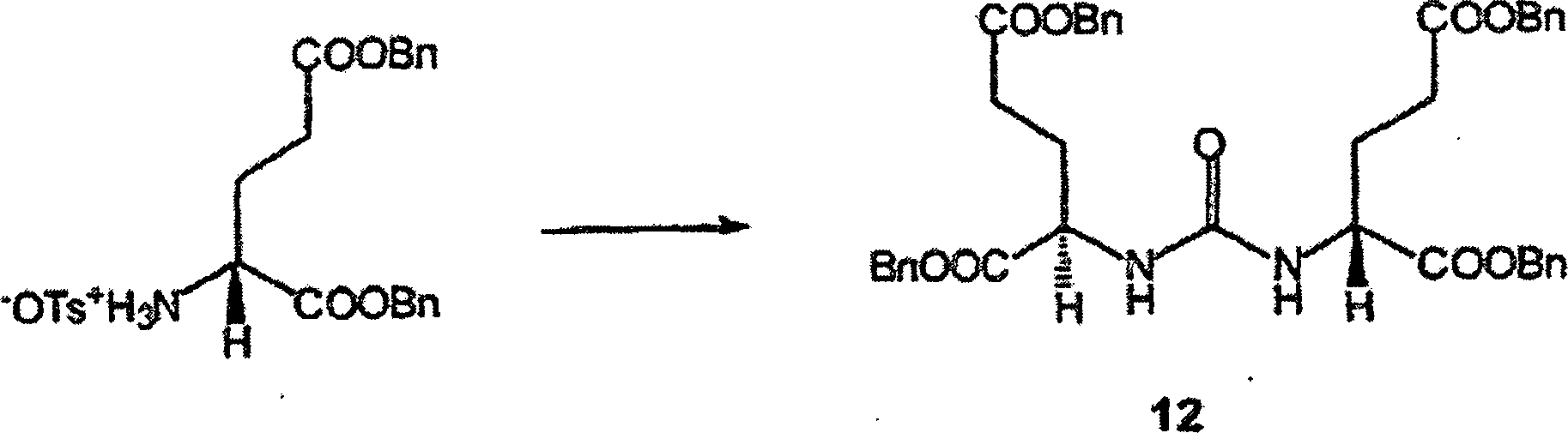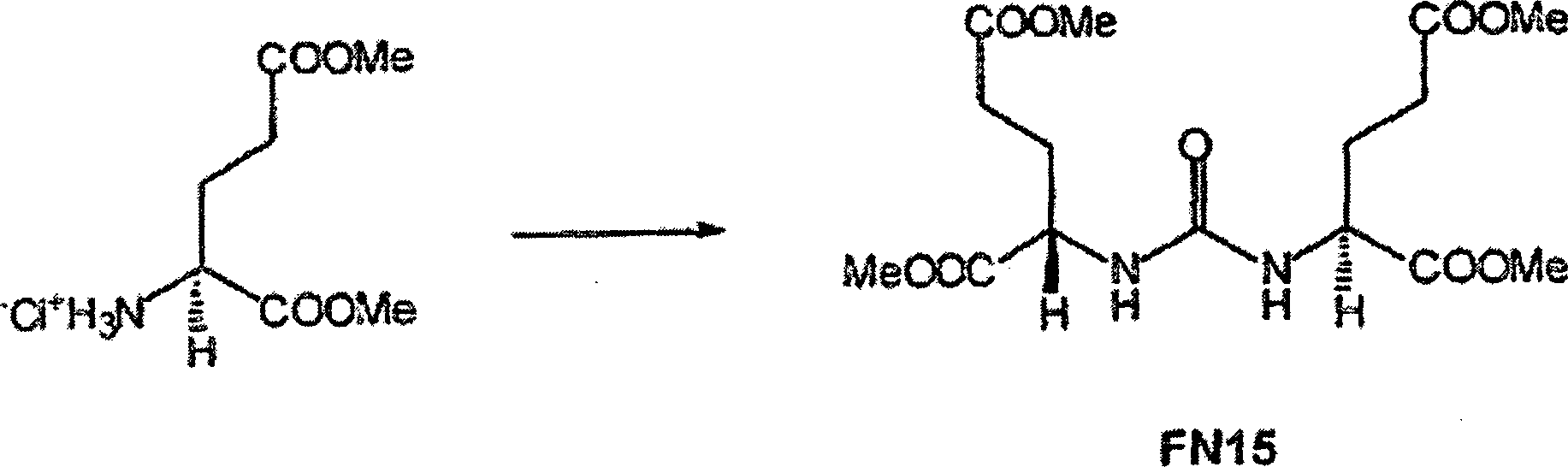-
Hintergrund
der Erfindung
-
Glutamat
ist ein bedeutender erregender Neurotransmitter im zentralen Nervensystem
von Säugetieren.
Die Neurotransmitter-Aktivität
von Glutamat wird primär
durch Liganden-gesteuerte Ionenkanäle vermittelt. Die Beobachtung,
dass Glutamat auch durch zweite Boten vermittelte Reaktionen induziert,
hat zu der Entdeckung einer eigenen Gruppe von an G-Proteine gekoppelten
Glutamat-Rezeptoren geführt,
welche als metabotropische Rezeptoren (mGluRs) bezeichnet werden.
Schoepp und Conn, Trends Pharmacol. Sci. 14: 13–20 (1993). Die erste beschriebene
Aktivität
der metabotropischen Glutamat-Rezeptoren
war Inositol-Phospholipid(PI)-Hydrolyse. Nicoletti et al., J. Neurochem.
46: 40–46
(1986) und Sugiyama et al., Nature 325: 531–533 (1987). Molekulare Klonierungstechniken
haben eine große
Familie von metabotropischen Rezeptoren mit verschiedenen Transduktionsmechanismen,
Expressionsmustern und Empfindlichkeiten gegenüber Glutamat-Agonisten offenbart.
Schoepp und Conn, supra.
-
Konsistent
mit der für
die metabotropischen Rezeptoren beobachteten molekularen Heterogenität haben
elektrophysiologische Studien diverse Rollen für diese Rezeptoren in synaptischer
Plastizität,
präsynaptischer
Inhibierung und Regulierung von Zellenerregbarkeit durch Ionenkanal-Modulation
nahegelegt. Bashir et al., Nature 363: 347–363 (1993); Linden et al.,
Neuron 7: 81–89
(1991); Baskys und Malenka, J. Physiol. (Lond.) 444: 687–701 (1991);
Charpak et al., Nature 347: 765–767
(1990); und Lester und Jahr, Neuron 5: 741–749 (1990). Jedoch sind die
spezifischen mGluR-Rezeptoren, welche diese zellulären Funktionen
vermitteln, im Großen
und Ganzen undefiniert.
-
Anzeichen
für eine
physiologische Rolle spezifischer mGluR-Subtypen sind aus Arbeiten mit selektiven
Agonisten und Antagonisten der Rezeptoren abgeleitet worden. Zum
Beispiel ist (1S,3R)-1-Aminocyclopentan-1,3-dicarbonsäure (ACPD)
ein selektiver und potenter Aktivator der mGluR1-, mGluR2-, mGluR3-
und mGluR5-Rezeptoren. Masu et al., Nature 349: 760–765 (1991);
Abe et al., J. Biol. Chem. 267: 13361–13368 (1992); Tanabe et al.,
Neuron 8: 169–179
(1992); und Tanabe et al., J. Neurosci. 13: 1372–1378 (1993). Es ist gezeigt
worden, dass L-2-Amino-4-phosphonobuttersäure (L-AP4) mGluR4 und mGluR6
aktiviert. Id., Thomsen et al., Eur. J. Pharmacol. 227: 361–362 (1992);
Nakajima et al., J. Biol. Chem. 268: 11868–11873 (1993). L-AP4 inhibiert
Transmitter-Freisetzung
und spannungs-abhängigen
Kalziumeintritt in ausgewählte
Gehirn- und Rückenmarks-Neuronen.
Koerner und Cotman, Brain Res. 216: 192–198 (1981); Trombley und Westbrook,
J. Neurosci. 12: 2043–2050
(1992); und Sahara und Westbrook, J. Neurosci. 13: 3041–3050 (1993).
In retinalen bipolaren Neuronen jedoch aktivieren postsynaptische
L-AP4-Rezeptoren Aphosphodiesterase. Nawy und Jahr, Nature 346:
269–271
(1990).
-
Es
können
innerhalb der gleichen Gruppe von Neuronen multiple mGluR-Subtypen
vorhanden sein. Da die zelluläre
und subzelluläre
Lokalisierung von spezifischen mGluRs wichtig sein kann bei der
Bildung eingehender Sinnesinformation, ist es wichtig, andere Rezeptoren
der mGluR-Gruppe zu identifizieren. Wenn sie einmal identifiziert
sind, können
spezifische Agonisten und Antagonisten hergestellt werden, um die
mit dem Rezeptor verbundenen Reaktionen zu modulieren. Recht überraschend
identifiziert die vorliegende Erfindung einen L-AP4-sensitiven Rezeptor,
welcher Transmitter-Freisetzung in Neuronen moduliert, welche weder mGluR4
noch mGluR6 exprimieren, und erfüllt
andere damit in Zusammenhang stehende Bedürfnisse.
-
Wie
bereits erwähnt
sind die metabotropischen Glutamat-Rezeptoren (mGluRs) eine heterogene
Familie von G-Protein-verbundenen
Rezeptoren, welche mit multiplen zweiten-Boten-Systemen gekoppelt sind. Diese umfassen
die negative Modulierung von Adenylatcyclase, Aktivierung von Phosphoinositid-spezifischer Phospholipase
C, und Modulierung von Ionenkanal-Strömen [Science, 1992, 258, 597;
Trends in Pharmacol. Sci. 1993, 14, 13; J. Med. Chem. 1995, 1417].
Drei Typen von mGluR-Rezeptoren sind identifiziert worden. Die Gruppe
I-Rezeptoren koppeln an Phosphoinositid-Hydrolyse und umfassen mGluR1 und mGluR5; Gruppe II-Rezeptoren
sind mit der Inhibierung der Bildung von cyclischem Adenosin 5'-Monophosphat (cAMP)
gekoppelt und umfassen mGluR2 und mGluR3. Gruppe III-Rezeptoren (mGluR4,
mGluR6, mGluR7 und
mGluR8) koppeln negativ an cAMP. Jeder der
mGluR-Subtypen ist damit auf der Basis seiner Pharmakologie und
Sequenzhomologie differenziert. Übermäßige Aktivierung
von Glutamat-Rezeptoren oder Störungen
in den zellulären Mechanismen,
welche gegen die möglichen
nachteiligen Konsequenzen von physiologischer Glutamat-Rezeptor-Aktivierung
schützen,
sind mit der Pathogenese einer Vielzahl von neurologischen Störungen in Verbindung
gebracht worden. Diese Störungen
umfassen Epilepsie, Ischämie,
Trauma des zentralen Nervensystems, neuropathischen Schmerz und
chronische neurodegenerative Krankheiten. Aufgrund der allgegenwärtigen Verteilung
von glutamatergen Synapsen haben mGluRs das Potential, an einer
großen
Vielfalt von Funktionen im zentralen Nervensystem von Säugetieren
teilzunehmen. Zusätzlich
existiert aufgrund der großen
Diversität
und heterogenen Verteilung der mGluR-Subtypen die Gelegenheit, hochselektive
Wirkstoffe zu entwickeln, welche eine begrenzte Anzahl von Funktionen
des zentralen Nervensystems von Säugetieren beeinflussen. Die
mGluRs stellen daher neue Targets für die Entwicklung von therapeutischen
Mitteln bereit, welche eine dramatische Auswirkung auf die Behandlung
einer großen
Vielfalt von psychiatrischen und neurologischen Störungen haben
könnte.
-
Ischämie, eine örtliche
begrenzte Gewebsanämie,
welche aus der Behinderung des Einflusses von arteriellem Blut resultiert,
kann erheblichen Schaden hervorrufen, wenn sie im Gehirn oder zentralen
Nervensystem auftritt. Zentrales Gewebe des zentralen Nervensystems,
und zu einem geringeren Ausmaß Gewebe
des peripheren Nervensystems, weist schlechte Instandsetzungseigenschaften
auf. Daher verursacht Schaden an Nervengewebe erhebliche permanente
Behinderung und stellt einen häufigen
Todesgrund dar. Schaden an Nervengewebe kann auf viele Arten und
Weisen auftreten, nicht nur durch Ischämie in zerebrovaskulären Unfällen, sondern
auch bei zerebralen Durchblutungsstörungen, Episoden absoluter
und relativer Hypoxie, bei metabolischen Störungen und verschiedenen Formen
von Traumata.
-
Globale
Ischämie
tritt unter Bedingungen auf, in welchen Blutfluss zum gesamten Gehirn
für eine
Zeitspanne aufhört, wie
es etwa als Folge von Herzstillstand auftreten kann. Fokale Ischämie tritt
unter Bedingungen auf, in welchen ein Abschnitt des Gehirns von
seiner normalen Blutzufuhr abgeschnitten ist, wie es etwa als Folge
von thromboembolytischer Okklusion eines zerebralen Gefäßes, traumatischer
Kopfverletzung, Ödemen
und Gehirntumoren auftreten kann. In Gebieten fokaler Ischämie oder
Schaden gibt es einen Kern mit ausgeprägterem Schaden, umgeben von
einer perifokalen Penumbra von geringerem Schaden. Dies kommt daher,
dass die Neuronen in der Penumbra eine zeitlang Homeostase aufrecht
erhalten können,
wodurch sie potentiell besser mittels pharmakologischer Mittel gerettet
werden können.
-
Sowohl
globale als auch fokale ischämische
Zustände
weisen das Potential auf, weit ausgedehnten neuronalen Schaden zu
erzeugen, selbst wenn der ischämische
Zustand vorübergehend
ist. Obwohl eine permanente neuronale Verletzung in dem anfänglichen
Gemisch auf das Aufhören
des Blutflusses im Gehirn folgend auftreten kann, tritt der Schaden
bei globaler und fokaler Ischämie über Stunden
oder selbst Tage nach Einsetzen der Ischämie auf. Viel dieses neuronalen
Schadens wird der Glutamat-Toxizität und sekundären Konsequenzen
von Reperfusion des Gewebes zugeschrieben, wie etwa der Freisetzung
von vasoaktiven Produkten durch beschädigtes Endothel und die Freisetzung
von zytotoxischen Produkten von dem beschädigtem Gewebe, einschließlich freier
Radikale, Leukotrienen und dergleichen.
-
Glutamat-Neurotoxizität, welches
ein bedeutender Faktor bei ischämischer
neuronaler Verletzung ist, scheint mit der Aufnahme des oxidativen
Metabolismus zu beginnen und tritt damit sowohl während reversibler Ischämie als
auch während der
Erholung auf. Es sind viele Versuche unternommen worden, dieses
Problem zu vermeiden durch Blockierung der verschiedenen Rezeptoren,
einschließlich
NMDA-Rezeptoren, AMPA-Rezeptoren, Kainate-Rezeptoren und MGR-Rezeptoren,
welche durch Glutamat stimuliert werden und auch sehr stark in nach
dem Einsetzen von globaler oder fokaler Ischämie auftretendem Nervenzellentod
involviert sind. Wenn Ischämie
auftritt, wie etwa während
eines Schlags oder Herzanfalls, kommt es zu einer übermäßigen Freisetzung
von endogenem Glutamat, was zu der Überstimulierung von NMDA-Rezeptoren,
AMPA-Rezeptoren, Kainate-Rezeptoren und MGR-Rezeptoren führt. Wechselwirkung
des Glutamats mit diesen Rezeptoren verursacht, dass sich der Ionenkanal,
welcher mit diesen Rezeptoren in Verbindung steht, öffnet, und
so einen Fluss von Kationen durch die Zellmembran erlaubt. Dieser
Ionenfluss, insbesondere von Ca2+ in die
Zellen, spielt eine wichtige Rolle bei Nervenzelltod.
-
Prostatakrebs
ist jetzt die führende
Krebsform bei Männern
und die zweithäufigste
Ursache für
Tod durch Krebs bei Männern.
Es wird geschätzt,
dass 1993 mehr als 165.000 neue Fälle von Prostatakrebs diagnostiziert
wurden, und mehr als 35.000 Männer
starben in dem Jahr an Prostatakrebs. Zusätzlich ist das Auftreten von
Prostatakrebs seit 1981 um 50% gestiegen, und die Mortalität aufgrund
dieser Krankheit ist fortdauernd gestiegen. Zuvor sind die meisten
Männer
an anderen Krankheiten oder Erkrankungen gestorben, bevor sie an
ihrem Prostatakrebs starben. Wir stehen jetzt einer erhöhten Morbidität von Prostatakrebs
gegenüber, da
Männer
länger
leben und die Krankheit die Gelegenheit erhält, fortzuschreiten. Gegenwärtige Therapien
für Prostatakrebs
legen ihren Schwerpunkt exklusiv auf die Verringerung der Dihydrotestosteron-Gehalte,
um das Wachstum von Prostatakrebs zu verringern oder zu verhindern.
Zusätzlich
zu der Verwendung von digitaler rektaler Untersuchung und transrektaler
Ultrasonografie wird bei der Diagnose von Prostatakrebs häufig die Prostata-spezifische
Antigen(PSA)-Konzentration verwendet.
-
PSA
ist ein Protein, welches von Prostatazellen erzeugt wird, und kommt
im Blut von Männern,
welche Prostatakrebs haben, häufig
in erhöhten
Gehalten vor. Es ist gezeigt worden, dass PSA mit der Tumorbelastung
korreliert, als ein Indikator metastatischer Involvierung dient
und einen Parameter bereitstellt, um die Reaktion auf Operation,
Bestrahlung und Androgen-Ersatz-Therapie bei Prostatakrebspatienten
zu verfolgen. Es sollte zur Kenntnis genommen werden, dass Prostata-spezifisches
Antigen (PSA) ein völlig
unterschiedliches Protein ist als Prostata-spezifisches Membran-Antigen (PSMA).
Die beiden Proteine weisen verschiedene Strukturen und Funktionen
auf und sollten nicht aufgrund ihrer ähnlichen Nomenklatur miteinander
verwechselt werden.
-
1993
wurde von der molekularen Klonierung eines Prostata-spezifischen Membran-Antigens
(PSMA) als einem potenziellen Prostatakarzinom-Marker berichtet
und die Hypothese aufgestellt, es könnte als ein Target für Abbildung
und zytotoxische Behandlungsmodalitäten für Prostatakrebs dienen. Antikörper gegen PSMA
sind beschrieben und klinisch hinsichtlich der Diagnose und Behandlung
von Prostatakrebs untersucht worden. Insbesondere sind Indium-111-markierte
PSMA-Antikörper
beschrieben und hinsichtlich der Diagnose von Prostatakrebs untersucht
worden, und Yttrium-markierte PSMA-Antikörper sind beschrieben und mit
Hinblick auf die Behandlung von Prostatakrebs untersucht worden.
-
PSMA
wird im duktalen Epithel der Prostata exprimiert und kommt in seminalem
Plasma, Prostatafluid und Urin vor. 1996 hat man herausgefunden,
dass die Expression von PSMA cDNA tatsächlich die Aktivität von NAALADase
vermittelt. Dies ist völlig
unerwartet, da bis vor kurzem Forschung bezüglich NAALADase auf seine Rolle
im Gehirn und seine Wirkung auf Neurotransmitter beschränkt war,
während
PSMA beschrieben und in Hinblick auf die Diagnose und Therapie von
Prostatakrebs untersucht wurde.
-
Das
Dipeptid NAAG ist ein reichlich vorhandenes, für das Nervensystem spezifisches
Peptid, welches in synaptischen Vesikeln vorkommt und bei neuronaler
Stimulierung in mehreren Systemen freigesetzt wird. Als eine bedeutende
peptidische Komponente des Gehirns kommt NAAG in Gehalten vor, welche
mit denen des bedeutenden inhibierenden Neurotransmitters Gamma-Aminobutansäure (GADA)
vergleichbar sind. Obgleich NAAG erstmals 1964 isoliert wurde, gab
es wenig Aktivität
zur Aufklärung
seiner Rolle im zentralen Nervensystem von Säugetieren, bis die schädliche Natur
von überschüssigem Glutamat
bei einer Vielfalt von Krankheitszuständen offenkundig wurde. Aufgrund
seiner strukturellen Ähnlichkeit
mit Glutamat ist für
NAAG vorgeschlagen worden, es habe eine Vielfalt von Rollen ähnlich denen
des Glutamats selbst, einschließlich
der Funktion als ein Neurotransmitter oder ein Co-Transmitter, Neuromodulator
oder als ein Vorgänger
des Neurotransmitters Glutamat. NAAG hat erregende Reaktionen sowohl
in vitro als auch in vivo hervorgerufen, ist jedoch signifikant
weniger potent als Glutamat.
-
1988
wurde ein Gehirnenzym, NAALADase, identifiziert, welches NAAG zu
N-Acetylaspartat (NAA) und Glutamat hydrolisiert. NAALADase, welche
ihren Namen von ihrer strukturellen Spezifität für N-acetylierte saure Dipeptide
ableitet, ist eine Membran-gebundene Metallopeptidase mit einer
detanurierten molekularen Masse von 94 kDa[x], welche NAAG zu N-Acetylaspartat
(NAA) und Glutamat katabolisiert. Es ist gezeigt worden, dass [3H]NAAG in vivo durch ein Enzym mit den pharmakologischen
Eigenschaften von NAALADase abgebaut wird, was unterstützt, dass
NAALADase in dem Metabolismus von endogenem NAAG eine Rolle spielt.
-
Ratten-NAALADase-Aktivität ist extensiv
charakterisiert worden und zeigt eine hohe Affinität für Hydrolyse
ihres putativen Substrats NAAG, mit Km = 140 nM. Neulich ist auch
gezeigt worden, dass NAALADase das nicht-acetylierte Peptid, Aspartylglutamat,
mit hoher Affinität
spaltet. Von der Forschung ist auch gefunden worden, dass das Enzym
Membran-gebunden ist, von Chloridionen stimuliert wird, und durch
polyvalente Kationen-Chelatoren inhibiert wird, was nahe legt, dass
es eine Metallopeptidase ist.
-
In
Tieren ist NAALADase in synaptischen Plasma-Membranen angereichert
und ist örtlich
primär
auf neurales Gewebe und die Nieren beschränkt. NAALADase ist nicht in
großen
Mengen in Säugetier-Leber, -Herz,
-Bauchspeicheldrüse
oder -Milz gefunden wurde. Untersuchung von NAAG und NAALADase ist
für mehrere
verschiedene menschliche und tierische pathologische Zustände durchgeführt worden.
Es ist gezeigt worden, dass intra-hippocampale Injektionen von NAAG
verlängerte
Anfall-Aktivität
hervorrufen. In jüngerer Zeit
ist berichtet worden, dass Ratten, welche genetisch für epileptische
Anfälle
anfällig
sind, eine anhaltende Erhöhung
ihres Grundniveaus von NAALADase-Aktivität aufweisen. Diese Beobachtungen
sind mit der Hypothese konsistent, dass eine erhöhte Verfügbarkeit von synaptischem Glutamat
die Empfindlichkeit für
Anfälle erhöht, und
legen nahe, dass NAALADase-Inhibitoren anti-epileptische Aktivität bereitstellen könnten.
-
NAAG
und NAALADase sind auch in Verbindung gebracht worden mit der Pathogenese
von ALS und der pathologisch ähnlichen
Tierkrankheit, die als vererbliche hündische spinale Muskelatrophie
bezeichnet wird (Hereditary Canine Spinal Muscular Atrophy HCSMA)
genannt. Es ist gezeigt worden, dass Konzentrationen von NAAG und
seinen Metaboliten – NAA,
Glutamat und Aspartat – in
dem zerebrospinalen Fluid von ALS-Patienten und HCSMA-Hunden zwei-
bis dreifach erhöht
sind.
-
Zusätzlich ist
die Aktivität
von NAALADase in post-mortem Rückenmarksgewebe
von ALS-Patienten und HCSMA-Hunden signifikant erhöht (zwei-
bis dreifach). Obwohl dies sehr stark spekulativ ist, könnten NAALADase-Inhibitoren
beim Beschränken
der Progression von ALS klinisch nützlich sein, wenn ein erhöhter Metabolismus
von NAAG für
die Veränderungen
der CSF-Gehalte dieser sauren Aminosäuren und Peptide verantwortlich
ist. Abnormalitäten
bei NAAG-Gehalten
und NAALADase-Aktivität
sind auch bei post-mortem schizophrenischem Gehirn dokumentiert
worden, spezifisch in den präfrontalen
und limbischen Gehirnregionen, was die Wichtigkeit einer Untersuchung
des Metabolismus von NAAG in der Pathophysiologie von Schizophrenie
unterstreicht. Die Identifizierung und Reinigung von NAALADase führte zu
dem Vorschlag einer weiteren Rolle für NAAG: spezifisch, dass das
Dipeptid als eine Speicherform synaptischen Glutamats dienen könnte.
-
Nur
wenige NAALADase-Inhibitoren sind identifiziert worden, und diejenigen,
welche identifiziert wurden, sind nur in nicht-klinischer neurologischer
Forschung verwendet worden. Beispiele für solche Inhibitoren umfassen
allgemeine Metallopeptidase-Inhibitoren, wie etwa o-Phenanthrolen,
Metall-Chelatoren, wie etwa EGTA und EDTA, und Peptid-Analoge, wie etwa
Quisqualinsäure
und beta-NAAG.
-
Beschreibung
der Erfindung
-
Bestimmte
Ausführungsformen
der vorliegenden Erfindung betreffen neue Liganden für metabotropische
Glutamat-Rezeptoren
und Zusammensetzungen, welche die Liganden umfassen. Die pharmazeutischen Zusammensetzungen
können
verwendet werden, um Glutamat-Rezeptor-gesteuerte Zellen zu beeinflussen, einschließlich Neuronen
und Gliazellen in dem zentralen Nervensystem.
-
In
weiteren Ausführungsformen
besteht die vorliegende Erfindung aus Inhibitoren der NAALADase-Enzymaktivität und diese
umfassende Zusammensetzungen. Zusätzliche Ausführungsformen
der vorliegenden Erfindung bestehen aus Verfahren zum Behandeln
von Glutamat-Abnormalitäten
und damit in Verbindung stehender Schädigung von Nervengewebe in
einem Tier durch Inhibierung von NAALADase-Enzym mit den zuvor genannten
Inhibitoren oder Zusammensetzungen davon.
-
Kurze Beschreibung
der Figuren
-
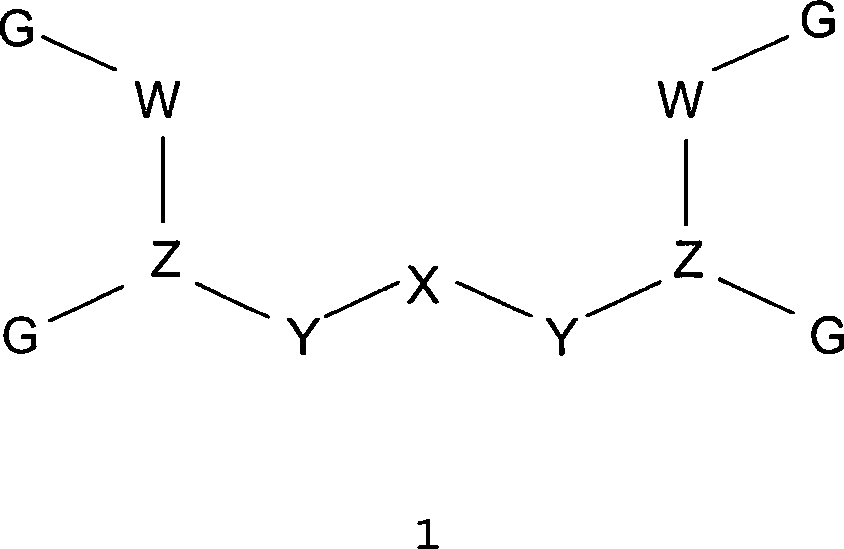
1 stellt
bestimmte bevorzugte Ausführungsformen
der Verbindungen gemäß der vorliegenden
Erfindung dar.
-
2 zeigt
die Wirkungen von sechs Verbindungen gemäß der vorliegenden Erfindung
auf die Aktivität
von NAALADase.
-
3 zeigt
die Wirkung einer Verbindung gemäß der vorliegenden
Erfindung auf sechs Subtypen von metabotropischen Glutamat-Rezeptoren
(mGluRs).
-
4 zeigt
die Wirkung einer Verbindung gemäß der vorliegenden
Erfindung auf einen einzelnen Subtypus von metabotropischem Glutamat-Rezeptor.
-
5 zeigt
die Wirkungen einer Verbindung gemäß der vorliegenden Erfindung
und 2-(Phosphomethyl)pentandisäure (PMPA)
auf die Aktivität
von NAAG-Peptidase in Rattenhirn-Membranen.
-
6 zeigt
die Wirkungen einer Verbindung gemäß der vorliegenden Erfindung,
PMPA und Quis auf die Aktivität
von Ratten-NAAG-Peptidase.
-
7 zeigt
die Wirkungen von fünf
erfindungsgemäßen Verbindungen
auf die Aktivität
von Ratten-NAAG-Peptidase.
-
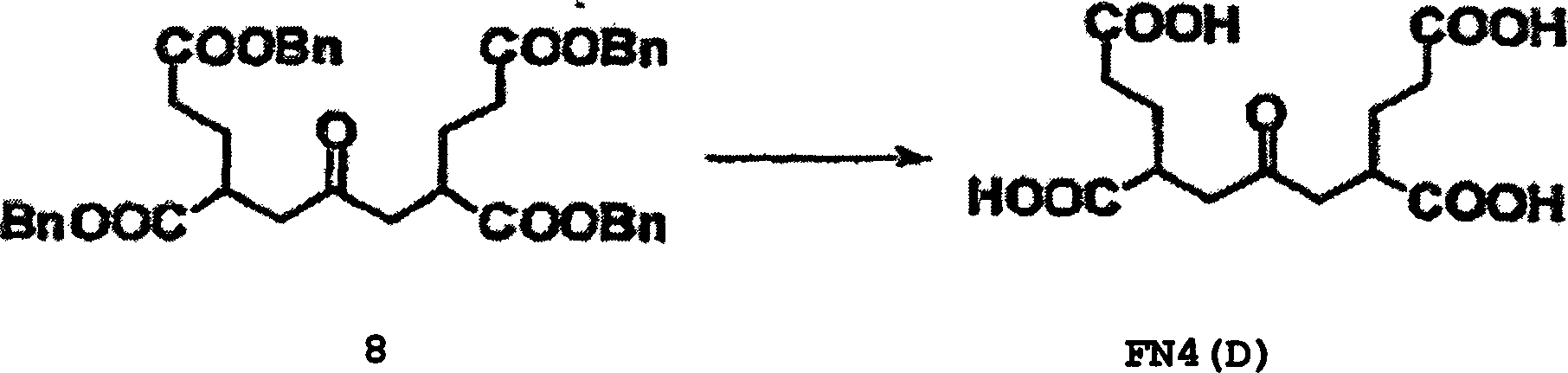
8 zeigt
die Wirkungen einer erfindungsgemäßen Verbindung, PMPA und Quis
auf die Aktivität
von Prostata-spezifischem Membran-Antigen (PMSA).
-
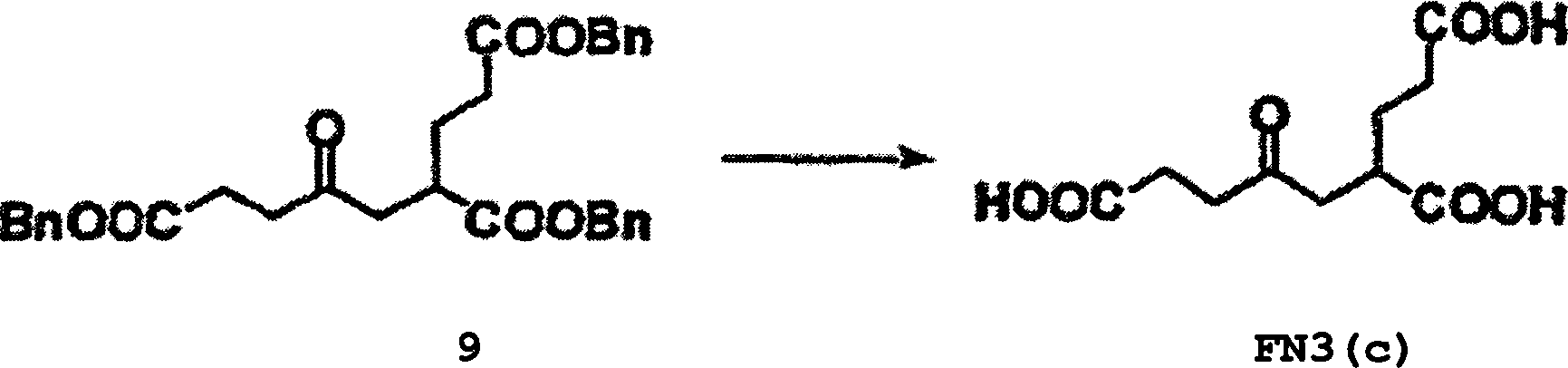
9 zeigt
die Wirkungen zweier erfindungsgemäßer Verbindungen auf die Aktivität von Ratten-NAAG-Peptidase.
-
10 zeigt bestimmte erfindungsgemäße Verbindungen,
ihre Aktivität
gegen NAAG-Peptidase und ihre Aktivität gegen bestimmte metabotropische
Glutamat-Rezeptoren.
-
11 zeigt bestimmte Verbindungen gemäß der vorliegenden
Erfindung, ihre Aktivität
gegen NAAG-Peptidase und ihre Aktivität gegen bestimmte metabotropische
Glutamat-Rezeptoren.
-
12 zeigt bestimmte Verbindungen gemäß der vorliegenden
Erfindung, ihre Aktivität
gegen NAAG-Peptidase und ihre Aktivität gegen bestimmte metabotropische
Glutamat-Rezeptoren.
-
13 zeigt bei geringer Vergrößerung die antiangiogene Wirkung
einer Verbindung gemäß der vorliegenden
Erfindung auf ein Glioblastom-Xenotransplantat.
-
14 zeigt bei hoher Vergrößerung die antiangiogene Wirkung
einer Verbindung gemäß der vorliegenden
Erfindung auf ein Glioblastom-Xenotransplantat.
-
15 stellt eine Auftragung von Tumorvolumen gegen
Behandlungsdauer für
Glioblastom-Xenotransplantate
dar, welche mit einer erfindungsgemäßen Verbindung bei 10 und 100 μM behandelt
wurden, dargestellt gegenüber
einem Vergleich.
-
Detaillierte
Beschreibung der Erfindung
-
Bis
dato sind alle herkömmlich
verwendeten Agonisten und Antagonisten, welche in biologischen Studien
der mGluRs verwendet werden, Aminosäuren, welche oft einen strukturell
versteiften Glutamat-ähnlichen Kern
verkörpern
[Neuropharmacology, 36, 1–11
(1997); Neuropharmacology 37, 1–12
(1998); Neuropharmacology 35, 1661–1672 (1996); J. Med. Chem.
38, 1417 (1995); J. Med. Chem. 41, 347 (1998); Current Pharmaceutical
Design, 1, 355 (1995)]. Während unserer
Bemühungen,
potente und selektive Liganden zu identifizieren, welche auf diese
Rezeptoren wirken, haben wir mGluR3-selektive
Agonisten entdeckt, welche nur Säuregruppen
enthalten.
-
Unsere
Studien begannen mit dem Dipeptid N-Acetyl-L-aspartat-L-glutamat (NAAG), einem Dipeptid, welches
im Gehirn relativ reichlich vorkommt, und von welchem geglaubt wird,
dass es als ein Transmitter oder Co-Transmitter im zentralen Nervensystem
wirkt, sehr wie Glutamat selbst. NAAG zeigt sowohl in vitro als
auch in vivo Erreger-Eigenschaften,
ist aber weniger aktiv als Glutamat und stellt eine Speicherform
von Glutamat dar. In Studien, in welchen mit mGluR1-6 transfizierte
Zelllinien verwendet werden, wurde gefunden, dass NAAG den mGluR3-Rezeptor selektiv aktiviert, mit einem
EC50-Wert im Bereich von 65 ± 20 μM [J. Neurochem. 69,
174 (1997)]. Neulich wurde ein Gehirnenzym identifiziert, welches
für die
Spaltung von N-acetylierten α-verbundenen
sauren Dipeptiden spezifisch ist, und demzufolge wurde das Enzym
NAALADase benannt [J. Biol. Chem., 1987, 262, 14498].
-
-
NAAG
und NAALADase sind mit mehreren pathologischen Zuständen, welche
mit Glutamat-Abnormalitäten
und Neurotoxizität
in Zusammenhang stehen, in Verbindung gebracht worden. Zum Beispiel
ist gezeigt worden, dass intra-hippocampale Injektionen von NAAG
verlängerte
Anfalls-Aktivität
hervorrufen [Proc. Natl. Acad. Sci. USA 80 (1983), 1116–1119].
Es ist auch berichtet worden, dass Ratten, welche genetisch für epileptische
Anfälle
anfällig
sind, eine anhaltende Erhöhung
ihrer Grundgehalte an NAALADase-Aktivität aufweisen [Brain Research,
593 (1992), 140–143].
Diese Ergebnisse unterstützen
die Hypothese, dass die erhöhte
Erhältlichkeit
von synaptischem Glutamat die Empfindlichkeit für Anfälle erhöht. Als ein Ergebnis davon
ist vorgeschlagen worden, dass NAALADase-Inhibitoren wirksame anti-epileptische
Therapien bereitstellen könnten.
-
NAAG
und NAALADase sind auch mit der Pathogenese von ALS (amyotrophischer
Lateralsklerose) [Brain Research, 556 (1991), 151–156] in
Verbindung gebracht worden. Von daher könnten NAALADase-Inhibitoren
klinisch nützlich
sein bei der Beschränkung
der Progression von ALS, wenn ein erhöhter Metabolismus von NAAG
für Änderungen
des CSF-Gehalts dieser sauren Aminosäuren verantwortlich ist [siehe
Ann. Neurol. 28 (1990), 18–25].
-
Bestimmte
Phosphonat-Analoge von NAAG, wie etwa 2-(Phosphonomethyl)-pentandisäure, wirken Berichten
zufolge als potente Inhibitoren von NAALADase [J. Med. Chem. 39,
619 (1996)]. Interessanterweise haben wir gefunden, dass während diese
Verbindung Berichten gemäß wenig
in Richtung Rezeptoraktivität zeigt,
sie tatsächlich
als ein Agonist auf mGluR3 wirkt.
-
Erfindungsgemäße Verbindungen
-
Auf
der Basis dieses unerwarteten Ergebnisses haben wir die Aktivität von damit
in Zusammenhang stehenden Analogen erforscht. Als Teil unserer Designstrategie
haben wir beschlossen, die Aktivität von solchen NAAG-ähnlichen
Analogen zu erforschen, denen die Amid-Bindung zwischen dem Asp-
und Glu-Rest (die standardgemäße Ketomethylen-Typ-Substitution) fehlt,
aber aus welchen auch die N-Acetyl-Gruppe entfernt wurde, da diese besondere
Gruppe Berichten zufolge kein absolutes Erfordernis für NAALADase-Aktivität ist.
-
Demgemäss wurde
eine Serie von Verbindungen hergestellt und hinsichtlich mGluR-Aktivität untersucht.
Von den synthetisierten Verbindungen erwies sich die Verbindung,
welche eine von den zwei Pentandisäuregruppen (A) flankierte Acetongruppe
enthält,
als interessant, da sie signifikante mGluR3-Aktivität beibehielt.
-
-
Basierend
auf dieser Beobachtung haben wir uns entschieden, die Aktivität von Verbindungen
zu erforschen, in welchen die zentrale Carbonylgruppe von A durch
P(O)OH, CHOH, und R3CHOH ersetzt war, mit dem
Gedanken, dass diese Verbindung nicht nur als ein mGluR3-selektiver
Ligand agieren könnte,
sondern dass sie auch als NAALADase-Inhibitor fungieren könnte. Die
Phosphorverbindung (B) war als ein NAALADase-Inhibitor besonders potent, mit einer
IC50 von 4 nM. Zusätzliche Daten zu den neuen
Verbindungen werden hierin bereitgestellt.
-
-
In
bestimmten Ausführungsformen
sind die Verbindungen gemäß der vorliegenden
Erfindung durch die verallgemeinerte Struktur 1 dargestellt:
wobei X ausgewählt ist
aus der Gruppe bestehend aus -C(O)-, -C(S)-, -P(O)(OR)-, -P(O)(NR
2)-, -P(NR)(OR)-, -P(NR)(NR
2)-,
-C(R)(OR)-, -C(R)(SR)-, -C(NR)-, -NR(O)-, und -N(OR)-;
Y für jedes
einzelne Auftreten unabhängig
ausgewählt
ist aus der Gruppe bestehend aus (CR
2)
n, (NR)
n und einer
Bindung;
Z für
jedes einzelne Auftreten unabhängig
ausgewählt
ist aus der Gruppe bestehend aus C(R), C(NR
2), C(NHAcyl);
W
für jedes
einzelne Auftreten unabhängig
ausgewählt
ist aus der Gruppe bestehend aus (CR
2)
m und einer Bindung;
G für jedes
einzelne Auftreten unabhängig
ausgewählt
ist aus der Gruppe bestehend aus -COOH, -C(O)NHOH, -C(O)NH
2, -C(S)SH, -SO
3H,
-SO
2H, -SOH, -S(O)
2NH
2, -P(O)(OH)
2 und
-P(OH)
2;
R für jedes einzelne Auftreten
unabhängig
ausgewählt
ist aus der Gruppe bestehend aus H, Alkyl, Heteroalkyl, Aryl, Heteroaryl
und Aralkyl; und auch umfassend eine negative Ladung für Fälle, in
denen R an ein Heteroatom gebunden ist; und m und n ganze Zahlen
sind, welche für
jedes einzelne Auftreten unabhängig
ausgewählt sind
aus dem Bereich von 0 bis 3 inklusive.
-
In
bestimmten Ausführungsformen
sind die Verbindungen gemäß der vorliegenden
Erfindung durch die verallgemeinerte Struktur 1 und die begleitenden
Definitionen dargestellt, wobei X ausgewählt ist aus der Gruppe bestehend
aus -P(O)(OR)- und -P(O)(NR2)-.
-
In
bestimmten Ausführungsformen
sind die Verbindungen gemäß der vorliegenden
Erfindung durch die verallgemeinerte Struktur 1 und die begleitenden
Definitionen dargestellt, wobei Y gleich (CR2)n ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei Y gleich (CR2)n ist; und n gleich 1 ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei Z gleich C(R) ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei W gleich (CR2)m ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei W gleich (CR2)m ist; und m gleich 2 ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei G unabhängig
für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH, -C(O)NHOH, -C(O)NH2, -SO3H, -SO2H, -S(O)2NH2 und -P(O)(OH)2.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei G für
jedes Auftreten unabhängig
ausgewählt
ist aus der Gruppe bestehend aus -COOH, -C(O)NHOH, -SO3H,
und -P(O)(OH)2.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei G unabhängig
für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH und -C(O)NHOH.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei G gleich -COOH ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei m und n ganze Zahlen sind, welche für jedes
Auftreten unabhängig
ausgewählt
sind aus dem Bereich von 1 bis 2 inklusive.
-
In
bestimmten Ausführungsformen
sind die Verbindungen gemäß der vorliegenden
Erfindung durch die verallgemeinerte Struktur 1 und die begleitenden
Definitionen dargestellt, wobei X ausgewählt ist aus der Gruppe bestehend
aus -C(O)-, -C(S)-, -C(NR)-, -C(R)(OR)-, -C(R)(SR)-, -P(O)(OR)-,
-P(O)(NR2)-, -P(NR)(OR)- und -P(NR)(NR2)-; und Y gleich (CR2)n ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -C(O)-, -C(S)-, -C(NR)-, -C(R)(OR)-,
-C(R)(SR)-, -P(O)(OR)-, -P(O)(NR2)-, -P(NR)(OR)- und -P(NR)(NR2)-; Y gleich (CR2)n ist; und Z unabhängig für jedes Auftreten ausgewählt ist
aus der Gruppe bestehend aus C(R), C(NR2)
und C(NHAcyl).
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -C(O)-, -C(S)-, -C(NR)-, -C(R)(OR)-,
-C(R)(SR)-, -P(O)(OR)-, -P(O)(NR2)-, -P(NR)(OR)- und -P(NR)(NR2)-; Y gleich (CR2)n ist; Z unabhängig für jedes Auftreten ausgewählt ist
aus der Gruppe bestehend aus C(R), C(NR2)
und C(NHAcyl); und W gleich (CR2)m ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -C(O)-, -C(S)-, -C(NR)-, -C(R)(OR)-,
-C(R)(SR)-, -P(O)(OR)-, -P(O)(NR2)-, -P(NR)(OR)- und -P(NR)(NR2)-; Y gleich (CR2)n ist; Z unabhängig für jedes Auftreten ausgewählt ist
aus der Gruppe bestehend aus C(R), C(NR2)
und C(NHAcyl); W gleich (CR2)m ist;
und G unabhängig
für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH, -C(O)NHOH, -C(O)NH2, -SO3H, -SO2H, -S(O)2NH2 und -P(O)(OH)2.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -C(O)-, -C(S)-, -C(NR)-, -C(R)(OR)-,
-C(R)(SR)-, -P(O)(OR)-, -P(O)(NR2)-, -P(NR)(OR)- und -P(NR)(NR2)-; Y gleich (CR2)n ist; Z unabhängig für jedes Auftreten ausgewählt ist
aus der Gruppe bestehend aus C(R), C(NR2)
und C(NHAcyl); W gleich (CR2)m ist;
G unabhängig
für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH, -C(O)NHOH, -C(O)NH2, -SO3H, -SO2H, -S(O)2NH2 und -P(O)(OH)2;
und m und n ganze Zahlen sind, welche für jedes Auftreten unabhängig ausgewählt sind
aus dem Bereich von 1 bis 2 inklusive.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -P(O)(OR)- und -P(O)(NR2)-; und Y gleich (CR2)n ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -P(O)(OR)- und -P(O)(NR2)-; Y gleich (CR2)n ist; und Z gleich C(R) ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -P(O)(OR)- und -P(O)(NR2)-; und Y gleich (CR2)n ist; Z gleich C(R) ist; und W gleich (CR2)m ist.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -P(O)(OR)- und -P(O)(NR2)-; und Y gleich (CR2)n ist; Z gleich C(R) ist; W gleich (CR2)m ist; und G unabhängig für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH und -C(O)NHOH.
-
In
bestimmten Ausführungsformen
sind die erfindungsgemäßen Verbindungen
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei X ausgewählt
ist aus der Gruppe bestehend aus -P(O)(OR)- und -P(O)(NR2)-; Y gleich (CR2)n ist; Z gleich C(R) ist; W gleich (CR2)m ist; G unabhängig für jedes
Auftreten ausgewählt
ist aus der Gruppe bestehend aus -COOH und -C(O)NHOH; und m und
n ganze Zahlen sind, welche für
jedes Auftreten unabhängig
ausgewählt
sind aus dem Bereich von 1 bis 2 inklusive.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Ligand für einen metabotropischen Glutamat-Rezeptor
ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Agonist eines metabotropischen
Glutamat-Rezeptors ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Antagonist eines metabotropischen
Glutamat-Rezeptors ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Ligand für einen einzelnen Subtypus
eines metabotropischen Glutamat-Rezeptors ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Agonist eines einzelnen Subtypus
eines metabotropischen Glutamat-Rezeptors ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Antagonist eines einzelnen Subtypus
eines metabotropischen Glutamat-Rezeptors ist.
-
In
bestimmten Ausführungsformen
ist eine erfindungsgemäße Verbindung
durch die verallgemeinerte Struktur 1 und die begleitenden Definitionen
dargestellt, wobei die Verbindung ein Inhibitor von NAALADase ist.
-
Ein
weiterer Aspekt der vorliegenden Erfindung betrifft Verfahren zum
Behandeln von Ischämie,
insbesondere globaler und fokaler Ischämie, unter Verwendung von Zusammensetzungen,
welche die Enzymaktivität
von N-acetylierter
alpha-verbundener saurer Dipeptidase (NAALADase) in Menschen und
warmblütigen Tieren
inhibieren. Bestimmte Verbindungen, welche durch die verallgemeinerte
Struktur 1 dargestellt sind, sind Inhibitoren für NAALADase. Fachleute auf
diesem Gebiet werden fähig
sein, unter Verwendung von nicht mehr als Routineexperimenten herauszufinden,
welche Verbindungen gemäß der vorliegenden
Erfindung Antagonisten von NAALADase sind.
-
NAALADase
ist ein Enzym, welches eine Membran-gebundene Metalloprotease ist,
welche das Dipeptid, N-Acetyl-L-aspartat-L-glutamat
(NAAG) hydrolisiert, um Glutamat und N-Acetylaspartat zu erzielen. Die erfindungsgemäßen Verfahren
umfassen die Verwendung von Phosphinsäure-Derivate enthaltenden Zusammensetzungen,
welche NAALADase-Enzym-Aktivität inhibieren
und welche sich zur Behandlung von Ischämie als nützlich erwiesen haben. Die
Aminosäure
L-Glutamat ist ein
Neurotransmitter, welcher schnelle neuronale Erregung in einer Vielzahl
von Synapsen in dem zentralen Nervensystem (CNS) vermittelt. Wenn
es einmal in die Synapse freigesetzt wurde, kann L-Glutamat den
N-Methyl-D-Aspartat
(NMDA)Rezeptor stimulieren, einen Subtypus eines erregenden Aminosäurerezeptors.
Es ist entdeckt worden, dass übermäßige Aktivierung
des NMDA- Rezeptors
in eine Vielzahl von akuten sowie chronischen neuropathologischen
Prozessen involviert ist, wie etwa Ischämie, Epilepsie und Huntington's Krankheit. Daher
sind beachtliche Anstrengungen auf das Auffinden neuer therapeutischer
Mittel schwerpunktmäßig gerichtet
worden, um die postsynaptischen Wirkungen von L-Glutamat, welche
durch den NMDA-Rezeptor vermittelt werden, zu antagonisieren.
-
Bestimmte
Ausführungsformen
der vorliegenden Erfindung bestehen aus einem Verfahren zum Behandeln
von Ischämie,
welches den Schritt der Verabreichung eines NAALADase-Inhibitors und pharmazeutisch
akzeptablen Trägers
für den
NAALADase-Inhibitor an ein an Ischämie leidendes Tier umfasst.
In Verfahren zur Behandlung von Schlaganfall, insbesondere akutem
ischämischen
Schlaganfall, und globaler Ischämie,
welche durch Ertrinken, Kopftrauma usw. verursacht ist, kann ein
NAALADase-Inhibitor mit einem oder mehreren Mitteln zusammen verabreicht
werden, welche bei der Verringerung des Risikos von Schlaganfall aktiv
sind, wie etwa Aspirin oder Ticlopidin (vorzugsweise Ticlopidin,
für welches
gezeigt wurde, dass es das Risiko eines zweiten ischämischen
Ereignisses reduziert). Co-Verabreichung kann in der Form einer
einzigen Formulierung (welche beispielsweise einen NAALADase-Inhibitor
und Ticlopidin mit pharmazeutisch akzeptablen Exzipienten (Arzneimittelträger) kombiniert,
optional die beiden aktiven Inhaltsstoffe in verschiedenen Exzipienten-Gemischen
auftrennt, welche ausgelegt sind, ihre jeweiligen Freisetzungsraten
und -zeitspannen unabhängig
zu steuern) oder durch unabhängige
Verabreichung von separaten Formulierungen, welche die aktiven Mittel
enthalten, durchgeführt
werden.
-
Wenn
erwünscht,
kann die zu verabreichende pharmazeutische Zusammensetzung auch
geringe Mengen an nicht-toxischen Hilfssubstanzen enthalten, wie
etwa Benetzungs- oder Emulgiermitteln, pH-puffernden Mitteln und
dergleichen, wie etwa beispielsweise Natriumacetat, Sorbitanmonolaurat,
Triethanolaminoleat, etc.
-
Die
NAALADase-Inhibitoren gemäß der vorliegenden
Erfindung werden im Allgemeinen als eine pharmazeutische Zusammensetzung
verabreicht, welche einen pharmazeutischen Exzipienten in Kombination
mit dem Inhibitor umfasst. Der Gehalt des Wirkstoffs in einer Formulierung
kann innerhalb des vollen Bereichs variieren, wie er von Fachleuten
auf dem Gebiet verwendet wird, nämlich
von etwa 0,01 Gewichtsprozent (Gew.-%) bis etwa 99 Gew.-% des Wirkstoffs,
basierend auf der Gesamtformulierung und etwa 0,01 Gew.-% bis 99,99
Gew.-% Exzipient. Vorzugsweise wird die Formulierung etwa 3,5 bis
60 Gew.-% des NAALADase-Inhibitors umfassen, wobei der Rest aus
geeigneten pharmazeutischen Exzipienten besteht.
-
Definitionen
-
Aus
Gründen
der Bequemlichkeit sind vor weiterer Beschreibung der vorliegenden
Erfindung bestimmte in der Beschreibung, den Beispielen und angehängten Ansprüchen verwendete
Terme hier aufgelistet.
-
"NAALADase", wie hierin verwendet,
bezieht sich auf N-acetylierte
alpha-verbundene saure Dipeptidase (N-Acetylatd Alpha-Linked Acidic
dipeptidase). Das Enzym wurde ursprünglich benannt nach seiner
Substratspezifität
für Hydrolyse
von N-acetylierten alpha-verbundenen sauren Dipeptiden. Gegenwärtig ist
bekannt, dass das Enzym einen breiteren Bereich von Substratspezifität aufweist
als ursprünglich
entdeckt, insbesondere dass das Enzym N-Acetylierung oder Alpha-Bindung nicht
erfordert. Damit umfasst "NAALADase", wie hierin verwendet,
andere in der Literatur verwendete Namen, wie etwa NAAG-hydrolisierendes
Enzym und NAALA-Dipeptidase.
-
Der
Begriff "Inhibierung" betrifft im Kontext
der Enzym-Inhibierung
reversible Enzyminhibierung, wie etwa kompetitive, unkompetitive
und nicht-kompetitive Inhibierung. Dies kann experimentell durch
die Wirkungen des Inhibitors auf die Reaktionskinetik des Enzyms
unterschieden werden, welche im Hinblick auf die grundlegende Michaelis-Menten-Geschwindigkeitsgleichung
analysiert werden kann. Kompetitive Inhibierung tritt auf, wenn
der Inhibitor sich mit dem freien Enzym auf solche Weise kombinieren
kann, dass er mit dem normalen Substrat um Bindung an die aktive
Stelle konkurriert. Ein kompetitiver Inhibitor reagiert reversibel
mit dem Enzym, um einen Enzym-Inhibitor-Komplex [EI] zu bilden.
Dem Michaelis-Menten-Formalismus folgend können wir die Inhibitor-Konstante,
K[i] als die Dissoziationskonstante des Enzym-Inhibitor-Komplexes
definieren. Damit ist gemäß dem oben
Gesagten und wie hierin verwendet K[i] im wesentlichen ein Maß für die Affinität zwischen
einem Molekül
und seinem Rezeptor, oder in Bezug auf die vorliegende Erfindung
zwischen den vorliegenden erfindungsgemäßen Verbindungen und dem zu
inhibierenden Enzym. Es sollte zur Kenntnis genommen werden, dass
IC50 ein damit zusammenhängender
Begriff ist, welcher verwendet wird, wenn die Konzentration oder
Menge einer Verbindung definiert wird, welche erforderlich ist,
um eine 50%ige Inhibierung des Zielenzyms zu verursachen.
-
Der
Begriff "Ischämie" betrifft örtlich beschränkte Gewebsanämie aufgrund
von Behinderung des Einflusses von arteriellem Blut. Globale Ischämie tritt
auf unter Bedingungen, in welchen Blutfluss zum gesamten Gehirn
für eine
Zeitspanne aussetzt, wie es etwa als Ergebnis eines Herzstillstands
auftreten kann. Fokale Ischämie
tritt auf unter Bedingungen, in welchen ein Abschnitt des Gehirns
von seiner normalen Blutzufuhr abgeschnitten ist, wie es etwa als
Ergebnis von tromboembolytischer Okklusion eines zerebralen Gefäßes, traumatischer
Kopfverletzung, Ödemen
und Gehirntumoren auftreten kann.
-
Der
Begriff "Nervengewebe" betrifft die verschiedenen
Komponenten, welche das Nervensystem ausbilden, einschließlich Neuronen,
neuralen Unterstützungszellen,
Glia, Schwann-Zellen, das darin enthaltene und diese Strukturen
versorgende Gefäßsystem,
das zentrale Nervensystem, das Gehirn, das Stammhirn, das Rückenmark,
die Verbindungsstelle zwischen dem zentralen Nervensystem mit dem
peripheren Nervensystem, das periphere Nervensystem und damit verbundene
Strukturen.
-
Der
Begriff "Nervenfunktion" betrifft die verschiedenen
Funktionen des Nervensystems und seiner Teile, welche sich bei der
Wahrnehmung der Umgebung, Bewusstsein darüber, Homeostase dazu und Wechselwirkung
mit ihr manifestieren, wie beispielsweise durch die Fähigkeit
gezeigt, Aktivitäten
des täglichen
Lebens, Arbeit, Denkfähigkeit
und Sprache auszuführen.
-
Der
Begriff "Nervenbeschädigungen" betrifft Schaden
an Nervengewebe, welcher Gehirn- und Nervengewebeschaden und -zerstörung, gesamt
oder in Teilen, und sich daraus ergebende Morbidität, Behinderung,
neurologische Defizite und Tod umfasst. Nervöse Beschädigung kann verschiedene Ursprünge haben, einschließlich Ischämie, Hypoxie,
zerebrovaskulärer
Unfall, metabolisch, toxisch, neurotoxisch, Trauma, Operation, iatrogen,
Druck, Masseneffekt, Blutung, thermal, chemisch, Bestrahlung, Vasospasmus,
neurodegenerative Krankheit, neurodegenerativer Prozess, Infektion,
Parkinsonsche Krankheit, amyotrophische Lateralsklerose, Myelinations/Demyelinations-Prozesse,
Epilepsie, kognitive Störungen,
Glutamat-Abnormalitäten und
ihrer sekundären
Effekten.
-
Der
Begriff "Glutamat-Abnormalitäten" betrifft einen jeglichen
Zustand, eine jegliche Krankheit oder Störung, welche Glutamat involviert,
und umfasst, jedoch nicht beschränkt
ist, auf die oben aufgelisteten Nervenbeschädigungen.
-
Der
Begriff "Glutamat-Modulator" betrifft eine jegliche
Zusammensetzung von Stoff, allein oder in Kombination mit einem
weiteren Mittel, welcher den Gehalt an Glutamat in einem Tier, einschließlich einem
Menschen, beeinflusst.
-
Der
Begriff "neuroprotektiv" ist eine Wirkung,
welche Nervenbeschädigung
verringert, aufhält
oder verbessert und für
Nervengewebe, welches Nervenbeschädigung erlitten hat, schützend, wieder
erweckend oder wieder belebend ist.
-
Der
Begriff "Behandlung" betrifft einen jeglichen
Prozess, eine jegliche Aktivität,
Anwendung, Therapie oder dergleichen, bei der ein Tier, einschließlich eines Menschen,
medizinische Hilfe mit der Absicht erhält, den Zustand des Tiers direkt
oder indirekt zu verbessern. Das Verfahren gemäß der vorliegenden Erfindung zum
Behandeln globaler Ischämie
umfasst, einem Subjekt, von welchem erwartet wird, dass es einen
positiven Nutzen davon davonträgt,
eine wirksame Menge eines NAALADase-Inhibitors intern zu verabreichen.
Dosen dieses Isomers, welche in den vorliegenden Verfahren und pharmazeutischen
Zusammensetzungen umfasst sind, sind eine wirksame, nicht-toxische Menge. Fachleuten
auf diesem Gebiet ist es unter Verwendung von routinemäßigen klinischen
Tests möglich,
optimale Dosen zu bestimmen. Die gewünschte Dosis wird einem Subjekt
von einem bis sechs oder mehr Male täglich oral, rektal, parenteral
oder topisch verabreicht und kann auf eine höhere anfängliche Menge folgen, welche
als eine Bolus-Dosis verabreicht wurde.
-
Der
Begriff "Nukleophil" ist im Fachbereich
bekannt und bedeutet, wie hierin verwendet, eine chemische Gruppe
mit einem reaktiven Elektronenpaar.
-
Der
Begriff "Elektrophil" ist im Fachbereich
bekannt und betrifft chemische Gruppen, welche ein Elektronenpaar
von einem wie oben definierten Nukleophil akzeptieren können. Elektrophile
Gruppen, welche in dem Verfahren gemäß der vorliegenden Erfindung
nützlich
sind, umfassen Halogenide und Sulfonate.
-
Der
Begriff "elektronenabziehende
Gruppe" ist im Fachbereich
bekannt und bezieht sich auf die Neigung eines Substituenten, Valenzelektronen
von benachbarten Atomen anzuziehen, d.h. der Substituent ist bezüglich benachbarter
Atome elektronegativ. Eine Quantifizierung des Ausmaßes der elektronenabziehenden
Fähigkeit
ist durch die Hammett Sigma(s)-Konstante gegeben. Diese wohl bekannte
Konstante wird in vielen Literaturstellen beschrieben, beispielsweise
im J. March, Advanced Organic Chemistry, McGraw Hill Book Company,
New York, (1977 Ausgabe), Seiten 251–259. Die Hammett-Konstanten-Werte
sind im Allgemeinen für
elektronenspendende Gruppen negativ (s[P] = –0,66 für NH2)
und positiv für
elektronenabziehende Gruppen (s[P] = 0,78 für eine Nitro-Gruppe), wobei
s[P] para-Substitution anzeigt. Beispiele für elektronenabziehende Gruppen
umfassen Nitro, Keton, Aldehyd, Sulfonyl, Trifluormethyl, -CN, Chlorid
und dergleichen. Beispiele für
elektronenspendende Gruppen umfassen Amino, Methoxy und dergleichen.
-
Der
Begriff "Alkyl" betrifft das Radikal
gesättigter
aliphatischer Gruppen, einschließlich geradkettiger Alkyl-Gruppen, verzweigtkettiger
Alkyl-Gruppen, Cycloalkyl-(alizyklischer)Gruppen,
Alkyl-substituierter Cycloalkylgruppen, und Cycloalkyl-substierter
Alkylgruppen. In bestimmten Ausführungsformen
weist ein geradkettiges oder verzweigtkettiges Alkyl 30 oder weniger
Kohlenstoffatome in seinem Grundgerüst auf (z.B. C1-C30 für
gerade Ketten, C3-C30 für verzweigte
Ketten) und weiter bevorzugt 20 oder weniger. Ähnlich haben bevorzugte Cycloalkyle
von 3–10
Kohlenstoffatome in ihrer Ringstruktur und weiter bevorzugt 5, 6
oder 7 Kohlenstoffe in der Ringstruktur.
-
Außerdem soll
der Begriff "Alkyl" (oder "niederes Alkyl"), wie durch die
Beschreibung und Ansprüche hindurch
verwendet, sowohl "unsubstituierte
Alkyle" wie auch "substituierte Alkyle" umfassen, wobei
letztere sich auf Alkyl-Gruppen mit Substituenten beziehen, welche
ein Wasserstoff an einem oder mehreren Kohlenstoffen des Kohlenwasserstoff-Grundgerüsts ersetzen.
Solche Substituenten können
beispielsweise ein Halogen, ein Hydroxyl, ein Carbonyl (wie etwa
ein Carboxyl, einen Ester, ein Formyl oder ein Keton), ein Thiocarbonyl
(wie etwa einen Thioester, ein Thioacetat oder ein Thioformat),
ein Alkoxyl, ein Phosphoryl, ein Phosphonat, ein Phosphinat, ein
Amino, ein Amido, ein Amidin, ein Imin, ein Cyano, ein Nitro, ein
Azido, ein Sulfhydryl, ein Alkylthio, ein Sulfat, ein Sulfonat,
ein Sulfamoyl, ein Sulfonamido, ein Sulfonyl, ein Heterocyclyl,
ein Aralkyl oder eine aromatische oder eine heteroaromatische Gruppe
umfassen. Es wird von Fachleuten auf diesem Gebiet verstanden werden,
dass die an der Kohlenwasserstoffkette substituierten Gruppen selbst
gegebenenfalls substituiert sein können. Beispielsweise können die
Substituenten eines substituierten Alkyls substituierte und unsubstituierte
Formen von Amino, Azido, Imino, Amido, Phosphoryl (einschließlich Phosphonat und
Phosphinat), Sulfonyl (einschließlich Sulfat, Sulfonamido,
Sulfamoyl und Sulfonat) und Silylgruppen, sowie Ether, Alkylthios,
Carbonyle, (einschließlich
Ketone, Aldehyde, Carboxylate und Ester), -CF3,
-CN und dergleichen umfassen. Beispiele für substituierte Alkyle sind
unterstehend beschrieben. Cycloalkyle können weiter mit Alkyl, Alkenyl,
Alkoxy, Alkylthio, Aminoalkyl, Carbonyl-substituiertem Alkyl, -CF3, -CN und dergleichen substituiert sein.
-
Der
Begriff "Aralkyl", wie hierin verwendet,
betrifft eine Alkylgruppe, welche mit einer Arylgruppe (z.B. einer
aromatischen oder heteroaromatischen Gruppe) substituiert ist.
-
Die
Begriffe "Alkenyl" und "Alkinyl" betreffen ungesättigte aliphatische
Gruppen, welche in Länge
und möglicher
Substitution den oben beschriebenen Alkylgruppen analog sind, aber
welche wenigstens eine Doppel- bzw. Dreifachbindung enthalten.
-
Wenn
die Anzahl an Kohlenstoffatomen nicht anderweitig spezifiziert ist,
bedeutet "niederes
Alkyl", wie hierein
verwendet, eine Alkylgruppe, wie oben definiert, aber mit von 1
bis zu 10 Kohlenstoffatomen, weiter bevorzugt von 1 bis 6 Kohlenstoffatomen
in seiner Grundgerüststruktur.
Dementsprechend haben "niederes Alkenyl" und "niederes Alkinyl" ähnliche Kettenlängen. Bevorzugte
Alkylgruppen sind niedere Alkyle. In bestimmten Ausführungsformen
ist ein hierin als Alkyl bezeichneter Substituent ein niederes Alkyl.
-
Der
Begriff "Aryl", wie hierin verwendet,
umfasst 5-, 6- und
7-gliedrige aromatische Einzel-Ring-Gruppen, welche von 0 bis 4
Heteroatome umfassen können,
beispielsweise Benzol, Pyrrol, Furan, Thiophen, Imidazol, Oxazol,
Thiazol, Triazol, Pyrazol, Pyridin, Pyrazin, Pyridazin und Pyrimidin
und dergleichen. Diese Arylgruppen mit Heteroatomen in der Ringstruktur
können
auch als "Arylheterocyclen" oder "Heteroaromaten" bezeichnet sein.
Der aromatische Ring kann an einer oder mehreren Ringpositionen
mit Substituenten wie oben beschrieben substituiert sein, beispielsweise
Halogen, Azid, Alkyl, Aralkyl, Alkenyl, Alkinyl, Cycloalkyl, Hydroxyl,
Amino, Nitro, Sulfhydryl, Imino, Amino, Phosphonat, Phosphinat,
Carbonyl, Carboxyl, Silyl, Ether, Alkylthio, Sulfonyl, Sulfonamido,
Keton, Aldehyd, Ester, Heterocyclyl, aromatischen oder heteroaromatischen
Gruppen, -CF3, -CN oder dergleichen. Der
Begriff "Aryl" umfasst auch polycyclische
Ringsysteme mit zwei oder mehr zyklischen Ringen, in welchen zwei
oder mehr Kohlenstoffatome zwei benachbarten Ringen gemeinsam sind (die
Ringe sind "annelierte
Ringe"), wobei wenigstens
einer der Ringe aromatisch ist, z.B. können die anderen zyklischen
Ringe Cycloalkyle, Cycloalkenyle, Cycloalkinyle, Aryle und/oder
Heterocyclyle sein.
-
Die
Abkürzungen
Me, Et, Ph, Tf, Nf, Ts, Ms stellen Methyl, Ethyl, Phenyl, Trifluormethansulfonyl,
Nonafluorbutansulfonyl, p-Toluolsulfonyl bzw. Methansulfonyl dar.
Eine ausführlichere
Liste der von organischen Chemikern durchschnittlichen Fachwissens
verwendeten Abkürzungen
erscheint in der ersten Ausgabe jedes Bandes des Journal of Organic
Chemistry. Diese Liste wird typischerweise in einer Tabelle dargestellt,
welche den Titel "Standard
List of Abbreviations" trägt. Die
in dieser Liste enthaltenen Abkürzungen
und alle von organischen Chemikern durchschnittlichen Könnens verwendeten
Abkürzungen
sind hierin durch Bezugnahme aufgenommen.
-
Die
Begriffe ortho, meta und para beziehen sich auf 1,2-, 1,3- bzw.
1,4-disubstituierte Benzole. Zum Beispiel sind die Namen 1,2-Dimethylbenzol
und ortho-Dimethylbenzol synonym.
-
Die
Begriffe "Heterocyclyl" oder "heterocyclische Gruppe" betreffen 3- bis
10-gliedrige Ringstrukturen, bevorzugter 3- bis 7-gliedrige Ringe,
deren Ringstrukturen 1 bis 4 Heteroatome umfassen. Heterocyclen
können
auch Polycyclen sein. Heterocyclylgruppen umfassen beispielsweise
Thiophen, Thioanthren, Furan, Pyran, Isobenzofuran, Chromen, Xanthen,
Phenoxathiin, Pyrrol, Imidazol, Pyrazol, Isothiazol, Isoxazol, Pyridin, Pyrazin,
Pyrimidin, Pyridazin, Indolizin, Isoindol, Indol, Indazol, Purin,
Chinolizin, Isochinolin, Chinolin, Phthalazin, Naphthyridin, Chinoxalin,
Chinazolin, Cinnolin, Pteridin, Carbazol, Carbolin, Phenanthridin,
Acridin, Pyrimidin, Phenanthrolin, Phenazin, Phenarsazin, Phenothiazin,
Furazan, Phenoxazin, Pyrrolidin, Oxolan, Thiolan, Oxazol, Piperidin,
Piperazin, Morpholin, Lactone, Lactame, wie etwa Azetidinone und
Pyrrolidinone, Sulfame, Sulfone und dergleichen. Der heterocyclische
Ring kann an einer oder mehr Positionen mit solchen Substituenten
wie oben beschrieben substituiert sein, wie beispielsweise Halogen,
Alkyl, Aralkyl, Alkenyl, Alkinyl, Cycloalkyl, Hydroxyl, Amino, Nitro,
Sulfhydryl, Imino, Amido, Phosphonat, Phosphinat, Carbonyl, Carboxyl,
Silyl, Ether, Alkylthio, Sulfonyl, Keton, Aldehyd, Ester, einem
Heterocyclyl, einer aromatischen oder heteroaromatischen Gruppe,
-CF3, -CN oder dergleichen.
-
Die
Begriffe "Polycyclyl" oder "polycyclische Gruppe" betrifft zwei oder
mehr Ringe (z.B. Cycloalkyle, Cycloalkenyle, Cycloalkinyle, Aryle
und/oder Heterocyclyle), in welchen zwei oder mehr Kohlenstoffe
zwei benachbarten Ringen gemeinsam sind, z.B. sind die Ringe "annelierte Ringe". Ringe, welche durch
nicht-benachbarte Atome verbunden sind, werden als "verbrückte Ringe" bezeichnet. Jeder
dieser Ringe des Polycyclus kann mit Substituenten wie den oben
beschriebenen substituiert sein, wie beispielsweise Halogen, Alkyl, Aralkyl,
Alkenyl, Alkinyl, Cycloalkyl, Hydroxyl, Amino, Nitro, Sulfhydryl,
Imino, Amido, Phosphonat, Phosphinat, Carbonyl, Carboxyl, Silyl,
Ether, Alkylthio, Sulfonyl, Keton, Aldehyd, Ester, einem Heterocyclyl,
einer aromatischen oder heteroaromatischen Gruppe, -CF3,
-CN oder dergleichen.
-
Der
Begriff "Carbocyclus" betrifft, wie hierin
verwendet, einen aromatischen oder nicht-aromatischen Ring, in welchem
jedes Atom des Rings Kohlenstoff ist.
-
Der
Begriff "Heteroatom", wie hierin verwendet,
bedeutet ein Atom eines jeglichen Elements ausser Kohlenstoff oder
Wasserstoff. Bevorzugte Heteroatome sind Stickstoff, Sauerstoff,
Schwefel und Phosphor.
-
Wie
hierin verwendet bedeutet der Begriff "Nitro" -NO2; der Begriff "Halogen" bezeichnet -F, -Cl,
-Br oder -I; der Begriff "Sulhydryl" bedeutet -SH; der
Begriff "Hydroxyl" bedeutet -OH; und
der Begriff "Sulfonyl" bedeutet -SO2-.
-
Die
Begriffe "Amin" und "Amino" sind im Fachbereich
anerkannt und beziehen sich sowohl auf unsubstituierte als auch
substituierte Amine, z.B. eine Gruppe, welche durch die allgemeine
Formel dargestellt werden kann:
wobei R
9,
R
10 und R'
10 unabhängig voneinander
einen Wasserstoff, ein Alkyl, ein Alkenyl, -(CH
2)
m-R
8 bedeuten oder
R
9 und R
10 zusammen
mit dem N-Atom, an welches sie gebunden sind, einen Heterocyclus
mit von 4 bis 8 Atomen in der Ringstruktur vervollständigen;
R
8 ein Aryl, ein Cycloalkyl, ein Cycloalkenyl,
einen Heterocyclus oder einen Polycyclus darstellt; und m 0 oder
eine ganze Zahl im Bereich von 1 bis 8 ist. In bevorzugten Ausführungsformen
kann nur eines von R
9 oder R
10 ein
Carbonyl sein, z.B. bilden R
9, R
10 und der Stickstoff zusammen nicht ein
Imid. In noch weiter bevorzugten Ausführungsformen stellen jedes
von R
9 und R
10 (und
optional R'
10) unabhängig
voneinander einen Wasserstoff, ein Alkyl, ein Alkenyl oder -(CH
2)
m-R
8 dar.
Damit bedeutet der Begriff "Alkylamin", wie hierin verwendet,
eine Amingruppe, wie oben definiert, mit einem daran gebundenen substituierten
oder unsubstituierten Alkyl, d.h. wenigstens eines von R
9 und R
10 ist eine
Alkylgruppe.
-
Der
Begriff "Acylamino" ist im Fachbereich
anerkannt und betrifft eine Gruppe, welche durch die allgemeine
Formel dargestellt werden kann:
wobei R
9 wie
oben definiert ist und R'
11 einen Wasserstoff, ein Alkyl, ein Alkenyl
oder -(CH
2)
m-R
8 darstellt, wobei m und R
8 wie
oben definiert sind.
-
Der
Begriff "Amido" ist im Fachbereich
anerkannt als ein amino-substituiertes Carbonyl und umfasst eine
Gruppe, welche durch die allgemeine Formel dargestellt werden kann:
wobei R
9,
R
10 wie oben definiert sind. Bestimmte Ausführungsformen
des Amids umfassen keine Imide, welche instabil sein können.
-
Der
Begriff "Alkylthio" bezieht sich auf
eine Alkylgruppe, wie oben definiert, welche daran ein Schwefel-Radikal
gebunden hat. In bestimmten Ausführungsformen
ist die "Alkylthio"-Gruppe dargestellt
durch eines von -S-Alkyl, -S-Alkenyl, S-Alkinyl und -S-(CH2)m-R8,
wobei m und R8 wie oben definiert sind.
Repräsentative Alkylthio-Gruppen
umfassen Methylthio, Ethylthio und dergleichen.
-
Der
Begriff "Carbonyl" ist im Fachbereich
anerkannt und umfasst solche Gruppen, wie sie durch die allgemeine
Formel dargestellt werden können:
wobei X eine Bindung ist
oder einen Sauerstoff oder ein Schwefel darstellt, und R
11 einen Wasserstoff, ein Alkyl, ein Alkenyl,
-(CH
2)
m-R
8 oder ein pharmazeutisch akzeptables Salz
darstellt, R'
11 einen Wasserstoff, ein Alkyl, ein Alkenyl
oder -(CH
2)
m-R
8 darstellt, wobei m und R
8 wie
oben definiert sind. Wenn X ein Sauerstoff ist und R
11 oder
R'
11 nicht
Wasserstoff sind, stellt die Formel einen "Ester" dar. Wenn X ein Sauerstoff ist und
R
11 wie oben definiert ist, wird die Gruppe
hierin als eine Carboxylgruppe bezeichnet, und besonders wenn R
11 ein Wasserstoff ist, stellt die Formel
eine "Carbonsäure" dar. Wenn X ein
Sauerstoff ist und R'
11 Wasserstoff ist, stellt die Formel ein "Format" dar. Im Allgemeinen
stellt die Formel, wenn das Sauerstoffatom der obigen Formel durch
Schwefel ersetzt ist, eine "Thiolcarbonyl"-Gruppe dar. Wenn
X ein Schwefel ist und R
11 oder R'
11 nicht Wasserstoff
sind, stellt die Formel einen "Thiolester" dar. Wenn X ein
Schwefel ist und R
11 Wasserstoff ist, stellt die
Formel eine "Thiolcarbonsäure" dar. Wenn X ein
Schwefel ist und R'
11 Wasserstoff ist, stellt die Formel ein "Thiolformat" dar. Auf der anderen
Seite stellt die obige Formel, wenn X eine Bindung ist und R
11 nicht Wasserstoff ist, eine "Keto"-Gruppe dar. Wenn X eine Bindung ist
und R
11 Wasserstoff ist, stellt die obige
Formel eine "Aldehyd"-Gruppe dar.
-
Die
Begriffe "Alkoxyl" oder "Alkoxy", wie hierin verwendet,
beziehen sich auf eine Alkylgruppe, wie oben definiert, mit einem
daran gebundenen Sauerstoffradikal. Repräsentative Alkoxylgruppen umfassen
Methoxy, Ethoxy, Propyloxy, tert-Butoxy
und dergleichen. Ein "Ether" besteht aus zwei
kovalent durch ein Sauerstoff verbundene Kohlenwasserstoffe. Demgemäss ist oder ähnelt der
Substituent eines Alkyls, welcher das Alkyl zu einem Ether macht,
einem Alkoxyl, wie es dargestellt werden kann durch eines von -O-Alkyl,
-O-Alkenyl, O-Alkinyl, -O-(CH2)m-R8, wobei m und R8 oben
beschrieben sind.
-
Der
Begriff "Sulfonat" ist im Fachbereich
anerkannt und umfasst eine Gruppe, welche durch die allgemeine Formel
dargestellt werden kann:
in welcher R
41 ein
Elektronenpaar, Wasserstoff, Alkyl, Cycloalkyl oder Aryl ist.
-
Die
Begriffe Triflyl, Tosyl, Mesyl, und Nonaflyl sind im Fachbereich
anerkannt und beziehen sich auf Trifluormethansulfonyl-, p-Toluolsulfonyl-,
Methansulfonyl- bzw.
Nonafluorbutansulfonyl-Gruppen. Die Begriffe Triflat, Tosylat, Mesylat
und Nonaflat sind im Fachbereich anerkannt und betreffen Trifluormethansulfonatester-,
p-Toluolsulfonatester-,
Methansulfonatester- und Nonafluorbutansulfonatester-funktionelle
Gruppen bzw. Moleküle,
welche diese Gruppen enthalten.
-
Der
Begriff "Sulfat" ist im Fachbereich
anerkannt und umfasst eine Gruppe, welche durch die allgemeine Formel
darstellt werden kann:
in welcher R
41 wie
oben definiert ist.
-
Der
Begriff "Sulfonamido" ist im Fachbereich
anerkannt und umfasst eine Gruppe, welche durch die allgemeine Formel
dargestellt werden kann:
in welcher R
9 und
R'
11 wie
oben definiert sind.
-
Der
Begriff "Sulfamoyl" ist im Fachbereich
anerkannt und umfasst eine Gruppe, welche durch die allgemeine Formel
dargestellt werden kann:
in welcher R
9 und
R
10 definiert sind.
-
Die
Begriffe "Sulfoxido" oder "Sulfinyl", wie hierin verwendet,
beziehen sich auf eine Gruppe, welche durch die allgemeine Formel
dargestellt werden kann:
in welcher R
44 aus
der Gruppe ausgewählt
ist, welche besteht aus Wasserstoff, Alkyl, Alkenyl, Alkinyl, Cycloalkyl,
Heterocyclyl, Aralkyl oder Aryl.
-
Ein "Phosphoryl" kann im Allgemeinen
durch die Formel dargestellt werden:
wobei Q
1 S
oder O darstellt und R
46 Wasserstoff, ein
niederes Alkyl oder ein Aryl darstellt. Bei Verwendung zur Substitution
beispielsweise eines Alkyls kann die Phosphoryl-Gruppe des Phosphorylalkyls
durch die allgemeine Formel dargestellt werden:
wobei Q
1 S
oder O darstellt und jedes R
46 unabhängig Wasserstoff,
ein niederes Alkyl oder ein Aryl darstellt, Q
2 O,
S oder N darstellt. Wenn Q
1 ein S ist, ist
die Phosphorylgruppe ein "Phosphorothioat".
-
Ein "Phosphoramidit" kann durch die allgemeine
Formel dargestellt werden:
wobei R
9 und
R
10 wie oben definiert sind und Q
2 O, S oder N darstellen.
-
Ein "Phosphonamidit" kann in der allgemeinen
Formel dargestellt werden:
wobei R
9 und
R
10 wie oben definiert sind, Q
2 O,
S oder N darstellt und R
48 ein niederes
Alkyl oder ein Aryl darstellt, und Q
2 O,
S oder N darstellt.
-
Ein "Selenoalkyl" bezieht sich auf
eine Alkylgruppe mit einer daran gebundenen substituierten Seleno-Gruppe.
Beispielhafte "Selenoether", welche an dem Alkyl
substituiert sein können,
sind ausgewählt
aus einem von -Se-Alkyl, -Se-Alkenyl, -Se-Alkinyl, und -Se-(CH2)m-R8,
wobei m und R8 oben definiert sind.
-
Analoge
Substitutionen können
durchgeführt
werden an Alkenyl- und Alkinyl-Gruppen, um beispielsweise Aminoalkenyle,
Aminoalkinyle, Amidoalkenyle, Amidoalkinyle, Iminoalkenyle, Iminoalkinyle,
Thioalkenyle, Thioalkinyle, Carbonyl-substituierte Alkenyle oder
Alkinyle herzustellen.
-
Der
Ausdruck "Schutzgruppe", wie hierin verwendet,
bedeutet temporäre
Modifikationen einer potentiell reaktiven funktionellen Gruppe,
welche sie vor unerwünschten
chemischen Transformationen schützt.
Beispiele solcher Schutzgruppen umfassen Ester von Carbonsäuren, Silylether
von Alkoholen und Acetale und Ketale von Aldehyden bzw. Ketonen.
Das Gebiet der Schutzgruppenchemie ist in einem Review dargestellt worden
(Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis,
2. Auflage; Wiley: New York, 1991).
-
Es
ist zu verstehen, dass "Substitution" oder "substituiert mit" die implizite Voraussetzung
umfasst, dass eine solche Substitution mit der erlaubten Valenz
des substituierten Atoms und des Substituenten im Einklang ist,
und dass die Substitution zu einer stabilen Verbindung führt, welche
z.B. nicht spontan einer Transformation, wie etwa durch Umordnung,
Ringschluss, Eliminierung, etc. unterliegt.
-
Wie
hierin verwendet, ist beabsichtigt, dass der Begriff "substituiert" alle erlaubten Substituenten
von organischen Verbindungen umfassen. Unter einem breiten Gesichtspunkt
umfassen die erlaubten Substituenten acyclische und cyclische, verzweigte
und unverzweigte, carbocyclische und heterocyclische, aromatische und
nicht-aromatische Substituenten von organischen Verbindungen. Beispielhafte
Substituenten umfassen beispielsweise die oben hierin beschriebenen.
Die erlaubten Substituenten können
einer oder mehrere sein und die gleichen oder verschiedene für geeignete
organische Verbindungen. Zu Zwecken dieser Erfindung können die
Heteroatome, wie etwa Stickstoff, Wasserstoff-Substituenten und/oder
jegliche erlaubte Substituenten organischer Verbindungen, welche
hierin beschrieben sind, aufweisen, welche zu den Valenzen des Heteroatoms
passen. Diese Erfindung soll nicht in irgendeiner Art und Weise
durch die erlaubten Substituenten organischer Verbindungen eingeschränkt sein.
-
Ein "polares Lösungsmittel" bedeutet ein Lösungsmittel
mit einem Dipolmoment (ε)
von 2,9 oder mehr, wie etwa DMF, THF, Ethylenglykoldimethylether,
DMSO, Aceton, Acetonitril, Methanol, Ethanol, Isopropanol, n-Propanol,
t-Butanol oder 2-Methoxyethylether. Bevorzugte Lösungsmittel sind DMF, Diglyme
und Acetonitril.
-
Ein "aprotisches Lösungsmittel" bedeutet ein Lösungsmittel,
welches kein Wasserstoffbindungsdonor ist. Beispiele für solche
Lösungsmittel
sind Acetonitril, Toluol, DMF, Diglyme, THF oder DMSO.
-
Ein "polares, aprotisches
Lösungsmittel" bedeutet ein Lösungsmittel,
welches einen Dipolmoment (ε) von
2,9 hat, und kein Wasserstoffbindungsdonor ist, z.B. DMF, Acetonitril,
DMSO und THF.
-
Zu
Zwecken dieser Erfindung sind die chemischen Elemente gemäß dem Periodensystem
der Elemente, CAS-Version, Handbuch der Chemie und Physik, 67. Auflage,
1986–87,
Innenumschlag identifiziert. Auch zu Zwecken dieser Erfindung soll
der Begriff "Kohlenwasserstoff" alle erlaubten Verbindungen
mit wenigstens einem Wasserstoff und einem Kohlenstoffatom umfassen.
Unter einem breiten Gesichtspunkt umfassen die erlaubten Kohlenwasserstoffe
acyclische und cyclische, verzweigte und unverzweigte, carbocyclische und
heterocyclische, aromatische und nicht- aromatische organische Verbindungen,
welche substituiert oder unsubstituiert sein können.
-
Pharmazeutische
Zusammensetzungen
-
Unter
einem weiteren Aspekt stellt die vorliegende Verbindung pharmazeutisch
akzeptable Zusammensetzungen bereit, welche eine therapeutisch wirksame
Menge einer oder mehr der oben beschriebenen Verbindungen umfassen,
formuliert zusammen mit einem oder mehr pharmazeutisch akzeptablen
Trägern (Additiven)
und/oder Verdünnungsmitteln.
Wie im Detail untenstehend beschrieben, können die pharmazeutischen Zusammensetzungen
gemäß der vorliegenden
Erfindung speziell für
die Verabreichung in fester oder flüssiger Form formuliert sein,
einschließlich
der, welche an die folgenden angepasst sind: (1) orale Verabreichung,
z.B. Arzneitränke
(wässrige
oder nicht-wässrige Lösungen oder
Suspensionen), Tabletten, Bolus, Pulver, Granulate, Pasten zur Anwendung
auf der Zunge; (2) parenterale Verabreichung, z.B. durch subkutane, intramuskuläre oder
intravenöse
Injektion, beispielsweise als eine sterile Lösung oder Suspension; (3) topische
Applikation, z.B. als eine Creme, Salbe oder Spray, welche auf die
Haut aufgebracht werden; oder (4) intravaginal oder intrarektal,
z.B. als ein Pessar, eine Creme oder ein Schaum.
-
Der
Ausdruck "therapeutisch
wirksame Menge",
wie hierin verwendet, bedeutet die Menge einer Verbindung, eines
Materials oder einer Zusammensetzung, welche eine Verbindung gemäß der vorliegenden
Erfindung umfasst, welche wirksam darin ist, eine gewünschte therapeutische
Wirkung in wenigstens einer Sub-Population von Zellen in einem Tier zu
erzeugen, in einem vernünftigen
Nutzen-/Risiko-Verhältnis, wie
es auf eine jegliche medizinische Behandlung anwendbar ist.
-
Der
Ausdruck "pharmazeutisch
akzeptabel" wird
hierin verwendet, um Bezug zu nehmen auf diejenigen Verbindungen,
Materialien, Zusammensetzungen und/oder Dosierungsformen, welche
innerhalb des Bereichs verlässlicher
medizinischer Beurteilung geeignet sind, in Kontakt mit den Geweben
von Menschen und Tieren ohne übermäßige Toxizität, Irritation,
allergische Reaktion oder ein anderes Problem oder eine andere Komplikation
verwendet zu werden, entsprechend einem vernünftigen Nutzen-/Risiko-Verhältnis.
-
Der
Ausdruck "pharmazeutisch
akzeptabler Träger", wie hierin verwendet,
bedeutet ein pharmazeutisch akzeptables Material, Vehikel oder eine
pharmazeutisch akzeptable Zusammensetzung, wie etwa einen flüssigen oder
festen Füllstoff,
Verdünnungsmittel,
Exzipient, Lösungsmittel
oder einkapselndes Material, welcher involviert ist im Tragen oder
Transportieren der betreffenden Verbindung von einem Organ, oder
Körperabschnitt,
zu einem anderen Organ oder Körperabschnitt.
Jeder Träger
muss "akzeptabel" in dem Sinn sein, dass
er kompatibel mit den anderen Inhaltsstoffen der Formulierung und
für den
Patienten nicht schädlich
ist. Einige Beispiele für
Materialien, welche als pharmazeutisch akzeptable Träger dienen
können,
umfassen: (1) Zucker, wie etwa Lactose, Glucose und Sucrose; (2)
Stärken,
wie etwa Maisstärke
und Kartoffelstärke;
(3) Cellulose und seine Derivate, wie etwa Natriumcarboxymethylcellulose,
Ethylcellulose und Celluloseacetat; (4) pulverförmiges Tragacanth; (5) Malz;
(6) Gelatine; (7) Talk; (8) Exzipienten, wie etwa Kakaobutter und
Zäpfchenwachse;
(9) Öle,
wie etwa Erdnussöl,
Baumwollsamenöl,
Distelöl,
Sesamöl,
Olivenöl,
Maisöl
und Sojaöl; (10)
Glycole, wie etwa Propylenglycol; (11) Polyole, wie etwa Glycerin,
Sorbitol, Mannitol und Polyethylenglycol; (12) Ester, wie etwa Ethyloleat
und Ethyllaurat; (13) Agar; (14) puffernde Mittel, wie etwa Magnesiumhydroxid
und Aluminiumhydroxid; (15) Alginsäure; (16) Pyrogen-freies Wasser;
(17) isotonische Salzlösung;
(18) Ringer'sche
Lösung;
(19) Ethylalkohol; (20) Phosphatpufferlösungen; und (21) andere nicht
toxische, kompatible Substanzen, welche in pharmazeutischen Formulierungen
verwendet werden.
-
Wie
oben ausgeführt,
können
bestimmte Ausführungsformen
der vorliegenden Verbindungen eine basische funktionelle Gruppe,
wie etwa Amino oder Alkylamino enthalten und sind daher fähig, pharmazeutisch akzeptable
Salze mit pharmazeutisch akzeptablen Säuren zu bilden. Der Begriff "pharmazeutisch akzeptable Salze" bezieht sich in
dieser Hinsicht auf die relativ nicht-toxischen, anorganischen und
organischen Säureadditionssalze
von erfindungsgemäßen Verbindungen.
Diese Salze können
in situ während
der endgültigen
Isolierung und Reinigung der erfindungsgemäßen Verbindungen hergestellt
werden, oder durch separates Umsetzen einer gereinigten erfindungsgemäßen Verbindung
in ihrer freien Basenform mit einer geeigneten organischen oder
anorganischen Säure
und Isolieren des Salzes, welches derart gebildet wurde. Repräsentative Salze
umfassen die Hydrobromid-, Hydrochlorid-, Sulfat-, Bisulfat-, Phosphat-,
Nitrat-, Acetat-, Valerat-, Oleat-, Palmitat-, Stearat-, Laurat-,
Benzoat-, Lactat-, Phosphat-, Tosylat-, Citrat-, Maleat-, Fumarat-,
Succinat-, Tartrat-, Naphthylat-, Mesylat-, Glucoheptonat-, Lactobionat-,
und Laurylsulfonat- Salze
und dergleichen. (Siehe beispielsweise Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci.
66: 1–19)
-
Die
pharmazeutisch akzeptablen Salze der in Frage stehenden Verbindungen
umfassen die herkömmlichen
nicht-toxischen Salze oder quarternären Ammoniumsalze der Verbindungen,
z.B. von nicht-toxischen organischen oder anorganischen Säuren. Z.B.
umfassen solche herkömmlichen
nicht-toxischen Salze diejenigen, welche von anorganischen Säuren abgeleitet
sind, wie etwa Chlorwasserstoff, Bromwasserstoff, Schwefelsäure, Sulfamsäure, Phosphorsäure, Salpetersäure und
dergleichen; und die Salze, welche hergestellt sind aus organischen
Säuren
wie etwa Essigsäure,
Propionsäure,
Bernsteinsäure,
Glycolsäure,
Stearinsäure, Milchsäure, Äpfelsäure, Weinsäure, Zitronensäure, Ascorbinsäure, Palmitinsäure, Maleinsäure, Hydroxymaleinsäure, Phenylessigsäure, Glutaminsäure, Benzoesäure, Salicylsäure, Sulfanilsäure, 2-Acetoxybenzoesäure, Fumarsäure, Toluolsulfonsäure, Methansulfonsäure, Ethandisulfonsäure, Oxalsäure, Isethionsäure und
dergleichen.
-
In
anderen Fällen
können
die erfindungsgemäßen Verbindungen
eine oder mehrere saure funktionelle Gruppen enthalten und daher
fähig sein,
pharmazeutisch akzeptable Salze mit pharmazeutisch akzeptablen Basen
zu bilden. Der Begriff "pharmazeutisch
akzeptable Salze" betrifft
in diesen Fällen
die relativ nicht-toxischen, anorganischen und organischen Basenadditionssalze
von erfindungsgemäßen Verbindungen.
Diese Salze können
gleichfalls in situ während
der endgültigen
Isolierung und Reinigung der Verbindungen hergestellt werden, oder
durch separates Umsetzen der gereinigten Verbindung in ihrer freien
Säureform
mit einer geeigneten Base, wie etwa dem Hydroxid, Carbonat oder
Bicarbonat eines pharmazeutisch akzeptablen Metallkations, mit Ammoniak,
oder mit einem pharmazeutisch akzeptablen organischen primären, sekundären oder
tertiären
Amin. Repräsentative
Alkali- oder Erdalkalisalze umfassen die Lithium-, Natrium-, Kalium-,
Calcium-, Magnesium- und Aluminiumsalze und dergleichen. Repräsentative
organische Amine, welche für
die Bildung von Basenadditionssalzen nützlich sind, umfassen Ethylamin,
Diethylamin, Ethylendiamin, Ethanolamin, Diethanolamin, Piperazin
und dergleichen. (Siehe beispielsweise Berge et al., supra)
-
Benetzungsmittel,
Emulgatoren und Schmiermittel, wie etwa Natriumlaurylsulfat und
Magnesiumstearat, sowie farbgebende Mittel, freisetzende Mittel,
Beschichtungsmittel, süßende, geschmackgebende
und parfümierende
Mittel, Konservierungsstoffe und Antioxidantien können auch
in den Zusammensetzungen vorliegen.
-
Beispiele
für pharmazeutisch
akzeptable Antioxidantien umfassen: (1) wasserlösliche Antioxidantien, wie
etwa Ascorbinsäure,
Cysteinhydrochlorid, Natriumbisulfat, Nariummetabisulfit, Natriumsulfit
und dergleichen; (2) öllösliche Antioxidantien,
wie etwa Ascorbylpalmitat, butyliertes Hydroxyanisol (BHA), butyliertes
Hydroxytoluol (BHT), Lecithin, Propylgallat, alpha-Tocopherol und
dergleichen; und (3) Metall-Chelierungsmittel, wie etwa Zitronensäure, Ethylendiamintetraessigsäure (EDTA),
Sorbitol, Weinsäure,
Phosphorsäure
und dergleichen.
-
Verabreichung
der Adenosin-Antagonisten und -Agonisten zur Verwendung in dem erfindungsgemäßen Verfahren
kann über
einen jeglichen der akzeptierten Verabreichungsmodi erfolgen. Diese
Verfahren umfassen, sind jedoch nicht beschränkt auf, orale, parenterale,
transdermale, intraartikuläre
und auf andere Weise systemische Verabreichung. Orale Verabreichung
wird bevorzugt. Die Verbindungen werden in einer therapeutisch wirksamen
Menge entweder alleine oder in Kombination mit einem geeigneten
pharmazeutisch akzeptablen Träger
oder Exzipienten verabreicht.
-
In
Abhängigkeit
von dem beabsichtigten Verabreichungsmodus kann der Adenosin-Antagonist
oder -Agonist der Wahl in einer jeglichen pharmazeutisch akzeptablen
Dosierungsform aufgenommen sein, wie etwa beispielsweise Tabletten,
Transdermalpflaster, Pillen, Kapseln, Pulvern, Flüssigkeiten,
Suspensionen, Emulsionen, Aerosolen oder dergleichen, vorzugsweise
in Einheitsdosierungsformen, welche geeignet sind für einzelne
Verabreichung von präzisen
Dosierungen, oder Langzeitfreisetzungsdosierungsformen für kontinuierliche,
gesteuerte Verabreichung. Vorzugsweise wird die Dosierungsform einen
pharmazeutisch akzeptablen Exzipienten umfassen und kann zusätzlich andere
medizinische Mittel, pharmazeutische Mittel, Träger, Hilfsstoffe und dergleichen
enthalten.
-
Für feste
Dosierungsformen umfassen nicht-toxische Träger, sind jedoch nicht beschränkt auf
beispielsweise Mannitol, Lactose, Stärke, Magnesiumstearat, Natriumsaccharin,
die Polyalkylenglycole, Talk, Cellulose, Glucose, Sucrose und Magnesiumcarbonat
pharmazeutischer Qualität.
Flüssige
pharmazeutisch verabreichbare Dosierungsformen können beispielsweise eine Lösung oder
Suspension eines aktiven Adenosin-Agens und optional pharmazeutische
Hilfsmittel in einem Träger
umfassen, wie etwa beispielsweise Wasser, Salzlösungs-wässriger Dextrose, Glycerol,
Ethanol und dergleichen, um dadurch eine Lösung oder Suspension zu bilden.
Wenn erwünscht,
kann die zu verabreichende pharmazeutische Zusammensetzung auch geringere
Mengen nicht-toxischer Hilfssubstanzen umfassen, wie etwa Benetzungs-
oder Emulgiermittel, pH-puffernde Mittel und dergleichen. Typische
Beispiele für
solche Hilfsmittel sind Natriumacetat, Sorbitanmonolaurat, Triethanolamin,
Natriumacetat, Triethanolaminoleat, etc. Verfahren zum Herstellen
solcher Dosierungsformen sind bekannt, oder werden Fachleuten in
diesem Bereich offensichtlich sein; siehe beispielsweise: Remington's Pharmaceutical
Sciences, Mack Publishing Company, Easton, Pa., 16. Auflage, 1980.
Die Zusammensetzung der zu verabreichenden Formulierung wird auf
jeden Fall eine Menge des aktiven Adenosin-Agens in einer zur Behandlung
wirksamen Menge enthalten.
-
Parenterale
Verabreichung ist im Allgemeinen charakterisiert durch Injektion,
entweder subkutan, intramuskulär
oder intravenös.
Injektionen können
hergestellt werden in herkömmlichen
Formen, entweder als flüssige
Lösungen
oder Suspension, festen Formen, welche zur Bildung von Lösung oder
Suspension in Flüssigkeit
vor Injektion geeignet sind, oder als Emulsionen. Geeignete Exzipienten
sind beispielsweise Wasser, Salzlösung, Dextrose, Glycerin, Ethanol
und dergleichen. Zusätzlich
können
die zu verabreichenden injizierbaren pharmazeutischen Zusammensetzungen
auch geringere Mengen nicht-toxischer Hilfssubstanzen enthalten,
wie etwa Benetzungs- oder Emulgiermittel, pH-puffernde Mittel und
dergleichen.
-
Die
Menge an aktivem Adenosin-Antagonisten oder -Agonist, welche verabreicht
wird, wird natürlich von
dem zu behandelnden Subjekt abhängen,
der Schwere und Natur des Leidens, der Art der Verabreichung, der
Potenz und der Pharmakodynamik des besonderen Mittels und der Beurteilung
des verschreibenden Arztes. Die therapeutisch wirksame Dosierung
zur Verwendung in dieser Erfindung wird jedoch im Allgemeinen im
Bereich von etwa 0,01 mu g/kg (Körpergewicht)
bis 5 mg/kg sein.
-
Erfindungsgemäße Formulierungen
umfassen solche, welche geeignet sind für orale, nasale, topische (einschließlich bukkale
und sublinguale), rektale, vaginale und/oder parenterale Verabreichung.
Die Formulierungen können
bequem in Einheitsdosierungsform vorgelegt werden und können durch
jegliche im Bereich der Pharmazie bekannte Verfahren hergestellt
werden. Die Menge an aktivem Inhaltsstoff, welcher mit einem Trägermaterial
kombiniert werden kann, um eine Einzeldosierungsform herzustellen,
wird in Abhängigkeit
von dem behandelten Wirt variieren, und der besonderen Art der Verabreichung.
Die Menge an aktivem Inhaltsstoff, welcher mit einem Trägermaterial
kombiniert werden kann, um eine Einzeldosierungsform herzustellen, wird
im Allgemeinen diejenige Menge der Verbindung sein, welche eine
therapeutische Wirkung hervorruft. Im Allgemeinen wird bezogen auf
100% diese Menge im Bereich von etwa 1% bis etwa 99% aktiver Inhaltsstoff, vorzugsweise
von etwa 5% bis etwa 70%, am meisten bevorzugt von etwa 10% bis
etwa 30% liegen.
-
Verfahren
zum Herstellen dieser Formulierungen oder Zusammensetzungen umfassen
den Schritt, eine erfindungsgemäße Verbindung
mit dem Träger
und optional einem oder mehreren begleitenden Inhaltsstoffen in
Kontakt zu bringen. Im Allgemeinen werden die Formulierungen hergestellt
durch gleichförmiges
und inniges Inkontaktbringen einer Verbindung gemäß der vorliegenden
Erfindung mit flüssigen
Trägern,
oder fein aufgespalteten festen Trägern, oder beiden, und dann,
falls nötig,
Formen des Produkts.
-
Erfindungsgemäße Formulierungen,
welche für
orale Verabreichung geeignet sind, können in der Form von Kapseln,
Gelatinekapseln, Pillen, Tabletten, Lutschtabletten (unter Verwendung
einer mit Geschmack versehenen Grundlage, üblicherweise Sucrose und Akazie
oder Tragacanth), Pulvern, Granulaten oder als eine Lösung oder
eine Suspension in einer wässrigen
oder nicht-wässrigen
Flüssigkeit
oder als eine Öl-in-Wasser-
oder Wasser-in-Öl-Flüssigemulsion
oder als ein Elixier oder Sirup oder als Pastillen (unter Verwendung
einer inerten Grundlage, wie etwa Gelatine und Glycerin oder Sucrose
und Akazie) und/oder als Mundspülungen
und dergleichen vorliegen, wobei jede eine vorbestimmte Menge einer
erfindungsgemäßen Verbindung
als einen aktiven Inhaltsstoff enthält. Eine Verbindung gemäß der vorliegenden
Erfindung kann auch als ein Bolus, Electuarium oder eine Paste verabreicht
werden.
-
In
festen Dosierungsformen der Erfindung für orale Verabreichung (Kapseln,
Tabletten, Pillen, Dragees, Pulvern, Granulaten und dergleichen)
wird der aktive Inhaltsstoff mit einem oder mehreren pharmazeutisch
akzeptablen Trägern
gemischt, wie etwa Natriumcitrat oder Dicalciumphosphat und/oder
einem jeglichen der folgenden: (1) Füllstoffen oder Streckmitteln,
wie etwa Stärken,
Lactose, Sucrose, Glucose, Mannitol und/oder Kieselsäure; (2)
Bindemitteln, wie etwa beispielsweise Carboxymethylcellulose, Alginate,
Gelatine, Polyvinylpyrrolidon, Sucrose und/oder Akazie; (3) Feuchthaltemittel,
wie etwa Glycerin; (4) auflösende
Mittel, wie etwa Agar Agar, Calciumcarbonat, Kartoffel- oder Tapiokastärke, Alginsäure, bestimmte
Silikate und Natriumcarbonat; (5) Lösungs-verzögernde Mittel, wie etwa Paraffin;
(6) Absorptionsbeschleuniger, wie etwa quarternäre Ammoniumverbindungen; (7)
Benetzungsmittel, wie etwa beispielsweise Cetylalkohol und Glycerinmonostearat;
(8) Absorptionsmittel, wie etwa Kaolin und Bentonit-Ton; (9) Schmiermittel,
wie etwa Talk, Calciumstearat, Magnesiumstearat, feste Polyethylenglycole,
Natriumlaurylsulfat und Gemische davon; und (10) farbgebende Mittel.
Im Falle von Kapseln, Tabletten und Pillen können die pharmazeutischen Zusammensetzungen
auch puffernde Mittel umfassen. Feste Zusammensetzungen eines ähnlichen
Typs können
auch als Füllstoffe
in weichen und hart-gefüllten
Gelatinekapseln verwendet werden, unter Verwendung solcher Exzipienten
wie Lactose oder Milchzuckern, sowie Polyethylenglycolen hohen Molekulargewichts
und dergleichen.
-
Eine
Tablette kann hergestellt werden durch Pressen oder Gießen, optional
mit einem oder mehreren begleitenden Inhaltsstoffen. Gepresste Tabletten
können
hergestellt werden unter Verwendung von Bindemittel (z.B. Gelatine
oder Hydroxypropylmethylcellulose), Schmiermittel, inertem Verdünnungsmittel,
Konservierungsstoff, auflösendem
Mittel (z.B. Natriumstärkeglycolat
oder quer-vernetzter Natriumcarboxymethylcellulose), oberflächenaktivem
oder dispergierendem Mittel. Gegossene Tabletten können hergestellt
werden durch Gießen
in einer geeigneten Maschine von einem Gemisch der pulverförmigen Verbindung,
welche mit einem inerten flüssigen
Verdünnungsmittel
angefeuchtet ist.
-
Die
Tabletten und anderen festen Dosierungsformen der erfindungsgemäßen pharmazeutischen
Zusammensetzungen, wie etwa Dragees, Kapseln, Pillen und Granulate,
können
optional eingekerbt oder mit Beschichtungen und Hüllen hergestellt
werden, wie etwa enterischen Beschichtungen und anderen Beschichtungen,
welche im Bereich der Herstellung von Pharmazeutika wohl bekannt
sind. Sie können
auch derart formuliert werden, dass sie langsame oder gesteuerte
Freisetzung des aktiven Inhaltsstoffs darin bereitstellen, unter Verwendung
von beispielsweise Hydroxypropylmethylcellulose in variierenden
Anteilen, um das erwünschte Freisetzungsprofil
bereitzustellen, anderen Polymermatrizes, Liposomen und/oder Mikrocentrosomen.
Sie können
sterilisiert werden beispielsweise durch Filtration durch einen
Bakterien zurückhaltenden
Filter oder durch Einarbeiten von Sterilisierungsmitteln in der
Form von sterilen festen Zusammensetzungen, welche in sterilem Wasser
aufgelöst
werden können
oder einem anderen sterilen, injizierbaren Medium, unmittelbar vor Verwendung.
Diese Zusammensetzungen können
auch optional trübende
Mittel enthalten, und sie können
von einer derartigen Zusammensetzung sein, dass sie den aktiven
Inhaltsstoff bzw. die aktiven Inhaltsstoffe nur oder vorzugsweise
in einem bestimmten Abschnitt des gastrointestinalen Trakts, optional
in einer verzögerten Art
und Weise, freisetzen. Beispiele für einbettende Zusammensetzungen,
welche verwendet werden können, umfassen
polymere Substanzen und Wachse. Der aktive Inhaltsstoff kann auch
in mikro eingekapselter Form vorliegen, geeignetenfalls, mit einem
oder mehr der oben beschriebenen Exzipienten.
-
Flüssige Dosierungsformen
für orale
Verabreichung der erfindungsgemäßen Verbindungen
umfassen pharmazeutisch akzeptable Emulsionen, Mikroemulsionen,
Lösungen,
Suspensionen, Sirups und Elixiere. Zusätzlich zu dem aktiven Inhaltsstoff
können
die flüssigen
Dosierungsformen inerte Verdünnungsmittel,
welche häufig
im Fachbereich verwendet werden, enthalten, wie beispielsweise etwa
Wasser oder andere Lösungsmittel,
solubilisierende Mittel und Emulgatoren, wie etwa Ethylalkohol,
Isopropylalkohol, Ethylcarbonat, Ethylacetat, Benzylalkohol, Benzylbenzoat,
Propylenglycol, 1,3-Butylenglycol, Öle (insbesondere Baumwollsamen-,
Walnuss-, Mais-, Keim-, Oliven-, Rizinus- und Sesamöle), Glycerin, Tetrahydrofurylalkohol,
Polyethylenglycole und Fettsäureester
von Sorbitan und Gemische davon.
-
Neben
inerten Verdünnungsmitteln
können
die oralen Zusammensetzungen auch Hilfsstoffe wie etwa Benetzungsmittel,
emulgierende und suspendierende Mittel, süßende, geschmackgebende, farbgebende,
parfümierende
und konservierende Mittel umfassen.
-
Suspensionen
können
zusätzlich
zu den aktiven Verbindungen Suspendiermittel enthalten, wie beispielsweise
etwa ethoxylierte Isostearylalkohole, Polyoxyethylsorbitol und Sorbitanester,
mikrokristalline Cellulose, Aluminiummetahydroxid, Bentonit, Agar
Agar und Tragacanth und Gemische davon.
-
Formulierungen
der pharmazeutischen Zusammensetzungen gemäß der Erfindung für rektale
und vaginale Verabreichung können
vorgelegt werden als ein Zäpfchen,
welches hergestellt werden kann durch Mischen einer oder mehr der
erfindungsgemäßen Verbindungen
mit einem oder mehreren geeigneten, nicht reizenden Exzipienten
oder Trägern,
welche beispielsweise Kakaobutter, Polyethylenglycol, ein Zäpfchenwachs oder
ein Salicylat umfassen und wobei das Zäpfchen bei Raumtemperatur fest
ist, jedoch flüssig
bei Körpertemperatur
und daher im Rektum oder der Vaginalhöhle schmelzen und die aktive
Verbindung freisetzen wird.
-
Erfindungsgemäße Formulierungen,
welche für
vaginale Verabreichung geeignet sind, umfassen auch Pessare, Tampons,
Cremes, Gele, Pasten, Schäume
oder Spray-Formulierungen,
welche im Fachbereich als geeignet bekannte Träger enthalten.
-
Dosierungsformen
für die
topische oder transdermale Verabreichung einer erfindungsgemäßen Verbindung
umfassen Pulver, Sprays, Salben, Pasten, Cremes, Lotionen, Gele,
Lösungen,
Pflaster und Inhalationsmittel. Die aktive Verbindung kann unter
sterilen Bedingungen mit einem pharmazeutisch akzeptablen Träger und
mit jeglichen Konservierungsstoffen, Puffern oder Treibmitteln,
welche erforderlich sein können,
gemischt werden.
-
Die
Salben, Pasten, Cremes und Gele können zusätzlich zu der erfindungsgemäßen aktiven
Verbindung Exzipienten, wie etwa tierische und pflanzliche Fette, Öle, Wachse,
Paraffine, Stärke,
Tragacanth, Cellulosederivate, Polyethylenglycole, Silikone, Bentonite,
Kieselsäure,
Talk und Zinkoxid oder Gemische davon enthalten.
-
Pulver
und Sprays können
zusätzlich
zu einer erfindungsgemäßen Verbindung
Exzipienten enthalten wie etwa Lactose, Talk, Kieselsäure, Aluminiumhydroxid,
Calciumsilikate und Polyamidpulver oder Gemische dieser Substanzen.
Sprays können
zusätzlich übliche Treibmittel
enthalten, wie etwa Chlorfluorkohlenwasserstoffe und flüchtige unsubstituierte
Kohlenwasserstoffe, wie etwa Butan und Propan.
-
Transdermalpflaster
haben den zusätzlichen
Vorteil, gesteuerte Abgabe einer erfindungsgemäßen Verbindung an den Körper bereitzustellen.
Solche Dosierungsformen können
hergestellt werden durch Auflösen
oder Dispergieren der Verbindung in dem richtigen Medium. Absorptionsverstärker können ebenfalls
verwendet werden, um den Fluß der
Verbindung durch die Haut zu erhöhen.
Die Rate eines solchen Flusses kann gesteuert werden entweder durch
Bereitstellen einer ratensteuernden Membran oder durch Dispergieren
der Verbindung in einer Polymermatrix oder einem Gel.
-
Ophthalmische
Formulierungen, Augensalben, Pulver, Lösungen und dergleichen sollen
ebenfalls in den Umfang der vorliegenden Erfindung fallen.
-
Pharmazeutische
Zusammensetzungen gemäß dieser
Erfindung, welche für
parenterale Verabreichung geeignet sind, umfassen eine oder mehrere
erfindungsgemäße Verbindungen
in Kombination mit einer oder mehreren pharmazeutisch akzeptablen,
sterilen, isotonischen, wässrigen
oder nicht-wässrigen
Lösungen,
Dispersionen, Suspensionen oder Emulsionen, oder sterilen Pulvern,
welche unmittelbar vor Verwendung in sterile injizierbare Lösungen oder
Dispersionen rekonstitutiert werden können, welche Antioxidantien,
Puffer, Bacteriostatica, gelöste
Stoffe, welche die Formulierung mit dem Blut des beabsichtigten
Empfängers
isotonisch machen, oder suspendierende oder verdickende Mittel enthalten
können.
-
Beispiele
für geeignete
wässrige
und nicht-wässrige
Träger,
welche in den pharmazeutischen Zusammensetzungen der Erfindung verwendet
werden können,
umfassen Wasser, Ethanol, Polyole (wie etwa Glycerin, Propylenglycol,
Polyethylenglycol und dergleichen) und geeignete Mischungen davon,
pflanzliche Öle, wie
etwa Olivenöl,
und injizierbare organische Ester, wie etwa Ethyloleat. Geeignete
Fluidität
kann beispielsweise aufrechterhalten werden durch die Verwendung
von Beschichtungsmaterialien, wie etwa Lecithin, durch die Aufrechterhaltung
der erforderlichen Partikelgröße im Falle
von Dispersionen und durch die Verwendung von oberflächenaktiven
Substanzen.
-
Diese
Zusammensetzungen können
auch Hilfsstoffe wie etwa Konservierungsstoffe, Benetzungsmittel,
Emulgiermittel und Dispergiermittel enthalten. Verhinderung der
Aktivität
von Mikroorganismen auf die fraglichen Verbindungen kann sichergestellt
werden durch die Einfügung
verschiedener antibakterieller und Antipilz-Mittel, z.B. Paraben,
Chlorbutanol, Phenolsorbinsäure,
und dergleichen. Es kann auch wünschenswert sein,
isotonische Mittel wie etwa Zucker, Natriumchlorid und dergleichen
in die Zusammensetzungen einzufügen.
Zusätzlich
kann verlängerte
Absorption der injizierbaren pharmazeutischen Form bewirkt werden
durch die Einfügung
von Mitteln, welche Absorption verzögern, wie etwa Aluminiummonostearat
und Gelatine.
-
Um
die Wirkung eines Wirkstoffs zu verlängern, ist es in einigen Fällen wünschenswert,
die Absorption des Wirkstoffs von subkutaner oder intramuskulärer Injektion
zu verlangsamen. Dies kann erreicht werden durch die Verwendung
einer flüssigen
Suspension kristallinen oder amorphen Materials mit schlechter Wasserlöslichkeit.
Die Absorptionsrate des Wirkstoffs hängt dann von der Rate seiner
Auflösung
ab, welche wiederum von Kristallgröße und kristalliner Form abhängen kann.
Alternativ wird verzögerte
Absorption einer parenteral verabreichten Wirkstoffform erreicht
durch Auflösen
oder Suspendieren des Wirkstoffs in einem Öl-Vehikel.
-
Injizierbare
Depotformen werden hergestellt durch Bilden von Mikroeinkapselungsmatrizes
der fraglichen Verbindungen in bioabbaubaren Polymeren, wie etwa
Polylactid-Polyglycolid.
In Abhängigkeit
von dem Verhältnis
von Wirkstoff zu Polymer und der Natur des besonderen verwendeten
Polymers kann die Rate der Wirkstofffreisetzung gesteuert werden.
Beispiele für
andere bioabbaubare Polymere umfassen Poly(orthoester) und Poly(anhydride).
Injizierbare Depotformulierungen werden auch hergestellt durch Einfangen
des Wirkstoffs in Liposomen oder Mikroemulsionen, welche mit Körpergewebe
kompatibel sind.
-
Wenn
die erfindungsgemäßen Verbindungen
als Pharmazeutica Menschen und Tieren verabreicht werden, können sie
per se gegeben werden oder als eine pharmazeutische Zusammensetzung,
welche beispielsweise 0,1 bis 99,5% (weiter bevorzugt 0,5 bis 90%)
an aktivem Inhaltsstoff in Kombination mit einem pharmazeutisch
akzeptablen Träger
enthält.
-
Die
Präparationen
der vorliegenden Erfindung können
oral, parenteral, topisch oder rektal gegeben werden. Sie werden
natürlich
in Formen gegeben, welche für
jeden Verabreichungsweg geeignet sind. Beispielsweise werden sie
in Tabletten- oder Kapselform, durch Injektion, Inhalation, Augenlotion,
Salbe, Zäpfchen
etc. verabreicht, Verabreichung durch Injektion, Infusion oder Inhalation;
topisch durch Lotion oder Salbe; und rektal durch Zäpfchen.
Orale Verabreichungen sind bevorzugt.
-
Die
Ausdrücke "parenterale Verabreichung" und "parenteral verabreicht", wie hierin verwendet,
bedeuten Verarbreichungsmodi mit Ausnahme von enteraler und topischer
Verabreichung, üblicherweise
durch Injektion, und sie umfassen ohne Beschränkung intravenöse, intramuskuläre, intraarterielle,
intrathekale, intrakapsuläre,
intraorbitale, intrakardiale, intradermale, intraperitoneale, transtracheale,
subkutane, subkutikulare, intraartikulare, subkapusuläre, subarachnoide,
intraspinale und intrasternale Injektion und Infusion.
-
Die
Ausdrücke "systemische Verabreichung", "systemisch verabreicht", "periphere Verabreichung" und "peripher verabreicht", wie hierin verwendet,
bedeuten die Verabreichung einer Verbindung, eines Wirkstoffs oder
anderen Materials anders als direkt in das zentrale Nervensystem,
derart, dass sie in das System des Patienten eintritt und damit
Metabolismus und anderen ähnlichen
Prozessen unterworfen ist, beispielsweise subkutane Verabreichung.
-
Diese
Verbindungen können
Menschen und anderen Tieren zur Therapie über einen jeglichen geeigneten
Verabreichungsweg verabreicht werden, einschließlich oral, nasal, wie beispielsweise
durch ein Spray, rektal, intravaginal, parenteral, intrazysternär und topisch,
wie durch Pulver, Salben oder Tropfen, einschließlich bukkal und sublingual.
-
Unabhängig von
dem ausgewählten
Verabreichungsweg werden die erfindungsgemäßen Verbindungen, welche in
einer geeigneten hydrierten Form verwendet werden können, und/oder
die pharmazeutischen Zusammensetzungen gemäß der vorliegenden Erfindung
in pharmazeutisch akzeptable Dosierungsformen durch herkömmliche,
Fachleuten auf diesem Gebiet bekannte Verfahren formuliert.
-
Tatsächliche
Dosierungsniveaus der aktiven Inhaltsstoffe in den erfindungsgemäßen pharmazeutischen
Zusammensetzungen können
variiert werden, um eine Menge des aktiven Inhaltsstoffs zu erhalten,
welcher wirksam ist, um die gewünschte
therapeutische Reaktion für
einen besonderen Patienten, eine besondere Zusammensetzung und einen
besonderen Verabreichungsmodus zu erzielen, ohne für den Patienten
toxisch zu sein.
-
Das
ausgewählte
Dosierungsniveau wird von einer Vielzahl von Faktoren abhängen, einschließlich der Aktivität der besonderen
erfindungsgemäßen Verbindung,
welche verwendet wird, oder des Esters, Salzes oder Amids davon,
dem Verabreichungsweg, der Verabreichungszeit, der Ausscheidungsrate
der besonderen verwendeten Verbindung, der Behandlungsdauer, anderen
Wirkstoffen, Verbindungen und/oder Materialien, welche in Kombination
mit der besonderen verwendeten Verbindung verwendet werden, dem Alter,
Geschlecht, Gewicht, Zustand, der allgemeinen Gesundheit und vorausgehenden
medizinischen Geschichte des behandelten Patienten und ähnlichen
im Bereich der Medizin wohl bekannten Faktoren.
-
Ein
Arzt oder Tierarzt mit durchschnittlichem Fachwissen kann ohne weiteres
die wirksame Menge der erforderlichen pharmazeutischen Zusammensetzung
bestimmen und verschreiben. Beispielsweise könnte der Arzt oder Tierarzt
mit Dosen der erfindungsgemäßen Verbindungen
beginnen, welche in der pharmazeutischen Zusammensetzung in geringeren
Gehalten verwendet sind, als sie erforderlich sind, um die gewünschte therapeutische
Wirkung zu erzielen, und nach und nach die Dosierung erhöhen, bis
die gewünschte
Wirkung erreicht wird.
-
Im
Allgemeinen ist eine geeignete tägliche
Dosis einer erfindungsgemäßen Verbindung
die Menge der Verbindung, welches die geringste Dosis darstellt,
welche wirksam ist, um eine therapeutische Wirkung hervorzurufen.
Eine solche wirksame Dosis hängt
allgemein von den oben beschriebenen Faktoren ab. Allgemein liegen
intravenöse,
intrazerebroventrikulare und subkutane Dosen der erfindungsgemäßen Verbindungen
für einen
Patienten, wenn sie für
die indizierten analgesischen Wirkungen verwendet werden, im Bereich
von etwa 0,0001 bis etwa 100 mg pro kg Körpergewicht pro Tag.
-
Wenn
gewünscht,
kann die wirksame tägliche
Dosis der aktiven Verbindung als zwei, drei, vier, fünf, sechs
oder mehr Sub-Dosen verabreicht werden, welche separat in geeigneten
Intervallen über
den Tag verteilt, optional in Einheitsdosierungsformen, verabreichten
werden.
-
Während es
möglich
ist, dass eine erfindungsgemäße Verbindung
allein verabreicht wird, ist es bevorzugt, die Verbindung als eine
pharmazeutische Formulierung (Zusammensetzung) zu verabreichen.
-
Unter
einem weiteren Aspekt stellt die vorliegende Erfindung pharmazeutisch
akzeptable Zusammensetzungen bereit, welche eine therapeutisch wirksame
Menge einer oder mehr der fraglichen Verbindungen, wie oben beschrieben,
umfassen, formuliert zusammen mit einem oder mehreren pharmazeutisch
akzeptablen Trägern
(Additiven) und/oder Verdünnungsmitteln.
Wie untenstehend im Detail beschrieben, können die pharmazeutischen Zusammensetzungen
gemäß der vorliegenden
Erfindung speziell für
Verabreichung in fester oder flüssiger
Form formuliert werden, einschließlich derer, welche hergerichtet
sind für
folgendes: (1) orale Verabreichung, z.B. Arzneitränke (wässrige oder
nicht-wässrige Lösungen oder
Suspensionen), Tabletten, Bolus, Pulver, Granulate, Pasten zur Auftragung
auf die Zunge; (2) parenterale Verabreichung, z.B. durch subkutane,
intramuskuläre
oder intravenöse
Injektion, als beispielsweise eine sterile Lösung oder Suspension; (3) topische
Anwendung, z.B. als eine Creme, Salbe oder Spray, welche auf die
Haut aufgebracht werden; oder (4) intravaginal oder intrarektal,
z.B. als ein Pessar, eine Creme oder ein Schaum.
-
Die
erfindungsgemäßen Verbindungen
können
zur Verabreichung in einer jeglichen bequemen Art und Weise zur
Verwendung in Human- oder Veterinärmedizin formuliert werden,
in Analogie zur anderen Pharmazeutika.
-
Der
Begriff "Behandlung" soll auch Prophylaxe,
Therapie und Heilung umfassen.
-
Der
diese Behandlung empfangene Patient ist ein jegliches bedürftiges
Tier, einschließlich
Primaten, insbesondere Menschen, und anderen Säugetiere, wie etwa Pferden,
Rindern, Schweinen und Schafen; und Geflügel und Haustiere im Allgemeinen.
-
Die
erfindungsgemäße Verbindung
kann als solche oder in Gemischen mit pharmazeutisch akzeptablen
Trägern
verabreicht werden und kann auch in Verbindung mit antimikrobiellen
Mitteln, wie etwa Penicillinen, Cephalosporinen, Aminoglycosiden
und Glycopeptiden verabreicht werden. Verbindende Therapie umfasst
damit sequentielle, simultane und separate Verabreichung der aktiven
Verbindung in einer Art und Weise, dass die therapeutischen Wirkungen
der zuerst verabreichten noch nicht ganz abgeklungen sind, wenn
die nachfolgende verabreicht wird.
-
Die
Zugabe der aktiven erfindungsgemäßen Verbindung
zu Tierfutter wird vorzugsweise erreicht durch Herstellen eines
geeigneten Futtervorgemischs, welches die aktive Verbindung in einer
wirksamen Menge enthält,
und Einbringen des Vorgemischs in die gesamte Ration.
-
Alternativ
kann ein Zwischenkonzentrat oder Futterzusatz, welches/r den aktiven
Inhaltsstoff enthält, in
das Futter eingemischt werden. Die Art und Weise, auf welche solche
Futtervorgemische und vollständige Rationen
hergestellt werden und verabreicht werden können, sind in Nachschlagebüchern beschrieben
(wie etwa "Applied
Animal Nutrition",
W. H. Freedman und CO., San Francisco, USA, 1969 oder "Livestock Feeds and
Feeding" O und B
books, Corvallis, Ore., USA, 1977).
-
Kombinatorische
Bibliotheken
-
In
der gegenwärtigen Ära der Wirkstoffentwicklung
spielt Hochdurchsatz-Screening von Tausenden bis Millionen von Verbindungen
eine Schlüsselrolle.
Hochdurchsatz-Screening beinhaltet im Allgemeinen Automatisierung
und Robotereinsatz, um zu ermöglichen,
dass diese Tausenden bis Millionen von Verbindungen in einem oder
mehr Bioassays in einer relativ kurzen Zeitperiode getestet werden.
Diese Screening-Technik hoher Kapazität erfordert enorme Mengen an "Rohmaterialien" mit immenser molekularer
Vielfalt, um zur Verfügung
stehende Kapazität
zu füllen.
Demgemäss
wird kombinatorische Chemie eine bedeutende Rolle dabei spielen,
diese Anforderung für
neue Moleküle
zum Screenen zu erfüllen.
Wenn einmal "Leads" unter Verwendung
von Hochdurchsatz-Screening-Techniken identifiziert sind, wird kombinatorische
Chemie vorteilhaft verwendet, um diese anfänglichen Leads zu optimieren
(welche Analoge/Varianten in dem gleichen Hochdurchsatz-Screening-Assay
getestet werden, welches das anfängliche
Lead identifiziert hat).
-
Eine
kombinatorische Bibliothek für
die Zwecke der vorliegenden Erfindung ist ein Gemisch von chemisch
verwandten Verbindungen, welche zusammen auf eine gewünschte Eigenschaft
hin gescreent werden können;
diese Bibliotheken können
in Lösung
oder kovalent an einen festen Träger
gekoppelt sein. Die Herstellung von vielen verwandten Verbindungen
in einer einzelnen Reaktion verringert und vereinfacht die Anzahl von
Screening- Prozessen,
welche ausgeführt
werden müssen,
sehr stark. Screening im Hinblick auf die geeignete biologische,
pharmazeutische, agrochemische oder physikalische Eigenschaft kann
mittels herkömmlicher
Verfahren durchgeführt
werden.
-
Vielfalt
in einer Bibliothek kann auf einer Vielzahl von unterschiedlichen
Niveaus erzeugt werden. Beispielsweise können die Substrat-Arylgruppen,
welche in einer kombinatorischen Herangehensweise verwendet werden,
hinsichtlich der Kern-Arylgruppe vielfältig sein, z.B. eine Variation
in Bezug auf die Ringstruktur, und/oder sie können bezüglich der anderen Substituenten
variieren.
-
Eine
Vielzahl von Techniken ist im Fachbereich erhältlich zum Erzeugen kombinatorischer
Bibliotheken kleiner organischer Moleküle. Siehe beispielsweise Blondelle
et al. (1995) Trends Anal. Chem. 14: 83; die Affymax U.S. Patente
5,359,115 und 5,362,899; das Ellman U.S. Patent 5,288,514; die Still
et al. PCT-Veröffentlichung
WO 94/08051; Chen et al. (1994) JACS 116: 2661; Kerr et al. (1993)
JACS 115: 252; PCT-Veröffentlichungen
WO 92/10092, WO 93/09668 und WO 91/07087; und die Lerner et al.
PCT-Veröffentlichung
WO 93/20242). Demgemäss
kann eine Vielzahl von Bibliotheken im Bereich von etwa 16 bis 1.000.000
oder mehr Diversomeren synthetisiert und auf eine besondere Aktivität oder Eigenschaft
hin gescreent werden.
-
In
einer beispielhaften Ausführungsform
kann eine Bibliothek substituierter Diversomere synthetisiert werden
unter Verwendung der in Frage stehenden Reaktionen, welche angepasst
sind an die Techniken, welche in der Still et al. PCT-Veröffentlichung
WO 94/08051 beschrieben sind, z.B. Anknüpfung an ein Polymerkügelchen
durch eine hydrolysierbare oder photolysierbare Gruppe, z.B. angeordnet
an einer der Substratpositionen. Gemäß der Still et al. Technik
ist die Bibliothek auf einem Satz von Kügelchen synthetisiert, wobei jedes
Kügelchen
einen Satz von Tags (Anhängseln)
umfasst, welcher das besondere Diversomer auf dem Kügelchen
identifiziert. In einer Ausführungsform,
welche besonders geeignet ist zur Entdeckung von Enzyminhibitoren,
können
die Kügelchen
auf der Oberfläche
einer permeablen Membran verteilt werden und die Diversomere von
dem Kügelchen
durch Lyse des Kügelchen-Linkers freigesetzt
werden. Das Diversomer von jedem Kügelchen wird durch die Membran
hin zu einer Assayzone diffundieren, wo es mit einem Enzymassay
wechselwirken wird. Detaillierte Beschreibungen einer Anzahl von
kombinatorischen Methodologien sind nachfolgend bereitgestellt.
-
A) Direkte Charakterisierung
-
Ein
wachsender Trend auf dem Gebiet kombinatorischer Chemie besteht
im Ausnutzen der Empfindlichkeit von Techniken wie beispielsweise
etwa Massenspektrometrie (MS), welche verwendet werden können, um
sub-femtomolare Mengen einer Verbindung zu charakterisieren und
die chemische Konstitution einer aus einer kombinatorischen Bibliothek
ausgewählten
Verbindung direkt zu bestimmen. Z.B. können, wenn die Bibliothek auf
einer unlöslichen
Trägermatrix
bereitgestellt ist, diskrete Populationen von Verbindungen als erstes
von dem Träger
freigesetzt und mittels MS charakterisiert werden. In anderen Ausführungsformen
können als
Teil der MS-Probenvorbereitungstechnik MS-Techniken wie MALDI verwendet
werden, um eine Verbindung von der Matrix zu befreien, insbesondere
dort, wo ursprünglich
eine labile Bindung verwendet wird, um die Verbindung an die Matrix
zu binden. Z.B. kann ein aus einer Bibliothek ausgewähltes Kügelchen
in einem MALDI-Schritt bestrahlt werden, um das Diversomer aus der
Matrix zu befreien und um das Diversomer für die MS-Analyse zu ionisieren.
-
B) Multipin-Synthese
-
Die
Bibliotheken des fraglichen Verfahrens können Multipin-Bibliothek-Format
annehmen. Kurz gefasst haben Geysen und Mitarbeiter (Geysen et al.
(1984) PNAS 81: 3998–4002)
ein Verfahren eingeführt
zum Herstellen von Verbindungsbibliotheken durch eine parallele
Synthese auf Polyacrylsäure-gerasterten
Polyethylen-Stiften, welche im Mikrotiterplattenformat angeordnet
sind. Die Geysen-Technik kann verwendet werden, um Tausende von
Verbindungen pro Woche zu synthetisieren und zu screenen, unter
Verwendung des Multipin-Verfahrens, und die gebundenen Verbindungen
können
in vielen Assays wieder verwendet werden. Es können auch geeignete Linker-Gruppen
an die Stifte angegliedert werden, so dass die Verbindungen nach Synthese
von den Trägern
zum Untersuchen von Reinheit und zur weiteren Evaluierung abgespalten
werden können
(c.f., Bray et al. (1990) Tetrahedron Lett 31: 5811–5814; Valerio
et al. (1991) Anal Biochem 197: 168–177; Bray et al. (1991) Tetrahedron
Lett 32: 6163–6166).
-
C) Teilen-Koppeln-Rekombinieren
-
In
einer weiteren Ausführungsform
kann eine abgewandelte Verbindungsbibliothek auf einem Satz von Kügelchen
unter Verwendung der Strategie des Teilen-Koppeln-Rekombinierens bereitgestellt
werden (siehe z.B. Houghten (1985) PNAS 82: 5131–5134 und U.S. Patente 4,631,2111
5,440,016; 5,480,971). Kurz gesagt, wie der Name bereits impliziert,
werden bei jedem Syntheseschritt, bei welchem Degenerierung in die
Bibliothek eingeführt
wird, die Kügelchen
in separate Gruppen eingeteilt, welche gleich sind zu der Anzahl
von verschiedenen Substituenten, welche an einer bestimmten Position
in der Bibliothek zugefügt
werden sollen, wobei die verschiedenen Substituenten in separaten
Reaktionen gekoppelt werden, und die Kügelchen in einen Pool für die nächste Iteration
rekombiniert werden.
-
In
einer Ausführungsform
kann die Teilen-Koppeln-Rekombinieren-Strategie
ausgeführt
werden unter Verwendung eines analogen Ansatzes zu der sogenannten "Teebeutel"-Methode, welche zuerst von Houghten entwickelt
wurde, wobei die Verbindungssynthese auf Harz stattfindet, welches
in porösen
Polypropylenbeuteln versiegelt ist (Houghten et al. (1986) PNAS
82: 5131–5135).
Substituenten werden an die Verbindungs-tragenden Harze gekoppelt
durch Plazieren der Beutel in geeigneten Reaktionslösungen,
während
alle gemeinsamen Schritte, wie etwa Harzwaschen und Schutzgruppenentfernung
simultan in einem Reaktionsbehälter ausgeführt werden.
Am Ende der Synthese enthält
jeder Beutel eine einzelne Verbindung.
-
D) Kombinatorische Bibliotheken
durch licht-geleitete, räumlich
adressierbare parallele chemische Synthese
-
Ein
Modell einer kombinatorischer Synthese, bei welcher die Identität einer
Verbindung durch ihre Anordnung auf einem Synthesesubstrat gegeben
ist, wird als räumlich
adressierbare Synthese bezeichnet. In einer Ausführungsform wird der kombinatorische
Prozess durchgeführt
durch Steuerung der Addition eines chemischen Reagenzes an spezifische
Orte auf einem festen Träger
(Dower et al. (1991) Annu Rep Med Chem 26: 271–280; Fodor, S. P. A. (1991)
Science 251: 767; Pirrung et al. (1992) U.S. Patent 5,143,854; Jacobs
et al. (1994) Trends Biotechnol 12: 19–26). Die räumliche Auflösung der
Photolithographie ermöglicht
Miniaturisierung. Diese Technik kann mit Hilfe der Verwendung von
Schutz-/Schutzgruppen-Entfernungsreaktionen
mit photolabilen Schutzgruppen durchgeführt werden.
-
Die
Schlüsselpunkte
dieser Technologie sind aufgezeigt in Gallop et al. (1994) J Med
Chem 37: 1233–1251.
Ein Synthesesubstrat wird durch die kovalente Anbindung von photolabilen
Nitroveratryloxycarbonyl(NVOC)-geschützten Amino-Linkern oder anderen
photolabilen Linkern für
die Kopplung hergestellt. Licht wird verwendet, um selektiv eine
spezifische Reaktion des Syntheseträgers zum Koppeln zu aktivieren.
Entfernen der photolabilen Schutzgruppen durch Licht (Schutzgruppenentfernung)
führt zur
Aktivierung von ausgewählten
Gegenden. Nach Aktivierung wird das erste eines Satzes von Aminosäureanalogen,
von denen jedes eine photolabile Schutzgruppe am Aminoterminus trägt, der
gesamten Oberfläche
ausgesetzt. Kopplung findet nur in Regionen statt, welche im vorangehenden
Schritt Licht ausgesetzt wurden. Die Reaktion wird gestoppt, die
Platten gewaschen und das Substrat wieder durch eine zweite Maske
beleuchtet, wodurch eine unterschiedliche Region zur Reaktion mit
einem zweiten geschützten
Baublock aktiviert wird. Das Muster von Masken und die Reihenfolge
von Reaktanden definieren die Produkte und ihre Orte. Da dieser
Prozess photolithographische Techniken einsetzt, ist die Anzahl
von Verbindungen, die synthetisiert werden können, lediglich durch die Anzahl
von Synthesestellen begrenzt, welche mit geeigneter Auflösung adressiert
werden können.
Die Position jeder Verbindung ist präzise bekannt; daher können ihre
Wechselwirkungen mit anderen Molekülen direkt untersucht werden.
-
In
einer licht-gesteuerten chemischen Synthese hängen die Produkte von dem Muster
der Beleuchtung und von der Reihenfolge der Addition von Reaktanden
ab. Durch Variation der lithographischen Muster können viele
verschiedene Sätze
von Testverbindungen simultan synthetisiert werden; dieses Charakteristikum
führt zu
der Erzeugung von vielen verschiedenen Maskierungsstrategien.
-
E) Kodierte kombinatorische
Bibliotheken
-
In
einer weiteren Ausführungsform
kann das in Frage stehende Verfahren eine Verbindungsbibliothek verwenden,
welche mit einem kodierten Tag-System ausgestattet ist. Eine neuere
Verbesserung bei der Identifizierung von aktiven Verbindungen aus
kombinatorischen Bibliotheken verwendet chemische Indexsysteme, welche
Tags verwenden, welche die Reaktionsschritte, welche ein gegebenes
Kügelchen
durchlaufen hat, einzigartig kodieren und durch Rückschluss
die Struktur, die es trägt.
Konzeptuell ahmt dieser Ansatz Phagen-Display-Bibliotheken nach,
bei denen Aktivität
sich von exprimierten Peptiden ableitet, aber die Strukturen der
aktiven Peptide von der korrespondierenden Genom-DNA-Sequenz abgeleitet
werden. Die erste Kodierung von synthetischen kombinatorischen Bibliotheken
verwendeten DNA als den Code. Eine Vielzahl anderer Formen der Kodierung
sind berichtet worden, einschließlich Kodierung mit sequenzierbaren
Bio-Oligomeren (z.B. Oligonucleotiden und Peptiden) und binärem Kodieren
mit zusätzlichen
nicht-sequenzierbaren
Tags.
-
1) Tagging mit sequenzierbaren
Bio-Oligomeren
-
Das
Prinzip der Verwendung von Oligonucleotiden zum Kodieren kombinatorischer
synthetischer Bibliotheken wurde 1992 beschrieben (Brenner et al.
(1992) PNAS 89: 5381–5383),
und ein Beispiel für
eine solche Bibliothek erschien im folgenden Jahr (Needles et al.
(1993) PNAS 90: 10700–10704).
Eine kombinatorische Bibliothek von nominal 77 (=
823.543) Peptiden, bestehend aus allen Kombinationen von Arg, Gln,
Phe, Lys, Val, D-Val und Thr (Drei-Buchstaben-Aminosäurecode), von denen jedes durch
ein spezifisches Dinucleotid (TA, TC, CT, AT, TT, CA bzw. AC) kodiert
war, wurde durch eine Serie von alternierenden Runden von Peptid-
und Oligonucleotid-Synthese auf festem Träger hergestellt. In dieser
Arbeit wurde die Amin-bindende Funktionalität auf dem Kügelchen speziell für Peptid-
oder Oligonucleotid-Synthese differenziert durch simultanes Vor-Inkubieren der Kügelchen
mit Reagenzien, welche geschützte
OH-Gruppen für
die Oligonucleotidsynthese und geschützte NH2-Gruppen
für die
Peptidsynthese erzeugen (hier in einem Verhältnis von 1:20). Nach Beendigung
bestand jeder Tag aus 69-meren, von denen 14 Einheiten den Code
trugen. Die Kügelchen-gebundene
Bibliothek wurde mit einem fluoreszierend markierten Antikörper inkubiert,
und die Kügelchen,
welche gebundene Antikörper
enthielten und stark fluoreszierten, wurden mittels Fluoreszenz-aktiviertem
Zellensortieren (FACS) geerntet. Die DNA-Tags wurden mittels PCR
amplifiziert und sequenziert, und die vorhergesagten Peptide wurden
synthetisiert. Gemäß solchen Techniken
können
Verbindungsbibliotheken zur Verwendung in dem in Frage stehenden
Verfahren abgeleitet werden, wo die Oligonucleotid-Sequenz des Tags
die aufeinanderfolgenden kombinatorischen Reaktionen identifizieren,
welche ein besonderes Kügelchen durchlief,
und daher die Identität
der Verbindung auf dem Kügelchen
liefert.
-
Die
Verwendung von Oligonucleotid-Tags erlaubt besonders empfindliche
Tag-Analyse. Dennoch erfordert das Verfahren sorgfältige Auswahl
von orthogonalen Sätzen
von Schutzgruppen, welche erforderlich sind zum Alternieren von
Co-Synthese des Tags und des Bibliothekmitglieds. Darüber hinaus
kann die chemische Labilität
des Tags, insbesondere der Phosphat- und Zucker-Anomeren-Verknüpfungen,
die Auswahl an Reagenzien und Bedingungen limitieren, welche für die Synthese
von nicht-oligomeren Bibliotheken verwendet werden können. In
bestimmten Ausführungsformen
verwenden die Bibliotheken Linker, welche selektives Entfernen des
Testverbindungsbibliotheksmitglieds für Assay erlauben.
-
Peptide
sind ebenfalls verwendet worden zum Taggen von Molekülen für kombinatorische
Bibliotheken. Zwei beispielhafte Ansätze sind im Fachbereich beschrieben,
von welchen beide verzweigte Linker an fester Phase verwenden, an
welchen Kodierungs- und Liganden-Stränge alternierend ausgearbeitet
werden. In dem ersten Ansatz (Kerr JM et al. (1993) J. Am. Chem.
Soc. 115: 2529–2531)
wird Orthogonalität
bei der Synthese erreicht durch Verwendung von Säure-labilem Schutz für den kodierenden Strang und
Basen-labilem Schutz für
den Verbindungsstrang.
-
In
einem alternativen Ansatz (Nikolaiev et al. (1993) Pept. Res. 6:
161–170)
werden verzweigte Linker verwendet, so dass die kodierende Einheit
und die Testverbindung beide an die gleiche funktionelle Gruppe auf
dem Harz gebunden werden können.
In einer Ausführungsform
kann ein spaltbarer Linker zwischen den Verzweigungspunkt und das
Kügelchen
platziert werden, so dass Spaltung ein Molekül freisetzt, welches sowohl
Codes als auch die Verbindung enthält (Ptek et al. (1991) Tetrahedron
Lett 32: 3891–3894).
In einer weiteren Ausführungsform
kann der spaltbare Linker derart platziert werden, dass die Testverbindung
selektiv von dem Kügelchen
getrennt werden kann, und so den Code zurücklässt. Das letzte Konstrukt ist
besonders wertvoll, da es Screenen der Testverbindung ohne potentielle
Interferenz der kodierenden Gruppen erlaubt. Beispiele auf diesem
Gebiet von unabhängiger
Spaltung und Sequenzierung von Peptid-Bibliotheksmitgliedern und ihren entsprechenden
Tags hat bestätigt,
dass die Tags die Peptidstruktur genau vorhersagen können.
-
2) Nicht-sequenzierbares
Tagging: Binäres
Kodieren
-
Eine
alternative Form der Kodierung der Testverbindungsbibliothek verwendet
einen Satz von nicht-sequenzierbaren
elektrophoren Tagging-Molekülen,
welche als ein binärer
Code verwendet werden (Ohlmeyer et al. (1993) PNAS 90: 10922–10926).
Beispiele für
Tags sind halogenaromatische Alkylether, welche als ihre Trimethylsilylether,
welche in kleiner als femtomolaren Konzentrationen durch Elektroneneinfanggaschromatographie
(electron capture gas chromatography ECGC)) detektierbar sind. Variationen
in der Länge der
Alkylkette sowie der Natur und Position der Halogen-Aromaten-Substituenten erlauben
die Synthese von wenigstens 40 solcher Tags, welche prinzipiell
240 (z.B. mehr als 1012)
verschiedene Moleküle
kodieren können. In
dem ursprünglichen
Bericht (Ohlmeyer et al., supra) wurden die Tags an etwa 1% der
zur Verfügung
stehenden Amingruppen einer Peptidbibliothek über einen photospaltbaren o-Nitrobenzyl-Linker
gebunden. Dieser Ansatz ist bequem, wenn kombinatorische Bibliotheken
von Peptid-ähnlichen
oder anderen Amin-enthaltenden Molekülen hergestellt werden. Ein
vielfältigeres
System ist jedoch entwickelt worden, welches Kodierung von im wesentlichen
einer jeglichen kombinatorischen Bibliothek erlaubt. Hier wird die
Verbindung über
den photospaltbaren Linker an den festen Träger gebunden, und der Tag wird über einen
Catecholether-Linker über
Carben-Insertion an die Kügelchenmatrix
gebunden (Nestler et al. (1994) J. Org. Chem. 59: 4723–4724). Diese
orthogonale Befestigungsstrategie erlaubt die selektive Entfernung
von Bibliotheksmitgliedern zum Assay in Lösung und nachfolgendes Dekodieren
durch ECGC nach oxidativem Entfernen des Tag-Satzes.
-
Obwohl
mehrere Amid-verknüpfte
Bibliotheken im Fachbereich binäres
Kodieren verwenden, wobei die elektrophoren Tags an Amin-Gruppen
gebunden sind, stellt Binden dieser Tags direkt an die Kügelchenmatrix
weitaus größere Vielfalt
bei den Strukturen bereit, welche in kodierten kombinatorischen
Bibliotheken hergestellt werden können. Auf diese Art und Weise
gebunden sind die Tags und ihr Linker fast so unreaktiv wie die
Kügelchenmatrix
selbst. Zwei binär
kodierte kombinatorische Bibliotheken sind veröffentlicht worden, in denen
die elektrophoren Tags direkt an die feste Phase gebunden sind (Ohlmeyer
et al. (1995) PNAS 92: 6027–6031),
und sie liefern eine Richtlinie zum Erzeugen der in Frage stehenden
Verbindungsbibliothek. Beide Bibliotheken wurden unter Verwendung
einer orthogonalen Bindungsstrategie konstruiert, bei welcher das
Bibliotheksmitglied durch einen photolabilen Linker an den festen
Träger
gebunden war und die Tags über
einen nur durch starke Oxidation spaltbaren Linker gebunden waren.
Da die Bibliotheksmitglieder wiederholt partiell von dem festen
Träger
photoeluiert werden können,
können
Bibliotheksmitglieder in vielen Assays verwendet werden. Sukzessive
Photoelution erlaubt auch eine iterative Screening-Strategie mit
sehr hohem Durchsatz: Als Erstes werden viele Kügelchen in 96-Well-Microtiter-Platten
platziert; als Zweites werden Verbindungen partiell entfernt und
auf Assay-Platten transferiert; als Drittes identifiziert ein Metallbindungsassay
die aktiven Wells; als Viertes werden die entsprechenden Kügelchen
einzeln auf neue Microtiter-Platten Array-mäßig angeordnet; als Fünftes werden
einzelne aktive Verbindungen identifiziert; und als Sechstes werden
die Strukturen dekodiert.
-
Beispiele
-
Nachdem
die Erfindung nun allgemein beschrieben ist, wird sie leichter durch
Bezugnahme auf die folgenden Beispiele verstanden, welche lediglich
zu Zwecken der Veranschaulichung bestimmter Aspekte und Ausführungsformen
der vorliegenden Erfindung beigefügt sind und die Erfindung nicht
einschränken
sollen.
-
Allgemeine, in den Beispielen
verwendete Verfahren
-
NMR-Spektren
wurden bei Protonenfrequenzen von 270 und 300 MHz aufgenommen, unter
Verwendung von CDCl3 als Lösungsmittel,
soweit nicht anders angegeben. 1H chemische
Verschiebungen werden relativ zu Me4Si ("TMS"; δ = 0,00 ppm)
oder CHCl3 (δ = 7,26 ppm) als interne Standards
berichtet; 31P chemische Verschiebungen
sind relativ zu externer wässriger
85% H3PO4 (δ = 0,00 ppm);
und 13C chemische Verschiebungen sind relativ
zu CHCl3 (δ = 77,00 ppm) oder TMS (δ = 0,00 ppm)
als internen Standards. Massenspektren wurden im Elektronen-Stoß-Ionisierungs-Modus
bei 70 eV aufgenommen. Optische Rotationen wurden bei Raumtemperatur
gemessen.
-
Beispiel
1 Synthese
von racemischem Diester 2
-
Trockenes
Natriumhypophosphit (hergestellt aus Natriumhydrophosphitmonohydrat
durch azeotrope Destillation mit Toluol in vacuo bei 50°C) (940 mg,
10,8 mmol) wurde in 60 mL trockenem CH2Cl2 suspendiert und auf 0°C abgekühlt, dann wurden Triethylamin
(2,50 mL, 18,9 mmol) und Chlortrimethylsilan (2,32 mL, 18,4 mmol)
zugegeben. Nach 5 Minuten wurde Verbindung I (500 mg, 1,54 mmol)
in 5 mL trockenem CH2Cl2 zugegeben.
Das Gemisch wurde bei RT 24 h lang gerührt, bevor 1 N HCl (20 mL)
zugegeben wurde. Das Reaktionsgemisch wurde mit CH2Cl2 (3 × 40
mL) extrahiert, und die vereinigten organischen Phasen getrocknet (MgSO4). Nach Aufkonzentrierung wurde der Rückstand
in 6 mL trockenem CH2Cl2 und
0,6 mL Pyridin gelöst und
auf 0°C
abgekühlt,
dann wurde Trimethylacetylchlorid (0,3 mL, 2,25 mmol) zugegeben,
gefolgt von Benzylalkohol (0,21 mL, 1,8 mmol). Das Gemisch wurde
bei 0°C
bis RT 2 h lang gerührt,
dann mit Ether verdünnt. Die
organische Lage wurde mit 1 N HCl (10 mL), H2O
(10 mL) und Salzlösung
(10 mL) gewaschen, getrocknet (MgSO4) und
auf konzentriert. Flash-Chromatographie über Silicagel mit CHCl3-MeOH (30:1) als Eluent ergab racemische
Verbindung II (657 mg, 89%) als ein farbloses Öl: 1H
NMR (CDCl3, 300 MHz) δ 7,22 (d, JH,P =
555 Hz, 0,6H), 7,19 (d, JH,P = 550 Hz, 0,4H),
7,39–7,34
(m, 15H), 5,15–4,95
(m, 6H), 2,98 (m, 1H), 2,47–2,17
(m, 3H), 2,14–1,79
(m, 3H); 31P NMR (CDCl3,
121 MHz) δ 36,06,
35,08; 13C NMR (CDCl3,
75 MHz) δ 173,46, 173,39,
172,13, 135,70, 135,30, 128,73, 128,60, 128,58, 128,53, 128,46,
128,43, 128,38, 128,33, 128,26, 128,20, 67,88, 67,79, 67,07, 66,99,
66,46, 38,05, 38,02, 31,25, 30,02, 28,29, 28,13.
-
Beispiel
2 Synthese
von racemischem Tetraester 3
-
Zu
einer Lösung
von II (210 mg, 0,44 mmol) in 5 mL trockenem THF wurde Natriumhydrid
(15 mg, 60% Dispersion in Öl,
0,44 mmol) zugegeben, gefolgt von Verbindung I (140 mg, 0,44 mmol)
bei 0°C,
und das Gemisch wurde bei RT 2 h lang gerührt, bevor 1 N HCl zugegeben
wurde. Das Reaktionsgemisch wurde mit CH2Cl2 extrahiert (3 × 20 ml). Die vereinigten organischen
Lagen wurden getrocknet (MgSO4) und auf
konzentriert. Flash-Chromatographie über Silicagel mit Ethylacetat-Hexanen
(1:1) als Eluent ergab Verbindung 3 (45 mg, 13%) als ein farbloses Öl: 1H NMR (CDCl3, 300
MHz) δ 7,34–7,27 (m,
25H), 5,06–4,89
(m, 10H), 2,83 (m, 2H), 2,33–2,14
(m, 6H), 2,00–1,60
(m, 6H); 31P NMR (CDCl3,
121 MHz) δ –28,51, –28,90, –29,34; 13C NMR (CDCl3, 75
MHz) δ 173,80,
172,19, 136,27, 135,78, 135,50, 128,60, 128,56, 128,42, 128,33,
128,28, 128,24, 128,19, 128,14, 66,91, 66,87, 66,80, 66,37, 66,10,
66,02, 38,65, 38,51, 31,71, 30,32, 31,24, 30,67, 30,57, 30,04, 28,85,
28,69.
-
Beispiel
3 Synthese
von racemischer Tetrasäure
FN-6
-
Zu
einer Lösung
von 3 (42 mg, 0,52 mmol) in tert-Butanol wurden 30 mg 20% Pd(OH)2/C (Aldrich, ≤ 50% H2O)
zugegeben, und dann wurde das Gemisch unter 70 psi Wasserstoff 24
h lang hydriert. Der Katalysator wurde mittels Filtration durch
Celit entfernt, und das Filtrat auf konzentriert. Der Rückstand
wurde in 5 ml Wasser gelöst
und lyophilisiert, um 17 mg (92%) FN-6 als einen weißen Feststoff
zu ergeben: 1H NMR (D2O, 300
MHz) δ 2,77
(m, 2H), 2,44 (t, J = 7,3 Hz, 4H), 2,20 (dt, J = 13,2, 11,3 Hz,
2H), 2,00–1,82
(m, 6H); 31P NMR (D2O,
121 MHz) 6 –33,12; 13C NMR (D2O, 75
MHz) δ 179,69, 179,61,
178,61, 39,89, 32,70, 32,28, 31,49, 29,68, 29,58, 29,52, 29,42.
-
Beispiel
4 Synthese
von Diester 5
-
Verbindung
4 (130 mg, 0,33 mmol) wurde in 2 mL trockenem CH2Cl2 und 0,2 mL Pyridin gelöst und auf 0°C abgekühlt, dann
wurde Trimethylacetylchlorid (80 μL,
0,6 mmol) zugegeben, gefolgt von (R)-(+)-1-Phenyl-1-butanol (65
mg, 0,44 mmol). Das Gemisch wurde bei 0°C bis RT 2 h lang gerührt, dann
mit Ether verdünnt,
und das Reaktionsgemisch wurde mit 1 N HCl (10 mL), H2O
(10 mL), und Salzlösung
(10 mL) gewaschen, getrocknet (MgSO4) und
auf konzentriert. Flash-Chromatographie über Silicagel
mit CHCl3-MeOH (30:1) als Eluent ergab 5
(164 mg, 94%) als ein Gemisch von 4 Diastereomeren. 1H
NMR (CDCl3, 300 MHz) δ 7,45–7,28 (m, 15H), 7,26 (d, JH,P = 558 Hz, 0,24H), 7,23 (d, JH,P =
554 Hz, 0,16H), 6,91 (d, JH,P = 552 Hz,
0,60H), 5,37–5,24
(m, 1H), 5,17–5,03
(m, 4H), 3,05–2,68
(m, 1H), 2,56–1,60
(m, 8H), 1,50–1,20
(m, 2H), 0,98–0,91
(m, 3H); 31P NMR (CDCl3,
121 MHz) δ 34,88,
34,10, 32,22, 31,32.
-
Beispiel
5 Synthese
von Tetraestern 6
-
Zu
einer Lösung
von 5 (220 mg, 0,44 mmol) in 5 mL trockenem THF wurde Natriumhydrid
(15 mg, 60% Dispersion in Öl,
0,44 mmol) zugegeben, gefolgt von Verbindung 1 (See Beispiel 1;
140 mg, 0,44 mmol) bei 0°C,
und das Gemisch wurde bei RT 2 h lang gerührt. 1 N HCl wurde zugegeben
und das Gemisch mit CH2Cl2 extrahiert
(3 × 20
mL). Die vereinigten organischen Phasen wurden getrocknet (MgSO4) und auf konzentriert. Flash-Chromatographie über Silicagel
mit CHCl3-MeOH (30:1) als Eluent ergab eine
Gemisch von 4 Isomeren (110 mg, 27%). 31P
NMR zeigte 4 Peaks: δ –29.91, –30.21, –30.37,
and –30.66.
Sorgsame präparative TLC-Trennung
(Ethylacetat-Hexane 2:3) ergab homogene Verbindungen 6a–d.
6a:
Rf 0.60 (Ethylacetat-Hexane 1:1); [α]D +12.1° (c
0,7, CHCl3); 1H
NMR (CDCl3, 300 MHz) δ 7,37–7,28 (m, 25H), 5,30 (dt, J
= 8,7, 7,2 Hz, 1H), 5,20–4,97
(m, 8H), 2,91 (m, 1H), 2,56 (m, 1H), 2,39–2,22 (m, 3H), 2,10–1,60 (m,
11H), 1,35–1,09
(m, 2H), 0,87 (t, J = 7,2 Hz, 3H); 31P NMR
(CDCl3, 121 MHz) δ –29,90; 13C
NMR (CDCl3, 75 MHz) δ 173,93, 173,86, 173,78, 173,70,
172,23, 172,12, 140,98, 135,78, 135,55, 128,59, 128,51, 128,46, 128,25,
128,23, 128,17, 126,55, 77,19, 77,10, 66,82, 66,76, 66,31, 66,23,
40,45, 40,38, 38,67, 38,63, 38,59, 38,56, 32,18, 31,30, 31,09, 30,93,
30,11, 29,66, 28,84, 28,69, 28,64, 28,48, 18,58, 13,68.
6b:
Rf 0,58 (Ethylacetat-Hexane 1:1); [α]D +12,9° (c
0,35, CHCl3); 1H
NMR (CDCl3, 300 MHz) δ 7,37–7,28 (m, 25H), 5,30 (dt, J
= 9,0, 6,6 Hz, 1H), 5,21–4,99
(m, 8H), 2,99 (m, 1H), 2,55 (m, 1H), 2,41–2,20 (m, 3H), 2,10–1,62 (m,
11H), 1,40–1,20
(m, 2H), 0,86 (t, J = 7,2 Hz, 3H); 31P NMR
(CDCl3, 121 MHz) δ –30,32; 13C
NMR (CDCl3, 75 MHz) δ 173,94, 173,90, 173,87, 173,81,
172,25, 172,15, 141,01, 135,82, 135,58, 135,52, 128,62, 128,57, 128,54,
128,49, 128,32, 128,28, 128,26, 128,24, 128,22, 128,21, 128,19,
126,58, 77,20, 77,16, 66,82, 66,79, 66,33, 66,25, 40,44, 40,37,
38,70, 38,67, 38,43, 38,38, 32,25, 31,90, 31,34, 31,13, 31,00, 30,75,
29,70, 28,81, 28,67, 28,53, 28,38, 18,55, 13,71.
6c: Rf 0,57 (Ethylacetat-Hexane 1:1); [α]D 0° (c
0,42, CHCl3); 1H
NMR (CDCl3, 300 MHz) δ 7,35–7,20 (m, 25H), 5,29 (dt, J
= 9,3, 6,9 Hz, 1H), 5,17–4,94
(m, 8H), 2,91 (m, 1H), 2,45–2,25
(m, 3H), 2,20–1,20
(m, 14H), 0,84 (t, J = 7,5 Hz, 3H); 31P
NMR (CDCl3, 121 MHz) δ –30,11; 13C
NMR (CDCl3, 75 MHz) δ 173,84, 173,74, 173,70, 173,62,
172,25, 172,10, 140,84, 135,80, 135,60, 135,49, 128,62, 128,53,
128,41, 128,32, 128,24, 128,21, 126,68, 77,24, 77,15, 66,87, 66,79,
66,33, 66,25, 40,27, 40,21, 38,86, 38,83, 38,46, 38,41, 31,95, 31,62,
31,30, 31,19, 30,80, 30,39, 29,70, 28,64, 28,53, 28,48, 28,40, 18,63,
13,73.
6d: Rf 0,55 (Ethylacetat-Hexane
1:1); [α]D –2,8° (c 0,42,
CHCl3); 1H NMR (CDCl3, 300 MHz) δ 7,41–7,20 (m, 25H), 5,31 (dt, J
= 9,3, 6,9 Hz, 1H), 5,23–4,98
(m, 8H), 2,97 (m, 1H), 2,50–2,17
(m, 4H), 2,15–1,82
(m, 6H), 1,80–1,50
(m, 5H), 1,45–1,20
(m, 2H), 0,85 (t, J = 7,2 Hz, 3H); 31P NMR
(CDCl3, 121 MHz) δ –30,57; 13C
NMR (CDCl3, 75 MHz) δ 173,77, 173,70, 173,66, 173,59,
172,18, 172,06, 140,81, 135,77, 135,50, 135,46, 128,59, 128,53,
128,51, 128,41, 128,30, 128,23, 128,19, 126,58, 77,16, 77,05, 66,82,
66,31, 66,24, 40,29, 40,23, 38,79, 38,30, 38,24, 31,96, 31,44, 31,34,
31,14, 30,81, 30,20, 28,95, 28,80, 28,69, 28,53, 18,54, 13,69.
-
Beispiel
6 Synthese
von optisch reinen Stereoisomeren von Tetrasäure FN-6(F)
-
Das
oben für
die Synthese von FN-6(F) umrissene Syntheseverfahren (siehe Beispiel
3) wurde für
die Herstellung von optisch reinen Stereoisomeren FN-6a–d verwendet.
FN-6a
(Ausbeute 94%): weißer
Feststoff; 1H NMR (D20,
300 MHz) δ 2,76
(m, 2H), 2,45 (t, J = 7,5 Hz, 4H), 2,14 (dt, J = 12,9, 9,6 Hz, 2H),
2,00–1,89
(m, 4H), 1,83 (dt, J = 14,4, 4,2 Hz, 2H); 31P
NMR (D2O, 121 MHz) δ –37,63; 13C
NMR (D2O, 75 MHz) δ 179,95, 178,74, 40,16, 32,55
(d, J1,P = 90,23 Hz), 32,38, 29,67 (d, J3,P = 11,48 Hz).
FN-6b (Ausbeute 96%):
weißer
Feststoff; 1H NMR (D20,
300 MHz) δ 2,76
(m, 2H), 2,45 (t, J = 7,8 Hz, 4H), 2,13 (dt, J = 12,6, 10,5 Hz,
2H), 2,04–1,89
(m, 4H), 1,83 (dt, J = 15,0, 4,2 Hz, 2H); 31P
NMR (D2O, 121 MHz) δ –36,42; 13C
NMR (D2O, 75 MHz) δ 180,07, 178,76, 40,10, 32,67
(d, J1,P = 90,75 Hz), 32,40, 29,57 (d, J3,P = 11,25 Hz).
FN-6c (Ausbeute 99%):
weißer
Feststoff; 1H NMR (D20,
300 MHz) δ 2,76
(m, 2H), 2,45 (t, J = 7,5 Hz, 4H), 2,09 (dt, J = 12,6, 9,0 Hz, 2H),
2,04–1,88
(m, 4H), 1,79 (dt, J = 15,0, 4,2 Hz, 2H); 31P
NMR (D2O, 121 MHz) δ –37,96; 13C
NMR (D2O, 75 MHz) δ 179,88, 178,72, 40,13, 32,46
(d, J1,P = 90,75 Hz), 32,36, 29,66 (d, J3,P = 12 Hz).
FN-6d (Ausbeute 96%):
weißer
Feststoff; 1H NMR (D20,
300 MHz) δ 2,76
(m, 2H), 2,45 (t, J = 7,5 Hz, 4H), 2,14 (dt, J = 13,2, 9,6 Hz, 2H),
2,00–1,86
(m, 4H), 1,85 (dt, J = 14,7, 3,6 Hz, 2H); 31P
NMR (D2O, 121 MHz) δ –35,56; 13C
NMR (D2O, 75 MHz) δ 179,88, 178,72, 40,13, 32,46
(d, J1,P = 90,75 Hz), 32,37, 29,66 (d, J3,P = 12 Hz),
-
-
Zu
einer Lösung
von 4 (400 mg, 1,23 mmol) in Nitromethan wurden 0,1 mL Triton B
(40% Lösung
in Methanol) zugegeben. Das Gemisch wurde bei Raumtemperatur 5 h
lang gerührt.
Das Nitromethan wurde bei vermindertem Druck entfernt. Flash-Chromatographie über Silicagel
mit Ethylacetat-Hexanen (8:1 bis 3:1) als Eluent ergab Verbindungen
5 (200 mg, 42%) und 6 (150 mg, 17%) als farbloses Öl.
5:
IR (Film) ν 1730
cm–1, 1H NMR (CDCl3, 300
MHz) δ 7,40–7,28 (m,
10H), 5,13 (s, 2H), 5,10 (s, 2H), 4,35 (m, 2H), 2,45–2,15 (m,
4H), 2,08–1,82
(m, 2H); 13C NMR (CDCl3,
75 MHz) δ 173,52,
172,22, 135,67, 135,32, 128,66, 128,57, 128,51, 128,32, 128,28,
73,01, 66,89, 66,48, 41,40, 31,41, 28,95, 26,94.
6: IR (Film) ν 1731 cm–1, 1H NMR (CDCl3, 300
MHz) δ 7,39–7,20 (m,
20H), 5,15–5,02
(m, 8H), 4,53 (m, 1H), 2,52–2,22
(m, 6H), 2,08–1,75
(m, 8H); 13C NMR (CDCl3,
75 MHz) δ 173,34,
172,10, 135,70, 135,37, 128,57, 128,40, 128,29, 128,27, 84,35, 66,87,
66,44, 41,17, 41,06, 35,83, 35,62, 31,32, 27,55, 27,41.
-
-
Zu
einer Lösung
von Verbindung 5 (58 mg, 0,15 mmol) und Benzylacrylat (25 mg, 0,15
mmol) in 1 mL trockenem CH2Cl2 wurde
eine katalytische Menge Triton B (40% Lösung in Methanol) zugegeben.
Das Gemisch wurde bei Raumtemperatur 4 h lang gerührt. Das
Lösungsmittel
wurde unter vermindertem Druck entfernt, und Flash-Chromatographie über Silicagel
mit Ethylacetat-Hexanen (5:1) als Eluent ergab Verbindung 7 (75
mg, 91%). 1H NMR (CDCl3,
300 MHz) δ 7,40–7,25 (m,
15H), 5,20–5,03
(m, 6H), 4,64–4,46
(m, 1H), 2,55–1,80
(m, 11H), 13C NMR (CDCl3,
75 MHz) δ 173,63,
172,10, 171,34, 135,67, 135,49, 128,60, 128,55, 128,41, 128,31,
128,27, 85,67, 85,03, 66,86, 66,67, 66,44, 41,17, 35,41, 35,32,
31,35, 30,07, 29,96, 29,07, 28,36, 27,48, 26,64.
-
-
Zu
einer Lösung
von Verbindung 8 (54 mg, 0,076 mmol) in 2 mL trockenem CH2Cl2 wurden 18 uL
Triethylamin zugegeben, nachdem 10 min lang bei Raumtemperatur gerührt wurde,
wurden 80 mg CATP zugegeben. Das resultierende Gemisch wurde weitere
4 h lang gerührt.
Dann wurden 10 mL Ether zugegeben, und es wurde durch Celit filtriert
und mit Ether gewaschen. Das Filtrat wurde auf konzentriert. Chromatographie
lieferte 8 (30 mg, 58%): 1H NMR (CDCl3, 300 MHz) δ 7,40–7,22 (m, 20H), 5,15–5,01 (m,
8H), 3,00–2,79
(m, 4H), 2,50–2,30
(m, 6H), 2,00–1,78
(m, 4H); 13C NMR (CDCl3,
75 MHz) δ 205,98,
205,73, 174,20, 174,03, 172,45, 172,40, 135,78, 135,68, 128,53,
128,51, 128,22, 128,16, 66,58, 66,33, 43,95, 39,27, 31,62, 31,59,
29,68, 26,66, 26,59.
-
-
Zu
einer Lösung
von Verbindung 7 (70 mg, 0,13 mmol) in 5 mL trockenem CH2Cl2 wurden 30 uL
Triethylamin zugegeben, nachdem 10 min lang bei Raumtemperatur gerührt wurde,
wurden 120 mg CATP zugegeben. Das resultierende Gemisch wurde weitere
4 h lang gerührt,
dann wurden 20 mL Ether zugegeben, und es wurde durch Celit filtriert
und mit Ether gewaschen. Das Filtrat wurde auf konzentriert. Chromatographie
lieferte 9 (40 mg, 60%). 1H NMR (CDCl3, 300 MHz) δ 7,40–7,25 (m, 15H), 5,16–5,05 (m,
6H), 3,02–2,90
(m, 2H), 2,80–2,50
(m, 5H), 2,36 (t, J = 7,2 Hz, 2H), 2,00–1,89 (m, 2H); 13C
NMR (CDCl3, 75 MHz) δ 206,30, 174,20, 172,43, 172,36,
135,76, 135,70, 128,51, 128,49, 128,20, 128,17, 128,15, 66,57, 66,47,
66,33, 43,92, 39,33, 37,06, 31,62, 27,85, 26,65.
-
-
Zu
einer Lösung
von 9 (40 mg, 0,078 mmol) in tert-Butanol wurden 30 mg 20% Pd(OH)2/C (Aldrich, ≤ 50% H2O)
zugegeben, und das Gemisch wurde unter 1 atm Wasserstoff 24 h lang
hydriert. Der Katalysator wurde mittels Filtration durch Celit entfernt,
und das Filtrat auf konzentriert. Der Rückstand wurde in 5 ml Wasser
gelöst
und lyophilisiert, um 18 mg (94%) FN3(c) als einen Sirup zu ergeben.
FN3(c): 1H NMR (CDCl3,
300 MHz) δ 3,03–2,74 (m,
5H), 2,59 (t, J = 6,0 Hz, 2H), 2,44 (t, J = 1,2 Hz, 2H), 1,96–1,76 (m,
2H).
-
-
Zu
einer Lösung
von 8 (24 mg, 0,034 mmol) in tert-Butanol wurden 10 mg 20% Pd(OH)2/C (Aldrich, ≤ 50% H2O)
zugegeben, und das Gemisch wurde unter 1 atm Wasserstoff 24 h lang
hydriert. Der Katalysator wurde mittels Filtration durch Celit entfernt
und das Filtrat auf konzentriert. Der Rückstand wurde in 5 ml Wasser
gelöst
und lyophilisiert, um 10 mg (93%) FN4(D) als einen Sirup zu ergeben.
FN4(D): 1H NMR (CDCl3,
300 MHz) δ 3,03–2,72 (m,
6H), 2,45 (t, J = 7,5 Hz, 4H), 1,96–1,75 (m, 4H).
-
-
Zu
einer Suspension von Dibenzyl-L-glutamat-tosylat (1 g, 2 mmol) in
20 mL of CH2Cl2 wurde
Triphosgen (110 mg, 0,37 mmol) zugegeben. Das Gemisch wurde auf –78°C abgekühlt und
Et3N (0,53 mL, 4 mmol) zugegeben. Das Reaktionsgemisch
wurde sich auf Raumtemperatur erwärmen lassen und wurde weitere
2 h lang gerührt.
Das Reaktionsgemisch wurde mit 100 mL EtOAc verdünnt und mit H2O
(3 × 30
mL), Salzlösung (30
mL) gewaschen und über
MgSO4 getrocknet. Das Lösungsmittel wurde abgedampft
und der Rückstand
in 5 mL CH2Cl2 gelöst, und
es wurde Hexan im Überschuss
zugegeben. Der weiße
Feststoff 10 wurde gesammelt (600 mg, 88%). 1H
NMR (CDCl3, 300 MHz) δ 7,40–7,25 (m, 20H), 5,17 (br s,
2H), 5,13 (br s, 4H), 5,08 (s, 4H), 4,53 (dt, J = 8,1, 4,8 Hz, 2H),
2,41 (m, 4H), 2,19 (m, 2H), 1,97 (m, 2H); 13C
NMR (CDCl3, 75 MHz) δ 172,70, 172,51, 156,53, 135,67,
135,12, 128,51, 128,46, 128,33, 128,14, 67,18, 66,38, 52,47, 30,17,
27,84.
-
-
Zu
einer Lösung
von 10 (510 mg, 0,75 mmol) in tert-Butanol wurden 200 mg 20% Pd(OH)2/C (Aldrich, ≤ 50% H2O)
zugegeben, und das Gemisch wurde unter 1 atm Wasserstoff 24 h lang
hydriert. Der Katalysator wurde mittels Filtration durch Celit entfernt
und das Filtrat auf konzentriert. Der Rückstand wurde in 15 ml Wasser
gelöst
und lyophilisiert, um 235 mg (98%) FN11 als einen weißen Feststoff
zu ergeben: [α]D –16.4° (c 0,5, H2O); 1H NMR (D2O, 300 MHz) δ 4,26 (br s, 2H), 2,51 (t, J
= 9,9 Hz, 4H), 2,18 (m, 2H), 1,98 (m, 2H); 13C
NMR (D2O, 75 MHz) δ 178,59, 177,59, 160,55, 53,91,
31,36, 27,51.
-
-
Zu
einer Suspension von Dibenzyl-L-glutamat-tosylat (1 g, 2 mmol) in
20 mL CH2Cl2 wurde
Triphosgen (200 mg, 0,66 mmol) zugegeben. Das Gemisch wurde auf –78°C abgekühlt, dann
wurde Et3N (0,60 mL, 4 mmol) zugegeben.
Das Reaktionsgemisch wurde 2 h lang bei –78°C gerührt, dann sich auf Raumtemperatur erwärmen lassen
und mit 100 mL EtOAc verdünnt
und mit H2O (30 mL), 1 N HCl (30 mL) und
Salzlösung
(30 mL) gewaschen. Das Gemisch wurde über MgSO4 getrocknet.
Das Lösungsmittel
wurde abgedampft und der Rückstand
auf Silicagel mit Hexanen/EtOAc (2:1) chromatographiert, um Isyocyanat
(230 mg) zu ergeben. Das Isycyanat (230 mg, 0,65 mmol) wurde in
5 mL CH2Cl2 gelöst, und
Dibenzyl-L-glutamat (213 mg, 0,65 mmol) bei 0°C zugegeben. Das Reaktionsgemisch
wurde 24 h lang bei Raumtemperatur gerührt. Hexan wurde im Überschuss
zugegeben. Der weiße
Feststoff 11 wurde gesammelt (440 mg, 99% aus Isycyanat). 1H NMR (CDCl3, 300
MHz) δ 7,35–7,25 (m,
20H), 5,51 (d, J = 7,8 Hz, 2H), 5,11 (s, 4H), 5,06 (s, 4H), 4,52
(dt, J = 7,8, 5,4 Hz, 2H), 2,41 (m, 4H), 2,19 (m, 2H), 1,98 (m,
2H); 13C NMR (CDCl3,
75 MHz) δ 172,79,
172,68, 156,78, 135,75, 135,21, 128,57, 128,53, 128,38, 128,23,
128,19, 67,22, 66,42, 52,53, 30,25, 27,88.
-
-
Dem
in Beispiel 14 umrissenen allgemeinen Verfahren wurde für die Herstellung
von FN16 aus 11 gefolgt. FN16: 1H NMR (CDCl3, 300 MHz) δ 4,24 (m, 2H), 2,45 (t, J =
7,2 Hz, 4H), 2,15 (m, 2H), 1,95 (m, 2H); 13C NMR
(D20, 75 MHz) δ 178,48, 177,40, 160,34, 53,80,
31,34, 27,62.
-
-
Dem
in Beispiel 13 umrissenen allgemeinen Verfahren wurde für die Herstellung
von 12 aus Dibenzyl-D-glutamat-tosylat gefolgt.
-
-
Dem
in Beispiel 14 umrissenen allgemeinen Verfahren wurde für die Herstellung
von FN13 aus 12 gefolgt. FN13: [α]D +17,1° (c
0,84 H2O); 1H NMR
(D2O, 300 MHz) und 13C
NMR (D20, 75 MHz) waren die gleichen wie
für FN11.
-
-
Zu
einer Suspension von Dimethyl-L-glutamat-chlorid (2,1 g, 10 mmol)
in 40 mL CH2Cl2 wurde
Triphosgen (1 g, 3,3 mmol) zugegeben. Das Gemisch wurde auf –78°C abgekühlt, dann
wurde Et3N (3 mL, 20 mmol) zugegeben. Das
Reaktionsgemisch wurde 0,5 h lang bei –78°C gerührt, dann sich auf Raumtemperatur
erwärmen
lassen und mit 100 mL EtOAc verdünnt
und mit H2O (30 mL), 1 N HCl (30 mL) und
Salzlösung
(30 mL) gewaschen. Das Gemisch wurde über MgSO4 getrocknet.
Das Lösungsmittel
wurde abgedampft und der Rückstand
auf Silicagel mit Hexanen/EtOAc (2:1) chromatographiert, um das
entsprechende Isyocyanat (870 mg) zu ergeben. Das Isocyanat (870
mg, 4,3 mmol) wurde in 10 mL CH2Cl2 gelöst,
und es wurde Dimethyl-L-glutamat
(752 mg, 4,3 mmol) bei 0°C
zugegeben. Das Reaktionsgemisch wurde 24 h lang bei Raumtemperatur
gerührt.
Hexan wurde im Überschuss
zugegeben und der weiße
Feststoff FN15 gesammelt (1,50 g, 92% aus Isycyanat). 1H
NMR (CDCl3, 300 MHz) δ 5,47 (d, J = 7,8 Hz, 2H), 4,50
(dt, J = 8,1, 5,1 Hz, 2H), 3,75 (s, 6H), 3,67 (s, 6H), 2,42 (m,
4H), 2,17 (m, 2H), 1,96 (m, 2H); 13C NMR
(CDCl3, 75 MHz) δ 173,47, 173,36, 156,71, 52,45,
52,40, 51,79, 30,03, 27,92.
-
-
Zu
einer Suspension von Dimethyl-L-glutamat (575 mg, 3,28 mmol) in
15 mL CH2Cl2 wurde
Triphosgen (187 mg, 1,64 mmol) zugegeben. Das Gemisch wurde auf –78°C abgekühlt, dann
wurde Et3N (0,43 mL, 3,28 mmol) zugegeben.
Das Reaktionsgemisch wurde eine kurze Zeit lang bei –78°C gerührt, dann
sich über
15 h auf Raumtemperatur erwärmen
lassen. Das Gemisch wurde dann mit 100 mL EtOAc verdünnt und
mit H2O (30 mL), 1 N HCl (30 mL) und Salzlösung (30
mL) gewaschen. Das Gemisch wurde über MgSO4 getrocknet.
Das Lösungsmittel
wurde abgedampft und der Rückstand
auf Silicagel mit Hexanen/EtOAc (3:1→1:1) chromatographiert, um
FN18 zu ergeben (410 mg, 64%). 1H NMR (CDCl3, 300 MHz) δ 5,08 (d, J = 7,8 Hz, 2H), 4,50
(m, 2H), 3,78 (s, 6H), 3,68 (s, 6H), 2,46 (m, 4H), 2,25 (m, 2H),
2,12 (m, 2H); 13C NMR (CDCl3,
75 MHz) δ 182,89, 173,47,
172,89, 56,19, 52,57, 51,82, 29,87, 27,40.
-
Beispiel 21
-
Antitumor-Aktivität von FN11
-
Implantierte
Xenotransplantate wurden durch s.c. Inokulation mit 2 × 106 U-87 Glioblastom-Multiform-Zellen, gemischt
mit 0,5 mg Matrigel gebildet. FN11 wurde in PBS gelöst zu einer
Konzentration von entweder 10 oder 100 μM, und Injektionen (0,1 ml)
in die Basis der s.c. implantierten Xenotransplantate wurden einmal
täglich
vorgenommen. Die Startvolumina der Tumore betrugen etwa 250 mm3. Die Ergebnisse dieses Protokolls sind
in 15 dargestellt.
-
Beispiel 22
-
Antiangiogenese-Aktivität von FN11
-
Vergleichstumore
und Tumore, welche unter den in Beispiel 21 beschriebenen Bedingungen
gewachsen waren, wurden am Ende von 7 Tagen geerntet, in Formalin
fixiert und zum Sektionieren in Paraffin eingebettet. Gewebe wurde
immunohistochemisch für
von Willebrand's
Faktor (vWF) gefärbt,
um ein Maß für die Vaskularisierung
zu erhalten. 13 und 14 stellen
die Ergebnisse dieses Protokolls für den Tumor dar, welcher mit
FN11 bei 100 μM
behandelt wurde. 13 wurde bei geringer Vergrößerung aufgenommen
(100 ×),
wohingegen 14 bei hoher Vergrößerung (400 ×) aufgenommen
wurde. Die Figuren zeigen ein Überwiegen
von avaskulären
und niedrig-vaskulären
Bereichen, zumeist kleine Gefäße, in dem
mit FN11 bei 100 μM
behandelten Tumor.
-
Beispiel 23
-
Studien von
neuroprotektiven Eigenschaften von FN-Verbindungen
-
Das
Testen von neuroprotektiven Wirkungen von Liganden, welche auf metabotropische
Glutamat-Rezeptoren wirken, erfordert die Verwendung von spezifischen
Modellen, welche es erlauben, die Wirkung dieser Verbindungen zu
visualisieren. Wie in mehreren Veröffentlichungen gezeigt wurde,
erfordert Schutz gegen Excitotoxizität (welche durch NMDA-Applikation
induziert werden kann) oft die Gegenwart von Gliazellen und kann
in Kulturen, welche nur neuronale Zellen enthalten, nicht gezeigt
werden. Es wird angenommen, dass die neuroprotektive Wirkung von
Gruppe II mGluR Agonisten von einer Wirkung auf mGluR3 oder mGluR2
Rezeptoren, welche an Gliazellen lokalisiert sind, resultiert, was
die Freisetzung von neurotrophischen Faktoren von diesen Gliazellen
induziert.
-
Wir
haben die Validität
dieser Feststellungen unter Verwendung von drei Modellen von MMDA-Toxizität in Kulturen
von kortikalen Neuronen, welche aus Mäuseföten hergestellt wurden, getestet.
Das erste Modell (A) besteht aus der Verwendung einer Kultur, welche
Neuronen ohne Gliazellen enthält.
NMDA (75 μM)
wird 10 Minuten lang ohne oder mit der getesteten Verbindung appliziert.
Dann wird das Kulturmedium ersetzt und die Zellen 24 Stunden lang
belassen, um Toxizität
zu entwickeln. Das zweite Modell (B) beinhaltet die Verwendung von
kortikalen Neuronen von Mäusen,
welche auf einer Lage von konfluenten Gliazellen (zumeist Astrozyten),
welche aus zerebralem Cortex von neugeborenen Mäusen hergestellt ist, ausgesät sind.
Die Inkubationen sind identisch wie in Modell A.
-
Das
dritte Modell (C) trennt die neuronalen und glialen Kulturen. Gliale
Kulturen werden mit der getesteten neuroprotektiven Verbindung behandelt,
um die Freisetzung von protektiven Faktoren in das Medium zu induzieren.
Parallel dazu werden Kulturen von kortikalen Neuronen (ohne Glia)
10 Minuten lang mit NMDA (20 μM)
behandelt. Dann wird NMDA ausgewaschen und die Neuronen mit dem
Medium behandelt, welches von den behandelten Gliazellen gesammelt
wurde. In allen Fällen
wird Toxizität
24 Stunden nach NMDA-Behandlung
durch Messungen von Lactatdehydrogenase(LDH)- Aktivität, welche sich während dieser
Zeitspanne in dem Medium angesammelt hat, untersucht.
-
Der
Vergleich der drei Modelle ist in Säulenauftragung 1 dargestellt.
Wie erwartet, war im Modell A ABHxD-I nicht fähig, gegen NMDA-Toxizität zu schützen. Im
Gegensatz dazu produzierte ABHxD-I in Gegenwart von Gliazellen (Modell
B) einen bedeutenden Schutz, wodurch die toxische Wirkung von NMDA
zu über
40% verringert wurde. Dies legte nahe, dass ABHxD-I tatsächlich die
Freisetzung von protektiven Faktoren von Gliazellen induzieren kann.
Diese Möglichkeit
wurde in Modell C getestet. Wie in Säulenauftragung 1c gezeigt, war
ABHxD-I bei separater Inkubation mit Gliazellen so wirksam wie in
Modell B. Es sollte zur Kenntnis genommen werden, dass Medium von
unbehandelten Gliazellen nicht neuroprotektiv war. Dies bestätigt die
Hypothese, dass die Wirkung von ABHxD-I durch die Freisetzung von
neuroprotektiven Faktoren von Gliazellen vermittelt wird. Wichtiger
noch zeigt dies an, dass die neuroprotektive Wirkung von ABHxD-I
nicht von der Gegenwart des Wirkstoffs während des toxischen Ereignisses
abhängt,
sondern dass der Wirkstoff wirksam sein kann, wenn er nach dem toxischen
Stimulus zugegeben wird.
-
Der
Vergleich von Modellen B und C zeigt, dass beide Modelle die indirekte
neuroprotektive Wirkung von ABHxD-I wiederspiegeln, welche durch
die Handlung des Wirkstoffs an auf Gliazellen angeordneten Rezeptoren
vermittelt ist. Dies unterstreicht die Rolle von Gliazellen in Mechanismen
von Hirnschädigung
und identifiziert diese Zellen als ein Target von möglicher
therapeutischer Intervention. Von einem praktischen Gesichtspunkt
aus erscheint zu dem Zweck des Testens von Verbindungen, welche über diesen Mechanismus wirken,
Modell C Modell B überlegen,
da die Variabilität
der Messungen in Modell C verringert ist. Dies wird wiedergespiegelt
durch eine zweifache Verringerung der Standardabweichung für die berechneten
Mittelwerte. Dieser Unterschied rührt aller Wahrscheinlichkeit
nach von der Natur der LDH-Messungen her. In Modell C kann LDH nur
von absterbenden Neuronen freigesetzt werden und spiegelt damit
direkt die Anzahl von toten Zellen wieder. Im Gegensatz dazu können in
Modell B LDH-Gehalte durch LDH-Entweichen von Gliazellen beeinflusst
sein. Wenn die Anzahl von Gliazellen verglichen mit Neuronen groß ist, kann
selbst ein geringes Entweichen zu bedeutenden Veränderungen
der LDH-Gehalte führen.
Es ist bekannt, dass Gliazellen gegenüber NMDA-Toxizität nicht
sensitiv sind, so dass solche Erhöhungen nicht NMDA-Toxizität wiederspiegeln,
sondern vielmehr eine zufällige
Variation, welches in jedem lebenden System auftritt. Damit wird
in Modell B LDH-Freisetzung
von sterbenden Neuronen vor einem höheren Hintergrund von variablen
Vergleichsgehalten gemessen, welches das Signal-zu-Rauschen-Verhältnis herabsetzt
und die zufällige
Variabilität
von Messungen erhöht.
In unseren Studien haben wir anfänglich
Modell B (wie in Säulenauftragungen
2 und 3 zu sehen) verwendet, jedoch dann zu Modell C (Säulenauftragungen
4 und 5) gewechselt, welches verlässlichere Messungen lieferte.
-
Testen von FN-Verbindungen
-
Anfänglich wurden
FN-Verbindungen im Modell B getestet. Säulenauftragung 2 zeigt die
Wirkungen von FN1–FN8,
welche bei 300 μM
Konzentration verwendet wurden. Nur FN1(A) und FN6(F) zeigen etwa
30% Neuroprotektion. Von den FN6-Enantiomeren,
welche bei 100 μM
Konzentration verwendet wurden (Säulenauftragung 2) erscheinen
die 6/1- und 6/4-Verbindungen
wirksamer.
-
FN6(F)-Enantiomere
wurden auch in Modell C (Säulenauftragung
4) getestet. Wie in Modell B waren die 6/1- und 6/4-Verbindungen
wirksamer, wobei der Schutz etwa 30% erreichte. Die Guilford-Verbindung
PMPA, welche FN6(F) ähnlich
ist, war weniger wirksam. Wir haben auch mehrere neuere FN-Verbindungen
im Modell C getestet. Wie in Säulenauftragung
5 gezeigt, zeigte von diesen Verbindungen nur FN13 eine neuroprotektive
Wirkung, welche sich auf etwa 20% Schutz belief.
-
Zusammenfassung
-
Die
stärksten
Wirkungen – etwa
30% – werden
von FN1(A) und FN6(F) gezeigt. Diese Wirkungen sind geringer als
die bei ABHxD-I beobachtete Neuroprotektion, aber besser als die
Wirkung von PMPA. Die getesteten Verbindungen können größere Wirksamkeit in Modellen
von Toxizität
mit einer geringeren nekrotischen Komponente und einer größeren apoptotischen
Komponente zeigen.
-
Säulenauftragungen
für Beispiel
23
-
Säulenauftragung
1: Vergleich der neuroprotektiven Wirkung von ABHxD-I in drei Modellen
von NMDA-Toxizität
zeigt ein Fehlen einer Wirkung in Kulturen reiner Neuronen ohne
Glia (A), während
signifikanter Schutz in dem Modell beobachtet wird, in welchem gemischte
kortikal-neuronale-gliale Kulturen verwendet werden (B) und wenn
Neuronen mit Medium von Gliazellen gerettet werden, welche der schützenden
Verbindung ausgesetzt wurden (C). "Control" bezieht sich auf Zellen, welche nur
mit NMDA (ohne die schützende Verbindung)
behandelt wurden. ABHxD-I wurde in 10 μM Konzentration verwendet. Säulen stehen
für Mittelwerte
und Standardabweichungen von 16 bis 30 Messungen.
-
-
Säulenauftragung
2: Wirkung von FN Verbindungen auf NMDA Toxizität in gemischten Kulturen von kortikal-neuronalen-glialen Zellen (Modell
B). FN Verbindungen waren 300 μM. "Control" bezieht sich auf
Zellen, welche nur mit NMDA (ohne die schützende Verbindung) behandelt
wurden. ABHxD-I (10 μM)
wurde als positiver Vergleich verwendet. Säulen stehen für Mittelwerte
und Standardabweichungen von 8–40
Messungen.
-
-
Säulenauftragung
3: Wirkung von FN6(F) Enantiomeren auf NMDA Toxizität in gemischten
Kulturen von kortikal-neuronalen-glialen
Zellen (Modell B). FN Verbindungen waren 100 μM. "Control" bezieht sich auf Zellen, welche nur
mit NMDA (ohne die schützende
Verbindung) behandelt wurden. ABHxD-I (10 μM) wurde als positiver Vergleich
verwendet. Säulen
stehen für
Mittelwerte und Standardabweichungen von 8–11 Messungen.
-
-
Säulenauftragung
4: Wirkung von FN6(F) Enantiomeren auf NMDA Toxizität in kortikal-neuronalen Zellen,
die Medium von mit der schützenden
Verbindung behandelten Gliazellen ausgesetzt wurden (Modell C).
FN Verbindungen waren 100 μM. "Control" bezieht sich auf
Zellen, welche nur mit NMDA (und Medium von unbehandelter Glia)
behandelt wurden. ABHxD-I (10 μM)
wurde als positiver Vergleich verwendet. Ergebnisse mit der Guilford
Verbindung PMPA sind zum Vergleich dargestellt. Säulen stehen
für Mittelwerte
und Standardabweichungen von 12–30
Messungen.
-
-
Säulenauftragung
5: Wirkung von FN Verbindungen auf NMDA Toxizität in kortikal-neuronalen Zellen, die
Medium von mit Wirkstoff behandelten Gliazellen ausgesetzt wurden
(Modell C). FN Verbindungen waren 100 μM. "Control" bezieht sich auf Zellen, welche nur
mit NMDA (und Medium von unbehandelter Glia) behandelt wurden. ABHxD-I
(10 μM)
wurde als positiver Vergleich verwendet. Säulen stehen für Mittelwerte
und Standardabweichungen von 8–61
Messungen.
-
Äquivalente
-
Fachleute
auf diesem Gebiet werden viele Äquivalente
zu den spezifischen Ausführungsformen
der hierin beschriebenen Erfindung erkennen oder unter Verwendung
von nicht mehr als Routineexperimenten auffinden können. Solche Äquivalente
sollen durch die folgenden Ansprüche
umfasst sein.

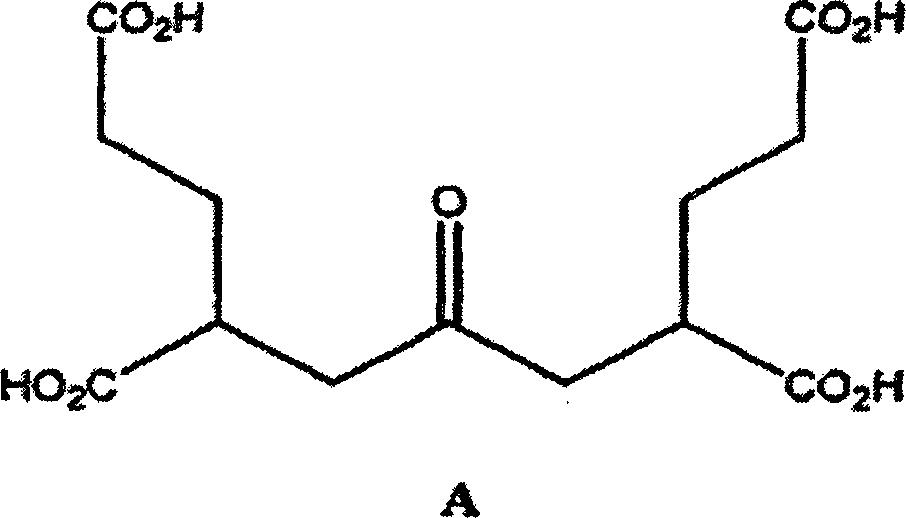


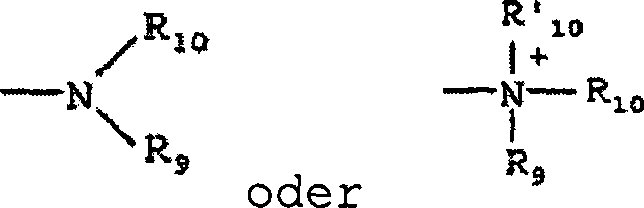
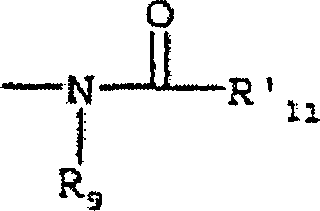







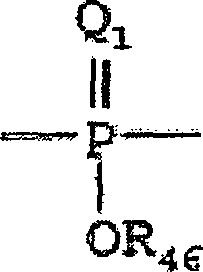 wobei Q1 S oder O darstellt und jedes R46 unabhängig Wasserstoff, ein niederes Alkyl oder ein Aryl darstellt, Q2 O, S oder N darstellt. Wenn Q1 ein S ist, ist die Phosphorylgruppe ein "Phosphorothioat".
wobei Q1 S oder O darstellt und jedes R46 unabhängig Wasserstoff, ein niederes Alkyl oder ein Aryl darstellt, Q2 O, S oder N darstellt. Wenn Q1 ein S ist, ist die Phosphorylgruppe ein "Phosphorothioat".