CN1805965A - 趋化因子受体活性调节剂杂环基环戊基四氢异喹啉和杂环基环戊基四氢吡啶并吡啶 - Google Patents
趋化因子受体活性调节剂杂环基环戊基四氢异喹啉和杂环基环戊基四氢吡啶并吡啶 Download PDFInfo
- Publication number
- CN1805965A CN1805965A CNA2004800165371A CN200480016537A CN1805965A CN 1805965 A CN1805965 A CN 1805965A CN A2004800165371 A CNA2004800165371 A CN A2004800165371A CN 200480016537 A CN200480016537 A CN 200480016537A CN 1805965 A CN1805965 A CN 1805965A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- compound
- hydroxyl
- cor
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/10—Anthelmintics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Virology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Pulmonology (AREA)
- Oncology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Communicable Diseases (AREA)
- Hematology (AREA)
- Dermatology (AREA)
- Rheumatology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Molecular Biology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- AIDS & HIV (AREA)
- Obesity (AREA)
- Biotechnology (AREA)
Abstract
本发明涉及式(I)化合物,其中R1、R2、R3、R4、R5、R6、R7、R8、R9、X、n和虚线为本文的定义,该化合物可用作趋化因子受体活性调节剂。具体地讲,这些化合物可用作趋化因子受体CCR-2的调节剂。
Description
发明背景
趋化因子属于一个家族的小分子量的(70-120个氨基酸)促炎性细胞因子,具有有效的趋化活性。趋化因子是许多不同细胞释放的趋化性细胞因子,以吸引各种细胞(例如单核细胞、巨噬细胞、T细胞、嗜曙红细胞、嗜碱细胞和嗜中性白细胞)至炎症位置(综述参见Schall,Cytokine,3,165-183(1991)和Murphy,
Rev.Immun.,12,593-633(1994))。这些分子最初通过四个保守的半胱氨酸定义,根据第一对的两个半胱氨酸的排列分为两个亚家族。在CXC趋化因子家族(包括IL-8、GROα、NAP-2和IP-10)中,这两个半胱氨酸被一个氨基酸隔开,而在CC-趋化因子家族(包括RANTES、MCP-1、MCP-2、MCP-3、MIP-1α、MIP-1β和嗜伊红粒细胞趋化蛋白)中,这两个残基是相邻的。
α-趋化因子(例如白介素-8(IL-8)、嗜中性白细胞激活蛋白-2(NAP-2)和黑素瘤生长刺激活性蛋白(MGSA))主要趋化嗜中性白细胞的,而β-趋化因子(例如RANTES、MIP-1α、MIP-1β、单核细胞趋化蛋白-1(MCP-1)、MCP-2、MCP-3和嗜伊红粒细胞趋化蛋白)趋化巨噬细胞、单核细胞、T细胞、嗜曙红细胞和嗜碱细胞(Deng等,
Nature,381,661-666(1996))。
趋化因子由许多不同类型的细胞分泌,结合白细胞和其它细胞的特异性G蛋白偶联受体(GPCR)(综述参见Horuk,
Trends Pharm. Sci.,15,159-165(1994))。这些趋化因子受体构成GPCR的亚家族,目前,该亚家族包括15个表征的成员和许多孤儿。与混杂趋化剂(例如C5a、fMLP、PAF和LTB4)的受体不同,趋化因子受体在白细胞子集上表达有更高的选择性。因此,特异性趋化因子的产生提供了募集特定白细胞子集的的机制。
在结合它们的同源配体时,趋化因子受体将细胞内信号通过相关三聚G蛋白转导,导致细胞内钙浓度快速提高。至少有7种结合β-趋化因子或者对其敏感的人受体,具体特征形式如下:CCR-1(或“CKR-1”或“CC-CKR-1”)[MIP-1α、MIP-1β、MCP-3、RANTES](Ben-Barruch等,
J.Biol.Chem.,270,22123-22128(1995);Beote等,Cell,72,415-425(1993));CCR-2A和CCR-2B(或“CKR-2A”/“CKR-2A”或“CC-CKR-2A”或“CC-CKR-2A”)[MCP-1、MCP-2、MCP-3、MCP-4];CCR-3(或“CKR-3”或“CC-CKR-3”)[Eotaxin、Eotaxin 2、RANTES、MCP-2、MCP-3](Rollins等,
Blood,90,908-928(1997));CCR-4(或“CKR-4”或“CC-CKR-4”)[MIP-1α、RANTES、MCP-1](Rollins等,
Blood,90,908-928(1997));CCR-5(或“CKR-5”或“CC-CKR-5”)[MIP-1α、RANTES、MIP-1β](Sanson等,Biochemistry,35,3362-3367(1996));Duffy血型抗原[RANTES、MCP-1](Chaudhun等,
J.Biol.Chem.,269,7835-7838(1994))。β-趋化因子还包括嗜伊红粒细胞趋化蛋白、MIP(“巨噬细胞炎性蛋白”)、MCP(“单核细胞趋化蛋白”)和RANTES(“调节激活正常T细胞表达和分泌的因子”)。
趋化因子受体(例如CCR-1、CCR-2、CCR-2A、CCR-2B、CCR-3、CCR-4、CCR-5、CXCR-3、CXCR-4)涉及作为炎性免疫调节性病症和疾病(包括哮喘、鼻炎和过敏性疾病)以及自身免疫性病变(例如类风湿性关节炎)和动脉粥样硬化的重要介质。在CCR-5基因中32个碱基对纯合性缺失的人似乎对类风湿性关节炎的易感性较小(Gomez等,
Arthritis & Rheumatism,42,989-992(1999))。有关嗜曙红细胞在变应性炎症的作用的综述参见Kita,H.等,
J.Exp.Med.183,2421-2426(1996)。有关趋化因子在变应性炎症的作用的综述参见Lustger,A.D.
New England J.Med.,338(7),426-445(1998)。
一个趋化因子子集是单核细胞和巨噬细胞的有效趋化剂。表征最充分的是MCP-1(单核细胞趋化蛋白-1),它的主要受体是CCR2。MCP-1是各种物种(包括啮齿动物和人)的不同细胞类型响应炎症刺激物而产生的,刺激单核细胞、淋巴细胞子集的趋化性。具体地讲,MCP-1的产生与单核细胞和巨噬细胞在炎症位置浸润有关。在小鼠进行同源重组使MCP-1或CCR2缺失,导致响应巯基醋酸酯注射和单核细胞增多性李氏菌(Listeria monocytogenes)注射的单核细胞募集显著减弱(Lu等,
J.Exp.Med.,187,601-608(1998);Kurihara等,
J.Exp.Med.,186,1757-1762(1997);Boring等,
J.Clin.Invest.,100,2552-2561(1997);Kuziel等,
Proc.Natl.Acad.Sci.,94,12053-12058(1997))。此外,这些小鼠中单核细胞浸润到肉芽肿性病变位置的数量减少,所述病变通过注射血吸虫(schistosomal)或分枝杆菌抗原诱发(Boring等,
J.Clin.Invest.,100,2552-2561(1997);Warmington等,
Am J.Path.,154,1407-1416(1999))。这些情况表明MCP-1诱导性CCR2激活在使单核细胞募集到炎症位置中起主要作用,对这种活性的拮抗作用将对免疫反应产生足够的抑制作用,从而对免疫性炎性疾病和自身免疫性疾病提供有益的治疗。
所以,调节趋化因子受体(例如CCR-2受体)的药物可用于这样的病症和疾病。
另外,募集单核细胞至血管壁的炎性病变位置是致动脉粥样化斑形成的主要原因。在高胆固醇血症的血管壁损伤后,MCP-1由内皮细胞和内膜平滑肌细胞产生、分泌。募集到损伤位置的单核细胞响应释放的MCP-1,浸润血管壁后分化为泡沫细胞。几个研究小组已经证实在回交APO-E-/-、LDL-R-/-或Apo B转基因小鼠的MCP-1-/-或CCR2-/-小鼠(供给高脂肪食物)中主动脉病变大小、巨噬细胞含量和坏死减少(Boring等,
Nature,394,894-897(1998);Gosling等,J.Clin.Invest.,103,773-778(1999))。因此,CCR2拮抗剂可以通过减少单核细胞在动脉壁募集和分化,从而抑制动脉粥样硬化病变形成以及病理进展。
发明概述
本发明进一步涉及这样的化合物:它们是趋化因子受体活性调节剂,可用于预防或治疗某些炎性免疫调节性病症和疾病、过敏性疾病、特应性病症(包括变应性鼻炎、皮炎、结膜炎和哮喘)以及自身免疫性病变(例如类风湿性关节炎)和动脉粥样硬化。本发明还涉及包含这些化合物的药用组合物以及以及这些化合物和组合物在预防或治疗涉及趋化因子受体的疾病中的用途。
发明详述
本发明涉及下式I化合物或者其药学上可接受的盐或单独的非对映异构体:
式I
X选自C、N、O、S和SO2;
Y为N或C;
R1选自以下基团:
氢、-C1-6烷基、-C0-6烷基-O-C1-6烷基、-C0-6烷基-S-C1-6烷基、-(C0-6烷基)-(C3-7环烷基)-(C0-6烷基)、羟基、杂环、-CN、-NR12R12、-NR12COR13、-NR12SO2R14、-COR11、-CONR12R12和苯基,
其中R11独立选自以下基团:羟基、氢、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R12选自以下基团:氢、C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R13选自以下基团:氢、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R14选自以下基团:羟基、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中所述烷基和环烷基未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(f)C1-3烷基,
(g)-O-C1-3烷基,
(h)-COR11,
(i)-SO2R14,
(j)-NHCOCH3,
(k)-NHSO2CH3,
(l)-杂环,
(m)=O,
(n)-CN,
其中所述苯基和杂环未被取代或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基和三氟甲基;
R2选自以下基团:
(a)氢,
(b)羟基,
(c)卤基,
(d)C1-3烷基,其中所述烷基未被取代或者被1-6个独立选自氟和羟基的取代基取代,
(e)-NR12R12,
(f)-COR11,
(g)-CONR12R12,
(h)-NR12COR13,
(i)-OCONR12R12,
(j)-NR12CONR12R12,
(k)-杂环,
(l)-CN,
(m)-NR12-SO2-NR12R12,
(n)-NR12-SO2-R14,
(o)-SO2-NR12R12,
(p)=O,其中R2通过双键与环连接;
在Y为N时,R3为氧或不存在;
在Y为C时,R3选自以下基团:
(a)氢,
(b)羟基,
(c)卤基,
(d)C1-3烷基,其中所述烷基未被取代或者被1-6个独立选自以下的取代基取代:氟、羟基和-COR11,
(e)-NR12R12,
(f)-COR11,
(g)-CONR12R12,
(h)-NR12COR13,
(i)-OCONR12R12,
(j)-NR12CONR12R12,
(k)-杂环,
(l)-CN,
(m)-NR12-SO2-NR12R12,
(n)-NR12-SO2-R14,
(o)-SO2-NR12R12,
(p)硝基;
R4选自以下基团:
(a)氢,
(b)C1-6烷基,
(c)三氟甲基,
(d)三氟甲氧基,
(e)氯,
(f)氟,
(g)溴,
(h)苯基;
R5选自以下基团:
(a)C1-6烷基,其中烷基可以是未被取代的,或者被1-6个氟取代,以及任选被羟基取代,
(b)-O-C2-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(c)-CO-C1-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(d)-S-C1-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(e)-吡啶基,它可以是未被取代的或者被一个或多个选自以下的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(f)氟,
(g)氯,
(h)溴,
(i)-C4-6环烷基,
(j)-O-C4-6环烷基,
(k)苯基,它可以是未被取代的或者被一个或多个选自以下的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(l)-O-苯基,它可以是未被取代的或者被一个或多个选自以下的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(m)-C3-6环烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(n)-O-C3-6环烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(o)-杂环,
(p)-CN,
(q)-COR11;
R6选自以下基团:
(a)氢,
(b)C1-6烷基,
(c)三氟甲基,
(d)氟,
(e)氯,
(f)溴;
R7选自以下基团:
不存在(X=O时)、氢、(C0-6烷基)-苯基、(C0-6烷基)-杂环、(C0-6烷基)-C3-7环烷基、(C0-6烷基)-COR11、(C0-6烷基)-(烯基)-COR11、(C0-6烷基)-SO3H、(C0-6烷基)-W-C0-4烷基、(C0-6烷基)-CONR12-苯基和(C0-6烷基)-CONR15-V-COR11,或者在X为O、S或SO2时,R7不存在,
其中V为C1-6烷基或苯基,W选自单键、-O-、-S-、-SO-、-SO2-、-CO-、-CO2-、-CONR12-和-NR12-,R15可以为氢、C1-4烷基,或者R15通过1-5个碳的接头连接V的一个碳构成环,C0-6烷基未被取代或被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基,
其中所述苯基、杂环、环烷基和C0-4烷基未被取代或被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-C0-3-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12,
(j)-C0-3-杂环,
或者,所述苯基和杂环可以与另一个杂环稠合,另一个杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基,
其中烯基未被取代或者被1-3个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)C1-3烷基,
(d)苯基,
(e)杂环;
R8选自以下基团:
(a)氢,
(b)在X为O、S、SO2或N时或者分别连接R7和R10的碳之间为双键时,R8不存在,
(c)羟基,
(d)C1-6烷基,
(e)C1-6烷基-羟基,
(f)-O-C1-3烷基,
(g)-COR11,
(h)-CONR12R12,
(i)-CN;
或者R7和R8接合在一起构成选自以下的环:
(a)1H-茚,
(b)2,3-二氢-1H-茚,
(c)2,3-二氢-苯并呋喃,
(d)1,3-二氢-异苯并呋喃,
(e)2,3-二氢-苯并噻吩,
(f)1,3-二氢-异苯并噻吩,
(g)6H-环戊二烯并[d]异噁唑-3-醇,
(h)环戊烷,
(i)环己烷,
其中所构成的环可以是未被取代的或者被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-C0-3-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12,
(j)-C0-3-杂环,
或者,R7与R9或R8与R10可以结合在一起构成苯环或杂环,其中所述环未被取代或被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12;
R9和R10独立选自以下基团:
(a)氢,
(b)羟基,
(c)C1-6烷基,
(d)C1-6烷基-COR11,
(e)C1-6烷基-羟基,
(f)-O-C1-3烷基,
(g)=O,这时R9或R10通过双键连接环,
(h)卤基;
n选自0、1和2;
虚线为单键或双键。
本发明另一实施方案包括下式Ia化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、Y和n为上文中的定义,
R16和R17独立选自以下基团:
(a)氢,
(b)卤基,
(c)三氟甲基,
(d)羟基,
(e)C1-3烷基,
(f)-O-C1-3烷基,
(g)-C0-3-CO2H,
(h)-C0-3-CO2C1-3烷基,
(i)-CN,
(j)-C0-3-杂环,
或者,R16和R17接合在一起构成与苯环稠合的杂环,杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基。
本发明另一实施方案还包括下式Ib化合物或者其药学上可接受的盐或单独的非对映异构体:

其中虚线为单键或双键,R1、R2、R3、R5、R9、R16、R17、Y和n为上文中的定义。
本发明又一实施方案包括下式Ic化合物或者其药学上可接受的盐或单独的非对映异构体:
其中R1、R2、R3、R5、R9、R16、R17、Y和n为上文中的定义,H为杂环。
本发明另一实施方案包括下式Id化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、R11、Y、W和n为上文中的定义,
其中C1-4碳链可以是未被取代的或者被1-4个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基
或者C1-4碳链可以包含在C3-7环烷基环中。
本发明的再一实施方案包括下式Ie化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、R16、R17、X、Y和n为上文中的定义;虚线为单键或双键;o为1或2;A、B和D可以独立选自C、N、O或S,在X、A、B、D均为C且o=2时,构成苯环,或者在至少X、A、B和D之一为N、O或S而不为C时,构成杂环。
本发明又一实施方案包括下式If化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R7、R9、R10、Y和n为上文中的定义;X为N或O,在X为O时,R7不存在。
本发明另一实施方案包括下式Ig化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R5、R9、R16、R17和Y为上文中的定义,
或者R16和R17接合在一起构成与苯环稠合的杂环,杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基。
本发明的再一实施方案包括下式Ih化合物或者其药学上可接受的盐或单独的非对映异构体:
其中虚线为单键或双键,R1、R5、R9、R16、R17和Y为上文中的定义。
本发明的再一实施方案包括下式Ii化合物或者其药学上可接受的盐或单独的非对映异构体:
其中R1、R5、R9、R16、R17和Y为上文中的定义,H为杂环。
本发明又一实施方案包括下式Ii化合物或者其药学上可接受的盐或单独的非对映异构体:
其中R1、R5、R9、R11、Y和W为上文中的定义,C1-4碳链可以是未被取代的或者被1-4个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基。
本发明另一实施方案包括下式Ik化合物或者其药学上可接受的盐或单独的非对映异构体:
式Ik
其中R1、R5、R9、R10和Y为上文中的定义。
本发明的又一实施方案中,X为C、O或N。
在本发明另一个实施方案中,X为C或O。
在本发明另一个实施方案中,R1以下基团:-C1-6烷基、-C0-6烷基-O-C1-6烷基和-(C0-6烷基)-(C3-7环烷基)-(C0-6烷基),所述烷基和环烷基未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(f)C1-3烷基,
(g)-O-C1-3烷基,
(h)-COR11,
(i)-CN,
(j)-NR12R12,
(k)-CONR12R12。
在本发明的另一方面,R1选自以下基团:
(1)-C1-6烷基,它未被取代或者被1-6个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(e)-COR11,
(2)-C0-6烷基-O-C1-6烷基-,它未被取代或者被1-6个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)-COR11,
(3)-(C3-5环烷基)-(C0-6烷基),它未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(e)-COR11。
在本发明的再一方面,R1选自以下基团:
(a)C1-6烷基,
(b)羟基取代的C1-6烷基,
(c)被1-6个氟取代的C1-6烷基。
在本发明的又一方面,R1选自以下基团:
(a)-CH(CH3)2,
(b)-CH(OH)CH3,
(c)-CH2CF3。
在本发明的另一方面,R2选自以下基团:
(a)羟基,
(b)氢,
(c)=O,其中R2通过双键与环连接。
在本发明的另一方面,R2为氢。
在本发明的又一方面,在Y为N时,R3不存在或为O(得到N-氧化物)。
在本发明的又一方面,在Y为N时,R3不存在。
在本发明的又一方面,在Y为C时,R3选自以下基团:
(a)氢,
(b)卤基,
(c)羟基,
(d)C1-3烷基,其中所述烷基未被取代或者被1-6个独立选自氟和羟基的取代基取代,
(e)-COR11,
(f)-CONR12R12,
(g)-杂环,
(h)-NR12-SO2-NR12R12,
(i)-NR12-SO2-R14,
(j)-SO2-NR12R12,
(k)-硝基,
(l)-NR12R12。
在本发明的另一方面,在Y为C时,R3为氢。
在本发明的另一方面,R4为氢。
在本发明的另一方面,R5选自以下基团:
(a)被1-6个氟取代的C1-6烷基,
(b)被1-6个氟取代的-O-C1-6烷基,
(c)氯,
(d)溴,
(e)苯基。
在本发明的另一方面,R5选自以下基团:
(a)三氟甲基,
(b)三氟甲氧基,
(c)氯,
(d)溴,
(e)苯基。
在本发明的另一方面,R5为三氟甲基。
在本发明的另一方面,R6为氢。
在本发明的另一方面,R7为苯基、杂环、C3-7环烷基、C1-6烷基、-COR11和-CONH-V-COR11,
其中V选自C1-6烷基或苯基,
其中所述苯基、杂环、C3-7环烷基、C1-6烷基未被取代或被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-COR11,
(g)-CN,
(h)-杂环,
(i)-CONR12R12。
在本发明的另一方面,当X不是O时,R7为苯基、杂环、C1-4烷基、-COR11和-CONH-V-COR11,V选自C1-6烷基或苯基,
其中所述苯基、杂环和C1-4烷基未被取代或被1-3个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)C1-3烷基,
(d)-O-C1-3烷基,
(e)-COR11,
(f)-杂环。
在本发明的另一方面,当X为C时,R7选自以下基团:

在本发明的另一方面,在X为C时,R8选自以下基团:
(a)氢,
(b)羟基,
(c)-CN,
(d)-F。
在本发明的另一方面,R7和R8可以结合在一起构成选自以下的环:
(a)1H-茚,
(b)2,3-二氢-1H-茚,
其中所构成的环可以是未被取代的或者被1-3个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)C1-3烷基,
(d)-O-C1-3烷基,
(e)-COR11,
(f)-杂环。
在本发明的另一方面,R9和R10独立选自以下基团:
(a)氢,
(b)羟基,
(c)-CH3,
(d)-O-CH3,
(e)=O(其中R9和/或R10通过双键与环连接)。
在本发明又一方面,n=1或2。
本发明的代表性化合物包括实施例的化合物以及它们的药学上可接受的盐和单独的非对映异构体。
本发明化合物至少在环戊基的1位和3位有两个不对称中心。可能存在其它不对称中心,这取决于分子上不同取代基的性质。所有这样的不对称中心将独立产生两个旋光异构体,本发明范围包括所有可能旋光异构体和非对映异构体的混合物形式以及纯净或部分纯净化合物形式。本发明化合物一方面的绝对构型是如下的构型,其中环戊基上的取代基(酰胺单元和胺单元)是顺式:
本发明化合物另一方面的绝对构型是如下定向的构型:
其中连接胺取代基的碳指定为(R)绝对构型,连接酰胺亚单元的碳可以根据R1的优先级指定为(S)或(R)绝对构型。例如R为异丙基时,则连接酰胺亚单元的碳的绝对立体化学构型将会是(S),因为酰胺单元和胺单元在环戊基上优先为顺式排列。
本领域已知可以通过适当修改本文公开的方法单独合成非对映异构体和对映异构体或者色谱分离。它们的绝对立体构型可以通过结晶产物或结晶中间体的x射线晶体照相确定,如果需要,结晶产物或结晶中间体也可用包含已知绝对构型的不对称中心的试剂衍生。
正如本领域熟练技术人员理解的那样,本文使用的卤基或卤素包括氯、氟、溴和碘。
本文使用的“烷基”是指没有双键或三键的直链、支链或环状结构。因此,C1-6烷基是指含有1、2、3、4、5或6个碳的直链或直链基团,这样的C1-6烷基特别包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基和己基。“环烷基”是部分或全部构成三个或三个以上原子的环的烷基。C0或C0烷基是指共价键。
本文使用的术语“杂环”包括以下基团:苯并咪唑基、苯并呋喃基、苯并呋咱基、苯并吡唑基、苯并三唑基、苯并噻吩基、苯并噁唑基、咔唑基、咔啉基、肉啉基、呋喃基、咪唑基、二氢吲哚基、吲哚基、吲嗪基(indolazinyl)、吲唑基、异苯并呋喃基、异吲哚基、异喹啉基、异噻唑基、异噁唑基、萘并吡啶基、噁二唑基、噁唑基、氧杂环丁烷基、吡喃基、吡嗪基、吡唑基、哒嗪基、吡啶并吡啶基、哒嗪基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、喹喔啉基、四氢吡喃基、四唑基、四唑并吡啶基、噻二唑基、噻唑基、噻吩基、三唑基、氮杂环丁烷基、1,4-二噁烷、六氢吖庚因基、哌嗪基、哌啶基、吡咯烷基、吗啉基、硫代吗啉基、二氢苯并咪唑基、二氢苯并呋喃基、二氢苯并噻吩基、二氢苯并噁唑基、二氢呋喃基、二氢咪唑基、二氢吲哚基、二氢异噁唑基、二氢异噻唑基、二氢噁二唑基、二氢噁唑基、二氢吡嗪基、二氢吡唑基、二氢吡啶基、二氢嘧啶基、二氢吡咯基、二氢喹啉基、二氢四唑基、二氢噻二唑基、二氢噻唑基、二氢噻吩基、二氢三唑基、二氢氮杂环丁烷基、亚甲二氧基苯甲酰基、四氢呋喃基和四氢噻吩基以及它们的N-氧化物。
本文使用的术语“药学上可接受的”是指这样的化合物、原料、组合物和/或剂型:在合理的医学判断内,适合与人及动物的组织接触,而没有过度的毒性、刺激、过敏反应、或其它问题或并发症,具有合理的受益/风险比。
本文使用的“药学上可接受的盐”是指将母体化合物修饰制备其酸盐或碱盐得到的衍生物。药学上可接受的盐的例子包括但不限于碱性残基(例如胺)的无机酸盐或有机酸盐;酸性残基(例如羧酸)的碱盐或有机盐等。药学上可接受的盐包括用例如无毒无机酸或有机酸形成的母体化合物的常规无毒盐或季铵盐。举例来讲,这样的常规无毒盐包括无机酸衍生的盐,例如盐酸盐、氢溴酸盐、硫酸盐、氨基磺酸盐、磷酸盐、硝酸盐等;用有机酸制备的盐,例如乙酸盐、丙酸盐、琥珀酸盐、乙醇酸盐、硬脂酸盐、乳酸盐、苹果酸盐、酒石酸盐、柠檬酸盐、抗坏血酸盐、扑酸盐、马来酸盐、羟基马来酸盐、苯基乙酸盐、谷氨酸盐、苯甲酸盐、水杨酸盐、对氨基苯磺酸盐、2-乙酰氧基苯甲酸盐、富马酸盐、甲苯磺酸盐、甲磺酸盐、乙烷二磺酸盐、草酸盐、羟乙磺酸盐等。
本发明药学上可接受的盐可以用包含碱性或酸性部分的母体化合物通过常规化学方法制备。通常,这样的盐可以如下制备:使上述化合物的游离酸或游离碱形式与化学计量量的适当碱或酸在水、有机溶剂或两者的混合物中反应,通常使用非水性介质,例如乙醚、乙酸乙酯、乙醇、异丙醇或乙腈。合适的盐可参见例如Remington′sPharmaceutical Sciences,第17版,Mack Publishing Company,Easton,PA,1985,p.1418。
利用实施例和此处公开的化合物举例说明本发明。
本发明的具体化合物包括选自以下的化合物:实施例的标题化合物以及它们的药学上可接受的盐和单独的非对映异构体。
主题化合物可用于调节需要这种调节的患者的趋化因子受体活性的方法,该方法包括给予有效量的化合物。
本发明涉及前述化合物作为趋化因子受体活性调节剂的用途。具体地讲,这些化合物可用作趋化因子受体(特别是CCR-2)的调节剂。
本发明化合物作为趋化因子受体活性调节剂的效力可以通过本领域已知的方法证实,例如以下文献公开的趋化因子结合分析法:VanRiper等,
J.Exp.Med.,
177,851-856(1993),很容易改进用于测量CCR-2结合作用。
CCR-2结合分析中受体亲和力如下确定:检测125I-MCP-1结合到不同细胞类型(包括单核细胞、THP-1细胞)内源CCR-2受体的抑制作用,或者克隆受体在真核细胞外源表达后检测125I-MCP-1结合的抑制作用。将细胞悬浮于结合缓冲液(50mM HEPES,pH 7.2,5mMMgCl2,1mM CaCl2和0.50%BSA),在室温加入受试化合物或DMSO和125I-MCP-1,放置1h让其结合。然后用GFB滤膜收集细胞,用含500mM NaCl的25mM HEPES缓冲液洗涤,定量结合125I-MCP-1的细胞。
在趋化性分析中,用缺失T细胞的PBMC进行趋化作用,所述PBMC从静脉全血或白细胞分离血分离,Ficoll-Hypaque离心提纯,然后用神经氨糖酸苷酶处理的绵羊红细胞形成玫瑰花结。分离完成后,将细胞用含0.1mg/ml BSA的HBSS洗涤,以1×107细胞/ml悬浮。在暗处、37℃下,将细胞用2μM Calcien-AM(Molecular Probes)荧光标记30min。将标记的细胞洗涤两次,并以5×106细胞/ml悬浮于RPMI1640(含L-谷氨酰胺、0.1mg/ml BSA,不含酚红)。将相同培养基稀释的10ng/ml MCP-1(Peprotech)或者仅仅是培养基加入下部的孔(27μl)。用DMSO或不同浓度的受试化合物预温育15min后,将单核细胞(150,000细胞)加入滤膜上面(30μl)。将相同浓度的受试化合物或DMSO加入底部孔以防止扩散稀释。在37℃、5%CO2温育60min后,撤去滤膜,顶部用含0.1mg/ml BSA的HBSS洗涤除去没有迁移到滤膜的细胞。确定没有趋化剂存在下的自发迁移作用(化学运动性)。
具体地讲,下文实施例的化合物在上述分析中具有结合CCR-2受体的活性,通常IC50小于约1μM。这样的结果表明化合物具有作为趋化因子受体活性调节剂的内在活性。
哺乳动物趋化因子受体提供了干扰或促进哺乳动物(例如人)的嗜曙红细胞和/或淋巴细胞功能的靶。抑制或促进趋化因子受体功能的化合物尤其可用于治疗性调节嗜曙红细胞和/或淋巴细胞功能。因此,抑制或促进趋化因子受体功能的化合物可用于治疗、预防、改善、控制多种以下疾病或降低其患病风险:炎性免疫调节性病症和疾病、过敏性疾病、特应性病症(包括变应性鼻炎、皮炎、结膜炎和哮喘)以及自身免疫性病变(例如类风湿性关节炎)和动脉粥样硬化。
例如,可以给予抑制哺乳动物趋化因子受体(例如人趋化因子受体)的一种或多种功能的本发明化合以抑制(即减轻或预防)炎症。由此,一种或多种炎性过程(例如白细胞游出、趋化性、胞吐作用(例如酶、组胺的胞吐作用)或炎性介质释放被抑制。
除了灵长目(例如人)外,许多其它哺乳动物也可根据本发明方法治疗。例如,还可治疗包括但不限于牛、绵羊、山羊、马、狗、猫、豚鼠、大鼠或其它牛科、羊科、马科、犬科、猫科、啮齿动物或鼠科物种的哺乳动物。然而,所述方法还可用于其它物种,例如鸟类(例如鸡)。
与炎症和感染有关的疾病和病症可以用本发明化合物治疗。在一个实施方案中,所述疾病或病症是需要抑制或促进淋巴细胞作用以调节炎症反应的疾病或病症。
可以用趋化因子受体功能抑制剂治疗的人或其它物种的疾病或病症包括但不限于:炎性或过敏性疾病和病症,包括呼吸道过敏性疾病例如哮喘(尤其是支气管哮喘)、变应性鼻炎、过敏性肺病、过敏性肺炎、嗜曙红细胞性肺炎(例如Loeffler综合症、慢性嗜曙红细胞性肺炎)、延迟型过敏反应、间质性肺炎(ILD)(例如特发性肺纤维化或与类风湿性关节炎、系统性红斑狼疮、强直性脊柱炎、系统性硬化、Sjogren综合症、多肌炎或皮肌炎相关的ILD);全身性过敏或超敏反应、药物变态反应(例如对青霉素、头孢菌素的变态反应)、昆虫蜇刺性过敏;自身免疫性疾病,例如类风湿性关节炎、银屑病关节炎、多发性硬化、系统性红斑狼疮、重症肌无力、青少年糖尿病;肾小球肾炎、自身免疫性甲状腺炎、贝切特氏综合征;移植排斥(例如移植术中的排斥),包括同种异体移植物的排斥或移植物抗宿主病;炎性肠病,例如克罗恩氏病和溃疡性结肠炎;脊柱关节病;硬皮病;银屑癣(包括T细胞介导性银屑癣)和炎性皮肤病(例如皮炎、湿疹、特应性皮炎、变应性接触性皮炎、荨麻疹);血管炎(例如坏死性血管炎、皮肤血管炎和过敏性血管炎);嗜曙红细胞性肌炎、嗜曙红细胞性筋膜炎;白细胞浸润皮肤或器官的癌症。还可以治疗需要抑制不良炎性反应的其它疾病或病症,包括但不限于再灌注损伤、动脉粥样硬化、某些血液恶性肿瘤、细胞因子诱导性毒性(例如脓毒性休克、内毒素性休克)、多肌炎、皮肌炎。
可以用趋化因子受体功能调节剂治疗的人或其它物种的疾病或病症包括但不限于:免疫抑制(例如患免疫缺陷综合症如AIDS或其它病毒感染的个体的免疫抑制,放疗、化疗、自身免疫性疾病的治疗或药物治疗(例如皮质类固醇疗法)引起的个体免疫抑制;受体功能先天性缺陷或其它原因的免疫抑制;感染疾病,例如寄生虫病,包括但不限于蠕虫感染,例如线虫(鞭虫病、蛲虫病、蛔虫病、钩虫病、类圆线虫病、旋毛虫病、丝虫病)、吸虫(血吸虫病、支睾吸虫病)、绦虫(包虫病、绦虫病、囊尾幼虫病)、内脏的虫、内脏幼虫移行症(visceral larva migraines)(例如弓蛔虫)、嗜曙红细胞性肠胃炎(例如Anisaki sp.、Phocanema sp.)和皮肤幼虫移行症(cutaneous larvamigraines)(巴西钩口线虫、犬钩虫)。另外,如果预计给予足够化合物,通过诱导趋化因子受体内化使得细胞表面上的受体表达减少或者以导致细胞迁移方向错误的方式给予化合物,则可考虑通过增强趋化因子受体功能治疗上述炎性、过敏性和自身免疫性疾病。
因此,本发明化合物可用于治疗、预防、改善、控制多种以下疾病或降低其患病风险:炎性免疫调节性病症和疾病、过敏性疾病、特应性病症以及自身免疫性病变。在一个特别的实施方案中,本发明涉及使用主题化合物治疗、预防、改善、控制自身免疫性疾病(例如类风湿性关节炎或银屑病关节炎)或降低其患病风险。
在另一方面,本发明可以用于评价趋化因子受体(包括CCR-2)的推定的具体激动剂或拮抗剂。据此,本发明涉及上述化合物在筛选分析的准备和实施中的用途,所述分析用于评价调节趋化因子受体活性的化合物。例如,本发明化合物可用于分离受体突变体,受体突变体是寻找更有效化合物的优异筛选工具。此外,本发明化合物可通过例如竞争性抑制作用证实或确定其它化合物与趋化因子受体的结合位点。本发明化合物还可用于评价趋化因子受体(包括CCR-2)的推定的具体调节剂。正如本领域所理解那样,彻底评价以上趋化因子受体的具体激动剂和拮抗剂受制于缺乏可用的非肽酰(抗代谢)化合物,该类化合物对这些受体具有高结合亲和力。因此,本发明化合物是未来用于这些目的的商品。
本发明进一步涉及制备药物的方法,所述药物包含本发明化合物以及药用载体或稀释剂,用于调节人和动物的趋化因子受体活性。
本发明进一步涉及使用本发明化合物治疗、预防、改善、控制反转录病毒(尤其是疱疹病毒或人免疫缺陷病毒(HIV))感染或降低其感染风险以及治疗随后的病理疾病(例如AIDS)或延迟其发作。治疗AIDS或者预防或治疗HIV感染定义为包括但不限于治疗各种HIV感染状态:AIDS、有症状或无症状的ARC(AIDS相关综合征)、以及实际或潜在接触HIV。例如,在怀疑过去由于例如输血、器官移植、体液交换、咬伤、意外针刺而接触HTV,或者在外科手术时接触患者血液后,可用本发明化合物治疗HIV感染。
在本发明一个方面,主题化合物可用于抑制趋化因子与靶细胞趋化因子受体(例如CCR-2)结合的方法,该方法包括使靶细胞与一定量的化合物接触,所述量的化合物可有效抑制趋化因子与趋化因子受体的结合。
以上方法治疗的患者是需要调节趋化因子受体活性的哺乳动物例如人,包括男性和女性。本文使用的“调节”包括拮抗、激动、部分拮抗、反向激动和/或部分激动。在本发明一方面,调节是指拮抗趋化因子受体活性。术语“治疗有效量”是指主题化合物的用量将引起组织、系统、动物或人的生物学或医学反应,这种反应是研究者、兽医、医生或其他临床医师所寻找的。
本文使用的术语“组合物”包括含有特定量特定成份的产品以及以特定量特定成分组合而直接或间接得到的任何产品。“药学上可接受的”是指载体、稀释剂或赋形剂必须与制剂其它成分相容,而且不会危害其接受者。
术语“给予”化合物应该理解为将本发明化合物提供给需要治疗的个体。
本文使用的术语“治疗”是指治疗性及预防性治疗上述疾病。
关于在烷基、环烷基、苯基、杂环或部分其它化学基团上取代作用的术语“取代的”包括被指定的取代基单取代和多取代,并且在任何指定的化学基团上的所述单取代或多取代应该是化学上所允许的。
应当理解的是分子中某个特定位置的取代基的定义独立于该取代基在分子中其它位置的定义。所以,例如当R3=1-5个R12(在别处定义)取代的烷基时,各个R12独立选自其可能选择的基团;即各个R12可以与任何其它R12相同或不相同。
术语“任选取代的”包括取代和未取代的。因此,例如任选取代的芳基可能为五氟苯基或苯基。
调节趋化因子受体活性并由此治疗、预防、改善、控制炎性免疫调节性病症和疾病(包括哮喘和过敏性疾病)、自身免疫性病变(例如类风湿性关节炎)和动脉粥样硬化以及上述病症或者降低其患病风险的联合疗法通过本发明化合物和已知具有这样效用的其它化合物的联合药物说明。
例如在治疗、预防、改善、控制炎症或降低患炎症风险中,本发明化合物可以与抗炎药或镇痛药联合应用,例如鸦片激动剂、脂肪氧合酶抑制剂(例如5-脂肪氧合酶抑制剂)、环加氧酶抑制剂(例如环加氧酶-2抑制剂)、白介素抑制剂(例如白介素-1抑制剂)、NMDA拮抗剂、一氧化氮抑制剂、一氧化氮合成抑制剂、非甾族抗炎药或细胞因子抑制性抗炎药,例如联合以下化合物:醋氨酚、阿斯匹林、可待因、依那西普(embrel)、芬太尼、布洛芬、消炎痛、酮咯酸、吗啡、萘普生、非那西汀、吡罗昔康、甾族镇痛药、舒芬太尼、舒林酸、替尼达普等。类似地,本发明化合物可以与以下药物联合应用:疼痛缓解剂;增效剂,例如咖啡因、H2-拮抗剂、二甲基硅油、氢氧化铝或氢氧化镁;减充血剂,例如苯肾上腺素、苯丙醇胺、伪麻黄碱、羟甲唑啉、肾上腺素(ephinephrine)、萘唑啉、赛洛唑啉、丙己君(propylhexedrine)或左旋脱氧麻黄碱;镇咳药,例如可待因、氢可酮、咳美芬、咳必清或右美沙芬;利尿药;镇静性或非镇静性抗组胺药。
同样地,本发明化合物还可以与其它药物联合使用,所述其它药物用于治疗/预防/抑制或改善可用本发明化合物治疗的疾病或病症。这样的其它药物可以其常规用药途径和用量与本发明化合物同时或序贯给药。本发明化合物与一种或多种其它药物同时使用时,通常使用包含这样的其它药物和本发明化合物的组合物。因此,本发明药用组合物包括这样的组合物:除了本发明化合物外,还含有一种或多种其它活性成分。
可以与本发明化合物联合使用的其它活性成分(可以单独或以同一药用组合物给药)的例子包括但不限于:(a)VLA-4拮抗剂,例如US5,510,332、WO95/15973、WO96/01644、WO96/06108、WO96/20216、WO96/22966、WO96/31206、WO96/40781、WO97/03094、WO97/02289、WO98/42656、WO98/53814、WO98/53817、WO98/53818、WO98/54207和WO98/58902介绍的拮抗剂;(b)甾族化合物,例如倍氯米松、甲基强的松龙、倍他米松、强的松、地塞米松和氢化可的松;(c)免疫抑制剂,例如环孢菌素、他克莫司、雷帕霉素和其它FK-506型免疫抑制剂;(d)抗组胺药(H1-组胺拮抗剂),例如溴苯那敏、氯苯吡胺、右氯苯那敏、曲普利啶、氯马斯汀、苯海拉明、二苯拉林、曲吡那敏、羟嗪、甲地嗪、普鲁米近、三甲泼拉嗪、阿扎他定、赛庚啶、安他唑啉、非尼腊明、吡拉明、阿司咪唑、特非那定、氯雷他定、地洛他定、西替立嗪、非索非那定、脱碳乙氧基氯雷他定等;(e)非甾族抗哮喘药,例如β2-激动剂(特布他林、奥西那林、非诺特罗、新异丙肾上腺素、舒喘宁、比托特罗和吡布特罗)、茶碱、色甘酸钠、阿托品、异丙托溴铵、白细胞三烯拮抗剂(鲁司特、孟鲁司特、普仑司特、伊拉司特、泊比司特、SKB-106,203)、白细胞三烯生物合成抑制剂(齐留通、BAY-1005);(f)非甾族抗炎药(NSAID),例如丙酸衍生物(阿明洛芬、苯噁洛芬、布氯酸、卡洛芬、联苯丁酮酸、非诺洛芬、氟洛芬、氟联苯丙酸、布洛芬、吲哚洛芬、酮洛芬、米洛芬、萘普生、奥沙普秦、吡洛芬、普拉洛芬、舒洛芬、噻洛芬酸和硫噁洛芬)、乙酸衍生物(消炎痛、阿西美辛、阿氯芬酸、环氯茚酸、双氯芬酸、芬氯酸、芬克洛酸、双苯噻酸、呋罗芬酸、异丁芬酸、伊索克酸、oxpinac、舒林酸、硫平酸、托美丁、齐多美辛和佐美酸)、芬那酸衍生物(氟芬那酸、甲氯芬那酸、甲芬那酸、尼氟灭酸和托芬那酸)、联苯基甲酸衍生物(二氟苯水杨酸和氟苯柳)、昔康类药物(伊索昔康、吡罗昔康、舒多昔康和替诺昔康)、水杨酸盐(乙酰水杨酸、柳氮磺胺吡啶)和吡唑啉酮类(阿扎丙宗、苄哌立隆(bezpiperylon)、非普拉宗、莫非布宗、羟基保泰松、苯丁唑酮);(g)环加氧酶-2(COX-2)抑制剂;(h)磷酸二酯酶IV型(PDE-IV)抑制剂;(i)趋化因子受体(尤其是CCR-1、CCR-2、CCR-3、CXCR-3和CCR-5)的其它拮抗剂;(j)降胆固醇药物,例如HMG-CoA还原酶抑制剂(洛伐他汀、辛伐他汀和普伐他汀、氟伐他汀、阿托伐他汀、罗苏伐他汀以及其它他汀)、螯合剂(消胆胺和考来替泊)、胆固醇吸收抑制剂(依泽替米贝)、烟酸、非诺贝酸衍生物(吉非罗齐、氯贝丁酯、非诺贝特和苯扎贝特)和普罗布考;(k)抗糖尿病药,例如胰岛素、磺酰脲类、双胍(甲福明)、α-葡糖苷酶抑制剂(阿卡波糖)和格列酮类(曲格列酮和吡格列酮);(l)β干扰素制剂(β-1α干扰素、β-1β干扰素);(m)其它化合物(例如5-氨基水杨酸及其前体药物)、抗代谢药(例如硫唑嘌呤和6-巯基嘌呤)以及癌症的细胞毒素性化疗药物。
本发明化合物与第二种活性成分的重量比例可以是变化的,取决于各个成分的有效剂量。通常,使用各个活性成分的有效剂量。由此举例来讲,当本发明化合物与NSAID联合使用时,本发明化合物与NSAID的重量比例通常为约1000∶1至约1∶1000,或为约200∶1至约1∶200。本发明化合物和其它活性成分的组合通常在上述范围,但是在所有情况下,应当使用各活性成分的有效剂量。
在这样的联合应用中,本发明化合物和其它活性药物可以单独或结合给予。另外,一种成分的给予可以在其它药物之前、同时或之后给予。
本发明化合物可以通过如下途径给予:口服、胃肠外(例如肌内、腹膜内、静脉内、ICV、脑池内注射或输注、皮下注射、或者移植)、吸入喷雾、鼻内、阴道、直肠、舌下或局部,并且可以单独或共同配制为包含载体、佐剂和溶媒的合适剂量单位的制剂,所述载体、佐剂和溶媒是常规无毒的、药学上可接受的,并且适合各个给药途径。除了治疗温血动物(例如小鼠、大鼠、马、牛、绵羊、狗、猫、猴子等)外,本发明化合物还可有效用于人。
给予本发明化合物的药用组合物可以方便地呈现为剂量单位剂型,可以通过药学领域公知的任何方法制备。所有方法包括将活性成分与一种或多种辅助成分构成的载体结合。通常,如下制备药用组合物:将活性成分与液体载体、微细固体载体或这两者均匀、紧密地结合,如果需要,再将产品成形为所需制剂。在药用组合物中,活性目标化合物的含量足以对疾病的过程或病症产生所需作用。本文使用的术语“组合物”包括含有特定量特定成份的产品以及以特定量特定成分组合而直接或间接得到的任何产品。
包含活性成分的药用组合物可以为适合口服的剂型,例如为片剂、糖锭、锭剂、水性或油性混悬剂、可分散的散剂或颗粒剂、乳剂、硬质或软质胶囊剂、或者糖浆剂或酏剂。口服组合物可以根据制备药用组合物领域的任何已知方法制备,为了获得药学上美观、适口的制剂,这样的组合物可以包含一种或多种选自以下的试剂:甜味剂、调味剂、着色剂和防腐剂。片剂包含活性成分和药学上可接受的无毒赋形剂,所述赋形剂是适合制备片剂的赋形剂。这些赋形剂可以为例如惰性稀释剂,例如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;成粒剂和崩解剂,例如玉米淀粉或海藻酸;粘合剂,例如淀粉、明胶或阿拉伯树胶;润滑剂,例如硬脂酸镁、硬脂酸或滑石粉。片剂可以是无涂层的,或者它们可以通过已知技术涂覆以延迟在胃肠道崩解、吸收,由此提供长期的持续作用。例如可以使用时间延迟原料(例如单硬脂酸甘油酯或二硬脂酸甘油酯)。它们也可以通过美国专利4,256,108、4,166,452和4,265,874介绍的技术涂覆,形成用于控制释放的渗透性治疗片剂。
口服制剂还可以为硬质明胶胶囊剂,其中活性成分与惰性固体稀释剂(例如碳酸钙、磷酸钙或高岭土)混合;或者为软质明胶胶囊剂,其中活性成分与水或油性介质(例如花生油、液体石蜡或橄榄油)混合。
水性混悬剂包含活性原料和适合制备水性混悬剂的赋形剂。这样的赋形剂有悬浮剂,例如羧甲基纤维素钠、甲基纤维素、羟基-丙基甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、黄蓍胶和阿拉伯树胶;分散剂或润湿剂可以为天然存在的磷脂,例如卵磷脂;烯化氧与脂肪酸的缩合产物,例如硬脂酸聚氧乙烯;环氧乙烷与长链脂肪醇的缩合产物,例如十七乙烯氧基十六醇(heptadecaethyleneoxycetanol);或者环氧乙烷与偏酯(由脂肪酸和己糖醇衍生)的缩合产物,例如聚氧乙烯山梨糖醇单油酸酯;环氧乙烷与偏酯(由脂肪酸和己糖醇酐衍生)的缩合产物,例如聚乙烯山梨糖醇酐单油酸酯。水性混悬剂还可包含一种或多种防腐剂(例如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯)、一种或多种着色剂、一种或多种调味剂以及一种或多种甜味剂(例如蔗糖或糖精)。
可将活性成分悬浮于植物油(例如花生油、橄榄油、芝麻油或椰子油)或矿物油(例如液体石蜡)配制油性混悬剂。油性混悬剂可包含增稠剂,例如蜂蜡、固体石蜡或鲸蜡醇。可以加入甜味剂(例如上述的甜味剂)和调味剂得到适口的口服制剂。这些组合物可以通过加入抗氧剂(例如抗坏血酸)防腐。
可分散的散剂和颗粒剂适合通过加入水制备水性混悬剂,得到活性成分与分散剂或润湿剂、悬浮剂以及一种或多种防腐剂的混合物。合适的分散剂或润湿剂和悬浮剂已在上文中举例说明。还可以包含其它赋形剂,例如甜味剂、调味剂和着色剂。
本发明药用组合物还可以为水包油的乳剂形式。油相可以为植物油(例如橄榄油或花生油)或矿物油(例如液体石蜡)或它们的混合物。合适的乳化剂可以为天然存在的树胶(例如阿拉伯树胶或黄蓍胶)、天然存在的磷脂(例如大豆、卵磷脂)、衍生自脂肪酸和己糖醇酐的酯或偏酯(例如山梨糖醇酐单油酸酯)以及所述偏酯与环氧乙烷的缩合产物(例如聚氧乙烯山梨糖醇酐单油酸酯)。乳剂还可以包含甜味剂和调味剂。
糖浆剂和酏剂可以用甜味剂(例如甘油、丙二醇、山梨糖醇或蔗糖)配制。这样的制剂还可以包含镇痛剂、防腐剂、调味剂和着色剂。
药用组合物可以为无菌注射水性或油性混悬剂。这种混悬剂可以用上述合适的分散剂或润湿剂以及悬浮剂根据已知工艺配制。无菌注射制剂还可以为无毒胃肠外可接受的稀释剂或溶剂中的无菌注射溶液剂或混悬剂,例如在1,3-丁二醇中的溶液剂。在可接受的溶媒和溶剂中可以使用水、林格氏溶液和等渗氯化钠溶液。另外,无菌不挥发油通常用作溶剂或悬浮介质。用于此目的时,可以使用任何温和的不挥发油,包括合成的甘油单酯或甘油二酯。另外,脂肪酸(例如油酸)可用于注射制剂。
本发明化合物还可以栓剂形式直肠给药。这些组合物可以如下制备:将药物与合适的无刺激赋形剂混合物,所述赋形剂在常温时为固体,在直肠温度为液体,因此将在直肠熔化释放药物。这样的原料有可可油或聚乙二醇。
对于局部用药,使用包含本发明化合物的乳膏剂、软膏剂、凝胶剂、溶液剂或混悬剂等。(对于这种应用来说,局部应用应该包括漱口剂和含漱剂)
本发明药用组合物和方法可以进一步包含上述其它治疗活性化合物,这些化合物通常用于治疗上述病理疾病。
在治疗、预防、改善、控制需要调节趋化因子受体的疾病或者降低其风险时,适当的剂量水平通常为每天约0.01-500mg/kg患者体重,可以分为单剂或多剂给药。剂量水平为每天约0.1至约250mg/kg;或为每天约0.5至约100mg/kg。合适的剂量水平可以为约0.01-250mg/kg/天、约0.05-100mg/kg/天或约0.1-50mg/kg/天。在此范围内,剂量可以为0.05-0.5、0.5-5或5-50mg/kg/天。对于口服给药,组合物可以为包含1.0-1000mg活性成分、2.0-500、3.0-200、或者1、5、10、15、20、25、30、50、75、100、125、150、175、200、250、300、400、500、600、750、800、900和/或1000mg活性成分的片剂,用于对需要治疗的患者调节症状。化合物的用法为每天1-4次,优选每天1-2次。
但是,应当理解的是对于任何特定患者的具体剂量水平和给药频率可能是不同的,将取决于各种因素,包括所用具体化合物的活性、该化合物的代谢稳定性及作用时间长短、患者的年龄、体重、一般健康状况、性别和饮食、给药模式和给药时间、排泄速率、药物组合、具体疾病的严重程度以及患者正在进行的治疗。
几种制备本发明化合物的方法在以下的流程和实施例中说明。初始原料是市售的、通过已知方法制备的或者按照本文介绍的方法制备。
流程1图示了用于制备含1,1,3-三取代的环戊烷骨架的本发明化合物1-5的一个主要途径。根据此途径,使酮酸1-1(按照流程2A、2B、2C和2D制备)与胺1-2(按照流程3A-G制备)偶合。此反应可以通过不同的方式完成,包括首先用试剂例如乙二酰氯将酸转化为它的酰氯,然后与胺1-2在碱(例如三乙胺)存在下化合。用例如NaB(OAc)3H或NaBH3CN作为还原剂,使1-3与胺1-4进行还原性胺化反应,得到趋化因子受体调节剂1-5。化合物1-9(可以根据流程1介绍的化学过程合成)为立体异构体混合物(Eliel,E.E.,Wilen,S.H.,Stereochemistryof Organic Compounds,John Wiley & Sons,Inc.,New York)。具体地讲,获得的化合物1-5通常为顺式异构体和反式异构体的混合物。1-1为单一立体异构体(1-1a)时,只能获得1-5的2种可能的异构体(顺式和反式);它们可以通过多种方法分离,包括制备型TLC、快速色谱法、MPLC、或HPLC(使用手性固定相的色谱柱)。1-1为外消旋时,能获得1-5的全部4种可能异构体。它们也可通过HPLC(使用手性固定相的色谱柱)或上述方法的组合分离。外消旋的1-1的合成方法在流程2A中详细介绍,而手性1-1a的合成方法在流程2B和2C中介绍。
此外,化合物1-5本身可以被修饰而得到新的趋化因子受体调节剂1-5.1。例如化合物1-5的酯官能团可以水解为相应的羧酸,相应的羧酸也可用作趋化因子受体调节剂。
流程1

如流程1A所示,可以在多种条件下(包括三乙酰氧基硼氢化钠或氰基硼氢化钠),将酮-酯1-6用胺1-4还原性胺化形成氨基酯1-7。将酯1-7用烷基化剂(例如烷基氯、烷基溴、烷基碘)在合适碱(例如双(三甲硅烷基)氨基锂)存在下烷基化,得到中间体酯1-8。通常,上述转化形成的酯为1,3-顺-和1,3-反-非对映异构体的混合物,可以用柱色谱法分离为两个相应的非对映异构体。类似的非对映异构体分离也可以在以后实现,在酯1-8水解性裂解得到相应的酸1-9后进行。水解很容易在常规条件(包括氢氧化锂、氢氧化钠或氢氧化钾)于室温至高温下完成,这取决于酯基团及取代基R1的性质。可以利用顺式-非对映异构体酸比其反式差向异构体的溶解性小,用不同的溶剂结晶分离这些非对映异构体。
然后在标准酰胺键形成性反应条件下,包括碳化二亚胺试剂(例如DCC、EDC)以及催化剂(例如DMAP、HOAT或HOBT),用酸1-9和四氢异喹啉衍生物1-2制备式1-5化合物。
流程1A
中间体1-3还可以通过手性HPLC拆分得到1-3a和1-3b(流程1B)。然后可制备得到顺/反异构体1-5a和1-5b。
流程1B
流程2A图示了用于制备中间体1-1和中间体1-6的一个主要途径。根据此途径,在标准条件下酯化3-氧代环戊烷甲酸(2-1)(可以用以下的已知方法合成)(Stetter,H.,Kuhlman,H.,Liebigs Ann.Chim.,1979,944))。R18为叔丁基时,可以如下制备相应的酯1-6:使合适的醇,在此例中为叔丁醇,与酸2-1在硫酸存在下反应。可以通过许多方式实现2-1中氧代基团的保护(Greene,T.,Wuts,P.G.M.,ProtectiveGroups in Organic Chemistry,John Wiley & Sons,Inc.,New York,NY 1991)。可用试剂原甲酸三甲酯在合适的溶剂(例如二氯甲烷和甲醇)中于酸性催化剂存在下引入特别合适的二甲缩醛保护基团。或者,R18为甲基时,可以用原甲酸三甲酯和酸性催化剂(例如对甲苯磺酸)将酸2-1直接转化为2-3。在合适碱(例如二异丙基氨基锂)存在下,用烷基化剂(例如烷基氯、烷基溴或烷基碘)烷基化酯2-3得到中间体2-4。2-4的酯保护基团可以用许多方法脱去,这取决于酯的性质。甲酯(R18=甲基)可以在酸或碱存在下于室温或高温水解,而叔丁酯(R18=叔丁基)在酸性条件下很容易裂解。在这些条件下,二甲缩醛保护被同时脱去得到1-1。
流程2A

中间体1-1可以通过不同的方法(包括流程2B和2C介绍的方法)制备为单一的立体异构体(1-1a)。根据流程2B,外消旋的1-1可以转化为它的苄基酯。有许多方法可以实现这种酯化,其中一种是先用例如乙二酰氯将1-1转化为相应的酰氯,然后用苄基醇在碱(例如三乙胺)存在下处理。外消旋的苄基酯2-5可以通过手性制备型HPLC分离得到单一立体异构体2-5a。可以通过多种方法脱去苄基得到手性酮酸1-1a。一种便利的方法是在催化剂例如Pd/C存在下进行氢化。
流程2B

根据流程2C,手性酮酸中间体1-1a可以用市售光学纯氨基酸2-6制备。羧酸基团的保护可以通过多种方法实现。R18为甲基时,用甲醇在酸催化剂(例如HCl)存在下处理完成酯化。用Boc2O处理以保护2-7的胺基团。用烷基化剂(例如烷基氯、烷基溴或烷基碘)在适当碱(例如双(三甲硅烷基)氨基锂)存在下立体选择性烷基化酯2-8得到中间体2-9。在催化剂(例如Pd/C)存在下氢化得到2-10。根据R18基团在标准条件下水解酯获得2-11。例如R18为甲基时(甲酯),可以用碱(例如氢氧化钠、氢氧化锂或氢氧化钾)处理完成水解,可加热或不加热。Boc保护基团可以在标准酸性条件(用含HCl的溶剂(例如二噁烷)或者用TFA)下脱去。可以通过多种方法氧化2-12得到1-1a(如果组成的R1是非手性的,则1-1a为单一立体异构体,如果组成的R1有一个手性中心,则1-1a为立体异构体的混合物),包括依次用NBS、甲醇钠处理的方法。
流程2C

按照2D所示流程,在强碱(例如二异丙基氨基锂)存在下用酯2-3(R18为苄基或叔丁基)得到的烯醇化物可以与醛(R1aCHO)或酮(R1aR2aCO)反应,得到合适羟基烷基取代的中间体2-4.1。所得羟基可以用不同方法保护,包括用醋酸酐在碱(例如三乙胺)存在下处理得到中间体2-4.2。再次在适合具体保护基团的条件下脱去酯保护基团。在为叔丁酯(R18为叔丁基)时,在酸性条件下脱去保护。后者通常也引起缩醛保护基团的裂解,酮酸1-1.1可用一锅法由此制备。可以按照前述方法(略微变化以适应1-1.1中保护的羟基)转化为最终趋化因子活性调节剂1-9。
流程2D
胺1-2可以用流程3A-3G所示的多种方法制备。5-氮杂四氢异喹啉片段可以根据MarCoux,J-F.等(J.Chem.Lett.,2000,2(15),2339-2341)的文献方法制备。或者,它可以如流程3A所示方法制备。将化合物3-1(通常从市场购买获得)溴化(Br2,AcOH)得到3-2。金属卤素交换(NaH,叔丁基锂)后用DMF处理,得到醛3-3。可用甲酸钠、羟胺盐酸盐和甲酸将醛基转化为腈。所得腈3-4可以用三氯氧化磷处理得到2-氯吡啶3-5。可用丙二酸二烷基酯的钠盐置换氯基。用氢和Raney Ni还原3-6的腈基伴随环化,获得化合物3-7。根据不同的酯用不同的方法进行脱羧反应。在流程3A的情况下,用TFA对叔丁酯进行脱羧作用得到3-8。还原反应(BH3),然后用Boc2O保护所得胺得到3-9,可以方便地提纯3-9。可以用多种方法脱去Boc保护基团得到1-2a,包括用无水HCl在二噁烷或部分其它溶剂中处理的方法。
流程3A

1-2a类化合物还可以根据流程3B制备。市售3-10可以用甲基碘在碱(例如K2CO3)存在下甲基化得到3-11。与保护的哌啶酮在NH3/甲醇存在下进行环加成反应,获得5-氮杂四氢异喹啉3-12(R10c可以是不同的保护基团,例如苄基或苯甲酰基)。用氢和催化剂(例如Pd/C)氢化化合物3-12的硝基得到3-13。形成重氮盐后与硫酸加热,得到5-氮杂-7-羟基四氢异喹啉3-14。根据R10c的不同性质而用不同的方法脱去保护基团R10c。在R10c为苄基时,可以在HCl和催化剂(例如Pd/C)存在下氢化。在R10c为苯甲酰基时,可以通过加热浓HCl溶液水解。可以用Boc2O在3-15上方便地引入Boc保护基团得到3-16。然后可以结合不同的R10d得到醚化合物(参见流程3C)。最终,用HCl或TFA脱去所得化合物3-17的Boc保护基团,得到1-2b。或者,化合物3-14本身可以转化为醚化合物(根据流程3C)。可以如上所述脱去所得醚3-18的R10c,转化为化合物3-19。
流程3B
流程3B中的5-氮杂-7-羟基四氢异喹啉3-14和3-16可以转化为不同的醚化合物(参见流程3C)。可以用烷基卤和碱(例如K2CO3、NaOH或NaH)制备烷基醚,得到化合物3-19和3-22。三氟甲基醚可以如下制备:开始生成乙黄原酸甲酯(NaH,CS2;MeI),然后依次用1,3-二溴-5,5-二甲基乙内酰脲(或NBS)和HF/吡啶溶液处理得到3-20。芳基醚可以用许多方法制备,包括与芳基硼酸在醋酸铜(II)和三乙胺存在下反应的方法,得到化合物3-21。
流程3C

化合物1-2c(R5为卤化物(IVc))可以根据流程3D制备。化合物3-13可以根据典型方法通过重氮盐转化为卤化物3-22。或者,可以使用适当保护的哌啶酮的已知环加成反应。脱去保护基团R10c可以通过前述方法完成。
流程3D
此外,在结合为高级中间体后,片段1-2c可以被进一步修饰以制备含7-芳基-5-氮杂四氢异喹啉的类似物。这可以如下完成:使5-氮杂-7-卤基四氢异喹啉中间体与芳基硼酸(或芳基锡烷)偶合,通过过渡金属催化剂例如Pd(OAc)2调节。
流程3E-3G图示四氢异喹啉胺组分的制备方法。结合为1-5酰胺部分的四氢异喹啉通常在不同位置包含一个或两个取代基。这些化合物大部分不能购买得到,但是可以通过合成得到,流程3F和3G展示了代表性的合成例子。
流程3E图示了合成简单四氢异喹啉(1-2d)的例子。根据此流程,将市售4-三氟甲基苯基乙腈(3-23)在Ra-Ni存在下氢化得到相应的胺(3-24),然后用三氟醋酸酐与所述胺反应。所得酰胺(3-25)用甲醛在硫酸存在下处理得到环状化合物(3-26),将其进一步转化为四氢异喹啉(1-2d)。
流程3E

许多5-取代的四氢异喹啉也可用3-26制备(流程3F)。在5位碘化得到中间体3-28。在钯(0)催化条件下转化为氰基化合物(3-29)后,酰胺裂解为胺3-30,可以将3-30通过两个步骤以高收率转化为氨基酯1-2e。碘化合物3-28也可如实验部分所示转化为其它化合物。还可以在装配最终骨架(1-5)后,修饰这些取代基以制备新的1-5.1类趋化因子调节剂(参见流程1)。一个修饰例子是水解甲酯(1-2e的)制备相应的甲酸(参见实施例)。
流程3F

杂环的7-取代四氢异喹啉可以利用市售四氢异喹啉获得。如流程3G所示,将四氢异喹啉(3-32)用硝酸钾在浓硫酸存在下硝化。将7-硝基-四氢异喹啉3-33用三氟醋酸酐处理以保护胺,然后将所得酰胺用10%钯碳在50psi氢压力下氢化获得苯胺衍生物3-34。碱水解获得7-氨基取代的四氢异喹啉(3-35),它可用于其它CCR2拮抗剂或四氢异喹啉衍生物的合成。将3-35用氯甲酸苄基酯在有机碱(例如三乙胺或二异丙基乙胺)存在下保护,获得氨基甲酸酯3-37。此中间体可以用于制备四唑和取代的四唑(例如1-2g)。将中间体3-37用三氟醋酸酐处理生成三氟乙酰基保护的酰胺,然后将其如下转化为三氟甲基取代的四唑:使其与三苯基膦在回流加热下反应15h,然后与叠氮化钠在DMF中于室温反应。用10%钯碳在氢气氛下氢化获得杂环取代的四氢异喹啉1-2g。
或者,3-34可以如下直接衍生为三唑3-36:与N,N-二甲基甲酰胺连氮(N,N-dimethylformamide azine)在催化量酸(例如甲苯磺酸)存在下加热至回流24-48h。此中间体的碱性水解获得胺组分1-2f。
流程3G

结合到本发明化合物酰胺部分的四氢异喹啉的其它例子以及它们的合成方法在实验部分进一步介绍。
胺1-4由不同的来源获得。大部分是市售的,部分是文献已知的并可以根据公开的方法制备,部分按照本文介绍的方法制备。由于它们的结构和制备方法是多种多样的,在此部分只介绍两个流程;可以在实验部分找到胺1-4的不同合成方法。流程4A展示了一种合成4-芳基取代的哌啶的方法。烯醇三氟甲基磺酸酯4-1(根据Wustrow,D.J.,Wise,L.D.,Synthesis,(1991),993-995制备)可以与硼酸4-2按照Wustrow和Wise介绍的方法偶合。4-3中烯烃的氢化可以用氢在催化剂(例如Pd(OH)2/C)存在下实现。脱去Boc保护基团可以用标准酸性条件(例如HCl的二噁烷溶液或TFA/DCM)实现,获得哌啶1-4.1。
流程4A
流程4B展示了合成胺1-4的另一个例子。首先将市售醇(4-5)用甲烷磺酰氯磺酰化得到中间体4-6,然后用四唑直接取代得到杂芳基哌啶4-7。在标准条件下脱去Boc保护基团,得到胺盐酸盐4-8。
流程4B
流程5图示了合成趋化因子受体调节剂的另一种主要途径。根据此途径,使用肽偶合试剂(例如EDC)使中间体2-11(流程2C介绍的)与胺1-2(流程1介绍的)缩合得到5-1。将Boc保护基团在标准条件下(例如含HCl的溶剂,例如二噁烷)脱去,然后在还原剂(例如三乙酰氧基硼氢化钠)存在下,用二醛5-3处理所得胺5-2,导致双重还原性烷基化,伴随环化得到1-5.2。与流程1一样,进一步修饰(例如水解1-5.2的酯基团)可以得到新的趋化因子受体调节剂1-5.3。
流程5
流程6图示了一种制备二醛5-3的方法。根据此途径,依次用例如臭氧、甲硫醚处理使环烯6-1氧化性裂解,得到二醛。或者,二醛5-3用中间体臭氧化物6-2替代,6-2可以直接用于双重还原性胺化反应,得到1-5.2。
流程6

在某些情况下,为了促进反应或者避免不需要的反应产物,可以调整上述反应流程的顺序。以下实施例仅用于进一步举例说明,并不是对本发明公开内容的限制。
通常用旋转蒸发器在减压下浓缩溶液。快速色谱法使用硅胶(230-400目)。MPLC是指中压液相色谱法,除非另有说明,否则使用硅胶固定相。除非另有说明,否则NMR光谱用CDCl3溶液获得。偶合常数(J)单位为赫兹(Hz)。缩写:三乙胺(TEA),N,N-二异丙基乙胺(DIEA),饱和水溶液(sat′d),室温(rt),小时(h),分钟(min)。
以下的代表性方法用于制备下文实施例中使用的化合物,可以替代下文实施例使用的不能购买得到的化合物。
在某些情况下,为了促进反应或者避免不需要的反应产物,可以调整上述反应流程的顺序。以下实施例仅用于进一步举例说明,并不是对本发明公开内容的限制。
通常用旋转蒸发器在减压下浓缩溶液。快速色谱法使用硅胶(230-400目)。除非另有说明,否则NMR光谱用CDCl3溶液获得。偶合常数(J)单位为赫兹(Hz)。缩写:三乙胺(TEA),N,N-二异丙基乙胺(DIEA),饱和水溶液(sat′d),室温(rt),小时(h),分钟(min)。
以下的代表性方法用于制备下文实施例中使用的化合物,可以替代下文实施例使用的不能购买得到的化合物。
中间体1
步骤A:

将4-三氟甲基苯基乙腈(10g,49mmol)的乙醇(100mL)和氢氧化铵(20mL 29.3%水溶液)溶液用Raney镍(1g)氢化16h。通过硅藻土过滤除去催化剂,将滤液蒸发至干。将纯残余物滴加到冷(0℃)三氟醋酸酐(25mL,180mmol),在0℃搅拌所得混合物30min。将反应混合物倾在冰(250mL)上,搅拌所得混合物30min,然后过滤移出沉淀,风干得到白色固体产物(13.4g,90%)。
步骤B:

步骤A产物(13.4g,44.0mmol)和多聚甲醛(2g,50mmol)的混合物中一次性加入浓硫酸(90mL)和冰醋酸(60mL)的混合物,在室温搅拌所得混合物16h。将反应混合物倾在冰和水(1L)的混合物上,用乙酸乙酯(3×150mL)萃取;合并的乙酸乙酯层用水(3×500mL)、饱和碳酸氢钠(200mL)和饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤后真空蒸发。残余物用二氧化硅柱色谱法提纯(用10%Et2O/己烷洗脱)得到产物(8.29g,60%)。
步骤C:

步骤B制备的三氟乙酰胺(8.29g,26.0mmol)的乙醇(200mL)溶液中加入碳酸钾(20g,150mmol)的水(50mL)溶液,回流搅拌所得混合物1h。旋转蒸发除去乙醇,将水(150mL)加入残余物。用CH2Cl2(3×100mL)萃取,合并的二氯甲烷层用饱和氯化钠(100mL)洗涤,用硫酸钠干燥,过滤后真空蒸发得到产物(5.2g,91%);1H NMR 500MHz(CDCl3)δ=1.81(1H,br s),2.84(2H,d,J=6.0Hz),3.15(2H,t,J=6.0Hz),4.05(2H,s),7.19(1H,d,J=8.0Hz),7.27(1H,s),7.37(1H,d,J=8.0Hz)。
中间体2
步骤A:
5-三氟甲基-2-吡啶醇(51.0g,307mmol)和乙酸钠(26.2g,319mmol)的冰醋酸(200mL)溶液中加入溴(16.7mL,325mmol),将所得混合物在80℃加热2.5h。让反应物冷却至室温,然后减压蒸发。残余物用饱和碳酸氢钠溶液中和,用乙酸乙酯(3×200mL)萃取。合并有机层,用硫酸镁干燥,过滤,真空蒸发获得74.45g(98.7%)粗产物。1H NMR(400MHz,CDCl3)δ8.04(d,J=2.6Hz,1H),7.89(m,1H)。
步骤B:
在氮气氛下,将取代的吡啶(步骤A,48.8g,202mmol)分为小批量加入NaH(8.9g,220mmol)的无水THF(500mL)悬浮液。在中间体加入完毕后,冷却反应混合物至-78℃,用注射器滴加叔丁基锂(260mL,444mmol)。在搅拌5min后,缓慢加入DMF(50mL,707mmol)以维持温度低于-50℃。然后将所得混合物搅拌10h,让其升至室温。混合物用2N HCl猝灭,然后用乙酸乙酯(1000mL)稀释。分离出有机层,用盐水洗涤,用硫酸镁干燥,真空蒸发。所需产物从乙酸乙酯和己烷析出,过滤获得浅褐色固体(28.55g,73.8%)。1H NMR(500MHz,CD3OD)δ10.13(s,1H),8.21(s,2H)。
步骤C:

将步骤B的中间体(18g,95mmol)、甲酸钠(7.1g,105mmol)、羟胺盐酸盐(7.3g,110mmol)和甲酸(150mL)的混合物在室温搅拌2h,然后回流过夜。冷却反应混合物,在室温静置7天。将反应物倾入水中,用乙酸乙酯(3x)萃取。合并的有机层用水(2x)、饱和NaHCO3和盐水洗涤,用硫酸钠干燥,过滤,真空浓缩获得所需褐色粉末状产物(17.84g,89.8%)。1H NMR(400MHz,CD3OD)δ8.37(d,J=2.7Hz,1H),8.19(q,J=0.7Hz,0.3??/Hz,1H)。
步骤D:
三氯氧化磷(13.4mL,144mmol)和苯醌(8.7mL,73.4mmol)的混合物中加入步骤C的产物(24.6g,131mmol),将所得混合物回流3h。冷却反应物至100℃,然后缓慢加入水(70mL)。进一步冷却混合物至室温,用饱和碳酸氢钠溶液小心中和。水层用乙酸乙酯(3x)萃取,合并有机层,用硫酸镁干燥,过滤,真空蒸发。粗产物用快速色谱法提纯获得(23.5g,87.0%)所需化合物。1H NMR(500MHz,CDCl3)δ8.88(d,J=2.0Hz,1H),8.26(d,J=2.5Hz,1H)。
步骤E:

在氮气氛下,用注射器向NaH(7.8g,200mmol)的THF(100mL)悬浮液滴加丙二酸叔丁酯甲酯(20mL,120mmol)的无水THF(100mL)溶液。搅拌反应混合物0.5h,然后用注射器缓慢加入步骤D制备的中间体(20.1g,97.6mmol)的THF(200mL)溶液。在室温搅拌反应物过夜,然后用氯化铵饱和溶液猝灭。分离出有机层,水层用乙酸乙酯(3x)萃取。合并的有机层用水(3x)洗涤,用硫酸钠干燥,过滤,真空蒸发。快速色谱法获得31.76g(94.6%)所需纯净产物。1H NMR(500MHz,CDCl3)δ9.03(d,J=1.5Hz,1H),8.25(d,J=2.0Hz,1H),5.25(s,1H),3.86(s,3H),1.52(s,9H)。
步骤F:

将Raney Ni(1g)和步骤E产物(18.2g,52.9mmol)的乙醇(130mL)悬浮液置于Parr装置,在40psi氢化过夜。悬浮液通过硅藻土过滤,真空蒸发滤液获得16.35g(97.8%)粗产物。1H NMR(500MHz,CDCl3)δ8.83(s,1H),7.89(s,1H),7.82(s,1H),4.83(d,J=16Hz,1H),4.72(s,1H),4.49(d,J=16Hz,1H),1.45(s,9H)。
步骤G:

步骤F产物(16g,51mmol)的DCM(60mL)混合物中加入TFA(30mL),在室温搅拌所得混合物0.5h。减压蒸发溶液,将残余物溶于DCM。缓慢加入饱和碳酸氢钠溶液中和混合物,移出有机层。水溶液用DCM(4x)萃取,然后合并所有有机层,用硫酸钠干燥,过滤,真空蒸发获得10.42g(95.2%)所需产物。1H NMR(400MHz,CDCl3)δ8.81(s,1H),7.78(s,1H),7.30(s,1H),4.63(s,2H),3.90(s,2H)。
步骤H:

步骤G产物(18.0g,83.3mmol)的THF(50mL)溶液中加入1.0M甲硼烷的THF溶液(417mL,420mmol),在室温搅拌所得溶液过夜。减压蒸发溶液,然后将残余物用1%HCl/MeOH溶液处理,将所得混合物在50℃加热过夜,以分解甲硼烷络合物。用酸的甲醇溶液重复处理两次以确保消除甲硼烷络合物。将此反应粗产物立即用于下一反应。
将以上粗产物(83.3mmol,假定100%转化)和DIEA(43mL,250mmol)的DCM溶液用二碳酸二叔丁酯(36.4g,167mmol)处理,所得混合物在室温搅拌过夜。溶液用饱和碳酸氢钠溶液、水和盐水洗涤。合并水层,再用DCM(2x)洗涤。然后用硫酸钠干燥合并的有机层,过滤,蒸发至干。粗产物用快速色谱法和MPLC提纯获得(11.89g,47.2%,最后两个步骤)黄色固体。
1H NMR(500MHz,CDCl3)δ8.69(s,1H),7.66(s,1H),4.67(s,2H),3.79(t,J=6.0Hz,2H),3.08(t,J=5.5Hz,2H),1.51(s,9H)。
步骤I:
将步骤H的产物(11.89g)用4M HCl的二噁烷溶液处理。在室温搅拌溶液2h,然后真空蒸发获得黄色粉末状中间体2(10.85g,99%)。LC-MS C9H10F3N2[M+H+]计算值202.07,实测值203.0。
中间体3
步骤A:

将3-氧代环戊烷-甲酸甲酯(20g,160mmol)和原甲酸三甲酯(85mL,780mmol)的甲醇溶液用催化量对甲苯磺酸(3.00g,15.6mmol)处理,在室温搅拌所得溶液4h。减压蒸发溶剂,然后将残余物溶于乙醚(600mL)。溶液用饱和碳酸氢钠(2×200mL)、水(150mL)和盐水(200mL)洗涤,用无水硫酸钠干燥,过滤,然后如前所述蒸发溶剂。快速柱色谱法提纯(洗脱剂:25%乙醚/戊烷)获得21.52g(73%)所需澄清油状产物。1H NMR(500MHz,CDCl3)δ3.68(s,3H),3.21(d,J=9.9Hz,6H),2.89(p,J=8.5Hz,1H),2.14-2.05(m,2H),2.02-1.80(m,4H)。
步骤B:
火焰干燥的500mL圆底烧瓶中装入150mL无水THF,然后在氮气氛下用丙酮/干冰浴冷却至-78℃。用注射器将二异丙基胺(19.2mL,137mmol)加入冷却的溶剂,然后缓慢加入2.5M正丁基锂的己烷溶液(55mL,140mmol)。在搅拌5min后,用注射器滴加步骤A的甲基缩酮中间体3(21.52g,114.4mmol)的50mL THF溶液,在-78℃搅拌所得混合物2h。然后用注射器滴加2-碘丙烷(34.3mL,343mmol),搅拌所得混合物过夜,让其缓慢升至室温。用10%柠檬酸溶液猝灭反应物,分离有机物。水层用乙醚(3×150mL)萃取,合并所有的有机物,用无水硫酸钠干燥,过滤,减压蒸发。粗产物用快速柱(flask column)提纯(洗脱剂:20%乙醚/戊烷)获得16.74g(64%)所需产物。1H NMR(400MHz,CDCl3)δ3.69(s,3H),3.18(d,J=20.5Hz,6H),2.57(d,J=13.9Hz,1H),2.29-2.20(m,1H),1.90(p,J=6.8Hz,1H),1.88-1.80(m,2H),1.69-1.61(m,2H),0.89(dd,J=11.9Hz,6.8Hz,6H)。
步骤C:
将酯(中间体3步骤B介绍的,16.7g,72.7mmol)的乙醇(30mL)溶液用5M NaOH(55mL)处理,将所得混合物加热至回流3天。然后冷却混合物至室温,用浓盐酸酸化。减压蒸发有机溶剂,水层用DCM(5×100mL)萃取。合并有机萃取液,用无水硫酸镁干燥,过滤,真空蒸发获得粗制的黄色油状3-氧代环戊烷甲酸(11.07g,90%)。1HNMR(500MHz,CDCl3)δ2.70(d,J=18.1Hz,1H),2.44-2.39(m,1H),2.30-2.15(m,2H),2.14(dd,J=18.1,1.0Hz,1H),2.06(p,J=6.9Hz,1H),1.98(m,1H),0.98(dd,J=11.4,6.9Hz,6H)。
步骤D:
方法A:
酸(中间体3步骤C介绍的,2.00g,11.8mmol)的DCM(50mL)溶液中加入乙二酰氯(1.54mL,17.6mmol),然后加入2滴DMF。在室温搅拌溶液80min,然后减压蒸发。将残余物溶于DCM(2mL),用注射器加入到准备的中间体1(2.36g,11.8mmol)和三乙胺(2.13mL,15.3mmol)的DCM(40mL)溶液。在室温搅拌所得混合物18h,然后用水(25mL)猝灭。分离有机物,用1N HCl、饱和碳酸氢钠和盐水洗涤,用无水硫酸镁干燥,过滤,然后蒸发。粗产物用MPLC提纯(洗脱剂:60%乙酸乙酯/己烷)获得中间体3(3.18g,77%)。1H NMR(500MHz,CDCl3)δ7.46(d,J=7.3Hz,1H),7.39(s,1H),7.29(d,J=7.7Hz,1H),4.81(m,2H),3.93(m,1H),3.82(m,1H),2.94(m,3H),2.54(m,1H),2.43(d,J=8.5Hz,1H),2.32(m,2H),2.26(p,J=6.6Hz,1H),2.16(m,1H),0.93(dd,J=19.7Hz,6.8Hz,6H)。LC-MS C19H23F3NO2计算值353.16,实测值:[M+H+]354.25。
方法B:
将步骤C制备的酸中间体3(1.0g,5.9mmol)、中间体1(1.18g,5.88mmol)、DMAP(71mg,0.59mmol)、N,N-二异丙基乙胺(1.02mL,5.88mmol)和二氯甲烷(20mL)的混合物用1-[3-(二甲基氨基)丙基]-3-乙基碳化二亚胺盐酸盐(EDC,2.25g,11.7mmol)处理,在室温搅拌过夜。反应混合物用二氯甲烷(30mL)稀释,用水(2×20mL)、盐水(1×30mL)洗涤,用无水硫酸钠干燥,蒸发溶剂。MPLC提纯(洗脱剂:60%乙酸乙酯/己烷)获得纯净化合物1.08g(52%)。LC-MS C19H23F3NO2计算值353.16,实测值[M+H+]354.25。
中间体4
酸(中间体3步骤C介绍的,540mg,3.20mmol)的DCM(50mL)溶液中加入乙二酰氯(0.834mL,9.60mmol),然后加入2滴DMF。在室温搅拌溶液80min,然后减压蒸发。将残余物溶于DCM(2mL),用注射器加入准备的中间体2(880mg,3.20mmol)和三乙胺(0.820mL,6.50mmol)的DCM(20mL)溶液。在室温搅拌所得混合物18h,然后用水(25mL)猝灭。分离有机物,用饱和碳酸氢钠和盐水洗涤,用无水硫酸钠干燥,过滤,然后蒸发。粗产物用MPLC提纯(用阶段梯度洗脱剂:0-70%乙酸乙酯/己烷)获得中间体2(720mg,64%)。ESI-MS计算值C18H21F3N2O2:354.16;实测值355(M+H)
中间体5
通过手性分离将中间体4拆分为它的单独的对映异构体,采用配备制备型ChiralPak AD柱的HPLC。如下完成分离:每次注射100mg,洗脱剂采用25%异丙醇和75%庚烷,流速9mL/min。
中间体6

步骤A:
方法A:
将3-氧代-环戊烷甲酸(Stetter,H.,Kuhlmann,H.Liebigs Ann.Chem.,1979,7,944-9)(5.72g,44.6mmol)的二氯甲烷(30mL)溶液用N,N′-二异丙基-O-叔丁基-异脲(21.2mL,89.3mmol)处理,在室温搅拌反应混合物过夜。滤除沉淀N,N′-二异丙基脲,真空浓缩滤液,蒸馏提纯残余物(bp:125-129℃,18mmHg)获得4.74g(58%)纯净产物。1H NMR(500MHz,CDCl3):δ3.02(p,J=7.8Hz,1H),2.05-2.50(m,6H),1.45(s,9H)。13C NMR(125MHz,CDCl3):δ217.00,173.47,80.99,41.88,41.14,27.94,26.57。
方法B:
2L圆底RBF中装入无水硫酸镁(113g,940mmol),加入二氯甲烷(940mL)。在搅拌下,将悬浮液用浓硫酸(12.5mL,235mmol)处理,然后在15min内加入3-氧代-环戊烷甲酸(30.1g,235mmol)。在搅拌15min后,加入叔丁醇(87g,1.2mol)。用塞子封闭反应容器以助保留异丁烯,在室温搅拌72h。通过硅藻土垫滤出固体,将滤液体积减至约500mL,用饱和碳酸氢钠溶液(2×150mL)洗涤。用无水硫酸镁干燥有机相,过滤,减压蒸馏除去溶剂(180mmHg)。粗产物通过蒸馏提纯获得39.12g(90%)纯净产物。
步骤B:
将3-氧代环戊烷甲酸叔丁酯(11.54g,62.64mmol)的二氯甲烷(200mL)溶液用原甲酸三甲酯(41.4mL,251mmol)在对甲苯磺酸(400mg)存在下处理,在室温搅拌48h。将黑色反应混合物倾在饱和碳酸氢钠溶液上,粗产物用二氯甲烷萃取。用无水硫酸镁干燥合并的有机萃取液,真空除去溶剂,蒸馏提纯粗产物(bp.:104℃,4mmHg)获得12.32g(85%)所需产物。1H NMR(500MHz,CDCl3):δ3.21(s,3H),3.20(s,3H),2.80(m,1H),2.10-1.80(bm,6H),1.46(s,9H)。13C NMR(125MHz,CDCl3):δ174.9,111.2,80.3,67.8,49.2,42.5,37.4,33.8,28.3,22.0。
步骤C:
火焰干燥的500mL圆底烧瓶中装入100mL无水THF,然后在氮气氛下用丙酮/干冰浴冷却至-78℃,用注射器将二异丙基胺(7.9mL,56mmol)加入冷却的溶剂,然后缓慢加入2.5M正丁基锂的己烷溶液(22.6mL,56.45mmol)。在搅拌5min后,用注射器滴加缩醛(中间体6步骤B介绍的,10.0g,43.4mmol)的50mL THF,在-78℃搅拌所得混合物2h。然后用注射器滴加乙醛(7.3mL,130mmol),在-78℃搅拌所得混合物2h。将混合物倾入10%柠檬酸溶液(300mL)中猝灭反应物,然后用二氯甲烷(2×150mL)萃取。合并有机物,用无水硫酸镁干燥,过滤,减压蒸发。在反应或后续处理时,部分缩醛水解为酮,因此,将粗制的混合物直接用于下一步骤无需再提纯。
步骤D:
将粗制的中间体(中间体6步骤C介绍的,56.45mmol,假设步骤C转化率100%)用10%三氟乙酸的二氯甲烷溶液处理,在室温搅拌所得混合物过夜。真空浓缩反应物,然后用水稀释,用二氯甲烷萃取。合并有机物,用无水硫酸镁干燥,过滤,减压蒸发获得8.04g(83%)粗产物,直接使用无需再提纯。
步骤E:

酸(中间体6步骤D介绍的,300mg,1.74mmol)、中间体2(486,1.74mmol)、HOAt(237mg,1.74mmol)、N,N-二异丙基乙胺(0.606mL,3.48mmol)和二氯甲烷(15mL)的混合物用1-[3-(二甲基氨基)丙基]-3-乙基碳化二亚胺盐酸盐(EDC,667mg,3.48mmol)处理,在室温搅拌5天。反应混合物用二氯甲烷(30mL)稀释,用水(20mL)、盐水(20mL)洗涤,用无水硫酸钠干燥,过滤,真空蒸发溶剂。通过制备型板(洗脱剂:100%乙酸乙酯)提纯获得中间体6260mg(42%)。
中间体7
步骤A

将2-氟-4-三氟甲基苯基乙腈(10g,49mmol)的乙醇(100mL)和氢氧化铵(20mL 29.3%水溶液)溶液利用Raney镍(1g)氢化16h。通过硅藻土过滤除去催化剂,将滤液蒸发至干。将纯残余物滴加到冷(0℃)三氟醋酸酐(25mL,180mmol),在0℃搅拌所得混合物30min。将反应混合物倾在冰(250g)上,搅拌所得混合物30min,然后过滤移出沉淀,风干得到白色固体产物(13.4g,90%);1H NMR 500MHz(CDCl3)δ=3.02(2H,t,J=7.0Hz),3.66(2H,q,J=6.6Hz),6.44(1H,brs),7.34(2H,m),7.41(1H,d,J=7.8Hz)。
步骤B

步骤A产物(13g,44mmol)和多聚甲醛(2.0g,48mmol)的混合物中一次性加入浓硫酸(90mL)和冰醋酸(60mL)的混合物,在室温搅拌所得混合物16h。将反应混合物倾在冰和水(1L)的混合物上,用乙酸乙酯(3×150mL)萃取;合并的乙酸乙酯层用水(3×500mL)、饱和碳酸氢钠(200mL)和饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤后真空蒸发。残余物用二氧化硅柱色谱法提纯(用10%Et2O/己烷洗脱)得到产物(8.29g,60%);1H NMR 500MHz(CDCl3)δ=3.01(2H,m),3.91和3.97(2H,t,J=6.2Hz),4.83和4.88(2H,s),7.21-7.28(3H,m)。
步骤C

步骤B制备的三氟乙酰胺(8.3g,26mmol)的乙醇(200mL)溶液中加入碳酸钾(20g,150mmol)的水(50mL)溶液,回流搅拌所得混合物1h。减压除去乙醇,将水(150mL)加入残余物,用CH2Cl2(3×100mL)萃取。合并的二氯甲烷层用饱和NaCl(100mL)洗涤,用硫酸钠干燥,过滤后真空蒸发,得到产物(5.2g,91%);1H NMR 500MHz(CDCl3)δ=1.74(1H,br s),2.78(2H,d,J=6.0Hz),3.17(2H,t,J=6.0Hz),4.05(2H,s),7.04-7.14(3H,m)。
中间体8

步骤A

将三颈圆底烧瓶装上加液漏斗和冷凝器,加入锌粉(2.45g,37.4mmol),用火焰干燥。在冷却后,用氮气净化系统,加入6mL THF,然后加入1,2-二溴乙烷(0.298mL,3.46mmol)。将混合物用加热枪加热至剧烈回流,回流搅拌~30s(观测到气体逸出),然后冷却至室温。重复加热、冷却两次。然后加入氯代三甲基硅烷(0.402mL,3.17mmol),在室温搅拌混合物20min。在约1min内加入N-叔丁氧基羰基-4-碘-哌啶(已知的:Billotte,S.Synlett(1998),379.,8.97g,28.8mmol)的15mL THF溶液。在50℃搅拌反应混合物1.5h,然后冷却至室温。其间,在氮气氛下将三-2-呋喃基膦(267mg,1.15mmol)和三(二亚苄基丙酮)-二钯(0)氯仿加合物(298mg,0.288mmol)的混合物溶于6mLTHF,在室温搅拌15min,加入有机锌溶液。然后加入2-溴嘧啶(5.50g,34.6mmol)的58mL THF和20mL N,N-二甲基乙酰胺溶液。将反应混合物加热至80℃,搅拌3.5h,然后冷却至室温,搅拌36h。通过硅藻土过滤反应混合物,滤饼用乙酸乙酯洗涤。滤液进一步用乙酸乙酯稀释,用饱和碳酸氢钠溶液洗涤。水层再次用乙酸乙酯萃取,合并有机层,用水洗涤两次,用盐水洗涤一次。有机相用无水硫酸镁干燥,过滤,然后浓缩。用快速色谱法提纯(二氧化硅,阶段梯度:25%乙酸乙酯/己烷,40%乙酸乙酯/己烷,60%乙酸乙酯/己烷,80%乙酸乙酯/己烷,100%乙酸乙酯)获得4.92g纯净4-(2-嘧啶基)-哌啶产物(65%)。1H NMR(500MHz,CDCl3):δ8.70(d,J=5.0Hz,2H),7.16(app t,J=4.5Hz,1H),4.24(br s,2H),3.05(m,1H),2.89(br m,2H),2.01(br d,J=13Hz,2H),1.84(dq,J=4.5,12.5Hz,2H),1.49(s,9H)。
步骤B
将步骤A制备的N-叔丁氧基羰基哌啶(4.64g,17.6mmol)溶于4NHCl的二噁烷溶液(50mL),在室温搅拌2.25h。浓缩反应混合物获得4.16g哌啶盐酸盐(100%),不需要再提纯。1H NMR(500MHz,CD3OD):δ8.95(d,J=5.5Hz,2H),7.60(t,J=5.0Hz,1H),3.53(dt,J=13,3.5Hz,2H),3.35(tt,J=4.0,11.0Hz,1H),3.20(brt,J=13.8Hz,2H),2.30(br d,J=14.0Hz,2H),2.11-2.20(m,2H);ESI-MS C9H13N3的计算值:163;实测值:164(M+H)。
中间体9

此中间体按照用于中间体8的方法制备,但是用4-溴嘧啶替代2-溴嘧啶。LC-MS C9H13N3计算值163.28,实测值[M+H]+64。
中间体10
4-(1H-1,2,4-三唑-1-基)哌啶盐酸盐

步骤A
4-羟基哌啶-1-甲酸叔丁酯
向4-羟基哌啶(60.8g)的二氯甲烷(500mL)搅拌溶液非常缓慢地加入二碳酸二叔丁酯(19g,0.55mol)的二氯甲烷(500mL)溶液。在加入后,加入耗时1h,在室温搅拌所得混合物5h。然后将混合物用饱和NaHCO3、3N HCl和盐水洗涤,干燥,然后蒸发得到稠油状4-羟基哌啶-1-甲酸叔丁酯(90g)。
步骤B:4-[(甲基磺酰基)氧基]哌啶-1-甲酸叔丁酯
在0℃,向4-羟基哌啶-1-甲酸叔丁酯(21.1g,100mmol)和三乙胺(22mL)的二氯甲烷(250mL)搅拌溶液中缓慢加入甲烷磺酰氯(9.0mL,1.1eq.)。再搅拌所得混合物1h,在此期间形成白色固体。然后将混合物用3N HCl洗涤,用Na2SO4干燥,然后蒸发得到白色固体4-[(甲基磺酰基)氧基]哌啶-1-甲酸叔丁酯(29.2g)。1H NMR(400MHz,CDCl3):δ4.92-4.87(m,1H),3.75-3.69(m,2H),3.34-3.28(m,2H),3.05(s,3H),2.01-1.94(m,2H),1.87-1.78(m,2H)。
步骤C:4-(1H-1,2,4-三唑-1-基)哌啶盐酸盐
在室温向4-[(甲基磺酰基)氧基]哌啶-1-甲酸叔丁酯(5.9g,21mmol)和1,2,4-三唑(1.8g,25mmol eq.)的DMF搅拌溶液中加入氢化钠(60%分散于矿物油,1.0g,25mmol)。在60℃搅拌混合物5天,TLC证实没有剩下初始甲磺酸酯。将混合物倾入冰水中,用乙酸乙酯(3x)萃取。干燥有机层,蒸发,用二氧化硅快速柱提纯(用0-10%甲醇/乙酸乙酯洗脱)得到白色固体4-(1H-1,2,4-三唑-1-基)哌啶-1-甲酸叔丁酯。然后将固体用氯化氢的二噁烷(4N,10mL)处理2h。然后将混合物蒸发除去大部分二噁烷得到白色固体,将其用乙酸乙酯洗涤得到所需4-(1H-1,2,4-三唑-1-基)哌啶盐酸盐(5.55g)。1H NMR(300MHz,CD3OD):δ10.00(s,1H),8.97(s,1H),5.10-5.00(m,1H),3.63-3.58(br.d,2H),3.33-3.26(br.d,2H),2.50-2.30(m,4H)。
以下中间体10-16按照类似于中间体10的方法制备,使用4-[(甲基磺酰基)氧基]哌啶-1-甲酸叔丁酯和合适的杂环。
中间体11
4-(1H-吡唑-1-基)哌啶盐酸盐
用吡唑按照中间体10的方法制备。
中间体12
4-(1H-咪唑-1-基)哌啶盐酸盐

用咪唑按照中间体10的方法制备:1H NMR(400MHz,CD3OD):δ9.18(s,1H),7.86(s,1H),7.65(s,1H),4.9-4.8(隐藏在CD3OD峰下,1H),3.61-3.61(br.d.,2H),3.33-3.26(m,2H),2.49-2.45(br.d,2H),2.39-2.28(m,2H)。
中间体13
4-(1H-1,2,3-三唑-1-基)哌啶盐酸盐

用1,2,3-三唑按照中间体10的方法制备。
4-(1H-1,2,3-三唑-1-基)哌啶盐酸盐:1H NMR(400MHz,CD3OD):δ8.77(s,1H),8.54(s,1H),5.26-5.19(m,1H),3.65-3.59(m,2H),3.37-3.29(m,2H),2.60-2.54(m,2H),2.50-2.39(m,2H)。
中间体14
4-(2H-1,2,3-三唑-2-基)哌啶盐酸盐
用1,2,3-三唑按照中间体10的方法制备。
4-(2H-1,2,3-三唑-2-基)哌啶盐酸盐:1H NMR(400MHz,CD3OD):δ7.72(s,2H),4.94-4.87(m,1H),3.54-3.48(m,2H),3.28-3.22(m,2H),2.46-2.32(m,4H)。
中间体15
4-(1H-四唑-1-基)哌啶盐酸盐

用四唑按照中间体10的方法制备。
4-(1H-四唑-1-基)哌啶盐酸盐:1H NMR(400MHz,CD3OD):δ8.77(s,1H),5.30-5.23(m,1H),3.58-3.53(m,2H),3.35-3.29(m,2H),2.58-2.2.52(m,2H),2.48-2.38(m,2H)。
中间体16
4-(2H-四唑-2-基)哌啶盐酸盐

用四唑按照中间体10的方法制备。
4-(2H-四唑-2-基)哌啶盐酸盐:1H NMR(400MHz,CD3OD):δ9.32(s,1H),5.08-5.00(m,1H),3.61-3.57(m,2H),3.33-3.28(m,2H),2.52-2.47(m,2H),2.42-2.32(m,2H)。
中间体17

用5-甲基四唑按照中间体10的方法制备。
1H NMR(400MHz,CD3OD):δ5.08-5.00(m,1H),3.61-3.57(m,2H),3.33-3.28(m,2H),2.52-2.47(m,2H),2.42-2.32(m,2H),1.68(s,3H)。
中间体18
步骤A
将羟基胺盐酸盐(8.26g,119mmol)和三乙胺(16.6mL,119mmol)在50mL DMSO中混合。过滤悬浮液以除去三乙胺盐酸盐,滤饼用THF洗涤。将滤液部分浓缩除去THF。然后将市售1-叔丁氧基羰基-4-氰基哌啶(5.0g,24mmol)加入以上DMSO溶液,在75℃搅拌所得反应混合物3h,然后在室温搅拌过夜。反应混合物用乙酸乙酯稀释,用水洗涤。水层再次用乙酸乙酯萃取,合并的有机层用水洗涤四次,用盐水洗涤一次。用无水硫酸镁干燥有机层,过滤,然后浓缩得到3.51g产物。
步骤B
将步骤A中间体(1.02g,4.19mmol)的20mL THF溶液用硫羰基二咪唑(897mg,5.03mmol)处理,随后观测到气体释放和放热。在室温搅拌反应混合物1h,然后转移到硅胶#60(20g)的180mL 5∶1CHCl3/甲醇悬浮液中。在室温搅拌反应混合物5天,然后过滤,浓缩。MPLC提纯(二氧化硅,50%乙酸乙酯/己烷)获得143mg噻二唑酮。
Boc中间体1H NMR(500MHz,CD3OD):δ4.16(m,2H),2.86(t,J=11.5Hz,2H),2.77(tt,J=4.0,11.0Hz,1H),1.98(dd,J=2.0,13.0Hz,2H),1.73(dq,J=4.5,12.0Hz,2H),1.47(s,9H)。
将Boc中间体(139mg,0.487mmol)溶于4N HCl/二噁烷(5mL),在室温搅拌1.5h。浓缩反应混合物得到94.3mg哌啶盐酸盐产物。
中间体19
4-(1H-吡唑-3-基)哌啶
步骤A:4-(1H-吡唑-3-基)吡啶
4-乙酰基吡啶(75mL,0.68mol)和甲酸乙酯(109mL)的无水苯(1L)混合物中加入甲醇钠(73g),将所得混合物回流18h。冷却混合物,从粘性固体(在反应中形成)倾析出苯。将粗产物溶于水(700mL),加入肼二盐酸盐,在室温搅拌所得混合物2h。加入5N NaOH溶解混合物。过滤移出沉淀,干燥得到4-(1H-吡唑-3-基)吡啶(35g)。
步骤B:1-苄基-4-(1H-吡唑-3-基)-1,2,3,6-四氢吡啶
4-(1H-吡唑-3-基)吡啶(9.6g)的2-丙醇(60mL)热(80℃)溶液中加入苄基溴(20mL,2.5eq.),回流加热所得混合物10min。在冰浴中冷却后,过滤沉淀,再用2-丙醇洗涤,风干。在0℃将固体悬浮于乙醇,在30min内分几批加入硼氢化钠(13g),再次搅拌混合物30min。小心加入水猝灭反应物,蒸发除去乙醇,残余物在二氯甲烷和水间分配。用硫酸镁干燥有机层,过滤,然后蒸发得到1-苄基-4-(1H-吡唑-3-基)-1,2,3,6-四氢吡啶(16g)。
步骤C:4-(1H-吡唑-3-基)哌啶
将1-苄基-4-(1H-吡唑-3-基)-1,2,3,6-四氢吡啶溶液(16g)用钯碳(10%,1g)在40psi下氢化过夜。通过硅藻土过滤除去催化剂,蒸发滤液。NMR证实产物为1-苄基-4-(1H-吡唑-3-基)-哌啶(16g)。
1-苄基-4-(1H-吡唑-3-基)-哌啶(16g)和甲酸(30mL)的乙醇(400mL)溶液中加入钯碳(10%,2g),在室温搅拌所得混合物过夜。过滤除去催化剂,蒸发滤液。通过加入二碳酸二叔丁酯(2eq.)和三乙胺(1.5eq.)的二氯甲烷溶液提纯产物,得到Boc保护的中间体。蒸发后通过二氧化硅柱色谱法提纯(用20-40%乙酸乙酯/己烷洗脱),得到纯净4-(1H-吡唑-3-基)哌啶-1-甲酸叔丁酯。然后将Boc中间体用HCl的甲醇溶液处理,得到4-(1H-吡唑-3-基)哌啶盐酸盐(3.5g)。因为形成二-Boc产物而损失原料,没有收集二-Boc产物。1H NMR(400MHz,CDCl3):δ8.00(s,2H),3.48(br.d,J=13Hz,2H),3.28-3.20(m,1H),3.13(br.t,J=13Hz,2H),2.23(br.d,J=14Hz,2H),1.97-1.85(m,2H)。
中间体20

步骤A:
向4-溴苯乙胺氢溴酸盐(25g,89mmol)、吡啶(36mL,445mmol)和CH2Cl2(100mL)的冷(0℃)混合物滴加三氟醋酸酐(18.8mL,133mmol)。在加入完毕后,在室温搅拌混合物48h,然后倾在冰(500g)上。混合物用CH2Cl2(4×100mL)萃取,合并的二氯甲烷层用1N HCl(4×100mL)、饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤后真空蒸发,得到产物(26.13g,100%);1H NMR 500MHz(CDCl3)δ=2.86(2H,t,J=7.1Hz),3.59(2H,q,J=6.6Hz),6.57(1H,br s),7.09(2H,d,J=8.5Hz),7.43(2H,d,J=8.5Hz)。
步骤B:

步骤A产物(26g,88mmol)和多聚甲醛(5.6g,130mmol)的混合物中一次性加入浓硫酸(130mL)和冰醋酸(195mL)的混合物,在室温搅拌所得混合物17h。将反应混合物倾在冰/水(1.5L)上,用乙酸乙酯(3×300mL)萃取,合并的乙酸乙酯层用水(2×600mL)、饱和NaHCO3(300mL)和饱和NaCl(150mL)洗涤,用硫酸镁干燥,过滤后真空蒸发。将残余物溶于乙醇(450mL),加入碳酸钾(60g,434mmol)的水(150ml)溶液。将混合物加热至回流1h,冷却,然后真空蒸发。将水(500mL)加入残余物,用CH2Cl2(3×300mL)萃取;合并的二氯甲烷层用水(500mL)、饱和氯化钠(150mL)洗涤,用硫酸钠干燥,过滤,真空浓缩。残余物用二氧化硅柱色谱法提纯(用含0.5%NH4OH的5%CH3OH/CH2Cl2洗脱)得到产物(10g,54%);1H NMR 500MHz(CDCl3)δ=1.77(1H,br s),2.77(2H,d,J=6.0Hz),3.11(2H,t,J=6.0Hz),3.97(2H,s),6.95(1H,d,J=8.0Hz),7.15(1H,s)7.23(1H,dd,J=1.2和8.2Hz)。
中间体21
顺式外消旋物
步骤A
在氮气氛下,3-环戊烯-1-甲酸(Org.Synth.75,p195-200,1998)(31.5g,281mmol)的无水N,N-二甲基甲酰胺(300mL)溶液中加入碳酸钾(97g,710mmol)和碘甲烷(35mL,560mmol)。在室温搅拌所得混合物16h,然后倾入水(1L)中,用乙醚(3×400mL)萃取。合并的乙醚层用水(3×500mL)、饱和NaCl(200mL)洗涤,用硫酸镁干燥,过滤,真空浓缩,得到34g(96%)。
1H NMR(CDCl3,500MHz):δ5.64(s,2H),3.68(s,3H),3.11(五重峰,J=8.5Hz,1H),2.63(d,J=8.3Hz,4H)。
步骤B
在氮气氛下,向二异丙基胺(34.4mL,250mmol)的无水四氢呋喃(250mL)冷(-78℃)溶液中缓慢加入正丁基锂(100mL 2.5M的己烷溶液,250mmol),在-78℃搅拌所得混合物10min。此混合物中加入3-环戊烯甲酸甲酯(25.8g,200mmol),再搅拌15min后,加入2-碘丙烷(41mL,410mmol),在-78℃搅拌混合物30min,然后让其升至+4℃,在该温度静置72h。将反应混合物倾入5%柠檬酸(700mL)中,用乙醚(3×300mL)萃取。合并的乙醚层用水(2×500mL)、饱和NaCl(1×100mL)洗涤,用硫酸镁干燥,过滤,真空浓缩。残余物通过真空蒸馏(50℃;5mmHg)提纯获得28.9g(86%)产物。
1H NMR(CDCl3,500MHz):δ5.54(s,2H),3.67(s,3H),2.85(d,J=15.1Hz,2H),2.30(dd,J 14.9Hz,2H),2.07(t,J=6.6Hz,1H),0.82(d,J=6.6Hz,6H)。
步骤C
在氮气氛下,用双头针向硼烷-甲硫醚(20mL,200mmol)的无水四氢呋喃(100mL)冷(0℃)溶液加入步骤B制备的环戊烯酯(28.9g,172mmol)溶液。在加入完毕后,在室温搅拌反应混合物20h。在冰浴中冷却混合物,滴加氢氧化钠(60mL 3N溶液,181mmol),然后加入30%过氧化氢(65mL),在40℃搅拌所得混合物1h。将混合物倾入水(600mL)中,用乙醚(3×200mL)萃取,合并的乙醚层用水(3×500mL)、饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤,真空浓缩。残余物用二氧化硅柱色谱法提纯(用20%EtOAc/己烷洗脱)得到18.5g(58%)产物。
步骤D

在氮气氛下,向乙二酰氯(55mL,110mmol)的无水二氯甲烷(300mL)(-78℃)溶液滴加二甲亚砜(15.5mL,219mmol),在-78℃搅拌所得混合物10min。用双头针向此混合物加入步骤C产物(18.5g,100mmol)的无水二氯甲烷(100mL)溶液。在-78℃搅拌反应混合物15min,然后加入三乙胺(69mL,500mmol),让所得混合物在2h内升至室温。反应混合物用水(500mL)、饱和NaCl(150mL)洗涤,用硫酸镁干燥,过滤,真空浓缩,得到18g产物,产物直接用于下一步无需再提纯。
步骤E
在氮气氛下,步骤D制备的环戊酮(18g,98mmol)的无水1,2-二氯乙烷(500mL)溶液中加入4-(4-氟苯基)哌啶盐酸盐(25g,120mmol)、二异丙基乙胺(20.4mL,116mmol)、三乙酰氧基硼氢化钠(112g,531mmol)和4分子筛(粉末状10g)。在室温搅拌混合物48h,然后用二氯甲烷(500mL)稀释,通过硅藻土过滤。滤液用饱和碳酸氢钠溶液(500mL)、水(500mL)、饱和NaCl(200mL)洗涤,用硫酸钠干燥,过滤,真空浓缩得到28g(82%)产物。产物直接用于下一步无需再提纯。
步骤F
1-异丙基-3-(4-(4-氟苯基)哌啶-1-基)环戊烷甲酸

步骤E制备的环戊烷甲酯(28g,81mmol)的乙醇(500mL)溶液中加入氢氧化钾(30g,540mmol)的水(100mL)溶液,回流加热所得混合物18h。真空浓缩冷却的混合物以除去乙醇,将水(200mL)加入残余物。混合物用乙醚(3×200mL)萃取,加入浓盐酸中和水层。混合物用9/1氯仿/2-丙醇(3×150mL)的混合物萃取,合并的有机萃取液用硫酸钠干燥,过滤,真空浓缩。残余物中加入丙酮(70mL),将混合物短暂加热至回流,然后在+5℃静置16h。将丙酮从白色固体倾析除去,干燥余下的固体,得到11.5g(43%)产物,它是顺式和反式异构体的混合物(10∶1)。
ESI-MS C20H28FNO2的计算值:333;实测值:334(M+H)。
中间体22

将2,5-二甲基-3-吡咯啉(3.128g,32.19mmol)溶于三乙胺(8.97mL,64.4mmol),冷却至0℃。滴加苄氧甲酰氯(10.11mL,70.83mmol)的最小量的二氯甲烷溶液。缓慢加热反应混合物至室温,搅拌48h。反应物用饱和碳酸氢钠溶液(150mL)猝灭,然后将有机层用饱和碳酸氢钠溶液(2×100mL)和盐水(1×100mL)洗涤,用硫酸镁干燥,过滤,浓缩。通过硅胶快速柱色谱法提纯(用5%EtOAc/己烷至10%EtOAc/己烷的梯度溶剂系统),以52%收率获得中间体1(3.844g)。ESI-MSC14H17NO2计算值:231.29,实测值232(M+H)。
中间体23
方法A:
步骤A
将(1S)-(+)-2-氮杂二环[2.2.1]庚-5-烯-3-酮(10.3g,94.4mmol)、乙酸乙酯(200mL)和10%Pd/C(0.5g)的混合物在室温氢化。在24h后,过滤反应混合物,蒸发剩下10.4g(100%)产物,将其溶于250mL甲醇和HCl(12M,6mL)。在室温搅拌所得混合物,直到反应完成(72h)。蒸发甲醇,然后高真空干燥,获得乳白色固体标题化合物(16.0g,96%)。1H NMR(500MHz,D2O):δ3.70(s,3H),3.01(m,1H),2.38(m,1H),2.16-1.73(m,6H)。
步骤B
在室温向步骤A中间体(10.2g,56.8mmol)的无水二氯甲烷(200mL)悬浮液加入二苯基甲亚胺(10.2g,56.8mmol),搅拌所得混合物24h。过滤反应混合物,蒸发滤液剩下黄色油状物,用乙醚(100mL)研磨,过滤后蒸发。将此操作重复两次以确保产物没有氯化铵杂质。将所得油状物充分真空干燥获得标题化合物(18.03g,>100%),不需要再提纯。1H NMR(500MHz,CDCl3):δ7.5-7.18(m,10H),3.75(m,1H),3.7(s,3H),2.78(m,1H),2.26-1.71(m,6H)。
步骤C
二异丙基氨基锂溶液(在-78℃,用二异丙基胺(7.7g,76mmol)和正丁基锂(30.4mL,2.5M的己烷溶液,76mmol)在四氢呋喃(120mL)溶液中制备)中加入步骤B的酯(18.0g,58.6mmol)。搅拌所得暗红色溶液20min,然后用2-碘丙烷(14.9g,88.0mmol)猝灭。将反应混合物在3h内逐步加热至0℃,在此温度再保持3h。反应物用水猝灭,用乙酸乙酯萃取。有机层用水、盐水洗涤,干燥(无水硫酸镁),浓缩获得油状物。粗制Schiff碱(20.0g)的四氢呋喃(100mL)溶液中加入HCl(5.0mL,12M)。在室温搅拌所得反应混合物3h。在除去所有挥发分后,将盐酸盐溶于二氯甲烷(250mL),加入饱和碳酸氢钠溶液(250mL)和二碳酸二叔丁酯(26.0g,1.4Eq.)。将所得混合物在室温剧烈搅拌过夜。分离出有机层,用水、盐水洗涤,干燥(无水硫酸镁),浓缩获得油状物。快速柱色谱法提纯(洗脱剂:己烷/乙酸乙酯19∶1)得到所需产物(4.91g,30%)。1H NMR(500MHz,CDCl3):4.79(br,1H),4.01(m,1H),3.71(s,3H),2.18-1.60(m,6H),1.44(s,9H),0.87(d,J=6.9Hz,3H),0.86(d,J=6.9Hz,3H)。
步骤D

步骤C的酯(4.91g,17.2mmol)的甲醇(100mL)溶液中加入LiOH(3.6g,85mmol)的水(20mL)和四氢呋喃(10mL)溶液。将所得混合物在80℃加热直到反应完成(18h)。真空除去甲醇,将粗产物用水/乙酸乙酯(200mL,1∶4)溶解,冷却至0℃。将混合物酸度调节至pH 6。分离出乙酸乙酯层,用水、盐水洗涤,干燥(无水硫酸镁),浓缩获得油状物。快速柱色谱法提纯(洗脱剂:己烷/乙酸乙酯1∶1+2%AcOH)得到中间体11(3.9g,84%)。1H NMR(500MHz,CDCl3):11.36(br,1H),6.49(br,1H),4.83(m,1H),3.71(s,3H),2.30-1.55(m,6H),1.46(s,9H),0.94(d,J=6.9Hz,3H),0.933(d,J=6.9Hz,3H)。
方法B:
步骤A:

通过典型方法将市售(1R,4S)-4-氨基环戊-2-烯-1-甲酸转化为它的甲酯盐酸盐。
步骤B:

步骤A的胺(6.31g,35.5mmol)的丙酮(40mL)和水(20mL)悬浮液中分批加入固体NaHCO3(6.6g,78mmol)。在5min后,加入二碳酸二叔丁酯(8.5g,39mmol)的丙酮(60mL)溶液,在室温搅拌反应混合物。在3h后,真空除去丙酮,残余物在乙醚(500mL)和饱和碳酸氢钠水溶液(120mL)间分配。乙醚层进一步用NaHCO3水溶液(1×100mL)、盐水(1×100mL)洗涤,用无水Na2SO4干燥,浓缩,用快速色谱法提纯(15%乙酸乙酯/己烷)获得产物(7.25g,85%)。
步骤C:

在-78℃,向双(三甲硅烷基)氨基锂(10.4g,62.1mmol)的四氢呋喃(100mL)溶液在10min内加入步骤B的中间体(6.71g,27.8mmol)的四氢呋喃(10mL)溶液。在-78℃搅拌所得溶液30min,然后一次性加入异丙基碘(3.3mL,33mmol)。让反应物升至-25℃,在此温度保持过夜。然后将反应物用饱和氯化铵水溶液(250mL)猝灭。分离出有机层,水层进一步用乙醚(3×100mL)萃取。合并的有机层用盐水(1×100mL)洗涤,用无水Na2SO4干燥,过滤,浓缩,用快速色谱法提纯(5-10%乙酸乙酯/己烷)得到澄清油状产物(5.66g,72%)(顺/反=4.3/1)。1H NMR(500MHz,CDCl3)顺式异构体:δ5.79(s,2H),4.75(m,1H),3.72(s,3H),2.28-2.20(m,2H),2.0(dd,J=15,4Hz,1H),1.45(s,9H),0.85(d,J=6.6Hz,3H),0.81(d,J=7Hz,3H)。
步骤D:

步骤C产物(1.6g,5.7mmol)的四氢呋喃(50mL)、甲醇(50mL)和水(10mL)溶液中加入LiOH一水化合物(400mg),将反应物加热至回流过夜,直到TLC显示完全反应为止。真空除去有机溶剂,水层用乙醚(1x)洗涤,然后用浓HCl缓慢酸化直到pH达到4。所得悬浮液用CH2Cl2(3x)萃取。用无水硫酸镁干燥合并的有机层,过滤,浓缩得到黄色泡沫状固体产物(两种顺/反异构体的混合物)(1.5g)。在加热下将此固体溶于乙酸乙酯(2mL),用己烷(50mL)稀释得到澄清溶液。让此溶液缓慢冷却至室温1h,然后在冰箱维持-25℃过夜。反式异构体与部分所需顺式异构体一起结晶(总共500mg)。收集母液,浓缩得到标题化合物(1g,66%,仅有顺式异构体)。1H NMR(500MHz,CDCl3)顺式异构体:δ5.80(m,2H),4.80(m,1H),2.40-2.20(m,2H),2.15-2.0(m,1H),1.5(m,9H),1.0-0.8(m,3H)。
步骤E:

步骤D产物(1g)的乙醇(30mL)溶液中加入10%Pd/C(100mg),所得混合物用Parr装置在50 lb氢压力下搅拌过夜。混合物通过硅藻土过滤,真空浓缩获得标题化合物(1g,99%)。1H NMR(500MHz,CDCl3):11.36(br,1H),6.49(br,1H),4.83(m,1H),3.71(s,3H),2.30-1.55(m,6H),1.46(s,9H),0.94(d,J=6.9Hz,3H),0.933(d,J=6.9Hz,3H)。
中间体24

步骤A

将中间体2(4.6g,16mmol)和中间体23(4.0g,14mmol)首先与甲苯(3×50mL)共沸蒸馏干燥,置于高真空下30min。在氮气氛下,依次加入4-二甲基氨基吡啶(1.08g,8.60mmol)、无水二氯甲烷(40mL)和二异丙基乙胺(7.0mL,40mmol)。在中间体溶解后,加入溴-三-吡咯烷基-磷鎓六氟磷酸盐(6.80g,14.3mmol),随后立即加入二异丙基乙胺(7.0mL,40mmol)。在室温搅拌反应混合物过夜,然后用饱和NaHCO3猝灭。水层再次用二氯甲烷(3×50mL)洗涤,合并有机层,用硫酸钠干燥,过滤,真空蒸发。粗产物用快速色谱法提纯(阶段梯度0-60%,乙酸乙酯/己烷)获得黄色泡沫状产物(4.80g,74%)。1H NMR(500MHz,CDCl3)δ8.72(s,1H),7.70(s,1H),4.88(br d,J=17.0Hz,1H),4.78(d,J=17.6Hz,1H),4.04-3.84(m,2H),3.52(br s,1H),3.12(br t,J=5.6Hz,1H),2.32-2.06(m,3H),1.98-1.70(m,4H),1.64-1.54(m,1H),1.44(s,9H),0.92-0.82(m,6H)。LC-MS C23H32F3N3O3计算值455.24,实测值[M+H]+456.2。
步骤B
将步骤A产物(1.2g,2.6mmol)溶于4M HCl/二噁烷(50mL),在室温搅拌所得溶液1h。真空蒸发反应混合物获得白色粉末状产物(904mg,97%)。LC-MS C18H24F3N3O计算值355.20,实测值[M+H]+356.2。
中间体25
步骤A

烧瓶中加入中间体23(1.1g,4.0mmol)、中间体1(0.944g,4.00mmol)、溴-三-吡咯烷基-磷鎓六氟磷酸盐(1.85g,4.00mmol)、DMAP(0.29g,2.4mmol)、DIEA(2.77mL,16mmol)和DCM(20mL)。在氮气氛下搅拌所得混合物36h。将全部混合物应用到硅胶柱上,用20%EtOAc/己烷洗脱。获得的所需Boc-酰胺为树胶状固体(1.5g,82%)。ESI-MS C24H33F3N2O3的计算值:454;实测值:455(M+H)。
步骤B

将步骤A的Boc氨基酰胺用10mL 4N HCl/二噁烷处理1h。蒸发反应混合物,真空干燥产物。获得黄色固体中间体25(1.2g)。ESI-MSC19H25F3N2O的计算值:354;实测值:355(M+H)。
中间体26

步骤A
装有中间体1(10g,50mmol)的烧瓶中加入30mL 70%硝酸。在0℃冷却混合物,在30min内加入30mL浓硫酸。在室温搅拌所得溶液过夜,倾入冰-水混合物中,在0℃用固体LiOH-H2O调节至pH>10。在剧烈搅拌下,加入二碳酸二叔丁酯(21.8g,100mmol)的500mLDCM溶液。搅拌混合物30min,分离出有机层,水层用DCM(2×200mL)萃取。合并的萃取液用水(500mL)洗涤,用硫酸钠干燥,然后蒸发。粗产物用快速色谱法提纯(硅胶,20%EtOAc/己烷)获得白色固体标题化合物(17.0g,98%)。1H NMR(400MHz,CDCl3)δ8.05(s,1H),7.62(s,1H),4.72(s,2H),3.67(t,J=6.0Hz,2H),3.13(t,J=6.0Hz,2H),1.49(s,9H)。
步骤B
将以上步骤A的中间体(17.0g)溶于100mL 4M HCl/二噁烷,搅拌1h,蒸发,真空干燥。获得白色固体中间体26。1H NMR(400MHz,CD3OD)δ8.75(s,1H),8.00(s,1H),2.58(s,2H),3.57(t,J=6.0Hz,2H),3.42(t,J=6.0Hz,2H)。
中间体27

步骤A

烧瓶中加入中间体27(1.10g,4.00mmol)、中间体23(1.15g,4.00mmol)、溴-三-吡咯烷基-磷鎓六氟磷酸盐(1.85g,4.00mmol)、DMAP(0.29g,2.4mmol)、DIEA(2.7mL,16mmol)和DCM(20mL)。在氮气氛下搅拌所得混合物36h。将全部混合物应用到硅胶柱上,用20%EtOAc/己烷洗脱获得树胶状固体标题化合物(1.5g,75%)。ESI-MSC24H32F3N3O5计算值:499;实测值:500(M+H)。
步骤B

将以上步骤的偶合产物(1.5g)用10mL 4N HCl/二噁烷处理1h,蒸发,高真空干燥获得黄色固体标题化合物(1.2g)。1H NMR(400MHz,CD3OD)δ8.20(s,1H),7.95(宽峰,1H),4.98(s,2H),4.00(dd,2H),3.90(t,2H),3.68(m,1H),3.45(m,3H),3.20(s,2H),2.15-2.50(m,3H),1.80-2.10(m,2H),1.80(m,2H),0.90(m,6H)。ESI-MS的计算值C19H24F3N3O3:399;实测值:400(M+H)。
中间体28

步骤A

向N-三氟乙酰基-7-三氟甲基-1,2,3,4-四氢异喹啉(中间体1步骤B,6.0g,20mmol)、NIS(6.9g,30mmol)和TFA(15mL)的搅拌混合物滴加浓硫酸(1.5mL)。形成大量固体。在室温搅拌混合物过夜,倾入冰-水混合物中,用乙酸乙酯(3x)萃取。合并的有机相用水和盐水洗涤,用硫酸钠干燥,蒸发。残余物用硅胶提纯(用10%EtOAc/己烷洗脱)。合并的部分用饱和NaHSO3洗涤,用硫酸钠干燥,蒸发,真空干燥获得白色固体标题化合物(5.0g)。1H NMR(400MHz,CDCl3)δ8.02(d,J=2.5Hz,1H),7.42(d,j=3.0Hz,1H),4.85,4.79(ss,2H),3.95,3.90(tt,J=1.5,1.5Hz,2H),2.97(m,2H)。ESI-MSC12H8F6INO计算值:423;实测值:424(M+H)。
步骤B

碘化合物(步骤A,4.2g,10mmol)、氰化锌(2.3g,20mmol)、四-三苯基膦(phosphene)钯(0)络合物(0.4g)和50mL DMF的混合物用氮气净化几次,然后在85℃、氮气氛下加热过夜。LC-MS证实完全转化。过滤除去不溶性物质。将滤液用水稀释,用乙酸乙酯(3x)萃取。合并乙酸乙酯层,通过硅藻土过滤,然后用水洗涤,用硫酸钠干燥,然后蒸发。残余物用硅胶提纯(用10%EtOAc/Hex洗脱)获得白色固体标题化合物(2.5g)。1H NMR(400MHz,CDCl3)δ7.85(d,J=2.1Hz,1H),7.65(d,J=2.6Hz,1H),4.91,4.86(ss,2H),4.00(m,2H),3.25(m,2H)。ESI-MS C13H8F6N2O的计算值:323;实测值:323(M+H)。
步骤C

将步骤B的酰胺中间体(500mg,1.55mmol)、碳酸钾(1.5g)、乙醇(20mL)和水(0.5mL)的混合物在80℃加热直到TLC证实完全裂解。蒸发溶剂,用水稀释,用DCM(3x)萃取,用硫酸钠干燥,蒸发,然后真空干燥。获得白色固体标题产物(0.41g)。ESI-MS C11H9F3N2计算值:226;实测值:227(M+H)。
步骤D
将以上步骤C的氰基中间体(1.9g,8.5mmol)与50mL浓HCl水溶液回流48h。LC-MS证实完全水解。冷却混合物至室温,过滤收集所得沉淀,用浓aq.HCl洗涤。在高真空干燥后获得所需产物的盐酸盐(1.75g,73%)。1H NMR(CD3OD,400MHz):δ8.20(s,1H),7.80(s,1H),4.51(s,2H),3.55(m,4H)。LC-MS C11H10F3NO2计算值245,实测值[M+H]+246。
步骤E
以上步骤D的氨基酸盐酸盐(1.75g,6.25mmol)的50mL甲醇悬浮液中缓慢加入乙酰氯纯溶液(5mL)。将所得混合物回流直到LC-MS证实完全酯化(~3h),然后蒸发溶剂,高真空干燥获得白色固体标题化合物(1.85g,100%)。1H NMR(CD3OD,400MHz):8.19(s,1H),7.82(s,1H),4.50(s,2H),3.94(s,3H),3.53(s,4H)。
LC-MS C12H12F3NO2计算值259,实测值[M+H]+260。
中间体29

方法A:
步骤A:

将H2SO4(conc.,15.3g,8.30mL,156mmol)滴加到硫酸镁(75g,620mmol)的DCM(650mL)剧烈搅拌悬浮液。搅拌混合物0.5h,然后加入已知的环戊酮-3-甲酸酯(20.0g,156mmol),接着加入叔丁醇(58g,780mmol)。将反应容器紧密地密封,在室温搅拌混合物过夜。第二天早晨再加入30mL叔丁醇。再次将反应容器紧密地密封,搅拌反应混合物过周末。反应混合物通过硅藻土过滤。滤液用2N NaOH洗涤。水层再次用DCM洗涤。合并有机层,依次用水、盐水洗涤,用无水硫酸镁干燥,过滤,然后浓缩获得19.9g(69%)3-氧代环戊烷甲酸叔丁酯。反应进程通过TLC监测,采用50%乙酸乙酯/己烷,用茴香醛染剂染色(SM和产物染上紫色)。
1H NMR(500MHz,CDCl3):3.02(p,J=7.8Hz,1H),2.05-2.50(m,6H),1.45(s,9H)。13C NMR(125MHz,CDCl3):217.00,173.47,80.99,41.88,41.14,27.94,26.57。
步骤B:

3-氧代环戊烷甲酸叔丁酯(19.8g,107mmol)的1∶1DCM/甲醇(150mL)溶液中依次加入原甲酸三甲酯(46.8mL,428mmol)、TsOH·H2O(~0.5g)。在室温搅拌反应混合物2h。然后再加入TsOH·H2O(~0.25g),搅拌反应混合物过夜。在室温浓缩反应混合物,将所得残余物溶于乙醚,依次用饱和碳酸氢钠溶液、盐水洗涤。用无水硫酸镁干燥乙醚层,过滤,然后浓缩。用快速色谱法提纯(二氧化硅,15%乙酸乙酯/己烷)得到22.2g(90%)3,3-二甲氧基环戊烷甲酸叔丁酯。1H NMR(500MHz,CDCl3):3.21(s,3H),3.20(s,3H),2.80(m,1H),2.10-1.80(bm,6H),1.46(s,9H)。13C NMR(125MHz,CDCl3):174.9,111.2,80.3,67.8,49.2,42.5,37.4,33.8,28.3,22.0。
步骤C:
在10min内,向LDA(1.5M的环己烷溶液,41mL,61mmol)的THF(150mL)冷(-78℃)溶液滴加3,3-二甲氧基环戊烷甲酸叔丁酯(9.37g,40.7mmol)的25mL THF溶液。在-78℃搅拌所得混合物30min,然后滴加2-碘丙烷(16.3mL,163mmol)。在搅拌10min后,让反应混合物升至室温。在搅拌过夜后,将反应混合物用乙醚稀释,用盐水洗涤。用无水硫酸镁干燥乙醚层,过滤,然后浓缩。在真空储存粗产物过夜,用MPLC提纯(二氧化硅,20%乙酸乙酯/己烷)得到8.32g 1-异丙基-3,3-二甲氧基环戊烷甲酸叔丁酯(75%)。
1H NMR(500MHz,CDCl3)δ3.21(s,3H),3.18(s,3H),2.56(appd,J=14Hz,1H),2.26(m,1H),1.78-1.89(m,3
步骤D:

将1-异丙基-3,3-二甲氧基环戊烷甲酸叔丁酯(8.32g,30.5mmol)溶于4N无水HCl/二噁烷(50mL),加入水(10mL)。在室温搅拌反应混合物过夜,然后浓缩。将残余物溶于DCM,用无水硫酸镁干燥,过滤,然后浓缩得到5.44g 1-异丙基-3-氧代环戊烷甲酸(直接使用无需提纯)。
1H NMR(500MHz,CDCl3)δ2.70(d,J=18.1Hz,1H),2.44-2.39(m,1H),2.30-2.15(m,2H),2.14(dd,J=18.1,1.0Hz,1H),2.06(p,J=6.9Hz,1H),1.98(m,1H),0.98(dd,J=11.4,6.9Hz,6H)。
步骤E:
将1-异丙基-3-氧代环戊烷甲酸(5.44g,32.0mmol)的DCM(75mL)冷(0℃)溶液依次用乙二酰氯(8.36mL,95.9mmol)、3滴DMF处理。让反应混合物升至室温,搅拌1.75h。然后浓缩反应混合物,真空储存30min。将所得酰氯溶于DCM(75mL),冷却至0℃,依次用苄基醇(8.28mL,80.0mmol)、三乙胺(8.92mL,64.0mmol,滴加)处理。然后加入约100mg DMAP,加热反应混合物至室温,搅拌2h。反应混合物用DCM稀释,用1N HCl溶液、饱和碳酸氢钠溶液和盐水洗涤。用无水硫酸镁干燥有机层,过滤,然后浓缩。MPLC提纯(二氧化硅,50%乙酸乙酯/己烷)得到6.11g(73%)1-异丙基-3-氧代环戊烷甲酸苄基酯。1H NMR(CDCl3,500MHz):δ7.36(m,5H),5.17(d,J=2.5Hz,2H),2.85(d,J=18.5Hz,1H),2.48(m,1H),2.29(dd,J=10.0,3.0Hz,1H),1.98-2.23(m,3H),1.93(m,1H),0.95(m,6H)。通过手性HPLC拆分外消旋产物:用chiralcel OD柱,用15%2-丙醇/己烷洗脱(100mg/注射;用编程的Gilson HPLC系统完成)。获得2.11g所需较快洗脱的异构体(1S)-1-异丙基-3-氧代环戊烷甲酸苄基酯。
步骤F:

将(1S)-1-异丙基-3-氧代环戊烷甲酸苄基酯(1.27g,4.88mmol)与Pd/C(10%Degussa,500mg)在20mL甲醇中混合,在氢气氛下(气罐)搅拌2h。反应仅进行了一部分(~30%转化),因此过滤反应混合物,另外加入Pd/C(500mg),在氢气氛下搅拌混合物5h。由于反应现已接近完成,通过硅藻土过滤反应混合物,浓缩获得704mg(1S)-1-异丙基-3-氧代环戊烷甲酸,无需进一步提纯。注意因为在手性分离后获得的酯肯定因杂质而中毒,所以使用了大量催化剂。这是该特定样品的特殊情况。通常催化剂的用量少很多。1H NMR与以上外消旋酸一致(步骤D)。
方法B:

(1S,3R)-3-[(叔丁氧基羰基)氨基]-1-异丙基环戊烷甲酸(7.46g,27.5mmol)的二噁烷(10mL)溶液中加入4N HCl的二噁烷溶液(30mL)。在室温搅拌反应混合物2h,然后真空浓缩得到相应的氨基酸盐白色固体。然后将此固体溶于CH2Cl2(100mL),加入固体NaHCO3(7.0g,82.5mmol)。在冷却至0℃后,将NBS(20.0g,110mmol)的CH2Cl2(200mL)溶液在4h内缓慢加入反应物。在加入后,将反应物真空浓缩至干,然后溶于乙醇(100mL)。乙醇溶液中加入NaOMe(4.45g,82.5mmol),将反应物加热至回流。回流1h后,将反应物冷却至0℃,加入2N硫酸水溶液(50mL)。在室温搅拌混合物1h,然后真空浓缩至约60mL。剩下的混合物在水(150mL)和乙酸乙酯(100mL)间分配。水层进一步用乙酸乙酯萃取两次。合并有机层,用无水硫酸镁干燥,浓缩,用快速色谱法提纯(二氧化硅,乙酸乙酯/己烷)得到(1S)-1-异丙基-3-氧代环戊烷甲酸(3.00g,64%)。
中间体30
反式外消旋物
步骤A:
4-[3-(乙氧基羰基)苯基]哌啶-1-甲酸叔丁酯(48g,220mmol)的氯仿(900mL)搅拌溶液中加入氧化钌(IV)水合物(6.0g,45mmol),然后加入高碘酸钠(150g,700mmol)的水(900mL)溶液。在室温搅拌所得不均匀反应混合物11天,然后通过硅藻土短柱过滤。将有机层移出,水层用DCM萃取两次。合并的有机层用10%硫代硫酸钠的水溶液洗涤两次,用盐水洗涤一次。溶液用硫酸镁干燥,过滤,减压浓缩。产物用快速色谱法提纯(硅胶,20%EA/己烷)得到22.5g(64.8mmol)4-[3-(乙氧基羰基)苯基]-2-氧代哌啶-1-甲酸叔丁酯(29%)。
ESI-MS C19H25NO5计算值:347.17;实测值370.1(M+Na)
步骤B:
将双(三甲硅烷基)氨基钾(14g,71mmol)与300mL THF在1000mL火焰干燥的圆底烧瓶中混合,将所得混合物冷却至-78℃。将溶于150mL THF的4-[3-(乙氧基羰基)苯基]-2-氧代哌啶-1-甲酸叔丁酯(22.5g,64.8mmol)通过加液漏斗缓慢加入混合物,在-78℃搅拌所得反应混合物30min。然后滴加甲基碘(12.1mL,195mmol),将反应混合物在-78℃搅拌4h,然后将其加热至室温过夜。用饱和氯化铵猝灭反应物,用乙醚萃取3次。合并的乙醚层用盐水洗涤,用硫酸镁干燥,过滤,然后减压浓缩。产物用快速色谱法提纯(10-20%EA/己烷)得到6.1g反式外消旋物4-[3-(乙氧基羰基)苯基]-3-甲基-2-氧代哌啶-1-甲酸叔丁酯(26%)。
ESI-MS C20H27NO5计算值:361.19;实测值384.25(M+Na)。
步骤C:
将步骤B的产物(6.1g,17mmol)溶于4.0M HCl/二噁烷,在室温搅拌2h,然后减压浓缩得到所需橙色固体产物,在下一步中直接使用无需再提纯。
ESI-MS C15H19NO3计算值:261.14;实测值262.1(M+H)。
步骤D:
反式外消旋物
将以上步骤的产物(全部数量~17mmol)溶于THF(100mL),滴加2.0M甲硼烷-甲硫醚的THF溶液(31mL,62mmol)。在室温搅拌所得溶液4h,然后在4℃储存72h。减压除去溶剂,将所得残余物溶于0.5M HCl(水溶液~38%)的乙醇溶液。将溶液加热至50℃,搅拌4h。除去溶剂,重复以上操作以确保硼烷络合物分解。除去溶剂,产物用MPLC提纯(0-15%(10%NH4OH/MeOH)/DCM)得到所需产物,纯度为80%。将粗产物溶于DCM(100mL),用二碳酸二叔丁酯(2.95g,13.5mmol)、二异丙基乙胺(2.30mL,13.5mmol)和DMAP(10mg)处理。在室温搅拌所得反应混合物过夜,然后用DCM稀释,用1N饱和碳酸氢钠水溶液和盐水洗涤。用硫酸镁干燥有机层,过滤,减压浓缩。中间体用MPLC提纯(0-40%EA/己烷)。将所得无色油状物溶于4.0M HCl/二噁烷,在室温搅拌所得反应混合物1.5h。浓缩反应混合物至干得到2.13g(7.52mmol)所需盐酸盐。
ESI-MS C15H21NO2计算值:247.16;实测值248.15(M+H)
实施例1
中间体3(150mg,0.425mmol)、4-乙氧甲酰基哌啶(125mg,0.425mmol)和DCM(25mL)的溶液中加入分子筛(4)和NaBH(OAc)3(450mg,2.12mmol)。在室温搅拌反应混合物18h,然后通过硅藻土过滤,用饱和NaHCO3稀释,用DCM(3x)萃取。用硫酸钠干燥合并的有机层,用制备型TLC(3/96.7/0.3,MeOH/DCM/NH4OH)提纯获得实施例1(220mg,97.8%)。LC-MS C27H38F3N2O3[M+H+]计算值495.28,实测值495.25。
许多化合物用不同的胺按照实施例1的方法制备。在下表中总结这些化合物。
表1(实施例2-6)
实施例7

将实施例1产物(185mg,0.349mmol)、5N NaOH(200μL,1.04mmol)和MeOH(5mL)的混合物在60℃加热3h,然后加入4N HCl/二噁烷溶液中和碱。浓缩反应物溶液,用反相HPLC提纯获得实施例7(115mg,65.7%)。LC-MS C25H34F3N2O3[M+H+]计算值467.24,实测值467.35。
实施例8-12用实施例2-6作为初始原料按照实施例7的方法制备。在下表中总结这些化合物。
表2(实施例8-12)

实施例13
中间体4(50mg,0.14mmol)的二氯甲烷(20mL)溶液中加入1-乙氧基羰基哌嗪(23mg,0.14mmol)。在加入粉末状4分子筛(25mg)后,加入三乙酰氧基硼氢化钠(180mg,0.84mmol),搅拌反应混合物过夜。混合物用二氯甲烷稀释,用饱和碳酸氢钠水溶液洗涤,用硫酸钠干燥,真空浓缩。粗产物用制备型TLC(7/92.3/.7,甲醇/二氯甲烷/氢氧化铵)提纯获得实施例13,为4种非对映异构体的混合物(55mg,79%)。LC-MS:MW计算值496.27,实测值497.3。
许多化合物按照实施例13的方法制备,其中用不同的哌嗪替代1-乙氧基羰基哌嗪。这些化合物(全部制备为4种非对映异构体的混合物)在下表中总结。
表3(实施例14-16)
| 实施例 | R | 分子式 | 计算值[M+H+] | 实测值[M+H+] |
实施例17

中间体4(50mg,0.14mmol)的二氯甲烷(20mL)溶液中加入4-苯甲酰基哌啶盐酸盐(32mg,0.14mmol)和N,N-二异丙基乙胺(73μL,0.42mmol)。在加入4粉末状分子筛(25mg)后,加入三乙酰氧基硼氢化钠(180mg,0.84mmol),搅拌反应混合物过夜。混合物用二氯甲烷萃取,用碳酸氢钠洗涤,用硫酸钠干燥,真空浓缩。粗产物用制备型TLC(7/92.3/.7,甲醇/二氯甲烷/氢氧化铵)提纯获得实施例17(30mg,43%),为4种非对映异构体的混合物。
ESI-MS:计算值MW:527.28,实测值528.25。
实施例18

将中间体4(176mg,0.5mmol)、螺接哌啶(为HCl盐,115mg,0.6mmol)、DIEA(100mg,0.8mmol)、分子筛(4,200mg)、三乙酰氧基硼氢化钠(212mg,1.0mmol)和二氯甲烷(10mL)的混合物搅拌过夜。用饱和碳酸钠水溶液猝灭反应物。通过硅藻土过滤除去固体。粗产物萃取到二氯甲烷中,用制备型TLC(1000微米,10%[aq.NH4OH/MeOH 1/9]/DCM)提纯。获得为顺式和反式外消旋异构体混合物的标题化合物(155mg,63%)。LC-MS C26H35F3N4O2计算值:492;实测值:493(M+H)。
C.ZHOU
实施例19

中间体5(50mg,0.14mmol)和哌啶(28μL,0.28mmol)的DCM(10mL)搅拌溶液中加入4粉末状分子筛(50mg)和三乙酰氧基硼氢化钠(150mg,0.71mmol)。在室温搅拌所得溶液3天,然后通过硅藻土过滤,用饱和碳酸氢钠水溶液和盐水洗涤。用硫酸钠干燥有机层,过滤,然后减压浓缩得到粗制的油状物,用制备型TLC(0.5%NH4OH/4.5%MeOH/95%DCM)提纯得到16mg无色固体,为2种非对映异构体的混合物。向小部分游离碱加入2N HCl的乙醚溶液,转化为它的盐酸盐。
ESI-MS C23H32F3N3O计算值:423.25;实测值424(M+H)。
几个其它实施例根据实施例19介绍的方法制备,但是用不同取代的哌啶作为胺组分替代哌啶。表4列出了这些实施例。
表4(实施例20-28)

实施例29和实施例30
将实施例22制备的游离碱产物(55mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用25%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收27mg较快洗脱的非对映异构体(实施例29)和20mg较慢洗脱的非对映异构体(实施例30)。
实施例29:ESI-MS C26H36F3N3O3的计算值:495.27;实测值496(M+H)。
实施例30:ESI-MS C26H36F3N3O3的计算值:495.27;实测值496(M+H)。
实施例31
将实施例29(10mg,0.018mmol)溶于甲醇(1mL)和THF(1mL)的混合物,用氢氧化锂一水化合物(5mg,0.12mmol)的水(1mL)溶液处理。在室温搅拌所得溶液18h,然后减压浓缩。产物用反相HPLC(C18,20-100%MeCN/H2O)提纯,通过加入2N HCl的乙醚溶液转化为盐酸盐,得到6.8mg产物(70%)。
ESI-MS C24H32F3N3O3的计算值:467.24;实测值468(M+H)。
实施例32
将实施例30(10mg,0.018mmol)溶于甲醇(1mL)和THF(1mL)的混合物,用氢氧化锂一水化合物(5mg,0.12mmol)的水(1mL)溶液处理。在室温搅拌所得溶液18h,然后减压浓缩。产物用反相HPLC提纯(C18,20-100%MeCN/H2O),通过加入2N HCl的乙醚溶液转化为盐酸盐,得到3.8mg产物(39%)。
ESI-MS C24H32F3N3O3的计算值:467.24;实测值468(M+H)。
实施例33
实施例30(15mg,0.026mmol)的THF(2mL)溶液中加入三乙基硼氢化锂(1.0M的THF溶液,150μL,0.15mmol)。在室温18h后,再次加入三乙基硼氢化锂(100μL),搅拌所得混合物24h,然后减压浓缩。将所得残余物溶于DCM,依次用饱和碳酸氢钠水溶液、1N HCl水溶液和盐水洗涤。用Na2SO4干燥有机层,过滤,依次用2N HCl/乙醚、己烷处理,减压浓缩得到1.5mg所需产物的盐酸盐(11%)。
ESI-MS C24H34F3N3O2的计算值:453.26;实测值454(M+H)。
实施例34和实施例35

将实施例20制备的游离碱产物(60mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用25%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收26mg较快洗脱的非对映异构体(实施例34)和17mg较慢洗脱的非对映异构体(实施例35)。
实施例34:ESI-MS C25H35F3N4O2的计算值:480.27;实测值481(M+H)。
实施例35:ESI-MS C25H35F3N4O2的计算值:480.27;实测值481(M+H)。
实施例36和实施例37
将实施例22制备的游离碱产物(54mg)拆分为2个含两种非对映异构体的混合物,采用配置ChiralCel OD柱的HPLC,用13%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收33mg较快洗脱的非对映异构体(实施例36)和10mg较慢洗脱的非对映异构体(实施例37)。
实施例36:ESI-MS C26H36F3N3O3的计算值:495.27;实测值496(M+H)。
实施例37:ESI-MS C26H36F3N3O3的计算值:495.27;实测值496(M+H)。
实施例38-41

将实施例24制备的游离碱产物(40mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用20%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收15mg较快洗脱的非对映异构体(实施例38)、1.5mg非对映异构体2(实施例39)、7mg非对映异构体3(实施例40)以及6mg最慢洗脱的非对映异构体(实施例41)。
实施例38:ESI-MS C24H34F3N3O的计算值:437.27;实测值438(M+H)。
实施例39:ESI-MS C24H34F3N3O的计算值:437.27;实测值438(M+H)。
实施例40:ESI-MS C24H34F3N3O的计算值:437.27;实测值438(M+H)。
实施例41:ESI-MS C24H34F3N3O的计算值:437.27;实测值438(M+H)。
实施例42-46

将实施例25制备的游离碱产物(40mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用20%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。拆分所有6种非对映异构体,但是使重叠流出的峰2和峰6混合在一起,得到4种纯非对映异构体和一个含两种非对映异构体的混合物。
实施例42:峰1:ESI-MS C25H36F3N3O的计算值:451.28;实测值452(M+H)。
实施例43:峰2/6:ESI-MS C25H36F3N3O的计算值:451.28;实测值452(M+H)。
实施例44:峰3:ESI-MS C25H36F3N3O的计算值:451.28;实测值452(M+H)。
实施例45:峰4:ESI-MS C25H36F3N3O的计算值:451.28;实测值452(M+H)。
实施例46:峰5:ESI-MS C25H36F3N3O的计算值:451.28;实测值452(M+H)。
实施例47和实施例48
将实施例27制备的游离碱产物(20mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用25%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收7mg较快洗脱的非对映异构体(实施例47)和6mg较慢洗脱的非对映异构体(实施例48)。
实施例47:ESI-MS C23H32F3N3O2的计算值:439.24;实测值440(M+H)。
实施例48:ESI-MS C23H32F3N3O2的计算值:439.24;实测值440(M+H)。
实施例49

中间体5(50mg,0.14mmol)和吗啉(25μL,0.28mmol)的DCM(10mL)搅拌溶液中加入4粉末状分子筛(50mg)和三乙酰氧基硼氢化钠(150mg,0.71mmol)。在室温搅拌所得溶液3天,然后通过硅藻土过滤,用饱和碳酸氢钠水溶液和盐水洗涤。用硫酸钠干燥有机层,过滤,然后减压浓缩得到粗制油状物,用制备型TLC(0.5%NH4OH/4.5%MeOH/95%DCM)提纯得到57mg无色固体,为2种非对映异构体的混合物。向小部分游离碱加入2N HCl的乙醚溶液,转化为它的盐酸盐。
ESI-MS C22H30F3N3O2的计算值:425.23;实测值426(M+H)。
几个其它实施例根据实施例49介绍的方法制备,但是用不同的取代吗啉作为胺组分替代吗啉。表5列出了实施例。
表5(实施例50-53)
实施例53和实施例54
将实施例50制备的游离碱产物(60mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用20%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收26mg较快洗脱的非对映异构体(实施例53)和27mg较慢洗脱的非对映异构体(实施例54)。
实施例53:ESI-MS C24H34F3N3O2的计算值:453.26;实测值454(M+H)。
实施例54:ESI-MS C24H34F3N3O2的计算值:453.26;实测值454(M+H)。
实施例55和实施例56

将实施例52制备的游离碱产物(48mg)拆分为它的单独非对映异构体,采用配置ChiralCel OD柱的HPLC,用25%乙醇/己烷洗脱。通过加入2N HCl的乙醚溶液,将各个化合物转化为相应的盐酸盐。回收18mg较快洗脱的非对映异构体(实施例53)和23mg较慢洗脱的非对映异构体(实施例54)。
实施例55:ESI-MS C23H30F3N3O2的计算值:437.23;实测值438(M+H)。
实施例56:ESI-MS C23H30F3N3O2的计算值:437.23;实测值438(M+H)。
实施例57
中间体5(50mg,0.14mmol)和吡咯烷(23μL,0.28mmol)的DCM(10mL)搅拌溶液中加入4粉末状分子筛(50mg)和三乙酰氧基硼氢化钠(150mg,0.71mmol)。在室温搅拌所得溶液3天,然后通过硅藻土过滤,用饱和碳酸氢钠水溶液和盐水洗涤。用硫酸钠干燥有机层,过滤,然后减压浓缩,得到粗制油状物,用制备型TLC提纯(0.5%NH4OH/4.5%MeOH/95%DCM)得到15mg较快流出的非对映异构体和35.5mg较慢流出的异构体,两种异构体均为无色固体。向小部分游离碱加入2N HCl的乙醚溶液,转化为它的盐酸盐。
顶部:ESI-MS C22H30F3N3O的计算值:409.23;实测值410(M+H)。
底部:ESI-MS C22H30F3N3O的计算值:409.23;实测值410(M+H)。
几个其它实施例根据实施例57介绍的方法制备,但是用不同的取代吡咯烷作为胺组分替代吡咯烷。表6列出了实施例。
表6(实施例58-62)
实施例63
将中间体3(55mg,0.15mmol)、(1S,4S)-(-)-2-(4-氟苯基)-2,5-二氮杂双环[2.2.1]庚烷(为HBr盐,85mg,0.3mmol)、DIEA(65mg,0.5mmol)、分子筛(4,250mg)、三乙酰氧基硼氢化钠(212mg,1.0mmol)和二氯甲烷(5mL)的混合物搅拌过夜。用饱和碳酸钠水溶液猝灭反应物。通过硅藻土过滤除去固体。粗产物萃取到二氯甲烷中,用制备型TLC(1000微米,5%[aq.NH4OH/MeOH 1/9]/DCM)提纯。获得为顺式和反式外消旋异构体混合物的标题化合物(75mg,95%)。LC-MSC30H35F4N3O的计算值:529;实测值:530(M+H)。
实施例64
将中间体3(55mg,0.15mmol)、(1S,4S)-(-)-2-(2-甲基)-2,5-二氮杂双环[2.2.1]庚烷(为马来酸盐,100mg,0.3mmol)、DIEA(65mg,0.5mmol)、分子筛(4,250mg)和三乙酰氧基硼氢化钠(212mg,1.0mmol)的二氯甲烷(5mL)混合物搅拌过夜。用饱和碳酸钠水溶液猝灭反应物。通过硅藻土过滤除去固体。粗产物萃取到二氯甲烷中,用制备型TLC(1000微米,5%[aq.NH4OH/MeOH 1/9]/DCM)提纯。获得为顺式和反式外消旋异构体混合物的标题化合物(52mg,66%)。LC-MS C31H38F3N3O的计算值:525;实测值:526(M+H)。
实施例65
步骤A:
将54g(0.29mol)(2-氨基噻唑-4-基)乙酸乙酯和50g(0.276mol)二苯基甲亚胺的纯混合物在190℃搅拌5h,然后在室温冷却,用100mL二氯甲烷稀释。将全部混合物转移到硅胶柱上,用20%EtOAc/己烷洗脱。获得浅黄色固体标题化合物(70g,69%收率)。1H MR(300MHz,CDCl3):δ1.26(t,3H),3.74(s,2H),4.15(q,2H),6.87(s,1H),77.25-7.86(m,10H);质谱(NH3-CI):m/z 351(M+1)。
步骤B:
在室温,向步骤A的35g(0.10Mol)Schiff碱酯、顺-1,3-二氯-2-丁烯(13mL,0.11Mol)和500mL DME的混合物中分批加入固体NaH(60%油,10.0g,0.25Mol)。搅拌所得混合物2天,倾入2000mL冰-水中,用1500mL乙醚萃取。乙醚层用水(3×500mL)洗涤,用硫酸钠干燥,然后蒸发。FC(硅胶,5%EtOAc/己烷)获得油状标题化合物(24g,59%)。1H NMR(300MHz,CDCl3):1.20(t,3H),2.87(d,2H),3.19(d,2H),4.14(q,2H),5.29(s,2H),6.71(s,1H),7.26-7.81(m,10H)。质谱(NH3-CI):m/z 403(M+1)。
步骤C:
将24.0g(0.059mol)步骤B的环戊烯Schiff碱溶于100mL 4NHCl/二噁烷。在1h后,加入1.8mL水。搅拌混合物3h,蒸发至干。将残余物溶于100mL CH2Cl2,加入15mL DIEA。将全部混合物倾倒在硅胶柱上,用20%EtOAc/己烷洗脱除去二苯甲酮,然后用40%EtOAc/己烷洗脱得到浅黄色固体标题化合物(12.0g,85%)。1H NMR(300MHz,CDCl3):δ1.19(t,3H),2.79(d,12H),3.15(d,2H),4.13(q,2H),5.66(s,2H),5.82(宽峰,2H),6.19(s,1H)。
步骤D:
将12g(50mmol)步骤C的氨基噻唑、28g(130mmol)二碳酸二叔丁酯、0.6g DMAP和250mL DCM的混合物搅拌过夜,然后蒸发。在硅胶快速色谱法提纯(10%EtOAc/己烷)后,获得黄色油状标题化合物(21.0g,96%)。1H NMR(300MHz,CDCl3):δ1.18(t,3H),1.49(d,18H),2.88(d,2H),3.18(d,2H),4.13(q,2H),5.65(s,2H),6.83(s,1H)。质谱(NH3-CI):m/z 439(M+1)。
步骤E:
在-78℃,向13.1g(30.0mmol)步骤D的酯的50mL无水乙醚溶液滴加BH3.DMS的THF溶液(14mL,24mmol)。撤去冷却浴,在室温搅拌混合物3h,用250mL DCM稀释,加入25g乙酸钠和55gPCC。搅拌混合物过夜。将全部混合物应用到硅胶柱,依次用10%EtOAc/己烷、30%EtOAc/己烷洗脱。获得两种组分。较快洗脱的异构体(黄色油状物,6.0g)被鉴定为标题化合物。1H NMR(300MHz,CDCl3):1.21(t,3H),1.50(s,18H),2.33(t,2H),2.42-2.70(m,2H),2.78-3.10(dd,2H),4.18(q,3H),6.88(s,1H)。质谱(NH3-CI):m/z 455(M+1)。
步骤F:
以上的较慢洗脱的组分被证实为标题化合物(树胶状物质,1.80g)。1H NMR(300MHz,CDCl3):(t,3H),1.46(s,9H),2.27(3,2H),2.38-2.62(m,2H),2.64-3.00(dd,2H),4.11(q,2H),6.66(s,1H)。质谱(NH3-CI):m/z 355(M+1)。
步骤G:
在室温,将步骤F的1.40g(4.00mmol)酮酯和0.82g(13mmol)氢氧化锂一水化合物在20mL MeOH和2mL水溶液中的混合物搅拌过夜。将全部混合物应用到硅胶柱,用10%MeOH/CH2Cl2洗脱。真空蒸发获得浅黄色固体。获得1.30g蓬松固体标题产物。1H NMR(300MHz,CDCl3):δ(t,9H),2.10-3.20(m,8H),6.60(s,1H)。
步骤H:

将步骤G制备的酮酸(1.0g,3.0mmol)、中间体1(为HCl盐,0.66g,3.0mmol)、EDC(0.95g,5.0mmol)和二氯甲烷(10mL)的混合物搅拌2天。混合物用二氯甲烷稀释,用1N aq.HCl洗涤,用硫酸钠干燥,然后蒸发。残余物用制备型TLC提纯(1500微米,10%[aq.NH4OH/MeOH 1/9]/DCM),获得黄色固体标题产物(0.52g,34%)。
LC-MS C25H27F3N2O4S的计算值:508;实测值:509(M+H)。
步骤I:
将步骤H产物(510mg,1.0mmol)与TFA(10mL)混合30min。除去TFA,残余物用制备型TLC提纯(10%[aq.NH4OH/MeOH1/9]/DCM)获得所需白色固体产物(223mg,55%)。LC-MSC20H19F3N2O2S的计算值:408;实测值:409(M+H)。
步骤J:
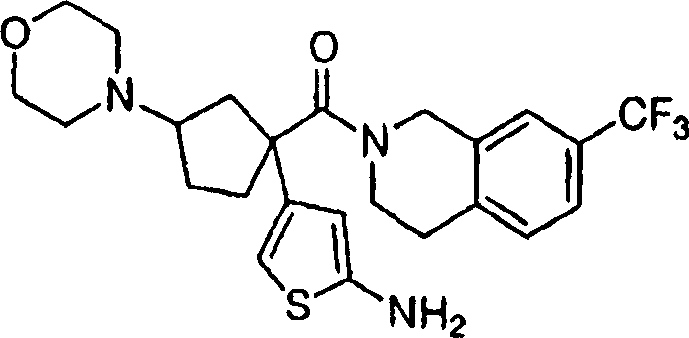
将步骤I的产物(210mg,0.50mmol)、吗啉(440mg,5.0mmol)、分子筛(4,500mg)和三乙酰氧基硼氢化钠(420mg,2.0mmol)的二氯甲烷(15mL)的混合物搅拌过夜。用饱和碳酸钠水溶液猝灭反应物。通过硅藻土过滤除去固体。粗产物萃取到二氯甲烷中,用制备型TLC提纯(1000微米,10%[aq.NH4OH/MeOH 1/9]/DCM)。获得为顺式和反式外消旋异构体混合物的标题化合物(200mg,84%)。LC-MSC24H28F3N3O2S的计算值:479;实测值:480(M+H)。
实施例66
将中间体3(70mg,0.20mmol)、顺-2,6-二甲基吗啉(23μL,0.20mmol)、分子筛(4μL,200mg)、三乙酰氧基硼氢化钠(210mg,0.99mmol)和DCM的混合物搅拌5天。反应混合物用DCM稀释,过滤,用sat.aq.NaHCO3、水和盐水洗涤。DCM层用硫酸钠干燥,过滤,浓缩。残余物用制备型TLC提纯(1000微米)(用3%[aq.NH4OH/MeOH1/9]/DCM展开)获得为游离碱的最终标题化合物。用4N HCl/二噁烷处理生成相应的盐酸盐(62.2mg)。ESI-MS C25H35F3N2O2的计算值:452.27;实测值:453(M+H)。
用HPLC(AD柱,5%EtOH/己烷)将非对映异构体分离为一个含两种顺式非对映异构体的混合物和两个单一的非对映异构体。
实施例67

实施例68用中间体3和(1S,4S)-(+)-2-氮杂-5-氧杂双环[2.2.1]庚烷盐酸盐按照实施例66的方法制备。将顺式和反式异构体用制备型TLC拆分(4/95.6/0.4,MeOH/DCM/NH4OH)。顶部斑点:ESI-MSC24H31F3N2O2的计算值:436.23;实测值:437(M+H)。底部斑点:ESI-MS C24H31F3N2O2的计算值:436.23;实测值:437(M+H)。
实施例68

实施例68用哌啶和中间体3按照实施例49的方法制备。ESI-MSC24H33F3N2O的计算值:422.25;实测值:423(M+H)。
实施例69

将中间体22(563mg,2.43mmol)溶于二氯甲烷,冷却至-78℃。将臭氧通入溶液中直到出现持续的蓝色。然后使氮气通过溶液直到蓝色消失。将Na2SO4加入以上搅拌溶液。将反应混合物过滤到装有中间体23的烧瓶。将二氯甲烷(20mL)、三乙胺(136μL,0.974mmol)和NaB(OAc)3H(929mg,4.38mmol)加入反应烧瓶。在室温、氮气氛下搅拌反应混合物2h,然后用二氯甲烷稀释,用饱和碳酸氢钠溶液(2×100mL)和盐水(1×100mL)洗涤。有机层用硫酸镁干燥,过滤,然后浓缩。将粗产物分为三批,装载到三个离子交换柱。用40%MeOH/己烷(100mL)冲洗除去杂质。然后将所需产物用30%2N NH3的MeOH溶液从色谱柱洗脱,再用二氯甲烷稀释。实施例69(131mg,0.223mmol,46%收率)用制备型TLC提纯(用32%EtOAc/hex)。然后将非对映异构体通过制备型TLC分离(用45%EtOAc/hex)(顶部异构体:20mg,0.034mmol,7%收率;中部异构体:45mg,0.076mmol,16%收率;底部异构体:54mg,0.092mmol,19%收率)。ESI-MSC32H41F3N4O3计算值:586.69,实测值587(M+H)。
几种其它的哌嗪和高哌嗪根据实施例69的相同方法制备。表7总结了这些化合物。
表7(实施例70-72)

实施例73

以上化合物用中间体5和4,5,6,7-四氢-1H-吡唑并[4,3-]吡啶按照实施例49的方法制备。ESI-MS C24H30F3N5O的计算值:461.24;实测值462(M+H)。
实施例74
以上化合物用中间体5和1,4-二氮杂庚环-5-酮按照实施例49的方法制备。ESI-MS C23H31F3N4O2的计算值:452.24;实测值453(M+H)。
实施例75
以上化合物用中间体5和2,3,4,5-四氢-1,4-苯并吖庚因按照实施例57的方法制备。顶部斑点:ESI-MS C27H32F3N3O2的计算值:487.24;实测值488(M+H)。底部斑点:ESI-MS C27H32F3N3O2的计算值:487.24;实测值488(M+H)。
实施例76
将碳载钯催化剂(顶部:6mg,中部:18mg,底部:20mg)加入分别装有实施例70三种非对映异构体之一的三个烧瓶(顶部:29.6mg,中部:89.9mg,底部:102.3mg)。将混合物溶于MeOH(3-6mL),反应容器中反复通入氢气。在室温、氢气氛下搅拌反应物4.5h,然后通过Acrodisc针筒过滤器(用0.45μm PTFE膜)。将化合物76(顶部异构体:16.0mg,0.0354mmol,71%收率;中部异构体:42.7mg,0.0943mmol,62%收率;底部异构体:34.5mg,0.0762mmol,44%收率)用制备型TLC分离(用10%[NH4OH/MeOH(1∶9)]/二氯甲烷溶剂系统)。ESI-MS C24H35F3N4O计算值:452.56,实测值453(M+H)。
实施例77

将实施例76(TLC的中部立体异构体,11mg,0.024mmol)溶于二氯甲烷(3mL),冷却至0℃。将氯甲酸乙酯(5μL,0.05mmol)、三乙胺(10μL,0.07mmol)和催化量DMAP(1-2mg)加入反应烧瓶。将反应物加热至室温,在氮气氛下搅拌2h。此后,用DCM稀释反应物,用饱和碳酸氢钠溶液(1×25mL)和盐水(1×25mL)洗涤。用硫酸镁干燥有机层,过滤,然后浓缩。实施例77(4.2mg,0.0080mmol,33%收率)通过制备型TLC提纯(用2%[NH4OH/MeOH(1∶9)]/二氯甲烷溶剂系统)。ESI-MS C27H39F3N4O3计算值:524.62,实测值:525(M+H)。
实施例78

将实施例76(TLC的中部立体异构体,11mg,0.024mmol)溶于二氯甲烷(3mL),冷却至0℃。将醋酸酐(11μL,0.12mmol)、三乙胺(23μL,0.17mmol)和催化量DMAP(1-2mg)加入反应烧瓶。在氮气氛下,将反应物加热至室温,搅拌2.5h。然后将反应物用DCM稀释,用饱和碳酸氢钠溶液(1×25mL)和盐水(1×25mL)洗涤。用硫酸镁干燥有机层,过滤,然后浓缩。实施例78(5.3mg,0.011mmol,45%收率)通过制备型TLC提纯(用2%[NH4OH/MeOH(1∶9)]/二氯甲烷溶剂系统)。ESI-MS C26H37F3N4O2计算值:494.59,实测值:495(M+H)。
实施例79

将二氯甲烷(2-3mL/烧瓶)加入三个分别装有实施例77的三种非对映异构体的烧瓶(顶部:16mg,0.035mmol,中部:11mg,0.023mmol,底部:14mg,0.031mmol)。将异氰酸甲酯(顶部:21μL,0.35mmol,中部:14μL,0.23mmol,底部:19μL,0.31mmol)加入各个反应容器。将各反应物搅拌3h,浓缩后用制备型TLC(2%[NH4OH/MeOH(1∶9)]/二氯甲烷溶剂系统)分离实施例79(顶部异构体:12.8mg,0.0251mmol,72%收率;中部异构体:10.4mg,0.0204mmol,89%收率;底部异构体:11.1mg,0.0218mmol,70%收率)。ESI-MS C26H38F3N5O2计算值:509.61,实测值:510(M+H)。
实施例80
步骤A
将顺-羟基-D-脯氨酸(2.98g,22.7mmol)的甲醇(20mL)溶液用亚硫酰氯(1.78mL,24.4mmol)处理,在室温搅拌1h。然后将反应混合物加热至65℃过夜。蒸发挥发分得到粗制的甲酯(4.0816g),将其溶于二氯甲烷(50mL),加入二异丙基乙胺(9.59mL,55.1mmol)。冷却反应混合物至0℃,用注射器加入纯氯甲酸苄基酯(3.71mL,26.0mmol)。在0℃搅拌30min后,撤去冷却浴。将反应物倾入柠檬酸水溶液(10%,50mL)中猝灭,将产物萃取到二氯甲烷。合并的有机相再次用盐水洗涤,用无水硫酸钠干燥,真空除去溶剂。粗产物用快速色谱法提纯(硅胶,乙酸乙酯∶己烷/4∶6)获得4.0717g(69%)纯净产物。1H NMR(500MHz,CDCl3):δ7.35m(5H),5.21(d,J=12.6Hz,2H),5.10(d,J=13Hz,1H),5.06(d,J=12.35Hz,1H),4.45(m,2H),3.80(s,3H),2.35(m,1H),2.15(m,1H)。
步骤B
将前一步骤的醇(2.63g,9.42mmol)和二异丙基乙胺(3.28mL,18.8mmol)的二氯甲烷40mL溶液冷却至-78℃,用注射器加入三氟甲磺酸叔丁基二甲基甲硅烷酯(2.596mL,11.30mmol)。在-78℃搅拌反应混合物2h,倾在50mL饱和碳酸氢钠溶液上猝灭。分离出有机相,用硫酸镁干燥,真空除去挥发分。残余物(6.0g)进一步用柱色谱法提纯(硅胶,乙酸乙酯∶己烷/1∶4)获得2.8355g(76%)纯净产物。1HNMR(500MHz,CDCl3):7.35m(5H),5.20(m=2H),4.45(m,2H),3.70(m,4H),3.45(m,1H),2.23(m,2H)0.87(bs,9H),0.05(m,6H)。
步骤C
在0℃,将前一步骤的酯(2.83g,7.19mmol)的THF(10mL)溶液用硼氢化锂(250mg,11.5mmol)处理,在室温搅拌反应混合物72h。然后用乙醚稀释,用水和1M磷酸溶液洗涤。干燥合并的有机萃取液(硫酸钠),真空除去溶剂。粗产物进一步用柱色谱法提纯(硅胶,乙酸乙酯∶己烷/3∶7)获得1.5636g(60%)纯净产物。LC MS:C19H31NO4Si计算值365.20,实测值366.25[M+H]+。
步骤D

将前一步骤的醇(1.56g,4.27mmol)和二异丙基乙胺(1.49mL,8.53mmol)的二氯甲烷(20mL)溶液用甲烷磺酰氯(496μL,6.41mmol)在0℃处理。搅拌冷的反应混合物15min,然后在室温搅拌30min。用二氯甲烷稀释,用水洗涤,用硫酸镁干燥。真空除去溶剂,获得的粗产物立即用于下一步骤。LC MS:C20H33NSO6Si计算值443.18,实测值444.25[M+H]+。
步骤E
将前一步骤的TBS-乙醚(1.82g,4.10mmol)的THF(50mL)溶液用四丁基氟化铵(4.51mL,1M THF溶液)处理。在室温搅拌反应混合物直到LC MS分析显示完全除去TBS基团(约15min)。然后冷却反应混合物至0℃,加入氢化钠(180mg,60%悬浮液,45.1mmol)。在室温连续搅拌24h。将反应物倾在水上猝灭,粗产物萃取到乙酸乙酯。真空除去溶剂,粗产物用快速色谱法提纯(二氯甲烷∶乙醚/7∶3)获得595mg(59%)所需产物。1H NMR(500MHz,CDCl3):7.38(m,5H),5.30(m,2H),4.52(m,2H),3.80(m,2H),3.40(m,2H),1.70(m,2H)。
步骤F

将前一步骤的CBZ-胺(590mg,2.53mmol)的乙醇(30mL)溶液在Pd/C(142mg,10%)存在下于气罐压力氢化。LC监测反应中初始原料的消失和甲苯的消失。滤除催化剂,蒸发滤液至干,剩下321mg(94%)所需产物。
步骤G

将中间体3(120mg,0.340mmol)、前一步骤的胺盐酸盐(46mg,0.34mmol)、二异丙基乙胺(60μL,0.34mmol)和4分子筛(碾碎的,360mg)的二氯乙烷(10mL)溶液用三乙酰氧基硼氢化钠处理,在室温搅拌24h。将反应物倾在饱和碳酸氢钠溶液上猝灭,粗产物用二氯甲烷萃取。真空除去挥发分,制备型TLC(乙酸乙酯∶乙醇∶氢氧化铵90∶8∶2)处理得到纯净产物(118mg,80%)。LC MS:C24H31N2F3O2计算值436.23,实测值437.20[M+H]+。
实施例81
中间体21(150mg,0.43mmol)的二氯甲烷(10mL)溶液中加入EDC(414mg,2.16mmol)、HOAt(59mg,0.43mmol)和中间体1(87mg,0.43mmol),在室温搅拌所得混合物4天。用水猝灭反应物,用20mL二氯甲烷稀释。分离出有机层,水层用DCM(2×20mL)萃取。合并有机物,用硫酸钠干燥,过滤后减压蒸发。残余物用制备型TLC提纯(洗脱剂:10%甲醇∶89.5%二氯甲烷∶0.5%NH4OH)获得40mg(17%)纯净的所需终产物,为两种顺式异构体的混合物。LC-MS C30H36N2OF4计算值516.28,实测值[M+H]+517。
实施例82

将嘧啶基哌啶盐酸盐(中间体8)(67mg,0.34mmol)与中间体4(100mg,0.28mmol)、三乙胺(46μL,0.35mmol)、4粉末状分子筛(100mg)在DCM中混合。在室温15min后,加入三乙酰氧基硼氢化钠(240mg,1.13mmol),搅拌所得混合物3天,然后通过硅藻土过滤,用DCM稀释,用饱和碳酸氢钠和盐水洗涤。用硫酸镁干燥有机层,过滤,减压浓缩得到粗制油状物,用制备型TLC提纯(硅胶,0.3%NH4OH/2.7%MeOH/97%DCM)得到110mg无色油状物。通过ChiralPak AD柱HPLC(用30%异丙醇/己烷洗脱)拆分各个非对映异构体得到2种单一非对映异构体和一个含2种其它非对映异构体的混合物。
第一个峰10mg:ESI-MS C28H35F3N4O的计算值:500.28;实测值504(M+H)。
第二个峰11mg:ESI-MS C28H35F3N4O的计算值:500.28;实测值504(M+H)。
第三个峰7.0mg ESI-MS C28H35F3N4O的计算值:500.28;实测值504(M+H)。
实施例83-91
几种其它实施例按照实施例82的类似方法制备,使用不同的哌啶中间体。以下是这些实施例(83-91)。
实施例92

此产物按照实施例81的类似方法制备,但是中间体21用市售4-氰基-4-苯基哌啶替代。粗产物用制备型TLC提纯(洗脱剂:5%甲醇∶94.5%DCM∶0.5%NH4OH)获得实施例92,为4种异构体的混合物。LC-MS C31H36F3N3O计算值523.28,实测值[M+H]+524。
实施例93

此产物按照实施例81的类似方法制备,但是中间体21用市售4-苯基哌啶替代。粗产物用制备型TLC提纯(洗脱剂:10%甲醇∶89.5%DCM∶0.5%NH4OH)获得实施例93,为4种异构体的混合物。LC-MSC30H37F3N2O计算值498.29,实测值[M+H]+499。
实施例94
步骤A
环戊酮甲酸(中间体3步骤C的)(2.3g,14mmol)的二氯甲烷(70mL)溶液中加入乙二酰氯(1.8mL,21mmol),然后加入2滴DMF。在室温搅拌溶液80min,然后减压蒸发。将残余物溶于DCM(20mL),用注射器加入准备好的中间体3(3.0g,14mmol)和三乙胺(2.1mL,15mmol)的DCM(50mL)溶液。在室温搅拌所得混合物18h,然后用水(25mL)猝灭。分离有机物,用1N HCl、饱和碳酸氢钠溶液和盐水洗涤,用无水硫酸镁干燥,过滤,然后蒸发。残余物用Biotage Flash40提纯(洗脱剂:40%乙酸乙酯/60%己烷)获得2.2g(43%)标题化合物。
步骤B
将步骤A的产物(200mg,0.54mmol)、市售4-氟苯基哌啶盐酸盐(120mg,0.54mmol)、二异丙基乙胺(94μL,0.54mmol)和碾碎的分子筛(4,100mg)的二氯甲烷(10mL)溶液用三乙酰氧基硼氢化钠(343mg,1.60mmol)处理,在室温搅拌过夜。用饱和碳酸氢钠(10mL)猝灭反应物,用10mL DCM稀释。分离出有机层,水溶液用二氯甲烷(2×5mL)洗涤。合并有机物,用无水硫酸钠干燥,过滤后蒸发。残余物用制备型TLC提纯(洗脱剂:8%乙醇∶90%乙酸乙酯∶2%NH4OH)获得两种异构体,分别是未知绝对立体构型的较高洗脱的异构体(25mg,5%)和较低洗脱的异构体(37.2mg,7.6%)。LC-MS C30H35N2OF5计算值534.28,实测值[M+H]+535。
实施例95

此产物按照实施例94的类似方法制备,但是4-氟苯基哌啶盐酸盐用中间体9替代。粗产物用制备型TLC提纯(洗脱剂:90%乙酸乙酯∶8%乙醇∶2%NH4OH)获得450mg(66%)标题产物,为4种异构体的混合物。LC-MS C28H34N4OF4计算值518.27,实测值[M+H]+519。
几种其它实施例按照实施例94的类似方法制备,其中采用不同的哌啶中间体。以下是这些实施例(96-104)。
实施例105
中间体21(150mg,0.43mmol)的二氯甲烷(10mL)溶液中加入EDC(170mg,0.86mmol)、HOAt(59mg,0.43mmol)和中间体21(91mg,0.43mmol),在室温搅拌所得混合物3天。用水猝灭反应物,用20mL二氯甲烷稀释。分离出有机层,水层用DCM(2×20mL)萃取。合并有机物,用硫酸钠干燥,过滤后减压蒸发。残余物用制备型TLC提纯(洗脱剂:7%乙醇∶92%二氯甲烷∶1.0%NH4OH)获得121mg(50%)所需终产物,为两种顺式异构体的混合物。LC-MS C29H36BrFN2O计算值526.28,实测值[M+H]+527和[(M+2)+H]+529。
实施例106
实施例105(100mg,0.210mmol)、苯基硼酸(30mg,0.23mmol)、甲苯(1.4mL)和MeOH(0.6mL)溶液中加入Na2CO3(80mg,0.74mmol)和Pd(PPh3)2Cl2(8mg,0.01mmol)的H2O(0.4mL)溶液。将反应混合物在高压管中于80℃加热12h,然后通过硅藻土过滤,浓缩至干。浓缩物用DCM稀释,用1N NaOH溶液(3x)洗涤,用硫酸钠干燥,真空浓缩。粗产物用制备型TLC提纯(洗脱剂:5/94.5/0.5,MeOH/DCM/NH4OH)获得实施例106(63.3mg,63.3%),为2种顺式异构体混合物。LC-MS C35H41FN2O计算值524.29,实测值[M+H]+525.4。
实施例107
此产物按照实施例106的类似方法制备,但是苯基硼酸用4。吡啶基硼酸替代。粗产物用制备型TLC提纯(洗脱剂:10/89/1.0,MeOH/DCM/NH4OH)获得实施例107,为两种顺式异构体的混合物。LC-MS C34H40FN3O计算值525.26,实测值[M+H]+526.3。
实施例108

此产物按照实施例106的类似方法制备,但是苯基硼酸用3-吡啶基硼酸替代。粗产物用制备型TLC提纯(洗脱剂:10/89/1.0,MeOH/DCM/NH4OH)获得实施例108,为两种顺式异构体的混合物。LC-MS C34H40FN3O计算值525.26,实测值[M+H]+526.3。
实施例109
此产物按照实施例106的类似方法制备,但是苯基硼酸用2-吡啶基硼酸替代。粗产物用制备型TLC提纯(洗脱剂:10/89/1.0,MeOH/DCM/NH4OH)获得实施例109,为两种顺式异构体的混合物。LC-MS C34H40FN3O计算值525.26,实测值[M+H]+526.3。
实施例110
步骤A
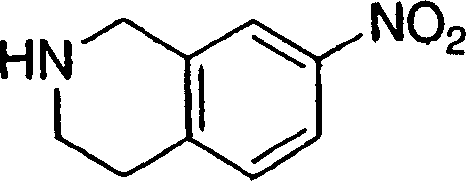
冰冷浓硫酸中滴加1,2,3,4-四氢异喹啉(23mL,170mmol),然后以温度上升不超过5℃的速率加入硝酸钾(18.8g,186mmol)。在加入完毕后,在室温搅拌混合物18h,然后倾在冰(700g)和NH4OH(150mL)的搅拌混合物上。混合物用CHCl3(3×300mL)萃取。合并的CHCl3层用饱和NaCl(200mL)洗涤,用硫酸钠干燥,过滤,真空浓缩。将残余物溶于乙醇(130mL),在冰浴冷却下加入浓盐酸(22mL)。过滤除去沉淀,用甲醇重结晶得到产物(12.45g,34%);1H NMR500MHz(DMSO-d6)δ=3.13(2H,t,J=6.2Hz),3.35(2H,t,J=6.2Hz),4.35(2H,s),7.50(1H,d,J=8.5Hz),8.07(1H,dd,J=2.3和8.5Hz),8.19(1H,d,J=2.3Hz)10.02(2H,brs)。
步骤B

步骤A产物(12g,58mmol)、吡啶(23.7mL,293mmol)的无水CH2Cl2(50mL)冷(0℃)悬浮液中滴加三氟醋酸酐(12mL,87mmol),在室温搅拌所得混合物18h。将反应混合物倾在冰(500g)上,用CH2Cl2(4×150mL)萃取。合并的二氯甲烷层用1N HCl(4×100mL)、饱和NaCl(100mL)洗涤,用硫酸钠干燥,过滤后真空蒸发得到产物(15.92g,89%);1H NMR 500MHz(CDCl3)δ=3.07(2H,m),3.91和3.94(2H,t,J=6.2Hz),4.85和4.88(2H,s),7.36(1H,dd,J=8.7和11.9Hz),8.07(1H,dd,J=2.3和8.5Hz),8.01-8.08(2H m)。
步骤C
在50psi下,将步骤B产物(16g,58mmol)的乙醇(200mL)溶液在Parr装置中通过PtO2氢化,直到停止吸收氢。通过硅藻土过滤除去催化剂,真空浓缩滤液得到产物(14.2g,100%);1H NMR 500MHz(CDCl3)δ=2.84(2H,t,J=5.7Hz),3.35(2H,br s),3.80和3.84(2H,t,J=6.0Hz),4.64和4.69(2H,s),6.45(1H,d,J=10.0Hz),6.57(1H,td,J=2.4和8.5Hz),6.95(1H,d,J=8.5Hz)。
步骤D

步骤C产物(2.0g,8.7mmol)的甲苯(50mL)溶液中加入N,N-二甲基甲酰胺连氮(2.3g,16mmol)和一刮勺端的甲苯磺酸,回流加热所得混合物24h。真空浓缩混合物,残余物在CH2Cl2(50mL)和水(50mL)间分配。分离出有机层,用饱和碳酸氢钠(25mL)和饱和NaCl(25mL)洗涤,用硫酸镁干燥,过滤,真空浓缩。残余物用CH2Cl2(4mL)研磨,过滤,干燥得到产物(1.0g,39%);1H NMR 500MHz(CDCl3)=3.05(2H,t,J=5.7Hz),3.90和3.95(2H,t,J=6.0Hz),4.83和4.89(2H,s),7.20(1H,d),7.26(1H,t),7.38(1H,t)8.45(2H,s)。
步骤E

步骤D产物(1.0g,3.4mmol)的乙醇(50mL)溶液中加入碳酸钾(2.3g,17mmol)的水(10mL)溶液,回流加热所得混合物90min。真空浓缩冷反应混合物,固体残余物用CH2Cl2(3×10mL)萃取。真空蒸发合并的CH2Cl2层,残余物用二氧化硅柱色谱法提纯(用含0.5%NH4OH的10%CH3OH/CH2Cl2洗脱)得到产物(550mg,82%);1H NMR500MHz(CDCl3)δ=2.69(1H,br s),2.85(2H,t,J=5.9Hz),3.17(2H,t,J=6.0Hz),4.07(2H,s),7.20(1H,d,J=1.8Hz),7.14(1H,dd,J=1.8和8.0Hz),7.23(1H,d,J=8.0Hz),8.43(2H,s)。
步骤F
此产物按照实施例81的类似方法制备,但是中间体1用步骤E产物替代。粗产物用制备型TLC提纯(洗脱剂:10%甲醇∶89.5%DCM∶0.5%NH4OH)获得实施例30,为两种顺式异构体的混合物。LC-MSC31H38FN5O计算值515.31,实测值[M+H]+516。
实施例111

步骤A

实施例110步骤C制备的三氟乙酰胺(7.5g,31mmol)的乙醇(200mL)溶液中加入碳酸钾(17g,120mmol)的水(50mL)溶液,回流加热所得混合物90min。真空浓缩冷反应混合物,将残余物用水(200mL)稀释。混合物用CH2Cl2(3×100mL)萃取。合并的CH2Cl2层用硫酸钠干燥,过滤,浓缩得到产物(3.76g,83%);1H NMR 500MHz(CDCl3)δ=2.69(2H,t,J=6.0Hz),3.11(2H,t,J=6.0Hz),3.45(2H,br s),3.92(2H,s),6.36(1H,d,J=2.3Hz),6.52(1H,dd,J=2.3和8.0Hz),6.89(1H,d,J=8.0Hz)。
步骤B
步骤A产物(3.76g,25.4mmol)的四氢呋喃(100mL)溶液中加入三乙胺(4.24mL,30.5mmol)、氯甲酸苄基酯(4.0mL,28mmol),在室温搅拌所得混合物4h。将乙酸乙酯(100mL)加入反应混合物,整体用水(250mL)、5%柠檬酸溶液(150mL)、饱和碳酸氢钠(150mL)和饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤,真空浓缩得到产物(7.2g,100%)。
步骤C:

在冰浴冷却下,步骤B产物(7.2g,25mmol)和吡啶(5.1mL,64mmol)的CH2Cl2(150mL)溶液中加入三氟醋酸酐(5.38mL,38.1mmol),在室温搅拌所得混合物5h。将混合物倾在冰(150g)上,用CH2Cl2(4×100mL)萃取。合并的二氯甲烷层用1N HCl(4×75mL)、饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤后蒸发。将残余物溶于CCl4(200mL),加入三苯基膦(10g,38mmol),回流加热所得混合物15h,冷却,真空浓缩。将残余物溶于无水N,N-二甲基甲酰胺(150mL),将此溶液加入叠氮化钠(1.65g,25.4mmol)的无水N,N-二甲基甲酰胺(75mL)溶液,在室温搅拌所得混合物3h。将反应混合物倾入水(500mL)中,用Et2O(3×100mL)萃取。合并的Et2O层用水(2×250mL)、饱和NaCl(100mL)洗涤,用硫酸镁干燥,过滤,真空浓缩。残余物用二氧化硅柱色谱法提纯(用10%EtOAc/己烷至20%EtOAc/己烷梯度洗脱)得到产物(3.4g,33%);1H NMR 500MHz(CDCl3)δ=2.99(2H,br m),3.80(2H,t,J=6.0Hz),4.75(2H,s),5.21(2H,s),7.20-7.45(8H,m)。
步骤D:
在通入氮气下,步骤C产物(3.4g,8.4mmol)的甲醇(100mL)溶液)中加入10%钯碳(500mg),在氢气罐下搅拌所得混合物5h。通过硅藻土过滤除去催化剂,真空蒸发滤液得到产物(2.2g,97%);1H NMR500MHz(CDCl3)δ=2.33(1H,brs),2.91(2H,t,J=6.0Hz),3.19(2H,t,J=6.0Hz),4.08(2H,s),7.14(1H,d,1.8Hz),7.22(1H,dd,J=1.8和8.2Hz),7.31(1H,d,J=8.2Hz)。
步骤E:
此产物按照实施例81的类似方法制备,但是中间体1用步骤D产物替代。粗产物用制备型TLC提纯(洗脱剂:10%甲醇:89.5%DCM:0.5%NH4OH)获得实施例31,为两种顺式异构体的混合物。LC-MSC31H36F3N6O计算值584.29,实测值[M+H]+585。
实施例112

此产物按照实施例81的类似方法制备,但是中间体1用市售6,7-二乙氧基-四氢异喹啉替代。粗产物用制备型TLC提纯(洗脱剂:5%乙醇∶94%DCM∶1.0%NH4OH)获得实施例32,为两种顺式异构体的混合物。LC-MS C33H45FN2O3计算值536.34,实测值[M+H]+537。
实施例113

步骤A

此产物按照中间体1的类似方法制备,但是2-氟-4-三氟甲基苯基乙腈用3,4-二-氟甲基苯基乙腈替代。LC-MS C9H9F2N计算值169.07,实测值[M+H]+170.1
步骤B

此产物按照实施例81的类似方法制备,但是中间体1用步骤A产物替代。粗产物用制备型TLC提纯(洗脱剂:6%乙醇∶93%DCM∶1.0%NH4OH)获得实施例33,为两种顺式异构体的混合物。LC-MSC29H35F3N2O计算值484.27,实测值[M+H]+485。
实施例114

此产物按照实施例82的类似方法制备,但是中间体8用市售4-羟基-4-苯基哌啶替代。粗产物用制备型TLC提纯(洗脱剂:10%甲醇∶89%DCM∶1.0%NH4OH)获得实施例114,为4种异构体的混合物。LC-MS C30H37F3N2O2计算值514.28,实测值[M+H]+515。
实施例115和116

实施例115 实施例116
将中间体5(100mg,0.22mmol)、4-氟苯基哌啶盐酸盐(57mg,0.26mmol)、二异丙基乙胺(45μL,0.26mmol)和碎分子筛(4,50mg)的二氯乙烷(5mL)溶液用三乙酰氧基硼氢化钠(233mg,1.10mmol)处理,在室温搅拌过夜。用饱和碳酸氢钠(10mL)猝灭反应物,用10mLDCE稀释。分离出有机层,水层用二氯甲烷(2×5mL)洗涤。合并有机物,用无水硫酸钠干燥,过滤后蒸发。残余物用制备型TLC提纯(洗脱剂:0.5%NH4OH∶5%MeOH∶94.5%CH2Cl2)获得72.3mg(63%)终产物,为两种非对映异构体的混合物。用制备型ChiralCel OD柱HPLC完成分离,利用15%乙醇和85%己烷洗脱剂洗脱,流速9mL/min。获得不需要的反式异构体(35mg,50%)和所需顺式异构体(25mg,36%)。总收率86%。两种产物的LC-MS:C29H35N3OF4计算值517.28,实测值[M+H]+518.3。
实施例115:1H NMR(第1种异构体,不需要)(500MHz,CDCl3)δ8.73(s,1H),7.69(s,1H),7.20(app dd,J=5.5,8.6Hz,2H)6.98(app t,J=8.6Hz,2H),[4.88(d,J=17.5Hz,1H),4.80(d,J=17.5Hz,1H)(ABx q)]4.05-3.96(m,1H),3.88(app dt,J=5.9,13.1Hz,1H),3.27(br d,J=11.4Hz,1H),3.12(brt,J=5.6Hz,3H),2.83(dd,J=5.7,11.9Hz,1H),2.52-2.44(m,1H),2.43-2.34(m,1H),2.16-1.95(m,6H),1.86-1.74(m,4H),1.45(br t,J=10.0,3H),1.03(d,J=6.6Hz,3H),0.83(d,J=6.6Hz,3H)。
实施例116:1H NMR(第2种异构体,所需的)(500MHz,CDCl3)δ8.72(s,1H),7.69(s,1H),7.19(app dd,J=5.5,8.6Hz,2H)6.98(app t,J=8.6Hz,2H),[4.94(br s,1H),4.69(br d,J=17.6Hz,1H)(ABx q)]4.05-3.80(m,1H),3.20-3.08(m,4H),2.68(dd,J=6.6,12.8Hz,1H),2.52-2.43(m,2H),2.28(dd,J=7.3,12.8Hz,1H),2.14-2.00(m,3H),1.97-1.87(m,2H),1.83(br d,J=12.8Hz,2H)1.75(br d,12.4Hz,2H),1.56-1.48(m,1H),1.42-1.34(m,1H),1.01(d,J=6.5Hz,3H),0.80(d,J=6.6Hz,3H)。
实施例117
将嘧啶基哌啶盐酸盐(中间体8,133mg,0.564mmol)与中间体5(100mg,0.282mmol)、DIEA(240μL,1.40mmol)和4粉末状分子筛(200mg)在DCM中混合。在室温15min后,加入三乙酰氧基硼氢化钠(300mg,1.41mmol),搅拌所得混合物3天,然后通过硅藻土过滤,用DCM稀释,用饱和碳酸氢钠和盐水洗涤。用硫酸钠干燥有机层,过滤,减压浓缩得到粗制油状物,用制备型TLC提纯(硅胶,0.5%NH4OH/4.5%MeOH/95%DCM)得到126mg无色油状物。通过HPLC拆分顺/反异构体(采用ChiralPak OD柱,用20%乙醇/己烷洗脱)得到57mg反式异构体和45mg顺式异构体。
第一个峰57mg:ESI-MS C27H34F3N5O的计算值:501.27;实测值502(M+H)。
第二个峰45mg:ESI-MS C27H34F3N5O的计算值:501.27;实测值502(M+H)。
实施例118-129

几种其它实施例按照实施例117的类似方法制备,其中采用不同的哌啶中间体。以下是这些实施例(118-129)。
实施例130
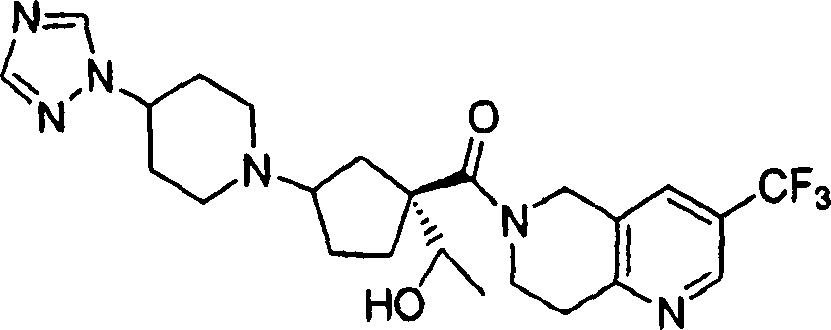
步骤A

此酮通过拆分外消旋的中间体6获得,其中采用ChiralCel OD制备柱,用15%乙醇/己烷洗脱,9.0mL/min。在类似分析条件下(1.0mL/min),较快洗脱的对映异构体的保留时间是11.25min。LC MSC17H19F3N2O3计算值356.13,实测值357.05[M+H]+。
步骤B

最终化合物用本实施例步骤A中较快洗脱的酮和中间体10按照实施例19的方法合成。相应的顺式-和反式-非对映异构体的混合物通过制备型TLC分离。LC MS C24H31F3N6O2计算值492.25,实测值493.30[M+H]+。
实施例131

本化合物用实施例130步骤A制备的酮和中间体9按照实施例19的方法合成。LC MS C26H32F3N5O2计算值503.25,实测值504.25[M+H]+。
实施例132

步骤A
此酮按照中间体6介绍的方法合成,但是在中间体6步骤C中用甲氧基甲基氯替代乙醛。相应的对映异构体通过ChiralCel OD制备柱HPLC(洗脱剂:己烷∶乙醇/85∶15,9.0mL/min)分离。LC MSC26H32F3N5O2计算值503.25,实测值504.25[M+H]+。
步骤B
最终化合物用步骤A的酮和4-(4-氟苯基)哌啶按照实施例19的方法制备。相应的异构体通过制备型TLC(洗脱剂:乙酸乙酯∶乙醇∶氢氧化铵/90∶9∶1)获得。LC MS C26H32F3N5O2计算值503.25,实测值504.70[M+H]+。
实施例133
步骤A

2-(三氟甲基)丙烯酸(20.0g,143mmol)和苄基醇(14.8mL,142mmol)的DCM(150mL)混合物中分批加入EDC(40.93g,214.2mmol)。搅拌反应混合物2h,用DCM稀释,用水和盐水洗涤,用硫酸钠干燥,过滤,浓缩。残余物用快速柱色谱法提纯(硅胶,5%EtOAc/己烷)获得粘性油状产物(13.7g,42%)。1H-NMR(400MHz,CDCl3)δ7.36-7.43(m,5H),6.78(d,1H),6.48(d,1H),5.32(s,2H)。
步骤B

在氮气氛下,火焰干燥的烧瓶中加入乙酸2-[(三甲硅烷基)甲基]-2-丙烯-1-基酯(12.18mL,57.35mmol)和实施例52步骤A的中间体(13.2g,57.4mmol)和四(三苯基膦)钯(0)(13.3g,11.5mmol)的THF(200mL)。将反应混合物回流过夜,用DCM(150mL)稀释,过滤,然后浓缩。残余物用快速柱色谱法提纯(硅胶,100%己烷-2.5%EtOAc/己烷-5%EtOAc/己烷)获得产物(11.68g,71.7%)。1H-NMR(400MHz,CDCl3)δ7.33-7.42(m,5H),5.23(s,2H),4.93(m,2H),3.03(m,1H),2.78(m,1H),2.36-2.52(m,3H),2.12-2.24(m,1H)。
步骤C
将步骤B产物(11.6g,40.8mmol)的DCM(150mL)溶液冷却至-78℃,通入氮气至饱和。将臭氧通入反应混合物直到溶液变成蓝色,然后将三苯基膦(12.8g,49.0mmol)加入混合物。搅拌反应混合物过夜,然后减压蒸发。残余物用快速柱色谱法提纯(硅胶,30%EtOAc/己烷)获得标题化合物(8.49g,72.7%)。1H-NMR(400MHz,CDCl3)δ7.33-7.42(m,5H),5.26(s,2H),2.92(m,1H),2.65(m,1H),2.35-2.54(m,4H)。
步骤D

步骤C中间体(1.00g,3.49mmol)的乙醇(60mL)溶液中加入Pd-C(10%,100mg)。将反应混合物加入Parr振荡器,在50lb H2压力下振荡1.5h。溶液用甲醇稀释,通过硅藻土过滤,然后真空蒸发获得黄色油状酸(692mg,100%)。LC-MS C7H7F3O3计算值:196.03;实测值:197(M+H)。
步骤E

步骤D产物(684mg,3.49mmol)、EDC(2.01g,10.5mmol)、中间体2(916mg,3.84mmol)和DCM(30mL)的混合物中加入DIEA(670μL,3.84mmol),在室温搅拌所得溶液过夜。反应混合物用DCM稀释,用水和盐水洗涤,用硫酸钠干燥,过滤后真空蒸发。残余物用柱色谱法提纯(硅胶,50%EtOAc/己烷)获得标题化合物。1H-NMR(400MHz,CDCl3)δ8.75(s,1H),7.71(s,1H),4.86(d,J=5.5Hz,2H),3.91-4.08(m,2H),3.18(t,J=5.0Hz,2H),2.98(s,2H),2.64-2.85(m,2H),2.43-2.56(m,2H)。LC-MS C16H14F6N2O2的计算值:380.10;实测值:381(M+H)。
步骤F

步骤E化合物(100mg,0.263mmol)、4-苯基哌啶盐酸盐(52mg,0.26mmol)、分子筛(4,180mg)、DIEA(46μL,0.26mmol)和DCM(5mL)的混合物中加入三乙酰氧基硼氢化钠(167mg,0.789mmol),在室温搅拌所得混合物过夜。用DCM稀释反应物,通过硅藻土过滤,减压蒸发。残余物用制备型TLC提纯(1000微米,洗脱剂:0.4%NH4OH水溶液∶4%MeOH∶95.6%DCM)获得为游离碱的顺式-和反式-异构体的混合物。顺式-和反式-异构体通过制备型手性HPLC(手性OD柱,洗脱剂:5%EtOH/己烷)分离获得终产物,为所需顺式异构体的标题化合物。用4N HCl/二噁烷处理生成盐酸盐(20.4mg)。1H-NMR(400MHz,CDCl3)δ8.74(s,1H),7.70(s,1H),7.20-7.35(m,5H),4.85(m,2H),3.99(m,2H),3.14-3.24(m,5H),2.46-2.56(m,3H),2.02-2.22(m,5H),1.70-1.91(m,5H)。LC-MS C27H29F6N3O的计算值:525.22;实测值:526(M+H)。
实施例134

步骤A
在-78℃,向环已烷(15mL)的100mL二氯甲烷搅拌溶液通入臭氧直到出现浅蓝色。通过氮气流除去过量臭氧,然后加入60mL甲硫醚。让混合物静置过夜,用硫酸钠干燥。低真空除去溶剂和DMS。粗产物直接用于下一步骤无需再提纯。
步骤B
将中间体27(135mg,0.3mmol)、步骤A的1,6-己烷-二醛(~100mg)、分子筛(4,50mg)、DIEA(130mg,1.0mmol)的DCM(10mL粗产物)的混合物搅拌5min。然后加入三乙酰氧基硼氢化钠(424mg,2.0mmol)。搅拌所得混合物2h,用Na2CO3饱和水溶液猝灭,过滤,用DCM洗涤。分离滤液,水溶液用DCM萃取。合并的DCM层用硫酸钠干燥,蒸发。残余物用制备型TLC提纯(1000微米)(用10%[aq.NH4OH/MeOH 1/9]/DCM展开)获得为游离碱的标题化合物终产物。用4M HCl/二噁烷处理形成盐酸盐(42mg)。ESI-MS C25H34F3N3O3的计算值:481;实测值:482(M+H)。
实施例135

将实施例134(35mg)、Pd/C(5%,5mg)和甲醇(20mL)的混合物在Parr装置上氢化1h。过滤除去催化剂。蒸发滤液获得白色固体标题产物(32mg)。ESI-MS C25H36F3N3O的计算值:451;实测值:452(M+H)。
实施例136

步骤A
在-78℃,向环庚烯(5mL)的50mL二氯甲烷搅拌溶液通入臭氧直到出现浅蓝色。通过氮气流除去过量臭氧,然后加入20mL甲硫醚。让混合物静置过夜,用硫酸钠干燥。低真空除去溶剂和DMS。粗产物直接用于下一步骤无需再提纯。
步骤B
将中间体27(135mg,0.3mmol)、步骤A的1,7-庚烷-二醛(~100mg)、分子筛(4,50mg)、DIEA(130mg,1.0mmol)和DCM(10mL粗产物)的混合物搅拌5min。然后加入三乙酰氧基硼氢化钠(424mg,2.0mmol)。搅拌所得混合物2h,用Na2CO3饱和水溶液猝灭,过滤,用DCM洗涤。分离滤液,水溶液用DCM萃取。合并的DCM层用硫酸钠干燥,蒸发。残余物用制备型TLC(1000微米)(用10%[aq.NH4OH/MeOH 1/9]/DCM展开)提纯获得为游离碱的标题化合物终产物。用4M HCl/二噁烷处理形成盐酸盐(50mg)。ESI-MSC26H36F3N3O3的计算值:485;实测值:486(M+H)。
实施例137

步骤A

将5-降冰片烯-2-甲酸(5.4g,40mmol)、苄基醇(4.3g,40mmol)、EDAC.HCl(9.5g,50mmol)、DIEA(5.2g,40mmol)称量加入烧瓶。加入50mL二氯甲烷。搅拌混合物过夜,用2M aq.HCl、水和碳酸钠饱和水溶液洗涤,用硫酸钠干燥,蒸发。残余物用MPLC(10%EtOAc/己烷)提纯。获得为外型和内型异构体混合物的标题化合物(5.2g)。
步骤B

在-78℃,向5-降冰片烯-2-甲酸苄基酯(1.2g,5mmol)的50mL二氯甲烷搅拌溶液通入臭氧直到出现浅蓝色。通过氮气流除去过量臭氧,然后加入20mL甲硫醚。让混合物静置过夜,用硫酸钠干燥。低真空除去溶剂和DMS。粗产物直接用于下一步骤无需再提纯。
步骤C

中间体27(86mg,0.2mmol)、步骤B的二-醛酯(~200mg)、分子筛(4,500mg)、DIEA(130mg,1.0mmol)和DCM(10mL粗制物)的混合物搅拌5min。然后加入三乙酰氧基硼氢化钠(420mg,2.0mmol)。搅拌所得混合物2h,用Na2CO3饱和水溶液猝灭,过滤,用DCM洗涤。分离滤液,水溶液用DCM萃取。合并的DCM层用硫酸钠干燥,蒸发。残余物用制备型TLC(1000微米)(用10%[aq.NH4OH/MeOH 1/9]/DCM展开)提纯获得为游离碱的标题化合物终产物。用4N HCl/二噁烷处理生成相应的盐酸盐(62mg)。ESI-MSC34H40F3N3O5的计算值:627;实测值:428(M+H)。
步骤D

将步骤C氨基酯(60mg)、Pd/C(5%,100mg)和甲醇(20mL)的混合物用Parr装置在50lb氢气氛下氢化1h。过滤除去催化剂。蒸发滤液,残余物用制备型TLC(用甲醇展开)提纯获得白色固体标题产物(18mg)。ESI-MS C27H36F3N3O的计算值:507;实测值:508(M+H)。
实施例138
步骤A
将中间体26(为盐酸盐,1.2g,4.0mmol)、中间体23(1.1g,4.0mmol)、PyBrOP(1.9g,4.0mmol)、DMAP(0.29g,2.4mmol)、DIEA(2.8mL,16mmol)和10mL二氯甲烷的混合物在室温搅拌过夜。将全部混合物应用到硅胶柱,用20%乙酸乙酯/己烷洗脱。获得白色固体标题化合物(1.7g,83%)。LC-MS C26H35F3N2O5计算值512,实测值[M+H-100]+413。
步骤B
将以上步骤A的酰胺(1.7g,3.3mmol)与20mL 4M HCl/二噁烷混合。搅拌所得溶液1h,蒸发,高真空干燥获得白色固体标题产物(1.45g,100%)。LC-MS C21H27F3N2O3计算值412,实测值[M+H]+413。
步骤C
将以上步骤B的中间体(140mg,0.30mmol)、戊二醛(50%H2O,120mg,0.60mmol)、分子筛(4,1500mg)、DIEA(52mg,0.40mmol)和DCM(10mL)的混合物搅拌5min。然后加入三乙酰氧基硼氢化钠(212mg,1.00mmol)。搅拌所得混合物1h,用Na2CO3饱和水溶液猝灭,过滤,用DCM洗涤。分离滤液,水溶液用DCM萃取。合并的DCM层用硫酸钠干燥,然后蒸发。残余物用制备型TLC(1000微米)(用10%[aq.NH4OH/MeOH 1/9]/DCM展开)提纯获得为游离碱的标题化合物终产物。用4N HCl/二噁烷处理生成相应的盐酸盐(80mg)。ESI-MS C26H35F3N2O3的计算值:480;实测值:481(M+H)。
实施例139

将实施例138(45mg,0.090mmol)、氢氧化锂一水化合物(50mg),水(0.1mL)和甲醇(1.0mL)的混合物在室温搅拌过夜,将全部混合物装载到制备型TLC上,用甲醇展开。获得白色固体标题化合物(34mg)。LC-MS C25H33F3N2O3计算值466,实测值[M+H]+467。
实施例140

将实施例138步骤B的中间体(140mg,0.30mmol)、实施例136步骤A的1,6-己烷二醛(~100mg)、分子筛(4,500mg)、DIEA(130mg,1.00mmol)和DCM(20mL)的混合物搅拌5min。然后加入三乙酰氧基硼氢化钠(424mg,2.00mmol)。搅拌所得混合物1h,用Na2CO3饱和水溶液猝灭,过滤,用DCM洗涤。分离滤液,水溶液用DCM萃取。合并的DCM层用硫酸钠干燥,蒸发。残余物用制备型TLC(1000微米)(用10%[aq.NH4OH/MeOH 1/9]/DCM展开)提纯获得为游离碱的标题化合物终产物。用4M HCl/二噁烷处理生成相应的盐酸盐(75mg)。ESI-MS C27H37F3N2O3的计算值:494;实测值:495(M+H)。
实施例141

将实施例140(20mg,0.04mmol)、氢氧化锂一水化合物(50mg)、水(0.1mL)和甲醇(1.0mL)的混合物在室温搅拌过夜,将全部混合物装载到制备型TLC上,用甲醇展开。获得白色固体标题化合物(15mg)。LC-MS C26H35F3N2O3计算值480,实测值[M+H]+481。
实施例142
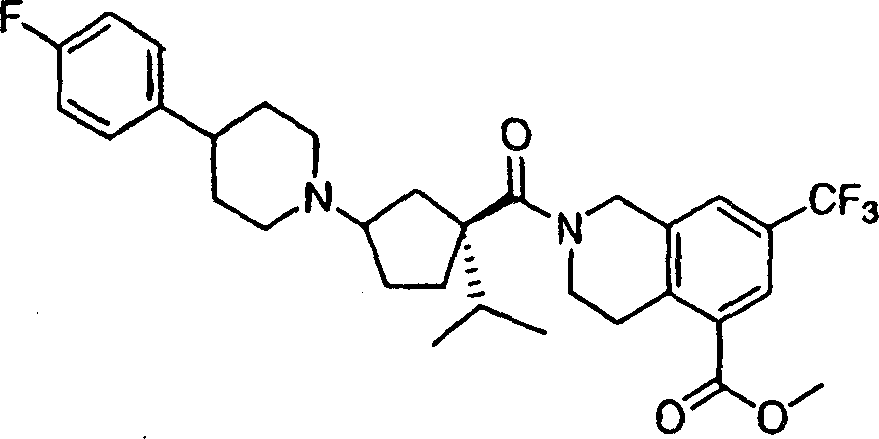
步骤A

中间体29(190mg,1.1mmol)的1mL二氯甲烷搅拌溶液中加入乙二酰氯(2M,0.70mL,1.4mmol)的二氯甲烷溶液,然后加入恒量DMF。在室温搅拌混合物30min。然后真空蒸发除去溶剂和过量试剂。将残余物溶于1mL二氯甲烷,加入中间体2(295mg,1.00mmol)和DIEA(260mg,2.0mmol)的二氯甲烷(2mL)溶液中。搅拌反应物2h.。将全部混合物装载到制备型TLC板(1000微米),用10%MeOH/DCM展开。获得黄色固体标题化合物(300mg)。LC-MSC21H24F3NO4计算值411,实测值[M+H]+412。
步骤B
将以上步骤A的酮(300mg,0.73mmol)、4-氟苯基哌啶盐酸盐(214mg,1.00mmol)、DIEA(129mg,1.00mmol)、分子筛(4,500mg)、三乙酰氧基硼氢化钠(212mg,1.00mmol)和10mL二氯甲烷的混合物在室温搅拌过周末,用碳酸钠饱和水溶液猝灭,用二氯甲烷萃取,用制备型TLC(1000微米,用10%MeOH/DCM洗脱)提纯。获得标题化合物的顺式和反式异构体混合物(270mg)。LC-MSC32H38F4N2O3计算值574,实测值[M+H]+575。
实施例143

将实施例142(22mg,0.04mmol)、氢氧化锂一水化合物(30mg)和MeOH/H2O(9/1,0.5mL)的混合物在60℃搅拌2h,将全部混合物装载到制备型TLC板,用甲醇展开。获得白色固体标题化合物(12mg)。LC-MS C31H34F4N2O3计算值560,实测值[M+H]+561。
实施例144

步骤A:
将中间体29(100mg,0.588mmol)溶于DCM(20mL),依次用乙二酰氯(153μL,1.76mmol)和DMF(1滴)处理。在室温搅拌所得溶液2h,然后浓缩至于,高真空干燥30min。将所得残余物溶于DCM(5mL),滴加到中间体1(177mg,0.882mmol)的DCM(5mL)和三乙胺(5mL)搅拌溶液中。在室温搅拌所得反应混合物过夜,然后用DCM稀释,用碳酸氢钠、1N HCl水溶液和盐水洗涤。用硫酸镁干燥有机层,过滤,然后减压浓缩得到230mg所需粗产物,直接用于下一步无需再提纯。
步骤B:
将前一步骤的产物(115mg,0.326mmol)、3-哌啶-4-基苯甲酸乙酯盐酸盐(131mg,0.489mmol)、DIEA(83μL,0.49mmol)、分子筛(4,100mg)、三乙酰氧基硼氢化钠(346mg,1.63mmol)和10mL二氯甲烷的混合物在室温搅拌过周末,用碳酸氢钠饱和水溶液猝灭,用二氯甲烷萃取,用制备型TLC提纯(硅胶,用40%THF/己烷洗脱)。获得标题化合物的顺式和反式异构体混合物(200mg)。LC-MSC33H41F3N2O3的计算值:570.3;实测值[M+H]+571.6。用ChiralCelOD柱(用10%乙醇/己烷洗脱)拆分各个立体异构体:
峰1:LC-MS C33H41F3N2O3的计算值:570.3;实测值[M+H]+571.6。
峰2:LC-MS C33H41F3N2O3的计算值:570.3;实测值[M+H]+571.6。
实施例145

将实施例144峰2(80mg,)、氢氧化锂一水化合物(30mg)和EtOH/H2O(4/1,4mL)的混合物在室温搅拌过夜。产物用反相HPLC提纯,用常见方法转化为盐酸盐。获得白色固体标题化合物(45mg)。LC-MS C31H37F3N2O3计算值:542.28;实测值[M+H]+543。
实施例146

将实施例144峰1(78mg,)、氢氧化锂一水化合物(30mg)和EtOH/H2O(4/1,4mL)的混合物在室温搅拌过夜。产物用反相HPLC提纯,用常见方法转化为盐酸盐。获得白色固体标题化合物(49mg)。LC-MS C31H37F3N2O3计算值:542.28;实测值[M+H]+543。
实施例147
实施例147按照实施例144的方法制备,但是用中间体30替代3-哌啶-4-基苯甲酸乙酯盐酸盐。所得4种立体异构体用手性色谱法分离。
实施例148

实施例148按照实施例145的方法制备,但是用实施例147的一种立体异构体替代实施例144的一种产物。
实施例149

实施例149按照实施例145的方法制备,但是用实施例147的一种立体异构体替代实施例144的一种产物。
实施例150

实施例150按照实施例145的方法制备,但是用实施例147的一种立体异构体替代实施例144的一种产物。
实施例151
实施例151按照实施例145的方法制备,但是用实施例147的一种立体异构体替代实施例144的一种产物。
Claims (20)
1.一种下式I的化合物或者其药学上可接受的盐或单独的非对映异构体:

式I
其中:
X选自C、N、O、S和SO2;
Y为N或C;
R1选自以下基团:
氢、-C1-6烷基、-C0-6烷基-O-C1-6烷基、-C0-6烷基-S-C1-6烷基、-(C0-6烷基)-(C3-7环烷基)-(C0-6烷基)、羟基、杂环、-CN、-NR12R12、-NR12COR13、-NR12SO2R14、-COR11、-CONR12R12和苯基,
其中R11独立选自以下基团:羟基、氢、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R12选自以下基团:氢、C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R13选自以下基团:氢、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中R14选自以下基团:羟基、C1-6烷基、-O-C1-6烷基、苄基、苯基和C3-6环烷基,所述烷基、苯基、苄基和环烷基可以是未被取代的或者被1-3个独立选自以下的取代基取代:卤基、羟基、C1-3烷基、C1-3烷氧基、-CO2H、-CO2-C1-6烷基和三氟甲基,
其中所述烷基和环烷基未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(f)C1-3烷基,
(g)-O-C1-3烷基,
(h)-COR11,
(i)-SO2R14,
(j)-NHCOCH3,
(k)-NHSO2CH3,
(l)-杂环,
(m)=O,
(n)-CN,
其中所述苯基和杂环未被取代或者被1-3个独立选自以下的取代
基取代:卤基、羟基、C1-3烷基、C1-3烷氧基和三氟甲基;R2选自以下基团:
(a)氢,
(b)羟基,
(c)卤基,
(d)C1-3烷基,其中所述烷基未被取代或者被1-6个独立选自氟和羟基的取代基取代,
(e)-NR12R12
(f)-COR11,
(g)-CONR12R12,
(h)-NR12COR13,
(i)-OCONR12R12,
(j)-NR12CONR12R12,
(k)-杂环,
(l)-CN,
(m)-NR12-SO2-NR12R12,
(n)-NR12-SO2-R14,
(o)-SO2-NR12R12,
(p)=O,其中R2通过双键与环连接;
在Y为N时,R3为氧或不存在;
在Y为C时,R3选自以下基团:
(a)氢,
(b)羟基,
(c)卤基,
(d)C1-3烷基,其中所述烷基未被取代或者被1-6个独立选自以下的取代基取代:氟、羟基和-COR11,
(e)-NR12R12,
(f)-COR11,
(g)-CONR12R12,
(h)-NR12COR13,
(i)-OCONR12R12,
(j)-NR12CONR12R12,
(k)-杂环,
(l)-CN,
(m)-NR12-SO2-NR12R12,
(n)-NR12-SO2-R14,
(o)-SO2-NR12R12,
(p)硝基;
R4选自以下基团:
(a)氢,
(b)C1-6烷基,
(c)三氟甲基,
(d)三氟甲氧基,
(e)氯,
(f)氟,
(g)溴,
(h)苯基;
R5选自以下基团:
(a)C1-6烷基,其中烷基可以是未被取代的,或者被1-6个氟取代,以及任选被羟基取代,
(b)-O-C1-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(c)-CO-C1-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(d)-S-C1-6烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(e)-吡啶基,它可以是未被取代的或者被一个或多个选自以的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(f)氟,
(g)氯,
(h)溴,
(i)-C4-6环烷基,
(j)-O-C4-6环烷基,
(k)苯基,它可以是未被取代的或者被一个或多个选自以下的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(l)-O-苯基,它可以是未被取代的或者被一个或多个选自以下的取代基取代:卤基、三氟甲基、C1-4烷基和COR11,
(m)-C3-6环烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(n)-O-C3-6环烷基,其中烷基可以是未被取代的或者被1-6个氟取代,
(o)-杂环,
(p)-CN,
(q)-COR11;
R6选自以下基团:
(a)氢,
(b)C1-6烷基,
(c)三氟甲基,
(d)氟,
(e)氯,
(f)溴;
R7选自以下基团:
氢、(C0-6烷基)-苯基、(C0-6烷基)-杂环、(C0-6烷基)-C3-7环烷基、(C0-6烷基)-COR11、(C0-6烷基)-(烯基)-COR11、(C0-6烷基)-SO3H、(C0-6烷基)-W-C0-4烷基、(C0-6烷基)-CONR12-苯基和(C0-6烷基)-CONR15-V-COR11,或者在X为O、S或SO2时,R7不存在,
其中V为C1-6烷基或苯基,
其中W选自单键、-O-、-S-、-SO-、-SO2-、-CO-、-CO2-、-CONR12-和-NR12-,
其中R15可以为氢、C1-4烷基,或者R15通过1-5个碳的接头连接V的一个碳构成环,
其中所述C0-6烷基未被取代或被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基,
其中所述苯基、杂环、环烷基和C0-4烷基未被取代或被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-C0-3-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12,
(j)-C0-3-杂环,
或者,所述苯基和杂环可以与另一个杂环稠合,另一个杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基,
其中烯基未被取代或者被1-3个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)C1-3烷基,
(d)苯基,
(e)杂环;
R8选自以下基团:
(a)氢,
(b)在X为O、S、SO2或N时或者分别连接R7和R10的碳之间为双键时,R8不存在,
(c)羟基,
(d)C1-6烷基,
(e)C1-6烷基-羟基,
(f)-O-C1-3烷基,
(g)-COR11,
(h)-CONR12R12,
(i)-CN;
或者R7和R8接合在一起构成选自以下的环:
(a)1H-茚,
(b)2,3-二氢-1H-茚,
(c)2,3-二氢-苯并呋喃,
(d)1,3-二氢-异苯并呋喃,
(e)2,3-二氢-苯并噻吩,
(f)1,3-二氢-异苯并噻吩,
(g)6H-环戊二烯并[d]异噁唑-3-醇,
(h)环戊烷,
(i)环己烷,
其中所构成的环可以是未被取代的或者被1-5个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-C0-3-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12,
(j)-C0-3-杂环,
或者,R7与R9或R8与R10可以结合在一起构成苯环或杂环,其中所述环未被取代或被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)羟基,
(d)C1-3烷基,
(e)-O-C1-3烷基,
(f)-COR11,
(g)-CN,
(h)-NR12R12,
(i)-CONR12R12;
R9和R10独立选自以下基团:
(a)氢,
(b)羟基,
(c)C1-6烷基,
(d)C1-6烷基-COR11,
(e)C1-6烷基-羟基,
(f)-O-C1-3烷基,
(g)=O,这时R9或R10通过双键与环连接,
(h)卤基;
n选自0、1和2;
虚线为单键或双键。
2.权利要求1的化合物,其为具有式Ia结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、Y和n为权利要求1中的定义,R16和R17独立选自以下基团:
(a)氢,
(b)卤基,
(c)三氟甲基,
(d)羟基,
(e)C1-3烷基,
(f)-O-C1-3烷基,
(g)-C0-3-CO2H,
(h)-C0-3-CO2C1-3烷基,
(i)-CN,
(j)-C0-3-杂环,
或者,R16和R17接合在一起构成与苯环稠合的杂环,杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基。
3.权利要求1的化合物,其为具有式Ib结构的化合物或者其药学上可接受的盐或单独的非对映异构体::

其中虚线为单键或双键,R1、R2、R3、R5、R9、R16、R17、Y和n为权利要求1中的定义。
4.权利要求1的化合物,其为具有式Ic结构的化合物或者其药学上可接受的盐或单独的非对映异构体::
其中R1、R2、R3、R5、R9、R16、R17、Y和n为权利要求1中的定义,H为杂环。
5.权利要求1的化合物,其为具有式Id结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、R11、Y、W和n为权利要求1中的定义,
其中所述C1-4碳链可以是未被取代的或者被1-4个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基
或者所述C1-4碳链可以包含在C3-7环烷基环中。
6.权利要求1的化合物,其为具有式Ie结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R9、R16、R17、X、Y和n为权利要求1中的定义;虚线为单键或双键;o为1或2;A、B和D可以独立选自C、N、O或S,在X、A、B、D均为C且o=2时,它们构成苯环,或者在至少X、A、B和D之一为N、O或S而不为C时,它们构成杂环。
7.权利要求1的化合物,其为具有式If结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R2、R3、R5、R7、R9、R10、Y和n为权利要求1中的定义;X为N或O,在X为O时,R7不存在。
8.权利要求1的化合物,其为具有式Ig结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中R1、R5、R9、R16、R17和Y为权利要求1中的定义,
或者R16和R17接合在一起构成与苯环稠合的杂环,杂环本身可以是未被取代的或者被1-2个独立选自以下的取代基取代:羟基、卤基、-COR11和-C1-3烷基。
9.权利要求1的化合物,其为具有式Ih结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

其中虚线为单键或双键,R1、R5、R9、R16、R17和Y为上文中的定义。
10.权利要求1的化合物,其为具有式Ii结构的化合物或者其药学上可接受的盐或单独的非对映异构体:
其中R1、R5、R9、R16、R17和Y为上文中的定义,H为杂环。
10.权利要求1的化合物,其为具有式Ij结构的化合物或者其药学上可接受的盐或单独的非对映异构体:
其中R1、R5、R9、R11、Y和W为上文中的定义,其中所述C1-4碳链可以是未被取代的或者被1-4个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-C0-6烷基,
(d)-O-C1-3烷基,
(e)三氟甲基,
(f)-C0-2烷基-苯基。
11.权利要求1的化合物,其为具有式Ik结构的化合物或者其药学上可接受的盐或单独的非对映异构体:

式Ik
其中R1、R5、R9、R10和Y为上文中的定义。
12.权利要求1的化合物,其中R1选自以下基团:-C1-6烷基、-C0-6烷基-O-C1-6烷基和-(C0-6烷基)-(C3-7环烷基)-(C0-6烷基),所述烷基和环烷基未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(f)C1-3烷基,
(g)-O-C1-3烷基,
(h)-COR11,
(i)-CN,
(j)-NR12R12,
(k)-CONR12R12。
13.权利要求1的化合物,其中R1选自以下基团:
(l)-C1-6烷基,它未被取代或者被1-6个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(e)-COR11,
(2)-C0-6烷基-O-C1-6烷基-,它未被取代或者被1-6个独立选自以下的取代基取代:
(a)卤基,
(b)三氟甲基,
(c)-COR11,
(3)-(C3-5环烷基)-(C0-6烷基),它未被取代或者被1-7个独立选自以下的取代基取代:
(a)卤基,
(b)羟基,
(c)-O-C1-3烷基,
(d)三氟甲基,
(e)-COR11。
14.权利要求13的化合物,其中R1选自以下基团:
(a)C1-6烷基,
(b)羟基取代的C1-6烷基,
(c)被1-6个氟取代的C1-6烷基。
15.权利要求14的化合物,其中R1选自以下基团:
(a)-CH(CH3)2,
(b)-CH(OH)CH3,
(c)-CH2CF3。
16.一种药用组合物,该组合物包含惰性载体和权利要求1的化合物。
17.一种调节哺乳动物趋化因子受体活性的方法,该方法包括给予有效量的权利要求1的化合物。
18.一种治疗、改善、控制或减轻炎性免疫调节性病症或疾病的方法,该方法包括给予患者有效量的权利要求1的化合物。
19.一种治疗、改善、控制或减轻类风湿性关节炎的方法,该方法包括给予患者有效量的权利要求1的化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US46367303P | 2003-04-17 | 2003-04-17 | |
| US60/463,673 | 2003-04-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1805965A true CN1805965A (zh) | 2006-07-19 |
| CN100384856C CN100384856C (zh) | 2008-04-30 |
Family
ID=33310806
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004800165371A Expired - Fee Related CN100384856C (zh) | 2003-04-17 | 2004-04-14 | 趋化因子受体活性调节剂杂环基环戊基四氢异喹啉和杂环基环戊基四氢吡啶并吡啶 |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1622916A4 (zh) |
| JP (1) | JP2006523704A (zh) |
| CN (1) | CN100384856C (zh) |
| AU (1) | AU2004232939A1 (zh) |
| CA (1) | CA2521625A1 (zh) |
| WO (1) | WO2004094371A2 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649056A (zh) * | 2011-05-10 | 2014-03-19 | 吉利德科学公司 | 作为离子通道调节剂的稠合杂环化合物 |
| CN117342958A (zh) * | 2023-09-12 | 2024-01-05 | 武汉理工大学 | 一种埋底界面材料及其制备得到的柔性钙钛矿太阳能电池 |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2521625A1 (en) * | 2003-04-17 | 2004-11-04 | Merck & Co., Inc. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
| US7598243B2 (en) | 2003-04-17 | 2009-10-06 | Merck & Co., Inc. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
| TWI350168B (en) | 2004-05-07 | 2011-10-11 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| WO2007053499A2 (en) | 2005-11-01 | 2007-05-10 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of ccr2 |
| WO2007053498A1 (en) | 2005-11-01 | 2007-05-10 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of ccr2 |
| BRPI0710512A2 (pt) * | 2006-04-20 | 2012-06-05 | Hoffmann La Roche | moduladores derivados de diazepan de receptores de quimiocinas |
| WO2008108445A1 (ja) | 2007-03-07 | 2008-09-12 | Takeda Pharmaceutical Company Limited | ベンゾオキサゼピン誘導体およびその用途 |
| US8952034B2 (en) | 2009-07-27 | 2015-02-10 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| AR080374A1 (es) | 2010-03-05 | 2012-04-04 | Sanofi Aventis | Procedimiento para la preparcion de 2-(ciclohexilmetil)-n-(2-((2s)-1-metilpirrolidin-2-il) etil)- 1,2,3,4-tetrahidroisoquinolin-7- sulfonamida |
| BR112012033402A2 (pt) | 2010-07-02 | 2017-01-24 | Gilead Sciences Inc | moduladores de canais de íons conforme os compostos heterocíclicos fundidos |
| NO3175985T3 (zh) | 2011-07-01 | 2018-04-28 | ||
| TW201837023A (zh) | 2011-07-01 | 2018-10-16 | 美商基利科學股份有限公司 | 作為離子通道調節劑之稠合雜環化合物 |
| ES2904252T3 (es) | 2015-05-21 | 2022-04-04 | Chemocentryx Inc | Moduladores de CCR2 |
| JP2019527232A (ja) | 2016-08-01 | 2019-09-26 | アプティニックス インコーポレイテッド | スピロ−ラクタムnmda修飾因子及びこれを用いた方法 |
| SG10202101055VA (en) | 2016-08-01 | 2021-03-30 | Aptinyx Inc | Spiro-lactam nmda receptor modulators and uses thereof |
| MX385336B (es) | 2016-08-01 | 2025-03-18 | Aptinyx Inc | Moduladores del receptor nmda espiro-lactam y uso de los mismos. |
| CN118267479A (zh) | 2017-09-25 | 2024-07-02 | 凯莫森特里克斯股份有限公司 | 使用趋化因子受体2(ccr2)拮抗剂和pd-1/pd-l1抑制剂的联合治疗 |
| IL275898B2 (en) | 2018-01-08 | 2025-05-01 | Chemocentryx Inc | Methods for treating solid tumors with CCR2 antagonists |
| US20190269664A1 (en) | 2018-01-08 | 2019-09-05 | Chemocentryx, Inc. | Methods of treating solid tumors with ccr2 antagonists |
| CN112204031B (zh) * | 2018-01-31 | 2024-05-24 | 元羿生物科技(香港)有限公司 | 螺-内酰胺nmda受体调节剂及其用途 |
| CN111056955B (zh) * | 2019-12-16 | 2021-05-25 | 中国科学院大连化学物理研究所 | 一种环己烯制备己二胺的方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1318811B1 (en) * | 2000-08-17 | 2006-08-30 | Merck & Co., Inc. | Cyclopentyl modulators of chemokine receptor activity |
| EP1501507B1 (en) * | 2002-04-29 | 2008-05-28 | Merck & Co., Inc. | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity |
| JP4459807B2 (ja) * | 2002-04-29 | 2010-04-28 | メルク エンド カムパニー インコーポレーテッド | テトラヒドロピラニルシクロペンチルテトラヒドロイソキノリン系のケモカイン受容体活性調節剤 |
| CA2521625A1 (en) * | 2003-04-17 | 2004-11-04 | Merck & Co., Inc. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
-
2004
- 2004-04-14 CA CA002521625A patent/CA2521625A1/en not_active Abandoned
- 2004-04-14 WO PCT/US2004/011463 patent/WO2004094371A2/en not_active Ceased
- 2004-04-14 CN CNB2004800165371A patent/CN100384856C/zh not_active Expired - Fee Related
- 2004-04-14 AU AU2004232939A patent/AU2004232939A1/en not_active Abandoned
- 2004-04-14 JP JP2006510018A patent/JP2006523704A/ja active Pending
- 2004-04-14 EP EP04750112A patent/EP1622916A4/en not_active Withdrawn
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649056A (zh) * | 2011-05-10 | 2014-03-19 | 吉利德科学公司 | 作为离子通道调节剂的稠合杂环化合物 |
| CN103649056B (zh) * | 2011-05-10 | 2016-04-27 | 吉利德科学公司 | 作为离子通道调节剂的稠合杂环化合物 |
| CN117342958A (zh) * | 2023-09-12 | 2024-01-05 | 武汉理工大学 | 一种埋底界面材料及其制备得到的柔性钙钛矿太阳能电池 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2521625A1 (en) | 2004-11-04 |
| EP1622916A4 (en) | 2008-11-05 |
| WO2004094371A3 (en) | 2005-03-24 |
| WO2004094371A2 (en) | 2004-11-04 |
| EP1622916A2 (en) | 2006-02-08 |
| AU2004232939A1 (en) | 2004-11-04 |
| CN100384856C (zh) | 2008-04-30 |
| JP2006523704A (ja) | 2006-10-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1805965A (zh) | 趋化因子受体活性调节剂杂环基环戊基四氢异喹啉和杂环基环戊基四氢吡啶并吡啶 | |
| CN1802187A (zh) | 趋化因子受体的苯并噁嗪基-酰胺基环戊基-杂环调节剂 | |
| US6248755B1 (en) | Pyrrolidine modulators of chemokine receptor activity | |
| CN1956975A (zh) | 趋化因子受体活性的氨基环戊基杂环和碳环调节剂 | |
| CN1331591A (zh) | 用作趋化因子受体5调节剂的氮杂二环烷烃 | |
| CN1913778A (zh) | 趋化因子受体活性的氨基环戊基吡啶并吡嗪酮调节剂 | |
| US7491737B2 (en) | Heterarylpiperidine modulators of chemokine receptor activity | |
| CN1826334A (zh) | 趋化因子受体活性的四氢吡喃基环戊基杂环酰胺调节剂 | |
| CN1835944A (zh) | 作为ccr-5拮抗剂的喹啉酰胺衍生物 | |
| CN1918145A (zh) | 作为趋化因子受体活性调节剂的氨基杂环类化合物 | |
| CN1897941A (zh) | 趋化因子受体活性的烷基氨基、芳基氨基以及氨磺酰基环戊基酰胺调节剂 | |
| US7410961B2 (en) | 2,6-disubstituted piperiddines as modulators | |
| US20070238723A1 (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| US20060183731A1 (en) | 7 and 8 membered heterocyclic cyclopentyl benzylamide modulators of chemokine receptor activity | |
| US7598243B2 (en) | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity | |
| US7091211B2 (en) | Cyclopentyl modulators of chemokine receptor activity | |
| US7230008B2 (en) | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity | |
| US7589085B2 (en) | Tetrahydropyran heterocyclic cyclopentyl heteroaryl modulators of chemokine receptor activity | |
| CN1946696A (zh) | 四氢吡喃基环戊基1-取代的和1,1-二取代的四氢异喹啉趋化因子受体活性调节剂 | |
| CN1976702A (zh) | 作为趋化因子受体调节剂的3-氨基环戊烷甲酰胺类 | |
| CN1738820A (zh) | 新的二氮杂双环芳基衍生物 | |
| HK1106501A (zh) | 作為趨化因子受體調節劑的3-氨基環戊烷甲酰胺類 | |
| HK1103957A (zh) | 作為趨化因子受體調節劑的3-氨基環戊烷甲酰胺類 | |
| HK1096390A (zh) | 作为ccr-5拮抗剂的喹啉酰胺衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080430 Termination date: 20100414 |