-
Gebiet der Erfindung
-
Die vorliegende Erfindung betrifft
chemische Verfahren zur Herstellung von Verbindungen, die bei der Behandlung
verschiedener medizinischer Beschwerden, einschließlich Atmungsbeschwerden,
Augenleiden, Magen-Darm-Beschwerden, nasaler Dekongestion, Bluthochdruck,
Migräne,
Beschwerden in Verbindung mit der Aktivität des sympathischen Nervensystems
und dem Missbrauch von Substanzen nützlich sind. Insbesondere sind
die Verfahren dieser Erfindung nützlich
zur Herstellung von 2-Amino-2-imidazolin-Derivaten,
Guanidin-Derivaten und 2-Amino-3,4,5,6-tetrahydropryrimidin-Derivaten.
-
Hintergrund
der Erfindung
-
Die vorliegende Erfindung betrifft
Verfahren zur Herstellung von – 2-Amino-2-imidazolin-Derivaten, Guanidin-Derivaten
und 2-Amino-3,4,5,6-tetrahydropryrimidin-Derivaten (alle hierin zusammenfassend
als "2-Amino-2-Derivate" beschrieben). Solche Derivate sind nützlich für die Behandlung
zahlreicher medizinischer Beschwerden, einschließlich z. B. Atmungsbeschwerden,
Augenleiden, Magen-Darm-Beschwerden, nasaler Dekongestion, Bluthochdruck,
Migräne,
Beschwerden in Verbindung mit der Aktivität des sympathischen Nervensystems
und dem Missbrauch von Substanzen. Eines der am meisten bekannten
von diesen Derivaten ist Clonidin, ein alpha-2-Adrenorezeptor-Agonist und ein blutdrucksenkendes
Mittel. Iopidin ist ebenfalls ein bekannter alpha-2-Adrenorezeptor-Agonist,
der bei der Senkung des Augeninnendrucks von Nutzen ist;
-
Clonidin, das in dem US-Patent Nr.
3 202 660 (1965) von Boehringer, Ing., offenbart ist, Iopidin, das
in dem US-Patent Nr. 4 517 199 (1985) von Alcon offenbart ist; Timmermans,
P.B.M.W.M., de Jonge, A., Thoolen, M.J.M.C., Wilffert, B., Batink,
H., van Zwieten, P. A., "Quantitative Relationships between α-Adrenergic
Activity and Binding Affinity of α-Adrenoceptor (Agonists
and Antagonists", Journal of Medicinal Chemistry, Bd. 27 (1984),
SS. 495–503;
Physician's Desk Reference (50. Ausg., 1996).
-
Therapeutische Indikationen von alpha-2-Adrenorezeptor-Agonisten
wurden in der Literatur erörtert. Ruffolo,
R. R., Nichols, A. J. Stadel, J. M. und Hieble, J. P.; "Pharmacological
and Therapeutic Applications of Alpha-2-Adrenoceptor Subtypes",
Annual Review of Pharmacology & Toxicology,
Bd. 32 (1993) SS. 243–279.
-
Weitere Informationen hinsichtlich
alpha-adrenergen Rezeptoren, Agonisten und Antagonisten sind allemein
in den folgenden Referenzen offenbart: Timmermans, P.B.M.W.M., Chiu,
A. T. und Thoolen, M.J.M.C., "12,1 α-Adrenergic Receptors", Comprehensive
Medicinal Chemistry, Bd. 3, Membranes & Receptors, P. G. Sammes & J. B. Taylor,
Hrsg., Pergamon Press (1990), SS. 133–185; Timmermans, P.B.M.W.M.,
und van Zwieten, P. A., "α-Adrenoreceptor
Agonists and Antagonists", Drugs of the Future, Bd. 9, Nr. 1 (Januar
1984), SS. 41–55;
Megens, A.A.H.P., Leysen, J. E., Awouters, F.H.L., und Niemegeers,
C.J.E., "Further Validation of in vivo and in vitro Pharmacological
Procedures for Assessing the α1 und α2-Selectivity of Test Compounds: (2) α-Adrenoreceptor
Agonists", European Journal of Pharmacology, Bd. 129 (1986), SS.
57–64;
Timmermans, P.B.M.W.M., deJonge, A., Thoolen, M.J.M.C., Wilffert,
B., Batink, H., vanZwieten, P. A., "Quantitative Relationships between α-Adrenergic
Activity and Binding Affinity of α-Adrenoceptor Agonists
and Antagonists", Journal of Medicinal Chemistry, Bd. 27 (1984),
SS. 495–503;
vanMeel, J.C.A., deJonge, A., Timmermans, P.B.M.W.M., und vanZwieten,
P. A., „Selectivity
of Some Alpha Adrenoceptor Agonists for Peripheral Alpha-1 und Alpha- 2-Adrenoreceptors
in the Normotensive Rat", The Journal of Pharmacology and Experimental
Therapeutics, Bd. 219, Nr. 3 (1981), SS. 760–767; Chapleo, C. B., et al.,
"Effect of 1,4-Dioxanyl Substitution on the Adrenergic Activity
of Some Standard α-Adrenoceptor Agents",
European Journal of Medicinal Chemistry, Bd. 24 (1989), SS. 619– 622; Chapleo,
C. B., et al., "Heteroaromatic Analogues of the α2-Adrenoreceptor
Partial Agonist Clondine", Journal of Medicinal Chemistry, Bd. 32
(1989), SS. 1627–1630;
Clare, K. A., Scrutton, M. C. und Thompson, N. T., "Effects of α2-Adrenoceptor
Agonists and of Related Compounds on Aggregation of, and on Adenylate
Cyclase Activity in Human Platelets", British Journal of Pharmacology,
Bd. 82 (1984), SS. 467–476; US-Patent
Nr. 3 890 319, erteilt an Danielewicz, Snarey and Thomas, 17. Juni
1975; US-Patent Nr. 5 091 528, erteilt an Gluchowski, 25. Febr.
1992; US-Patent Nr. 5 478 858, erteilt an Cupps and Bogdan, 26.
Dez. 1995; und US-Patent Nr. 5 541 210, erteilt an Cupps und Bogdan,
30. Juli 1996.
-
Im Fachbereich wurden die 2-Amino-2-Derivate
gemäß zahlreichen
verschiedenen Verfahren synthetisiert. US-Patent Nr. 4 398 028,
erteilt an Neumann am 9. Aug. 1983; Chapleo, C., et al., "Heteroaromatic
Analogues of the α2-Adrenoreceptor Partial Agonist Clonidine",
Journal of Medicinal Chemistry, Bd. 32 (1989), SS 1627–1630; US-Patent
Nr. 5 130 441, erteilt an Gluchowski, 14. Juli 1992; US-Patent Nr.
5 478 858, erteilt an Cupps und Bogdan, 26. Dez. 1995.
-
Zum Beispiel beinhaltet die Synthese
von Clonidin-Analoga die Umsetzung von 2-Thiomethyl-2-imidazolin mit einem aromatischen
primären
Amin in Gegenwart einer großen Überschussmenge
an Pyridin. Jedoch führt
die Literatur sehr geringe Ausbeuten in dieser Umsetzung an; siehe
Chapleo, C., et al., "Heteroaromatic Analogues of the α2-Adrenoreceptor
Partial Agonist Clondine", Journal of Medicinal Chemistry, Bd. 32
(1989), SS. 1627–1630.
-
Altnerative Synthesen von 2-Amino-2-Derivaten
wurden durchgeführt.
Allerdings sind bei diesen Synthesen die zeitraubenden, kostspieligen
Mehrfachschritte, die von diesen Synthesen verlangt werden, und/oder
die Verwendung von Quecksilber- oder anderen Übergangsmetallreagenzien von
weiterem Nachteil, die zum Vorliegen toxischer Verunreinigungen
führen
können.
US-Patent Nr. 4 398 028, erteilt an Neumann am 9. Aug. 1983; US-Patent
Nr. 5 478 858, erteilt an Cupps und Bogdan. am 26. Dez. 1995.
-
Weiterhin wurden andere synthetische
Herstellungen von 2-Amino-2-Derivaten durchgeführt. Das US-Patent Nr. 5 130
441, erteilt an Gluchowski am 14. Juli 1992. Gluchowski stellte
fest, dass die Ausbeuten bei der Bildung von 2-Amino-2-imidazolinen
gegenüber
der Chapleo-Verfahrensweise verbessert werden könnten durch Kuppeln eines aromatischen
primären
Amins mit Imidazolinsulfonsäure.
Allerdings waren die Ausbeuteverbesserungen bei Gluchowski nur mäßig. Weiterhin
erforderte diese Synthese die weniger ergiebige Herstellung einer
Imidazolinsulfonsäure-Zwischenverbindung.
-
Es ist im Fachbereich offensichtlich,
dass höhere
Ausbeuten erbringende, wirtschaftlichere Verfahren zur Herstellung
von 2-Amino-2-Derivaten vorteilhaft wären. Es wurde überraschenderweise
festgestellt, die Nachteile der in der Literatur zu findenden Synthesen
dieser Verbindungen durch Kuppeln eines primären Amins oder von dessen Salzen
mit einer acylierten 2-thiosubstituierten-2-Imidazolin, -Amidin
oder -Tetrahydropyrimidin-Zwischenverbindung in Gegenwart einer
Protonenquelle unter Erhalt des gewünschten 2-Amino-2-Derivats
in einem Schritt überwunden
werden können.
Die Ausbeuten in dieser Umsetzung sind wesentlich höher als
die bei Chapleo berichteten. Die Umsetzung ist auch vorteilhafter
als die Verfahrensweisen von Neumann und Cupps, weil sie langwierige
Synthesen überwindet
und die Verwendung von Übergangsmetallreagenzien
umgeht.
-
Ferner behebt die vorliegende Erfindung
den Mangel der Gluchowski-Synthese. Die vorliegende Erfindung verwendet
nicht eine Sulfonsäure,
sondern vielmehr ein acyliertes 2-thiosubstituiertes-2-Imidazolin, -Amidin
oder -Tetrahydropyrimidin als Zwischenverbindung bei der Synthese
von 2-Amino-2-Derivaten. Allgemein sind acylierte 2-thiosubstituierte-2-Imidazoline
bekannt. Allerdings liefern die bekannten Synthesen von acylierten
2-Thiomethyl-2-imidazolinen niedrige Ausbeuten. Kohn, H., et al.,
"Synthesis and Pharmacolagical Activity of Substituted Imidazolinethiones
and Thioimidazolines", Journal of Medicinal Chemistry, Bd. 20 (1977),
SS. 158–160;
Kohn, H., et al., "Syntheses and Spectral Properties of Substituted
Imidazolidones and Imidazolines", Journal of Organic Chemistry,
Bd. 42 (1977), SS. 941–948.
Es wurde überraschenderweise
festgestellt, dass acylierte 2-thiosubstituierte-2-Imidazoline,
-Amidine, und -Tetrahydropyrimidine in einer Zweistufen-Eintopfreaktion
in hohen Ausbeuten hergestellt werden können. Diese Verfahrensweise
macht die Synthese von acylierten 2-thiosubstituierten-2-Derivaten
ergiebiger, einfacher und weniger zeitraubend als die Verfahrensweise
in der Kohn-Referenz.
-
Es wurde daher nun festgestellt,
dass 2-Amino-2-imidazolir-, Guanidin- und 2- Amino-3,4,5,6-tetrahydropyrimidin-Derivate
in geeigneter Weise in hohen Ausbeuten durch Herstellen des entsprechenden
acylierten 2-thiosubstituierten-2-Derivats in einem Zweistufen-Eintopf-Verfahren
in hohen Ausbeuten und durch weiteres Umsetzen dieses isolierten
Derivats mit dem geeigneten Amin oder dessen Salzen in Gegenwart
einer Protonenquelle synthetisiert werden können. Das vorliegende Verfahren
ermöglicht
die Herstellung von 2-Amino-2-imidazolinen, Guanidinen und 2-Amino-3,4,5,6-tetrahydropyrimidinen
unter Reaktionsbedingungen, die die Notwendigkeit langdauernder,
kostspieliger oder wenig ergiebiger Mehrfachschritte und hochtoxischer
Reaktanten eliminiert.
-
Dieses Verfahren ermöglicht verbesserte
Ausbeuten und Produktreinheit und sorgt für eine zusätzliche Synthese-Flexibilität für die Herstellung
dieser Klassen von Molekülen.
-
Insbesondere stellen die bevorzugten
Verfahren der vorliegenden Erfindung eine neue Methodik bereit,
die besonders für
das Scale-up bzw. die Maßstabsvergrößerung und
die Herstellung von 2-Amino-2-Derivaten geeignet ist. Die Verfahren
benutzen kommerziell verfügbare,
kostengünstige
Ausgangsmaterialien. Die acylierte 2-thiosubstituierte-2-Imidazolin-, -Amidin-
oder -Tetrahydropyrimidin-Zwischenverbindung und das entsprechende
2-Amino-2-Derivat kann häufig
durch direkte Präzipitation
erhalten werden, wodurch die typischen Extraktions- und Verdampfungsprozeduren
umgangen werden, die in den in der Literatur aufgeführten Verfahren
anzutreffen sind.
-
Zusammenfassung
der Erfindung
-
Die vorliegende Erfindung liefert
ein Verfahren zur Herstellung eines 2-Amino-2-imidazolin-, Guanidin- und 2-Amino-3,4,5,6-tetrahydropryrimidin-Derivats
mit einer allgemeinen Struktur:
oder den Tautomeren
hiervon, worin:
-
- (a) R1 Methyl, Ethyl,
eine Methylengruppe, verbunden mit R2 über eine
Einfach bindung, sodass R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (b) R2 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R1 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (c) Z Alkyl oder ein gesättigter,
ungesättigter
oder aromatischer, monozyklischer oder polyzyklischer Carbozyklus
oder Heterozyklus ist, enthaltend ein oder mehrere aus O, N oder
S gewählte
Heteroatome; und
- (d) R4 ein oder mehrere Substituenten
an Z ist, umfassend unabhängig
Wasserstoff, Alkoxy, Alkylthio, Alkyl, Alkenyl, Amino, Carboxyl,
Cyano, Halogen, Hydroxy, Nitro und Thiol;
- (e) oder ein Salz oder pharmazeutisch annehmbares Salz hiervon;
wobei das Verfahren die folgenden Schritte umfasst:
-
(I) Herstellen einer Zwischenverbindung
mit der allgemeinen Struktur:
worin:
-
- (a) R aus der Gruppe gewählt ist, bestehend aus Methyl,
Ethyl und Benzyl;
- (b) R1 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R2 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (c) R2 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R1 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (d) R3 -O-R5 oder
-R6 ist;
- (e) R5 aus der Alkyl, Methyl, Ethyl,
Benzyl, tert-Butyl und Phenyl umfassenden Gruppe gewählt ist;
und
- (f) R6 aus der Methyl, Ethyl, tert-Butyl
und Phenyl umfassenden Gruppe gewählt ist; aus einem Thioharnstoff
mit der allgemeinen Struktur:
worin:
- (a) R1 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R2 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (b) R2 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R1 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
in einer Zweistufen-Eintopfreaktion durch:
- a) Alkylieren des Thioharnstoffs unter Verwendung eines Alkylierungsmittels
zur Bildung eines 2-thiosubstituierten-2-Imidazolins, eines Amidins
oder 2-thiosubstituiertem-3,4,5,6-Tetrahydropyrimidins;
- b) Acylieren des 2-thiosubstituierten-2-Imidazolins, Amidins
oder 2-thiosubstituiertem-3,4,5,6-Tetrahydropyrimidins aus Schritt
(I)(a) mit einem Acylierungsmittel in Gegenwart einer Base; und
-
(II) Kuppeln der Zwischenverbindung
aus Schritt (I) mit einem Amin oder seinem Salz der Struktur:
in Gegenwart einer
organischen Säure.
-
Genaue Beschreibung
der Erfindung
-
Die vorliegende Erfindung betrifft
Verfahren zur Herstellung von 2-Amino-2-imidazolin-, Guanidin- und 2-Amino-3,4,5,6-tetrahydropyrimidin-Derivaten.
Solche 2-Amino-2-Derivate
sind nützlich
für die
Behandlung verschiedener medizinischer Beschwerden, einschließlich Atembeschwerden,
Augenleiden, Magen-Darm-Beschwerden, nasaler Dekongestion, Bluthochdruck,
Migräne,
Beschwerden in Verbindung mit der Aktivität des sympathischen Nervensystems
und dem Missbrauch von Substanzen. Wenn die gemäß diesen Verfahren hergestellten
Verbindungen zur Behandlung solcher Beschwerden verwendet werden,
müssen
sie pharmazeutisch annehmbar sein. Wie hierin verwendet, ist eine
solche "pharmazeutisch annehmbare" Komponente eine solche, die für die Verwendung
bei Menschen und/oder Tieren ohne unangemessene schädliche Nebenwirkungen
(wie Toxizität,
Reizung und allergische Reaktion) in Übereinstimmung mit einem vernünftigen
Nutzen/Risiko-Verhältnis
geeignet ist. Solche pharmazeutisch annehmbaren Formen schließen Salze,
biohydrolysierbare Ester und Solvate ein.
-
Die gemäß den Verfahren der vorliegenden
Erfindung hergestellten 2-Amino-2-Derivate können auch als Zwischenverbindungen
zur Herstellung anderer 2-Amino-2-Derivate verwendet werden. Das heißt, die
hergestellten Verbindungen können
weiter umgesetzt werden unter Anwendung bekannter Chemien, um andere aktive
Analoga zu erhalten.
-
Definitionen
und Gebrauch von Ausdrücken
-
Das Folgende ist eine Auflistung
von Definitionen für
hierin verwendete Ausdrücke:
-
Wie hierin verwendet, bedeutet "Acylierungsmittel"
ein Reagens, das zum Acylieren eines Stickstoffatoms zur Bildung
eines Carbamats oder eines Amids, vorzugsweise eines Carbamats,
geeignet ist. Bevorzugte Acylierungsmittel schließen Di-tert-butyldicarbonat,
Diethylpyrocarbonat, Dimethylpyrocarbonat, Methylchlorformiat, Ethylchlorformiat,
Benzylchlorformiat, Allylchlorformiat, Phenylchlorformiat, Acetylchlorid,
Propionylchlorid, Essigsäureanhydrid,
Propionsäureanhydrid,
Trimethylacetylchlorid, Trimethylessigsäureanhydrid und Benzoylchlorid
ein. Stärker
bevorzugte Acylierungsmittel sind Ditert-butyldicarbonat, Dimethylpyrocarbonat und
Methylchlorformiat. Das am stärksten
bevorzugte Acylierungsmittel ist Methylchlorformiat.
-
Wie hierin verwendet, bedeutet "Alkenyl"
einen Kohlenwasserstoffsubstituenten mit einer oder mehreren Doppelbindungen,
geradkettig oder verzweigtkettig, unsubstituiert oder substituiert.
-
Wie hierin verwendet, bedeutet "Alkoxy"
einen Substituenten der Struktur Q-O-, worin Q Alkyl oder Alkenyl
ist.
-
Wie hierin verwendet, bedeutet "Alkyl"
einen gesättigten
Kohlenwasserstoff substituenten, gerad- oder verzweigtkettig, unsubstituiert
oder substituiert.
-
Wie hierin verwendet, bedeutet "Alkylierungsmittel"
ein Reagens, das zum Alkylieren eines Heteroatoms, wie Schwefel,
geeignet ist. Bevorzugte Alkylierungsmittel schließen Methyliodid,
Methylbromid, Methylchlorid, Dimethylsulfat, Ethyliodid, Ethylbromid,
Ethylchlorid, Diethylsulfat und Benzylbromid ein. Stärker bevorzugte
Alkylierungsmittel schließen
Methyliodid, Methylbromid, Dimethylsulfat, Ethyliodid und Diethylsulfat ein.
Die am stärksten
bevorzugten Alkylierungsmittel sind Methyliodid und Dimethylsulfat.
-
Wie hierin verwendet, bedeutet "Alkylthio"
einen Substituenten der Struktur Q-S-, worin Q Alkyl oder Alkenyl
ist.
-
Wie hierin verwendet, bedeutet „Base"
ein basisches Reagens, das einer Reaktionsmischung hinzugefügt wird,
um die Acylierung von Stickstoff mit Hilfe eines Acylierungsmittels
zu erleichtern. Basen schließen Stickstoffbasen
und anorganische Basen ein. Bevorzugte Basen schließen jene
ein, die leicht filtrierbare oder anderweitig entfernbare Salze
aufweisen. Insbesondere schließen
bevorzugte Basen N,N-Diisopropylethylamin, Triethylamin, Trimethylamin,
4-Dimethylaminopyridin, Pyridin, Kaliumcarbonat, Natriumcarbonat,
Kaliumbicarbonat und Natriumbicarbonat ein. Die stärker bevorzugten
Basen sind Triethylamin, Trimethylamin und Kaliumcarbonat. Die am
stärksten
bevorzugte Base ist Kaliumcarbonat.
-
Wie hierin verwendet, bedeutet "carbozyklischer
Ring" einen gesättigten,
ungesättigten
oder aromatischen Kohlenwasserstoffringrest. Carbozyklische Ringe
sind monozyklische oder verschmolzene, überbrückte oder spiropolyzyklische
Ringsysteme. Monozyklische Ringe enthalten 3 bis 9 Atome, vorzugsweise
4 bis 7 Atome, und am meisten bevorzugt 5 oder 6 Atome. Polyzyklische
Ringe enthalten 7 bis 17 Atome, vorzugsweise 7 bis 14 Atome, und
am meisten bevorzugt 9 oder 10 Atome.
-
Wie hierin verwendet, ist "Ether-Lösungsmittel"
ein Lösungsmittel,
bei dem zwei Alkylgruppen mit einem Sauerstoff verbunden sind, einschließlich jener,
bei welchen die Alkylgruppen und das Sauerstoffatom Teil des Rings
sind. Bevorzugte Ether-Lösungsmittel
schließen
Diethylether, Methyl-tert-butylether, Tetrahydrofuran und Isopropylether
ein. Stärker
bevorzugte Ether-Lösungsmittel
schließen
Methyl-tert-butylether und Isopropylether ein. Das am meisten bevorzugte
Ether-Lösungsmittel
ist Methyl-tert-butylether.
-
Wie hierin verwendet, sind „Halogenkohlenwasserstoff-Lösungsmittel"
Lösungsmittel,
bei denen eines oder mehrere Halogene an eine Kohlenstoffkette gebunden
sind. Bevorzugte Kohlenwasserstoff-Lösungsmittel schließen Dichlormethan,
Ethylendichlorid, Chloroform und Kohlenstofftetrachlorid ein. Stärker bevorzugt sind
Dichlormethan und Ethylendichlorid. Noch stärker bevorzugt ist Ethylendichlorid.
-
Wie hierin verwendet, ist „Halogen"
ein Chlor-, Brom-, Fluor- oder Iodatomrest. Brom, Chlor und Fluor sind
bevorzugte Halogene.
-
Wie hierin verwendet, ist „heterozylischer
Ring" ein gesättigter,
ungesättigter
oder aromatischer Ringrest, der aus Kohlenstoffatomen und einem
oder mehreren Heteroatomen in dem Ring aufgebaut ist. Heterozylische
Ringe sind monozyklisch oder sind verschmolzene, überbrückte oder
spiropolyzyklische Ringsysteme. Monozyklische Ringe enthalten 3
bis 9 Atome, vorzugsweise 4 bis 7 Atome, und am meisten bevorzugt
5 oder 6 Atome. Polyzyklische Ringe enthalten 7 bis 17 Atome, vorzugsweise
7 bis 14 Atome, und am meisten bevorzugt 9 oder 10 Atome.
-
Wie hierin verwendet, ist "Methylen"
ein -CH2-Rest.
-
Wie hierin verwendet, ist "organische
Säure"
eine organische Carbonsäure,
wie Ameisensäure,
Essigsäure,
Chloressigsäure,
Dichloressigsäure,
Propionsäure,
Benzoesäure,
Maleinsäure,
Fumarsäure,
Bernsteinsäure
und Weinsäure.
Bevorzugte organische Säuren
schließen
Essigsäure,
Propionsäure
und Chloressigsäure
ein. Die am meisten bevorzugte organische Säure ist Essigsäure.
-
Wie hierin verwendet, ist "polares,
aprotisches Lösungsmittel"
ein Lösungsmittel,
das die Eigenschaft einer hohen Polarität besitzt, aber dennoch nicht
die Fähigkeit
besitzt, ein Proton abzugeben. Bevorzugte polare aprotische Lösungsmittel
schließen
Acetonitril, Methylethylketon, N,N-Dimethylformamid, N,N-Dimethylacetamid,
N-Methylpyrrolidinon und Methylsulfoxid ein. Die am meisten bevorzugten
polaren, aprotischen Lösungsmittel
sind Acetonitril und N,N-Dimethylacetamid.
-
Wie hierin verwendet, ist "protisches
Lösungsmittel"
ein Lösungsmittel,
das ein Wasserstoffatom enthält,
das an ein Sauerstoff- oder Stickstoffatom gebunden ist. Bevorzugte
protische Lösungsmittel
schließen Methanol,
Ethanol, 2-Propanol, Butanol, sec- Butanol und Isoamylalkohol ein. Die
am meisten bevorzugten protischen Lösungsmittel sind Ethanol und
Methanol.
-
Wie oben stehend definiert und wie
hierin verwendet, können
Substituentengruppen selbst substituiert sein. Eine derartige Substitution
kann mit einem oder mehreren Substituenten erfolgen. Solche Substituenten schließen die
bei C. Hansch und A. Leo, Substituent Constants for Correlation
Analysis in Chemistry and Biology (1979), ein, die hierin durch
den Bezug mit eingeschlossen sind. Bevorzugte Substituenten schließen (zum
Beispiel) Alkyl, Alkenyl, Alkoxy, Hydroxy, Oxo, Amino, Aminoalkyl
(z. B. Aminomethyl etc.), Cyano, Halogen, Alkoxy, Alkoxyacyl (z.
B. Carboethoxy etc.), Thiol, Aryl, Cycloalkyl, Heteroaryl, Heterocycloalkyl
(z. B. Piperidinyl, Morpholinyl, Pyrrolidinyl etc.), Imino, Thioxo,
Hydroxyalkyl, Aryloxy, Arylalkyl und Kombinationen davon ein.
-
Unter Anwendung
des vorliegenden Verfahrens hergestellte Verbindungen
-
Die durch die Verfahren dieser Erfindung
hergestellten Verbindungen umfassen beliebige einer Vielzahl von
Heteroaryl-2-amino-2-imidazolinen, Guanidinen und 2-Amino-3,4,5,6-tetrahydropryrimidinen.
Diese Verbindungen sind von der folgenden allgemeinen Struktur:
oder der Tautomeren
hiervon, worin:
-
- (a) R1 Methyl, Ethyl,
eine Methylengruppe, verbunden mit R2 über eine
Einfachbindung, sodass R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (b) R2 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R1 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R, über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist;
- (c) Z Alkyl oder ein gesättigter,
ungesättigter
oder aromatischer, monozyklischer oder polyzyklischer Carbozyklus
oder Heterozyklus ist, enthaltend ein oder mehrere aus O, N oder
S gewählte
Heteroatome; und
- (d) R4 ein oder mehrere Substituenten
an Z ist, umfassend unabhängig
Wasserstoff, Alkoxy, Alkylthio, Alkyl, Alkenyl, Amino, Carboxyl,
Cyano, Halogen, Hydroxy, Nitro und Thiol;
- (e) oder ein Salz oder pharmazeutisch annehmbares Salz hiervon.
-
Wie hierin verwendet, sind R1 und R2 weiter bevorzugt
Methylengruppen, die entweder über
eine Einzelbindung oder eine andere Methylengruppe miteinander verbunden
sind unter Bildung eines fünfgliedrigern bzw.
eines sechsgliedrigen Rings. Weiterhin ist es auch mehr bevorzugt,
dass nur R1 oder R2 Methyl
sind, wobei der andere Substituent Ethyl ist. R1 und
R2 sind am meisten bevorzugt Methylengruppen,
die über
eine Einzelbindung miteinander verbunden sind unter Bildung eines
fünfgliedrigen
Rings.
-
Wie hierin verwendet, ist Z weiter
bevorzugt ein aromatischer monozyklischer oder polyzyklischer Ring.
Wenn Z monozyklisch ist, ist Z vorzugsweise ein fünf- oder
sechsgliedriger Ring und am meisten bevorzugt ein sechsgliedriger
Ring. Wenn Z polyzyklisch ist, ist Z vorzugsweise ein entweder mit
einem oder zwei fünf-
oder sechsgliedrigen Ringen verschmolzener sechsgliedriger Ring.
Wenn Z polyzyklisch ist, ist Z am meisten bevorzugt ein mit einem
fünfgliedrigen
Ring verschmolzener sechsgliedriger Ring.
-
Wie hierin verwendet, ist R4 vorzugsweise Wasserstoff, Alkoxy, Alkylthio,
Alkyl, Alkenyl, Amino, Carboxyl, Cyano, Halogen, Hydroxy, Nitro
oder Thiol. R4 ist weiter bevorzugt Wasserstoff,
Cyano, Alkoxy, Alkylthio, Amino, C1-C4-Alkyl, C1-C4-Alkenyl, Halogen oder Hydroxy. R4 ist am meisten bevorzugt Wasserstoff, Cyano, Alkoxy,
C1-C4-Alkyl, C1-C4-Alkenyl oder Halogen.
-
Wo die unter Anwendung der vorliegenden
Verfahren synthetisierten Verbindungen als Zwischenverbindungen
verwendet werden, können
Gruppen, wie Amine, Imine oder Alkohole, durch allgemein im Fachbereich
bekannte Verfahren funktionalisiert werden.
-
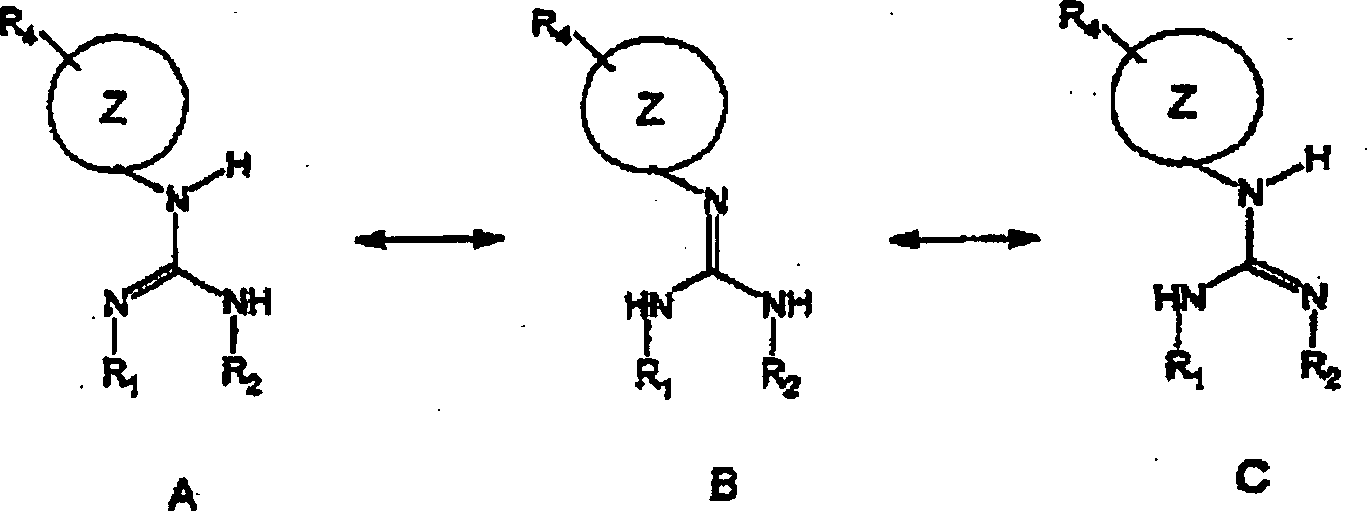
Der Durchschnittsfachmann wird erkennen,
dass tautomere Formen bei bestimmten Verbindungen der Erfindung
vorliegen. Wenn das Tautomer A des Moleküls angeführt wird, versteht es sich,
die Tautomeren B und C des Moleküls
einzuschließen,
obwohl dies nicht speziell beschrieben ist. Zur Veranschaulichung:
-
Beispiele für Verbindungen, die unter Anwendung
des Verfahrens der vorliegenden Erfindung hergestellt werden können, sind
unten stehend gezeigt. Diese Verbindungen dienen lediglich der Erläuterung
und sind auf keinen Fall eine erschöpfende Auflistung der Möglichkeiten.
-
-
-
Die oben stehenden Moleküle sind
in den folgenden Quellen beschrieben: Alinidin, offenbart in dem US-Patent
Nr. 3 708 485 (1973) von Boehringer, Ing; Iopidin, offenbart in
dem US-Patent Nr. 4 517 199 (1985) von Alcon; Brimonidin, offenbart
in dem deutschen Patent Nr. 2 538 620 von Pfizer; Clonidin, offenbart
in dem US-Patent Nr. 3 202 660 (1965) von Boehringer, Ing.; Indanazolin,
offenbart in dem US-Patent Nr. 3 882 229 von Nordmark; Moxonidin,
US-Patent Nr. 4 323 570 (1982) von Beiersdorf Aktiengesellschaft;
Tiamendin, offenbart in dem US-Patent Nr. 3 758 476 (1973) von Hoechst;
Tizanidin, offenbart in dem US-Patent Nr. 3 843 668 (1974) von Wander-Sandoz;
Tolonidin, offenbart in dem US-Patent Nr. 3 236 857 (1966) von Boehringer, Ing.;
Tramazolin, offenbart in dem deutschen Patent Nr. 1 191 381 (1965)
von Thomae.
-
Herstellungsverfahren
-
Allgemein umfassen die Verfahren
der vorliegenden Erfindung die neue Synthese einer acylierten 2-thiosubstituierten-2-Imidazolin-,
Amidin- oder -3,4,5,6-Tetrahydropyrimidin-Zwischenverbindung (im
Folgenden als "acylierte Zwischenverbindung" beschrieben), gefolgt
von einem Kuppeln der acylierten Zwischenverbindung mit einem geeigneten
Amin oder von dessen Salzen in Gegenwart einer organischen Säure. Die
in der Synthese verwendete acylierte Zwischenverbindung wird herkömmlicherweise
in einer neuen Zweistufen-Eintopfreaktion aus dem entsprechenden
Thioharnstoff in hohen Ausbeuten hergestellt.
-
Dieses Verfahren wird durch das folgende
allgemeine Schema veranschaulicht:
-
In dem oben stehenden allgemeinen
Schema:
-
- (a) ist R1 Methyl,
Ethyl, eine Methylengruppe, verbunden mit R2 über eine
Einfachbindung, so dass R1 und R2 einen fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring
bilden;
- (b) ist R2 Methyl, Ethyl, eine Methylengruppe,
verbunden mit R1 über eine Einfachbindung, sodass
R1 und R2 einen
fünfgliedrigen
Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine
andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden; und
-
R1 und R2 sind weiter bevorzugt entweder über eine
Einfachbindung oder eine andere Methylengruppe miteinander verbundene
Methylengruppen unter Bildung eines fünfgliedrigen bzw. sechsgliedrigen
Rings. R1 und R2 sind
am meisten bevorzugt Methylengruppen, die aneinander durch eine
Einfachbindung unter Bildung eines fünfgliedrigen Ringes gebunden
sind.
-
In dem oben stehenden allgemeinen
Schema, ist R ein Alkyl oder aromatischer Substituent, abgeleitet von
dem in dem Verfahren verwendeten Akylierungsmittel. R ist vorzugsweise
ein Methyl-, Ethyl- oder Benzylrest. R ist am meisten bevorzugt
ein Methylrest.
-
In dem oben stehenden allgemeinen
Schema ist R3 abgeleitet von dem in dem
Verfahren verwendeten Acylierungsmittel. R3 kann
-O-R
5 oder -R6 sein, worin R5 und
R6 ebenfalls von dem in dem Verfahren verwendeten
Acylierungsmittel abgeleitet sind. R3 ist
vorzugsweise -O-R5, R5 ist
vorzugsweise ein Allyl-, Methyl-, Ethyl, Benzyl-, tert-Butyl- oder Phenylrest.
R5 ist am meisten bevorzugt ein Methylrest.
R6 ist vorzugsweise ein Methyl-, Ethyl-,
tert-Butyl- oder Phenylrest.
-
In dem oben stehenden allgemeinen
Schema ist Z ein Alkyl oder gesättigter,
ungesättigter
oder aromatischer, monozyklischer oder polyzyklischer Carbozyklus
oder Heterozyklus mit einem oder mehreren Heteroatomen, gewählt aus
O, N oder S. Z ist vorzugsweise ein aromatischer monozyklischer
oder polyzyklischer Ring. Wenn Z monozyklisch ist, ist Z vorzugsweise
ein fünf-
oder sechsgliedriger Ring, und am meisten bevorzugt ein sechsgliedriger
Ring. Wenn Z polyzyklisch ist, ist Z am meisten bevorzugt ein sechsgliedriger
Ring, verschmolzen entweder mit einem oder zwei fünf- oder
sechsgliedrigen Ringen. Wenn Z polyzyklisch ist, ist Z am meisten
bevorzugt ein mit einem fünfgliedrigen
Ring verschmolzener sechsgliedriger Ring.
-
In dem oben stehenden allgemeinen
Schema ist R4 einer oder mehrere Substituenten
auf Z, umfassend unabhängig
Wasserstoff, Alkoxy, Alkylthio, Alkyl, Alkenyl, Amino, Carboxyl,
Cyano, Halogen, Hydroxy, Nitro und Thiol. R4 ist
weiter bevorzugt Wasserstoff, Cyano, Alkoxy, Alkylthio, Amino, C1-C4-Alkyl, C1-C4-Alkenyl, Halogen
oder Hydroxy. R4 ist am meisten bevorzugt
Wasserstoff, Cyano, Alkoxy, C1-C4-Alkyl, C1-C4-Alkenyl oder Halogen.
-
In dem oben stehenden allgemeinen
Schema wird ein Thioharnstoff mit einem Alkylierungsmittel in einem
Lösungsmittel
umgesetzt, das die Alkylierungsreaktion vonstatten gehen lässt. Stärker bevorzugte
Alkylierungsmittel schließen
Methyliodid, Methylbromid und Dimethylsulfat, Ethyliodid und Diethylsulfat
ein. Die am meisten bevorzugten Alkylierungsmittel sind Methyliodid
und Dimethylsulfat. Bevorzugte Lösungsmittel
schließen
Ester-Lösungsmittel
(wie zum Beispiel Butylacetat, Ethylacetat oder Methylacetat, vorzugsweise
Ethylacetat), Ether-Lösungsmittel,
protische Lösungsmittel
und polare, aprotische Lösungsmittel
ein. Stärker
bevorzugte Lösungsmittel
schließen
Ether-Lösungsmittel,
protische Lösungsmittel
und polare aprotisch Lösungsmittel
ein. Die am meisten bevorzugten Lösungsmittel sind protische
Lösungsmittel.
Das am meisten bevorzugte Lösungsmittel
ist Ethanol. Die Mischung bei einer Temperatur vorzugsweise zwischen
etwa 0°C
und etwa 150°C,
weiter bevorzugt zwischen Umgebungstemperatur und etwa 100°C, und am
meisten bevorzugt zwischen etwa 30°C und etwa 70°C ablaufen
gelassen.
-
Die so erhaltene thiosubstituierte
Verbindung kann durch Verfahren isoliert werden, die Fachleuten
auf dem Gebiet offensichtlich sind, wie die Anwendung von Verfahren
einschließlich
Extraktions-, Lösungsmittelverdampfungs-,
Destillations- oder Kristallisationsverfahrettsweisen. Am meisten
bevorzugt wird das 2-thio-2-substituierte Derivat weiter in dem
gleichen Gefäß und in
dem gleichen Lösungsmittel
ohne Isolierung umgesetzt. Vor der weiteren Umsetzung des 2-thio-2-substituierten
Derivats wird die Reaktionsmischung vorzugsweise von etwa –10°C bis etwa
75°C, weiter
bevorzugt von etwa 0°C
bis etwa 40°C,
und am meisten bevorzugt auf Umgebungstemperatur abgekühlt.
-
Das 2-thio-2-substituierte Derivat
wird danach mit einem Acylierungsmittel in Gegenwart einer Base
in einem beliebigen Lösungsmittel
umgesetzt, das die Reaktion vonstatten gehen lässt. Bevorzugte Acylierungsmittel
schließen
Di-tert-dicarbonat, Diethylpyrocarbonat, Dimethylpyrocarbonat, Methylchlorformiat,
Ethylchlorformiat, Allylchloformiat, Phenylchlorformiat, Acetylchlorid,
Propionylchlorid, Essigsäureanhydrid,
Propionsäureanhydrid,
Trimethylacetylchlorid, Trimethylessigsäureanhydrid und Benzoylchlorid
ein. Stärker
bevorzugte Acylierungsmittel sind Di-tert-butyldicarbonat, Dimethylpyrocarbonat;
Methylchlorformiat und Essigsäureanhydrid.
Das am meisten bevorzugte Acylierungsmittel ist Methylchlorformiat.
Bevorzugte Basen schließen jene
ein, die leicht filtrierbare oder auf andere Weise entfernbare Salze
aufweisen. Insbesondere sind die stärker bevorzugten Basen Triethylamin
und Kaliumcarbonat. Die am meisten bevorzugte Base ist Kaliumcarbonat.
Bevorzugte Lösungsmittel
schließen
Ester-Lösungsmittel
(wie zum Beispiel Butylacetat, Ethylacetat oder Methylacetat, vorzugsweise
Ethylacetat), Ether-Lösungsmittel,
protische Lösungsmittel
und polare aprotische Lösungsmittel
ein. Stärker
bevorzugte Lösungsmittel
schließen
Ether-Lösungsmittel,
protische Lösungsmittel (insbesondere
Ethanol und Isopropanol) und polare aprotische Lösungsmittel (insbesondere N,N-Dunetyhlacetamid)
ein. Die am stärksten
bevorzugten Lösungsmittel
sind protische Lösungsmittel.
Das am meisten bevorzugte Lösungsmittel
ist Ethanol. Die Base wird vorzugsweise der Reaktionsmischung zuerst
hinzugegeben, gefolgt von dem Ayclierungsmittel, wobei die Temperatur
der Mischung vorzugsweise zwischen etwa 0°C bis etwa 50°C, weiter
bevorzugt zwischen etwa 20°C
bis etwa 35°C
gehalten wird. Die Reaktion wird bei einer Temperatur von vorzugsweise
zwischen etwa 20°C
bis etwa 60°C,
weiter bevorzugt zwischen etwa 40°C
bis etwa 55°C
ablaufen gelassen.
-
Nach Beendigung der Umsetzung kann
die so acylierte Zwischenverbindung durch Fachleuten auf dem Gebiet
bekannte Verfahren isoliert werden, wie Verfahren einschließlich Extraktions-,
Lösungsmittelverdampfungs-,
Destillations- oder Kristallisationsverfahrensweisen. Weiter bevorzugt
wird die Reaktionsmischung filtriert, und die Nebenproduktsalze
zu entfernen, bei einer Temperatur zwischen etwa 30°C bis etwa 70°C, weiter
bevorzugt zwischen etwa 50°C
bis etwa 60°C.
Die Nebenproduktsalze werden danach vorzugsweise mit Ester-Lösungsmitteln
(wie zum Beispiel Butylacetat, Ethylacetat oder Methylacetat, vorzugsweise Ethylacetat),
protischen Lösungsmitteln
oder polaren aprotischen Lösungsmitteln,
weiter bevorzugt mit einem protischen oder Ester-Lösungsmittel,
gewaschen. Nach der Filtration kann die so erhaltene acylierte Zwischenverbindung
durch Fachleuten auf dem Gebiet bekannte Verfahren isoliert werden,
wie unter Anwendung von Verfahren einschließlich Extraktions-, Lösungsmittelverdampfungs-,
Destillations- oder Kristallisationsverfahrensweisen. Vorzugsweise
wird das Produkt als Feststoff durch Abkühlen des Filtrats auf eine
Temperatur von etwa –30°C bis Umgebungstemperatur,
weiter bevorzugt von etwa –20°C bis etwa
0°C, isoliert.
Der so erhaltene Feststoff wird filtriert und mit einem protischen
oder Ester-Lösungsmittel
gewaschen, der auf etwa –30°C bis Umgebungstemperatur,
weiter bevorzugt auf etwa –20°C bis etwa
20°C vorgekühlt wurde.
Der Feststoff wird vorzugsweise durch Fachleuten auf dem Gebiet
bekannte Verfahren getrocknet.
-
Die acylierte Zwischenverbindung
kann danach weiter mit dem passenden Amin oder von dessen Salzen
in einem protischen Lösungsmittel
oder einem polaren aprotischen Lösungsmittel
oder Mischungen davon in Gegenwart einer organischen Säure umgesetzt
werden. Die acylierte Zwischenverbindung kann auch weiter mit dem
passenden Amin oder von dessen Salzen in einer Lösung der organischen Säure allein
umgesetzt werden. Bevorzugte Säuren
schließen
Ameisensäure,
Essigsäure,
Chloressigsäure,
Dichloressigsäure,
Propionsäure,
Benzoesäure,
Maleinsäure,
Fumarsäure,
Bernsteinsäure
oder Weinsäure
ein. Die bevorzugten protischen Lösungsmittel schließen Methanol
und Ethanol ein. Das am meisten bevorzugte polare aprotische Lösungsmittel
ist Acetonitril. Die Reaktion wird vorzugsweise bei einer Temperatur
zwischen Umgebungstemperatur und etwa 150°C, weiter bevorzugt zwischen
etwa 40°C
bis etwa 100°C,
und noch weiter bevorzugt zwischen etwa 55°C bis etwa 80°C durchgeführt. In
einigen Fällen,
wo R3 nicht leicht zu entfernen ist, kann
es notwendig sein, eine anorganische Säure, wie HCl oder HBr, zusätzliche
Mengen einer organischen Säure, oder
ein protischeres Lösungsmittel,
zuzusetzen und/oder eine verstärkte
Erwärmung
auf die Reaktionsmischung anzuwenden, um die Abspaltung der Acylgruppe
zu erleichtern. Fachleute auf dem Gebiet werden erkennen, dass die
Hydrolyse dieser Acylgruppe auch ebenso unter basischen Bedingungen
erreicht werden kann. Nach Beendigung der Umsetzung kann das so
erhaltene 2-Amino-2-Derivat durch Fachleuten auf dem Gebiet bekannte
Verfahren isoliert werden, wie Verfahren einschließlich Extraktions-,
Lösungsmittelverdampfungs-,
Destillations- oder Kristallisationsverfahrensweisen. Fachleute
auf dem Gebiet werden auch erkennen, dass verschiedene Säuren in
den Endstufen des Verfahrens zugesetzt werden können zur Bildung verschiedener
Salzformen, welche die Isolierung und Handhabung erleichtern können.
-
Die folgenden nicht-einschränkenden
Beispiele veranschaulichen die Verfahren der vorliegenden Erfindung:
-
-
a. 2-Thiomethyl-2-imidazolin:
-
2-Imidazolidinethion (150 Gramm,
1,5 Mol) und N,N-Dimethylacetamid (1,1 l) werden in einem Rundkolben
vereinigt. Hierzu wird Dimethylsulfat (213 Gramm, 1,7 Mol) bei Umgebungstemperatur
hinzugefügt. Diese
Reaktionsmasse wird zwei Stunden lang umgerührt.
-
b. N-Carbomethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) wird in einem Eisbad gekühlt,
und dieser umgerührten
Lösung wird
Triethylamin (376 Gramm, 3,7 Mol) tropfenweise hinzugegeben. Zu
dieser Mischung wird Methylchlorformiat (166,7 Gramm, 1,8 Mol) tropfenweise
hinzugegeben. Nach Beendigung der Zugabe wird die Reaktionsmischung
sich auf Umgebungstemperatur erwärmen
gelassen. Nach 5 Stunden Rühren
wird die Mischung in kaltes Wasser (3 l) geschüttet. Das Produkt wird in Ethylacetat
(4 × 2,5
l) extrahiert. Die vereinigten Extrakte werden mit kaltem Wasser,
Kochsalzlösung,
gewaschen, über
Natriumsulfat getrocknet und unter vermindertem Druck konzentriert.
Das Trocknen des Rückstands
unter Vakuum ergibt das gewünschte
N-Carbomethoxy-2-thiomethyl-2-imidazolin.
-
c. 4-(2-Imidazolinylamino)-1,3,2-benzothiadiazol-acetatsalz:
-
N-Carbomethoxy-2-thiomethyl-2-imidazolin
(19,2 Gramm, 11 mMol) und 4-Amino-2,1,3-benzothiadiazol (11,1 Gramm, 73 mMol)
werden in einer 10%igen Lösung
von Eisessig in 2-Propanol (500 ml) gelöst. Die resultierende Lösung wird
bis fast zum Refluxieren (90– 95°C) 19 Stunden
lang erwärmt.
Die Mischung wird unter vermindertem Druck konzentriert, erneut
in 2-Propanol gelöst
und erneut präzipitiert
unter Erhalt von 4-(2-Imidazolinylamino)-1,3,2-benzothiadiazol-acetatsalz.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
2-Imidazolidinethion (50 Gramm) und
absolutes Ethanol (400 ml) werden unter Rühren zusammengebracht. Methyliodid
(43 ml, 1,4 Äqu.)
wird rasch der Rührmischung
hinzugegeben. Die Reaktionsmischung wird dann auf 35°C erwärmt, bis
die Bildung von 2-Thiomethyl-2-imidazolin
abgeschlossen ist.
-
b. N-Carbomethoxy-2-thiomethyl-2-imidazolin:
-
Kaliumcarbonat (101 Gramm) wird der
Mischung in dem oben stehenden Schritt (a) zugegeben, gefolgt von
der Zugabe von Methylchlorformiat (42 ml) unter Rühren. Nach
45 Minuten wird die Reaktionsmischung auf 55°C erwärmt und die unlöslichen
Salze werden abfiltriert. Diese Salze werden mit absolutem Ethanol
gewaschen. Das Filtrat (und Ethanol-Waschflüssigkeit) werden auf –20°C abgekühlt und
das Endprodukt wird durch Filtration isoliert. Das Endprodukt wird
mit kaltem (–20°C) absolutem
Ethanol gewaschen. Das Produkt wird über Nacht unter Vakuum bei
Raumtemperatur getrocknet unter Erhalt von N-Carbomethoxy-2-thiomethyl-2-imidazolin.
-
c. 2-[(2,6-Dichlorphenyl)amino]-2-imidazolin-acetatsalz:
-
2,6-Dichloranilin (2 g) und N-Carbomethoxy-2-thiomethyl-2-imidazolin
(2,68 g) werden in Eisessig (40 ml) gelöst und die Reaktionsmasse wird
bei 65 bis 75°C
umgerührt,
bis der Kupplungsschritt beendet ist. Die Reaktionsmischung wird
mit 70% Methanol-Wasser (40 ml) verdünnt, zum Refluxieren gebracht,
bis die Entschützung
vollständig
ist, und danach unter vermindertem Druck konzentriert, wodurch ein öliger Rückstand erhalten
wird. Die Zusetzung von Ethylacetat zu dem öligen Rückstand präzipitiert die Verunreinigungen,
die durch Filtration abgetrennt werden. Die Konzentrierung des Filtrats
liefert 2-[(2,6-Dichlorphenyl)amino]-2-imidazolin
als ein Acetatsalz.
-
-
a. N,N-Dimethyl-(2-thiomethyl)amidin:
-
Zu 1,3-Dimethyl-2-thioharnstoff (50
Gramm, 480 mMol) wird absolutes Ethanol (400 ml) unter Rühren hinzugegeben.
Indomethan (43 ml, 690 mMol) wird rasch zugesetzt. Die Reaktionsmischung
wird auf 30–35°C erwärmt und
umgerührt,
bis die Bildung von N,N'-Dimethyl-(2-thiomethyl)amidin
vollendet ist.
-
b. N,N'-Dimethyl-(N-methoxycarbonyl-2-thiomethyl)amidin:
-
Kaliumcarbonat (101 Gramm) wird der
Mischung des obigen Schritts (a) zugegeben. Methylchlorformiat (42
ml, 540 mMol) wird danach zugegeben. Nach 1 Stunde wird die Reaktionsmischung
auf 55°C
erwärmt und
die unlöslichen
Salze werden filtriert. Die Salze werden mit Ethanol (100 ml) gewaschen.
Das Filtrat (und Ethanol-Waschflüssigkeit)
werden auf –20°C gekühlt und
das Endprodukt wird durch Filtration isoliert. Das Endprodukt wird
mit 100 ml kaltem (–20°C) absolutem
Ethanol gewaschen. Das 2-Thiomethylamidin
wird über Nacht
unter Vakuum bei Umgebungstemperatur getrocknet.
-
c. N,N'-Dimethyl-N''-(8-methylchinolin-7-yl)guanidin-acetatsalz:
-
Die oben in Schritt (b) hergestellte
Zwischenverbindung wird mit 0,7 Äguivalenten
von 7- Amino-8-methylchinolin
(hergestellt in dem US-Patent Nr. 5 576 437, erteilt an Cupps und
Bogdan, 19. Nov. 1996 (hierin durch den Bezug darauf eingeschlossen)
in einer 10 %igen Lösung
Eisessig in Ethanol (2 l) vereinigt. Die Mischung wird bis zum Refluxieren
erwärmt,
und nachdem das Ausgangs-Amin aufgebraucht wurde, wird die Mischung
mit deaktiviertem Kohlenstoff entfärbt. Das Produkt wird auf Umgebungstemperatur
gekühlt,
filtriert, getrocknet und aus Acetonitril und Wasser umkristallisiert.
Nach dem Trocknen unter einem hohen Vakuum wird N,N'-Dimethyl-N''-(8-methylchinolin-7-yl)guanidinacetatsalz
erhalten.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
Dimethylsulfat (111 ml) wird langsam
einer umgerührten
Lösung
von 2-Imidazolidinethion (120 g) in Isopropanol (750 ml) bei Umgebungstemperatur
zugegeben. Die Reaktionsmischung wird auf 70°C erwärmt, bis die Bildung von 2-Thiomethyl-2-imidazolinhydrosulfat
abgeschlossen ist.
-
b. N-Carboethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) wird auf Umgebungstemperatur abkühlen gelassen, woraufhin Natriumcarbonat
(249 g) zugegeben werden, gefolgt von der Zugabe von Ethylchlorformiat
(168 ml). Die Reaktionsmischung wird bei 40°C bis zur Vervollständigung
umgerührt,
im Anschluss wird die Reaktionsmischung auf 55°C erwärmt und die heiße Mischung
wird filtriert, um die unlöslichen
Salze zu entfernen. Diese Salze werden mit kaltem Isopropanol gewaschen.
Das Filtrat (und die Waschlösung)
wird auf –20°C gekühlt und
2 Stunden lang umgerührt.
Der erhaltene Feststoff wird abfiltriert und mit kaltem Wasser und
danach mit kaltem absolutem Ethanol gewaschen. Das Produkt wird
unter Vakuum bei Raumtemperatur getrocknet, wodurch N-Carboethoxy-2-thiomethyl-2-imidazolin als Feststoff
erhalten wird.
-
c. 6-(2-Imidazolinylamino)-4,5,8-trimethylchinolin-hydrochloridsalz:
-
6-Amino-4,5,8-trimethylchinolin (115,5
g) (wie in Beispiel 2 der gleichzeitig anhängigen US-Patentanmeldung Serien-Nr.
08/169 343 hergestellt) und N-Carboethoxy-2-thiomethyl-2-imidazolin (140
g) werden in 10 %ige Chloressigsäure
in Methanol (2,8 l, w/w) gelöst
und bei 65°C
bis zur Vollendung umgerührt.
Die Reaktionsmischung wird auf Raumtemperatur gekühlt und
HCl wird hinzugegeben. Die Reaktionsmischung wid 2 Stunden lang
umgerührt
und danch auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert. Das
so erhaltene Rohprodukt wird filtriert, aus Ethanol/Wasser umkristallisiert
und getrocknet (Vakuum, 40°C), wodurch
das gewünschte
gereinigte 6-(2-Imidazolinylamino)-4,5,8-trimethylchinolin-hydrochloridsalz
erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
2-Imidazolidinethion (68 Gramm, 670
mMol) und N,N-Dimethylacetamid (700 ml) werden in einem Rundkolben
zusammengebracht. Dazu wird Iodmethan (50 ml, 810 mMol) bei Umgebungstemperatur
gegeben. Diese Reaktion wird 2 Stunden lang umrühren gelassen.
-
b. N-Carboethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung wird in einem
Eisbad gekühlt
und dieser umgerührten
Lösung
wird Triethylamin (250 ml, 1,8 Mol) tropfenweise hinzugegeben. Dieser
Mischung wird Ethylchlorformiat (81 ml, 850 mMol) tropfenweise bei –5°C zugegeben.
Fünf Minuten
nach Beendigung der Zugabe wird die Reaktionsmischung auf Umgebungstemperatur
sich erwärmen
gelassen und wird danach 6 Stunden lang umgerührt. Die Reaktionsmasse wird
in eiskaltes Wasser (4 l) geschüttet.
Das Produkt wird in Ethylacetat (4 × 2,5 l) extrahiert. Die vereinten
Extrakte werden mit kaltem Wasser (3 × 2 l), Kochsalzlösung (2
l), gewaschen, über
Natriumsulfat getrocknet und unter vermindertem Druck konzentriert,
wodurch ein öliger
Rückstand
erhalten wird. Durch Trocknen des Rückstands unter Vakuum wird
das gewünschte
N-Carboethoxy-2-thiomethyl-2-imidazolin erhalten.
-
c. 2-(1',3'-Benzodioxolyl-5'-amino)imidazolin:
-
N-Carboethoxy-2-thiomethyl-2-imidazolin
(22,5 g, 104 mMol) wird in Methanol (1 l) gelöst und es wird Eisessig (12
ml, 208 mMol) hinzugegeben. Die Reaktionsmischung wird 10 Minuten
lang umrühren
gelassen. Dieser Lösung
wird 3,4-(Methylendioxy)anilin (14,3 Gramm, 104 mMol) zugegeben
und die Reaktionsmasse wird bei Umgebungstemperatur umrühren gelassen,
bis die Reaktion abgeschlossen ist. Das Lösungsmittel wird unter vermindertem
Druck entfernt. Das Rohprodukt wird in Ethylacetat extrahiert, getrocknet
und abgedampft unter Erhalt von 2-(1',3'-Benzodioxolyl-5'-amino)imidazolin.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
2-Imidazolidinethion (10,2 Gramm,
100 mMol) und Dichlormethan (400 ml) werden in einem Rundkolben,
welcher mit einem Refluxkühler
ausgerüstet
ist, unter Rühren
zusammengebracht. Indomethan (8,7 ml, 140 mMol) wird rasch hinzugegeben.
Die Reaktionsmischung wird auf 30°C–35°C erwärmt, bis
die Bildung von 2-Thiomethyl-2-imidazolin
abgeschlossen ist.
-
b. N-t-Butoxycarbonyl-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) weiter oben wird auf Umgebungstemperatur abkühlen gelassen. Triethylamin
wird danach der Rührmischung
(14 ml) hinzugegeben. Dieser Lösung
wird bei Raumtemperatur 4-Dimethylaminopyridin (12,2 Gramm. 100
mMol) und danach Di-tert-butyldicarbonat (65,4 Gramm, 300 mMol)
zugegeben. Die Reaktionsmasse wird 6 Stunden lang umrühren gelassen.
Das Lösungsmittel
wird unter vermindertem Druck entfernt, wobei ein Feststoff zurückbleibt,
welcher weiter unter Vakuum getrocknet wird. Das Rohmaterial wird
in Ethylacetat extrahiert, mit Wasser gewaschen, getrocknet und
verdampft unter Erhalt des reinen N-t-Butoxycarbonyl-2-thiomethyl-2-imidazolin.
-
c. 5-(2-Imidazolinylamino)-benzimidazol-hydrobromidsalz:
-
5-Aminobenzimidazol (5 Gramm, 38
mMol) und N-t-Butoxycarbonyl-2-thiomethyl-2-imidazolin (9,4 Gramm, 42 mMol) werden
in 10%iger Essigsäure
in Methanol (400 ml) gelöst
und 24 Stunden lang bei Umgebungstemperatur umgerührt. Dieser
Lösung
wird 30 %iges HBr/AcOH (100 ml) zugegeben und die Reaktionsmasse
wird weitere 4 Stunden lang umgerührt. Die resultierende Lösung wird
unter vermindertem Druck konzentriert, erneut in Methanol gelöst und aus
Methanol/Diethylether umkristallisiert unter Erhalt des gewünschten
Produkts als Hydrobromidsalz.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
In einem Druckreaktor wird Methylbromid
(32 g) langsam einer Lösung
von 2-Imidazolidinethion
(17 g) in Methylethylketon (175 ml) unter Rühren bei Umgebungstemperatur
hinzugegeben. Die Reaktionsmischung wird auf 65°C unter Druck erwärmt, bis
die Bildung von 2-Methylthio-2-imidazolinhydrobromid abgeschlossen ist.
-
b. N-Carboethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) wird auf Umgebungstemperatur abkühlen gelassen und das überschüssige Methylbromid
wird freigesetzt und eingeschlossen. Dieser Mischung wird Natriumcarbonat (26,5
g) zugegeben, gefolgt von der Zugabe von Diethylpyrocarbonat (42
ml). Die Reaktionsmischung wird bei 40°C bis zur Vervollständigung
umgerührt,
woraufhin die Reaktionsmischung auf 55°C erwärmt wird und die heiße Lösung filtriert
wird, um die unlöslichen
Salze zu entfernen. Diese Salze werden mit kaltem absolutem Ethanol
gewaschen. Das Filtrat (und Ethanol-Waschflüssigkeit) wird auf –20°C gekühlt und
2. Stunden lang umgerührt.
Der erhaltene Feststoff wird filtriert und mit kaltem Wasser und
danach mit kaltem absolutem Ethanol gewaschen. Das Produkt wird
unter Vakuum bei Raumtemperatur getrocknet, wodurch N-Carboethoxy-2-thiomethyl-2-imidazolin
als Feststoff erhalten wird.
-
c. 5-(2-Imidazolinylamino)-4-methyl-1,3-benzodioxol-hydrochloridsalz:
-
5-Amino-4-methyl-1,3-benzodioxol
(13,25 g) (wie in der Anmeldung Serien-Nr. 08/-478 708 hergestellt) und N-Carboethoxy-2-thiomethyl-2-imidazolin
(20 g) werden in 10 %iger Propionsäure in Isoamylalkohol (325
ml, w/w) gelöst
und die Reaktionsmischung wird bei 65°C bis zur Vervollständigung
umgerührt.
Die Reaktionsmischung wird auf Raumtemperatur gekühlt und
HCl-Gas (13 g) wid langsam zugeführt
Die Mischung wird für
weitere 2 Stunden umgerührt,
woraufhin sie dann auf –20°C gekühlt wird
und umgerührt
wird, bis das Produkt präzipitiert.
Das erhaltene Rohprodukt wird filtriert, aus Methanol/-Diethylether umkristallisiert
und getrocknet (Vakuum, 40°C),
wodurch das gewünschte
gereinigte 5-(2-Imidazolinylamino-4-methyl-1,3-benzodioxol-hydrochloridsalz
erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
2-Imidazolidinethion (50 Gramm) und
absolutes Ethanol (400 ml) werden unter Rühren vereinigt. Methyliodid
(43 ml, 1,4 Äqu.)
wird rasch der Rührmischung
zugegeben. Die Reaktionsmischung wird danach auf 35°C erwärmt, bis
die Bildung von 2-Thiomethyl-2-imidazolin
abgeschlossen ist.
-
b. N-Carbomethoxy-2-thiomethyl-2-imidazolin:
-
Kaliumcarbonat (101 Gramm) wird der
Mischung in Schritt (a) weiter oben hinzugegeben, gefolgt von der
Zugabe von Methylchlorformiat (42 ml) unter gleichzeitigem Rühren. Nach
45 Minuten wird die Reaktionsmischung auf 55°C erwärmt und die unlöslichen
Salze werden abfiltriert. Diese Salze werden mit absolutem Ethanol
gewaschen. Das Filtrat (und Ethanol-Waschflüssigkeit) werden auf –20°C gekühlt und
das Endprodukt wird auf einem Filter isoliert. Das Endprodukt wird
mit kaltem (–20°C) absolutem
Ethanol gewaschen. Das Produkt wird über Nacht unter Vakuum bei
Raumtemperatur getrocknet, wodurch N-Carbomethoxy-2-thiomethyl-2-imidazolin
erhalten wird.
-
c. 4-Ethyl-5-(2-imidazolinylamino)benzimidazol-maleatsalz:
-
Das N-Carbomethoxy-2-thiomethyl-2-imidazolin
(23,8 Gramm, 140 mMol) wird mit 5-Amino-4-ethylbenzimidazol (20 Gramm,
124 mMol) (hergestellt durch Entschützen der tert-Butoxycarbonyl-Schutzgruppe
einer Zwischenverbindung, hergestellt in dem US-Patent Nr. 5 478
858, erteilt an Cupps und Bogdan am 26. Dez. 1995 (hierin durch
den Bezug eingeschlossen) unter standardmäßigen Entschützungsbedingungen,
die in dem Fachbereich bekannt sind) und einer 10%igen Lösung von
Essigsäure
in Ethanol (500 ml) in einem mit einem Refluxkühler ausgerüsteten Kolben vereinigt. Diese
Mischung wird 1 Stunde lang umgerührt. Die Mischung wird danach
bei 65°C
12 Stunden lang erwärmt.
Zu diesem Zeitpunkt wird die Reaktionsmasse auf Umgebungstemperatur
gekühlt
und Maleinsäure
(48 Gramm, 410 mMol) wird zugegeben. Die resultierende Mischung
wird zwei Stunden lang umgerührt
und danach auf 0°C
gekühlt.
Die Mischung wird umgerührt,
bis das Produkt vollständig
präzipitiert
(ungefähr
1 Stunde), woraufhin die Mischung filtriert wird. Das Rohprodukt
wir mit kaltem Ethanol gewaschen und danach aus Acetonitril/Wasser
umkristalliert unter Erhalt von 4-Ethyl-5-(2-imidazolinylamino)benzimidazol-maleatsalz.
-
-
a. 2-Thiomethyl-3,4,5,6-tetrahydropyrimidin:
-
Methyliodid (75 ml) wird langsam
einer umgerührten
Lösung
von 3,4,5,6-Tetrahydro-2-pyrimidinthiol (100
g) in Ethanol (600 ml) bei Umgebungstemperatur zugegeben. Die Reaktionsmischung
wird auf 40°C
erwärmt,
bis die Bildung von 2-Thiomethyl-3,4,5,6- tetrahydropyrimidin-hydrochlorid abgeschlossen
ist.
-
b. N-3-Carbomethoxy-2-thiomethyl-4,5,6-tetrahydropyrimidin:
-
Die Mischung vom obigen Schritt (a)
wird auf Umgebungstemperatur gekühlt
und Kaliumcarbonat (178 g) wird zugegeben, gefolgt von der Zugabe
von Methylchlorformiat (73,2 ml) unter gleichzeitigem Umrühren. Die
Reaktionsmischung wird bei 40°C
bis zur Vervollständigung
umgerührt,
woraufhin die Reaktionsmischung auf 55°C erwärmt wird und die heiße Lösung filtriert
wird, um die unlöslichen
Salze zu entfernen. Diese Salze werden mit kaltem absolutem Ethanol
gewaschen. Das Filtrat (und Ethanol-Waschflüssigkeit) wird auf –20°C gekühlt und
2 Stunden lang umgerührt.
Der erhaltene Feststoff wird filtriert und mit kaltem Wasser und
danach mit kaltem absolutem Ethanol gewaschen. Das Produkt wird
unter Vakuum bei Raumtemperatur getrocknet, wodurch N-3-Carbomethoxy-2-thiomethyl-4,5,6-tetrahydropyrimidin
erhalten wird.
-
c. 2-(5-Methyl-6-chinoxalinylamino)-3,4,5,6-tetrahydropyrimidin:
-
6-Amino-5-methylchinoxalin (73,8
g) (wie in der gleichzeitig anhängigen
US-Patent-Anmeldung
Serien-Nr. 08/478 708 hergestellt) und N-3-Carbomethoxy-2-thiomethyl-4,5,6-tetrahydropyrimidin
(113,5 g) werden in 10 %iger Essigsäure in Ethanol (1,1 l) gelöst und bei
65°C bis
zu Vervollständigung
gelöst.
Die Reaktionsmischung wird auf Umgebungstemperatur gekühlt, und
es wird Fumarsäure
zugegeben (189 g). Die Mischung wird 2 Stunden lang umgerührt und
danach auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert
ist. Das so erhaltene Rohprodukt wird aus Acetonitril/Wasser umkristallisiert,
wodurch das gewünschte,
gereinigte 2-(5-Methyl-6-chinoxalinylamino)-3,4,5,6-tetrahydropyrimidin-fumaratsalz
erhalten wird.
-
-
a. 2-Thioethyl-2-imidazolin-hydrosulfat:
-
Diethylsulfat (47,75 ml) wird langsam
einer Lösung
von 2-Imidazolidinethion (30 g) in Isopropanol (250 ml) unter Rühren bei
Umgebungstemperatur zugegeben. Die Reaktionsmischung wird auf 50°C erwärmt, bis die
Bildung von 2-Thioethyl-2-imidazolin-hydrosulfat abgeschlossen ist.
-
b. N-tert-Butoxycarbonyl-2-thioethyl-2-imidazolin:
-
Die Reaktionsmischung in Schritt
(a) wird auf Umgebungstemperatur abkühlen gelassen, woraufhin Triethylamin
(105 ml) und danach Di-tert-butyldicarbonat (74,25 ml) zugegeben
werden. Die Reaktionsmischung wird auf 55°C erwärmt und bis zum Abschluss umgerührt. Die
Reaktionsmischung wird danach heiß filtriert, unter Entfernung
der unlöslichen
Salze. Diese Salze werden mit kaltem Isopropanol gewaschen. Das Filtrat
(und Isopropanol-Waschflüssigkeit)
wird auf –20°C gekühlt und
2 Stunden lang umgerührt.
Der erhaltene Feststoff wird filtriert und mit Wasser und danach
mit kaltem absolutem Ethanol gewaschen. Das Produkt wird unter Vakuum
bei Raumtemperatur getrocknet, wodurch N-tert-Butoxycarbonyl-2-thioethyl-2-imidazolin
als Feststoff erhalten wird.
-
c. 5-(2-Imidazolinylamino)-4-methoxybenzothiazol-succinatsalz:
-
5-Amino-4-methoxybenzothiazol (18
g) (wie in Beispiel 5 der gleichzeitig anhängigen US-Patent-Anmeldung Serien-Nr. 60/031756
hergestellt) und N-tert-Butoxycarbonyl-2-thioethyl-2-imidazolin (32,5 g) werden in
10%iger Chloressigsäure
in N-Methylpyrrolidinon (390 ml, w/w) gelöst. Die Mischung wird danach
bei 50°C umgerührt, bis
die Reaktion abgeschlossen ist. Die Mischung wird auf Umgebungstemperatur
gekühlt
und es wird Bernsteinsäure
(47,5 g) zugegeben, und die Mischung wird weitere 4 Stunden lang
umgerührt.
Die erhaltene Lösung
wird auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert.
Das Rohprodukt wird danach filtriert und aus Ethanol/Wasser umkristallisiert,
wodurch das gewünschte,
gereinigte Salz von 5-(2-Imidazolinylamino)-4-methoxybenzothiazol erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin-hydrobromid:
-
In einem Druckreaktor wird Methylbromid
(20,5 g) langsam einer umgerührten
Lösung
von 2-Imidazolidinethion (15 g) in Methyl-tert-butylether (120 ml)
bei Umgebungstemperatur hinzugegeben. Die Reaktionsmischung wird
auf 70°C
unter Druck erwärmt,
bis die Bildung von 2-Thiomethyl-2-imidazolinhydrobromid abgeschlossen
ist.
-
b. N-Carbomethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) wird auf Umgebungstemperatur abkühlen gelassen und das überschüssige Methylbromid
wird freigesetzt und eingeschlossen. Dieser Mischung wird Triethylamin
(53,2 ml) zugegeben, gefolgt von der Zugabe von Methylchlorformiat
(13,6 ml). Die Reaktionsmischung wird bei 40°C bis zum Abschluss umgerührt, woraufhin
die Reaktionsmischung auf 55°C
erwärmt
wird und die heiße
Lösung filtriert
wird, um die unlöslichen
Salze zu entfernen. Diese Salze werden mit kaltem Methyl-tert-butylether
gewaschen. Das Filtrat (und Waschflüssigkeit) wird auf –20°C gekühlt und
2 Stunden lang umgerührt.
Der erhaltene Feststoff wird filtriert und mit kaltem Wasser und
danach mit kaltem absolutem Ethanol gewaschen. Das Produkt wird
unter Vakuum über
Nacht bei Raumtemperatur getrocknet, wodurch N-Carbomethoxy-2-thiomethyl-2-imidazolin
als Feststoff erhalten wird.
-
c. 7-Ethyl-6-(2-imidazolinylamino)indazol-citratsalz:
-
6-Amino-7-ethylindazol (9,9 g) (wie
in Beispiel 1 der gleichzeitig anhängigen Anmeldung Serien-Nr. 60/031
740 hergestellt) und N-Carbomethoxy-2-thiomethyl-2-imidazolin (14
g) werden in 10 %iger Essigsäure in
Isopropanol (210 ml, w/w) gelöst
und bei 60°C
bis zum Abschluss der Reaktion umgerührt. Die Reaktionsmisch ung
wird auf Raumtemperatur gekühlt
und Citronensäure
(41,5 g) wird zugegeben. Die resultierende Mischung wird 2 Stunden
lang umgerührt.
Die Lösung
wird auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert
ist. Das so erhaltene Rohprodukt wird filtriert, aus Ethanol/Wasser
umkristallisiert und getrocknet (Vakuum, 40°C), wodurch das gewünschte gereinigte
7-Ethyl-6-(2-imidazolinylamino)imidazol-citratsalz erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin-hydroiodid:
-
Methyliodid (91 ml) wird langsam
einer Lösung
von 2-Imidazolinethion (210 g) in Ethanol (1,2 l) unter Rühren bei
Umgebungstemperatur zugegeben. Die Reaktionsmischung wird auf 40°C erwärmt, bis
die Bildung von 2-Thiomethyl-2-imidazolin-hydroiodid abgeschlossen
ist.
-
b. N-Carbomethoxy-2-thiomethyl-2-imidazolin:
-
Die Reaktionsmischung von Schritt
(a) wird auf Umgebungstemperatur abkühlen gelassen, woraufhin Kaliumcarbonat
(426 g) zugegeben wird, gefolgt von der Zugabe von Dimethylpyrocarbonat
(442 ml). Die Reaktionsmischung wird auf 55°C erwärmt und bis zum Abschluss umgerührt. Die
heiße
Lösung
wird filtriert, um die unlöslichen
Salze zu entfernen. Diese Salze werden mit kaltem absolutem Ethanol
gewaschen. Das Filtrat (und Ethanol-Waschflüssigkeit) wird auf –20°C gekühlt und
2 Stunden lang umgerührt.
Der erhaltene Feststoff wird filtriert und mit kaltem Wasser und
danach mit kaltem absolutem Ethanol gewaschen. Das Produkt wird unter
Vakuum bei Raumtemperatur getrocknet, wodurch N-Carbomethoxy-2-thiomethyl-2-imidazolin
als Feststoff erhalten wird.
-
c. 3-Cyano-6-(2-imidazolinylamino)-7-methylindol-hydrobromidsalz:
-
6-Amino-3-cyano-7-methylindol (34
g) (wie in Beispiel 3 der gleichzeitig anhängigen US-Patentanmeldung Serien-Nr. 60/031 774
hergestellt) und N-Carbomethoxy-2-thiomethyl-2-imidazolin (38 g) werden in 10%ige Chloressigsäure in N,N-Dimethylformamid
(480 ml, w/w) gelöst
und bei 50°C
umgerührt,
bis die Reaktion abgeschlossen ist. Diese Lösung wird auf Umgebungstemperatur
gekühlt
und es wird 30%iges HBr in Essigsäure (140 ml) hinzugegeben,
und die Mischung wird weitere 4 Stunden lang umgerührt. Die
resultierende Lösung
wird auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert
ist. Das Rohprodukt wird filtriert und aus Ethanol/Wasser umkristallisiert,
wodurch das gewünschte
gereinigte 3-Cyano-6-(2-imidazolinylamino)-7-methylindol-hydrobromidsalz
erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
Dimethylsulfat (200 ml) wird einer
umgerührten
Lösung
von 2-Imidazolidinethion (200 g) in Ethanol (2 l) zugegeben. Die
Reaktionsmischung wird auf 75°C
erwärmt,
bis die Bildung von 2-Thiomethyl-2-imidazolin abgeschlossen ist.
-
b. N-Propionyl-2-thiomethyl-2-imidazolin:
-
Die Mischung in Schritt (a) wird
auf Umgebungstemperatur gekühlt,
und dieser umgerührten
Lösung wird
Triethylamin (1,36 l), gefolgt von Propionsäureanhydrid (360 ml) hinzugegeben.
Die Reaktionsmischung wird bis zum Abschluss umgerührt, woraufhin
die Mischung auf 50°C
erwärmt
wird. Die heiße
Lösung
wird filtriert, um die unlöslichen
Salze zu entfernen und die Salze werden mit kaltem absolutem Ethanol
gewaschen. Die vereinigten Filtrate werden auf –20°C gekühlt und 2 Stunden lang umgerührt. Der
erhaltene Feststoff wird filtriert und mit kaltem Wasser und danach
mit kaltem absolutem Ethanol gewaschen. Das Produkt wird unter Vakuum
bei Raumtemperatur getrocknet, wodurch N-Propionyl-2-thiomethyl-2-imidazolin
als Feststoff erhalten wird.
-
c. 4-Methyl-5-(2-imidazolinylamino)benzimidazol-acetatsalz:
-
N-Propionyl-2-thiomethyl-2-imidazolin
(206,4 Gramm) wird mit 5-Amino-4-methylbenzimidazol (147 g) (hergestellt
durch Entschützen
der tert-Butoxycarbonyl-Schutzgruppe einer Zwischenverbindung, hergestellt in
dem US-Patent Nr. 5 478 858, erteilt an Cupps und Bogdan am 26.
Dez. 1995 (hierin durch den Bezug darauf eingeschlossen) unter standardmäßigen Entschützungsbedingungen,
die in dem Fachbereich bekannt sind) in einer 10%igen Lösung von
Essigsäure
in Ethanol (3 l) vereinigt. Die Mischung wird auf 60°C bis zum
Abschluss der Reaktion erwärmt.
Die Mischung wird danach auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert
ist. Das so erhaltene Rohprodukt wird filtriert, aus Ethanol/Wasser
umkristallisiert und getrocknet (Vakuum, Umgebungstemperatur), wodurch
das gewünchte,
gereinigte 4-Methyl-5-(2-imidazolinylamino)benzimidazol-acetatsalz
erhalten wird.
-
-
a. 2-Thiomethyl-2-imidazolin:
-
Dimethylsulfat (693 g) wird einer
umgerührten
Mischung von 2-Imidazolidinethion (500 g) in Dimethylacetamid (5
l) hinzugegeben. Die Reaktionsmischung wird bei Umgebungstemperatur
umgerührt,
bis die Bildung von 2-Thiomethyl-2-imidazolin abgeschlossen ist.
-
b. N-Benzoyl-2-thiomethyl-2-imidazolin:
-
Die Mischung in Schritt (a) wird
auf Umgebungstemperatur gekühlt
und dieser umgerührten
Lösung wird
Triethylamin (2,47 kg), gefolgt von Benzoylchlorid (964 g) zugegeben.
Die Reaktionsmischung wird bei Umgebungstemperatur bis zum Abschluss
umgerührt.
Die Reaktionsmischung wird mit kaltem Wasser versetzt und das Präzipitat,
das sich bildet, wird filtriert und zweimal mit kaltem Wasser gespült. Der
erhaltene Feststoff wird getrocknet (Vakuum, Umgebungstemperatur),
wodurch N-Benzoyl-2-thiomethyl-2-imidazolin
erhalten wird.
-
c. 7-Methyl-6-(2-Imidazolinylamino)indazol-hydrochlorid:
-
N-Benzoyl-2-thiomethyl-2-imidazolin
(264 g) wird mit 6-Amino-7-methylindazol (147 g) (wie in Beispiel 2
der gleichzeitig anhängigen
US-Patent-Anmeldung Serien-Nr. 60/-031 740 hergestellt) in einer 10%igen
Lösung
von Essigsäure
in Ethanol (3 l) vereinigt. Die Mischung wird bis zum Abschluss
auf 60°C
erwärmt.
Die Reaktionsmischung wird auf Raumtemperatur gekühlt und
es wird Chlorwasserstoffgas (128 g) hinzugefügt. Die Mischung wird bei Umgebungstemperatur
2 Stunden lang umgerührt,
danach auf –20°C gekühlt und
umgerührt,
bis das Produkt vollständig
präzipitiert
ist. Das Rohprodukt wird aus Methyl-tert-butylether/Methanol umkristallisiert,
wodurch das gewünschte,
gereinigte 7-Methyl-6-(2-imidazolinylamino)indazol-hydrochloridsalz
erhalten wird
-
-
[(4-Nitrophenyl)amino]-2-imidazolin-acetatsalz
-
4-Nitroanilin (2 g) und N-Methoxycarbonyl-2-thiomethyl-2-imidazolin
(3,15 g) (hergestellt wie in Beispiel 2 beschrieben) werden in Eisessig
(40 ml) gelöst
und die Reaktionsmischung wird bei 60 bis 70°C umgerührt, bis der Kupplungsschritt
abgeschlossen ist. Die Reaktionsmischung wird mit Methanol (20 ml)
verdünnt, zum
Refluxieren gebracht, bis die Entschützung vollendet ist, und danach
unter vermindertem Druck konzentriert. Der resultierende Rückstand
wird aus Ethylacetat und Hexan kristallisiert, wodurch [(4-Nitrophenyl)amino]-2-imidazolin-monoacetatsalz
erhalten wird.
-
-
N-(4,5-Dihydro-1H-imidazol-2-yl)-7-cyano-4-methyl-1H-benzimidazol-5-aminschwefelsäuresalz:
-
5-Amino-7-cyano-4-methylbenzimidazol
(4 g) wird hergestellt, indem eine heterogene Lösung von 7-Cyano-4-methyl-5-nitrobenzimidazol
(0,91 g, 0,0045 Mol) und 10% Pd/C (100 mg) in Methanol (200 ml)
mit einer Atmosphäre
von H2 (1 atm, Ballon) 14 h lang behandelt
wird. Die resultierende Mischung wird durch Celit filtriert und
mittels Rotationsverdampfung konzentriert, wodurch ein gelber Rückstand
entsteht. Dieser Rückstand
wird chromatographiert (Silicagel, 95 : 5 Ethylacetat : Methanol),
wodurch 5-Amino-7-cyano-4-methylbenzimidazol
erhalten wird. 5-Amino-7-cyano-4-methylbenzimidazol und N-Methoxycarbonyl-2-thiomethyl-2-imidazolin
(4,45 g, 1,1 Äqu.)
(hergestellt wie in Beispiel 2 beschrieben) werden in Acetonitril
(100 ml) und Eisessig (10 ml) gelöst und die Reaktionsmischung
wird bei 70°C
umgerührt,
bis der Kupplungsschritt abgeschlossen ist. Die Reaktionsmischung
wird mit Methanol (50 ml) verdünnt
und zum Refluxieren gebracht, bis die Entschützung abgeschlossen ist, und
danach wird Acetonitril unter vermindertem Druck abgedampft. Die erhaltene
Essigsäurelösung wird
in Wasser (9,25 ml) gelöst
und die resultierende Mischung wird auf 0°C gekühlt. Ein 5-molare wässrige Lösung von
H2SO4 (5,1 ml) wird
tropfenweise der kalten Mischung zugegeben. Die Lösung wird
danach auf 65°C
erwärmt
und es wird absolutes Ethanol zugegeben, bis eine Eintrübung festgestellt
wird. Die Mischung wird sich auf Raumtemperatur einstellen gelassen
und danach auf 5°C
abgekühlt. Der
erhaltene Feststoff wird filtriert, mit Ethanol gewaschen und getrocknet,
wodurch N-(4,5-Dihydro-1H-imidazol-2-yl)-7-cyano-4-methyl-1H-benzimidazol-5-amin
als ihr Schwefelsäuresalz
erhalten wird.
-
-
a. 4-Methyl-5-amino-7-fluorbenzimidazolhydrochlorid:
-
1-tert-Butoxycarbonyl-4-methyl-5-amino-7-fluorbenzimidazol
(1 g) (hergestellt wie in dem US-Patent Nr. 5 478 858, erteilt an
Cupps und Bogdan am 26. Dez. 1995, beschrieben, hierin durch den
Bezug darauf eingeschlossen) und 6 N HCl (10 ml) werden vereinigt
und bis zum Refluxieren unter Rühren
erwärmt.
Nach Beendigung der t-BOC-Gruppen-Entschützung wird die Reaktionsmischung
unter vermindertem Druck konzentriert und getrocknet, wodurch 4-Methyl-5-amino-7-fluorbenzimidazol-hydrochlorid
erhalten wird.
-
b. 4-Methyl-7-fluor-5-(2-imidazolinylamino)-benzimidazoldihydrochlorid:
-
Dem in Schritt a erhaltenen Feststoff
wird N-Methoxycarbonyl-2-thiomethyl-2-imidazolin (0,78 g) und Eisessig
(20 ml) zugegeben. Die Mischung wird bei 60 bis 70°C gerührt, bis
der Kupplungsschritt abgeschlossen ist. Die Reaktionsmischung wird
danach mit Methanol (10 ml) verdünnt,
zum Refluxieren gebracht, bis die Entschützung abgeschlossen ist, und
danach unter vermindertem Druck konzentriert. Der resultierende
Rückstand
wird mit methanolischer HCl (20 ml) verdünnt und dann mit wasserfreiem
Ether behandelt, um 4-Methyl-7-fluor-5-(2-imidazolinylamino)benzimidazoldihydrochlorid
zu präzipitieren.


























 worin: (a) R aus der Gruppe gewählt ist, bestehend aus Methyl, Ethyl und Benzyl; (b) R1 Methyl, Ethyl, eine Methylengruppe, verbunden mit R2 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (c) R2 Methyl, Ethyl, eine Methylengruppe, verbunden mit R1 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (d) R3 -O-R5 oder -R6 ist; (e) R5 aus der aus Methyl, Ethyl, Benzyl und tert-Butyl bestehenden Gruppe gewählt ist; und (f) R6 aus der aus Methyl, Ethyl, tert-Butyl und Phenyl bestehenden Gruppe gewählt ist; aus einem Thioharnstoff mit der allgemeinen Struktur:
worin: (a) R aus der Gruppe gewählt ist, bestehend aus Methyl, Ethyl und Benzyl; (b) R1 Methyl, Ethyl, eine Methylengruppe, verbunden mit R2 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (c) R2 Methyl, Ethyl, eine Methylengruppe, verbunden mit R1 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (d) R3 -O-R5 oder -R6 ist; (e) R5 aus der aus Methyl, Ethyl, Benzyl und tert-Butyl bestehenden Gruppe gewählt ist; und (f) R6 aus der aus Methyl, Ethyl, tert-Butyl und Phenyl bestehenden Gruppe gewählt ist; aus einem Thioharnstoff mit der allgemeinen Struktur: worin: (a) R1 Methyl, Ethyl, eine Methylengruppe, verbunden mit R2 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (b) R2 Methyl, Ethyl, eine Methylengruppe, verbunden mit R1 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; in einer Zweistufen-Eintopfreaktion durch: a) Alkylieren des Thioharnstoffs unter Verwendung eines Alkylierungsmittels zur Bildung eines 2-thiosubstituierten-2-Imidazolins, 2-Thioalkyl-2-guanidins oder 2-Thioalkyl-3,4,5,6-tetrahydro-pyrimidins; b) Acylieren des 2-thiosubstituierten-2-Imidazolins, 2-Thioalkyl-2-guanidins oder 2-Thioalkyl-3,4,5,6-tetrahydro-pyrimidins aus Schritt (I)(a) mit einem Acyliexungsmittel in Gegenwart einer Base; und (II) Kuppeln der Zwischenverbindung aus Schritt (I) mit einem Amin der Struktur:
worin: (a) R1 Methyl, Ethyl, eine Methylengruppe, verbunden mit R2 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R2 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; (b) R2 Methyl, Ethyl, eine Methylengruppe, verbunden mit R1 über eine Einfachbindung, sodass R1 und R2 einen fünfgliedrigen Ring bilden, oder eine Methylengruppe, verbunden mit R1 über eine andere Methylengruppe, sodass R1 und R2 einen sechsgliedrigen Ring bilden, ist; in einer Zweistufen-Eintopfreaktion durch: a) Alkylieren des Thioharnstoffs unter Verwendung eines Alkylierungsmittels zur Bildung eines 2-thiosubstituierten-2-Imidazolins, 2-Thioalkyl-2-guanidins oder 2-Thioalkyl-3,4,5,6-tetrahydro-pyrimidins; b) Acylieren des 2-thiosubstituierten-2-Imidazolins, 2-Thioalkyl-2-guanidins oder 2-Thioalkyl-3,4,5,6-tetrahydro-pyrimidins aus Schritt (I)(a) mit einem Acyliexungsmittel in Gegenwart einer Base; und (II) Kuppeln der Zwischenverbindung aus Schritt (I) mit einem Amin der Struktur: in Gegenwart einer organischen Säure.
in Gegenwart einer organischen Säure.