-
Diese
Anmeldung beansprucht den Nutzen unter 35 USC §119(e) der amerikanischen vorläufigen Anmeldenummern
60/338,881 und 60/380,148, eingereicht am 6. November 2001 bzw.
5. Mai 2002, und USSN 10/139,786, eingereicht am 5. Mai 2002.
-
Diese
Erfindung bezieht sich auf Fasern. In einem Aspekt bezieht sich
die Erfindung auf Fasern, die isotaktische Copolymere aus Propylen
und einem oder mehreren ungesättigten
Comonomere umfassen. In noch einem anderen Aspekt bezieht sich die
Erfindung auf Verfahren zur Verwendung und auf Artikel, die aus diesen
isotaktischen Proypylen-Copolymeren hergestellt werden.
-
Polypropylen
in seinen vielen und mannigfaltigen Arten, ist seit langem ein etabliertes
Haupterzeugnis der Polymerindustrie. Abhängig von dessen Art weist es
eine Anzahl von wünschenswerten
Eigenschaften auf, beinhaltend Zähigkeit
(gemessen durch irgendeine der zahlreichen Schlagversuche, zum Beispiel
gekerbter Izod, Fallbolzenprüfung
etc.), Steifheit (gemessen durch irgendeine der zahlreiche Modultests,
wie zum Beispiel Young's),
Klarheit, chemische Beständigkeit
und Hitzebeständigkeit.
Oft wird eine besondere Kombination von Eigenschaften gewünscht, die
ein Ausgleichen der verschiedenen Eigenschaften gegeneinander erfordert
(zum Beispiel Steifheit gegen Zähigkeit).
Bei Anwendungen, die gute Verarbeitungseigenschaften erfordern (zum
Beispiel Fasern), hat das Polypropylen bevorzugt eine relativ enge
Polydispersität
oder Molekulargewichtsverteilung (MWD), z.B. weniger als 3,5.
-
Viele
Arten von Fasern und Geweben wurden aus Thermoplasten, beinhaltend
Polypropylen, wie oben erwähnt,
hergestellt. Die Eigenschaften der Fasern und Gewebe sind, zumindest
teilweise, eine Funktion des/der Polymers/Polymere aus denen und
des/der Verfahren(s), durch das sie hergestellt werden. Repräsentativ
für diese
verschiedenen Polymere, Faser- und Gewebetypen, und Verfahren zur
Herstellung der Fasern und Gewebe, sind solche, die in USP 4,076,698,
4,644,045, 4,830,907, 4,909,975, 4,578,414, 4,842,922, 4,990,204,
5,112,686, 5,322,728, 4,425,393, 5,068,141 und 6,190,768, beschrieben
sind.
-
Kristallines
Polypropylen hat eine isotaktische Struktur und wird leicht mittels
eines Ziegler-Natta (Z-N)- oder eines Metallocenkatalysators hergestellt.
Während
Metallocenkatalysatoren effektiv sind zur Herstellung von Propylenhomo-
und -copolymeren mit einem hohen isotaktischen Index und einer relativ
engen MWD, ist es relativ schwierig, Propylenhomo- oder -copolymere
mit hohem Mw, zum Beispiel über 300.000, wirtschaftlich
mit einem Metallocenkatalysator herzustellen, besonders in einem
Lösungsverfahren.
Darüber hinaus
behält
die Industrie ein andauerndes Interesse an neuen Polypropylenpolymeren,
besonders an solchen zur Verwendung in Faseranwendungen, bei.
-
In
einer ersten Ausführungsform
ist die Erfindung eine Faser, die ein isotaktisches Copolymer aus
Propylen und einem oder mehreren ungesättigten Comonomeren umfasst.
Diese Copolymere sind dadurch gekennzeichnet, dass sie mindestens
60 Gew.-% an Einheiten, die aus Propylen stammen, und zwischen 0,1
und 40 Gew.-% an Einheiten, die aus dem ungesättigten Comonomer stammen,
aufweisen. Diese Copolymere sind auch dadurch gekennzeichnet, dass
sie mindestens eine der folgenden Eigenschaften haben: (i) 13C NMR-Peaks entsprechend einem Regiofehler
bei etwa 14,6 und etwa 15,7 ppm, wobei die Peaks etwa die gleiche
Intensität
haben, (ii) ein B-Wert größer als
1,4, wenn der Comonomergehalt, d.h. die Einheiten des Copolymers,
die aus dem/den ungesättigten
Comonomer(en) stammen, mindestens 3 Gew.-% ist, (iii) ein Schiefe-Index,
Six, größer als –1,20, (iv)
eine DSC-Kurve mit einer Tme, die im Wesentlichen
die gleiche bleibt, und einer Tmax, die
fällt,
wenn die Menge an Comonomer in dem Copolymer, d.h. an Einheiten,
die aus dem/den ungesättigten
Comonomer(en) stammen, erhöht
wird, und (v) ein Röntgendiagramm,
das mehr Gamma-förmige
Kristalle anzeigt als ein vergleichbares Copolymer, das mit einem
Ziegler-Natta (Z-N)-Katalysator hergestellt wird. Typischerweise
sind die Polymere dieser Ausführungsform
durch mindestens zwei dieser Eigenschaften gekennzeichnet. Gewisse
Polymere dieser Ausführungsform
sind durch mindestens drei dieser Eigenschaften gekennzeichnet,
während
andere Polymere dieser Ausführungsform
durch mindestens vier oder sogar alle fünf dieser Eigenschaften gekennzeichnet
sind.
-
Im
Hinblick auf die Röntgeneigenschaft
des obigen Unterparagraphen (v), bedeutet ein „vergleichbares" Copolymer eines,
das die gleiche Monomerzusammensetzung innerhalb 10 Gew.-% und das
gleiche Mw innerhalb 10 Gew.-% aufweist.
Wenn zum Beispiel das Propylen/Ethylen/1-Hexen-Copolymer 9 Gew.-%
Ethylen und 1 Gew.-% 1-Hexen
und ein Mw von 250.000 aufweist, dann würde ein vergleichbares Polymer,
das mit einem Ziegler-Natta-Katalysator hergestellt wurde, von 8,1-9,9
Gew.-% Ethylen, 0,9-1,1 Gew.-% 1-Hexen und ein Mw zwischen 225.000
und 275.000 aufweisen.
-
In
einer zweiten Ausführungsform
ist die Erfindung eine Faser, die eine Mischung aus zwei oder mehr Polymeren
umfasst, in der mindestens ein Polymer mindestens eines von (i)
den Propylen/ungesättigten
Comonomercopolymeren, die in der ersten Ausführungsform beschrieben sind,
ist (gelegentlich bezeichnet als „P/E*-Copolymer" oder durch einen ähnlichen
Ausdruck). Die Menge der jeweiligen Komponente in der Mischung kann
stark variieren. Die Mischung kann jeglichen Gewichtsanteil an beiden
Komponenten, bezogen auf das Gesamtgewicht des Blends, enthalten
und die Mischung kann entweder homo- oder heterophasig sein. Für den letzteren
Fall kann das Copolymer der ersten oder zweiten Ausführungsform
dieser Erfindung entweder die kontinuierliche oder die diskontinuierliche
(d.h. dispergierte) Phase sein. Zum Zweck dieser Offenbarung, beziehen
sich „P/E-Copolymer" und ähnliche
Ausdrücke
auf ein Propylen/Ethylen-Copolymer abweichend von einem P/E*-Copolymer.
P/E-Copolymere umfassen
50 Gew.-% oder mehr Propylen, während
Ethylen-Propylen-Copolymere
(EP-Copolymere) 51 Gew.-% oder mehr Ethylen umfassen. „Umfassen
... Propylen", „umfassen
... Ethylen" und ähnliche
Ausdrücke,
wie sie hier verwendet werden, meinen Einheiten, die aus Propylen,
Ethylen oder dergleichen stammen, im Gegensatz zu den Verbindungen
selbst.
-
Die
anderen Polymere in der Mischung dieser zweiten Ausführungsform
sind irgendein Polymer abweichend von einem P/E*-Polymer. Typischerweise
und bevorzugt ist/sind diese(s) Polymer(e) ein Polyolefin, wie zum
Beispiel ein oder mehrere von einem Polyethylen, Ethylen/α-Olefin,
Butylen/α-Olefin,
Ethylen/Styrol und dergleichen. Die Mischung kann irgendein Gewichtsanteil
beider Komponenten, bezogen auf das Gesamtgewicht der Mischung,
enthalten und die Mischung kann entweder homo- oder heterophasig
sein. Im letzteren Fall kann das Propylen-Homopolymer entweder die
kontinuierliche oder die dispergierte Phase sein.
-
Andere
erfindungsgemäße Ausführungsformen
beinhalten Artikel, die aus den erfindungsgemäßen Fasern hergestellt werden,
und die Verfahren zur Herstellung dieser Artikel. Erfindungsgemäße Fasern
finden ihre Anwendungen zum Beispiel als Membrane zur chemischen
Trennung, Staubmasken, Teppiche, elastische Gewebe, synthetisches
Haar, Hygieneprodukte (zum Beispiel Windeln, Produkte für Frauen,
Inkontinenzartikel für
Erwachsene, Tücher),
Kleidung, wie zum Beispiel Athletik-Sportbekleidung und bügelfreie
und sich der Form anpassende Kleidung, Schienbeinschoner, Polstermöbel und
medizinische Anwendungen, wie zum Beispiel Binden und sterilisierbare
medizinische Kleidung und Instrument-Hüllen, beinhaltend Chirurgenmasken, Kittel,
Binden und Verpackungsprodukte für
medizinische Instrumente.
-
1 stellt
die ungewöhnliche
Comonomerverteilung eines Propylen/Ethylen (P/E*)-Copolymers dar, das
mit einem metall-zentrierten Katalysator mit Heteroaryl-Ligand hergestellt
wird.
-
2A und 2B zeigen
einen Vergleich der DSC-Aufheizkurven des Propylen/Ethylen (P/E)-Copolymers
von Vergleichsbeispiel 1 bzw. von dem P/E*-Copolymer von Beispiel 2.
-
3 zeigt
einen Vergleich der Tg-Daten eines P/E*-Copolymers und eines herkömmlich Ziegler-Natta
(Z-N)-katalysierten P/E-Copolymers bei äquivalenter Kristallinität.
-
4 zeigt
einen Vergleich der Tg-Daten eines P/E*-Copolymers und eines herkömmlichen,
mit einem Katalysator mit gezwungener Geometne (CGC) hergestellten,
P/E-Copolymers bei gleichem Ethylengehalt.
-
5 zeigt
einen Vergleich einer TREF-Kurve für ein herkömmliche Metallocenkatalysiertes
P/E-Copolymer und für
ein P/E*-Copolymer.
-
6 zeigt
das 13C NMR-Spektrum des P/E*-Copolymer-Produkts
von Beispiel 2, das mit Katalysator G hergestellt wird.
-
7 zeigt
das 13C NMR-Spektrum des P/E-Copolymer-Produkts
von Vergleichsbeispiel 1, das mittels Metallocenkatalysator E hergestellt
wird, wobei das Spektrum keine Regiofehler-Peaks in der Region um
15 ppm aufweist.
-
8 ist
ein Balkendiagramm, in dem die Elastizität von ungestreckten P/E*-Copolymeren mit verschiedenen
herkömmlichen
ungestreckten thermoplastischen Elastomeren verglichen wird.
-
9 ist
ein Balkendiagramm, in dem die Elastizität von vorgestreckten P/E*-Copolymeren mit verschiedenen
herkömmlichen
vorgestreckten thermoplastischen Elastomeren verglichen wird.
-
10A–10J zeigen die chemischen Strukturen verschiedener
Katalysatoren.
-
11 zeigt einen Vergleich des Schiefe-Index für ein P/E*-Copolymer
und verschiedene P/E-Copolymere.
-
12 vergleicht die Schmelzendothermen von Proben
8 und 22a von Beispiel 9.
-
13 zeigt die Verschiebung der Peak-Schmelztemperatur
zu geringerer Temperatur für
das P/E*-Copolymer von Beispiel 9.
-
14 ist ein Diagramm der Temperatur bei der annäherungsweise
1 Prozent Kristallinität
in den DSC-Proben von Beispiel 9 bestehen bleibt.
-
15 zeigt die Varianz relativ zum ersten Moment
der Schmelzendotherme als eine Funktion der Schmelzwärme von
verschiedenen Proben von Beispiel 9.
-
16 zeigt den maximalen Wärmefluss normalisiert auf die
Schmelzwärme
als eine Funktion der Schmelzwärme
für verschiedene
Proben von Beispiel 9.
-
17 zeigt, dass die Rate, bei der der letzte Teil
Kristallinität
in P/E*-Polymeren verschwindet, signifikant geringer ist als für Metallocen-katalysierte
Polymere.
-
18 zeigt dass bei einem ähnlichen prozentualem Anteil
an Kristallinität
eine P/E*-Faser
eine bessere Elastizität
hat als eine Metallocen-katalysierte E/O-Faser.
-
19 zeigt die Festigkeit für drei verschiedene Garne,
die in Beispiel 11 getestet wurden.
-
20 zeigt die Bruchdehnung für drei verschieden Garne, die
in Beispiel 11 getestet wurden.
-
21 ist ein Endothermen-Diagramm der Proben, die
in Tabelle 11 aufgeführt
sind.
-
Polydispersität
-
Die
Polydispersität
der P/E*-Polymere, die in den erfindungsgemäßen Fasern verwendet werden,
liegt typischerweise zwischen 2 und 6. „Enge Polydispersität", „enge Molekulargewichtsverteilung", „enge MWD" und ähnliche
Ausdrücke
bedeuten ein Verhältnis
(Mw/Mn) von gewichtsmittlerem
Molekulargewicht (Mw) zu zahlenmittlerem
Molekulargewicht (Mn) von weniger als 3,5,
bevorzugt von weniger als 3,0, stärker bevorzugt von weniger
als 2,8, stärker
bevorzugt von weniger als 2,5 und am stärksten bevorzugt von weniger
als 2,3. Polymere zur Verwendung in Faseranwendungen haben typischerweise
eine enge Polydispersität.
-
Mischungen,
umfassend die P/E-Polymere, können,
abhängig
von dem Molekulargewicht der anderen Komponenten der Mischung, eine
höhere
Polydispersität
haben. Mischungen, die unter Verwendung irgendeiner der Mehr-Reaktor-Verfahren,
die in der vorliegenden Erfindung offenbart sind, hergestellt werden,
können insbesondere
einen breiten Bereich von Polydispersitäten von 2 angefangen bis 100
oder höher
haben. Mw/Mn von
solchen Mischungen liegt bevorzugt zwischen 2 und 50, stärker bevorzugt
zwischen 2 und 20, am stärksten
bevorzugt zwischen 2 und 10.
-
Gelpermeationschromatographie
-
Die
Molekulargewichtsverteilung der kristallisierbaren Propylenpolymere
wird mittels Gelpermeationschromatographie (GPC) auf einer Polymer
Laboratories PL-GPC-220 Hochtemperaturchromatographie-Einheit mit
vier linearen „mixed
bed"-Säulen (Polymer
Laboratories (20-Mikrometer Partikelgröße)) bestimmt. Die Ofentemperatur
liegt bei 160 °C,
wobei die heiße
Zone des Autosamplers bei 160 °C
und die warme Zone bei 145 °C
liegt. Das Lösungsmittel
ist 1,2,4-Trichlorbenzol und enthalten 200 ppm, 2,6-Di-t-butyl-4-methylphenol. Die
Flussrate ist 1,0 Milliliter/Minute und die Einspritzgröße ist 100
Mikroliter. Es werden etwa 0,2 gewichtsprozentige Lösungen der
Proben für
das Einspritzen hergestellt, indem die Probe in Stickstoff-gespültem 1,2,4-Trichlorbenzol,
enthaltend 200 ppm 2,6-Di-t-butyl-4-methylphenol, 2,5 Stunden bei
160 °C unter
leichtem Rühren gelöst wird.
-
Die
Molekulargewichtsbestimmung wird mittels zehn Polystyrol-Standards
mit enger Molekulargewichtsverteilung (von Polymer Laboratories,
EasiCal PS1 im Bereich von 580-7.500.000 g/Mol) in Verbindung mit
ihren Elutionsvolumina abgeleitet. Die äquivalenten Polypropylen-Molekulargewichte
werden mittels geeigneter Mark-Houwink-Coeffizienten
für Polypropylen
(wie beschrieben bei Th. G. Scholte, N. L. J. Meijerink, H. M. Schoffeleers
und A. M. G. Brands, J. Appl. Polym. Sci., 29, 3763-3782 (1984)) und
für Polystyrol
(wie beschrieben bei E. P. Otocka, R. J. Roe, N. Y. Hellman, P.
M. Muglia, Macromolecules, 4, 507 (1971)) in der Mark-Houwink-Gleichung
bestimmt: {N} = KMa wobei Kpp = 1,90 E-04, app =
0,725 und Kps = 1,26E-04, aps = 0,702.
-
Dynamische
Differenzkalorimetrie
-
Dynamische
Differenzkalorimetrie (DSC) ist eine bekannte Technik, die verwendet
werden kann, um das Schmelzen und die Kristallisation von halbkristallinen
Polymeren zu untersuchen. Die allgemeinen Prinzipien der DSC-Messungen
und Anwendungen von DSC zur Untersuchung semikristalliner Polymere
werden in Standardschriften beschrieben (zum Beispiel E. A. Turi,
Herausgeber, Thermal Characterization of Polymeric Materials, Academic
Press, 1981).
-
Gewisse
Copolymere, die in der Praxis dieser Erfindung verwendet werden,
sind durch eine DSC-Kurve mit einer Tme,
die bei steigender Menge an ungesättigten Comonomer in dem Copolymer
im Wesentlichen die gleiche bleibt, und einer Tmax,
die bei steigender Menge an ungesättigten Comonomer in dem Copolymer fällt, gekennzeichnet.
Tme meint die Temperatur, bei der das Schmelzen
beendet ist. Tmax meint die Peak-Schmelztemperatur.
-
Dynamische
Differenzkalorimetrie (DSC)-Analyse wird mittels eines Geräts Q1000
DSC von TA Instruments, Inc. bestimmt. Die Kalibrierung der DSC
wird wie folgt ausgeführt.
Zunächst
wird eine Basislinie erhalten, indem die DSC von –90 °C auf 290 °C ohne irgendeine
Probe in dem Aluminium-DSC-Pfännchen
gefahren wird. Anschließend
werden 7 Milligram einer frischen Indiumprobe analysiert, indem
die Probe auf 180 °C
geheizt, die Probe auf 140 °C
bei einer Abkühlrate
von 10 °C/Minute
abgekühlt,
und anschließend
die Probe isotherm bei 140 °C
für eine
Minute gehalten wird. Anschließend
wird die Probe von 140 °C
auf 180 °C
bei einer Aufheizrate von 10 ° C/Min
aufgeheizt. Die Schmelzwärme
und der Onset des Schmelzens der Indiumprobe werden bestimmt und
dahingehend überprüft, dass
sie innerhalb 0,5 °C
ausgehend von 156,6 °C
für den
Onset des Schmelzens und innerhalb 0,5 J/g ausgehend von 28,71 J/g
für die
Schmelzwärme
liegen. Anschließend wird
deionisiertes Wasser analysiert, indem ein kleiner Tropfen einer
frischen Probe in dem DSC-Pfännchen von
25 °C auf –30 °C bei einer
Abkühlrate
von 10 °C/Minute
abgekühlt
wird. Die Probe wird isotherm bei –30 °C 2 Minuten gehalten und auf
30 °C mit
einer Heizrate von 10 °C/Min
aufgeheizt. Der Onset des Schmelzens wird bestimmt und dahingehend überprüft, dass
er innerhalb 0,5 °C
ausgehend von 0 °C
ist.
-
Die
Polypropylenproben werden zu einem dünnen Film bei einer Temperatur
von 190 ° C
gepresst. 5 bis 8 mg der Probe werden ausgewogen und in das DSC-Pfännchen platziert.
Der Deckel wird auf das Pfännchen
gequetscht, um eine geschlossene Atmosphäre sicherzustellen. Das Probenpfännchen wird
in die DSC-Zelle platziert und mit einer Heizrate von etwa 100 °C/Min auf
eine Temperatur von etwa 30 °C über der Schmelztemperatur
aufgeheizt. Die Probe wird bei dieser Temperatur etwa 3 Minuten
gehalten. Anschließend wird
die Probe mit einer Rate von 10 °C/Min
auf –40 °C abgekühlt, und
isotherm bei dieser Temperatur 3 Minuten gehalten. Anschließend wird
die Probe mit einer Rate von 10 °C/Min
bis zum kompletten Schmelzen aufgeheizt. Die resultierenden Enthalpie-Kurven
werden auf die Peak-Schmelztemperatur, den Onset und die Peakkristallisations-Temperatur,
Schmelzwärme
und Kristallisationswärme,
Tme und auf jegliche andere DSC-Analyse
von Interesse, analysiert.
-
B-Wert
-
„Hoher
B-Wert" und ähnliche
Ausdrücke
bedeuten, dass die Ethyleneinheiten eines Copolymers aus Propylen
und Ethylen, oder eines Copolymers aus Propylen, Ethylen und mindestens
einem ungesättigten
Comonomer entlang der Polymerkette in einer nicht-zufälligen Weise
verteilt sind. B-Werte reichen von 0 bis 2, wobei 1 eine perfekt
zufällige
Verteilung der Comonomereinheiten kennzeichnet. Je höher der
B-Wert, desto alternierender ist die Comonomerverteilung in dem
Copolymer. Je niedriger der B-Wert ist, desto blockiger oder angehäufter ist
die Comonomerverteilung in dem Copolymer. Die hohen B-Werte der
erfindungsgemäßen Polymere
sind typischerweise mindestens 1,3, bevorzugt mindestens 1,4, stärker bevorzugt
mindestens 1,5 und am stärksten
bevorzugt mindestens 1,7. Der B-Wert wird wie folgt berechnet.
-
B
ist für
ein Propylen/Ethylencopolymer definiert als:
wobei f(EP + PE) = die Summe
der EP- und PE-Diaden-Anteile ist; und Fe und Fp = der molare Anteil
an Ethylen bzw. Propylen im Copolymer ist. B-Werte können für andere
Copolymere in einer analogen Weise durch die Zuweisung der jeweiligen
Copolymerdiaden berechnet werden. Zum Beispiel wird für die Berechnung
des B-Werts für ein Propylen/1-Octen-Copolymer
die folgende Gleichung verwendet:
-
-
Für Propylenpolymere,
die mit einem Metallocenkatalysator hergestellt werden, liegen die
B-Werte typischerweise zwischen 1,1 und 1,3. Für Propylenpolymere, die mit
einem Katalysator mit gezwungener Geometrie hergestellt werden,
liegen die B-Werte typischerweise zwischen 0,9 und 1,0. Im Gegensatz
dazu sind die B-Werte der erfindungsgemäßen Propylenpolymere, die typischerweise
mit einem aktivierten Nicht- Metallocen,
Metall-zentriertem Katalysator mit Heteroaryl-Ligand, hergestellt
werden, über
1,4, typischerweise zwischen 1,5 und 1,85. Das bedeutet wiederum,
dass für
irgendein P/E*-Copolymer nicht nur die Propylenblocklänge relativ
kurz für
einen gegebenen Prozentanteil von Ethylen ist, sondern dass lange
Sequenzen von 3 oder mehr sequenziellen Ethyleninsertionen in dem
Copolymer sehr selten, wenn überhaupt,
vorhanden sind, ausser wenn der Ethylengehalt des Polymers sehr
hoch ist. 1 und die Daten der folgenden
Tabellen sind veranschaulichend. Die Katalysatoren sind aktivierte
Nicht-Metallocen-, Metall-zentrierte Katalysatoren mit Heteroaryl-Ligand,
mit denen P/E*-Polymere hergestellt werden. Der Katalysator E ist
ein Metallocenkatalysator, und dieser lieferte nicht die P/E*-Polymere.
Interessanterweise blieben die B-Werte der P/E*-Polymere auch für Polymere
mit relativ hohen Mengen, zum Beispiel > 30 Mol-% Ethylen, hoch.
-
Tabelle
A: B-Werte von ausgewählten
Propylenpolymeren
-
Katalysator
I ist Dimethylamidoboran-bis-η5-(2-methyl-4-naphtylinden-1-yl)zirconium-η4-1,4-diphenyl-1,3-butadien.
-
Tabelle
B: B-Werte von ausgewählten
Propylen/Ethylencopolymeren
-
Die
unten beschriebenen Verfahren können
verwendet werden, um Propyleninterpolymere aus Ethylen und gegebenenfalls
C4-C20-alpha-Olefinen
mit einem relativ breiten Schmelzpunkt in einer DSC-Aufheizkurve
herzustellen. Ohne dass es gewünscht
wird, dass an irgendeiner speziellen Theorie der Funktionsweise festgehalten
wird, wird angenommen, dass die hohen B-Werte für die neuen Propylen/Ethylen-Interpolymere und
das Verfahren zu ihrer Herstellung zu einer Ethylenverteilung innerhalb
der Polymerketten führt,
die zu einem breiten Schmelzverhalten führt. In den 2A und 2B wird
zum Beispiel ein relativ enger Schmelzpeak für ein Propylen/Ethylen-Copolymer
beobachtet, das mittels eines Metallocens als ein Vergleichsbeispiel (Vergleichsbeispiel
1) hergestellt wurde, während
der Schmelzpeak für
ein ähnliches
Copolymer aus Propylen und Ethylen, das gemäß der hier angeführten Lehren
hergestellt wurde, einen breiten Schmelzpunkt aufweist. Solch ein
breites Schmelzverhalten ist nützlich
bei Anwendungen, die zum Beispiel eine relativ niedrige Heißsiegel-Anfangs-Temperatur
oder ein breites Heißklebe-
und/oder Heisssiegelfenster erfordern.
-
Thermische
Eigenschaften
-
3 und 4 veranschaulichen
ferner die thermischen Eigenschaften der P/E*-Polymere, die in der Praxis dieser Erfindung
verwendet werden. 3 veranschaulicht, dass die
P/E*-Polymere eine höhere Glasübergangstemperatur
(Tg) haben als vergleichbare Metallocen-katalysierte Propylenpolymere
bei einer vergleichbaren Kristallinität. Das bedeutet, dass die P/E*-Copolymere
wahrscheinlich bessere Kriechbeständigkeit aufweisen als konventionelle
Metallocen-katalysierte Propylen-Copolymere. Darüber hinaus zeigen die Tmax-Daten in Tabelle A, dass die P/E*-Copolymere
einen niedrigeren Schmelzpunkt bei gleicher Kristallinität als ein
Metallocen-katalysiertes Propylen-Copolymer haben. Dies bedeutet
wiederum, dass die P/E*-Polymere wahrscheinlich besser zu verarbeiten
(d.h. weniger Aufheizen erfordern) als herkömmliche Metallocen-katalysierte
Propylenpolymere sind.
-
4 veranschaulicht,
dass die P/E*-Polymere auch einen niedrigen Tg bei einem äquivalenten
Ethylengehalt haben als ein Propylenpolymer, das mit einem Katalysator
mit gezwungener Geometrie (CGC) hergestellt wurde, und das wiederum
bedeutet, dass die P/E*-Polymere wahrscheinlich eine bessere Tieftemperaturfestigkeit aufweisen
als die CGC-Propylenpolymere, was die P/E*-Polymere zu attraktiven
Kandidaten für Lebensmittelverpackungs-Anwendungen
macht.
-
Elutionsfraktionierung
bei steigender Temperatur
-
Die
Bestimmung von kristallisierbarer Sequenzlängenverteilung kann im präparativen
Maßstab
durch Elutionsfraktioniertung bei steigender Temperatur (TREF) erreicht
werden. Die relativen Massen der einzelnen Fraktionen können verwendet
werden als eine Basis zum Schätzen
einer kontinuierlicheren Verteilung. L. Wild, et al., Journal of
Polymer Science: Polymer. Physics Ed., 20, 441 (1982), verkleinerte
die Probengröße und fügte einen
Massendetektor hinzu, um eine kontinuierliche Repräsentation
der Verteilung als eine Funktion der Elutionstemperatur herzustellen.
Diese verkleinerte Version, analytische Elutionsfraktionierung bei
steigender Temperatur (ATREF), betrifft nicht die aktuelle Isolierung
von Fraktionen, sondern mehr die genaue Bestimmung der Gewichtsverteilungen
der Fraktionen.
-
Während TREF
ursprünglich
bei Copolymeren aus Ethylen und höheren α-Olefinen angewendet wurde,
kann es auch für
die Analyse von Copolymeren aus Propylen mit Ethylen (oder höheren α-Olefinen)
verwendet werden. Die Analyse von Copolymeren aus Propylen erfordert
höhere
Temperaturen zur Lösung
und Kristallisation von reinem isotaktischen Polypropylen, allerdings
eluieren die meisten Copolymerisationsprodukte von Interesse bei ähnlichen
Temperaturen wie sie für
Copolymere aus Ethylen beobachtet werden. Die folgende Tabelle ist
eine Zusammenfassung der Bedingungen, die für die Analyse der Copolymere
aus Propylen verwendet wurden. Wenn nicht anders gekennzeichnet,
stimmen die Bedingungen für
TREF mit denen von Wild, et al., ibid, und Hazlitt, Journal of Applied
Polymer Science: Appl. Polym. Symp., 45, 25 (1990) überein.
-
Tabelle
C: Parameter bei TREF
-
Die
Daten, die aus TREF erhalten werden, werden wiedergegeben als ein
normalisiertes Diagramm der Gewichtsfraktion als eine Funktion der
Elutionstemperatur. Der Trennungsmechanismus ist analog zu dem der
Copolymere aus Ethylen, wobei der molare Gehalt der kristallisierbaren
Komponente (Ethylen) der primäre Faktor
ist, der die Elutionstemperatur bestimmt. In dem Fall der Copolymere
aus Propylen ist es der molare Gehalt an isotaktischen Propylen-Einheiten,
der primär
die Elutionstemperatur bestimmt. 5 stellt
den typischen Verteilungstyp dar, den man für ein Propylen/Ethylencopolymer,
das mit einem Metallocen-Polymer und einem Beispiel von einem P/E*-Copolymer
gemacht wurde, erwarten würde.
-
Die
Form der Metallocenkurve in 5 ist
typisch für
ein homogenes Copolymer. Die Form entsteht durch den inhärenten zufälligen Einbau
von Comonomer. Eine markante Charakteristik der Kurvenform ist die Schwanzbildung
(tailing) bei niedriger Elutionstemperatur verglichen mit der Schärfe oder
Steilheit der Kurve bei den höheren
Elutionstemperaturen. Eine Statistik, die diesen Typ der Asymmetrie
wiedergibt, ist die Schiefe. Gleichung 1 beschreibt den Schiefe-Index,
Six, als ein Maß für diese Asymmetrie mathematisch.
-
-
Der
Wert, TMax, wird definiert als die Temperatur
der größten Gewichtsfraktion,
die zwischen 50 und 90 °C
in der TREF-Kurve eluiert. Ti und wi sind die Elutionstemperatur bzw. Gewichtsfraktion
einer beliebigen i-ten Fraktion in der TREF-Verteilung. Die Verteilungen
wurden normalisiert (die Summe der wi ist
gleich 100 %) im Hinblick auf die Gesamtfläche der Kurve, die über 30 °C eluiert.
Der Index gibt also nur die Form des kristallisierten Polymers wieder,
und irgendwelches unkristallisiertes Polymer (Polymer, das bei oder
unter 30 °C
immer noch in Lösung
ist) wurde aus der Berechnung, die in Gleichung 1 gezeigt wird,
weggelassen.
-
Polymerdefinitionen
und Beschreibungen
-
„Polymer" meint eine makromolekulare
Verbindung, die durch Polymerisation von Monomeren des gleichen
oder verschiedenen Typs hergestellt wird. „Polymer" beinhaltet Homopolymere, Copolymere,
Terpolymere, Interpolymere und so weiter. Der Ausdruck „Interpolymer" meint ein Polymer,
das durch die Polymerisation von mindestens zwei Arten von Monomeren
oder Comonomeren hergestellt wird. Er beinhaltet, ist aber nicht
darauf beschränkt,
Copolymere (die sich gewöhnlicherweise
auf Polymere beziehen, die aus zwei verschiedenen Arten von Monomeren
oder Comonomeren hergestellt werden, obwohl sie oft synonym mit „Interpolymer" verwendet werden,
die sich auf Polymere beziehen, die aus drei oder mehr verschiedenen
Typen von Monomeren oder Comonomeren hergestellt werden), Terpolymere
(die sich gewöhnlicherweise
auf Polymere beziehen, die aus drei verschiedenen Typen von Monomeren
oder Comonomeren hergestellt werden), Tetrapolymeren (die sich gewöhnlicherweise
auf Polymere beziehen, die aus vier verschiedenen Typen von Monomeren
oder Comonomeren hergestellt werden) und dergleichen. Die Ausdrücke „Monomer" oder „Comonomer" werden synonym verwendet
und beziehen sich auf jegliche Verbindung mit einer polymerisierbaren
Gruppe, die in einen Reaktor gegeben wird, um ein Polymer herzustellen.
In diesen Umständen,
in denen ein Polymer dadurch beschrieben wird, dass es eine oder mehrere
Monomere umfasst, zum Beispiel ein Polymer, das Propylen und Ethylen
umfasst, umfasst das Polymer natürlich
Einheiten, die aus den Monomeren, stammen, zum Beispiel -CH2-CH2-, und nicht
das Monomer selbst, zum Beispiel CH2=CH2.
-
"Metallocen-katalysiertes
Polymer" oder ein ähnlicher
Ausdruck meint irgendein Polymer, das in der Anwesenheit eines Metallocenkatalysators
hergestellt wird. „Polymer
katalysiert mit einem Katalysator mit gezwungener Geometrie", „CGC-katalysiertes Polymer" oder ein ähnlicher
Ausdruck meint irgendein Polymer, das in der Anwesenheit eines Katalysators
mit gezwungener Geometrie hergestellt wird. „Ziegler-Natta-katalysiertes Polymer", „Z-N-katalysiertes
Polymer" oder ein ähnlicher
Ausdruck meint irgendein Polymer, das in der Anwesenheit eines Ziegler-Natta-Katalysators
hergestellt wird. „Metallocen" meint eine Metall-enthaltende Verbindung
mit mindestens einer substituierten oder unsubstituierten Cyclopentadienylgruppe,
die an das Metall gebunden ist. „Katalysator mit gezwungener
Geometrie" oder „CGC", wie hier verwendet,
hat die gleiche Bedeutung wie der Ausdruck in USP 5,272,236 und
USP 5,278,272 definiert und beschrieben ist.
-
„Statistisches
Copolymer" meint
ein Copolymer, in dem das Monomer zufällig über die Polymerkette verteilt
ist.
-
„Propylen-Homopolymer" und ähnliche
Ausdrücke
meinen ein Polymer, das allein oder im Wesentlichen allein aus Einheiten
besteht, die aus Propylen stammen. „Palypropylen-Copolymer" und ähnliche
Ausdrücke
meinen ein Polymer umfassend Einheiten, die aus Propylen und Ethylen
und/oder einem oder mehreren ungesättigten Comonomeren stammen.
Der Ausdruck „Copolymer" beinhaltet Terpolymere,
Tetrapolymere etc.
-
Die
ungesättigten
Comonomere, die in der Praxis dieser Erfindung verwendet werden,
beinhalten C4-20-α-Olefine, besonders C4-12-α-Olefine,
wie zum Beispiel 1-Buten, 1-Penten,
1-Hexen, 4-Methyl-1-penten, 1-Hepten, 1-Octen, 1-Decen, 1-Dodecen
und dergleichen; C4-20-Diolefine, bevorzugt
1,3-Butadien, 1,3-Pentadien, Norbornadien, 5-Ethyliden-2-norbornen (ENB) und Dicyclopentadien;
C8-40-vinylaromatische Verbindungen beinhalten
Styrol, o-, m- und p-Methylstyrol, Divinylbenzol, Vinylbiphenyl,
Vinylnaphthalin; und Halogen-substituierte C8-40-vinylaromatische Verbindungen,
wie zum Beispiel Chlorstyrol und Fluorstyrol. Zum Zwecke dieser
Erfindung sind Ethylen und Propylen nicht in der Definition von
ungesättigten
Comonomeren beinhaltet.
-
Die
Propylen-Copolymere, die in der Praxis dieser Erfindung verwendet
werden, umfassen typischerweise Einheiten, die aus Propylen stammen,
in einer Menge von mindestens 60, bevorzugt mindestens 80 und stärker bevorzugt
mindestens 85 Gew.-% bezogen auf das Copolymer. Die typische Menge
an Einheiten in Propylen/Ethylen-Copolymeren,
die aus Ethylen stammen, ist mindestens 0,1, bevorzugt mindestens
1 und stärker
bevorzugt mindestens 5 Gew.-%, und die maximale Menge an Einheiten,
die aus Ethylen stammen und in diesen Copolymeren zugegen sind, überschreitet
typischerweise nicht 35, überschreitet
bevorzugt nicht 30 und überschreitet
stärker
bevorzugt nicht 20 Gew.-% bezogen auf das Copolymer. Die Menge an
Einheiten, die aus dem/den ungesättigten
Comonomer(en) stammen, ist, wenn überhaupt zugegen, mindestens
0,01, bevorzugt mindestens 1 und stärker bevorzugt mindestens 5
Gew.-%, und die typische maximale Menge an Einheiten, die aus dem/den
ungesättigten
Comonomer(en) stammen, überschreitet
typischerweise nicht 35, überschreitet
bevorzugt nicht 30 und überschreitet
stärker
bevorzugt nicht 20 Gew.-% bezogen auf das Copolymer. Die kombinierte
Gesamtmenge an Einheiten, die aus Ethylen und irgendeinem ungesättigten
Comonomer stammt, überschreitet
typischerweise nicht 40, überschreitet
bevorzugt nicht 30 und überschreitet
stärker
bevorzugt nicht 20 Gew.-% bezogen auf das Copolymer.
-
Die
Copolymere, die in der Praxis dieser Erfindung verwendet werden
und Propylen und ein oder mehrere ungesättigte Comonomere (abweichend
von Ethylen) umfassen, umfassen typischerweise auch Einheiten, die
aus Propylen stammen, in einer Menge von mindestens 60, bevorzugt
mindestens 70 und stärker
bevorzugt mindestens 80 Gew.-% bezogen auf das Copolymer. Das/die
eine oder mehreren ungesättigte(n)
Comonomer(e) des Copolymers umfassen mindestens 0,1, bevorzugt mindestens
1 und stärker
bevorzugt mindestens 3 Gewichtsprozent, und die typische maximale
Menge an ungesättigtem
Comonomer überschreitet nicht
40, und überschreitet
bevorzugt bevorzugt nicht 30 Gew.-% bezogen auf das Copolymer.
-
13C
NMR
-
Die
P/E*-Polymere, die in der Praxis dieser Erfindung verwendet werden,
sind ferner dadurch gekennzeichnet, dass sie im Wesentlichen isotaktische
Propylensequenzen haben. „Im
Wesentlichen isotaktische Propylensequenzen" und ähnliche Ausdrücke meinen,
dass die Sequenzen eine isotaktische Triade (mm), gemessen mittels 13CNMR, von größer als 0,85, bevorzugt größer als
0,90, stärker
bevorzugt größer als
0,92 und am stärksten
bevorzugt größer als
0,93 haben. Isotaktische Triaden sind wohl bekannt im Fachgebiet
und sind zum Beispiel in USP 5,504,172 und WO 00/01745 beschrieben,
die sich auf die isotaktische Sequenzen im Sinne von einer Triadeneinheit
in der molekularen Copolymerkette beziehen, bestimmt mittels 13C NMR-Spektren. Die NMR-Spektren werden
wie folgt bestimmt.
-
13C NMR-Spektroskopie ist eine von einer
Vielzahl von Techniken, die im Fachgebiet für die Messung von Comonomereinbau
in ein Polymer bekannt sind. Ein Beispiel für diese Technik zur Bestimmung
von Comonomergehalt von Ethylen/α-Olefin-Copolymeren ist bei
Randall (Journal of Macromolecular Science, Reviews in Macromolecular
Chemistry and Physics, C29 (2 & 3),
201 – 317
(1989)) beschrieben. Das normale Vorgehen zur Bestimmung des Comonomergehalts
von einem Olefininterpolymer umfasst Aufnahme des 13C NMR-Spektrums
unter Bedingungen, bei denen die Intensität der Peaks, die den verschiedenen
Kohlenstoffen in der Probe entsprechen, direkt proportional zu der
Gesamtzahl von beitragenden Kernen in der Probe ist. Verfahren,
um diese Proportionalität
sicherzustellen, sind im Fachgebiet bekannt und müssen genügend Zeit zur
Relaxation nach einem Puls lassen, und umfassen die Verwendung von
gepulsten Entkopplungstechniken, Relaxationsreagenzien und dergleichen.
Die relative Intensität
von einem Peak oder einer Gruppe von Peaks wird in der Praxis von
einem Computer-erzeugten Integral erhalten. Nach Erhalten des Spektrums
und Integrieren der Peaks werden die Peaks, die mit dem Comonomer
in Verbindung stehen, zugeordnet. Die Zuordnung kann unter Bezugnahme
auf bekannte Spektren oder auf Literatur, oder durch Synthese und
Analyse von Modellverbindungen, oder durch die Verwendung von Isotopenmarkiertem
Comonomer gemacht werden. Der molare Anteil an Comonomer kann über das
Verhältnis
der Integrale, die der Zahl von Molen des Comonomers entsprechen,
zu den Integralen, die der Anzahl von Molen aller Monomere in dem
Interpolymer entsprechen, bestimmt werden, wie zum Beispiel bei
Randall beschrieben.
-
Die
Daten werden mittels eines Varian UNITY Plus 400 MHz-NMR-Spektrometers
entsprechend einer 13C-Resonanzfrequenz
von 100,4 MHz erfasst. Erfassungsparameter werden ausgewählt, um
quantitative 13C-Datenerfassung in der Gegenwart
von dem Relaxationsreagenz sicherzustellen. Die Daten werden mittels gepulster 1H-Entkopplung, 4000 Transienten pro Datendatei,
einem 7 Sek.-Pulsrepetitionsdelay,
Spektralbreite von 24,200 Hz und einer Dateigröße von 32K Datenpunkten erfasst,
wobei der Probenkopf auf 130 °C
erhitzt wird. Die Probe wird durch Zugabe von annähernd 3
ml einer 50/50-Mischung von Tetrachlorethand2/Orthodichlorbenzol,
das heißt
0,025 M in Chromacetylacetonat (Relaxationsreagenz) zu 0,4 g Probe
in ein 10 mm NMR-Röhrchen
hergestellt. Der Gasraum des Röhrchens
wird durch Verdrängung
mit reinem Stickstoff von Sauerstoff befreit. Die Probe wird gelöst und homogenisiert,
indem das Röhrchen
und dessen Inhalte auf 150 °C
unter periodischem Rückfluss
aufgeheizt werden, initiiert durch eine Heissluftpistole.
-
Nach
Erfassen der Daten werden die chemischen Verschiebungen intern auf
die mmmm-Pentade bei 21,90 ppm bezogen.
-
Für Propylen/Ethylen-Copolymere
wird das folgende Verfahren verwendet, um den Anteil an Ethylen in
dem Polymer zu berechnen. Integrale Regionen werden wie folgt bestimmt:
-
Tabelle
D: Integrale Region zur Bestimmung von % Ethylen
-
Region
D wird berechnet als D = P × (G × Q)/2.
Region E = R + Q + (G × Q)/2.
-
Tabelle E: Berechnung
der Region D
-
- PPP = (F + A – 0,5
D)/2
- PPE = D
- EPE = C
- EEE = (E – 0,5
G)/2
- PEE = G
- PEP = H
- Mole P = Summe der P-zentrierten Triaden
- Mole E = Summe der E-zentrierten Triaden
- Mole P = (B + 2A)/2
- Mole E = (E + G + 0,5B + H)/2
-
C2-Werte
werden aus dem Durchschnitt der zwei obigen Verfahren (Triadensummation
und algebraisch) berechnet, obwohl die zwei gewöhnlich nicht variieren.
-
Die
Molfraktion von Propyleninsertionen, die in Regiofehlern resultiert,
wird berechnet als die halbe Summe der zwei Methyl-Gruppen, die
bei 14.6 und 15,7 ppm auftreten, geteilt durch die Zahl der Methyl-Gruppen
bei 14-22 ppm, die zu Propylen gerechnet werden können. Die
Molprozent der Regiofehler-Peaks ist die Molfraktion mal 100.
-
Isotaktizität auf dem
Triadenlevel (mm) wird bestimmt aus den Integralen der mm-Triade (22,70-21,28 ppm),
der mr-Triade (21,28-20,67 ppm) und der rr-Triade (20,67-19,74). Die mm-Isotaktizität wird bestimmt durch
Teilen der Intensität
der mm-Triade durch die Summe der mm-, mr- und rr-Triaden. Bei Ethylen-Copolymeren
wird die mr-Region
korrigiert, indem das Integral zwischen 37,5-39 ppm abgezogen wird.
Für Copolymere
mit anderen Monomeren, die Peaks in den Regionen von den mm-, mr- und rr-Triaden erzeugen,
werden die Integrale für
diese Regionen auf ähnliche
Weise durch Abziehen der Intensität des störenden Peaks mittels Standard-NMR-Techniken
korrigiert, wenn die Peaks einmal identifiziert worden sind. Das
kann zum Beispiel erreicht werden durch Analyse einer Serie von
Copolymeren mit verschiedenen Levels an Monomereinbau, durch Literaturarbeit,
durch isotope Markierung oder andere Mittel, die im Fachgebiet bekannt
sind.
-
Die 13C NMR-Peaks, die einem Regiofehler bei
14,6 und 15,7 ppm entsprechen, werden für das Ergebnis von stereoselektiven
2,1-Insertionsfehlern von Propyleneinheiten in die wachsende Polymerkette
gehalten. In einem typischen P/E*-Polymer haben diese Peaks etwa die gleiche
Intensität
und sie repräsentieren 0,02
bis 7 Molprozent der Propyleninsertionen in die Homopolymer- oder
Copolymerkette. Für
einige Ausführungsformen
repräsentieren
sie 0,005 bis 20 Mol-% oder mehr der Propyleninsertionen. Im Allgemeinen
führen höhere Level
an Regiofehlern zu einer Erniedrigung des Schmelzpunkts und des
Moduls des Polymers, während
geringere Level zu einem höheren
Schmelzpunkt und einem höheren
Modul des Polymers führen.
-
Die
Beschaffenheit der und das Level an Comonomeren abweichend von Propylen
steuern auch den Schmelzpunkt und den Modul des Copolymers. In irgendeiner
besonderen Anwendung kann es wünschenswert
sein, entweder einen hohen oder einen niedrigen Schmelzpunkt oder
einen hohen Modul oder einen niedrigen Modul zu haben. Das Level
an Regiofehlern kann durch mehrere Mittel gesteuert werden, beinhaltend die
Polymerisationstemperatur, die Konzentration von Propylen und anderen
Monomeren in dem Verfahren, die Art der (Co)Monomere und andere
Faktoren. Verschiedene individuelle Katalysatorstrukturen können von sich
aus inhärent
mehr oder weniger Regiofehler als andere Katalysatoren erzeugen.
Zum Beispiel hat das Propylen-Homopolymer, das mit Katalysator G
hergestellt wurde, in der obigen Tabelle A, einen höheren Grad an
Regiofehler und einen niedrigeren Schmelzpunkt als das Propylen-Homopolymer,
das mit Katalysator H hergestellt wurde und einen höheren Schmelzpunkt
hat. Falls ein Polymer mit höherem
Schmelzpunkt (oder höherem
Modul) gewünscht
wird, dann ist es bevorzugt, dass es weniger als 3 Mol-% der Propyleninsertionen Regiofehler
aufweist, stärker
bevorzugt weniger als 1,5 Mol-% der Propyleninsertionen, und noch
mehr bevorzugt weniger als 1,0 Mol-% der Propyleninsertionen und
am stärksten
bevorzugt weniger als 0,5 Mol-% der Propyleninsertionen. Falls ein
Polymer mit einem niedrigeren Schmelzpunkt (oder Modul) gewünscht wird, dann
ist es bevorzugt, dass es mehr als 3 Mol-% der Propyleninsertionen
Regiofehler aufweist, stärker
bevorzugt mehr als 5 Mol-% der Propyleninsertionen, noch stärker bevorzugt
mehr als 6 Mol-% der Propyleninsertionen, und am meisten bevorzugt
mehr als 10 Mol-% der Propyleninsertionen.
-
Der
Fachmann wird verstehen, dass der Mol-Anteil an Regiofehlern für ein P/E*-Polymer, das eine Komponente
einer Mischung ist, sich auf den Mol-Anteil an Regiofehler der speziellen
P/E*-Polymerkomponente der Mischung bezieht, und nicht auf einen
Mol-Anteil der gesamten Mischung.
-
Der
Vergleich von mehreren 13C NMR-Spektren
veranschaulicht ferner die einzigartigen Regiofehler des P/E*-Polymers. 6 ist
das 13C NMR-Spektrum des Propylen-Ethylen-Copolymers
aus Beispiel 2 und es zeigt einen hohen Grad an Isotaktizität und die
gleichen Regiofehler des Propylen-Homopolymers aus 7. Die
Anwesenheit des Ethylen-Comonomers schließt das Auftreten von diesen
einzigartigen Regiofehlern nicht aus. Das 13C
NMR-Spektrum in 7 ist das des Propylen-Ethylen-Copolymer-Produkts von Vergleichsbeispiel
1, das mittels eines Metallocenkatalysators hergestellt wurde. Dieses
Spektrum zeigt nicht den Regiofehler (bei etwa 15 ppm), der charakteristisch
ist für
die P/E*-Polymere.
-
Schmelzflussrate (MFR)
-
Die
Propylen-Homo- und Copolymere, die in der Praxis dieser Erfindung
verwendet werden, weisen typischerweise ein MFR von mindestens 1,
bevorzugt von mindestens 2, stärker
bevorzugt von mindestens 5 und am stärksten bevorzugt von mindestens
10 auf. Das maximale MFR übersteigt
typischerweise nicht 1.500, bevorzugt übersteigt es nicht 1000 und
stärker
bevorzugt übersteigt
es nicht 400, stärker
bevorzugt übersteigt es
nicht 1000 und am stärksten
bevorzugt übersteigt
es nicht 50. Das MFR für
Propylen-Homopolymere und Copolymere aus Propylen und Ethylen und/oder
einem oder mehreren C4-C20-α-Olefinen
wird gemäß ASTM D-1238,
Bedingung L (2,16 kg, 230 Grad C) gemessen.
-
Propylen-Copolymere
-
Die
Propylencopolymere von besonderem Interesse, die in der Praxis dieser
Erfindung verwendet werden, beinhalten Propylen/Ethylen, Propylen/1-Buten,
Propylen/1-Hexen, Propylen/4-Methyl-1-penten, Propylen/1-Octen,
Propylen/Ethylen/1-Buten, Propylen/Ethylen/ENB, Propylen/Ethylen/1-Hexen,
Propylen/Ethylen/1-Octen, Propylen/Styrol und Propylen/Ethylen/Styrol.
-
Katalysatordefinitionen
und -beschreibungen
-
Die
P/E*-Polymere, die in der Praxis dieser Erfindung verwendet werden,
werden mittels eines Metall-zentrierten Katalysators mit Heteroaryl-Ligand
in Kombination mit einem oder mehreren Aktivatoren, z.B. einem Alumoxan,
hergestellt. In gewissen Ausführungsformen
ist das Metall eines oder mehrere von Hafnium und Zircon.
-
Genauer
gesagt wurde in gewissen Ausführungsformen
von Katalysatoren gefunden, dass die Verwendung von einem Hafniummetall
im Vergleich zu einem Zirconmetall für Katalysatoren mit Heteroaryl-Liganden
bevorzugt ist. Ein breiter Bereich von zusätzlichen Ligandsubstituenten
kann die erhöhte
katalytische Leistung begünstigen.
Die Katalysatoren sind in gewissen Ausführungsformen Zusammensetzungen,
umfassend den Liganden und Metallprecursor, und können ggf.
zusätzlich
einen Aktivator, Kombination von Aktivatoren oder ein Aktivatorpaket
beinhalten.
-
Die
Katalysatoren, die verwendet werden, um P/E*-Polymere herzustellen,
beinhalten zusätzlich
Katalysatoren, umfassend zusätzliche
Ligand-Hafnium-Komplexe, zusätzliche
Ligand-Zircon-Komplexe und ggf. Aktivatoren, die die Polymensations-
und Copolymerisations-Reaktionen, besonders mit Monomeren wie Olefinen,
Diolefinen oder anderen ungesättigten
Verbindungen, katalysieren. Zirconkomplexe, Hafniumkomplexe, Zusammensetzungen
oder Verbindungen, die die offenbarten Liganden verwenden, sind
innerhalb des breiten Bereichs der Katalysatoren, die nützlich in
der Praxis dieser Erfindung sind. Die Metall-Ligand-Komplexe können in
neutralem oder geladenem Zustand vorliegen. Das Ligand-zu-Metall-Verhältnis kann
auch variieren, wobei das exakte Verhältnis abhängt von der Art des Liganden
und des Metall-Ligandkomplexes. Der Metall-Ligandkomplex oder die
Komplexe können
verschiedene Formen annehmen, zum Beispiel können sie monomer, dimer oder
von noch höherer
Ordnung sein.
-
„Nicht-Metallocen" meint, dass das
Metall des Katalysators nicht an einen substituierten oder unsubstituierten
Cyclopentadienylring gebunden ist. Representative Nicht-Metallocen,
metallzentrierte Katalysatoren mit Heteroaryl-Ligand werden in den
vorläufigen
U.S.-Patentanmeldungen Nr. 60/246,781, eingereicht am 7. November
2000 und 60/301,666, eingereicht am 28. Juni 2001, beschrieben.
-
„Nicht-Metallocen,
metall-zentrierter Katalysator mit Heteroaryl-Ligand", wie hier verwendet,
meint, dass der Katalysator von dem Liganden stammt, der in Formel
I beschrieben ist. „Heteroaryl", wie er in diesem Ausdruck
verwendet wird, beinhaltet substituiertes Heteroaryl.
-
Der
Ausdruck „charaktensiert
durch die Formel",
wie er hier verwendet wird, soll nicht einschränkend wirken und wird in der
gleichen Weise verwendet wie „umfassend" gemeinhin verwendet
wird. Der Ausdruck „unabhängig ausgewählt" wird hier verwendet,
um anzuzeigen, dass die R-Gruppen, z.B. R1,
R2, R3, R4 und R5 identisch
oder unterschiedlich sein können
(z.B. können
R1, R2, R3, R4 und R5 alle substituierte Alkyle sein oder R1 und R2 können ein
substituiertes Alkyl sein und R3 kann ein
Aryl sein etc.). Verwendung des Singulars beinhaltet die Verwendung
des Plurals und umgekehrt (z.B. beinhaltet ein Hexanlösungsmittel
Hexane). Eine R genannte Gruppe hat im Allgemeinen die Struktur,
wie sie R-Gruppen mit diesem Namen im Fachgebiet bekanntermaßen entsprechen.
Die Ausdrücke „Verbindung" und „Komplex" sind im Allgemeinen
synonym verwendet in der Spezifikation, allerdings können Fachleute
gewisse Verbindungen als Komplexe und umgekehrt erkennen. Zum Zwecke
der Darstellung werden gewisse repräsentative Gruppen hierin definiert.
Diese Definitionen sollen zur Unterstützung und Darstellung sein
und nicht die Definitionen, wie sie dem Fachmann bekannt sind, ausschließen.
-
„Hydrocarbyl" bezieht sich auf
univalente Hydrocarbylreste enthaltend 1 bis 30 Kohlenstoffatome,
bevorzugt 1 bis 24 Kohlenstoffatome, am stärksten bevorzugt 1 bis 12 Kohlenstoffatome
beinhaltend verzweigte oder unverzweigte gesättigte oder ungesättigte Spezies,
wie zum Beispiel Alkylgruppen, Alkenylgruppen, Arylgruppen und dergleichen. „Substituiertes
Hydrocarbyl" bezieht
sich auf Hydrocarbyl, das mit einer oder mehreren Substituenten-Gruppen
substituiert ist und die Ausdrücke „Heteroatomenthaltendes
Hydrocarbyl" und „Heterohydrocarbyl" bezieht sich auf
Hydrocarbyl, in dem mindestens ein Kohlenstoffatom durch ein Heteroatom
ersetzt ist.
-
Der
Ausdruck „Alkyl", bezieht sich hierin
auf einen verzweigten oder unverzweigten, gesättigten oder ungesättigten,
azyklischen Kohlenwasserstoffrest. Geeignete Alkylreste beinhalten
zum Beispiel Methyl, Ethyl, n-Propyl, i-Propyl, 2-Propenyl (oder
Allyl), Vinyl, n-Butyl, t-Butyl, i-Butyl (oder 2-Methylpropyl) etc.
In besonderen Ausführungsformen
weisen die Alkyle zwischen 1 und 200 Kohlenstoffatome, zwischen
1 und 50 Kohlenstoffatome oder zwischen 1 und 20 Kohlenstoffatome
auf.
-
„Substituiertes
Alkyl" bezieht sich
auf ein Alkyl wie gerade beschrieben, in dem ein oder mehrere Wasserstoffatome,
die an irgendeinen Kohlenstoff des Alkyls gebunden sind, durch eine
andere Gruppe ersetzt sind, wie zum Beispiel ein Halogen, Aryl,
substituiertes Aryl, Cycloalkyl, substituiertes Cycloalkyl, Heterocycloalkyl,
substituiertes Heterocycloalkyl, Halogen, Alkylhalogene (z.B. CF3), Hydroxy, Amino, Phosphido, Alkoxy, Amino,
Thio, Nitro und Kombinationen davon. Geeignete substituierte Alkyle
beinhalten zum Beispiel Benzyl, Trifluormethyl und dergleichen.
-
Der
Ausdruck „Heteroalkyl" bezieht sich auf
ein Alkyl wie oben beschrieben, in dem ein oder mehrere Kohlenstoffatome
an irgendeinem Kohlenstoff des Alkyls durch ein Heteroatom ersetzt
sind, ausgewählt
aus der Gruppe bestehend aus N, O, P, B, S, Si, Sb, Al, Sn, As,
Se und Ge. Diese gleiche Liste an Heteroatomen ist für diese
ganze Beschreibung hindurch verwendbar. Die Bindung zwischen dem
Kohlenstoffatom und dem Heteroatom kann gesättigt oder ungesättigt sein.
Ein Alkyl substituiert mit einem Heterocycloalkyl, substituiertem
Heterocycloalkyl, Heteroaryl, substituiertem Heteroaryl, Alkoxy,
Aryloxy, Boryl, Phospino, Amino, Silyl, Thio oder Seleno, ist also
innerhalb des Schutzbereichs des Ausdrucks Heteroalkyl. Geeignete
Heteroalkyle beinhalten Cyano, Benzoyl, 2-Pyridyl, 2-Furyl und dergleichen.
-
Der
Ausdruck „Cycloalkyl", bezieht sich hierin
auf einen gesättigten
oder ungesättigten
zyklischen, nicht-aromatischen Kohlenwasserstoffrest mit einem einzelnen
Ring oder vielfach kondensierten Ringen. Geeignete Cykloalkylradikale
beinhalten zum Beispiel Cyclopentyl, Cyclohexyl, Cyclooctenyl, Bicyclooctyl
etc. In besonderen Ausführungsformen
weisen die Cycloalkyle zwischen 3 und 200 Kohlenstoffatome, zwischen
3 und 50 Kohlenstoffatome oder zwischen 3 und 20 Kohlenstoffatome
auf.
-
„Substituiertes
Cycloalkyl" bezieht
sich auf Cycloalkyl, wie gerade beschrieben, beinhaltend Cycloalkyle,
in denen ein oder mehrere Wasserstoffatome an irgendeinem Kohlenstoffatom
des Cycloalkyls durch eine andere Gruppe ersetzt sind, wie zum Beispiel
ein Halogenalkyl, substituiertes Alkyl, Aryl, substituiertes Aryl, Cycloalkyl,
substituiertes Cycloalkyl, Heterocycloalkyl, substituiertes Heterocycloalkyl,
Heteroaryl, substituiertes Heteroaryl, Alkoxy, Aryloxy, Boryl, Phosphino,
Amino, Silyl, Thio, Seleno und Kombinationen davon. Geeignete substituierte
Cycloalkylreste beinhalten zum Beispiel 4-Dimethylaminocyclohexyl,
4,5-Dibromocyclohept-4-enyl und dergleichen.
-
Der
Ausdruck „Heterocycloalkyl", bezieht sich hierin
auf einen Cycloalkylrest, wie beschrieben, allerdings einen, in
dem ein oder mehrere oder alle Kohlenstoffatome des gesättigten
oder ungesättigten
zyklischen Rests ersetzt sind durch ein Heteroatom, wie zum Beispiel
Stickstoff, Phosphor, Sauerstoff, Schwefel, Silicium, Germanium,
Selen oder Bor. Geeignete Heterocycloalkyle beinhalten zum Beispiel
Piperazinyl, Morpholinyl, Tetrahydropyranyl, Tetrahydrofuranyl,
Piperidinyl, Pyrrolidinyl, Oxazolinyl und dergleichen.
-
„Substituiertes
Heterocycloalkyl" bezieht
sich auf Heterocycloalkyl wie gerade beschrieben, beinhaltend Heterocycloalkyl,
in dem ein oder mehrere Wasserstoffatome an irgendeinem Atom des
Heterocycloalkyls durch eine andere Gruppe ersetzt wird, wie zum
Beispiel ein Halogen, Alkyl, substituiertes Alkyl, Aryl, substituiertes
Aryl, Heteroaryl, substituiertes Heteroaryl, Alkoxy, Aryloxy, Boryl,
Phosphino, Amino, Silyl, Thio, Seleno und Kombinationen davon. Geeignete
substituierte Heterocycloalkylreste beinhalten zum Beispiel N-Methylpiperazinyl,
3-Dimethylaminomorpholinyl und dergleichen.
-
Der
Ausdruck „Aryl", bezieht sich hierin
auf einen aromatischen Substituenten, der aus einem einzelnen aromatischen
Ring oder mehreren aromatischen Ringen bestehen kann, die miteinander
verbunden sind, nämlich
kovalent verbunden sind oder verbunden sind über eine gemeinsame Gruppe,
wie zum Beispiel über eine
Methylen- oder Ethylengruppe.
Der/die aromatische(n) Ring(e) können
u.a. Phenyl, Naphthyl, Anthracenyl und Biphenyl beinhalten. In besonderen
Ausführungsformen
haben die Aryle zwischen 1 und 200 Kohlenstoffatome, zwischen 1
und 50 Kohlenstoffatome oder zwischen 1 und 20 Kohlenstoffatome.
-
„Substituiertes
Aryl" bezieht sich
auf Aryl, wie gerade beschrieben, in dem ein oder mehrere Wasserstoffatome,
die an irgendein Kohlenstoffatom gebunden sind, durch eine oder
mehrere funktionelle Gruppen ersetzt werden, wie zum Beispiel Alkyl,
substituiertes Alkyl, Cycloalkyl, substituiertes Cycloalkyl, Heterocycloalkyl,
substituiertes Heterocycloalkyl, Halogen, Alkylhalogen (z.B. CF3), Hydroxy, Amino, Phosphido, Alkoxy, Amino,
Thio, Nitro und sowohl gesättigte
als auch ungesättigte
zyklische Kohlenwasserstoffe, die mit den/dem aromatischen Ringen)
kovalent verbunden sind, nämlich
kovalent verbunden sind oder verbunden sind über eine gemeinsame Gruppe,
wie zum Beispiel einer Methylen- oder Ethylengruppe. Die gebräuchlichen
Verbindungsgruppen können
ein Carbonyl wie in Benzophenon, oder Sauerstoff wie in Diphenylether,
oder Stickstoff wie in Diphenylamin sein.
-
Der
Ausdruck „Heteroaryl", wie er hier verwendet
wird, bezieht sich auf aromatische oder ungesättigte Ringe, in denen ein
oder mehrere Kohlenstoffatome des/der aromatischen Rings/Ringe ersetzt
werden durch (ein) Heteroatom(e), wie zum Beispiel Stickstoff, Sauerstoff,
Bor, Selen, Phosphor, Silicium oder Schwefel. Heteroaryl bezieht
sich auf Strukturen, die ein einzelner aromatischer Ring, mehrere
aromatische Ring(e) oder einen oder mehrere aromatische Ringe, die
an einen oder mehrere nicht- aromatische
Ring(e) gekoppelt sind, sein können.
In Strukturen mit mehreren Ringen können die Ringe miteinander
verbunden sein, nämlich
kovalent verbunden sein oder verbunden sein über eine gemeinsame Gruppe,
wie zum Beispiel einer Methylen- oder Ethylengruppe. Die gemeinsame
Verbindungsgruppe kann auch ein Carbonyl, wie in Phenylpyridylketon sein.
Wie hierin verwendet, werden Ringe, wie zum Beispiel Thiophen, Pyridin,
Isoxazol, Pyrazol, Pyrrol, Furan etc. oder Benzo-verbundene Analoga
dieser Ringe durch den Ausdruck „Heteroaryl" definiert.
-
„Substituiertes
Heteroaryl" bezieht
sich auf Heteroaryl wie gerade beschrieben, beinhaltend Heteroaryl,
in dem ein oder mehrere Wasserstoffatome, die zu irgendeinem Atom
der Heteroarylgruppe gebunden sind, durch eine andere Gruppe ersetzt
sind, wie zum Beispiel ein Halogen, Alkyl, substituiertes Alkyl,
Aryl, substituiertes Aryl, Heteroaryl, substituiertes Heteroaryl,
Alkoxy, Aryloxy, Boryl, Phosphino, Amino, Silyl, Thio, Seleno und
Kombinationen davon. Geeignete substituierte Heteroarylreste beinhalten
zum Beispiel 4-N,N-Dimethylaminopyridin.
-
Der
Ausdruck „Alkoxy", bezieht sich hierin
auf den -OZ1-Rest, wobei Z1 ausgewählt ist
aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl,
substituiertem Cycloalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Silylgruppen und Kombinationen davon wie hierin beschrieben. Geeignete
Alkoxyreste beinhalten zum Beispiel Methoxy, Ethoxy, Benzyloxy,
t-Butoxy etc. Ein verwandter Ausdruck ist „Aryloxy", wobei Z1 ausgewählt ist
aus der Gruppe bestehend aus Aryl, substituiertem Aryl, Heteroaryl,
substituiertem Heteroaryl und Kombinationen davon. Beispiele für geeignete
Aryloxyreste beinhalten Phenoxy, substituiertes Phenoxy, 2-Pyridinoxy,
8-Quinalinoxy und
dergleichen.
-
Der
Ausdruck „Silyl", wie er hierin verwendet
wird, bezieht sich auf den -SiZ1Z2Z3-Rest, wobei jeweils Z1, Z2 und Z3 unabhängig
voneinander ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, Heterocycloalkyl, heterozyklischem Rest, Aryl,
substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxy,
Aryloxy, Amino, Silyl und Kombinationen davon.
-
Der
Ausdruck „Boryl", wie er hierin verwendet
wird, bezieht sich auf die -BZ1Z2-Gruppe, wobei jeweils Z1 und
Z2 unabhängig
voneinander ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, Heterocycloalkyl, heterozyklischem Rest, Aryl,
substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxy,
Aryloxy, Amino, Silyl und Kombinationen davon.
-
Der
Ausdruck „Phosphino", wie er hierin verwendet
wird, bezieht sich auf die Gruppe -PZ1Z2, wobei jeweils Z1 und
Z2 unabhängig
voneinander ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, substituiertem oder
unsubstituiertem Alkyl, Cycloalkyl, Heterocycloalkyl, heterozyklischem
Rest, Aryl, substituiertem Aryl, Heteroaryl, Silyl, Alkoxy, Aryloxy,
Amino und Kombinationen davon.
-
Der
Ausdruck „Phosphin", wie er hierin verwendet
wird, bezieht sich auf die Gruppe: PZ1Z2Z3, wobei jeweils
Z1, Z3 und Z2 unabhängig
voneinander ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, substituiertem oder
unsubstituiertem Alkyl, Cycloalkyl, Heterocycloalkyl, heterozyklischem
Rest, Aryl, substituiertem Aryl, Heteroaryl, Silyl, Alkoxy, Aryloxy,
Amino und Kombinationen davon.
-
Der
Ausdruck „Amino", bezieht sich hierin
auf die Gruppe: NZ1Z2,
wobei jeweils Z1 und Z2 unabhängig voneinander
ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl,
substituiertem Heteroaryl, Alkoxy, Aryloxy, Silyl und Kombinationen
davon.
-
Der
Ausdruck „Amin", bezieht sich hierin
auf die Gruppe: NZ1Z2Z3, wobei jeweils Z1,
Z2 und Z2 unabhängig voneinander
ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl (einschließlich Pyridine), substituiertem
Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxy, Aryloxy, Silyl
und Kombinationen davon.
-
Der
Ausdruck „Thio", bezieht sich hierin
auf die Gruppe -SZ1, wobei Z1 ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heterocycloalkyl, substituiertem
Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl, substituiertem
Heteroaryl, Alkoxy, Aryloxy, Silyl und Kombinationen davon.
-
Der
Ausdruck „Seleno", bezieht sich hierin
auf die Gruppe -SeZ1, wobei Z1 ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl,
substituiertem Heteroaryl, Alkoxy, Aryloxy, Silyl und Kombinationen
davon.
-
Der
Ausdruck „gesättigt" bezieht sich auf
den Mangel von Doppel- und Dreifachbindungen zwischen den Atomen
einer Restgruppe, wie zum Beispiel Ethyl, Cyclohexyl, Pyrrolidinyl
und dergleichen.
-
Der
Ausdruck „ungesättigt" bezieht sich auf
die Anwesenheit einer oder mehrerer Doppel- und/oder Dreifachbindungen
zwischen Atomen einer Restgruppe, wie zum Beispiel Vinyl, Acetylid,
Oxazolinyl, Cyclohexenyl, Acetyl und dergleichen.
-
Liganden
-
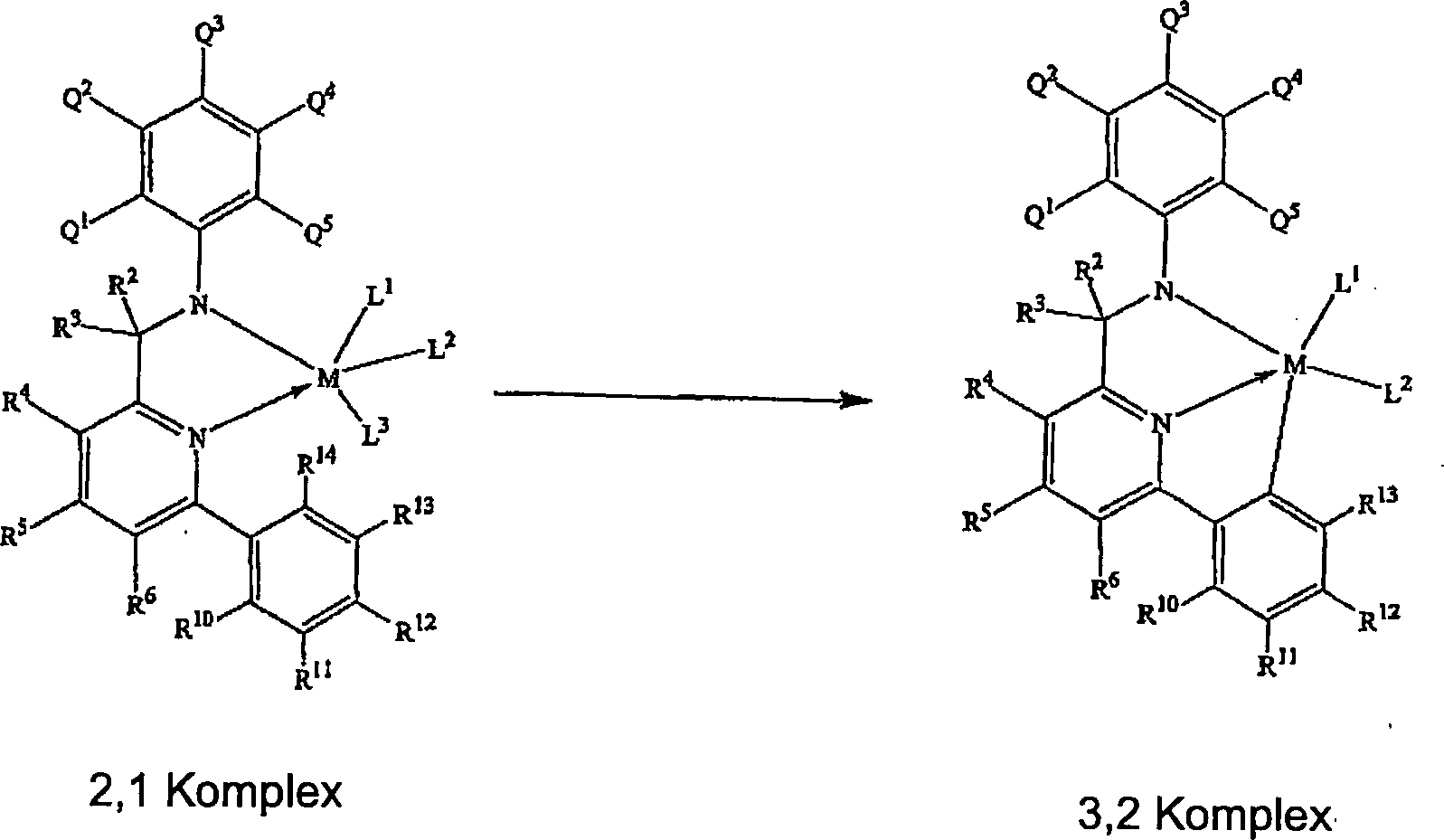
Geeignete
Liganden, die in den Katalysatoren, die verwendet werden um P/E*-Polymere herzustellen, nützlich sind
und in der Praxis dieser Erfindung verwendet werden, können in
breiter Form als monoanionische Liganden mit einem Amin und einer
Heteroaryl- oder substituierten Heteroarylgruppe charakterisiert
werden. Die Liganden dieser Katalysatoren werden zum Zwecke dieser
Erfindung als Nicht-Metallocen-Liganden
bezeichnet und können
durch die folgende allgemeine Formel charakterisiert werden:
wobei R
1 sehr
allgemein ausgewählt
ist aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem
Cycloalkyl, Heteroalkyl, substituiertem Heteroaryl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroalkyl,
substituiertem Heteroaryl und Kombinationen davon. In vielen Ausführungsformen
ist R
1 ein Ring mit von 4-8 Atomen in dem
Ring im Allgemeinen ausgewählt
aus der Gruppe bestehend aus substituiertem Cycloalkyl, substituiertem
Heterocycloalkyl, substituiertem Aryl und substituiertem Heteroaryl,
so dass R
1 durch die allgemeine Formel charakterisiert
werden kann:
wobei Q
1 und
Q
5 Ringsubstituenten orthoständig zu
Atom E sind, wobei E ausgewählt
ist aus der Gruppe bestehend aus Kohlenstoff und Stickstoff und
wobei mindestens einer von Q
1 oder Q
5 sperrig ist (dadurch definiert, dass sie
mindestens 2 Atome aufweisen). Q
1 und Q
5 sind unabhängig voneinander ausgewählt aus
der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl,
substituiertem Cycloalkyl, Aryl, substituiertem Aryl und Silyl,
allerdings unter der Voraussetzung, dass Q
1 und
Q
5 nicht beide Methyl sind. Q''
q repräsentiert zusätzliche
mögliche
Substituenten an dem Ring, wobei q 1, 2, 3, 4 oder 5 ist und Q'' ausgewählt ist aus der Gruppe bestehend
aus Wasserstoff, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem
Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl,
substituiertem Heteroaryl, Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino,
Amino, Thio, Seleno, Halogenid, Nitro und Kombinationen davon. T
ist eine Verbindungsgruppe ausgewählt aus der Gruppe bestehend
aus -CR
2R
3- und
-SiR
2R
3-, wobei
R
2 und R
3 unabhängig voneinander
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl,
Heterocycloalkyl, substituiertem Heterocycloalkyl, Aryl, substituiertem
Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxyl, Aryloxyl,
Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid, Nitro und
Kombinationen davon. J" ist
im Allgemeinen ausgewählt
aus der Gruppe bestehend aus Heteroaryl und substituiertem Heteroaryl,
wobei besondere Ausführungsformen
für besondere
Reaktionen hierin beschrieben sind.
-
In
einer spezifischeren Ausführungsform
können
geeignete Nicht-Metallocen-Liganden durch die folgende allgemeine
Formel charakterisiert werden:
wobei R
1 und
T wie oben definiert sind und jeweils R
4,
R
5, R
6 und R
7 unabhängig
voneinander ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno,
Halogenid, Nitro und Kombinationen davon. Gegebenenfalls können irgendwelche
Kombinationen aus R
1, R
2,
R
3 und R
4 miteinander
in einer Ringstruktur verbunden sein.
-
In
gewissen spezifischeren Ausführungsformen
können
die Liganden durch die folgende allgemeine Formel charakterisiert
werden:
wobei
Q
1, Q
5, R
2, R
3, R
4,
R
5, R
6 und R
7 alle wie oben definiert sind. Q
2, Q
3 und Q
4 sind unabhängig voneinander ausgewählt aus
der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl,
Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno,
Nitro und Kombinationen davon.
-
In
anderen spezifischeren Ausführungsformen
können
geeignete Liganden durch die folgende allgemeine Formel charakterisiert
werden:
wobei R
1,
R
2, R
3, R
4, R
5 und R
6 wie oben definiert sind. In dieser Ausführungsform
wurde der Substituent R
7 ersetzt durch eine
Aryl oder substituierte Arylgruppe, wobei R
10,
R
11, R
12 und R
13 unabhängig
voneinander ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Halogen, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno,
Nitro und Kombinationen davon; ggf. können zwei oder mehr R
10-, R
11-, R
12- und R
13-Gruppen
verbunden sein, um ein verbundenes Ringsystem mit von 3-50 Nicht-Wasserstoffatomen
zu bilden. R
14 ist ausgewählt aus
der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl,
Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid,
Nitro und Kombinationen davon.
-
In
noch spezifischeren Ausführungsformen
können
die Liganden durch die allgemeine Formel charaktensiert werden:
wobei R
2-R
6, R
10-R
14 und
Q
1-Q
5 alle wie oben
definiert sind.
-
In
gewissen Ausführungsformen
ist R2 bevorzugt Wasserstoff. Weiterhin
ist bevorzugt, wenn jeweils R4 und R5 Wasserstoff ist und R6 entweder
Wasserstoff ist oder mit R7 verbunden ist,
um ein verbundenes Ringsystem zu bilden. Auch ist es bevorzugt,
wenn R3 ausgewählt ist aus der Gruppe bestehend
aus Benzyl, Phenyl, 2-Biphenyl, t-Butyl, 2-Dimethylaminophenyl (2-(NMe2)-C6H4-), 2-Methoxyphenyl
(2-MeO-C6H4-), Anthracenyl,
Mesityl, 2-Pyridyl, 3,5-Dimethylphenyl, o-Tolyl, 9-Phenanthrenyl.
Es ist auch bevorzugt, wenn R1 ausgewählt ist
aus der Gruppe bestehend aus Mesityl, 4-Isopropylphenyl (4-Pri-C6H4-), Naphthyl,
3,5-(CF3)2-C6H3-, 2-Me-naphtyl,
2,6-(Pri)2-C6H3-, 2-Biphenyl,
2-Me-4-MeO-C6H3-; 2-But-C6H4-,
2,5-(But)2-C6H3-, 2-Pri-6-Me-C6H3-; 2-But-6-Me-C6H3-, 2,6-Et2-C6H3-, 2-sec-Butyl-6-Et-C6H3-. Es ist auch
bevorzugt, wenn R7 ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Phenyl, Naphthyl, Methyl,
Anthracenyl, 9-Phenanthrenyl,
Mesityl, 3,5-(CFa)2-C6H3-, 2-CF3-C6H4-, 4-CF3-C6H4-,
3,5-F2-C6H3-, 4-F-C6H4-, 2,4-F2-C6H3-,
4-(NMe2)-C6H4-, 3-MeO-C6H4-, 4-MeO-C6H4-, 3,5-Me2-C6H3-, o-Tolyl, 2,6-F2-C6H3-
oder wenn R7 zusammen mit R6 verbunden ist,
um ein verbundenes Ringsystem zu bilden, z.B. Chinolin.
-
Gegebenenfalls
können
auch zwei oder mehr R4-, R5-,
R6-, R7-Gruppen
verbunden sein, um ein verbundenes Ringsystem mit von 3-50 Nicht-Wasserstoffatomen
zusätzlich
zu dem Pyridinring zu bilden, z.B. um eine Chinolingruppe zu erzeugen.
In diesen Ausführungsformen
ist R3 ausgewählt aus der Gruppe bestehend aus
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
primären
und sekundären
Alkylgruppen und -PY2, wobei Y ausgewählt ist
aus der Gruppe bestehend aus Aryl, substituiertem Aryl, Heteroaryl
und substituiertem Heteroaryl.
-
Gegebenenfalls
können
innerhalb der obigen Formeln IV und V, R6 und
R10 verbunden sein, um ein Ringsystem mit
von 5-50 Nicht-Wasserstoffatomen zu bilden. Zum Beispiel hat der
Ring 5 Atome im Rückgrat des
Ringes, der mit anderen Atomen substituiert sein kann oder nicht,
wenn R6 und R10 zusammen
ein Methylen bilden. Der Ring hat zum Beispiel auch 6 Atome im Rückgrat des
Rings, der mit anderen Atomen substituiert sein kann oder nicht,
wenn R6 und R10 zusammen
ein Ethylen bilden. Ringsubstituenten können ausgewählt sein aus der Gruppe bestehend
aus Halogen, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem
Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl,
substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl,
substituiertem Heteroaryl, Alkoxy, Aryloxy, Silyl, Boryl, Phosphino,
Amino, Thio, Seleno, Nitro und Kombinationen davon.
-
In
gewissen Ausführungsformen
sind die Liganden neue Verbindungen. Ein Beispiel der neuen Ligandverbindungen
beinhaltet solche Verbindungen, die allgemein durch obige Formel
(III) charakterisiert sind, wobei R2 ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Aryl und substituiertem
Aryl; und R3 ein Phosphino ist, charakterisiert
durch die Formel -PZ1Z2,
wobei Z1 und Z2 jeweils
unabhängig
voneinander ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, substituiertem oder
unsubstituiertem Alkyl, Cycloalkyl, Heterocycloalkyl, heterozyklischem
Rest, Aryl, substituiertem Aryl, Heteroaryl, Silyl, Alkoxy, Aryloxy,
Amino und Kombinationen davon. Besonders bevorzugte Ausführungsformen
dieser Verbindungen beinhalten solche, wo Z1 und
Z2 jeweils unabhängig voneinander ausgewählt sind
aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl,
Heterocycloalkyl, Aryl und substituiertem Aryl; und spezifischer
Phenyl; wo Q1, Q3 und
Q5 jeweils ausgewählt ist aus der Gruppe bestehend
aus Alkyl und substituiertem Alkyl und jeweils Q2 und
Q4 ausgewählt ist aus Wasserstoff; und
wo R4, R5, R6 und R7 jeweils
Wasserstoff ist.
-
Die
Liganden können
hergestellt werden mittels bekannter Verfahren. Vergleiche zum Beispiel
Advanced Organic Chemistry, March, Wiley, New York 1992 (4. Auflage).
-
Spezifischerweise
können
die erfindungsgemäßen Liganden
mittels des zweistufigen Verfahrens, das in Schema 1 gezeigt ist,
hergestellt werden.
-
-
Schema 1
-
In
Schema 1 stellt * ein chirales Zentrum dar, wenn R
2 und
R
3 nicht identisch sind; die R-Gruppen haben
also die gleichen Definitionen wie oben. Im Allgemeinen ist R
3M
2 ein Nukleophil,
wie zum Beispiel ein Alkylierungs- oder Arylierungs- oder Hydrierungsreagenz,
und M
2 ist ein Metall, wie zum Beispiel
ein Hauptgruppenmetall, oder ein Metalloid, wie zum Beispiel Bor.
Das Alkylierungs-, Arylierungs- oder Hydrierungsreagenz kann ein
Grignard-, Alkyl-, Aryllithium- oder Borhydridreagenz sein. In Stufe
2 des Schemas 1 wird als erstes die Verwendung des Komplexierungsreagenz
eingesetzt. Bevorzugt wird, wie im Fall von Schema 1, Magnesiumbromid
als Komplexierungsreagenz verwendet. Die Rolle des Komplexierungsreagenzes
ist es, das Nukleophil, R
3M
2,
selektiv an den Iminkohlenstoff zu lenken. Wenn die Anwesenheit
von funktionellen Gruppen diesen synthetischen Ansatz verhindert,
können
alternative Synthesestrategien eingesetzt werden. Zum Beispiel können Liganden,
bei denen R
3 = Phosphino ist, gemäß den Lehren
von USP 6,034,240 und 6,043,363 hergestellt werden. Zusätzlich können in
Stufe 2 Tetraalkylhafnium-Verbindungen oder tetra-substituierte
Alkyl-Hafniumverbindungen oder Tetra-Arylhafnium-Verbindungen oder
tetra-substituierte Arylhafnium-Verbindungen eingesetzt werden,
gemäß den Lehren
von USP 6,103,657. Schema 2 beschreibt ferner ein Syntheseverfahren:
- *
= chirales Zentrum, wenn R2 nicht gleich
R3 ist
-
Schema 2
-
In
Schema 2 können
h = 1 oder 2 und die Bromionen an dem Magnesium gebunden sein oder
nicht. Der Effekt der Komplexierung liegt darin, den anschließenden nukleophilen
Angriff von R3M2 auf
den Iminkohlenstoff zu richten. Die Komplexierung kann also zu einer
selektiveren Reaktion führen,
wodurch die Ausbeute an gewünschten
zusätzlichen
Liganden gesteigert werden kann. Mittels dieser Technik ist die
Selektivität
im Allgemeinen größer als
50 %, stärker
bevorzugt größer als
70% und noch stärker
bevorzugt größer als
80 %. Die Komplexierung kann besonders nützlich sein zur Herstellung
von Arrays zusätzlicher
Liganden von der Art, wie sie in der Erfindung offenbart sind, wobei
R3 eine Variable in der Herstellung des
zusätzlichen
Ligandenarrays ist. Wie in Schema 2 durch * gezeigt wird, führt dieser
Ansatz auch zu der Bildung eines chiralen Zentrums bei den erfindungsgemäßen zusätzlichen
Liganden, wenn R2 und R3 unterschiedlich
sind. Unter manchen Umständen
kann R3M2 erfolgreich an das Imin in Abwesenheit
des Komplexierungsreagenzes addiert werden. Zusätzliche Liganden, die chiral
sind, können
für gewisse
Olefinpolymerisations-Reaktionen
wichtig sein, vor allem für
die, die zu einem stereospezifischen Polymer führen, vergleiche hierzu „Stereospecific
Olefin Polymerization with Chiral Metallocene Catalysts", Brintzinger, et
al., Angew. Chem. Int. Ed. Engl., 1995, Bd. 34, S. 1143-1170, und
die darin befindlichen Referenzen; Bercaw et al., J. Am. Chem. Soc.,
1999, Bd. 121, 564-573; und Bercaw et al., J. Am. Chem. Soc., 1996,
Bd. 118, 11988-11989.
-
In
der Praxis von High-Throughput-Verfahren oder kombinatorischer Materialwissenschaft
kann die Einführung
von Diversität
wichtig für
die Konzipierung von Bibliotheken oder Arrays sein. Die Syntheseschemata,
die hierin erörtert
werden, gestatten dem Fachmann, Diversität in Liganden einzuführen, was
helfen kann die Auswahl eines besonderen Liganden für eine besondere
Polymerisationsreaktion zu optimieren. Stufe 1 (vergleiche Schema
1) kann zum Beispiel mit irgendeiner Kombination aus Pyridinen und
Anilinen wie hier gezeigt, durchgeführt werden.
-
Zusammensetzungen
-
Ist
der gewünschte
Ligand einmal gebildet, kann er mit einem Metallatom, Ion, einer
Verbindung oder anderen Metallprecursor-Verbindung kombiniert werden.
In einigen Anwendungen werden die Liganden mit einer Metallverbindung
oder -precursor kombiniert und das Produkt einer solchen Kombination
wird nicht bestimmt, falls sich ein Produkt bildet. Zum Beispiel
kann der Ligand in einen Reaktionsgefäß gleichzeitig mit dem Metall
oder der Metallprecursorverbindung und den Reagenzien, Aktivatoren,
Fängern
etc. zugegeben werden. Der Ligand kann zusätzlich vor der Zugabe oder
nach der Zugabe des Metallprecursors, z.B. durch eine Deprotonierungsreaktion
oder irgendeine andere Modifikation, modifiziert werden.
-
In
den Formeln I, II, III, IV und V können die Metallprecursorverbindungen
durch die allgemeine Formel Hf(L)n charakterisiert
werden, wobei L unabhängig
ausgewählt
wird aus der Gruppe bestehend aus Halogeniden (F, Cl, Br, I), Alkyl,
substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl,
substituiertem Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Hydroxy, Boryl, Silyl, Amino, Amin, Hydrido, Allyl,
Dien, Seleno, Phosphino, Phosphin, Carboxylaten, Thio, 1,3-Dionaten,
Oxalaten, Carbonaten, Nitraten, Sulfaten und Kombinationen davon.
n ist 1, 2, 3, 4, 5 oder 6. Die Hafniumprecursor können monomer,
dimer oder von höheren
Ordnungen sein. Es ist wohl bekannt, dass Hafniummetall typischerweise
mit einer gewissen Menge an Zircon verunreinigt ist. Die Erfindung
verwendet also als reines Hafnium, ein kommerziell sinnvolles Hafnium.
Spezifische Beispiele geeigneter Hafniumprecursor beinhalten, sind
aber nicht darauf beschränkt,
HfCl4, Hf(CH2Ph)4, Hf(CH2CMe3)4, Hf(CH2SiMe3)4,
Hf (CH2Ph)3Cl, Hf(CH2CMe3)3Cl,
Hf(CH2SiMe3)3Cl, Hf(CH2Ph)2Cl2, Hf(CH2CMe3)2Cl2, Hf (CH2SiMe3)2Cl2,
Hf(NMe2)4, Hf(NEt2)4 und Hf(N(SiMe3)2)2Cl2. Lewis-Basen-Addukte dieser Beispiele sind
auch geeignet als Hafniumprecursor, zum Beispiel sind Ether, Amine,
Thioether, Phosphine und dergleichen geeignete Lewis-Basen.
-
In
den Formeln IV und V kann der Metallprecursor durch die allgemeine
Formel M(L)n charakterisiert werden, wobei
M Hafnium oder Zircon ist und L jeweils unabhängig voneinander ausgewählt ist
aus der Gruppe bestehend aus Halogenid (F, Cl, Br, I), Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Hydroxy, Boryl, Silyl, Amino, Amin, Hydrido, Allyl, Dien,
Seleno, Phosphino, Phosphin, Carboxylaten, Thio, 1,3-Dionaten, Oxalaten,
Carbonaten, Nitraten, Sulfaten und Kombinationen davon. n ist typischerweise
4. Es ist wohl bekannt, dass Hafniummetall typischerweise mit einer
gewissen Menge Zircon verunreinigt ist. In der Praxis wird also
reines Hafnium oder Zircon verwendet, wie es kommerziell sinnvoll
ist. Spezifische Beispiele von geeignetem Hafnium- und Zirconprecursor
beinhalten, sind aber nicht darauf beschränkt, HfCl4,
Hf(CH2Ph)4, Hf(CH2CMe3)4,
Hf(CH2SiMe3)4, Hf(CH2Ph)3Cl, Hf(CH2CMe3)3Cl, Hf(CH2SiMe3)3Cl,
Hf(CH2Ph)2Cl2, Hf(CH2CMe3)2Cl2,
Hf(CH2SiMe3)2Cl2, Hf(NMe2)4, Hf(NEt2)4 und Hf(N(SiMe3)2)2Cl2; ZrCl4, Zr(CH2Ph)4, Zr(CH2CMe3)4,
Zr (CH2SiMe3)4, Zr(CH2Ph)3Cl, Zr(CH2CMe3)3Cl, Zr(CH2SiMe3)3Cl,
Zr(CH2Ph)2Cl2, Zr (CH2CMe3)2Cl2,
Zr(CH2SiMe3)2Cl2, Zr(NMe2)4, Zr(NEt2)4, Zr(NMe2)2Cl2, Zr(NEt2)2Cl2 und
Zr(N(SiMe3)2)2Cl2. Lewis-Basen-Addukte
dieser Beispiele sind auch geeignet als Hafniumprecursor, zum Beispiel
sind Ether, Amine, Thioether, Phosphine und dergleichen geeignete
Lewis-Basen.
-
Das
Verhältnis
Ligand-zu-Metallprecursor-Verbindung liegt typischerweise im Bereich
von 0,01:1 bis 100:1, stärker
bevorzugt im Bereich von 0,1:1 bis 10:1.
-
Metall-Ligand-Komplexe
-
Im
Allgemeinen wird der Ligand mit einer geeigneten Metallprecursor-Verbindung
gemischt, vor oder gleichzeitig mit dem in Kontakt Bringen der Mischung
mit den Reagenzien (z.B. Monomeren). Wenn der Ligand mit der Metallprecursor-Verbindung
gemischt wird, kann ein Metall-Ligandkomplex gebildet werden, der
ein Katalysator sein kann oder noch aktiviert werden muss, um ein
Katalysator zu werden. Die Metall-Ligand-Komplexe, die hier erörtert werden,
werden als 2,1-Komplexe oder 3,2-Komplexe
bezeichnet, wobei die erste Zahl die Zahl der koordinierenden Atome
bezeichnet und die zweite Zahl die Ladung des Metalls darstellt.
Die 2,1-Komplexe weisen entsprechend zwei koordinierende Atome und
eine einzelne anionische Ladung auf. Andere Ausführungsformen sind solche Komplexe,
die ein allgemeines 3,2-Koordinationsschema
zu einem Metallzentrum aufweisen, wobei 3,2 sich auf einen Liganden
bezieht, der drei Koordinationsstellen am Metall besetzt, wobei
zwei von diesen Stellen anionisch sind und die verbleibende Stelle
eine neutrale Lewis-Basenartige Koordination ist.
-
Wenn
man zunächst
auf die 2,1-Nicht-Metallocen-Metall-Ligandkomplexe schaut, können die
Metall-Ligandkomplexe durch die folgende allgemeine Formel charakterisiert
werden:
wobei T, J'', R
1, L und
n wie vorher definiert sind; und x 1 oder 2 ist. Das J''-Heteroaryl kann auch über eine dative
Bindung gebunden sein oder nicht, wird allerdings als Bindung gezeichnet.
Spezifischer können
die Nicht-Metallocenkomplexe durch die Formel charaktersiert werden:
wobei R
1,
T, R
4, R
5, R
6, R
7, L und n wie
vorher definiert sind; und x 1 oder 2 ist. In einer bevorzugten
Ausführungsform
ist x = 1 und n = 3. Zusätzlich
können
auch Lewis-Basen-Addukte
dieser Metall-Ligandkomplexe verwendet werden, zum Beispiel sind
Ether, Amine, Thioether, Phosphine und dergleichen geeignet als
Lewis-Basen.
-
Insbesondere
können
die Nicht-Metallocen-Metall-Ligandkomplexe durch die allgemeine
Formel charakterisiert werden:
wobei die Variablen im Allgemeinen
wie oben definiert sind. Q
2, Q
3,
Q
4, R
2, R
3, R
4, R
5,
R
6 und R
7 sind also
zum Beispiel unabhängig
voneinander ausgewählt
aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno,
Nitro und Kombinationen davon; ggf. können zwei oder mehr R
4-, R
5-, R
6-, R
7-Gruppen verbunden
sein, um ein verbundenes Ringsystem mit von 3-50 Nicht-Wasserstoffatomen zusätzlich zu
dem Pyridinring zu bilden, zum Beispiel um eine Chinolingruppe zu
erzeugen. Gegebenenfalls kann auch irgendeine Kombination von R
2, R
3 und R
4 miteinander zu einer Ringstruktur verbunden
werden; Q
1 und Q
5 sind
ausgewählt
aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl,
substituiertem Cycloalkyl, Aryl, substituiertem Aryl, unter der
Voraussetzung, dass Q
1 und Q* nicht beide
Methyl sind; und L jeweils unabhängig
voneinander ausgewählt
ist aus der Gruppe bestehend aus Halogenid, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Hydroxy, Boryl, Silyl, Amino, Amin, Hydrido, Allyl,
Dien, Seleno, Phosphino, Phosphin, Carboxylaten, Thio, 1,3-Dionaten,
Oxalaten, Carbonaten, Nitraten, Sulfaten und Kombinationen davon;
n ist 1, 2, 3, 4, 5 oder 6; und x = 1 oder 2.
-
In
anderen Ausführungsformen
können
die 2,1-Metall-Ligandkomplexe durch die allgemeine Formel charakterisiert
werden:
wobei die Variablen im Allgemeinen
wie oben definiert sind.
-
In
noch anderen Ausführungsformen
können
die 2,1-Metall-Ligandkomplexe durch die allgemeine Formel charakterisiert
werden:
wobei die Variablen im Allgemein
wie oben definiert sind.
-
Die
spezifischeren Ausführungsformen
der Nicht-Metallocen-Metall-Ligandkomplexe aus Formeln VI, VII,
VIII, IX und X werden oben erklärt
mit Bezug auf die Eigenheiten, die für die Liganden und Metallprecursor beschrieben
sind. Spezifische Beispiele von 2,1-Metall-Ligandkomplexen beinhalten,
sind aber nicht beschränkt
auf:
wobei
L, n und x wie oben definiert sind (z.B. x = 1 oder 2) und Ph =
Phenyl ist. In bevorzugten Ausführungsformen
ist x = 1 und n = 3. Darüber
hinaus ist in bevorzugten Ausführungsformen
L ausgewählt
aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Aryl,
substituiertem Aryl oder Amino.
-
Nun
zu den 3,2-Metall-Ligand-Nicht-Metallocenkomplexen; die Metall-Ligandkomplexe
können
durch die allgemeine Formel charakterisiert werden:
wobei M Zircon oder Hafnium
ist;
R
1 und T sind oben definiert;
J''' ist
ausgewählt
aus der Gruppe der substituierten Heteroaryle mit 2 Atomen, die
an das Metall M gebunden sind, wobei mindestens eines dieser 2 Atome
ein Heteroatom ist und wobei ein Atom von J''' über eine dative Bindung, das
andere über
eine kovalente Bindung an M gebunden ist; und
L
1 und
L
2 sind unabhängig voneinander ausgewählt aus
der Gruppe bestehend aus Halogenid, Alkyl, substituiertem Alkyl,
Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Hydroxy, Boryl, Silyl, Amino, Amin, Hydrido, Allyl,
Dien, Seleno, Phosphino, Phosphin, Carboxylaten, Thio, 1,3-Dionaten,
Oxalaten, Carbonaten, Nitraten, Sulfaten und Kombinationen davon.
-
Spezifischer
können
die 3,2-Metall-Liganden mit Nicht-Metallocenkomplexen durch die
allgemeine Formel charakterisiert werden:
wobei M Zircon oder Hafnium
ist;
T, R
1, R
4,
R
5, R
6, L
1 und L
2 oben definiert
sind; und
E'' entweder Kohlenstoff
oder Stickstoff ist und ein Teil einer zyklischen Aryl-, substituierten
Aryl-, Heteroaryl- oder substituierten Heteroarylgruppe ist.
-
Insbesondere
können
die 3,2-Metall-Ligand-Nicht-Metallocenkomplexe durch die allgemeine
Formel charakterisiert werden:
wobei M Zircon oder Hafnium
ist; und
T, R
1, R
4,
R
5, R
6, R
10, R
11, R
12, R
13, L
1 und L
2 oben definiert
sind.
-
Insbesondere
können
die 3,2-Metall-Ligand-Nicht-Metallocenkomplexe durch die allgemeine
Formel charaktensiert werden:
wobei M Zircon oder Hafnium
ist; und
T, R
1, R
4,
R
5, R
6, R
10, R
11, R
12, R
13, Q
1, Q
2, Q
3,
Q
4, Q
5, L
1 und L
2 oben definiert
sind.
-
Die
spezifischeren Ausführungsformen
der Metall-Ligandkomplexe aus den Formeln XI, XII, XIII und XIV
werden mit Bezug auf die Eigenheiten, die für die Liganden und Metallprecursor
beschrieben sind, bereits erklärt.
-
In
den obigen Formeln sind R10, R11,
R12 und R13 unabhängig voneinander
ausgewählt
aus der Gruppe bestehend aus Wasserstoff, Halogen, Alkyl, substituiertem
Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem
Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl,
Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl,
Alkoxy, Aryloxy, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Nitro
und Kombinationen davon; ggf. können
zwei oder mehr R10-, R11-,
R12- und
R13-Gruppen verbunden sein, um ein verbundenes
Ringsystem mit von 3-50 Nicht-Wasserstoffatomen
zu bilden.
-
Zusätzlich sind
auch Lewis-Basen-Addukte der Metall-Ligandkomplexe in den obigen
Formeln geeignet, zum Beispiel sind Ether, Amine, Thioether, Phosphine
und dergleichen geeignete Lewis-Basen.
-
Die
Metallligandkomplexe können
durch dem Fachmann bekannte Techniken gebildet werden. In einigen
Ausführungsformen
ist R
14 Wasserstoff und die Metall-Ligandkomplexe werden
durch eine Metallierungsreaktion (in situ oder nicht), wie weiter
unten in Schema 3 gezeigt, gebildet:
-
Schema 3
-
In
Schema 3 ist R14 Wasserstoff (aber vergleiche
weiter oben die gesamte Definition für R14 in
anderen Ausführungsformen).
Die Metallisierungsreaktion, um den 2,1-Komplex auf der linken Seite
zu dem 3,2-Komplex auf der rechten Seite umzuwandeln, kann über eine
Vielzahl von Mechanismen geschehen, wahrscheinlich abhängig von
den Substituenten, die für
L1, L2 und L3 und die anderen Substituenten, wie zum
Beispiel Q1-Q5, R2-R6, R10 bis
R13 ausgewählt wurden. In einer Ausführungsform
kann die Reaktion durch Aufheizen des 2,1-Komplexes auf eine Temperatur über etwa
100 °C voranschreiten,
wenn L1, L2 und
L3 jeweils N(CH3)2 sind. In dieser Ausführungsform bleiben L1 und L2 wahrscheinlich
N(CH3)2 in dem 3,2-Komplex.
In einer anderen Ausführungsform,
in der L1, L2 und
L3 jeweils N(CH3)2 ist, kann die Reaktion voranschreiten,
in dem ein Reagenz aus der 13. Gruppe (wie unten beschrieben) zu
dem 2,1-Komplex bei einer geeigneten Temperatur (zum Beispiel Raumtemperatur)
zugegeben wird. Als Reagenz der 13. Gruppe ist zu diesem Zweck Diisobutylaluminiumhydrid,
Triisobutylaluminium oder Trimethylaluminium bevorzugt. In dieser
Ausführungsform
werden L1 und L2 typischwerweise
in die Liganden umgewandelt (z.B Alkyl oder Hydrid), die aus dem
Reagenz der 13. Gruppe stammen (z.B. sind bei Trimethylaluminium,
L1 und L2 jeweils
CH3 im 3,2-Komplex). Der 2,1-Komplex in Schema
3 wird durch die oben erörterten
Verfahren gebildet.
-
In
einer alternativen Ausführungsform,
möglicherweise
außerhalb
des Schutzbereichs von Schema 3, ist es für die Herstellung von isotaktischem
Polypropylen derzeit bevorzugt, dass R14 entweder
Wasserstoff oder Methyl ist.
-
Spezifische
Beispiele von 3,2-Komplexen beinhalten:
-
Verschiedene
Referenzen offenbaren Metallkomplexe, die ähnlich zu sein scheinen; vergleiche
zum Beispiel USP 6,103,657 und USP 5,637,660. Allerdings stellen
gewisse Ausführungsformen
hierin unerwartet verbesserte Polymerisationseigenschaften verglichen
mit den Ausführungsformen,
die in diesen Referenzen offenbart sind, bereit (z.B. höhere Aktivität und/oder
höhere
Polymerisationstemperaturen und/oder höhere Comonomer-Einbau und/oder
kristalline Polymere aufgrund eines hohen Anteils von Stereoselektivität während der
Polymerisation). Insbesondere ist die Aktivität der Hafniummetall-Katalysatoren,
wie in gewissen Beispielen hierin gezeigt, bei Weitem besser als
die der Zirconkatalysatoren.
-
Die
Ligandenkomplexe oder Katalysatoren können auf einem organischen
oder anorganischen Träger geträgert sein.
Geeignete Träger
beinhalten Siliciumdioxide, Aluminiumoxide, Tonerden, Zeolite, Magnesiumchlorid,
Polyethylenglycole, Polystyrole, Polyester, Polyamide, Peptide und
dergleichen. Polymere Träger
können
vernetzt sein oder nicht. Auf ähnliche
Weise können
die Ligandenkomplexe oder Katalysatoren auf ähnliche, dem Fachmann bekannte,
Träger
geträgert
sein. Zusätzlich
können
die Katalysatoren mit anderen Katalysatoren in einem einzelnen Reaktor
kombiniert werden und/oder in einer Serie von Reaktoren (parallel
oder in Serie) eingesetzt werden, um Mischungen von Polymerprodukten
zu bilden. Geträgerte
Katalysatoren stellen typischerweise P/E*-Copolymere mit einer MWD
her, die größer ist
als von solchen Polymeren, die mit ungeträgerten Katalysatoren hergestellt
werden, obwohl diese MWDs typischerweise kleiner als 6, bevorzugt kleiner
als 5 und stärker
bevorzugt kleiner als 4 sind.
-
Die
Metallkomplexe werden durch Kombination mit einem aktivierenden
Cokatalysator oder durch Verwendung einer Aktivierungstechnik katalytisch
aktiv gemacht. Geeignete Aktivierungs-Cokatalysatoren beinhalten
neutrale Lewis-Säuren,
wie zum Beispiel Alumoxan (modifiziert und unmodifiziert), C1-30-Hydrocarbyl-substituierte Verbindungen
der 13. Gruppe, besonders Tri(hydrocarbyl)aluminium- oder Tri(hydrocarbyl) borverbindungen
und halogenierte (inklusive perhalogenierte) Derivate davon mit
von 1 bis 10 Kohlenstoffen in jeder Hydrocarbyl- oder halogenierten
Hydrocarbylgruppe, noch spezieller perfluorierte Tri(aryl)borverbindungen
und im stärksten
Maße Tris(pentafluorphenyl)boran;
nicht-polymere, kompatible, nicht-koordinierende Ionenbildende Verbindungen
(beinhaltend die Verwendung von solchen Verbindungen unter oxidierenden
Bedingungen), besonders die Verwendung von Ammonium-, Phosphonium-,
Oxonium-, Carbonium-, Silylium- oder Sulfoniumsalze kompatibler,
nicht-koordinierender Anionen, oder Ferroceniumsalzen kompatibler, nicht-koordinierender Anionen;
Bulkelektrolyse (weiter unten noch genauer erklärt); und Kombinationen der vorangehenden
Aktivierungs-Cokatalysatoren und -techniken. Die vorangehenden Aktivierungs-Cokatalysatoren
und Aktivierungstechniken wurden bereits in den folgenden Referenzen
unter Bezug auf verschiedene Metallkomplexe gelehrt: USP 5,153,157,
5,064,802, 5,721,185 und 5,350,723, und EP-A-277,003 und -A-468,651 (äquivalent
zu USSN 07/547,718).
-
Das
Alumoxan, das als ein Aktivierungs-Cokatalysator verwendet wird,
hat die Formel (R4 x(CH3)yAlO)n, wobei
R4 ein lineares verzweigtes oder zyklisches
C1 bis C6- Hydrocarbyl ist,
x von 0 bis 1, y von 1 bis 0 und n eine ganze Zahl von 3 bis inklusive
25, ist. Die bevorzugten Alumoxankomponenten, bezeichnet als modifizierte
Methylaluminoxane, sind solche, bei denen R4 ein
lineares, verzweigtes oder zyklisches C3 bis
C9-Hydrocarbyl ist, x von 0,15 bis 0,50,
y von 0,85 bis 0,5 und n eine ganze Zahl zwischen 4 und inklusive
20 ist; noch stärker
bevorzugt ist R4 Isobutyl, Tertiärbutyl oder
n-Octyl, x von 0,2 bis 0,4, y von 0,8 bis 0,6 und n eine ganze Zahl
zwischen 4 und inklusive 15. Mischungen der oben genannten Alumoxane
können
auch eingesetzt werden.
-
Am
stärksten
bevorzugt ist das Alumoxan der Formel (R4 x(CH3)yAlO)n, wobei R4 Isobutyl
oder Tertiärbutyl
ist, x etwa 0,25, y etwa 0,75 und n von 6 bis 8 ist.
-
Besonders
bevorzugte Alumoxane sind die sogenannten modifizierten Alumoxane,
bevorzugt modifizierte Methylalumoxane (MMAO), die vollständig in
Alkan-Lösungsmitteln,
zum Beispiel in Heptan, löslich
sind und sehr wenig, wenn überhaupt,
Trialkylaluminium beinhalten können.
Eine Technik zur Herstellung solcher modifizierten Alumoxane ist
in US-Patent Nr. 5,041,584 offenbart (auf welches hiermit Bezug
genommen wird). Alumoxane, die als ein Aktivierungs-Cokatalysator
nützlich
sind, können
auch, wie in USP 4,542,199; 4,544,762; 4,960,878; 5,015,749; 5,041,583
und 5,041,585 offenbart, hergestellt werden. Verschiedene Alumoxane
können
kommerziell erhalten werden, zum Beispiel von Akzo-Nobel Corporation,
und beinhalten MMAO-3A, MMAO-12 und PMAO-IP.
-
Kombinationen
von neutralen Lewis-Säuren,
besonders die Kombination von einer Trialkylaluminiumverbindung
mit von 1 bis 4 Kohlenstoffen in jeder Alkylgruppe und einer halogenierten
Tri(hydrocarbyl)bor-Verbindung mit von 1 bis 10 Kohlenstoffen in
jeder Hydrocarbylgruppe, besonders Tris(pentafluorphenyl)boran und
Kombinationen von neutralen Lewis-Säuren, besonders Tris(pentafluorophenyl)boran,
mit nicht-polymeren,
kompatiblen nicht-koordinierenden Ionen-bildenden Verbindungen,
sind auch nützlich
als Aktivierungs-Cokatalysatoren.
-
Geeignete
Ionen-bildende Verbindungen, die als Cokatalysatoren nützlich sind,
umfassen ein Kation, das eine Bronsted-Säure ist und ein Proton abgeben
kann und ein kompatibles, nicht-koordinierendes Anion, A–.
Der Ausdruck „nicht-koordinierend", wie er hier verwendet
wird, meint ein Anion oder eine Substanz, die entweder nicht mit
einem Precursorkomplex, der ein Metall der Gruppe 4 enthält und das
davon stammende katalytische Derivat koordiniert, oder die nur schwach
zu solchen Komplexen koordiniert und dabei ausreichend labil bleibt,
um durch eine neutrale Lewis-Base ersetzt zu werden.
-
Ein
nicht-koordinierendes Anion kennzeichnet im Speziellen ein Anion,
das, wenn es als ein Ladungsausgleichsanion in einem kationischen
Metallkomplex wirkt, nicht einen anionischen Substituenten oder
ein Fragment davon zu dem Kation überträgt und dabei neutrale Komplexe
bildet. „Kompatible
Anionen" sind Anionen,
die nicht neutral werden, wenn der anfangs gebildete Komplex sich
zersetzt, und die bei der erwünschten
anschließenden
Polymerisation oder anderen Verwendungen des Komplexes nicht stören.
-
Bevorzugte
Anionen sind solche, die einen einzelnen Koordinationskomplex enthalten,
umfassend ein ladungstragendes Metall oder einen Metalloidkern,
wobei das Anion geeignet ist, die Ladung der aktiven Katalysatorspezies
(des Metallkations) auszugleichen, die gebildet werden können, wenn
die beiden Komponenten kombiniert werden. Das Anion sollte auch
ausreichend labil sein, um durch olefinisch, diolefinisch und acetylenisch
ungesättigte
Verbindungen oder andere neutrale Lewis-Basen, wie zum Beispiel
Ether oder Nitrile, ersetzt zu werden. Geeignete Metalle beinhalten,
sind aber nicht darauf beschränkt,
Aluminium, Gold und Platin. Geeignete Metalloide beinhalten, sind
aber nicht darauf beschränkt,
Bor, Phosphor und Silicium. Verbindungen, die Anionen enthalten,
die Koordinationskomplexe umfassen, die ein einzelnes Metall- oder
Metalloidatom enthalten, sind natürlich wohl bekannt und viele,
besonders solche Verbindungen, die ein einzelnes Boratom in dem
anionischen Teil enthalten, sind kommerziell erhältlich.
-
In
einer Ausführungsform
können
die aktivierenden Cokatalysatoren durch die folgende allgemeine Formel
dargestellt werden: [L*-H]+ d[Ad–] wobei:
L*
eine neutrale Lewis-Base ist;
[L*-H]+ eine
Bronsted-Säure
ist;
Ad– ein
nicht-koordinierendes, kompatibles Anion mit einer Ladung von d– ist,
und
d eine ganze Zahl von 1 bis 3 ist.
-
Stärker bevorzugt
entspricht Ad– der
Formel: [M'k+Qn']d–,
wobei:
k eine ganze Zahl von 1 bis 3 ist;
n' eine ganze Zahl
von 2 bis 6 ist;
n'-k
= d;
M' ein
Element ausgewählt
aus der 13. Gruppe des Periodensystems der Elemente ist; und
Q
bei jeglichem Auftreten unabhängig
ausgewählt
ist aus Hydrid, Dialkylamid, Halogenid, Hydrocarbyl, Hydrocarbyloxy,
Halogen-substituierten Hydrocarbyl, Halogensubstituierten Hydrocarbyloxy-
und Halogen-substituierten Silylhydrocarbylresten (beinhaltend perhalogenierten
Hydrocarbyl-, perhalogenierten Hydrocarbyloxy- und perhalogenierten
Silylhydrocarbylresten), und Q bis zu 20 Kohlenstoffe aufweist,
unter der Voraussetzung, dass bei nicht mehr als einem Auftreten
Q Halogenid ist. Beispiele von geeigneten Q-Gruppen aus Hydrocarbyloxid
sind in USP 5,296,433 offenbart.
-
In
einer stärker
bevorzugten Ausführungsform
ist d eins, d.h. das Gegenion hat eine einzelne negative Ladung
und ist A–.
Aktivierungs-Cokatalysatoren, die Bor umfassen und besonders nützlich bei
der Herstellung der Katalysatoren sind, können durch die folgende allgemeine
Formel dargestellt werden: [L*-H]+[BQ4]– wobei:
[L*-H]+ wie zuvor definiert ist;
B Bor in
der Oxidationsstufe 3 ist; und
Q eine Hydrocarbyl-, Hydrocarbyloxy-,
fluorierte Hydrocarbyl-, fluorierte Hydrocarbyloxy- oder fluorierte
Silylhydrocarbylgruppe von bis zu 20 Nicht-Wasserstoffatomen ist, unter der Voraussetzung,
dass Q maximal einmal Hydrocarbyl ist. Am stärksten bevorzugt ist Q bei
jeglichem Auftreten eine fluorierte Arylgruppe, besonders eine Pentafluorophenylgruppe.
-
Veranschaulichende,
aber nicht einschränkende
Beispiele für
Borverbindungen, die als Aktivierungskatalysator zur Herstellung
von den Katalysatoren verwendet werden können, sind tri-substituierte
Ammoniumsalze, wie zum Beispiel:
Triethylammoniumtetraphenylborat,
N,N-Dimethylaniliniumtetraphenylborat,
Tripropylammoniumtetrakis(pentafluorophenyl)borat,
N,N-Dimethylanilinium-n-butyltris(pentafluorophenyl)borat,
Triethylammoniumtetrakis(2,3,4,6-tetrafluorophenyl)borat,
N,N-Diethylaniliniumtetrakis(2,3,4,6-tetrafluorophenyl)borat,
und
N,N-Dimethyl-2,4,6-trimethylaniliniumtetrakis(2,3,4,6-tetrafluorophenyl)borat;
Dialkylammoniumsalze
wie zum Beispiel:
Di-(i-propyl)ammoniumtetrakis(pentafluorophenyl)borat,
und
Dicyclohexylammoniumtetrakis(pentafluorophenyl)borat;
trisubstituierte
Phosphoniumsalze wie zum Beispiel:
Triphenylphosphoniumtetrakis(pentafluorophenyl)borat,
Tri(o-tolyl)phosphoniumtetrakis(pentafluorophenyl)borat,
und
Tri(2,6-dimethylphenyl)phosphoniumtetrakis(pentafluorophenyl)borat;
disubstituierte
Oxoniumsalze wie zum Beispiel:
Diphenyloxoniumtetrakis(pentafluorophenyl)borat,
Di(o-tolyl)oxoniumtetrakis(pentafluorophenyl)borat,
und
Di(2,6-dimethylphenyl)oxoniumtetrakis(pentafluorophenyl)borat;
disubstituierte
Sulfoniumsalze wie zum Beispiel:
Diphenylsulfoniumtetrakis(pentafluorophenyl)borat,
Di(o-tolyl)sulfoniumtetrakis(pentafluorophenyl)borat,
und
Di(2,6-dimethylphenyl)sulfoniumtetrakis(pentafluorophenyl)borat.
-
Bevorzugte
[L*-H]+-Kationen sind N,N-Dimethylanilinium
und Tributylammonium.
-
Ein
anderer geeigneter Ionen-bildender Aktivierungscokatalysator umfasst
ein Salz aus einem kationischem Oxidationsmittel und einem nicht-koordinierenden,
kompatiblen Anion dargestellt durch die Formel: (Oxe+)d(Ad–)e wobei:
Oxe+ ein
kationisches Oxidationsmittel mit einer Ladung von e+ ist;
e
eine ganze Zahl von 1 bis 3 ist ; und
Ad– und
d bereits definiert sind.
-
Beispiele
für kationische
Oxidationsmittel beinhalten: Ferrocenium, Hydrocarbylsubstituiertes
Ferrocenium, Ag+ oder Pb+2.
Bevorzugte Ausführungsformen
von Ad– sind
solche Anionen, wie sie bereits im Hinblick auf die Bronstedt-Säure-haltigen
Aktivierungs-Cokatalysatoren, insbesondere Tetrakis(pentafluorophenyl)borat
definiert worden sind.
-
Ein
anderer geeigneter Ionen-bildender Aktivierungs-Cokatalysator umfasst
eine Verbindung, die ein Salz aus einem Carbeniumion und einem nicht-koordinierenden,
kompatiblen Anion ist, das durch die Formel dargestellt wird: ©+A– wobei:
©+ ein C1-20-Carbeniumion
ist; und
A– bereits definiert ist.
-
Ein
bevorzugtes Carbeniumion ist das Tritylkation, d.h. Triphenylmethylium.
-
Ein
weiterer geeigneter Ionen-bildender, aktivierender Cokatalysator
umfasst eine Verbindung, die ein Salz aus einem Silyliumion und
einem nicht-koordinierenden, kompatiblen Anion ist, die durch die
Formel dargestellt wird: R3Si(X')q +A– wobei:
R
ein C1-10-Hydrocarbyl ist und X', q und A– bereits
definiert sind.
-
Bevorzugte
Aktivierungs-Cokatalysatoren aus Silyliumsalz sind Trimethylsilyliumtetrakis(pentafluorophenyl)borat,
Triethylsilylium(tetrakispentafluor) phenylborat und Ether-substituierte
Addukte davon. Silyliumsalze wurden bereits ganz allgemein offenbart
in J. Chem Soc. Chem. Comm., 1993, 383-384, sowie von Lambert, J.
B., et al., Organometallics, 1994, 13, 2430-2443.
-
Gewisse
Komplexe von Alkoholen, Mercaptanen, Silanolen und Oximen mit Tris
(pentafluorphenyl)boran sind auch effektive Katalysatoraktivatoren
und können
gemäß der vorliegenden
Erfindung verwendet werden. Solche Cokatalysatoren sind in USP 5,296,433
offenbart.
-
Die
Technik der Bulkelektrolyse schließt die elektrochemische Oxidation
des Metallkomplexes unter Elektrolysebedingungen in der Anwesenheit
eines Hilfselektrolyts, der ein nicht-koordinierendes, inertes Anion umfasst,
ein. In der Technik werden Lösungsmittel,
Hilfselektrolyte und elektrolytische Potenziale für die Elektrolyse
so verwendet, dass Elektrolysebeiprodukte, die den Metallkomplex
katalytisch inaktiv machen würden, im
Wesentlichen nicht während
der Reaktion gebildet werden. Insbesondere sind geeignete Lösungsmittel
Materialien, die unter den Bedingungen der Elektrolyse (im Allgemeinen
bei Temperaturen von 0 bis 100 C) flüssig sind, den Hilfselektrolyt
lösen können und
inert sind. „Inerte
Lösungsmittel" sind solche, die
unter den Reaktionsbedingungen, die für die Elektrolyse eingesetzt
werden, nicht reduziert oder oxidiert werden. Es ist im Allgemeinen
möglich,
im Hinblick auf die gewünschte
Elektrolysereaktion ein Lösungsmittel
und einen Hilfselektrolyten auszuwählen, die von dem elektrischen
Potenzial, das für
die gewünschte
Elektrolyse verwendet wird, unbeeinträchtigt bleiben. Bevorzugte
Lösungsmittel
beinhalten Difluorbenzol (alle Isomere), Dimethoxyethan (DME) und
Mischungen davon.
-
Die
Elektrolyse kann in einer elektrolytischen Standard-Zelle, die eine
Anode und eine Kathode (auch bezeichnet als die Arbeitselektrode
bzw. die Gegenelektrode) enthält,
durchgeführt
werden. Geeignete Materialien zum Aufbau der Zelle sind Glas, Kunststoff,
Keramik und mit Glas beschichtetes Metall. Die Elektroden werden
aus inerten leitenden Materialien hergestellt, womit leitende Materialien
gemeint sind, die unbeeinflusst von der Reaktionsmischung oder den
Reaktionsbedingungen sind. Platin oder Paladium sind bevorzugte
inerte leitende Materialien. Normalerweise teilt eine ionenpermeable
Membran, wie zum Beispiel eine feine Glasfritte die Zelle in zwei
separate Bereiche, den Arbeitselektrodenbereich und den Gegenelektrodenbereich.
Die Arbeitselektrode wird in ein Reaktionsmedium getaucht, das den
Metallkomplex, der aktiviert werden soll, Lösungsmittel, Hilfselektrolyt
und jegliche anderen Materialien, die erwünscht sind, um die Elektrolyse
abzumildern oder den resultierenden Komplex zu stabilisieren, umfasst.
Die Gegenelektrode wird in eine Mischung aus dem Lösungsmittel
und dem Hilfselektrolyt getaucht. Die erwünschte Spannung kann mittels
theoretischen Berechnungen oder experimentell durch Abtasten der
Zelle mittels einer Referenzelektrode, zum Beispiel einer in den
Zellelektrolyt eingetauchten Silberelektrode, bestimmt werden. Der
Hintergrundzellstrom, die Stromaufnahme in Abwesenheit der erwünschten
Elektrolyse wird auch bestimmt. Die Elektrolyse ist abgeschlossen, wenn
der Strom von dem erwünschten
Level auf das Hintergrundlevel abfällt. Auf diese Weise kann die
vollständige
Umsetzung des anfänglichen
Metallkomplexes leicht bestimmt werden.
-
Geeignete
Hilfselektrolyte sind Salze, umfassend ein Kation und ein kompatibles,
nicht-koordinierendes
Anion, A–.
Bevorzugte Hilfselektrolyte sind Salze gemäß der Formel: G+A– wobei:
G+ ein Kation ist, das nicht-reaktiv gegenüber dem
Ausgangskomplex und dem resultierenden Komplex ist, und
A– wie
bereits definiert ist.
-
Beispiele
von Kationen, G+, beinhalten Tetrahydrocarbyl-substituierte
Ammonium- oder Phosphoniumkationen mit bis zu 40 Nicht-Wasserstoffatomen.
Bevorzugte Kationen sind die Tetra-n-butylammonium- und Tetraethylammonium-Kationen.
-
Während der
Aktivierung der efindungsgemäßen Komplexe
durch Bulkelektrolyse durchwandert das Kation des Hilfselektrolyten
zu der Gegenelektrode und A– wandert zu der Arbeitselektrode,
um das Anion des resultierenden oxidierten Produkts zu werden. Entweder
das Lösungsmittel
oder das Kation des Hilfselektrolyten wird an der Gegenelektrode
in äquimolarer
Menge zu der Menge an oxidiertem Metallkomplex, der an der Arbeitselektrode
gebildet wird, reduziert. Bevorzugte Hilfselektrolyte sind Tetrahydrocarbylammoniumsalze von
Tetrakis(perfluoraryl)boraten mit von 1 bis 10 Kohlenstoffen in
jeder Hydrocarbyl- oder Perfluorarylgruppe, besonders Tetra-n-butylammoniumtetrakis(pentafluorphenyl)borat.
-
Eine
weitere erforschte elektrochemische Technik zur Erzeugung von Aktivierungs-Cokatalysatoren ist die
Elektrolyse einer Disilanverbindung in der Gegenwart einer Quelle
von einem nicht-koordinierenden kompatiblen Anion. Diese Technik
ist in USP 5,625,087 genauer offenbart und beansprucht.
-
Die
vorhergehenden Aktivierungstechniken und Ionen-bildenden Cokatalysatoren
werden auch bevorzugt in Kombination mit einer Tri(hydrocarbyl)aluminium-
oder Tri(hydrocarbyl)boranverbindung mit von 1 bis 4 Kohlenstoffen
in jeder Hydrocarbylgruppe verwendet.
-
Das
bevorzugte molare Verhältnis
von Katalysator/Cokatalysator, das eingesetzt wird, reicht von 1:10.000
bis 100:1, stärker
bevorzugt von 1:5000 bis 10:1, am stärksten bevorzugt von 1:100
bis 1:1. In einer Ausführungsform
kann der Cokatalysator in Kombination mit einer Tri(hydrocarbyl)aluminiumverbindung
mit von 1 bis 10 Kohlenstoffen in jeder Hydrocarbylgruppe verwendet
werden. Mischungen von Aktivierungs-Cokatalysatoren können auch
eingesetzt werden. Es ist möglich,
diese Aluminiumverbindungen aufgrund deren vorteilhafter Fähigkeit,
Verunreinigungen, wie zum Beispiel Sauerstoff, Wasser und Aldehyde,
aus der Polymerisationsmischung zu beseitigen, zu nutzen. Bevorzugte
Aluminiumverbindungen beinhalten Trialkylaluminiumverbindungen mit
von 1 bis 6 Kohlenstoffen in jeder Alkylgruppe, besonders diese,
worin die Alkylgruppen Methyl, Ethyl, Propyl, Isopropyl, n-Butyl,
Isobutyl, Pentyl, Neopentyl oder Isopentyl sind. Das molare Verhältnis von
Metallkomplex zu Aluminiumverbindung liegt bevorzugt von 1:10.000
bis 100:1, stärker bevorzugt
von 1:1000 bis 10:1, am stärksten
bevorzugt von 1:500 bis 1:1. Ein äußerst bevorzugter Aktivierungs-Cokatalysator aus
Boran umfasst eine starke Lewis-Säure, besonders Tris(pentafluorphenyl)boran.
-
In
manchen Ausführungsformen
können
zwei oder mehrere verschiedene Katalysatoren, umfassend die Verwendung
von gemischten Katalysatoren, eingesetzt werden. Zusätzlich zu
einem Nicht-Metallocen, metall-zentriertem Katalysator mit Heteroaryl-Ligand
kann, wenn eine Vielzahl von Katalysatoren verwendet wird, irgendein
Katalysator, der ein oder mehrere Olefinmonomere copolymerisieren
kann, um ein Interpolymer oder Homopolymer herzustellen, in Ausführungsformen
der Erfindung in Verbindung mit einem Nicht-metallocen, metall-zentrierten
Katalysator mit Heteroaryl-Liganden verwendet werden. Für gewisse
Ausführungsformen sollten
bevorzugt zusätzliche
Auswahlkriterien, wie zum Beispiel die Fähigkeit ein bestimmtes Molekulargewicht
aufbauen und/oder Comonomer einbauen zu können, in Betracht gezogen werden.
Zwei oder mehrere Nicht-Metallocen, metall-zentrierte Katalysatoren
mit Heteroaryl-Liganden, die unterschiedliche Substituenten haben,
können
in der Praxis gewisser Ausführungsformen,
die hierin offenbart sind, verwendet werden. Geeignete Katalysatoren,
die in Verbindung mit den Nicht-metallocen metall-zentrierten Katalysatoren
mit Heteroaryl-Liganden, die hierin offenbart sind, verwendet werden
können,
beinhalten, sind aber nicht darauf beschränkt, Ziegler-Natta-Katalysatoren,
Metallocenkatalysatoren und Katalysatoren mit gezwungener Geometrie
und Variationen eines oder mehrerer von diesen. Sie beinhalten jegliche
bekannte und derzeit unbekannte Katalysatoren für die Olefinpolymerisation.
Es versteht sich, dass sich der Ausdruck „Katalysator", wie er hier verwendet
wird, auf eine Metall-haltige Verbindung bezieht, die zusammen mit
einem Aktivierungs-Cokatalysator verwendet wird, um ein Katalysatorsystem
zu bilden. Der Katalysator, wie er hier verwendet wird, ist für gewöhnlich katalytisch
inaktiv ohne einen Cokatalysator oder eine andere Aktivierungstechnik.
Allerdings sind nicht alle geeigneten Katalysatoren ohne einen Cokatalysator
katalytisch inaktiv.
-
Eine
geeignete Klasse von Katalysatoren sind Katalysatoren mit gezwungener
Geometrie, die in USP 5,064,802, 5,132,380, 5,703,187 und 6,034,021;
EP 0 468 651 und 0 514 828;
und WO 93/19104 und 95/00526 offenbart sind. Eine andere geeignete
Klasse von Katalysatoren sind die Metallocenkatalysatoren, die in
USP 5,044,438, 5,057,475, 5,096,867 und 5,324,800 offenbart sind.
Die Katalysatoren mit gezwungener Geometrie können als Metallocenkatalysatoren
angesehen werden und beide werden manchmal im Fachgebiet als Single-Site-Katalysatoren
bezeichnet.
-
Eine
andere geeignete Klasse von Katalysatoren sind substituierte Indenyl-haltige
Metallkomplexe, wie sie in USP 5,965,756 und 6,015,868 offenbart
sind. Andere Katalysatoren sind in den gleichzeitig anhängigen Anmeldungen
USSN 09/230,185, 09/715,380, 60/215,456, 60/170,175 und 60/393,862
offenbart. Diese Katalysatoren neigen dazu, die Fähigkeit
Polymere mit höherem
Molekulargewicht herzustellen. Weitere Katalysatoren, Cokatalysatoren,
Katalysatorsysteme, und Aktivierungstechniken, die verwendet werden
können, beinhalten
solche, die in WO 96/23010, 99/14250, 98/41529 und 97/42241; bei
Scollard, et al., in J. Am. Chem. Soc 1996, 118, 10008-10009;
EP 0 468 537 B1 ;
WO 97/22635; in
EP
0 949 278 A2 , 0 949 279 A2 und 1 063 244 A2; USP 5,408,017,
5,767,208 und 5,907,021; WO 88/05792, 88/05793 und 93/25590; USP
5,599,761 und 5,218,071; WO 90/07526; USP 5,972,822, 6,074,977,
6,013,819, 5,296,433, 4,874,880, 5,198,401, 5,621,127, 5,703,257,
5,728,855, 5,731,253, 5,710,224, 5,883,204, 5,504,049, 5,962,714,
5,965,677 und 5,427,991; WO 93/21238, 94/03506, 93/21242, 94/00500,
96/00244 und 98/50392; bei Wang, et al., Organometallics 1998, 17,
3149-3151; Younkin, et al., Science 2000, 287, 460-462; Chen und
Marks, Chem. Rev. 2000, 100, 1391-1434; Alt und Koppl, Chem. Rev.
2000, 100, 1205-1221; Resconi, et al., Chem. Rev. 2000, 100, 1253-1345;
Ittel, et al., Chem. Rev. 2000, 100, 1169-1203; Coates, Chem. Rev.,
2000, 100, 1223-1251; in USP 5,093,415, 6,303,719 und 5,874,505;
und WO 96/13530 offenbart sind. Solche Katalysatoren, Cokatalysatoren
und Katalysatorsysteme, welche in USSN 09/230,185 und 09/715,380
und USP 5,965,756 und 6,150,297 offenbart sind, sind auch nützlich.
-
Verfahrensbeschreibung
-
Die
P/E*-Polymere, die in der Praxis dieser Erfindung verwendet werden,
können
durch jegliches geeignetes Verfahren hergestellt werden. In einer
Ausführungsform
werden die Verfahrensreagenzien, d.h. (i) Propylen (ii) Ethylen
und/oder eine oder mehrere ungesättigte
Comonomere, (iii) Katalysator und (iv) gegebenenfalls Lösungsmittel
und/oder ein Molekulargewichtsregler (z.B. Wasserstoff), einem einzelnen Reaktionsgefäß jeglicher
geeigneter Ausführung,
z.B. ein gerührter
Behälter,
Schleifenreaktor, Wirbelschichtreaktor etc. zugeführt. Die
Verfahrensreagenzien werden innerhalb des Reaktionsgefässes unter
geeigneten Bedingungen (z.B. Lösung,
Fällung,
Gasphase, Suspension, Hockdruck) in Kontakt gebracht, um das gewünschte Polymer zu
bilden und die Produktion aus dem Reaktor wird anschließend für die Aufarbeitung
nach der Reaktion entnommen. Die ganze Produktion kann auf einmal
aus dem Reaktor gewonnen werden (wie im Fall eines Single-Pass oder
Batch-Reaktors)
oder sie kann in Form eines „Bleed
Stream", der nur
einen Teil, typischerweise einen kleineren Teil der Reaktionsmasse
bildet, entnommen werden (wie im Fall eines kontinuierlichen Verfahrensreaktors-,
bei dem ein Produktstrom mit der gleichen Rate abgeleitet wird,
mit der die Reagenzien zugegeben werden, um die Polymerisation bei
einer stationären
Zustand zu halten. „Reaktionsmasse" meint den Inhalt
innerhalb eines Reaktors, typischerweise während oder im Anschluss an
die Polymerisation. Die Reaktionsmasse beinhaltet Reaktanden, gegebenenfalls
Lösungsmittel,
Katalysator und Produkte und Nebenprodukte. Das entnommene Lösungsmittel
und die unreagierten Monomere können
in den Reaktionsgefäß zurückgewonnen
werden.
-
Die
Polymerisationsbedingungen, bei denen der Reaktor betrieben wird,
sind ähnlich
denen bei der Polymerisation von Polypropylen, bei der ein bekannter
konventioneller Ziegler-Natta-Katalysator verwendet wird. Typischerweise
wird eine Lösungspolymerisation
von Propylen bei einer Polymerisationstemperatur zwischen –50 bis
200, bevorzugt zwischen –10
und 150 °C
und stärker
bevorzugt zwischen 20 bis 150 °C
und am stärksten
bevorzugt zwischen 80 und 150 °C
durchgeführt,
und der Polymerisationsdruck liegt typischerweise zwischen Atmosphärendruck
bis 7, bevorzugt zwischen 0,2 und 5 MPa. Falls Wasserstoff zugegen
ist, dann liegt er gewöhnlich
bei einem Partialdruck (wie er in dem Gasphasenbereich der Polymerisation
gemessen wird) bei 0,1 kPa bis 5 MPa, bevorzugt zwischen 1 kPa bis
3 MPa. Gasphasen-Suspensions- und andere Polymerisationsmodelle
verwenden Bedingungen wie für
solche Modelle üblich
sind. Bei Gasphasen- oder Fällungs-Polymerisationsverfahren
ist es wünschenswert,
die Polymerisation bei einer Temperatur unterhalb des Schmelzpunktes
des Polymers durchzuführen.
-
Für die Propylen/Ethylen-Copolymerverfahren,
die hierin beschrieben sind und gegebenenfalls zusätzliches
ungesättigtes
Monomer enthalten, liegt das Gewichtsverhältnis von Propylen zu Ethylen
im Zufluss zu den Reaktoren bevorzugt im Bereich von 10.000: 1 bis
1:10, stärker
bevorzugt von 1.000:1 bis 1:1 und noch stärker bevorzugt von 500:1 bis
3:1. Für
die Propylen/C4-20-α-Olefin-Copolymerverfahren der
vorliegenden Erfindung liegt das Gewichtsverhältnis von Propylen zu C4-20-α-Olefin
in dem Zufluss im Bereich von 10.000: 1 bis 1:20, stärker bevorzugt
von 1.000:1 bis 1:1 und noch stärker
bevorzugt von 1.000:1 bis 3:1.
-
Die
Aufarbeitung der erhaltenen Reaktionsmasse aus dem Polymerisationsgefäß nach der
Reaktion im Reaktor beinhaltet typischerweise die Deaktivierung
des Katalysators, Entfernen von Katalysatorrest, Trocknen des Produkts
und dergleichen. Das erhaltene Polymer ist dann fertig zum Lagern
und/oder zur Verwendung.
-
Die
P/E*-Polymere, die in einem einzelnen Reaktionsgefäß gemäß dieser
Erfindung hergestellt werden, haben den erwünschten MFR, enge MWD, 13C NMR-Peaks bei 14,6 und 15,7 ppm (die
Peaks haben annähernd
die gleiche Intensität),
gegebenenfalls hohen B-Wert und die anderen sie definierenden Eigenschaften.
Falls jedoch eine breitere MWD erwünscht ist, z.B. eine MWD von
zwischen 2,5 und 3,5 oder noch höher, ohne
dass sich die anderen definierenden Eigenschaften der Propylen-Copolymere wesentlich ändern, wird das
Copolymer bevorzugt in einem System von mehreren Reaktoren hergestellt.
In Systemen mit mehreren Reaktoren kann eine MWD so breit wie 15,
stärker
bevorzugt 10, am stärksten
bevorzugt 4-8 hergestellt werden.
-
In
einer Ausführungsform
umfassen die Monomere Propylen und mindestens ein Olefin, ausgewählt aus
der Gruppe bestehend aus C4-10-α-Olefinen,
besonders 1-Buten, 1-Hexen und 1-Octen, und die Schmelzflussrate
(MFR) des Interpolymers liegt bevorzugt im Bereich von 0,1 bis 500,
stärker
bevorzugt im Bereich von 0,1 bis 100, noch stärker bevorzugt von 0,2 bis
80, am stärksten
bevorzugt im Bereich von 0,3-50. In einigen Ausführungsformen können die
Nicht-Metallocen Katalysatoren, die in der Praxis der Erfindung,
wie sie hier beschrieben ist, in Verbindung mit mindestens einem
zusätzlichen
homogenen oder heterogenen Polymerisationskatalysator in separaten
Reaktoren, die miteinander in Serie oder parallel verbunden sind,
genutzt werden, um Polymermischungen mit wünschenswerten Eigenschaften
herzustellen. Ein Beispiel für
so ein Verfahren ist in WO 94/00500, äquivalent zu USSN 07/904,770
sowie in USSN 08/10958, eingereicht am 29. Januar 1993, offenbart.
Inbegriffen in diesen Ausführungsformen
ist die Verwendung von zwei verschiedenen Nicht-Metallocen, metallzentrierten
Katalysatoren mit Heteroaryl-Ligand.
-
Das
Katalysatorsystem kann als ein homogener Katalysator hergestellt
werden durch Zugabe von den erforderlichen Komponenten in ein Lösungsmittel,
in dem die Polymerisation durch Lösungspolymerisations-Verfahren
durchgeführt
wird. Das Katalysatorsystem kann auch als ein heterogener Katalysator
hergestellt und eingesetzt werden, indem die erforderlichen Komponenten
auf ein Katalysator-Trägermaterial,
wie zum Beispiel Silicagel, Aluminiumoxid oder anderes geeignetes
anorganisches Trägermaterial,
adsorbiert werden. Wenn es in heterogener oder geträgerter Form
hergestellt wird, ist es bevorzugt, Siliciumdioxid als das Trägermaterial
zu verwenden. Die heterogene Form des Katalysatorsystems kann in
einer Fällungs-
oder Gasphasenpolymerisation eingesetzt werden. Als praktische Beschränkung findet
die Fällungspolymerisation
in flüssigen
Verdünnungsmitteln,
in denen das Polymerprodukt im Wesentlichen unlöslich ist, statt. Das Verdünnungsmittel
für Fällungspolymerisation
ist bevorzugt ein oder mehrere Kohlenwasserstoffe mit weniger als
5 Kohlenstoffatomen. Falls erwünscht,
können
gesättigte
Kohlenwasserstoff(e), wie zum Beispiel Ethan, Propan oder Butan,
ganz oder teilweise als Verdünnungsmittel
verwendet werden. Gleichermaßen
können
auch das α-Olefin-Comonomer oder eine
Mischung aus verschiedenen α-Olefin-Comonomeren
ganz oder teilweise als Verdünnungsmittel
verwendet werden. Am stärksten
bevorzugt umfasst der Hauptteil des Verdünnungsmittels mindestens das α-Olefin-Monomer
oder die Monomere, die polymerisiert werden sollen.
-
Lösungspolymerisations-Bedingungen
benötigen
ein Lösungsmittel
für die
jeweiligen Komponenten der Reaktion. Bevorzugte Lösungsmittel
beinhalten, sind aber nicht darauf beschränkt, Mineralöle und die
verschiedenen Kohlenwasserstoffe, die bei Reaktionstemperaturen
und Reaktionsdrücken
flüssig
sind. Erläuternde
Beispiele nützlicher
Lösungsmittel
beinhalten, sind aber nicht darauf beschränkt, Alkane, wie zum Beispiel Pentan,
Isopentan, Hexan, Heptan, Octan und Nonan, sowie Mischungen aus
Alkanen beinhaltend Kerosin und Isopar ETM,
erhältlich
von Exxon Chemicals Inc.; Cycloalkane, wie zum Beispiel Cyclopentan,
Cyclohexan und Methylcyclohexan; und Aromaten, wie zum Beispiel
Benzol, Toluol, Xylole, Ethylbenzol und Diethylbenzol.
-
Die
einzelnen Bestandteile sowie die Katalysatorkomponenten sollten
zu jeder Zeit vor Sauerstoff und Feuchtigkeit geschützt werden.
Daher sollten die Katalysatorkomponenten und Katalysatoren in einer
sauerstofffreien und feuchtigkeitsfreien Atmosphäre hergestellt und aufbewahrt
werden. Bevorzugt werden daher die Reaktionen in Gegenwart von einem
trockenen Inertgas, wie zum Beispiel Stickstoff oder Argon, durchgeführt.
-
Die
Polymerisation kann als ein Batch- oder kontinuierliches Polymerisationsverfahren
durchgeführt werden.
Bevorzugt wird ein kontinuierlicher Prozess, bei dessen Ablauf Katalysatoren,
Lösungsmittel
oder Verdünnungsmittel
(falls eingesetzt) und Comonomere (oder Monomer) kontinuierlich
der Reaktionszone zugeführt
werden und Polymerprodukt kontinuierlich daraus entfernt wird. Die
Polymerisationsbedingungen zur Herstellung der Interpolymere gemäß den erfindungsgemäßen Ausführungsformen
sind im Allgemeinen solche, die in Lösungspolymerisations-Verfahren
nützlich
sind, obwohl die Anwendung nicht darauf beschränkt ist. Es wird auch angenommen,
dass Gasphasen- und Fällungspolymerisations-Verfahren
nützlich
sind, vorausgesetzt, dass einwandfreie Katalysatoren und Polymerisationsbedingungen
eingesetzt werden.
-
In
manchen Ausführungsformen
wird die Polymerisation in einem kontinuierlichen Lösungspolymerisationssystem
durchgeführt,
das zwei oder mehrere Reaktoren, die in Serie oder parallel geschaltet
sind, umfasst. Eine Katalysatorlösung,
umfassend einen Nicht-Metallocen, metallzentrierten Katalysator
mit Heteroaryl-Ligand wie bereits beschrieben, wird verwendet, um
Propylen und gegebenenfalls zusätzliche
Olefinmonomere in mindestens einem Reaktor zu polymerisieren. In
einem Reaktor wird ein Produkt mit relativ hohem Molekulargewicht
(Mw von 100.000 bis über 1.000.000, stärker bevorzugt
200.000 bis 500.000) gebildet, während in
dem zweiten oder folgenden Reaktor(en) ein Produkt relativ geringer
Molekulargewicht (Mw 2.000 bis 300.000)
gebildet wird. Das Endprodukt ist eine Mischung der zwei Reaktorausflüsse, die
vor der Entgasung kombiniert werden, um eine einheitliche Mischung
der zwei Polymerprodukte zu ergeben. Ein solches Zweireaktor/Zweikatalysatorverfahren
erlaubt die Herstellung von Produkten mit maßgeschneiderten Eigenschaften.
-
In
einer Ausführungsform
sind die Reaktoren in Serie geschaltet, d.h. der Ausfluss aus dem
ersten Reaktor wird in den zweiten Reaktor eingebracht und frisches)
Monomer (e), Lösungsmittel
und Wasserstoff wird dem zweiten Reaktor zugegeben. Die Reaktorbedingungen
sind so angepasst, dass das Gewichtsverhältnis von Polymer, das in dem
ersten Reaktor hergestellt wird, zu dem das in dem zweiten Reaktor
hergestellt wird, typischerweise von 20:80 bis 80:20 liegt. Bevorzugt
liegt das Gewichtsverhältnis
von Polymer, das in dem ersten Reaktor hergestellt wird zu dem,
das in dem zweiten Reaktor hergestellt wird, von 25:75 bis 72:25,
stärker bevorzugt
von 30:70 bis 70:30.
-
Ein
repräsentatives
Beispiel einer Serienpolymerisation ist die Herstellung von P/E*-Copolymer, bei der
in dem ersten Reaktor Propylen, Ethylen, Lösungsmittel und Katalysator
unter Lösungsphasenbedingungen
in Kontakt gebracht werden, so dass Propylen und Ethylen copolymerisieren,
um ein Propylen-Copolymer zu bilden. Der Nicht-Metallocen, metallzentrierte
Katalysator mit Heteroaryl-Ligand ist allerdings hochaktiv und setzt
unter geeigneten Bedingungen 50 % oder mehr des Propylens und fast
das gesamte Ethylen zu dem Copolymer um. Folglich ist, wenn der
Inhalt, d.h. die Reaktionsmasse, des ersten Reaktors in den zweiten
Reaktor überführt wird,
das meiste, wenn nicht alles Polymer, das in dem zweiten Reaktor
hergestellt wird, Propylenhomopolymer, da Ethylencomonmer in dem
zweiten Reaktor abwesend oder nahezu abwesend ist. Der Fachmann
versteht, dass viele Variationen dieses Themas möglich sind, indem Ethylen durch
eine oder mehrere ungesättigte
Comonomere ersetzt wird oder indem Ethylen in Kombination mit einem
oder mehreren ungesättigen
Comonomeren verwendet wird; indem ein zweiter, aber ähnlicher
Katalysator in dem zweiten Reaktor verwendet wird; etc.
-
In
einer Ausführungsform
enthält
der zweite Reaktor in einem Serienpolymerisations-Verfahren einen im
Fachgebiet bekannten heterogenen Ziegler-Natta-Katalysator oder
Chromkatalysator. Beispiele für
Ziegler-Natta-Katalysatoren beinhalten, sind aber nicht darauf beschränkt, titanbasierte
Katalysatoren, geträgert auf
MgCl2, und umfassen zusätzlich Aluminiumverbindungen,
die mindestens eine Aluminium-Alkylbindung enthalten. Geeignete
Ziegler-Natta-Katalysatoren und ihre Herstellung beinhalten, sind
aber nicht darauf beschränkt
solche, die in USP 4,612,300, 4,330,646 und 5,869,575 offenbart
sind. In einer anderen Ausführungsform
der vorliegenden Erfindung enthält
der zweite Reaktor in einem Serienpolymerisations-Verfahren einen Katalysator
mit gezwungener Geometrie oder einen Bis-Cp-Metallocenkatalysator.
-
In
einer anderen Ausführungsform
werden die Propylen/Ethylencopolymere in hoher Ausbeute und Produktivität hergestellt.
Das Verfahren, das eingesetzt wird, um diese Copolymere herzustellen,
kann entweder ein Lösungs-
oder Fällungsverfahren
sein, die beide im Fachgebiet bekannt sind. Kaminski, J. Poly. Sci. Band
23, Seiten 2151-64 (1985) berichteten über die Verwendung von einem
löslichen
Bis(cyclopentadienyl) zirkondimethyl-Alumoxan-Katalysatorsystem
zur Lösungspolymerisation
von Propylen/Ethylen (PE)-Elastomeren. USP 5,229,478 offenbart ein
Fällungspolymerisations-Verfahren,
das ähnliche
Bis(cyclopentadienyl)-zirkonbasierte Katalysatorsysteme verwendet.
-
Das
folgende Verfahren kann durchgeführt
werden, um ein P/E*-Copolymer zu erhalten: In einem gerührten Behälterreaktor
wird Propylenmonomer kontinuierlich zusammen mit Lösungsmittel
und Ethylenmonomer eingeführt.
Der Reaktor enthält
eine flüssige
Phase, die im Wesentlichen aus Ethylen- und Propylenmonomeren zusammen
mit irgendeinem Lösungsmittel
oder zusätzlichen
Verdünnungsmittel
besteht. Falls erwünscht,
kann auch eine kleine Menge von einem „H"-verzweigungsinduzierenden Dien, wie
zum Beispiel. Norbornadien, 1,7-Octadien oder 1,9-Decadien zugegeben
werden. Ein Nicht-Metallocen, metallzentrierter Katalysator mit
Heteroaryl-Ligand und geeignete Cokatalysatoren werden kontinuierlich
in die flüssige
Phase des Reaktors eingeführt.
Die Reaktortemperatur und der Reaktordruck können über das Einstellen des Verhältnisses
von Lösungsmittels/Monomer,
der Katalysatorzugabe-Rate sowie durch Kühl- oder Heizschlangen oder
-mäntel
oder beidem gesteuert werden. Die Polymerisationsrate wird durch
die Rate der Zugabe an Katalysator gesteuert. Der Ethylengehalt
des Polymerprodukts wird über
das Verhältnis
von Ethylen zu Propylen in dem Reaktor gesteuert, das mittels Einstellen
der jeweiligen Zuflussrate dieser Komponenten in den Reaktor gesteuert
wird. Das Molekulargewicht des Polymerprodukts wird gegebenenfalls
durch Steuerung anderer Polymerisationsvariablen, wie zum Beispiel
der Temperatur, Monomerkonzentration oder durch einen Strom von Wasserstoff,
der in den Reaktor eingeführt
wird, wie es im Fachgebiet bekannt ist, gesteuert. Der Reaktorabfluss
wird mit einem Reagenz, das den Katalysator deaktiviert, wie z.B.
Wasser, in Kontakt gebracht. Die Polymerlösung wird gegebenenfalls geheizt
und das Polymerprodukt wird durch Abtrennen von nicht umgesetzten,
gasförmigen
Ethylen und Propylen sowie von restlichem Lösungsmittel oder Verdünnungsmittel
unter reduziertem Druck, erhalten und falls nötig wird Entgasung in einem
Gerät,
wie zum Beispiel in einem Entgasungsextruder oder einem anderen
Entgasungsgerät,
das bei reduziertem Druck betrieben wird, durchgeführt. Für ein Lösungspolymerisationsverfahren,
insbesondere für
eine kontinuierliche Lösungspolymerisation,
liegen bevorzugte Bereiche der Propylenkonzentration bei einem Gleichgewichtszustand
von 0,05 Gew.-% des gesamten Reaktorinhalts bis 50 Gew.-% des gesamten
Reaktorinhalts, stärker
bevorzugt von 0,5 Gew.-% des gesamten Reaktorinhalts bis 30 Gew.-%
des gesamten Reaktorinhalts und am stärksten bevorzugt von 1 Gew.-%
des gesamten Reaktorinhalts bis 25 Gew.-% des gesamten Reaktorinhalts.
Der bevorzugte Bereich an Polymerkonzentration (andererseits bekannt
als Prozentanteil an Feststoffen) liegt von 3 Gew.-% des Reaktorinhalts
bis 45 Gew.-% des Reaktorinhalts oder höher, stärker bevorzugt von 10 % des
Reaktorinhalts bis 40 % des Reaktorinhalts und am stärksten bevorzugt
von 15 % des Reaktorinhalts bis 40 % des Reaktorinhalts.
-
In
einem kontinuierlichen Verfahren liegt die mittlere Verweildauer
des Katalysators und des Polymers in dem Reaktor im Allgemeinen
bei 5 Minuten bis 8 Stunden und bevorzugt bei 10 Minuten bis 6 Stunden,
stärker
bevorzugt bei 10 Minuten bis 1 Stunde.
-
In
einigen Ausführungsformen
wird Ethylen zu dem Reaktionsgefäß in einer
Menge zugegeben, um einen Differentialdruck über dem kombinierten Dampfdruck
von Propylen und Dienmonomeren zu halten. Der Ethylengehalt des
Polymers wird durch das Verhältnis
von Ethylendifferentialdruck zu Gesamtdruck innerhalb des Reaktors
bestimmt. Im Allgemeinen wird das Polymerisationsverfahren bei einem
Ethylendruck von 70 bis 7000 kPa (10 bis 1000 psi), am stärksten bevorzugt
von 30 bis 600 kPa (40 bis 800 psi) durchgeführt. Die Polymerisation wird
im Allgemeinen bei einer Temperatur von 25 bis 250 °C, bevorzugt
von 75 bis 200 °C
und am meisten bevorzugt von größer als
95 bis 200 °C
durchgeführt.
-
In
einer anderen Ausführungsform
umfasst ein Verfahren zur Herstellung eines Propylenhomopolymers
oder Interpolymers aus Propylen mit mindestens einem zusätzlichen
olefinischen Monomer, ausgewählt aus
Ethylen oder C4-20 α-Olefin, die folgenden Schritte:
1) Kontrollierte Zugabe eines Nicht-Metallocen, metallzentrierten Katalysators
mit Heteroaryl-Ligand, der einen Cokatalysator und gegebenenfalls
eine Fängerkomponente
beinhaltet, in einen Reaktor ; 2) kontinuierliche Beschickung des
Reaktors mit Propylen und gegebenenfalls einem oder mehreren zusätzlichen
olefinischen Monomeren, unabhängig
voneinander ausgewählt aus
Ethylen oder C4-20 α-Olefinen, gegebenenfalls mit
einem Lösungsmittel
oder Verdünnungsmittel
und gegebenenfalls mit einer bestimmten Menge an H2;
und 3) erhalten des Polymerprodukts. Das Verfahren ist bevorzugt
ein kontinuierliches Lösungsverfahren.
Die Cokatalysatoren und gegebenenfalls die Fängerkomponenten können in
dem neuen Verfahren unabhängig
voneinander mit der Katalysatorkomponente gemischt werden, bevor
sie in den Reaktor eingeführt
werden oder sie können
jeweils unabhängig
voneinander in separaten Strömen
in den Reaktor zugeführt
werden, was zu einer Aktivierung direkt im Reaktor führt. Fängerkomponenten
sind im Fachgebiet bekannt und beinhalten, sind aber nicht darauf
beschränkt,
Alkylaluminiumverbindungen, beinhaltend Alumoxane. Beispiele für Fänger beinhalten,
sind aber nicht beschränkt
auf, Trimethylaluminium, Triethylaluminium, Triisobutylaluminium,
Trioctylaluminium, Methylalumoxan (MAO) und andere Alumoxane, beinhaltend,
aber nicht darauf beschränkt,
MMAO-3A, MMAO-7, PMAO-IP (alle erhältlich bei Akzo Nobel).
-
Wie
bereits erwähnt,
kann das oben beschriebene Verfahren auch gegebenenfalls mehr als
einen Reaktor verwenden. Die Verwendung von einem zweiten Reaktor
ist besonders nützlich
in solchen Ausführungsformen,
bei denen ein zusätzlicher
Katalysator, besonders ein Ziegler-Natta oder Chromkatalysator oder
ein Metallocenkatalysator. besonders ein CGC, eingesetzt wird. Der
zweite Reaktor beinhaltet typischerweise den zusätzlichen Katalysator.
-
Durch
einwandfreie Auswahl der Verfahrensbedingungen inklusive der Katalysatorauswahl
können Polymere
mit maßgeschneiderten
Eigenschaften hergestellt werden. Für ein Lösungspolymensationsverfahren,
insbesondere eine kontinuierliche Lösungspolymerisation, liegen
bevorzugte Bereiche der Ethylenkonzentration bei einem Gleichgewichtszustand
von weniger als 0,02 Gew.-% des gesamten Reaktorinhalts bis 5 Gew.-%
des gesamten Reaktorinhalts und der bevorzugte Bereich der Polymerkonzentration
liegt von 10 Gew.-% des Reaktorinhalts bis 45 % des Reaktorinhalts
oder höher.
-
Im
Allgemeinen sinkt die Katalysatoreffizienz (ausgedrückt im Sinne
von Gramm Polymer pro Gramm Übergangsmetall)
bei steigender Temperatur und sinkender Ethylenkonzentration. Zusätzlich sinkt
das Molekulargewicht des Polymerprodukts im Allgemeinen bei ansteigender
Reaktortemperatur und sinkt mit sinkender Propylen- und Ethylenkonzentration.
Das Molekulargewicht des Polyolefins kann auch über die Zugabe von Kettentransferverbindungen,
insbesondere durch die Zugabe von H2, gesteuert
werden.
-
Anwendungen
-
Die
isotaktischen Propylenhomo- und -copolymere, die in dem erfindungsgemäßen Fasern
verwendet werden, haben viele nützliche
Anwendungen. Repräsentative
Beispiele beinhalten Mono- und Multifilamentfasern, Stapelfasern,
Bindefasern, Spinnvlies- oder schmelzzerstäubte Fasern (beispielsweise
unter Verwendung von Systemen wie in USP 4,430,563, 4,663,220, 4,668,566
oder 4,322,027 offenbart), sowohl gewebte Gewebe als auch Vliese,
Gurtband-Monofilament, kontinuierliches Filament (z.B. zur Verwendung
in Bekleidung und Polstermaterialien) und Strukturen, die aus solchen
Fasern hergestellt werden (beinhaltend z.B. Mischungen dieser Fasern
mit anderen Fasern, wie zum Beispiel PET oder Baumwolle. Stapel-
und Filamentfasern können
in den endgültigen
Faserdurchmesser direkt schmelzgesponnen werden ohne zusätzliches
Ziehen oder sie können
in einem größeren Durchmesser
schmelzgesponnen werden und anschließend heiß oder kalt zu dem gewünschten
Durchmesser mittels konventionellen Faserziehtechniken gezogen werden.
-
Die
Polymere, die in der Praxis dieser Erfindung verwendet werden, weisen
auch exzellente Elastizität auf. 8 zeigt
die Vergleichsergebnisse von zwei erfindungsgemäßen Polymeren bei unterschiedlichen Ethylengehalten
gegenüber
einem Metallocen-katalysierten Propylenpolymer, einem Polyethylen-Elastomer, einem
Ethylen-Styrol-Elastomer und einem hydrierten Styrol-Ethylen-Buten-Styrol-Block-Elastomer. Das erfindungsgemäße Polymer
ist nicht nur gut verglichen mit jedem dieser Elastomere, sondern
ist besonders gut verglichen mit dem Metallocenkatalysierten Propylenpolymer
(eine Steigerung des Ethylengehalts um dreißig Prozent in dem Metallocen-katalysierten
Propylenpolymer war erforderlich, um eine vergleichbare Elastizität zu erhalten).
Von noch größerem Interesse
ist, dass in 9 die Vergleichsergebnisse
dieser gleichen Elastomere, nachdem sie gedehnt worden sind, gezeigt
sind. Nicht nur die Elastizität
der erfindungsgemäßen Polymere
war signifikant erhöht,
sondern sogar ein Vergleich mit den hydrierten Styrol-Ethylen-Buten-Styrol-Block-Elastomeren
war positiv. Die Daten für
diese Figuren sind in Beispiel 10 gezeigt.
-
Ob
Vordehnen wünschenswert
ist oder nicht, hängt
von der Anwendung ab. Zum Beispiel können die erfindungsgemäßen Elastomeren
Propylenhomo- und -copolymere, die thermoplastischen Triblock-Elastomere
als die Filamentschicht in dem gestreckt gebundenem (stretch bond)
Laminatverfahren aus USP 6,323,389 ersetzen. Die Filamentschicht
würde gedehnt
werden, bevorzugt nur einmal, bevor sie zwischen die zwei Spinnvliesschichten
eingebracht wird. In einem alternativen Beispiel können die
erfindungsgemäßen elastomeren
Polymere die elastische Schicht in dem eingeschnürt gebundenen (necked bonded)
Laminatverfahren aus USP 5,910,224 ersetzen. Ein gewisses Vordehnen
des Propylenpolymers kann bevorzugt sein.
-
Die
erfindungsgemäßen Polymere
können
entweder allein oder in Verbindung mit einem oder mehreren anderen
Polymeren (entweder erfindungsgemäßen Polymeren oder nicht erfindungsgemäßen Polymeren),
falls erwünscht
oder notwendig, mit verschiedenen Additiven, wie zum Beispiel Antioxidantien,
Ultraviolett absorbierenden Reagenzien, Antistatika, Nukleierungsmitteln,
Gleitmitteln, Flammschutzmitteln, Antiblockmitteln, Farbmitteln,
anorganischen oder organischen Füllstoffen
oder dergleichen gemischt werden.
-
Elastische
Fasern, die Polyolefine umfassen, sind bekannt zum Beispiel aus
USP 5,272,236, 5,278,272, 5,322,728, 5,380,810, 5,472,775, 5,645,542,
6,140,442 und 6,225,243. Die erfindungsgemäßen Polymere können grundsätzlich auf
die gleiche Weise zur Herstellung und Verwendung von elastischen
Fasern verwendet werden wie bekannte Polyolefine. In dieser Beziehung
können
die erfindungsgemäßen Polymere funktionelle
Gruppen, wie zum Beispiel Carbonyl-, Sulfid-, Silanreste etc. beinhalten
und können
vernetzt oder nicht vernetzt sein. Im Falle von vernetzten Polymeren
können
die Polymere mittels bekannter Techniken und Materialien vernetzt
werden, voraussetzend, dass nicht alle Vernetzungstechniken und
Materialien bei allen Polyolefinen wirksam sind, z.B. sind Peroxid,
azo- und elektromagnetische Bestrahlungstechniken (wie zum Beispiel
Elektronenstrahl, UV, IR und sichtbares Licht), alle, zumindest
zu einem bestimmten Grad, bei Polyethylenen wirksam, allerdings
sind nur einige von diesen, z.B. Elektronenstrahl, bei Polyproylenen
wirksam und auch dann nicht notwendigerweise im gleichen Ausmaß wie bei
Polyethylenen. Die Verwendung von Additiven, Promotoren etc. kann
wie erwünscht
eingesetzt werden.
-
„Faser" meint ein Material,
bei dem das Verhältnis
von Länge-
zu Durchmesser typischerweise größer als
10 ist. Der Faserdurchmesser kann gemessen und angegeben werden
in einer Vielzahl von Arten. Im Allgemeinen wird der Faserdurchmesser
in Denier pro Filament gemessen. Denier ist ein Ausdruck aus dem
Textilbereich, der definiert ist als Gramm der Faser pro 9000 Meter
Faserlänge.
Monofilament bezieht sich im Allgemeinen auf einen extrudierten
Strang mit einem Filament von größer als
15 Denier, gewöhnlich
von größer als
30. Eine feine Denierfaser bezeichnet im Allgemeinen eine Faser
mit 15 Denier oder weniger. Mikrodenier (aka Mikrofaser) bezeichnet
im Allgemeinen eine Faser mit einem Durchmesser von nicht mehr als
100 Mikrometer.
-
„Filamentfaser" oder „Monofilamentfaser" meint einen kontinuierlichen
Materialstrang von undefinierter (d.h. nicht vorbestimmter) Länge, im
Gegensatz zu einer „Stapelfaser", die einen diskontinuierlichen
Materialstrang einer bestimmten Länge (d.h. einen Strang der
geschnitten oder andersartig in Segmente einer vorbestimmten Länge geteilt
wurde) bezeichnet.
-
„Elastisch" bedeutet, dass eine
Faser mindestens 50 Prozent seiner gedehnten Länge nach dem ersten Ziehen
und nach dem vierten bei 100 % Dehnung (Verdopplung der Länge) wiedererlangt.
Elastizität
kann auch als die „permanente
Verformung" der
Faser beschrieben werden. Permanente Verformung ist das Gegenteil
von Elastizität.
Eine Faser wird bis zu einem gewissen Punkt gedehnt und anschließend zu
der ursprünglichen
Position vor dem Dehnen entlastet und anschließend wieder gedehnt. Der Punkt,
bei dem die Faser beginnt eine Last zu ziehen, wird als die prozentuale
permanente Verformung bezeichnet. „Elastische Materialien" werden im Fachgebiet
auch als „Elastomere" und „Elastomer" bezeichnet. Elastisches
Material (manchmal bezeichnet als ein elastischer Artikel) beinhaltet
das Polyolefinpolymer selbst sowie, aber nicht darauf beschränkt, das
Polyolefinpolymer in der Form einer Faser, eines Films, eines Streifens,
eines Bandes, eines Bändchens,
eines Bogens, einer Beschichtung, einer Form und dergleichen. Das
bevorzugte elastische Material ist eine Faser. Das elastische Material
kann entweder ausgehärtet
oder ungehärtet,
bestrahlt oder unbestrahlt und/oder vernetzt oder unvernetzt sein.
-
„Nichtelastisches
Material" meint
ein Material, z.B. eine Faser, die nicht elastisch ist, wie oben
definiert.
-
„Homofile
Faser", „monolithische
Faser" und ähnliche
Ausdrücke
meinen eine Faser, die eine einzelne Polymerregion oder Domäne aufweist
und nicht irgendwelche anderen verschiedenen Polymerregionen aufweist
(im Gegensatz zu Bikomponentenfasern).
-
„Bikomponentenfaser" meint eine Faser,
die zwei oder mehrere verschiedene Polymerregionen oder Domänen aufweist.
Bikomponentenfasern sind auch bekannt als konjugierte Fasern oder
Multikomponentenfasern. Die Polymere unterscheiden sich gewöhnlicherweise
voneinander, obwohl zwei oder mehr Komponenten das gleiche Polymer
umfassen können.
Die Polymere sind in im Wesentichen verschiedenen Zonen über den
Querschnitt der Bikomponentenfaser angeordnet und erstrecken sich
gewöhnlicherweise
kontinuierlich entlang der Länge
der Bikomponentenfaser. Die Konfiguration einer Bikomponentenfaser
kann zum Beispiel eine Hülle/Kernanordnung
(in der ein Polymer umgeben ist von einem anderen), eine Seite-an-Seite-Anordnung,
eine Kuchenanordnung oder eine „Inseln-in-der-See"-Anordnung sein.
Bikomponentenfasern sind ferner beschrieben in USP 6,225,243, 6,140,442,
5,382,400, 5,336,552 und 5,108,820.
-
„Schmelzzerstäubte Fasern" sind Fasern, die
durch Extrusion einer geschmolzenen thermoplastischen Polymerzusammensetzung
durch eine Vielzahl von feinen, gewöhnlich kreisrunden Düsenkapillaren
als geschmolzene Fäden
oder Filamente in zusammenlaufende Hochgeschwindigkeitsgasströme (z.B.
Luft), die dazu beitragen, dass die Fäden oder Filamente auf verringerte
Durchmesser gebracht werden, hergestellt werden. Die Filamente oder
Fäden werden
durch die Hochgeschwindigkeitsgasströme transportiert und auf eine Sammeloberfläche abgeschieden,
um ein Gewebe aus zufällig
verteilten Fasern mit durchschnittlichen Durchmessern, im Allgemeinen
kleiner als 10 Mikrometer, zu bilden.
-
„Schmelzgesponnene
Fasern" sind Fasern,
die durch Schmelzen mindestens eines Polymers und anschließendem Ziehen
der Faser in der Schmelze zu einem Durchmesser (oder einer anderen
Querschnittsform) von weniger als dem Durchmesser (oder einer anderen
Querschnittsform) der Düse,
gebildet werden.
-
„Spinnvliesfasern" sind Fasern, die
durch Extrusion einer geschmolzenen thermoplastischen Polymerzusammensetzung
als Filamente durch eine Vielzahl von feinen, gewöhnlich kreisrunden,
Düsenkapillaren
einer Spinndüse
hergestellt werden. Der Durchmesser der extrudierten Filamente wird
rasch reduziert und die Filamente werden anschließend auf
einer Sammeloberfläche
abgeschieden, um ein Gewebe aus zufällig verteilten Fasern mit
im Allgemeinen durchschnittlichen Durchmessern zwischen 7 und 30 μm (Mikrometer)
zu bilden.
-
„Vlies" meint ein Flor oder
Gewebe mit einer Struktur einzelner Fasern oder Fäden, die
zufällig übereinander
gelegt sind, allerdings nicht in einer identifizierbaren Art und
Weise, wie es bei Maschenware der Fall ist. Die elastische Faser
der vorliegenden Erfindung kann eingesetzt werden, um Vliesstrukturen
sowie Kompositstrukturen elastischer Vliesgewebe in Kombination
mit nicht elastischen Materialien herzustellen.
-
Die
P/E*-Polymere können
mit anderen Polymeren gemischt werden, um, unter anderem, nützliche Fasern
zu bilden. Geeignete Polymere zum Mischen mit den P/E*-Polymeren sind kommerziell
erhältlich
bei einer Vielzahl von Anbietern und beinhalten, sind aber nicht
darauf beschränkt,
andere Polyolefine, wie zum Beispiel ein Ethylenpolymer (z.B. Polyethylen
geringer Dichte (LDPE), ULDPE, Polyethylen mittlerer Dichte (MDPE),
LLDPE, HDPE, homogen verzweigtes lineares Ethylenpolymer, im Wesentichen
lineares Ethylenpolymer, durch Pfropfung modifiziertes Ethylenpolymer,
Ethylen-Styrol-Interpolymere (ESI), Ethylenvinylacetat-Interpolymer, Ethylenacrylsäure-Interpolymer,
Ethylenethylacetat-Interpolymer, Ethylenmethacrylsäure-Interpolymer,
Ethylenmethacrylsäure-Ionomer
und dergleichen), Polycarbonat, Polystyrol, herkömmliches Polyproylen (z.B.
Homopolymer-Polypropylen, Polyproylen-Copolymer, statistisches Blockpolypropylen- Interpolymer und
dergleichen), thermoplastisches Polyurethan, Polyamid, Polymilchsäure-Interpolymer,
thermoplastisches Blockpolymer (z.B. Styrol-Butadien-Copolymer, Styrol-Butadien-Styrol-Triblock-Copolymer,
Styrol-Ethylen-Butylen-Styrol-Triblock-Copolymer
und dergleichen), Polyetherblock-Copolymer (z.B. PEBAX), Copolyesterpolymer,
Polyester/Polyetherblockpolymere (z.B. HYTEL), Ethylen-Kohlenmonoxid-Interpolymer
(z.B. Ethylen/Kohlenmonoxid (ECO)-Copolymer, Ethylen/Acrylsäure/Kohlenmonoxid
(EAACO)-Terpolymer, Ethylen/Methacrylsäure/Kohlenmonoxid (EMAACO)-Terpolymer,
Ethylen/Vinylacetat/Kohlenmonoxid (EVACO)-Terpolymer und Styrol/Kohlenmonoxid
(SCO)), Polyethylenterephthalat (PET), chloriertes Polyethylen und
dergleichen und Mischungen davon. In anderen Worten, das isotaktische
Propylenhomo- und/oder -copolymer, wie es in der Praxis dieser Erfindung
verwendet wird, kann mit zwei oder mehreren Polyolefinen gemischt
werden oder mit einem oder mehreren Polyolefinen und/oder mit einem
oder mehreren Polymeren, abweichend von einem Polyolefin gemischt
werden. Für
den Fall, dass das isotaktische Propylenhomo- und/oder -copolymer, das
in der Praxis dieser Erfindung verwendet wird, eine Mischung aus
einem oder mehreren Polyolefinen mit einem oder mehreren Polymeren
abweichend von einem Polyolefin ist, dann umfassen die Polyolefine
mindestens 1, bevorzugt mindestens 50 und stärker bevorzugt mindestens 90
Gew.-% des Gesamtgewichts der Mischung.
-
In
einer Ausführungsform
sind ein oder mehrere P/E*-Polymere mit einem herkömmlichen
Polyproylenpolymer gemischt. Geeignete herkömmliche Polyproylen-Polymere für die Verwendung
in der Erfindung, inklusive statistischer Block-Propylen-Ethylenpolymere,
sind von einer Vielzahl von Herstellern, wie zum Beispiel Montell
Polyolefins and Exxon Chemical Company, erhältlich. Geeignete herkömmliche
Polyproylen-Polymere von Exxon werden unter den Bezeichnungen ESCORENE
and ACHIEVE vertrieben.
-
Geeignete,
durch Pfropfung modifizierte Polymere, die als Mischpolymere in
der Praxis der Erfindung nützlich
sind, sind wohl bekannt im Fachgebiet und beinhalten die verschiedenen
Ethylenpolymere, die ein Maleinsäureanhydrid-Rest
und/oder einen anderen Carbonyl-enthaltenden ethylenisch ungesättigten
organischen Rest tragen.
-
Repräsentative,
durch Pfropfung modifizierte Polymere werden in USP 5,883,188 beschrieben,
zum Beispiel ein homogen verzweigtes, durch Pfropfung mit Maleinsäureanhydrid
modifiziertes Ethylenpolymer.
-
Geeignete
Polymilchsäure
(PLA)-Polymere zur Verwendung als Mischpolymere in der Praxis dieser Erfindung
sind wohl bekannt in der Literatur (z.B. siehe D.M. Bigg et al., „Effect
of Copolymer Ratio on the Crystallinity and Properties of Polylactic
Acid Copolymers" (Einfluss
des Copolymerverhältnisses
auf die Kristallinität
und Eigenschaften von Milchsäurecopolymeren),
ANTEC' 96, Seiten
2028-2039; WO 90/01521;
EP
0 515203A und
EP
0 748 846 A2 . Geeignete Polymilchsäurepolymere werden kommerziell
von Cargill Dow unter der Bezeichnung EcoPLA vertrieben.
-
Geeignete
thermoplastische Polyurethanpolymere zur Verwendung als Mischpolymere
in der Praxis dieser Erfindung sind kommerziell erhältlich bei
The Dow Chemical Company unter der Bezeichnung PELLETHANE.
-
Geeignete
Polyolefin-Kohlenmonoxid-Interpolymere zur Verwendung als Mischpolymere
in der Praxis dieser Erfindung können
mittels wohl bekannten radikalischen Hochdruck-Polymerisationsverfahren
hergestellt werden. Allerdings können
sie auch mittels traditioneller Ziegler-Natta-Katalyse oder so genannter
homogener Katalysatorsysteme, wie zum Beispiel solche, die oben
beschrieben und auf die oben Bezug genommen wurde, hergestellt werden.
-
Geeignete
radikalisch initiierte Hochdruck-Carbonyl enthaltende Ethylenpolymere,
wie zum Beispiel Ethylenacrylsäure-Interpolymere,
zur Verwendung als Mischpolymere in der Praxis dieser Erfindung
können durch
irgendeine Technik, die im Fachgebiet bekannt ist, hergestellt werden,
beinhaltend die Verfahren, die von Thomson und Waples in USP 3,520,861,
4,988,781; 4,599,392 und 5,384,373 gelehrt werden.
-
Geeignete
Ethylenvinylacetat-Interpolymere zur Verwendung als Mischpolymere
in der Praxis dieser Erfindung sind kommerziell erhältlich bei
verschiedenen Anbietern, inklusive Exxon Chemical Company und Du
Pont Chemical Company.
-
Geeignete
Ethylen/Alkylacrylat-Interpolymere zur Verwendung als Mischpolymere
in der Praxis dieser Erfindung sind kommerziell von verschiedenen
Anbietern erhältlich.
Geeignete Ethylenacrylsäure-Interpolymere
zur Verwendung als Mischpolymere in der Praxis dieser Erfindung
sind kommerziell bei The Dow Chemical Company unter der Bezeichnung
PRIMACOR erhältlich.
Geeignete Ethylen/Methacrylsäure-Interpolymere
zur Verwendung als Mischpolymere in der Praxis dieser Erfindung
sind kommerziell bei Du Pont Chemical Company unter der Bezeichnung
NUCREL erhältlich.
-
Chloriertes
Polyethylen (CPE), insbesondere chlorierte, im Wesentichen lineare
Ethylenpolymere zur Verwendung als Mischpolymere in der Praxis dieser
Erfindung können
durch Chlorierung von Polyethylen gemäß wohl bekannten Techniken
hergestellt werden. Chloriertes Polyethylen umfasst bevorzugt gleich
oder mehr als 30 Gew.-% Chlor. Geeignete chlorierte Polyethylene
zur Verwendung als Mischpolymere in der Praxis dieser Erfindung
sind kommerziell bei The Dow Chemical Company unter der Bezeichnung
TYRIN erhältlich.
-
Bikomponentenfasern
können
auch aus den Propylenhomopolymeren und den erfindungsgemäßen P/E*-Copolymeren
hergestellt werden. Solche Bikomponentenfasern weisen das Polyproylenpolymer
der vorliegenden Erfindung in mindestens einem Teil der Faser auf.
In einer Hülle/Kern-Bikomponentenfaser
(d.h. einer, in der die Hülle
den Kern umgibt) kann das Polyproylen zum Beispiel entweder in der
Hülle oder
dem Kern sein. Verschiedene erfindungsgemäße Polyproylenpolymere können auch
unabhängig
voneinander als die Hülle
und der Kern in der gleichen Faser bevorzugt, wenn beide Komponenten
elastisch sind und insbesondere wenn die Hüllenkompomente einen niedrigeren
Schmelzpunkt als die Kernkomponente hat, verwendet werden. Andere
Bikomponentenfasertypen sind ebenfalls innerhalb des Schutzbereichs
der Erfindung und beinhalten zum Beispiel Strukturen, wie Seite-an-Seite konjugierte
Fasern (d.h. Fasern mit separaten Polymerregionen, wobei das Polyolefin
der vorliegenden Erfindung mindestens eine Region der Faser umfasst).
-
Die
Form der Faser ist nicht beschränkt.
Zum Beispiel hat eine typische Faser eine runde Querschnittsform,
allerdings haben Fasern manchmal auch verschiedene Formen, wie zum
Beispiel eine dreiflüglige
Struktur oder eine flache Form (d.h. eine „bandartige") Form. Die Ausführungsformen
der erfindungsgemäßen Faser
sind nicht auf die Faserform beschränkt.
-
Die
Fasern, elastisch oder nicht elastisch, können mit anderen Fasern, wie
zum Beispiel PET, Nylon, Baumwolle, KevlarTM etc.
verwendet werden, um elastische und nicht elastische Gewebe herzustellen.
-
Gewebe,
die aus den erfindungsgemäßen elastischen
und/oder nicht elastischen Fasern hergestellt werden, beinhalten
gewebte Gewebe, Vliesstoffe und Maschenware. Vliesstoffe können mittels
verschiedenartiger Varianten hergestellt werden, z.B. wasserstrahlverfestigtes
(oder hydrodynamisch verhaktes) Vlies, wie in USP 3,485,706 und
4,939,016 offenbart, Kartieren und thermisches Verbinden von Stapelfasern;
Spinnverbinden kontinuierlicher Fasern in einem kontinuierlichen
Schritt; oder durch Schmelzzerstäuben
von Fasern in ein Gewebe und anschließendes Calandrieren oder thermisches
Binden des erhaltenen Gewebes. Diese verschiedenen Herstellungstechniken
für Vliesstoffe
sind dem Fachmann wohl bekannt und die Offenbarung ist nicht auf
irgendein besonderes Verfahren beschränkt. Andere Strukturen, die
aus solchen Fasern hergestellt werden, sind auch innerhalb des Schutzbereichs
der Erfindung beinhaltet und beinhalten z.B. Mischungen solcher
neuen Fasern mit anderen Fasern (z.B. Polyethylenterephthalat oder
Baumwolle).
-
Hergestellte
Artikel, die unter Verwendung der elastischen und/oder nicht elastischen
Fasern und Gewebe dieser Erfindung hergestellt werden können, beinhalten
Kompositartikel (z.B. Windeln), die elastische Anteile aufweisen.
Zum Beispiel werden elastische Anteile typischerweise in Teile des
Bundes einer Windel eingearbeitet, um zu verhindern, dass die Windel
rutscht und in Teile der Beinbänder
um undichte Stellen zu verhindern (wie gezeigt in USP 4,381,781).
Oft fördern
die elastischen Anteile eine bessere Passform und/oder Verschlusssysteme
für eine
gute Kombination aus Komfort und Sicherheit. Durch die erfindungsgemäßen elastischen
Fasern und Gewebe können
auch Strukturen hergestellt werden, die Elastizität mit Atmungsaktivität vereinen.
Zum Beispiel können
die erfindungsgemäßen elastischen
Fasern, Gewebe und/oder Filme in Strukturen eingearbeitet werden,
wie sie in der vorläufigen
US-Patentanmeldung
60/083 784, eingereicht am 1. Mai 1998, offenbart sind.
-
Die
erfindungsgemäßen elastischen
Fasern und Gewebe können
auch in verschiedene Strukturen, wie sie in USP 2,957,512 beschrieben
sind, verwendet werden. Zum Beispiel kann die Schicht 50 der Struktur (d.h.
die elastische Komponente), die in USP '512 beschrieben sind, durch die erfindungsgemäßen elastischen Fasern
und Gewebe ersetzt werden, besonders dort, wo flache, gewirkte,
gekräuselte,
gequetschte etc. nicht elastische Materialien zur elastischen Struktur
verwendet werden. Die Befestigung der erfindungsgemäßen elastischen
Fasern und/oder Gewebe an nicht elastische Fasern oder Gewebe und
andere Strukturen kann mittels Schmelzkleben oder mit Klebstoffen
erreicht werden. Zusammengezogene oder gekräuselte elastische Strukturen
können
aus den erfindungsgemäßen elastischen
Fasern und/oder Geweben und nicht elastischen Komponenten durch
Falten der nicht elastischen Komponente (wie beschrieben in USP '512) vor der Befestigung,
Vordehnen der elastischen Komponente, vor der Befestigung oder Wärmeschrumpfen
der elastischen Komponente nach Befestigung, hergestellt werden.
-
Kontinuierliche
elastische Filamente, wie sie hierin beschrieben sind, können auch
in gewobenen Anwendungen, wo hohe Elastizität erwünscht ist, verwendet werden.
-
USP
5,037,416 beschreibt die Vorteile einer sich anpassenden Deckfolie
unter Verwendung elastischer Bänder
(vergleiche Teil 19 in USP '416).
Die erfindungsgemäßen elastischen
Fasern können
in der Funktion von Teil 19 in USP '416 dienen oder können in Gewebeform verwendet
werden, um die gewünschte
Elastizität
bereitzustellen.
-
Die
erfindungsgemäßen elastischen
Fasern können
auch eine schmelzzerstäubte
elastische Komponente, wie in Referenz 6 der Zeichnungen in USP
4,879,170 beschrieben, sein.
-
Es
können
auch elastische Platten aus den hier beschriebenen erfindungsgemäßen elastischen
Fasern und Geweben hergestellt werden und können zum Beispiel als Teile
18, 20, 14 und/oder 26 der USP 4,940,464 verwendet werden. Die hier
beschriebenen erfindungsgemäßen elastischen
Fasern und Gewebe können
auch als elastische Komponenten von Kompositseitenwänden (wie
z.B. Schicht 86 in '464)
verwendet werden.
-
Die
erfindungsgemäßen elastischen
Materialien können
auch durchlässig
oder „atmungsaktiv" ausgerüstet werden
mittels jeglichem bekannten Verfahren, beinhaltend Öffnen, Längsschneiden,
Mikroperforieren, Mischen mit Fasern oder Schäumen, Zugabe von Füllstoffen
oder dergleichen und Kombinationen davon. Beispiele solcher Verfahren
beinhalten USP 3,156,242, 3,881,489, 3,989,867 und 5,085,654.
-
Die
isotaktischen Propylenhomo- und -copolymere, die in der Praxis dieser
Erfindung verwendet werden, können
nicht nur mit einem oder mehreren anderen Polymeren gemischt werden,
sondern sie können auch
mit verschiedenen Additiven, wie zum Beispiel Nukleierungsmittel,
Clarifyern, Steifigkeits- und/oder Kristallisationsraten beeinflussende
Mitteln gemischt werden. Diese Mittel werden auf herkömmliche
Art und Weise und in herkömmlichen
Mengen verwendet.
-
Die
P* und P/E*-Polymere können
Behandlungen nach der Reaktion unterworfen werden, wie z.B. Vernetzen,
Brechen der Viskosität
und dergleichen. Brechen der Viskosität ist besonderes nützlich um
die Viskosität
von Polymeren mit hohem Molekulargewicht zu reduzieren. Diese Nachbehandlungen
werden auch auf herkömmliche
Art und Weise angewendet.
-
Die
folgenden Beispiele sind angegeben, um verschiedene Ausführungsformen
der Erfindung darzulegen. Sie beabsichtigen nicht die Erfindung
zu beschränken,
wenn sie hierin andersartig beschrieben und beansprucht sind. Alle
numerischen Werte sind Zirka-Werte. Wenn ein numerischer Bereich
angegeben ist, versteht sich, dass die Ausführungsformen außerhalb
des Bereichs immer noch innerhalb des Schutzbereichs der Erfindung
liegen, wenn nicht anders angegeben. In den folgenden Beispielen
wurden verschiedene Polymere anhand einer Vielzahl an Verfahren
charakterisiert. Leistungsdaten dieser Polymere wurden auch erhalten.
Die meisten Verfahren oder Tests wurden gemäß einem ASTM-Standard, falls
anwendbar, oder mittels bekannter Verfahren durchgeführt. Alle
Teile und Prozentangaben sind auf das Gewicht bezogen, wenn nicht
anders angegeben. Die 12A–12J stellen die chemischen Strukturen verschiedener
Katalysatoren, die in den folgenden Beispielen beschrieben sind,
dar.
-
Spezifische
Ausführungsformen
-
Tetrahydrofuran
(THF), Diethylether, Toluol, Hexan und ISOPAR E (erhältlich bei
Exxon Chemicals) werden nach Reinigung mit reinem, trockenem Stickstoff
unter Durchlauf durch Doppel-Säulen,
die mit aktiviertem Aluminiumoxid und Aluminiumoxid geträgerten gemischten
Metalloxidkatalysator (Q-5-Katalysator, erhältlich bei Engelhard Corp.)
beladen sind, verwendet. Alle Synthesen und die Handhabung der Katalysatorkomponenten
werden unter Verwendung von streng getrockneten und deoxygenierten
Lösungsmitteln
unter Stickstoff- oder Argon-Inertgasatmosphäre entweder in einer Glove
Box, unter Hochvakuum oder mit Schlenktechniken, wenn nicht anders
gekennzeichnet, durchgeführt.
MMAO-3A, PMAO und PMAO-IP können
von Akzo-Nobel-Corporation erworben werden.
-
Synthese von (C5Me4SiMe2NtBu)Ti(η4-1,3-Pentadien) (Katalysator A)
-
Katalysator
A kann gemäß Beispiel
17 in USP 5,556,928 synthetisiert werden.
-
Synthese von Dimethylsilyl(2-methyl-s-indacenyl)(t-butylamido)titan1,3-pentadien
(Katalysator B)
-
Katalysator
B kann gemäß Beispiel
23 in USP 5,965,756 synthetisiert werden.
-
Synthese
von N-(1,1-Dimenthylethyl)-1,1-di-p-tolyl-1-((1,2,3,3a,7a-η)-3-(1,3-dihydro-2H-isoindol-2-yl)-1H-inden-1-yl)silanaminato-(2-)-N-)dimethyltitan
(Katalysator C) (1)
Herstellung von Dichlor(N-(1,1,-dimethylethyl)-1,1-di(p-tolyl)-1-((1,2,3,3a,7a-η)-3-(1,3-dihydro-2H-isoindol-2-yl)-1H-inden-1-yl)silanaminato-(2-)-N-)-titan
-
(A)
Herstellung von N-(tert-butyl)-N-(1,1-p-tolyl)-1-(3-(1,3-dihydro-2H-isoindol-2-yl)-1H-indenyl)silyl)amin:
-
1,279
g (5,35 mmol) 1-(1H-3-indenyl)-1-(2,3-dihydro-1H-isoindolinyl)lithiumsalz,
gelöst
in 20 ml THF, werden zu 1,70 g (5,35 mmol) N-(terf-butyl)-N-(1-chlor-1,1(di(3-p-tolyl)
silylamin, gelöst
in 20 ml THF, gegeben. Nach der Zugabe wird die Reaktionsmischung
9 Stunden gerührt
und anschließend
das Lösungsmittel
unter reduziertem Druck entfernt. Der Rückstand wird mit 40 ml Hexan
extrahiert und abfiltriert. Das Lösungsmittel wird unter reduziertem
Druck entfernt und es werden 2,806 Produkt als ein grauer Feststoff
erhalten.
1H (C6D6) δ:
1,10 (s, 9H), 2,01 (s, 3H), 2,08 (s, 3H), 4,12 (d, 1H, 3JH-H = 1,5 Hz); 4,39 (d, 1H, 2JH-H = 11,1 Hz), 4,57 (d, 1H, 2JH-H = 11,7 Hz), 5,55 (d, 1H, 3JH-H = 2,1 Hz), 6,9-7,22 (m, 10H), 7,56 (d,
1H, 3JH-H = 7,8
Hz), 7,62 (d, 1H, 3JH-H =
6,9 Hz), 7,67 (d, 1H, 3JH-H =
7,8 Hz), 7,83 (d, 1H, 3JH-H =
7,8 Hz).
13C {1H}
(C6D6) δ: 21,37,
21,43, 33,78, 41,09, 50,05, 56,56, 104,28, 120,98, 122,46, 123,84,
124,71, 124,84, 126,98, 128,29, 128,52, 129,05 132,99, 133,68, 135,08,
135,90, 136,01, 138,89, 139,05, 139,09, 141,27, 146,39, 148,48.
-
(B) Herstellung von N-(terf-butyl)-N-(1,1-p-tolyl)-1-(1,3-dihydro-2H-isoindol-2-yl)-1H-indenyl)silyl)amindilithiumsalz:
-
7,4
ml einer 1,6 M n-BuLi-Lösung
werden zu einer Lösung
aus 2,726 g (5,61 mmol) N-(ferf-butyl)-N-(1,1-p-tolyl)-1-(3-(1,3-dihydro-2H-isoindol-2-yl)-1H-indenyl)silyl)amin
in 50 ml Hexan zugegeben. Während
der Zugabe von n-BuLi erscheint ein gelber Niederschlag. Nach 6
Stunden Rühren
wird der gelbe Niederschlag auf einer Fritte gesammelt, mit 2 × 25 ml
Hexan gewaschen und unter reduziertem Druck getrocknet. Es werden
2,262 g des Produkts in Form eines gelben Pulvers erhalten.
1H (C6D6) δ: 1,17 (s,
9H), 2,30 (s, 6H), 4,51 (s, 4H), 6,21 (s, 1H), 6,47 (m, 2H), 6,97
(d, 4H, 3JH-H =
8,1 Hz), 7,15 (m, 2H), 7,23 (m, 2H), 7,50 (m, 1H), 7,81 (d, 4H, 3JH-H = 7,8 Hz),
8,07 (d, 1H, 3JH-H =
7,2 Hz). 13C {1H}
(C6D6) δ: 21,65,
38,83, 52,46, 59,82, 95,33, 112,93, 114,15, 115,78, 118,29, 122,05,
122,60, 124,16, 124,78, 126,94, 127,30, 133,06, 134,75, 137,30,
141,98, 148,17.
-
(C) Herstellung von Dichlor(N-(1,1,-dimethylethyl)-1,1-di-(p-tolyl)-1-((1,2,3,3a,7a-η)-3-(1,3-dihydro-2H-isoindol-2-yl)-1H-inden-1-yl)silanaminato-(2-)-N-)-titan:
-
In
der Trockenbox werden 1,552 g (4,19 mmol) TiCl3(THF)3 in 20 ml THF suspendiert. Zu dieser Lösung werden
2,206 g (4,19 mmol) N-(terf-butyl)-N-(1,1-p-tolyl)-1-(1,3-dihydro-2H-isoindol-2-yl)-1H-indenyl)silyl)amindilithiumsalz,
gelöst
in 30 ml THF, innerhalb 1 Minute zugegeben. Die Lösung wird
anschließend
60 Minuten gerührt.
Danach werden 0,76 g PbCl2 (2,75 mmol) zugegeben
und die Lösung
60 Minuten gerührt. THF
wird anschließend
unter reduziertem Druck entfernt. Der Rückstand wird zunächst mit
60 ml Methylenchlorid extrahiert und abfiltriert. Das Lösungsmittel
wird unter reduziertem Druck entfernt, wobei ein schwarzer kristalliner
Feststoff zurückbleibt.
Hexan (30 ml) wird zugegeben und die schwarze Suspension wird 10
Stunden gerührt.
Die Feststoffe werden auf einer Fritte gesammelt, mit 30 ml Hexan
gewaschen und unter reduziertem Druck getrocknet. Man erhält 2,23
g des gewünschten
Produkts in Form eines tief violetten Feststoffs.
1H
(THF-d8) δ:
1,40 (s, 9H), 2,46 (s, 3H), 2,48 (s, 3H), 5,07 (d, 2H, 2JH-H = 12,3 Hz), 5,45 (d, 2H,2JH-H = 12,6 Hz), 5,93 (s, 1H), 6,95 (d, 1H,3JH-H = 9,0 Hz),
7,08 (d, 1H, 3JH-H =
7,8 Hz), 7,15-7,4 (m, 9H), 7,76 (d, 1H, 3JH-H = 7,8 Hz), 7,82 (d, 1H, 3JH-H = 7,5 Hz), 8,05 (d, 1H, 3JH-H = 8,7 Hz). 13C
{1H} (THF-d8) δ: 21,71,
21,76, 33,38, 56,87, 61,41, 94,5, 107,95, 122,86, 125,77, 126,68,
127,84, 127,92, 128,40, 128,49, 129,36, 129,79, 131,23, 131,29, 135,79,
136,43, 136,73, 141,02, 141,22, 150,14.
-
(2)
Herstellung von (N-(1,1,-dimethylethyl)-1,1-di-p-tolyl-1-((1,2,3,3a,7a-η)-3-(1,3-dihydro-2H-isoindol-2-yl)-1H-inden-1-yl)silanaminato-(2-)-N-)-dimethyltitan:
-
In
der Trockenbox werden 0,5 g Dichlor(N-(1,1,-dimethylethyl)-1,1-di-p-tolyl-1-((1,2,3,3a,7a-η)-3-(1,3-dihydro-2H-isoindol-2-yl)-1H-inden-1-yl)silanaminato-(2-)-N-)-titan-Komplex (0,79
mmol) in 30 ml Diethylether gelöst.
Zu dieser Lösung
werden 1,14 ml (1,6 mmol) MeLi (1,6 M in Ether) unter Rühren über einen
Zeitraum von 1 Minute zugetropft. Nachdem die Zugabe von MeLi beendet
ist, wird die Lösung
1,5 Stunden gerührt.
Diethylether wird unter reduziertem Druck entfernt und der Rückstand
mit 45 ml Hexan extrahiert. Hexan wird unter reduziertem Druck entfernt
und man erhält
ein rotes kristallines Material. Dieser Feststoff wird in etwa 7
ml Toluol und 25 ml Hexan gelöst,
abfiltriert und die Lösung
wurde 2 Tage in den Eisschrank (–27 °C) gestellt. Das Lösungsmittel
wird anschließend
dekantiert und die resultierenden Kristalle mit kaltem Hexan gewaschen
und unter reduziertem Druck getrocknet. Man erhält 156 mg Produkt.
1H (C6D6) δ: 0,25 (s,
3H), 0,99 (3H), 1,72 (s, 9H), 2,12 (s, 3H), 2,15 (s, 3H), 4,53 (d,
2H, 2JH-H = 11,7
Hz), 4,83 (d, 2H, 2JH-H =
11,7 Hz), 5,68 (s, 1H), 6,72 (dd, 1H, 3JH-H = 8,6 Hz, 3JH-H = 6,6 Hz), 6,9-7,2 (m, 11H), 7,30 (d,
1H, 3JH-H = 8,6
Hz), 7,71 (d, 1H, 3JH-H =
8,5 Hz), 7,93 (d, 1H, 3JH-H =
7,8 Hz), 8,11 (d, 1H, 3JH-H =
7,8 Hz). 13C {1H} (C6D6) δ: 21,45,
21,52, 35,30, 50,83, 56,03, 56,66, 57,65, 83,80, 105,64, 122,69,
124,51, 124,56, 125,06, 125,35, 127,33, 128,98, 129,06, 129,22,
133,51, 134,02, 134,62, 136,49, 136,84, 137,69, 139,72, 139,87, 143,84.
-
Synthese von (1H-cyclopenta[1]phenanthren-2-yl)dimethyl(t-butylamido)silantitandimethyl
(Katalysator D)
-
Katalysator
D kann gemäß Beispiel
2 in USP 6,150,297 synthetisiert werden.
-
Synthese von rac-[Dimethylsilylbis(1-(2-methyl-4-phenyl)indenyl)]zirkon(1,4-diphenyl-1,3-butadien) (Katalysator
E)
-
Katalysator
E kann gemäß Beispiel
15 in USP 5,616,664 synthetisiert werden.
-
Synthese von rac-[1,2-Ethandiylbis(1-indenyl)]zirkon(1,4-diphenyl-1,3-butadien)
(Katalysator F)
-
Katalysator
F kann gemäß Beispiel
11 in USP 5,616,664 synthetisiert werden.
-
Synthese von
Katalysator G
-
Hafniumfetrakisdimefhylamin.
Die Reaktion wird in einer Trockenbox durchgeführt. Ein 500 ml Rundkolben,
der einen Rührfisch
enthält,
wird mit 200 ml Toluol und LiNMe2 (21 g,
95 %, 0,39 mol) befüllt.
HfCl4 (29,9 g, 0,093 mol) wird langsam innerhalb
2 h zugegeben. Die Temperatur erreicht 55 °C. Die Mischung wird über Nacht
bei Umgebungstemperatur gerührt.
LiCl wird abfiltriert. Toluol wird vorsichtig aus dem Produkt abdestilliert.
Die endgültige
Reinigung wird durch Destillation über ein Vakuumsübergangsstück, an das
ein kalter Auffangkolben angebracht ist (–78 °C), erhalten. Dieses Verfahren
wird außerhalb
der Trockenbox an einer Schlenk-Line durchgeführt. Das Material hat einen Übergangspunkt
bei 110-120 °C
bei 300-600 μm.
19,2 g eines weißen
Feststoffs werden gesammelt.
-
2-Formyl-6-naphthylpyridin.
In einer Trockenbox werden Naphthylboronsäure (9,12 g, 53,0 mmol) und Na2CO3 (11,64 g, 110
mmol) in 290 ml entgasten 4:1 H2O/MeOH gelöst. Diese
Lösung
wird zu einer Lösung aus
8 g (43 mmol) 2-Brom-6-formylpyridin und 810 mg (0,7 mmol) Pd(PPh3)4 in 290 ml entgastem
Toluol gegeben. Der befüllte
Reaktor wird aus der Trockenbox entnommen und ist dabei mit Stickstoff überlagert
und wird an die N2-Hausleitung angeschlossen.
Die zweiphasige Lösung
wird kräftig gerührt und
auf 70 °C
4 h erhitzt. Beim Abkühlen
auf Raumtemperatur separiert sich die organische Phase. Die wässrige Schicht
wird mit 3 × 75
ml Et2O gewaschen. Die kombinierten organischen
Extrakte werden mit 3 × 100
ml H2O und 1 × 100 ml gesättigter
Kochsalzlösung
gewaschen und über
Na2SO4 getrocknet.
Nach Entfernen der flüchtigen
Stoffe im Vakuum wird das erhaltene hellgelbe Öl mittels Trituration mit Hexan
gereinigt. Das isolierte Material wird aus heißer Hexanlösung rekristallisiert und letztlich
werden 8,75 g erhalten, 87 % Ausbeute. Schmelzpunkt 65-66 °C.
1H NMR (CDCI3) δ: 7,2-8,3
(m, 10H), 10,25 (s, 1H) ppm. 13C NMR (CDCl3) 120,3, 125,64, 125,8, 126,6, 127,26, 128,23,
129,00, 129,74, 130,00, 131,39, 134,42, 137,67, 137,97, 153,07,
160,33, 194,23 ppm.
-
6-Naphthylpyridin-2-(2,6-diisopropylphenyl)imin:
Ein trockener 500 ml 3-Halsrundkolben
wird mit einer Lösung
aus 5,57 g (23,9 mmol) 2-Formyl-6-naphthylpyridin und 4,81 g (27,1 mmol)
2,6-Diisopropylanilin in 238 ml wasserfreiem THF, das 3 Å Molekularsieb
(6 g) und 80 mg p-TsOH enthält,
befüllt.
Die Beschickung des Reaktors wird unter N2 durchgeführt. Der
Reaktor wird mit einem Rückflusskühler, einem
mechanischen Überkopfrührer und
einem Thermoelement ausgerüstet.
Die Mischung wird unter N2 12 h unter Rückfluss
erhitzt. Nach Filtration und Entfernung der flüchtigen Stoffe im Vakuum wird
das rohe braune Öl
mit Hexan trituriert. Das Produkt wird abfiltriert und mit kaltem
Hexan gespült.
Der leicht cremefarbene Feststoff wiegt 6,42 g. Keine weitere Reinigung
wird durchgeführt.
Schmelzpunkt 142-144 °C.
1H NMR (CDCl3) δ: 1,3 (d,
12H) 3,14 (m, 2H) 7,26 (m, 3H) 7,5-7,6 (m, 5H), 7,75-7,8 (m, 3H),
8,02 (m, 1H) 8,48 (m, 2H) ppm. 13C NMR (CDCl3) 23,96, 28,5, 119,93, 123,50, 124,93, 125,88,
125,94, 126,49, 127,04, 127,24, 128,18, 128,94, 129,7, 131,58, 134,5,
137,56, 137,63, 138,34, 148,93, 154,83, 159,66, 163,86 ppm.
-
(6-Naphfhyl-2-pyridyl-N-(2,6-diisopropylphenyl)benzylamin:
Ein 250 ml 3-Halskolben, ausgerüstet
mit mechanischem Rührer
und einem N2-Einlass, wird mit 6-Naphthylpyridin-2-(2,6-diisopropylphenyl)imin
(6,19 mg, 15,8 mmol) und 80 ml wasserfreiem entgasten Et2O befüllt.
Die Lösung
wird auf –78 °C gekühlt, während eine
Lösung
von Phenyllithium (13,15 ml, 1,8 M in Cyclohexan, 23,7 mmol) innerhalb
10 min zugetropft wird. Nachdem die Lösung innerhalb 1 h auf Raumtemperatur
erwärmt
wird, wird sie bei Raumtemperatur 12 Stunden gerührt. Die Reaktion wird dann mit
~50 ml wässriger
NH4Cl gequencht. Die organische Schicht
wird abgetrennt, mit gesättigter
Kochsalzlösung
und Wasser gewaschen und anschließend über Na2SO4 getrocknet. Mittels dem Biotage-Chromatographiesystem
(Column # FKO-1107-19073, 5 % THF/95 % Hexan) wird das Produkt als
ein farbloses Öl
isoliert. Die Chromatographie wird durchgeführt, indem das rohe Öl in 50
ml Hexan gelöst
wird. Die Reinigung erfolgt in 2 × ~25 ml Ansätzen, wobei
bei jedem Lauf etwa die Hälfte
der Hexanstammlösung
verwendet wird. 7 g Öl
werden isoliert (93 % Ausbeute).
1H
NMR (CDCl3) δ: 0,90 (d, 12H) 3,0 (m, 2H),
4,86 (s, 1H), 5,16 (s, 1H), 7,00 (m, 3H), 7,1-7,6 (m, 12H), 7,8-7,88 (m,
2H), 7,91-7,99 (d, 1H) ppm. 13C NMR (CDCl3) 24,58, 28,30, 70,02, 121,14, 123,62, 123,76,
123,95, 125,71, 126,32, 126,55, 126,74, 127,45, 128,04, 128,74,
129,47, 131,66, 134,49, 137,4, 138,95, 142,68, 143,02, 143,89, 159,36,
162,22 ppm.
-
Katalysator
G-(Nme2)3: Die Reaktion
wird in einer Trockenbox durchgeführt. Ein 100 ml Rundkolben wird
mit Hf(Nme2)4 (2,5
g, 5,33 mmol), 30 ml Pentan und einem Rührfisch befüllt. Das Amin 1 wird in 40
ml Pentan gelöst
und anschließend
unter Rühren
der Hf(NMe2)4-Lösung zugegeben.
Die Mischung wird bei Umgebungstemperatur 16 h gerührt (über Nacht).
Der hellgelbe Feststoff wird abfiltriert und mit kaltem Pentan gespült. Das
Trockengewicht des Pulvers ist 2,45 g. Eine zweite Menge mit einem
Gewicht von 0,63 g wird aus dem Filtrat gewonnen. Die Gesamtausbeute
ist 74 %.
1H NMR (C6D6) δ:
0,39 (d, 3H, J = 6,77 Hz), 1,36 (d, 3H, J = 6,9 Hz), 1,65 (d, 3H,
J = 6,68 Hz), 1,76 (d, 3H, J = 6,78 Hz), 2,34 (br s, 6H), 2,80 (br
s, 6H), 2,95 (br s, 6H), 3,42 (m, 1H, J = 6,8 Hz), 3,78 (m, 1H,
J = 6,78 Hz), 6,06 (s, 1H), 6,78 (m, 2H), 6,94 (m, 1H), 7,1-7,4
(m, 13H), 7,8 (m, 2H) ppm.
-
Katalysator
G: Die Reaktion wird innerhalb einer Trockenbox durchgeführt. Ein
100 ml Rundhalskolben wird mit 70 ml Pentan und 15 ml einer 2,0
M Trimethylaluminiumlösung
in Hexan befüllt.
Die Lösung
wird auf –40 °C gekühlt. Die
Hafniumtrisamid-Verbindung aus der vorherigen Reaktion (1,07 g,
1,28 mmol) wird in kleinen Teilen innerhalb 5-10 Minuten zugegeben.
Nach der Zugabe bildet sich ein weißer gallertartiger Rückstand.
Nach 45-60 Minuten wird die Reaktion gelb, wobei ein feines gelbes
Pulver aus der Mischung ausfällt. Nach
einer gesamten Reaktionsdauer von 2,5 h wird die Mischung filtriert
und 615 mg Katalysator G werden als ein helles gelbes Pulver isoliert.
Keine weitere Reinigung wird durchgeführt.
1H
NMR (C6D6) δ: 0,51 (d,
3H, J = 6,73 Hz), 0,79 (s, 3H), 1,07 (s, 3H), 1,28 (d, 3H, J = 6,73
Hz), 1,53 (m, 6H), 3,37 (m, 1H J = 6,75 Hz), 3,96 (m, 1H, J = 6,73
Hz), 6,05 (s, 1H), 6,50 (d, 1H, J = 7,75 Hz), 6,92 (t, 1H, J = 7,93 Hz),
7,1-7,59 (m, 12H), 7,6 (d, 1H), 7,8-8,0 (m, 2H), 8,3 (m, 1H), 8,69
(d, 1H, J = 7,65 Hz) ppm.
-
Synthese von
Katalysator H
-
27
ml (43,2 mmol) einer 1,6 M n-BuLi-Lösung in Hexan werden einer
Lösung
aus 9-Bromphenanthren (10,36
mg, 41 mmol) in 132 ml wasserfreiem, entgastem Et2O,
das auf –40 °C gekühlt ist,
unter Stickstoff zugegeben. Die Lösung wird im Strudel gemischt
und man lässt
das Gemisch bei –40 °C 3 Stunden
reagieren, während
dessen aus der Lösung
farblose Kristalle ausfallen. Das 9-Phenanthrenyllithium wird als
eine Aufschlämmung
zu einer gut durchmischten Lösung
aus 6-Naphthylpyridin-2-(2,6-diisopropylphenyl)imin
(10,6 g; 27,04 mmol) in 130 ml Et2O, das
auf –40 °C gekühlt ist,
gegeben. Nachdem die Lösung
innerhalb 1 h auf Umgebungstemperatur erwärmt wurde, wird sie bei Umgebungstemperatur
2 Stunden gerührt.
Die Reaktion wird anschließend
mit wässriger
NH4Cl-Lösung
gequencht und einer wässrig/organischen
Aufarbeitung unterworfen. Die organischen Waschlösungen werden vereint und über Na2SO4 getrocknet.
Nach Entfernen der flüchtigen
Stoffe an einem Rotationsverdampfer fällt aus der Lösung das
Produkt aus. Die isolierten Feststoffe werden mit kaltem Hexan gespült. Das
Material wird bei 70 °C
unter Vakuum mit Hausvakuum über
Nacht getrocknet. Das getrocknete Material wird als ein weißer Feststoff
isoliert und wiegt 12,3 g entsprechend einer Ausbeute von 80 %.
Eine zweite Menge mit einem Gewicht von 0,37 g wird isoliert. Schmelzpunkt
166-168 °C.
1H NMR (C6D6) δ:
1,08 (dd, 12H) 3,43 (m, 2H), 5,47 (m, 1H), 6,16 (d, 1H), 7,0-7,8
(m, 14H), 8,2 (d, 1H), 8,5-8,6 (m, 4H), ppm. 13C
NMR (CDCl3) 24,68, 28,22, 68,87, 120,56,
122,89, 123,63, 123,73, 124,07, 124,1, 125,5, 125,59, 126,24, 126,42,
126,52, 126,76, 126,83, 126,9, 127,05,127,14, 128,0, 128,55, 129,49,
129,55, 130,67, 130,71, 131,52, 131,55, 132,24, 134,39, 137,57,
143,31, 159,1, 162 ppm.
-
Katalysator
H-(Nme2)3: In einer
Trockenbox werden sechs unterschiedliche, mit Teflonschraubdeckeln verschließbaren Druckrohrreaktoren
aus Glas jeweils mit Hf(NMe2)4 (1,55
g, 4,37 mmol, insgesamt 9,3 g, 26,2 mmol), 10 ml Toluol und dem
Ligand, isoliert aus dem obigen Schritt (2,1 g, 3,68 mmol, insgesamt
12,6 g, 22,1 mmol), befüllt.
Die dicht verschlossenen Reaktoren werden aus der Trockenbox entfernt
und in einem Heizblock, dessen Temperatur auf 125 °C eingestellt
ist, gestellt. Die Reaktorrohre werden über Nacht geheizt (~16 h).
Die abgekühlten
Rohre werden in die Trockenbox überführt und
der Inhalt der Reaktorrohre in einem 500 ml Rundkolben vereint.
Der Kolben wird evakuiert, um das Dimethylamin und Toluol zu entfernen.
Der hellgelbe/grüne
Feststoff der übrig
bleibt, wird mit ~125 ml kaltem Pentan gespült und filtriert. Man erhält 13,6
g eines hellgelben Pulvers entsprechend einer Ausbeute von 65 %.
-
Katalysator
H: Die Reaktion wird in einer Trockenbox durchgeführt. Ein
500 ml Gefäß wird mit
250 ml Pentan und dem Hafniumamid, das in dem gerade eben beschriebenen
Schritt isoliert wurde (13,6 g, 15,5 mmol), befüllt. Die Mischung wird auf –40 °C abgekühlt. 70
ml einer 2,0 M Trimethylaluminiumlösung (140 mmol) in Hexan werden
langsam zu der gerührten
Mischung zugegeben. Nach 3 h wird die Reaktion gelb, wobei ein feines
Pulver aus der Mischung ausfällt.
Die Mischung wird anschließend
auf –40 °C abgekühlt und
filtriert. Das anfangs gesammelte Produkt wird mit 2 × 60 ml
kaltem Pentan gespült.
10,24 g Katalysator H werden isoliert (84 % Ausbeute) mit einer
Reinheit von > 99
% entsprechend 1H NMR.
-
Synthese von Armeeniumborat[methylbis(hydriertes
Talgalkyl)ammoniumtetrakis (pentafluorphenyl)borat]
-
Armeeniumborat
kann aus ARMEEN® M2HT
(erhältlich
bei Akzo-Nobel), HCl und Li[B(C6F5)4] gemäß Beispiel
2 in USP 5,919,983 hergestellt werden.
-
Generell 1 Gallone kontinuierliches
Lösungs-Propylen/Ethylen
-
Copolymerisationsverfahren
-
Ein
1 Gallonen-Reaktor, ausgestattet mit einem Mantel zur Temperaturkontrolle
und einem internen Thermoelement, wird mit gereinigtem Toluol-Lösungsmittel,
Ethylen, Wasserstoff und Propylen beschickt. Der Lösungsmittelzufluss
in den Reaktor wird mittels eines Massenfluss-Steuergeräts gemessen.
Eine in ihrer Geschwindigkeit variable Diaphragmapumpe steuert die
Lösungsmittelflussrate
und erhöht
den Lösungsmitteldruck
zum Reaktor. Der Propylenzufluss wird mittels eines Massenflussmeters
gemessen und der Fluss wird über
eine in ihrer Geschwindigkeit variablen Diaphragmapumpe gesteuert.
Am Ausfluss der Pumpe befindet sich ein Zuspeisestrom, um einen
Spülfluss
für das
Katalysator-Injektionsrohr und den Reaktorrührer bereitzustellen. Das verbleibende
Lösungsmittel
wird mit Ethylen und Wasserstoff vereint und in den Reaktor abgegeben.
Der Ethylenstrom wird mit einem Massenflussmeter gemessen und mit
einem Research Control Ventil gesteuert. Ein Massenfluss-Steuergerät wird verwendet,
um Wasserstoff in den Ethylenstrom am Auslass des Ethylenkontrollventils
abzugeben. Die Temperatur des Lösungsmittels/Monomers
wird mittels eines Wärmeaustauschers
gesteuert, bevor sie in den Reaktor treten. Dieser Strom tritt am
Boden des Reaktors ein. Die Katalysatorkomponentenlösungen werden
mittels Pumpen und Massenflussmeter abgemessen und werden mit dem
Katalysator-Spüllösungsmittel
vereint. Dieser Strom tritt am Boden des Reaktors ein, allerdings
an einem anderen Eingang als der Monomerstrom. Der Reaktor wird
voll mit Flüssigkeit
bei 3,5 MPa (500 psig) am Druckanzeiger unter starkem Rühren betrieben.
Der Verfahrensfluss geht am Boden hinein und kommt am oberen Ende
heraus. Alle aus dem Reaktor heraustretende Rohre sind dampfbeheizt
und isoliert. Die Polymerisation wird mit der Zugabe einer kleinen
Menge Wasser beendet und andere Additive und Stabilisatoren können an
diesem Punkt zugegeben werden. Der Strom fließt durch einen statischen Mischer
und einen Wärmeaustauscher,
um das Lösungsmittel/das
Polymer-Gemisch aufzuheizen. Das Lösungsmittel und die nicht umgesetzten
Monomere werden bei reduziertem Druck entfernt und das Produkt wird
durch Extrusion mittels eines Entgasungsextruders erhalten. Der
extrudierte Strang wird unter Wasser gekühlt und in Granulat gehackt. Die
Bedienung des Reaktors wird mit einem Verfahrenssteuerungscomputer
gesteuert.
-
Beispiel 1
-
Propylen/Ethylen Polymerisation
-
Verwendung Metallocen
Katalysator E (Vergleich)
-
Das
allgemeine Verfahren für
die kontinuierliche Lösungspolymerisation
einer Gallone, wie sie oben beschrieben wurde, wurde eingesetzt.
Es wurde eine Katalysatorlösung,
die 2,6 ppm Zr aus Katalysator E enthält, hergestellt, und einem
4 l Katalysator-Lagergefäß zugegeben.
Diese Lösung
wurde in einem kontinuierlichen Strom mit einem kontinuierlichen
Strom einer Armeeniumtetrakis(pentafluorphenyl)borat-Lösung in
Toluol und einem kontinuierlichen Strom einer PMAO-IP Lösung in
Toluol vereint, so dass sich ein Verhältnis von insgesamt Ti:B:Al
von 1:1,2:30 ergibt. Die aktivierte Katalysatorlösung wurde kontinuierlich in
den Reaktor mit einer Rate zugeführt,
die ausreicht, um die Reaktortemperatur bei annähernd 80 °C und einer Polymerproduktionsrate
von annähernd
3 Pfund pro Stunde beizubehalten. Die Polymerlösung wurde kontinuierlich über den Reaktorausgang
entfernt und mit einer Lösung,
die 100 ppm Wasser pro Teil Polymerlösung und Polymerstabilisatoren
(d.h. 1000 ppm Irgaphos 168 und 1000 ppm Irganox 1010 pro Teil Polymer)
enthält
in Kontakt gebracht. Die resultierenden Ausgangsströme wurden
gemischt, in einem Wärmeaustauscher
erhitzt und die Mischung wurde in einen Separator eingeleitet, wo
das geschmolzene Polymer vom Lösungsmittel
und den nicht umgesetzten Monomeren abgetrennt wurde. Das resultierende
geschmolzene Polymer wurde extrudiert und in Granulat gehackt, nachdem
es in einem Wasserbad abgekühlt
war. In diesem Beispiel betrug das Propylen zu Ethylen-Verhältnis 22,0.
Produktproben wurden in Zeitabständen
von 1 Stunde gesammelt, wobei nach dieser Zeit die Schmelzflussrate
für jede
Probe bestimmt wurde. 7 ist ein 13C
NMR von Vergleichsbeispiel 1 und es zeigt die Abwesenheit von Regio-Fehler
Peaks in der Region um 15 ppm.
-
Beispiele 2-6
-
Beispiele
2-6 wurden vergleichbar zu Beispiel 1 durchgeführt, es sei denn, es ist in
den Tabellen 2-6-1 und 2-6-2 weiter unten anders gekennzeichnet.
Katalysator E wird zu Vergleichszwecken aufgelistet. 6 zeigt
das 13C NMR-Spektrum des Propylen/Ethylen-Copolymerprodukts
aus Beispiel 2. 2A und 2B zeigen
einen Vergleich der DSC-Aufheizkurven der Propylen/Ethylen-Copolymere
von Vergleichsbeispiel 1 und Beispiel 2.
-
Tabelle
2-6-1 – Polymerisationsbedingungen
-
Tabelle
2-6-2 – Monomerumsetzung
und Aktivität
-
Tabelle
2-6-3 – Zusammenfassung
der Daten der Polymeranalyse
-
Tabelle
2-6-4 – Fortsetzung
der Zusammenfassung der Daten der Polymeranalyse
-
Tabelle
2-6-5 – Fortsetzung
der Zusammenfassung der Daten der Polymeranalyse
-
Tabelle
2-6-6 – Fortsetzung
der Zusammenfassung der Daten der Polymeranalyse
-
Beispiel 7
-
Dieses
Beispiel zeigt die Berechnungen von B-Werten für gewisse Beispiele, die hierin
offenbart sind. Das Polymer aus Vergleichsbeispiel 1 wird wie folgt
analysiert. Die Daten wurden mittels eines Varian UNITY Plus 400
MHz NMR Spektrometers entsprechend einer 13C
Resonanzfrequenz von 100,4 MHz erfasst. Erfassungsparameter wurden
ausgewählt,
um quantitative 13C Datenerfassung in Anwesenheit
des Relaxationsmittels zu gewährleisten.
Die Daten wurden mittels 1H-Protonenentkopplung,
4000 Transienten pro Datendatei, einem 7 Sek.-Pulsrepetitionsdelay, Spektralbreite
von 24,200 Hz und einer Dateigröße von 32K
Datenpunkten erfasst, wobei der Probenkopf auf 130 °C erhitzt
wurde. Die Probe wurde durch Zugabe von annähernd 3 ml einer 50/50-Mischung
von Tetrachlorethand2/Orthodichlorbenzol, das heißt 0,025
M in Chromacetylacetonat (Relaxationsreagenz) zu 0,4 g Probe in
ein 10 mm NMR-Röhrchen
hergestellt. Der Gasraum des Röhrchens wird
durch Verdrängung
mit reinem Stickstoff von Sauerstoff befreit. Die Probe wurde gelöst und homogenisiert,
indem das Röhrchen
und dessen Inhalte auf 150 °C
unter periodischem Rückfluss
aufgeheizt werden, initiiert durch eine Heissluftpistole.
-
Nach
Datenerfassung wurden die chemischen Verschiebungen intern auf die
mmmm Pentade bei 21,90 ppm bezogen.
-
Für Propylen/Ethylen-Copolymere
wird das folgende Verfahren verwendet, um den Prozentanteil an Ethylen
in dem Polymer zu berechnen. Integrale Regionen werden wie folgt
bestimmt:
-
Tabelle
7-1: Integrale Regionen zur Berechnung des Prozentanteils Ethylen
-
Region
D wird wie folgt berechnet: D = P × (G × Q)/2.
-
Region
E wird wie folgt berechnet: E = R + Q + (G × Q)/2.
-
Die
Triaden werden wie folgt berechnet:
-
Tabelle 7-2: Triadenberechnung
-
- PPP = (F + A – 0,5D)/2
- PPE = D
- EPE = C
- EEE = (E – 0,5G)/2
- PEE = G
- PEP = H
- Mole P = (B + 2A)/2
- Mole E = (E + G + 0,5B + H)/2
-
Für dieses
Beispiel wird der Mol-%-Anteil an Ethylen zu 13,6 mol-% berechnet.
-
Für dieses
Beispiel werden die Triaden Molfraktionen wie folgt berechnet:
-
Tabelle 7-3: Triadenmole-Berechnung
-
- PPP = 0,6706
- PPE = 0,1722
- EPE = 0,0224
- EEE = 0,0097
- PEE = 0,0442
- EPE = 0,0811
-
Daraus
wird der B-Wert berechnet zu (0,172 + 0,022 + 0,044 + 0,081)/2 (,136 × ,864)
= 1,36.
-
Auf ähnliche
Weise werden die B-Werte für
die folgenden Beispiele berechnet zu: Tabelle
7-4: B-Wert Berechnung
-
Beispiel 8
-
Tabelle
8 ist eine Zusammenfassung, die den Schiefe-Index, Six,
für erfindungsgemäße Proben
und Proben des Standes der Technik zeigt. Alle Proben wurden hergestellt
und gemessen wie in Tabelle C in der Beschreibung der bevorzugten
Ausführungsformen
beschrieben und mit „Parameter
bei TREF", betitelt
ist. Die erfindungsgemäßen Copolymere
haben einen Schiefe-Index von größer als
(–1,2).
Die Ergebnisse aus Tabelle 8 werden in 11 grafisch
dargestellt.
-
Die
erfindungsgemäßen Beispiele
zeigen ungewöhnliche
und unerwartete Ergebnisse, wenn sie mittels TREF untersucht werden.
Die Verteilungen neigen dazu, einen großen Elutionstemperaturbereich
zu bedecken, während
sie gleichzeitig einen markanten engen Peak ergeben. Zusätzlich ist über einen
weiten Bereich des Ethyleneinbaus die Peaktemperatur, Tmax,
nahe bei 60 °C
bis 65 °C.
Im Stand der Technik bewegt sich dieser Peak für ähnliche Levels an Ethyleneinbau
zu höheren
Elutionstemperaturen mit geringerem Ethyleneinbau.
-
Für herkömmliche
Metallocen-Katalysatoren wird die annähernde Beziehung zwischen der
Molfraktion von Propylen, Xp, zu der TREF
Elutionstemperatur für
das Peakmaximum, Tmax, durch folgende Formel
angegeben: Logc(Xp)= –289/(273
+ Tmax) + 0,74
-
Für die erfindungsgemäßen Copolymere
ist der natürliche
Logarithmus der Molfraktion an Propylen, LnP, größer als der von herkömmlichen
Metallocenen, wie in dieser Gleichung gezeigt: LnP > –289/(273 + Tmax)
+ 0,75
-
Tabelle
8: Zusammenfassung der Schiefe-Index-Ergebnisse
-
Beispiel 9
-
Die
DSC-Analyse zeigt, dass Propylen/Ethylen-Copolymere, die mittels
eines Lösungspolymerisations-Verfahrens
unter Verwendung eines Nicht-Metallocen, metallzentrierten Katalysators
mit Pyridalaminligand hergestellt werden, ein Schmelzverhalten aufweisen,
das sich in erstaunlicherweise von Propylen/Ethylen-Copolymeren, die
mittels im Fachgebiet bekannten Metallocen-Polymerisations-Verfahren hergestellt
werden, unterscheidet. Das unterschiedliche Schmelzverhalten dieser
Copolymere im Vergleich zu dem der im Fachgebiet bekannten Copolymere,
zeigt nicht nur die Neuheit dieser Materialien, sondern kann auch
verwendet werden, um gewisse Vorteile dieser Materialien für einige
Anwendungen abzuleiten. Die neuen Aspekte des Schmelzverhaltens
dieser Copolymere und ihr damit verbundener Nutzen werden weiter
unten diskutiert, nachdem zunächst
das DSC-Analyseverfahren beschrieben wird.
-
Jegliche
flüchtigen
Materialien (z.B. Lösungsmittel
oder Monomer) werden aus dem Polymer vor der DSC-Analyse entfernt.
Eine kleine Menge Polymer, typischerweise fünf bis fünfzehn Milligramm, wird sorgfältig in
ein Aluminium-DSC-Pfännchen
mit Deckel eingewogen. Es sind entweder hermetisch- oder standardartige Pfännchen geeignet.
Das Pfännchen,
das die Probe enthält,
wird anschließend
auf einer Seite der DSC-Zelle platziert, wobei ein leeres Pfännchen mit
Deckel auf die Referenzseite der DSC-Zelle platziert wird. Die DSC-Zelle
wird anschließend
geschlossen, wobei ein langsamer Reinigungsstrom aus Stickstoffgas
während des
Tests durch die Zelle strömt.
Anschließend
wird die Probe einer programmierten Temperatursequenz unterworfen,
die typischerweise sowohl isotherme Segmente als auch Segmente aufweist,
bei der die Temperatur so eingestellt ist, dass sie mit einer konstanten
Rate ansteigt oder fällt.
Die Ergebnisse, die hier gezeigt werden, wurden alle mittels wärmeflussartigen
DSC-Geräten,
hergestellt von TA Instruments (z.B. Modell 2910 DSC), erhalten.
Die Messungsprinzipien, denen die Wärmefluss-DSC unterliegt, werden
auf Seite 16 von Turi (siehe dort) beschrieben. Die primären Signale,
die durch solche Instrumente erzeugt werden, sind Temperatur (Einheiten: °C) und differenzieller
Wärmefluss
(Einheiten: Watt) in die Probe oder aus der Probe heraus (d.h. bezogen
auf die Referenz) als Funktion der abgelaufenen Zeit. Schmelzen
ist endotherm und bedingt ein Übermaß an Wärmefluss
in die Probe bezogen auf die Referenz, wohingegen Kristallisation
exotherm ist und ein Übermaß an Wärmefluss
aus der Probe heraus bedingt. Diese Instrumente werden mittels Indium
und anderen Standards, die innerhalb eines engen Bereichs schmelzen,
kalibriert. Die Kalibrierung gewährleistet,
dass die Temperaturskala korrekt ist, und die einwandfreie Korrektur
von unvermeidbaren Hitzeverlusten.
-
Temperaturprogramme
für DSC-Analyse
von semikristallinen Polymeren umfassen mehrere Schritte. Obwohl
die Temperaturprogramme, die verwendet werden, um die hier gezeigten
Daten zu erzeugen sich in einigen Details unterschieden, wurden
die kritischen Schritte durchwegs konstant gehalten. Der erste Schritt besteht
aus einem anfänglichen
Heizen bis zu einer Temperatur, die ausreicht, um die Probe vollständig aufzuschmelzen;
für Polypropylen-Homopolymere
und Copolymere liegt diese bei 210 °C oder höher. Dieser erste Schritt hilft
auch, um einen hervorragenden thermischen Kontakt der Polymerprobe
mit dem Pfännchen
zu gewährleisten.
Obwohl sich Details dieses ersten Schritts für die hier gezeigten Daten
unterschieden – zum Beispiel
in der Aufheizrate der oberen Temperatur und der Haltezeit bei der
oberen Temperatur – war
die Wahl in jedem Fall ausreichend, um das prinzipielle Ziel dieses
Schritts, nämlich
alle Proben auf einen gemeinsamen vollständig geschmolzenen Startpunkt
mit gutem thermischen Kontakt zu bringen. Der zweite Schritt umfasst Abkühlen bei
einer konstanten Rate von 10 °C/min
von einer oberen Temperatur von mindestens 210 °C auf eine niedrigere Temperatur
von 0 °C
oder weniger. Die niedrigere Temperatur wird so gewählt, dass
sie bei oder leicht unter der Glasübergangstemperatur des jeweiligen
Propylenpolymers liegt. Die Kristallisationsrate wird sehr langsam
bei der Glasübergangstemperatur;
daher hat zusätzliches
Abkühlen
eine geringe Wirkung auf das Ausmaß der Kristallisation. Dieser
zweite Schritt dient dazu, eine Standardkristallisationsbedingung vor
der sich anschließenden
Untersuchung des Schmelzverhaltens bereitzustellen. Nach einem kurzen
Halt bei dieser geringeren Temperaturgrenze, typischerweise ein
bis drei Minuten, wird der dritte Schritt eingeleitet. Der dritte
Schritt umfasst Aufheizen der Probe von einer Temperatur von 0 °C oder niedriger
(d.h. die Endtemperatur des vorherigen Schritts) auf 210 °C oder höher bei
einer konstanten Rate von 10 °C/min.
Dieser dritte Schritt dient dazu, eine Standard-Schmelzbedingung
bereitzustellen, wenn eine Standard-Kristallisationsbedingung vorausgegangen
ist. Alle hier gezeigten Ergebnisse des Schmelzverhaltens wurden
aus diesem dritten Schritt, d.h. aus dem zweiten Schmelzen der Probe,
erhalten.
-
Die
von der DSC ausgegebenen Daten bestehen aus Zeit(s), Temperatur
(°C) und
Wärmefluss
(Watt). Anschließende
Schritte bei der Analyse von Schmelzendothermen sind wie folgt.
Zunächst
wird der Wärmefluss
durch die Probenmasse geteilt, um einen spezifischen Wärmefluss
(Einheit: W/g) zu erhalten. Zweitens wird eine Basislinie angelegt
und von dem spezifischen Wärmefluss
subtrahiert, um einen um die Basislinie korrigierten Wärmefluss
zu erhalten. Für
die hier gezeigten Analysen wird eine geradlinige Basislinie verwendet.
Die untere Temperaturgrenze für
die Basislinie wird als ein Punkt auf der Seite der höheren Temperatur des
Glasübergangs
gewählt.
Für die
obere Temperaturgrenze für
die Basislinie wird eine Temperatur 5-10 °C über der Temperatur, die bei
der Beendigung der Schmelzendotherme vorliegt, ausgewählt. Obwohl
eine geradlinige Basislinie theoretisch nicht exakt ist, bietet
sie eine leichtere Handhabung und bessere Konsistenz der Analyse
und der damit eingefügte
Fehler ist relativ gering für
Proben mit spezifischen Schmelzwärmen
von 15-20 Joule pro Gramm oder höher.
Der Einsatz einer geradlinigen Basislinie anstatt einer theoretischeren
korrekten Basislinie beeinflusst nicht wesentlich die Ergebnisse
oder Aussagen, die weiter unten gezeigt werden, obwohl erwartet
wird, dass sich die feinen Details der Ergebnisse mit einer anderen
Vorschrift für
die instrumentelle Basislinie ändern.
-
Es
gibt eine Vielzahl von Größen, die
aus den DSC-Schmelzdaten extrahiert werden können. Größen, die besonders nützlich sind,
um Unterschiede oder Ähnlichkeiten
zwischen verschiedenen Polymeren zu zeigen, sind: (1) die Peakschmelztemperatur,
Tmax (°C),
die die Temperatur ist, bei der der um die Basislinie korrigierte
Wärmefluss
ein Maximum ist. (hier ist das Übereinkommen,
dass der Wärmefluss
in die Probe positiv ist); (2) die spezifische Schmelzwärme, Δhm (J/g), die die Fläche unter der Schmelzendotherme
darstellt, die durch Integrieren des um die Basislinie korrigierten
Wärmeflusses
(dq/dt) (W/g) über
die Zeit zwischen den Basisliniengrenzen erhalten wird; (3) der
spezifische Wärmefluss
(dq/dt)max (W/g) bei der Peakschmelztemperatur;
(4) der Peak-spezifische Wärmefluss,
genormt auf die spezifische Schmelzwärme {(dq/dt)max/Δhm} (s–1); (5) der erste Moment
T1 der Schmelzendotherme, definiert und
berechnet wie unten beschrieben; (6) die Varianz V1 (°C2) der Schmelzendotherme bezogen auf den
ersten Moment T1, definiert und berechnet
wie unten beschrieben; und (7) die Quadratwurzel der Varianz, V1 1/2 (°C), die ein
Maß für die Breite
der Schmelzendotherme ist.
-
Die
Schmelzendothermen als eine Verteilung anzusehen ist ein nützlicher
Weg, deren Breite zu quantifizieren. Die Quantität, die verteilt ist als eine
Funktion der Temperatur, ist der um die Basislinie korrigierte Wärmefluss
(dq/dt). Dass diese auch eine Verteilung der Temperatur ist, wird
anhand der Kettenregel für
Differenzialrechnen erklärt
(dq/dt) = (dq/Dt)(Dt/dt), wobei (Dt/dt) die Heizrate ist. Die Standarddefinition
des ersten Moments T1 dieser Verteilung
wird durch die folgende Gleichung angegeben, bei der die Integration
zwischen den Grenzen der Basislinie durchgeführt wird. Alle Integrationen
werden am zuverlässigsten
als (dq/dt) über die
Zeit durchgeführt,
im Gegensatz zu der Alternative (dq/Dt) über die Temperatur. In der
folgenden Gleichung sind (dq/dt) und T der spezifische Wärmefluss
und die Temperatur zur Zeit t.
-
-
Die
Varianz V
1 im Verhältnis zu dem ersten Moment
ist dann standardmäßig definiert
-
Sowohl
V1 als auch V1 1/2 sind ein Maß für die Breite der Schmelzendotherme.
-
Ergebnisse
der DSC-Analysen, sowohl von den erfindungsgemäßen als auch von den Vergleichspolymeren,
sind in Tabelle 9-1 gezeigt. Alle Proben sind Propylen/Ethylen-Copolymere. Polymere
1-12 wurden mittels Katalysator H in einem Lösungsverfahren hergestellt.
Polymere 13 bis 21 wurden mit Katalysator E in einem Lösungsverfahren
hergestellt. Eine Idee für
die Präzision
der experimentellen Methode und des Datenanalyseverfahrens bekommt
man durch die Wiederholungen (Polymere 15 und 17) und durch die
Konsistenz der Ergebnisse der Polymerreihen, die unter annähernd identischen
Bedingungen hergestellt wurden (Polymere 1 bis 3, 6 bis 8 und 9-12).
Die Unterschiede im Schmelzverhalten lassen sich am leichtesten
anhand der Figuren erkennen. 12 vergleicht
die Schmelzendothermen der Proben 4 und 17a. Diese zwei Propylen/Ethylen.-Copolymere
haben annähernd äquivalente
Schmelzwärmen
und Molprozent-Gehalte an Ethylen mit etwa 71 J/g und 8 mol-%. Allerdings
unterscheidet sich, trotz dieser Ähnlichkeiten das Schmelzverhalten des
erfindungsgemäßen Copolymers
(Probe 4) überraschenderweise
von dem des Vergleichscopolymers (Probe 17a). Die Schmelzendotherme
von Beispiel 4 verschiebt sich zu geringeren Temperaturen und ist
signifikant verbreitert, wenn man bei äquivalenter Schmelzwärme vergleicht.
Diese Veränderungen
im Schmelzverhalten sind einzigartig und charakteristisch für die erfindungsgemäßen Copolymere.
-
Der
Vergleich bei äquivalenten
Schmelzwärmen
ist besonders bedeutsam und relevant. Das liegt daran, weil äquivalente
Schmelzwärmen
ein annähernd
gleiches Level an Kristallinität
bedeuten, was wiederum bedeutet, dass die Moduli bei Raumtemperatur ähnlich sein
sollten.
-
Daher
besitzen die erfindungsgemäßen Copolymere
bei einem gegebenen Modul oder einer gegebenen Steifigkeit nützlicherweise
verbreiterte Schmelzbereiche im Vergleich zu typischen nicht erfindungsgemäßen Copolymeren.
-
13 bis 17,
die aus den Ergebnissen in Tabelle 9-1 stammen, stellen ferner die
Unterschiede im Schmelzverhalten der erfindungsgemäßen Copolymere
heraus verglichen mit den typischen Copolymeren. Bei allen fünf Figuren
sind die Quantitäten
als Funktion der Schmelzwärme
aufgetragen, die, wie oben beschrieben, eine besonders bedeutsame
und relevante Basis ist, um Vergleiche untereinander zu machen und Nutzen
abzuleiten. Für
diese Diagramme wurden die Daten in zwei Serien geteilt, bezogen
auf den Katalysatortyp, entweder Metallocen- oder Nicht-Metallocen-Typ, der verwendet
wird, um das Polymer herzustellen.
-
13 zeigt, wie sich die Peakschmelztemperatur für die erfindungsgemäßen Copolymere
zu geringerer Temperatur verschiebt. All die Änderungen im Schmelzverhalten,
von dem diese Verschiebung der Peakschmelztemperatur nur ein Beispiel
ist, bedeuten, dass Unterschiede in der kristallinen Struktur auf
dem Level von kristallinen Lamellen oder andersartiger primärer kristalliner
Elemente bestehen. Solche Unterschiede in der kristallinen Struktur
können
wiederum am vernünftigsten
auf Unterschiede in der Mikrostruktur zurückgeführt werden, zum Beispiel die
unterschiedliche Art von Fehlinsertionsfehlern oder die höheren B-Werte,
die die erfindungsgemäßen Polymere
kennzeichnen. Ungeachtet der exakten Natur der mikrostrukturellen
Merkmale, die zu den Veränderungen
im Schmelzverhalten führen,
sind die Veränderungen
an und für
sich ein Beweis, dass die erfindungsgemäßen Copolymere neue Zusammensetzungen
sind.
-
14, die ein Diagramm der Temperatur T1%c ist,
bei der annähernd
1 % Restkristallinität
vorliegt, zeigt einen anderen überraschenden
Aspekt des Schmelzverhaltens der erfindungsgemäßen Copolymere. Der Faktor,
der verwendet wird, um spezifische Schmelzwärme in nominelle Gewichtsprozent
Kristallinität
umzurechnen, ist 165 J/g = 100 Gew.-% Kristallinität. (Die
Verwendung von einem anderen Umrechnungsfaktor könnte Details der Ergebnisse
verändern,
allerdings nicht substanzielle Ergebnisse.) Mit diesem Umrechnungsfaktor
wird die Gesamtkristallinität
einer Probe (Einheiten: Gew.-% Kristallinität) berechnet zu 100 % mal Δhm geteilt durch 165 J/g. Und bei diesem Umrechnungsfaktor
entspricht 1 % Restkristallinität
1,65 J/g. Deshalb ist T1%c definiert als
die obere Grenze für
die partielle Integration der Schmelzendotherme, so dass Δhm minus dem partiellen Integral gleich 1,65
J/g ist, wobei die gleiche untere Grenze und Basislinie verwendet
werden für
diese partielle Integration wie für die vollständige Integration. Überraschenderweise
verschiebt sich diese Temperatur bei 1 % Restkristallinität für Nicht-Metallocen-katalysierte
Copolymere verglichen mit Metallocen-katalysierten Copolymeren nach
unten langsamer bei ansteigendem Ethylengehalt (d.h. bei abnehmender Schmelzwärme). Dieses
Verhalten von T1%c ist ähnlich zu dem der End- Schmelztemperatur
Tme.
-
15, die die Varianz relativ zu dem ersten Moment
der Schmelzendotherme als eine Funktion der Schmelzwärme zeigt,
zeigt direkt die größere Breite
der Schmelzendotherme für
die erfindungsgemäßen Copolymere.
-
16, die den maximalen Wärmefluss, normiert auf die
Schmelzwärme,
als eine Fuktion der Schmelzwärme
zeigt, zeigt ferner die Verbreiterung der Schmelzendothermen. Das
liegt daran, weil bei gleicher Schmelzwärme ein geringerer Peakwert
bedeutet, dass die Verteilung verbreitert sein muss, um die gleiche
Fläche
zu ergeben. Wenn man die Form dieser Schmelzkurven grob annähert als
ein Dreieck, für
dass die Fläche
durch die Formel einhalb mal der Basis mal der Höhe gegeben ist, dann ist b1/b2
= h2/h1. Die erfindungsgemäßen Copolymere
zeigen mehr als einen vierfachen Abfall in der Höhe, was bedeutet, dass die
Breite signifikant ansteigt.
-
17 stellt einen nützlichen Aspekt des breiteren
Schmelzbereichs der erfindungsgemäßen Polymere dar, nämlich dass
die Rate, bei der der letzte Anteil an Kristallinität verschwindet
(Einheiten: Gewichtsprozent Kristallinität pro °C), signifikant geringer ist
als die für
Metallocen-Polymere.
-
Die
Daten in Tabelle 9-2 zeigen im praktischen Sinne die Nützlichkeit
dieser Verbreiterung der Schmelzendothermen. Einträge in Tabelle
9-2 stellen dar: (1) das Ausmaß,
bei dem eine größere Schmelzfraktion
bei geringeren Temperaturen auftritt, was wichtig ist für Heißsiegel-
und Verbindungsanwendungen und das für die erfindungsgemäßen Polymere
größer ist;
und (2) das Ausmaß,
bei dem Kristallinität
bei höheren
Temperaturen verbleibt, und die Rate, bei der der letzte Anteil
an Kristallinität
verschwindet, was für
die Herstellungsschritte, wie zum Beispiel Thermoformen, Schäumen, Blasformen
und dergleichen, wichtig sein kann, wobei in beiden Fällen das
Ausmaß für die erfindungsgemäßen Copolymere
größer ist. Tabelle
9-1 - Schmelzergebnisse aus DSC
- *Proben –1 bis –12 mit Katalysator H und –13 bis –19 mit
Katalysator E.
- **Einheiten von Rf: Gewichts-% Kristallinität pro °C
-
Tabelle
9-2: Verbreiterung der Schmelzendothermen
-
Beispiel 10
-
8 und 9 zeigen
gewisse Elastizitätsdaten
für die
erfindungsgemäßen Polymere
und gewisse Vergleichspolymere. Tabelle 10-1 beschreibt die Elastomere.
-
Die
Testproben waren formgepresste Probenkörper. Polymergranulat wurde
bei 190 °C
in 1,5 mm dicke Schichten formgepresst. Die Schichten wurden abgekühlt, indem
sie jeweils zwischen zwei Platten bei 25C unter leichtem Druck gelegt
wurden. Die Probenkörper
ließ man
vor den Untersuchungen für
mindestens sieben Tage bei Umgebungsbedingungen altern.
-
Zug „Hundeknochen" wurden aus den Schichten
mittels eines Messers, das gemäß ASTM D-1708
geformt war, herausgestanzt. Das erste Schmelzverhalten wurde gemessen,
indem ein 5 bis 10 mg schweres Stück aus dem gealterten Probenkörper herausgeschnitten
wurde. Der Probenkörper
wurde in ein Aluminiumpfännchen
gelegt und in einem dynamischen Differenzkalorimeter, hergestellt
von TA Instruments Inc., analysiert. Das Aufheizen erstreckte sich
von –50
auf 230 C bei 10 C/Minute.
-
Die
Probenkörper
wurden unter uniaxialer Zugspannung mit einem mechanischen Testgerät (Instron Corp.),
ausgerüstet
mit pneumatischen Probenhaltern, untersucht. Die Spannungsrate bei
der Ausführung
war 400 % pro Minute. Sowohl Bruchspannung als auch multizyklische
Belastungs-Spannungs-Darstellungen wurden verwendet. Im Falle der
multizyklischen Belastung wurden die Probenkörper bei verschiedenen Spannungen
(100 bis 900 %) für
bis zu 10 Zyklen belastet und entlastet. Es wurde keine Pause zwischen
den nacheinander folgenden Zyklen eingelegt.
-
Tabelle
10-2 zeigt die Daten in 8 und 9. Die
Daten in Tabelle 10-2 zeigen die Elastizität gegenüber der Kristallinität für die gleiche
Gruppe von Elastomeren. Kristallinität ist eine wichtige Eigenschaft
in elastomeren Anwendungen, weil sie zu einem großen Maß die Steifigkeit
(d.h. den Modul) eines Materials steuert. Die ursprünglichen
oder ungedehnten Metallocen-Polypropylene weisen eine Elastizität gegenüber der
Kristallinität,
die ähnlich
zu homogenem Polyethylen und Ethylen-Styrol-Elastomeren ist, auf, während die erfindungsgemäßen elastomeren
Polymere höhere
Elastizitäten
bei gleicher Kristallinität
aufweisen. Sogar nach Vordehnen weisen die erfindungsgemäßen elastomeren
Polymere weiterhin bessere Elastizität bei der gleichen Kristallinität auf im
Vergleich zu den Metallocen-Propylenen als auch zu dem homogenen
Polyethylen und Ethylen-Styrol-Elastomeren. Die erfindungsgemäßen Polymere
zeigen nach Dehnen ähnliche
Elastizität wie
auch SEBS-Polymere in kommerzieller Qualität, z.B. Krayton G-1657.
-
-
Tabelle
10-2 Elastizitätswerte
der Daten in Figuren 8 und 9
-
Beispiel 11
-
In
einer Serie von Spinnversuchen wurden verschiedene P/E*- und Vergleichsfasern
hergestellt, von denen Elastizität,
Festigkeits/Dehnungs-Gleichgewicht und thermisches Verhalten (Haftfestigkeit)
gemessen wurden.
-
Elastizität
-
Zwei
einzelne Filamentfasern wurden hergestellt. Eine Faser (52,2 Denier)
wurde mit einem P/E*-Copolymer (11 Gew.-% Ethylen, Prozentanteil
Kristallinität
von 15,3, MFR 2, Dichte 0,865 g/cm2 und
einen Schmelzpunkt von 67 C) und die andere Faser (52,5 Denier)
wurde aus einem Metallocen-katalysierten Ethylen-Octen-Copolymer
hergestellt (Prozentanteil Kristallinität von 13,2, MI 2 und eine Dichte
von 0,87 g/cm3). Die Elastizität der Fasern
wurde mittels ASTM D1774 – elastischer
Rückverformung
gemessen. Gemäß diesem
Test ist die Elastizität
um so besser, je geringer die unwiderbringliche Dehnung ist (wenn
die unwiderbringliche Dehnung Null ist, ist die Rückverformung
100 %). Die Ergebnisse sind in 18 gezeigt.
-
18 zeigt, dass bei ähnlichem Prozentanteil Kristallinität die Faser,
die aus dem P/E*-Polymer hergestellt wurde, bessere Elastizität zeigte
als die Faser, die aus dem Metallocen-katalysierten Ethylen-Octen-Copolymer
hergestellt wurde. Die bessere Elastizität bedeutet, dass die Faser,
die aus dem P/E*-Copolymer hergestellt wird, bessere elastische
Rückverformung
für Vliesstoffe
bereitstellt, und dies wiederum bedeutet, dass die Gewebe dünner sein
können
und sich besser dem Körper
anpassen können
(für erhöhten Komfort).
Die Faser kann entweder als Spinnvlies- oder in stabiler Form vorliegen,
wobei ein repräsentativer
Vliesstoff, in dem die Faser verwendet wird, eine Windel ist, z.B.
als Deckfolie oder Rückfolie
(Dehnen ohne nachzugeben).
-
Die
bessere Elastizität
bedeutet auch, dass die Faser, die aus dem P/E*-Polymer hergestellt
ist, auch die Elastizität
eines Teppichs erhöht,
wenn er entweder nur aus P/E*-Polymer hergestellt wird oder wenn
er aus einer Mischung von P/E*-Polmyer und Propylen-Homopolymer
hergestellt wird. Diese erwünschte
Elastizität
erlaubt es, dass die Fasern unter einer Last (z.B. wenn darauf gegangen
wird) sich dehnen, ohne nachzugeben und dann rückverformen, ohne übereinander
zu liegen zu kommen. Darüber
hinaus verbessert das P/E*-Polymer die Haptik, z.B. das Tastgefühl der Faser
verglichen zu der Faser, die aus einem Ethylen-Octen-Copolymer hergestellt
wird.
-
Die
bessere Elastizität
bedeutet auch, dass die Faser, die aus dem P/E*-Polymer hergestellt
wird, falls vernetzt, wünschenswert
ist für
die Verwendung zur Herstellung von dehnbarer Kleidung. Nochmal,
Gewebe, die aus diesen Fasern hergestellt werden, können in
einem dünneren
Format erzeugt werden, was wiederum eine bessere Anpassung an den
Körper
und höheren
Komfort bedeutet.
-
Festigkeits-/Dehnungsgleichgewicht
-
Drei
monolytische Fasern mit 2 Denier (pro Filament, d.h. 2 dpf) wurden
hergestellt mittels eines 55 Düsenlochpunkts.
Alle Polymere, aus denen die Fasern hergestellt wurden, hatten einen
MFR von 25. Zwei der Fasern wurde mit einem P/E*-Copolymer mit einer
MWD zwischen 2 und 3 hergestellt. Das erste Polymer enthielt 3 Gew.-%
Ethylen und hatte einen Prozentanteil an Kristallinität von 48.
Das zweite Polymer enthielt 5 Gew.-% Ethylen und einen Prozentanteil
an Kristallinität
von 43. Die dritte Faser wurde aus einem Ziegler-Natta katalysierten
Propylen-Homopolymer hergestellt und hatte einen Prozentanteil an
Kristallinität
von 59. Die Fasern wurden als ein Bündel (d.h. als Garn) mittels
ASTM D2256 untersucht.
-
Das
Festigkeits- und Bruchdehnungs-Gleichgewicht der drei Garne werden
in 19 und 20 gezeigt.
Das Garn, das aus den Fasern, die aus dem P/E*-Polymeren hergestellt
wurden, hergestellt wurde, weist ein besseres Gleichgewicht dieser
Eigenschaften auf als das Garn, das aus den Fasern hergestellt wurde,
die aus dem Propylenhomopolymer hergestellt wurden. Das letztere
hat hohe Festigkeit bei geringer Bruchdehnung, während das erstere geringere
Festigkeit und höhere
Bruchdehnung zeigt. Dieses verbesserte Gleichgewicht stellt eine
bessere Dehnbarkeit bereit, wenn die Fasern in nicht gewobenen Anwendungen,
z.B. Babywindeln und Hygieneprodukte für Frauen, verwendet werden.
-
Thermisches
Verhalten
-
Drei
Bikomponentenfasern mit 2 Denier (pro Filament, d.h. 2 dpf) wurden
hergestellt. Der Kern jeder Faser war PET und die Hülle war
entweder ein P/E*-Polymer, gemischt mit mit Maleinsäureanhydrid
gepfropften Polypropylen, oder ein Ziegler-Nattakatalysiertes Polyethylen-Polymer,
gemischt mit, mit Maleinsäureanhydrid,
gepfropften Polyethylen. Die Bikomponentenfasern wurden geschnitten
und gekräuselt
und anschließend
mit Zellulose gemischt, um eine nicht gewebte Rückschicht zu erzeugen.
-
Zwei
der Fasern wurden mit P/E*-Copolymer mit einem MFR von 25, hergestellt.
Das erste Polymeren enthielt 3 Gew.-% Ethylen und hatte einen Prozentanteil
an Kristallinität
von 48. Das zweite Polymer enthielt 5 Gew.-% Ethylen und einen Prozentanteil
an Kristallinität
von 43. Die dritte Faser wurde aus einem Ziegler-Natta katalysierten
Ethylen-Octen-Copolymer mit einer Dichte von 0,93 g/cm3,
einem MI von 18 und einem Prozentanteil an Kristallinität von 53,
hergestellt. Dupont DSC-910 wurde verwendet, um die thermischen Übergangstemperaturen
und Übergangswärmen für die Probenserie
unter Stickstoff zu messen. Um die vorherige thermische Vorgeschichte
zu eliminieren, wurden die Proben zuerst auf etwa 200 C aufgeheizt.
Aufheiz- und Abkühlkurven
wurden bei 10 C/min aufgezeichnet. Die Klebkraft wurde mittels 1 × 152,4
mm (6 (inch) Vlies-Streifen mit 50,8 mm (2 inch) Breite und einer
Dehnrate von 25,4 mm (1 inch) pro Minute gemessen. Die Ergebnisse
sind in Tabelle 11 gezeigt.
-
-
Die
niedrige Schmelztemperatur des P/E*-Polymers und der breite Schmelzbereich
stellen eine bessere Klebkraft bereit als das Ethylen-Octen-Copolymer. 21 ist ein Endothermendiagramm dieser Proben und
es zeigt die Breite des Schmelzbereichs eines P/E*-Polymers im Vergleich
zu dem Ethylen-Octen-Copolymer. Die Breite des Schmelzbereichs korreliert
mit dem Haftenster der Faser. Die Klebkraft ist auf die Peak-Schmelztemperatur,
wie in Tabelle 11 gezeigt, bezogen.
-
Das
thermische Verhalten der P/E*-Polymere erlaubt deren Verwendung
in niederschmelzenden Klebfaseranwendungen und Vliesstoffanwendungen,
bei denen Haftenster und Klebkraft (z.B. geringe Initiationstemperatur,
breites Haftenster und hohe Haftfestigkeit) wichtig sind. Anwendungen
für Fasern,
die aus diesen P/E*-Polymeren
hergestellt werden, beinhalten eine Bindemittel-Faser für absorbierende
Kerne, Verstärkung des
Faserbarts von einem Teppich, Bindemittel für Polstermaterial und dergleichen.
Fasern, die aus diesen Materialien hergestellt werden, können auch
gemischt und/oder geschmolzen werden mit Ziegler-Natta katalysierten
Propylenfasern zu dreidimensionalen „High-Loft" Vliesmaterialien.





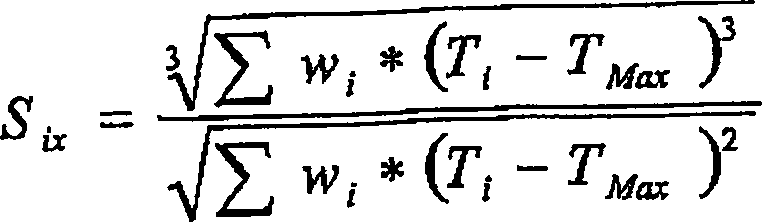

 wobei Q1 und Q5 Ringsubstituenten orthoständig zu Atom E sind, wobei E ausgewählt ist aus der Gruppe bestehend aus Kohlenstoff und Stickstoff und wobei mindestens einer von Q1 oder Q5 sperrig ist (dadurch definiert, dass sie mindestens 2 Atome aufweisen). Q1 und Q5 sind unabhängig voneinander ausgewählt aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Aryl, substituiertem Aryl und Silyl, allerdings unter der Voraussetzung, dass Q1 und Q5 nicht beide Methyl sind. Q''q repräsentiert zusätzliche mögliche Substituenten an dem Ring, wobei q 1, 2, 3, 4 oder 5 ist und Q'' ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid, Nitro und Kombinationen davon. T ist eine Verbindungsgruppe ausgewählt aus der Gruppe bestehend aus -CR2R3- und -SiR2R3-, wobei R2 und R3 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid, Nitro und Kombinationen davon. J" ist im Allgemeinen ausgewählt aus der Gruppe bestehend aus Heteroaryl und substituiertem Heteroaryl, wobei besondere Ausführungsformen für besondere Reaktionen hierin beschrieben sind.
wobei Q1 und Q5 Ringsubstituenten orthoständig zu Atom E sind, wobei E ausgewählt ist aus der Gruppe bestehend aus Kohlenstoff und Stickstoff und wobei mindestens einer von Q1 oder Q5 sperrig ist (dadurch definiert, dass sie mindestens 2 Atome aufweisen). Q1 und Q5 sind unabhängig voneinander ausgewählt aus der Gruppe bestehend aus Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Aryl, substituiertem Aryl und Silyl, allerdings unter der Voraussetzung, dass Q1 und Q5 nicht beide Methyl sind. Q''q repräsentiert zusätzliche mögliche Substituenten an dem Ring, wobei q 1, 2, 3, 4 oder 5 ist und Q'' ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid, Nitro und Kombinationen davon. T ist eine Verbindungsgruppe ausgewählt aus der Gruppe bestehend aus -CR2R3- und -SiR2R3-, wobei R2 und R3 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Alkyl, substituiertem Alkyl, Cycloalkyl, substituiertem Cycloalkyl, Heteroalkyl, substituiertem Heteroalkyl, Heterocycloalkyl, substituiertem Heterocycloalkyl, Aryl, substituiertem Aryl, Heteroaryl, substituiertem Heteroaryl, Alkoxyl, Aryloxyl, Silyl, Boryl, Phosphino, Amino, Thio, Seleno, Halogenid, Nitro und Kombinationen davon. J" ist im Allgemeinen ausgewählt aus der Gruppe bestehend aus Heteroaryl und substituiertem Heteroaryl, wobei besondere Ausführungsformen für besondere Reaktionen hierin beschrieben sind.




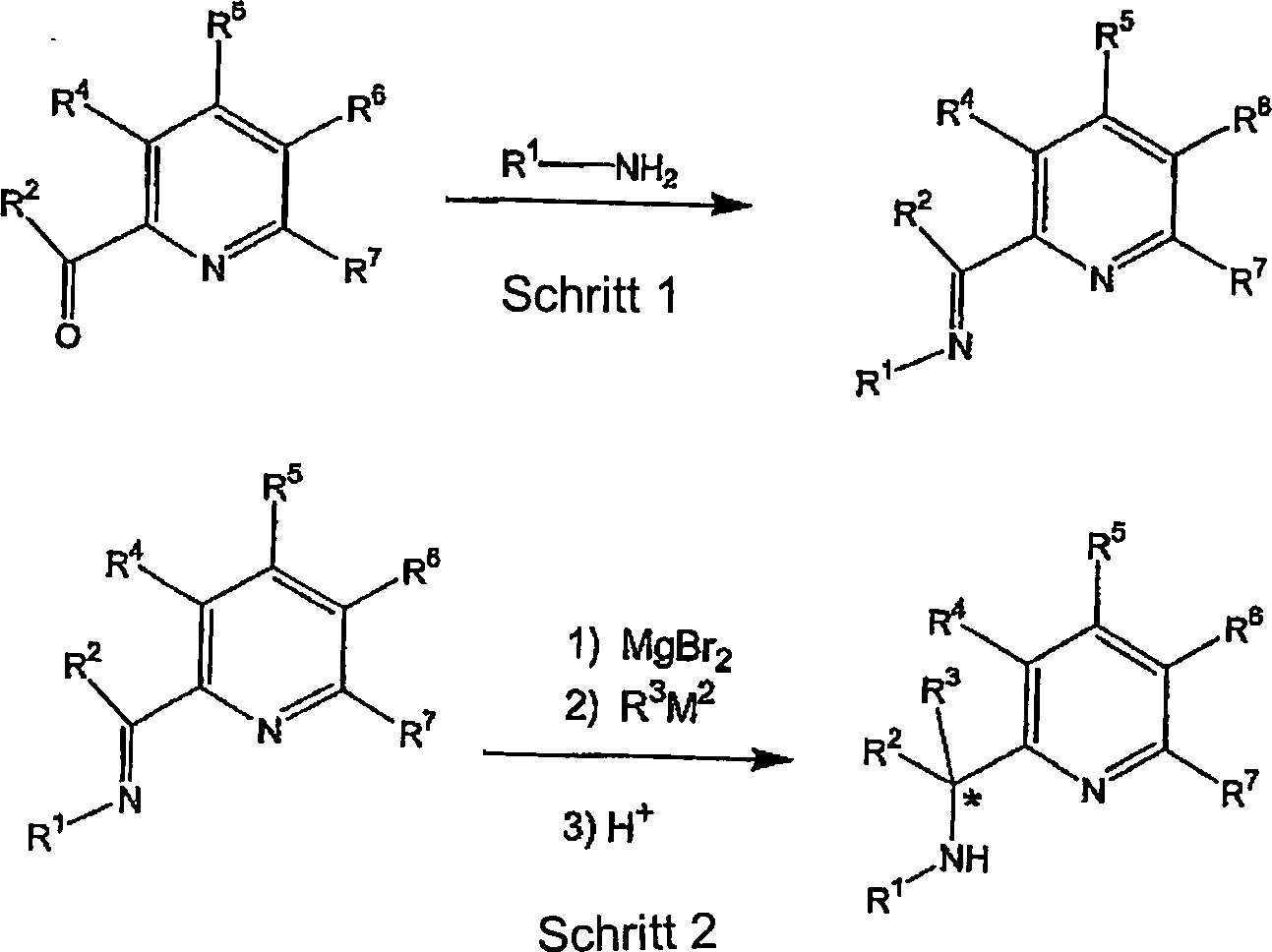

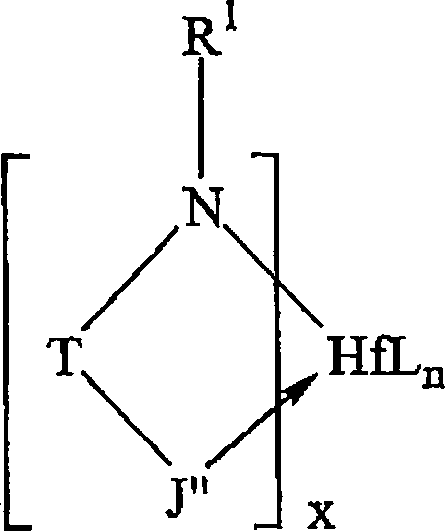 wobei R1, T, R4, R5, R6, R7, L und n wie vorher definiert sind; und x 1 oder 2 ist. In einer bevorzugten Ausführungsform ist x = 1 und n = 3. Zusätzlich können auch Lewis-Basen-Addukte dieser Metall-Ligandkomplexe verwendet werden, zum Beispiel sind Ether, Amine, Thioether, Phosphine und dergleichen geeignet als Lewis-Basen.
wobei R1, T, R4, R5, R6, R7, L und n wie vorher definiert sind; und x 1 oder 2 ist. In einer bevorzugten Ausführungsform ist x = 1 und n = 3. Zusätzlich können auch Lewis-Basen-Addukte dieser Metall-Ligandkomplexe verwendet werden, zum Beispiel sind Ether, Amine, Thioether, Phosphine und dergleichen geeignet als Lewis-Basen.