CN1257166C - 制备具有抗组胺活性的三环化合物的方法 - Google Patents
制备具有抗组胺活性的三环化合物的方法 Download PDFInfo
- Publication number
- CN1257166C CN1257166C CNB998159670A CN99815967A CN1257166C CN 1257166 C CN1257166 C CN 1257166C CN B998159670 A CNB998159670 A CN B998159670A CN 99815967 A CN99815967 A CN 99815967A CN 1257166 C CN1257166 C CN 1257166C
- Authority
- CN
- China
- Prior art keywords
- alkyl
- formula
- compound
- substituted
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Steroid Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
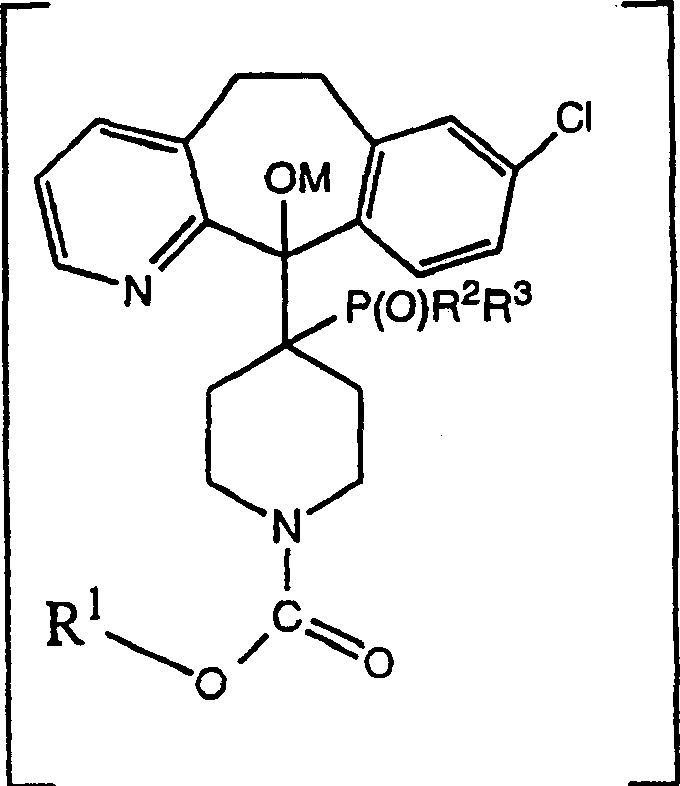
本发明公开了制备式(I)化合物的方法,其中R1选自:烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代,所述方法包括如下步骤:(a)将式(A)的酮与式(B)的负碳离子反应,其中R1如上所定义,R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基;(b)将步骤(a)的反应混合物用质子化试剂处理形成式(C)的β-羟基中间体,其中R1、R2和R3如上所定义;然后(c)将β-羟基中间体热分解形成式(I)化合物。由该方法制备的化合物具有抗组胺活性,例如氯雷他定。本发明还公开了通过进行上述方法然后将产物转变成脱乙氧羰基氯雷他定来制备脱乙氧羰基氯雷他定的方法。本发明还公开了式(II)和(III)的新的中间体,其中R1、R2和R3如上所定义。
Description
发明背景
本发明提供了制备抗组胺药物的改进的方法和新的中间体。具体地讲,本发明的方法和中间体可用于制备U.S.4,282,233中所公开的氯雷他定以及U.S.4,659,716中所公开的脱乙氧羰基氯雷他定(8-氯-6,11-二氢-11-(4-亚哌啶基)-5H-苯并[5,6]环庚三烯并[1,2-b]吡啶)(“DCL”)。
U.S.4,659,716公开了如下制备氯雷他定和DCL的方法:

根据该方法,将三环酮(1)与从4-氯-N-甲基哌啶衍生的格氏试剂偶联。将由该加成反应形成的醇(2)在酸性条件下脱水生成化合物(3)。然后将化合物(3)与氯甲酸乙酯进行Von Braun反应生成氯雷他定。DCL可以通过氯雷他定的脱烷氧羰基化反应制得。该方法有许多严重的缺点。与酮(1)进行格氏反应所需的卤化物4-氯-N-甲基哌啶必需通过5步合成制得,并且在高于室温的温度下是不稳定的。酮(1)与N-甲基-哌啶-4-基氯化镁的生成醇(2)的反应收率不高(约60%),其原因是发生了共轭加成(即,向吡啶环加成)和还原。将醇(2)脱水生成化合物(3)是一个很敏感的反应,会发生化合物(3)的异构化。从化合物(3)生成氯雷他定的Von Braun反应会产生有毒的氯甲烷气体副产物。在排放入大气之前必需将该有毒的副产物进行化学分解。本发明排除了这些问题。
发明概述
本发明提供了制备下式化合物的方法:

其中R1选自:烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代,所述方法包括如下步骤:
(a)将下式的酮
与下式的负碳离子反应

其中R1如上所定义,R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基;
(b)将步骤(a)的反应混合物用质子化试剂处理形成下式的β-羟基中间体

其中R1、R2和R3如上所定义;然后
(c)将β-羟基中间体热分解形成式(I)化合物。
本发明还提供了制备下式化合物的方法:
所述方法包括如下步骤:
(a)将下式的酮

与下式的负碳离子反应

其中R1选自:烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代;R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基;
(b)将步骤(a)的反应混合物用质子化试剂处理形成下式的β-羟基中间体
其中R1、R2和R3如上所定义;
(c)将β-羟基中间体热分解形成下式化合物

其中R1如上所定义;然后
(d)将式(I)化合物转变成式(IV)化合物。
在制备化合物(IV)(DCL)的特别优选的实施方案中,上述方法中的R1选自易于在酸性条件下除去的基团,优选烷基,首选叔丁基。
本发明还提供了具有如下结构式的新的中间体

其中R1、R2和R3如上所定义。
本发明还提供了具有如下结构式的新的中间体

其中R1、R2和R3如上所定义。
发明详述
本文所用的术语“烷基”是指1至6个碳原子的直链或支链烃基链。
“链烯基”是指含有至少一个碳碳双键的1至6个碳原子的直链或支链烃基链。
“链炔基”是指含有至少一个碳碳叁键的1至6个碳原子的直链或支链烃基链。
“芳基”是指含有至少一个芳环的碳环基团(例如,苯基或萘基)。
“芳烷基”是指式芳基-R-的基团,其中R是烷基。
“环烷基”是指3至6个碳原子的非芳香性的碳环。
“环烷基烷基”是指式环烷基-R-的基团,其中R是烷基。
“卤素”是指氟、氯、溴或碘基团。
“取代的苯基”是指被选自卤素、-OH、烷基、烷氧基或-CF3的取代基取代的苯基。
“取代的环烷基”是指被选自卤素、-OH、烷基、烷氧基或-CF3的取代基取代的环烷基。
“取代的环烷基烷基”是指其中的环烷基部分被选自卤素、-OH、烷基、烷氧基或-CF3的取代基取代了的环烷基烷基。
本方法是对制备氯雷他定和相关化合物的现有技术方法的重要改进。一个明显的优点是可以以高的收率和纯度将原料酮以“单釜法”(即,无需分离β-羟基中间体)转变成所需产物(例如,氯雷他定)。本方法所用的负碳离子从热稳定的式(III)化合物衍生得到,所述式(III)化合物比U.S.4,659,716中所公开的热不稳定的4-氯-N-甲基哌啶稳定得多。此外,式(III)化合物可以从吡啶通过两步反应以大约70%的收率得到,这比制备现有技术中所用的4-氯-N-甲基哌啶所需的5步反应简单得多。
R1优选是烷基,更优选乙基或叔丁基,首选乙基。
R2和R3优选是烷基,更优选甲基、乙基、异丙基或叔丁基,最优选乙基。
当R1、R2和R3被取代时,取代基的数量优选是1至3个。
本发明方法的步骤(a)中所用的负碳离子优选通过将上述的式(III)化合物用强碱在适宜的非质子有机溶剂中处理生成。优选所述的碱是有机锂。适宜的碱的例子包括但不仅限于二异丙基氨基锂(“LDA”);正丁基锂;叔丁基锂;仲丁基锂和二乙基氨基锂。首选LDA。用于生成负碳离子和进行步骤(a)所用的适宜的非质子有机溶剂的例子包括但不仅限于:二甲苯;四氢呋喃(“THF”);乙醚;乙二醇二甲醚;叔丁基甲基醚;二甘醇二甲醚;苯;甲苯及其混合物。优选步骤(a)中所用的溶剂是醚,首选THF。在特别优选的实施方案中,步骤(a)在醚溶剂与沸点更高的非醚溶剂的混合物中进行。步骤(a)优选在-150℃至+10℃的温度下进行,更优选-80℃至-10℃,首选-40℃至-20℃。优选式(III)化合物以及由其生成的负碳离子的用量相对于酮而言为1至3当量、更优选1至1.5当量,最优选1.05至1.1当量。
本方法的步骤(b)通过向步骤(a)的反应混合物中加入质子化试剂来完成。不受任何理论的束缚,确信负碳离子和三环酮在步骤(a)中反应形成了如下醇盐中间体,该中间体是不稳定的:
M=碱金属原子,例如锂
加入质子化试剂将该不稳定的醇盐转变成β-羟基中间体。优选质子化试剂的用量相对于酮而言为1至3当量,更优选1至1.5当量,最优选1.05至1.1当量。质子化试剂优选是水或酸。当质子化试剂是酸时,本方法可以不用分离β-羟基中间体。用作质子化试剂的适宜的酸包括但不仅限于,无机酸的醇溶液(例如无水硫酸的甲醇溶液和盐酸的甲醇溶液)、磺酸和C1至C6链烷酸。优选酸是无水的。特别优选乙酸。当用酸、优选无水的酸终止反应时,步骤(a)的溶剂(优选低沸点的醚溶剂)可以通过加入高沸点的溶剂并将混合物加热至高于低沸点溶剂之沸点的温度来除去。热分解步骤(c)可以通过将该混合物回流、优选在110至160℃、更优选110至140℃、首选130至140℃下回流来完成,以生成化合物(I)。高沸点溶剂优选具有110至160℃、更优选110至140℃、最优选130至140℃的沸点。适宜的高沸点溶剂包括但不仅限于二甲苯、氯苯、烷烃和醇。化合物(I)优选通过蒸除高沸点溶剂,将残余物溶于乙腈,蒸馏至干然后用乙腈重结晶进行回收。该重结晶可以有效地除去唯一发现的杂质,即少量的三环酮原料和用于生成负碳离子的膦酸酯和氧化膦。
如果使用水作为质子化试剂,优选将β-羟基中间体进行分离,该中间体极易溶于水。因此,当向步骤(a)的反应混合物中加入水时,可将形成的有机相和水相分离,然后可以从水相中回收β-羟基中间体。回收优选通过将水溶液用无机盐(例如,碳酸钾)在可以溶解β-羟基中间体的有机溶剂(例如,THF)的存在下处理来完成。β-羟基中间体可以通过重结晶进行纯化。然后可以通过将纯化的β-羟基中间体在高沸点溶剂中的悬浮液回流完成步骤(c)。然后可以通过蒸除溶剂并用适宜的溶剂(例如,乙腈)重结晶回收化合物(I)。
当DCL是所需的产物时,可以进行上述的方法以得到式(I)化合物,然后通过常规方法将式(I)化合物转变成DCL。例如,可以通过用强碱例如KOH或NaOH处理或用酸如三氟乙酸、硫酸水溶液或对甲苯磺酸处理将式(I)化合物转变成DCL。当DCL是所需产物时,R1优选选自易于在酸性条件下除去的基团,优选烷基,最优选叔丁基。
式(III)化合物按照如下流程所示进行制备:

如上所示,将吡啶先用氯甲酸酯C1-COOR1(其中R1如上所定义)在适宜的溶剂(优选乙腈)中处理,然后用化合物
6a(其中R2和R3如上所定义,R4是烷基,优选甲基或乙基,特别优选乙基)处理。由此得到化合物
6和少量的化合物7。然后将化合物
6和
7的混合物用适宜的金属催化剂、优选钯进行催化氢化生成所需的式(III)化合物和少量的化合物
8,式(III)化合物的1,2-异构体。化合物(III)可以通过蒸馏进行纯化除去低沸点的化合物
8。如果所需的式(III)化合物带有对催化氢化敏感的R1基团,则该化合物可以从其中R1是乙基的式(III)化合物通过用酸或碱脱乙氧羰基化然后将形成的化合物与带有所需R1基团的氯甲酸酯C1-COOR1反应进行制备。
包括在化合物
6a范围内的化合物可以购买到,或者是可以通过已知方法制备的已知化合物。如果化合物
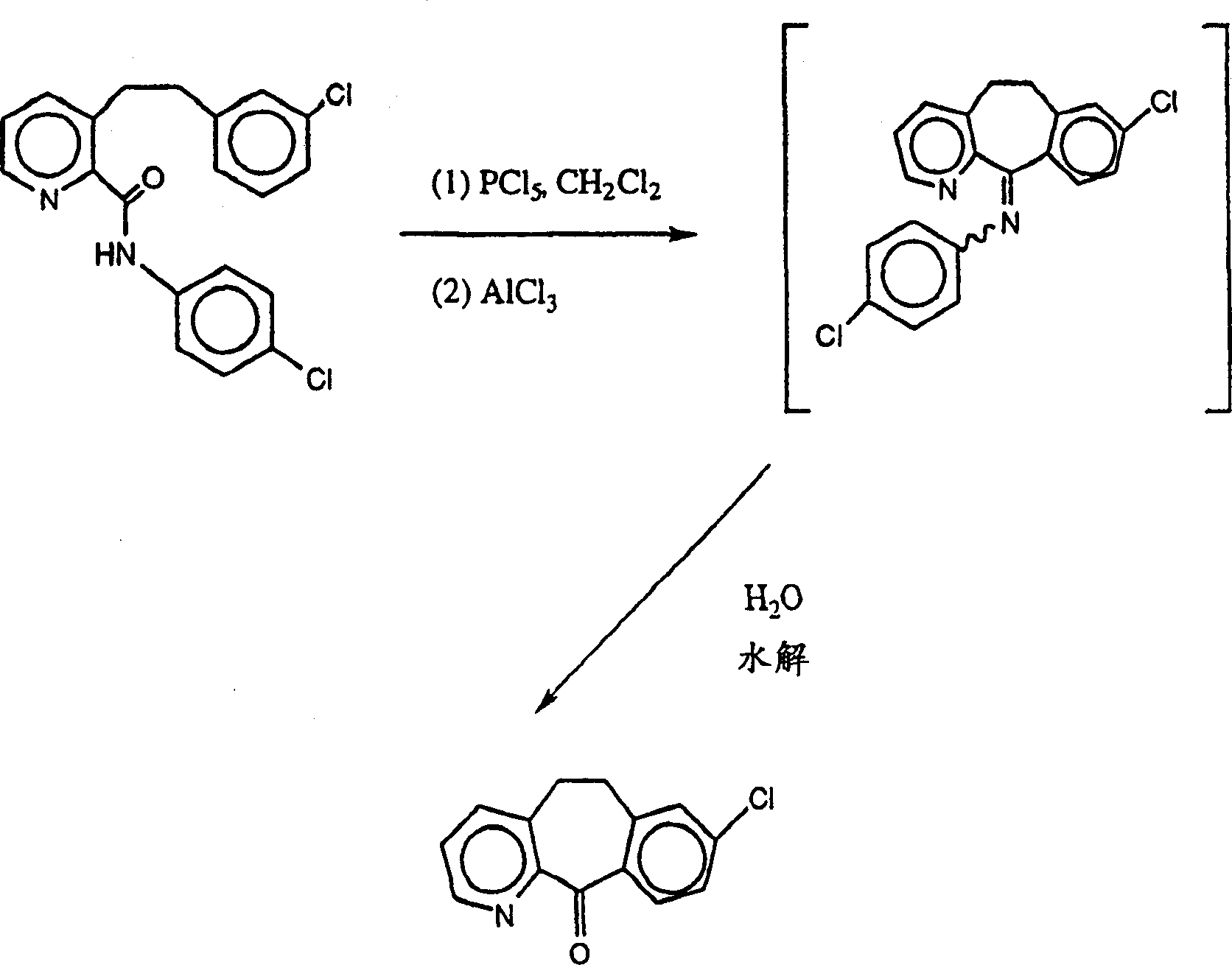
6a的取代基不完全相同,则该化合物可以通过将一摩尔PCl3或PBr3与一摩尔乙醇或甲醇反应将一个卤素基团用甲氧基或乙氧基取代,然后将形成的化合物依次与一摩尔醇或格氏试剂反应将剩余的两个卤素基团依次用所需的R2和R3基团取代。本方法所用的三环酮可以按照本领域已知的方法制备,例如通过U.S.4,659,716、U.S.4,731,447或PCT公开号WO96/31478(1996年10月10日公开)中公开的方法制备。或者,三环酮可以按照如下流程制备:
如以上流程图所示,将吡啶化合物
9与苯胺在钯催化剂例如Pd(OAc)2或PdCl2、一氧化碳、碱例如1,8-二氮杂双环-[5.4.0]十一-7-烯(“DBU”)或二异丙基乙基胺和选自乙二醇二甲醚、2-甲氧基乙醚和三甘醇二甲醚的醚的存在下反应形成酰胺化合物
10。形成酰胺化合物
10的反应优选在约45℃至90℃的温度和约40至100psi的压力下在适宜的溶剂例如甲苯或氯苯中进行。将酰胺化合物
10与碘-取代的化合物
11在强碱例如二异丙基氨基锂的存在下在适宜的溶剂例如THF中反应形成化合物
12。将化合物
12与CH3I和碱例如NaH反应形成甲基化的化合物
13。将化合物
13通过与格氏试剂例如2-甲氧基苯基溴化镁反应环化形成所需的酮。
或者,三环酮可以按照如下流程制备:

如以上流程图所示,将酰胺化合物
10a与化合物
14在强碱例如二异丙基氨基锂的存在下、在适宜的溶剂例如THF中反应形成化合物
15。将化合物
15用脱水剂和超酸例如P2O5/CF3SO3H或脱水剂和Lewis酸例如PCl5/AlCl3或POCl3/ZnCl2处理进行环化,然后将反应产物
16水解形成所需的三环酮。
酰胺化合物
10a可以按照如下所示进行制备:
R5=苯基或对氯苯基
如上所示,将3-甲基吡啶甲酸与有机碱例如三乙胺反应,然后与酰氯(例如新戊酰氯)或氯甲酸酯(例如C2H5OCOCl)在适宜的溶剂例如二氯甲烷中、在约-30℃至0℃的温度下反应生成混合酸酐。于-30℃至0℃的温度下向混合物中加入NH2R5或其在适宜溶剂中的溶液形成酰胺化合物
10a。
以下实施例说明了上述的发明,但所述实施例不应看作是对本发明范围的限定。本发明范围内的其它试剂和类似方法对于本领域技术人员是显而易见的。
制备例A
向3-甲基-吡啶甲酸(400g;2.92mol)的二氯甲烷(“DCM”)(1600ml)悬浮液中加入三乙胺(406.4ml;2.92mol)。将溶液冷却至-20℃,滴加氯甲酸乙酯(278.8ml;2.92mol)并保持反应温度在-10至-20℃之间。将反应混合物在该温度下继续搅拌2小时。然后滴加4-氯苯胺(372.1g;2.92mol)的二氯甲烷(150ml)溶液并保持内温在-10和-20℃之间。于该温度下继续搅拌2小时,然后将反应液升温至室温。加入水(400ml)终止反应。分液,将有机层用水(400ml)洗涤。通过真空蒸除溶剂将反应混合物浓缩至大约800ml。加入异丙醇(400ml)并减压蒸除1体积溶剂。加入异丙醇(400ml)并再次减压蒸除溶剂,浓缩至800ml。加入异丙醇(1200ml)并将混合物加热至70至80℃之间。将混合物冷却至60℃并加入晶种。将混合物在该温度下保持30分钟,然后冷却到0至-5℃并在该温度下保持1小时。滤出产物,在泵上用冰冷的异丙醇洗涤。将产物于60℃下在真空烘箱中干燥得到655.8g酰胺2(91%)。
制备例B
于-25℃下向酰胺2(150g;0.61mol)的THF(750ml)溶液中加入2.5M丁基锂的己烷溶液(486ml;1.22mol),同时保持内温在-20和-30℃之间。将混合物于-25℃搅拌1小时,然后在55分钟内滴加3-氯苄基氯,同样保持内温在-20和-30℃之间。将反应混合物于-25℃搅拌1小时,然后升温至室温。加入水(300ml),将形成的混合物搅拌30分钟。分液并将水层用乙酸乙酯(150ml)萃取。将合并的有机相真空蒸发至干并将残余物用异丙醇结晶(900ml)得到205.5g酰胺3(91%)。
制备例C
用20分钟的时间于5℃下向五氯化磷(95%,26.6g;0.121mol)的二氯甲烷(60ml)溶液中滴加N-(4-氯苯基)-3-[2-(3-氯苯基)乙基]-2-吡啶甲酰胺(30g;0.081mol)的二氯甲烷(60ml)溶液。将形成的混合物于5至10℃搅拌1小时,然后在30分钟内升温至室温。在45分钟内分4份加入氯化铝(43.1g;0.323mol)并保持反应温度在30℃以下。将混合物搅拌1小时,然后倒在冰(300g)上。通过蒸馏从混合物中除去二氯甲烷,然后将剩余的水溶液于80℃加热1小时。加入柠檬酸三钠二水合物(70g;0.24mol),然后加入氢氧化钠水溶液(10M,140ml)将pH调至7。加入甲苯(150ml),然后加入马来酸酐(12.0g 0.122mol)的甲苯(50ml)溶液。将形成的混合物搅拌30分钟,然后用氢氧化钠水溶液(10M,60ml)将水相的pH调至12。将混合物加热至70℃然后分液。将水相进一步用甲苯(2×90ml)萃取并将合并的有机层用水(90ml)洗涤。HPLC分析表明酮产物的溶液收率为95%。将产物混合物用甲苯/己烷重结晶得到黄白色固体状所需的三环酮(13.96g,71%)。
实施例1
A.
N-乙氧羰基-1,4-二氢吡啶-4-膦酸二乙酯:
将吡啶(16.1L,0.2kmol)的乙腈(160L)溶液冷却至-10℃。加入氯甲酸乙酯(19.1L,0.2kmol),加入的速度为使温度不超过0℃。然后将悬浮液于-10至0℃搅拌2小时以确保N-酰化完全。将反应物冷却至-30℃然后加入亚磷酸三乙酯(34.3L,0.2kmol),加入的速度为使温度不超过-20℃(在-30至-20℃之间)。将溶液在该温度下搅拌2小时,然后在数小时内升温至室温(或过夜)。在Sihi真空下蒸除乙腈至温度不超过80℃,然后将残余的液体冷却至30℃。加入二氯甲烷(140L),然后加入水(60L)和浓盐酸(10L)的混合物。将混合物室温搅拌1小时,然后将下层的有机相抽吸到分液漏斗中并用水(100L)洗涤。在抽吸回原来的(用水洗涤的)容器中后,将其用1%碳酸钾溶液洗涤,最后用水洗至中性。然后蒸除二氯甲烷得到粗品(约92%纯度)N-乙氧羰基-1,4-二氢吡啶-4-膦酸二乙酯,其中含有约5%1,2-异构体和痕量的磷酸三乙酯(由相应的亚磷酸酯氧化生成)。得到约48.4kg浅黄色液体,将其在氮气下保存,因其对空气略微不稳定。
B.
N-乙氧羰基哌啶-4-膦酸二乙酯
将上述粗品膦酸酯(24kg)溶于甲醇(64L)然后加入到氢化器中。然后将催化剂(5%Pd/C浆液,3.46kg)加入甲醇(14L)中并用7.5L溶剂冲洗到氢化器内。将混合物在10巴/25℃下氢化直至摄取完全(约10小时),然后放空氢气并用氮气净化。滤除催化剂并将催化剂用甲醇(25L)洗涤。然后蒸除甲醇,以定量的收率得到N-乙氧羰基哌啶-4-膦酸二乙酯(
5a),其中含有约5%1,2-异构体。将该物质通过高真空蒸馏进行纯化,b.p.185-90℃/1.5毫巴。1,2-异构体集中在蒸馏的前馏份中。
实施例2

氯雷他定:
不分离β-羟基膦酸酯中间体:
将正丁基锂己烷溶液(318mL,1.6M,0.509mol)于-70℃下加入到二异丙基胺(53.1g,0.526mol)的二甲苯:THF(265:525mL)溶液中。将形成的溶液于-70至-60℃下搅拌1小时,然后加入按照实施例1的方法制备的N-乙氧羰基哌啶-4-膦酸二乙酯(150g,0.489mol,纯度96%,其余的为1,2-异构体)的二甲苯(150ml)溶液。将该悬浮液于-70至-60℃搅拌0.5小时,然后在约1小时内升温至-20℃。用约0.5小时的时间加入如上所示的8-氯酮(111.3g,0.457mol)的二甲苯∶THF(120mL∶240ml)溶液,同时保持温度不超过-10℃。形成深绿色的溶液。将其在-20至-10℃下搅拌1小时。加入乙酸(36g,0.6mol)终止反应。此时的TLC分析[二氯甲烷∶甲苯∶甲醇∶33%氨水100∶40∶20∶1]显示了β-羟基膦酸酯以及少量未反应的酮。(Rf约0.2)。
补加二甲苯(450ml)然后蒸除溶剂直至溶液的温度达到135至140℃。然后将其回流1小时,将样品通过TLC进行分析直至β-羟基膦酸酯全部消失。将溶液冷却至60℃然后加入水(500ml)。分液,将有机溶液用5%甲酸(2×200ml)和水洗涤至中性。氯雷他定的溶液收率(毛细管GC-HP-5柱,80℃/3分钟至265℃)约为90%(残留有约3至5%的酮以及痕量的对应于该酮的醇,还检测到了少量的膦酸酯)。蒸除二甲苯并将残余物溶于乙腈(500ml)然后蒸馏至干。将该过程重复一次以完全除去二甲苯,因为二甲苯可以溶解氯雷他定。最后将粗产物(204.6g)用乙腈(460ml)重结晶并于60℃真空干燥。纯净氯雷他定的收率为130.3g(74.5%)(TLC分析为纯净,毛细管GC分析纯度为99.99%)。可以通过浓缩和冷却从母液中分离另一批纯净的氯雷他定(3.6g,2.0%)。总收率为133.9g(76.5%)。
实施例3
氯雷他定:
分离β-羟基膦酸酯中间体
完全按照以上实施例2的描述以150g(0.619mol)三环8-氯酮的规模进行反应。用水(800mL)代替乙酸终止反应。分液并将水相真空加热至约45℃除去THF。将β-羟基膦酸酯的溶液用甲苯(2×200mL)洗涤。然后依次加入THF(1L)和碳酸钾(200g)。分出含有产物的THF层并减压蒸除溶剂得到330g胶状固体,将其溶于甲醇(700ml)。将该溶液搅拌过夜然后冷却至约-30℃。过滤得到187.7g(56%)白色的β-羟基膦酸酯。如需要,还可以从甲醇母液中得到部分物质(约50g,15%)。总收率为约70%。该物质含有约4-7%牢固残留的水。
β-羟基膦酸酯的热解
将按照上述方法分离的β-羟基膦酸酯(79g,0.147mol)的二甲苯(320ml)悬浮液在迪安-斯塔克装置中回流并收集到约3mL水。将混合物继续回流2小时使热分解进行完全。将溶液冷却至60℃,用200mL水处理然后分液(水相的pH值约为5)。将有机相用热水洗涤数次,然后减压蒸除二甲苯。将残余物溶于乙腈(200ml)然后蒸发至干。重复该过程,最后将残余物的油用乙腈(270ml)重结晶并于60℃真空干燥。纯净氯雷他定的收率为43.44g(80%,其余的为无水β-羟基膦酸酯)。在浓缩和冷却时从母液中得到第二批产物(3.5g,6.4%)。
实施例4

A.
N-乙氧羰基-1,4-二氢吡啶-4-膦酸二甲酯的制备
将吡啶(316g,4.0mol)的乙腈(4.0L)溶液冷却至0℃,然后在约1.25小时内加入氯甲酸乙酯(434g,4.0mol),同时保持温度不超过5℃。将形成的黄色悬浮液于-5℃至0℃搅拌1.5小时。然后将其冷却至-25℃并在1小时内加入亚磷酸三甲酯(496.3g,4.0mol),保持温度不超过-20℃。将其升温至室温过夜。于低于70℃的温度下减压蒸除溶剂,将得到的油溶于二氯甲烷(2.8L)并依次用水(2L)、5%HCl(2L)、2%K2CO3溶液(1L)和水(2L)洗涤至中性pH。然后将溶液减压浓缩得到黄色油(645g,62%),经毛细管GC分析,其为N-乙氧羰基-1,4-二氢吡啶-4-膦酸二甲酯和N-乙氧羰基-1,2-二氢吡啶-2-膦酸二甲酯的1∶1的混合物。将该物质直接用于随后的氢化,因为在尝试蒸馏时造成了分解。
B.
氢化成N-乙氧羰基哌啶-4-膦酸二甲酯
将上述物质在甲醇(1600ml)中用5%Pd/C(80g 50%用水润湿的)在室温和15巴的氢气下进行氢化。氢化完全后,将溶液在15巴氢气下搅拌20小时。放空并洗净氢气后,滤除催化剂然后减压蒸除溶剂以定量收率得到N-乙氧羰基哌啶-4-膦酸二甲酯和N-乙氧羰基哌啶-2-膦酸二甲酯的1∶1的混合物。将该混合物通过分馏进行分离,1,4-异构体集中在后馏份(b.p.130-40℃/1毫巴)中。由此得到130g纯度为98%的N-乙氧羰基哌啶-4-膦酸二甲酯。
实施例5
通过将243mL(0.389mol)1.6M正丁基锂的己烷溶液于-20℃至-5℃下加入到二异丙基胺(40.8g,0.404mol)的二甲苯:THF(210:360ml)溶液中生成二异丙基氨基锂(LDA)溶液。在该温度下搅拌0.75小时后,用0.5小时的时间在-30℃至-20℃的温度下加入膦酸二甲酯(100g,0.377mol)的二甲苯(105ml)溶液。将溶液在该温度下搅拌1.5小时,然后用0.75小时的时间在-30℃至-20℃的温度下加入8-氯-三环-酮(85.0g,0.349mol)的THF(255ml)溶液。使其在0.75小时内升温至-10℃,然后用乙酸(38g,0.633mol)终止反应。将黄色的溶液加热至回流,加入二甲苯(170ml)并蒸除THF/己烷(约910ml)至溶液的温度为134℃。将其回流1.5小时,冷却至80℃,加入水(340ml)。将其继续回流1小时然后分液(通过用二氯甲烷萃取从水相中回收得到22%未分解的β-羟基膦酸酯中间体)。将二甲苯相用水洗涤然后减压蒸除溶剂。将残余物用乙腈重结晶,分两批得到94.7g(71%收率)纯净的氯雷他定。
虽然结合上述具体的实施方案对本发明进行了描述,但对其进行的多种替换、修饰和改变对于本领域普通技术人员是显而易见的。所有这些替换、修饰和改变均包括在本发明的实质和范围之内。
Claims (15)
1.制备下式化合物的方法:

其中R1选自:烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代,所述方法包括如下步骤:
(a)将下式的酮

与下式的负碳离子反应

其中R1如上所定义,R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基;
(b)将步骤(a)的反应混合物用质子化试剂处理形成下式的β-羟基中间体
其中R1、R2和R3如上所定义;
(c)将质子化了的步骤(b)产物热分解形成式(I)化合物。
2.权利要求1的方法,其中R1是烷基。
3.权利要求1的方法,其中R1是乙基。
4.权利要求3的方法,其中RA是烷基。
5.权利要求4的方法,其中R2和R3均是-ORA。
6.权利要求5的方法,其中R2和R3均是-OC2H5。
7.权利要求6的方法,其中的负碳离子是通过将下式化合物与有机锂碱反应形成的

8.权利要求7的方法,其中的有机锂碱是二异丙基氨基锂。
9.制备下式化合物的方法:

所述方法包括如下步骤:
(a)将下式的酮

与下式的负碳离子反应

其中R1选自:烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代;R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基;
(b)将步骤(a)的反应混合物用质子化试剂处理形成下式的β-羟基中间体;

其中R1、R2和R3如上所定义;
(c)将质子化了的步骤(b)产物热分解形成下式化合物
其中R1如上所定义;然后
(d)将式(I)化合物转变成式(IV)化合物。
10.权利要求9的方法,其中R1是烷基。
11.权利要求10的方法,其中R1是叔丁基。
12.权利要求11的方法,其中RA是烷基,步骤(d)是通过将式(I)化合物用酸处理完成的。
13.权利要求12的方法,其中R2和R3均是-OC2H5。
14.下式化合物
其中R1选自:除了乙基之外的烷基、链烯基、链炔基、芳基、芳烷基、环烷基和环烷基烷基,R1任选地被选自卤素、-OH、烷基、烷氧基或-CF3的取代基所取代;R2和R3彼此独立地选自-ORA和-RA,其中RA是烷基、苯基、取代的苯基、环烷基、取代的环烷基、环烷基烷基或取代的环烷基烷基。
15.权利要求14的化合物,其中R1是除了乙基之外的烷基并且RA是烷基。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21617098A | 1998-12-18 | 1998-12-18 | |
| US09/216170 | 1998-12-18 | ||
| US09/216,170 | 1998-12-18 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1334810A CN1334810A (zh) | 2002-02-06 |
| CN1257166C true CN1257166C (zh) | 2006-05-24 |
Family
ID=22805995
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB998159670A Expired - Lifetime CN1257166C (zh) | 1998-12-18 | 1999-12-16 | 制备具有抗组胺活性的三环化合物的方法 |
Country Status (16)
| Country | Link |
|---|---|
| EP (1) | EP1140899B1 (zh) |
| JP (1) | JP4558942B2 (zh) |
| CN (1) | CN1257166C (zh) |
| AR (1) | AR021715A1 (zh) |
| AT (1) | ATE249453T1 (zh) |
| AU (1) | AU2157100A (zh) |
| CA (1) | CA2355050C (zh) |
| CO (1) | CO5251403A1 (zh) |
| DE (1) | DE69911252T2 (zh) |
| ES (1) | ES2207329T3 (zh) |
| HK (1) | HK1039334B (zh) |
| MY (1) | MY119697A (zh) |
| PE (1) | PE20001334A1 (zh) |
| TW (1) | TWI225056B (zh) |
| WO (1) | WO2000037457A1 (zh) |
| ZA (1) | ZA200104631B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113135893A (zh) * | 2021-06-21 | 2021-07-20 | 北京鑫开元医药科技有限公司 | 苯并环庚烷并吡啶化合物、其制备方法及其用途 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20012308A1 (it) * | 2001-11-05 | 2003-05-05 | Zambon Spa | Processo per la preparazione dell'estere etilico dell'acido 4-(8-cloro-5,6-diidro-11h-benzo-5,67-cicloepta-1,2-b-piridin-11-ilidene)-1-piper |
| CN106478595B (zh) * | 2016-09-18 | 2019-05-21 | 西安交通大学 | 氯雷他定晶型及其制备方法和用途 |
| CN113135899B (zh) * | 2021-06-21 | 2021-11-23 | 北京鑫开元医药科技有限公司 | 苯并环庚烷并吡啶化合物、其制备方法及其用途 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4282233B1 (en) * | 1980-06-19 | 2000-09-05 | Schering Corp | Antihistaminic 11-(4-piperidylidene)-5h-benzoÄ5,6Ü-cyclohepta-Ä1,2Ü-pyridines |
| US4355036A (en) * | 1980-06-19 | 1982-10-19 | Schering Corporation | Tricyclic-substituted piperidine antihistamines |
| EP0208855B1 (en) * | 1985-05-13 | 1991-03-06 | Schering Corporation | process for preparing piperidylidene dihydrodibenzo(a,d)cycloheptenes and aza derivatives thereof, compounds obtained by such process and the use of such compounds for preparing useful pharmaceutical compositions |
-
1999
- 1999-12-16 AR ARP990106458A patent/AR021715A1/es not_active Application Discontinuation
- 1999-12-16 CA CA002355050A patent/CA2355050C/en not_active Expired - Lifetime
- 1999-12-16 HK HK02100883.8A patent/HK1039334B/zh not_active IP Right Cessation
- 1999-12-16 JP JP2000589529A patent/JP4558942B2/ja not_active Expired - Fee Related
- 1999-12-16 DE DE69911252T patent/DE69911252T2/de not_active Expired - Lifetime
- 1999-12-16 AT AT99965895T patent/ATE249453T1/de not_active IP Right Cessation
- 1999-12-16 PE PE1999001278A patent/PE20001334A1/es not_active Application Discontinuation
- 1999-12-16 AU AU21571/00A patent/AU2157100A/en not_active Abandoned
- 1999-12-16 TW TW088122098A patent/TWI225056B/zh not_active IP Right Cessation
- 1999-12-16 WO PCT/US1999/027936 patent/WO2000037457A1/en not_active Ceased
- 1999-12-16 ES ES99965895T patent/ES2207329T3/es not_active Expired - Lifetime
- 1999-12-16 CN CNB998159670A patent/CN1257166C/zh not_active Expired - Lifetime
- 1999-12-16 EP EP99965895A patent/EP1140899B1/en not_active Expired - Lifetime
- 1999-12-16 CO CO99078840A patent/CO5251403A1/es not_active Application Discontinuation
- 1999-12-17 MY MYPI99005547A patent/MY119697A/en unknown
-
2001
- 2001-06-06 ZA ZA200104631A patent/ZA200104631B/en unknown
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113135893A (zh) * | 2021-06-21 | 2021-07-20 | 北京鑫开元医药科技有限公司 | 苯并环庚烷并吡啶化合物、其制备方法及其用途 |
| CN113135893B (zh) * | 2021-06-21 | 2022-02-11 | 北京鑫开元医药科技有限公司 | 苯并环庚烷并吡啶化合物、其制备方法及其用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4558942B2 (ja) | 2010-10-06 |
| JP2002533334A (ja) | 2002-10-08 |
| ZA200104631B (en) | 2002-09-06 |
| AU2157100A (en) | 2000-07-12 |
| WO2000037457A1 (en) | 2000-06-29 |
| MY119697A (en) | 2005-06-30 |
| DE69911252D1 (de) | 2003-10-16 |
| PE20001334A1 (es) | 2000-12-01 |
| CA2355050C (en) | 2007-04-17 |
| TWI225056B (en) | 2004-12-11 |
| CO5251403A1 (es) | 2003-02-28 |
| HK1039334B (zh) | 2003-12-05 |
| CA2355050A1 (en) | 2000-06-29 |
| EP1140899A1 (en) | 2001-10-10 |
| ATE249453T1 (de) | 2003-09-15 |
| DE69911252T2 (de) | 2004-07-01 |
| ES2207329T3 (es) | 2004-05-16 |
| HK1039334A1 (zh) | 2002-04-19 |
| CN1334810A (zh) | 2002-02-06 |
| AR021715A1 (es) | 2002-07-31 |
| EP1140899B1 (en) | 2003-09-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1582272A (zh) | 制备5-(3-氰基苯基)-3-甲酰基苯甲酸化合物的方法 | |
| JP4048119B2 (ja) | 2−(4−クロロベンゾイルアミノ)−3−[2(1h)−キノールリノン−4−イル]プロピオン酸の製造方法 | |
| KR100794091B1 (ko) | 2-에톡시-3-(4-히드록시페닐)프로판산 및 그의 유도체의 (s)-거울상이성질체의 제조 방법 | |
| CN1257166C (zh) | 制备具有抗组胺活性的三环化合物的方法 | |
| JP2004504319A (ja) | 4−フェニルピペリジン誘導体の新規製造方法 | |
| KR100881617B1 (ko) | 아토바스타틴 제조를 위한 중간체 및 그의 제조방법 | |
| JP6148351B2 (ja) | 置換ピロリジン−2−カルボキサミドの不斉合成 | |
| JPH032134B2 (zh) | ||
| CN1180353A (zh) | 2-取代的苯并[b]噻吩类化合物及其中间体的制备方法 | |
| JP4874122B2 (ja) | トルテロジンを得るための方法 | |
| CN1128989A (zh) | N-取代的氮杂环羧酸及其酯 | |
| JPH0345077B2 (zh) | ||
| US6271378B1 (en) | Process for preparing tricyclic compounds having antihistaminic activity | |
| CN1599736A (zh) | 4-(8-氯-5,6-二氢-11H-苯并[5,6]环庚并[1,2-b]吡啶-11-亚基)-1-哌啶羧酸乙基酯(氯雷他定)的制备方法 | |
| CN1086699C (zh) | 合成苯并[b]噻吩的方法 | |
| CN1093853C (zh) | 制备(s)-4-氨基-庚-5,6-二烯酸的新方法及其中间体 | |
| JP4157361B2 (ja) | 9−スピロフルオレン化合物の製造方法 | |
| HK1048983A1 (zh) | 邻氯甲基苯基二羟乙酸衍生物的制备方法 | |
| JP2001302658A (ja) | 3−イソクロマノン類の製造方法 | |
| JP3819444B2 (ja) | 1−アルキルヘキサハイドロアゼピン−4−オンの製造法 | |
| JP4564135B2 (ja) | 高純度フェノチアジン化合物とその製造方法、およびその中間体の製造方法、並びにその中間体の原料の水和物と新規結晶 | |
| JP3774601B2 (ja) | フェニルピペリジン類の製造法 | |
| JPH08325228A (ja) | ビシクロヘキサンアミン誘導体の製造法 | |
| JP3569877B2 (ja) | m−置換−α−ヒドロキシメチルスチレン誘導体の製造法および3−クロロ−α−ブロモスチレン | |
| CN1668597A (zh) | 生产2,3,6-三烷基-8-氟-4-喹啉衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| C10 | Entry into substantive examination | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C56 | Change in the name or address of the patentee |
Owner name: MSD CORP. Free format text: FORMER NAME: SCHERING CORP (US) |
|
| CP01 | Change in the name or title of a patent holder |
Address after: New jersey, USA Patentee after: MERCK SHARP & DOHME Corp. Address before: New jersey, USA Patentee before: SCHERING Corp. |
|
| CX01 | Expiry of patent term |
Granted publication date: 20060524 |
|
| CX01 | Expiry of patent term |