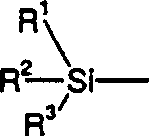-
Die Erfindung betrifft das Gebiet
von Vitamin D-Vorstufen, wie (3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion und insbesondere
ein Verfahren zur Herstellung solcher Derivate.
-
Die Verbindung (3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion
(die Verbindung der nachstehenden Formel II) ist ein Schlüsselzwischenprodukt
bei der Herstellung von 1a,25-Dihydroxy-l6-en-23-in-cholecalciferol
(die Verbindung der nachstehenden Formel III), und 1α-Fluor-25-hydroxy-16,23-dien-26,27-bishomo-20-epi-cholecalciferol
(die Verbindung der nachstehenden Formel IV) sowie anderer Vitamin
D-Analoge. Die Herstellung dieser Verbindung wurde ursprünglich von
Daniewski, A. R. und Kiegiel, J., J. Org. Chem., 53: 5334 (1988)
mitgeteilt. Die Synthese bezog stereoselektive 1,4-Reduktion des
Ketons der Formel I (siehe nachstehend) in Gegenwart von 38 Molprozent
tert-Butylkupfer(I)-Katalysator (P3) und anschließende Bromierung
des erhaltenen Enolats [Daniewski, A. R. und Kiegiel, J., Synth.
Commun., 18: 115(1988)] ein. Die Verbindung der Formel II wurde
in einer Ausbeute von 57% nach Reinigung durch Kieselgelchromatographie
und Kristallisation isoliert. Jedoch ist der tert-Butylkupferkatalysator
instabil, wodurch dieses Verfahren schwierig zu reproduzieren war
und eine genaue Temperatursteuerung erforderte.
-
-
Es ist bekannt, dass Reduzieren des
Ketons der Formel I das trans-Hydrindanderivat der Formel 5 erzeugt,
dessen Kohlenstoffgerüst,
obwohl ziemlich ähnlich
in der Struktur von vielen Naturprodukten, einschließlich Vitamin
D und Steroide, schwierig zu synthetisieren ist, da thermodynamisch
das entsprechende cis-Isomer (wie die Verbindung der Formel 6) stark
begünstigt
ist.
-
-
Die Reduktion der Verbindung der
Formel I mit Diisobutylaluminiumhydrid („DIBALH") in Gegenwart von
MeCu als Katalysator (Pl) war kaum befriedigend, weil ein 4 : 3
: 3-Gemisch der Verbindungen der Formel 5, 6 beziehungsweise 7 entsteht.
Somit waren Stereo- und Regioselektivität nur 55% (das Verhältnis der
Verbindungen der Formeln 5 zu 6 war 4 : 3), beziehungsweise 70%
(das Verhältnis
der Verbindungen der Formeln 5 plus 6 zu 7 war 7: i). Die Stereoselektivität erhöhte sich
auf: 66% mit n-Butylkupfer
(Katalysator P2) und 90% mit tert-Butylkupferkatalysator. Jedoch
verblieb die Regioselektivität
eigentlich unverändert
und die Verbindung der Formel 7 wurde im All-gemeinen in einer Ausbeute von 30% isoliert.
Folglich übersteigt
die isolierte Ausbeute des gewünschten
Bromketons der Formel II niemals 57%, nachdem die Reaktion mit Brom
gestoppt wurde, selbst wenn eine hohe Stereoselektivität (90%)
mit dem tert-Butylkupfer (Katalysator P3) erzielt wird.
-
Tabelle
1. Stereo- und Regioselektivitäten
bei der Reduktion von Keton (I) unter Verwendung bekannter Katalysatoren.
-
Darüber hinaus wird durch die thermische
Instabilität
von tert-Butylkupfer, aufgrund seiner Tendenz unter Bildung von
Isobutylen und Kupferhydrid (β-Eliminierung
zu unterliegen, diese Reaktion schwer reproduzierbar, da das so
gebildete Kupferhydrid eine nicht steroselektive 1,4-Reduktion katalysiert.
Folglich gab es auf dem Fachgebiet einen Bedarf für einen
ausgezeichneten Katalysator, um diese Reaktion auszuführen.
-
Die vorliegende Erfindung stellt
ein Verfahren zur Herstellung der Verbindung der Formel II (3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion
bereit. Dieses Verfahren umfasst Reduzieren von (S)-(+)-2,3,5,6,7,7a-Hexahydro-7a-methylinden-l,5-dion
und Behandeln des Reduktats mit einem Brom enthaltenden Elektrophil.
Das (S)-(+)-2,3,5,6,7,7a-Hexahydro-7a-methylinden-l,5-dion wird
durch Reduktion mit Diisobutylaluminiumhydrid („DIBALH") und Hexamethylphosphorsäuretriamid
(„HMPA")
in Gegenwart eines Katalysators der Formels R-Cu, worin R
darstellt
und R
1, R
2 und R
3 jeweils unabhängig ausgewählt sind aus der Gruppe, bestehend
aus C
1-7-Alkyl,
Phenyl, Phenyl, substituiert mit mindestens einer C
1-4-Alkylgruppe, Benzyl
oder Benzyl, substituiert mit mindestens einer C
1-4-Alkylgruppe,
reduziert. Das so gebildete Reduktat wird mit einem Brom enthaltenden
Elektrophil unter Gewinnung von (3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-roden-l,5-dion
behandelt.
-
Wenn nach dem erfindungsgemäßen Verfahren
hergestellt, wurde die Verbindung der Formel II [(3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion]
aus der Verbindung der Formel I [(S)-(+)-2,3,5,6,7,7a-Hexahydro-7a-methylinden-l,5-dion]
in 70%iger Ausbeute erzeugt, was um 13% höher ist als durch die vorangehenden
Verfahren erhalten.
-
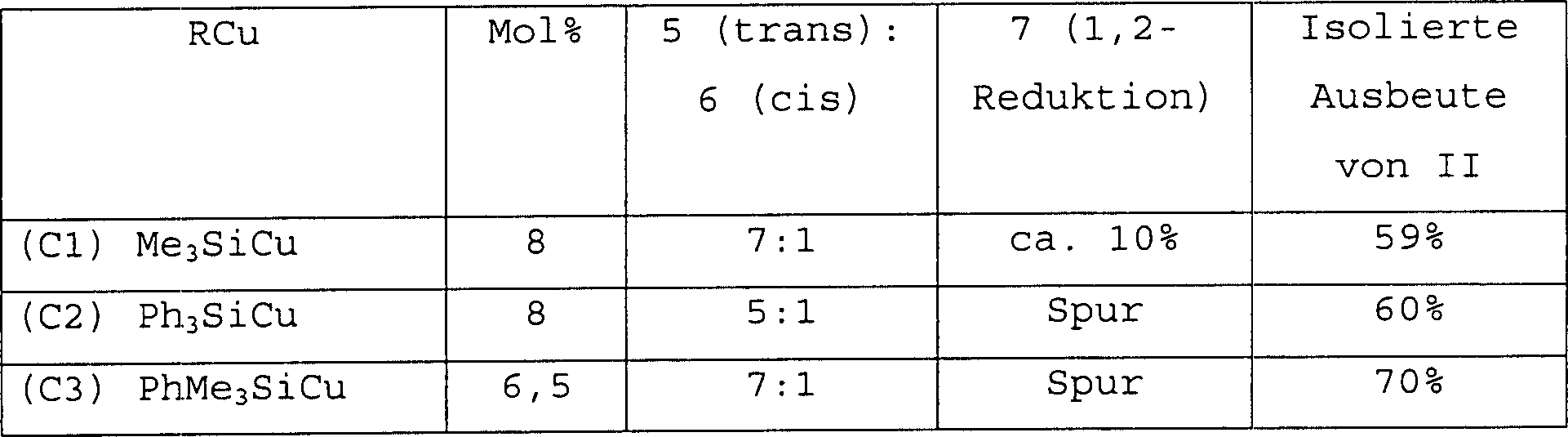
Es wurde gefunden, dass Silylkupferkatalysatoren,
verglichen mit bekannten Kupferkatalysatoren, überlegene Regioselektivität bereitstellen.
Trimethylsilylkupfer (Katalysator Cl) ist, verglichen mit dem bekannten
tert-Butylkupfer (Katalysator P3), bei der 1,4-Reduktion des Ketons
der Formel I wirksam und stabil, wahrscheinlich weil in einem Silylkupferkatalysator
eine β-Eliminierung
aufgrund der hohen Energie einer Kohlenstoff-Siliciumdoppelbindung
nicht bevorzugt ist. Durch Verwenden der erfindungsgemäßen Kupfersilylkatalysatoren
kann die Katalysatorbeladung, bezogen auf bekannte Kupferkatalysatoren,
vermindert werden. Obwohl die Steroselektivität (85%) unter Verwendung von
erfindungsgemäßem Katalysator
Trimethylsilylkupfer niedriger war als mit dem bekannten tert-Butylkupferkatalysator
(Katalysator P3) (90%) erreicht, zeigt Trimethylsilylkupfer höhere Regioselektivität (90%)
als das wie in Tabelle 1 gezeigte tert-Butylkupfer (Katalysator
P3) (70%). Im Ergebnis wurde das gewünschte Bromketon der Verbindung
der Formel II in 59%iger Ausbeute durch Einkristallisation isoliert.
Die Regioselektivität
wurde weiterhin mit Triphenylsilylkupfer (Katalysator C2) auf > 95% verbessert. Die
Anwendung von erfindungsgemäßem Katalysator
Dimethylphenylsilylkupfer (Katalysator C3) ergab eine sehr hohe
isolierte Ausbeute der Verbindung der Formel II (70%). Die Stereo-
und Regioselektivitäten
mit diesem Katalysator waren 85% beziehungsweise > 95%.
-
Tabelle
2: Stereo- und Regioselektivitäten
bei der Reduktion. von Keton (I) unter Verwendung von erfindungsgemäßen Katalysatoren.
-
Die Erfindung betrifft somit ein
verbessertes Verfahren für
die Herstellung von (3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-lH-finden-l,5-dion
der Formel II aus dem Keton der Formel I. Ein Reaktionschema erläutert dieses
nachstehende Verfahren. Dieses neue Verfahren ist reproduzierbar
und die Verbindung der Formel II kann in Ausbeuten von 70% erhalten
werden. Reaktionsschema
worin Si Silicium darstellt, X Brom, Chlor,
Jod oder Cyano darstellt, R
1, R
2 und
R
3 jeweils unabhängig C
1-7-Alkyl, Phenyl
oder Phenyl oder Benzyl, substituiert mit einer oder mehreren Alkylgruppen
mit 1–4
Kohlenstoffatomen, darstellt und R
4 Methyl,
Butyl oder sec-Butyl darstellt.
-
Wie hierin verwendet, bedeutet „C1-7-Alkyl" eine Alkylgruppe mit 1–7 Kohlenstoffatomen,
einschließlich beispielsweise
Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl. Die Begriffe „Phenyl
oder Benzyl, substituiert mit Alkyl mit 1–4 Kohlenstoffatomen" schließen beispielsweise
Methylphenyl, Methylbenzyl, Ethylphenyl, Ethylbenzyl, Propylphenyl,
Propylbenzyl, Butylphenyl, Butylbenzyl, Isopropylphenyl, Isopropylbenzyl,
Isobutylphenyl, Isobutylbenzyl ein.
-
Die Disilanverbindungen der Formel
A, die in dem erfindungsgemäßen Verfahren
angewendet werden, sind bekannte Verbindungen oder können gemäß bekannten
Verfahren hergestellt werden. Insbesondere wurde das Dimethylphenylsilylkupfer
(Katalysator C3) in-situ aus 1,2-Diphenyltetramethyldisilan, Methyllithium und
Kupfer-I-jodid wie in dem vorstehenden Reaktionsschema gezeigt,
hergestellt. Das 1,2-Diphenyltetramethyldisilan wurde mit einer
unterstöchiometrischen
Menge Methyllithium behandelt, um vollständigen Verbrauch des Methyllithiums
zu sichern, unter Gewinnung von Dimethylphenylsiliyllithium (inertes
Phenyltrimethylsilan wird auch hergestellt). Die so gebildetete
Silyllithiumverbindung wird dann mit einer unterstöchiometrischen
Menge Kupfer-I-jodid behandelt, unter Gewinnung des gewünschten
Dimethylphenylsilylkupfers (Katalysator C3). Überschüssiges Kupfer-I-jodid wird
zu Kupferhydrid umgewandelt, welches eine nicht stereoselektive
1,4-Reduktion katalysiert.
-
R4Li Reagenzien
oder Reaktanten, die vorstehend angewendet wurden, sind bekannte
Verbindungen oder können
gemäß bekannten
Verfahren hergestellt werden. Beispielhaft für die Reagenzien R4Li
sind Methyllithium, Butyllithium.
-
In ähnlicher Weise sind die Reagenzien
CuX, worin X Brom, Chlor, Jod oder Cyano darstellt, bekannte Verbindungen
und schließen
Kufper-I-jodid, Kupfer-I-bromid ein.
-
Die Hydridreduktion der Formel I
wird unter Anwenden eines Reduktionsmittels, wie beispielsweise
Diisobutylaluminiumhydrid, ausgeführt.
-
Die Lösungsmittel, die in dem erfindungsgemäßen Verfahren
angewendet werden können,
schließen beispielsweise
Hexamethylphosphorsäuretriamid
(HMPA) und äquivalente
Lösungsmittel,
einzeln oder in Kombination mit anderen kompatiblen Lösungsmitteln
ein.
-
Die in dem erfindungsgemäßen Verfahren
angewendeten Elektrophile schließen beispielsweise Brom, N-Bromsuccinimid,
1,3-Dibrom-5,5-dimethylhydantoin, das für die Herstellung der Verbindung
der Formel II anzuwenden ist, ein.
-
Das erfindungsgemäße Verfahren wird im Allgemeinen
bei Temperaturen ausgeführt,
die alle im Bereich von 0° bis –78°C liegen.
Während
die Reaktionstemperaturen nicht annähernd kritisch sind, sind niedere Temperaturen
allgemein bevorzugt, um gute Ausbeuten zu erreichen.
-
Die gewünschte Verbindung der Formel
II wird gemäß bekannten
Methoden und Verfahren gewonnen und, vorzugsweise wie hierin in
Beispiel 1 beschrieben, ausgeführt.
-
Die nachstehenden Beispiele beschreiben
die Erfindung weiterhin.
-
Beispiel
1
Herstellung von Brom-Keton der Formel II [(3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion]
-
Ein 2 Liter-Dreihalskolben, ausgestattet
mit einem mechanischen Rührer,
Thermometer, Septum, Tropftrichter und Stickstoffeinlass, wurde
mit Stickstoff für
45 Minuten gespült
und dann mit 13,7 g (50,6 mMol) 1,2-Diphenyltetramethyldisilan,
320 ml Tetrahydrofuran und 120 ml Hexamethylphosphor säuretriamid
beschickt. Nach Kühlen
auf –5°C mit: einem
Eis-Aceton-Bad,
wurden 31 ml (43,4 mMol) 1,4 M Methyllithium in Diethylether zugegeben.
-
Das erhaltene dunkelbraune Gemisch
wurde bei –5°C bis 0°C für 0,5 Stunden
gerührt.
Nach Kühlen auf –60°C wurden
7,60 g (40,0 mMol) Kupfer-I-jodid in einer Portion zugegeben und
der Trichter wurde mit 50 ml Tetrahydrofuran gespült. Das
Reaktionsgemisch wurde langsam innerhalb 25 Minuten auf –35°C erwärmt, dann
für 0,5
Stunden bei –35°C bis –40°C gerührt. Während jener
Zeit fielen schwarze Feststoffe aus und der Überstand änderte sich zu fast farblos
oder schwach gelb.
-
Nach Kühlen auf –70°C wurde eine Lösung von
Diisobutylaluminiumhydrid [gesondert hergestellt durch Zugeben von
132 ml (732 mMol) Diisobutylaluminiumhydrid (rein) zu einem Gemisch
von 100 ml Tetrahydrofuran und 176 ml Hexamethylphosphorsäuretriamid
bei –10°C] unter
Halten der Temperatur des Gemisches unter –68°C über eine Kanüle gegeben.
Nach Kühlen
auf –70°C wurde eine
Lösung
von 100 g (609 mMol) des Ketons der Formel I in einem Gemisch von
100 ml Hexamethylphosphorsäuretriamid
und 100 ml Tetrahydrofuran innerhalb einer Stunde unter Halten der
Temperatur des Reaktionsgemisches bei –70°C bis –68°C zugegeben. Unmittelbar nach
der Zugabe der Verbindung der Formel I zeigte Dünnschichtchromatographieanalyse
vollständige
Reaktion an. Dann wurden 60 ml (1,16 mMol) Brom innerhalb 10 Minuten
zugegeben. Eine Exothermie folgte, die die Temperatur des Gemisches
auf –20°C anhob.
Nach Rühren
bei –20°C für 5 Minuten
wurde das Reaktionsgemisch in 1,3 kg Eiswasser, enthaltend 80 ml
Schwefelsäure,
gegossen.
-
Das erhaltene Gemisch wurde 20 Minuten
gerührt.
Der Reaktionskolben wurde mit insgesamt 100 ml Wasser gespült und die
Waschlaugen wurden zu dem gestoppten Gemisch vereinigt. Das erhaltene
Gemisch wurde mit 1,5 1 + 1,0 1 = 2,5 l Essigsäureethylester extrahiert und
die vereinigten organischen Schichten wurden mit 2 × 500 ml
= 1 l 5%iger Schwefelsäure
und dann mit 150 ml gesättigter
wässriger
Natriumbicarbonatlösung
gewaschen.
-
Die erhaltene Emulsion wurde durch
eine Lage von pulverförmigem
Kieselgelfiltriermittel filtriert und die organische Schicht wurde
abgetrennt, über
Natriumsulfat getrocknet und zur Trockne auf konzentriert. Der Rückstand
wurde unter Hochvakuum getrocknet und dann mit 250 ml Hexan verrieben
und der Überstand
wurde durch Abdekantieren entfernt. Der Rückstand wurde erneut mit 50
ml Hexan verrieben. Anschließend
wurde der Überstand
durch Dekantieren entfernt. Leer erhaltene Feststoff wurde unter
vermindertem Druck getrocknet, um das zurückbleibende Hexan zu entfernen
und mit 250 ml Diethylether verrieben. Nach Kühlen in einem Gefrierschrank
für 5 Minuten,
wurde der Feststoff durch Filtration gesammelt, mit kaltem Diethylether
gewaschen und trocken gesaugt, unter Gewinnung von 106 g (70,9%)
der Formel II ((3aR,4S,7aS)-4-Bromoctahydro-7a-methyl-1H-inden-l,5-dion) als einen
weißen
Feststoff.
-
Das Verfahren wurde kontrolliert
durch: Dünnschichtchromatographie
(1 : 1 Hexan : Essigsäureethylester,
Kurzwellen UV Nachweis und Phosphomolybdänsäureanfärbung, Rf I
= 0,6 und Rf I I = 0 , 7 5) und 1H NMR (CDCl3) .