = 11.7, 3.6 Hz, 2H), 4.37 - 4.33 (m,2H), 4.33 - 4.26 (m, 1H), 3.82 (dd, J = 11.7, 6.4 Hz, 2H);M/Z: 502, 504, 506[M+H]+, ESI+, RT = 3.56 (S4)。
路线16的方案
步骤16.a: 2-[(6-氯-5-氟吡啶-3-基)氧基]乙酸叔丁酯
向6-氯-5-氟吡啶-3-醇(4.90 g, 33.2 mmol)在DMF (50 mL)中的溶液中加入2-溴乙酸叔丁酯(4.5 mL, 34.9 mmol)和K2CO3 (13.8 g, 0.0996 mol),并将得到的混合物在65℃搅拌2 h。将反应混合物冷却至室温,悬浮于EtOAc (100 mL)中,并用H2O (2×50 mL)和盐水(50 mL)洗涤。将合并的有机萃取物经Na2SO4干燥并在真空中浓缩以提供为棕色油的标题化合物(9.00 g, 32.7 mmol, 98%收率);1H NMR (500 MHz, 氯仿-d) δ 7.91 (d, J= 2.6 Hz, 1H), 7.07 (dd, J = 9.1, 2.6 Hz, 1H), 4.55 (s, 2H), 1.53 - 1.39 (m,9H);M/Z: 262, 264 [M+H]+, ESI+, RT = 1.00 min (S2)。
步骤16.b: 2-[(6-氯-5-氟吡啶-3-基)氧基]乙酸
将4 M HCl/1,4-二氧杂环己烷(25 mL, 98.0 mmol)加入2-[(6-氯-5-氟吡啶-3-基)氧基]乙酸叔丁酯(9.00 g, 32.7 mmol)中并将得到的混合物在室温搅拌2 h。加入另外一部分的4 M HCl/1,4-二氧杂环己烷(25 mL, 98.0 mmol)并将反应混合物在50℃搅拌5h。将反应混合物在真空中浓缩,然后使用Et2O和庚烷研磨。将得到的沉淀物在真空下过滤以提供为灰白色固体的标题化合物(6.48 g, 31.2 mmol, 96%收率);1H NMR (500 MHz,DMSO-d 6) δ 13.22 (s, 1H), 8.07 (d, J = 2.6 Hz, 1H), 7.76 (dd, J = 10.4, 2.6Hz, 1H), 4.85 (s, 2H);M/Z: 206, 208 [M+H]+, ESI+, RT = 0.60 min (S2)。
实施例10 (步骤16.c): 2-[(6-氯-5-氟-3-吡啶基)氧基]-N-[反式-2-[5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺

将2-[(6-氯-5-氟吡啶-3-基)氧基]乙酸(34 mg, 0.163 mmol)在THF (2 mL)中的溶液冷却至0℃,并用氯甲酸异丁酯(20 μL, 0.155 mmol)和NMM (18 μL, 0.163 mmol)处理。将反应搅拌15 min,然后逐滴加入反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-胺(50 mg, 0.163 mmol, 中间体16, 描述在路线12中)在THF (1 mL)中的溶液。将反应在室温搅拌1 h,用H2O (2滴)淬灭并在真空中浓缩。通过制备型HPLC (方法3)纯化,提供为白色粉末的标题化合物(37 mg, 0.0745 mmol, 46%收率);1H NMR (500 MHz, 氯仿-d) δ 8.03 (d, J = 2.6 Hz, 1H), 7.17 (dd, J = 8.8,2.6 Hz, 1H), 6.78 (d, J = 8.4 Hz, 1H), 5.96 (s, 1H), 4.77 - 4.67 (m, 1H),4.57 (s, 2H), 4.49 - 4.41 (m, 2H), 4.39 - 4.28 (m, 1H), 3.85 (dd, J = 11.7,6.4 Hz, 2H), 3.44 - 3.32 (m, 1H), 2.95 - 2.84 (m, 2H), 2.78 - 2.66 (m, 2H);M/ Z: 497, 499 [M+H]+, ESI+, RT = 3.15 (S4)。
II 生物学测定
HEK-ATF4高内涵成像测定
在HEK-ATF4高内涵成像测定中测试实施例化合物,以评估它们的阻止衣霉素诱导的ISR的药理学效力。将野生型HEK293细胞以每孔12,000个细胞的密度铺板于384-孔成像测定板中的生长培养基(含有DMEM/F12、10% FBS、2mM L-谷氨酰胺、100 U/mL青霉素- 100μg/mL链霉素)中并在37℃、5% CO2下温育。24小时后,将培养基更换为每孔50 μl测定培养基(DMEM/F12、0.3% FBS、2mM L-谷氨酰胺、100 U/mL青霉素- 100 μg/mL链霉素)。将实施例化合物在二甲亚砜(DMSO)中连续稀释,点样至中间板中,并用含有3.3 μM衣霉素的测定培养基预稀释,以产生最终测定浓度的11倍过量。除了实施例化合物测试区域之外,所述平板还含有多个用于测定归一化目的的对照孔、含有衣霉素但不含实施例化合物的孔(高对照)、以及既不含实施例化合物又不含衣霉素的孔(低对照)。通过将5 μl从中间板转移进测定板中开始测定,随后在37℃、5% CO2下温育6小时。随后,将细胞固定(4% PFA/PBS,在室温20分钟)并进行间接ATF4免疫荧光染色(一级抗体兔抗ATF4,克隆D4B8,Cell SignalingTechnologies;二级抗体Alexa Fluor 488山羊抗-兔IgG (H+L),ThermofisherScientific)。使用Hoechst染料(Thermofisher Scientific)将细胞核染色,并在配备405nm和488nm激发的Opera Phenix High Content成像平台上对平板成像。最后,使用基于脚本的算法分析图像。主要读数HEK-ATF4监测细胞核和细胞质之间的ATF4信号比。衣霉素诱导总体ATF4比率信号的增加,这被调节ISR的实施例化合物所阻止。此外,将对应于健康细胞的染色细胞核的数目计数,衍生出HEK-CellCount读数。该读数充当内部毒性对照。本文的实施例化合物在CellCount中没有产生显著的减少。
测试的实施例化合物的活性提供于如下表3中:
+++ = IC50 1-500 nM;++ = IC50 >500-2000 nM;+ = IC50 >2000-15000 nM。
表3

方案-使用体外Rapid ICE通过尾电流记录测量对hERG通道的影响
使用Rapid ICE(快速离子通道电生理学)测定,在用在诱导型启动子下的hERGcDNA稳定转染的重组HEK293细胞系中评估实施例化合物在抑制人ERG钾通道(hERG)尾电流中的效力。Rapid ICE是一种利用QPatch HTX系统(Sophion Bioscience A/S)的自动化膜片箝测定。简而言之,在补充了10% FBS、1% 非必需氨基酸、1% 丙酮酸钠、2 mM l-谷氨酰胺、15 μg/mL杀稻瘟素和100 μg/mL潮霉素的最低必需培养基中培养诱导型HEK hERG细胞。通过在记录前加入10 μg/mL四环素持续24、48或72 h,获得hERG通道表达诱导。
在实验当天,将细胞用TrypLE脱离并准备加载到仪器上。将细胞重新悬浮在7 mL含有25 mM Hepes和大豆胰蛋白酶抑制剂的无血清培养基中,并立即放入机器的细胞储存罐中。细胞外缓冲液的组成为(mM):NaCl 137,KCl 4,CaCl2 1.8,MgCl2 1.0,d-葡萄糖10,N-2-羟基乙基哌嗪-N′-2-乙磺酸(HEPES) 10,用1 M NaOH调至pH 7.4。细胞内溶液的组成为(mM):KCl 130,MgCl2 1.0,乙二醇-双(β-氨基乙基醚)-N, N, N′, N′-四乙酸(EGTA) 5,MgATP 5,HEPES 10,用1 M KOH调至pH 7.2。电压方案包括以下步骤:从-80至-50 mV持续200 ms的步骤,+20 mV持续4.8 s的步骤,至-50 mV持续5 s的步骤,然后是至-80 mV的保持电位的步骤。将实施例化合物溶解在DMSO中并在细胞外缓冲液中稀释以达到在0.3% DMSO中的最终测试浓度(0.3、3和30 μM)。在实验过程中连续运行并记录电压方案。然后将对应于在细胞外缓冲液中的0.3% DMSO的媒介物施用3分钟,随后施用一式三份的实施例化合物。标准组合暴露时间为5分钟。使用从四个连续电压脉冲记录的尾电流振幅值的平均值如下计算实施例化合物对每个细胞的影响:计算与媒介物预处理相比的残余电流(%对照)。将数据报告为每个测试浓度的% 抑制,并使用QPatch软件估计IC50值。测试了至少两个细胞,并且如果结果出现分歧,则测试更多。
表5
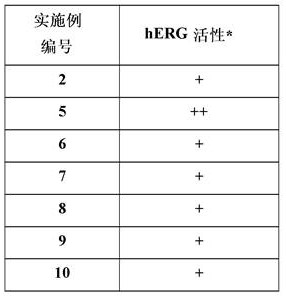
*hERG活性分类:+++ = IC50 1-1000 nM;++ = IC50 >1000-5000 nM;+ = IC50 >5000 nM。
参考文献


Claims (26)
1.式(I)的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体

其中
Ra1、Ra2、Ra3、Ra4、Ra5、Ra6独立地选自H、卤素、C1-4烷基和A2a,其中C1-4烷基任选地被一个或多个取代基取代,所述取代基选自卤素、OH和O-C1-3烷基,其中所述取代基相同或不同,且
前提条件是,Ra1、Ra2、Ra3、Ra4、Ra5、Ra6中的仅一个是A2a;
A1是C5亚环烷基、C5亚环烯基或含有氮环原子的5元亚杂环基,前提条件是,用星号标记的环A1的环原子是碳原子,其中A1任选地被一个或多个相同或不同的R4取代;
每个R4独立地是氧代(=O),其中所述环是至少部分饱和的、硫代(=S),其中所述环是至少部分饱和的、卤素、CN、OR5或C1-6烷基,其中C1-6烷基任选地被一个或多个相同或不同的卤素取代;
R5是H或C1-6烷基,其中C1-6烷基任选地被一个或多个相同或不同的卤素取代;
A2是R6a或A2a;
R6a是OR6a1、SR6a1、N(R6a1R6a2)、C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个取代基取代,所述取代基选自卤素、OR6a3、CN和A2a,其中所述取代基相同或不同;
R6a1、R6a2独立地选自H、C1-6烷基、C2-6烯基、C2-6炔基和A2a,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个取代基取代,所述取代基选自卤素、CN、OR6a3、A2a和OA2a,其中所述取代基相同或不同;
R6a3是H或C1-4烷基,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;
A2a是苯基、C3-7环烷基、C4-12二环烷基或3-7元杂环基,其中A2a任选地被一个或多个相同或不同的R6取代;
每个R6独立地是R6b、OH、OR6b、卤素或CN,其中R6b是环丙基、C1-6烷基、C2-6烯基或C2-6炔基,其中R6b任选地被一个或多个相同或不同的卤素取代;或
两个R6连接以与它们所连接的原子一起形成环A2b;
A2b是苯基、C3-7环烷基或3-7元杂环基,其中A2b任选地被一个或多个相同或不同的R7取代;
每个R7独立地是C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代;
R1是H或C1-4烷基,优选H,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;
R2是H、F或C1-4烷基,其中C1-4烷基任选地被一个或多个相同或不同的卤素取代;且
R3是A3、C1-6烷基、C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的R8取代;或
R2和R3连接以与它们所连接的氧原子和碳原子一起形成环A3a,其中A3a是7-12元杂二环基,其中7-12元杂二环基任选地被一个或多个相同或不同的R10取代;
R2a是H或F,优选H;
每个R8独立地是卤素、CN、C(O)OR9、OR9、C(O)R9、C(O)N(R9R9a)、S(O)2N(R9R9a)、S(O)N(R9R9a)、S(O)2R9、S(O)R9、N(R9)S(O)2N(R9aR9b)、SR9、N(R9R9a)、NO2、OC(O)R9、N(R9)C(O)R9a、N(R9)SO2R9a、N(R9)S(O)R9a、N(R9)C(O)N(R9aR9b)、N(R9)C(O)OR9a、OC(O)N(R9R9a)或A3;
R9、R9a、R9b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素、或一个OH、或一个OC1-4烷基、或一个A3取代;
每个A3独立地是苯基、萘基、C3-7环烷基、3-7元杂环基或7-12元杂二环基,其中A3任选地被一个或多个相同或不同的R10取代;
每个R10独立地是卤素、CN、C(O)OR11、OR11、C(O)R11、C(O)N(R11R11a)、S(O)2N(R11R11a)、S(O)N(R11R11a)、S(O)2R11、S(O)R11、N(R11)S(O)2N(R11aR11b)、SR11、N(R11R11a)、NO2、OC(O)R11、N(R11)C(O)R11a、N(R11)S(O)2R11a、N(R11)S(O)R11a、N(R11)C(O)OR11a、N(R11)C(O)N(R11aR11b)、OC(O)N(R11R11a)、氧代(=O),其中所述环是至少部分饱和的、C1-6烷基,C2-6烯基或C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的R12取代;
R11、R11a、R11b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代;
每个R12独立地是卤素、CN、C(O)OR13、OR13、C(O)R13、C(O)N(R13R13a)、S(O)2N(R13R13a)、S(O)N(R13R13a)、S(O)2R13、S(O)R13、N(R13)S(O)2N(R13aR13b)、SR13、N(R13R13a)、NO2、OC(O)R13、N(R13)C(O)R13a、N(R13)SO2R13a、N(R13)S(O)R13a、N(R13)C(O)N(R13aR13b)、N(R13)C(O)OR13a或OC(O)N(R13R13a);
R13、R13a、R13b独立地选自H、C1-6烷基、C2-6烯基和C2-6炔基,其中C1-6烷基、C2-6烯基和C2-6炔基任选地被一个或多个相同或不同的卤素取代。
2.权利要求1所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中Ra1、Ra2、Ra3、Ra4、Ra5、Ra6是H。
3.权利要求1或2所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A1是含有氮环原子的5元亚杂环基,且其中A1任选地被一个或多个相同或不同的R4取代。
4.权利要求1-3中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A1是含有氮环原子的5元亚杂环基,其选自以下二价杂环:噁二唑、咪唑、咪唑烷、吡唑和三唑,优选三唑或噁二唑,更优选噁二唑,且其中A1任选地被一个或多个相同或不同的R4取代。
5.权利要求1-4中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A1是未被取代的或被一个或两个相同或不同的R4取代,优选地A1是未被取代的。
6.权利要求1-5中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R4是氧代,其中所述环是至少部分饱和的。
7.权利要求1-5中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A1是
8.权利要求1-7中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A2是R6a。
9.权利要求8所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R6a是OR6a1且R6a1优选地是A2a或C1-6烷基,所述C1-6烷基任选地被一个或多个卤素和/或一个A2a和/或一个OR6a3取代;或R6a是C1-6烷基,所述C1-6烷基任选地被一个或多个卤素和/或一个A2a和/或一个OR6a3取代。
10.权利要求8或9所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R6a是OR6a1且R6a1优选地是C1-6烷基,所述C1-6烷基任选地被一个或多个F和/或一个OR6a3取代;或R6a是C1-6烷基,所述C1-6烷基任选地被一个或多个卤素和/或一个OR6a3取代。
11.权利要求1-7中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A2是A2a。
12.权利要求11所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A2a是苯基、环丁基、氮杂环丁基或5-6元芳族杂环基,且其中A2a任选地被一个或多个相同或不同的R6取代。
13.权利要求1-9、11和12中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A2a被一个或两个相同或不同的R6取代。
14.权利要求1-9、11-13中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中每个R6独立地是F、Cl、CF3、OCH3、OCF3、CH3、CH2CH3或环丙基。
15.权利要求1-14中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R2是H。
16.权利要求1-15中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R3是A3。
17.权利要求1-16中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A3是苯基、吡啶基、吡嗪基、嘧啶基、环丙基、环丁基或环己基,且其中A3任选地被一个或多个相同或不同的R10取代。
18.权利要求1-17中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中A3被一个或两个相同或不同的R10取代。
19.权利要求1-14中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R2和R3与它们所连接的氧和碳原子连接在一起以形成二氢苯并吡喃环,其中所述环任选地被一个或多个相同或不同的R10取代,优选地所述环被一个或两个R10取代。
20.权利要求1-19中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中R10独立地是F、Cl、Br、CHF2、CF3、OCF3、CH=O、CH2OH或CH3。
21.权利要求1-20中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中所述化合物是
2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4-氯苯基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[顺式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-[1-(4-氯苯基)-1H-1,2,3-三唑-4-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-[5-(4,4,4-三氟丁氧基)-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-(4-氯-3-氟苯氧基)-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;
2-[(6-氯-5-氟-3-吡啶基)氧基]-N-[反式-2-[5-[顺式-3-(三氟甲氧基)环丁基]-1,3,4-噁二唑-2-基]-1,3-二氧杂环己烷-5-基]乙酰胺;
2-[3-氯-4-(二氟甲基)苯氧基]-N-[反式-2-{5-[3-(三氟甲氧基)氮杂环丁烷-1-基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺;或
2-(3,4-二氯苯氧基)-N-[反式-2-{5-[2-(三氟甲氧基)乙氧基]-1,3,4-噁二唑-2-基}-1,3-二氧杂环己烷-5-基]乙酰胺。
22.权利要求1-21中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其中所述化合物具有式(Ia)

23.药物组合物,其包含至少一种如在权利要求1-22中的任一项中所定义的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体以及药学上可接受的载体,任选地与一种或多种其它生物活性化合物或药物组合物组合。
24.权利要求1-22中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体,其用作药物。
25.权利要求1-22中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或权利要求23所述的药物组合物,其用于治疗或预防一种或多种与整合应激反应有关的疾病或障碍的方法。
26.权利要求1-22中的任一项所述的化合物或其药学上可接受的盐、溶剂化物、水合物、互变异构体或立体异构体或权利要求23所述的药物组合物,其用于治疗或预防一种或多种疾病或障碍的方法,所述疾病或障碍选自脑白质营养不良、智力障碍综合征、神经变性疾病和障碍、肿瘤性疾病、感染性疾病、炎性疾病、肌肉骨骼疾病、代谢疾病、眼疾病,以及选自器官纤维化、慢性和急性肝病、慢性和急性肺病、慢性和急性肾病、心肌梗塞、心血管疾病、心律失常、动脉粥样硬化、脊髓损伤、缺血性中风和神经性疼痛的疾病。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20162329 | 2020-03-11 | ||
| EP20162329.5 | 2020-03-11 | ||
| PCT/EP2021/056023 WO2021180774A1 (en) | 2020-03-11 | 2021-03-10 | Modulators of the integrated stress response pathway |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN115190813A true CN115190813A (zh) | 2022-10-14 |
| CN115190813B CN115190813B (zh) | 2024-10-15 |
Family
ID=69804610
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202180020265.6A Active CN115190813B (zh) | 2020-03-11 | 2021-03-10 | 整合应激反应途径的调节剂 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US12441720B2 (zh) |
| EP (1) | EP4117780B1 (zh) |
| JP (1) | JP2023517944A (zh) |
| KR (1) | KR20220151635A (zh) |
| CN (1) | CN115190813B (zh) |
| AU (1) | AU2021236284B2 (zh) |
| BR (1) | BR112022014706A2 (zh) |
| CA (1) | CA3165813A1 (zh) |
| IL (1) | IL296220A (zh) |
| MX (1) | MX2022011143A (zh) |
| WO (1) | WO2021180774A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024109736A1 (zh) * | 2022-11-21 | 2024-05-30 | 深圳众格生物科技有限公司 | 一种化合物、包含其的药物组合物及其合成方法和用途 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2021012904A (es) * | 2019-04-23 | 2022-01-18 | Evotec Int Gmbh | Moduladores de la via de respuesta al estres integrada. |
| US20230391763A1 (en) | 2020-10-22 | 2023-12-07 | Evotec International Gmbh | Modulators of the integrated stress response pathway |
| JP2023546224A (ja) | 2020-10-22 | 2023-11-01 | エヴォテック・インターナショナル・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 統合ストレス応答経路のモジュレーター |
| MX2023004623A (es) | 2020-10-22 | 2023-05-12 | Evotec Int Gmbh | Moduladores de la via integrada de respuesta al estres. |
| DE102023120315A1 (de) * | 2023-07-31 | 2025-02-06 | Rheinisch-Westfälische Technische Hochschule Aachen, abgekürzt RWTH Aachen, Körperschaft des öffentlichen Rechts | Verfahren zur Herstellung von Pyrrolidon-substituierten Polyolen |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019032743A1 (en) * | 2017-08-09 | 2019-02-14 | Denali Therapeutics Inc. | COMPOUNDS, COMPOSITIONS AND METHODS |
| WO2019046779A1 (en) * | 2017-09-01 | 2019-03-07 | Denali Therapeutics Inc. | COMPOUNDS, COMPOSITIONS AND METHODS |
| WO2019090081A1 (en) * | 2017-11-02 | 2019-05-09 | Calico Life Sciences Llc | Modulators of the integrated stress pathway |
| US20190142806A1 (en) * | 2016-05-05 | 2019-05-16 | Calico Life Sciences Llc | Modulators of the integrated stress pathway |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69835518T2 (de) | 1997-06-12 | 2007-08-09 | Aventis Pharma Ltd., West Malling | Imidazolyl-cyclische acetale |
| JP6806562B2 (ja) | 2013-03-15 | 2021-01-06 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | eIF2α経路の調節因子 |
| TW201808888A (zh) | 2016-05-05 | 2018-03-16 | 嘉來克生命科學有限責任公司 | 整合應激途徑之調節劑 |
| TW201808903A (zh) | 2016-05-05 | 2018-03-16 | 嘉來克生命科學有限責任公司 | 整合應激途徑之調節劑 |
| TWI763668B (zh) | 2016-05-05 | 2022-05-11 | 美商嘉來克生命科學有限責任公司 | 整合應激途徑之調節劑 |
| AU2017279027A1 (en) | 2016-06-08 | 2018-12-20 | Glaxosmithkline Intellectual Property Development Limited | Chemical Compounds |
| CA3026982A1 (en) | 2016-06-08 | 2017-12-14 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds as atf4 pathway inhibitors |
| WO2018225093A1 (en) | 2017-06-07 | 2018-12-13 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds as atf4 pathway inhibitors |
| US20210145771A1 (en) | 2017-07-03 | 2021-05-20 | Glaxosmithkline Intellectual Property Development Limited | N-(3-(2-(4-chlorophenoxy)acetamido)bicyclo[1.1.1] pentan-1-yl)-2-cyclobutane-1- carboxamide derivatives and related compounds as atf4 inhibitors for treating cancer and other diseases |
| CN110896634A (zh) | 2017-07-03 | 2020-03-20 | 葛兰素史密斯克莱知识产权发展有限公司 | 作为atf4抑制剂用于治疗癌症和其它疾病的2-(4-氯苯氧基)-n-((1-(2-(4-氯苯氧基)乙炔氮杂环丁烷-3-基)甲基)乙酰胺衍生物和相关化合物 |
| US11939320B2 (en) | 2017-11-02 | 2024-03-26 | Abbvie Inc. | Modulators of the integrated stress pathway |
| UY37956A (es) | 2017-11-02 | 2019-05-31 | Abbvie Inc | Moduladores de la vía de estrés integrada |
| UY37957A (es) | 2017-11-02 | 2019-05-31 | Abbvie Inc | Moduladores de la vía de estrés integrada |
| UY37958A (es) | 2017-11-02 | 2019-05-31 | Abbvie Inc | Moduladores de la vía de estrés integrada |
| EP3704125B1 (en) | 2017-11-02 | 2026-03-11 | Calico Life Sciences LLC | Modulators of the integrated stress pathway |
| EP3704098B1 (en) | 2017-11-02 | 2024-01-24 | Calico Life Sciences LLC | Modulators of the integrated stress pathway |
| CA3080804A1 (en) | 2017-11-02 | 2019-05-09 | Calico Life Sciences Llc | Modulators of the integrated stress pathway |
| EP3704096B1 (en) | 2017-11-02 | 2026-04-22 | Calico Life Sciences LLC | Modulators of the integrated stress pathway |
| WO2019118785A2 (en) | 2017-12-13 | 2019-06-20 | Praxis Biotech LLC | Inhibitors of integrated stress response pathway |
| WO2019183589A1 (en) | 2018-03-23 | 2019-09-26 | Denali Therapeutics Inc. | Modulators of eukaryotic initiation factor 2 |
| WO2019193541A1 (en) | 2018-04-06 | 2019-10-10 | Glaxosmithkline Intellectual Property Development Limited | Bicyclic aromatic ring derivatives of formula (i) as atf4 inhibitors |
| WO2019193540A1 (en) | 2018-04-06 | 2019-10-10 | Glaxosmithkline Intellectual Property Development Limited | Heteroaryl derivatives of formula (i) as atf4 inhibitors |
| WO2019236710A1 (en) | 2018-06-05 | 2019-12-12 | Praxis Biotech LLC | Inhibitors of integrated stress response pathway |
| JP2021529814A (ja) | 2018-07-09 | 2021-11-04 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 化学化合物 |
| WO2020031107A1 (en) | 2018-08-08 | 2020-02-13 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| TWI877863B (zh) | 2018-10-11 | 2025-03-21 | 美商嘉來克生命科學有限責任公司 | 整合應激路徑之前藥調節劑 |
| MA54953A (fr) | 2019-02-13 | 2021-12-22 | Denali Therapeutics Inc | Composés, compositions et procédés |
| US12091392B2 (en) | 2019-02-13 | 2024-09-17 | Denali Therapeutics Inc. | Compounds, compositions and methods |
| US20200270232A1 (en) | 2019-02-25 | 2020-08-27 | Praxis Biotech LLC | Inhibitors of integrated stress response pathway |
| US20220177456A1 (en) | 2019-03-06 | 2022-06-09 | Denali Therapeutics Inc. | Compounds, compositions and methods |
| MX2021012904A (es) | 2019-04-23 | 2022-01-18 | Evotec Int Gmbh | Moduladores de la via de respuesta al estres integrada. |
| CA3137213A1 (en) | 2019-04-23 | 2020-10-29 | Evotec International Gmbh | Modulators of the integrated stress response pathway |
| MX2021013197A (es) | 2019-04-30 | 2022-02-24 | Calico Life Sciences Llc | Cicloalquilos sustituidos como moduladores de la vía integrada del estrés. |
| AR118836A1 (es) | 2019-04-30 | 2021-11-03 | Calico Life Sciences Llc | Moduladores de la vía de tensión integrada |
| MX2021015210A (es) | 2019-06-12 | 2022-01-18 | Praxis Biotech LLC | Inhibidores de la via de respuesta al estres integrada. |
| JP2022536663A (ja) | 2019-06-12 | 2022-08-18 | プラクシス バイオテック エルエルシー | 統合的ストレス応答経路のモジュレーター |
| AU2021213289A1 (en) | 2020-01-28 | 2022-07-07 | Evotec International Gmbh | Modulators of the integrated stress response pathway |
| US20230391763A1 (en) | 2020-10-22 | 2023-12-07 | Evotec International Gmbh | Modulators of the integrated stress response pathway |
| JP2023546224A (ja) | 2020-10-22 | 2023-11-01 | エヴォテック・インターナショナル・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 統合ストレス応答経路のモジュレーター |
| MX2023004623A (es) | 2020-10-22 | 2023-05-12 | Evotec Int Gmbh | Moduladores de la via integrada de respuesta al estres. |
-
2021
- 2021-03-10 MX MX2022011143A patent/MX2022011143A/es unknown
- 2021-03-10 US US17/906,014 patent/US12441720B2/en active Active
- 2021-03-10 WO PCT/EP2021/056023 patent/WO2021180774A1/en not_active Ceased
- 2021-03-10 BR BR112022014706A patent/BR112022014706A2/pt unknown
- 2021-03-10 AU AU2021236284A patent/AU2021236284B2/en active Active
- 2021-03-10 CN CN202180020265.6A patent/CN115190813B/zh active Active
- 2021-03-10 KR KR1020227033901A patent/KR20220151635A/ko active Pending
- 2021-03-10 JP JP2022554500A patent/JP2023517944A/ja active Pending
- 2021-03-10 CA CA3165813A patent/CA3165813A1/en active Pending
- 2021-03-10 EP EP21710001.5A patent/EP4117780B1/en active Active
- 2021-03-10 IL IL296220A patent/IL296220A/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20190142806A1 (en) * | 2016-05-05 | 2019-05-16 | Calico Life Sciences Llc | Modulators of the integrated stress pathway |
| WO2019032743A1 (en) * | 2017-08-09 | 2019-02-14 | Denali Therapeutics Inc. | COMPOUNDS, COMPOSITIONS AND METHODS |
| WO2019046779A1 (en) * | 2017-09-01 | 2019-03-07 | Denali Therapeutics Inc. | COMPOUNDS, COMPOSITIONS AND METHODS |
| WO2019090081A1 (en) * | 2017-11-02 | 2019-05-09 | Calico Life Sciences Llc | Modulators of the integrated stress pathway |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024109736A1 (zh) * | 2022-11-21 | 2024-05-30 | 深圳众格生物科技有限公司 | 一种化合物、包含其的药物组合物及其合成方法和用途 |
| US12612392B2 (en) | 2022-11-21 | 2026-04-28 | Shenzhen Zhongge Biological Technology Co., Ltd. | Compound, pharmaceutical compositions containing same, synthesis method therefor and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN115190813B (zh) | 2024-10-15 |
| WO2021180774A1 (en) | 2021-09-16 |
| US12441720B2 (en) | 2025-10-14 |
| MX2022011143A (es) | 2022-10-13 |
| CA3165813A1 (en) | 2021-09-16 |
| KR20220151635A (ko) | 2022-11-15 |
| EP4117780B1 (en) | 2026-05-06 |
| BR112022014706A2 (pt) | 2022-10-11 |
| EP4117780A1 (en) | 2023-01-18 |
| JP2023517944A (ja) | 2023-04-27 |
| IL296220A (en) | 2022-11-01 |
| US20230129907A1 (en) | 2023-04-27 |
| AU2021236284A1 (en) | 2022-08-18 |
| AU2021236284B2 (en) | 2026-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN115190813B (zh) | 整合应激反应途径的调节剂 | |
| JP7590343B2 (ja) | 統合ストレス応答経路のモジュレーター | |
| JP7518324B2 (ja) | Rho関連プロテインキナーゼのモジュレーター | |
| JP7699595B2 (ja) | 統合的ストレス応答経路の調節因子 | |
| JP7588089B2 (ja) | 統合ストレス応答経路のモジュレーター | |
| KR20230110510A (ko) | 통합 스트레스 반응 경로의 조절제 | |
| KR20230110511A (ko) | 통합 스트레스 반응 경로의 조절제 | |
| HK40079271A (zh) | 整合应激反应途径的调节剂 | |
| CN116964047B (zh) | 整合应激反应途径的调节剂 | |
| HK40079271B (zh) | 整合应激反应途径的调节剂 | |
| EA048763B1 (ru) | Модулятор пути интегрированного ответа на стресс и его применение | |
| HK40068295A (zh) | 整合应激反应途径的调节剂 | |
| HK40068816A (zh) | 整合应激反应路径的调节剂 | |
| HK40073491A (zh) | 整合应激反应途径的调节剂 | |
| EA046988B1 (ru) | Модуляторы пути интегрированной реакции на стресс | |
| EA051483B1 (ru) | Модуляторы пути интегрированного ответа на стресс | |
| EA048846B1 (ru) | Модуляторы пути интегрированного ответа на стресс |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 40079271 Country of ref document: HK |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant |