CN1909902A - 用于治疗糖尿病的作为ppar调节剂的三唑、噁二唑和噻二唑衍生物 - Google Patents
用于治疗糖尿病的作为ppar调节剂的三唑、噁二唑和噻二唑衍生物 Download PDFInfo
- Publication number
- CN1909902A CN1909902A CNA2004800383003A CN200480038300A CN1909902A CN 1909902 A CN1909902 A CN 1909902A CN A2004800383003 A CNA2004800383003 A CN A2004800383003A CN 200480038300 A CN200480038300 A CN 200480038300A CN 1909902 A CN1909902 A CN 1909902A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- compound
- aryl
- hydrogen
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
- C07D271/107—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles with two aryl or substituted aryl radicals attached in positions 2 and 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
- C07D285/02—Thiadiazoles; Hydrogenated thiadiazoles
- C07D285/04—Thiadiazoles; Hydrogenated thiadiazoles not condensed with other rings
- C07D285/12—1,3,4-Thiadiazoles; Hydrogenated 1,3,4-thiadiazoles
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Pyrrole Compounds (AREA)
Abstract
本发明涉及以下结构式I表示的化合物,其中(a)X选自单键、O、S、S(O) 2和N;(b)U为脂族连接基;(c)Y选自O、C、S、NH和单键;(d)W为N、O或S;(e)E为C(R3)(R4)A或A且其中A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺。
Description
发明背景
过氧化物增殖物激活受体(PPARs)为核激素受体超家族的成员,为介导配体依赖型转录激活和阻遏的多种不同的蛋白质。PPAR分为三种亚型:PPARα、PPARγ和PPARδ。
各种同种型的表达概况明显互不相同,其中PPARα主要表达于但不限于肝脏;PPARγ主要表达于动物脂肪组织,PPARδ其表达无处不在。对PPAR各同种型和配体的研究揭示了其对与胰岛素抗性和糖尿病以及脂类疾病(如高脂血症和血脂异常症)相关过程的调节作用。PPARγ激动剂(如吡格列酮)可用于治疗非胰岛素依赖型糖尿病。这类PPARγ激动剂与胰岛素敏化作用相关。
PPARα激动剂(如非诺贝特)可用于治疗高脂血症。尽管没有临床证据显示PPARδ激动剂用于人类,许多临床前的研究表明PPARδ激动剂可用于治疗糖尿病和脂类疾病。
包括代谢综合征(肥胖、胰岛素抗性、高脂血症、高血压和动脉粥样硬化)的疾病的流行继续呈现增长的势头。需要新型的药物来解决患者的临床需要。
已表明PPARδ作为可能的治疗手段用于调节许多与代谢综合征和粥样动脉硬化相关的许多参数。例如对于肥胖的未患糖尿病的恒河猴,PPARδ激动剂降低了循环的甘油三酯和LDL的水平,减少了基础胰岛素的水平并且增加了HDL的水平(Oliver,W.R.等,Proc NatlAcad Sci 98:5306-5311;2001)。认为使用PPARδ激动剂产生胰岛素敏化作用的原因部分归于减少了myocellular脂类(Dressel,U.等,MolEndocrinol 17:2477-2493;2003)。
此外,认为动脉粥样硬化是血脂异常导致的疾病并可并发炎性疾病。对于大多数炎症、感染和组织损伤,产生C-反应蛋白(CRP)为急性期的部分反应。在诊断上其为轻度炎症的标志。血浆CRP水平高于3mg/L预示着患冠状动脉疾病的高危险性(J.Clin.Invest 111:1085-1812,2003)。
人们认为PPARδ激动剂介导消炎作用。实际上认为,用PPARδ激动剂处理LPS-刺激的巨噬细胞,结果发现降低了iNOS、IL-12和IL-6的表达(Welch,J.S.等,Proc Natl Acad Sci 100:6712-67172003)。
特别希望当活性药物选择性调节PPAR受体亚型时,提供特别需要的药理作用。在某些情况下,希望活性药物选择性调节多于一种PPAR受体亚型以得到所需的药理作用。
发明概述
本发明涉及以下结构式I表示的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:
其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0-4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)W为N、O或S;
(g)Y选自C、O、S、NH和单键;
(h)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(i)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(j)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(k)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(1)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(m)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(n)R32选自氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(o)----为任选的键,以在指示位置形成双键。
本发明的另一个实施方案为式Ia的化合物:

本发明的另一个实施方案为式Ib的化合物:

其中W1为O或S。
本发明的另一个实施方案为式Ic的化合物:

本发明的另一个实施方案为式II的化合物:
及其立体异构体、药学上可接受的盐、溶剂合物和水合物,其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0-4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基被1-4个各自独立选自R30的取代基取代;
(f)Y选自C、O、S、NH和单键;
(g)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
本发明的另一个实施方案为式III的化合物:
及其立体异构体、药学上可接受的盐、溶剂合物和水合物,
其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0-4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)Y选自O、S、NH、C和单键;
(g)E为C(R3)(R4)A;其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;条件是当Y为O时,R4选自C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、环烷基和芳基-烷基各自任选被1-3个独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
在一个实施方案中,本发明还涉及药物组合物,所述药物组合物包含至少一种本发明的混合物或其药学上可接受的盐、溶剂合物、水合物或立体异构体以及药学上可接受的载体。
在另一个实施方案中,本发明涉及一种选择性调节PPARδ受体的方法,所述方法包括将所述受体与至少一种结构式I表示的化合物或其药学上可接受的盐、溶剂合物、水合物或立体异构体接触。
在另一个实施方案中,本发明涉及一种调节PPARα、β、γ和/或δ受体中的一种或多种的方法。
在另一个实施方案中,本发明涉及一种制备结构式I表示的化合物的方法。
认为本发明的化合物能够有效治疗和/或预防代谢疾病、II型糖尿病、高血糖、高脂血、肥胖、coagaulopathy,、高血压、动脉粥样硬化和其他与代谢疾病相关的疾病以及心血管疾病。本发明的化合还可用于降低纤维蛋白原、提高HDL水平、治疗肾病、控制所需的体重、治疗demyelinating疾病、治疗特定的病毒感染以及治疗肝病。此外,与目前用于治疗这类病症的化合物相比,本发明的化合物产生较少的临床副作用。
发明详述
用于描述本发明的术语具有如下含义。
本文所用的术语″脂族连接基″或″脂族基团″表示仅由碳和氢组成的非芳族基团并且可任选包含一个和多个不饱和单元,如双和/或叁键(本文中也称作″烯基″和″炔基″)。脂族或脂族基团可为直链、支链(这两种情况本文中也称作″烷基″)或环状的(本文中也称作″环烷基)。当脂族基团为直链或支链时,通常包含约1-约10个碳原子,更通常的情况是包含约1-约6个碳原子。当脂族基团为环状时,包含约3-约1O个碳原子,更通常的情况是包含约3-约7个碳原子。脂族基团优选为C1-C10直链或支链烷基(即完全饱和的脂族基团),更优选C1-C6直链或支链烷基。实例包括但不限于甲基、乙基、丙基、正丙基、异丙基、正丁基、仲丁基和叔丁基。其他实例包括但不限于环丙基、环戊基、环己基、环戊基、环己基等。这类脂族连接基任选被1-4个各自独立选自R30的取代基取代。可优选所述脂族连接基被0-2个各自独立选自R30的取代基取代。此外,可优选所述脂族连接基的一个碳被O、NH或S置换。最后,可优选所述脂族连接基为纯粹的烷基,即没有碳原子被O、NH或S置换。
除非另有说明,否则术语″烷基″是指直链或支链饱和构型的指定碳原子数目的那些烷基。本文所用的“C0烷基”是指不存在碳,因而代表键。″烷基″的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基和叔丁基、戊基、己基、异戊基等。如上定义的烷基可任选被如上述实施方案中提到的指定数目的取代基取代。本文所用的术语“氧代烷基”是指被“=O”取代基取代的指定碳原子数目的烷基。
术语″烯基″是指直链或支链构型的具有特定碳原子数目并且具有至少一个碳-碳双键的烃链,该碳-碳双键可存在于链上的任何位置,例如乙烯基、丙烯基、丁烯基、戊烯基、乙烯基、烯丙基(alkyl)、2-丁烯基等。如上定义的烯基可任选被指定数目的如上述实施方案中提到的取代基取代。
术语″炔基″指直链或支链构型的具有特定碳原子数目并且具有至少一个碳-碳叁键的烃链,该碳-碳叁键可存在于链上的任何位置。炔基的实例有乙炔基。如上定义的炔基可任选被指定数目的如上述实施方案中提到的取代基取代。
术语“杂烷基”是指具有特定碳原子数目的直链或支链的烃链,其中至少一个碳原子被选自O、N和S的杂原子置换。
术语″环烷基″是指包含一个或多个3-12个碳原子(通常为3-7个碳原子)的环的饱和或部分饱和的碳环。环烷基的实例包括但不限于环丙基、环丁基、环戊基、环己基和环庚基等。“环烷基芳基”是指与环烷基稠合的芳基,“环烷基芳基-烷基”是指环烷基芳基通过烷基与母体分子连接。如上定义的环烷基可任选被指定数目的如上述实施方案中提到的取代基取代。
术语″卤素″是指氟、氯、溴和碘。
术语″卤代烷基″为被一个或多个选自氟、氯、溴和碘的卤素原子取代的C1-C6烷基。卤代烷基的实例为三氟甲基(CF3)。
术语″烷氧基″表示通过桥氧连接的指定碳原子数的烷基,例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基、戊氧基等。如上定义的烷氧基可任选被指定数目的如上述实施方案中提到的取代基取代。
术语″卤代烷氧基″表示通过桥氧连接C1-C6卤代烷基,例如OCF3。如上定义的卤代烷氧基可任选被指定数目的如上述实施方案中提到的取代基取代。
术语″芳基″包括碳环芳环体系(例如苯基)、稠合的多环芳环体系(例如萘基和蒽基)以及与碳环非芳环体系稠合的芳环体系(例如1,2,3,4-四氢萘基)。如上定义的“芳基”可任选被指定数目的如上述实施方案中提到的取代基取代。
术语“芳基烷基”是指通过烷基与母体分子连接的芳基烷基,该芳基烷基还可任选被被指定数目的如上述实施方案中提到的取代基取代。当芳基烷基为芳基C0烷基时,则所述芳基直接与母体分子连接。同样,芳基杂烷基是指通过杂烷基与母体分子相连的芳基。
术语“酰基”是指烷基羰基、芳基羰基和杂芳基羰基类。
本文所用的术语″杂芳基″为具有至少一个杂原子如N、S或O的芳环体系,包括含有一个或多个选自O、N和S的杂原子的5-14个碳原子的单环、双环或三环芳环。如上定义的″杂芳基″可任选被指定数目的如上述实施方案中提到的取代基取代。杂芳基的实例包括但不限于呋喃基、吲哚基、噻吩基、噻唑基、咪唑基、异唑基(isoxazoyl)、唑基、吡唑基、吡咯基、吡嗪基、吡啶基、嘧啶基(pyrimidyl)、嘧啶基(pyrimidinyl)、嘌呤基、噌啉基、苯并呋喃基、苯并噻吩基、苯并三唑基、苯并唑基、喹啉基、异唑基(isoxazolyl)、异喹啉基等。术语“杂芳基烷基”是指通过杂芳基烷基的烷基部分与母体分子连接的杂芳基。
术语″杂环烷基″是指包含一个或多个O、N或S的非芳环,包括含有一个或多个选自O、N或S的杂原子的5-14个碳原子的单环、双环或三环非芳环。如上定义的″杂环烷基″可任选被指定数目的如上述实施方案中提到的取代基取代。杂环烷基的实例包括但不限于吗啉基、哌啶基、哌嗪基、吡咯烷基和硫代吗啉基。本文所用的烷基包括完全饱和的直链和直链的烃基。
当“----”作为任选的键在五元杂环中形成双键时,可能的杂环包括:

本文所用的短语“选择性调节”是指化合物对规定的PPAR受体的EC50较其他PPAR受体亚型的EC50至少低10倍。
已提议将PPARδ与控制基础和应激产生的抗炎性作用的选择性协阻抑物(BCL-6)联合使用或分开使用(Lee,C-H.等,Science 302:453-4572003)。认为PPARδ激动剂可用于缓和其他炎症如类风湿性关节炎发作时的关节和相关组织的炎症、相关的自身免疫性疾病、骨关节炎以及种种其他炎性疾病、克罗恩氏病和牛皮癣。
当结构式I表示的化合物具有多于一个手性取代基时,该化合物可以非对映异构形式存在。可通过本领域技术人员公知的方法将非对映异构对分离,例如色谱法或结晶法,各对映异构对中的单个对映体可使用本领域技术人员熟悉的方法分离。本发明包括结构式I的化合物的各种非对映异构体及其混合物。
某些结构式I的化合物可以不同的可分离的稳定构象形式存在。由于围绕不对称单键的旋转受阻(如由于空间位阻或环张力引起)产生的扭曲不对称,使得可分离出不同的构象异构体。本发明包括结构式I的化合物的各构象异构体及其混合物。
某些结构式I的化合物可以两性离子的形式存在,因此本发明包括结构式I的化合物的各两性离子形式及其化合物。
″药学上可接受的盐″是指认为可用于临床和/或兽用的结构式I的化合物的盐。典型的药学上可接受的盐包括那些通过本发明的化合物与无机或有机酸或有机或无机碱反应制备的盐。这类盐分别被称作酸加成盐和碱加成盐。应当认识到,形成本发明任何盐的一部分的特定反荷离子并不关键,只要整体的盐是可药用的,并且反荷离子不给作为整体的盐带来不良性质即可。这些盐可通过本领域技术人员公知的方法制备。
术语“活性组分”是指结构式I化合物以及这些化合物的异构体、盐、溶剂合物和水合物。
术语“药学上可接受”是指载体、稀释剂、赋形剂和盐与组合物的其他组分在药学上相容。本发明药物组合物可通过本领域已知的方法使用众所周知且易于获得的组分制得。
“预防”是指降低接受者发生或发展任何本文所述病症的可能性。术语“预防”特别用于易患特定疾病的患者。
“治疗”是指调节疾病或病症,并防止或缓和其进一步发展或者改善与该疾病或病症有关的综合征。
“药物有效量”是指将在组织、系统或哺乳动物中引起生物或医疗反应的活性成分的量。这样的量可以出于预防目的给予认为易于发展成疾病或病症的患者。当出于预防目的对患者给药时,这样的量还可以有效地预防病症或减轻病症的严重程度。这样的量包括足以调节选择的PPAR受体或预防或调节疾病或病症的量。通常,式I化合物的有效量为0.02-5000mg/天。优选有效量为1-1,500mg/天。优选剂量为1-1,000mg/天。最优选的剂量为1-100mg/天。
所需的剂量可为单剂量或以适当的间隔给药的分剂量形式。
“哺乳动物”是哺乳类动物个体。哺乳类动物包括人、猴子、黑猩猩、大猩猩、牛、猪、马、羊、狗、猫、小鼠和大鼠。
最优选对人给药。本发明化合物和组合物可用于治疗和/或预防心血管疾病、提高血清HDL胆固醇水平、降低血清甘油三酯水平和降低血清LDL胆固醇水平。高甘油三酯和LDL水平以及低HDL水平是发展成心脏病、中风和循环系统病症和疾病的危险因素。
此外,本发明的组合物和化合物可降低患者发生意外心脏疾病的可能性。医生将知道如何确定会受益于本发明化合物和组合物给药的人。
本发明化合物和组合物还可用于治疗和/或预防肥胖症。
此外,本发明化合物和组合物可用于治疗和/或预防非胰岛素依赖性糖尿病(NIDDM),同时减轻患者的体重或不使其体重增加。另外,本发明化合物和组合物可用于治疗或预防急性或暂时性胰岛素敏感性病症例如在手术、创伤、心肌梗塞等后发生的这样的病症。医生将知道如何确定会受益于本发明化合物和组合物给药的人。
本发明还提供了治疗和/或预防人或非人哺乳动物中高血糖的方法,包括给予有此需要的患有高血糖的人或非人哺乳动物有效量的如上定义的活性成分。
本发明还涉及上述式I化合物在制备用于治疗PPAR受体介导的病症的药物中的应用。
治疗有效量的结构式I化合物可用于制备用于下述方面的药物:治疗哺乳动物,特别是人的代谢综合征、糖尿病,肥胖症,降低甘油三酯水平,降低血清LDL水平,提高血浆高密度脂蛋白水平,和治疗、预防或减小发展成动脉粥样硬化的危险性,以及预防或降低首次发生或后来发生动脉粥样硬化性疾病的危险性。一般情况下,治疗有效量的本发明化合物通常将患者的血清甘油三酯水平降低约20%或更多,并提高患者血清HDL水平。HDL水平优选被提高约30%或更多。此外,用于预防或治疗NIDDM的治疗有效量的化合物通常将患者的血糖水平、更具体来说HbAlc降低约0.7%或更多。
当在本文中使用时,代谢综合征包括前驱糖尿病性胰岛素抗性综合征及其所带来的并发症、胰岛素抗性、非胰岛素依赖性糖尿病、异常脂血症、高血糖肥胖症、凝血病、高血压以及与糖尿病有关的其他并发症。本文所述的方法和治疗包括治疗上述病症,并且还包括治疗和/或预防任一种或任意组合的下列病症:前驱糖尿病性胰岛素抗性综合征、其所带来的并发症、胰岛素抗性、II型或非胰岛素依赖性糖尿病、异常脂血症、高血糖、肥胖症和与糖尿病有关的并发症,包括心血管疾病、尤其是动脉粥样硬化。此外,本文所述方法和治疗包括以上所述的这些,以及包括治疗和/或预防任一种或任意组合的下列病症:成人呼吸窘迫综合征、类风湿性关节炎、脱髓鞘疾病、Chrohne’疾病、哮喘、系统性红斑狼疮、牛皮癣和粘液囊炎。
按照与本文详述相同的一般方式配制和给予本发明组合物。根据所需的治疗目标,本发明化合物可以有效地单独使用或者与一种或多种另外的活性剂联合使用。联合治疗包括给予含有结构式I化合物、其立体异构体、盐、溶剂合物和/或水合物(“活性成分”)和一种或多种另外的活性剂的单一剂型药物组合物,以及将活性成分与各种活性剂在分开的药物制剂中给予。例如,活性成分与胰岛素促分泌剂例如双胍类、噻唑烷二酮类、磺酰脲类、胰岛素或α-葡糖苷酶抑制剂可以在单一口服剂型组合物例如片剂或胶囊中对患者给药,或者各药物在分开的口服制剂中给药。当使用分开的制剂时,活性成分与一种或多种另外的活性剂可基本上同时给药,即并行给药,或者在错开的时间给药,即顺序给药;应当理解,联合治疗包括所有这些方案。
联合治疗或预防动脉粥样硬化的一个实例可以是这样的,其中将活性成分与一种或多种下列活性剂联合给药:抗高脂血剂;血浆HDL增高剂;抗高胆固醇血剂、贝特类药物(fibrates)、维生素、阿司匹林等。如上所述,可将活性成分与一种以上的另外的活性剂联合给药。
联合治疗的另一实例是用于治疗糖尿病和相关病症,其中可将活性成分与例如磺酰脲类、双胍类、噻唑烷二酮类、α-葡糖苷酶抑制剂、其他胰岛素促分泌剂、胰岛素以及上述用于治疗动脉粥样硬化的活性剂有效地联合使用。
本发明的活性成分具有有价值的药理性质,并且可用于包含治疗有效量的本发明的活性成分和一种或多种可药用赋形剂的药物组合物中。赋形剂是惰性物质,例如但不限于载体、稀释剂、填充剂、矫味剂、甜味剂、润滑剂、助溶剂、悬浮剂、润湿剂、粘合剂、崩解剂、包封材料和其他常规辅助剂。适当的制剂取决于所选的给药途径。药物制剂一般含有约1-约99%重量的本发明的活性组分。
药物制剂优选是单位剂型。“单位剂型”是含有适于对受治疗人或其他哺乳动物给药的单位剂量的物理不连续单位。例如,单位剂型可以是胶囊或片剂,或多个胶囊或片剂。“单位剂量”是预先确定的本发明活性化合物与一种或多种可药用赋形剂的量,这样的剂量经计算确定能够产生所需疗效。根据所涉及的特定治疗,活性组分在单位剂型中的量可以在约0.1-约1500毫克或更高的范围内改变或调节。可优选单位剂量为约1mg-约1000mg。
使用本发明化合物的给药方案由医疗或兽医领域技术人员根据多种因素选择,这些因素包括但不限于受治疗者的种类、年龄、体重、性别和身体状况、所治疗病症的严重程度、给药途径、受治疗者的代谢水平和排泄功能、所用的剂型、所用的特定化合物及其盐等。
含有结构式I化合物或其盐的组合物最好以单位剂型提供,优选给予包含约1-约500mg的单位剂型,当然应当容易地理解到,实际给予的一种或多种结构式I的化合物的量由医生视所有的相关情况而定。
本发明化合物优选以单次日剂量给药,或者总的日剂量每天分两次、三次或更多次给药。当给药是经透皮形式进行时,给药当然是连续的。
本发明药物组合物的合适的给药途径包括例如经口、眼、直肠、粘膜、局部或肠给药;非胃肠道给药(推注或滴注),包括肌内、皮下、髓内,以及鞘内、直接的心室内、静脉内、腹膜内、鼻内或眼内注射给药。本发明化合物还可以在靶向给药系统中给药,例如在包被有内皮细胞特异性抗体的脂质体中给药。
固体形式的制剂包括粉剂、片剂和胶囊剂。
无菌液体制剂包括混悬剂、乳剂、糖浆剂和酏剂。
本发明的药物组合物可以本身已知的方法(例如常规的混合、溶解、成粒、包糖衣、磨细、乳化、包囊、包埋或冻干法)制备。
以下的药物制剂1和2仅用于举例说明,而不是以任何方式限制本发明的范围。
制剂1
使用以下成分制备硬明胶胶囊:
量
(mg/胶囊)
活性成分 250
干燥的淀粉 200
硬脂酸镁 10
总计 460mg
制剂2
使用以下成分制备片剂:
量
(mg/片剂)
活性成分 250
微晶纤维素 400
热解法二氧化硅 10
硬脂酸 5
总计 665mg
将各组分混合并压制成每片665mg的片剂。
在本发明化合物的另一个实施方案中,所述化合物经放射性标记(例如用碳-14)或氚化。这样的放射标记或氚化的化合物可用作体外测定的参考标准以鉴定新的选择性PPAR受体激动剂。
本发明的化合物可用于调节胰岛素分泌并用作研究手段。优选本发明范围内的某些化合物和条件。以表格形式列出的以下条件、发明实施方案和化合物特征可独立组合以得到各种优选的化合物和方法条件。以下列出的本发明的实施方案并非以任何方式限制本发明的范围。
式I化合物的某些优选的特征为:
(a)R3为甲基;
(b)R4为氢;
(c)R3和R4均为氢;
(d)R3和R4均为甲基;
(e)A为羧基;
(f)X为-O-;
(g)X为-S-;
(h)X为单键;
(i)U为CH(R30);
(j)U为CH2CH(R30);
(k)U为CH2CH(R30)CH2;
(l)U为CH2N(R30)CH2;
(m)U为CH2OCH2;
(n)U为CH2CH2CH2;
(o)U为CH2;
(p)U为CH2NHCH2;
(q)U为CH2N(CH3)CH2;
(r)U为CH2N(CH(CH3)2)CH2;
(s)U为CH2N(CH2CH2CH3)CH2;
(t)U为CH2N(CH2CH3)CH2;
(u)W为N;
(v)W为O;
(w)W为S;
(x)R30为CH3;
(y)R30为苯基;
(z)R30为CH2CH2CH2CH3;
(aa)R30为CH2CH2CF3;
(bb)R30为CH2CH=CH2;
(cc)R30为CH(CH3)2;
(dd)R30为CH2CH2CH3;
(ee)R30为CH2CH3;
(ff)R9为甲基;
(gg)R9为氢;
(hh)R9为C1-C3烷基;
(ii)R8为甲基;
(jj)R8和R9均为氢;
(kk)R10为CF3;
(ll)R10为卤代烷基;
(mm)R10为卤代烷氧基;
(nn)R11为氢;
(oo)R10和R11均为氢;
(pp)R11为卤代烷基;
(qq)R10和R11结合形成稠合的双环;
(rr)R10和R11与其连接的苯基一起形成萘基取代基;
(ss)R1为任选取代的C2-C3芳基烷基;
(tt)R1为取代的C2芳基烷基;
(uu)R1为C1-C8杂烷基;
(vv)R1为杂烷基,其中一个碳原子被氧置换;
(ww)R1为杂烷基,其中两个碳原子被氧置换;
(xx)R1被一个R1’取代;
(yy)R1为C1-C3烯基;
(zz)R1为C1-C4氧代烷基;
(aaa)R1为C1-C4烷基;
(bbb)R32为氢;
(ccc)五元环中的----均在式I指示的位置形成双键;
(ddd)V为键;
(eee)V为C1-C3烷基;
(fff)V为CH2;
(ggg)V为CH2CH2;
(hhh)V为CH2CH2CH2;
(iii)Y为O;
(jjj)Y为S;
(kkk)Y为C;
(lll)Y为C、NH或键;
(mmm)E为C(R3)(R4)A;
(nnn)R3为氢;
(ooo)R3为C1-C2烷基;
(ppp)R4为C1-C2烷基;
(qqq)R3和R4均为氢;
(rrr)R3和R4均为甲基;
(sss)A为COOH;
(ttt)脂族连接基为饱和的;
(uuu)脂族连接基被C1-C3烷基取代;
(vvv)脂族连接基被1-3个各自独立选自R30的取代基取代;
(www)脂族连接基被1-2个各自独立选自R30的取代基取代;
(xxx)脂族连接基为C1-C3烷基;
(yyy)脂族连接基为C1-C2烷基;
(zzz)脂族连接基为C1-C3烷基且一个碳被-O-置换;
(aaaa)式IV的化合物:
IV;
(bbbb)结构式V的化合物:
(cccc)结构式VI的化合物:

(dddd)结构式VII的化合物:

(eeee)结构式VIII的化合物:

(ffff)结构式VIIIa的化合物:

VIIIa;
(gggg)结构式IX的化合物:
(hhhh)结构式X的化合物:

(iiii)结构式XI的化合物:

(jjjj)结构式XII的化合物:

(kkkk)结构式XIII的化合物:

XIII;
(llll)芳基为苯基;
(mmmm)芳基为萘基;
(nnnn)选择性调节δ受体的式I的化合物;
(oooo)一种如本文所述的为调节γ受体和δ受体的PPAR共激动剂(coagaonist)的活性成分;
(pppp)一种如本文所述的用于治疗心血管疾病的活性成分;
(qqqq)一种如本文所述的用于治疗代谢疾病的活性成分;
(rrrr)一种用于控制肥胖的活性成分;
(ssss)一种用于治疗糖尿病的活性成分;
(tttt)一种为PPAR受体激动剂的活性成分;
(uuuu)一种式I的化合物,所述化合物选自2-甲基-2-{4-[3-(5-萘-2-基甲基-2H-[1,2,4]三唑-3-基)-苯氧基}丙酸。
流程1
式I的+/-异构体
式I的化合物通过将碳酰肼(carknyl hydrazone)1a与腈1b反应或者通过碳酰肼与腈1c反应来得到中间体2来制备。形成三唑核2的反应在强碱(如钠、锂或钾的醇盐或者钠、锂或钾的氢氧化物)存在下、在介于室温和混合物的回流温度之间的反应温度下、在极性质子溶剂如低分子量链烷醇(例如甲醇、乙醇、正丙醇或异丙醇)中进行。在碱(例如钠、锂或钾的碳酸氢盐或碳酸盐)存在下、在介于室温和混合物的回流温度之间的反应温度下、在极性质子惰性溶剂(例如乙腈、丙酮或DMF)中用伯或仲烷基卤R-Z将化合物2烷基化,其中R的定义包括R32和R1的定义。如果Z为Cl或Br,可加入催化性的KI将R-Z原位置换为活性更高的烷基化试剂R-I,从而有利于彻底烷基化。用R烷基化得到区域专一性的混合物并且从而得到式I的旋光异构体。
由于通过手性色谱将式I的外消旋化合物拆分并分离成纯的异构体,因此有时需要对式I的外消旋混合物进行官能团调整,以使该混合物在手性色谱过程中得到最佳分离。本发明预期并且为有机反应操作领域技术人员所理解的官能团调节包括在可行的条件下将羧酸衍生物转化为低分子量的酯(如甲酯或乙酯)或转化为具有空间位阻的酯(如叔丁酯)。或者如合成领域技术人员所理解,在可行的情况下,在极性质子溶剂(如甲醇或乙醇)中用钠、锂或钾的氢氧化物皂化低分子量的酯或者在弱极性质子惰性溶剂(二氯甲烷或二氯乙烷)中用TFA水解具有位阻的酯(叔丁酯)将酯官能团调整为游离羧酸。
其他官能团处理包括在可行的情况下用低分子量的链烷二醇和强酸(例如tosic acid和硫酸)将酮或醛转化为缩酮和缩醛。或者用强酸和水合的低级链烷醇或用BBr3的二氯甲烷或二氯乙烷溶液将缩醛和缩酮(如果存在)转化为相应的羰基化合物。
用于上述流程的原料通常可购自商品,特别是腈化合物1c和1b可购自商品。其他式1c和1b的腈可按照有机合成领域公知的方法由以下原料制得:1)将市售或已知的羧酸转化为伯酰胺,随后用Vilsmeier试剂脱水,或2)将市售或已知的醛(carboxaldehyde)转化为肟,随后用Vilsmeier试剂脱水。
式1a和1d的腙在碱(如吡啶、三乙胺或钠、锂、钾的碳酸氢盐或碳酸盐)的存在下,由相应的羧酸或酰氯和肼在二氯甲烷或二氯乙烷中制备。制备式1a或1d的产物的特别有用的方法为Xu,Yanping等人在J.Med.Chem.46(24)(2003)5121-5124页中提出的方法,其中使用市售或已知的羧酸,按照类似的反应合成1a或1d。
制备1(化合物1)
实施例2A(化合物2A)

将化合物1或制备1的化合物[Xu等人,Loc.Sit.(2003)](1.34g,4mmol)溶于甲醇(30ml)中。往该溶液中加入4-甲基苄腈(1.04g,8mmol),随后加入甲醇钠(75mg)。在搅拌下将反应物加热回流(约80℃)24小时。用乙酸乙酯(50ml)稀释。用水(3×60ml)洗涤乙酸乙酯,经硫酸钠干燥并用旋转蒸发器浓缩,得到油状的残余物。残余物通过硅胶柱纯化,得到油状的实施例2A的化合物(670mg)。
m/z:M+1450。
实施例2B-2D
按照实施例2A的方法,使用下表所示的适合的腈,由制备1的化合物得到下表所示的合成实施例2B-2D的化合物。

| 实施例 | R | 使用的腈 | (m/z)M+1 |
| 2B | 苯乙基 | 2-苯基丙腈 | 450 |
| 2C | 萘基甲基 | 2-萘基乙腈 | 486 |
| 2D | 3-(4-氯苯基)-3-乙氧基丙基 | 4-(4-氯苯基)-4-乙氧基丁腈 | 542 |
实施例3A

将实施例2A的化合物(120mg)溶于50%TFA-二氯甲烷(5ml)。在室温下将该化合物搅拌18小时。用旋转蒸发器除去溶剂并在高真空下干燥残余物,得到油状物(101mg)。
m/z:394(M+1)。
实施例3B-3D
按照实施例3A的方法,使用适当的实施例2B-2D的叔丁酯进行TFA介导的水解得到下表所示的合成实施例3B-3C的化合物。

实施例4A
往实施例2A的化合物(70mg)的无水DMF(1.5ml)溶液中加入2-乙氧基乙基溴(0.1ml),随后加入无水粉末状碳酸钾。在50℃下将反应混合物加热搅拌18小时。用乙酸乙酯(30ml)稀释并用水(3×30ml)洗涤乙酸乙酯。用硫酸钠干燥乙酸乙酯层,并用旋转蒸发器浓缩,得到油状残余物。残余物通过硅胶柱纯化,得到油状的约40-60实施例4A的区域异构混合物(62mg)。
m/z:522(M+1)。
实施例4B-4F
按照实施例4A的方法,使用下表所示的适合的烷基溴,由实施例2A的化合物得到下表所示的合成实施例4B-4F的化合物。
| 实施例 | R | 使用的烷基溴 | (m/z)M+1 |
| 4B | 2-(2-甲氧基)乙氧基乙基 | 2-(2-甲氧基)-乙氧基乙基溴 | 552 |
| 4C | 3-四氢吡喃氧基丙基 | 3-四氢吡喃氧基丙基溴 | 592 |
| 4D | 6-四氢吡喃氧基己基 | 6-四氢吡喃氧基己基溴 | 634 |
| 4E | 4-叔丁基苄基 | 4-叔丁基苄基溴 | 596 |
| 4F | 2-氧代丁基 | 1-溴丁烷-2-酮 | 520 |
实施例5A

将实施例4A的三唑混合物(61mg)溶于50%TFA-二氯甲烷(4ml)。将该混合物在室温下搅拌4小时。用旋转蒸发器除去溶剂并在高真空下干燥残余物,得到油状的实施例5A的区域异构混合物(30mg)。m/z:466(M+1)。
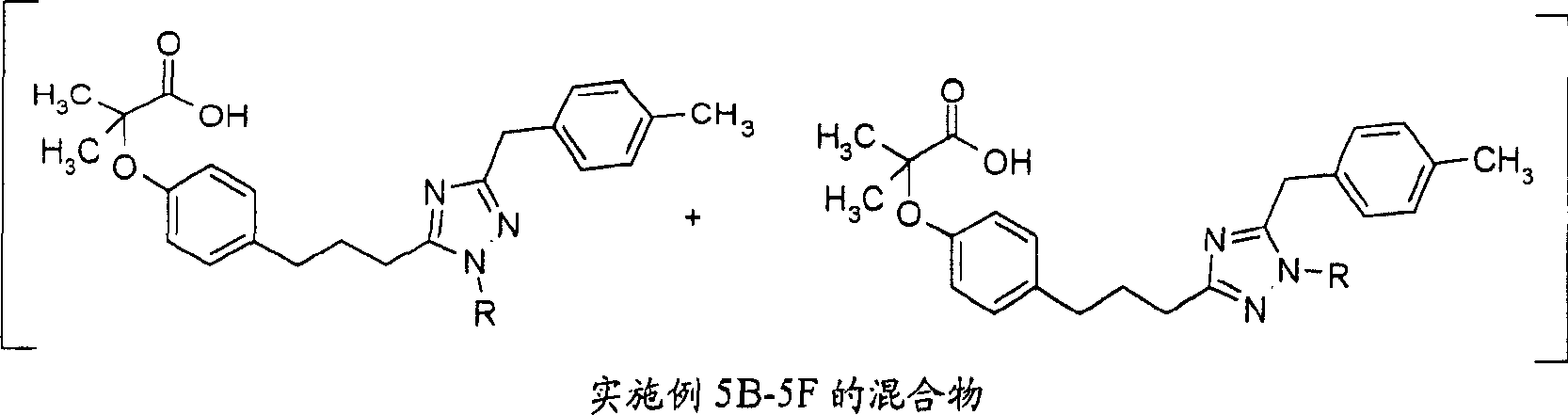
实施例5B-5F
按照实施例5A的方法,使用实施例4E-4C的叔丁酯进行TFA介导的水解,得到下表所示的合成实施例5B-5F的化合物。
实施例6和7
往实施例5A制得的混合物(70mg)的甲醇(25ml)溶液中加入浓硫酸。将该反应混合物在室温下搅拌18小时。除去大部分溶剂并用乙酸乙酯(30ml)稀释残余物。用水(3×30ml)洗涤乙酸乙酯层,经硫酸钠干燥,用旋转蒸发器浓缩,得到油状残余物(71mg)。所得残余物通过手性HPLC柱纯化,得到纯的实施例6和7的化合物。
实施例6:(15mg),m/z:480(M+1)。

实施例7:(19mg),m/z:480(M+1)。

实施例8

将实施例6的三唑酯(15mg)溶于甲醇(2ml)。往该溶液中加入2N氢氧化钠水溶液(1ml)。在室温下将该混合物搅拌2小时。用旋转蒸发器蒸发溶剂并将残余物溶解于水(5ml)。用0.1M盐酸将该溶液酸化至pH为约3,得到乳状溶液。用二氯甲烷(3×10ml)萃取该混合物。用硫酸钠干燥合并的二氯甲烷层,用旋转蒸发器浓缩,随后真空干燥得到实施例8的产物(11mg)。
m/z:466(M+1)。
实施例9
按照实施例8的方法,使用实施例6的酯进行氢氧化钠介导的水解,得到合成实施例9的化合物。m/z:466(M+1)。
实施例9a
使用实施例3A和流程I的通用方法得到合成实施例9a的化合物。m/z:436.3。
实施例10
2-甲基-2-(4-{3-[5-(4-三氟甲基-苯基)-2H-[1,2,4]三唑-3-基]-丙基}-苯氧基)-丙酸叔丁酯
往2-[4-(3-肼基羰基-丙基)-苯氧基]-2-甲基-丙酸叔丁酯(0.5g,1.5mmol)的甲醇(5ml)溶液中加入4-(三氟甲基)苄腈(0.51g,3.0mol)和叔丁醇钾(0.023g,0.21mmol)。将该混合物在回流下搅拌过夜,加入另外的0.2当量的叔丁醇钾,将反应物搅拌24小时。用水猝灭粗产物,真空除去甲醇,用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到0.32g(44%)浅黄色油状的实施例10的标题化合物。MS数据(ES+)m/z 490.6[M+H]。
实施例11
2-甲基-2-(4-{3-[5-(4-三氟甲基-苯基)-2H-[1,2,4]三唑-3-基]-丙基}-苯氧基)-丙酸

往实施例10产物(0.65g,1.33mmol)的二氯甲烷(6.5ml)溶液中加入TFA(0.31ml,3.98mmol)。在室温下将该混合物搅拌过夜。通过TLC分析表明仍存在大量的原料。真空除去溶剂和残留的TFA。通过快速柱层析(己烷/乙酸乙酯2∶1和1∶1)纯化粗产物,得到0.16g(28%)白色固体状的实施例10的产物,回收0.4g原料。MS数据(ES+)m/z434.3[M+H]。
流程2

R30a为R30或H。
流程2的通用方法
往上述市售原料(1.0mmol)的乙酸乙酯(5ml/mmol)溶液中加入BnBr(1.1mmol)和Ag2O(1.1mmol)。将该反应物回流过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。通过快速层析(己烷和己烷/乙酸乙酯20%)纯化粗产物,得到流程2式A化合物。
将流程2式A的苄基衍生物(1.0mmol)溶于乙醇(0.8M)。随后加入一水合肼(3.0mmol)。在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥并浓缩有机层。无需进一步纯化直接使用粗产物。往流程2式B的酰肼化合物(1.0mmol)的甲醇(2.4M)溶液中加入4-(三氟甲基)苄腈(2.0mol)和叔丁醇钾(0.6mmol)。将该混合物回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇,用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到流程2式C化合物。

三唑烷基化:往流程2的相应的三唑衍生物C(1.0mmol)的THF(5ml/mmol)溶液中加入粉状KOH(2.2mmol)、R-I(Br)(2.0mmol)和Bu4NBr(0.2mmol)。在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯7∶1和4∶1)纯化粗产物,得到流程2式D化合物。
往流程2式D的三唑化合物(1.0mmol)的乙醇(5ml/mmol)溶液中加入Pd(C)(10-20%重量)和NH4 +COO-(10-20mmol)。在80℃下将该混合物搅拌过夜。用TLC监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。采用Biotage(己烷/乙酸乙酯1∶1)纯化粗产物,得到流程2式E化合物。
流程2A
往流程2a的相应的三唑衍生物(C)(1.0mmol)的DMSO(20ml/mmol)溶液中加入碳酸钾(1.1mmol)和4-氟-硝基苯(1.0mmol),在90℃下将混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用冰水猝灭反应混合物并加入DCM。再用20%的DCM/甲醇萃取。合并有机层并经硫酸镁干燥,真空浓缩。用快速层析(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程2A式D化合物。
往流程2a式D的三唑衍生物(1.0mmol)的乙醇(5ml/mmol)溶液中加入Pd(C)(10%重量)。在氢气气氛下将该混合物搅拌过夜。用TLC监测反应进行情况。该溶液经Celite硅藻土过滤并真空除去溶剂。该粗产物(流程2A式E的产物)无需进一步纯化直接使用。
在0℃下往流程2A式E的相应的三唑衍生物(1.0mmol)在混合溶剂(THF/CH3COOH 8∶1)(8ml/mmol)的溶液中加入2N HCl溶液(10.0mmol),在该温度下将该化合物搅拌5分钟。随后依次加入NaNO2(1.0mmol)的水溶液(0.32M)和3%H2O2溶液。在0℃下将该反应混合物搅拌30分钟,随后在室温下搅拌1小时。接着加入乙酸乙酯并用水萃取该混合物。有机层经硫酸镁干燥并真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程2A式F化合物(产率17%)。
往流程2A式F化合物(1.0mmol)的乙醇(5ml/mmol)溶液中加入Pd(C)(10-20%重量)和NH4 +COO-(10-20mmol)。在80℃下将该混合物搅拌过夜。用TLC监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。用Biotage(己烷/乙酸乙酯1∶1)纯化粗产物,得到流程2a式F化合物。

Mitsounobu反应:往流程2的三唑衍生物E(1.0mmol)的THF/DCM混合物(1∶1)(10ml/mmol)溶液中加入headpiece(2mmol)、Bu3P(2.0mmol)和TMAD(2.0mmol)。在室温下将该混合物搅拌10-20分钟。用TLC监测反应进行情况。减压除去溶剂,随后加入水和乙醚。分离有机层并与2N NaOH溶液搅拌10分钟。萃取该混合物,用硫酸镁干燥有机层,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程2式F化合物。
往流程2的三唑衍生物F(1.0mmol)的乙醇/THF混合物(1∶1)(10ml/mmol)溶液中加入2N KOH溶液(10mmol),在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。减压除去溶剂,随后加入水和乙酸乙酯。加入1N HCl溶液直到pH为5-7。萃取该混合物,用水洗涤有机层,分离,经硫酸镁干燥,真空浓缩。得到流程2式G化合物。
实施例13的合成
制备2

往羟乙酸甲酯(50g,0.56mol)的乙酸乙酯(300ml)溶液中加入BnBr(72.7ml,0.61mol,1.1当量)和Ag2O(141.6g,0.61mol,1.1当量),在搅拌下将该混合物回流过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。使油状物通过快速硅胶柱并吸附于其上,在快速硅胶柱(500g)的顶部加入20%乙酸乙酯/己烷进行洗脱,得到75.8g(75%产率)无色油状物。
制备3

将制备2的产物(7.5g,41.6mmol)溶于乙醇(100ml)。随后加入一水合肼(6.05ml,124.9mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监视反应的进行情况。减压除去溶剂,将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩,粗产物无需纯化直接使用。
制备4

往制备3得到的产物的甲醇(100ml)溶液中加入4-(三氟甲基)苄腈(14.2g,83.2mmol)和叔丁醇钾(2.8g,25mmol),将混合物搅拌回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取有机层。分离有机层,经硫酸镁干燥,真空浓缩。采用快速层析(己烷/乙酸乙酯4∶1)纯化粗产物,得到11.2g(产率81%)白色固体状的制备4的产物。
制备5

往制备4得到的产物(1.0g,3.0mmol)的THF(15ml)溶液中加入粉状KOH(0.37g,6.6mmol)、EtI(0.48ml,6.0mmol)和Bu4NBr(0.19g,0.6mmol),将该混合物在室温下搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用快速层析(己烷/乙酸乙酯9∶1)纯化粗产物,得到1.09g(产率99%)白色固体状的制备5的产物。
制备6
往制备5得到的产物(1.05g,2.9mmol)的乙醇(25ml)溶液中加入Pd(C)(20%重量)和NH4 +COO-(1.83g,10.0mmol),在80℃下将该混合物搅拌6小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。反应物经Celite硅藻土过滤并减压除去溶剂,得到0.88g白色固体状的制备6的产物,无需进一步纯化直接使用。
制备7

往制备6得到的产物(0.88g,2.9mmol)的THF/DCM混合物(1∶1)(15+15ml)溶液中加入headpiece(1.38g,5.8mmol)、Bu3P(1.44ml,5.8mmol)和TMAD(0.99g,5.8mmol),将该混合物在室温下搅拌1小时。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂,随后加入水和乙醚。分离有机层并与2N NaOH溶液一起搅拌10分钟。萃取该混合物,有机层经硫酸镁干燥,真空浓缩。采用Biotage[40+S](己烷/乙酸乙酯9∶1)纯化粗产物,得到0.99g(产率70%)白色固体状的制备7的产物。MS数据(ES+)m/z 492.3[M+H]。残留350mg含杂化合物(B部分)。
实施例13

往制备7得到的产物(0.98g,1.99mmol)的乙醇/THF混合物(1∶1)(10+10ml)溶液中加入2N KOH溶液(10ml,19.9mmol),将该混合物在室温下搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂,随后加入水和乙醚,将该化合物搅拌2分钟。分离有机层并在水相中加入1N HCl溶液直到pH为5-7。滤出白色固体沉淀并用水洗涤。真空干燥,得到0.80g(87%产率)实施例13的产物(纯度97%)。MS数据(ES+)m/z 464.2[M+H]。
按照与制备实施例13产物的相同方法制备实施例12和14-34的产物。



流程3

流程3的通用方法
往溴代酯衍生物(流程3式H化合物)(1.2mmol)的MeCN(5ml/mmol)溶液中加入碳酸钾(3.0mmol)和流程3式A化合物(1.0mmol)。将该混合物搅拌回流过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。减压除去溶剂,随后加入水和乙酸乙酯。萃取该混合物,用硫酸镁干燥有机层,真空浓缩。采用快速层析(己烷/乙酸乙酯6∶1)纯化粗产物,得到流程3式B的化合物。
将流程3式B的酯化合物(1.0mmol)溶于乙醇(0.8M)。随后加入一水合肼(3.0mmol)。在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩。粗产物无需纯化直接使用。往流程C式3的酰肼化合物(1.0mmol)的乙醇溶液(2.4M)中加入4-(三氟甲基)苄腈(2.0mol)和叔丁醇钾(0.6mmol)。将该混合物搅拌回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇,用乙酸乙酯萃取有机层。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯6∶1)纯化粗产物,得到无色油状的流程3式F的化合物。
往流程3式F三唑衍生物(1.0mmol)的乙醇/THF混合物(1∶1)(10ml/mmol)溶液中加入2N KOH溶液(10mmol)。在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。真空除去溶剂,随后加入水和乙酸乙酯。加入1N HCl溶液直到pH为5-7。萃取该混合物,用水洗涤有机层,分离,经硫酸镁干燥,真空浓缩。得到白色固体状的流程3式G的化合物。
实施例36的合成
制备8
往市售溴代酯衍生物(2.54ml,26.8mmol)的MeCN(100ml)溶液中加入碳酸钾(9.23g,66.9mmol)和headpiece(5.0g,22.3mmol),将该混合物搅拌回流过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂,随后加入水和乙酸乙酯。萃取该混合物,用硫酸镁干燥有机层并真空浓缩。采用快速层析(己烷/乙酸乙酯19∶1)纯化粗产物,得到无色油状的制备8的产物(6.59g)。
制备9

将制备8(6.59g,22.3mmol)的产物溶于乙醇(30ml)。随后加入一水合肼(4.32ml,89.2mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩。制备9的粗产物无需进一步纯化直接使用。
制备10

往制备9的酰肼(3.22g,10.9mmol)的乙醇(50ml)溶液中加入4-(三氟甲基)苄腈(4.72g,27.6mmol)和叔丁醇钾(0.93g,8.3mmol),将该混合物搅拌回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取有机层。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1至6∶1)纯化粗产物,得到白色固体状的制备10的产物(得到2.28g,两个步骤的产率为47%)。
实施例36
往制备10得到的产物(200mg,0.45mmol)的乙醇/THF混合物(1∶1)(8ml)溶液中加入2N KOH溶液(2.7ml,4.5mmol),在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。真空除去溶剂,随后加入水和乙酸乙酯。加入1N HCl溶液直到pH为5-7。萃取该混合物,用水洗涤有机层,分离,经硫酸镁干燥,真空浓缩。得到白色固体状的实施例36的产物(164mg,产率87%)。
按照与制备实施例36产物的相同方法,制备实施例35、37和38的化合物。


流程4

流程4的通用方法
往流程4式A化合物(1.5mmol)的甲醇(5ml)溶液中加入4-(三氟甲基)苄腈(3.0mol)和叔丁醇钾(0.21mmol),将该混合物搅拌回流过夜。加入另外的0.2当量的叔丁醇钾,并将该反应物搅拌24小时。用水猝灭粗产物,真空除去甲醇,用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到流程4式B化合物。
R=烷基
三唑烷基化:往流程4式B相应三唑化合物(1.0mmol)的THF(5ml/mmol)溶液中加入粉状KOH(2.2mmol)、R-I(Br)(2.0mmol)和Bu4NBr(0.2mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。用Biotage(己烷/乙酸乙酯7∶1和4∶1)纯化粗产物,得到流程4式C化合物。
R=芳基(Ph和p-CF3-Ph)
偶合反应:在氮气气氛下,往流程4式B相应三唑衍生物(1.2mmol)的无水二烷溶液中加入芳基碘(1.0mmol)、碳酸钾(2.0mmol)、Cu(OAc)2(0.01mmol)和反式-1,2-二氨基环己烷(0.07mmol)。在110℃下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。反应混合物经Celite硅藻土过滤,用水萃取有机层。用乙酸乙酯萃取水层(2次),分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程4式C化合物。
流程4a
R=芳基(o-F-Ph)
往式B相应三唑化合物(1.0mmol)的DMSO(20ml/mmol)溶液中加入碳酸钾(1.1mmol)和3,4-二氟-硝基苯(1.0mmol),在90℃下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用冰水猝灭粗产物,随后加入DCM。用20%DCM/甲醇进行第二次萃取。合并有机层,经硫酸镁干燥,真空浓缩。用快速层析(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程4a式Ba化合物。
往三唑衍生物(1.0mmol)的醇(5ml/mmol)溶液中加入Pd(C)(10%重量)。在氢气气氛下将该混合物搅拌过夜。用TLC监测反应进行情况。溶液混合物经Celite硅藻土过滤并减压除去溶剂,粗产物可未经纯化直接使用。
在0℃下往流程4a式Bb相应三唑化合物(1.0mmol)的混合溶剂(THF/CH3COOH 8∶1)(8ml/mmol)溶液中加入2N HCl溶液(10.0mmol)。在该温度下将混合物搅拌5分钟,加入NaNO2(1.0mmol)的水溶液(0.32M),随后加入3%H2O2溶液(0.56ml)。在0℃下将反应混合物搅拌30分钟,随后在室温下搅拌1小时。接着加入乙酸乙酯并用水萃取混合物。用硫酸镁干燥有机层并真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程4a式C化合物。
往流程4式C化合物(1.0mmol)的二氯甲烷(5ml)溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到流程4式D化合物。
往流程4式D相应三唑化合物(1.0mmol)的乙醇(8ml/mmol)溶液中加入I2(2.0mmol)和AgSO4(2.0mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。用Biotage(己烷/乙酸乙酯7∶1和4∶1)纯化粗产物,得到流程4式E化合物。
在氮气气氛,往流程4式E相应三唑化合物(1.0mmol)的无水二烷溶液中(10ml/mmol)加入MeB(OH)2(3.0mmol)、CsF(3.0mmol)和PdCl3(dppf)(0.16mmol)。在80℃下将混合物搅拌过夜。用LC/MS监测反应进行情况。冷却反应物,经Celite硅藻土过滤,随后真空除去溶剂。用快速层析(己烷/乙酸乙酯7∶1和4∶1的混合物)纯化粗产物,得到流程4式F化合物。
往流程4式F化合物(1.0mmol)的二氯甲烷(5ml)溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到流程4式G化合物。
实施例47的合成
制备11

往上述A(0.5g,1.5mmol)的甲醇(5ml)溶液中加入4-(三氟甲基)苄腈(0.51g,3.0mol)和叔丁醇钾(0.023g,0.21mmol)并将该混合物回流过夜。加入另外的0.2当量的叔丁醇钾,将反应物搅拌24小时。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取有机层。分离有机层,经硫酸镁干燥,真空浓缩。用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到0.32g(44%)浅黄色油状的制备11的产物。MS数据(ES+)m/z 490.6[M+H]。
制备12

往制备11得到的产物(0.75g,1.5mmol)的THF(5ml)溶液中加入粉状KOH(0.14g,2.45mmol)、MeI(0.14ml,2.3mmol)和Bu4NBr(0.05g,0.15mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。用快速层析(己烷/乙酸乙酯9∶1)纯化粗产物,得到0.51g(产率66%)白色固体状的制备12的产物。
制备13

往制备12得到的产物(0.25g,0.5mmol)的乙醇(5ml)溶液中加入I2(0.2g,0.8mmol)和AgSO4(0.25g,0.8mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到0.15g(48%产率)白色固体状的制备13的产物。
制备14
在氮气气氛下,往制备13的产物(0.15g,0.24mmol)的无水二烷(2ml)溶液中加入MeB(OH)2(0.04g,0.71mmol)、CsF(0.11g,0.71mmol)和PdCl2(dppf)(0.03g,0.04mmol),将混合物在80℃下搅拌过夜。用LC/MS监测反应进行情况。冷却反应物并随后通过Celite硅藻土过滤,真空除去溶剂。采用快速层析(己烷/乙酸乙酯4∶1混合物)纯化粗产物,得到0.09g(72%产率)白色固体状的制备14的产物。
实施例47

往制备14得到的产物(0.09g,0.17mmol)在二氯甲烷(5ml)中的溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到0.05g(92%产率)固体形式的实施例47的产物。
按照制备实施例47的类似方法,制备实施例39-46和48-52的化合物。



流程5

流程5中的R为R1或R32。
流程5的通用方法
往流程5式A化合物(1.0mmol)的乙醇(8ml/mmol)溶液中加入I2(1.1mmol)和AgSO4(1.1mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到流程5式B化合物。
在氮气气氛下,往流程5式B化合物(1.0mmol)的无水二烷(10ml/mmol)溶液中加入MeB(OH)2(3.0mmol)、CsF(3.0mmol)和PdCl2(dppf)(0.16mmol),将混合物在80℃下搅拌过夜。用LC/MS监测反应进行情况。将反应物冷却并经Celite硅藻土过滤,随后真空除去溶剂。用快速层析(己烷/乙酸乙酯19∶1混合物)纯化粗产物,得到流程5式C化合物。
将流程5式C化合物(1.0mmol)溶于乙醇(0.8M)。随后加入一水合肼(3.0mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩,随后粗产物无需进一步纯化直接使用。往流程5式D酰肼化合物(1.0mmol)的甲醇(2.4M)溶液中加入4-(三氟甲基)苄腈(2.0mol)和叔丁醇钾(0.6mmol)。将该混合物回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇,用乙酸乙酯萃取有机层。分离有机层,经硫酸镁干燥,真空浓缩。用Biotage(己烷/乙酸乙酯4∶1和2∶1)纯化粗产物,得到流程5式E化合物。
往流程5式E化合物(1.0mmol)的二氯甲烷(5ml)溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到纯净固体形式的流程5式F化合物。
往流程5式E相应三唑衍生物(1.0mmol)的THF(5ml/mmol)溶液中加入粉状KOH(2.2mmol)、R-I(Br)(2.0mmol)和Bu4NBr(0.2mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次),分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯7∶1和4∶1)纯化粗产物,得到白色固体状的流程5式G化合物。
往流程5式G化合物(1.0mmol)的二氯甲烷(5ml)溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到固体形式的流程5式H化合物。
实施例54的合成
制备15
往相应三唑衍生物(上述化合物A)(20g,64.4mmol)的乙醇(65ml)溶液中加入I2(18.2g,71.4mmol)和AgSO4(22.2g,71.4mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯9∶1)纯化粗产物,得到白色固体状的制备15的产物(m=15.8g,产率56%)。
制备16

在氮气气氛下,往制备15得到的产物(15.8g,36.4mmol)的无水二烷(40ml)溶液中加入MeB(OH)2(3.48g,58.2mmol)、CsF(13.8g,91.0mmol)和PdCl2(dppf)(4.75g,5.82mmol),将混合物在80℃下搅拌过夜。用LC/MS监测反应进行情况。冷却反应物并随后通过Celite硅藻土过滤,真空除去溶剂。采用快速层析(己烷/乙酸乙酯19∶l混合物)纯化粗产物,得到7.75g(66%产率)白色固体状的制备16的产物。
制备17

将制备16的产物(7.75g,24.04mmol)溶于乙醇(50ml)。随后加入一水合肼(4.66ml,96.2mmol)。在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯9∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩。制备17的产物无需进一步纯化直接使用。
制备18

往制备17得到的产物(24.04mmol)的乙醇(50ml)溶液中加入4-(三氟甲基)苄腈(8.2g,48.08mmol)和叔丁醇钾(1.62g,14.42mmol),将该混合物搅拌回流48小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。用快速层析(己烷/乙酸乙酯4∶1和2∶1)纯化粗产物,得到2.5g(产率22%)制备18的产物。
制备19

往制备18得到的产物(0.25g,0.52mmol)的THF(5ml)溶液中加入粉状KOH(0.065g,1.16mmol)、PrI(0.10ml,1.04mmol)和Bu4NBr(0.033g,0.10mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。用快速层析(己烷/乙酸乙酯9∶1)纯化粗产物,得到0.13g(产率48%)制备19的产物。
实施例54

往制备19得到的产物(0.09g,0.17mmol)的二氯甲烷(5ml)溶液中加入TFA(过量),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。真空除去溶剂和残余的TFA。将残余物溶于DCM并用碳酸氢钠饱和溶液洗涤。用硫酸镁干燥有机层并真空浓缩。得到0.06g(产率72%)固体形式的实施例54的产物。
按照制备实施例54的类似方法,制备实施例53的化合物。
流程6

流程6中的R为R1或R32。
流程6a

流程
6b
流程6的通用方法
往羟乙酸甲酯(50g,0.56mol)的乙酸乙酯(300ml)溶液中加入BnBr(72.7ml,0.61mol,1.1当量)和Ag2O(141.6g,0.61mol,1.1当量),在搅拌下将该混合物回流过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。使油状物通过快速硅胶柱并吸附于其上,在快速硅胶柱(500g)的顶部加入20%乙酸乙酯/己烷进行洗脱,得到75.8g(产率75%)流程6式A化合物。
将流程6式A苄基衍生物(1.0mmol)溶于乙醇(0.8M),随后加入一水合肼(3.0mmol)。在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩。粗产物无需纯化直接使用。往流程6式B酰肼化合物(1.0mmol)的甲醇溶液(2.4M)中加入4-(三氟甲基)苄腈(2.0mol)和叔丁醇钾(0.6mmol),随后将该混合物回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到流程6式C化合物。
R=Me或MeOCH2CH2
三唑烷基化:往流程6式C相应三唑化合物(1.0mmol)的THF(5ml/mmol)溶液中加入粉状KOH(2.2mmol)、R-I(Br)(2.0mmol)和Bu4NBr(0.2mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到流程6式D化合物。
往流程6式D三唑化合物(1.0mmol)的乙醇(5ml/mmol)溶液中加入Pd(C)(10-20%重量)和NH4 +COO-(10-20mmol),在80℃下将该混合物搅拌过夜。用TLC监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。采用Biotage(己烷/乙酸乙酯1∶1)纯化粗产物,得到流程6式E化合物。
在0℃下往流程6式E三唑衍生物(1.0mmol)的无水DMF(10ml)溶液中加入NaH(2.1mmol,60%),将该混合物在室温下搅拌15分钟,冷却至0℃,加入苄基溴(流程6化合物F)(1.5mmol)的无水DMF(5ml)溶液。在室温下将该反应混合物搅拌30分钟,加入甲醇,随后加入水。真空除去甲醇,用乙酸乙酯萃取水层。有机层用水和盐水洗涤,经硫酸镁干燥,过滤并减压浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到流程6式G化合物。
往流程6式G三唑衍生物(1.0mmol)的乙醇/THF混合物(1∶1)(10ml/mmol)溶液中加入2N KOH溶液(10mmol),在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。减压除去溶剂,随后加入水和乙酸乙酯。加入1N HCl溶液直到pH为5-7。萃取该混合物,用水洗涤有机层,分离,经硫酸镁干燥,真空浓缩。得到白色固体状的流程6式H化合物产物。
实施例55的合成
制备19

往羟乙酸甲酯(50g,0.56mol)的乙酸乙酯(300ml)溶液中加入BnBr(72.7ml,0.61mol,1.1当量)和Ag2O(141.6g,0.61mol,1.1当量),在搅拌下将该混合物回流过夜。用TLC(己烷/乙酸乙酯1∶1)监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。将所得油状物通过快速硅胶柱并吸附于其上。在快速硅胶柱(500g)的顶部加入20%乙酸乙酯/己烷进行洗脱,得到75.8g(产率75%)无色油状的制备19的产物。
制备20

将制备19的产物(7.5g,41.6mmol)溶于乙醇(100ml)。随后加入一水合肼(6.05ml,124.9mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。干燥有机层并浓缩。制备20的产物无需进一步纯化直接使用。
制备21

往制备20得到的产物的甲醇(100ml)溶液中加入4-(三氟甲基)苄腈(14.2g,83.2mmol)和叔丁醇钾(2.8g,25mmol),将该混合物回流24小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物,真空除去甲醇。用乙酸乙酯萃取水层。分离有机层,经硫酸镁干燥,真空浓缩。采用快速层析(己烷/乙酸乙酯4∶1)纯化粗产物,得到11.2g(81%产率)白色固体状的制备21的产物。
制备22
往制备21得到的产物(1.0g,3.0mmol)的THF(15ml)溶液中加入粉状KOH(0.27g,4.8mmol)、MeI(0.28ml,4.5mmol)和Bu4NBr(0.09g,0.3mmol),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。用水猝灭粗产物并加入乙酸乙酯。用乙酸乙酯萃取水层(2次)。分离有机层,经硫酸镁干燥,真空浓缩。采用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到制备22的产物(1.04g)。
制备23
往制备22得到的产物(1.04g,3.0mmol)的乙醇(15ml)溶液中加入Pd(C)(10-20%重量)和NH4 +COO-(1.89g,30mmol),在80℃下将该混合物搅拌过夜。用TLC监测反应进行情况。反应物经Celite硅藻土过滤并真空除去溶剂。采用Biotage(己烷/乙酸乙酯1∶1)纯化粗产物,得到制备23的产物(0.59g,产率76%)。
制备24

在0℃下往制备23得到的产物(0.28g,1.09mmol)的无水DMF(10ml)溶液中加入NaH(0.091g,2.29mmol,60%),将该混合物在室温下搅拌15分钟。再次冷却至0℃并加入在无水DMF(5ml)中的苄基溴化合物A(0.68g,1.63mmol),在室温下将反应混合物搅拌30分钟。加入甲醇,随后加入水。真空除去甲醇,用乙酸乙酯萃取水层。有机层用水和盐水洗涤,经硫酸镁干燥,过滤并减压浓缩。用Biotage(己烷/乙酸乙酯4∶1)纯化粗产物,得到白色固体状的制备24的产物(0.39g,产率70%)。
实施例55

往制备24得到的产物(0.1g,0.21mmol)的乙醇/THF混合物(1∶1)(2ml)溶液中加入2N KOH溶液(1.05ml,2.1mmol),在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。减压除去溶剂,随后加入水和乙酸乙酯。加入1N HCl直到pH为5-7。萃取该混合物,用水洗涤有机层,分离,经硫酸镁干燥,真空浓缩。得到白色固体状的实施例55的产物(65mg,产率69%)。
按照与制备实施例55类似的方法,制备实施例56的产物。
流程7
制备25

在0℃下将氢化钠(369mg,9.2mmol)在DMF(25ml)中制成浆状物,随后一次性加入4-甲氧基苯酚(1.15g,9.2mmol)。搅拌30分钟后,一次性加入2-溴丙酸乙酯(1.391g,7.7mmol),将反应混合物在室温下静置过夜。用1N HCl猝灭该反应混合物,随后用乙酸乙酯萃取。合并有机相,经硫酸镁干燥,蒸发。采用硅胶层析纯化残余物(30%乙酸乙酯/己烷),得到983.1mg透明粘稠油状的制备25的化合物(57.2%)。MS=225(M+H+);242(M+NH4 +)。
制备26

将制备26的产物(983mg,4.4mmol)溶于甲醇(10ml),往该混合物中加入1N NaOH(10ml)。在室温下搅拌12小时。随后蒸发溶剂,将残余物溶解于1N HCl(20ml)并用乙酸乙酯萃取。合并有机相,经硫酸镁干燥,蒸发。无需进一步纯化直接使用该粗产物(767.5mg,89%)。MS=195(M-H-);214(M+NH4 +)。
制备27

将制备26的产物(100mg,0.5mmol)溶于二氯甲烷(10ml)并加入EDC(117mg,0.6mmol),随后加入HOBT(83mg,0.6mmol),搅拌5分钟。接着一次性加入4-(三氟甲基)苯甲酰肼(104mg,0.5mmol),随后加入吡啶(0.124ml,1.5mmol),在室温下搅拌过夜。随后用饱和碳酸氢钠溶液和1N HCl洗涤该反应混合物。有机层经硫酸镁干燥并蒸发。采用硅胶层析(50%乙酸乙酯/己烷)纯化残余物,得到127.8mg白色固体状的制备27的产物(65.6%)。MS=383(M+H+);381(M-H-)。
制备28
将制备27的产物(127mg,0.33mmol)溶于甲苯(5ml)。往其中加入Lawesson试剂(134mg,0.33mmol),将反应混合物加热回流2小时。随后冷却反应物并用20ml乙酸乙酯稀释。用饱和碳酸氢盐溶液洗涤,有机层经硫酸镁干燥并蒸发。采用硅胶层析(15%乙酸乙酯/己烷)纯化残余物,得到103.8mg白色固体状的制备28的产物(82.2%)。MS=381(M+H+)。
制备29
将制备28的产物(100mg,0.26mmol)溶于二氯甲烷(5ml)。随后将该混合物冷却至0℃,加入三溴化硼(0.788ml,0.79mmol)。2小时后,用饱和碳酸氢盐猝灭该反应,用20ml乙酸乙酯萃取。有机相经硫酸镁干燥并蒸发。得到87.2mg灰白色固体状的制备29的产物(98.4%)。MS=337&338(M+H+)。
制备30

将2-(4-羟基-2-甲基-苯氧基)-2-甲基-丙酸乙酯(73.6mg,0.31mmol)溶于DMF。往该混合物中加入氢化钠(12.4mg,0.31mmol)并在室温下搅拌5分钟。随后一次性加入制备29的产物(87mg,0.26mmol),将反应继续进行2小时。用饱和氯化铵猝灭该反应并用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。采用硅胶层析(15%乙酸乙酯/己烷)纯化残余物,得到59.7mg透明油状的制备30的产物(46.8%)。MS=495(M+H+)。
实施例57

外消旋
将制剂30的产物(59mg,0.12mmol)溶于甲醇(2ml)。往该混合物中加入1N NaOH溶液(2ml)。12小时后用1N HCl溶液使反应物pH为6并用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。有机层经硫酸镁干燥并蒸发。先采用硅胶层析(50%乙酸乙酯/己烷)随后采用反相HPLC(40-90%梯度@140ml/min 30分钟,柱50×250mm,mm,C18Symmetry柱)纯化该残余物,得到7.1mg白色固体状的实施例57的产物(12.7%)。MS=467(M+H+)。
流程8
制备31
在配有短径回流冷凝器的圆底烧瓶中加入4-(三氟甲基)苯甲酰肼(5g,24.5mmol),往其中加入原甲酸三甲酯(4ml,36.7mmol),随后加入对甲苯磺酸(46.6mmg,0.24mmol)。加热至100℃并蒸除甲醇。2小时后,不再收集到甲醇,将体系的压力降低至300微米以除去原甲酸三甲酯。将残余固体冷却并溶于乙酸乙酯。用饱和碳酸氢盐溶液洗涤,合并有机相,经硫酸镁干燥,蒸发。粗产物(5.2g,99%)无需进一步纯化直接使用。MS=215(M+H+)。
制备32
将制剂31(500mg,2.3mmol)的产物溶于THF并冷却至-78℃。往该混合物中加入叔丁基锂(1.5ml,1.7M溶液)并搅拌30分钟。随后加入乙醛(0.157ml,2.8mmol),在6小时内让反应混合物缓慢升至室温。随后用饱和氯化铵溶液猝灭该反应并用乙酸乙酯萃取。经硫酸镁干燥合并的有机层并蒸发。采用硅胶层析(20%乙酸乙酯/己烷)纯化该残余物,得到24mg白色固体状的制备32的产物(3.9%)。
制备33

将制剂32的产物(24mg,0.09mmol)溶于甲苯(2ml)并加入2-(4-羟基-2-甲基-苯氧基)-2-甲基-丙酸乙酯(26.5mg,0.11mmol)。用氮气往该混合物中鼓泡15分钟。随后加入ADDP(35.1mg,0.135mmol),加入Bu3P(0.034ml,0.135mmol),在室温下将该反应混合物搅拌18小时。随后,用20ml乙酸乙酯稀释该反应混合物并用盐水洗涤。有机层经硫酸镁干燥并蒸发。采用硅胶层析(20%乙酸乙酯/己烷)纯化该残余物,得到24.4mg白色固体状的制备33的产物(54.9%)。MS=479(M+H+)。
实施例58

外消旋
将制备33的产物(59mg,0.12mmol)溶于甲醇(2ml)。往其中加入1N NaOH溶液(2ml)。12小时后用1N HCl溶液使反应混合物的pH=6并用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。先采用硅胶层析(50%乙酸乙酯/己烷)随后采用反相HPLC(40-90%梯度@140ml/分钟,30分钟,50×250mm,mm,C18 Symmetry柱上)纯化该残余物,得到8.6mg白色固体状的实施例58的产物(9.5%)。MS=451(M+H+)。
实施例59
本领域普通技术人员按照与实施例57相同的方法,使用2-(4-羟基甲基-苯氧基)-2-甲基-丙酸乙酯(162mg,0.67mmol)作为亲核试剂,2-溴甲基-5-(4-三氟甲基-苯基)-[1,3,4]噻二唑(200mg,0.62mmol)作为亲电试剂(如制备30所述),随后按照实施例57中描述的方法进行皂化,可制备实施例59的化合物。采用硅胶层析(20%乙酸乙酯/己烷)纯化后,得到粘稠油状的实施例59的产物60.2mg(21.5%)。MS=451(M-H-)。
实施例60
本领域普通技术人员按照与实施例57相同的方法,使用2-甲基-2-(4-甲基氨基甲基-苯氧基)-丙酸乙酯(171mg,0.67mmol)作为亲核试剂,2-溴甲基-5-(4-三氟甲基-苯基)-[1,3,4]噻二唑(200mg,0.62mmol)作为亲电试剂(如制备30所述),随后按照实施例57中描述的方法进行皂化,可得到实施例60的化合物。采用硅胶层析(10%甲醇/二氯甲烷)纯化后,得到75.3mg(26.3%)粘稠油状的实施例60的产物。
MS=464(M-H-)。
实施例61
本领域普通技术人员按照与实施例57相同的方法,使用2-(4-乙基氨基甲基-苯氧基)-2-甲基-丙酸乙酯(180.6mg,0.67mmol)作为亲核试剂,2-溴甲基-5-(4-三氟甲基-苯基)-[1,3,4]噻二唑(200mg,0.62mmol)作为亲电试剂(如制备30所述),随后按照实施例57中描述的方法进行皂化,可得到实施例61的化合物。采用硅胶层析(10%甲醇/二氯甲烷)纯化后,得到24.3mg(8.3%)粘稠油状的实施例61的产物。MS=478(M-H-)。
制备34
将制剂31的产物(500mg,2.3mmol)溶于THF并冷却至-78℃。往其中加入叔丁基锂(1.5ml,1.7M溶液)并搅拌30分钟。随后加入丙醛(0.202ml,2.8mmol),在6小时内让反应混合物缓慢升至室温。随后用饱和氯化铵溶液猝灭该反应,再用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。采用硅胶层析(20%乙酸乙酯/己烷)纯化该残余物,得到白色固体状的99.8mg制备34的产物(15.7%)。
制备35
将制剂34的产物(99mg,0.36mmol)溶于甲苯(5ml)并加入2-(4-羟基-2-甲基-苯氧基)-2-甲基-丙酸乙酯(105mg,0.43mmol)。用氮气往该混合物中鼓泡15分钟。随后加入ADDP(139mg,0.54mmol),再加入Bu3P(111.5mg,0.54mmol),在室温下将反应混合物搅拌18小时。随后,用20ml乙酸乙酯稀释反应混合物并用盐水洗涤。有机层经硫酸镁干燥并蒸发。采用硅胶层析(20%乙酸乙酯/己烷)纯化该残余物,得到63.0mg白色固体状的制备35的产物(34.8%)。MS=493(M+H+)。
实施例62

外消旋
将制备35的产物(63mg,0.13mmol)溶于甲醇(2ml)。往该混合物中加入1N NaOH溶液(2ml)。12小时后用1N HCl溶液使反应混合物的pH=6并用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。采用硅胶层析(50%乙酸乙酯/己烷)随后采用反相HPLC(40-90%梯度@140ml/分钟,30分钟,50×250mm,mm,C18 Symmetry柱)纯化该残余物,得到9.7mg白色固体状的实施例62的化合物(16.3%)。MS=465(M+H+)。
流程9

制备36

在15分钟内往0℃的酰肼(上述化合物1)(13.7g,76mmol)在二氯甲烷(200ml)和吡啶(20ml)中的溶液中加入对三氟甲基苯甲酰氯,将反应物搅拌30分钟。随后倾入5N HCl(200ml)中。用乙醚(250ml)和乙酸乙酯(250ml)萃取该溶液。依次用H2O(200ml)和盐水(200ml)洗涤合并的萃取液,经硫酸钠干燥、过滤并浓缩。将粗制的半固体与10%乙醚/己烷一起研磨,沉淀出所需的白色固体状的制备36的产物(23.4g,88%)。1H NMR(CDCl3)δ10.64(s,1H),10.07(s,1H),8.09(m,2H),7.93(m,2H),7.40(m,5H),4.64(s,2H),4.11(s,2H)。
制备37

将制备36的产物(12.0g,34.1mmol)和Lawesson试剂(16.5g,40.9mmol)的甲苯(200ml)溶液加热回流4小时,随后冷却至室温,倾入水(400ml)中。用乙醚(200ml)和乙酸乙酯(400ml)萃取该混合物。用盐水(250ml)洗涤合并的有机萃取液、经硫酸钠干燥、过滤并浓缩。采用MPLC(0%-10%-15%-20%乙酸乙酯/己烷,梯度洗脱)纯化粗产物,得到透明油状的制备37的产物(11.2g,93%)。1H NMR(CDCl3)δ8.10(d,J=8.4Hz,2H),7.75(d,J=8.4Hz,2H),7.37(m,5H),4.99(s,2H),4.70(s,2H)。
制备38

在15分钟内往0℃的制备37的产物(9.8g,27.9mmol)的二氯甲烷(120ml)溶液中加入BBr3溶液(42ml,1M的二氯甲烷溶液,42mmol)。反应在0℃下继续进行15分钟后,将反应混合物倾入半饱和的NaHCO3(500ml)中。用乙醚(250ml)和乙酸乙酯(300ml)萃取混合物。用盐水(250ml)洗涤合并的有机萃取液,经硫酸钠干燥,过滤并浓缩,得到白色固体。将粗产物与10%乙醚/己烷一起研磨,沉淀出所需的白色固体状的制备38的产物(7.0g,>95%)。1H NMR(CDCl3)δ8.09(d,J=8.4Hx,2H),7.60(d,J=8.4Hz,2H),5.16(s,2H),2.40-2.60(br s,1H)。
制备39

让氮气鼓泡通过制备38的产物(0.125g,0.48mmol)与苯酚化合物2(0.119g,0.50mmol)在甲苯(5ml)中的溶液,历时10分钟。将该溶液冷却至0℃,加入三正丁基膦(0.180ml,0.72mmol),随后加入(1,1’)-偶氮二羰基-二哌啶(ADDP)(0.182g,0.72mmol)。5分钟后,让该反应混合物升至室温并搅拌16小时。随后将该反应混合物倾入半饱和NaHCO3(25ml)中。用乙醚(25ml)和乙酸乙酯(25ml)萃取该混合物。用盐水(25ml)洗涤合并的有机萃取液,经硫酸钠干燥,过滤并浓缩。通过MPLC(0%-5%-8%-12%乙酸乙酯/己烷)纯化粗产物,得到白色泡沫形式的制备39的产物(0.138g,x%)。1H NMR(CDCl3)δ8.11(d,J=8.4Hz,2H),7.77(d,J=8.4Hz,1H),6.72(s,1H),6.65(m,1H),6.56(d,J=3.2Hz,1H),5.48(s,2H),4.27(q,J=7.2Hz,2H),2.19(s,3H),1.55(s,6H),1.32(t,J=7.2Hz,3H)。
实施例63
将制备39的产物(0.138g,0.29mmol)在乙醇(5ml)和2N NaOH(1ml)中的溶液在40℃下加热1小时。将该混合物倾入1N HCl(20ml)并用乙醚(20ml)和乙酸乙酯(2×20ml)萃取。用盐水(25ml)洗涤合并的有机萃取液,经硫酸钠干燥过滤并浓缩。采用硅胶层析(35%乙酸乙酯/2%HOAc的己烷溶液)纯化粗产物,得到白色固体状的实施例59的产物(0.102g,77%)。LRMS 453.1(M++H)。
制备40
按照制备39的方法进行该反应。将制备38的产物(0.125g,0.48mmol)和苯酚化合物3(如上所述)(0.112g,0.50mmol)反应,得到白色泡沫状的制备40的化合物(0.124g,55%)。1H NMR(CDCl3)δ8.08(d,J=8.2Hz,2H),7.74(d,J=8.2Hz,2H),6.89(m,2H),6.75(m,2H),5.47(s,2H),4.23(q,J=7.2Hz,2H),1.52(s,6H),1.27(t,J=7.2Hz,3H)。
实施例64
按照实施例63的方法进行该反应。将制备40的产物(0.124g,0.27mmol)水解,得到白色泡沫状的实施例60的产物(0.082g,69%)。LRMS439.1(M++H)。
流程10

制备41
将氯化氢气体鼓泡通过0℃的对三氟甲基苄腈(5.50g,32.1mmol)在乙醇(9.4ml,161mmol)和甲苯(50ml)中的溶液,历时30分钟。除去冷却浴,将溶液在室温下搁置16小时。将该混合物浓缩,得到中间体亚氨酸盐(imidate salt),将该盐加至邻二甲苯(165ml)中。加入酰肼化合物1(制备36的方法)(5.78g,32.1mmol)的邻二甲苯(10ml)溶液,在30分钟内滴加Et3N(4.47ml,32.1mmol),将溶液搅拌2小时,加热回流2.5小时,冷却至室温并保持16小时。将该溶液倾入半饱和的NaHCO3(250ml)中并用乙酸乙酯(2×200ml)萃取。用盐水(250ml)洗涤合并的有机相,经硫酸钠干燥,过滤并浓缩。采用硅胶层析(0%-15%-20%-25%-35%的乙酸乙酯/己烷)纯化粗产物,得到白色固体状的制备41的产物(0.53g,5%)。1H NMR(CDCl3)δ7.79(d,J=8.0Hz,2H),7.72(d,J=8.0Hz,2H),7.30-7.40(m,5H),4.83(s,2H),4.70(s,2H)。
制备42
将40℃的制备41产物(0.53g,1.59mmol)和10%Pd-C(0.10g)在甲醇(30ml)和THF(10ml)中的溶液暴露于40psi的H2(气体)中24小时。用N2(气体)冲洗该混合物,随后经Celite硅藻土过滤,得到的澄清溶液,经浓缩,得到白色固体状的制备42的产物(0.24g,62%)。1HNMR(CDCl3)δ8.18(d,J=8.0Hz,2H),7.70(d,J=8.0Hz,2H),5.00(s,2H)。
制备43

按照制备39的方法,由制备42的产物(0.050g,0.20mmol)和酚2(0.051g,0.21mmol)制得制备43的产物,得到澄清油状的制备43的产物(0.072g,76%)。1H NMR(CDCl3)δ8.20(d,J=8.4Hz,2H),7.78(d,J=8.4Hz,2H),6.85(s,1H),6.68(m,2H),5.27(s,2H),4.25(q,J=7.2Hz,2H),2.22(s,3H),1.54(s,6H),1.28(t,J=7.2Hz,3H)。
实施例65
按照实施例63的方法,由制备43的产物制备实施例65的产物,得到白色固体状的实施例65的产物(0.070g,>95%)。LRMS 455.1(M++H)。
按照流程4的方法制备实施例66的产物,按照流程2的方法制备实施例67的产物。

按照流程2中描述的方法制备实施例68-73的产物,按照流程6a和/或6b中描述的方法制备实施例74-76的产物。


流程11

流程11的通用方法
在装有标准蒸馏装置的圆底烧瓶中将酰肼化合物A(100mmol)悬浮于原甲酸三甲酯(150mmol)和对甲苯磺酸一水合物(1.5mmol)中。加热至90℃直到生成沉淀,随后加热至120℃。蒸除甲醇。用HPLC和TLC(己烷/乙酸乙酯1∶1)监测反应进行的情况。将黄色油状物溶于乙酸乙酯中,并用水、盐水洗涤,经硫酸镁干燥并真空浓缩。采用硅胶层析(己烷/乙酸乙酯9∶1-7∶3)纯化粗产物,得到流程11式B化合物。
在氮气气氛下往冷却的(-78℃)二唑化合物B(10mmol)的THF(33ml)溶液中滴加nBuLi(1.6M的己烷溶液,10mmol)。40分钟后,加入MgBr2·乙醚(10mmol),将冷却浴升至-45℃并在-45℃下将得到的浆状物再搅拌1.5小时。加入醛(9mmol)的THF(11ml)溶液,将反应温度升至-20℃并在该温度下再搅拌2.5小时。用HLPC和TLC(己烷/乙酸乙酯8∶2)监视反应的进行情况。用氯化铵溶液猝灭该粗产物并加入乙酸乙酯。水层用乙酸乙酯萃取两次。分离有机层,用水、盐水洗涤有机层,经硫酸镁干燥,过滤并真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到流程11式C化合物。
Mitsounobu反应:在0℃下往已经脱气数次的二唑C(1.0mmol)和酚(化合物D)(2mmol)的甲苯(20ml)溶液中加入Bu3P(2.0mmol)和ADDP(2.0mmol)。在室温下将该混合物搅拌过夜。用TLC监测反应进行情况。真空除去溶剂,与乙醚一起研磨,滤除得到的沉淀。用2NNaOH溶液、水和盐水洗涤滤液,经硫酸镁干燥并真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到流程11式E的化合物。
水解:往二唑C(1.0mmol)在乙醇/THF混合物(1∶1)(40ml)的溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)和HPLC监测反应进行情况。加入乙酸乙酯和水,加入6N HCl溶液将pH调整至6。分离两相,水层用乙酸乙酯萃取两次,用水、盐水洗涤有机层,经硫酸镁干燥,过滤并真空浓缩。用ISCO(己烷-TFA 0.05%/丙酮9∶1-85∶15)纯化粗产物,得到流程11式F的酸化合物。
Head H2的制备

将对苄氧基苯酚(300mmol)溶于甲腈(750ml),分次加入Cs2CO3(360mmol),随后加入溴衍生物。得到的混合物回流过夜。冷却,通过Celite硅藻土垫过滤,真空浓缩。采用硅胶层析(己烷/乙酸乙酯8∶2)纯化,得到化合物H1,产率为94%。
往H1(282mmol)的乙醇(940ml)溶液中加入Pd-C(10%重量)。在室温、氮气气氛下搅拌该混合物。经Celite硅藻土垫过滤,除去溶剂并采用Biotage(己烷/乙酸乙酯7∶3)纯化,得到化合物H2,产率为64%。
Head H4的制备

将原料醛(45mmol)溶于THF∶乙醇(95∶5ml)中,冷却至0℃,分次加入NaBH4,直到通过TLC未检测到原料。在0℃下用1N HCl猝灭该反应物并加入乙酸乙酯。将pH调整至6。分离两相,用乙酸乙酯萃取两次。用水、盐水洗涤,经硫酸镁干燥、过滤并真空浓缩。采用硅胶层析,得到醇H3,产率为95%。
往H3(42.7mmol)的乙醇(430ml)溶液中加入Pd-C(10%重量)。在室温、氮气气氛下将该混合物搅拌6小时。经Celite硅藻土垫过滤,除去溶剂并采用硅胶层析(己烷/乙酸乙酯7∶3)纯化,得到化合物H4,产率为76%。
Head H7的制备
将硫醇(100mmol)溶于甲醇(170ml),冷却至0℃并分批加入I2(0.5mmol)和NaHCO3(2.8mmol)。得到的混合物在室温下搅拌过夜。减压除去溶剂,采用硅胶层析纯化残余物,得到化合物H5,产率83%。
将二硫化物H5(70mmol)溶于甲腈(110ml),加入Cs2CO3(3.5mmol),搅拌10分钟,随后加入溴衍生物(210mmol)。回流过夜。用TLC(己烷/乙酸乙酯7∶3)监测反应进行情况。冷却至室温,经Celite硅藻土垫过滤,真空浓缩,采用硅胶层析(己烷-己烷/乙酸乙酯4∶1)纯化残余物,得到化合物H6,产率为51%。
将H6(6mmol)溶于THF∶乙醇(16∶5ml),在0℃下分次加入NaBH4直到未检出原料。在0℃下用1N HCl溶液猝灭反应物并加入乙酸乙酯。将pH调整至4。分离两相,用乙酸乙酯萃取。用水、盐水洗涤,经硫酸镁干燥,过滤并浓缩。采用硅胶层析(己烷-己烷/乙酸乙酯95∶5)纯化,得到H7,产率为99%。
制备52
在装有标准蒸馏装置的圆底烧瓶中将酰肼(5g)悬浮于原甲酸三甲酯(4.0ml)和对甲苯磺酸一水合物(47mg)中。加热至90℃直到观察到沉淀形成,随后加热至120℃。蒸除甲醇,得到黄色油状物。将该黄色油状物溶于乙酸乙酯并用水、盐水洗涤,经硫酸镁干燥并真空浓缩。采用硅胶柱色谱纯化粗产物(己烷/乙酸乙酯9∶1-7∶3),得到白色固体状的制备52的产物(95%)。
制备53

在氮气气氛下往冷却至-78℃的制备52的产物(500mg)的THF(20ml)溶液中滴加nBuLi(1.51ml,1.7M的戊烷溶液)。40分钟后,加入MgBr2·乙醚(10mmol),将冷却浴升温至-45℃,在-45℃下将得到的浆状物再搅拌1.5小时,加入乙醛(0.157ml)的THF(10ml)溶液,将反应温度升至-20℃并在该温度下再搅拌2.5小时。用氯化铵溶液猝灭粗物质并加入乙酸乙酯。水层用乙酸乙酯萃取两次。分离有机层,用水、盐水洗涤,并用硫酸镁干燥,过滤并真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到制备53的产物(22%)。
制备54
Mitsounobu反应:在0℃下往已经脱气数次的制备53的产物(24mg)与酚(26.5mg)的甲苯(2ml)溶液中加入Bu3P(0.035ml)和ADDP(35mg)。在室温下将该混合物搅拌过夜。真空除去溶剂,残余物与乙醚一起研磨,滤除得到的沉淀。用2N NaOH溶液洗涤滤液,用水、盐水洗涤,经硫酸镁干燥,真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到制备54的产物(55%)。
实施例77
往制备54的产物(24mg)的乙醇/THF混合物(1∶1)(4ml)溶液加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。加入乙酸乙酯和水,加入1N HCl溶液将pH调节至6。分离两相,水层用乙酸乙酯萃取两次,用水、盐水洗涤有机层,经硫酸镁干燥,过滤并真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到实施例77的产物(90%)。
按照与制备实施例77类似的方法制备实施例78、81、84、85和88-90的产物。


流程12

流程12的通用方法
往酚A(10mmol)与碳酸钾在甲腈(10ml)中的悬浮液中加入市售的酯原料(10mmol)。将混合物回流过夜。用TLC(己烷/乙酸乙酯9∶1)监测反应进行情况。冷却、经Celite硅藻土垫过滤并真空除去溶剂。采用硅胶层析(己烷-己烷/乙酸乙酯9∶1)纯化粗产物,得到流程12式B化合物。
将甲酯B(10mmol)溶于乙醇(12.5ml)。随后加入一水合肼(40mmol)。在室温下将该混合物搅拌过夜。用TLC监测反应进行情况(己烷/乙酸乙酯9∶1)。真空除去溶剂。将残余物溶解于乙酸乙酯并用水洗涤。有机层经硫酸镁干燥并浓缩。粗产物无需纯化直接使用。
将酰肼C(10mmol)溶于THF(25ml)。随后在0℃下加入三乙胺,将所得溶液搅拌5分钟,随后在-30℃下滴加酰氯(11mmol)。在该温度下搅拌混合物45分钟,随后在室温下搅拌过夜。采用HPLC和TLC(己烷/乙酸乙酯1∶1)监视反应的进行情况。用水猝灭该粗产物,加入乙酸乙酯。分离有机层,经硫酸镁干燥,真空浓缩。采用硅胶层析(己烷/乙酸乙酯9∶1-7.3)纯化粗产物,得到流程12式D化合物。
将化合物D(10mmol)溶于甲苯(100ml),加入Lawesson试剂(20.0mmol)。将混合物回流4小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。真空除去溶剂。采用硅胶层析(己烷-己烷/乙酸乙酯85∶15)纯化残余物,得到流程12式E化合物。
往噻二唑E(1.0mmol)的乙醇/THF混合物(1∶1)(40ml)溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)和HPLC监测反应进行情况。加入乙酸乙酯和水,加入6N HCl溶液将pH调节至6。分离两相,水层用乙酸乙酯萃取两次,用水、盐水洗涤有机相,经硫酸镁干燥,过滤并真空干燥。采用ISCO(己烷-TFA 0.05%/丙酮9∶1-85∶15)纯化粗产物,得到流程12的酸F。
实施例91的实验方法
制备55
往酚A(224mg)与碳酸钾在甲腈(10ml)中的悬浮液中加入市售原料(167mg)。将混合物回流过夜。冷却,经Celite硅藻土垫过滤并真空除去溶剂。采用硅胶层析纯化粗产物(己烷-己烷/乙酸乙酯9∶1),得到制备55的产物,产率95%。
制备56

将制备55的产物(300mg)溶于乙醇(12.5ml)。随后加入一水合肼(190mg)。在室温下将该混合物搅拌过夜。真空除去溶剂,将残余物溶解于乙酸乙酯并用水洗涤。有机层经硫酸镁干燥并浓缩。粗产物无需进一步纯化直接使用。
制备57
将制备56的产物(300mg)溶于THF(25ml)。在0℃下加入三乙胺并将所得溶液搅拌5分钟,随后在-30℃下滴加市售的酰氯(160mg)。在该温度下将该混合物搅拌45分钟,随后在室温下搅拌过夜。用水猝灭粗产物并加入乙酸乙酯。分离有机层,经硫酸镁干燥,真空浓缩。采用硅胶层析(己烷/乙酸乙酯9∶1-7.3)纯化粗产物,得到制备57的产物,两步的产率为60%。
制备58
将制备57的产物(127mg)溶于甲苯(5ml)并加入Lawesson试剂(134mg)。将混合物回流4小时。减压除去溶剂,采用硅胶层析(己烷-己烷/乙酸乙酯85∶15)纯化残余物,得到制备58的产物,产率60%。
实施例91

往制备58产物(24mg)的乙醇/THF混合物(1∶1)(4ml)溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。加入乙酸乙酯和水,加入1N HCl溶液将pH调节至6。分离两相,用乙酸乙酯萃取水相两次,有机相用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用硅胶层析纯化粗产物,得到实施例91的产物(90%)。
使用类似于制备实施例91的方法制备实施例94的产物。

流程13

流程13的实验方法
在装有标准蒸馏装置的圆底烧瓶中加入酰肼(100mmol)、原甲酸三甲酯(150mmol)和对甲苯磺酸一水合物(1.5mmol)。加热至90℃直到得到沉淀,随后加热至120℃。蒸除甲醇后得到油状物。采用HPLC和THC(己烷/乙酸乙酯1∶1)监测反应的进行情况。将该油状物溶于乙酸乙酯,用水、盐水洗涤,经硫酸镁干燥,真空浓缩。采用硅胶层析(己烷/乙酸乙酯9∶1-7∶3)纯化粗产物,得到流程13式A的二唑化合物。
在氮气气氛下往冷却至-78℃的二唑(10mmol)的THF(33ml)溶液中滴加nBuLi(1.6M的己烷溶液,10mmol)。40分钟后,加入MgBr2·乙醚(10mmol),将冷却浴升温至-45℃,在-45℃下将得到的浆状物再搅拌1.5小时。加入醛(9mmol)的THF(11ml)溶液,将反应温度升至-20℃,在该温度下再搅拌2.5小时。采用HLPC和TLC(己烷/乙酸乙酯8∶2)监测反应的进行情况。用氯化铵溶液猝灭该粗产物并加入乙酸乙酯。水层用乙酸乙酯萃取两次。分离有机层,用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到流程13式B化合物。
往二唑B(1.0mmol)与溴衍生物C(1.1mmol)的甲腈(2.5ml)溶液中加入Cs2CO3(1.3mmol),在室温下将所得的混合物搅拌过夜。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。经Celite硅藻土垫过滤。真空除去溶剂。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到流程13式D化合物。
往二唑D(1.0mmol)的乙醇/THF混合物(1∶1)(40ml)溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)和HPLC监测反应进行情况。加入乙酸乙酯和水,加入6N HCl溶液将pH调节至6。分离两相,用乙酸乙酯萃取水相两次,有机相用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用ISCO(己烷-TFA 0.05%/丙酮9∶1-85∶15)纯化粗产物,得到流程13的酸E。
溴衍生物的制备
Head H9的制备
将酚(154mmol)溶于乙醇(513ml),加入碳酸钾(600mmol)和硫酸镁,随后加入溴衍生物(231mmol)。将得到的混合物回流过夜。冷却,经Celite硅藻土垫过滤,真空浓缩。采用硅胶层析(己烷/乙酸乙酯8∶2)纯化,得到化合物H8,产率53%。
在0℃下将H8(13mmol)溶于二氯甲烷(130ml),分批加入CBr4,随后分批加入PPh3。在0℃下将混合物搅拌2小时。用TLC(己烷/乙酸乙酯4∶1)监测反应进行情况。除去溶剂。采用硅胶层析(己烷/乙酸乙酯9∶1)纯化,得到H9,产率95%。
实施例97的实验方法
制备59

在装有标准蒸馏装置的圆底烧瓶中将酰肼(5g)悬浮于原甲酸三甲酯(4.0ml)和对甲苯磺酸一水合物(47mg)中。加热至90℃直到观察到沉淀形成,随后加热至120℃。蒸除甲醇得到黄色油状物。将所述黄色油状物溶于乙酸乙酯并用水、盐水洗涤,经硫酸镁干燥并真空浓缩。采用硅胶层析(己烷/乙酸乙酯9∶1-7∶3)纯化粗产物,得到白色固体状的制备59的产物(95%)。
制备60
在氮气气氛下往冷却至-78℃的二唑(500mg)的THF(20ml)溶液中滴加nBuLi(1.51ml,1.7M的戊烷溶液)。40分钟后,加入MgBr2·乙醚(10mmol),将冷却浴升温至-45℃,在-45℃下将所得的浆状物继续搅拌1.5小时。加入丁醛(0.157ml)的THF(10ml)溶液。将反应温度升至-20℃并在该温度下继续搅拌2.5小时。用氯化铵溶液猝灭该粗产物并加入乙酸乙酯。水层用乙酸乙酯萃取两次。分离有机层,用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到制备60的产物(25%)。
制备61

往制备60的产物(80mg)和溴衍生物(217mg)的甲腈(5ml)溶液中加入Cs2CO3(213mg),在室温下将得到的混合物搅拌过夜。经Celite硅藻土垫过滤。真空除去溶剂。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化,得到制备61的产物,产率90%。
实施例97

往制备61的产物(24mg)的乙醇/THF混合物(1∶1)(4ml)溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。加入乙酸乙酯和水,加入1N HCl溶液将pH调节至6。分离两相,用乙酸乙酯萃取水相两次,有机相用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用硅胶层析纯化粗产物,得到实施例97的产物(85%)。
采用类似于制备实施例97的方法制备实施例100的产物。

流程14
流程14的实验方法
对于W=O:
市售可得的酰肼(1当量)在DCM中制成浆状物,随后依次加入酰氯(1.1当量)和三乙胺(2当量)。在氮气气氛和室温下搅拌12小时。随后用1N HCl猝灭反应并用DCM萃取。有机层经硫酸镁干燥并蒸发。用乙酸乙酯重结晶得到的固体。
将该物质溶于吡啶并加入甲苯磺酰氯(1.5当量),将混合物回流18小时。将反应物冷却,用乙酸乙酯稀释并用1N HCl、饱和碳酸氢盐和水洗涤。有机层经硫酸镁干燥并蒸发。采用柱色谱(20%乙酸乙酯/己烷)纯化残余物,得到流程14式A化合物。
将该物质溶于DCM并冷却至0℃,在10分钟内加入BBr3(3当量)。在2小时内让反应物升至室温。用乙酸乙酯稀释反应物,用饱和碳酸氢盐和1N HCl洗涤。有机层经硫酸镁干燥并蒸发。
将所得固体溶于DCM中并加入PBr3(1.2当量)。在室温下搅拌该混合物2小时,用水洗涤,有机层经硫酸镁干燥并蒸发,得到流程14式B化合物,该化合物无需纯化直接使用。
对于W=S:
酰肼(1当量)在DCM中制成浆状物,随后依次加入酰氯(1.1当量)和三乙胺(2当量)。在氮气气氛和室温下搅拌12小时。随后用1NHCl猝灭反应物并用DCM萃取。有机层经硫酸镁干燥并蒸发。在乙酸乙酯中重结晶该固体。
将该物质溶于吡啶并加入甲苯磺酰氯(1.5当量),将混合物回流18小时。将反应物冷却,用乙酸乙酯稀释并用1N HCl、饱和碳酸氢盐和水洗涤。有机层经硫酸镁干燥并蒸发。采用柱色谱(20%乙酸乙酯/己烷)纯化残余物,得到流程14式A化合物。
将该物质溶于DCM并冷却至0℃,在10分钟内加入BBr3(3当量)。在2小时内让反应物升至室温。用乙酸乙酯稀释反应物,用饱和碳酸氢盐和1N HCl洗涤。有机层经硫酸镁干燥并蒸发。
化合物D的合成:
将适合的胺headpiece(式C的化合物)溶于DMF并冷却至0℃。往其中加入NaH(1当量)并搅拌30分钟。加入化合物B(0.9当量)并让该反应物缓慢升至室温。在原料完全消耗后,用饱和氯化铵猝灭反应物并用乙酸乙酯萃取。有机层经硫酸镁干燥并蒸发。采用柱层析(20%乙酸乙酯/己烷)纯化残余物,得到流程14式D的化合物。
化合物E的合成:
往式D化合物(1.0mmol)的甲醇/THF混合物(1∶1)(40ml)溶液中加入1N NaOH溶液(1.5ml),在室温下将该混合物搅拌过夜。用TLC(己烷/乙酸乙酯1∶1)和HPLC监测反应进行情况。加入乙酸乙酯和水。加入1N HCl溶液将pH调节至6。分离两相,用乙酸乙酯萃取水相两次,有机相用水、盐水洗涤,经硫酸镁干燥,过滤并真空干燥。采用柱层析(30%乙酸乙酯/己烷)纯化残余物,得到产物E。
实施例104的实验方法
制备62

4-三氟甲基苯甲酰肼(benzhydrazine)(1g)在DCM(25ml)中制成浆状物,往该混合物中加入苄氧基乙酰氯(0.837ml),随后加入三乙胺(1.37ml)。在氮气气氛室温下搅拌12小时。随后用1N HCl猝灭反应,用DCM萃取。有机层经硫酸镁干燥并蒸发。在乙酸乙酯中重结晶固体(93%)。
将该物质溶于甲苯并加入Lawsen试剂(3g),将该混合物回流18小时。随后,冷却该反应并用乙酸乙酯稀释,用1N HCl、饱和碳酸氢盐和水洗涤。有机层经硫酸镁干燥并蒸发。采用柱色谱(20%乙酸乙酯/己烷)纯化该残余物,得到浅黄色固体状的制备62的产物(80%)。
制备63

将制备62的产物(100mg)溶于DCM(5ml)并冷却至0℃。在10分钟内往混合物中加入BBr3(0.788ml))。在2小时内让反应物升至室温。用乙酸乙酯稀释反应物并用饱和碳酸氢盐和1N HCl洗涤。有机相经硫酸镁干燥并蒸发,得到灰白色固体状的制备63的产物(98%)。
制备64
将headpiece(429mg)溶于乙腈(20ml)并冷却至0℃。往所得混合物中加入CsCO3(1g)和制备63的产物(500mg),随后让反应物缓慢升至室温。原料完全消耗后,用饱和氯化铵猝灭反应物并用乙酸乙酯萃取。有机相经硫酸镁干燥并蒸发。采用柱层析(20%乙酸乙酯/己烷)纯化残余物,得到制备64的产物(62%)。
实施例104

往制备64的产物(24mg)的乙醇/THF混合物(1∶1)(4ml)溶液中加入2N KOH溶液(1.5ml),在室温下将该混合物搅拌过夜。加入乙酸乙酯和水,加入1N HCl溶液将pH调节至6。分离两相,用乙酸乙酯萃取水相两次,有机相用水、盐水洗涤,经硫酸镁干燥,过滤并真空浓缩。采用硅胶层析(己烷/乙酸乙酯95∶5-85∶15)纯化粗产物,得到实施例104的产物(80%)。
采用类似于制备实施例104的方法制备实施例105-112的产物。


实施例113
实施例3C化合物的赖氨酸盐
往装有203mg实施例3c的小瓶中加入7ml甲醇,加热至约60℃。随后加入0.5ml L-赖氨酸(在水中,1摩尔等量)。冷却至室温。在氮气气氛下蒸发至干。加入7ml异丙醇后得到固体悬浮液。将小瓶加热至约80℃,加入水直到溶解(总计2ml)。让小瓶冷却至室温过夜,真空过滤分离固体。熔点:起始温度(Thermal:Onset)227.8℃
实施例114
将实施例3C的化合物(2.00g,0.00455m)在20.0ml IPA中制成浆状物并加热至70℃。将热的混浊/雾状溶液过滤并用1.5ml IPA洗涤,将得到的滤液加热至70℃。在另一个容器中,将L-赖氨酸(681mg,0.00466m,1.0当量;Aldrich 97%)在水(2.00ml)中制成浆状物,在50℃的水浴中短暂加热溶解为澄清的溶液。在70℃、35分钟内往游离酸溶液中滴加L-赖氨酸溶液。在70℃下将该粘稠、浆状物继续搅拌5小时,随后停止加热,让其冷至室温1小时。得到的浆状物在冰浴中继续冷却1小时,过滤,用冷IPA清洗,真空/40℃干燥过夜,得到白色晶体状的实施例3C产物的L-赖氨酸盐(2.44g,产率91%重量)。熔点227.6℃
实施例115
将实施例114的产物(0.50g,0.00087m)在甲醇(13ml)和水(3.0ml)中制成浆状物并加热至60℃。真空浓缩该溶液,除去约6.0g溶剂。在60℃下加热该溶液并在10分钟内滴加IPA(10ml),在加入过程中保持温度为60℃。再次浓缩溶液,除去约8.4g溶剂。随后在60℃、10分钟内滴加IPA(10ml)。真空浓缩该浆状物,除去约4.9g溶剂,在5分钟内加入最后量的IPA(5ml)(热)。在60℃下短暂加热浆状物,让其冷却至室温,随后在冰浴中冷却1小时。过滤,用IPA清洗,在40℃下真空干燥过夜,得到白色晶体产物(404mg;HPLC:以面积计算的产率为97.4%,以重量计算的产率为81%,HPLC:以面积计算的校正产率为86.6%)。熔点:起始温度220.19℃
实施例116
将实施例3C的产物的盐酸盐(2.00g,0.0043m)(可使用游离碱形式)在水(15ml)、二氯甲烷(15ml)和甲醇(5ml)中制成浆状物。加入5NNaOH(1.80ml,2.1当量)直到pH=11。分离各层,用另外的二氯甲烷(15ml)萃取水层。合并并丢弃有机层。往水相中加入新鲜的二氯甲烷(15ml),随后在剧烈搅拌下加入1N HCl(15ml)。分离各层,用额外的二氯甲烷(15ml)萃取水层并合并有机层。往混浊的有机层中加入乙酸乙酯(5ml)和甲醇(5ml)以防止盐酸盐过早结晶,随后用硫酸钠干燥有机层,过滤,清洗并真空浓缩,得到11g乳白色溶液。用原料作为晶种使该溶液结晶,在室温下搅拌使得产物结晶。过滤并用二氯甲烷清洗,随后真空/室温干燥过夜,得到白色双折射固体(669mg,以重量计算的产率为33%,经HPLC校正的产率为38%,HPLC:以面积计算的纯度为98.1%)。熔点:起始温度162.44℃
外消旋混合物的分离
实施例的外消旋混合物的各异构体通过HPLC分离。
| 外消旋实施例 | HPLC条件 | 保留时间(分钟)异构体1实施例 | 保留时间(分钟)异构体2实施例 |
| 68 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)梯度-20至60%B,15分钟 | 6.7实施例69 | 9.0实施例70 |
| 71 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)梯度-20至60%B,15分钟 | 9.9实施例72 | 17.1实施例73 |
| 78 | Chiralpak AD(250×20mm),己烷(A)/IPA(B)70/30等度模式 | 7.2实施例79 | 11.4实施例80 |
| 81 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)75/25等度模式 | 8.1实施例82 | 10.4实施例83 |
| 85 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)75/25等度模式 | 7.2实施例86 | 12.3实施例87 |
| 91 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/Ethanol(B)75/25梯度模式 | 11.5实施例92 | 18.1实施例93 |
| 94 | Chiralpak AD(250×20mm),己烷(A)/IPA(B)70/30等度模式 | 10.5实施例95 | 15.5实施例96 |
| 97 | Chiralpak AD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)95/5等度模式 | 15.7实施例98 | 18.7实施例99 |
| 100 | Chiralpak OD(250×4.6mm),己烷-TFA(0.05%)(A)/IPA(B)75/25等度模式 | 8.7实施例101 | 11.3实施例102 |
生物测定
结合与其转染实验
通过下述方法测定化合物调节PPARα受体的体外效力。使用SPA技术,用PPAR受体进行DNA-依赖性结合(ABCD结合)。使用氚标记的PPARα激动剂作为放射性配体以用本发明化合物产生置换曲线和得到IC50值。共转染测定是在CV-1细胞中进行的。报道质粒含有酰基CoA氧化酶(AOX)PPRE和虫萤光素酶报道cDNA的上游TK促进子。使用含有CMV促进子的质粒组成性地表达合适的PPAR。对于PPARα,CV-1细胞中的内源性PPARγ的干扰是个问题。为了消除这样的干扰,使用这样的GAL4嵌合系统,其中转染的PPAR的DNA结合域被GAL4的DNA结合域替代,并且使用GAL4应答元件来代替AOX PPRE。测定相对于PPARα激动剂参照分子的共转染效力。通过计算机拟合浓度-应答曲线,或者在某些情况下在一个高的激动剂浓度下(10μM)测定效力。
进行这些实验来评价本发明化合物结合和/或激活各种核转录因子,特别是huPPARα(“hu”是指“人”)的能力。这些实验提供了关于本发明化合物的效力和选择性的体外数据。此外,将本发明化合物的结合和共转染数据与作用于huPPARα的市售化合物的相应数据进行比较。
特别可用于调节PPAR受体的本发明化合物的结合和共转染效力值分别≤100nM和≥50%。
在HuapoAI转基因小鼠中评价甘油三酯减少和HDL胆固醇增加
研究了本发明化合物对人apoAI小鼠中的HDL和甘油三酯水平的作用。对于每种测试化合物,让7-8周龄的人apoAI转基因雄性小鼠(C57BL/6-tgn(apoal)lrub,Jackson Laboratory,Bar Harbor,ME)在单独的笼子中适应新环境2周,期间给它们提供可自由摄取的标准食物(Purina 5001)和水。适应新环境后,称重小鼠和食物,按照体重随机分配到测试组(n=5)中。使用29规格、1-1/2英寸弯曲的进料针(Popper & Sons)通过管饲法每日对小鼠给药,给药8天。用于对照、测试化合物和阳性对照(非诺贝特100mg/kg)的溶媒是含有0.25%吐温80(w/v)的1%羧甲基纤维素(w/v)。所有小鼠每天都是在早晨6-8点给药,给药体积是0.2ml。在结束之前,称重动物和食物,计算体重改变和食物摄取量。最后一次给药3小时后,用二氧化碳将小鼠安乐死,通过心脏穿刺取血(0.5-1.0ml)。处死后,将肝脏、心脏和附睾脂肪切下来,并称重。让血样凝结,通过离心从血液中分离出血清。
使用市售试剂(例如得自Sigma#339-1000和Roche#450061,分别用于测定甘油三酯和胆固醇),通过比色法测定胆固醇和甘油三酯。由公开文献(McGowan M.W.等人,Clin Chem 29:538-542,1983;AllainC.C.等人,Clin Chem 20:470-475,1974)对方法作些改进。使用200μl试剂以一式两份的方式测定甘油三酯和总胆固醇的市售标准样品、市售质量对照血浆和样本。将另一等分试样的样本加到含有200μl水的孔中,以给每一样本提供空白对照。将培养板在平板震动器上于室温培养,分别在500nm和540nm读取关于总胆固醇和甘油三酯的吸收度。阳性对照的值总是在预期范围内,样本的变异系数低于10%。为了将测定间变异减至最小,实验的所有样本同时测定。
分离出血清脂蛋白,通过偶联到在线检测系统上的快速蛋白液相色谱法(FPLC)定量测定胆固醇。将样本施加到Superose 6HR尺寸排阻柱(Amersham Pharmacia Biotech)上,用磷酸盐缓冲盐水-EDTA以0.5ml/分钟的速度洗脱。将胆固醇试剂(Roche Diagnostics Chol/HP704036)以0.16ml/分钟的速度与柱流出物经由T-连接混合,让该混合物流经浸在37℃水浴的15m×0.5mm id结合管道反应器。在液体流中在505nm监测在胆固醇存在下产生的有色产物,并将监测器中的模拟电压转化成用于收集和分析的数字信号。将相应于胆固醇浓度改变的电压改变对时间绘图,使用Perkin Elmer Turbochrome软件计算相应于极低密度脂蛋白(VLDL)、低密度脂蛋白(LDL)和高密度脂蛋白(HDL)的洗脱的曲线下面积。
将施用本发明化合物的小鼠中的甘油三酯血清水平与接受溶媒的小鼠进行比较,以鉴定可用于降低甘油三酯的化合物。一般情况下,施用30mg/kg的剂量后,与对照相比,甘油三酯下降大于或等于30%(百分之三十),表明化合物可特别用于降低甘油三酯水平。
将接受本发明化合物的小鼠中的HDL血清水平的增加百分比与接受溶媒的小鼠进行比较,以鉴定可特别用于提高HDL水平的本发明化合物。一般情况下,施用30mg/kg的剂量后,HDL水平增加大于或等于25%(百分之二十五),表明化合物可特别用于增加HDL水平。
选择既能降低甘油三酯水平,又能增加HDL水平的本发明化合物可是特别理想的。然而,能降低甘油三酯水平或增加HDL水平的化合物也符合要求。
评价db/db小鼠中的葡萄糖水平
测定给雄性db/db小鼠施用不同剂量水平的不同的本发明化合物和PPARγ激动剂rosiglitazone(BRL49653)或PPARα激动剂非诺贝特和对照物对血糖的影响。
将5周龄雄性糖尿病(db/db)小鼠[例如C57BlKs/j-m+/+Lepr(db),Jackson Laboratory,Bar Harbor,ME]或瘦的littermates以每个笼子6只的比例饲养,让它们一直能获得食物和水。适应新环境2周后,通过耳朵切迹标记确定小鼠的身份,称重,并经由尾静脉采血来测定初始葡萄糖水平。通过下述方法从未禁食的动物采集血液(100μl):将每只小鼠包在手巾中,用解剖刀切开尾巴的末梢,将血液从尾巴挤到肝素化的毛细管中。用凝胶分离器将血液排放到肝素化的微容器内,并保持在冰上。在4℃离心后获得了血浆,立即测定葡萄糖。当测定所有样本中的葡萄糖和甘油三酯时,将剩余血浆冷冻直至实验完成。根据初始葡萄糖水平和体重将动物分组。从接下来的第二天早晨开始,通过管饲法每天对动物给药,给药7天。治疗是用测试化合物(30mg/kg)、阳性对照剂(30mg/kg)或溶媒[1%羧甲基纤维素(w/v)/0.25%吐温80(w/v);0.3ml/小鼠]进行的。在第7天,给药3小时后,将小鼠称重并采血(尾静脉)。第7次给药24小时后(即第8天),再次对动物采血(尾静脉)。测定在第0、7和8天得自清醒动物的样本的葡萄糖。采血24小时后,称重动物,并进行最后一次给药。在第8天给药3小时后,通过吸入异氟烷将动物麻醉,并经由心脏穿刺获得血液(0.5-0.7ml)。将全血转移到血清分离器管中,用冰冷却并让其凝结。在4℃离心后获得血清,冷冻直至分析化合物水平。通过颈脱臼处死后,切下肝脏、心脏和附睾脂肪块并称重。
使用市售试剂,通过比色法测定葡萄糖。依据生产商的说明,由公开的文献(McGowan,M.W.,Artiss,J.D.,Strandbergh,D.R.&Zak,B.Clin Chem,20:470-5(1974)和Keston,A.葡萄糖的特异性比色酶分析试剂.Abstract of papers 129th Meeting ACS,31C(1956))对方法作些改进;并且根据每摩尔被分析物所释放的过氧化氢的摩尔数,与Trinder首先描述的显色反应结合起来(Trinder,P.使用具有另一氧受体的葡萄糖氧化酶测定血液中的葡萄糖.Ann Clin Biochem,6:24(1969))。所产生的染料的吸收度与样本中的被分析物线性相关。在我们的实验室中对该测定作进一步改变以用于96孔格式中。使用200μl试剂以一式两份的方式测定葡萄糖的市售标准样品、市售质量对照血浆和样本(2或5μl/孔)。将另一等分试样的样本吸移到第3个孔中,在200μl水中稀释,以给每一样本提供空白对照。将培养板在平板震动器(DPC Micormix 5)上于室温培养18分钟以测定葡萄糖,在平板读数器上在500nm读取吸收度。将样本吸收度与标准曲线作比较(对于葡萄糖是100-800)。质量对照样本的值总是在预期范围内,样本的变异系数低于10%。为了将测定间变异减至最小,实验的所有样本同时测定。
评价本发明化合物对Ay小鼠体重、脂肪质量、葡萄糖和胰岛素水平的影响
雌性Ay小鼠
将雌性Ay小鼠单独饲养,保持在标准条件下(22℃,12小时光照:黑暗循环),在整个实验期间让其能自由摄取食物和水。在20周龄时,根据体重和如通过DEXA扫描所估计的身体脂肪含量将小鼠随机分配到溶媒对照组和治疗组中(N=6)。然后在开始光照循环后1小时(例如约早晨7点)通过管饲法给予小鼠溶媒或本发明化合物(50mg/kg),给药18天。在实验期间每天测定体重。在第14天,将小鼠保持在单独的代谢室中,以进行能量消耗和燃料利用的间接量热法评估。在第18天,再次对小鼠进行DEXA扫描以测定治疗后的身体组成。
评价口服给药化合物18天后在体重、脂肪质量和瘦肉质量方面的结果,并鉴定哪些本发明化合物可特别用于保持理想体重和/或提高所需的瘦肉与脂肪质量比例。
间接量热法测定表明,在黑暗循环期间,在治疗的动物中呼吸商(RQ)有显著下降[0.864±0.013(对照)vs.0.803±0.007(治疗);p<0.001]。RQ下降是在动物活动(黑暗)循环期间脂肪利用增加的指示。此外,治疗的动物表现出显著高于对照动物的能量消耗速度,这表明本发明的化合物是特别符合要求。
雄性KK/Ay小鼠
将雄性KK/Ay小鼠单独饲养,保持在标准条件下(22℃,12小时光照:黑暗循环),在整个实验期间让其能自由摄取食物和水。在20周龄时,根据血浆葡萄糖水平将小鼠随机分配到溶媒对照组和治疗组中。然后在开始光照循环后1小时(早晨7点)通过管饲法给小鼠施用溶媒或本发明化合物(30mg/kg),给药14天。在第14天测定血浆葡萄糖、甘油三酯和胰岛素水平。
评价口服施用化合物14天在血浆葡萄糖、甘油三酯和胰岛素方面的结果,以鉴定出可能特别理想的本发明化合物。
证明降低LDL-胆固醇、总胆固醇和甘油三酯的作用的方法
在使用前,给重80-120g的雄性叙利亚仓鼠(Harlan SpragueDawley)提供高脂肪的富含胆固醇的饮食2-3周。在实验期间让动物自由摄取食物和水。在这些条件下,仓鼠变得血胆固醇过多,表现出180-280mg/dl的血浆胆固醇水平(喂养正常食物的仓鼠的总血浆胆固醇水平为100-150mg/dl)。使用GroupOptimizeV211.xls程序,根据其总胆固醇水平将具有高血浆胆固醇(180mg/dl以及以上)的仓鼠随机分配到治疗组中。
将本发明化合物溶解在含水溶媒(含有CMC和吐温80)中,这样每只仓鼠每天通过管饲法接受一次约1ml溶液,剂量为3和30mg/kg体重。将非诺贝特(Sigma Chemical,在相同载体中制成悬浮液)作为已知的α-激动剂对照以200mg/kg的剂量施用,空白对照是单独的溶媒。给药每天在早晨进行,进行14天。
定量测定血浆脂质:
在测试的最后一天,给药2小时后,在异氟烷麻醉下,从仓鼠的眶下静脉窦采血(400μl)。将血样收集到在冰浴中冷却的肝素化微离心管中。通过短暂离心从血细胞中分离出血浆样本。通过按照生产商的说明,在Monarch装置(Instrumentation Laboratory)上自动进行的酶测定来确定总胆固醇和甘油三酯。通过下述方法分离出血浆脂蛋白(VLDL、LDL和HDL):将25μl收集的血浆样本注射到FPLC系统中,用磷酸盐缓冲盐水以0.5ml/分钟的速度经由保持在室温的Superose 6 HR 10/30柱(Pharmacia)洗脱。通过将洗脱排放物与Cholesterol/HP试剂(例如Roche Lab System;以0.12ml/分钟的速度输注)在保持在37℃的编结反应线圈中进行柱后培养来完成所分离的血浆的检测和表征。所形成的颜色的强度与胆固醇浓度成比例,并在505nm通过光度法测定。
研究了施用本发明化合物14天的作用,这种作用是用相对于载体组LDL水平的下降百分比表示的。特别需要的化合物的LDL降低效力显著强于非诺贝特。与溶媒相比,将LDL降低大于或等于30%(百分之三十)的本发明化合物可能是特别希望的。
还研究了本发明化合物降低总胆固醇和甘油三酯的作用。将用本发明化合物治疗14天后关于总胆固醇和甘油三酯降低的数据与溶媒进行比较,以鉴定出特别理想的化合物。在相同实验条件下,已知对照化合物非诺贝特没有表现出显著效力。
证明PPAR调节剂减少血纤蛋白原的作用的方法
Zucker肥胖大鼠模型:
研究本发明化合物降低血纤蛋白原的作用的活体阶段(LifePhase)是相同化合物抗糖尿病研究的活体阶段操作的一部分。在治疗的最后一天(第14天),将动物置于手术麻醉下,通过心脏穿刺将~3ml血液收集到含有柠檬酸盐缓冲液的注射器中。将血样冷却,在4℃离心以分离出血浆,在进行血纤蛋白原测定之前于-70℃贮藏。
定量测定大鼠血浆血纤蛋白原:
按照生产商的说明,使用由凝聚装置构成的市售测定系统定量测定大鼠血浆血纤蛋白原。大体来说,从每份标本中取100μl血浆,用缓冲液进行1/20稀释。将稀释的血浆在37℃培养240秒。然后加入50微升凝固试剂凝血酶溶液(由仪器生产商以标准浓度提供)。由仪器监测凝固时间—相对于标准样本定量测定的血纤蛋白原浓度的函数。降低血纤蛋白原水平的能力大于溶媒的化合物可能是特别理想的。
本发明化合物还在Zucker大鼠中也产生了降低胆固醇和甘油三酯的作用。
证明本发明化合物抗体重增加和抗食欲作用的方法
在Zucker肥胖大鼠1或ZDF大鼠2模型中的14天实验:
在治疗之前,将年龄和体重差别不大的没有糖尿病的雄性Zucker肥胖大鼠(Charles River Laboratories,Wilmington,MA)或雄性ZDF大鼠(Genetic Models,Inc,Indianapolis,IN)适应新环境1周。在实验期间,让大鼠能自由摄取标准食物和水。
将本发明的化合物溶解在含水溶媒中,这样每只大鼠每天通过管饲法接受一次约1ml溶液,剂量为0.1、0.3、1和3mg/kg体重。将非诺贝特(Sigma Chemical,制成在同样载体中的悬浮液)—一种已知的α-激动剂,以300mg/kg的剂量施用,溶媒作为对照。给药在每天早晨进行,给药14天。在实验期间,监测体重和食物摄取量。使用该测定,确定本发明化合物使得体重显著下降。
说明PPARδ受体体内活性的方法
该方法具体可用于测定本发明化合物的体内PPARδ受体活化作用,以确定该化合物对于该受体同种型的体外活性明显高于PPATγ同种型。
在使用前至少一周给8-9周龄的雄性PPARα小鼠(129s4 SvJae-PPARa<tm1Gonz>mice;Jackson Laboratories)提供可自由摄取的标准食物(Purina 5001)和水。在整个实验期间提供可自由摄取的食物和水。使用GroupOptimizeV211.xls程序,小鼠按照体重随机分配到五个测试组中。
将本发明的化合物悬浮于1%(重量/体积)羧甲基纤维素和0.25%Tween 80的含水溶媒中,每天通过管饲法以每kg体重0.2-20mg的量给予每只小鼠约0.2ml的该溶液。每个实验的对照组的小鼠仅平行给予溶媒。每天早晨给予该剂量,共计7天。
给药的最后一天,用二氧化碳窒息3小时将小鼠安乐死。通过心脏抽取血液样品,置于含EDTA的微型离心管中并冷却于冰中。通过尸体剖检收集肝脏,在液氮中快速冷冻并贮存在-80℃下。为了从肝脏中分离RNA,将5-10mg冷冻的肝脏置于700μl 1x Nucleic AcidLysis溶液中(Applied Biosystems Inc.,Foster City,CA),使用手持组织浸渍器(Biospec Products Inc.,Bartlesville,OK)均化。组织匀浆经ABITissue预滤器(Applied Biosystems Inc.,Foster City,CA)过滤并收集在ABI 6100 Nucleic Acid prep station(Applied Biosystems Inc.,Foster City,CA)上的深孔板上。随后将经过滤的匀浆加载于RNA分离板上,接着在ABI 6100上进行RNA Tissue-Filter-DNA方法。分离的RNA用150μl无核糖核酸酶的水洗脱。为了进行定性分析,将9μl分离的RNA溶液加载在1%TBE琼脂糖凝胶上,用溴化乙锭荧光剂显影。
使用ABI High Capacity Archive Kit(Applied Biosystems Inc.,Foster City,CA)合成互补DNA(cDNA)。立刻按照生产商的方案(RTBuffer,dNTP,Random Primers,MultiScribe RT(50U/μl),无核糖核酸酶的水)制备2x反转录酶Master Mix。对于每个反应,将50μl 2x RTMaster Mix加入温育在循环变温器中的PCR管中的50μl分离的RNA中(在25℃下10分钟,随后在37℃下2小时)。以1∶100的比例将得到的cDNA制剂稀释在dH2O中分析实时PCR。cDNA的标准曲线以1∶20、1∶100、1∶400、1∶2000、1∶10,0000的比例稀释用于最后的定量分析。
用于小鼠Cyp4A1基因表达的实时PCR Master Mix混合物中包含:
1X Taqman Universal PCR Master Mix(Applied Biosystems Inc.,Foster City,CA)
6毫摩尔最终浓度的正向引物(Qiagen/Operon Technologies,Alameda,CA)
6毫摩尔最终浓度的反向引物(Qiagen/Operon Technologies,Alameda,CA)
0.15毫摩尔最终浓度的Probe(5’6-FAM和3’Tamra-Q;Qiagen/Operon Technologies,Alameda,CA);
无核糖核酸酶的水至10微升;
用于18S核糖体核糖核酸控制基因表达的实时PCR Master Mix混合物包含:
1X Taqman Universal PCR Master Mix(Applied Biosystems Inc.,Foster City,CA)
0.34毫摩尔Probe/Primer TaqManRibosomal RNA ControlReagents#4308329 Applied Biosystems Inc.,Foster City,CA)
无核糖核酸酶的水至10微升;
对于实时PCR分析,分别将6ul Master Mix溶液(Cyp4A1或18S)和4ul稀释的cDNA或标准曲线样品加入384孔板的各孔中(对于标准样品n=2;对于未知样品n=4)。使用ABI 7900HT标准通用RT-PCR循环方案进行反应。使用SDS 2.1(Applied Biosystems Inc.,FosterCity,CA)分析数据。按照标准曲线值自动计算各个样品的平均值和标准偏差。使用Microsoft Excel 2000计算5只小鼠组成的各组的平均值。用服用化合物组的平均值除以服用溶媒组的平均值。设定服用溶媒组的值为1.0来确定相对于服用溶媒组的诱导倍数(foldinduction),各组平均值倍数的变化表示为诱导倍数除以服用溶媒组的值(1.0).数据使用Jandel SigmaPlot 8.0记录。
猴子研究
有效性研究
本发明的化合物可在脂血异常(dyslipidemic)的恒河猴模型中试验。肥胖、未患糖尿病的恒河猴口服28天后,测定各剂量下的HDL-c升高并于处理前的水平比较。同时测定各剂量下的LDL胆固醇水平。测定C-反应蛋白水平并与处理前比较。
在非洲绿猴模型中式1化合物也表现出可提高血浆HDL-胆固醇水平,其作用方式类似于上述恒河猴模型。
对两组猴子进行逐渐加大剂量的研究,研究由以下步骤组成:1周基线测定,9周治疗(溶媒,式I化合物)和4周洗出。在基线测定期间,分成三组的猴子每日给予一次溶媒,共给予7天。每日给予一次在溶媒中的被测式I化合物,给予3周,随后以更高的浓度每日给予一次(剂量可为双倍),给予3周,随后以更高的浓度每日给予一次(剂量可为最近剂量的双倍),给予3周。治疗完后,每日一次给予两组猴子溶媒并继续监测6周。
使动物禁食一夜,随后给予镇静剂,在1(溶媒)、2、3、4、6、7、9、10、12和14周后测定体重和采集血液。
测定的各参数例如为:
体重
总血浆胆固醇
HDL
LDL
甘油三酯
胰岛素
葡萄糖
4、7和10周的PK参数(每种剂量的最后一周的血浆药物浓度)
ApoAI
ApoAII
ApoB
ApoCIII
肝酶(SGPT,SGOT,□GT)
完全血细胞计数
此外,在适当的情况下,还可进行其他测定,这些测定与上述研究设计一致。
等价物
虽然已经用优选的实施方案特别表明和描述了本发明,但是本领域技术人员应当理解,可在不背离由权利要求书所限定的本发明范围的情况下对其作不同形式和细节的改变。
Claims (84)
1.一种式I的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:
其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0- 4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-6-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)W为N、O或S;
(g)Y选自C、O、S、NH和单键;
(h)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(i)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(j)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(k)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(l)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(m)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(n)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(o)----为任选的键,以在指示位置形成双键。
2.权利要求1的化合物,其中所述化合物为式Ia的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:
其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0- 4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)Y选自C、NH和单键;
(g)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
3.权利要求1的化合物,其中所述化合物为式Ic的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:
其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0- 4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)Y选自C、O、S、NH和单键;
(g)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
4.权利要求1的化合物,其中所述化合物为式Ib的化合物:
其中W1为S或O。
5.权利要求5的化合物,其中W1为S。
6.权利要求5的化合物,其中W1为O。
7.权利要求4、5或6中任一项的化合物,其中当Y为O时,则U为C1-C4脂族连接基,其中C1-C4烷基的一个碳被O置换,并且其中所述脂族连接基任选被1-4个各自独立选自R30的取代基取代。
8.权利要求1、2、3或7中任一项的化合物,其中所述化合物为式III的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:

其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0- 4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基任选被1-4个各自独立选自R30的取代基取代;
(f)Y选自C、O、S、NH和单键;
(g)E为C(R3)(R4)A;其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
条件是当Y为O时,R4选自C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、环烷基和芳基-烷基各自任选被1-3个独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
9.权利要求1-8中任一项的化合物,其中X为O。
10.权利要求1-8中任一项的化合物,其中X为S。
11.权利要求1-10中任一项的化合物,其中Y为O。
12.权利要求1-10中任一项的化合物,其中Y为C。
13.权利要求1-10中任一项的化合物,其中Y为S。
14.权利要求1-13中任一项的化合物,其中所述五元环中的两个“----”各自为键,以在指示位置形成双键。
15.权利要求1-14中任一项的化合物,其中E为C(R3)(R4)A。
16.权利要求1-15中任一项的化合物,其中A为COOH。
17.权利要求1-16中任一项的化合物,其中R10为卤代烷基。
18.权利要求17的化合物,其中R10为CF3。
19.权利要求1-16中任一项的化合物,其中R10为卤代烷氧基。
20.权利要求1-16中任一项的化合物,其中R10和R11各自独立选自氢、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12”、C1-C6烷氧基、C1-C6卤代烷基和C1-C6卤代烷氧基。
21.权利要求1-16中任一项的化合物,其中R10和R11各自独立选自C1-C2烷基。
22.权利要求1-16中任一项的化合物,其中R10选自C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基和芳氧基。
23.权利要求1-22中任一项的化合物,其中R1为任选取代的C2-C3芳基烷基。
24.权利要求1-23中任一项的化合物,其中R8和R9各自独立选自氢和C1-C3烷基。
25.权利要求1-22和24中任一项的化合物,其中R1、V、R3和R4各自独立选自C1-C2烷基。
26.权利要求1-22和24中任一项的化合物,其中R1、R3和R4各自独立选自氢和C1-C2烷基。
27.权利要求1-24或26中任一项的化合物,其中V为键。
28.权利要求1-24中任一项的化合物,其中V选自C0-C1烷基。
29.权利要求1-28中任一项的化合物,其中U为C1-C3烷基。
30.权利要求1-29中任一项的化合物,其中U为饱和的。
31.权利要求1-22或24-30中任一项的化合物,其中R1为C1-C6杂烷基。
32.权利要求1-31中任一项的化合物,其中脂族连接基的一个碳被O置换。
33.权利要求1-31中任一项的化合物,其中U为一个碳被N置换的脂族连接基。
34.权利要求1-31中任一项的化合物,其中U为一个碳被S置换的脂族连接基。
35.权利要求1-34中任一项的化合物,其中所述脂族连接基被1-3个各自独立选自R30的取代基取代。
36.权利要求35的化合物,其中所述脂族连接基被1-2个各自独立选自R30的取代基取代。
37.权利要求1-36中任一项的化合物,其中R30各自独立选自C1-C6烷基。
38.权利要求1-37中任一项的化合物,其中R30各自独立选自C2-C3烷基。
39.权利要求1-36中任一项的化合物,其中R30独立选自芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基。
40.权利要求39的化合物,其中Y为O,E为-CH2COOH。
41.权利要求1-40中任一项的化合物,其中U被甲基取代。
42.权利要求1-41中任一项的化合物,其中U为亚甲基。
43.权利要求1-16和22-42中任一项的化合物,其中R10和R11结合形成稠合的六元环。
44.权利要求1-3、8-13、15-24、26和36-41中任一项的化合物,所述化合物用以下结构式IV表示:

45.权利要求1-3、8-13、15-24、26、36-41中任一项的化合物,所述化合物用以下结构式V表示:
46.权利要求1-3、8-13、15-24、26、36-41中任一项的化合物,所述化合物用以下结构式VI表示:
47.权利要求1-3、8-13、15-24、26、36-41中任一项的化合物,所述化合物用以下结构式VIII表示:
48.权利要求1-3、8-13、15-24、26、36-41中任一项的化合物,所述化合物用以下结构式IX表示:
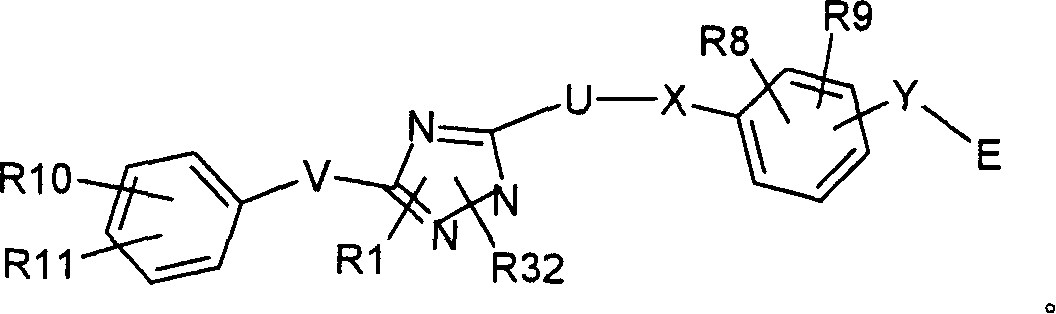
49.权利要求1-48中任一项的化合物,其中X和Y取代于1,4-位,使得X和Y互为对位取代。
50.权利要求1-48中任一项的化合物,其中X和Y取代于1,3-位,使得X和Y互为邻位取代。
51.权利要求1中任一项的化合物,其中所述化合物为下式的化合物或其药学上可接受的盐、溶剂合物或水合物:

52.权利要求1-50中任一项的化合物,其中X为键。
53.权利要求1中任一项的化合物,所述化合物为2-甲基-2-{4-[3-(5-萘-2-基甲基-2H-[1,2,4]三唑-3-基)-苯氧基}丙酸。
54.权利要求1-53中任一项的化合物,所述化合物为S构象。
55.权利要求1-53中任一项的化合物,所述化合物为R构象。
56.一种药物组合物,所述药物组合物包含作为活性成分的至少一种权利要求1-55中任一项的化合物和药学上可接受的载体或稀释剂。
57.一种调节过氧化物增殖物激活受体的方法,所述方法包括将所述受体与至少一种权利要求1-55中任一项的化合物接触的步骤。
58.一种用于治疗哺乳动物的糖尿病的方法,所述方法包括给予需要治疗的哺乳动物治疗有效量的至少一种权利要求1-55的化合物。
59.一种用于治疗哺乳动物的代谢疾病的方法,所述方法包括给予需要治疗的哺乳动物治疗有效量的至少一种权利要求1-55的化合物。
60.权利要求59的方法,其中所述需要治疗的哺乳动物经诊断患有代谢疾病。
61.一种用于选择性调节PPARδ受体的方法,所述方法包括给予有需要的哺乳动物权利要求1-55中任一项的化合物。
62.权利要求1-55中任一项的化合物在制造用于治疗和/或预防代谢疾病的药物中的用途。
63.一种用于治疗哺乳动物的动脉粥样硬化的方法,所述方法包括给予需要治疗的哺乳动物治疗有效量的至少一种权利要求1-55的化合物。
64.权利要求1-55中任一项的化合物,所述化合物用作药物。
65.一种用于治疗或预防需要治疗的哺乳动物的心血管疾病的发展的方法,所述方法包括给予治疗有效量的至少一种权利要求1-55中任一项的化合物。
66.权利要求65的方法,其中所述哺乳动物经诊断为需要这类治疗。
67.权利要求1-55中任一项的化合物,其中所述化合物经放射性标记。
68.本发明说明书中任一实施例所公开的化合物。
69.本发明说明书中公开的所有制备结构式I表示的化合物的方法。
70.权利要求1、4、5、6、9-13、15-24、26或36-41中任一项的化合物,所述化合物用结构式X表示:

71.权利要求1、5、7、9-13、15-24、26、36-41中任一项的化合物,所述化合物用结构式XI表示:
72.权利要求1、4、5、6、9-13、15-24、26或36-41中任一项的化合物,所述化合物用结构式XII表示:

73.权利要求1、5、7、9-13、15-24、26、36-41中任一项的化合物,所述化合物用结构式XIII表示:

74.权利要求45的化合物,其中所述化合物选自:

或其药学上可接受的盐、溶剂合物或水合物。
75.权利要求46的化合物,其中所述化合物选自:


或其药学上可接受的盐、溶剂合物或水合物。
76.权利要求46的化合物,其中所述化合物选自:

或其药学上可接受的盐、溶剂合物或水合物。
77.权利要求48的化合物,其中所述化合物选自:

或其药学上可接受的盐、溶剂合物或水合物。
78.权利要求70的化合物,其中所述化合物选自:
或其药学上可接受的盐、溶剂合物或水合物。
79.权利要求71的化合物,其中所述化合物选自:


或其药学上可接受的盐、溶剂合物或水合物。
80.权利要求72的化合物,其中所述化合物选自:
或其药学上可接受的盐、溶剂合物或水合物。
81.权利要求73的化合物,其中所述化合物选自:

或其药学上可接受的盐、溶剂合物或水合物。
82.一种药物制剂,所述药物制剂包含权利要求1-55、70-81、83或84中任一项的化合物和至少一种药学上可接受的赋形剂或载体。
83.权利要求1、2、3、8、12-26、39、40和43中任一项的化合物,所述化合物为下面结构式的化合物:

84.权利要求1中任一项的化合物,所述化合物为式Ia的化合物及其立体异构体、药学上可接受的盐、溶剂合物和水合物:

其中:
(a)R1选自氢、C1-C8烷基、C1-C8烯基、C1-C8杂烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C8烷基、C1-C8烯基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R1’的取代基取代;
(b)R1’、R26、R27、R28和R31各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烷基-COOR12、C1-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳氧基、芳基-C0- 4-烷基、杂芳基、杂环烷基、C(O)R13、COOR14、OC(O)R15、OS(O)2R16、N(R17)2、NR18C(O)R19、NR20SO2R21、SR22、S(O)R23、S(O)2R24和S(O)2N(R25)2;R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24和R25各自独立选自氢、C1-C6烷基和芳基;
(c)V选自C0-C8烷基和C1-4-杂烷基;
(d)X选自单键、O、S、S(O)2和N;
(e)U为脂族连接基,其中所述脂族连接基的一个碳原子任选被O、NH或S置换,并且其中这类脂族连接基被1-4个各自独立选自R30的取代基取代;
(f)Y选自C、O、S、NH和单键;
(g)E为C(R3)(R4)A或A且其中
(i)A选自羧基、四唑、C1-C6烷基腈、甲酰胺、磺酰胺和酰基磺酰胺;其中所述磺酰胺、酰基磺酰胺和四唑各自任选被一至两个独立选自R7的基团取代;
(ii)各R7独立选自氢、C1-C6卤代烷基、芳基C0-C4烷基和C1-C6烷基;
(iii)R3选自氢、C1-C5烷基和C1-C5烷氧基;和
(iv)R4选自H、C1-C5烷基、C1-C5烷氧基、芳氧基、C3-C6环烷基和芳基C0-C4烷基,且R3和R4任选结合形成C3-C4环烷基,其中所述烷基、烷氧基、芳氧基、环烷基和芳基-烷基各自任选被1-3个各自独立选自R26的取代基取代;
(h)R8选自氢、C1-C4烷基、C1-C4烯基和卤素;
(i)R9选自氢、C1-C4烷基、C1-C4烯基、卤素、芳基-C0-C4烷基、杂芳基、C1-C6烯丙基和OR29,其中所述芳基-C0-C4烷基、杂芳基各自任选被1-3个独立选自R27的取代基取代;R29选自氢和C1-C4烷基;
(j)R10、R11各自独立选自氢、羟基、氰基、硝基、卤素、氧代基、C1-C6烷基、C1-C6烯基、C1-C6烷基-COOR12”、C0-C6烷氧基、C1-C6卤代烷基、C1-C6卤代烷氧基、C3-C7环烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基、C3-C6环烷基芳基-C0-2-烷基、芳氧基、C(O)R13’、COOR14’、OC(O)R15’、OS(O)2R16’、N(R17’)2、NR18’C(O)R19’、NR20’SO2R21’、SR22’、S(O)R23’、S(O)2R24’和S(O)2N(R25’)2;其中所述芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个独立选自R28的取代基取代;其中R10和R11任选与其连接的苯基结合形成5-6元稠合的双环;
(k)R12’、R12”、R13’、R14’、R15’、R16’、R17’、R18’、R19’、R20’、R21’、R22’、R23’、R24’和R25’各自独立选自氢、C1-C6烷基和芳基;
(l)R30选自C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基,且其中所述C1-C6烷基、芳基-C0-4-烷基、芳基-C1-4-杂烷基、杂芳基-C0-4-烷基和C3-C6环烷基芳基-C0-2-烷基各自任选被1-3个各自独立选自R31的取代基取代;
(m)R32选自键、氢、卤素、C1-C6烷基、C1-C6卤代烷基和C1-C6氧代烷基;和
(n)----为任选的键,以在指示位置形成双键。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53232003P | 2003-12-22 | 2003-12-22 | |
| US60/532,320 | 2003-12-22 | ||
| US58656304P | 2004-07-09 | 2004-07-09 | |
| US60/586,563 | 2004-07-09 | ||
| EP04380159 | 2004-07-21 | ||
| EP04380159.6 | 2004-07-21 | ||
| EP04380158 | 2004-07-21 | ||
| EP04380158.8 | 2004-07-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1909902A true CN1909902A (zh) | 2007-02-07 |
Family
ID=34753767
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2004800383003A Pending CN1909902A (zh) | 2003-12-22 | 2004-12-21 | 用于治疗糖尿病的作为ppar调节剂的三唑、噁二唑和噻二唑衍生物 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7507832B2 (zh) |
| EP (1) | EP1725231A1 (zh) |
| JP (1) | JP2007515484A (zh) |
| CN (1) | CN1909902A (zh) |
| AU (1) | AU2004311909A1 (zh) |
| BR (1) | BRPI0417947A (zh) |
| CA (1) | CA2549385A1 (zh) |
| WO (1) | WO2005065683A1 (zh) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101863852A (zh) * | 2010-06-29 | 2010-10-20 | 沈阳航空航天大学 | 含1,3,4-噁二唑杂环结构芳香二元胺及其制备方法 |
| CN102076671A (zh) * | 2008-07-03 | 2011-05-25 | 安斯泰来制药有限公司 | 三唑衍生物或其盐 |
| CN102365273A (zh) * | 2009-01-28 | 2012-02-29 | 赛诺菲 | 噻二唑与*二唑衍生物及其制备与其治疗用途 |
| CN103467406A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 卤素取代的四氮唑羧酸类化合物、其制备方法和用途 |
| CN103467408A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 一类四氮唑羧酸类化合物及其用途 |
| CN103467409A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 取代的四氮唑羧酸类化合物及其用途 |
| CN103467405A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 一类四氮唑羧酸类化合物、其制备方法和用途 |
| CN103467407A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 四氮唑羧酸类化合物及其用途 |
| CN103467398A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其抗糖尿病用途 |
| CN103467399A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其用途 |
| CN103483281A (zh) * | 2013-09-03 | 2014-01-01 | 浙江医药高等专科学校 | 一类三唑酰胺类化合物、其制备方法和其抗糖尿病用途 |
| CN103483282A (zh) * | 2013-09-03 | 2014-01-01 | 浙江医药高等专科学校 | 苯基取代的三唑酰胺类化合物及用途 |
| CN103554043A (zh) * | 2013-09-03 | 2014-02-05 | 浙江医药高等专科学校 | 苯基取代的三唑酰胺类化合物及用途 |
| CN106632125A (zh) * | 2016-10-08 | 2017-05-10 | 李纪焕 | 一种烷基芳基取代噁二唑化合物的三组分合成方法 |
| US12220412B2 (en) | 2018-11-20 | 2025-02-11 | Sparrow Pharmaceuticals, Inc. | Methods for administering corticosteroids |
| US12329745B2 (en) | 2022-05-16 | 2025-06-17 | Sparrow Pharmaceuticals, Inc. | Methods and compositions for treating glucocorticoid excess |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7816385B2 (en) * | 2002-12-20 | 2010-10-19 | High Point Pharmaceuticals, Llc | Dimeric dicarboxylic acid derivatives, their preparation and use |
| JP2007284352A (ja) * | 2004-08-05 | 2007-11-01 | Dai Ichi Seiyaku Co Ltd | ピラゾール誘導体 |
| CN101014578B (zh) | 2004-09-16 | 2011-01-19 | 安斯泰来制药有限公司 | 三唑衍生物或其盐 |
| AU2006210503B2 (en) * | 2005-02-03 | 2009-09-03 | Irm Llc | Compounds and compositions as PPAR modulators |
| EP1937658A1 (en) | 2005-10-06 | 2008-07-02 | Sanofi-Aventis | 4-oxy-n-[1 ,3,4]-thiadiazol-2-yl-benzene sulfonamides, processes for their preparation and their use as pharmaceuticals |
| BRPI0712262A2 (pt) | 2006-05-24 | 2012-07-10 | Lilly Co Eli | agonistas de fxr |
| US8106077B2 (en) | 2006-05-24 | 2012-01-31 | Eli Lilly And Company | Compounds and methods for modulating FXR |
| MX2008015228A (es) * | 2006-05-30 | 2008-12-12 | Astrazeneca Ab | Acido 5-fenilamino-1,3,4-oxadiazol-2-ilcarbonilamino-4-fenoxi-cicl ohexanocarboxilico sustituido como inhibidor de diacilglicerol aciltransferasa de acetil coenzima a. |
| CN101460470B (zh) * | 2006-05-30 | 2011-05-18 | 阿斯利康(瑞典)有限公司 | 作为dgat1抑制剂的1,3,4-二唑衍生物 |
| WO2009086277A1 (en) | 2007-12-20 | 2009-07-09 | Envivo Pharmaceuticals, Inc. | Tetrasubstituted benzenes |
| JP2010018531A (ja) * | 2008-07-09 | 2010-01-28 | Sumitomo Seika Chem Co Ltd | 3−ベンジルオキシベンゼンチオールの製造方法 |
| EA022417B1 (ru) * | 2008-10-21 | 2015-12-30 | Саймабэй Терапевтикс, Инк. | Арильные агонисты рецептора gpr120 и их применение |
| FR2941457A1 (fr) * | 2009-01-28 | 2010-07-30 | Sanofi Aventis | Derives de thiadiazoles et d'oxadiazoles leur preparation et leur application en therapeutique |
| SG187103A1 (en) | 2010-07-16 | 2013-02-28 | Abbvie Inc | Phosphine ligands for catalytic reactions |
| US9255074B2 (en) | 2010-07-16 | 2016-02-09 | Abbvie Inc. | Process for preparing antiviral compounds |
| CA2805748A1 (en) | 2010-07-16 | 2012-01-19 | Shashank Shekhar | Process for preparing n-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1-(2h)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide |
| US8975443B2 (en) | 2010-07-16 | 2015-03-10 | Abbvie Inc. | Phosphine ligands for catalytic reactions |
| EP2612669A4 (en) | 2010-08-31 | 2014-05-14 | Snu R&Db Foundation | USING THE FÖTAL REPROGRAMMING OF A PPAR AGONIST |
| UA112418C2 (uk) | 2010-09-07 | 2016-09-12 | Астеллас Фарма Інк. | Терапевтичний болезаспокійливий засіб |
| JP2015131783A (ja) * | 2014-01-14 | 2015-07-23 | 株式会社日本ファインケム | アリール−1,2,4−トリアゾール誘導体の製造方法 |
| JP7113023B2 (ja) | 2017-03-30 | 2022-08-04 | ブリストル-マイヤーズ スクイブ カンパニー | 6-(シクロプロパンアミド)-4-((2-メトキシ-3-(1-メチル-1h-1,2,4-トリアゾール-3-イル)フェニル)アミノ)-n-(メチル-d3)ピリダジン-3-カルボキシアミドの製造方法 |
| KR102750722B1 (ko) * | 2022-03-16 | 2025-01-09 | 재단법인 대구경북첨단의료산업진흥재단 | 싸이아다이아졸 화합물 및 이를 포함하는 염증성 질환의 예방 또는 치료용 약학적 조성물 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3637672A (en) * | 1968-07-09 | 1972-01-25 | Osaka Seika Kogyo Kk | Azole compounds |
| DE4031158A1 (de) * | 1990-04-21 | 1991-10-24 | Bayer Ag | Aryloxycarbonylverbindungen |
| JPH05202038A (ja) * | 1992-01-30 | 1993-08-10 | Sumitomo Chem Co Ltd | アゾール誘導体、その製造法およびそれを有効成分とする除草剤 |
| AU6526896A (en) | 1995-07-22 | 1997-02-18 | Rhone-Poulenc Rorer Limited | Substituted aromatic compounds and their pharmaceutical use |
| GB0029974D0 (en) | 2000-12-08 | 2001-01-24 | Glaxo Group Ltd | Chemical compounds |
| US6875780B2 (en) | 2002-04-05 | 2005-04-05 | Warner-Lambert Company | Compounds that modulate PPAR activity and methods for their preparation |
-
2004
- 2004-12-21 CA CA002549385A patent/CA2549385A1/en not_active Abandoned
- 2004-12-21 AU AU2004311909A patent/AU2004311909A1/en not_active Abandoned
- 2004-12-21 WO PCT/US2004/039775 patent/WO2005065683A1/en not_active Ceased
- 2004-12-21 CN CNA2004800383003A patent/CN1909902A/zh active Pending
- 2004-12-21 EP EP04812321A patent/EP1725231A1/en not_active Withdrawn
- 2004-12-21 JP JP2006547018A patent/JP2007515484A/ja active Pending
- 2004-12-21 US US10/580,202 patent/US7507832B2/en not_active Expired - Fee Related
- 2004-12-21 BR BRPI0417947-1A patent/BRPI0417947A/pt not_active IP Right Cessation
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102076671A (zh) * | 2008-07-03 | 2011-05-25 | 安斯泰来制药有限公司 | 三唑衍生物或其盐 |
| US8377923B2 (en) | 2008-07-03 | 2013-02-19 | Astellas Pharma Inc. | Triazole derivative or salt thereof |
| CN102076671B (zh) * | 2008-07-03 | 2013-05-15 | 安斯泰来制药有限公司 | 三唑衍生物或其盐 |
| CN102365273A (zh) * | 2009-01-28 | 2012-02-29 | 赛诺菲 | 噻二唑与*二唑衍生物及其制备与其治疗用途 |
| CN101863852A (zh) * | 2010-06-29 | 2010-10-20 | 沈阳航空航天大学 | 含1,3,4-噁二唑杂环结构芳香二元胺及其制备方法 |
| CN101863852B (zh) * | 2010-06-29 | 2011-09-21 | 沈阳航空航天大学 | 含1,3,4-噁二唑杂环结构芳香二元胺及其制备方法 |
| CN103467399A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其用途 |
| CN103554043A (zh) * | 2013-09-03 | 2014-02-05 | 浙江医药高等专科学校 | 苯基取代的三唑酰胺类化合物及用途 |
| CN103467409A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 取代的四氮唑羧酸类化合物及其用途 |
| CN103467405A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 一类四氮唑羧酸类化合物、其制备方法和用途 |
| CN103467407A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 四氮唑羧酸类化合物及其用途 |
| CN103467398A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其抗糖尿病用途 |
| CN103467406A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 卤素取代的四氮唑羧酸类化合物、其制备方法和用途 |
| CN103483281A (zh) * | 2013-09-03 | 2014-01-01 | 浙江医药高等专科学校 | 一类三唑酰胺类化合物、其制备方法和其抗糖尿病用途 |
| CN103483282A (zh) * | 2013-09-03 | 2014-01-01 | 浙江医药高等专科学校 | 苯基取代的三唑酰胺类化合物及用途 |
| CN103467408A (zh) * | 2013-09-03 | 2013-12-25 | 浙江医药高等专科学校 | 一类四氮唑羧酸类化合物及其用途 |
| CN103467406B (zh) * | 2013-09-03 | 2014-12-17 | 浙江医药高等专科学校 | 卤素取代的四氮唑羧酸类化合物、其制备方法和用途 |
| CN103467399B (zh) * | 2013-09-03 | 2015-01-07 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其用途 |
| CN103467408B (zh) * | 2013-09-03 | 2015-01-21 | 浙江医药高等专科学校 | 一类四氮唑羧酸类化合物及其用途 |
| CN103467398B (zh) * | 2013-09-03 | 2015-01-21 | 浙江医药高等专科学校 | 三唑酰胺类化合物、其制备方法和其抗糖尿病用途 |
| CN103554043B (zh) * | 2013-09-03 | 2015-04-01 | 浙江医药高等专科学校 | 苯基取代的三唑酰胺类化合物及用途 |
| CN103467407B (zh) * | 2013-09-03 | 2015-04-22 | 浙江医药高等专科学校 | 四氮唑羧酸类化合物及其用途 |
| CN106632125A (zh) * | 2016-10-08 | 2017-05-10 | 李纪焕 | 一种烷基芳基取代噁二唑化合物的三组分合成方法 |
| US12220412B2 (en) | 2018-11-20 | 2025-02-11 | Sparrow Pharmaceuticals, Inc. | Methods for administering corticosteroids |
| US12329745B2 (en) | 2022-05-16 | 2025-06-17 | Sparrow Pharmaceuticals, Inc. | Methods and compositions for treating glucocorticoid excess |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005065683A1 (en) | 2005-07-21 |
| AU2004311909A1 (en) | 2005-07-21 |
| US7507832B2 (en) | 2009-03-24 |
| US20070112045A1 (en) | 2007-05-17 |
| BRPI0417947A (pt) | 2007-04-17 |
| JP2007515484A (ja) | 2007-06-14 |
| EP1725231A1 (en) | 2006-11-29 |
| CA2549385A1 (en) | 2005-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1909902A (zh) | 用于治疗糖尿病的作为ppar调节剂的三唑、噁二唑和噻二唑衍生物 | |
| CN1157368C (zh) | 人过氧化物酶体增殖剂活化受体(PPAR)α激动剂-取代苯基丙酸衍生物 | |
| CN1093124C (zh) | 对PPAR-γ具有激动剂活性的取代的4-羟基-苯基alcanoic acid衍生物 | |
| CN1293042C (zh) | 芳香族氨基酸衍生物及其药物组合物 | |
| CN1293076C (zh) | 过氧化物酶体增殖剂应答性受体δ的活化剂 | |
| CN1215059C (zh) | 新的杂环化合物及其盐和它们的医药用途 | |
| CN1058969C (zh) | 新的取代胍衍生物,其制备方法和其药物用途 | |
| CN1095839C (zh) | 4,5-二芳基噁唑衍生物 | |
| CN1235870C (zh) | 苯基杂烷基胺衍生物的新应用 | |
| CN1578659A (zh) | 过氧化物酶体增殖剂激活的受体(ppar)的调节剂 | |
| CN1649820A (zh) | 调节ppar活性的化合物及其制备方法 | |
| CN101048402A (zh) | 咔唑衍生物、其溶剂合物或其药学上允许的盐 | |
| CN1681763A (zh) | 化合物 | |
| CN101035774A (zh) | 用于治疗前列腺素d2介导的疾病的crth2受体活性调节剂 | |
| CN1678578A (zh) | 具有抗糖尿病活性的吲哚化合物 | |
| CN1544420A (zh) | 用取代杂环脲抑制raf激酶 | |
| CN1671659A (zh) | 新的取代吲哚 | |
| CN1638768A (zh) | 调节ppar活性的噻唑和噁唑衍生物 | |
| CN1147250A (zh) | 作为前列腺素i2激动剂的萘衍生物 | |
| CN1070173C (zh) | 用作药物的苯甲酰基胍衍生物 | |
| CN1674892A (zh) | 杂联芳基衍生物作为基质金属蛋白酶抑制剂 | |
| CN1761657A (zh) | Ep4受体拮抗剂 | |
| CN1780822A (zh) | 2,4-二氢-[1,2,4]三唑-3-硫酮衍生物作为酶髓过氧化物酶(mpo)抑制剂的用途 | |
| CN1558897A (zh) | 口服抗糖尿病剂 | |
| CN1182129C (zh) | 具有白细胞三烯拮抗作用的苯并吡喃衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Open date: 20070207 |