CN86106902A - 9-(取代硫基)-4H-吡啶并[1,2-a]嘧啶-4-酮衍生物 - Google Patents
9-(取代硫基)-4H-吡啶并[1,2-a]嘧啶-4-酮衍生物 Download PDFInfo
- Publication number
- CN86106902A CN86106902A CN86106902.1A CN86106902A CN86106902A CN 86106902 A CN86106902 A CN 86106902A CN 86106902 A CN86106902 A CN 86106902A CN 86106902 A CN86106902 A CN 86106902A
- Authority
- CN
- China
- Prior art keywords
- group
- compound
- formula
- alkyl
- gram
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
可用作抗溃疡药的9-(取代硫基)-4H-吡啶并[1,2-a]嘧啶-4-酮衍生物,结构式(I),其中n为0或1,R为-COR1,-CONR4R5或-CH2R6,R1为C1-C5烷基,C3-C7环烷基,丙烯基硫基,苯乙烯基,苯氧甲基,噻吩甲基,任意取代的C6-C10芳基,任意取代的苄基或任意取代的5元或6元杂环基,R2和R3各自为氢,C1-C6烷基,羧基,C2-C6烷氧羰基,或任意取代的苄氧羰基,R4和R5各自为氢,C1-C5烷基,C3-C7环烷基或任意取代的苯基,R6是任意取代的吡啶基或苯基。
Description
本发明涉及9-(取代硫基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮衍生物。本发明尤其针对已被发现对治疗消化性溃疡特别有效的9-(取代硫基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮衍生物的制备、用途及含有该类化合物的药物配方。
美国专利NO.4,022,897公开2-烷基-9-(取代氧)-4H-吡啶并〔1,2-a〕嘧啶-4-酮作为中枢神经系统兴奋剂,美国专利NO.4,122,274和4,209,620描述3-(1H-四唑-5基)-4H吡啶并〔1,2-a〕嘧啶-4-酮类化合物作为抗过敏药,美国专利NO.4,457,932公开用上述的3-(1H-四唑-5基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮类化合物作为抗溃疡药。
本发明的9-(取代硫基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮衍生物是具有取代硫基而无1H-四唑-5基的那些化合物,因此,与上述文献中公开的化合物完全不同。
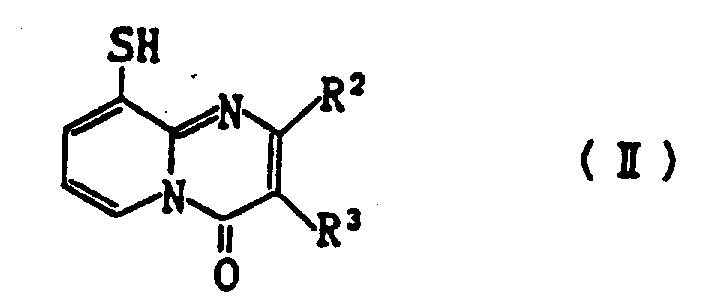
根据本发明,提供了式(Ⅰ)所示的9-(取代硫基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮衍生物或其盐。

(其中n为0或1;
R为-COR1,
或-CH2R6
R1为C1-C3烷基、C3-C7环烷基、烯丙硫基,苯乙烯基、苯氧甲基、噻吩甲基、可由选自C1-C3烷基、C3-C6碳原子的环烷基、C1-C3烷氧基、苯基-C1-C3烷氧基、卤素、硝基、C1-C3链烷磺酰基、C2-C5烷氧羰基、氰基、乙酰氧基、乙酰基、四唑基、三氟甲基和氨磺酰基中一个或多个基团取代的C6-C10芳基、可由选自C1-C3烷基、C1-C3烷氧基、囟素和硝基中一个或多个基团取代的苄基、或可由选自C1-C3烷基、C1-C3烷氧基和C3-C6环烷基中一个或多个基团取代的5元或6元杂环基团;
R2和R3各自为氢、C1-C5烷基、羧基、C2-C5烷氧羰基或可由选自C1-C3烷基、C1-C3烷氧基和卤素中一个或多个基团取代的苄氧羰基;
R4和R5各自为氧、C1-C5烷基、C3-C7环烷基或可由选自囟素、C1-C3烷基、C1-C3烷氧基、C2-C5烷氧羰基、硝基和三氟甲基中一个或多个基团取代的苯基;
R6为可由选自卤素、C1-C3烷基和C1-C3烷氧基中一个或多个基团取代的吡啶基或苯基。
本发明化合物具有极好的抗溃疡活性,对人体没有不良的作用。因此,本发明也提供一种抗溃疡配方,该配方含有至少与一种载体、稀释剂或赋形到结合的式(Ⅰ)所示化合物,它作为活性成份占重量的0.1到95%。
本发明也提供一种治疗消化性溃疡的方法,该法包含向病人给以药理学上有效剂量的式(Ⅰ)化合物。
本发明进一步提供式(Ⅰ)所示化合物的制备方法。它包括:
(A)将式(Ⅱ)所的化合物与式(Ⅲ)所示的化合物反应

得式(Ⅰa)所示的化合物。

(B)将式(Ⅱ)所示的化合物与N、N′-羰基二咪唑反应得式(Ⅳ)所示的化合物,

再将式(Ⅳ)化合物与式(Ⅴ)所示的化合物反应,
得式(Ⅰb)所示的化合物。

(C)将式(Ⅱ)所示的化合物与式(Ⅵ)所示的化合物反应,
得式(Ⅰc)所示的化合物,

最后氧化产物(Ⅰc),得式(Ⅰd)所示的9-(取代亚硫酰基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮

或(D)将式(Ⅱ)所示的化合物与式(Ⅶ)所示的化合物反应,
得式(Ⅰe)所示的化合物。

(其中X为囟素,羟基或羟基的活化酯基;R1、R2、R3、R4和R5如上述定义)。
这里所采用的术语“C1-C5烷基”是指直链或支链的饱和脂肪族烃基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲-丁基、叔-丁基、戊基、异戊基、新戊基、叔-戊基或1-甲基异丁基,其中甲基和乙基最好。
术语“C3-C7环烷基”包括环丙基、环丁基、环戊基、环乙基和环庚基。
术语“C6-C10芳基”包括苯基和萘基。
术语“C1-C3烷氧基”是指烷基部分含有C1-C3烷基部分的烷氧基,包括甲氧基、乙氧基和异丙氧基。术语“C2-C5烷氧羰基”是指烷基部分含有C1-C4烷基部分的烷氧羰基,包括甲氧羰基、乙氧羰基、丙氧羰基和丁氧羰基。
术语“C1-C3链烷磺酰基”包括甲磺酰基、乙磺酰基、丙磺酰基和异丙磺酰基。
术语“卤素”包括氟、氯、溴和碘。
术语“5-或6-元杂环基团”指含有一个或两个杂原子的5-或6-元环如氮杂、氧杂或硫杂,包括呋喃基、噻吩基、异恶唑基、恶唑基、噻唑基和吡啶基部分。
羟基活化酯包括无机酸酯如硫酸酯或磷酸酯和有机酸酯如甲磺酸酯、甲苯磺酸酯、乙氧碳酸酯和三氟甲磺酸酯。
制备化合物(Ⅰ)的方法将在下面详述。
方法A
本发明化合物(Ⅰa)可通过化合物(Ⅱ)与化合物(Ⅲ)反应制备。因而,将9-巯基-4H-吡啶并〔1,2-a〕嘧啶(Ⅱ)和异氰酸酯(Ⅲ)在约0℃到80℃温度范围内,最好在室温(10-30℃)反应1到10小时。
该反应通常在溶剂中进行,如二氯甲烷、1,2-二氯乙烷、氯仿、四氯化碳、环戊烷、环乙烷、正-己烷、苯、乙酸乙酯、乙腈、四氢呋喃、丙酮、甲乙酮等。
异氰酸酯(Ⅲ)可用相应的胺与光气〔Slocombe等.,J.Am.Chem.Soc.,72,1888(1950年)〕或与草酰氯〔Ulrich等.,J.org.Chem.,34,3200(1969年)〕反应制得。
方法B
化合物(Ⅰb)可通过化合物(Ⅱ)与N,N′-羰基二咪唑反应得咪唑基化合物(Ⅳ),然后再将化合物(Ⅳ)与胺(Ⅴ)反应。
首先,化合物(Ⅱ)的羰基化反应在约0℃到约100℃温度范围内,最好在恒温左右并在一合适的溶剂中进行,例如二氯甲烷、1,2-二氯乙烷,氯仿、四氯化碳、苯、四氢呋喃、乙腈、乙酸乙酯等。
其次,将得到的咪唑基化合物(Ⅳ)与胺(Ⅴ)反应,该反应从室温到约100℃温度范围内;并在上步反应反叙述的溶剂中进行。
方法C
化合物(Ⅰc)可通过化合物(Ⅱ)与卤化物(Ⅵ)反应制得。因而,将9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮(Ⅱ)与囟化物(Ⅵ)在碱存在下于一合适溶剂中在约10℃到约100℃温度范围内进行反应。
作为碱,举例有无机碱,例如碱金属或碱土金属氢氧化物如氢氧化钠、氢氧化钾和氢氧化钙;碱金属碳酸盐如碳酸钠和碳酸钾;有机碱如三乙胺、N-甲基吡咯烷、N-乙基哌啶,吗啉,吡啶,甲基吡啶和二甲基吡啶。溶剂的例子是甲醇、乙醇、异丙醇、丙醇、甲乙酮、乙醚、四氢呋喃、二甲基甲酰胺、二甲基乙酰胺和二甲基亚砜。
最后,上面得到的化合物(Ⅰc)在约-50℃到10℃温度范围内用过氧化物进行氧化。氧化反应可在一合适的溶剂如氯仿、二氯甲烷、1,2-二氯乙烷、四氧化碳、苯或甲苯中进行,根据所用过氧化物的特性,可对溶剂作适当的选择。过氧化物包括氢过氧化物如过氧化氢、乙基过氧化氢、叔-丁基过氧化氢和过酸如过乙酸、过苯甲酸和3-氯过苯甲酸。当使用氢过氧化物时,可通过加硫酸、盐酸、对-甲苯磺酸、甲磺酸或氯化铝来加速反应。
方法D
化合物(Ⅰe)可通过化合物(Ⅱ)与化合物(Ⅶ)反应制备。因而可将9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮(Ⅱ)与酰化剂(Ⅶ)进行反应。
酰化反应可在约0℃到80℃温度范围内,最好在室温左右,在碱存在下于一合适溶剂中按常规方式进行。溶液的例子是二氯甲烷、1,2-二氯乙烷、氯仿、环乙烷、正-己烷、苯、丙酮、乙腈、四氢呋喃等,方法C中列举的碱也可被用在方法D中。
或者,化合物(Ⅱ)与羧酸(Ⅶ)的酰化反应也可通过在一种脱水剂如Dcc等存在下,在一溶剂中进行反应而完成。进一步,与羧酸(Ⅶ)和氯碳酸低级烷基酯-三乙胺的酰化反应可在溶剂中于0℃到100℃下进行,这些反应均可以常规方式进行。
此外,化合物(Ⅰe)可通过化合物(Ⅰb)与酰基囟(Ⅶ)在路易斯酸(Lewis acid),例如碘化锌、氯化锌、硼酸三乙酯或氯化铝存在下进行反应制取,该反应可在一合适溶剂中如1,2-二氯乙烷、二氯甲烷或甲苯中,通过在所用溶剂的沸点温度附近加热的方法进行。
起始化合物(Ⅱ)的制备
起始化合物(Ⅱ)可按下列所示合成方法制备。
可用Saul Patail所著“巯基的化学”,Interscience出版,P163-269(1974年)中所叙述的任何一种合成方法引入硫巯基。把羟基转换成巯基将在下面举例说明。
原料合成方法(1)
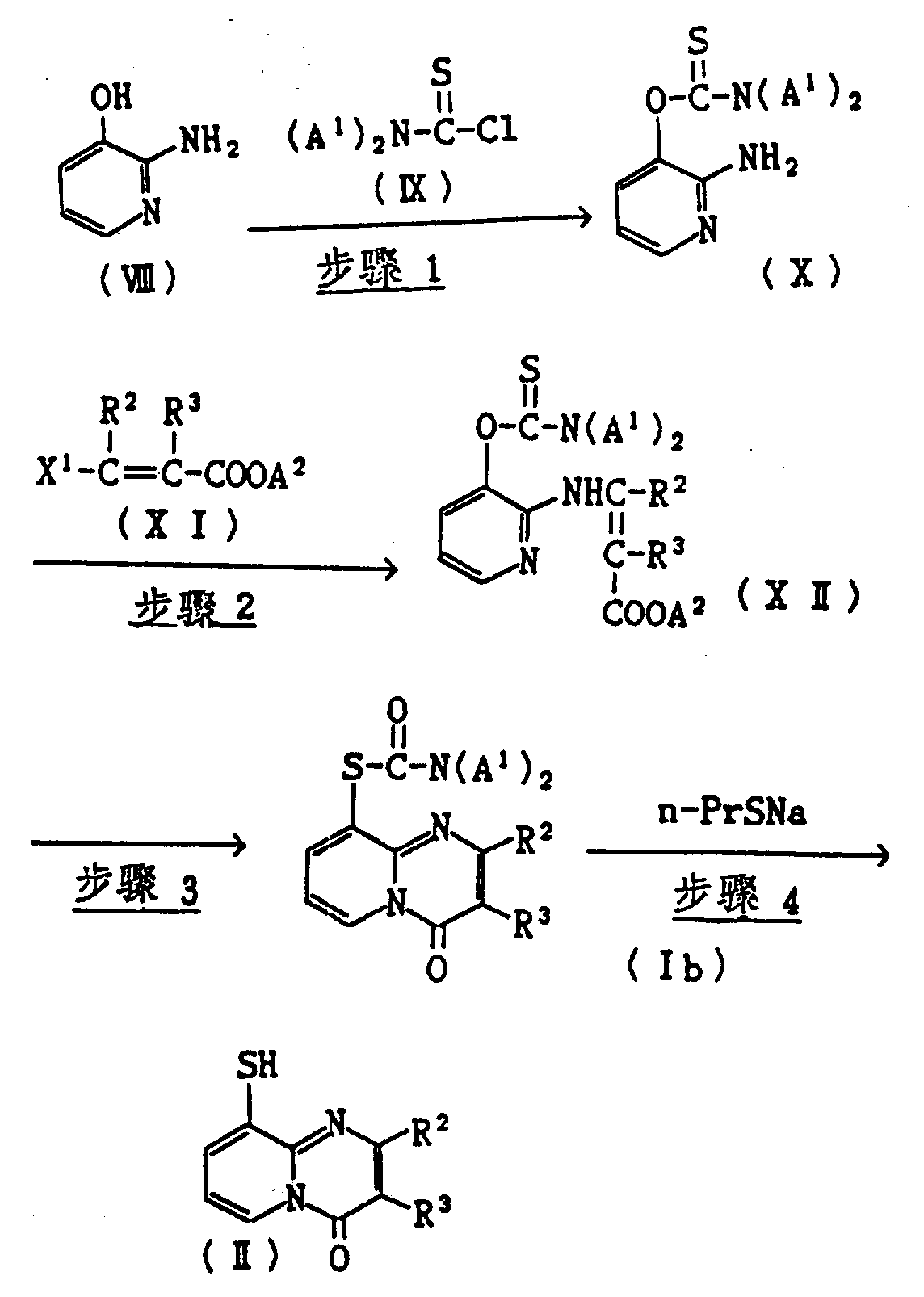
(其中A1和A2各为低级烷基,X1为一离去基团如囟素、氨基、低级烷氧基、低级烷硫基、甲磺酰氧基或甲苯磺酰氧基,R2和R3如上述定义。)
步骤1
将起始物2-氨基-3-羟基吡啶(Ⅷ)在碱存在下与硫代氨甲酰氯(Ⅸ)反应得化合物(Ⅹ)。此反应在0-80℃进行,最好在室温并在溶剂中例如丙酮、甲乙酮、乙酸乙酯、四氢呋喃、甲苯、乙醚、二恶烷、二甲基亚砜、二甲基甲酰胺或二甲基乙酰胺中进行。碱的例子是方法C的步骤1中所叙述的那些碱。
步骤2
将2-氨基-3-硫代氨甲酰基氧吡啶(Ⅹ)在从室温到约150℃温度范围内,最好在约100-120℃,与酯(Ⅺ)反应得烯胺(Ⅻ)。此反应可在溶剂中或没有溶剂存在下进行,如果使用溶剂,则包括非质子传递溶剂如乙醚、四氢呋喃、二恶烷、二甲基甲酰胺、苯和甲苯。如果需要,也可加方法C中所列举的碱来加速反应。
试剂(Ⅺ)可用R.M.Carlson等.,Tetrahdedron Letters,4819(1973年)和G.H.Posner等.,chem.Commun.907(1973年)中公开的方法或在这两篇文献中所引用的方法制备。
步骤3
由于使烯胺(Ⅻ)进行这步反应,所以通过闭环和同时进行O→S重排来制备混合物(Ⅰb),通过在溶剂中例如甲苯、二甲苯或沸点高于100℃,最好是高于200℃的二苯醚中回流,反应将在几分钟内结束。
步骤4
将化合物(Ⅰb)与正-丙基硫醇钠反应得化合物(Ⅱ)。反应在0到80℃,最好是0℃到室温在溶剂例如四氢呋喃、二恶烷、乙醇、甲苯、二甲基甲酰胺或二甲基亚砜中进行。
原料合成方法(2)

(其中A1、A3、R2、R3和X1如上述定义。)
步骤1
将2-氨基-3-羟基吡啶(Ⅷ)在酸性缩合剂存在下与β-氧化羧酸酯(ⅪⅤ)反应,得化合物(ⅩⅤ)。该反应在50℃到150℃范围,最好在80到120℃内进行。酸性缩合剂包括多磷酸、乙酸和丙酸等。如果需要,可以加任何溶剂如水、甲醇、乙醇、异丙醇、正-丁醇等。
β-氧化羧酸酯(ⅪⅤ)可用C.R.Hauser等.,Orga-nic Reactions,1,266(1942)中公开的方法制备。
或者,9-羟基-4H-吡啶并〔1,2-a〕嘧啶-4-酮(ⅩⅤ)也可用2-氨基-3-羟基吡啶(Ⅷ)与酯(Ⅺ)反应制得。反应按合成方法(1)步骤2进行。
步骤2
反应按合成方法(1)步骤1进行。
步骤3
反应在100到170℃于溶剂中如甲苯、二甲苯、苯甲醚、二甘醇成四氯乙烷,按合成方法(1)步骤3进行。
步骤4
反应按合成方法(1)步骤4进行。
本发明的目的化合物(Ⅰ)能被转换成其盐。根据取代基的种类和其它条件,(Ⅰ)可被转换成碱金属盐(如锂盐、钠盐、钾盐等)或碱土金属盐(如钙盐、镁盐等)。目的化合物(Ⅰ)最后能被更进一步转换成它的酸加成盐,能用于这种目的的酸包括无机酸如盐酸、氢溴酸和磷酸;有机酸如乙酸、草酸、马来酸、富马酸、柠檬酸、平果酸、乙二酸和琥珀酸。
本发明的目的化合物(Ⅰ)和/或它的盐对人或动物可以口服或胃肠外给药。例如,化合物(Ⅰ)可以片、颗粒、散剂、胶囊或液体剂型口服和以注射剂或栓剂形式胃肠外给药。
这些制剂可用稀释剂、粘合剂、崩解剂、润滑剂、稳定剂、悬浮剂、分散剂、助溶剂、防腐剂等按常规方法制备。
稀释剂例子包括乳糖、蔗糖、淀粉、纤维素、山梨醇等;粘合剂包括阿接伯胶、明胶、聚乙烯吡咯烷酮等;润滑剂包括硬脂酸镁、滑石粉、硅胶等。
当本发明化合物(Ⅰ)用于治疗成人消化性溃疡时,化合物(Ⅰ)的剂量为1-100毫克/公斤,每天1次或分次口服或胃肠外给药。
本发明将通过以下实施例、参考例和配方作更详细的说明。
Me=甲基:Et=乙基;n-pr=正-丙基;t-Bu=叔-丁基;CH2Cl2=二氯甲烷;CHCl3=氯仿1;AcOEt=乙酸乙酯;THF=四氢呋喃,K2CO3=碳酸钾。
DMF=二甲基甲酰胺;NaH=氢化钠(60%油状悬浮体);m-CPBA=3-氯过苯甲酸;(d)=分解点。
实施例1
3-乙氧羰基-9-(4-甲基苯-氨基甲酰基硫)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰa-1的制备

将0.35克4-甲基苯基异氰酸酯Ⅲ-1加到0.4克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1在10毫升干燥二氯甲烷的悬浮液中,得到的混合物于室温搅拌4小时,此时一旦反应物溶解后,反应混合物再次结晶。
然后将产物过滤,用乙酸乙酯洗并从乙酸乙酯中重结晶,得标题化合物Ⅰa-10.441克。
收率:72%
烷点:170-172℃
分析计算值:C19H17O4N3S
C,59.52;H,4.47;N,10.96;S,8.36(%)
实验值:C,59.52;H,4.45;N,10.84;S,8.47(%)。
实施例2-19
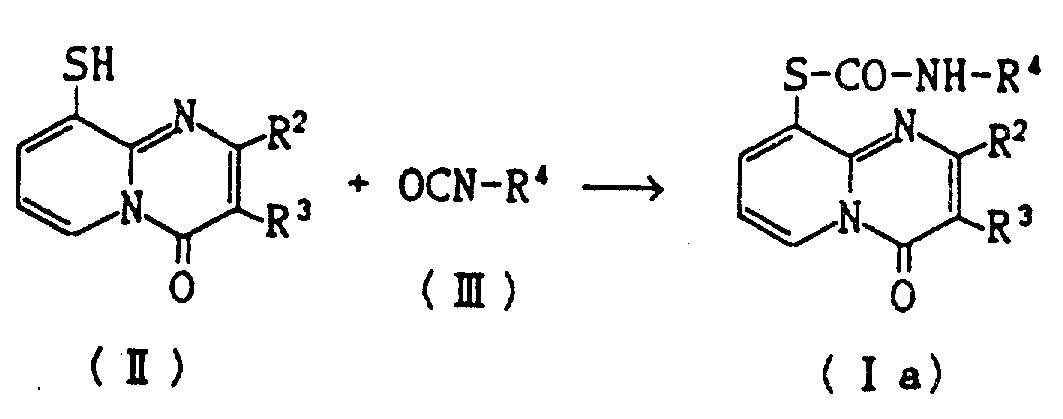
将异氰酸酯(Ⅲ)加到9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮(Ⅱ)在合适溶剂的悬浮液中,得到的混合物在室温搅拌约1-5小时。将析出的结晶过滤,用乙酸乙酯洗并从乙酸乙酯中重结晶得目的化合物(Ⅰa)。
表1指出制备化合物(Ⅰa)的反应条件(即反应物结构和用量、溶剂、反应时间等),以及产物(Ⅰa)的结构和它们的物理常数(即熔点和元素分析)。

实施例20
3-乙氧羰基-9-(N-乙基-N-苯基-氨基甲酰基硫)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰb-20的制备
将0.3克N,N′-羰基二咪唑于搅拌下加到0.413克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1在10毫升二氯甲烷的悬浮液中,得到的混合物搅拌1小时。将反应混合物与0.2克N-乙基苯胺Ⅴ-1混合并再搅拌16小时。反应结束后,减压蒸去溶剂,残余物在硅胶柱上用色谱法纯化,得0.34克标题化合物Ⅰb-20,为一粘稠油状物。
核磁共振(CDCl3)δ:1.11(3H,t,J=7Hz),1.27(3H,d,J=7Hz),3.22(2H,q,J=7Hz),4.15(2H,q,J=7Hz),6.80-7.80(8H,m),7.96(1H,s)。
质谱,M+(m/e):397
实施例21
(1)3-乙氧羰基-9-苄硫基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰc-21的制备

将0.3克苄基溴Ⅵ-Ⅰ和0.5克碳酸钾加到0.3克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1在4毫升无水二甲基甲酰胺的悬浮液中,得到混合物在室温搅拌5小时,用水将反应混合物稀释,过滤析出的结晶并将其溶于氯仿中氯仿溶液在硫酸钠上干燥,蒸发,得0.389克标题化合物Ⅰc-21。
收率:95.3%
熔点:173-174℃(从乙酸乙酯中重结晶)
分析计算值:C18H16O3N2S
C,63.51;H,4.74;N,8.23;S,9.42(%)
实验值:C,63.48;H,4.63;N,8.17;S,9.30(%)
(2)3-乙氧羰基-9-苄基亚硫酰基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰd-22的制备
将上述项(1)中提供的3-乙氧羰基-9-苄巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰc-21在25毫升氯仿中的溶剂冷到0℃,在搅拌下,加0.5克固态同一氯过苯甲酸,将混合物在同样温度下加六4小时,反应混合物依次用5%硫代硫酸钠水溶液、5%碳酸氢钠水溶液和水洗涤,将得到的混合物在无水硫酸钠上干燥后,把溶剂蒸发掉,残余物(1克)用快速色谱法进行纯化,用乙酸乙酯洗脱,将从洗脱液中制得的固体用乙酸乙酯重结晶,得标题化合物(Ⅰb-22)0.6克。
收率:81.9%
熔点:175-176℃
分析计算值:C18H16O1N2S
C,60.66;H,4.53;N,7.86;S,9.00(%)
实验值:C,60.72;H,4.64;N,7.75;S,8.80(%)
实施例22-23
(1)将氯化物(Ⅵ)和碳酸钾加到3-乙氧羰基-9-巯基-

4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1无水二甲基甲酸胺的悬浮液中,将混合物在室温搅拌数小时,反应混合物用水稀释,过滤析出的结晶并将它溶在CHCl3中,得到的混合物在无水硫酸钠上干燥,从混合物中蒸去溶剂得化合物(Ⅰc)。
表2指出制备化合物(Ⅰc)的反应条件(即反应物的结构和用量,溶剂,反应时间等),以及产物(Ⅰc)的结构和它们的物理常数(即熔点和元素分析)。
(2)在冷却和搅拌下,将间-氯过苯甲酸加到化合物(Ⅰc)的氯仿溶液中,将得到的混合物在同样温度下搅拌数小时,所应混合物依次用5%硫代硫酸钠水溶液、5%碳酸氢钠水溶液和水洗涤。得到的混合物经在无水硫酸钠上干燥之后,将溶剂蒸去,残余物用快速色谱法纯化,用醋酸乙酯洗脱,从洗脱液中得到的固体用醋酸乙酯重结晶,得化合物(Ⅰd)。
表3指出制备化合物(Ⅰd)的反应条件(即反应物的结构和用量、溶剂、反应时间等),以及产物(Ⅰd)的结构和它们的物理常数(即熔点和元素分析)。
实施例24
3-乙氧羰基-9-〔(4-正-丁氧基-苯甲酰基)硫基〕-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-2〕的制备
将0.7克4-正-丁氧基-苯甲酰氯在搅拌下加到0.8克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1、0.5克粉末状碳酸钾和25毫升丙酮的混合物中,将得到的混合物在室温搅拌2小时,过滤生成的沉淀并将它分散在水-氯仿中,氯仿层用饱和盐水洗涤,在无水硫酸钠上干燥并浓缩。将残余物(1.1克)在硅胶柱上用色谱法纯化,用乙酸乙酯洗脱,洗脱液浓缩得0.7克标题化合物。
收率:51%
分析计算值:C22H22O5N2S
C,61.96;H,5.20;N,6.57;S,7.52(%)

实验值:C,61.73;H,5.22;N,6.47;S,7.65(%)
实施例25-50

将一种合适的碱加到9-巯基-4H-吡啶并-〔1,2-a〕嘧啶-4-酮(Ⅱ)在合适溶剂的悬浮液或溶液中,然后再加酰基氯(Ⅶ),将生成的混合物在室温搅拌约1到5小时,反应混合物在真空中浓缩到干,将残余物分散在水-氯仿中,有机层用水洗涤、干燥并浓缩。残余物通过用溶剂重结晶或进行色谱纯化,从而得目的化合物(Ⅰe)。
表4指出制备化合物(Ⅰe)的反应条件(即反应物结构和用量,溶剂,反应时间等)以及产物(Ⅰe)的结构和物理常数。
实施例51
3-乙氧羰基-9-〔〔(5-甲基异噁唑-3-基)-羰基〕硫基〕-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-54的制备


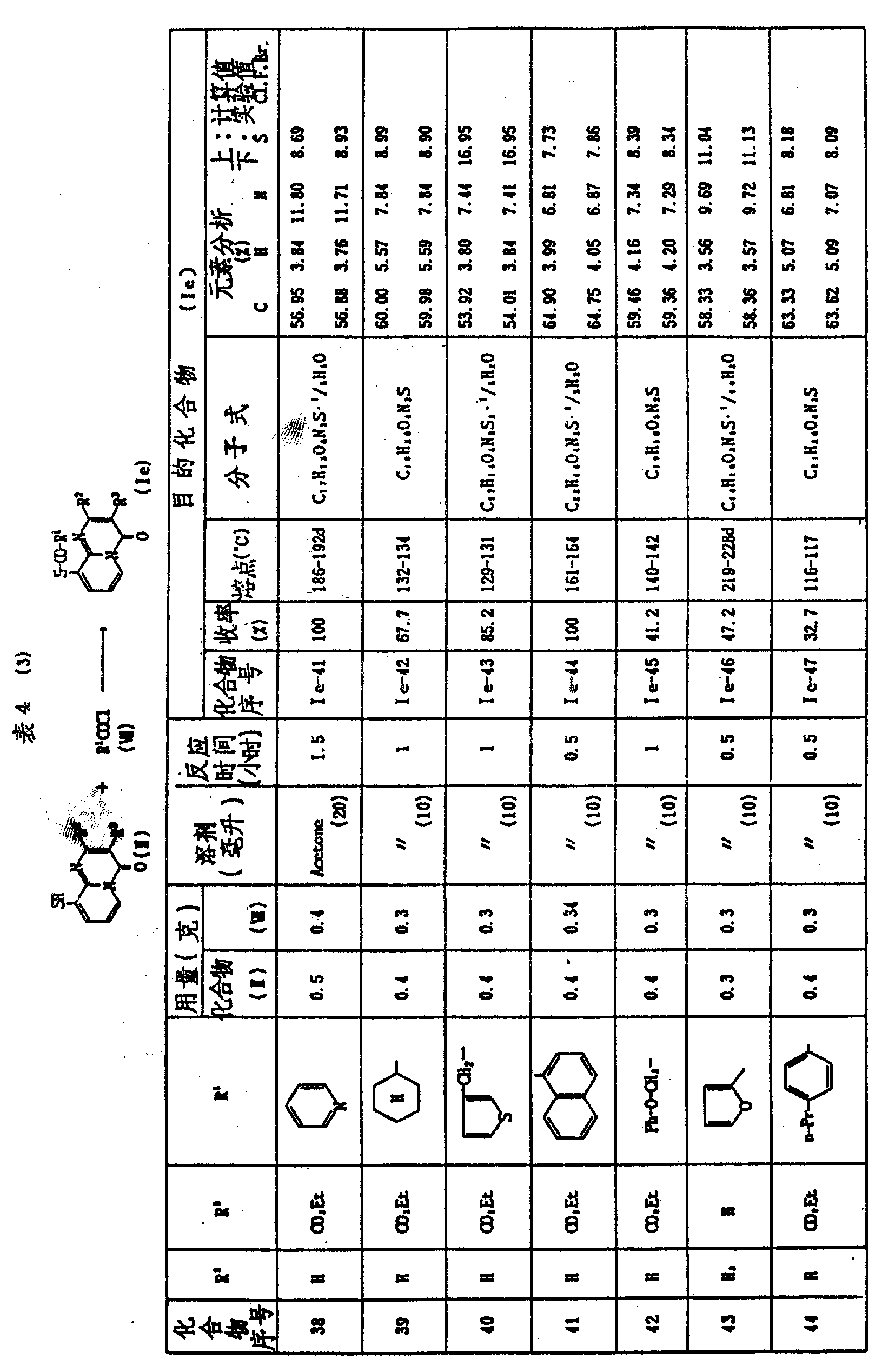

将0.39克亚硫酰氯加到0.23克3-羧基-5-甲基异噁唑在5毫升干燥苯的溶液中,然后加3滴二甲基甲酰胺。将生成的混合物在油浴上回流加热5小时,并真空浓缩。将残余物溶于10毫升丙酮,并与1.1克粉末状碳酸钾和0.4克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1混合,混合物在室温搅拌1小时,将反应混合物真空浓缩,残余物在硅胶柱上用色谱法纯化,用乙酸乙酯洗脱,将洗脱液浓缩得0.2克标题化合物。
收率:33.8%
熔点:147-150℃(从氯仿-正-己烷中重结晶)
分析计算值:C16H13O5N3S·1/2H2O
C,52.17;H,3.93;N,11.41;S,8.71(%)
实验值:C,52.42;H,3.69;N,11.32;S,8.87(%)
实施例52
3-乙氧羰基-9-〔〔(5-环丙基异噁唑-3-基羰基〕硫〕-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-55的制备
将0.39克亚硫酰氯加到0.28克3-羧基-5-环丙基异噁唑在5毫升干燥苯的溶液中,然后加3滴二甲基甲酰胺。将生成的混合物在油浴上回流内加热5小时,并真空浓缩。将残余物溶于10毫升丙酮,并与1.1克粉末状碳酸钾和0.4克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮混合,将生成的混合物在室温搅拌1小时并真空浓缩。将残余物溶于氯仿并过滤此溶液,以除去不溶解的物质,滤液真空浓缩到干,残余物从氯仿-正-己烷中重结晶得0.32克标题化合物Ⅰe-55。
收率:52%
熔点:189-191℃(分解)
C16H15O5N3S·1/5H2O
计算值:C,55.57;H,3.99;N,10.80;S,8.24(%)
实验值:C,55.53;H,3.99;N,10.72;S,8.17(%)
实施例53
3-乙氧羰基-9-(4-乙氧羰基-苯甲酰基)硫-4-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-56的制备

将0.52克(4.4毫摩尔)亚硫酰氯加到1.47克(2.4毫摩尔)4-乙氧羰基-苯甲酸、3滴二甲基甲酰胺和10毫升苯的混合物中。将得到的混合物在搅拌下回流加热3小时然后真空浓缩到干。将残余物溶于丙酮并与1.4克固体碳酸钾然后与0.5克(2毫摩尔)3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1混合,将生成的混合物在室温强烈搅拌20分钟。将反应混合物真空浓缩到干,并将残余物分散在氯仿-水中,有机层在硫酸钠上干燥并真空浓缩。将残余物在硅胶柱上用色谱法纯化,用乙酸乙酯洗脱,得标题化合物Ⅰe-560.9克。
收率:约100%
熔点:146-148℃(氯仿-正-己烷)
C21H18N2O6S
计算值:C,59.14;H,4.25;N,6.57;S,7.52;
实验值:C,59.92;H,4.23;N,6.55;S,7.44。
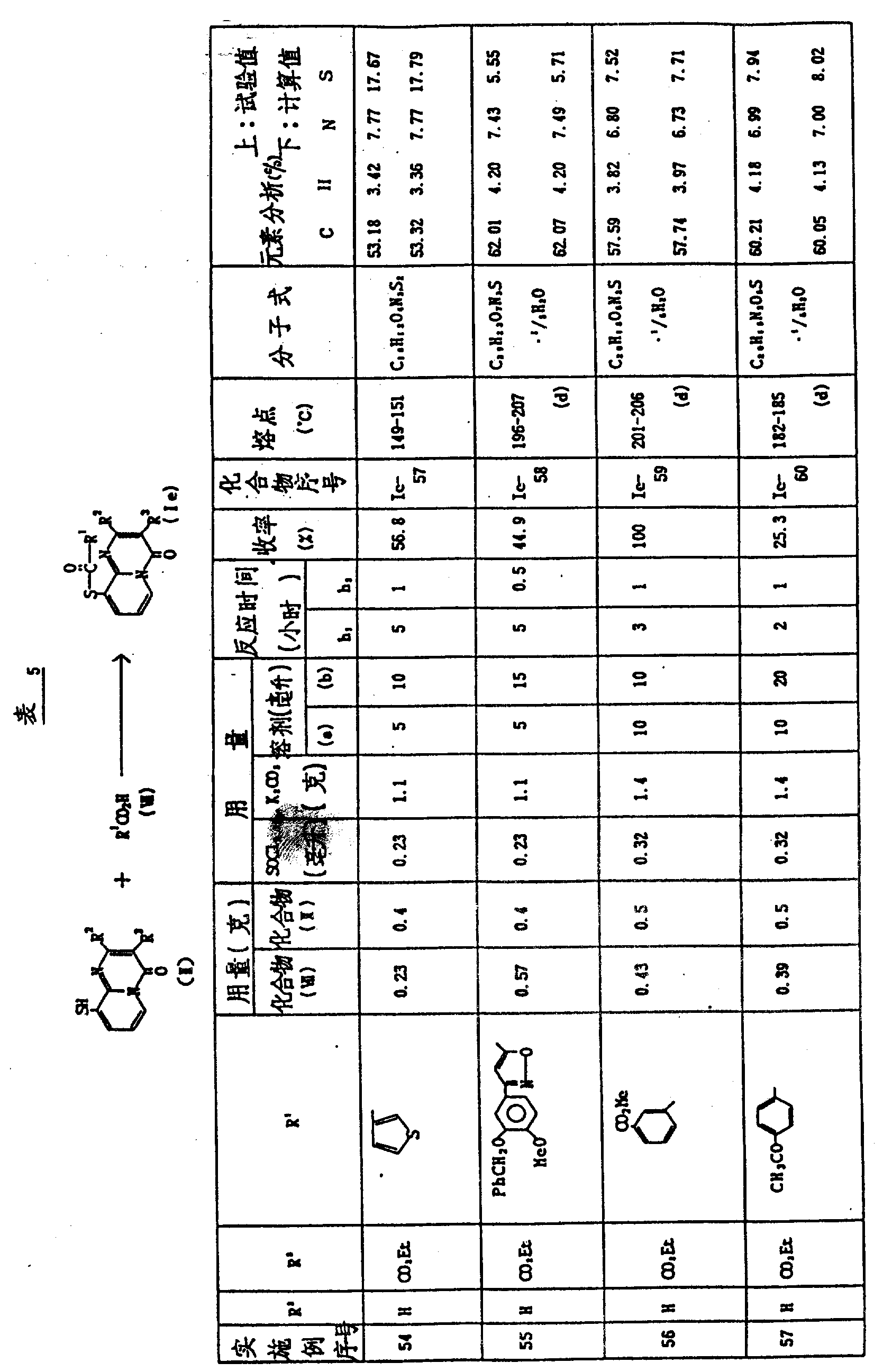
实施例54-57

将催化量的二甲基甲酰胺加到取代芳基-羧酸(Ⅶ)在一合适溶剂(溶剂a)的悬浮液或溶液中,然后加亚硫酰氯,得到的混合物回流加热约1-5小时(反应时间h1),然后真空浓缩到干。将残余物溶在合适的溶剂中(溶剂b),与合适的碱和3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕-嘧啶-4-酮(Ⅱ-Ⅰ)混合,得到的混合物在室温搅拌约0.5-3小时(反应时间h2),反应混合物真空浓缩到干,将残余物分散在氯仿-水中,有机层用水洗涤、干燥、浓缩,残余物可用合适的溶剂通过重结晶或在硅胶上用色谱法纯化。
表5指出反应条件(即反应物的结构和用量、溶剂、反应时间等)和产物(Ⅰe0的结构和物理常数。
实施例58
3-乙氧羰基-9-(4-氨磺酰基苯甲酰基)硫-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-61的制备
将0.29克三乙胺加到0.48克(2.4毫摩尔)的4-氨磺酰基苯甲酸在10毫升二甲基甲酰胺的溶液中,然后,再加入0.28克氯甲酸乙酯,该混合物在室温搅拌1小时,和0.5克固体3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1混合,该混合物在室温搅拌2小时,用100毫升水稀释并用醋酸乙酯振摇,有机层用水洗、干燥、真空浓缩,残留物用醋酸乙酯洗涤,得到0.68克标题化合物。
收率:78.5%
熔点:214-217℃(分解)(醋酸乙酯-四氯呋喃)
分析计算值:C18H15N3O2S2
C,49.87;H,3.49;N,9.70;S,14.80(%)
实验值:C,49.86;H,3.57;N,9.49;S,14.64(%)
实施例59-62
将合适的碱加入到取代的芳基羧酸(Ⅶ)在合适溶剂中的悬浮液或溶液中,再将氯甲酸乙酯加入,得到的混合物于0℃-室温搅拌约0.5-2小时(反应时间h1),将3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕-嘧啶-4-酮Ⅱ-1以固体形式或溶于适当的溶剂中的溶液形式加入到上面的混合物中,在0℃-100℃搅拌约0.5-5小时(反应时间h2),将反应混合物分散到水和适当的有机溶剂中,有机层用水洗,干燥,真空浓缩至干,残留物通过重结晶或在硅胶上用色谱法纯化,从而得到化合物(Ⅰe)
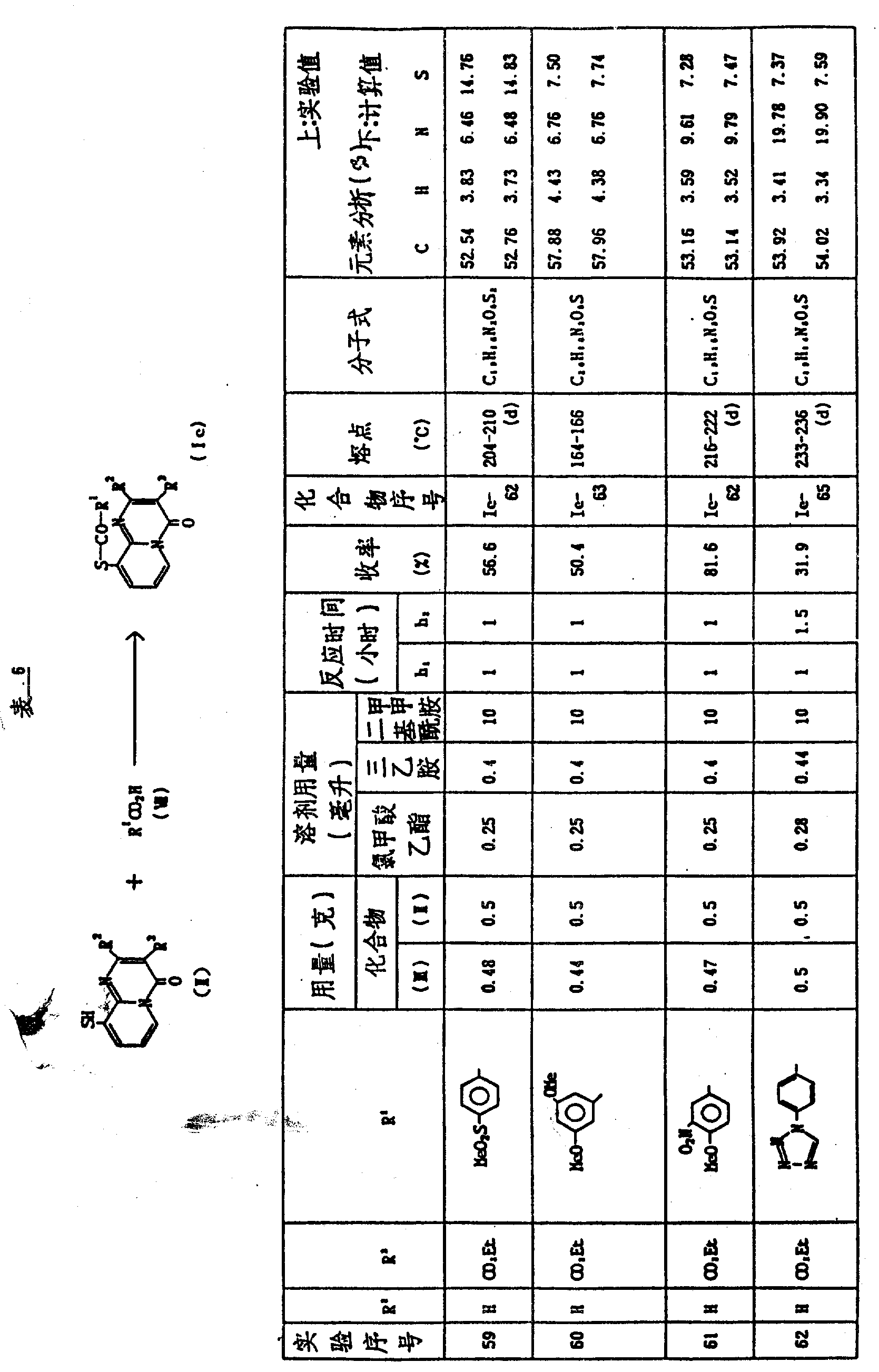
表6表示反应条件(即反应物的结构和用量溶剂,反应时间等)和产物(Ⅰe)的结构和物理常数。
实施例63
3-乙氧羰基-9-(4-甲氧羰基苯甲酰基)硫-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-66的制备
将23.8克4-甲氧羰基苯甲酰氯在搅拌下加入到30克3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1、2.49克粉状碳酸钾和500毫升丙酮的混合物中,得到的混合物于室温搅拌1小时,反应混合物真空浓缩至干,将残留物分散到水-氯仿中,氯仿层用饱和盐水洗涤,在硫酸钠上干燥,真空浓缩,残留物从醋酸乙酯中重结晶,得到43克标题化合物Ⅰe-66。
收率:86.9%
熔点:174-176℃
分析计算值:C20H16N2O6S
C,58.24;H,3.91;N,6.79;S,7.78(%)
实验值:C,58.39;H,4.00;N,6.81;S,7.84(%)。
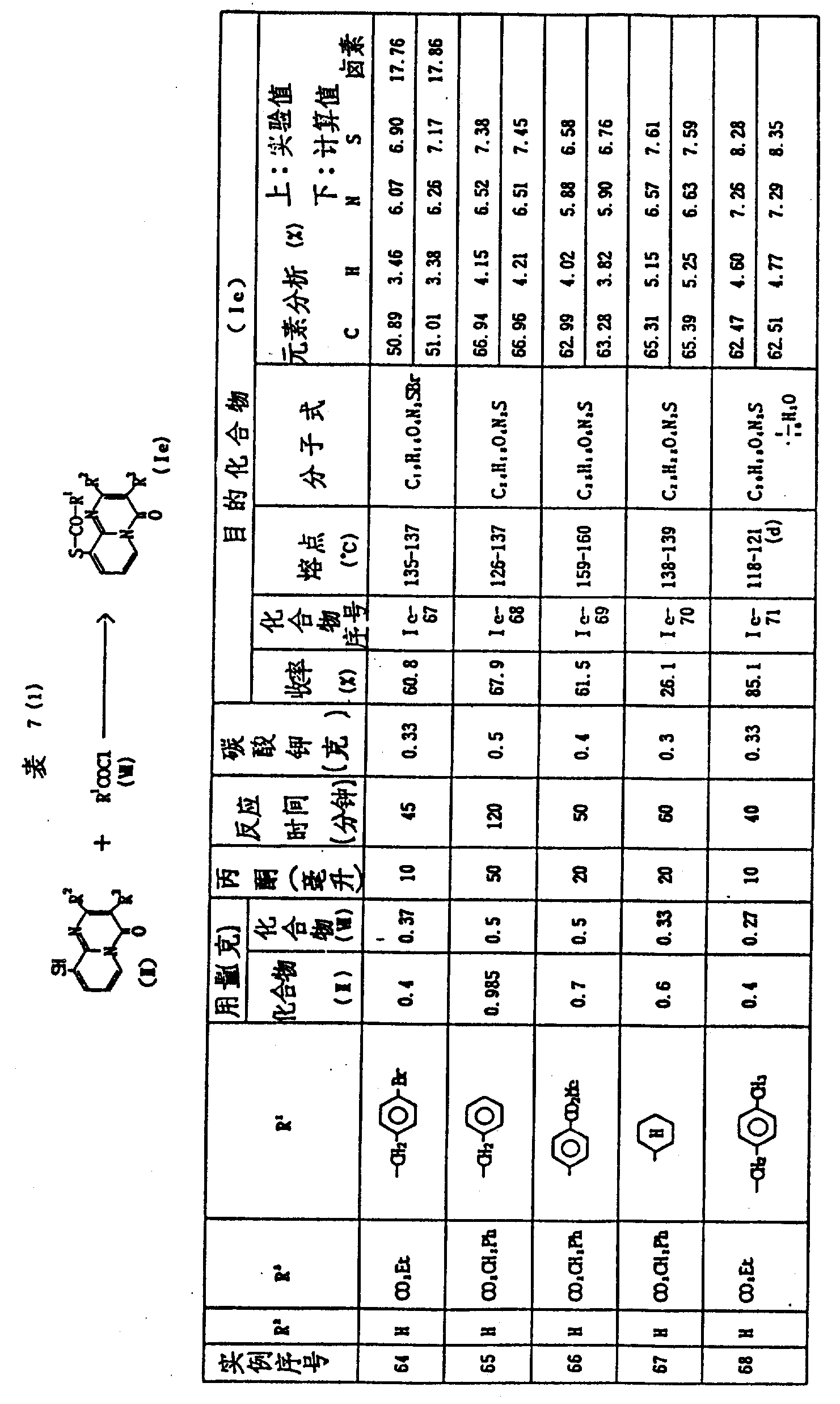
实施例64-74

将合适的碱加入到9-巯基-4H-吡啶并〔1,2-a〕-嘧啶-4-酮(Ⅱ)在适当溶剂中的悬浮液或溶液中,然后加入酰基氯(Ⅶ),该混合物在室温搅拌大约1-5小时,将反应混合物真空浓缩至干,残留物分散到水-氯仿中,有机层用水洗,干燥,真空浓缩,残留物以适当溶剂中重结晶成在硅胶上用色谱法进行纯化,从而得到产物(Ⅰe)。
表7表示反应条件和产物(Ⅰe)的结构和物理常数。
实施例75
9-(二甲基氨甲酰基)硫-4H-吡啶并〔1,2-a〕-嘧啶-4-酮Ⅰb-78的制备

1)6克2.2-二甲基-1.3-二噁烷-4.6-二酮和30毫升原甲酸乙酯的混合物在油浴上回流加热2小时,与8克2-氨基-3-二甲基硫代氨甲酰氧吡啶混合,在搅拌下再回流加热2小时,冷却后,过滤沉淀的结晶,用乙醚洗涤,得到6.4克2-(2,

2-二甲基-4,6-二氧-1,3-二噁烷-5-基亚(idene)甲基)氨基-3-二甲基硫代氨甲酰氧吡啶,产物从醋酸乙酯中重结晶。
熔点:223-225℃(分解)
分析计算值:C15H17O5N3S
C,51.27;H,4.88;N,11.96S,8.99(%)
实验值:C,51.29;H,4.78;N,11.84S,8.99(%)
2)将上面产物2.5克在100毫升道氏热载体A(道化学公司)中的溶液回流加热5分钟。
冷却后,该反应混合物在硅胶柱上用色谱法纯化,先用正己烷洗脱道氏热载体A,然后用醋酸乙酯洗脱,得1.4克标题化合物Ⅰb-78。
熔点:151-152℃(醋酸乙酯)
分析计算值:C11H11O2N3S
C,53.00;H,4.45;N,16.86;S,12.86(%)
实验值:C,52.89;H,4.36;N,16.74;S,12.83(%)
实施例76
9-(苄氧羰基)硫-3-羧基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe_80的制备

将0.872克9-(苄基羰基)硫-3-(4-甲氧基苄氧羰基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-79、2.5克苯甲醚和10毫升干燥的二氯甲烷混合物用冰水冷却,该混合物与5毫升三氟醋酸混合,在冰水冷却下搅拌3小时,使反应混合物真空浓缩至干,残留物从醋酸乙酯中重结晶,得到标题化合物Ⅰe-80。
分析计算值:C17H12O4N2S
C,59.99;H,3.55;N,8.23;S,9.42(%)
实验值:C,60.00;H,3.68;N,8.20;S,9.27(%)。
实施例77
3-羧基-9-(二甲基氨甲酰基)硫-4H-吡啶〔1,2-a〕嘧啶-4-酮Ⅰb-81的制备
0.508克9-(二甲基氨甲酰基)硫-3-(4-甲氧基苄氧羰基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰb-82、2.5克苯甲醚和5毫升干燥的二氯甲烷的混合物用冰水冷却,该混合物与5毫升三氟醋酸混合,在冰水冷却下搅拌3小时,反应混合物真空浓缩至干,残留物用乙醚洗涤,得0.32克粗制品,从95%乙醇中重结晶,得到标题化合物Ⅰb-81。
熔点:223-224℃
分析计算值:C12H11O4N3S
C,49.14;H,3.78;N,14.33S,10.93(%)
实值值:C,49.08;H,3.80;N,14.24S,10.98(%)
实施例78
9-巯基-3-(4-甲氧基苄氧羰基)-4H-吡啶并-〔1,2-a〕嘧啶-4-酮Ⅱ-2的制备

1)14.4克二(4-甲氧基苄基)-4-甲氧基-苄氧亚甲基丙二酸酯和5.9克2-氨基-3-(二甲基硫代氨甲酰基氧)吡啶X-1在搅拌下、于油浴上、在120℃加热1小时,反应混合物在硅胶柱上用色谱法纯化,用正己烷-醋酸乙鸱(1∶1容积/容积)洗脱,得13.3克3-二甲基硫代-氨甲酰基氧-2-〔2,2-双(4-甲氧基苄氧基)乙烯氨基〕吡啶。
熔点:117-118℃(醋酸乙酯-乙醚)
分析计算值:C28H29O7N3S
C,60.97;H,5.30;N,7.55S,5.81(%)
实验值:C,60.91;H,5.31;N,7.55;S,5.80(%)。
2)上述产物11克在100毫升二苯醚中的溶液回流加热半小时,冷却后,溶解该反应混合物,并在硅胶柱上用色谱法纯化,先用正己烷洗脱除去二苯醚,然后用醋酸乙酯洗脱,得4.3克9-二甲基氨甲酰基硫-3-(4-甲氧基苄氧羰基)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰb-83。
熔点:153-154℃(醋酸乙酯)
分析计算值:C20H19O5N3S
C,58.10;H,4.63;N,10.16
S.7.75(%)
实验值:C,57.98;H,4.75;N;10.07S.7.64(%)
3)在室温氮气流下,将1.13毫升正丙基硫醇滴加到0.5克氢化钠在50毫升四氢呋喃中的悬浮液中,得到的混合物搅拌1小时。然后,将4.3克由(2)得到的产物在300毫升合适溶剂中的溶液一次加入到上面的混合物中,该反应混合物在室温搅拌24小时,再与0.9克醋酸混合,真空浓缩至干,残留物分散到氯仿-水中,收集有机层,用水洗,干燥,真空浓缩,残留物用乙醚-醋酸乙酯洗涤,得1.8克标题化合物Ⅱ-2,为红棕色结晶。
熔点:230℃以上
实施例79
9-(苄基羰基)硫-3-乙氧基羰基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰe-84的制备

将65微升苯乙酰氯和0.13克碘化锌加到0.129克9-(二甲基氨甲酰基)硫-3-乙氧羰基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰb-85在5毫升1,2-二氯乙烷的溶液,使得到的混合物在氮气流下回流加热45分钟,冷却后,反应混合物用N-盐酸振摇,有机层用饱和盐水适当洗涤,干燥,真空浓缩,残留物在硅胶柱上用色谱法纯化,用醋酸乙酯洗脱,从而得到0.118克标题化合物Ⅰe-84。
收率:58%
熔点:126-128℃
分析计算值:C9H16N2O4S
C,61.94;H,4.38;N,7.60
实验值:C,62.09;H,4.29;N,7.48;S,8.64(%)。
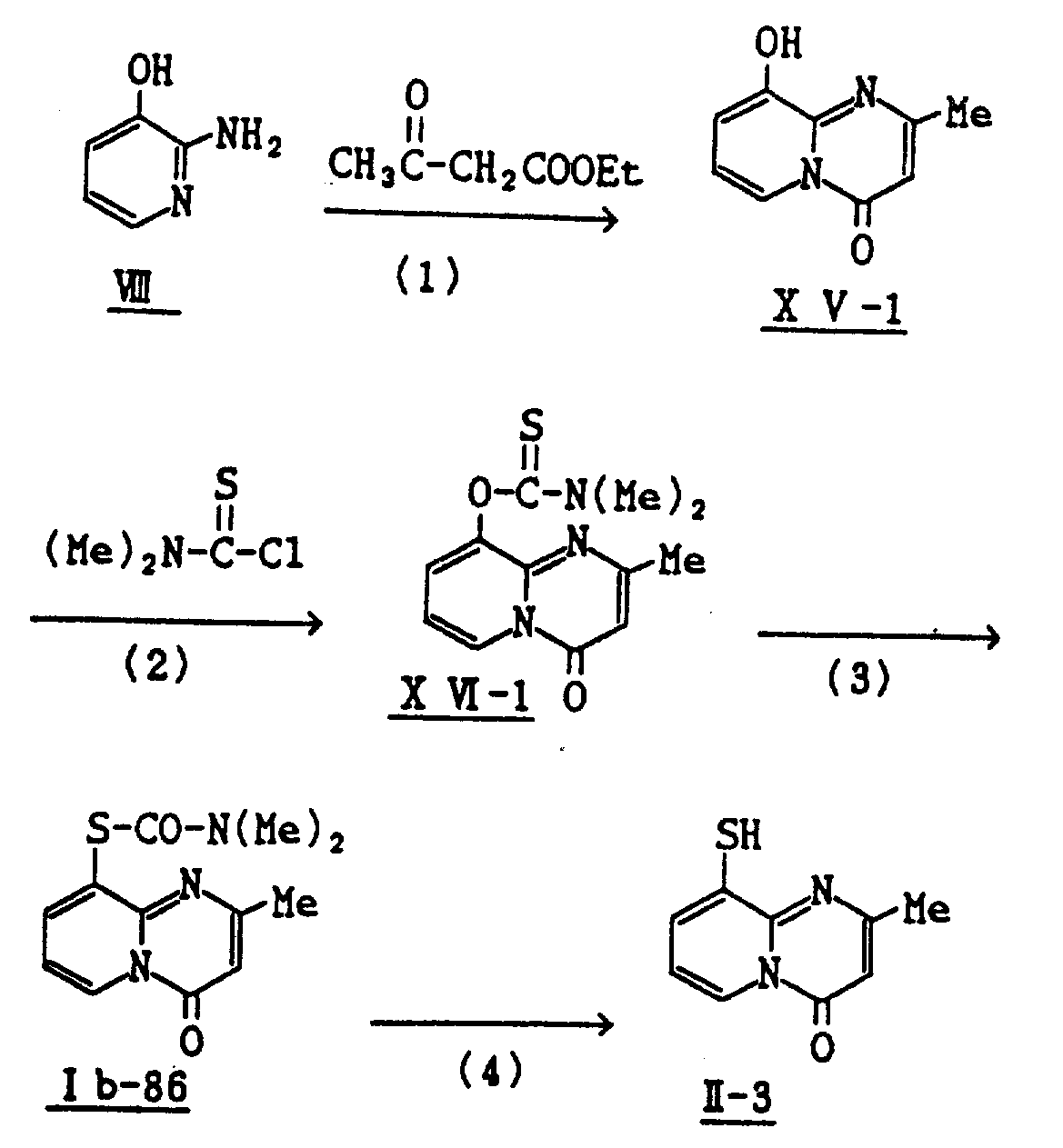
参考实例1

1)2-氨基-3-二甲基硫代氨甲酰基吡啶X-1的制备
将58克粉状的碳酸钾加到45克2-氨基-3-羟基吡啶Ⅷ在700毫升丙酮的溶液中,搅拌该混合物,在其中再加入49克二甲基硫代氨甲酰氯,该混合物在室温搅拌48小时,过滤除去不溶物,并用丙酮洗涤,将得到的溶剂和滤液合并,蒸发,残留物用硅胶柱色谱法纯化,从被醋酸乙酯洗脱的部分中得到44.2克标题化合物Ⅹ-1。
收率:54.8%
熔点:134-136℃(从醋酸乙酯中重结晶)
分析计算值:C8H11ON3S
C,48.71;H,5.62;N,21.30;S,16.25(%)
实验值:C,48.43;H,5.59;N,20.77;S.16.29(%)
2)2-〔2′,2′-双(乙氧羰基)乙烯氨基〕-3-(N′,N′-二甲基硫代氨甲酰基氧)吡啶Ⅻ-1的制备
13.7克上述(Ⅰ)项提供的化合物Ⅹ-1和14克乙氧基亚甲甲基丙二酸二乙酯Ⅺ-1在100℃加热1小时,该反应混合物通过硅胶色谱法纯化,从被苯和醋酸乙酯混合物(4∶1容积/容积%)洗脱部分中得到19.3克标题化合物Ⅻ-1。
熔点:91-92.5℃(从乙醚中重结晶)
分析计算值:C16H21O6N3S
C,52.30;H,5.76;N,11.44。S,8.73(%)
实验值:C,52.37;H,5.82;N,11.48;S,8.50(%)
3)3-乙氧羰基-9-二甲基氨甲酰基硫-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰb-85的制备
8克上述(2)项中提供的化合物Ⅻ-1在20毫升乙醚中的溶液,在油浴上加热回流30分钟,将该反应混合物倾入1升正乙烷中,过滤沉淀的结晶,用硅胶色谱法纯化,从被醋酸乙酯洗脱部分中得到4.0克标题化合物Ⅰb-85。
收率:53%
熔点:133-134℃(从醋酸乙酯中重结晶)
分析计算值:C14H16O4N3S
C,52.33;H,4.71;N,13.08;S,9.98(%)
实验值:C,52.13;H,4.66;N,12.99;
S,9.75(%)
4)3-乙氧羰基-9-巯基-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅱ-1的制备
在0℃和氮气流下,将0.6克正丙基硫醇滴加到0.3克氢化钠在30毫升无水四氢呋喃的悬浮液中,该混合物搅拌15分钟,将在上述(3)项中提供的Ⅰb=85化合物2.0克在100毫升无水四氢呋喃中的溶液加到上述混合物中,室温将被8小时,得到的混合物放置过夜,该反应混合物再与0.5克醋酸混合,并真空浓缩至干,残留物与大约150毫升水混合并搅拌,得到1.3克标题化合物Ⅱ-1。
收率:81%
熔点:151-152℃(从醋酸乙酯中重结晶)
分析计算值:C11H10O3N2S
C,52.79;H,4.03;N,11.19;S,12.81(%)
实验值:C,52.88;H,3.97;N,11.24;S.12.66(%)
参考实例2
1)9-羟基-2-甲基-4H-吡啶并〔1,2-a〕-嘧啶-4-酮Ⅺ-1的制备
将4毫升多磷酸加到1.1克2-氨基-3-羟基吡啶Ⅷ和1.1克乙酰乙酸乙酯的混合物中,该混合物在100℃加热下,搅拌4小时,将反应混合物倾入冰水中,得到的溶液用2N氢氧化钠水溶液调到PH4,然后,再用碳酸钠水溶液调节到PH=7,用氯仿振摇,将有机层用水洗,在无水硫酸钠上干燥,蒸去溶剂,得到0.9克标题化合物ⅩⅤ-1。
收率:55.5%
2)2-甲基-9-二甲基硫代氨甲酰基氧-4H-吡啶并〔1,2-a〕嘧啶-4-酮ⅩⅥ-1的制备
将0.3克碳酸钾和0.3克二甲基硫代氨甲酰氯加入到0.3克上述(1)项中提供的混合物ⅩⅤ-1和15毫升丙酮的混合物中,得到的混合物在室温搅拌8小时,过滤固体,并用丙酮洗涤,将洗液和滤液合并,浓缩,残留物用水洗,在五氧化二磷上干燥,得到0.3克标题化合物ⅩⅤ-1。
收率:66.9%
熔点:165-167℃(从醋酸乙酯中重结晶)
分析计算值:C12H13O2N3S
C,54.79;H,4.88;N,15.94;S,12.07(%)
实验值:C,54.74;H,4.98N,15.96;S,12.18(%)
核磁共振(CDCl3)δ:2.37(3H,S),3.46(6H,S)
3)2-甲基-9-二甲基氨甲酰基硫-4H-吡啶并-〔1,2-a〕嘧啶-4-酮Ⅰb-86的制备
将5.2克在上述(2)项中提供的化合物ⅩⅤ-1在40毫升道氏热载体A(道氏化学公司)中的悬浮液加热回流20分钟,将该反应混合物冷却,并倾入800毫升正己烷中,得到5.0克标题化合物Ⅰb-86
熔点:165-167℃(从醋酸乙酯中重结晶)
分析计算值:C12H13O2N3S
C,54.74;H,4.98;N,15.96;S,12.18(%)
实验值:C,54.78;H,4.90;N,15.94;S,12.10(%)
核磁共振(CDCl3)δ:2.43(3H,S),3.10(6H,S)
4)9-巯基-2-甲基-4H-吡啶并-〔1,2-a〕-嘧啶-4-酮Ⅱ-3的制备
使0.4克氢化钠(60%油分散)在20毫升无水四氯呋喃中的悬浮液在0℃冷却,将1.4毫升正丙基硫醇滴加入内,混合物搅拌20分钟,再将4.1克在上述(3)项中提供的化合物Ⅰb-86在130毫升无水四氢呋喃中的溶液加入到该混合物中,俟反应混合物冷却至室温,搅拌24小时,得到的混合物真空浓缩至干,残留物溶解在100毫升水中,与2克醋酸混合,过滤沉淀的结晶,干燥,得到3.0克标题化合物Ⅱ-3。
熔点:240-241℃(从氯仿中重晶)
分析计算值:C9H8ON2S
C,56.23;H,4.19;N,14.57;S.16.59(%)
实验值:C,56.11;H,4.11;N,14.52;S,16.59(%)
配方
3-乙氧羰基-9-(4-甲基苯基氨甲酰基硫)-4H-吡啶并〔1,2-a〕嘧啶-4-酮Ⅰa_1 25毫克
乳糖 100毫克
小麦淀粉 15毫克
明胶 5毫克
硬脂酸镁 5毫克
150毫克
上面所述的组份可装入胶囊或胶囊剂。
本发明的效果
实验
(对浸水应激反应溃疡的抗溃疡活性)
使标准雄性大鼠(体重:260-290克)在试验前禁食24小时,为了使受试大鼠产生抑制应激反应,把它们关在网状的金属丝笼子中,并浸入水浴(23℃)7小时,然后处死,取出每个大鼠的胃,沿胃大弯切开,测定在腺体部位上每一受损部分的长度,并将它们加合起来,与对照组的结果比较,计算对急性溃疡的抑制率。试验的化合物悬浮于5%阿拉伯胶的溶液中,在产生应激反应前30分钟口服给药。
试验用化合物
西咪替丁用作对照药物。
表示的方法
溃疡形成的抑制作用在71%以上者,用++表示
溃疡形成的抑制作用在51%-70%者,用+表示结果
表8表示实验的结果
表8

(续)表8

上面实验结果表明,本发明的化合物(Ⅰ)具有强的抗溃疡活性。所以,化合物(Ⅰ)对预防和治疗溃疡或抑制停药后溃疡复发是有效的。
勘误表 CPCH866542

Claims (5)
1、结构式(Ⅰ)的化合物或其盐
(其中n是0或1,
R为-COR1,
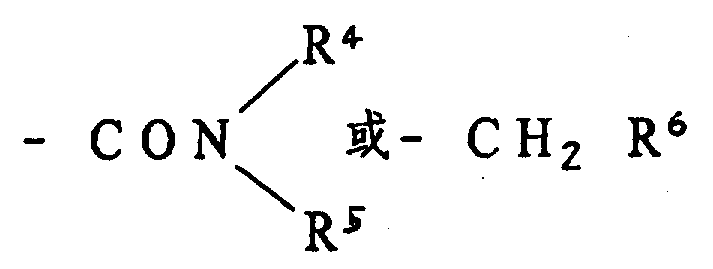
R1是C1-C5烷基、C3-C7环烷基、烯丙硫基、苯乙烯基、苯氧甲基、噻吩甲基、可由选自C1-C3烷基、C3-C6环烷基、C1-C3烷氧基苯基-C1-C3烷氧基、卤素、硝基、C1-C3-链烷磺酰基、C2-C5烷氧羰基、氰基、乙酰基、乙酰氧基、四唑基、三氟甲基和氨磺酸基基中一个或多个基团取代的C6-C10、可由选自C1-C3烷基、C1-C3烷氧基、卤素和硝基中一个或多个基团取代的苄基、或可由选自C1-C3烷基、C1-C3烷氧基和C3-C6环烷基中一个或多个基团取代的5元或6元杂环基团,
R2和R3各自为氢、C1-C5烷基、羧基,C2-C5烷氧羰基,或可由选自C1-C3烷基、C1-C3烷氧基和卤素中一个或多个基团取代的苄氧羰基,
R4和R5各自为氢、C1-C5烷基,C3-C7环烷基,或
可由选自卤素、C1-C3烷基、C1-C3烷氧基、C2-C5烷氧羰基、硝基和三氟甲基中一个或多个基团取代的苯基,
R6是可由选自囟素、C1-C3烷基和C1-C3烷氧基中一个或多个基团取代的吡啶基或苯基。
2、根据权利要求1的化合物,其中,R1是可由选自C1-C3烷基、C1-C3烷氧基、卤素和C2-C5-烷氧羰基中一个或多个基团取代的苯基。
3、根据权利要求2的化合物,其中,R2和R3各自为氢、C1-C3烷基、羧基或C2-C5烷氧羰基。
4、由药理学上有效剂量的权利要求1化合物和可用作医药的载体、稀释剂或赋形剂组成的药物组方。
5、制备权利要求1化合物的方法,其中包括:(A)结构式(Ⅱ)化合物(其中R2和R3如上述定义)和结构式(Ⅲ)化合物(其中R4如上述定义反应得到结构式(Ⅰa)化合物(其中R2,R3和R4如上述定义),
(B)化合物(Ⅱ)与N,N′-羰基二咪唑反应得到结构式(Ⅳ)化合物(其中R2和R3如上述定义),并且化合物Ⅳ与结构式(Ⅴ)化合物(其中R4和R5如上述定义)反应得结构式(Ⅰb)化合物(其中R4和R5如上述定义),

(C)化合物(Ⅱ)与结构式(Ⅵ)化合物反应(其中Hal是卤素,R6如上述定义)得到结构式(Ⅰc)化合物(其中R2,R3和R4如上述定义),最后,把产物(Ⅰc)氧化成结构式(Ⅰd)化合物(其中R2,R3和R6如上述定义),


或(D)化合物(Ⅱ)与结构式(Ⅶ)化合物(其中R1和Hal如上述定义)反应得到结构式(Ⅰe)化合物(其中R1,R2和R3如上述定义)。

Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP213181/1985 | 1985-09-25 | ||
| JP213181/85 | 1985-09-25 | ||
| JP21318185 | 1985-09-25 | ||
| JP11591/86 | 1986-01-21 | ||
| JP1159186 | 1986-01-21 | ||
| JP11591/1986 | 1986-01-21 | ||
| JP181159/86 | 1986-07-30 | ||
| JP18115986 | 1986-07-30 | ||
| JP181159/1986 | 1986-07-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN86106902A true CN86106902A (zh) | 1987-06-10 |
| CN1013447B CN1013447B (zh) | 1991-08-07 |
Family
ID=27279485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN86106902A Expired CN1013447B (zh) | 1985-09-25 | 1986-09-25 | 9-(取代硫基)-4H-吡啶并[1.2-a]嘧啶-4-酮衍生物的制备方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US4840953A (zh) |
| EP (1) | EP0218423B1 (zh) |
| KR (1) | KR930009011B1 (zh) |
| CN (1) | CN1013447B (zh) |
| AU (1) | AU588917B2 (zh) |
| CA (1) | CA1283411C (zh) |
| DE (1) | DE3670843D1 (zh) |
| DK (1) | DK169580B1 (zh) |
| ES (1) | ES2002023A6 (zh) |
| GB (1) | GB2181129B (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2668259B2 (ja) * | 1988-02-18 | 1997-10-27 | 塩野義製薬株式会社 | 複素環化合物および抗潰瘍剤 |
| KR101574332B1 (ko) | 2008-06-17 | 2015-12-08 | 재단법인 한국파스퇴르연구소 | 항감염성 화합물 |
| KR101828094B1 (ko) * | 2009-09-09 | 2018-03-22 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 제초제 피리미돈 유도체 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907798A (en) * | 1966-06-15 | 1975-09-23 | Sterling Drug Inc | Preparation of 4H-pyrido{8 1,2-a{9 pyrimidin-4-ones from cyclic alkylidene 2-pyridylaminomethylenemalonates |
| DE1670480C2 (de) * | 1966-11-02 | 1983-06-01 | Chinoin Gyógyszer és Vegyészeti Termékek Gyára R.T., 1045 Budapest | Pyrido[1,2-a]pyrimidinderivate, Verfahren zu deren Herstellung sowie diese Verbindungen enthaltende pharmazeutische Präparate |
| US3960847A (en) * | 1972-04-17 | 1976-06-01 | E. R. Squibb & Sons, Inc. | 9-Substituted-4-oxopyrido(1,2-α)pyrimidine-3-carboxylic acids and derivatives thereof |
| HU168541B (zh) * | 1973-11-24 | 1976-05-28 | ||
| US4022897A (en) * | 1974-03-04 | 1977-05-10 | E. R. Squibb & Sons, Inc. | CNS active compounds |
| US4122274A (en) * | 1977-05-25 | 1978-10-24 | Bristol-Myers Company | 3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones |
| US4209620A (en) * | 1978-06-19 | 1980-06-24 | Bristol-Myers Company | Substituted 3-(1H-tetrazol-5-yl)-4H-pyrido[1,2-a]pyrimidin-4-one derivatives having antiallergy activity |
| JPS5830760B2 (ja) * | 1980-10-09 | 1983-07-01 | 株式会社日立製作所 | プリント回路板の製法 |
| US4457932A (en) * | 1983-07-22 | 1984-07-03 | Bristol-Myers Company | Anti-ulcer agents |
-
1986
- 1986-09-24 CA CA000519033A patent/CA1283411C/en not_active Expired - Fee Related
- 1986-09-24 DK DK456986A patent/DK169580B1/da active
- 1986-09-24 AU AU63104/86A patent/AU588917B2/en not_active Ceased
- 1986-09-25 KR KR1019860008029A patent/KR930009011B1/ko not_active Expired - Fee Related
- 1986-09-25 ES ES8602449A patent/ES2002023A6/es not_active Expired
- 1986-09-25 EP EP86307404A patent/EP0218423B1/en not_active Expired - Lifetime
- 1986-09-25 DE DE8686307404T patent/DE3670843D1/de not_active Expired - Fee Related
- 1986-09-25 GB GB8623116A patent/GB2181129B/en not_active Expired
- 1986-09-25 CN CN86106902A patent/CN1013447B/zh not_active Expired
-
1988
- 1988-06-15 US US07/206,665 patent/US4840953A/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| AU6310486A (en) | 1987-03-26 |
| GB2181129B (en) | 1989-09-20 |
| EP0218423A2 (en) | 1987-04-15 |
| EP0218423B1 (en) | 1990-05-02 |
| CN1013447B (zh) | 1991-08-07 |
| KR870003105A (ko) | 1987-04-15 |
| DK456986D0 (da) | 1986-09-24 |
| EP0218423A3 (en) | 1988-01-20 |
| DE3670843D1 (de) | 1990-06-07 |
| KR930009011B1 (ko) | 1993-09-18 |
| US4840953A (en) | 1989-06-20 |
| ES2002023A6 (es) | 1988-07-01 |
| AU588917B2 (en) | 1989-09-28 |
| DK456986A (da) | 1987-03-26 |
| GB8623116D0 (en) | 1986-10-29 |
| GB2181129A (en) | 1987-04-15 |
| DK169580B1 (da) | 1994-12-12 |
| CA1283411C (en) | 1991-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1031570C (zh) | 吡唑并吡啶化合物的制备方法 | |
| CN1068879C (zh) | 二氢苯并呋喃 | |
| CN1046724C (zh) | 吡唑并吡啶化合物、含有它们的药物组合物、及其制法与用途 | |
| CN1038511C (zh) | 咪唑并吡啶及其制备方法和制药的应用 | |
| CN1055182A (zh) | N-(吡咯并《2,3-d》嘧啶-3-基酰基)-谷氨酸衍生物 | |
| CN1109056A (zh) | 咪唑并吡啶衍生物 | |
| CN87108354A (zh) | 6-苯并嗪基和6-苯并噻嗪基-2,3,4,5-四氢哒嗪-3-酮 | |
| CN1035255C (zh) | 杂三环衍生物 | |
| CN1138583A (zh) | 9-取代的2-(2-正烷氧基苯基)-嘌呤-6-酮类化合物 | |
| CN1205704A (zh) | 2,7-取代的八氢-吡咯并[1,2-a]吡嗪衍生物 | |
| CN1552714A (zh) | 2-取代苯基-5,7-二烃基-3,7-二氢吡咯[2,3-d]嘧啶-4-酮衍生物,其制备方法及其药物用途 | |
| CN1052482C (zh) | 喜树碱衍生物及其制备方法和抗肿瘤剂 | |
| CN1071665A (zh) | 唑烷酮衍生物 | |
| CN1341103A (zh) | 哒嗪-3-酮衍生物和含有该类化合物的药物 | |
| CN85108607A (zh) | 1,4-苯并噻嗪衍生物的生产和应用 | |
| CN1033585C (zh) | 1,2,4-三唑并(1,5-α)嘧啶衍生物的制法 | |
| CN1079745A (zh) | 新的9-氟-7-氧代-7H-吡啶并[1,2,3-d,e][1,4]苯并嗪-6-羧酸及其酯 | |
| CN1100425A (zh) | 噻唑并嘧啶衍生物 | |
| CN100347173C (zh) | 具有免疫调制活性的吡唑并喹啉 | |
| CN1993367A (zh) | 稠合的嘧啶衍生物和黄嘌呤氧化酶抑制剂 | |
| CN1171785A (zh) | 嘧啶衍生物 | |
| CN1049161A (zh) | 具有血清素2-受体拮抗活性的杂环化合物 | |
| CN1077955A (zh) | 杂环衍生物 | |
| CN86106902A (zh) | 9-(取代硫基)-4H-吡啶并[1,2-a]嘧啶-4-酮衍生物 | |
| CN1100419A (zh) | 3-喹啉基取代二氢吡啶类化合物及其制备方法与药用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C13 | Decision | ||
| C14 | Grant of patent or utility model | ||
| C19 | Lapse of patent right due to non-payment of the annual fee |