CN1265643A - 邻氨基苯甲酸类似物 - Google Patents
邻氨基苯甲酸类似物 Download PDFInfo
- Publication number
- CN1265643A CN1265643A CN98807943A CN98807943A CN1265643A CN 1265643 A CN1265643 A CN 1265643A CN 98807943 A CN98807943 A CN 98807943A CN 98807943 A CN98807943 A CN 98807943A CN 1265643 A CN1265643 A CN 1265643A
- Authority
- CN
- China
- Prior art keywords
- hydrogen
- amino
- aryl
- compound
- haloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/63—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/18—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/10—Systems containing only non-condensed rings with a five-membered ring the ring being unsaturated
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Pulmonology (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及式(Ⅰ)化合物或其药物学上可接受的盐,其中R1、R2和R3相互独立地是氢、硝基、氰基、C1-10卤代烷氧基、氨基、C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酸氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基和C6-12芳基;前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;R4、R5和R6相互独立地是氢、卤素、硝基、氰基、C1-10烷氧甲酰基、C1-10卤代烷氧基、氨基C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-10烷基和C6-12芳基;R7是氢、金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑、异噻唑、呋喃、噻吩、2H-吡咯、吡咯、2-吡咯啉、3-吡咯啉、咪唑、吡唑、1,2,3-噁二唑或1,2,3-三唑,它们通过钾通道和氯化物通道调节来治疗与平滑肌收缩有关的病症。
Description
本发明涉及一系列新的具有药物活性的由邻氨基苯甲酸衍生的酰胺(I),它们的制备方法,含有它们的药物组合物,以及它们在通过钾通道和氯化物通道调节来治疗与平滑肌收缩有关的病症方面的用途。这些病症包括但不限于尿失禁、哮喘、早产、过敏性肠道综合症、充血性心力衰竭、心绞痛和外周血管病。
钾通道调节一直处于对控制休止细胞膜潜能和影响细胞应激性的最新研究的最前沿。在几种最新的综述中存在各种不同的离散钾通道,并且根据其结构、功能、药理学性能和选通机理已经将它们完全归类[Rudy,B.Neuroscience 1988,25,729-749;Atwal,K.,《医药研究综述》(Medicinal Research Review)1992,12,569-591;Gopalakrischnan,M.等《药物发展研究》(Drug DevelopmentResearch)1995,I,391-406;Edwards,G.等Exp.Opin.Invest.Drugs 1996,5(11),1453-1464]。正在极力开发钾通道调节在心血管病症、代谢紊乱、中枢神经系统紊乱、支气管性气喘和膀胱过敏中的治疗潜能。
对氯化物通道的研究兴趣正在快步增长[Strange,K.等KidneyInternational 1995,48,994-1003;Franciolini,F.等Biochimica et Biophysica Acta 1990,247]。可能适合于氯化物通道调节的各种疾病状况包括支气管性气喘、心脏心率失常、胰囊性纤维变性和肾病。
Harita等在日本专利申请49-102692中公开了制备间取代的芳族酰胺羧酸衍生物的方法,并且同样是基于日本专利申请49-42465中的生产芳族肉桂酸(cinammic)衍生物的方法上。几个专利和专利申请主要集中到具有请求保护的抗变应性/止喘/抗组胺活性的试剂上:Sato等在日本专利申请57-179976中报道了一组邻氨基苯甲酸衍生物(强调Tranilast作为抗变应性剂)。Aoyanagi等在日本专利申请58-79436中要求保护这些邻氨基苯甲酸酯,该专利申请公开了制备邻氨基苯甲酸衍生物的制备方法;同样相关的是匈牙利专利HU200 996B,其强调制备几个Tranilast类似物的方法;Yukihiko在日本专利J 6 0019 754也提出制备邻氨基苯甲酸衍生物的方法,其中苯乙烯基部分被严格局限于烷氧基、羟基或酰氧基。
此外,Tsumoro等的日本专利J0 2218-654-A公开了一组用作反转录酶抑制剂的氨基苯甲酸衍生物。同样Tsumoro等的专利J6 0097-946-A公开了一系列取代的羧酰胺衍生物,其表现出作为白细胞三烯拮抗剂和磷脂酶抑制剂的活性。
发明描述
根据本发明,这里提供下式表示的化合物:
其中:
R1、R2和R3相互独立地是氢、硝基、氰基、C1-10卤代烷氧基、氨基、C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基(alkanoyl)、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基和C6-12芳基;前提条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、C1-10烷氧甲酰基(carboalkoxy)、C1-10卤代烷氧基、氨基C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-10烷基和C6-12芳基;
R7是氢、金属阳离子、乙酰氨基、烷氧基乙酰基(acetoyl)或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑、异噻唑、呋喃、噻吩、2H-吡咯、吡咯、2-吡咯啉、3-吡咯啉、咪唑、吡唑、1,2,3-噁二唑或1,2,3-三唑。
在本发明更优选的方面包括式(I),其中
R1、R2和R3相互独立地是氢、硝基、氰基、全卤代烷氧基、氨基、C1-10烷基氨基、C1-10二烷基氨基、C6-12芳基氨基、C1-10芳烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C6-12芳基亚磺酰氨基、C2-10烷基甲酰氨基、C6-12芳基甲酰氨基、C2-10链烷酰基、C6-12芳酰基、C2-22芳烷酰基、C1-10烷基磺酰基、C1-10全卤代磺酰基、C6-12芳基磺酰基、C2-22芳烷基磺酰基、C1-10羧基、C1-10卤代烷基;C1-10全卤代烷 基,芳基,卤代芳基,全卤代芳基,C1-10芳烷基部分,前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、烷氧甲酰基、全卤烷氧基、氨基、C1-10烷基氨基、C1-10二烷基氨基、芳基氨基、C1-10芳烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C6-12芳基亚磺酰氨基、C2-10烷基甲酰氨基、C6-12芳基甲酰氨基、C2-10链烷酰基、C6-12芳酰基、C2-22芳烷酰基、C1-10烷基磺酰基、C1-10全卤磺酰基、C6-12芳基磺酰基、C2-22芳烷基磺酰基、C1-10羧基、C1-10直链烷基、C1-10支链烷基、C3-10环烷基或双环烷基、C1-10卤代烷基、C1-10全卤烷基、C2-12链烯基(单烯属的或多烯属)、芳基、卤代芳基、全卤芳基、C1-10芳烷基;
R7是氢、碱金属阳离子、碱土金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑或异噻唑。
本发明更优选的方面包括式(1)化合物,其中:
R1、R2、R3、R4、R5和R6具有上述含义;
R7选自氢、金属阳离子、选自下列结构的部分: 其中:
其中:
R9、R10、R11和R12相互独立地是氢、C1-10直链烷基、C1-10支链烷基、C3-10环基或二环基、芳基或C1-10芳烷基;和
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑或异噻唑。
本发明最优选的方面是包括式(I)的化合物,其中R7可以是氢、或上述金属阳离子。
应当理解,当R1、R2、R3、R4、R5、R6、R7或通过X、Y和Z形成的环体系包括不对称碳原子时,式(I)化合物的定义包括具有下述活性的所有可能的立体异构体及其混合物。特别地,它包括具有标明活性的外消旋体及任何光学异构体。通过标准分离技术或对映体特定合成,可以制得纯净形态的光学异构体。应当理解,本发明包括式(I)化合物所有晶型。本发明的碱性化合物的药物学上可接受盐是那些由下列有机和无机酸衍生的:乳酸、柠檬酸、乙酸、酒石酸、富马酸、琥珀酸、马来酸、丙二酸、氢氯酸、氢溴酸、磷酸、硝酸、硫酸、甲磺酸和类似的可接受的已知酸。这里,如果R1、R2、R3、R4、R5、R6或通过X、Y和Z形成的环体系包括羧基,那么本发明中可以与碱例如碱金属(Na、K、Li)或碱土金属(Ca或Mg)形成该化合物的盐。
本发明也提供制备式(I)化合物的方法。制备方法示于反应式1至4中。
式(I)的异噁唑可以通过式(II)的化合物与适当的氧化腈(III)的氧化腈环加成反应以获得杂环化合物(IV)来制备的(反应式1)。如上所示,皂化反应获得中间产物羧酸(V)
反应式1其中R1、R2和R3与上述R1、R2和R3相同,并且A任选地选自基团R1、R2、R3、R4、R5、R6或R7。
式(I)的噁唑可以经适合的苯甲酰氯(VI)与异氰基乙酸甲酯(VII)进行缩合反应以获得杂环化合物(VIII)来制备的(反应式2)。如上所述,通过皂化反应获得中间产物羧酸(IV)
反应式2这里R1、R2和R3与上述R1、R2和R3相同。
在更一般的意义上,式(I)化合物也可以通过适合的活化杂或碳环烯(IX)与合适的偶合伙伴(X)反应获得一系列式(XI)化合物,其中活化中间产物的典型实例是M为邻三氟甲磺酸盐和M’是三烷基锡烷,或者M是溴化物或碘化物,而M是硼酸。
反应式3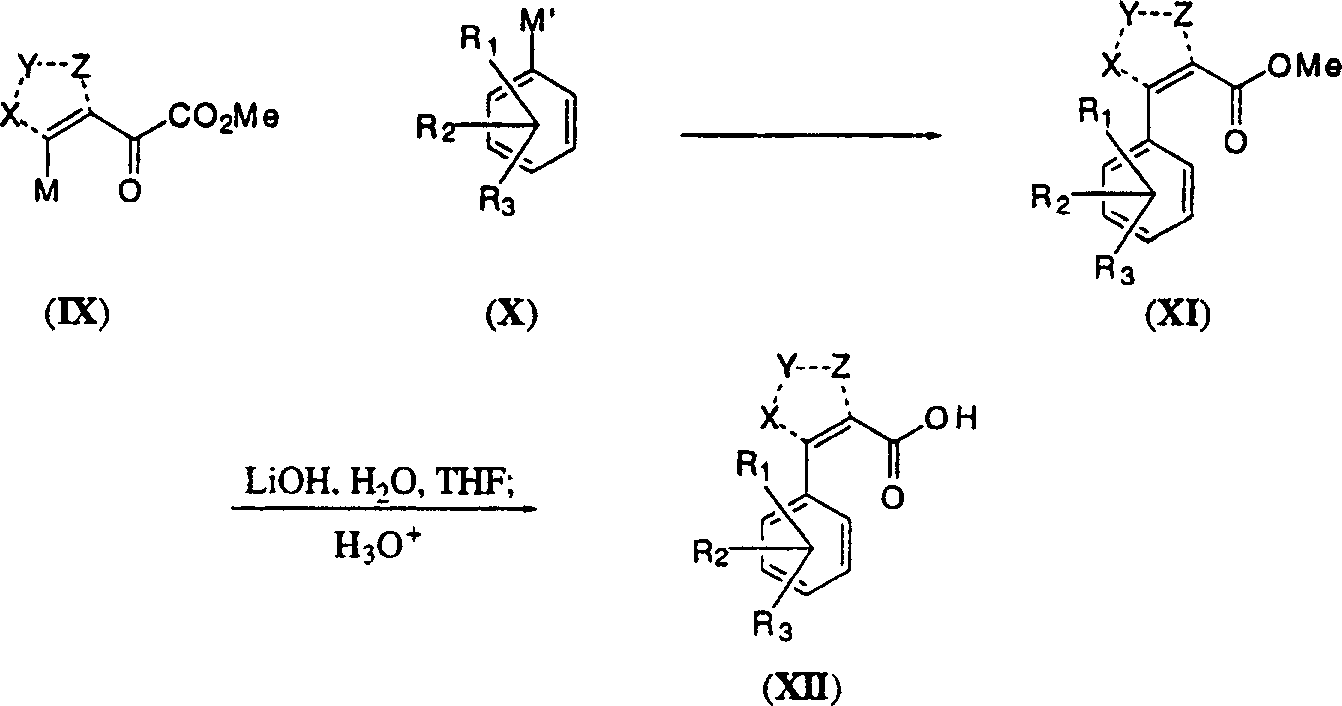 其中X、Y、X、R1、R2和R3与上述X、Y、Z、R1、R2和R3相同。如上所述,通过皂化反应将获得中间产物羧酸(XII)。
其中X、Y、X、R1、R2和R3与上述X、Y、Z、R1、R2和R3相同。如上所述,通过皂化反应将获得中间产物羧酸(XII)。
随后使用一种下面规定的偶合步骤(方法A:(COCl)2、cat.DMF、CH2Cl2,然后在邻氨基苯甲酸于氢氧化钠的溶液中加入该纯的酰氯;方法B:二异丙基碳化二亚胺、DMAP、CH2Cl2,然后加入邻氨基苯甲酸甲酯;或方法C:(COCl)2、cat.DMF、CH2Cl2,或SOCl2,随后用三乙胺和邻氨基苯甲酸甲酯处理该纯的酰基氯)使羧酸中间产物(V)、VI和(XII)偶合(反应式4)于式(XIII)的适当衍生的邻氨基苯甲酸的胺,获得式(I)的酰胺,如果使用酯(XIII),那么最终皂化获得游离酸(R7=H)。
反应式4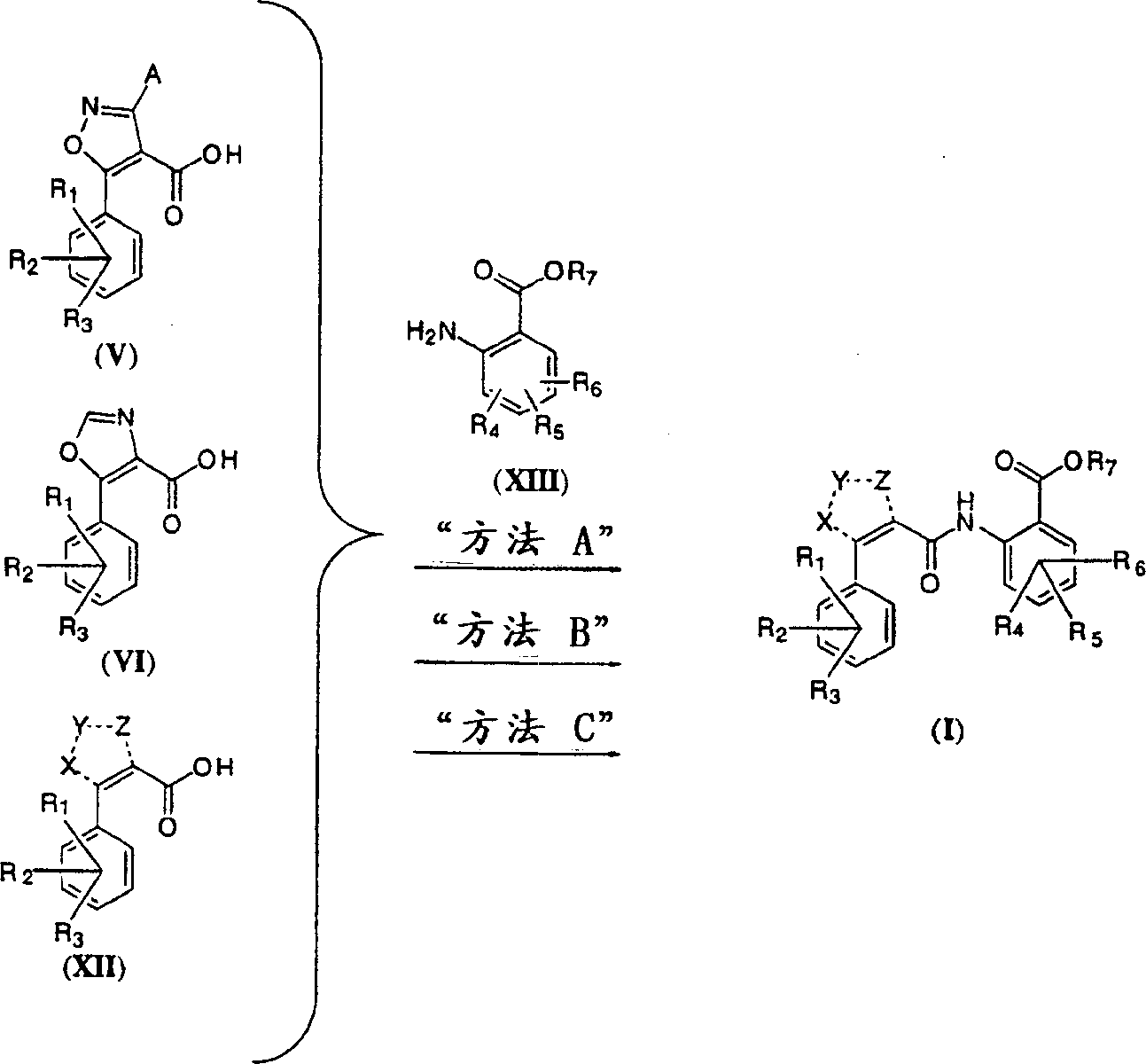
除CH2Cl2外,上述反应可以在低于环境温度下在非质子传递溶剂例如二乙醚、二氯乙烷、二噁烷或THF中进行。如果使用氢氧化钠作为碱,那么其它也有能力的无机碱是氢氧化锂或氢氧化钾等。同样三乙胺可以任选地被任何三烷基胺代替。
如上所述,已经发现式(1)化合物及其药物学上可接受的盐可以松弛平滑肌。因此,它们可以用于治疗与平滑肌收缩有关的紊乱,紊乱包括尿道平滑肌过度收缩(如尿失禁),或胃肠道平滑肌过度收缩(如过敏性大肠综合症),哮喘和脱发。另外,式(1)化合物为钾通道活化剂,用于治疗外周血管病、充血性心衰竭、中风、焦虑症、脑缺氧症及其它神经变性性紊乱。此外,式(1)和(II)化合物为氯化物通道封阻剂,用于治疗上述紊乱。
本发明化合物的特征在于它们在体内有效松弛平滑肌的性能。本发明化合物通过钾通道活化和/或氯化物通道封阻表现出它们松弛平滑肌的活性(表1)。对比化合物Tranilast未表现出有效或膀胱选择性平滑肌松弛性。
因此本发明提供药用组合物,其包括本发明化合物与药物学上可接受的载体组合或联合。更确切地说,本发明提供了包括有效剂量的本发明化合物以及药物学上可接受载体的药用组合物。
该组合物优选适于口服给药,但其也可采用其它方式给药,如对心衰患者非肠道给药。
为了获得给药的连贯性,本发明组合物优选为单位剂量形式。适当的单位剂量形式包括片剂、胶囊以及装于小囊或玻璃瓶中的粉剂。这些单位剂量形式可含有0.1至100毫克的本发明化合物,优选2至50毫克。更优选的单位剂量含有5至25毫克的本发明化合物。本发明化合物可以0.01至100毫克/千克的剂量范围口服给药,或优选0.1至10毫克/千克的剂量范围。这些组合物每日可给药1至6次,更常见为每日1至4次。
本发明组合物可用常用的赋形剂如填充剂、崩解剂、粘合剂、润滑剂、香味剂等来制成制剂。它们用常规的方式,如以类似于用于已知抗高血压剂、利尿剂及β-阻断剂的方式来制备。
本发明还提供了用作活性治疗物质的本发明化合物。式(I)化合物特别地用于诱导平滑肌松弛。
本发明还提供用作治疗哺乳动物包括人的平滑肌紊乱方法,其包括给患病的哺乳动物有效剂量的本发明化合物或药用组合物。
用下列实施例来说明而不是限制本发明代表性化合物的制备方法。
实施例
实施例1
2-{[2-(4-三氟甲基-苯基)-环戊-1-烯羰基]-氨基}-苯甲酸
步骤1)制备(邻-三氟甲基磺酰基)-环戊烯-2-酸甲酯
在0℃下,在2-甲酯基环戊酮(4.00mL,32.2毫摩尔)于1,2-二氯乙烷(50mL)的均匀溶液中加入三乙胺(5.84mL,41.9毫摩尔)。使获得的混合物保持在0℃下,通过注射泵在0.5小时向其中加入三氟甲烷磺酰酐(6.50mL,38.7毫摩尔)。完全完成加入之后,再保持该温度2小时,随后用EtOAc(50mL)稀释,经短的二氧化硅垫过滤,用30%EtOAc-己烷洗脱,浓缩并进行快速色谱纯化(使用10%EtOAc-己烷洗脱),获得6.14克(69%)浅黄色油:
1H NMR(CDCl3)δ3.79(s,3H),2.68-2.78(m,4H),1.88-2.08(m,2H).
步骤2)制备2-[4-(三氟甲基-苯基)]-环戊烯酸甲酯
在上述烯醇triflate(1.77克,6.44毫摩尔)、4-(三氟甲基)-苯基三甲基锡烷[Morlein,S.M.J.Organomet.Chem.1987,319,29-39](1.81克,5.86毫摩尔)和无水氯化锂(745毫克,17.6毫摩尔)在无水二噁烷的混合物中加入四-(三苯基膦)钯(0)(203毫克,0.176毫摩尔)。将获得的混合物加热至110℃,然后搅拌12小时。在冷却至室温之后,将反应混合物浓缩为淤浆,溶解在乙醚(100mL)中,经短的二氧化硅垫过滤,进一步浓缩,然后进行快速色谱纯化(使用5%乙醚-石油醚),获得1.155克(73%)透明的无色油:
1H NMR(CDCl3)δ7.50(ABq,4H),3.63(s,3H),2.85(m,4H),2.02(m,2H).
步骤3)制备2-[4-(三氟甲基-苯基)]-环戊烯酸
在室温下,在上述甲酯于THF(20毫升)的均匀溶液中加入1.00NLiOH(17.8mL,17.8毫摩尔)。将所获得的二相混合物剧烈搅拌16小时,随后通过旋转蒸发除去使用的挥发物。将剩余的含水溶液用乙醚洗涤(3×100mL),使用浓HCl(1.73mL)酸化至pH2,用乙醚(300mL)分配。然后含水相用固体NH4Cl饱和并再次萃取(2×150mL)。合并的有机萃取物经硫酸镁干燥,用Norite处理,经硅藻土过滤,并浓缩至固体。用乙醚-己烷研磨,随后过滤和真空干燥,获得1.38克(91%)纯白色固体。
1H NMR(DMSO-d6)δ12.37(s,1H),7.65(ABq,4H),2.75-2.91(m,4H),1.92-2.02(m,2H).
步骤4)制备2-{[2-(4-三氟甲基-苯基)-环戊-1-烯羰基]-氨基}-苯甲酸
在0℃下,在上述羧酸(500毫克,1.95毫摩尔)和无水DMF(4滴)于无水CH2Cl2(3mL)的不均匀混合物中滴加草酰氯(340μL,3.90毫摩尔)。将获得的混合物加热至室温,并搅拌2.5小时,随后浓缩为浅棕色油,并在真空中清除过剩的草酰氯。然后,在5℃下,在邻氨基苯甲酸(535毫克,3.90毫摩尔)于2.5N含水NaOH(3.12mL,7.80毫摩尔)的均匀溶液中加入纯的酰基氯,结果立即形成白色沉淀。然后将反应混合物加热至室温,随后用最低量的水稀释以便于搅拌,该搅拌持续进行1.5小时。通过加入浓盐酸(0.75mL)将混合物酸化至pH2,用2.0N盐酸稀释,并搅拌1.5小时。过滤悬浮液,随后用水洗涤,空气干燥,随后用MeOH重结晶,获得118毫克(16%)的纯白色晶体状固体:mp 225.1-225.9℃;1H NMR(DMSO-d6)d 13.51(br s,1H),11.28(s,1H),8.56(dd,1H),7.92(dd,1H),7.60(ABq,4H),7.12(ddd,1H),2.91(m,4H),2.03(m,2H);IR(KBr)3121,2966,1700,1662,1636,1586,1528,1450,1381,1321,1205,1163,1131,1066,1017,837,755,694cm-1;MS(m/z)375[M+].
元素分析C20H16F3NO3:
计算值:C 64.00;H 4.30;N 3.73.
实测值:C 63.79;H 4.08;N 3.57.
实施例2
2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸
步骤1)制备5-(4-三氟甲基-苯基)-噁唑-4-羧酸甲酯
在4-三氟甲基苯甲酰氯(8.42克,40.4毫摩尔)和三乙胺(12.3克,122毫摩尔)于无水THF中的混合物中加入异氰基乙酸甲酯(3.60克,36.3毫摩尔)。将获得的混合物在室温下搅拌72小时,随后通过旋转蒸发除去所有挥发物,将获得的残渣分配在EtOAc(300mL)和水(100mL)中。使用饱和NaHCO3(100mL)萃取有机相,用盐水(100mL)洗涤,经硫酸镁干燥,用Norite处理,经硅藻土过滤,然后浓缩,获得9.27克(94.2%)棕色固体,该固体无需进一步纯化便可用于下一步反应。
步骤2)制备5-(4-三氟甲基-苯基)-噁唑-4-羧酸
以类似于实施例1步骤3的方式,由商业上可获得的5-(4-三氟甲基-苯基)-噁唑-4-羧酸甲酯制备标题中间产物(83%)。
步骤3)2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸甲酯
以类似于实施例1步骤4的方式,由上述羧酸制备5-(4-三氟甲基-苯基)-噁唑-4-碳酰氯。以类似于实施例1步骤1的方式,由上述酰基氯和邻氨基苯甲酸甲酯制备标题中间产物。进行快速色谱纯化(用17%乙醚-己烷洗脱),获得白色固体(92%)。
步骤4)制备2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸
以类似于实施例1步骤3的方式,由上述甲酯制备标题化合物(45%):熔点231至232℃;
1H NMR(DMSO-d6)δ13.53(brs,1H),12.51(s,1H),8.74-8.78(m,2H),8.16(ABq,4H),8.02(dd,1H),7.62(ddd,1H),7.21(ddd,1H);IR(KBr)3449,3245,3080,3022,2647,2559,1669,1607,1585,1519,1466,1451,1410,1327,1261,1151,1126,1060,1017,993,880,848792,761,662cm-1;MS(m/z)376[M+].
元素分析:C18H11F3N2O4
计算值:C 57.46;H 2.95;N 7.45
实测值:C 57.62;H 3.19;N 7.01
实施例3
2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸锂盐
在氮气氛下,在室温下,在2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸(500毫克,1.33毫摩尔)和氢化锂粉的固体混合物中加入THF(15mL,从二苯甲酮羰游基钠中蒸馏出)。在回流下将不均匀的混合物加热40小时,获得大量白色沉淀,随后冷却至室温,用THF(85毫升)稀释,经硅藻土过滤,并浓缩至白色固体。然后该固体与乙醚(40mL,从二苯甲酮羰游基钠中蒸馏出)一起研磨72小时,获得细的白色悬浮液,在氮气氛中过滤,用乙醚(40mL)洗涤,最后在80℃下。在高真空下干燥,获得311毫克(61%)白色粉末:熔点356.3至357.3℃:
1H NMR(DMSO-d6)δ15.13(s,1H),8.69(s,1H),8.63(dd,1H),8.42(d,1H),8.01(dd,1H),7.89(d,1H),7.30(ddd,1H),7.00(ddd,1H);IR(KBr)3412,3140,3091,3062,2920,1651,1590,1527,1449,1375,1324,1160,1126,1073,1062901,883,780,759,680cm-1;MS(m/z)389[(M+Li)+].
元素分析:C18H10F3N2O4Li
计算值: C 56.51;H 2.62;N 7.33
实测值: C 56.02;H 2.44;N 7.28
实施例4
2-{[3-甲基-5-(4-三氟甲基-苯基)-异噁唑-4-羰基]-氨基}-苯甲酸
步骤1)制备3-[4-(三氟甲基-苯基)]-丙-2-酸甲酯
在三苯基膦(75.32克,287.1亳摩尔)和α,α,α-对-甲苯甲醛(10.00克,57.43毫摩尔)于CH2Cl2(250mL)中的均匀溶液中加入四溴化碳(47.62克,143.6毫摩尔)于CH2Cl2(50mL)中溶液。将反应混合物在0℃下搅拌5分钟,然后加热至室温,并再搅拌4.5小时,随后倾倒入剧烈搅拌的硅藻土(100克)于石油醚(1500mL)的淤浆中。然后,将获得的反应混合物搅拌0.5小时,经二氧化硅垫过滤,并浓缩至棕色油状物。将该油状物溶解在石油醚(150mL)中,用Norite处理,经硅藻土过滤,并浓缩以获得13.82克(73%)中间产物二溴链烯,其是透明的、无色油状物。
无需进一步纯化,将二溴链烯溶解在无色THF(115mL)中,冷却至-78℃。在1小时内,在该溶液中经注射器泵滴加2.5M丁基锂(18.4mL,46.0毫摩尔)。然后在-78℃下搅拌获得的混合物0.5小时,然后用氯甲酸甲酯(5.00mL,64.7毫摩尔),随后缓慢加热至室温,之后浓缩为油状残渣,并将其分配在乙醚(500mL)和水(250mL)中。用盐水(100mL)洗涤有机相,经硫酸镁干燥,用Norite处理,过滤、浓缩并进行快速色谱纯化(用2%乙醚-石油醚洗脱),获得3.13克(66%)白色固体:1H NMR(DMSO-d6)δ7.86(ABq,4H),3.79(s,3H)。
步骤2)制备3-甲基-5-(4-三氟甲基-苯基)-异噁唑-4-羧酸甲酯
在室温下,在3-[4-(三氟甲基-苯基)]-丙-2-酸甲酯(1.00克,4.38毫摩尔)和异氰酸苯酯(857μL,7.89毫摩尔)于无色苯(2.5mL)的均匀溶液中滴加硝基乙烷(315μL,4.38毫摩尔)和馏过的三乙胺(5滴)于苯(1.5mL)中的均匀溶液。将获得的混合物搅拌10分钟,这时产生沉淀。将反应混合物加热至回流12小时,随后冷却至室温,用乙醚稀释(100mL),过滤以除去所有固体。然后将滤液连续用1.0NNaOH(50mL)、水(50mL)和盐水(50mL)分配。经Na2SO4干燥有机相,用Norite处理,浓缩,然后进行快速色谱纯化(梯度洗脱:10∶15∶20%乙醚-石油醚),获得900毫克(72%)白色固体:1H NMR(DMSO-d6)δ8.00(Abq,4H),3.77(s,3H),2.44(s,3H)。
步骤3)制备3-甲基-5-(4-三氟甲基-苯基)-异噁唑-4-羧酸
在室温下,在上述甲酯(865毫克,3.19毫摩尔)于THF(10mL)的均匀溶液中加入1.00N LiOH(9.57mL,9.57毫摩尔)。将获得的二相混合物剧烈搅拌16小时,随后,通过旋转蒸发除去所有挥发物。剩余的水溶液用乙醚(3×50mL)洗涤,用浓HCl(0.93mL)酸化至pH2,并用乙醚(300mL)分配。然后用固体NH4Cl使含水相饱和,并再次萃取(2×150mL)。合并的有机萃取物经硫酸镁干燥,用Norite处理,经硅藻土过滤,浓缩至固体。与乙醚-己烷一起研制,随后过滤并在真空中干燥,获得735毫克(89%)白色结晶固体。
步骤4)制备2-{[3-甲基-5-(4-三氟甲基-苯基)-异噁唑-4-羰基]-氨基}-苯甲酸
在0℃下,在上述羧酸(1.00克,4.34毫摩尔)和无水DMF(2滴)于无水CH2Cl2(5mL)的不均匀混合物中滴加草酰氯(760μL,8.69毫摩尔)。将获得的混合物加热至室温,并搅拌2.5小时,随后浓缩为不均匀的黄色混合物,并在真空中清除过剩的草酰氯。然后,在5℃下,在邻氨基苯甲酸(1.19克,8.69毫摩尔)于2.5N含水NaOH(6.95mL,17.4毫摩尔)的均匀溶液中加入酰基氯,结果立即形成白色沉淀。然后将反应混合物加热至室温,随后用最低量的水稀释以便于搅拌,该搅拌持续进行1.5小时。通过加入浓盐酸(1.63mL)将混合物酸化至pH2,用2.0N盐酸稀释,并搅拌1.5小时。过滤悬浮液,随后用水洗涤,空气干燥,之后用MeOH重结晶。通过酸基团和(三甲基甲硅烷基)重氮甲烷的完全甲基化反应从未反应的原材料中分离出标题化合物。以类似于该实施例步骤2的方式,使分离的2-{[3-甲基-5-(4-三氟甲基-苯基)-异噁唑-4-羰基]-氨基}-苯甲酸甲酯皂化,获得标题化合物(60%):熔点204.6至205.5℃:
1H NMR(DMSO-d6)δ13.52(br s,1H),11.47(s,1H),8.42(d,1H),7.97(ABq,4H),7.93(dd,1H),7.64(ddd,1H),7.23(ddd,1H)2.47(s,3H);IR(KBr)3374,3118,2987,2656,1681,1659,1604,1585,1532,1441,1263,1166,1075,1014,905,847,762,719,701cm-1;MS(m/z)380[M+].
元素分析C19H13F3N2O4:
计算值:C 58.47;H 3.36;N 7.18.
实测值:C 58.22;H 3.24;N 7.02.
按照标准药学上可接受的测试方法用代表性化合物来测定本发明化合物的平滑肌舒张活性如下。
通过CO2窒息使Sprague-Dawley鼠(150-200克)昏迷,然后通过颈脱位对大鼠实施安死术。取出膀胱浸入包含下述组分(mM)的温(37℃)生理盐水水溶液(PSS)内:NaCl,118.4;4.7;KCl,4.7;CaCl2,2.5;MgSO4,4.7;H2O,1.2;NaHCO3,24.9;KH2PO4,1.2;葡萄糖,11.1;EDTA,0.023,用95%O2;2/5%CO2脱气;PH7.4。打开膀胱,然后切成宽1-2毫米,长7-10毫米肌条。随后将膀胱肌条于1.5克初静息张力下悬浮于10mL组织浴中。膀胱肌条用两个外科夹子固定,其中一个连在固定钩上,而另一连于等长肌力换能器上。用0.1μM卡巴胆碱激发之前,使通常显示出小的自发收缩的标本恢复1小时。然后洗去卡巴胆碱并使组织松弛至其静息作用张度。在1.5小时恢复期后,再向组织浴槽内加入15mM KCl。KCl浓度的增加导致叠加在基张力的小量增加之上的自发收缩的幅度有较大增加(收缩发生于预静息肌条上)。在该增强的收缩作用程度稳定之后,向组织浴内加入浓度递增的试验化合物或赋形剂。在30分钟激发时间的最后分钟测定化合物或赋形剂在各浓度下的收缩活性。
采用诱发抑制50%前药收缩活性所需要的浓度(IC50浓度)测定膀胱肌条所产生的等长肌力,IC50浓度根据浓度响应曲线计算得到。还测定了浓度小于或等于30μM的试验化合物的收缩活性的最大百分抑制率。
该研究结果示于表1中
表1
对分离的鼠膀胱肌条收缩的抑制和选择性的指示
IC50/μM IC50/μM IC50(A)/实施例# n 膀胱(B) n Aorta(A) IC50(B)1 5 11.04±4.04 -- -- --2 2 17.9±5.8 -- -- --3 4 17.5±12.4 2 7.35±0.25 0.424 2 11.9±3.95 -- -- --Tranilast§ 2 14.4±4.5 5 15.59±8.96 1.08§Tranilast是(E)-2-[3-(3,4-二甲氧基-苯基)-丙烯酰氨基]-苯甲酸。
*在30μM的百分抑制率
另外,按照Malmgrem描述的下列方法(A.Malmgrem,K.E.Andersson,C.Sjogren,P.O.Andersson,Effect ofPinacidil and Cromakalim(BRL 34915)on Bladder Function inRats with Detrusor Instability,J.Urol.142:1134,1989),测定了化合物抑制带有肥大膀胱的清醒雌鼠的肥大膀胱(迫肌)平滑肌的活性过度的能力,因此减轻了尿失禁。
使用Sprague-Dawley雌鼠,体重范围190至210克。每次准备25只动物。使膀胱肥大之后,每次实验用4至8只动物。
将化合物溶于PEG-200中,以5毫升/千克的体积胃插管或静脉内给药。初筛时,所有药物以10毫克/千克的任意剂量经口服施用给4只鼠为一组的各组别。
将动物用卤烷麻醉。中线切开,暴露膀胱和尿道,在有一不锈钢条(1毫米直径)存在下,在尿道近端周围扎上4-0丝带以产生部分闭塞。然后除去不锈钢条。将腹部区域用手术U型针封闭,给每只鼠施用150000单位的苄星青霉素C-R。让动物过6周以产生足够的膀胱肥大。6周之后,在卤烷麻醉下除去丝带,将带有环扣的导管(PE60)置于膀胱顶部内,用一囊带缝线系牢。将导管在皮肤下穿过,通过在颈背部开口取出。将切开的腹部缝合,使导管露出的一端封闭。为防止感染,给鼠注射苄星青霉素C-R(150000单位/鼠)。2天之后,将动物进行膀胱容积应力测试。将动物置于代谢箱(metabolic cages)中,将导管(用“T”形连接器)连接到Stathan应力传导器(P23Db型)和Harrard灌尿泵上。将连有一个力转换传导器(Grass FTO3)的塑料烧杯置于鼠箱下面以收集和记录尿量。让动物休息15至30分钟,然后在第一次膀胱容积压力检定期开始灌注盐水(以20毫升/小时连续灌注20分钟)。首次膀胱容积检定期后2小时,给鼠服用媒介物或试验化合物,1小时后进行第二次膀胱容积检定。
记录下列尿动力变量:
基础膀胱压力=膀胱容积压力检定期最低的膀胱压力
阀值压力=临排尿前的膀胱压力
排尿体积=排出的体积
排尿压力=排泄期间的峰压力
自发活性=充尿期间膀胱压力波动的平均幅度
结果的表达:
在化合物给药前后计算每个变量的平均值。对每个化合物,将其测得的变量的变化与治疗前的值相比较,以百分抑制率表示,也将数据进行两方向方差分析来确定在测定变量中的显著(p<0.05)变化。
活性的判断标准:
在鼠模型中发现的大多数特征是在充盈期间表现出的自发收缩。在口服或静脉注射10毫克/千克时抑制自发收缩至少50%的化合物被认为是有效的。
因此,本发明化合物对平滑肌收缩具有显著作用,并可用于治疗尿失紧,过敏性膀胱和肠疾病,哮喘,高血压、中风以及如上所述的类似病症,通过对需要这种治疗的病人口服,非胃肠道,或吸入给予钾通道激活和/或氯化物通道封阻本发明化合物而进行治疗。
Claims (12)
1.式(I)表示的化合物: 其中:
其中:
R1、R2和R3相互独立地是氢、硝基、氰基、C1-10卤代烷氧基、氨基、C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基和C6-12芳基;前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、C1-10烷氧甲酰基、C1-10卤代烷氧基、氨基C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-10烷基和C6-12芳基;
R7是氢、金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑、异噻唑、呋喃、噻吩、2H-吡咯、吡咯、2-吡咯啉、3-吡咯啉、咪唑、吡唑、1,2,3-噁二唑或1,2,3-三唑。
2.权利要求1的化合物,其中
R1、R2和R3相互独立地是氢、硝基、氰基、全卤代烷氧基、氨基、C1-10烷基氨基、C1-10二烷基氨基、C6-12芳基氨基、C1-10芳烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C6-12芳基亚磺酰氨基、C2-10烷基甲酰氨基、C6-12芳基甲酰氨基、C2-10链烷酰基、C6-12芳酰基、C2-22芳烷酰基、C1-10烷基磺酰基、C1-10全卤磺酰基、C6-12芳基磺酰基、C2-22芳烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-C10全卤代烷基、芳基,卤代芳基,全卤代芳基,C1-10芳烷基部分;前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、烷氧甲酰基、全卤烷氧基、氨基、C1-10烷基氨基、C1-10二烷基氨基、芳基氨基、C1-10芳烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C6-12芳基亚磺酰氨基、C2-10烷基甲酰氨基、C6-12芳基甲酰氨基、C2-10链烷酰基、C6-12芳酰基、C2-22芳烷酰基、C1-10烷基磺酰基、C1-10全卤磺酰基、C6-12芳基磺酰基、C2-22芳烷基磺酰基、C1-10羧基、C1-10直链烷基、C1-10支链烷基、C3-10环烷基或双环烷基、C1-10卤代烷基、C1-10全卤代烷基、C2-12链烯基(单烯属的或多烯属)、芳基、卤代芳基、全卤芳基、C1-10芳烷基;
R7是氢、碱金属阳离子、碱土金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑或异噻唑。
3.权利要求1的化合物,其中R7选自氢、金属阳离子、选自下列结构的部分: 其中:
其中:
R9、R10、R11和R12相互独立地是氢、C1-10直链烷基、C1-10支链烷基、C3-10环基或二环基、芳基或C1-10芳烷基;和
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑或异噻唑。
4.权利要求1的化合物,其中R7可以是氢或金属阳离子。
5.权利要求1的化合物,其是2-{[2-(4-三氟甲基-苯基)-环戊-1-烯羰基]-氨基}-苯甲酸。
6.权利要求1的化合物,其是2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸。
7.权利要求1的化合物,其是2-{[5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸锂盐。
8.权利要求1的化合物,其是2-{[3-甲基-5-(4-三氟甲基-苯基)-噁唑-4-羰基]-氨基}-苯甲酸。
9.药用组合物,其包括式(I)的化合物或其药物学上可接受的盐以及药物学上可接受的载体, 其中:
其中:
R1、R2和R3相互独立地是氢、硝基、氰基、C1-10卤代烷氧基、氨基、C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基和C6-12芳基;前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、C1-10烷氧甲酰基、C1-10卤代烷氧基、氨基C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-10烷基和C6-12芳基;
R7是氢、金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑、异噻唑、呋喃、噻吩、2H-吡咯、吡咯、2-吡咯啉、3-吡咯啉、咪唑、吡唑、1,2,3-噁二唑或1,2,3-三唑。
10.降低平滑肌不利影响的方法,其包括对需要此治疗的患者口服或非胃肠道施用式(I)化合物或其药物学上可接受的盐,其中
R1、R2和R3相互独立地是氢、硝基、氰基、C1-10卤代烷基、氨基、C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基和C6-12芳基;前体条件是:(1)R1、R2和R3不同时全是氢,和(2)当R1和R2是氢时,R3不可以是间-CF3;
R4、R5和R6相互独立地是氢、卤素、硝基、氰基、C1-10烷氧甲酰基、C1-10卤代烷氧基、氨基C1-10烷基氨基、磺基、氨磺酰基、C1-10烷基亚磺酰氨基、C2-10烷基甲酰氨基、C2-10链烷酰基、C1-10烷基磺酰基、C1-10卤代烷基磺酰基、C1-10羧基、C1-10卤代烷基、C1-10烷基和C6-12芳基;
R7是氢、金属阳离子、乙酰氨基、烷氧基乙酰基或在体内产生羧酸盐的相关部分;
X、Y和Z可以形成键合在碳骨架上的C3-13碳环、噁唑、异噁唑、噻唑、异噻唑、呋喃、噻吩、2H-吡咯、吡咯、2-吡咯啉、3-吡咯啉、咪唑、吡唑、1,2,3-噁二唑或1,2,3-三唑。
11.权利要求10的方法,其中平滑肌不利的收缩引起尿失禁。
12.权利要求10的方法,其中平滑肌不利的收缩引起过敏性肠道综合症。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90593097A | 1997-08-05 | 1997-08-05 | |
| US08/905,930 | 1997-08-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1265643A true CN1265643A (zh) | 2000-09-06 |
Family
ID=25421704
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN98807943A Pending CN1265643A (zh) | 1997-08-05 | 1998-08-03 | 邻氨基苯甲酸类似物 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP1003713A1 (zh) |
| JP (1) | JP2001513527A (zh) |
| CN (1) | CN1265643A (zh) |
| AU (1) | AU734786B2 (zh) |
| BR (1) | BR9811828A (zh) |
| CA (1) | CA2297409A1 (zh) |
| WO (1) | WO1999007670A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101378995B (zh) * | 2006-02-03 | 2013-05-08 | 波隆有限公司 | 水性液体的处理以及邻氨基苯甲酸的制备 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10061876A1 (de) | 2000-12-12 | 2002-06-20 | Aventis Pharma Gmbh | Arylierte Furan- und Thiophencarbonsäureamide, Verfahren zu ihrer Herstellung, ihre Verwendung als Medikament sowie sie enthaltende pharmazeutische Zubereitungen |
| WO2004047738A2 (en) * | 2002-11-22 | 2004-06-10 | Bristol-Myers Squibb Company | Arylcyclopropylcarboxylic amides as potassium channel openers |
| GB0319124D0 (en) * | 2003-08-14 | 2003-09-17 | Smithkline Beecham Corp | Chemical compounds |
| AU2007275301A1 (en) | 2006-07-20 | 2008-01-24 | Amgen Inc. | Substituted azole aromatic heterocycles as inhibitors of 11-beta-HSD-1 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5970654A (ja) * | 1982-10-15 | 1984-04-21 | Nippon Redarii Kk | アントラニル酸誘導体 |
| JPS6097946A (ja) * | 1983-11-01 | 1985-05-31 | Ono Pharmaceut Co Ltd | カルボキサミド誘導体 |
| GB9214120D0 (en) * | 1991-07-25 | 1992-08-12 | Ici Plc | Therapeutic amides |
| DK41193D0 (da) * | 1993-04-07 | 1993-04-07 | Neurosearch As | Ionkanalaabnere |
-
1998
- 1998-08-03 JP JP2000507206A patent/JP2001513527A/ja active Pending
- 1998-08-03 CA CA002297409A patent/CA2297409A1/en not_active Abandoned
- 1998-08-03 AU AU86795/98A patent/AU734786B2/en not_active Ceased
- 1998-08-03 EP EP98938221A patent/EP1003713A1/en not_active Withdrawn
- 1998-08-03 CN CN98807943A patent/CN1265643A/zh active Pending
- 1998-08-03 BR BR9811828-5A patent/BR9811828A/pt not_active Application Discontinuation
- 1998-08-03 WO PCT/US1998/015981 patent/WO1999007670A1/en not_active Ceased
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101378995B (zh) * | 2006-02-03 | 2013-05-08 | 波隆有限公司 | 水性液体的处理以及邻氨基苯甲酸的制备 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU8679598A (en) | 1999-03-01 |
| AU734786B2 (en) | 2001-06-21 |
| BR9811828A (pt) | 2000-08-15 |
| WO1999007670A1 (en) | 1999-02-18 |
| EP1003713A1 (en) | 2000-05-31 |
| JP2001513527A (ja) | 2001-09-04 |
| CA2297409A1 (en) | 1999-02-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6096770A (en) | Anthranilic acid analogs | |
| CN1592744A (zh) | 具有cb1拮抗、激动或部分激动活性的噻唑衍生物 | |
| JPWO2006059744A1 (ja) | ペルオキシソーム増殖剤活性化受容体δの活性化剤 | |
| JP2000515133A (ja) | 血糖降下性および脂質低下性の化合物 | |
| JP2006510600A (ja) | 2−チオヒダントインに由来する化合物及び治療におけるその使用 | |
| JPWO2002051820A1 (ja) | ジヒドロナフタレン誘導体化合物およびその化合物を有効成分とする薬剤 | |
| JP2022058610A (ja) | Dpアンタゴニスト | |
| CN1130361C (zh) | 异噁唑烷二酮化合物的制备方法 | |
| CN1265643A (zh) | 邻氨基苯甲酸类似物 | |
| JP2011500632A (ja) | Npyy2受容体モジュレーターとして有用なイミダゾリジン−2,4−ジオン(ヒダントイン)誘導体 | |
| AU8684598A (en) | Anthranilic acid analogs | |
| JP2948076B2 (ja) | 新規チアゾリジンジオン化合物 | |
| JP2005179281A (ja) | ビフェニル化合物 | |
| RU2295520C2 (ru) | ПРОИЗВОДНЫЕ ОКСАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕНСИБИЛИЗАТОРОВ ИНСУЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВИРУЮЩИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ PRARα И/ИЛИ PRARγ | |
| JPWO2007004733A1 (ja) | ペルオキシソーム増殖剤活性化受容体δの活性化剤 | |
| CN1259940A (zh) | 晶体Roxifiban | |
| AU727217B2 (en) | Heterocyclylmethylamino derivatives of cyclobutene-3,4- diones as potassium channel modulators | |
| MXPA00001255A (en) | Anthranilic acid analogs | |
| CN1245494A (zh) | 新的4-(1-哌嗪基)苯甲酸衍生物,其制备方法及其治疗用途 | |
| CN1417209A (zh) | 一类四氢喹啉酮哌啶类化合物及其制备方法和用途 | |
| RU2803243C2 (ru) | Антагонист dp | |
| JP7047954B2 (ja) | フェニル酢酸化合物を含有する医薬組成物 | |
| CN1286824C (zh) | 对甲磺酰基苯丙烯酸衍生物及其制备方法与抗炎作用 | |
| CN1221536C (zh) | 氰基芳基(或氰基杂芳基)-羰基-哌嗪基-嘧啶类衍生物、其制备方法和应用 | |
| CA2578332A1 (en) | Derivatives of pyrazoline, procedure for obtaining them and use thereof as therapeutic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |