-
Hintergrund der Erfindung
-
Die
vorliegende Erfindung betrifft Isochinolin-Derivate, Zusammensetzungen
und Medikamente, die diese enthalten, sowie Verfahren zur Herstellung
und Verwendung solcher Verbindungen, Zusammensetzungen und Medikamente.
Solche Isochinolin-Derivate sind von potentiellem therapeutischen
Nutzen in der Behandlung von Krankheiten, die mit unangemessener
oder pathologischer Angiogenese assoziiert sind.
-
Ein
wichtige große
Familie von Enzymen ist die Familie der Proteinkinaseenzyme. Gegenwärtig gibt es
ungefähr
500 verschiedene bekannte Proteinkinasen. Proteinkinasen dienen
zur Katalyse der Phosphorylierung einer Aminosäureseitenkette in verschiedenen
Proteinen durch den Transfer des γ-Phosphats
des ATP-Mg2+-Komplexes auf die Aminosäureseitenkette.
Diese Enzyme kontrollieren die Mehrheit der Signalprozesse innerhalb
der Zellen, wodurch Zellfunktion, Wachstum, Differenzierung und
Zerstörung
(Apoptose) durch reversible Phosphorylierung der Hydroxyl-Gruppen
von Serin-, Threonin- und Tyrosin-Resten in Proteinen gesteuert
wird. Untersuchungen haben gezeigt, daß Proteinkinasen Schlüsselregulatoren
vieler Zellfunktionen sind, einschließlich Signaltransduktion, transkriptionelle
Regulation, Zellmotilität
und Zellteilung. Für
einige Onkogene ist ebenfalls gezeigt worden, daß sie Proteinkinasen codieren,
was darauf hinweist, daß Kinasen
eine Rolle in der Onkogenese spielen. Diese Prozesse sind besonders
reguliert, oft durch komplexe ineinandergreifende Stoffwechselwege,
wobei jede Kinase selbst durch eine oder mehrere Kinasen reguliert
wird. Konsequenterweise kann eine abweichende oder unangemessene
Proteinkinaseataktivität
zum Entstehen von Krankheitszuständen,
die mit solch einer abweichenden Kinaseaktivität assoziiert sind, beitragen.
Aufgrund ihrer physiologischen Relevanz, Vielfältigkeit und Allgegenwart wurden
Proteinkinasen eine der wichtigsten und weitläufig untersuchtesten Enzymfamilie
der biochemischen und medizinischen Forschung.
-
Die
Proteinkinasefamilie von Enzymen wird typischerweise in zwei Hauptunterfamilien
klassifiziert: Protein-Tyrosin-Kinasen (PTK) und Protein-Serin/Threonin-Kinase,
basierend auf dem Aminosäurerest,
den sie phosphorylieren. Die Serin/Threonin-Kinase (PSTK) einschließlich cyclisches
AMP- und cyclisches GMP-abhängige
Proteinkinasen, Calcium- und Phospholipid-abhängige Proteinkinase, Calcium-
und Calmodulin-abhängige
Proteinkinasen, Caseinkinasen, Proteinkinasen des Zellteilungszykluses
und andere. Diese Kinasen sind normalerweise cytoplasmatisch oder
mit besonderen Zellfraktionen assoziiert, möglicherweise durch Ankerproteine.
Abweichende Protein-Serin/Threonin-Kinaseaktivität ist in einer Anzahl von Pathologien wie
rheumatoider Arthritis, Psoriasis, septischem Schock, Knochenverlust,
vielen Krebsarten und anderen proliferativen Krankheiten impliziert
oder angenommen worden. Entsprechend sind Serin/Threonin-Kinasen
und die Signaltransduktionswege, von denen sie ein Teil sind, wichtige
Ziele für
die Arzneistoffentwicklung. Die Tyrosinkinasen phosphorylieren Tyrosinreste.
Tyrosinkinasen spielen eine ebenso wichtige Rolle in der Zellregulation.
Diese Kinasen schließen
einige Rezeptoren für
Moleküle
wie Wachstumsfaktoren und Hormone ein, einschließlich epidermalen Wachstumsfaktorrezeptor,
Insulinrezeptor, Blutplättchen-abgeleiteter
Wachstumsfaktorrezeptor und andere. Untersuchungen haben darauf
hingewiesen, daß viele
Tyrosinkinasen Transmembranproteine sind, deren Rezeptordomänen auf
der Außenseite
von Zellen und Kinasedomänen
auf der Innenseite lokalisiert sind. Viel Arbeit erfolgt ebenfalls
zur Zeit, um ebenfalls Modulatoren von Tyrosinkinasen zu identifizieren.
-
Der
Prozeß der
Angiogenese ist die Entwicklung von neuen Blutgefäßen, im
allgemeinen Kapillargefäße, aus
einer vorher vorhandenen Gefäßanordnung.
Angiogenese wird definiert als mit sich bringend (i) Aktivierung
von Endothelzellen; (ii) zunehmende vaskuläre Permeabilität; (iii)
nachfolgende Auflösung
der Basalmembran und Extravasion von Plasmakomponenten, was zur
Bildung einer vorläufigen
extrazellulären
Fibringelmatrix führt;
(iv) Proliferation und Mobilisierung von Endothelzellen; (v) Neuordnung
von mobilisierten Endothelzellen zur Ausbildung funktioneller Kapillargefäße; (vi)
Ausbildung einer Gefäßschleife;
und (vii) Ablagerung der Basalmembran und Rekrutierung von perivaskulären Zellen,
um Gefäße neu auszubilden.
Normale Angiogenese wird während
des Gewebewachstums, von der embryonalen Entwicklung bis zur Reife,
aktiviert und tritt dann in eine Periode relativer Ruhe während des
Erwachsenenstadiums. Normale Angiogenese wird ebenfalls während der
Wundheilung und in bestimmten Stadien des weiblichen Reproduktionszyklusses
aktiviert. Unangemessene Angiogenese wurde mit mehreren Krankheitszuständen assoziiert,
einschließlich
verschiedener Retinopathien; ischämischer Krankheit; Atherosklerose;
chronische Entzündungsstörungen;
und Krebs. Die Rolle der Angiogenese in Krankheitszuständen wird
zum Beispiel in Fan et al., Trends in Pharmacol. Sci. 16: 54–66; Shawver
et al., DDT Bd. 2, Nr. 2, Februar 1997; Folkmann, 1995, Nature Medicine
1: 27–31 diskutiert.
-
Für das Wachstum
von soliden Tumoren in Krebs wurde gezeigt, daß dieses Angiogenese-abhängig ist.
Der Verlauf der Leukämie
sowie die Akkumulation von Fluid, das mit bösartigem Bauchwasser und pleuraler
Effusionen assoziiert ist, involvieren ebenfalls proangiogene Faktoren.
(Siehe J. Folkman, J. Natl. Cancer Inst., 1990, 82, 4–6.) Konsequenterweise
ist das Abzielen auf proangiogene Stoffwechselwege eine Strategie, die
weitläufig
verfolgt wird, um neue Therapeutika auf diesen Gebieten des großen, unerreichten
medizinischen Bedarfs bereitzustellen. Die Rolle von Tyrosinkinasen,
die an der Angiogenese und der Vaskulierung von soliden Tumoren
beteiligt sind, hat Interesse geweckt.
-
Bis
vor kurzem wurde das meiste Interesse auf diesem Gebiet auf Wachstumsfaktoren,
wie zum Beispiel Gefäßzell-Wachstumsfaktor
(VEGF) und seine Rezeptoren, die als Gefäßzelle-Wachstumsfaktorrezeptor(en)
(VEGFR) bezeichnet werden, fokussiert. Die Rollen von VEGF und VEGFRs,
die sie in der Vaskulisierung von soliden Tumoren im Verlauf von
hämatopoetischen
Krebsarten und in der Modulation der vaskulären Permeabilität spielen,
hat in der Wissenschaftsgemeinschaft großes Interesse geweckt. VEGF,
ein Polypeptid, ist Mitose-auslösend
für Endothelzellen
in vitro und stimuliert angiogene Antworten in vivo. VEGF wurde
ebenfalls mit unangemessener Angiogenese verbunden (H. M. Pinedo
et al., The Oncologist, Bd. 5, Nr. 90001, 1–2, April 2000). VEGFR(s) sind
Proteintyrosinkinasen (PTKs). PTKs katalysieren die Phosphorylierung
spezifischer Tyrosylreste in Proteinen, die an der Regulierung des
Zellwachstums und der Differentierung beteiligt sind. (A. F. Wilks,
Progress in Growth Factor Research, 1990, 2, 97–111; S. A. Courtneidge, Dev.
Supp. I, 1993, 57–64;
J. A. Cooper, Semin. Cell Biol., 1994, 5(6), 377–387; R. F. Paulson, Semin.
Immunol., 1995, 7(4), 267–277;
A. C. Chan, Curr. Opin. Immunol., 1996, 8(3), 394–401).
-
Drei
PTK-Rezeptoren für
VEGF wurden identifiziert: VEGFR-1 (Flt-1); VEGFR-2 (Flk-1 oder
KDR) und VEGFR-3 (Flt-4). Diese Rezeptoren sind an der Angiogenese
beteiligt und partizipieren an der Signaltransduktion (T. Mustonen
et al., J. Cell. Biol. 1995: 129: 895–898). Von besonderem Interesse
ist VEGFR-2, das eine Transmembranrezeptor-PTK ist, die primär in Endothelzellen
exprimiert wird. Die Aktivierung von VEGFR-2 durch VEGF ist ein
kritischer Schritt im Signaltransduktionsweg, der Tumorangiogenese
einleitet. Die VEGF-Expression kann konstitutiv für Tumorzellen
sein und kann ebenfalls in Antwort auf bestimmte Stimuli hochreguliert
werden. Ein solches Stimuli ist Hypoxie, bei der die VEGF-Expression
sowohl im Tumor als auch im assoziierten Wirtsgewebe hochreguliert
wird. Der VEGF-Ligand aktiviert VEGFR-2 durch Binden an dessen extrazelluläre VEGF-Bindungsstelle.
Dies führt
zu einer Rezeptordimerisierung von VEGFRs und Autophosphorylierung
von Tyrosinresten in der intrazellulären Kinasedomäne von VEGFR-2.
Die Kinasedomäne
führt zum
Transfer eines Phosphats von ATP auf die Tyrosinreste, wodurch Bindungsstellen
für Signalproteine stromabwärts von
VEGFR-2 bereitgestellt werden, was letztendlich zur Einleitung der
Angiogenese führt
(G. McMahon, The Oncologist, Bd. 5, Nr. 90001, 3–10, April 2000).
-
Agiopoieten
1 (Ang1), ein Ligand für
die Endothel-spezifische Rezeptortyrosinkinase TIE-2, ist ein neuer
angiogener Faktor (Davis et al., Cell, 1996, 87: 1161–1169; Partanen
et al., Mol. Cell Biol., 12: 1698–1707 (1992);
US-Patene Nrn. 5,521,073 ;
5,879,672 ;
5,877,020 und
6,030,831 ). Das Acronym TIE stellt "Tyrosinkinase-enthaltendes
Ig und EGF-Homolog-Domänen" dar. TIE wird verwendet,
um eine Klasse von Rezeptortyrosinkinasen zu identifizieren, die
exklusiv in vaskulären
Endothelzellen und frühen
hämatopoetischen
Zellen exprimiert werden. Typischerweise werden TIE-Rezeptorkinasen durch
die Gegenwart einer EGF-ähnlichen
Domäne
und einer Immunglobulin(IG)-ähnlichen
Domäne
gekennzeichnet, die aus extrazellulären Faltungseinheiten, die
durch Disulfidbrückenbindungen
innerhalb der Kette stabilisiert werden, bestehen (Partanen et al.,
Curr. Tropics Microbiol. Immunol., 1999, 237: 159–172). Im
Gegensatz zu VEGF, das während des
frühen
Stadiums der vaskulären
Entwicklung wirkt, wirkt Ang1 und sein Rezeptor TIE-2 in den späten Stadien
der vaskulären
Entwicklung, d. h. während
der vaskulären
Ummodulierung (Ummodulierung bezieht sich auf die Ausbildung eines
vaskulären
Lumens) und Reifung (Yancopoulos et al., Cell, 1998, 93: 661–664; K.
G. Peters, Circ. Res., 1998, 83(3): 342–3; Suri et al., Cell 87, 1171–1180 (1996)).
-
Konsequenterweise
würde zu
erwarten sein, daß die
Inhibierung von TIE-2 zur Unterbrechung des Ummodulierens und der
Reifung einer neuen Gefäßanordnung,
die durch Angiogenese eingeleitet wird, dienen könnte, wodurch der angiogene
Prozeß unterbrochen
wird. Des weiteren würde
die Inhibierung an der Kinasedomänebindungsstelle
von VEGFR-2 die Phosphorylierung von Tyrosinresten blockieren und
zum Unterbrechen der Einleitung der Angiogenese dienen. Wahrscheinlich
sollte Inhibierung von TIE-2 und/oder VEGFR-2 dann die Tumorangiogenese
unterbinden und zum Verzögern
oder Auslöschen
des Tumorwachstums dienen. Entsprechend könnte eine Behandlung für Krebs
oder anderen Störungen,
die mit einer unangemessenen Angiogenese assoziiert sind, bereitgestellt
werden.
-
EphB4
ist eine andere Rezeptortyrosinkinase, die ursprünglich in J. Biol. Chem. als
HTK (13. Mai 1994, 269(19): 14211–8) von B. D. Bennett et al.
beschrieben wurde. Die kürzliche
Beobachtung von vaskulären
Defekten in Ephrin-B2- und EphB4-Knockout-Mäusen weist stark darauf hin,
daß die
Wechselwirkung zwischen dem Ephrin-B2-Ligand und seinem erkennenden
EphB4-Rezeptor die
Grenzen von arteriellen-venösen Domänen definiert.
Ephrin-B2-Liganden
werden weitgehend in einigen anderen nicht-vaskulären Geweben wie
mesenchymalen Zellen, die an vaskuläre Endothelzellen angrenzen,
exprimiert, jedoch sind EphB4-Rezeptoren einzig in vaskulären Endothelzellen
lokalisiert. Nicht nur daß EphB4-Rezeptoren
durch ihre entsprechenden Ephrin-B2-Liganden aktiviert werden, die
ebenfalls Transmembranproteine sind, sondern auch EphB4-Rezeptoren
aktivieren ihre Ephrin-B2-Liganden. Embryonen, die heterozygot für das EphB4-Allel
sind, zeigen keine ersichtlichen Defekte im Vergleich zum Wildtyp.
Jedoch zeigen homozygote Embryos kardiovaskuläre Defekte durch Verzögerung des
endothelialen Zellwachstums und arretierter Herzentwicklung, und
embryonaler Letalität
mit hoher Häufigkeit.
Diese Ergebnisse zeigen eindeutig, daß der EphB4-Signalweg eine wesentliche Rolle in
der Vaskulogenese, Angiogenese und Gefäßreifung spielt, und daß diese
Ereignisse ebenfalls untrennbar mit Krebs und Atherosklerose verbunden
ist.
-
Die
Verbindungen der vorliegenden Erfindung besitzen Aktivitäten zu einer
oder mehreren der hier beschriebenen Tyrosinkinasen, insbesondere
zu denen ausgewählt
aus der Gruppe, die aus Tie-2, VEGFR-2 und EphB4-Protein besteht,
die in Krebsarten oder Atherosklerose impliziert sind, durch Inhibieren
oder Verhindern einer unangemessenen Angiogenese, einer Vaskulogenese,
Gefäßreifung
oder Zellmotilität.
WO 99/20608 und
WO 00/09495 offenbaren
Verbindungen, die Angiogenese inhibieren.
-
Kurze Zusammenfassung der Erfindung
-
In
einem Aspekt der vorliegenden Erfindung wird eine Verbindung der
Formel (I) bereitgestellt:
oder ein Salz oder ein Solvat
davon, worin:
eines von R
1 und R
2 H ist und das andere -NHCONHR
4 darstellt,
worin
R
4 eine Phenyl- oder Naphthyl-Gruppe (die
gegebenenfalls mit einem oder mehreren Substituenten substituiert
sein kann, die unabhängig
ausgewählt
sind aus -C
1-5-Alkyl, -C
1-5-Halogenalkyl,
-CH
2CH
2CH
2-, Halogen, C
1-6-Alkoxy,
C
1-6-Halogenalkoxy, OH, NO
2)
oder C
3-7-Cycloalkyl darstellt, oder R
4 zusammen mit dem NH, an das es gebunden
ist, eine Morpholino-Gruppe bildet, und
R
3 H
oder NHR
5 ist, worin R
5 H,
-Chinolinyl oder -Isochinolinyl, -(CONH)
p-Phenyl
(worin p 0 oder 1 ist und das Phenyl gegebenenfalls mit einem oder
mehreren Substituenten substituiert ist, die unabhängig ausgewählt sind
aus Halogen, -C
1-6-Alkyl, -C
1-6-Halogenalkyl,
-Morpholino, -SO
2NH
2,
Benzothiazol (gegebenenfalls mit Methyl substituiert)).
-
In
einem zweiten Aspekt der vorliegenden Erfindung wird eine pharmazeutische
Zusammensetzung bereitgestellt, die eine Verbindung der Formel (I)
oder ein Salz oder Solvat davon und einen oder mehrere aus pharmazeutisch
annehmbaren Trägern,
Verdünnungsstoffen
und Exzipienten umfaßt.
-
In
einem dritten Aspekt der vorliegenden Erfindung wird eine Verbindung
der Formel (I) oder ein Salz oder Solvat davon zur Verwendung in
der Therapie bereitgestellt.
-
Es
wird ein Verfahren zum Behandeln einer Störung in einem Säugetier
bereitgestellt, die durch mindestens eins aus unangemessener TIE-2-,
EphB4- und VEGFR-2-Aktivität vermittelt
wird, wobei das Verfahren das Verabreichen einer Verbindung der
Formel (I) oder eines Salzes, Solvats oder eines physiologisch funktionellen
Derivats davon (das nicht Teil der Erfindung bildet) an das Säugetier
umfaßt.
-
Es
wird die Verwendung einer Verbindung der Formel (I) oder eines Salzes
oder Solvats davon bei der Herstellung eines Medikaments zur Verwendung
in der Behandlung einer Störung,
die durch mindestens eins aus unangemessener TIE-2-, EphB4- und
VEGFR-2-Aktivität
vermittelt wird, bereitgestellt.
-
Es
wird ein Verfahren zur Behandlung einer Störung in einem Säugetier
bereitgestellt, die durch mindestens eins aus unangemessener TIE-2-,
EphB4- und VEGFR-2-Aktivität vermittelt
wird, wobei das Verfahren folgendes umfaßt: Verabreichen an das Säugetier
(i) einer Verbindung der Formel (I) oder eines Salzes, Solvats oder
physiologisch funktionellen Derivats davon und (ii) eines Mittels
zum Inhibieren einer Wachstumsfaktorrezeptorfunktion (bildet keinen
Teil der Erfindung).
-
Es
wird eine Verwendung einer Verbindung der Formel (I) oder eines
Salzes oder eines Solvats davon und eines Mittels zur Inhibierung
einer Wachstumsfaktorrezeptorfunktion bei der Herstellung eines
Medikaments zur Behandlung einer Störung bereitgestellt, die durch
mindestens eins aus unangemessener TIE-2-, EphB4- und VEGFR-2-Aktivität vermittelt
wird.
-
Es
wird ein Verfahren zum Behandeln einer Störung in einem Säugetier
bereitgestellt, die durch unangemessene Angiogenese gekennzeichnet
ist, wobei das Verfahren folgendes umfaßt: Verabreichen an das Säugetier
einer Verbindung der Formel (I) oder eines Salzes, Solvats oder
physiologisch funktionellen Derivats davon (bildet keinen Teil der
Erfindung).
-
In
einem vierten Aspekt der Erfindung wird die Verwendung einer Verbindung
der Formel (I) oder eines Salzes, Solvats oder physiologisch funktionellen
Derivats davon bei der Herstellung eines Medikaments zur Behandlung
einer unangemessenen Angiogenese bereitgestellt.
-
Ausführliche
Beschreibung der Erfindung
-
Wie
hier verwendet, bedeutet der Begriff "effektive Menge" diejenige Menge eines Arzneistoffs
oder pharmazeutischen Mittels, die die biologische oder medizinische
Antwort eines Gewebes, Systems, Tiers oder Menschen auslöst, die
zum Beispiel durch einen Forscher oder Kliniker begehrt wird. Des
weiteren bedeutet der Begriff "therapeutisch
effektive Menge" jedwede
Menge, die im Vergleich zu einem entsprechenden Probanden, der eine
solche Menge nicht erhalten hat, in einer verbesserten Behandlung,
Heilung, Prävention
oder Verbesserung einer Krankheit, Störung oder Nebenwirkung oder
in einer Abnahme der Rate des Fortschreitens einer Krankheit oder
Störung
resultiert. Der Begriff schließt
ebenfalls innerhalb seines Umfangs Mengen ein, die effektiv zum
Steuern einer normalen physiologischen Funktion sind.
-
Wie
hier verwendet, bezeichnet der Begriff "Alkyl" ein geradkettiges oder verzweigtkettiges
Kohlenwasserstoffradikal mit der spezifizierten Anzahl an Kohlenstoffatomen.
Beispiele für "Alkyl", wie hier verwendet,
schließen
uneingeschränkt
Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, t-Butyl,
n-Pentyl, Isopentyl und dgl. ein. Wie hier verwendet, bezeichnet
der Begriff "C1-6-Alkyl" eine
Alkyl-Gruppe, wie zuvor definiert, die mindestens 1 und höchstens
6 Kohlenstoffatome enthält.
Beispiele für
verzweigtkettige oder geradkettige "C1-6-Alkyl"-Gruppen, die nützlich in
der vorliegenden Erfindung sind, schließen uneingeschränkt Methyl,
Ethyl, n-Propyl, Isopropyl, Isobutyl, n-Butyl, t-Butyl, n-Pentyl
und Isopentyl ein.
-
Wie
hier verwendet, bezeichnet der Begriff "Halogen" Fluor (F), Chlor (Cl), Brom (Br) oder
Iod (I), und der Begriff "Halogen" bezeichnet die Halogenradikale
Fluor (-F), Chlor (-Cl), Brom (-Br) und Iod (-I).
-
Wie
hier verwendet, bezeichnet der Begriff "C1-6-Halogenalkyl" eine wie zuvor definierte
Alkyl-Gruppe, die mindestens 1 und höchstens 6 Kohlenstoffatome
enthält
und mit mindestens einer Halogen-Gruppe substituiert ist, wobei
das Halogen wie hier definiert ist. Beispiele für verzweigtkettige oder geradkettige "C1-6-Halogenalkyl"-Gruppen, die nützlich in
der vorliegenden Erfindung sind, schließen uneingeschränkt Methyl,
Ethyl, Propyl, Isopropyl, Isobutyl und n-Butyl ein, die unabhängig mit
einen oder mehreren Halogenen, zum Beispiel Fluor, Chlor, Brom und
Iod, substituiert sind.
-
Wie
hier verwendet, bezeichnet der Begriff "C3-7-Cycloalkyl" einen nicht-aromatischen
cyclischen Kohlenwasserstoffring mit 3 bis 7 Kohlenstoffatomen.
Beispiele für "C3-7-Cycloalkyl"-Gruppen schließen uneingeschränkt Cyclopropyl,
Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl ein.
-
Wie
hier verwendet, bezeichnet der Begriff "Alkoxy" die RaO-Gruppe,
worin Ra ein wie zuvor definiertes Alkyl
ist, und der Begriff "C1-6-Alkoxy" bezeichnet eine wie hier definierte
Alkoxy-Gruppe, worin der Alkylrest mindestens 1 und höchstens
6 Kohlenstoffatome enthält.
Beispiele für
C1-6-Alkoxy-Gruppen,
die nützlich
in der vorliegenden Erfindung sind, schließen uneingeschränkt Methoxy,
Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy und t-Butoxy ein.
-
Wie
hier verwendet, bezeichnet der Begriff "Halogenalkoxy" die Gruppe RaO-,
worin Ra ein wie zuvor definiertes Halogenalkyl
ist, und der Begriff "C1-6-Halogenalkoxy" bezeichnet eine wie hier definierte
Halogenalkoxy-Gruppe,
worin der Halogenalkoxyrest mindestens 1 und höchstens 6 Kohlenstoffatome
enthält.
Beispiele für
C1-6-Halogenalkoxy-Gruppen, die nützlich in
der vorliegenden Erfindung sind, schließen uneingeschränkt Trifluormethoxy
ein.
-
Wie
hier verwendet, bedeutet der Begriff "gegebenenfalls", daß die nachfolgend beschriebenen
Ereignisse auftreten können
oder nicht, und schließt
sowohl Ereignisse, die auftreten als auch Ereignisse, die nicht auftreten,
ein.
-
Wie
hier verwendet, bezeichnet der Begriff "physiologisch funktionelles Derivat" jedes pharmazeutisch annehmbare
Derivat einer Verbindung der vorliegenden Erfindung, zum Beispiel
einen Ester oder ein Amid, das, nach Verabreichung an ein Säugetier,
in der Lage ist, (direkt oder indirekt) eine Verbindung der vorliegenden
Erfindung oder ein aktives Stoffwechselprodukt bereitzustellen.
Solche Derivate sind den Fachleuten ohne unnötiges Experimentieren und mit
Verweis auf die Lehre von Burger's
Medicinal Chemistry And Drug Discovery, 5. Auflage, Bd. 1: Principles
and Practice, ersichtlich, das hier durch Verweis auf den Umfang
hinsichtlich gelehrter physiologisch funktioneller Derivate eingefügt ist.
-
Wie
hier verwendet, bezieht sich der Begriff "Solvat" auf einen Komplex von variabler Stöchiometrie, der
durch einen gelösten
Stoff (in dieser Erfindung eine Verbindung der Formel (I) oder ein
Salz oder ein physiologisch funktionelles Derivat davon) und einem
Lösungsmittel
gebildet wird. Solche Lösungsmittel
können zum
Zwecke der Erfindung nicht die biologische Aktivität des gelösten Stoffs
stören.
Beispiele für
geeignete Lösungsmittel
schließen
uneingeschränkt
Wasser, Methanol, Ethanol und Essigsäure ein. Vorzugsweise ist das
verwendete Lösungsmittel
ein pharmazeutisch annehmbares Lösungsmittel.
Beispiele für
geeignete pharmazeutisch annehmbare Lösungsmittel schließen uneingeschränkt Wasser,
Ethanol und Essigsäure
ein. Am meisten bevorzugt ist das verwendete Lösungsmittel Wasser.
-
Wie
hier verwendet, bezieht sich der Begriff "substituiert" auf eine Substitution mit dem/den bezeichneten
Substituent oder Substituenten, wobei mehrere Grade an Substitution,
sofern nicht anders angegeben, möglich
sind.
-
Wie
hier verwendet, bedeutet "eine
Verbindung der Erfindung" eine
Verbindung der Formel (I) oder ein Salz oder ein Solvat davon.
-
In
einer Ausführungsform
stellt R4 ein Phenyl-Gruppe (die gegebenenfalls
mit einem oder mehreren Substituenten substituiert sein kann, die
unabhängig
ausgewählt
sind aus -C1-6-Halogenalkyl, -CH2CH2CH2-, Halogen)
oder C3-7-Cycloalkyl dar.
-
In
einer Ausführungsform
ist R3 H oder NHR5,
worin R5 H, -Chinolinyl, -(CONH)p-Phenyl (worin p 0 oder 1 ist und das Phenyl
gegebenenfalls mit einem oder mehreren Substituenten substituiert
ist, die unabhängig ausgewählt sind
aus Halogen, -C1-6-Halogenalkyl, -Morpholino,
-SO2NH2, Benzothiazol
(mit Methyl substituiert)) ist.
-
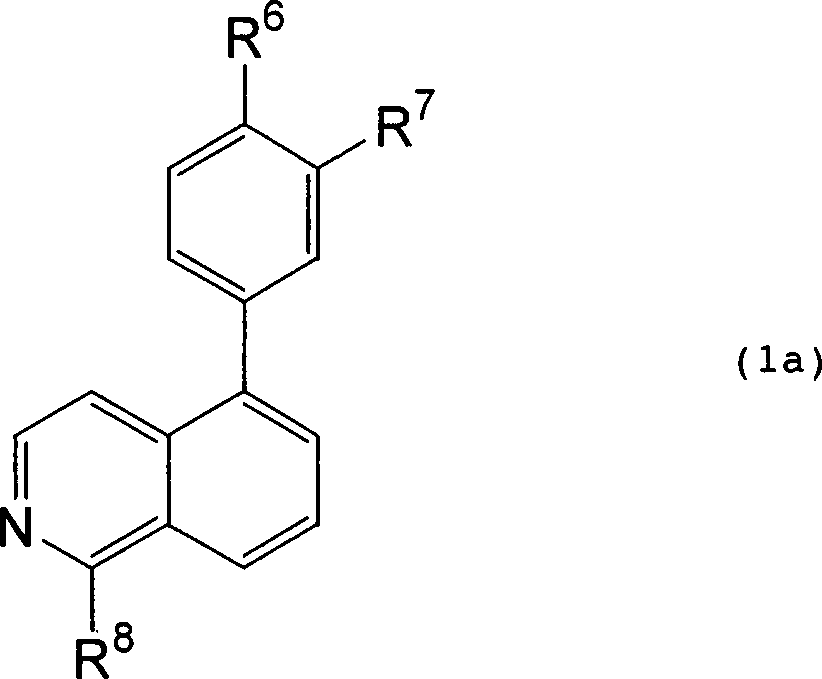
Vorzugsweise
ist die Verbindung die der Formel (1a):
worin:
eines von R
6 und R
7 H ist und
das andere -NHCONHR
9 darstellt;
worin
R
9 eine Phenyl-Gruppe (jedes davon kann
gegebenenfalls mit einem oder mehreren Substituenten substituiert
sein, die unabhängig
ausgewählt
sind aus -C
1-6-Halogenalkyl, -CH
2CH
2CH
2-,
Halogen) oder C
3-7-Cycloalkyl darstellt.
-
R8 ist H oder NHR10,
worin R10 H, Chinolinyl, -(CONH)p-Phenyl (worin p 0 oder 1 ist und das Phenyl gegebenenfalls
mit einem oder mehreren Substituenten substituiert ist, die unabhängig ausgewählt sind
aus Halogen, -C1-6-Halogenalkyl, -Morpholino,
-SO2NH2, Benzothiazol
(mit Methyl substituiert)) ist.
-
Vorzugsweise
stellt R9 ein mit Fluor und Trifluormethyl
disubstituiertes Phenyl dar.
-
Besonders
bevorzugt ist NHCONHR9:
-
-
Vorzugsweise
ist R10 H,
-
-
Spezifische
Beispiele für
Verbindungen der vorliegenden Erfindung schließen folgende ein:
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-isochinolin-5-ylphenyl)harnstoff,
1-Cyclohexyl-3-(3-isochinolin-5-ylphenyl)harnstoff,
1-[3-(1-Amino-isochinolin-5-yl)phenyl]-3-(2-fluor-5-trifluormethyl-phenyl)harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(5-{3-[3-(2-fluor-5-trifluormethyl-phenyl)-ureido]-phenyl}-isochinolin-1-yl)harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(chinolin-6-ylamino)-isochinolin-5-yl]-phenyl}harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-isochinolin-5-ylphenyl)harnstoff,
1-Indan-5-yl-3-(3-isochinolin-5-ylphenyl)harnstoff,
1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(4-morpholin-4-yl-phenylamino)-isochinolin-5-yl]-phenyl}harnstoff und
3-{5-[3-(3-Cyclohexyl-ureido)phenyl]-isochinolin-1-ylamino}benzolsulfonamid.
-
Typischerweise
sind die Salze der vorliegenden Erfindung pharmazeutisch annehmbare
Salze. Salze, die von dem Begriff "pharmazeutisch annehmbare Salze" umfaßt sind,
beziehen sich auf nicht-toxische Salze von Verbindungen dieser Erfindung.
Salze der Verbindungen der vorliegenden Erfindung können Säureadditionssalze,
die von einem Stickstoff auf einem Substituenten in der Verbindung
(I) abgeleitet sind, umfassen. Repräsentative Salze schließen die
folgenden Salze ein: Acetat, Benzolsulfonat, Benzoat, Bicarbonat,
Bisulfat, Bitartrat, Borat, Bromid, Calciumedetat, Camsylat, Carbonat,
Chlorid, Clavulanat, Citrat, Dihydrochlorid, Edetat, Edisylat, Estolat,
Esylat, Fumarat, Gluceptat, Gluconat, Glutamat, Glycollylarsanilat,
Hexylresorcinat, Hydrabamin, Hydrobromid, Hydrochlorid, Hydroxynaphthoat,
Iodid, Isethionat, Lactat, Lactobionat, Laurat, Malst, Malest, Mandelat,
Mesylat, Methylbromid, Methylnitrat, Methylsulfat, Monokaliummaleat,
Mucat, Napsylat, Nitrat, N-Methylglucamin,
Oxalat, Pamoat (Embonat), Palmitat, Pantothenat, Phosphat/Diphosphat,
Polygalacturonat, Kalium, Salicylat, Natrium, Stearat, Subacetat,
Succinat, Tannst, Tartrat, Teoclat, Tosylat, Triethiodid, Trimethylammonium
und Valerat. Andere Salze, die nicht pharmazeutisch annehmbar sind,
können
nützlich bei
der Herstellung von Verbindungen dieser Erfindung sein, und diese
bilden einen weiteren Aspekt der Erfindung.
-
Während es
möglich
ist, daß zur
Verwendung in der Therapie therapeutisch effektive Mengen einer Verbindung
der Formel (I) als auch Salze und Solvate davon als die Rohchemikalie
verabreicht werden können,
ist es möglich
den aktiven Bestandteil als eine pharmazeutische Zusammensetzung
anzubieten. Entsprechend stellt die Erfindung ferner pharmazeutische
Zusammensetzungen bereit, die therapeutisch effektive Mengen der
Verbindungen der Formel (I) und Salze und Solvate davon, und einen
und mehrere pharmazeutisch annehmbare Träger, Verdünnungsmittel oder Exzipienten
einschließen.
Die Verbindungen der Formel (I) und Salze und Solvate davon sind
wie oben beschrieben. Der Träger
(die Träger),
das Verdünnungsmittel
(die Verdünnungsmittel)
oder der Exzipient (die Exzipienten) muß (müssen) annehmbar im Sinne der
Kompatibilität mit
den anderen Bestandteilen der Formulierung und dürfen nicht schädlich für den Empfänger sein.
Gemäß einem
anderen Aspekt der Erfindung wird ebenfalls ein Verfahren zur Herstellung
einer pharmazeutischen Zusammensetzung bereitgestellt, das das Mischen
einer Verbindung der Formel (I) oder eines Salzes oder Solvats davon
mit einem oder mehreren pharmazeutisch annehmbaren Trägern, Verdünnungsmitteln
oder Exzipienten einschließt.
-
Pharmazeutische
Zusammensetzungen können
in Einheitsdosisformen dargeboten werden, die eine vorbestimmte
Menge des aktiven Bestandteils pro Einheitsdosis enthalten. Solch
eine Einheit kann zum Beispiel 0,5 mg bis 1 g enthalten, vorzugsweise
1 mg bis 700 mg, besonders bevorzugt 5 mg bis 100 mg der Verbindung
der Formel (I), abhängig
von dem zu behandelnden Zustand, dem Weg der Verabreichung und dem Alter,
Gewicht und Zustand des Patienten, oder die pharmazeutischen Zusammensetzungen
können
in Einheitsdosisformen dargeboten werden, die eine vorbestimmte
Menge des aktiven Bestandteils pro Einheitsdosis enthalten. Bevorzugte
Einheitsdosierungszusammensetzungen sind die, die eine tägliche Dosis
oder Subdosis, wie bereits oben vorgetragen, oder eine angemessene
Fraktion davon, eines aktiven Bestandteils enthalten. Außerdem können solche
pharmazeutischen Zusammensetzungen durch jedes auf dem Gebiet der Pharmazie
dem Fachmann bekannte Verfahren hergestellt werden.
-
Pharmazeutische
Zusammensetzungen können
für die
Verabreichung über
jeden angemessenen Weg angepaßt
sein, zum Beispiel über
den oralen (einschließlich
bukkalen oder sublingualen), rektalen, nasalen, toxischen (einschließlich bukkalen,
sublingualen oder transdermalen), vaginalen oder parenteralen (einschließlich subkutanen,
intramuskulären,
intravenösen
oder intradermalen) Weg. Solche Zusammensetzungen können durch
jede dem auf dem Gebiet der Pharmazie vertrauten Fachmann bekannten
Verfahren hergestellt werden, zum Beispiel durch das In-Kontakt-Bringen
des aktiven Bestandteils mit dem Träger (den Trägern) oder dem Exzipient (den
Exzipienten).
-
Pharmazeutische
Zusammensetzungen, die für
die orale Verabreichung angepaßt
sind, können
als diskrete Einheiten dargeboten werden, wie zum Beispiel als Kapseln
oder Tabletten; Pulver oder Granalien; Lösung oder Suspensionen in wäßrigen oder
nicht-wäßrigen Flüssigkeiten;
eßbare
Schäume
oder geschlagene Cremes; oder Öl-in-Wasser-Flüssigemulsionen
oder Wasser-in-Öl-Flüssigemulsionen.
-
Zum
Beispiel für
die orale Verabreichung in der Form einer Tablette oder Kapsel kann
die aktive Arzneistoffkomponente mit einem oralen, nicht-toxischen pharmazeutisch
annehmbaren inerten Träger
kombiniert sein, wie zum Beispiel Ethanol, Glycerin, Wasser und
dgl. Pulver werden durch Zerkleinern der Verbindung in eine geeignete
feine Größe und Mischen
mit einem ähnlich
zerkleinerten pharmazeutischen Träger hergestellt, wie zum Beispiel
einem eßbaren
Kohlenhydrat, wie zum Beispiel Stärke oder Mannit. Aromastoffe,
Konservierungsstoffe, Dispergiermittel und Farbstoffe können ebenfalls
vorliegen.
-
Kapseln
werden durch Herstellen des Pulvergemisches, wie oben beschrieben,
und Füllen
in geformte Gelatinehülsen
hergestellt. Gleitmittel und Schmiermittel, wie zum Beispiel kolloidale
Kieselerde, Talkum, Magnesiumstearat, Calciumstearat oder festes
Polyethylenglykol, können
dem Pulvergemisch vor dem Abfüllen zugesetzt
werden. Ein Spreng- oder Solubilisierungsmittel, wie zum Beispiel
Agar-Agar, Calciumcarbonat oder Natriumcarbonat, können ebenfalls
zugesetzt werden, um die Verfügbarkeit
des Medikaments, wenn die Kapsel aufgenommen wird, zu verbessern.
-
Des
weiteren, falls erwünscht
oder notwendig, können
geeignete Bindemittel, Schmiermittel, Sprengmittel und Farbstoffe
ebenfalls in das Gemisch eingebracht werden. Geeignete Bindemittel
schließen
Stärke, Gelatine,
natürliche
Zucker wie Glucose oder beta-Lactose, Maissüßstoffe, natürliche und
synthetische Kautschuks, wie Gummi arabicum, Tragacanthgummi oder
Natriumalginat, Carboxymethylcellulose, Polyethylenglykol, Wachse
und dgl. ein. Schmiermittel, die in diesen Dosierungsformen verwendet
werden, schließen
Natriumoleat, Natriumstearat, Magnesiumstearat, Natriumbenzoat,
Natriumacetat, Natriumchlorid und dgl. ein. Sprengmittel schließen uneingeschränkt Stärke, Methylcellulose,
Agar, Bentonit, Xanthangummi und dgl. ein. Tabletten werden zum
Beispiel durch Herstellen eines Pulvergemisches, granulierend oder
schlagend, Zugeben eines Schmier- und Sprengmittels und Verpressen
in Tabletten formuliert. Ein Pulvergemisch wird durch Mischen der
Verbindung, geeigneterweise zerkleinert, mit einem Verdünnungsmittel
oder einer Basis, wie oben beschrieben, und gegebenenfalls mit einem
Bindemittel, wie zum Beispiel Carboxymethylcellulose, einem Alginat,
Gelatine oder Polyvinylpyrrolidon, einem Auflösungsverzögerer, wie zum Beispiel Paraffin,
einem Abbaubeschleuniger, wie zum Beispiel ein quaternäres Salz,
und/oder einem Absorptionsmittel, wie zum Beispiel Bentonit, Kaolin
oder Dicalciumphosphat, hergestellt. Das Pulvergemisch kann durch
Befeuchten mit einem Bindemittel, wie zum Beispiel Sirup, Stärkepaste,
Akazienschleim oder Lösungen
von Cellulose-basierenden oder polymeren Materialien und Treiben
durch ein Drahtsieb granuliert werden. Als eine Alternative zur
Granulierung kann das Pulvergemisch durch die Tablettiermaschine
laufen gelassen werden, und das Ergebnis sind unvollkommen geformte
Rohlinge, die in Granalien zerbrochen sind. Die Granalien können durch
die Zugabe von Stearinsäure,
eines Stearinsalzes, Talkum oder Mineralöls eingeschmiert sein, um zu
verhindern, daß sie
an den Tabletten-formenden
Prägestempel
haften bleiben. Das eingeschmierte Gemisch wird dann in Tabletten
verpreßt.
Die Verbindungen der vorliegenden Erfindung können ebenfalls mit einem freifließenden inerten
Träger
kombiniert sein und direkt ohne das Durchführen der Granulierungs- oder
Schlagungsschritte in Tabletten verpreßt werden. Eine klare oder
undurchsichtige Schutzbeschichtung kann bereitgestellt werden, die
aus einer versiegelten Schicht von Schellack, einer Beschichtung
aus Zucker oder polymeren Materials und einer Polierbeschichtung
aus Wachs besteht. Farbstoffe können
zu diesen Beschichtungen hinzugegeben werden, um die verschiedenen
Einheitsdosierungen zu unterscheiden.
-
Orale
Flüssigkeiten,
wie zum Beispiel Lösungen,
Sirupe und Elixiere, können
in einer Dosierungseinheitsform hergestellt werden, so daß eine gegebene
Menge eine vorbestimmte Menge der Verbindung enthält. Sirupe
können
durch Auflösen
der Verbindung in eine geeignete mit Geschmack versehene Lösung hergestellt werden,
während
Elixiere durch die Verwendung eines nicht-toxischen akoholischen
Vehikels hergestellt werden. Suspensionen können durch Dispergieren der
Verbindung in einem nicht-toxischen Vehikel formuliert sein. Solubilisierungsmittel
und Emulgatoren, wie zum Beispiel ethoxylierte Isostearylalkohole
und Polyoxyethylensorbitester, Konservierungsmittel, Geschmackzusätze, wie
zum Beispiel Pfefferminzöl,
und natürliche Süßstoffe
oder Saccharin oder andere künstliche
Süßstoffe
und dgl. können
ebenfalls zugesetzt werden.
-
Wo
angebracht, können
die Dosierungseinheitsformulierungen für die orale Verabreichung mikroeingekapselt
sein. Die Formulierung kann ebenfalls hergerichtet sein, um die
Freisetzung zu verlängern
oder fortzusetzen, wie zum Beispiel durch Beschichten oder Einbetten
von partikulärem
Material in Polymeren, Wachs oder dgl.
-
Die
Verbindungen der Formel (I) und Salze, Solvate und physiologisch
funktionelle Derivate davon können
ebenfalls in der Form von Liposom-Übertragungssystemen,
wie zum Beispiel kleine unilammelare Vesikel, große unilammelare
Vesikel und multilammelare Vesikel, verabreicht werden. Liposome
können
aus einer Vielfalt von Phospholipiden gebildet werden, wie zum Beispiel
Cholesterin, Stearylamin oder Phosphatidylcholinen.
-
Die
Verbindungen der Formel (I) und Salze und Solvate davon können ebenfalls
durch Verwendung von monoklonalen Antikörpern als individuelle Träger zugeführt werden,
an die die Verbindungsmoleküle
gekoppelt sind. Die Verbindungen können ebenfalls mit löslichen
Polymeren als zieleingestellte Arzneimittelträger verknüpft sein. Solche Polymere können Polyvinylpyrrolidon,
Pyran-Copolymer, Polyhydroxypropylmethacrylamidphenol, Polyhydroxyethylaspartatamidphenol
oder Polyethylenoxidpolylysin, das mit Palmitoyl-Resten substituiert
ist, einschließen.
Des weiteren können
die Verbindungen an eine Klasse von bioabbaubaren Polymeren gekoppelt
sein, die nützlich
im Erreichen des kontrollierten Freisetzens eines Arzneistoffs sind,
wie zum Beispiel Polymilchsäure,
Polepsilon-caprolacton, Polyhydroxy buttersäure, Polyorthoester, Polyacetale,
Polydihydropyrane, Polycyanoacrylate und vernetzte oder amphipatische
Blockcopolymere von Hydrogelen.
-
Pharmazeutische
Zusammensetzungen, die für
die transdermale Verabreichung angepaßt sind, können als diskrete Pflaster
dargeboten werden, die beabsichtigt sind, den unmittelbaren Kontakt
mit der Epidermis des Empfängers
für eine
verlängerte
Zeit beizubehalten. Zum Beispiel kann der aktive Bestandteil von
dem Pflaster durch Iontophorese, wie allgemein beschrieben in Pharmaceutical
Research, 3(6), 318 (1986), zugeführt werden.
-
Pharmazeutische
Zusammensetzungen, die für
die topische Verabreichung angepaßt sind, können als Salben, Cremen, Suspensionen,
Lotionen, Puder, Lösungen,
Pasten, Gele, Sprays, Aerosole oder öle formuliert sein.
-
Zur
Behandlung des Auges oder anderer äußerer Gewebe, zum Beispiel
Mund und Haut, werden die Zusammensetzungen vorzugsweise als eine
topische Salbe oder Creme aufgetragen. Wenn in einer Salbe formuliert,
kann der aktive Bestandteil mit sowohl einer Paraffin-basierenden
oder einer wassermischbaren Salbenbasis eingesetzt werden. Alternativ
kann der aktive Bestandteil in einer Creme mit einer Öl-in-Wasser-Cremebasis
oder einer Wasser-in-Öl-Basis
formuliert sein.
-
Pharmazeutische
Zusammensetzungen, die für
die topische Verabreichung über
das Auge angepaßt sind,
schließen
Augentropfen ein, worin der aktive Bestandteil in einem geeigneten
Träger,
speziell einem wäßrigen Lösungsmittel,
aufgelöst
oder suspendiert wird.
-
Pharmazeutische
Zusammensetzungen, die für
die topische Verabreichung über
den Mund angepaßt sind,
schließen
Lutschtabletten, Pastillen und Mundwasser ein.
-
Pharmazeutische
Zusammensetzungen, die für
die rektale Verabreichung angepaßt sind, können als Suppositorien oder
als Klistieren dargeboten werden.
-
Pharmazeutische
Zusammensetzungen, die für
die nasale Verabreichung angepaßt
sind, worin der Träger
ein Feststoff ist, schließen
ein grobkörniges
Pulver ein, das eine Teilchengröße zum Beispiel
im Bereich von 20 bis 500 μm
hat, das auf ähnliche
Weise wie Schnupftabak eingenommen wird, verabreicht wird, d. h. durch
rasches Inhalieren über
den Nasengang aus einem Pulverbehälter, der nahe an die Nase
gehalten wird. Geeignete Formulierungen zur Verabreichung als ein
Nasenspray oder als Nasentropfen, worin der Träger ein Feststoff ist, schließen wäßrige oder Öl-Lösungen des
aktiven Bestandteils ein.
-
Pharmazeutische
Zusammensetzungen, die für
die Verabreichung durch Inhalation angepaßt sind, schließen feine
Partikelstäube
oder -nebel ein, die durch verschiedene Arten an dosierten, Dosis-druckverdichteten
Aerosolen, Nebulisatoren oder Insufflatoren erzeugt werden.
-
Pharmazeutische
Zusammensetzungen, die für
vaginale Verabreichung angepaßt
sind, können
als Pessare, Tampons, Cremen, Gele, Pasten, Schäume oder Spray-Formulierungen
vorliegen.
-
Pharmazeutische
Zusammensetzungen, die für
die parenterale Verabreichung angepaßt sind, schließen wäßrige und
nicht-wäßrige sterile
Injektionslösungen,
die Antioxidantien, Puffer, Bakteriostatika und gelöste Substanzen,
die die Zusammensetzung isotonisch mit dem Blut des beabsichtigten
Empfängers
machen, enthalten können;
und wäßrige und
nicht-wäßrige sterile
Suspensionen ein, die Suspendiermittel und Verdickungsmittel einschließen können. Die
Zusammensetzungen können
in Einheitsdosis- oder
Mehrfachdosisbehältern
vorliegen, zum Beispiel in versiegelten Ampullen und Fläschchen,
und können
in einem gefriergetrockneten (lyophilsierten) Zustand gelagert werden,
was nur die Zugabe eines sterilen Flüssigkeitsträgers unmittelbar vor der Verwendung
erfordert, wie zum Beispiel Wasser für Injektionen. Unvorbereitete
Injektionslösungen
und -suspensionen können
aus sterilen Pulvern, Granalien und Tabletten zubereitet werden.
-
Es
sollte selbstverständlich
sein, daß zusätzlich zu
den besonderen oben erwähnten
Bestandteilen die Formulierungen andere Mittel, die üblich auf
dem Gebiet mit Hinsicht auf die Art der infragekommenden Formulierung
sind, einschließen
können,
zum Beispiel solche geeignet für
die orale Verabreichung können
Aromastoffe einschließen.
-
Eine
therapeutisch effektive Menge einer Verbindung der vorliegenden
Erfindung wird von einer Anzahl von Faktoren abhängen, einschließlich zum
Beispiel dem Alter und dem Gewicht des Tiers, dem genauen Zustand,
der die Behandlung erfordert, und seiner Schwere, der Art der Formulierung
und dem Weg der Verabreichung, und unterliegt letztendlich der Ermessensfreiheit
des behandelnden Arztes oder Veterinärs. Jedoch wird eine effektive
Menge einer Verbindung der Formel (I) für die Behandlung von neoplastischem Wachstum,
zum Beispiel Darm- oder Brustkarzinoma, im allgemeinen im Bereich
von 0,1 bis 100 mg/kg Körpergewicht
des Empfängers
(Säugetier)
pro Tag sein, und gewöhnlicher
im Bereich von 1 bis 10 mg/kg Körpergewicht
pro Tag. Daher würde
die eigentliche Menge für
ein erwachsenes Säugetier
von 70 kg gewöhnlich von
70 bis 700 mg pro Tag sein, und diese Menge kann in einer einzelnen
Dosis pro Tag oder für
gewöhnlich in
einer Anzahl (wie zwei, drei, vier, fünf oder sechs) an Sub-Dosen
pro Tag gegeben werden, so daß die
gesamte Tagesdosis die gleiche ist. Eine effektive Menge eines Salzes
oder Solvats davon kann als ein Anteil der effektiven Menge der
Verbindung mit der Formel (I) alleine bestimmt werden. Es ist ersichtlich,
daß gleiche Dosierungen
angemessen für
die Behandlung der anderen Zustände,
auf die oben Bezug genommen wurde, sein würden.
-
Die
Verbindungen der vorliegenden Erfindung und ihre Salze und Solvate
davon können
alleine oder in Kombination mit anderen Therapeutika zur Behandlung
der oben genannten Zustände
eingesetzt werden. Insbesondere ist in der Antikrebstherapie eine
Kombination mit anderen Chemotherapeutika, hormonellen Mitteln oder
Antikörpermitteln
vorgesehen, sowie eine Kombination mit der chirurgischen Therapie
und Radiotherapie. Erfindungsgemäße Kombinationstherapien
umfassen daher die Verabreichung mindestens einer Verbindung der
Formel (I) oder eines pharmazeutisch annehmbaren Salzes oder Solvats
davon und die Verwendung mindestens einer anderen Krebsbehandlungsmethode.
Vorzugsweise umfassen die erfindungsgemäßen Kombinationstherapien die
Verabreichung mindestens einer Verbindung der Formel (I) oder eines
pharmazeutisch annehmbaren Salzes oder Solvats davon und mindestens
eines anderen pharmazeutisch wirksamen Mittels, vorzugsweise eines
antineoplastischen Mittels. Die Verbindung(en) der Formel (I) und
das/die andere(n) pharmazeutisch wirksame(n) Mittel können zusammen
oder getrennt verabreicht werden, und wenn getrennt verabreicht,
dann kann dies simultan oder in einer nacheinanderfolgenden Reihe
erfolgen. Die Mengen der Verbindung(en) der Formel (I) und des/der
anderen pharmazeutisch wirksame(n) Mittel(n) und die relative zeitliche
Koordinierung der Verabreichung wird ausgewählt, um den gewünschten
kombinierten therapeutischen Effekt zu erhalten.
-
Die
Verbindungen der Formel (I) oder Salze oder Solvate davon und mindestens
eine zusätzliche Krebsbehandlungstherapie
können
in Kombination, gleichzeitig oder sequentiell in jeder therapeutisch
angemessenen Kombination mit solch anderen Antikrebstherapien eingesetzt
werden. In einer Ausführungsform
ist die andere Antikrebstherapie mindestens eine zusätzliche
chemotherapeutische Therapie, die die Verabreichung mindestens eines
antineoplastischen Mittels einschließt. Die Verabreichung in Kombination
der Verbindung der Formel (I) oder Salzen oder Solvaten davon mit
anderen antineoplastischen Mitteln kann in Kombination gemäß der Erfindung
durch gleichzeitige Verabreichung in (1) eine einheitlichen pharmazeutischen Zusammensetzung,
die beide Verbindungen einschließt, oder (2) getrennte pharmazeutische
Zusammensetzungen, die jeweils eine der Verbindungen einschließt, sein.
Alternativ kann die Kombination getrennt in einer sequentiellen
Art und Weise verabreicht werden, worin ein antineoplastisches Mittel
zuerst verabreicht wird und das andere als zweites oder umgekehrt.
Solch eine sequentielle Verabreichung kann zeitlich nah oder zeitlich entfernt
vorliegen.
-
Antineoplastische
Mittel können
antineoplastische Effekte in einer Zellzyklus-spezifischen Art induzieren,
d. h. sie sind phasenspezifisch und wirken in einer spezifischen
Phase des Zellzykluses, oder binden DNA und wirken in einer Nicht-Zellzkylus-spezifischen
Art, d. h. sie sind Nicht-Zellzyklus-spezifisch
und agieren durch andere Mechanismen.
-
Antineoplastische
Mittel, die nützlich
in Kombinationen mit den Verbindungen der Formel (I) und Salzen
oder Solvaten davon sind, schließen die folgenden ein:
- (1) Zellzyklus-spezifische antineoplastische
Mittel, die uneingeschränkt
Diterpenoide, wie zum Beispiel Paclitaxel und sein Anologon Docetaxel;
Vincaalkaloide, wie zum Beispiel Vinblastin, Vincristin, Vindesin
und Vinorelbin; Epipodophyllotoxine, wie zum Beispiel Etoposid und
Teniposid; Fluorpyrimidine wie 5-Fluorouracil und Fluorodesoxyuridin;
Antimetabolite, wie zum Beispiel Allopurinol, Fludurabin, Methotrexat,
Cladrabin, Cytarabin, Mercaptopurin und Thioguanin; und Camptothecine,
wie zum Beispiel 9-Aminocamptothecin, Irinotecan, Topotecan, CPT-11
und verschiedene optische Formen von 7-(4-Methylpiperazino-methylen)-10,11-ethylendioxy-20-camptothecin
einschließen;
- (2) cytotoxische chemotherapeutische Mittel, die uneingeschränkt alkylierende
Mittel, wie zum Beispiel Melphalan, Chlorambucil, Cyclophosphamid,
Mechlorethamin, Hexymethylmelamin, Busulfan, Carmustin, Lomustin
und Dacarbazin; Antitumor-Antibiotika, wie zum Beispiel Doxorubicin,
Daunomycin, Epirubicin, Idarubicin, Mitomycin-C, Dacttinomycin und
Methramycin; und Platin-Koordinationskomplexe, wie zum Beispiel
Cisplatin, Carboplatin und Oxaliplatin einschließen; und
- (3) andere chemotherapeutische Mittel, die uneingeschränkt Anti-Östrogene, wie zum Beispiel
Tamoxifen, Toremifen, Raloxifen, Droloxifen und Iodoxyfen; Progesterone,
wie zum Beispiel Megestrolacetat; Aromatase-Inhibitoren, wie zum Beispiel Anastrozol,
Letrazol, Vorazol und Exemestan; Antiandrogene, wie zum Beispiel
Flutamid, Nilutamid, Bicalutamid und Cyproteronacetat; LHRH-Agonisten
und -Antagonisten, wie zum Beispiel Goserelinacetat und Luprolid,
Testosteron-5α-dihydroreduktase-Inhibitoren,
wie zum Beispiel Finasterid; Metalloprotease-Inhibitoren, wie zum
Beispiel Marimastat; Antiprogestogene; Inhibitoren der Urokinaseplasminogen-Aktivatorrezeptorfunktion;
Inhibitoren der Wachstumsfaktorfunktion, wie zum Beispiel Inhibitoren
der Funktion der Hepatocyten-Wachstumsfaktors; für erb-B2, erb-B4, epidermalen
Wachstumsfaktorrezeptor (EGFR), Blutplättchen-abgeleiteten Wachstumsfaktorrezeptor
(PDGFR), vaskulären endothelialen
Wachstumsfaktorrezeptor (VEGFR und EpHB4, TIE-2 (verschieden von
solchen VEGFR-, EpHB2- und TIE-2-Inhibitoren, die in der vorliegenden
Erfindung beschrieben sind)); und andere Tyrosinkinase-Inhibitoren
einschließen,
wie zum Beispiel Inhibitoren des CDK1 und CDK4-Inhibitoren.
-
Von
den Verbindungen der Formel (I) und deren Salze oder Solvate wird
angenommen, daß sie
eine Antikrebswirkung haben, als ein Ergebnis der Inhibierung der
Proteinkinase TIE-2, EpHB4 und/oder VEGFR-2 und ihres Effekts auf
ausgewählte
Zelllinien, deren Wachstum von TIE-2-, EpHB4- und/oder VEGFR-2-Proteinkinaseaktivität abhängig ist.
-
Die
vorliegende Erfindung stellt daher Verbindungen der Formel (I) und
pharmazeutisch annehmbare Salze oder Solvate davon zur Verwendung
in der medizinischen Therapie bereit, und im speziellen in der Behandlung
von Störungen,
die durch mindestens eines aus unangemessener TIE-2-, EpHB4- und
VEGFR-2-Aktivität
vermittelt werden.
-
Die
unangemessene TIE-2-, EphB4- und/oder VEGFR-2-Aktivität, auf die
hier Bezug genommen wird, ist jede TIE-2-, EpHB4- und/oder VEGFR-2-Aktivität, die von
der normalen TIE-2-, EpHB4- und/oder VEGFR-2-Aktivität, die in
einem bestimmten Säugetierprobanden
erwartet wird, abweicht. Eine unangemessene TIE-2-, EpHB4- und/oder
VEGFR-2-Aktivität
kann die Form von zum Beispiel einer abnormalen Aktivitätszunahme
oder einer Anomalie in der Zeitsteuerung und/oder Kontrolle der
TIE-2-, EpHB4- und/oder VEGFR-2-Aktivität annehmen.
Eine solche unangemessene Aktivität kann dann von zum Beispiel
einer Überexpression
oder Mutation der Proteinkinase resultieren, die zu einer unangemessenen
oder unkontrollierten Aktivierung führt. Des weiteren ist es selbstverständlich,
daß eine
unerwünschte
TIE-2-, EpHB4- und/oder
VEGFR-2-Aktivität
in einer abnormalen Quelle liegen kann, wie zum Beispiel einem bösartigen
Tumor. Das heißt, das
Level der TIE-2-, EpHB4-und/oder
VEGFR-2-Aktivität
muß nicht
abnormal sein, um als unangemessen erachtet zu werden, vielmehr
stammt die Aktivität
von einer abnormalen Quelle. In ähnlicher
Weise bezieht sich die unangemessene Angiogenese auf jede angiogene
Aktivität,
die von der normalen angiogenen Aktivität, die in einem bestimmten
Säugetierprobanden
erwartet wird, abweicht. Die unangemessene Angiogenese kann die Form
von zum Beispiel einer abnormalen Zunahme in der Aktivität oder Anomalie
in der Zeitsteuerung und/oder Kontrolle der angiogenen Aktivität annehmen.
Eine solche unangemessene Aktivität kann dann zum Beispiel aus
einer Überexpression
oder Mutation einer Proteinkinase resultieren, die zu einer unangemessenen
oder unkontrollierten Aktivierung führt. Des weiteren ist es selbstverständlich,
daß eine
unerwünschte
angiogene Aktivität
in einer abnormalen Quelle liegen kann, wie zum einem bösartigen
Tumor. Das heißt,
das Level der angiogenen Aktivität
muß nicht
abnormal sein, um als unangemessen angesehen zu werden, vielmehr
stammt die Aktivität
aus einer abnormalen Quelle.
-
Die
vorliegende Erfindung ist auf die Verbindungen der Erfindung zur
Verwendung in Verfahren zur Regulierung, Modulierung oder Inhibierung
von TIE-2, EpHB4 und/oder VEGFR-2 zur Prävention und/oder Behandlung
von Störungen,
die einer unregulierten TIE-2-, EpHB4- und/oder VEGFR-2-Aktivität zugeordnet
sind, gerichtet. Insbesondere können
Verbindungen der vorliegenden Erfindung ebenfalls in der Behandlung
von bestimmten Formen von Krebs verwendet werden. Des weiteren können die
Verbindungen der vorliegenden Erfindung verwendet werden, um zusätzliche
oder synergetische Effekte mit bestimmten existierenden Krebs-Chemotherapien
bereitzustellen, und/oder können
verwendet werden, um die Effektivität bestimmter existierender
Krebs-Chemotherapien und Bestrahlung wieder herzustellen.
-
Die
Verbindungen der vorliegenden Erfindung sind ebenfalls nützlich in
der Behandlung einer oder mehrerer Krankheiten, die Säugetiere
plagen, die durch zelluläre
Proliferation im Bereich der Störungen,
die mit Neovaskularisierung und/oder vaskulärer Permeabilität assoziiert
sind, gekennzeichnet sind, einschließlich Blutgefäß-proliverative
Störungen,
einschließlich
Arthritis und Restenose; fibrotische Störungen, einschließlich hepatische
Zirrhose und Atherosklerose; mesangio-proliverative Störungen,
einschließlich
Glomerulonephritis, diabetische Nephropathie, bösartige Nephrosklerose, thrombotische
Mikroangiopathie-Syndrome, Organtransplantatabstoßung und
Glomerulopathien; und Stoffwechselstörungen, einschließlich Psoriasis,
Diabetes mellitus, chronische Wundheilung, Entzündung und neurodegenerative
Krankheiten.
-
Ein
weiterer Aspekt ist ein Verfahren zur Behandlung eines Säugetiers,
das unter einer Störung
leidet, die durch mindestens eins aus unangemessener TIE-2-, EpHB4-
und/oder VEGFR-2-Aktivität
vermittelt wird, einschließlich
empfängliche
bösartige
Tumoren, das das Verabreichen einer Verbindung der Formel (I) oder eines
pharmazeutisch annehmbaren Salzes, Solvats oder eines physiologisch
funktionellen Derivats davon an das Subjekt einschließt. In einer
bevorzugten Ausführungsform
ist die Störung
Krebs (nicht Teil der Erfindung).
-
Ein
weiterer Aspekt ist ein Verfahren zur Behandlung eines Säugetiers,
das unter Krebs leidet, wobei das Verfahren das Verabreichen einer
Verbindung der Formel (I) oder eines pharmazeutisch annehmbaren Salzes
oder Solvats davon, oder eines physiologisch annehmbaren Derivats
davon an das Subjekt einschließt (nicht
Teil der Erfindung).
-
Ein
weiterer Aspekt ist die Verwendung einer Verbindung der Formel (I)
oder eines pharmazeutisch annehmbaren Salzes oder Solvats davon
bei der Herstellung eines Medikaments zur Behandlung einer Störung, die
durch mindestens eins aus unangemessener TIE-2-, EpHB4- und/oder
VEGFR-2-Aktivität gekennzeichnet
ist. In einer bevorzugten Ausführungsform
ist die Störung
Krebs.
-
Ein
weiterer Aspekt der vorliegenden Erfindung stellt die Verwendung
einer Verbindung der Formel (I) oder eine pharmazeutisch annehmbaren
Salzes oder Solvats davon bei der Herstellung eines Medikaments zur
Behandlung von Krebs und bösartigen
Tumoren bereit.
-
Das
Säugetier,
das der Behandlung mit einer Verbindung der vorliegenden Erfindung
bedarf, ist typischerweise ein Mensch.
-
In
einer anderen Ausführungsform
können
therapeutisch effektive Mengen der Verbindungen der Formel (I) oder
Salze oder Solvate davon und Mittel, die eine Wachstumsfaktorrezeptorfunktion
inhibieren, in Kombination an ein Säugetier zur Behandlung einer
Störung
verabreicht werden, die durch mindestens eins aus unangemessener
TIE-2-, EpHB4- und/oder VEGFR-2-Aktivität vermittelt
wird, zum Beispiel in der Behandlung von Krebs. Solche Wachstumsfaktorrezeptoren
schließen
zum Beispiel EGFR, PDGFR, erB2 und erbB4 ein. Wachstumsfaktorrezeptoren
und Mittel, die eine Wachstumsfaktorrezeptorfunktion inhibieren,
sind zum Beispiel in Kath, C. John, Exp. Opin. Ther. Patents (2000),
10(6): 803–818
und in Shawver et al., DDT, Bd. 2, Nr. 2, Februar 1997, beschrieben.
-
Die
Verbindungen der Formel (I) oder Salze oder Solvate davon und das
Mittel zur Inhibierung einer Wachstumsfaktorrezeptorfunktion können in
Kombination gleichzeitig oder sequentiell in jeder therapeutisch angemessenen
Kombination eingesetzt werden. Die Kombination kann in Kombination
gemäß der Erfindung durch
gleichzeitiges Verabreichen in (1) einer einheitlichen pharmazeutischen
Zusammensetzung, die beide Verbindungen einschließt, oder
(2) getrennten pharmazeutischen Zusammensetzungen, die jeweils eine
der Verbindungen einschließt,
eingesetzt werden. Alternativ kann die Kombination getrennt in einer
sequentiellen Art und Weise verabreicht werden, wobei eine Verbindung
zuerst verabreicht wird und die andere als zweite oder umgekehrt.
Solch eine sequentielle Verabreichung kann zeitlich nahe oder zeitlich
entfernt sein.
-
In
einem anderen Aspekt wird ein Verfahren zur Behandlung einer Störung in
einem Säugetier
bereitgestellt, die durch unangemessene Angiogenese vermittelt wird,
einschließlich:
Verabreichen einer Verbindung der Formel (I) oder eines Salzes,
Solvats oder physiologisch funktionellen Derivats davon an das Säugetier.
In einer Ausführungsform
besteht die unangemessene angiogene Aktivität aufgrund mindestens eins aus
unangemessener VEGFR-2-, EpHB4- oder TIE-2-Aktivität. In einer
anderen Ausführungsform
besteht die unangemessene Angiogenese aufgrund einer unangemessenen
VEGFR-2- und TIE-2-Aktivität.
In einer weiteren Ausführungsform
schließt
das Verfahren ferner das Verabreichen eines VEGFR-2-Inhibitors zusammen mit
den Verbindungen der Formel (I) oder Salzen, Solvaten oder physiologisch
funktionellen Derivaten davon ein. Vorzugsweise ist die Störung Krebs
(nicht bildender Teil der Erfindung).
-
In
einem anderen Aspekt wird die Verwendung einer Verbindung der Formel
(I) oder eines Salzes oder Solvats davon bei der Herstellung eines
Medikaments zur Verwendung in der Behandlung einer Störung in
einem Säugetier
bereitgestellt, die durch unangemessene Angiogenese gekennzeichnet
ist. In einer Ausführungsform
besteht die unangemessene angiogene Aktivität aufgrund mindestens eins
aus unangemessener VEGFR-2-, EpHB4-, VEGFR3- oder TIE-2-Aktivität. In einer
anderen Ausführungsform
besteht die unangemessene Angiogenese aufgrund einer unangemessenen
VEGFR-2-, EpHB4- und TIE-2-Aktivität. In einer
weiteren Ausführungsform
schließt
die Verwendung ferner die Verwendung eines VEGFR-2-Inhibitors zur
Herstellung des Medikaments ein.
-
Die
Kombination einer Verbindung der Formel (I) oder eines Salzes oder
Solvats davon, mit einem VEGFR-2-Inhibitor kann in Kombination gemäß der Erfindung
durch gleichzeitiges Verabreichen (1) einer einheitlichen pharmazeutischen
Zusammensetzung, die beide Verbindungen einschließt, oder
(2) getrennten pharmazeutischen Zusammensetzung, die jeweils eine
der Verbindungen einschließt,
eingesetzt werden. Alternativ kann die Kombination getrennt in einer
sequentiellen Art und Weise verabreicht werden, wobei eine Verbindung
zuerst verabreicht wird und die andere als zweite oder umgekehrt.
Solch eine sequentielle Verabreichung kann zeitlich nah oder zeitlich
entfernt sein.
-
Die
Verbindungen dieser Erfindung können
durch eine Vielzahl von Verfahren hergestellt werden, einschließlich Standardchemie.
Jede zuvor definierte Variante, soweit nicht anderweitig angegeben,
behält
weiterhin die zuvor definierte Bedeutung. Illustrative allgemeine
Syntheseverfahren werden nachfolgend dargelegt, und spezifische
Verbindungen der Erfindung werden dann in den Arbeitsbeispielen
hergestellt.
-
Die
Verbindungen der allgemeinen Formel (I) können durch Verfahren, die auf
dem Gebiet der organischen Synthese bekannt sind, wie im Teil durch
die nachfolgenden Syntheseschemata dargelegt, hergestellt werden.
In all den nachfolgend beschriebenen Schemata ist es selbstverständlich,
daß Schutzgruppen
für sensitive
oder reaktive Gruppen da eingesetzt werden, wo es gemäß den allgemeinen
Prinzipien der Chemie notwendig ist. Schutzgruppen werden gemäß den Standardverfahren
der organischen Synthese manipuliert (T. W. Green und P. G. M. Wuts
(1991), Protecting Groups in Organic Synthesis, John Wiley & Sons). Diese
Gruppen werden in einem passenden Arbeitsgang in der Verbindungssynthese
unter Verwendung von Verfahren, die den Fachleuten leicht ersichtlich
werden, entfernt. Die Auswahl der Verfahren sowie die Reaktionsbedingungen
und Reihenfolge ihrer Ausführung
wird übereinstimmend
mit der Herstellung der Verbindung der Formel (I) sein. Die Fachleute
werden erkennen, wenn ein Stereozentrum in den Verbindungen der
Formel (I) existiert. Entsprechend schließt die vorliegende Erfindung
beide mögliche
Stereoisomere ein, und schließt
nicht nur racemische Verbindungen sondern auch die individuellen
Enantiomere ein. Wenn eine Verbindung als ein einzelnes Enantiomer
erwünscht
ist, kann es durch stereospezifische Synthese und durch Trennung
des Endprodukts oder jeder konventionellen Zwischenstufe erhalten
werden. Trennen des Endprodukts, einer Zwischenstufe oder eines
Ausgangsmaterials kann durch jedes geeignete fachbekannte Verfahren
besorgt werden. Stereochemistry of Organic Compounds von E. L. Eliel,
S. H. Wilen und L. N. Mander (Wiley-Interscience, 1994).
-
Die
Verbindungen der Formel (I) können
gemäß den synthetischen
Sequenzen, die nachfolgend allgemein und durch illustrative Beispiele
dargestellt werden, hergestellt werden und ferner durch spezifische Beispiele
veranschaulicht werden.
-
-
Bestimmte
Ausführungsformen
der vorliegenden Erfindung werden nun durch Beispiele alleine veranschaulicht.
Die physikalischen Daten, die für
exemplarische Verbindungen angegeben sind, sind konsistent mit den
zugeordneten Strukturen dieser Verbindungen.
-
Beispiele
-
Wie
hier verwendet, sind die in diesem Verfahren, Schemata und Beispielen
verwendeten Symbole und Konventionen konsistent mit denen, die in
der wissenschaftlichen Gegenwartsliteratur, zum Beispiel dem Journal
of the American Chemical Society oder dem Journal of Biological
Chemistry verwendet werden. Standard-Ein-Buchstagen- oder Drei-Buchstaben-Abkürzungen
werden im allgemeinen verwendet, um Aminosäurereste, soweit nicht anders
vermerkt, zu bezeichnen, von denen angenommen wird, daß Sie in
der L-Konfiguration sind. Soweit nicht anders angegeben, wurden
alle Ausgangsmaterialien von kommerziellen Lieferanten erhalten,
und ohne weitere Aufreinigung verwendet. Speziell die folgenden
Abkürzungen
können
in den Beispielen für
die gesamte Beschreibung verwendet werden:
- g
- (Gramm)
- mg
- (Milligramm)
- l
- (Liter)
- ml
- (Milliliter)
- μl
- (Mikroliter)
- psi
- (Pfund pro Quadratinch)
- M
- (Molar)
- mM
- (Millimolar)
- i. v.
- (intravenös)
- Hz
- (Hertz)
- MHz
- (Megahertz)
- mol
- (Mol)
- mmol
- (Millimol)
- Rt
- (Raumtemperatur)
- min
- (Minuten)
- h
- (Stunden)
- Smp.
- (Schmelzpunkt)
- TLC
- (Dünnschichtchromatographie)
- Tr
- (Retentionszeit)
- RP
- (reverse Phase)
- MeOH
- (Methanol)
- i-PrOH
- (Isopropanol)
- TEA
- (Triethylamin)
- TFA
- (Trifluoressigsäure)
- TFAA
- (Trifluoressigsäureanhydrid)
- THF
- (Tetrahydrofuran)
- DMSO
- (Dimethylsulfoxid)
- AcOEt
- (Ethylacetat)
- DME
- (1,2-Dimethoxyethan)
- DCM
- (Dichlormethan)
- DCE
- (Dichlorethan)
- DMF
- (N,N-Dimethylformamid)
- DMPU
- (N,N'-Dimethylpropylenharnstoff)
- CDI
- (1,1-Carbonyldiimidazol)
- IBCF
- (Isobutylchlorformiat)
- HOAc
- (Essigsäure)
- HOSu
- (N-Hydroxysuccinimid)
- HOBt
- (1-Hydroxybenzotriazol)
- mCPBA
- (meta-Chlorperbenzoesäure)
- EDC
- (Ethylcarbodiimidhydrochlorid)
- BOC
- (tert-Butyloxycarbonyl)
- FMOC
- (9-Fluorenylmethoxycarbonyl)
- DCC
- (Dicyclohexylcarbodiimid)
- CBZ
- (Benzyloxycarbonyl)
- AC
- (Acetyl)
- atm
- (Atmosphäre)
- TMSE
- (2-(Trimethylsilyl)ethyl)
- TMS
- (Trimethylsilyl)
- TIPS
- (Triisopropylsilyl)
- TBS
- (t-Butyldimethylsilyl)
- DMAP
- (4-Dimethylaminopyridin)
- BSA
- (Rinderserumalbumin)
- ATP
- (Adenosintriphosphat)
- HRP
- (Meerrettichperoxidase)
- DMEM
- (Dulbecco's modifiziertes Eagle-Medium)
- HPLC
- (Hochleistungsflüssigkeitschromatographie)
- BOP
- (Bis(2-oxo-3-oxazolidinyl)phosphinchlorid)
- TRAF
- (Tetra-n-butylammoniumfluorid)
- HBTU
- (O-Benzotriazol-1-yl-N,N,N',N'-tetramethyluroniumhexafluorphosphat)
- HEPES
- (4-(2-Hydroxyethyl)-1-piperazinethansulfonsäure)
- DPPA
- (Diphenylphosphorylazid)
- fHNO3
- (geräuchertes
HNO3); und
- EDTA
- (Ethylendiamintetraessigsäure).
-
Alle
Verweise auf Ether beziehen sich auf Diethylether; Salzlösung bezieht
sich auf eine gesättigte wäßrige Lösung aus
NaCl. Soweit nicht anders angegeben, werden alle Temperaturen in °C (Grad Celsius) ausgedrückt. Alle
Reaktionen werden soweit nicht anders angegeben unter einer inerten
Atmosphäre
bei Raumtemperatur durchgeführt.
-
1H-NMR-Spektren wurden auf einem Varian VXR-300-,
einem Varian Unity-300-,
einem Varian Unity-400-Instrument, einem Brucker AVANCE-400 oder
einem General Electric QE-300 aufgenommen. Chemische Verschiebungen
werden in Teile pro Million (ppm, δ-Einheiten) ausgedrückt. Kupplungskonstanten
sind in Hertz(Hz)-Einheiten. "Splitting"-Muster beschreiben
die anscheinlichen Multiplizitäten
und werden als s (Singulett), d (Dublett), t (Triplett), q (Quartett),
qunit (Quintett), m (Multiplett) und br (breit) bezeichnet.
-
HPLC
wurde auf einem HPLC- oder Shimazu-HPLC-System mit den folgenden
Bedingungen aufgezeichnet. Säule:
50 × 4,6
mm (id) rostfreier Stahl, gepackt mit 5 μm Phenomenex Luna C-18; Flußrate: 2,0 ml/min;
mobile Phase: A-Phase = 50 mM Ammoniumacetat (pH 7,4), B-Phase =
Acetonitril, 0–0,5
min (A: 100%, B: 0%), 0,5–3,0
min (A: 100–0%,
B: 0–100%),
3,0–3,5
min (A: 0%, B: 100%), 3,5–3,7
min (A: 0–100%, B:
100–0%),
3,7–4,5
min (A: 100%, B: 0%); Detektion: UV 254 nm; Injektionsvolumen: 3 μl.
-
Niedrig-Auflösungsmassenspektren
(MS) wurden auf einem JOEL JMS-AX505HA-,
JOEL SC-102- oder einem SCIEX-APliii-Spektrometer aufgezeichnet; LC-MS
wurden auf einem Micromass 2MD und Waters 2690 aufgezeichnet; Hochauflösungs-MS
wurde unter Verwendung eines JOEL SX-102A-Spektrometers erhalten.
Alle Massenspektren wurden unter Elektrosprayionisation (ESI), chemischer
Ionisation (CI), Electron-Impact (EI) oder durch "fast atom bombadment"(FAB)-Verfahren aufgezeichnet.
Infrarot(IR)-Spektren wurden auf einem Nicolet 510 FT-IR-Spektrometer
unter Verwendung einer 1 mm-NaCl-Zelle
erhalten. Die meisten der Reaktionen wurden durch Dünnschichtchromatographie
auf 0,25 mm E. Merck Kieselgelplatten (60F-254) überwacht, und mit UV-Licht,
5% ethanolische Phosphomolybdänsäure oder
p-Anisaldehyd-Lösung sichtbar
gemacht. "Flash"-Säulenchromatographie
wurde auf Kieselgel (230–400
mesh, Merck) durchgeführt.
-
Beispiel 1
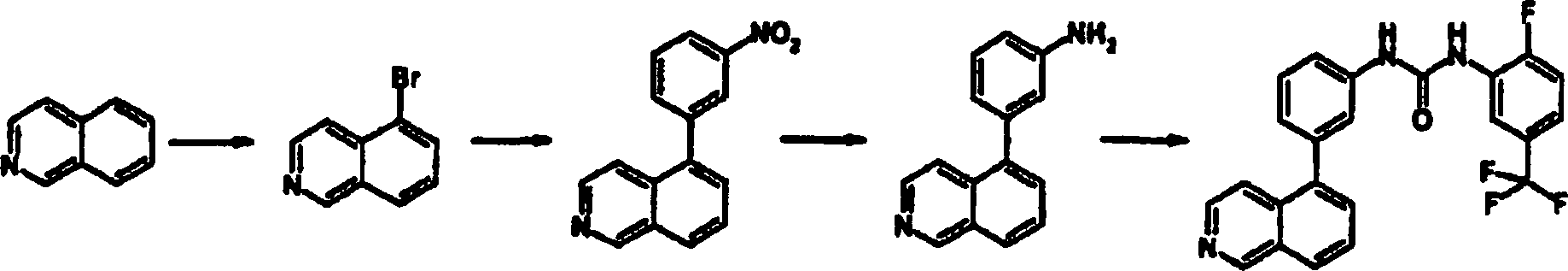
-
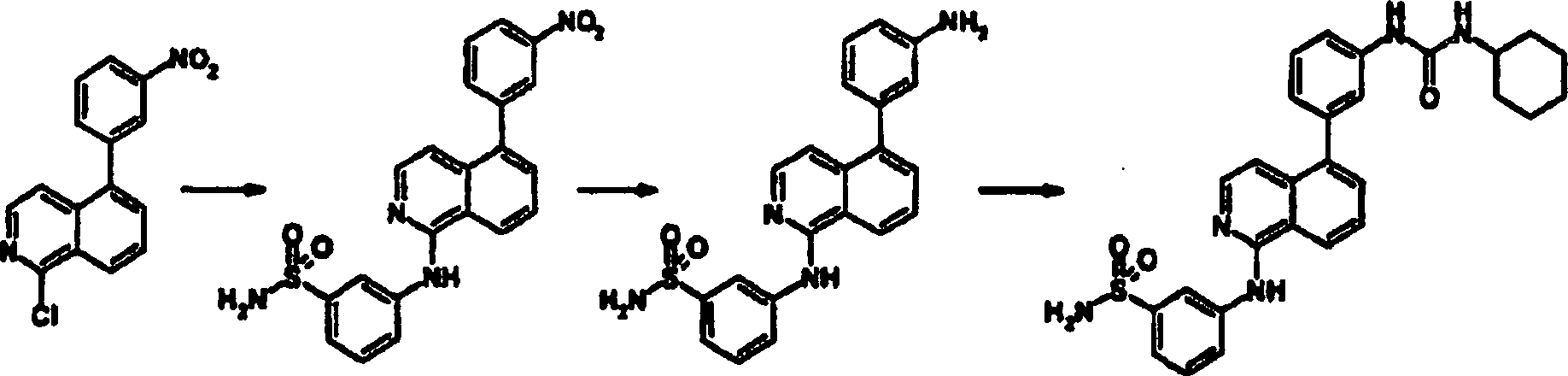
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-isochinolin-5-ylphenyl)harnstoff
-
a. 5-Bromisochinolin
-
Zu
einer Suspension aus AlCl3 (156,7 g, 1,18
mol) CH2Cl2 (500
ml) wurde eine Lösung
aus Isochinolin (605 mmol, 71 ml) in CH2Cl2 (100 ml) bei einer Rate zugetropft, so
daß die
Reaktion behutsam refluxiert wurde. Nach Zugabe wurde CH2Cl2 durch Destillation
entfernt. Der schwarze Rückstand
wurde bei 120°C
geschmolzen, und die Temperatur dann auf 100°C eingestellt. Zu dem Gemisch
wurde Br2 (31 ml, 605 mmol) über einen Zeitraum
von 2 h bei 100°C
zugetropft und für
30 Minuten bei der gleichen Temperatur gerührt, dann über Nacht bei 75°C gerührt. Die
Reaktion wurde auf Raumtemperatur abgekühlt, dann vorsichtig in Eiswasser
gegossen. Das wäßrige Gemisch
wurde mit aq. NaOH basifiziert und mit Ether extrahiert. Die organischen Schichten
wurden über
Na2SO4 getrocknet,
dann eingedampft. Sequenzaufreinigung über zweimalige SiO2-Säulenchromatographie
und Kristallisation (aus Hexan), ergab die Titelverbindung (34,5
g, 28%).
-
b. 5-(3-Nitrophenyl)isochinolin
-
Ein
Gemisch aus 5-Bromisochinolin (4,16 g, 20 mmol), Pd(PPh3)4 (290 mg, 0,25 mmol) und 3-Nitrophenylboronsäure (3,67
g, 22 mmol) wurden mit N2-Gas gespült und dann
Dioxan (200 ml), Ethanol (40 ml) und 2 M aq K2CO3 hinzugegeben. Das Gemisch wurde über Nacht
bei 100°C
gerührt.
Nach Abkühlen
wurde das Gemisch eingedampft, um Dioxan zu entfernen, und wurde dann
mit AcOEt extrahiert. Die organische Schicht wurde mit Salzlösung gewaschen,
dann über
Na2SO4 getrocknet.
Nach Eindampfen wurde der Rückstand
durch SiO2-Säulenchromatographie aufgereinigt,
um die Titelverbindung (4,65 g, 93%) zu ergeben.
-
c. 5-(3-Aminophenyl)isochinolin
-
Zu
einer Lösung
aus 5-(3-Nitrophenyl)isochinolin (751 mg, 3 mmol) wurde Zn-Pulver
(ca. 1 g) hinzugegeben und über
Nacht bei Raumtemperatur gerührt.
Unlösliche
Materialien wurden durch Filtration entfernt, dann eingedampft,
um das Lösungsmittel
zu entfernen. Der Rückstand
wurde mit AcOEt extrahiert. Die organische Schicht wurde mit aq.
NaHCO3 und Salzlösung gewaschen, dann über Na2SO4 getrocknet.
Nach Eindampfen wurde der Rückstand
durch SiO2-Säulenchromatographie und durch
SCX-SPE aufgereinigt, um die Titelverbindung (521 mg, 79%) zu ergeben.
-
d. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-isochinolin-5-ylphenyl)harnstoff
-
Zu
einer Lösung
aus 5-(3-Aminophenyl)isochinolin (30 mg, 0,136 mmol) in THF (1,5
ml) wurde 2-Fluor-5-(trifluormethyl)phenylisocyanat (24 μl, 0,16 mmol)
zugegeben, dann für
3 Tage bei Raumtemperatur gerührt.
Das Gemisch wurde durch SCX-SPE aufgereinigt und der gebildete Feststoff
dann mit MeOH gewaschen, um die Titelverbindung (61,2 mg, 98%) zu
ergeben.
-
Beispiel 2
-
-Cyclohexyl-3-(3-isochinolin-5-ylphenyl)harnstoff
-
Die
Titelverbindung wurde aus 5-(3-Aminophenyl)isochinolin und Cyclohexylisocyanat
wie in Beispiel 1d beschrieben hergestellt.
-
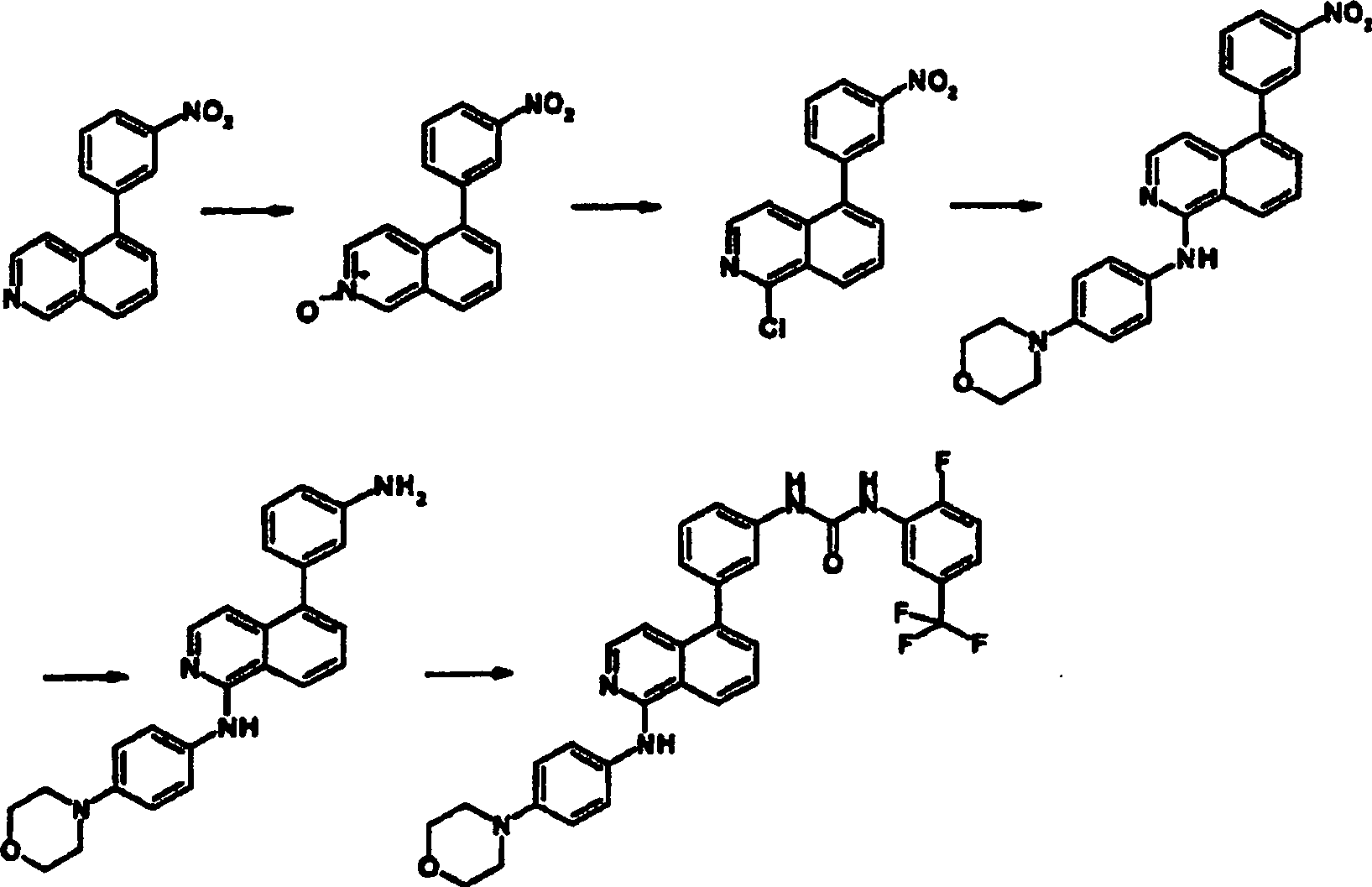
Beispiel 3 und 4
-
1-[3-(1-Amino-isochinolin-5-yl)phenyl]-3-(2-fluor-5-trifluormethyl-phenyl)harnstoff
(Beispiel 3) und 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(5-{3-[3-(2-fluor-5-trifluormethyl-phenyl)-ureido]-phenyl}-isochinolin-1-yl)harnstoff (Beispiel
4)
-
a. 5-Bromisochinolin-N-oxid
-
Zu
einer Lösung
aus 5-Bromisochinolin (20,8 g, 100 mmol) in CH2Cl2 (500 ml) wurde mCPBA (80% Test, 23,7 g,
110 mmol) hinzugegeben und über
Nacht bei 45°C
gerührt.
Nach Abkühlen
wurde das Gemisch mit Na2S2O3 abgeschreckt, dann mit CH2Cl2 extrahiert. Die organische Schicht wurde
mit aq. NaOH gewaschen, über
Na2SO4 getrocknet
und dann eingedampft. Sequenzkristallisation (aus CH2Cl2-Ether) ergab die Titelverbindung (20,1
g, 90%).
-
b. 5-Brom-1-chlorisochinolin
-
Zu
einer Lösung
aus 5-Bromisochinolin-N-oxid (20,1 g, 89,5 mmol) in CH2Cl2 (500 ml) wurde POCl3 (20
ml, 215 mmol) hinzugegeben und über
Nacht bei 45°C
gerührt.
Nach Abkühlen
wurde das gemischt eingedampft, um POCl3 zu
entfernen, dann Wasser hinzugegeben. Die Reaktion wurde mit CH2Cl2 extrahiert.
Die organische Schicht wurde mit aq. NaHCO3 gewaschen, über Na2SO4 getrocknet und
eingedampft. Der gebildete Feststoff wurde mit MeOH gewaschen, um
die Titelverbindung (16,1 g, 74%) zu ergeben.
-
c. 1-Amino-5-bromisochinolin
-
Eine
Suspension aus 5-Brom-1-chlorisochinolin (2,0 g, 8,25 mmol) in gesättigtem
NH3-MeOH (100 ml) wurde für 15 Tage
in einem Autoklaven auf 180°C
erhitzt. Nach Abkühlen
wurde das Lösungsmittel
durch Verdampfung entfernt. Der Rückstand wurde mit CH2Cl2 gewaschen, dann über SXC-SPE
aufgereinigt. Der gebildete Feststoff wurde mit Hexan gewaschen,
um die Titelverbindung (1,57 g, 85%) zu ergeben.
-
d. 1-Amino-5-(4-aminophenyl)isochinolin
-
Die
Titelverbindung wurde aus 1-Amino-5-bromisochinolin und 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)anilin
wie in Beispiel 1b beschrieben hergestellt.
-
e. 1-[3-(1-Amino-isochinolin-5-yl)phenyl]-3-(2-fluor-5- trifluormethyl-phenyl)harnstoff
und 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(5-{3-[3-(2-fluor-5-trifluormethyl-phenyl)-ureido]-phenyl}-isochinolin-1-yl)harnstoff
-
Die
Titelverbindungen wurde aus 1-Amino-5-(4-aminophenyl)isochinolin
wie in Beispiel 1d als Gemisch hergestellt. Diese Verbindungen wurden
durch Säulenchromatographie
aufgetrennt.
-
Beispiel 5
-
1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(chinolin-6-ylamino)isochinolin-5-yl]-phenyl}harnstoff
-
a. 5-Brom-1-(chinolin-6-yl)aminoisochinolin
-
Zu
einer Lösung
aus 5-Brom-1-chlorisochinolin (3,0 g, 12,4 mmol) in iPrOH (100 ml)
wurden 6-Aminochinolin (4,5 g, 31,2 mmol), 4 M HCl-Dioxan (5 ml)
und MeOH (15 ml) hinzugegeben und für 3 Tage bei 80°C gerührt. Nach
Abkühlen
wurde das Gemisch eingedampft, um das Lösungsmittel zu entfernen, dann
in AcOEt suspendiert. Das Gemisch wurde mit aq. NaHCO3 basifiziert,
und dann wurde das geformte Präzipitat
durch Filtration gesammelt und mit AcOEt gewaschen, um die Titelverbindung
(3,4 g, 78%) zu ergeben.
-
b. 5-(4-Aminophenyl)-1-(chinolin-6-yl)aminoisochinolin
-
Die
Titelverbindung wurde aus 5-Brom-1-(chinolin-6-yl)aminoisochinolin
und 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)anilin wie in
Beispiel 1b beschrieben hergestellt.
-
c. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(chinolin-6-ylamino)-isochinolin-5-yl]-phenyl}harnstoff
-
Die
Titelverbindung wurde aus 5-(4-Aminophenyl)-1-(chinolin-6-yl)aminoisochinolin
wie in Beispiel 1d beschrieben hergestellt.
-
Beispiel 6
-
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff
-
a. 1-[4-(6-Methyl-benzothiazol-2-yl)-phenyl]amino-5-bromisochinolin
-
Die
Titelverbindung wurde aus 5-Brom-1-chlorisochinolin und 4-(6-Methyl-benzothiazol-2-yl)-phenylamin
wie in Beispiel 4a beschrieben hergestellt.
-
b. [5-(4-Amino-phenyl)-isochinolin-1-yl]-[4-(6-methyl-benzothiazol-2-yl)-phenyl]-amin
-
Die
Titelverbindung wurde aus 1-[4-(6-Methyl-benzothiazol-2-yl)-phenyl]amino-5-bromisochinolin
und 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)anilin wie in
Beispiel 1b beschrieben hergestellt.
-
c. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff
-
Die
Titelverbindung wurde aus [5-(4-Amino-phenyl)-isochinolin-1-yl]-[4-(6-methyl-benzothiazol-2-yl)-phenyl]-amin
wie in Beispiel 1d beschrieben hergestellt.
-
Beispiel 7
-
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff
-
a. [5-(3-Amino-phenyl)-isochinolin-1-yl]-[4-(6-methyl-benzothiazol-2-yl)-phenyl]-amin
-
Die
Titelverbindung wurde aus 1-[4-(6-Methyl-benzothiazol-2-yl)-phenyl]amino-5-bromisochinolin
und 3-Aminophenylboronsäurehydrochlorid
wie in Beispiel 1b beschrieben hergestellt.
-
b. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(3-{1-[4-(6-methyl-benzothiazol-2-yl)-phenylamino]-isochinolin-5-yl}-phenyl)harnstoff
-
Die
Titelverbindung wurde aus [5-(3-Amino-phenyl)-isochinolin-1-yl]-[4-(6-methyl-benzothiazol-2-yl)-phenyl]-amin
wie in Beispiel 1d beschrieben hergestellt.
-
Beispiel 8
-
1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-isochinolin-5-ylphenyl)harnstoff
-
a. 5-(4-Aminophenyl)isochinolin
-
Die
Titelverbindung wurde aus 5-Bromisochinolin und 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)anilin
wie in Beispiel 1b beschrieben hergestellt.
-
b. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-(4-isochinolin-5-ylphenyl)harnstoff
-
Die
Titelverbindung wurde aus 5-(4-Aminophenyl)isochinolin wie in Beispiel
1d beschrieben hergestellt.
-
Beispiel 9
-
1-Indan-5-yl-3-(3-isochinolin-5-ylphenyl)harnstoff
-
Die
Titelverbindung wurde aus 5-(4-Aminophenyl)isochinolin und 5-Indanyl
wie in Beispiel 1d beschrieben hergestellt.
-
Beispiel 10
-
1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(4-morpholin-4-yl-phenylamino)-isochinolin-5-yl]-phenyl}harnstoff
-
a. 5-(3-Nitrophenyl)isochinolin-N-oxid
-
Zu
einer Lösung
aus Peressigsäure
(hergestellt aus 64 ml AcOH und 32 ml 30% H2O2) wurde 5-(3-Nitrophenyl)isochinolin (3,75
g, 15 mmol) hinzugegeben, dann über
Nacht bei 80°C
gerührt.
Nach Abkühlen
wurde das Gemisch mit AcOEt extrahiert. Die organische Schicht wurde
mit Wasser, aq. NaHCO3, aq. NaHSO3 und Salzlösung gewaschen, dann über Na2SO4 getrocknet.
Nach Eindampfen wurde der Rückstand
durch SiO2-Säulenchromatographie und Sequenzkristallisation
aus MeOH aufgereinigt, um die Titelverbindung (Ausbeute nicht bestimmt)
zu ergeben.
-
b. 1-Chlor-5-(3-nitrophenyl)isochinolin
-
Die
Titelverbindung wurde aus 5-(3-Nitrophenyl)isochinolin-N-oxid wie
in Beispiel 3b beschrieben hergestellt.
-
c. 1-[4-Morpholin-4-ylphenyl)amino]-5-(3-nitrophenyl)isochinolin
-
Die
Titelverbindung wurde aus 1-Chlor-5-(3-nitrophenyl)isochinolin und
4-Morpholin-4-ylanilin wie in Beispiel 4a beschrieben hergestellt.
-
d. 5-(3-Aminophenyl)-1-[4-(morpholin-4-ylphenyl)amino]isochinolin
-
Die
Titelverbindung wurde aus 1-[4-(Morpholin-4-ylphenyl)amino]-5-(3-nitrophenyl)isochinolin
wie in Beispiel 1c beschrieben hergestellt.
-
e. 1-(2-Fluor-5-trifluormethyl-phenyl)-3-{3-[1-(4-morpholin-4-yl-phenylamino)-isochinolin-5-yl]-phenyl}harnstoff
-
Die
Titelverbindung wurde aus 5-(3-Aminophenyl)-1-[4-(morpholin-4-ylphenyl)amino]isochinolin
wie in Beispiel 1d beschrieben hergestellt.
-
Beispiel 11
-
3-{5-[3-(3-Cyclohexyl-ureido)phenyl]-isochinolin-1-ylamino}benzolsulfonamid
-
a. 3-[5-(3-Nitrophenyl)isochinolin-1-ylamino]benzolsulfonamid
-
Die
Titelverbindung wurde aus 1-Chlor-5-(3-nitrophenyl)isochinolin und
3-Aminobenzolsulfonamid wie in Beispiel 4a beschrieben hergestellt.
-
b. 3-[5-(3-Aminophenyl)isochinolin-1-ylamino]benzolsulfonamid
-
Die
Titelverbindung wurde aus 3-[5-(3-Nitrophenyl)isochinolin-1-ylamino]benzolsulfonamid
und 3-Aminobenzolsulfonamid wie in Beispiel 1c beschrieben hergestellt.
-
c. 3-{5-[3-(3-Cyclohexyl-ureido)phenyl]-isochinolin-1-ylamino}benzolsulfonamid
-
Die
Titelverbindung wurde aus 3-[5-(3-Aminophenyl)isochinolin-1-ylamino]benzolsulfonamid
und Cyclohexylisocyanat wie in Beispiel 1d beschrieben hergestellt.
-
Biologische Daten
-
TIE-2-Enzymtest (TIE2-E)
-
Der
TIE-2-Enzymtest verwendet das LANCE-Verfahren (Wallac) und GST-TIE-2, Baculovirus-exprimierte
rekombinante Konstrukte der intrazellulären Domänen des humanen TIE-2 (Aminosäuren 762–1104, GenBank
Accession #: L06139), durch GST getaggt. Das Verfahren maß die Fähigkeit
des aufgereinigten Enzyms zum Transfer des γ-Phosphats vom ATP auf Tyrosin-Reste
in einem biotinylierten synthetischen Peptid, D1-15 (Biotin-C6- LEARLVAYEGWVAGKKK-Amid)
zu katalysieren. Diese Peptid-Phosphorylierung wurde unter Verwendung
des folgenden Verfahrens detektiert: für eine Enzym-Präaktivierung
wurde GST-TIE-2 für
30 min bei Raumtemperatur mit 2 mM ATP, 5 mM MgCl2 und
12,5 mM DTT in 22,5 mM HEPES-Puffer (pH 7,4) inkubiert. Präaktiviertes
GST-TIE-2 wurde für
30 Minuten bei Raumtemperatur in Platten mit 96 Vertiefungen mit
1 μM D1-15-Peptid,
80 μM ATP,
10 mM MgCl2, 0,1 mg/ml BSA und der Testverbindung
(verdünnt
aus einer 10 mM Stammlösung
in DMSO, DMSO-Endkonzentration war 2,4%) in 1 mM HEPES (pH 7,4)
inkubiert. Die Reaktion wurde durch Zugabe von EDTA (Endkonzentration
45 mM) beendet. Streptavidin-verknüpftes APC (Allophycocyanin,
Molecuar Probe) und Europium-markierter anti-phosphoryliertes Tyrosin-Antikörper (Wallac) wurden
dann bei einer Endkonzentration von 17 μg/Vertiefung bzw. 2,1 μg/Vertiefung
hinzugegeben. Das APC-Signal wurde unter Verwendung eines ARVO-"Multilabel"-Zählers (Wallac
Berthold, Japan) gemessen. Die Prozentinhibierung der Aktivität wurde
relativ zur Blindkontrollvertiefung berechnet. Die Konzentration
der Testverbindung, bei der 50% der Aktivität inhibiert wird (IC50), wurde unter Verwendung einer nicht-linearen
Regression (Levernberg-Marquardt) und der Gleichung y = Vmax (1 – x/(K +
x)) + Y2 interpoliert, wobei "K" dem IC50-Wert
gleich war. Die IC50-Werte wurden zu pIC50-Werte umgewandelt, d. h. –log IC50 in Molarkonzentration.
-
VEGF-R2-Enzymtest (VEGF-E)
-
Enzymtest:
-
Verbindungen
der vorliegenden Erfindung wurden auf ihre Aktivität, die Tyrosinkinase
des vaskulären endothelialen
Wachstumsfaktor 2 (VEGFR) zu inhibieren in Substrat-Phosphorylierungstests
getestet. Dieser Test untersucht die Fähigkeit niedrigmolekulare organischer
Verbindungen die Tyrosinphosphorylierung eines Peptid-Substrats
zu inhibieren.
-
Homogene Zeit-aufgelöste Fluoreszenz-(HTFR)-Test
-
Die
Substrat-Phosphorylierungstests verwendeten die katalytische Domäne von VEGFR-2,
die in Sf9-Insektenzellen als ein Amino-terminales GST-getaggtes Fusionsprotein
exprimiert wurde.
-
Autophosphorylierung
ermöglicht
Enzymen vor der Zugabe zu den Peptidsubstraten vollständig aktiviert
zu werden. Die Tests wurden unter Verwendung des Enzyms, das durch
Autophosphorylierung durch Präinkubation
in Puffer mit ATP und Magnesium aktiviert worden ist, durchgeführt. Das
aktivierte Enzym wird dann verdünnt
und zu der titrierten Verbindung und dem Substratgemisch hinzugegeben.
200 nM VEGFR-2-Enzym wurde für
20 Minuten bei Raumtemperatur durch Inkubierung des Enzym in Puffer
aktiviert, der 100 mM HEPES (pH 7,2), 75 μM ATP, 0,3 mM DTT, 0,1 mg/ml
BSA und 10 mM MgCl2 enthielt. Nach der Aktivierung
wurde VEGFR-2, 100-fach in zweimal-Verdünnungspuffer
verdünnt:
200 mM HEPES (pH 7,5), 0,2 mg/ml BSA, 0,6 mM DTT. 20 μl verdünnten Enzymgemischs
wurde zu 20 μl
eines 2×-Substratgemischs
(150 μM
ATP, 20 mM MgCl2, 0,72 μM biotinyliertes Peptid) in
den Testplatten hinzugegeben. Die Endtestbedingungen waren wie folgt:
100 mM HEPES (pH 7,2), 75 μM
ATP, 10 mM MgCl2, 0,1 mg/ml BSA, 0,3 mM
DTT, 0,36 μM
biotinyliertes Peptid und 1 nM VEGFR-2-Enzym. Die Testplatten wurden
für 1,5
Stunden bei Raumtemperatur inkubiert, bevor 30 μl von 100 mM EDTA zu den Vertiefungen
hinzugegeben wurde, um die Enzymreaktion zu beenden. 40 μl/Vertiefung
an HTRF-Gemisch wurde dann zu den Testplatten für die Detektion des phosphorylierten
Substrats hinzugegeben. Endtestbedingungen waren wie folgt: 100
mM HEPES (pH 7,2), 0,1 mg/ml BSA, 15 nM Streptavidin-markiertes
Allophycocyanin (PerkinElmer) und 1 nM Europium-markierter Anti-Phosphotyrosin-Antikörper (PerkinElmer).
Die Testplatten wurden unversiegelt stehengelassen und wurden in
einem Wallac "Multilabel"-Zähler 1420
(PerkinElmer) gezählt.
-
Die
Daten für
die Dosisantworten wurden als % Kontrolle, berechnet mit der Datenreduktionsformel 100·(U1 – C2)/(C1 – C2), gegen
Konzentration der Verbindung geplottet, wobei U der unbekannte Wert
ist, C1 der Durchschnittskontrollwert, erhalten aus DMSO, und C2
der Durchschnittskontrollwert, erhalten aus 0,05 M EDTA, ist. Die
Daten wurden an die Kurve angeglichen, die durch: y = ((Vmax·x)/(K
+ x)) beschrieben ist, wobei Vmax die obere Asymptote und K der
IC50-Wert ist. Die Resultate für jede Verbindung
wurden als pIC50 aufgezeichnet, berechnet
wie folgt: pIC50 = –Log10(K).
-
EphB4-Enzymtest
-
Der
EphB4-Enzymtest verwendete die "Scintillation
Proximity Assay"-Technologie, um die
Enzymaktivität
zu messen. Dieses Verfahren maß die
Fähigkeit
der aufgereinigten Enzyme, den Transfer des γ-Phosphats von ATP auf Tyrosinreste
in einem biotinylierten synthetischen Peptid zu katalysieren. Das
Peptid, das für
den EphB4-Enzymaktivitätstest
verwendet wurde, war Biotin-Ahx-MAHFENYEFFHAKKK-CONH2 (SEQ
ID NO: 1). Das verwendete Enzym war GST-EphB4, Baculovirus-exprimierte
rekombinante Konstrukte der intrazellulären Domänen des humanen EphB4 (Aminosäuren 600–617, BR
#: 21454), getaggt durch GST. Die Peptidphosphorylierung wurde unter
Verwendung des folgenden Verfahrens detektiert: für die Enzympräaktivierung
wurde 7,4 μM
GST-EphB4 für
30 Minuten bei Raumtemperatur mit 50 μM ATP und 10 mM MgCl2 in 30 mM HEPES-Puffer (pH 7,4) inkubiert.
Präaktiviertes
GST-EpHB4 wurde dann für
3 Stunden bei Raumtemperatur in Platten mit 96 Vertiefungen mit
6 μM Peptid,
1 μM ATP,
10 mM MgCl2, 0,1 mg/ml BSA, 5 μCi/ml P33,
1 mM DTT, 1 mM CHAPS, 5 mM KCl und 23–25 μM Testverbindung in 100 mM HEPES
(pH 7,4) inkubiert. Jede Reaktion wurde durch die Zugabe von 0,1
mg Streptavidin-SPA-Perlen in 100 mM EDTA/1x PBS, pH 7,2, terminiert.
Das Signal wurde unter Verwendung eines Wallac "Trilux"-Szintillationszählers (Wallac) gemessen. Die
Prozentinhibierung der Aktivität
wurde relativ zu positiven (C1) und negativen (C2) Kontrollvertiefungen
berechnet, unter Verwendung von 100·(1 – (U1 – C2)/(C1 – C2)). Die Konzentration der
Testverbindung wurde unter Verwendung der Gleichung y = ((Vmax·x)/(K
+ x)) + Y2 bestimmt, wobei "K" dem IC50 gleich
war. Die IC50-Werte wurden in pIC50-Werte
umgewandelt, d. h. –Log
IC50 in Molarkonzentration.
-
Die
Verbindungen der Beispiele zeigen alle inhibitorische Aktivität gegen
mindestens eine der drei getesteten Kinasen bei einem pIC von >5,0.