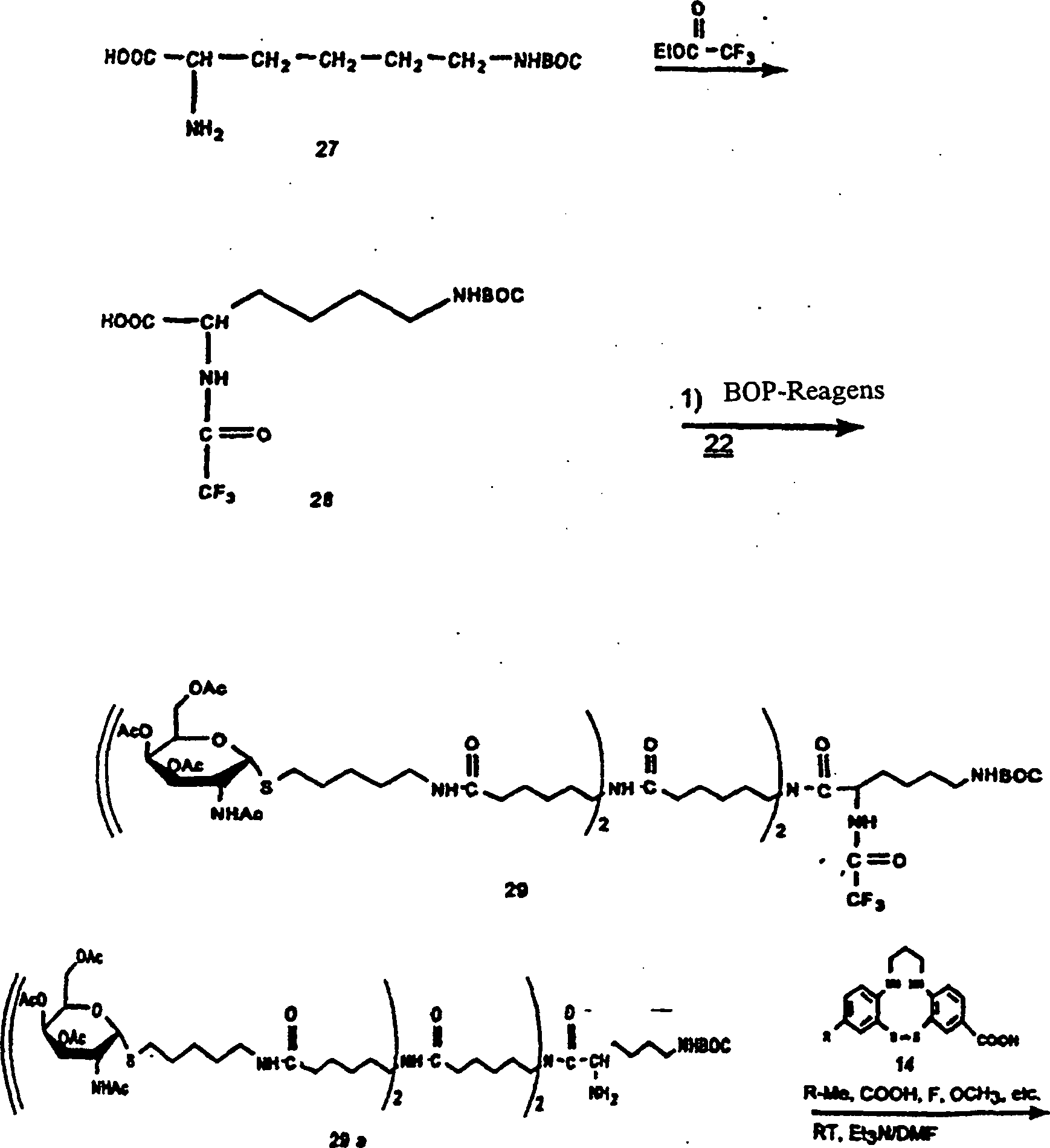
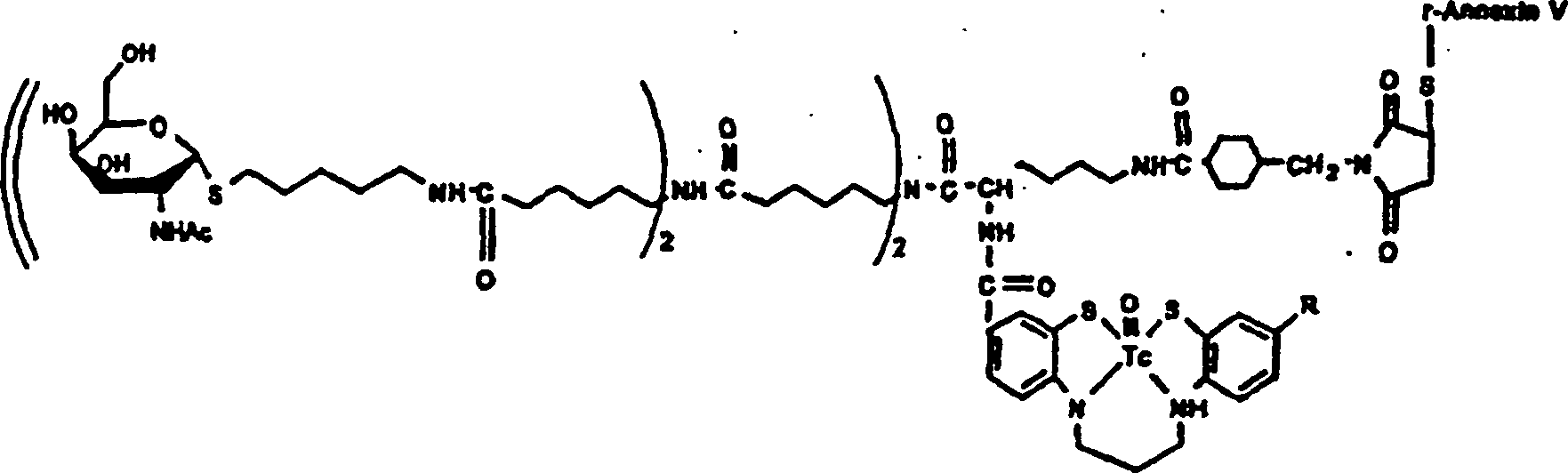
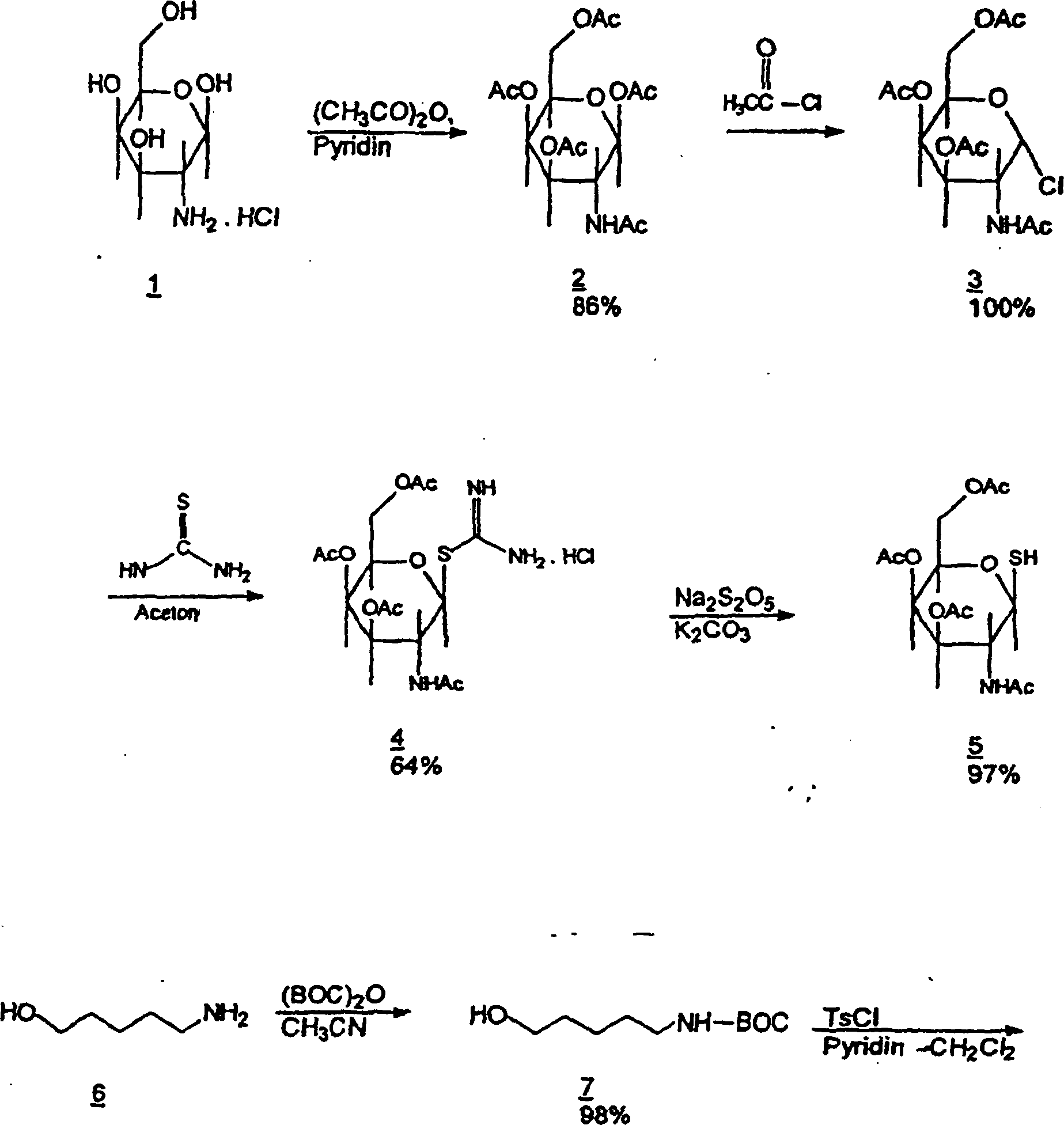
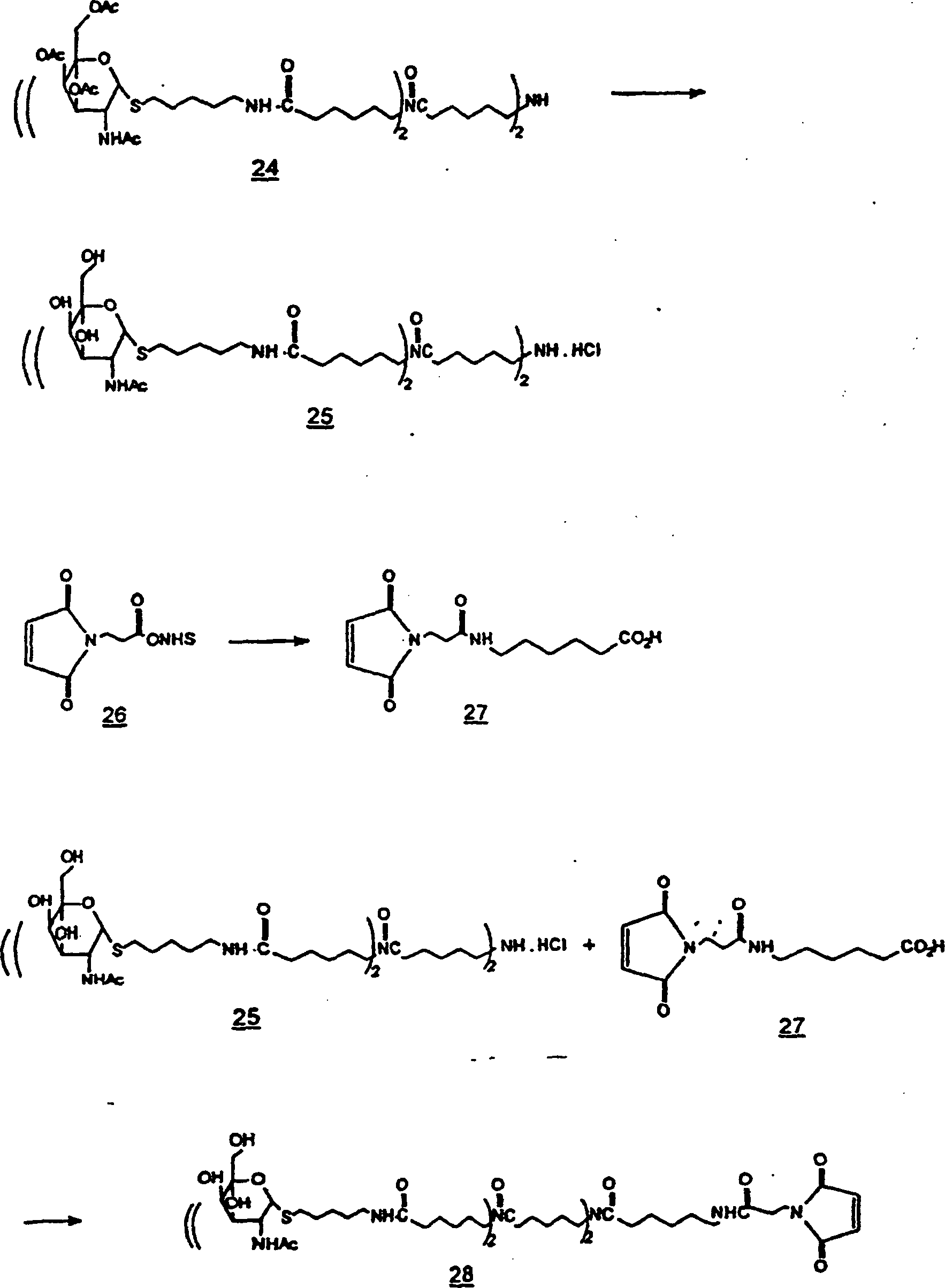
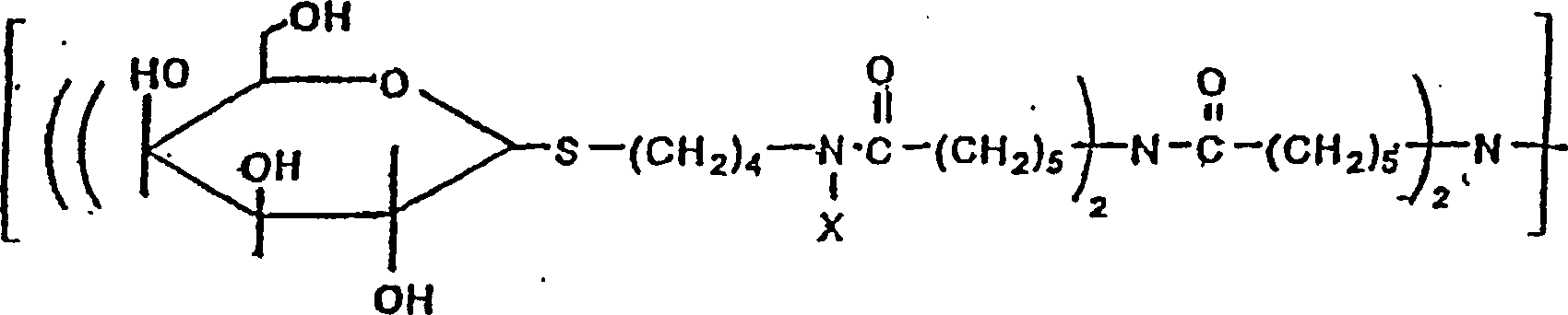-
Technisches
Gebiet
-
Die
vorliegende Erfindung ist gerichtet auf radioaktivmarkierte Annexine,
einschließlich
Annexin-Hexose-Konjugate wie beispielsweise Hexose-Einheit-Konjugate und deren
Bestandteile. Die vorliegende Erfindung ist auch allgemein gerichtet
auf Modifikationen der Annexin-Bestandteile für die Verwendung bei der Herstellung
von radioaktivmarkierten Annexinen. Im Zusammenhang mit der vorliegenden
Erfindung kommen auch Aufnahmeprotokolle in Betracht, welche die
Verabreichung von beispielsweise einem radioaktivmarkierten Annexin-Hexose-Einheit-Konjugat
einschließen.
Der Annexin-Bestandteil des Konjugats dient dazu, den radioaktivmarkierten
aktiven Bestandteil des Konjugats an die vaskuläre Thrombi-Zielstelle zu liefern. Der Hexose-Einheit-Bestandteil
des Konjugats erleichtert die schnelle Eliminierung der radioaktivmarkierten
Annexin-Hexose-Einheit-Konjugate von dem Kreislauf eines Empfängers.
-
Hintergrund
der Erfindung
-
Wenn
Patienten unter Brustschmerz, Herzklopfen oder irgendwelchen anderen
Symptomen einer Herzkranz-Verletzung oder -Erkrankung leiden, ist
die Gegenwart von vaskulären
Thromben in dem Herzen ein potentieller wesentlicher Komplikations-Faktor für die Behandlung.
Wenn ein Arzt nicht-invasiv bestimmen könnte, ob einer oder mehr bevorzugtere
vaskuläre
Thromben anwesend sind, und wenn sie anwesend sind, die Lage dieser
vaskulären
Thromben bestimmen könnte,
wäre eine
bessere Beurteilung der Behandlungsoptionen möglich. Darüber hinaus könnten, wenn
ein Arzt bestimmen könnte,
dass keine vaskulären
Thromben anwesend waren, wodurch eine potentielle Komplikation bei
der Behandlung eliminiert wird, kardiale Zustände sicherer und wirksamer
behandelt werden.
-
Die
bisherigen Techniken zur Bestimmung der Anwesenheit von vaskulären Thromben
sind invasiv und/oder beschwerlich und/oder scheitern daran, dass
solche Thromben mit guter Empfindlichkeit und Spezifität erfaßt werden.
Deshalb ist ein Aufnahme-Mittel, das nützlich ist bei der nicht-invasiven
Aufnahme von vaskulären
Thromben, wünschenswert.
-
Annexine
sind eine Klasse von Proteinen, die charakterisiert sind durch eine
Calcium-vermittelte Bindung an anionische Phospholipide. Anionische
Phospholipide sind um das etwas 20-fache höher verbunden mit aktivierten
Blutplättchen
als Blutplättchen
im Ruhezustand, und aktivierte Blutplättchen sind verbunden mit vaskulären Thromben.
-
Es
wurde gezeigt, dass radioiodiertes Annexin V vaskuläre Thromben
in vivo lokalisiert, jedoch weist es nicht optimale Aufnahme-Eigenschaften
auf, die möglicherweise
auf die ausgeprägte
Beta-Phase bei der Blut-Clearance zurückzuführen ist, infolge einer möglichen
Transjodierung und/oder eines metabolischen Abbaus mit Wiedereinbau
in Serum-Makromoleküle
oder Nicht-Zielgewebe. Darüber
hinaus setzten freies radioaktives Iod oder Iod-enthaltende metabolische
Abbauprodukte Nicht-Zielgewebe
insbesondere die Schilddrüse,
Radioaktivität
aus. Darüber
hinaus leiden die Iod-Radioisotope I-123, I-124 und I-131 mit aufnehmbaren Photonen
unter verschiedenen Nachteilen. Die Radioaktiv-Markierung mit I-Iod-123
mit ausgezeichneten Aufnahme-Eigenschaften ist teuer und schwierig
zu erhalten und ist deshalb für
eine breit gestreute Anwendung nicht geeignet. Iod-124 emittiert
Positronen und Hochenergie-Gamma-Photonen. Letztendlich hat Iod-131
eine partikluäre
Emission und seine Gamma-Emission ist in Bezug auf die Energie zu
hoch für
eine optimale Aufnahme. Folglich sind verbesserte radioaktivmarkierte
Annexin-Verbindungen wünschenswert.
-
Darüber hinaus
sind die herkömmliche
Aufnahme und Therapie oft geplagt von dem Problem, dass das allgemein
erreichbare Ziel-Verhältnis
niedrig ist. Das Ziel- Verhältnis wird
definiert als das Verhältnis
von verabreichter Dosis, welche das Ziel lokalisiert, zu der verabreichten
Dosis, die im Blut zirkuliert, oder das Verhältnis von verabreichter Dosis,
die das Ziel lokalisiert, zu verabreichter Dosis, die in das Knochenmark übergeht.
Eine Verbesserung des Target-Verhältnisses wird auch angestrebt.
Aus den oben genannten Gründen besteht
deshalb noch ein Bedarf an einem Produkt mit erhöhter Empfindlichkeit zur Aufnahme
von Thromben, um kleine Thromben aufzunehmen, wie beispielsweise
Carotid-Thromben oder intrakardiale Thromben und solche, die in
Herzkranz-Arterien nach einer Angioplastie oder während Herzinfarkten
oder in zerebralen Arterien während
einer Apoplexie zugegen sind. Die Entwicklung eines Mittels zur
Aufnahme eines radioaktivmarkierten Thrombus, das in der Lage ist,
intrakoronare Thromben zu erfassen, würde ein Durchbruch-Produkt
mit großer
klinischer und wirtschaftlicher Bedeutung verkörpern. Die vorliegende Erfindung
erfüllt
dieses Erfordernis und stellt weitere ähnliche Vorteile bereit.
-
Zusammenfassung
der Erfindung
-
Die
vorliegende Beschreibung stellt radioaktivmarkierte Annexin-Hexose-Einheit-Konjugate
und Verfahren zur Herstellung und Verwendung dieser Konjugate bereit.
Die vorliegende Erfindung stellt bereit einen Annexin-Bestandteil,
der in einer solchen Weise modifiziert ist, dass er eine zugängliche
Sulfhydryl-Gruppe bereitstellt, was dazu dient, die Aufnahme-Eigenschaften
von Annexin zu verbessern. Annexin enthaltende Konjugate der vorliegenden
Erfindung sind geeignet zur Radiomarkierung mit einem Diagnose-Aufnahmemittel. Konjugate
der vorliegenden Erfindung umfassen:
- – ein modifiziertes
Annexin, wobei die Modifikation eine zugängliche Sulfhydryl-Gruppe bereitstellt;
und
- – eine
Hexose-Einheit, die von einem Säuger-Leber-Rezeptor
erkannt wird, wobei die Hexose-Einheit an das Annexin konjugiert
ist.
-
Durch
die vorliegende Erfindung werden ebenso bereitgestellt radiomarkierte
modifizierte Annexin-Hexose-Einheit-Konjugate.
-
Ein
anderes bevorzugtes Konjugat der vorliegenden Erfindung schließt ein:
- – das
modifizierte Annexin; und
- – eine
Esterase-empfindliche NxSy-Chelat-Verbindung,
die an das Annexin konjugiert ist.
-
Ein
weiteres bevorzugtes Konjugat gemäß der vorliegenden Erfindung
schließt
ein:
- – ein
modifiziertes Annexin-Multimer; und
- – eine
NxSy-Chelat-Verbindung,
die an das Annexin konjugiert ist.
-
Es
versteht sich für
Fachleute in diesem technischen Bereich, dass in den Fällen, in
denen eine chelatisierende Verbindung eine Komponente des Konjugats
ist, irgendeines der oben angegebenen bevorzugten Konjugate weiter
ein Radionuklid einschließt,
das durch die chelatisierende Verbindung komplexiert wird.
-
Das
Annexin gemäß der vorliegenden
Erfindung ist ein modifiziertes Annexin. Der Ausdruck „ein modifiziertes
Annexin" wird um
eine Zahl von Aminosäuren
an dem C-Terminus oder dem N-Terminus verlängert. Die Aminosäure-Verlängerung
führt zu
einer Modifikation des Annexins, wodurch eine zugängliche
Sulfhydryl-Gruppe geschaffen wird. Ein bevorzugtes modifiziertes
Annexin zur Verwendung im Rahmen der vorliegenden Erfindung wird
verlängert
an dem N-Terminus. In bevorzugten Ausführungsformen der vorliegenden Erfindung
sorgt die Struktur des modifizierten Annexins für die Bildung einer endogenen
Radiomarkierungs-Chelatisierungs-Stelle (so dass dadurch die Notwendigkeit
für die
intermediäre
Produktion eines Amin-gerichteten aktiven Esters zur Erleichterung
der Radiomarkierung eliminiert wird). In einer weiteren bevorzugten
Ausführungsform
der vorliegenden Erfindung erleichtert die Struktur des modifizierten
Annexins die Produktion von Multimeren. Ein bevorzugtes Annexin
zur Verwendung im Rahmen der vorliegenden Erfindung ist Annexin
V. Auch ein bevorzugtes Annexin gemäß der vorliegenden Erfindung
ist ein modifiziertes Annexin. So ist ein besonders bevorzugtes
Annexin gemäß der vorliegenden
Erfindung ein modifiziertes Annexin V.
-
Das
modifizierte Annexin oder die Annexin-Multimer-Komponente des Konjugats
ermöglicht
ein schnelles Binden der Radiomarkierung an das Annexin, so dass
eine vereinfachte Markierungs-Prozedur für eine klinische Anwendung
geschaffen wird. In einigen Ausführungsformen
der vorliegenden Erfindung sorgt die Modifikation des Annexins für eine Beschleunigung
der Blut-Clearance, wodurch die Hintergrund-Radioaktivität des Blut-Pools verringert
wird.
-
Weiter
kann das modifizierte Annexin oder das Annexin-Multimer an eine
Hexose-Einheit konjugiert werden, wie beispielsweise ein Hexose-Cluster.
Bevorzugte Hexose-Einheiten zur Verwendung im Rahmen der vorliegenden
Erfindung schließen
Hexose-Cluster ein und inkorporieren ein Mehrfaches von 4 Galactosen. Ein
anderes bevorzugtes Hexose-Cluster zur Verwendung im Rahmen der
vorliegenden Erfindung inkorporiert ein Mehrfaches von 3 Galactosen.
Es versteht sich, dass die Prototyp-artige Hexose-Einheit ein Hexose-Cluster
wie beispielsweise ein Galactose-Cluster ist. Weiter weist ein Prototyp-mäßiger Galactose-Cluster
als Galactose-Komponente N-Acetylgalactosanmin
auf.
-
Die
Hexose-Einheit-Komponente kann an die Annexin-Komponente gebunden
sein und kann gegebenenfalls an eine chelatisierende Verbindungs-Komponente über ein
trifunktionelles Verknüpfungs-Molekül (Linker)
wie beispielsweise Lysin gebunden sein. Ein Verlängerungs-Molekül kann zwischen
der Hexose-Einheit und dem trifunktionellen Linker verwendet werden,
um die Bioverfügbarkeit
der Hexose-Einheit zu fördern. Diese
Ausführungsform
der vorliegenden Erfindung wird bevorzugt, wenn eine chelatisierende
Verbindung verwendet wird und die chelatisierende Verbindung durch
eine einzelne funktionelle Gruppe charakterisiert ist, die für eine Konjugation
verfügbar
und geeignet ist.
-
Wenn
eine chelatisierende Verbindungs-Komponente durch mehr als eine
für eine
Konjugation an andere Konjugat-Komponenten verfügbare und geeignete funktionelle
Gruppe gekennzeichnet ist, wird vorzugsweise eine Struktur wie die
folgende verwendet: Hexose-Einheit – bifunktionelles Verknüpfungs-Molekül (Linker) – chelatisierende
Verbindung – bifunktionelles
Verknüpfungs-Molekül (Linker) -Annexin.
-
Auch
wird in einigen anderen Ausführungsformen
der vorliegenden Erfindung eine Esterase-empfindliche Chelatisierungs-Komponente
verwendet und so eine beschleunigte Ausscheidung von Radioaktivität aus der
Leber geschaffen, wodurch die Hintergrund-Radioaktivität nahe dem
Herzen und den Lungen verringert wird.
-
Bevorzugte
diagnostische Radionuklide zur Verwendung in der praktischen Durchführung der
vorliegenden Erfindung sind Tc-99m, Re-186 und Re-188, wobei Tc-99m besonders bevorzugt
ist. Vaskuläre
Thromben, die in oder nahe dem Herzen lokalisiert sind, sind speziell
einer Bildgebung in Übereinstimmung
mit der vorliegenden Erfindung zugänglich.
-
Diese
und andere Aspekte der vorliegenden Erfindung werden offensichtlich
unter Bezugnahme auf die folgende Beschreibung und die beigefügten Figuren.
-
Kurze Beschreibung
der Figuren
-
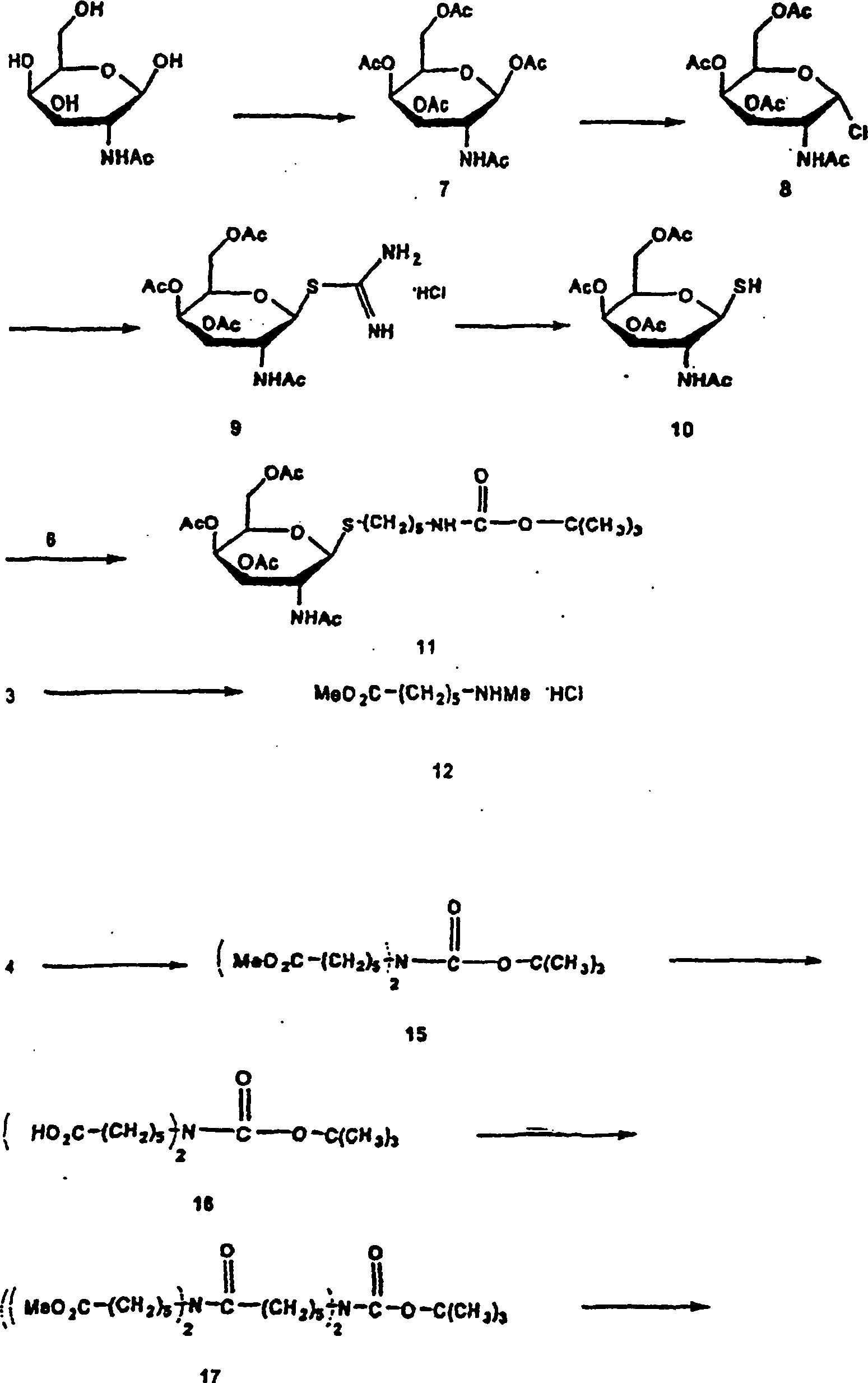
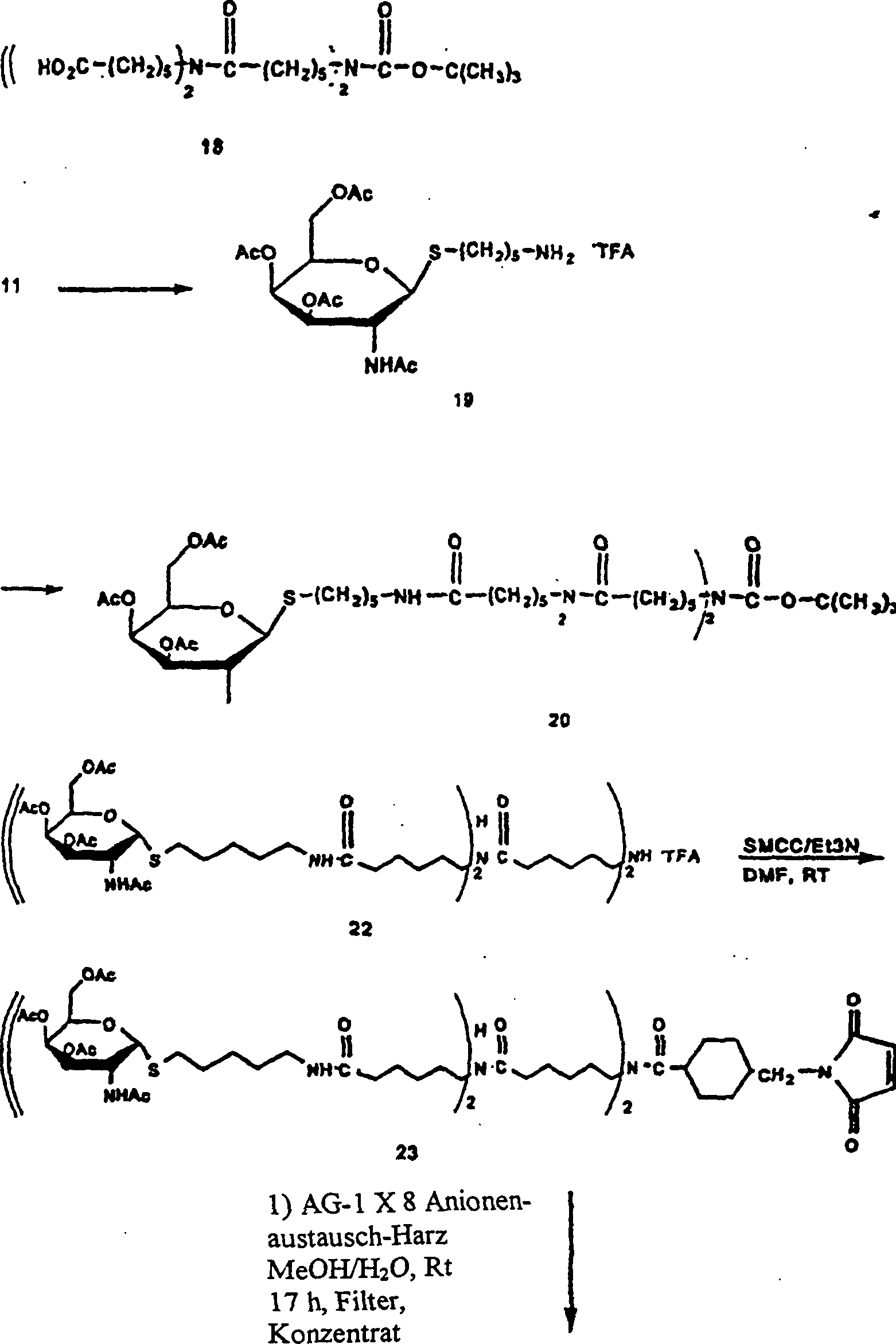
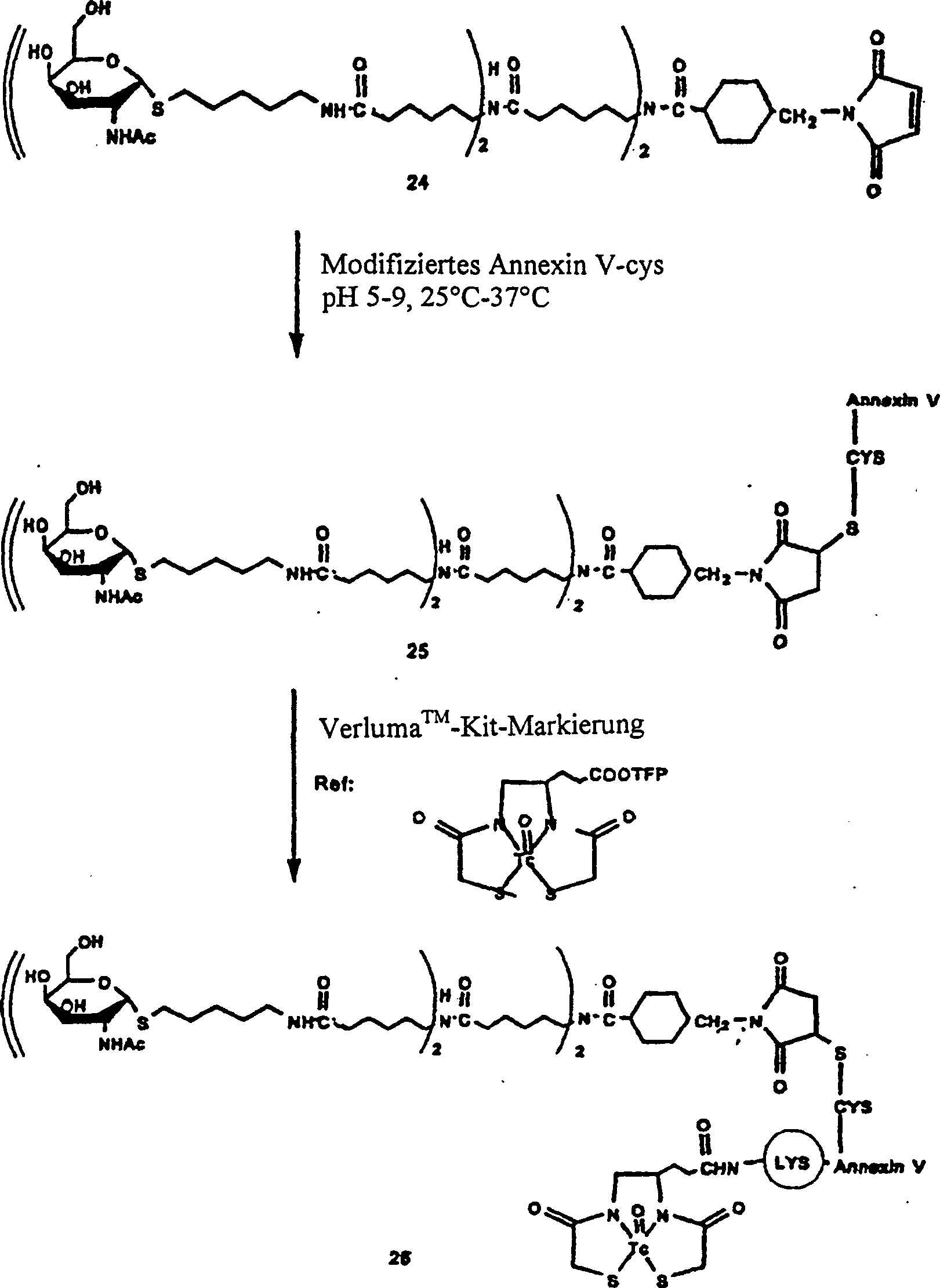
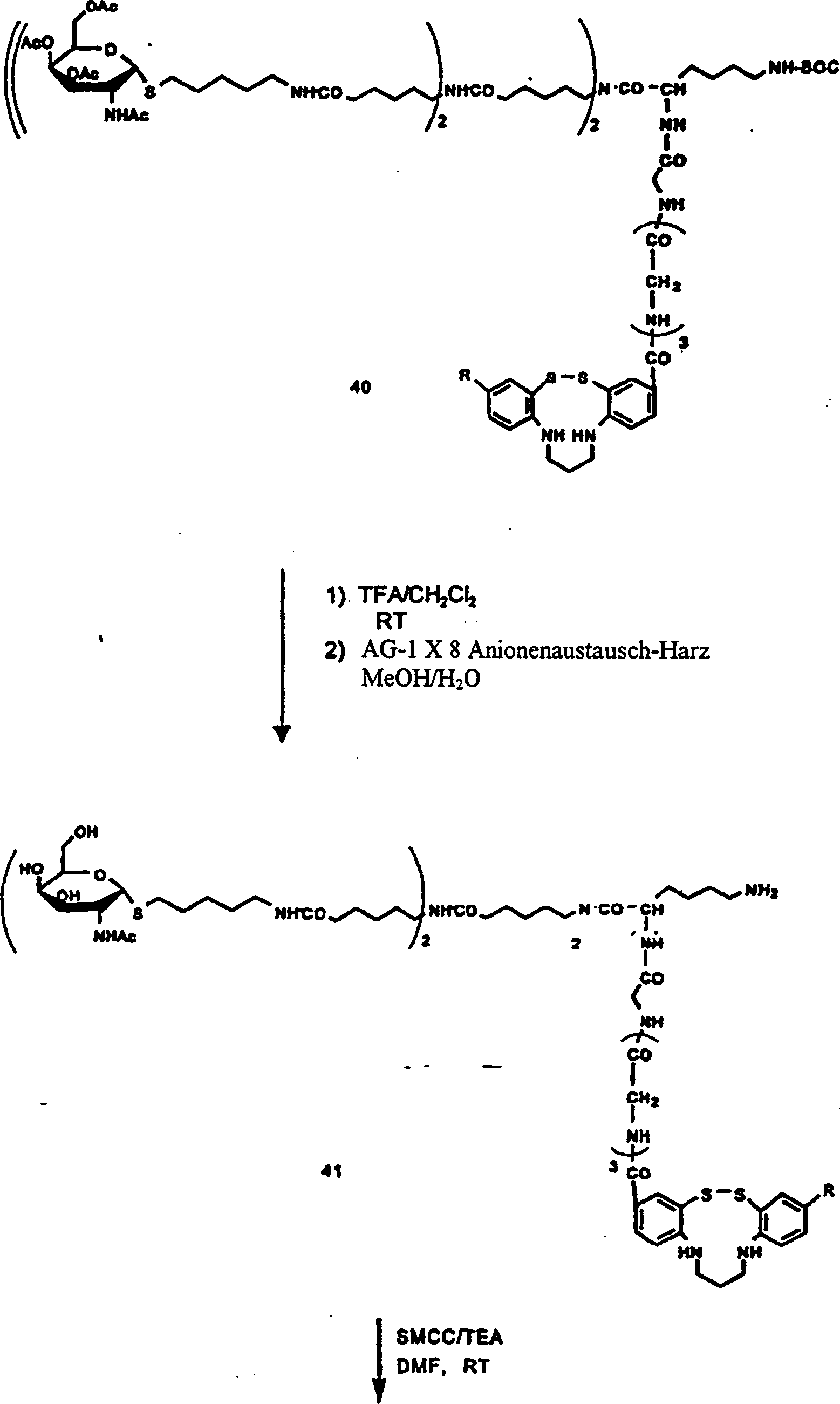
1 gibt schematisch ein
Verfahren zum Radiomarkieren von Annexin V wieder.
-
2 zeigt die Blut-Clearance
von Tc-99m-Annexin V (o) und von Tc-99m-Annexin V-Galactose (D).
-
3 zeigt schematisch die
Synthese von N, N'-Bis-(2-disulfidyl-4-carbonylphenyl-)
1,3-propyldiamin.
-
4 zeigt schematisch die
Synthese von N, N'-Bis-(disulfidyl-4-methylphenyl-)
gamma, gamma-diamino-isovalerat-N-hydroxysuccinimid.
-
5a und 5b zeigen schematisch die Synthese eines
acht-Galactose-Moleküle
enthaltenden Galactose-Clusters.
-
6 zeigt schematisch die
Synthese eines verlängerten
acht-Galactose-Einheiten enthaltenden Galactose-Clusters.
-
7 gibt schematisch eine
pET-12a-Plasmid-Map wieder.
-
8 gibt schematisch einen
pET-12a-PAP1, 3/7/94, Klon 1, wieder.
-
9 zeigt zeichnerisch ein
Konjugat, das ein Annexin-Dimer, eine chelatisierende Verbindung
und eine Hexose-Einheit einschließt.
-
Detaillierte
Beschreibung der Erfindung
-
Vor
der weiteren Beschreibung der Erfindung mag es hilfreich sein, Definitionen
bestimmter Begriffe anzugeben, die innerhalb der vorliegenden Offenbarung
verwendet werden.
-
Annexin:
Eine Klasse von Verbindungen, die gekennzeichnet ist durch das Vermögen, sich
mit hoher Affinität
an Membran-Lipide in Gegenwart von millimolaren Konzentrationen
von Calcium zu binden. Es wurde gezeigt, dass Annexine antikoagulative
Wirkungen zeigen, die durch das Binden von Annexinen an negativ
geladene Oberflächen-Phospholipide
vermittelt werden (z.B. an aktivierte Blutplättchen). Diese Annexin-Phospholipid-Bindung
blockiert – wie
angenommen wird – die
Aktivierung von Clotting-Faktoren (Verklumpungs-Faktoren) durch
derartige negativ geladene Oberflächen-Phospholipide. Vor der
Erkennung der Annexin-Molekül-Klasse
wurden Glieder dieser Klasse von Molekülen in der Literatur auch als
Plazenta-Antikoagulations-Proteine (z.B. PAP-1, 2, 3 und 4), Lipocortine,
Calpactine, vaskuläre
Antikoagulanzien (alpha und beta), Calphobindin I, Plazenta-Protein
4 (PP4), Endonexin II, Anchorin C II, Calcium-abhängiges Phospholipid-Bindungs-Protein
und dergleichen bezeichnet (verwiesen wird auf Crumpton et al.,
Nature 345, 212 (1990)). Annexin V ist ein Prototyp-artiges Annexin-Molekül, das in
der Beschreibung der vorliegenden Erfindung verwendet wird. Der
Begriff „Annexin" schließt natives
Annexin, das aus natürlichen
Quellen gereinigt wurde, z.B. aus Human-Plazenta, oder Annexin-Moleküle ein,
die eine native Sequenz enthalten, die durch beispielseise Genetic
Engineering, rekombinante Verfahren oder andere Mittel produziert
werden. Der Begriff „Annexin" schließt weiter
modifizierte Annexine ein, wie sie nachfolgend beschrieben werden,
die von irgendeiner Quelle abgeleitet oder aus irgendeiner Quelle
produziert werden.
-
Modifiziertes
Annexin: Ein Annexin-Molekül,
in dem die native Sequenz oder das Molekül in einer solchen Weise verändert wird,
ohne dass materiell die Membran-Bindungs-Affinität des Annexins
verändert
wird. Solche Annexine können
hergestellt werden durch chemische, Genetic Engineering oder rekombinante
Verfahrensweisen, wie sie Fachleuten in diesem technischen Bereich
bekannt sind. Die Modifikation kann eine Modifikation der Sequenz
durch den Zusatz von einigen Aminosäure-Resten und/oder einen Zusatz/eine
Streichung einer Aminosäure
an einer einzelnen Stelle auf der nativen oder einem Genetic Engineering
unterworfenen Sequenz einschließen.
Beispielsweise kann das Annexin am N-Terminus über den Zusatz von Aminosäure-Resten modifiziert
werden, worin wenigstens eine der Aminosäuren eine zugängliche
Sulfhydryl-Gruppe liefert. Die zugängliche Sulfhydryl-Gruppe oder
die zugänglichen
Sulfhydryl-Gruppen können
während
einer Konjugation verwendet werden oder bleiben für eine weitere
Konjugation verfügbar.
Der Begriff „modifiziertes Annexin" schließt Annexin-Multimere
ein.
-
Annexin-Multimer:
Eine Kombination aus zwei oder mehr monomeren modifizierten Annexin-Molekülen, wobei
die Komponenten des Multimers nativ oder rekombinant oder irgendeine
Kombination daraus sein können;
dies führt
zu einer ähnlichen
oder verbesserten Membran-Bindungs-Affinität, verglichen mit dem monomeren
Annexin. Ein aus bis zu etwa 20 modifizierten Annexinen aufgebautes
Monomer ist nützlich
für die vorliegende
Erfindung. Die bevorzugte Multimer-Zusammensetzung weist zwischen 2 und
etwa 10 modifizierten Annexinen auf. Ein Beispiel eines Annexin-Multimers
ist ein Annexin-Dimer, das aus 2 modifizierten Annexinen aufgebaut
sein kann, die über
Disulfid-Bindungen zwischen zugänglichen
Sulfhydryl-Gruppen an den modifizierten Annexinen verknüpft sind.
Das Annexin-Dimer
kann direkt als Fusions-Protein unter Verwendung bekannter Expressions-Systeme hergestellt
werden, wobei die beiden Annexin-Moleküle über einen Peptid-Linker durch die
zugänglichen
Sulfhydryl-Gruppen verbunden werden können. Ein dimeres Molekül könnte zusätzliche
funktionelle Stellen wie beispielsweise eine endogene Radiomarkierungs-Chelatisierungs-Stelle oder
eine zugängliche
Sulfhydryl-Gruppe
zum Binden eines oder mehrerer Hexose-Restes) enthalten, um ein Beispiel
zu nennen. Wie oben definiert, ist auch der Begriff „Annexin-Multimer" durch den Begriff „Annexin" abgedeckt.
-
Verlängerung:
Eine Reihe von Aminosäuren,
die an den N-Terminus des Annexin-Moleküls addiert werden, was eine
Sulfhydryl-Gruppe innerhalb von zehn Aminosäuren liefert, berechnet vom
N-Terminus. Vorzugsweise liegt die Sulfhydryl-Gruppe innerhalb von sechs Aminosäuren des
N-Terminus. Noch mehr bevorzugt ist die Sulfhydryl-Gruppe der zweite
Rest der N-terminalen Verlängerung
von sechs Aminosäuren.
Die Sulfhydryl-Gruppe wird vorzugsweise geliefert durch einen Cystein-Rest. Diese Addition
von Aminosäuren
liefert ein hochflexibles Mittel zum Binden funktioneller Einheiten
an dem terminalen Bereich des Annexins.
-
Radiomarkiertes
Annexin: Ein Annexin, das darin komplexiert ein Radionuklid aufweist.
-
Radiomarkierte
Annexin-Galactose: Ein Galactose-derivatisiertes Annexin, das darin
komplexiert ein Radionuklid aufweist.
-
Hexose-Einheit:
Eine Zusammensetzung aus einem oder mehreren sechs Kohlenstoffe
umfassenden Zucker-Rests (Hexose-Rest) oder auf solchen Einheiten
basierende Derivate, die durch Ashwell-Rezeptoren oder andere Leber-Rezeptoren
erkannt werden, wie beispielsweise den Mannose-/N-Acetylglucosamin-Rezeptor,
der mit Endothel-Zellen und/oder Kupffer-Zellen der Leber assoziiert
ist, oder durch den Mannose-6-phosphat-Rezeptor. Eine Hexose-Einheit
schließt
einen Hexose-Cluster ein. Eine Hexose-Einheit schließt auch
mehr als eine Hexose-Einheit ein, die an separate Stellen an andere
Moleküle
wie beispielsweise Annexin konjugiert ist.
-
Hexose-Cluster:
Ein Konstrukt, das eine Mehrzahl von Hexose-Einheiten (einschließlich Derivaten) aufweist,
die so konfiguriert sind, dass sie durch einen Leber-Rezeptor erkannt
werden. Solche Cluster sind vorzugsweise aus Hexose-Resten aufgebaut,
die in einer verzweigten Konfiguration verbunden sind (über eine Verbindungs-Einheit,
die ihrerseits an eine Verzweigungs-Struktur gebunden ist), und
sind an Annexin oder an andere Komponenten eines Hexose-Clusters,
der ein Konjugat enthält, über einen
einzelnen Befestigungspunkt (über
eine Bindungs-Einheit) gebunden. Vorzugsweise besteht das verzweigende
Netzwerk aus zwei oder drei gabelartigen Verzweigungen, d.h. besteht
aus 2, 4, 8, 16, 32 oder 64 Hexose-Resten oder besteht aus 3, 9,
27 oder 81 Hexose-Resten.
-
Galactose-Cluster:
Ein Konstrukt, das von etwa 3 bis etwa 64 Galactose-Resten aufweist,
die in einer verzweigten Konfiguration verbunden sind. Vorzugsweise
besteht das verzweigende Netzwerk aus zwei oder drei gabelartigen
Verzweigungen, d.h. besteht aus 2, 4, 8, 16, 32 oder 64 Galactose-Resten
oder besteht aus 3, 9, 27 oder 81 Galactose-Resten.
-
Radiomarkierter
Annexin-Galactose-Cluster: Ein Galactose-Cluster-derivatisiertes
Annexin mit einem darin komplexierten Radionuklid.
-
NxSy-Chelat-Verbindungen:
Wie in der vorliegenden Beschreibung definiert schließt der Begriff „NxSy-Chelat-Verbindung" bifunktionelle Chelatoren
ein, die in der Lage sind (i) koordinationsmäßig ein Metall oder Radiometall
zu binden; und (ii) kovalent an ein Annexin-Molekül gebunden
werden zu können.
Bevorzugte NxSy-Chelat-Verbindungen
weisen die Kerne N2S2 (allgemein
beschrieben in dem US-Patent Nr. 4,897,225 oder dem US-Patent Nr.
5,164,176 oder dem US-Patent Nr. 5,120,526), N3S
(allgemein beschrieben in dem US-Patent Nr. 4,965,392), N2S3 (allgemein beschrieben
in dem US-Patent Nr. 4,988,496), N2S4 (allgemein beschrieben in dem US-Patent Nr. 4,988,496),
N3S3 (allgemein
beschrieben in dem US-Patent Nr. 5,075,099) oder N4 (allgemein
beschrieben in dem US-Patent Nr. 4,963,688 und dem US-Patent Nr.
5,227,474) auf. Besonders bevorzugte NxSy-Chelat-Verbindungen haben N2S2- Kerne
und N3S-Kerne. Beispielhafte NxSy-Chelat-Verbindungen sind beschrieben in
der Druckschrift „Fritzberg
et al., Proc. Natl. Acad. Sci. USA, 85, 4024 – 29 (1988); in: Weber et al.,
Bioconj. Chem. 1, 431- 37 (1990) und in den darin zitierten Druckschriften,
um zwei Beispiele zu nennen. Für
die Zwecke der vorliegenden Beschreibung ist die Prototyp-artige
NxSy-Chelat-Verbindung
eine N2S2-Chelat-Verbindung. Eine
chelatisierende Verbindung, die ein Metall oder Radiometall komplexiert,
wird mit „Chelat" bezeichnet.
-
Esterase-empfindliche
chelatisierende Verbindung: Amid-Thiolat-Acetat-Ester-Chelat-Verbindungen der
NxSy-Familie, in
denen die Esterase-empfindliche chelatisierende Verbindung das Radionuklid-Metall
mit dem Annexin verbindet. Gemäß der vorliegenden
Definition schließt
der Begriff „Esterase-empfindliche
Gelatisierungs-Verbindungen" diejenigen ein,
die beschrieben sind in den US-Patenten Nrn. 5,112,953 und 5,175,257.
Die Esterase-empfindlichen chelatisierenden Verbindungen werden
während
des Metabolismus des radioaktivmarkierten Proteins in der Leber
gespalten und erzeugen Radioaktivität, die mit Cataboliten von vorteilhafter
Verteilung assoziiert ist, was zur Reduktion von Hintergrund-Radioaktivität in der
Nähe des
Herzens und der Lungen führt.
Eine bevorzugte Esterase-empfindliche chelatisierende Verbindung
ist das N3S-Seryl-Succinat, das beschrieben
ist in dem US-Patent Nr. 5,112,953.
-
Spaltbarer
Linker: Ein bifunktioneller Linker, der durch Leber-Enzym in der
Weise erkannt wird, dass der Linker enzymatisch abspaltbar ist und
einen hydrophilen Cataboliten erzeugt. In einigen bevorzugten Ausführungsformen
der vorliegenden Erfindung bindet der abspaltbare Linker die chelatisierende
Verbindung an die Hexose-Einheit.
Der abspaltbare Linker wirkt in Richtung auf eine Erhöhung der
renalen Exkretion und hepatobiliären
Exkretion und erhöht
so das Ziel-zu-Hintergrund-Verhältnis, was
einen frühen
Nachweis arterieller und venöser
Thromben ermöglicht.
Beispiele dieser Arten von Linker schließen ein Polymer wie beispielsweise Monosaccharide,
Polysaccharide, Polyaminosäuren,
Hydroxyalkylacrylamide, Polyethylenglycol-basierte hydrophile Polymere,
biologisch abbaubare Polymere, die eine Ether- oder Ester-Bindung
enthalten, sowie Dextran und Hemisuccinyl-Ester ein.
-
Konjugat:
Ein Konjugat umfasst chemische Konjugate (kovalent oder nicht-kovalent gebunden),
Fusionsproteine und dergleichen.
-
Für Fachleute
mit üblichem
Sachverstand in diesem technischen Bereich ist es offensichtlich,
dass im Rahmen der Anwendung Bezugnahmen auf Annexin modifiziertes
Annexin sowie ein Annexin-Multimer einschließen, solange nichts anderes
speziell angegeben ist oder aus dem Kontext offensichtlich ist.
Weiter werden Fachleute mit üblichem
Sachverstand in diesem technischen Bereich verstehen, dass trotz
der Tatsache, dass Galactose nachfolgend als repräsentatives
Beispiel beschrieben ist, irgendeine Bezugnahme auf Galactose auch
andere Hexose-Einheiten einschließt, wie beispielsweise N-Acetylgalactosamin,
sowie deren Cluster.
-
Die
vorliegende Erfindung ist gerichtet auf Annexin-Hexose-enthaltende
Konjugate und radiomarkierte Annexine, einschließlich radiomarkierter Annexin-Galactose-Cluster-Konjugate,
und deren Verwendung für
diagnostische Bildgebungs-Zwecke.
Radiomarkierte Annexine gemäß der vorliegenden
Offenbarung sind gekennzeichnet durch die folgenden Eigenschaften:
Schnelle Anbindung an Ziel-Zellen-Stellen, gekennzeichnet durch anionische
Phospholipide; kurze Halbwertszeit im Kreislauf; in-vivo-Stabilität gegen
metabolischen Abbau oder Radionuklid-Trennung von dem Chelat (in
den Fällen,
in denen im Rahmen der vorliegenden Erfindung ein Chelat verwendet
wird); und Eignung für
Verpackung in Form eines Kalt-Kits.
-
Radiomarkierte
Annexin-Galactose-Konjugate zeigen auch die oben genannten charakteristischen
Eigenschaften, wenn auch in unterschiedlichen Graden. Beispielsweise
zeigen radiomarkierte Annexin-Galactose-Konjugate eine kürzere Halbwertszeit
im Kreislauf als ihre radiomarkierten Annexin-Gegenstücke. Auch zeigen
radiomarkierte Annexin-Galactose-Konjugate allgemein eine niedrigere
Bindungsaffinität über Ziel-Stellen
als ihre radiomarkierten Annexin-Gegenstücke. Eine erfolgreiche diagnostische
Bildgebung schließt
sowohl eine Signal-Akkumulation an der Ziel-Stelle als auch eine
schnelle Eliminierung eines nicht auf das Ziel gerichteten Signals
ein. Folglich liefert eine beschleunigte Elimination der radiomarkierten
Annexin-Galactose-Konjugate vom Kreislauf eines Rezipienten eine
kennzeichnende Möglichkeit
zum Erhalt diagnostischer Bilder in einem kürzeren Zeitraum.
-
Darüber hinaus
verringert sich ein Ziel-Stellen-Signal über die Zeit als Ergebnis des
radioaktiven Zerfalls. Weiter verringert ein Metabolismus des Ziel-assoziierten
Materials ebenfalls das Target-Signal. Folglich erhöht eine
Bildgebung innerhalb eines kürzeren
Zeitraums das Verhältnis
Ziel : Nicht-Ziel und das gesamte Ziel-Signal, wodurch verbesserte
diagnostische Information gespeichert wird.
-
Radiomarkierte
Annexin-Galactose-Cluster-Konjugate gemäß der vorliegenden Erfindung
kombinieren wünschenswerte
Merkmale radiomarkierter Annexine und radiomarkierter Annexin-Galactose-Konjugate. Beispielsweise
zeigen radiomarkierte Annexin-Galactose-Cluster-Konjugate eine kürzere Halbwertszeit
im Kreislauf als ihre radiomarkierten Annexin-Gegenstücke. Auch
zeigen Radiomarkierte Annexin-Galactose-Cluster-Konjugate
allgemein eine höhere
Bindungs-Affinität
gegenüber
Ziel-Stellen als
ihre radiomarkierten Annexin-Galactose-Konjugat-Gegenstücke. Folglich
bieten die erhöhte
Eliminierung vom Kreislauf eines Rezipienten und die Aufrechterhaltung
einer erheblichen Bindungs-Affinität der radiomarkierten Annexin-Galactose-Cluster-Konjugate
gemäß der vorliegenden
Erfindung die Möglichkeit,
klare diagnostische Bilder in einer kürzeren Zeitspanne zu erhalten.
-
Eine
Ausführungsform
der vorliegenden Erfindung ist gerichtet auf Annexinenthaltende
Konjugate, die für
ein Radio-Labelling mit einem diagnostischen Bildgebungsmittel geeignet
sind und die einschließen:
- – ein
modifiziertes Annexin, wobei die Modifikation eine zugängliche
Sulfhydryl-Gruppe
hat; und
- – eine
Hexose-Einheit, die von einem Säuger-Leber-Rezeptor
erkannt wird, wobei die Hexose-Einheit an das Annexin konjugiert
ist.
-
Radiomarkierte
modifizierte Annexin-Hexose-Einheit-Konjugate, die für das Abbilden
vaskulärer Thrombi
geeignet sind, kommen ebenfalls in Betracht, wobei die radiomarkierten
Annexin-Hexose-Einheit-Konjugate ein modifiziertes Annexin, eine
Hexose-Einheit und weiter ein diagnostisches Radionuklid einschließen, das
an das modifizierte Annexin konjugiert ist.
-
Ein
anderes bevorzugtes Annexin-Konjugat gemäß der vorliegenden Erfindung
ist dasjenige, das zum Radiomarkieren mit einem diagnostischen Bildgebungsmittel
geeignet ist und einschließt:
- – ein
modifiziertes Annexin; und
- – eine
Esterase-empfindliche NxSy-Chelat-Verbindung,
die an das Annexin konjugiert ist.
-
Radiomarkierte
Annexin-Konjugate, die zum Abbilden vaskulärer Thrombi geeignet sind,
werden ebenfalls in Betracht gezogen, wobei die radiomarkierten
Annexin-Konjugate ein Annexin, eine Esterase-empfindliche NxSy-Chelat-Einheit
und weiter eine Hexose-Einheit und ein diagnostisches Radionuklid
einschließen, das
durch die chelatisierende Verbindung komplexiert wird.
-
Ein
weiteres bevorzugtes Konjugat gemäß der vorliegenden Erfindung
schließt
ein:
- – ein
modifiziertes Annexin-Multimer; und
- – eine
NxSy-Chelat-Verbindung,
die an das Annexin konjugiert ist.
-
Radiomarkierte
Annexin-Multimer-Konjugate, die zum Abbilden vaskulärer Thrombi
geeignet sind, werden ebenfalls in Betracht gezogen, wobei die radiomarkierten
Annexin-Multimer-Konjugate ein Annexin-Multimer, eine NxSy-Chelat-Verbindung
und weiter ein diagnostisches Radionuklid einschließen, das
durch die Chelat-Verbindung komplexiert wird.
-
Für das Sichtbarmachen
von vaskulären
Thromben, die mit einer Zahl von pathologischen Zuständen assoziiert
sind, wird ein radiomarkiertes Annexin wie beispielsweise ein Konjugat
aus einem Annexin, das mit einem abbildenden Radionuklid komplexiert
ist, wie beispielsweise Tc-99m, einem Empfänger verabreicht, für den eine
derartige Diagnose gewünscht
ist. Das radiomarkierte Annexin oder der Annexin-Teil des radiomarkierten
Konjugats lokalisiert sich schnell an Target-Stellen, die durch
negative geladene Oberflächen-Phospholipide
gekennzeichnet sind, wie beispielsweise vaskuläre Thromben. Das Radionuklid
wird nach seinem Vermögen
ausgewählt, über eine
der verschiedenen dafür
geeigneten Verfahrensweisen sichtbar gemacht zu werden, beispielsweise
für eine
Bildgebung mit einer Gamma-Kamera. Wegen der schnellen Anbindung
von Annexinen an die Target-Stelle und die kurze Halbwertszeit im
Serum (allgemein weniger als 30 Minuten) von Annexinen (die durch
Radiomarkierung nicht signifikant verlängert wird), läuft eine
Bildgebung dieser Ziel-Stellen
mit nur geringer Aussetzung der Nicht-Target-Stellen gegenüber Radioaktivität ab.
-
Eine
diagnostische Bildgebung ist abhängig
von dem Verhältnis
Signal zu Geräusch.
Verbesserungen, die entweder eine Akkumulation des Ziel-Signals
oder eine Verringerung des Geräusche
einschließen,
verstärken
die Wirksamkeit des Diagnose-Bildgebungs-Produkts.
Für eine
zielorientierte Bildgebung ist die Geräusch-Verringerung synonym mit einer Verringerung
der Hintergrund-Radioaktivität,
insbesondere der Blut-Pool-Aktivität. Annexine einschließlich Annexin
V werden durch die Leber schnell entfernt. Jedoch nur ein Bruchteil
der Aktivität
wird bei jedem Durchlauf des umlaufenden Konjugats durch die Leber
entfernt.
-
Um
die Hintergrund-Aktivität
noch effizienter aus dem Blut zu entfernen, könnte eine Verbesserung der Leber-Extraktion
der Annexin-enthaltenden Konjugate verwendet werden. Ein bevorzugtes
Verfahren zur Erhöhung
der Leber-Extraktion schließt
eine Derivatisierung des Annexin-enthaltenden Konjugats mit einer
Hexose-Einheit wie
beispielsweise Galactose ein, die von einem Leber-Rezeptor erkannt
wird. Die Wirksamkeit der Leber-Rezeptor-Extraktion der Hexose-Einheit,
die dadurch erkannt wird, resultiert zu einer erhöhten Entfernung
von dem derivatisierten Annexinenthaltenden Konjugat (z.B. Annexin-Galactose-Konjugat
pro Durchlauf durch die Leber, verglichen mit der Menge an genauso
entferntem nicht-derivatisertem Annexinenthaltendem Konjugat).
-
Eine
weitere Verbesserung des Verhältnisses
Signal zu Geräusch
kann erreicht werden durch Derivatisieren von radiomarkiertem Annexin
oder von einem Annexinenthaltenden radiomarkierten Konjugat mit
einem Cluster von Galactose-Molekülen, die von einem Leber-Rezeptor
erkannt werden. Die Effizienz einer Leber-Rezeptor-Extraktion der erkannten
Einheit führt
dadurch zu einer erhöhten
Entfernung des derivatisierten Annexin-enthaltenden Konjugats pro
Durchlauf durch die Leber (z.B. eines Annexin-Galactose-Cluster-Konjugats),
verglichen mit der Menge an underivatisertem Annexin-enthaltendem
Konjugat, das auf diesem Wege entfernt wird. Darüber hinaus können mehrere
Galactose-Reste, die in einem Cluster angeordnet sind, an die Annexin-Konjugat-Komponente über einen
einzelnen Bindungspunkt gebunden werden. In der Folge kann weniger
oder gar keine Reduktion der Annexin-Zielmolekül-Bindungsaffinität, die von einer Galactose-Cluster-Derivatisierung
resultiert, beobachtet werden, verglichen mit nicht in Form eines
Clusters angeordneten Annexin-Galactose-Konjugaten.
-
Annexine
liegen allgemein (wobei die am meisten bemerkenswerte Ausnahme Annexin
II ist) in Form einzelkettiger, nicht-glycosylierter Proteine mit
einem Molekulargewicht von etwa 33 bis 72 Kilodalton (kD) vor. Annexine
besitzen eine Anzahl biologischer Aktivitäten, die mit einer durch Calciumionen
vermittelten Bindung assoziiert sind.
-
Untersuchungen
haben gezeigt, dass sich Annexine mit hoher Affinität an anionische
Membran-Lipide in Gegenwart millimolarer Konzentrationen Calcium
binden. In der Gegenwart von Calcium haben diese Proteine eine besonders
hohe Affinität
gegenüber
negativ geladenen Phospholipiden wie beispielsweise Phosphatidylserin,
Phosphatidylglycerin, Phosphatidylsäure oder Phosphatidylinositol.
-
Hierzu
wird beispielsweise verwiesen auf „Funakoshi et al., Biochem.
26, 5572 – 5578
(1987)" und „Tait et
al., Biochem. 27, 6268 – 6272
(1988)". Derartige
negativ geladene Phospholipide sind mit vaskulären Thromben assoziiert (z.B.
sind auf der Oberfläche
aktivierter humaner Blutplättchen
angeordnet).
-
Annexine üben antikoagulatorische
Wirkungen aus. Eine Koagulations-Inhibition
wird vermittelt durch das Binden von Annexinen an negativ geladene
Oberflächen-Phospholipide
(z.B. wie sie auf der Oberfläche aktivierter
Blutplättchen
zugegen sind). Man geht davon aus, dass dieser Bindungsvorgang die
Aktivierung von Gerinnungsfaktoren (Clotting-Faktoren) durch derartige
negativ geladene Oberflächen-Phospholipide blockiert.
Annexine ordnen sich schnell an Ziel-Stellen an, die anionische
Phospholipide tragen, d.h. in einer Zeit von etwa 5 bis 30 Minuten,
abhängig
von deren Umlauf-Konzentrationen, bleiben jedoch im Serum für eine etwas
längere
Zeitdauer umlaufend (Halbwertszeit im Kreislauf < 30 min.). Das nachfolgende Beispiel
III diskutiert Ergebnisse von Bildgebungs-Experimenten, bei denen
vaskuläre
Thromben in planaren Bildern in einer mittleren Zeit (im Anschluss
an die Annexin-Verabreichung)
von 82 Minuten visualisiert wurden.
-
Aufgrund
dieser Eigenschaften können
Annexine oder an diagnostische oder therapeutische Mittel konjugierte
Annexine in Protokollen für
die in vivo-Diagnose oder -Behandlung von vaskulären Thromben verwendet werden,
die mit einer Anzahl von Indikationen assoziiert sind, wie beispielsweise
DVT (deep vein thrombosis), PE (pulmonary embolism), Herzinfarkt,
Vorhof-Flimmern (atrial fibrillation), Probleme mit prosthetischen
kardiovaskulären
Materialien, Hirnschlag (Apoplexie) und dergleichen. Andere Indikationen,
die mit einer Akkumulation aktivierter Blutplättchen assoziiert sind, für die die
Annexin-Konjugate gemäß der vorliegenden
Erfindung nützlich
sind, schließen
die folgenden ein: Abszess-Bildgebung, Restenose im Anschluss an eine
Ballon-Angioplastie (restenosis post balloon angioplasty; PCTA),
Entzündungen
von Gelenken (d.h. rheumatoide Arthritis), geschädigte Endothel-Zellen (d.h.
Alzheimer-Krankheit),
Abbilden von Gerinseln in cerebralen (Gehirn-)Arterien, Verschlüsse in peripheren
Arterien, Abbildung von Vorhof-Thrombosen und Abbildung von Koronar- und Carotis-Arterien-Thromben.
-
Über das
Abbilden von Blutplättchen
in vivo ist es auch wichtig, Blutplättchen-Populationen in den Bereichen klinische
Forschung, in vitro-Diagnoseforschung und Grundlagenforschung zu
charakterisieren. Von den Zell-Oberflächen-Markern, die derzeit zum
Charakterisieren von Blutplättchen
verfügbar
sind, sind viele nicht kreuzreaktiv zwischen Spezies und können alle
Plättchen
erkennen, sondern im Gegensatz dazu gerade nur die aktivierte Blutplättchen-Population.
Es wird angenommen, dass sich Annexin selektiv an aktivierte Blutplättchen in
vielen Spezies bindet. So können
Annexin-Konjugate gemäß der Erfindung
als ein alternativer Zell-Marker in den Gebieten Forschung und Diagnose
verwendet werden, wie beispielsweise in der Immunohisto-Chemie und
der Durchfluss-Cytometrie. Beispielsweise können Konjugate gemäß der vorliegenden
Erfindung dazu verwendet werden, aktivierte Blutplättchen in
fixierten Geweben/Tumoren, Blut-Ausstrichen, in Tieren mit Koagulopathien
und in situ in Blutplättchen-Aktivierungs-Assays
verwendet werden, und zwar in Response auf verschiedene chemische
oder infektiöse
Stimuli sowie zur Bestimmung aktivierter Blutplättchen im Blut, in Zellkultur-Assays
und in Blutplättchen-Response-Assays.
Von Fachleuten mit üblichem
Sachverstand in diesem technischen Bereich wird erkannt, dass die
Konjugate gemäß der vorliegenden
Erfindung nützlich für jeden
beliebigen Zweck oder jede beliebige Indikation sind, in der ein
Ansprechen oder Binden aktivierter Blutplättchen wünschenswert ist. Beispielhafte
diagnostische Protokolle und Experimente, in denen radiomarkierte
Annexine verwendet werden, werden nachfolgend angegeben, um weiter
diesen Aspekt der vorliegenden Erfindung aufzuklären.
-
Ein
Beispiel eines bevorzugten Annexins, das nützlich in der praktischen Durchführung der
vorliegenden Offenbarung ist, ist Annexin V, das von Bohn im Jahre
1979 aus Human-Plazenta, einer reichen Quelle von Annexinen, isoliert
wurde und mit Plazenta-Protein 4 (PP4) bezeichnet wurde. Annexin
V wurde in E. coli exprimiert (Iwasaki et al., J. Biochem., Band
102, Nummer 5, 1261 – 1273
(1987)). Auch wurde ein cDNS-Klon voller Länge von Annexin V erhalten
und in Expressions-Vektoren subkloniert, wodurch die Produktion
von Fusions-Proteinen erleichtert wurde, die Annexin V enthalten
(siehe allgemein dazu Tait et al., J. Biol. Chem. 270 (37), 21594 – 21599
(1995)). Annexin V besteht aus vier Domänen (vier nebeneinander vorliegende,
nicht perfekte Wiederholungen von etwa 75 Aminosäure-Resten; „Funakoshi
et al., Biochem. 26, 8087 – 8092
(1987)", worin jede
Domain aus 5 alpha-Helices besteht. Von der Seite erscheint das
Annexin V-Molekül
kronenartig mit wenigstens vier Calcium-Bindungsstellen auf seiner
konvexen Oberfläche,
durch die Annexin-Phospholipid-Wechselwirkungen
vermittelt werden. Andere Annexin-Moleküle sind ebenfalls nützlich in
der praktischen Durchführung
der vorliegenden Erfindung, und die auf Annexin V bezogenen Diskussionen
der obigen Beschreibung gelten allgemein für Annexin-Moleküle.
-
Von
den Annexinen hat Annexin V die stärkste Bindungsaffinität (Kd < 10–10M)
gegenüber
Phospholipid-Vesikeln, die 80 % Phosphatidylcholin und 20 % Phosphatidylserin
enthalten, unter Bedingungen, die denen von Plasma-Flüssigkeit
und extracellulärer
Flüssigkeit
vergleichbar sind (1,2 mM ionisiertes Calcium; 0,15 M Ionenstärke). Diese
Bindung ist reversibel und Calcium-abhängig. Der Grad einer Annexin-Bindung
an Phopsholipide kann quantitativ erfaßt werden durch Fluoreszenz-Quenchen, wie dies
beschrieben wurde von „Taft
et al., J. Biol. Chem. 264, 7944 – 7949 (1989)".
-
Da
Annexin V eine Mehrzahl von Calcium-Bindungsstellen aufweist und
da ein Binden von Annexin V an negativ geladene Phospholipide durch
Calcium vermittelt wird, kann ein im Engineering behandeltes Molekül, das aus
einer oder mehreren einzelnen Annexin V-Domänen besteht, in Bildgebungs-Protokollen
gemäß der vorliegenden
Offenbarung verwendet werden. Auch kann das Annexin-Molekül an einer
Position oder Positionen, die von den Domain-Grenzen verschieden
sind, verteilt werden und so ein dem Engineering unterworfenes Molekül liefern,
das einer Calciumvermittelten Bindung von anionischen Phospholipiden
fähig ist. Auch
kann Annexin V an einem oder mehreren Aminosäure-Resten) verändert werden,
solange die Affinität von
Annexin V für
anionische Phospholipide nicht signifikant verschlechtert wird.
Beispielsweise kann der Cystein-Aminosäure-Rest (Position 316) von
Annexin V entweder gestrichen werden oder durch Alanin oder andere,
keine Schwefel enthaltende Aminosäuren ersetzt werden, wie dies
Fachleuten mit üblichem
Sachverstand in diesem technischen Bereich bekannt ist. Dabei wird
beim Markieren ein monomer radiomarkiertes Annexin V hergestellt.
-
In
einem anderen Beispiel kann ein natives Annexin an dem N-terminalen
Ende dadurch modifiziert werden, dass man Aminosäure-Reste addiert und so für eine zugängliche
Sulfhydryl-Gruppe oder zugängliche Sulfhydryl-Gruppen
sorgt. Dies kann bewirkt werden mit der Addition von wenigstens
einem einzelnen Cystein-Rest nahe dem N-terminalen Ende. Die zugänglichen
Sulfhydryl-Gruppen können
nach einer anfänglichen
Konjugation verfügbar
sein oder der weiteren Konjugation dienen, abhängig von der verwendeten Ausführungsform
der Erfindung.
-
Ein
bevorzugtes modifiziertes Annexin-Molekül, wie es in der vorliegenden
Beschreibung und in den Patentansprüchen definiert ist, ist eine
monomere Form von Annexin V mit einer N-terminalen Verlängerung einer
Aminosäure-Sequenz.
Die Sequenz wird so gewählt,
dass sie Aminosäuren
enthält,
die einer Aminosäure
benachbart sind, die eine zugängliche
Sulfhydryl-Gruppe enthält,
wie beispielsweise Cystein. Die gewählte Sequenz verbessert die
Verhältnisse
Ziel-Organ zu normalem Organ und das überlegene Gerinsel-Bildgebungs-Potential.
Die Verwendung von hydrophilen Aminosäuren in der Sequenz wie beispielsweise
Serin, Glycin, Threonin, Aspartat, Glutamat und andere erleichtert
die Ausscheidung der Chelat-Produkte des Catabolismus über die
Nieren im Anschluß an
eine Aufnahme des radiomarkierten Annexins in normalen Organen wie beispielsweise
der Leber. Eine bevorzugte Aminosäure-Sequenz, die dem N-terminalen
Ende von Annexin V zuaddiert wird, ist Ala-Cys-Asp-His-Ser-Met.
Ein Vorteil dieser speziellen Konfiguration ist, dass mit dem Cystein-Rest
nahe dem N-terminalen Ende die Sulfhydryl-Gruppe und die benachbarten
Amid-Gruppen eine stabile Chelatisierung eines Radionuklids erlauben.
Beispielsweise kann ein Radionuklid wie beispielsweise Technetium
in dem nun gebildeten stabilen N3S-Chelat
verwendet werden, und zwar zur Verwendung beim endogenen Radiomarkieren
des Annexin-Moleküls.
-
So
wird es durch die vorliegende Erfindung in Betracht gezogen, dass
Tc-99m direkt über
eine spezielle, dem Bio-Engineering unterworfene Stelle an Annexin
V gebunden werden kann, und zwar ohne die Notwendigkeit einer inzwischen
ablaufenden Produktion einer Amin-gerichteten aktiven Ester-Chelatisierungs-Verbindung für die Radiomarkierung.
Wie oben festgestellt, kann zum Erleichtern eines direkten Markierens
die modifizierte Annexin V-N-Terminus-Sequenz aus Ala-Cys-Asp-His-Ser-... usw.
bestehen, um ein Beispiel zu nennen. Ein definiertes Chelat-System durch eine
endogene Protein-Sequenz kann in den Fällen erreicht werden, in denen
die benachbarten Amid-Donor-Atome von Aminosäure-Resten sich um die Basis
der Metall-Oxo-Bindung in einer quadratisch pyramidalen Anordnung
herumwickeln. So sorgt diese Anordnung für ein stabiles Chelatisieren, ähnlich dem
stabilen Chealtisieren, das erhalten wird, wenn man ein N3S-Chelat in einem vorgebildeten Markierungs-Verfahren
verwendet. In einer bevorzugten Ausführungsform liegt der Cystein-Rest
nahe dem N-terminalen Ende, und der Sulfhydryl-Rest des Cysteins
und die benachbarten Amid-Gruppen von den anderen Amniosäuren innerhalb
des modifizierten Annexins bilden eine stabile endogene MAG3-ähnliche
Chelatisierungs-Struktur.
Für die
endogene Chelatisierung ist Technetium-99m das bevorzugte Radionuklid.
-
Andere
Modell-Peptide, mit denen die Sequenz des N-terminalen Endes eines
modifizierten Annexin V variiert werden kann (beispielsweise die
Sequenz von George et al., in: Proc. Natl. Acad. Sci. USA 92, 8358 – 8362 (1995)),
können
hergetellt und verwendet werden. Diese Peptide werden denselben
Bedingungen einer in vitro-Markierung
mit Tc-99m unterworfen, wie sie oben beschrieben wurden. Peptide
mit geeigneten charakteristischen Markierungs-Eigenschaften können gewählt werden,
und die Sequenz kann in das rekombinante Annexin V-Molekül eingearbeitet
werden. Ein Erfolg wird angezeigt durch eine Herstellung eines Derivats
mit spezieller Aktivität,
die genauso hoch ist wie oder höher
ist als diejenige des vorliegenden Derivats, wie berichtet wurde
von „Stratton
et al., in: Circulation 92, 3113 – 3121 (1995), das stabil in
vitro und in vivo ist, das seine Membran-Bindungsaktivität und Thrombus-Bindungsaktivität beibehält und das
nicht nachteilig verändert
wird in Bezug auf die Clearance im Blut oder die Bio-Verteilung.
Wenn die oben angegebenen Kriterien erfüllt werden, wird das modifizierte
Annexin V-Molekül
oder das Multimer aus dem modifizierten Annexin V-Molekül in Übereinstimmung
mit der Erfindung verwendet.
-
Eine
andere Ausführungsform
der vorliegenden Erfindung ist ein Annexin-Multimer. Ein Annexin-Multimer ist aus
zwei oder mehr modifizierten Annexin-Molekülen aufgebaut, die über Disulfid-Brücken zwischen einer
oder mehreren der zugänglichen
Sulfhydryl-Gruppen der jeweiligen Annexine miteinander verbunden werden.
Das Prototyp-artige und bevorzugte Annexin-Multimer ist ein Annexin-Dimer.
Es ist beabsichtigt, dass das Dimer eine höhere Aktivität gegenüber der
Blutplättchen-Membran aufweist,
was von einer niedrigeren Dissoziationsrate herrührt. Das Dimer kann dann an
der Target-Stelle in einer höheren
Konzentration ausgeschüttet
werden oder länger
in dem Thrombus in vivo gehalten werden, wodurch das Target zu Hintergrund-Verhältnis zu
jedem gegebenen Zeitpunkt verbessert wird.
-
Das
Annexin-Multimer sowie das Annexin-Dimer können als Fusionprotein hergestellt
werden. Beispielsweise kann dies bewirkt werden durch Verwendung
bekannter Expressions-Systeme mit den modifizierten Annexinen, worin
die modifizierten Annexine mit einem Peptid-Linker über die
zugänglichen
Sulfhydryl-Gruppen
miteinander verbunden werden.
-
Ein
multimeres Molekül
kann auch weitere funktionelle Stellen enthalten, wie beispielsweise
eine endogene radiomarkierte Chelatisierungs-Stelle, wie sie oben
genannt wurde, oder eine freie Sulfhydryl-Stelle, um das Binden
einer Hexose-Einheit möglich
zu machen. Beispielsweise kann ein Annexin-Dimer produziert werden,
das eine Hexose-Einheit und eine NxSy-Chelatisierungs-Verbindung einschließt. Diese
Verbindung kann dann zum Erzeugen stabilisierter Monomere für weitere Radiomarkierungs-Verfahren über Reduktion
der Disulfid-Bindungen des Dimers verwendet werden.
-
Um
die Hintergrund-Radiomarkierungs-Aktivität zu senken, können Annexine
(einschließlich
modifizierte Annexine und deren Multimere) mit Hexose-Einheiten
oder Einheiten auf Hexose-Basis derivatisiert werden. Noch spezieller
können
Annexine derivatisiert werden, so dass sie eine oder mehrere Hexosen
eingearbeitet enthalten (z.B. sechs Kohlenstoffatome aufweisende
Zucker-Einheiten), die von Ashwell-Rezeptoren oder anderen Leber-Rezeptoren
erkannt werden, wie beispielsweise dem Mannose-/N-Acetylglucosamin-Rezeptor,
der mit Endothel-Zellen und/oder Kupffer-Zellen der Leber assoziiert
ist, oder dem Mannose-6-phosphat-Rezeptor. Beispielhaft für solche
Hexosen sind Galactose, Mannose, Mannose-6-phosphat, N-Acetylglucosamin,
Pentamannosylphosphat und dergleichen. Andere Einheiten, die von
Ashwell-Rezeptoren
erkannt werden, einschließlich
Glucose, N-Galactosamin, N-Acetylgalactosamin,
Thioglycoside von Galactose und allgemein D-Galactoside und Glucoside
und dergleichen können
ebenfalls in der praktischen Durchführung der vorliegenden Erfindung
verwendet werden. Damit schließen
Hexose-Einheiten, die in der praktischen Durchführung der vorliegenden Erfindung
nützlich
sind, eine Vielzahl von Galactose-, Mannose- und Glucose-Zuckern
ein, die von Leber-Rezeptoren erkannt werden. Es versteht sich,
dass Galactose und Galactose-Cluster von dem Begriff „Hexose-Einheit" umfaßt werden.
Weiter sollte auch erkannt werden, dass die Verwendung des Begriffs „Galactose" in der Diskussion
so verstanden wird, dass er nicht nur Galactose umfaßt, sondern auch
N-Galactosamin, N-Acetylgalactosamin, Thioglycoside von Galactose
und allgemein auch D-Galactoside. Der Begriff „Mannose" wird in der Weise verstanden, dass
er Mannose, Mannose-6-phosphat, Pentamannosylphosphat und dergleichen
einschließt.
Der Begriff „Glucose" wird so verstanden,
dass er Glucose, Glucoside und dergleichen umfaßt. Bevorzugte Hexose-Einheiten sind Hexose,
Zucker, wie beispielsweise Galactose und Galactose-Cluster. Für die Zwecke
der Erfindung ist die am meisten bevorzugte Galactose N-Acetylgalactosamin.
-
Wie
oben festgestellt wurde, ist Galactose die Prototyp-artige Hexose,
die für
die Zwecke dieser Beschreibung verwendet wird. Eine Galactosethioglycosid-Konjugation
an ein Protein wird vorzugsweise bewirkt in Übereinstimmung mit den Lehren
von Lee et al. „2-Imino-2-methoxyethyl-1-thioglycosides.
New Reagents for Attaching Sugars to Proteins", Biochemistry 15 (18), 3956, (1976)". Ein weiteres nützliches
Galactosethioglycosid-Konjugations-Verfahren wird beschrieben in
Drantz et al., „Attachment
of Thioglycosides to Proteins: Enhancement of Liver Membrane Binding", Biochemistry 15
(18), 3963 (1976)".
Eine Annexin-Galactose-Konjugation wird auch in den nachfolgenden
Beispielen diskutiert.
-
In
einer Ausführungsform
der vorliegenden Erfindung liegt die Zahl der Galactose-Reste, die
an das Annexin-Konjugat gebunden sind, im Bereich von 1 bis zur
Maximalzahl von Galactose-Einheiten, die nicht signifikant die Bindungsaffinität von Annexin
gegenüber
seinem Ziel-Molekül
verringern; siehe Beispiel IV. Beispielsweise ist eine Galactose-Derivatisierung,
die wenigstens 20 % der nativen Annexin-Bindungs-Aktivität bewahrt, bevorzugt, wobei
die Bewahrung von wenigstens 50 % der nativen Annexin-Bindungs-Aktivität noch mehr
bevorzugt ist. Die theoretisch mögliche
Maximalzahl von Galactose-Resten, die an dem Annexin-Molekül angeordnet
sind, ist 22 (d.h. die Zahl von Lysin-Resten innerhalb der Annexin-Struktur).
Eine beispielhafte Zahl von Galactose-Resten an radiomarkierten
Annexin-Galactose-Konjugaten gemäß der vorliegenden
Erfindung liegt im Bereich von 1 und etwa 5.
-
In
einer anderen Ausführungsform
der vorliegenden Erfindung werden vorzugsweise Hexose-Cluster bei
der praktischen Durchführung
der vorliegenden Erfindung verwendet. Galactose-Cluster sind die
Prototyp-artigen Hexose-Cluster, die für die Zwecke der vorliegenden
Beschreibung verwendet werden. Ein die Galactose, N-Acetylgalactosamin
enthaltender Cluster ist ein besonders bevorzugter Galactose-Cluster gemäß der vorliegenden
Erfindung.
-
Der
Aufbau von Hexose-Clustern gemäß der vorliegenden
Erfindung wird durchgeführt
unter Berücksichtigung
der folgenden Kriterien, wie dies im Kontext des Aufbaus eines Galactose-Clusters
beschrieben wird:
- 1) Zahl von Galactose-Resten
in einem Cluster;
- 2) Entfernung zwischen Galactose-Einheiten in dem Cluster; und
- 3) Entfernung zwischen Galactose-Cluster und der Annexin-Konjugat-Komponente.
-
Hinblick
auf das Kriterium Nr. 1 gibt die Literatur an, dass Galactose-Rezeptoren auf der
Oberfläche eines
Human-Hepatozyten als Heterotrimere und vielleicht als Bis-Heterotrimere
gruppiert sind. Siehe beispielsweise „Hardy et al., Biochemistry
24, 22 – 28
(1985)". Für eine optimale
Affinität
gegenüber
solchen Rezeptoren wird im Rahmen der vorliegenden Erfindung in
Betracht gezogen, dass jeder Galactose-Cluster vorzugsweise wenigstens
drei Galactose-Reste enthalten sollte. Allgemein gilt, dass die
Neigung für
den Cluster, dass er durch Leber-Rezeptoren erkannt wird, um so
größer ist,
je größer die
Zahl von Galactose-Resten in dem Cluster ist.
-
Eine
erhöhte
Galactose-Cluster-Größe kann
das Binden des Annexins an ein Target-Molekül stören. Wenn eine signifikante
Störung
der Annexin-Bindung an ein Ziel-Molekül beobachtet wird (eine Verringerung auf < 20 % des nativen
Annexin-Bindungs-Vermögens), kann
ein engeres Linker-Molekül
zwischen den beiden Einheiten verwendet werden, oder es sollten
derart große
Cluster nicht in radiomarkierten Annexin-Galactose-Cluster-Konjugaten
gemäß der vorliegenden
Erfindung verwendet werden.
-
Im
Hinblick auf das obige Kriterium Nr. 2 sind die Galactose-Rezeptoren
innerhalb jedes Trimers voneinander um Entfernungen von 15, 22 und
25 Angström
entfernt. Folglich kommt es im Rahmen der vorliegenden Erfindung
in Betracht, dass die Galactose-Reste innerhalb eines Clusters vorzugsweise
durch flexible Linker- Molekül voneinander
getrennt sind, was die Trennung um wenigstens 25 Angström erlaubt.
Der Abstand zwischen Galactose-Resten ist wahrscheinlicherweise
wichtiger, wenn die Zahl von Galactose-Resten klein ist. Bei größeren Konstrukten
tritt eher ein passendes Abstandhalten in Bezug auf Galactose-Reste
ein, die nicht unmittelbare Nachbarn sind (d.h. Zucker-Reste, die
weiter voneinander entfernt sind als diejenigen, die unmittelbare
Nachbarn sind). Wenn eine mittlere Bindungslänge von 1,5 Angström angenommen
wird, sind bevorzugte Galactose-Cluster gemäß der vorliegenden Erfindung
gekennzeichnet durch eine Trennung von benachbarten Galactose-Resten
von etwa 10 Bindungslängen
oder mehr. Andere bevorzugte Konstrukte schließen Galactose-Cluster ein,
die gekennzeichnet sind durch eine Trennung benachbarter Zucker-Reste
um etwa 25 Bindungslängen
oder mehr.
-
Betreffend
Kriterium Nr. 3 sollte die Entfernung zwischen dem Annexin und dem
Galactose-Cluster ausreichend sein, um irgendwelche nachteiligen
sterischen Effekte auf eine Bindung von Annexin an ein Ziel-Molekül zu vermeiden,
die durch die Größe oder
Orientierung des Galactose-Clusters hervorgerufen werden. Diese
Entfernung ist vorzugsweise größer als
etwa 7 Bindungslängen
oder etwa 10 Angström.
Sofern nötig,
wird ein Verlängerungs-Molekül zwischen
dem Galactose-Cluster und dem Linker eingearbeitet (der den Galactose-Cluster
und die Annexin-Komponente verbindet), oder zwischen dem Annexin
und dem Linker, um die erforderliche Entfernung einzustellen.
-
Zwar
scheinen die vorstehend erwähnten
Parameter optimal für
Galactose zu sein; es sollte jedoch angemerkt werden, dass diese
Faktoren bei anderen Hexosen oder Mischungen daraus schwanken können, die
sich an dieselben Rezeptoren binden können oder nicht oder die sich
in unterschiedlicher Weise binden können. Unterstellt man die Lehren
dieser Anmeldung als gegeben, kann ein Fachmann mit üblichen
Sachverstand in diesem technischen Bereich unter Verwendung verfügbarer Synthese-Verfahren ein Annexin
und ein aktives Mittel an andere Hexose-Cluster binden und kann
solche Konstrukte identifizieren, die optimale Leistungseigenschaften
liefern.
-
Beliebige
verzweigte Zucker-Strukturen, die die Kriterien erfüllen, die
oben beschrieben wurden, können
in der praktischen Durchführung
der vorliegenden Erfindung verwendet werden. Bevorzugte Galactose-Cluster
gemäß der vorliegenden
Erfindung weisen die folgenden Strukturen auf:
worin
X vorzugsweise H oder Methyl ist, was zu Galactose-Clustern führt, die
4, 8, 16 und 32 Galactose-Reste enthalten. Eine weitere Wiederholung
des Verzweigungs-Schemas
ermöglicht
eine Verlängerung
des Galactose-Clusters dahingehend, dass dieser 32, 64 usw. Galactose-Reste
einschließt.
Darüber
hinaus kann die Linker-Einheit zwischen den Hexose-Resten selbst
und der Verzweigungs-Struktur (gezeigt als -S-(CH
2)
4-NX-) hinsichtlich ihrer Länge variabel
sein.
-
Alternative
Verzweigungs-Strukturen können
auch im Aufbau der Galactose-Cluster
in Übereinstimmung
mit der vorliegenden Erfindung verwendet werden. Beispielsweise
können
andere Konstrukte, bei denen das Verzweigen zu einer Verdoppelung
der Zahl von Galactose-Resten führt,
verwendet werden. Außerdem kommen
im Rahmen der vorliegenden Erfindung auch Konstrukte in Betracht,
bei denen ein Verzweigen zu einer Verdreifachung oder anderen passenden
Vervielfachungen der Zahl von Galactose-Resten führt.
-
Eine
weitere potentielle Verzweigungs-Konstruktion basiert auf dem Molekül Bishomotris: (HO-CH
2)
3-C-NH
2.
Das eine Sulfhydryl-Gruppe enthaltende Derivat dieses Moleküls kann
ebenfalls verwendet werden. In dieser Ausführungsform der Erfindung wird
jeder Arm des Bishomotris-Moleküls
verlängert
und mit einer Carbonsäure
terminiert: (HO
2C-(CH
2)
y-Z-(CH
2)
3-C-NH
2), worin Z
für S oder
O steht und y im Bereich von 1 bis etwa 10 liegt. Für diese
Ausführungsform
der vorliegenden Erfindung ist ein bevorzugter Galactose-Cluster
gekennzeichnet durch die folgenden Strukturen:
worin
X vorzugsweise H oder Methyl ist, y im Bereich von 1 bis etwa 10
liegt und Z für
O oder S steht. Die oben gezeigten Strukturen tragen 3, 9 bzw. 27
Galactose-Reste. Eine weitere Wiederholung des Verzweigens erlaubt
eine Erweiterung bei Einschluß von
81 usw. Galactose-Resten.
-
Auch
kann X eine Niederalkyl-Einheit (aufgebaut aus 2 bis 12 Kohlenstoffatomen)
sein, die von Methyl verschieden ist, wie beispielsweise Ethyl,
t-Butyl und dergleichen.
X kann auch eine Niederalkyl-Gruppe sein, die ein Heteroatom trägt wie beispielsweise
eine Niederalkyl-Säure,
ein Niederalkyl-Ester, ein Niederalkyl-Aldehyd, ein Niederalkyl-Keton oder
ein Niederalkyl-Ether.
-
Es
ist intendiert, dass an eine Hexose-Einheit konjugiertes Annexin
V durch die Leber schneller aus dem Blut beseitigt wird, was es
ermöglicht,
bald nach der Verabreichung eines radiomarkierten Mittels ein optimales
Verhältnis
Ziel zu Hintergrund zu erreichen. Beispielsweise kann ein Galactose-Cluster
wie beispielsweise ein N-Acetylgalactosamin verwendet werden, um
das oben beschriebene Ergebnis zu erreichen. Die Bindung des Clusters
erfolgt nahe dem N-terminalen Ende des modifizierten Annexins, um
nicht die Bindung der Annexin-Einheit an Thromben zu beeinträchtigen.
-
Wie
früher
angegeben, können
Annexin-Moleküle
durch Addition von etwa 2 bis etwa 6 terminalen Aminosäure-Resten
modifiziert werden, um die Konjugations-Reaktion zwischen dem Annexin-Molekül und der
Hexose-Einheit oder zwischen dem Annexin-Molekül und entweder einem Linker-Molekül oder einer
Hexose-Einheit zu erleichtern oder um die Bildung eines Annexin-Multimers
zu erleichtern. Beispielsweise können
terminale Aminosäure-Reste
addiert werden, um eine Sulfhydryl-Gruppe bereitzustellen oder um
eine Gruppe bereitzustellen, die für eine Derivatisierung an eine
Maleimid-Gruppe befähigt
ist, wobei derartige Sulfhydryl- und Maleimid-Gruppen für die Konjugations-Reaktion
verfügbar
sind. Diese Modifikation kann über
Verfahrensweisen der Protein-Chemie oder über eine Produktion eines passenden
Fusions-Proteins oder andere, hierfür nützliche Verfahrensweisen erfolgen.
-
In
einer Ausführungsform
der Erfindung wird ein rekombinantes Annexin-Molekül an dem N-terminalen Ende
durch Addition von Aminosäure-Resten
modifiziert, mit der Maßgabe,
dass wenigstens eine der Aminosäuren
eine zugängliche
Sulfhydryl-Gruppe liefert. Beispielsweise kann dies bewirkt werden
mit der Addition wenigstens eines einzelnen Cystein-Rests nahe dem
N-terminalen Ende. Ein bevorzugtes modifiziertes Annexin, wie es
in der vorliegenden Beschreibung und in den Patentansprüchen definiert
ist, ist eine Monomer-Form von Annexin V mit einer N-terminalen Verlängerung
der bevorzugten Aminosäure-Sequenz
aus Ala-Cys-Asp-His-Ser-Met.
Ein Vorteil dieser Konfiguration ist, dass mit dem Cystein-Rest
nahe dem N-terminalen
Ende die Sulfhydryl-Gruppe und die benachbarten Amid-Gruppen für die Chelatisierung
des Technetiums sorgen, was zu einer N3S-artigen
stabilen chelatiserenden Verbindung führt, und zwar für ein direktes
Markieren. Es ist im Rahmen der vorliegenden Erfindung beabsichtigt,
dass die benachbarten Aminosäure-Donor-Atome von Aminosäure-Einheiten
sich um die Basis der Metall-Oxo-Bindung in einer quadratisch pyramidalen
Anordnung winden, wodurch Stabilität erreicht wird, und zwar ähnlich einer
stabilen Chelatisierung, wie sie durch Verwendung einer N3S-Chelat-Verbindung
erreicht wird.
-
In
einer anderen Ausführungsform
der Erfindung wird ein modifiziertes Annexin-Molekül produziert, das
einen einzelnen Cystein-Rest als zweiten Rest in einer N-terminalen
Verlängerung
aus sechs Aminosäuren
enthält.
In dieser Ausführungsform
wurde der einzelne natürliche
innere Cystein-Rest zu einer keinen Schwefel enthaltenden Aminosäure mutiert,
wie beispielsweise Alanin oder Serin. Ein Verfahren zum Mutieren des
Cystein-Rests wird in Beispiel VIII geliefert. Diese Aminosäure-Verlängerung
liefert ein hochgradig flexibles Mittel zum Binden funktioneller
Einheiten an den N-terminalen Bereich eines Annexins wie beispielsweise von
Annexin V. Die Verfahrensweisen, die in der vorliegenden Beschreibung
beschrieben werden, produzieren erfolgreich monomere und dimere
modifizierte Annexin V-Moleküle.
Es ist offensichtlich, dass die Proteine im wesentlichen rein sind
und das erwartete Molekulargewicht aufweisen. Darüber hinaus
kann irgendeine Produktion eines modifizierten Annexin V-Dimers
quantitativ in das Monomer überführt werden,
und zwar durch Behandlung mit 2-Mercaptoethanol, was das Vorhandensein
der erwarteten Disulfid-Bindung bestätigt.
-
Es
wurden Messungen der Membran-Bindungseigenschaften von modifiziertem
Annexin und dem modifizierten Annexin-Dimer in einem Konkurrenz-Assay
gegen natives monomeres Annexin V durchgeführt. Das Dimer ist 6-fach potenter
als monomeres Annexin V in der Verdrängung von markiertem Ligand
von der Membran. Pharmacokinetische Untersuchungen, die in der vorliegenden
Beschreibung offenbart sind, haben gezeigt, dass das erhöhte Molekulargewicht
des modifizierten Annexin-Dimers
die Geschwindigkeit seines Verschwindens aus dem Blut nicht verlangsamt.
Tatsächlich
wird das modifizierte Annexin V-Dimer aus dem Blut mit ungefähr derselben
Geschwindigkeit wie monomeres Annexin V des Wild-Typs entfernt.
Siehe dazu die nachfolgende Tabelle 1.
-
Tabelle
1 Pharmacokinetische
Daten von mit
125I jodiertem modifiziertem
Annexin V-Dimer und Annexin V in Mäusen
-
Die
Offenbarung der vorliegenden Erfindung zeigt, dass das dimere Molekül von Annexin
V wahrscheinlich eine höhere
Affinität
zu der Blutplättchen-Membran
aufweist, und zwar höchstwahrscheinlich
aufgrund einer langsameren Dissoziationsgeschwindigkeit. Damit kann
dieses Molekül
in höherer
Konzentration aufgenommen oder länger
in dem Thrombus in vivo gehalten werden, wodurch das Verhältnis Target-zu-Hintergrund
zu jedem gegebenen Zeitpunkt verbessert wird.
-
Radionuklide,
die nützlich
im Rahmen der vorliegenden Erfindung sind, schließen penetrierende
Photonen-Emitter einschließlich
gamma-Emitter und Röntgenstrahl-Emitter
ein. Diese Strahlen begleiten eine nukleare Transformation wie beispielsweise
das Einfangen von Elektronen, eine beta-Emission und einen isomeren Übergang.
Radionuklide, die nützlich
sind schließen
solche mit Photonen zwischen 80 und 400 keV und Prositronen-Produzenten,
511 keV-Zerstrahlungs-Photonen und annehmbare Strahlungsdosis aufgrund
von absorbierten Photonenteilchen und deren Halbwertszeit ein. Radionuklide,
die geeignet zur Verwendung im Rahmen der vorliegenden Erfindung
sind, sind in diesem technischen Bereich bekannt und schließen ein: 18F, 69Cu, 186Re, 188Re, 100Pd, 212Bi, 212Pb 109Pd 67CU, 67Ca, 68Ga, 99mTc, 94Tc, 95Ru, 105Ru, 99Rh, 105Rh, 111In, 123I, 125I, 153Sm, 177Lu, 170Lu, 189Pt, 193Pt, 199Au, 197Hg und dergleichen.
-
Tc-99m
ist ein bevorzugtes Radionuklid für die praktische Durchführung der
vorliegenden Erfindung. Tc-99m wurde stabil an Annexin V in Übereinstimmung
mit der vorliegenden Erfindung sowohl bei niedriger als auch bei
hoher spezifischer Aktivität
gebunden (0,53 μCi/μg – 1,012 μCi/μg). Adäquate radiochemische
Ausbeuten und gute radiochemische Reinheiten wurden erhalten. Aktivierte
Blutplättchen-Bindungs-Untersuchungen
wurden ebenfalls durchgeführt
und die radiomarkierten Annexin V-Konjugate banden sich gut an aktivierte Blutplättchen.
-
In
einer anderen Ausführungsform
der vorliegenden Erfindung kann eine NXSy-Chelat-Verbindung
an ein Annexin-Hexose-Konjugat, ein Annexin-Multimer oder ein modifiziertes
Annexin gebunden werden, um ein Radiomarkieren zu erleichtern. NxSy-Chelat-Verbindungen
schließen
chelatisierende Verbindungen ein, die in der Lage sind, sich koordinativ
an ein Metall oder Radio-Metall zu binden und sich kovalent an ein
Annexin-Molekül
zu binden. Bevorzugte NxSy-Chelat-Verbindungen
haben die N2S2-Kerne (allgemein
beschrieben in dem US-Patent Nr. 4,897,225 oder dem US-Patent Nr.
5,164,176 oder dem US-Patent Nr. 5,120,526), den N3S-Kern
(allgemein beschrieben in dem US-Patent Nr. 4,965,392), den N2S3-Kern (allgemein
beschrieben in dem US-Patent
Nr. 4,988,496), den N2S4-Kern
(allgemein beschrieben in dem US-Patent Nr. 4,988,496), den N3S3-Kern (allgemein
beschrieben in dem US-Patent Nr. 5,075,099) und den N4-Kern
(allgemein beschrieben in dem US-Patent Nr. 4,963,688 und dem US-Patent Nr. 5,227,474).
Besonders bevorzugte NxSy-Chelat-Verbindungen
haben den N2S2-Kern
und den N3S-Kern. N2S2- und N3S-Chelat-Verbindungen
sind in diesem technischen Bereich bekannt. Beispielsweise sind
bevorzugte N2S2-Verbindungen
beschrieben in dem US-Patent Nr. 4,897,225, und bevorzugte N3S-Chelat-Verbindungen
sind allgemein beschrieben in dem US-Patent Nr. 4,965,392 und in
dem US-Patent Nr. 5,112, 953.
-
Die
N2S2-Chelat-Verbindungen
sind bifunktionelle Diamid-Chelatoren und bifunktionelle Dimercaptid-Chelatoren
der NxSy-Familie,
die in der Lage sind, stabil ein Radionuklid über zwei Stickstoff-Atome und zwei
Schwefel-Atome zu komplexieren, die in passender Weise angeordnet
sind. N2S2-Chelat-Verbindungen sind
allgemein beschrieben in dem US-Patent Nr. 4,897,225.
-
Bevorzugte
Chelat-Verbindungen mit einem N
2S
2-Kern schließen auch Diamindimercaptoamid-Chelat-Verbindungen
ein, die das folgende Biphenyl-Grundgerüst aufweisen und die allgemein
beschrieben sind in der US-Patentanmeldung mit dem amtlichen Aktenzeichen
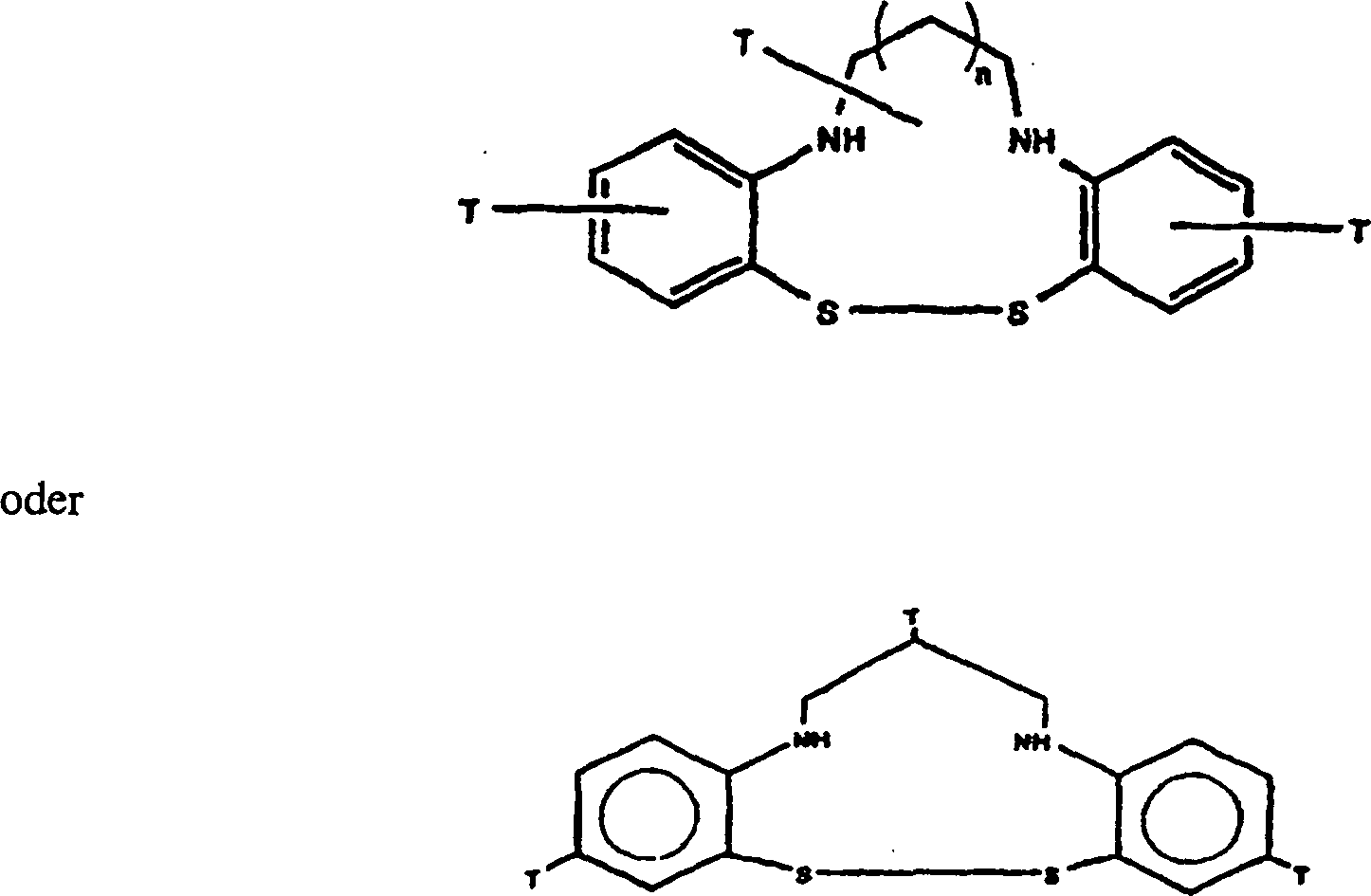
Nr. 08/463,232:
worin
n = 0 bis 1 ist und ein oder mehrere T-Substituenten eine funktionelle
Gruppe einschließen,
die verfügbar und
geeignet für
eine Konjugation mit einer anderen Konjugat-Komponente ist. Solche
beispielhaften funktionellen Gruppen schließen hydrophile Gruppen ein,
die nützlich
für eine
Ausscheidung über
die Niere sind, im Gegensatz zu einer hepatobiliären Ausscheidung.
-
Die
N3S-Chelat-Verbindungen sind bifunktionelle
Triamid-mercaptid-Chelatoren
der NxSy-Familie,
die in der Lage sind, ein Radionuklid stabil durch drei Stickstoff-Atome
und ein Schwefel-Atom zu komplexieren, die in passender Weise positioniert
sind. Bevorzugte N35-Chelat-Verbindungen
sind beschrieben in dem US-Patent
Nr. 4,965,392 und dem US-Patent Nr. 5,091,514.
-
Die
vorliegende Erfindung betrifft diese stabile Chelat-Technologie
zur Ausnutzung des Thrombus-Auffinde-Vermögens von Annexin-Molekülen. Dadurch
wird ein Bildgebungs-Mittel bereitgestellt, das in der Lage ist,
vaskuläre
Thromben in vivo schnell zu visualisieren. Die radiomarkierten Annexine,
einschließlich
radiomarkierter Annexin-Galactose-Konjugate und radiomarkierter
Annexin-Galactose-Cluster-Konjugate
gemäß der vorliegenden
Erfindung können
dazu verwendet werden, Thrombus-Bilder zu erhalten, die den hohen
Wert der Hintergrund-Aktivität
reduzieren oder eliminieren, der von metabolisch abgebautem radiomarkiertem
Konjugat resultiert. Die radiomarkierten Annexine einschließlich radiomarkierter
Annexin-Galactose-Konjugate und
Radiomarkierter Annexin-Galactose-Cluster-Konjugate vermeiden auch
eine klinisch nicht annehmbare Toxizität gegenüber Nicht-Zielzellen-Stellen.
Mit Tc-99m radiomarkiertes
Annexin V zeigte bessere Ergebnisse als I-123 in Untersuchungen
an Schweinen, wie im nachfolgenden Beispiel III beschrieben wird.
-
Die
Radiomarkierung von Annexin V mit einem Radionuklid unter Verwendung
einer N2S2-Chelat-Verbindung
oder N3S3-Chelat-Verbindung
kann durchgeführt
werden unter Verwendung entweder eines vorher gebildeten oder eines
nachher gebildeten Ansatzes. Mit anderen Worten: Das Radionuklid
wird entweder innerhalb der Chelat-Verbindung vor (vorgebildet) oder im
Anschluß an
(nachgebildet) die Konjugation der Chelat-Verbindung an Annexin
V komplexiert. Der vorgeformte Ansatz ist bevorzugt, und geeignete
Verfahrensweisen dafür
sind in den Beispielen I, II, IV und XV beschrieben. Weitere Untersuchungen
zeigten an, dass der Vorform-Radiomarkierungs-Ansatz zu zwei radiometrischen Peaks
bei der HPLC-Analyse führte.
Das Auftreten von zwei Peaks resultiert aus Cystein-konjugiertem
Annexin V sowie aus Lysinkonjugiertem Annexin V. So kann die Cystein-Aminosäure von
Annexin V entweder gestrichen oder durch eine andere, keinen Schwefel enthaltende
Amiosäure
ersetzt werden und führt
so zu einem monomer markierten Annexin V; siehe dazu Beispiel IX.
Die Verwendung des Vorform-Ansatzes zum Radiomarkieren des modifizierten
Annexin V führt
auch zu einem monomer markierten Annexin V. Die Konjugation kann
auftreten über
die zugängliche
Sulfhydryl-Gruppe eines modifizierten Annexins dazu über den
Lysin-Rest des modifizierten Annexins. Auch kann eine Hexose-Einheit- Konjugation an das
Annexin über
chemische Verfahrensweisen entweder vor der Konjugation (nachgeformter
Ansatz) oder im Anschluß an
eine Konjugation (vorgeformter Ansatz) einer Chelat-Verbindung an
das Annexin erfolgen. Eine chemische Konjugation einer Hexose-Einheit
wird vorzugsweise vor der Konjugation der Chelat-Verbindung durchgeführt.
-
Für Ausführungsformen
der vorliegenden Erfindung, in denen es erwünscht ist, dass die chelatisierenden
Verbindungen eine schnelle Komplexierung von Radionuklid bei Raumtemperatur
zeigen, können
die folgenden Chelat-Verbindungen verwendet werden:
worin
n = 0 bis 1 ist und einer oder mehrere T-Substituenten eine funktionelle
Gruppe einschließen,
die für
eine Konjugation mit einer anderen Konjugat-Komponente verfügbar und
geeignet ist. In dem oben angegebenen Beispiel ist eine funktionelle
Gruppe wie beispielsweise ein Amin in der Lage, mit der Lysin-Carboxyl-Einheit oder
einem aktivierten Ester-Derivat davon zu reagieren. Alternativ kann
eine einen aktiven Ester tragende Chelat-Verbindung an eine funktionelle
Amino-Gruppe konjugiert werden, um ein Beispiel zu nennen.
-
In
einer weiteren Ausführungsform
der vorliegenden Erfindung kann auch eine Esterase-empfindliche Chelat-Verbindung
verwendet werden. Solche Ausführungsformen
schließen
Annexin-Chelat-Verbindungs-Konjugate ein, die modifizierte Annexin-Chelat-Verbindungs-Konjugate
einschließen,
sowie Multimer-Chelat-Verbindungs-Konjugate.
Eine Hexose-Einheit kann auch mit den oben angegebenen Ausführungsformen
der vorliegenden Erfindung verwendet werden.
-
Ein
Vorteil bei der Verwendung einer Esterase-empfindlichen Chelat-Verbindung ist, dass
die Verbindung während
des Metabolismus des radiomarkierten Proteins in der Leber gespalten
wird. Die Freisetzung der Verbindung erzeugt mit Radioaktivität assoziierte
Cataboliten mit vorteilhafter Distribution, was zur Verringerung
der Hintergrund-Radioaktivität
in der Nähe
des Herzens und der Lungen führt.
So ist eine Verwendung dieses Typs von Chelat-Verbindung die Bildgebung
des Herzens und der Lungen. Weiter ist ein anderes wünschenswertes
Merkmal dieser Art von Chelat, dass ein größerer Bruchteil des Technetium-99m
schnell durch die Leber freigesetzt wird und dann über die
Nieren ausgeschieden wird.
-
Mit
anderen Worten: Die Ester-Komponente dieser Chelat-Verbindung wird
während
des Metabolismus des markierten Proteins in der Leber gespalten,
was zu einer größeren Freisetzung
von Radioaktivität
aus der Leber in das Blut und eine Ausscheidung über die Nieren führt. Eine
bevorzugte Esterase-empfindliche Chelat-Verbindung ist eine solche, die ein
N3S-Seryl-Succinat enthält, wie sie beschrieben ist
in dem US-Patent Nr. 5,112,953.
-
Beispielsweise
kann eine Esterase-empfindliche Chelat-Verbindung der folgenden
Struktur mit Annexin verwendet werden, um die oben angegebenen Ergebnisse
zu erhalten:
worin R irgendeine Säure-labile
oder Basen-labile Schwefel-Schutzgruppe ist, wie sie im Stand der
Technik bekannt ist, beispielsweise eine Ethoxyethyl-Gruppe, und
R
1 ein aktivierter Ester ist, der N-Hydroxysuccimidat-tetrafluorphenyl
und Tetrafluorphenyl- und
Tetrafluorthiophenyl-Ester einschließt.
-
Ein
Fachmann mit üblichem
Sachverstand in diesem technischen Bereich ist mit der Natur und
Spezifität
funktioneller Einheiten vertraut, und die Natur und Spezifität werden
auch in der vorliegenden Beschreibung diskutiert.
-
In
Ausführungsformen
der vorliegenden Erfindung, in denen die Chelat-Verbindung gekennzeichnet ist durch
eine einzelne funktionelle Gruppe, die für die Konjugation verfügbar und
geeignet ist, werden das Annexin, die Chelat-Verbindung und Galactose-Cluster-Komponenten
vorzugsweise über
ein trifunktionelles Linker-Molekül verbunden.
Funktionelle Gruppen, die für
eine Konjugation verfügbar
sind, sind solche, die nicht durch sterische Hinderungen vor einer
Konjugation bewahrt werden. Funktionelle Gruppen, die für eine Konjugation
geeignet sind, sind solche Gruppen, die im chemischen Sinn in der
Lage sind, mit verfügbaren
funktionellen Gruppen zu reagieren, die mit anderen Konjugat-Komponenten
assoziiert sind. Darüber
hinaus stört eine
Konjugation geeigneter funktioneller Gruppen nicht wesentlich eine
notwendige Funktion der Komponente, mit der die funktionelle Gruppe
assoziiert ist. Beispielsweise ist eine funktionelle Gruppe, die
in der die Komplementarität
bestimmenden Region einer Antikörper-Ziel-Einheit
angeordnet ist, beispielsweise allgemein nicht „geeignet" für
eine Konjugation, da das Ziel-Bindungs-Vermögen des Antikörpers durch
eine solche Verbindung mit hoher Wahrscheinlichkeit wesentlich beeinträchtig wird.
in gleicher Weise ist eine funktionelle Gruppe, die innerhalb des Bindungsbereichs
des Annexins angeordnet ist, allgemein nicht „geeignet" für
eine Konjugation.
-
Nützliche
tifunktionelle Linker sind zugänglich
für ein
Binden mit funktionellen Gruppen, die auf den drei Konjugat-Komponenten
verfügbar
sind oder auf irgendwelchen Verlängerungs-Einheiten,
die beim Konjugat-Aufbau verwendet werden. Ein nützlicher trifunktioneller Linker
ist Lysin, in dem die funktionelle alpha-Aminogruppe, die funktionelle epsilon-Aminogruppe
und die funktionelle Carboxyl-Gruppe
verwendet werden. Ein Fachmann mit üblichem Sachverstand in diesem
Bereich könnte
andere tifunktionelle Linker identifizieren und diese in der praktischen
Durchführung
der vorliegenden Erfindung verwenden, wie dies weiter unten beschrieben
ist.
-
Abspaltbare
Linker können
im Rahmen der vorliegenden Erfindung verwendet werden. Solche Linker sind
bifunktionelle Einheiten, die in der Lage sind, sich mit dem trifunktionellen
Linker in der Weise zu verbinden, dass sie die chelatisierende Verbindung
mit dem trifunktionellen Linker verbinden, der seinerseits mit der Annexin-Komponente und der
Cluster-Komponente verbunden wird. Beispiele geeigneter abspaltbarer
Linker schließen
ein: Monosaccharide, Polysaccharide, Polyaminosäuren, Hydroxyalkylacrylamide
(z.B. HPMA), hydrophile Polymere auf Polyethylenglycol-Basis, biologisch
abbaubare Polymere, die eine Ether- oder Ester-Bindung enthalten,
sowie Dextran und Hemisuccinylester und dergleichen.
-
Ebenfalls
sind Verlängerungs-Moleküle, die
Rahmen der vorliegenden Erfindung nützlich sind, bifunktionelle
Einheiten, die in der Lage sind, sich entweder mit der Annexin-Komponente
oder dem Linker oder der Galactose-Cluster-Komponente und dem Linker
zu verbinden. Geeignete Verlängerungs-Moleküle schließen Aminocaproat-Einheiten,
HS-(CH2)nOOOH oder
eine aktivierte Ester-Form davon, worin n im Bereich von 2 etwa
5 liegt, und dergleichen ein. Ein Fachmann mit üblichem Sachverstand in diesem
technischen Bereich ist in der Lage, andere geeignete Verlängerungs-Moleküle zu identifizieren
und zu verwenden, wie sie in der vorliegenden Erfindung und in den
Ansprüchen
beschrieben sind. Alternativ dazu kann ein passend aufgebauter Linker
der Funktion des Verlängerungs-Moleküls dienen.
-
Auch
können
im Rahmen der vorliegenden Erfindung eine Bindung erleichternde
Einheiten verwendet werden. Solche Einheiten sind bifunktionell
und erleichtern ein Binden der Konjugat-Komponenten, z.B. Galactose-Cluster,
Annexin, Chelat-Verbindung, Linker und Verlängerungs-Molekül. Beispiele
solcher eine Bindung erleichternde Einheiten schließen Moleküle mit funktionellen
Harnstoff-Gruppen,
Moleküle
mit funktionellen Thioharnstoff-Gruppen, Succinat-Brücken, Maleimide
und dergleichen ein. Derartige eine Bindung erleichternde Einheiten
sind einer Identifikation und Verwendung durch Fachleute mit üblichem
Sachverstand in diesem technischen Bereich zugänglich.
-
Ein
Beispiel eines Linker-Verlängerungs-Moleküls zum Erleichtern
des Bindungs-Systems ist nachfolgend gezeigt: - CO-(CH2)5-NH-C(O
or S)-NH-CH-(CH2)4-NH-CO-(CH2)2-CO-(COOH) worin die alpha-Amingruppe
des Lysin-Linkers über
eine funktionelle Harnsstoff- oder Thioharnstoff-Gruppe an einen
Amino-Caproat-Spacer gebunden ist (der seinerseits an einen Galactose-Cluster
gebunden ist, der nicht gezeigt ist); die Lysin-Carboxylat-Gruppe für eine Bindung
zu einer Chelat-Verbindung (nicht gezeigt) verfügbar ist; und die epsilon-Aminogruppe
des Lysin-Linkers für
eine Bindung zu einem Lysin-Rest der Annexin-Komponente (nicht gezeigt) über eine
Succinat-Brücke
verfügbar
ist. Andere Aminosäure-Reste
der Annexin-Komponente wie beispielsweise Cystein können auch
für Bindungszwecke
verwendet werden. Alternativ dazu kann eine Maleimid-S(CH2)nCO-Bindungserleichterungs-Einheit-Verlängerungsmolekül-Kombination verwendet
werden, um den Zucker-Rest mit dem Lysin zu verbinden. Ein Beispiel
eines durch einen trifunktionellen Linker verbundenen Konjugats
wird in Beispiel VII diskutiert.
-
Wenn
die Chelat-Verbindungs-Komponente des Konjugats durch mehr als eine
funktionelle Gruppe gekennzeichnet ist, die für eine Konjugation verfügbar und
geeignet ist, kann der Galactose-Cluster mit der Chelat-Verbindungs-Komponente
verbunden werden, welche ihrerseits mit der Annexin-Komponente des Konjugats über zwei
oder mehr bifunktionelle Linker verbunden wird. Alternativ dazu
wird der Hexose-Cluster wie
beispielsweise N-Acetylgalactosamin mit der zugänglichen Sulfhydryl-Gruppe des modifizierten
Annexins verbunden, und die Chelat-Verbindungs-Komponente wird mit der Annexin-Komponente über den
Lysin-Rest an dem Annexin verbunden. In einer anderen möglichen
Konfiguration wird die Chelat-Verbindung mit einem Hexose-Cluster über einen
trifunktionellen Linker verbunden, worin der Hexose-Cluster, der an die
Chelat-Verbindung konjugiert ist, dann mit der zugänglichen
Sulfhydryl-Gruppe des modifizierten Annexins durch eine die Bindung
erleichternde Einheit gebunden wird. Die oben angegebene mögliche Konfiguration
kann auch einen abspaltbaren Linker zwischen der Chelat-Verbindung
und dem Linker einschließen,
der den Cluster an das Annexin bindet. Vorzugsweise wird die Annexin-Komponente
des Konjugats zuletzt in die Bildung eines Galactose-Cluster-Chelat-Verbindung-Annexin-Konjugats eingebunden.
Geeignete bifunktionelle Linker und Einbinde-Verfahrensweisen können von Fachleuten mit üblichem
Sachverstand in diesem technischen Bereich identifiziert und eingesetzt
werden. Ein Beispiel einer derartigen chelatisierenden Verbindung
und einer derartigen Konjugations-Verfahrensweise wird in Beispiel
V beschrieben.
-
Die
folgenden Variationen sind Beispiele der Reihenfolge, in der die
Komponenten (speziell das modifizierte Annexin, die Hexose-Einheit
und die chelatisierende Verbindung) aneinander konjugiert werden,
die im Rahmen der vorliegenden Erfindung in Betracht kommen. Es
ist für
einen Fachmann mit üblichem
Sachverstand in diesem technischen Bereich offenbar, dass in den
Fällen,
in denen ein Konjugat der vorliegenden Erfindung drei Komponenten
aufweist (z.B. ein Annexin, eine Hexose-Einheit und eine chelatisierende
Verbindung), eine Komponente an eine andere Komponente in einer
Vielzahl von Orientierungen gebunden werden kann. Beispielsweise
können
die Hexose-Einheit und eine chelatisierende Verbindung auf unterschiedlichen Seiten
an einem Annexin gebunden werden oder können aneinander gebunden werden,
und nur eines der Moleküle
wird direkt an das Annexin gebunden:
-
Die
Hexose-Einheit ist an das modifizierte Annexin über eine zugängliche
Sulfhydryl-Gruppe
oder mehrere derartige Gruppen gebunden, und die Chelat-Verbindung
ist an das modifizierte Annexin über
einen Lysin-Rest an dem modifizierten Annexin konjugiert; eine Chelat-Verbindung
ist an das modifizierte Annexin über
eine zugängliche
Sulfhydryl-Gruppe oder mehrere derartige Gruppen konjugiert und
die Hexose-Einheit ist an das modifizierte Annexin über einen
Lysin-Rest an dem modifizierten Annexin konjugiert; eine Chelat-Verbindung
ist an die Hexose-Einheit konjugiert, die ihrerseits an das modifizierte
Annexin über
eine zugängliche
Sulfhydryl-Gruppe
oder mehrere derartige Gruppen konjugiert ist; die Hexose-Einheit
ist an die Chelat-Verbindung konjugiert, die ihrerseits an das modifizierte
Annexin über
zugängliche
Sulfhydryl-Gruppen konjugiert ist; die Chelat-Verbindung ist an
die Hexose-Einheit konjugiert, die ihrerseits an das modifizierte
Annexin über
die Lysin-Reste
des modifizierten Annexins konjugiert ist, so dass sie die zugänglichen
Sulfhydryl-Gruppen für
eine weitere Konjugation verfügbar
läßt; die
Hexose-Einheit ist an die Chelat-Verbindung konjugiert, die ihrerseits
an das modifizierte Annexin über
Lysin-Reste konjugiert ist, wobei sie die zugänglichen Sulfhydryl-Gruppen
wieder verfügbar
für eine
weitere Konjugation läßt, und
andere ähnliche
Variationen.
-
An
die Hexose-Einheit und die Chelat-Verbindung über chemische Verfahren konjugiertes
Annexin kann entweder vor einer Komplexierung (dies ist der nachträglich gebildete
Ansatz) oder im Anschluß an
eine Komplexierung (dies ist der vorgeformte Ansatz) eines Radio-Metalls
mit der Chelat-Verbindung auftreten. In einer bevorzugten Ausführungsform
wird eine Hexose-Einheit an die Annexin-Komponente vor der Zugabe
der Radiomarkierung gebunden. Die chemische Konjugation wird vorzugsweise
durchgeführt
im Anschluß an
die Radio-Metall-Komplexierung, wenn nicht die Chelat-Verbindung, die in
dem Konjugat eingesetzt wird, in der Lage ist, das Radionuklid mit
schneller Kinetik bei Raumtemperatur zu binden. Annexin V wird mit
einem bildgebenden Radionuklid für
eine Verwendung im Rahmen der vorliegenden Erfindung radiomarkiert.
-
Radiomarkierte
Annexine, einschließlich
radiomarkierter Annexin-Galactose-Konjugate und radiomarkierter Annexin-Galactose-Cluster-Konjugate,
gemäß der vorliegenden
Erfindung bieten einen zusätzlichen Vorteil
gegenüber
früher
hergestellten, mit I-123 markierten Annexinen, und zwar dahingehend,
dass sie dafür zugänglich sind,
in Form eines kalten Kits abgepackt zu werden. Das bedeutet, dass
das Annexin oder Annexin-Galactose oder Annexin-Galactose-Cluster
oder Galactose-Cluster
und Chelat-Verbindungs-Komponenten einzeln in Röhrchen abgefüllt und
getrennt von der Tc-99m-Komponente zur Verfügung gestellt werden können (und
möglicherweise
auch getrennt voneinander in Röhrchen
abgefüllt
werden können).
-
Eine
Lyophilisierung und Abfüllung
der Konjugat-Komponenten in steriler, pyrogenfreier Umgebung kann
nach Verfahrensweisen bewirkt werden, die Fachleuten im Bereich
der Technik einer guten Herstellungspraxis bekannt sind, insbesondere
da solche Praktiken biologische Materialien betreffen.
-
Wenn
ein Patient, der ein Thrombus-Bild benötigt, identifiziert ist, kann
ein kaltes Kit geordert oder aus dem Lager geholt werden. Tc-99m
kann aus einer Radio-Apotheke
oder einer anderen Quelle dafür
erhalten werden. Der vorgebildete oder nachgebildete Prozeß der Chelatisierung/Komplexierung
wird durchgeführt. Das
radiomarkierte Annexin, das radiomarkierte Annexin-Galactose-Konjugat
oder das radiomarkierte Annexin-Galactose-Cluster-Konjugat wird
dem Patienten verabreicht, und der Patient wird anschließend einem
Bildgebungs-Verfahren unterzogen. Für radiomarkierte Annexin-Galactose-Konjugate
wird ein Annexin-Galactose-Konjugat vorzugsweise hergestellt und
in dem Kit in ein Röhrchen
abgefüllt.
Für radiomarkierte
Annexin-Galactose-Cluster-Konjugate wird ein Konjugat aus Chelat-Verbindung,
Annexin und Galactose-Cluster vorzugsweise hergestellt und in dem
Kit in Röhrchen
abgefüllt,
obwohl auch Annexin-Galactose-Cluster-Chelat-Verbindung- Zwei Komponenten-
oder andere Mehr-Komponenten-Kits verwendet werden können.
-
Radiomarkierte
Annexine einschließlich
radiomarkierter Annexin-Galactose-Konjugate oder radiomarkierte Annexin-Galactose-Cluster-Konjugate,
gemäß der vorliegenden
Erfindung werden in solchen Mengen verabreicht, dass man eine diagnostisch
wirksame Menge an Radionuklid an die Ziel-Stelle liefert. Passende verabreichte
Dosierungen hängen
von einer Vielzahl von Faktoren ab, die in weitem Umfang Patienten-spezifisch
sind, und Praktiker in diesem technischen Bereich werden solchen
Faktoren in der praktischen Anwendung der vorliegenden Erfindung
in Betracht ziehen. Die Komponenten des radiomarkierten Annexins
beeinflussen auch Dosis-Mengen
auf Wegen, die Praktikern in diesem technischen Bereich als feststellbar
bekannt sind oder routinemäßig feststellbar
sind. Im Allgemeinen wird radiomarkiertes Annexin einschließlich Radiomarkierten
Annexin-Galactose-Konjugats oder radiomarkierten Annexin-Galactose-Cluster-Konjugats
in großen
Säugern
in einer Dosis verabreicht, die zwischen etwa 0,3 und etwa 300 Microgramm/Kilogramm
Körpergewicht
des Empfängers
liegen, wobei etwa 3 bis etwa 10 Microgramm/Kilogramm Körpergewicht
bevorzugt sind, und zwar abhängig
von den physiologischen charakteristischen Eigenschaften des Patienten
und der involvierten oder vermuteten Krankheit.
-
Ein
Praktiker in diesem technischen Bereich ist in der Lage, eine passende
Dosis und einen passenden Verabreichungsweg für einen gegebenen Empfänger bei
einer gegebenen Krankheit zu identifizieren.
-
Radiomarkierte
Annexine einschließlich
radiomarkierter Annexin-Galactose-Konjugate oder radiomarkierter Annexin-Galactose-Cluster-Konjugate
gemäß der vorliegenden
Erfindung können
auf jede dafür
passende Weise verabreicht werden. Beispielsweise kann eine intravenöse Infusion
zur Verabreichung radiomarkierter Annexine angewendet werden. Andere
Verabreichungswege finden ebenfalls Gebrauch in der praktischen
Durchführung
der vorliegenden Erfindung. Beispielsweise sind zusätzliche
Verabreichungswege Injektion über
die Arterien (z.B. die Koronar-Arterie), ein intrakoronarer Weg,
ein intralymphatischer Weg, ein intrathecaler Weg, oder ein anderer
in einen Körper-Hohlraum
erfolgender Weg und dergleichen.
-
Nach
Verabreichung des Radionuklids und in Abhängigkeit von der Natur des
Radionuklids und dem Zweck der Verabreichung kann der Empfänger verschiedenen
Verfahrensschritten zum Nachweis radioaktiver Emissionen von der
Stelle oder den Stellen unterzogen werden, an denen sich das Radionuklid
lokalisiert. Beispielsweise werden Tc-99m enthaltende Konjugate
mit einer Gamma-Kamera bildmäßig erfaßt.
-
Die
Konjugate der vorliegenden Erfindung können in diagnostischen oder
therapeutischen Verfahren oder als Medikament oder zur Herstellung
eines Medikaments für
die Behandlung von z.B. einem vaskulären oder Herz-Zustand verwendet
werden.
-
Die
Erfindung wird weiter durch die Präsentation der folgenden Beispiele
beschrieben. Diese Beispiele werden zur Illustration der Erfindung
angeboten und nicht zu deren Beschränkung.
-
BEISPIEL I
-
Verfahren zum Radiomarkieren
eines Annexin-N2S2-Chelat-Konjugats
-
Annexin
V kann von einer Vielzahl von Gewebe-Extrakten isoliert werden,
beispielsweise von der Leber, der Lunge und der Plazenta, und zwar
in Übereinstimmung
mit Verfahrensweisen, wie sie beschrieben sind in der Druckschrift „Funakoshi
et al., Biochem. 26, 8087 – 8092
(1987)", der Druckschrift „Taft et
al., Biochem., 27, 6268 – 6276
(1988)" und dem
US-Patent Nr. 4,937,324, um einige Beispiele zu nennen. Darüber hinaus
kann Annexin V in E. coli exprimiert werden, wie dies beschrieben
wurde in der Druckschrift „Taft
et al., Archives of Biochemistry and Biophysics, 288, 141 – 144 (1991)".
-
Annexin
wurde mit Tc-99m unter Verwendung einer Diamid-dimercaptid-N2S2-Chelat-Verbindung
radiomarkiert, in Übereinstimmung
mit der Markierungs-Verfahrensweise
mit dem Onko-Trac® (auch bezeichnet als
VerlumaTM) Small Cell Lung Cancer Imaging
Kit, wie dies beschrieben ist in: "J. Nucleinsäure. Medizin., 32, 1445 – 1451 (1991)".
-
Ein
bevorzugtes Verfahren zum Radiomarkieren von Annexin V mit Tc-99m
stellt eine modifizierte Verfahrensweise mit dem Onko-Trac®-Kit
unter Verwendung von C-18-Baker-gereinigtem Tc-99m-N2S2-TFP dar. In dieser Verfahrensweise wurde
eine angesäuerte
Lösung
des aktiven Esters hergestellt durch Zusatz von 0,16 ml, 0,2 M Chlorwasserstoffsäure-Eisessig
(Verhältnis:
14 : 2) zu 0,6 ml 2,3,5,6-Tetrafluorphenyl-4,5-bis-(S-1-ethoxyethylmercaptoacetamido-)pentanoat
(0,3 mg; 0,0005 Mol frisch gelöst
in 0,9 ml Isopropylalkohol). Dann wurden 0,5 ml dieser Lösung zugesetzt
zu 1,1 ml Tc-99m-Gluconat (hergestellt aus 0,12 mg SnCl2 · 2 H2O, 5,0 mg Natriumgluconat bei pH 6,1 bis
6,3 und 100 mCi/ml [Tc-99m] Pertechnetat, d.h. der erste Schritt
in der Onko-Trac®-Kit-Markierungs-Verfahrensweise.
Die Reaktionsmischung wurde 15 Minuten lang auf 75°C erhitzt
und anschließend
auf Eis gekühlt.
Das resultierende Tc-99m-transchelatisierte
Tetrafluorphenyl-Aktivester-Derivat von Tc-99m-4,5-Bis-(thioacetamido-)pentanoat
wurde gegebenenfalls und vorzugsweise mit 2 ml Wasser verdünnt und
dadurch gereinigt, dass man die Reaktionsmischung auf eine konditionierte C-18-Kartusche
aufgab (J.T. Baker, Philipsberg, N.J.), mit 2,0 ml Wasser acht Mal
wusch, gefolgt von einem Schritt des Trocknens der Säule für 5 Minuten
und Eluieren mit 100 % Acetonitril. Das Lösungsmittel wurde unter einem
stetigen Strom von N2-Gas verdampft. Danach
wurden 0,15 ml Phosphat-gepufferte Kochsalz-Lösung (PBS), 0,15 ml Annexin
V in einer Konzentration von 2,35 mg/ml und 0,2 ml 0,2 M Bicarbonat
(pH 10,0) zur Konjugation an das Tc-99m-N2S2-Chelat zugesetzt. Nach 20 Minuten bei Raumtemperatur
wurde das Tc-99m-N2S2-Annexin
V-Konjugat gereinigt durch Durchlaufenlassen durch eine G-25-SEPHADEX®-(PD-10)-Säule (erhältlich von
der Firma Pharmacia, Piscataway, N.J.), die mit PBS equilibriert
worden war. Fraktionen (1,0 ml) wurden aufgefangen, und diejenigen
Fraktionen, die Annexin V enthielten, wurden gepoolt. Die Protein-Konzentration
wurde bestimmt durch UV-Absorption
bei 280 nm. Die Tc-99m-Annexin V-Konjugat-Lösung (300 – 350 μg) wurde verdünnt und
wurde zur Injektion in PBS gelagert, die Rinderserumalbumin (BSA)
in einer Endkonzentration von 15 bis 20 mg BSA/ml PBS enthielt.
-
BEISPIEL II
-
Verfahrensweise zum Radiomarkieren
eines Annexin-N3S-Chelat-Konjugats
-
S-Benzoylmercaptoacetylglycylglycylglycin
(S-Benzoyl-MAG3) wurde in Übereinstimmung
mit den Verfahrensweisen hergestellt, die in dem US-Patent Nr. 4,965,392
beschrieben sind. Danach wurden 25 μg S-Benzoylmercaptoacetylglycylglycylglycin
in 0,10 ml 1,0 M Carbonat-Puffer (pH 12) gelöst. Dann wurden 75 mCi Tc-99m-Pertechnetat
in etwa 1,0 ml zugesetzt, gefolgt von 1,0 mg frisch gelöstem Natriumdithionits
(10 mg/ml). Diese Mischung wurde auf 100°C ± 4 °C 3 Minuten lang erhitzt und
wurde dann in einem Eisbad 5 min abgekühlt und ergab so Tc-99m-MAG3, wie dies bestimmt wurde durch ITLC (CH3CN-Lösungsmittel),
Anionenaustausch-HPLC (Beckman AX, 10 Micron 0,01 M Na2SO4/0,01 M Na3PO4, pH 7,0) und Umkehrphasen-HPLC (Beckman
ODS, 5 Micron 2 % CH3CN/0,01 M Na3PO4, pH 7,0).
-
Der
Tc-99m-MAG3-Komplex in Carboxylat-Form wurde
dann verestert; 0,20 ml 1 N HCl, 0,30 ml 0,2 M Phopshat-Puffer (pH
6,0), 10,0 mg 2,3,5,6-Tetrafluorphenol (TFP) in 0,01 ml 90 % CH3CN und 12,5 mg EDC (1-(3-Dimethylaminopropyl-)3-ethylcarbodimid-hydrochlorid)
in 0,1 ml 90 % CH3CN wurden zusammengegeben,
und die Reaktanden wurden bei Raumtemperatur (25°C ± 2°C) 1 h lang gemischt. An dieser
Stelle erhielt man Tc-99m-MAG3-TFP-Ester,
wie bestimmt wurde durch ITLC (CH3CN als
Lösungsmittel),
Anionenaustausch-HPLC (Beckman AX, 10 Micron 0,01 M Na2SO4/0,01 M Na3PO4, pH 7,0) und Umkehrphasen-HPLC (Beckman
ODS, 5 Micron 34 % CH3CN/0,01 M Na3PO4, pH 7,0). Die
Präparation
wurde unter Verwendung einer C-18-Baker-Säule gereinigt. Die Reaktionsmischung
wurde gegebenenfalls und vorzugsweise mit 2 ml Wasser verdünnt und
auf die Säule
aufgegeben, zweimal mit Wasser gewaschen und danach achtmal mit
10 % C2H5OH/0,01
M Na3PO4, pH 7,0,
gewaschen. Das Produkt wurde mit CH3CN eluiert
und das Lösungsmittel wurde
vor der Konjugation mit Annexin V entfernt.
-
Die
Konjugation des aktiven Esters an Annexin V wurde durchgeführt durch
Zusatz von Annexin V in einem Phosphat-Puffer, pH 9,5, zu dem Tc-99m-MAG3-TFP-Ester.
Die Reaktion wurde für
wenigstens 30 Minuten durchgeführt,
und das gewünschte
radiomarkierte Annexin-Produkt wurde erhalten durch Durchlaufenlassen
durch eine PD-10-Gel-Filtrationssäule.
-
BEISPIEL III
-
Thrombus-Bildgebung
mit einem radiomarkierten Annexin
-
A. Vorbereitung der Tiere
- LA-Vaskular-Thromben
-
Auf
Fastendiät
gesetzte, 25 – 30
kg schwere Yorkshire-Schweine wurden sediert durch intramuskuläre Verabreichung
von Telazol (5 – 10
mg/kg) (im Handel erhältlich
von der Firma AVECO Co.) und mit im Handel erhältlichem Atropin (1 mg, Firma
Elkins-Sinn, Inc., Cherry Hills, N.J.). Surital (200 mg) (im Handel
erhältlich von
der Firma Abbott Laboratories) wurde intravenös zur Anästhesie verabreicht. Die Tiere
wurden intubiert und erhielten eine Inhalations-Anästhesie
von 1,5 – 2
% Halothan (im Handel erhältlich
von der Firma Abbott Laboratories, Abbott Park, IL) und O2 in ausreichender Menge, um einen Tiefenzustand
der Anästhesie
und physiologische Werte der arteriellen Blutgase zu erhalten. Eine
kontinuierliche elektrokardiographische Überwachung wurde unter Verwendung
von Alligator-Clip-Elektroden eingerichtet. Es wurde ein Einschnitt
im Nacken-Bereich durchgeführt,
und ein Acht-French-Katheter (Firma USCI Co., Billerica, MA) wurde
in der rechten gemeinen Carotis-Arterie zur Überwachung von Blutdruck und
Arterien-Blutgasen sowie zur Blutabnahme angebracht.
-
Die
Schweine wurden in eine rechtsseitige seitliche Decubitus-Position
gebracht, und es wurde eine laterale Thoracotomie durchgeführt, um
das Herz freizulegen. Der Einschnitt wurde mittels eines Thoracotomie-Retraktors
offengehalten. Das Pericardium wurde geöffnet, und der linke Vorhof-Anhang
wurde von dem linken Atrium mittels einer Vaskular-Quer-Klammer
isoliert. Mit Gummispitzen versehene Zangen wurden dazu verwendet,
um den Anhang vorsichtig zusammenzudrücken. Fünf Minuten später wurden
Ricinoleat (1 mg, Firma ICN Pharmaceuticals, Costa Mesa, CA) und
Thrombin (50 mg, Firma Johnson und Johnson Co., Arlington, TX) in
den linken Vorhof-Anhang (LAA; left artrial appendage) unter Verwendung
einer 27 Ga-Nadel injiziert. Die Quer-Klammer wurde 10 Minuten später entfernt.
-
Eine
Stunde nach der durch Zusammendrücken
bewirkten Verletzung wurde TC-99m-Annexin
V, das in Übereinstimmung
mit dem obigen Beispiel I hergestellt worden war, in Form einer
intravenösen
Bolus-Dosis in einer Ohr-Vene verabreicht. Die intravenöse Leitung
wurde dann mit Kochsalz-Lösung
gespült.
Bei 7 Tieren wurde mit I-125 markiertes Ovalbumin als nicht spezifisches
Kontrollmittel verabreicht, das herstellbar ist beispielsweise durch
die Verfahrensweise, die beschrieben wurde von „Fraker et al., Biochem. Biophys.
Res. Commun. 80: 849 – 857
(1978)". Kurz gesagt,
wurde mit I-125 radiomarkiertes Ovalbumin hergestellt durch das
Iodogen-Verfahren unter Verwendung von 600 μg Ovalbumin (Firma Sigma Chemical
Co., St. Louis, MO) und NaI-125 (2 mCi, 0,92 nMol).
-
Eine
Bildaufnahme wurde wie unten beschrieben durchgeführt. Am
Ende des experimentellen Vorgehens (allgemein nach etwa 150 Minute)
wurden die Tiere durch eine intravenöse Bolus-Dosis von 80 mEq KCl getötet, wobei
die Tiere nach wie vor unter allgemeiner Anästhesie lagen. Eine letzte
Blutprobe wurde für
die Zählrohr-Zählung abgenommen. Das Herz
wurde schnell herausoperiert, blutfrei gewaschen und in Proben für eine Zählrohr-Zählung verschnitten.
Weitere Proben der Carotis-Arterie, der Lunge, der Leber, der Milz,
der Muskeln und der Nieren wurden von einigen Tieren erhalten.
-
B. Kontroll-Experimente
-
Es
wurden 5 unterschiedliche Arten von Kontroll-Experimenten durchgeführt: Simulation
des offenen Brustkorbs, Simulation des geschlossenen Brustkorbs,
Ovalbumin, Indium-Blutplättchen
und nicht-spezifischer, mit Tc-99m markierter Antikörper.
-
- 1. Simulation des offenen Brustkorbs: In drei
Tieren wurde das Herz wie oben beschrieben freigelegt, jedoch wurde
der linke Vorhof nicht isoliert, zusammengedrückt oder mit einer Injektion
mit Ricinoleat/Thrombin versehen. Marker-Bilder mit einem Cobalt-Marker
wurden durchgeführt,
wie dies unten beschrieben ist, und das Tc-99m-Annexin V wurde 30 bis 60 Minuten nach
der Freilegung des linken Vorhof-Anhangs (LAA) injiziert. Die Bildaufnahme
und Probengewinnung waren identisch mit derjenigen, wie sie oben
im Teil A beschrieben wurden.
- 2. Simulation des geschlossenen Brustkorbs: In sieben Tieren
wurde eine intravenöse
Leitung ins Ohr eingerichtet. Es wurde keine Thoracotomie durchgeführt. Die
Sedierung und Anästhesie
waren identisch mit derjenigen, die oben im Teil A beschrieben wurde.
Das Tc-99m-Annexin V (mit bzw. ohne andere Kontroll-Radionuklide, wie
beispielsweise I-125-Ovalbumin oder In-111-Blutplättchen)
wurde verabreicht und die Bildgewinnung wurde durchgeführt.
- 3. Ovalbumin: I-125-Ovalbumin wurde in sieben Tieren als negatives
Kontroll-Protein verabreicht. Ovalbumin hat eine Molekulargröße, die ähnlich derjenigen
von Annexin ist, und zeigt eine geringfügig niedrigere Freisetzung
aus dem Blut.
- 4. Indium-Blutplättchen:
In-111-Blutplättchen-Markierung
wurde in sieben Tieren als positive Zählrohr-Zählungs-Markierung durchgeführt. Mit
In-111 radiomarkierte Blutplättchen
wurden in Übereinstimmung
mit der Verfahrensweise hergestellt, die beschrieben wurde von „Stratton
et al. in: Am. J. Cardiol. 47: 874 (1981)" und von „Stratton et al. in: Circulation,
69, 561 (1984)".
Eine Bildgewinnung wurde nicht versucht wegen der langen Serum-Halbwertszeit
der In-111-Blutplättchen.
- 5. Nicht-spezifischer Tc-markierter Antikörper. In einem einzelnen Experiment
wurde ein Thrombus im linken Atrium (LA) durch das oben beschriebene
Verfahren erzeugt, jedoch wurde Tc-99m-Annexin V nicht verabreicht.
Stattdessen wurde ein Tc-99m-Fab-Fragment
eines Antikörpers
verabreicht, der mit NR-LU-10 bezeichnet war. NR-LU-10 ist ein monoklonaler
IgG2b-Antikörper
mit einem Molekulargewicht von 150 kD, der ein etwa 40 kD großes Glycoprotein-Antigen
erkennt, das auf den meisten Carcinomen exprimiert wird. NR-LU-10
ist ein gut charakterisierter Pancarcinom-Antikörper, der sicher an über 565
Patienten in Human-Klinik-Versuchen verabreicht wurde. NR-LU-10-Fab wurde hergestellt
in Übereinstimmung
mit bekannten Verfahrensweisen und wurde in Übereinstimmung mit den Verfahrensweisen
radiomarkiert, die beschrieben sind in: „J. Nucl. Med., 32, 1445 – 1451 (1991)" und durch das modifizierte
C-18-Baker-Verfahren mit
gereinigtem Tc-99m-N2S2-TFP,
wie es beschrieben wurde in Beispiel I zur Herstellung von radiomarkiertem
Annexin V. Dieses Tc-99m-Fab-Konjugat
wurde als negativer Kontrollwert sowohl für die Zählrohr-Zählung als auch für die Bildgebung
bezeichnet.
-
C. Bildaufnahme
-
Ein
Cobalt-Marker wurde auf die freigelegte Oberfläche des linken Vorhof-Anhangs (LAA) aufgelegt und
mit einem Operationsband an Ort und Stelle gehalten, das an dem
Thoracotomie-Refraktor befestigt war. Das Band wurde so eingestellt,
dass sich der Marker allgemein mit dem LAA mit jedem Herzschlag-Zyklus
bewegte. Marker-Bilder wurden 10 Sekunden in jeder planaren Schicht
und 10 Sekunden in jeder tomographischen Schicht aufgenommen. Der
Cobalt-Marker wurde dann entfernt.
-
Eine
Starport-Kamera der Firma General Electric mit einem Colimator für allgemeine
Zwecke wurde verwendet, um die Tc-99m-Bilder aufzunehmen. Fünf minütige Planar-Aufnahmen
wurden der Reihe nach in der linken seitlichen Ansicht, bei 45 Grad
zum LAO und von hinten aufgenommen. Diesen folgte eine zehnminütige tomographische
Aufnahme. Der gesamte Satz von drei planaren und einer tomographischen
Aufnahme wurde für
insgesamt 5 Sätze
wiederholt. Es wurde dafür
gesorgt, das Schwein oder das Aufnahme-Gerüste während der gesamten Aufnahme-Sequenz nicht zu
bewegen.
-
Bilder
wurden auf einem Microdelta-Computer aufgezeichnet, der an ein VAX-Mainframe-System
angeschlossen war. Die Bilder wurden auf einem Band oder auf der
VAX-Festplatte gespeichert. Eine Planar-Bildanalyse bestand daraus,
zuerst das Bild mit dem Marker anzuschauen und die Marker-Position
auf dem Betrachtungs-Bildschirm
aufzuzeichnen. Das erste Bild, das nach der Injektion von Tc-99m-Annexin
V aufgenommen worden war, wurde dazu verwendet, um den Herz-Blut-Pool
zu definieren. Jedes nachfolgende Bild wurde angeschaut und unter
Verwendung des Markers und des anfänglich aufgezeichneten Blut-Pools
als Referenz analysiert. Jedes Bild wurde als positiv, ungewiß oder negativ
eingestuft.
-
Bei
dreizehn Tieren waren die Thromben im linken Vorhof gebildet worden,
und diese wurden abgebildet, wie dies oben beschrieben wurde. Zwölf Tieren
war Tc-99m Annexin V für
das Bildgebungs-Verfahren injiziert worden, und einem Tier war ein
nicht-spezifischer Tc-99m-Fab als Kontrolle injiziert worden. Ein
Bildgebungs-Verfahren
bei geschlossenem Brustkorb wurde an sieben Tieren ohne Verletzung
des Vorhofs durchgeführt,
und Simulationsexperimente am offenen Brustkorb wurden an drei Tieren
durchgeführt.
Bei Tieren mit Vorhof-Thromben waren alle planaren Aufnahmen, die
zu einem Zeitpunkt von weniger als 35 Minuten nach der Verabreichung
von Tc-99m-Annexin V aufgenommen worden waren, negativ. Neun der
planaren Bilder von Vorhof-Thromben, die zu einem Zeitpunkt von
mehr als 70 Minuten aufgenommen wurden, waren positiv, eine Aufnahme
war mehrdeutig und zwei waren negativ. Alle tomographischen Bilder
der Vorhof-Thromben waren entweder positiv (n = 10) oder mehrdeutig
(n = 2) bei Aufnahme-Zeiten von mehr als 2 h nach der Injektion. Die
mittlere Zeit im Anschluß an
die Verabreichung von Tc-99m-Annexin
V, bei der die planaren Bilder positiv wurden, betrug 82 Minuten
(35 – 135
Minuten).
-
Keines
der Kontroll-Tiere mit geschlossenem Brustkorb hatte positive Aufnahmen.
Eines der drei Tiere der Simulations-Versuche mit offenem Brustkorb
hatte ein positives Bild bei 85 Minuten. Man geht davon aus, dass
dieses falsche positive Ergebnis von der Produktion eines Thrombus
resultierte, der durch den chirurgischen Eingriff induziert worden
war.
-
Diese
Ergebnisse zeigen an, dass die intravenöse Verabreichung von Tc-99m-Annexin
V den Erhalt diagnostischer Bilder erlaubte, mit denen vaskulare
Vorhof-Thromben innerhalb einer kurzen Zeit nach der Verabreichung
des Konjugats identifiziert werden.
-
D. Proben-Entnahme
-
Proben
(sowohl Blut als auch Gewebe), die wie oben beschrieben entnommen
worden waren, wurden gewogen und in Röhrchen für eine unmittelbare Zählung von
Tc –99m
gegeben. Nachdem das Tc-99m zerfallen war (typischerweise zu einem
Zeitpunkt von 5 – 7
Tagen nach der Entnahme) wurden die Proben erneut getestet und so
die I-125-Zählwerte
erhalten. Jedes Röhrchen
wurde für
1 Minute gezählt.
Die Zählungen
wurden im Hinblick auf den Zerfall korrigiert, anschließend im
Hinblick auf das Gewicht korrigiert und mit der Einheit Zählwerte
pro Minute pro Gramm aufgezeichnet. Die Zählwerte pro Minute pro Gramm
für jede
Probe wurden dann normalisiert durch Teilen durch die Zählwerte
pro Minute pro Gramm der am Ende abgenommenen Blutprobe. Folglich
wurden die Ergebnisse für
jede der Probe berechnet in einem Verhältnis zu der zuletzt abgenommenen
Blutprobe, was einen sinnvollen Vergleich zwischen einzelnen Tieren
erlaubte. Diese Verfahrensweise wurde für alle Radionuklide in einem
gegebenen Experiment durchgeführt.
-
Die
Zählrohr-Zähl-Verhältniswerte
wurden für
eine Vielzahl von Gewebe-Proben
erhalten. Für
die verletzten Gewebe und Thromben wurden üblicherweise mehrereProben
von demselben Tier abgenommen. In diesen Fällen wird das Maximal-Verhältnis für irgendeine
Probe angegeben, wie auch der Mittelwert für alle abgenommenen Proben.
Der Maximal-Wert für
jedes Tier wurde über
alle Tiere gemittelt und wird angegeben als Maximal-Anx-V-Verhältnis.
-
Diese
Ergebnisse zeigen an, dass sich das Tc-99m-Annexin V vorzugsweise
in Atrium-(Vorhof-)Thromben und im verletzten linken Atrium sammelt,
wobei der höchste
Nicht-Target-Anordnungswert in der Niere auftritt. Der Konzentrationswert
der Niere ist wenigstens teilweise ein Indikator für die Ausscheidung von
Tc-99m-Annexin V über
den Nieren-Weg.
-
BEISPIEL IV
-
Radiomarkierte Annexin-Galactose-Konjugate
-
A. Herstellung von Annexin
V von Human-Plazenta und durch Expression in E. coli
-
Annexin
V wurde aus Human-Plazenta bis zu einer Endreinheit von 99 % gereinigt,
wie dies beschrieben wurde von „Funakoshi et al., in: Biochemistry,
26, 5572 – 5578
(1987)" und von „Tait et
al. in: Biochemistry, 27, 6268 – 6276
(1988)".
-
Alternativ
wurde Annexin V erhalten durch Expression in E. coli; dieses wurde
jedoch nicht in den folgenden Experimenten verwendet. Diese Expression
wurde wie folgt durchgeführt:
-
1.
Herstellung einer Massen-Fermentations-Überstandsflüssigkeit.
-
Es
wurden Standard-molekularbiologische Verfahrensweisen zum Exprimieren
von Annexin V in E. coli angewendet. Noch spezieller wurde die Protein-kodierende
Region der Annexin V-cDNS (siehe Funakoshi et al., a.a.O.) in die
NdeI/BamHI-Site des Plasmids pET-12a insertiert (Firma Novagen,
Madison, WI). Das ausgewählte
Plasmid wurde verwendet, um den E. coli-Stamm BL21 (DE3) (Firma
Novagen) zu transformieren.
-
Einzelne
Kolonien (Klone) wurden auf Luria Broth-Platten isoliert, die 50
g/ml Ampicillin oder Carbenicillin enthielten, wie dies diskutiert
ist in: „Sambrook
et al., Molecular Cloning Laboratory Manual, 2. Auflage, Cold Springs
Harbor Laboratory Press, (1989)".
Zur Produktion wurden Kulturen der ausgewählten Klone, die das gewünschte Plasmid
enthielten, über
Nacht bei 37°C
unter Schütteln
in "Terrific Broth" gezüchtet (siehe: "Tartof et al., Bethesda
Res. Lab. Focus, 9, 12 (1987)"),
die 50 μg/ml Carbenicillin
enthielt (im Handel erhältlich von
der Firma Sigma Chemical Co., St. Lous, Missouri). Die Kultur wurde
1 : 20 in demselben Medium verdünnt und
bei 37 °C
unter Schütteln
für 18
bis 24 h inkubiert. Die Bakterien wurden durch Zentrifugieren gewonnen, einmal
mit einem gleichen Volumen 0,05 M Tris-HCl, 0,15 M Natriumchlorid,
pH 8, gewaschen und als Pellets bei –20 °C gelagert. Die Pellets wurden
aufgetaut und erneut mit 10 – 15
% (w/v) kaltem 0,05 M Tris-HCl, 0,02 M Na4EDTA,
pH 7,2 suspendiert. Die Suspension wurde im Ultraschall 4 Minuten
auf Eis behandelt, zentrifugiert, und die überstehende Flüssigkeit
wurde bei –70 °C gelagert.
-
2.
Reinigung von Annexin V aus der Masse-Fermentations-Überstandsflüssigkeit: Nach dem Auftauen wurde
die überstehende
Flüssigkeit
unter Verwendung eines 0,1 um Hohllfaser-Filtermoduls filtriert
(erhältlich im
Handel von der Firma A/G Technology, Needham, MA) und die Leitfähigkeit
wurden mit konzentriertem Natriumchlorid auf einen Wert eingestellt,
der gleich war zu 0,02 M Tris-HCl, 0,5 M Natriumchlorid, pH B. Polyethylenimin-WP
(Firma J.T. Baker, Philipsberg, N.J.) wurde suspendiert in 0,02
M Tris-HCI, 0,5 M Natriumchlorid, pH 8, und 1 g Harz pro 10 ml Masse-Fermentations-Überstandsflüssigkeit
wurde der filtrierten Überstandsflüssigkeit
zugesetzt. Nach Rühren
für 30
Minuten wurde die überstehende
Flüssigkeit
entfernt, in 0,02 M Tris-HCI, pH 8, ausgetauscht und auf eine Q-SEPHAROSE®HP-Säule aufgegeben
(Firma Pharmacia, Piscataway, N.J.), die mit demselben Puffer equilibriert
worden war. Die Säule
wurde entwickelt mit einem Schritt-Gradienten von 0 bis 0,5 M Natriumchlorid.
Fraktionen wurden auf Annexin V getestet durch Größen-Ausschluß-HPLC,
und Fraktionen, die ein bei A280 absorbierendes
Material passender Größe enthielten, wurden
gepool. Das halbgereinigte Annexin V wurde in PBS ausgetauscht und
durch Ultrafiltration konzentriert. Gel-Filtration über SEPHACRYL®-100
HP (Firma Pharmacia), das in PBS equilibriert worden war, ergab das
gereinigte Protein. Die Protein-Konzentration wurde eingestellt
auf 1 mg/ml, steril bei 0,2 μm
filtriert und bei 2 bis 8°C
gelagert.
-
B. Derivatisierung von Annexin
V mit Galactose
-
Man
folgte der allgemeinen Verfahrensweise von „Lee et al., Biochemistry,
15, 6268 – 6276
(1976)".
-
a. Herstellung von 2-Imino-2-methoxyethyl-1-thio-D-galactopyranosid
-
Eine
1,0 M-Lösung
von Cyanomethyl-2,3,4,6-tetra-O-acetyl-1-thio-D-galactopyranosid (im Handel erhältlich von
der Firma Sigma Chemical Company, St. Louis, MO) wurde hergestellt
durch Lösen
von 1,83 g (4,5 mMol) in 4,5 ml wasserfreiem Methanol unter Erhitzen
auf 50 °C.
Die Lösung
wurde bei 50 °C
gehalten, und 0,1 ml (0,45 mMol) 25 % Natriummethoxid in Methanol
(im Handel erhältlich
von der Firma Aldrich Chemical Company, Milwaukee, WI) wurde unter
kontinuierlichem Rühren
zugesetzt. Nach 6 h bei 50 °C
wurde das Methanol unter verringertem Druck entfernt, und dies ergab
das Galactose-Methylimidat als farbloses, viskoses Öl.
-
b. Galactosylierung von
Annexin V
-
Das
Galactose-Methylimidat wurde mit Annexin V in Mol-Verhältnissen
300 1, 150 : 1 und 75 : 1 zusammengegeben. Für die Zusammengabe von 300
: 1 wurden 3,35 mg Galactosemethylimidat in Methanol in ein Öl unter
einem Strom von Stickstoff überführt. Dem Öl wurden
2,53 mg Annexin V in 0,05 M HEPES-Puffer (im Handel erhältlich von
der Firma Sigma Chemical Co., St. Louis, Missouri), pH 7,4, und
0,04 ml 0,5 M Natriumborat-Puffer, pH 8,5 zugesetzt. Die Lösung wurde
1 h bei Raumtemperatur durch Taumeln in dem Reaktionsgefäß gemischt,
und man ließ sie
dann bei Raumtemperatur über
Nacht (etwa 18 h) stehen. Die Reaktionsmischung wurde mit 0,4 ml
PBS verdünnt,
in ein Dialyse-Röhrchen überführt (Cut-Off
6.000 bis 8.000 MW) und wurde 24 h gegen PBS dialysiert. Nach Entfernen
des Materials aus dem Dialyse-Beutel wurde die Lösung durch ein 0,2 μm-Spritzen-Filter
filtriert. Die anderen Zusammengabe-Verhältnisse wurden in analoger
Weise behandelt.
-
C. Charakterisierung des
Galactose-derivatisierten Annexins V
-
Die
Protein-Konzentration wurde unter Verwendung von A280 von
0,6 für
eine 1 mg/ml-Lösung
von Annexin V bestimmt. Die Zahl der Galactose-Reste pro Molekül Annexin
V wurde bestimmt durch Messen der Gesamtzahl reaktiver Amine an
Annexin V vor und nach der Reaktion mit Galactosemethylimidat unter
Verwendung von Trinitrobenzolsulfonsäure, wie dies beschrieben wurde
von „Habeeb,
Analytical Biochemistry, 14, 328 – 336 (1966)".
-
-
Das
Vermögen
von Galactose-modifiziertem Annexin V, sich an aktivierte Blutplättchen zu
binden, wurde dadurch getestet, dass man sein Vermögen zum
Inhibieren des Bindens von unmodifiziertem, mit 125I-radiomarkiertem
Annexin V an frisch isolierte Human-Blutplättchen abschätzte, wobei
man der Verfahrensweise von „Thiagarajan
und Tail, J. Biol. Chem., 265, 17240 – 17243 (1990)" folgte. Die folgende
Tabelle zeigt die Ergebnisse des Konkurrenz-Assays und zwar sowohl
als absoluten Wert (linke Spalte) als auch als Wert normalisiert
auf 100 % für
unmodifiziertes Annexin V (rechte Spalte).
-
-
D. Herstellung von Tc-99m-Annexin
V-Galactose
-
Annexin
V-Galactose wurde radiomarkiert mit Technetium-99m unter Verwendung
einer Diamiddimercaptid-(N2S2-)Chelat-Verbindung,
wie dies beschrieben wurde durch „Kasina et al., J. Nucl. Med.,
32, 1445 – 1451
(1991)". Die vorgeformte
aktive Ester-Chelat-Verbindung wurde mit 2,0 ml Wasser verdünnt und
vor der Konjugation an Annexin V-Galactose unter Verwendung einer
modifizierten konditionieren C-18-Säule gereinigt ( im Handel erhältlich von
der Firma J.T. Baker) wie beschrieben in „Fritzberg et al., Proc. Natl.
Acad. Sci. USA, 85, 4025 – 4029
(1988)", achtmaliges
Waschen mit 2,0 ml Wasser, gefolgt von einem Schritt des Trocknens
der Säule
für 5 Minuten
und Eluieren mit 100 % Acetonitril. Das Lösungsmittel wurde unter einem
stetigen Strom von N2 entfernt. Anschließend wurden
0,15 ml PBS, 0,35 ml Annexin V-Galactose (4,7 : 1 = Galactose :
Annexin V) in einer Konzentration von 1,0 mg/ml und 0,2 ml 0,2 M
Bicarbonat-Puffer (pH 10,0) zur Konjugation an den Tc-99m-N2S2-TFP-Ester zugesetzt.
Nach 20 Minuten bei Raumtemperatur wurde das Tc-99m-Annexin V-Galactose-Konjugat
gereinigt durch Durchlaufenlassen durch eine G-25-SEPHADEX®-PD-10
Säule (im Handel
erhältlich
von der Firma Pharmacia), die mit PBS equilibriert worden war.
-
Fraktionen
von 1,0 ml wurden aufgefangen, und Fraktionen, die Annexin V enthielten,
wurden gepoolt. Die Protein-Konzentration wurde bestimmt durch UV-Absorption bei 280
nm. Die radiochemische Ausbeute des Konjugats betrug 35,3 %. Die
radiochemische Reinheit betrug 99,8 %, abgeschätzt durch sofortige Dünnschicht-Chromatographie auf
Silica-Gel-imprägnierten
Glasfaser-Platten, die entwickelt wurden in 12 % (w/v) wässriger
Trichloressigsäure.
Die ITLC-Platten wurden in zwei Hälften geschnitten und jede
Hälfte
wurde in einem Gamma-Counter oder einem Dosis-Calibrator gecounted. Das radiomarkierte
Annexin V-Galactose-Konjugat fiel am Anfangspunkt aus und alle Nicht-Protein-gebundene
Radioaktivität
wanderte mit der Lösungsmittelfront
in diesem Lösungsmittelsystem.
Die Tc-99m-Annexin V-Galactose-Konjugat-Lösung (300
bis 350 g) wurde verdünnt
und in PBS aufgehoben, das Rinderserumalbumin enthielt (im Handel
erhältlich
von der Firma Sigma Chemical Co.) und zwar in einer Endkonzentration
von 15 bis 20 mg/ml PBS vor der intravenösen Injektion.
-
E. Freisetzung von Tc-99m-Annexin
V und von Tc-99m-Annexin V-Galactose aus dem Blut
-
Ein
Tiermodell, wie es vorstehend in Beispiel III beschrieben worden
war, wurde verwendet zur Bewertung der Freisetzung der Moleküle aus dem
Blut. Für
N = 20 Schweine ist die Freisetzung aus dem Blut (
2) zweiphasig (biexponential):
| %ID/g
a = 0,0389 | t1/2
a = 9,6 min |
| %ID/g
b 0,0300 | t1/2
b = 46 min |
a + b = 0,0689; daher werden 57 % (0,0389/0,0689)
der injizierten Dosis/Gramm Blut in der schnelleren a-Phase freigesetzt,
und 43 % des ID/g wird in der langsameren b-Phase freigesetzt.
-
Die
Blut-Freisetzung von Tc-99m Annexin V-Galactose im Schwein (
2) ist ebenfalls zweiphasig (biexponentiell):
| %ID/g
a = 0,0313 | t1/2
a = 3,5 min |
| %ID/g
b = 0,0045 | t1/2
b = 160 min |
a + b = 0,0358, daher werden 87 % (0,0313/0,0358)
der injizierten Dosis/Gramm Blut sehr schnell in der frühen a-Phase
freigesetzt. Diese schnellere Freisetzung reduziert die Hintergrund-Radioaktivität in dem
Blut-Bereich, d.h. das Geräusch,
und verbessert daher das Verhältnis
Signal : Geräusch.
Folglich kann eine Thrombus-Abbildung zu kürzeren Zeitpunkten erreicht
werden.
-
BEISPIEL V
-
Herstellung
der Chelat-Verbindung
-
4,4-Diethoxycarbonylpropyl-1,3-dianilin:
Eine gerührte
Lösung
aus 2,065 g (1,25 Mo) Ethyl-4-aminobenzoat, 14,35 ml (0,125 Mol)
1,3-Di-iodpropan und 10,5 g (0,125 Mol) Natriumbicarbonat in 500
ml trockenem DMSO wird bei 100 °C
3 Stunden lang unter Stickstoff erhitzt. Nach dem Abkühlen wird
die Mischung unter Rühren
in 21 Eiswasser gegossen, und der resultierende Niederschlag wird
durch Filtration gesammelt. Der Niederschlag wird danach mit Eisessig
(14 × 75
ml) gewaschen, bis das gesamte anfängliche Ethyl-4-aminobenzoat
entfernt worden war. Nach dem Trocknen im Vakuum wird das so erhaltene
Produkt ohne weitere Reinigung im nächsten Schritt verwendet.
-
1,3-Di-(2-imino-6-ethoxycarbonylbenzthiazolyl-3-)propan:
Ammoniumthiocyanat (16,5 g; 0,217 Mol) wird einer mit einem Magneten
gerührten
Suspension von 4,4-Diethoxycarbonylpropyl-1,3-dianilin (10,0 g; 0,27
Mol) in 1.500 ml Eisessig zugesetzt. Eine Lösung aus Brom (34,6 g; 0,216
Mol) in 100 ml Eisessig wird danach tropfenweise der Suspension
unter Rühren
bei Raumtemperatur zugesetzt. Nachdem man die Reaktionsmischung über Nach
bei Raumtemperatur rührte,
wird das Dihydrobromid-Salz des Rohprodukts durch Filtration gesammelt
und getrocknet. Das Produkt wird isoliert durch Lösen des
Rohproduktes in heißem
Wasser, Einstellen auf einen basischen pH-Wert durch die Zugabe
von gesättigter
Natriumbicarbonat-Lösung, Sammeln
des Niederschlages durch Filtration und Trocknen im Vakuum.
-
N,N-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin:
Festes Natriumhydroxid (20,0 g; 0,357 Mol) wird einer Suspension
von 1,3-Di-(2-imino-6-ethoxycarbonylbenzthiazolyl-3-)propan
(1,0 g; 0,002 Mol) in 40 ml destilliertem Wasser zugesetzt, und
die resultierende Mischung wird 12 Stunden lang auf 120 ° C erhitzt.
Das vollständige
Lösen trat
nach 1 Stunde ein. Die Reaktionsmischung wurde danach in einem Eisbad
abgekühlt,
und der pH-Wert wurde mit 5,0 N Essigsäure auf 5,0 eingestellt. Die
wässrige
Lösung
wird danach mit drei 100 ml-Portionen Ethylacetat extrahiert. Die
vereinigten Ethylacetat-Extrakte werden über wasserfreiem Natriumsulfat
getrocknet, und das Trockenmittel wird abfiltriert. Nach der Entfernung
des Lösungsmittels
erhält
man das Produkt.
-
Bis
zu diesem Punkt ist die Synthese schematisch in 3 gezeigt.
-
N,N'-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin-mono-(methylaminocaproat-)Addukt:
Zu einer Mischung aus N,N'-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin
und 2 bis 3 Equivalenten von Methyl-6-aminohexanoat-hydrochlorid (hergestellt,
wie dies nachfolgend in Beispiel VI beschrieben ist) in Dimethylformamid
werden 10 Equivalente Triethylamin und anschließend 0,9 bis 1,0 Equivalente
BOP (Benzotriazol-1-yloxy-tris-(dimethylamino) phosphoniumhexafluorophosphat)
zugesetzt. Die Mischung wird 24 Stunden lang bei 15 bis 30 °C gerührt und
danach konzentriert. Der Rückstand
wird mit entionisiertem Wasser verdünnt, und der pH-Wert wird auf
etwa 2 mit einer 1 N wässrigen
Chlorwasserstoffsäure eingestellt.
Die Mischung wird erneut konzentriert. Der Rückstand wird an Silica-Gel
chromatographiert. Die Fraktionen der Chromatographie, die das Produkt
enthalten, werden vereinigt und konzentriert, und man erhält das Produkt.
-
Diese
Chelat-Verbindung ist zugänglich
für die
Verwendung mit einem geeigneten trifunktionellen Linker oder einem
Paar bifunktioneller Linker, um die radiomarkierten Annexin-Galactose-Cluster-Konjugate
der vorliegenden Erfindung herzustellen. Deren Verwendung mit einem
Paar bifunktioneller Linker wird in dem nachfolgenden Beispiel angesprochen.
-
BEISPIEL VI
-
Radiomarkierte Annexin-Galactose-Cluster-Konjugate
mit bifunktionellem Linker
-
A. Herstellung eines Acht-Galactose-Clusters
-
Diese
Vorgehensweise wird schematisch in 5 gezeigt.
-
Methvl-6-bromhexanoat:
In einen 1 Liter Rundkolben, der mit 20 g (102,5 mMol) 6-Bromhexansäure und
500 ml Methanol befällt
war, wurde 2 bis 3 Minuten lang Chlorwasserstoff-Gas eingeblasen.
Die Mischung wurde 4 Stunden lang bei Raumtemperatur gerührt und
konzentriert, und ergab 21,0 g des Produktes in Form eines gelben Öls (99 %): 1H-NMR (200 MHz, d6-DMSO);
3,57 (s, 3H), 3,51 (t, 2H), 2,30 (t, 2H), 1,78 (Pentett), 2H) und
1,62 bis 1,27 (m, 4H) ppm.
-
Methvl-6-aminohexanoat-hydrochlorid:
Einem 1 Liter-Rundkolben, der mit 40,0 g Aminocapronsäure befüllt war,
wurden 500 ml Methanol zugesetzt. Chlorwasserstoffgas wurde 5 Minuten
lang durch die Mischung durchgeblasen, und die Mischung wurde 5
Stunden lang bei Raumtemperatur gerührt. Die Mischung wurde danach
mittels Rotationsverdampfung und anschließend unter vollem Vakuumpumpen-Druck (< 0,1 mm Hg) konzentriert.
Dies ergab 55 g des Produktes als weißen Feststoff (99 %): 1H-NMR (200 MHz, CD3OD);
3,67 (s, 3H), 3,02 (t, 2H), 2,68 (s, 3H), 2,48 (t, 2H) und 2,03
bis 1,87 (Pentett, 2H) ppm.
-
Meth
1-6-(trifluoracetamido-)hexanoat: In einen 1 Liter-Rundkolben, der
mit 25,0 g (138 mMol) Methyl-6-aminohexanoat-hydrochlorid und 500
ml Methylenchlorid befällt
war, wurden 24 ml (170 mmol) Trifluoressigsäureanhydrid gegeben. Die Mischung
wurde in einem Eisbad abgekühlt
und 42 ml (301 mMol) Triethylamin wurden über einen Zeitraum von 25 bis
30 Minuten zugesetzt. Die Mischung wurde 2 Stunden lang bei 0 °C bis Raumtemperatur
gerührt
und danach konzentriert. Der Rückstand
wurde mit 150 ml Diethylether und 150 ml Petrolether verdünnt, und
die resultierende Lösung
wurde zuerst mit 1 N wässriger
HCl-Lösung
(3 × 150
ml) und danach mit gesättigter
wässriger
Natriumbicarbonat-Lösung
(3 × 150
ml) gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert, und ergab 32,9 g des Produktes in Form
eines schwach gelben Öls
(99 %): 1H-NMR (200 MHz, d6-DMSO);
9,39 (m, 1H), 3,57 (s, 3H), 3,14 (q, 2H), 2,29 (t, 2H), 1,60 bis
1,38 (m, 4H) und 1,32 bis 1,19 (m, 2H) ppm.
-
N,N'-Bis-(6-methoxycarbonylhexyl-)amin-hydrochlorid:
In einen 500 ml trockenen Rundkolben, der mit 12,0 g (50,0 mMol)
des sekundären
Amids Methyl-6-(trifluoracetamido-)hexanoat
und 250 ml trockenem Tetrahydrofuran befüllt war, wurden 2,2 g (55 mMol;
1,1 Equivalente) 60 % Natriumhydrid gegeben. Die Mischung wurde
30 Minuten lang bei Raumtemperatur gerührt. Danach wurden 10,25 g
(49,0 mMol; 0,98 Equivalente) des Alkylbromids, Methyl-6-bromhexanoat
zugesetzt. Die Mischung wurde 3 Stunden lang unter Rückfluß gerührt; zusätzlich wurden
5,80 g (27,7 mMol; 0,55 Equivalente) Methyl-6-bromhexanoat zugesetzt,
und die Mischung wurde 70 Stunden lang unter Rückfluß gerührt. Die Mischung wurde abgekühlt, mit
150 ml einer 1 N wässrigen
HCl verdünnt
und danach mit Ethylacetat (3 × 100
ml) extrahiert. Die organischen Extrakte wurde vereinigt, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wurde mit 200 ml Methanol
verdünnt
und danach mit 30 ml einer 10 N wässrigen Natriumhydroxid-Lösung behandelt.
Die Mischung wurde 18 Stunden lang bei Raumtemperatur gerührt und
danach konzentriert. Der Rückstand
wurde mit 200 ml entionisiertem Wasser verdünnt und mit 37 %iger konzentrierter
HCl auf einen pH-Wert von 1 bis 2 angesäuert. Die Lösung wurde mit Diethylether
(3 × 100
ml) gewaschen. Die wässrige
Phase wurde konzentriert. Der Rückstand
wurde mit 200 ml Methanol verdünnt
und erneut konzentriert. Der nachfolgende Rückstand wurde mit 250 ml Methanol
verdünnt,
und HCl-Gas wurde 2 bis 3 Minuten lang durchgeblasen, mit anschließendem Rühren bei
Raumtemperatur für
3 Stunden. Die Mischung wurde konzentriert. Der Rückstand
wurde mit 300 ml Methanol verdünnt
und filtriert, um anorganische Salze zu entfernen. Das Filtrat wurde
mit 3 g Aktivkohle durch ein Celite-Filter (hergestellt von der Firma 7.
T. Baker) filtriert und konzentriert. Der Rückstand, ein cremefarbiger
Feststoff, wurde aus 100 ml 2-Propanol erneut auskristallisiert,
und ergab 7,0 g des Produktes in Form eines weißen Feststoffes. Die Konzentration
des Filtrats und erneute Auskristallisation des Rückstandes
ergab eine zusätzliche
Ausbeute an 1,65 g des Produktes von insgesamt 8,65 g (56 %): 1H-NMR (200 MHz, d6-DMSO); 3,57
(s, 3H), 2,90 bis 2,73 (m, 4H), 2,30 (t, 4H); 1,67 bis 1,44 (m,
8H) und 1,37 bis 1,20 (m, 4H) ppm.
-
Methyl-4-methylaminobutyrat-hydrochlorid:
In einen 1 Liter-Rundkolben, der mit 30,0 g (195 mMol) 4-Methylaminobuttersäure und
500 ml Methanol befällt
war, wurde 1 bis 2 Minuten lang HCl-Gas durchgeblasen. Die Mischung
wurde 3 bis 4 Stunden lang bei Raumtemperatur gerührt und
danach konzentriert. Dies ergab 32,5 g des Produktes in Form eines
schaumigen, cremefarbenen Feststoffes (99 %): 1H-NMR
(200 MHz, CD3OD); 3,67 (s, 3H), 3,03 (t,
2H), 2,68 (s, 3H), 2,48 (t, 2H) und 2,03 bis 1,87 (Pentett), 2H)
ppm.
-
Methylaminobutanol:
In einen 1 Liter-Rundkolben, der mit 32,5 g (194 mMol) des Esters
Methyl-4-methylaminobutyrat-hydrochlorid befällt war, wurden über eine
Zeitdauer von 1 Stunde bei 0 °C
500 ml 1 M Boran in Tetrahydrofuran zugesetzt. Nachdem die Zugabe
beendet war, wurde die Mischung 20 Stunden lang unter Rückfluß erhitzt,
auf 0 °C
abgekühlt
und das überschüssige Boran
wurde durch sorgfältige
Zugabe von 100 ml Methanol zerstört.
Nachdem das gesamte Methanol zugegeben war, wurde die Mischung 1
Stunde lang bei Raumtemperatur gerührt und anschließend konzentriert.
Der Rückstand
wurde mit 400 ml Methanol verdünnt und
danach wurde HCl-Gas 5 Minuten lang in die Lösung eingeblasen. Die Mischung
wurde 16 Stunden lang unter Rückfluß erhitzt.
Die Mischung wurde abgekühlt,
konzentriert und danach mit 250 ml entionisiertem Wasser verdünnt. Das
Produkt wurde zunächst
in Form der freien Base freigesetzt durch Zugabe einer 10 N wässrigen
Natriumhydroxid-Lösung
auf einen pH-Wert von 9 bis 9,5, und danach durch Zugabe von 70
g eines AG-1 X-8-Anionenaustausch-Harzes
(Hydroxid-Form), im Handel erhältlich
von der Firma BioRad, Richmond, CA, und durch Rühren der Lösung für eine Zeitdauer von 2 Stunden.
Das Harz wurde abfiltriert und mit 150 ml entionisiertem Wasser
gewaschen. Die wässrigen
Filtrate wurden vereinigt und konzentriert. Der Rückstand
wurde mit 200 ml 2-Propanol verdünnt
und filtriert. Die gesammelten Feststoffe wurden mit 100 ml 2-Propanol gespült. Die
organischen Filtrate wurden vereinigt und konzentriert. Der Rückstand
wurde unter verringertem Druck destilliert und ergab 12,85 g des
Produktes in Form eines farblosen Öls (Siedepunkt: 68 °C bei 0,1
bis 0,2 mm Hg; 64 %): 1H-NMR (200 MHz, D2O); 3,52 (t, 2H), 2,56 (t, 2H), 2,31 (s,
3H) und 1,65 bis 1,43 (m, 4H) ppm.
-
4-(N-Methyltrifluoracetamido-)1-butanol:
In einen 250 ml-Rundhalskolben, der mit 10,0 g (96,9 mMol) des Amins
4-Methylaminobutanol in 100 ml trockenem Methanol befüllt war,
wurden 17,5 ml (147 mMol) Ethyltrifluoracetat gegeben. Die Mischung
wurde 24 Stunden lang bei Raumtemperatur gerührt und danach konzentriert,
ergab 18,55 g des Produktes in Form eines nahezu farblosen Öls (96 %): 1H-NMR (200 MHz, D2O); 3,63
und 3,50 (2 Tripletts, 4H), 3,20 und 3,05 (d und s, 3H), und 1,82
bis 1,47 (m, 4H) ppm.
-
1-(p-Tololnsulfonyloxy-)4-(N-methyltrifluoracetamido-)butan:
In einen trockenen 1 Liter-Rundhalskolben, der mit 17,0 g (85,4
mMol) des Alkohols (4-(N-methyltrifluoracetamido-)1-butanol
in 400 ml Methylenchlorid befüllt
war, wurden 17,1 g (89,7 mMol; 1,05 Equivalente) Toluolsulfonylchlorid
und anschließend
30 ml (213 mMol; 2,5 Equivalente) Triethylamin bei 0 °C über eine
Zeitdauer von 10 Minuten gegeben. Die Mischung wurde 15 Stunden
lang bei 0 °C
bis Raumtemperatur gerührt
und danach mit 5 % (v/v) wässriger
HCl (3 × 200
ml) gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde an Silica-Gel
chromatographiert, mit Hexan/Methylenchlorid (50 : 50) und danach
mit Methylenchlorid eluiert. Dies ergab 25,1 g des Produktes in
Form eines blassgelben Öls
(83 %): 1H-NMR (200 MHz, CDCl3);
7,80 (d, 2H), 7,37 (d, 2H), 4,07 (m, 2H), 3,41 (m, 3H), 3,09 und
2,98 (q und s, 3H), 2,45 (s, 3H) und 1,68 (m, 4H) ppm; TLC (Methylenchlorid),
Rf = 0,31.
-
1-S-(2,3,4,6-Tetra-O-acetyl-beta-D-galactopyranosyl-)2-thiopseudoharnstoffhydrobromid:
In einen 250 ml-Rundkolben, der mit 5,08 g (60,8 mMol; 1,09 Equivalente)
Thioharnstoff und 36 ml Aceton befüllt war, wurden 25,0 g (66,7
mMol) Tetra-acetyl-alpha-D-galactopyranosylbromid gegeben. Die Mischung
wurde 15 bis 20 Minuten unter Rückfluß gerührt und
danach auf Eis abgekühlt.
Die Mischung wurde durch einen Buchner-Trichter filtriert und mit
25 ml eiskaltem Aceton gespült.
Die Feststoffe wurden mit 50 ml Aceton behandelt, 15 Minuten lang
unter Rückfluß erhitzt,
auf Eis gekühlt
und filtriert. Die Feststoffe wurden mit 25 ml kaltem Aceton gespült, an der
Luft getrocknet und danach im Vakuum getrocknet. Dies ergab 22,6
g des Produktes in Form eines weißen Feststoffes (76 %): 1H-NMR (200 MHz, d6-DMSO);
9,4 bis 9,0 (breites d, 4H), 5,63 (d, 1H), 5,38 (d, 1H), 5,23 (dd,
1H), 5,09 (t, 1H), 4,40 (t, 1H), 4,04 (dd, 1H), 2,13 (s, 3H), 2,08
(s, 3H), 2,00 (s, 3H), 1,93 (s, 3H) ppm.
-
4-(N-Methylaminobutyl-)1-thio-beta-D-alactopyranosid:
In einen 500 ml-Rundkolben,
der mit 20,7 g (42,5 mMol; 1,07 Equivalente) des Thiopseudoharnstoffhydrobromids,
das wie oben beschrieben hergestellt worden war, in 70 ml entionisiertem
Wasser befüllt
war, wurden 6,4 g (46,3 mMol; 1,16 Equivalente) Kaliumcarbonat und
4,7 g (45,2 mMol; 1,13 Equivalente) Natriumbisulfit gegeben, und
danach wurden sofort 14,1 g (39,9 mMol; 1,0 Equivalente) des Tosylats,
1-(p-Toluolsulfonyloxy-)4-(N-methyltrifluoracetamido-)butan
in 70 ml Aceton zugegeben. Die Mischung wurde 16 Stunden lang bei
Raumtemperatur gerührt.
Die Mischung wurde mit 50 ml Kochsalzlösung verdünnt und mit Ethylacetat (3 × 200 ml)
extrahiert. Die organischen Extrakte wurden vereinigt, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wurde an Silica-Gel
chromatographiert, zuerst mit 75% Methylenchlorid/Hexan, danach
mit Methylenchlorid, danach mit 2 % Methanol/Methylenchlorid und
letztendlich mit 10 % Methanol/Methylenchlorid eluiert. Alkylierungsprodukt mit
unterschiedlichen Graden der Acetylierung enthaltende Fraktionen
wurden zusammengegeben und konzentriert. Der Rückstand wurde mit 250 ml Methanol
und 150 ml entionisiertem Wasser verdünnt und mit 110 g AG-1 X-8-Harz
(Hydroxid-Form; 2,6 m Equivalente/g Trockengewicht), im Handel erhältlich von
der Firma BioRad, behandelt. Die Mischung wurde 18 Stunden lang
bei Raumtemperatur gerührt.
Die Mischung wurde filtriert und das Harz wurde mit Methanol (2 × 150 ml)
gespült.
Die Filtrate wurden vereinigt und konzentriert. Dies ergab 6,1 g
des Produktes (54 %): 1H-NMR (200 MHz, D2O); 4,38 (d, 1H), 3,88 (d, 1H), 3,69 bis
3,41 (m, 5H), 2,82 bis 2,64 (m, 4H), 2,43 (s, 3H) und 1,68 bis 1,57
(4H) ppm.
-
N-BOC-Bis-methylester:
Zu 1,00 g (3,23 mMol) des Aminhydrochlorids N,N-Bis-(6-methoxycarbonylhexyl-)amin-hydrochlorid,
das wie oben beschrieben hergestellt worden war, wurden 1,5 ml (10,6
mMol) Triethylamin und danach 875 mg (3,55 mMol; 1,1 Equivalente)
BOC-ON 2-(tert-Butoxycarbonyloxyimino-)2-phenylacetonitril zugesetzt.
Die Mischung wurde 18 Stunden lang bei Raumtemperatur gerührt und
danach konzentriert. Der Rückstand
wurde mit 100 ml Ethylacetat verdünnt und mit 1 N wässriger
Chlorwasserstoffsäure
(3 × 50
ml) und anschließend
mit gesättigter
wässriger
Natriumbicarbonat-Lösung
(2 × 50
ml) gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde an Silica-Gel chromatographiert
und mit 15 % (Volumenprozent) Ethylacetat/Hexan eluiert. Die chromatrographierten
Fraktionen, die das Produkt enthielten, wurden vereinigt und konzentriert.
Dies ergab 990 mg des Produktes als nahezu farbloses Öl (83 %).
-
N-BOC-Bis-Säure: Zu
980 mg (2,62 mMol) des Diesters, der in dem vorhergehenden Schritt
hergestellt worden war, in 10 ml Methanol wurden 5,8 ml einer 1
N wässrigen
Natriumhydroxid-Lösung
(5,8 mMol) zugesetzt. Die Mischung wurde 16 Stunden lang bei Raumtemperatur
gerührt
und danach konzentriert. Der Rückstand
wurde mit 30 ml entionisiertem Wasser verdünnt und auf einen pH-Wert von
1,5 bis 2 angesäuert. Die
Mischung wurde mit Ethylacetat (6 × 50 ml ) extrahiert. Die organischen
Extrakte wurden über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wurde an einem Umkehr-Phasen-C-18-Silica-Gel chromatographiert,
das im Handel erhältlich
ist von der Firma J. 7. Baker, und mit 65 % Methanol/Wasser eluiert.
Die chromatographierten Fraktionen, die das Produkt enthielten,
wurden vereinigt und konzentriert. Dies ergab 851 mg des Produktes
als nahezu farbloses Öl
(94 %).
-
N-BOC-Tetramethylester:
Zu 825 mg (2,39 mMol) der Bis-Säure,
die wie oben beschrieben hergestellt worden war, in 35 ml trockenem
Dimethylformamid wurden 1,75 g (5,65 mMol; 2,36 Equivalente) des
Aminhydrochlorid N,N-Bis-(6-methoxycarbonylhexyl-)amin-hydrochlorid
und 3,0 ml Triethylamin und anschließend 2,4 g (5,4 mMol; 2,3 Equivalente)
BOP zugesetzt. Die Mischung wurde 17 Stunden lang bei Raumtemperatur
gerührt
und danach konzentriert. Der Rückstand
wurde mit 100 ml Ethylacetat verdünnt und mit 1 N Chlorwasserstoffsäure (3 × 50 ml)
und anschließend
mit wässriger
Natriumbicarbonat-Lösung
(2 × 50
ml) gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde an Silica-Gel
chromatographiert und mit Ethylacetat eluiert. Die chromatographierten
Fraktionen, die das Produkt enthielten, wurden vereinigt und konzentriert.
Dies ergab 1,63 g des Produktes als nahezu farbloses Öl (80 %).
-
N-BOC-Tetra-Säure: Einer
Lösung
aus 1,41 g (1,65 mMol) Tetramethylester, der wie oben beschrieben hergestellt
worden war, in 25 ml Methanol wurden 7,4 ml (7,4 mMol) 1 N wässrige Natriumhydroxid-Lösung zugesetzt.
Die Mischung wurde 22 Stunden lang bei Raumtemperatur gerührt und
danach konzentriert. Der Rückstand
wurde mit 30 ml entionisiertem Wasser verdünnt und mit 1 N wässriger
Chlorwasserstoffsäure
auf einen pH-Wert von 2 angesäuert.
Die Mischung wurde mit Ethylacetat/Isopropanol in einem Verhältnis von
3 : 1 (Volumenverhältnis)
(3 × 100
ml) extrahiert. Die organischen Extrakte wurden konzentriert. Der
Rückstand wurde
an einem Umkehr-Phasen-C-18-Silica-Gel chromatographiert, zunächst mit
Methanol/Wasser (50 : 50) (Volumenverhältnis) und letztendlich mit
Methanol/Wasser (75 : 25) eluiert. Die chromatographierten Fraktionen,
die das Produkt enthielten, wurden vereinigt und konzentriert. Dies
ergab 1,19 g des Produktes als farbloses Öl (90 %).
-
N-BOC-Octamethylester:
Einer Mischung aus 501 mg (0,626 mMol) der Tetra-Säure,
die wie oben beschrieben hergestellt worden war, und 30 ml trockenem
Dimethylformamid wurden 968 mg (3,12 mMol; 5,0 Equivalente) des
Aminhydrochlorids, N,N'-Bis-(6-methoxycarboxyhexyl-)amin-hydrochlorid
und 2,0 ml (14,2 mMol) Triethylamin und anschließend 1,22 g (2,76 mMol; 4,6
Equivalente) BOP zugesetzt. Die Mischung wurde 19 Stunden lang bei
Raumtemperatur gerührt
und danach konzentriert. Der Rückstand
wurde mit 75 ml Ethylacetat verdünnt
und mit 1 N wässriger
Chlorwasserstoffsäure
(2 × 50
ml) gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde an einem Umkehr-Phasen-C-18-Silica-Gel
chromatographiert, zunächst
mit Methanol/Wasser (60 : 40) und letztendlich mit Methanol/Wasser
(90 : 10) eluiert. Die chromatographierten Fraktionen, die das Produkt
enthielten, wurden vereinigt und konzentriert. Dies ergab 715 mg
des Produktes als farbloses Öl
(63 %).
-
N-BOC-Octa-Säure: Einer
Lösung
aus 715 mg (0,393 mMol) Octamethylester, der wie oben beschrieben
hergestellt worden war, in 20 ml Methanol wurden 6 ml 1 N wässrige Natriumhydroxid-Lösung (6
mMol) und 5 ml entionisiertes Wasser zugesetzt. Die Mischung wurde
16 Stunden lang bei Raumtemperatur gerührt und danach konzentriert.
Der Rückstand
wurde mit 20 ml entionisiertem Wasser verdünnt, und die Lösung wurde
auf einen pH-Wert von 1,5 bis 2,0 angesäuert. Die Mischung wurde konzentriert,
und der Rückstand
wurde an einem Umkehr-Phasen-C-18-Sillica-Gel chromatographiert,
zunächst
mit Methanol/Wasser (50 : 50) und letztendlich mit Methanol/Wasser
(80 : 20) eluiert. Die chromatographierten Fraktionen, die das Produkt
enthielten, wurden vereinigt und konzentriert. Dies ergab 647 mg
des Produktes als nahezu farbloses Öl (96 %).
-
Die
oben angegebene Vorgehensweise ist bestimmt für die Herstellung eines Galactose-Clusters
mit 8 Galactose-Resten. Die Version mit vier Galactose-Resten kann
in Übereinstimmung
mit dieser Vorgehensweise hergestellt werden, indem man von der
Tetra-Säure
zu dem Galactose-Derivatisierungs-Schritt, der nachfolgend für die 8-Galactose-Cluster
beschrieben ist, verfährt.
In ähnlicher
Weise können
16-, 32- etc. Galactose-Cluster-Konstrukte in Übereinstimmung mit der vorliegenden
Erfindung durch die Einführung
mehrerer Wiederholungen der Methylester- und Säure-Bildungs-Schritte hergestellt
werden. Noch spezieller werden die 16-Methylester-Konstrukte, die
16-Säure,
die 32-Methylester und so weiter im wesentlichen hergestellt, wie dies
oben für
die Tetra- und Octa-Formen beschrieben wurde. Wenn die erwünschte Anzahl
an Säure-Resten gebildet
ist, wird der Galactose-Derivatisierungs-Schritt durchgeführt, wobei
die Anteile der Bestandteile so eingestellt wird, dass sie mit der
Anzahl der Säure-Reste
in Einklang gebracht werden.
-
N-BOC-Octa-Galactosyl-Konstrukt:
Einer Mischung aus 161 mg (94 mMol) der Octa-Säure, die wie oben beschrieben
herstellt worden war, und 225 mg (906 Micromol; 9,64 Equivalente)
des Galactosamins, 4-N-(Methylaminobutyl-)1-thio-beta-D-galactopyranosid,
in 8 ml trockenem Dimethylformamid wurden 0,5 ml (3,54 mMol) Triethylamin
und anschließend
371 mg (839 Micromol; 8,4 Equivalente) BOP zugesetzt. Die Mischung
wurde 17 Stunden lang bei Raumtemperatur gerührt und danach konzentriert.
Der Rückstand
wurde an einem Umkehr-Phasen-C-18-Silica-Gel chromatographiert,
zunächst
mit Methanol/Wasser (40 : 60) und letztendlich mit Methanol/Wasser
(70 : 30) eluiert. Die chromatographierten Fraktionen, die das Produkt
enthielten, wurden vereinigt und konzentriert. Dies ergab 170 mg
des Produktes als nahezu farbloses Öl (47 %).
-
Octa-Galactosylamin:
170 mg des N-BOC-Octa-Galactosyl-Konstrukts, das wie oben beschrieben hergestellt
worden war, wurden 5 ml Trifluoressigsäure zugesetzt. Die Mischung
wurde 10 Minuten lang bei Raumtemperatur gerührt und danach konzentriert.
Der Rückstand
wurde mit 10 ml Methanol verdünnt
und erneut konzentriert. Der Rückstand
wurde ohne weitere Reinigung verwendet.
-
B. Herstellung der verlängerten
Galctose-Cluster
-
Diese
Verfahrensweise ist in 6 schematisch
gezeigt.
-
Methyl-6-(N-BOC-)aminocaproat:
Einer Mischung aus dem Aminhydrochlorid Methyl-6-aminohexanoat-hydrochlorid,
das wie oben beschrieben hergestellt worden war, wurden 1,1 Equivalente
BOC-ON und anschließend
2 bis 3 Equivalente Triethylamin zugesetzt. Die Mischung wurde 16
bis 24 Stunden lang bei 15 bis 30 °C gerührt und konzentriert. Der Rückstand
wurde in Ethylacetat gelöst
und mit 1 N wässriger
Chlorwasserstoffsäure
und danach mit gesättigter
wässriger
Natriumbicarbonat-Lösung
gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet,
filtriert und über
Reaktionsverdampfung bei verringertem Druck konzentriert. Der Rückstand
wurde an Silica-Gel chromatographiert und mit 25% Ethylacetat/Hexan
eluiert. Die chromatographierten Fraktionen, die das Produkt enthielten,
wurden vereinigt und konzentriert und man erhielt das Produkt.
-
6-(N-BOC-)aminocapronsäure: Zu
einer Lösung
des Methylesters Methyl-6-(N-BOC-)aminocaproat
in Methanol wurden 1,5 Equivalente einer 1 N wässrigen Natriumhydroxid-Lösung zugesetzt.
Die Mischung wurde 16 bis 24 Stunden lang bei 15 bis 30 °C gerührt und
danach konzentriert. Der Rückstand
wurde mit entionisiertem Wasser verdünnt und mit Ethylacetat extrahiert.
Die organischen Extrakte wurden vereinigt, über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde an Silica-Gel
chromatographiert, zunächst
mit 25 % Ethylacetat/Hexan und letztendlich mit 100 % Ethylacetat
eluiert. Die chromatographierten Fraktionen, die das Produkt enthielten,
wurden vereinigt und konzentriert und man erhielt das Produkt.
-
N-BOC-verlängertes
Octa-Galactosyl-Konstrukt: Zu einer Lösung des Octa-Galactosylamins,
das wie oben beschrieben hergestellt worden war, in Dimethylformamid
und 1,5 bis 3 Equivalenten 6-(N-BOC-)aminocapronsäure wurden
4 bis 6 Equivalente Triethylamin und anschließend 1,1 bis 1,5 Equivalente
BOP zugesetzt. Die Mischung wurde 4 bis 24 Stunden lang bei 15 bis
30 °C gerührt und
danach konzentriert. Der Rückstand
wurde mit entionisiertem Wasser verdünnt, und der pH-Wert wurde
durch Zugabe einer 1 N wässrigen Chlorwasserstoffsäure auf
1,5 bis 2,0 eingestellt. Die Mischung wurde mit Ethylacetat gewaschen.
Die wässrige
Phase wurde konzentriert und der Rückstand wurde an einem Umkehr-Phasen-C-18-Silica-Gel
chromatographiert, zunächst
mit Methanol/Wasser (50 : 50) und letztendlich mit Methanol/Wasser
(65 : 35) eluiert. Die chromatographierten Fraktionen, die das Produkt
enthielten, wurden vereinigt und konzentriert und man erhielt das
Produkt.
-
Amin-verlängertes
Octa-Galactosyl-Konstrukt: Dem N-BOC-geschützten Amin, das in dem vorhergehenden
Schritt hergestellt worden war, wurde Trifluoressigsäure zugesetzt.
Die Mischung wurde 10 Minuten lang bei 15 bis 30 °C gerührt und
danach konzentriert. Der Rückstand
wurde mit Methanol verdünnt
und erneut konzentriert. Man erhielt das Produkt, das ohne weitere
Reinigung verwendet wurde.
-
C. Konjugation
von Galactose-Cluster mit Chelat-Verbindung und Annexin-Bestandteilen
-
Octa-Galactosyl-Chelat-Verbindung-Konstrukt:
Einer Mischung aus Aminverlängertem
Octa-Galactosylamin, 1,2 Equivalenten N,N'-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin-mono-(methylaminocaproat-)Addukt
und 5 Equivalenten Triethylamin in Dimethylformamid wurden 1,1 Equivalente
BOP zugesetzt. Die Mischung wurde 2 bis 24 Stunden lang bei 15 bis
30 °C gerührt und
danach konzentriert. Der Rückstand
wurde mit entionisiertem Wasser gewaschen, und der pH-Wert wurde
durch Zugabe einer 1 N wässrigen
Chlorwasserstoffsäure
auf 2,5 eingestellt. Die Mischung wurde mit Ethylacetat gewaschen.
Die wässrige
Phase wurde konzentriert. Der Rückstand
wurde an einem Umkehr-Phasen-C-18-Silica-Gel chromatographiert.
Die Fraktionen, die das Produkt enthielten, wurden vereinigt und
konzentriert und man erhielt das Produkt.
-
Octa-Galactosvl-Chelat-Verbindung-Carbonsäure-Konstrukt:
Einer methanolischen Lösung
des Estertragenden Octa-Galactosyl-Chelat-Verbindung-Konstrukts, das vorangehend
hergestellt worden war, wurden 4 bis 5 Equivalente einer 1 N wässrigen
Natriumhydroxid-Lösung
zugesetzt. Die Mischung wurde 16 bis 24 Stunden lang bei 15 bis
30 °C gerührt. Die
Mischung wurde konzentriert und der Rückstand wurde mit entionisiertem
Wasser gewaschen. Der pH-Wert der resultierenden Lösung wurde
auf etwa 2,5 eingestellt, und die Lösung wurde erneut konzentriert.
Der Rückstand
wurde an einem Umkehr-Phasen-C-18-Silica-Gel chromatographiert.
Die Fraktionen, die das Produkt enthielten, wurden vereinigt und
konzentriert und man erhielt das Produkt.
-
Annexin
V-Chelat-Verbindung-Octa-Galactosvl-Konstrukt: Das Octa-Galactosyl-Chelat-Verbindung-Carbonsäure-Konstrukt
wird mit Annexin V in molaren Verhältnissen von 30:1, 15:1, 5:1
und 2:1 zusammengegeben. Die Konjugation von Annexin V wird durchgeführt durch
die Aktivierung der funktionellen Carbonsäure- Gruppe des Galactose-Cluster-Chelat-Verbindung-Konstrukts
mit Benzotriazol-1-yl-oxy-tris-(dimethylamino-)phosphoniumhexafuorphosphat
(BOP). Dem Galactose-Cluster-Chelat-Verbindung-Konstrukt
in DMF wird eine equimolare Konzentration an BOP in DMF zugesetzt.
Annexin V in 0,05 M HEPES-Puffer (im Handel erhältlich von der Firma Sigma
Chemical Co., St. Louis, MO), pH 7,4, wird der Reaktionsmischung
zugesetzt und anschließend
wird ein 0,5 M Borat-Puffer, pH 8,5, zugesetzt. Die DMF-Konzentration in
der Reaktionsmischung wird zwischen 5 bis 20 % gehalten. Die Mischung
wird 1 Stunde lang bei Raumtemperatur durch Rotieren des Reaktionsgefäßes gemischt,
und wird danach bei Raumtemperatur über Nacht (etwa 18 bis 24 Stunden)
stehen gelassen. Die Reaktionsmischung wird danach mit PBS verdünnt, in
einen Dialyseschlauch (Cut-Off: Molekulargewicht: 6.000 bis 8.000
MW) überführt, und
24 Stunden lang gegen PBS dialysiert. Nach dem Entfernen des Materials
aus dem Dialysebeutel wurde die Lösung durch ein 0,2 Micrometer
Spitzen-Filter filtriert. Alle anderen Zusammengabe-Verhältnisse
wurden in analoger Weise behandelt.
-
D. Charakterisierung des
Galactose-Cluster-Chelat-Verbindung-Annexin V-Koniugats
-
Die
Protein-Konzentration wird bestimmt unter Verwendung von A280 von 0,6 für eine 1 mg/ml-Lösung Annexin
V. Die Anzahl der Galactose-Reste pro Molekül Annexin V wird bestimmt durch
Messung der Gesamtzahl an reaktiven Aminen an Annexin V vor und
nach der Reaktion mit dem Galactosyl-Cluster-Chelat-Verbindung-Konjugat unter Verwendung
von Trinitrobenzolsulfonsäure,
wie dies beschrieben ist in „Habeeb,
Analytical Biochemistry, 14, 328 – 336 (1966)". Die Fähigkeit
von Galactose-Chelat-Verbindung-Annexin,
sich an aktivierte Blutplättchen
zu binden, wird dadurch bewertet, indem man seine Fähigkeit
bestimmt, die Bindung von unmodifiziertem, I-125-radiomarkiertem Annexin V an frisch
isolierte menschliche Blutplättchen
zu inhibierten, in Anlehnung an das Verfahren von „Thiagarajan
und Tait, J. Biol. Chem., 265, 17240 – 17243 (1990)".
-
E. Radiomarkierungs-Verfahren
zur Verwendung im nach-gebildeten Chelat-Konjugations-Verfahren
-
Verfahren
A: Zinngluconat-Kits wurden hergestellt, die 5 mg Natriumgluconat,
100 Microgramm Zinnchlorid, 1,0 mg (1 mg/ml) Galactose-Cluster-Chelat-Verbindung-Annexin V und 0,1
bis 1,0 mg Lactose enthielten. Der pH-Wert wurde unter Verwendung
von HCl, Essigsäure
oder NaOH zwischen 5 und 7 gehalten. Dem Zinngluconat-Kit wurden
1,0 ml Natriumpertechnetat (Tc-99m) mit einer spezifischen Aktivität von 50
mCi zugesetzt. Die Phiole wurde 5 bis 30 Minuten lang bei 25 bis
37°C inkubiert.
Der prozentuale Anteil an markiertem Konjugat, verbleibendem TcO4, und hydrolyisiertem reduziertem Technetium
wurde bestimmt durch ITLC in 12 % TCA als Entwicklungslösungsmittel.
-
Verfahren
B: Zinntartrat-Kits wurden in einer Vakuum-Phiole unter Stickstoff
hergestellt, die 0,5 ml Dinatriumtartrat (10 mg/ml) und 0,1 ml Zinnchlorid
(1,0 mg/ml in Ethanol) enthielten. Der pH-Wert der Lösung wurde
zwischen 5 und 7, vorzugsweise bei 6,0 gehalten. Dieser Zinntartrat-Lösung wurden
1,0 ml Natriumpertechnetat (50 mCi) zugesetzt, und man ließ die Lösung bei
Raumtemperatur stehen. Einer evakuierten Phiole wurden nacheinander
200 Microliter Natriumphosphat (0,5 M; pH 8 oder 10) und 1,0 ml
Galactose-Cluster-Chelat-Verbindung-Annexin V-Konjugat (1,0 mg/ml)
zugesetzt. Danach wurde Tc-99m-Tartrat (50 mCi) zugesetzt, und die
Phiole wurde 5 bis 30 Minuten lang bei 25 bis 37 °C inkubiert.
Der prozentuale Anteil an markiertem Konjugat, verbleibendem TcO4 und hydrolyisiertem reduziertem Technetium
wurde bestimmt durch ITLC in 12 % (w/v) Trichloressigsäure als
Entwicklungslösungsmittel.
-
Konstrukte,
die gemäß diesem
Beispiel hergestellt worden waren, wurden in Übereinstimmung mit den Verfahrensabläufen, die
oben angegeben wurden (Beispiel III und Beispiel IV (E)) getestet,
um die Nützlichkeit in
Anwendungen, beispielsweise bei der Darstellung von Blutgerinseln,
zu überprüfen.
-
BEISPIEL VII
-
Annexin-Galactose-Cluster-Konjugate
mit trifunktionellem Linker
-
A. Herstellung der Chelat-Verbindung
-
Die
Herstellung der Chelat-Verbindung N,N'-Bis-(2-disulfidyl-4-methylphenyl-)
gamma, gamma'-diamino-isovalerat-N-hydroxysuccinimid
ist schematisch in 4 gezeigt.
-
3-Iodmethvl-4-Iodbuttersäure: Einer
Lösung
aus 1,61 g (10mMol) 3-Hydroxymethyl-4-butanolid
(hergestellt durch das Verfahren von Kinoshita und Hirano, J. Heterocyclic
Chem., 29, 1025 (1992)) in 100 ml Kohlenstofftetrachlorid wurden
8 g (40 mMol) Iodtrimethylsilan zugesetzt. Die Reaktionsmischung
wurde 12 Stunden lang unter Stickstoff auf 50 °C erhitzt. Die Mischung wurde
mit Chloroform verdünnt
und mit Wasser (3 × 100
ml), 5 % wässriger
Natriumthiosulfat-Lösung
(100 ml), 10 % wässriger
Natriumbicarbonat-Lösung
und Kochsalzlösung
gewaschen. Die organische Schicht wurde über Magnesiumsulfat getrocknet,
filtriert und verdampft. Dies ergab das erwünschte Rohprodukt. Das Rohprodukt
wurde mittels Silica-Gel-Chromatographie (Ethylacetat/Hexan = 3
: 7 als Elutionsmittel) gereinigt, und man erhielt 3-Iodmethyl-4-Iodbutttersäure.
-
Ethyl-3-Iodmethyl-4-Iodbutyrat:
Eine Lösung
aus 2,831 g (8 mMol) 3-Iodmethyl-4-Iodbuttersäure in 80 ml
Ethanol wurde mit HCl-Gas bei 0 °C
gesättigt.
Nach dem Rühren
der Lösung
bei Raumtemperatur für
2 Tage wurde das Lösungsmittel
unter Vakuum entfernt, und der Rückstand
wurde in Dichlormethan gelöst.
Die Dichlormethan-Schicht wurde mit 10 % wässriger Natriumbicarbonat-Lösung (3 × 100 ml),
Wasser (1 × 100
ml) und Kochsalzlösung
gewaschen. Die abgetrennte Dichlormethan-Schicht wurde über Magnesiumsulfat
getrocknet, filtriert und verdampft und ergab Ethyl-3-Iodmethyl-4-Iodbutyrat.
-
Ethyl-gamma
gamma'-di-(4-methylanilino-)isovalerat:
Eine gerührte
Lösung
aus 7,5 g (70 mMol) 4-Toluidin, 2,764 g (7 mMol) Ethyl-3-Iodmethyl-4-Iodbutyrat
und 0,588 g (7 mMol) Natriumbicarbonat in 30 ml trockenem Dimethylsulfoxid
wurde 3 Stunden lang unter Stickstoff auf 100°C erhitzt. Die abgekühlte Mischung wurde
unter Rühren
auf 400 ml Eiswasser gegossen. Der resultierende Niederschlag wurde
durch Filtration gesammelt. Das verbleibende 4-Toluidin in dem Niederschlag
wurde durch mehrmaliges Waschen mit wässriger Essigsäure entfernt.
Das Produkt wurde erhalten durch Umkristallisation des gewaschenen
Niederschlags in Heptan.
-
Ethyl
gamma gamma'-[1,3-di-(2-imino-6-methylbenzthiazolyl-3-)]isovalerat:
Einer mit einem Magneten gerührten
Suspension aus 2,0 g (6,5 mMol) Ethyl-gamma, gamma'-di-(4-methylanilino-)isovalerat
in 250 ml Eisessig wurde Ammoniumthiocyanat (3,5 g; 0,046 Mol) zugesetzt,
und anschließend
wurde tropfenweise eine Lösung
aus Brom (7,27 g; 0,046 Mol) in 50 ml Eisessig zugesetzt. Nachdem
die Zugabe beendet war, wurde das Rühren über Nacht fortgesetzt. Der
gelbe Niederschlag aus Dihydrobromidsalz wurde filtriert und getrocknet.
Der trockene Feststoff wurde danach in heißem Wasser gelöst, und
die Benzothiazol-freie Base wurde freigesetzt mit einer gesättigten
Natriumbicarbonat-Lösung.
Der weiße
Feststsoff wurde filtriert und getrocknet, und man erhielt das Rohprodukt,
das ohne weitere Reinigung verwendet wurde.
-
N,N'-Bis-(2-disulfidyl-4-methylphenyl-)gamma,
gamma'-diaminoisovaleriansäure: Einer
Suspension aus Ethyl-gamma, gamma'-[1,3-di-(2-imino-6-methylbenzthiazolyl-3-)]isovalerat
in 40 ml destilliertem Wasser wurden feste Kaliumhydroxid-Pellets (20,0 g;
0,037 Mol) zugesetzt, und die resultierende Lösung wurde 15 bis 24 Stunden
auf 120 °C
erhitzt. Nach mehreren Stunden des Erhitzens wurde aus der Suspension
eine klare Lösung.
Die Reaktionsmischung wurde in einem Eisbad abgekühlt und
mit 5,0 N Essigsäure
auf einen pH-Wert von 5,0 angesäuert
und die wässrige
Lösung
wurde mit drei 100 ml-Portionen Ethylacetat extrahiert. Die vereinigten
Ethylacetat-Extrakte wurden über
wasserfreiem Natriumsulfat getrocknet und filtriert. Das Lösungsmittel
aus dem Filtrat wurde unter verringertem Druck entfernt, und man
erhielt das Rohprodukt. Dieses Rohprodukt wurde an einer Silica-Gel-Säule unter
Verwendung einer Mischung aus Ethylacetat : Hexan (20 : 80) mit
1 % Essigsäure
als eluierendes Lösungsmittel
chromatographiert und man erhielt das Produkt in Form eines kristallinen
gelben Feststoffs.
-
N,N'-Bis-(2-disulfidyl-4-methylphenyl-)gamma,
gamma'-diaminoisovalerat-N-hdroxysuccinimid: N,N'-Bis-(2-disulfidyl-4-methylphenyl-)gamma,
gamma'-diaminoisovaleriansäure wurde
mit N-Hydroxysuccimid (NHS) und Dicyclohexylcarbodiimid (DCC) entweder
in Tetrahydrofuran (THF) oder Dimethylformamid (DMF) bei Raumtemperatur
umgesetzt. Nach dem Rühren über Nacht
bei Raumtemperatur wurde das Lösungsmittel
entfernt, und das Rohprodukt wurde mittels Säulenchromatographie an Silica-Gel
gereinigt.
-
B. Konjugat-Bildung
-
Diese
Chelat-Verbindung ist zugänglich
für die
Verwendung mit einem geeigneten trifunktionellen Linker, um ein
radiomarkiertes Annexin-Galactose-Cluster-Konjugat der vorliegenden Erfindung,
wie dies nachfolgend beschrieben wird, zu bilden.
-
Im
Handel erhältliches
N-epsilon-t-BOC-Lysin (Firma Sigma Chemical Company) wurde unter
Verwendung von Trifluoressigsäureanhydrid
in sein N-alpha-Trifluoracetamid-Addukt
umgewandelt. Die Aktivierung der Carbonsäure-Funktion, beispielsweise
mit BOP (Benzotriazol-1-yloxy-tris-(dimethyhnino-) phosphoniumhexafluorphosphat),
im Handel erhältlich
von der Firma Aldrich Chemical Co., und die Reaktion der aktivierten Einheit
mit dem einzig vorhandenen Amin an einem Galactose-Cluster, z.B.
wie er oben beschrieben wurde, erfordert eine Galactose-Cluster-trifunktionelle
Linker-Spezies. Das alpha-Amin des trifunktionellen Lysin-Linker-Bestandteils
der Galctose-Cluster-trifunktionelle Linker-Spezies wird deblockiert
unter Verwendung von methanolischem Natriumhydroxid. Die Umsetzung
mit dem N-Hydroxysuccinimidester des Chelat-Verbindungsmoleküls, das
wie in Teil A dieses Beispiels beschrieben hergestellt worden war,
bringt eine Galactose-Cluster- Chelat-Verbindung
trifunktionelle Linker-Spezies hervor. Die Entfernung der Schutzgruppe
von dem epsilon-Amin des trifunktionellen Lysin-Linker-Bestandteils
unter Verwendung von Trifluoressigsäure und anschließender Umsetzung
mit Bernsteinsäureanhydrid
stellt eine verfügbare
Carbonsäure-Funktionalität bereit, durch
welche das Annexin im Anschluss an die folgende Aktivierung der
Carbonsäure
(z.B. mit BOP) konjugiert werden kann.
-
BEISPIEL VIII
-
Verfahren zur
Herstellung eines Zellexpressions-Klons für Annexin V
-
Ein
Stamm-Klon, HPAP 1.6, wird beschrieben in „Funakoshi et al., Primary
Structure of Human Placental Anticoagulant Protein, Biochemistry,
26, 8087 – 8092
(1987)". Eine Polymerase-Kettenreaktion
(PCR) wurde verwendet, um das Annexin-Gen aus dem λHPAP 1.6-Stammklon zu amplifizieren.
Der Sense-Primer(CAT ATG GCA CAG CTT CTCA) enthielt eine NdeL-Restriktionsstelle
(unterstrichen) und die ersten 16 Nucleotide der Annexin-Leader-Sequenz,
beginnende mit dem ATG-Start-Codon.
Der Antisense-Primer (GGA TCC TTA GTC ATC TTC TCC ACA) kodierte
das Ende der Kodierungs-Sequenz, ein Stop-Codon (fett gedruckt)
und die BamHI-Restriktionsstelle
(unterstrichen). Das PCR-Produkt und das Plasmid pET-12a (2), die von der Firma Novagen
(Palo Alto, Kalifornien) erhalten wurden, wurden jeweils mit NdeL
und BamHI verdaut und miteinander mit T4-DNS-Ligase verbunden. Ein
Teil der Bindungs-Lösung
wurde in einen E. coli-Wirts-Stamm transformiert und auf Agar-Nährplatten, die Ampicillin enthielten,
selektiert. Das Plasmid von dem resultierenden Klon wurde bezeichnet
als pET-12a-PAP1-E287G, 7/16/93, Klon 1. Eine Didesoxy-DNS-Sequenzanalyse
zeigte, dass dieses Plasmid DNS enthielt, welche mit der Wild-Typ-Annexin-Sequenz übereinstimmte,
ausgenommen zwei Mutationen [855 G → A (stumm); 860 A → G (wandelt
Glu-287 in Gly-287 um)].
-
Die
Sequenzänderungen,
die in pET-12a-PAP-E287G, 7/16/93, Klon 1 vorhanden waren, wurden
in der folgenden Weise korrigiert: Das Plasmid wurde mit den Restriktionsenzymen
SfuL und BamHI verdaut, die ein Fragment von etwa 240 Basenpaaren,
das beide Mutationen enthielt, herausschnitten. Das Fragment wurde
ersetzt durch ein 240 Basenpaar-Fragment von einem unabhängigen Klon,
von dem bekannt war, dass er die Wild-Typ-Sequenz enthielt. Die
DNSs wurden gebunden und in einen E. coli-Wirt transformiert. Die
E. coli-Kolonien, die das DNS-Plasmid enthielten, wurden auf Ampicillin
enthaltenden Agar-Nährplatten
selektiert. Der resultierende Klon beherbergte das Plasmid pET-12a-PAP1,
3/7/94, Klon 1 (3).
Die DNS-Sequenz bestätigte,
dass die Annexin-kodierende Sequenzierung auf dem Plasmid exakt
mit der Wild-Typ-Sequenz übereinstimmte.
-
Der
Wirts-Stamm BL21 (DE3), der von der Firma Novagen erhalten wurde,
wurde mit dem Plasmid pET-12a-PAP1, 3/7/94, Klon 1 transformiert.
Ein Glycerin-Vorrat
wurde hergestellt aus dem resultierenden Transformanten BL21 (DE3)(pET-12A-PAP1, 3/7/94,
Klon 1) und bei ≤–65 °C gelagert.
-
Das
Wachstum von E. coli-BL21 (DE3) (pET-12A-PAP1, 3/7/94, Klon 1) in
einer flüssigen
Kultur bei 37°C
führte
zu einer Anreicherung des Annexin V-Proteins in dem E. coli-Cytoplasma-Raum.
-
BEISPIEL IX
-
Verfahren zur Modifzierung
(Ersetzen einer Aminosäure)
von Annexin V
-
Das
Plasmid pET-12a ist oben in Beispiel VIII beschrieben Annexin-Aminosäure-Varianten-Gene
wurden zwischen den NdeL- und BamHI-Restriktionsstellen auf dem pET-12a-Plasmid
angeordnet.
-
Eine
unabhängige
Modifikation von Annexin V wurde durch zielgerichtete Aminosäure-Änderung durchgeführt, unter
Verwendung der PCR-Amplifikation. Um diese Änderung zu erreichen, wurden
Oligonucleotide erzeugt, welche in der Antisense-Richtung des 3'-Endes des Annexins eingebaut waren.
Die besondere Änderung
von Annexin V, die in der vorliegenden Erfindung beschrieben wird,
ist ein Ersatz eines Cystein-Restes (Position 316) durch Alanin
oder durch eine andere Nicht-Schwefel enthaltende Aminosäure, wie
beispielsweise Serin. Um diese Änderung
zu erreichen, wurde die Nucleotid-Sequenz geändert von TGT in GCA. Das Oligonucleotid
schließt
eine BamHI-Enzym-Restriktionsstelle und ein Stop-Codon ein.
-
Das
Sense-Oligonucleotid ist eingebunden in die T7-Promoter-Region des
pET-12a, genau stromaufwärts von
der NdeL-Restriktionsstelle oder des 5'-Endes von Annexin V. Die nachfolgend
angegebenen Sequenzen sind die Antisense- bzw. Sense-Oligonucleotid-Sequenz:
-
-
Die
Oligonucleotide N×168
und T7 wurden synthetisiert unter Verwendung eines DNS-Synthesizers, Modell
381A (Applied Biosystem Inc., Modell 381 A, Foster City, CA). Nachdem
die Synthese abgeschlossen war, wurde bei beiden Oligonucleotiden
die Schutzgruppe entfernt gemäß Anhang
5 des Protokolls des oben genannten Herstellers. Die Reinigung wurde
durchgeführt
unter Verwendung einer SephadexTM-G-25-Säule (Firma
Pharmacia, Uppsala, Schweden).
-
Die
PCR-Reaktionen wurden durchgeführt
unter Verwendung der UITmaTM-Polymerase (Firma
Perkin Elmer, Norwalk, CT). Die Reaktionen wurden durchgeführt unter
Verwendung eines etwa gleich langen pET-12a-Annexin V-Templates,
30 p Mol der Oligonucleotide T7 und N×168-Oligonucleotide und 0,4
mM jedes Nucleotids. Eine Überschichtung
mit Mineralöl
war eingeschlossen. Die Reaktionsansätze wurden in einem Coy-Temperaturwechsler,
Modell 110 P angeordnet und 5 Minuten lang bei 94°C inkubiert.
Etwa 2 Minuten nach Beginn der Inkubation wurden 0,5 Einheiten Polymerase
zugesetzt. Die Reaktionen wurden anschließend 30 Mal einem Zyklus bei
94 °C 30
Sekunden lang, bei 55 °C
1 Minute lang und bei 74 °C
1 Minute lang unterworfen. Die Reaktionen wurden danach 5 Minuten
lang bei 74 °C
inkubiert und danach bei 15 °C
aufgesaugt.
-
Das
resultierende Ala-substituierte Annexin-Gen wurde danach gereinigt
unter Verwendung eines DNS-Reinigungs-Kits (promega MagicTM, PCR Preps-DNS-Purification System, Madison, WI) gemäß dem Protokoll
des Herstellers.
-
Annexin-Al1a-
und pET-12a wurden danach mit NdeL- und BamHI-(Gibco/BRL, Gaithersburg, MD) Restriktionsenzymen
verdaut. Die Abbauprodukte wurden danach mittels Agarose-Gel-Elektrophorese
gereinigt. Geeignete Banden wurden herausgeschnitten und unter Verwendung
eines GeneClean II®-Kits (Bio 101, Inc.,
La Jolla, CA) gemäß dem Protokoll
des Herstellers gereinigt. Annexin-Ala und pET-12a wurden danach durch
T4-DNS-Ligase (Promega) 12 bis 18 Stunden lang bei 4 °C miteinander
verbunden.
-
Annexin-Ala-pET-12a
wurden in einen E. coli-Stamm, DH5∞ maximal wirksame kompetente
Zellen (Gibco/BRL), transformiert. Jeder andere geeignete Wirts-Stamm für das pET-12a-Plasmid
kann ebenso verwendet werden. Die transformierten E. coli-Zellen
wurden auf einem LB-Agar (GibcoBRL) angeordnet, der mit 100 μg/ml Ampicillin
(Firma International Biotechnologies Inc., New Haven, CT) angereichert
war, und 12 bis 18 Stunden bei 37 °C inkubiert. Eine geeignete
Kolonie wurde danach selektiert und in eine Terrific Broth (GibcoBRL)
eingebracht, die mit 100 μg/ml
Ampicillin (International Biotechnologies Inc.) angereichert worden
war.
-
Mehrere
Kolonien von den LB-Agarplatten wurden selektiert und expandiert.
Das DNS-Plasmid wurde gereinigt unter Verwendung des Promega-Wizard-Miniprep-Reinigungs-Kits.
Sobald das DNS-Plasmid gereinigt war, wurden die Kolonien mittels Sequenz-Analyse
gescreent, um zu bestätigen,
dass ein besonderer selektierter K1on die Aminosäure-Modifikation oder -Änderung
enthielt.
-
BEISPIEL X
-
Herstellung
und Radiomarkierung eines modifizierten Annexins
-
Das
modifizierte Annexin, bei dem die zugänglichen Sulfhydryl-Gruppen
an dem N-Ende des Annexin-Moleküls
angehängt
sind, können
hergestellt werden in Übereinstimmung
mit den Verfahren, die dargelegt sind in „Tanaka et al., Biochem.,
35, 922 – 929
(1996)", und zwar
wie folgt:
-
Herstellung-eines
N-terminal verlängerten
Annexins V (Annexin V-N-6; auch als modifiziertes Annexin bezeichnet):
Zuerst wurde eine mutanten-Annexin V-cDNS (pANXVC-S) konstruiert,
bei dem das Cys3 1 6-Codon ersetzt wurde durch ein Ser-Codon, eine NdeL-Stelle
vor dem Initiator Met-Codon eingeführt war, und eine BamHI-Stelle nach dem Stop-Codon
eingeführt
war. Diese wurde konstruiert durch PCR unter Verwendung einer Annexin
V-cDNS (pPAP-I-1.6; Funakoshi et al., Biochemistry, 26, 5572 – 5578 (1987);
Cookson et al., Genomics, 20, 463 – 467 (1994)) als Template
und zweier Oligonucleotide, nämlich 5'-g·gaa·ttc·cat·atg·gca·cag·gtt·ctc·aga·ggc·act·gtg-3' und 5'-cgc·gga·tcc·tta·gtc·atc·ttc·tcc·gga·gag·cag-3'. Diese DNS wurde
in die NdeL- und BamHI-Klonierungs-Stellen in dem pET-12a-Plasmid
(Novagen, Madison, WI) gebunden. Ein Oligonucleotid (5'-t·atg·gca·tgt·gac·cat·tc-3') und sein inverses
Komplement (5'-t·aga·atg·gtc·aca·tgc·ca-3') wurden hergestellt,
um sechs Aminosäure-Reste (Met-Ala-Cys-Asp-His-Ser)
zu kodieren, mit NdeL-kompatiblen Überhängen an beiden Enden. Die zwei
komplementären
Oligonucleotide wurden eingebunden und das Produkt wurde in pANXVC-S
gebunden, welches mit NdeL verdaut worden war, um das pANXVC-S-N6-Plasmid
herzustellen. Die DNS-Sequenzierung diese Plasmids bestätigte, dass
die beabsichtigten Mutationen richtig eingeführt waren.
-
Dieses
Plasmid (pANXVC-S-N6) wurde danach in einen Escherichia coli-Stamm K-38 transformiert, der
das pGP-1-Plasmid enthielt, und zwar zur Produktion von rekombinantem
Protein durch Expression von dem T7-Promotor des Vectors. Die Zellen
wurden gezüchtet
bei 30 °C
in 3 1 einer 2 %igen L-Brühe
in 50 mM Kaliumphosphat, pH 7,4, 35 μM Kanamycin. Nach 4 h (OD600 = 0,4) wurden die Zellen einem Hitzeschock
ausgesetzt, indem man die Kultur-Glaskolben 20 Minuten lang in ein
42 °C warmes
Wasserbad stellte und die Kultivierung wurde 2 h lang bei 37 °C fortgesetzt.
Die Zellen wurden danach geerntet und über Nacht bei –20 °C gefroren
gelagert.
-
Die
aufgetauten Zellen wurden in 100 ml einer Lösung aus 50 mM Tris-HCI, pH
8,0, 10 mM CaCl2, 0,1 mM DFP, das 1 mg Leupeptin
und 1 mg Pepstatin enthielt, suspendiert und 5 Minuten lang mit
Ultraschall behandelt. Das Zell-Lysat wurde mit 5 μlDNase (10
Einheiten/ml) für
10 Minuten bei Raumtemperatur behandelt. Danach wurden Pellets durch
Zentrifugieren gesammelt, und Annexin V-N6 wurde aus den Pellets
unter Rühren
für 1 h
bei 4 °C
in 70 ml einer Lösung
aus 50 mM Tris-HCI, pH 8,0, 8 M Harnstoff, 0,1 M NH4Cl,
10 m M EDTA, 0,1 m M DFP, das 1 mg Leupeptin und 1 mg Pepstation
enthielt, extrahiert. Der durch Zentrifugieren erhaltene Überstand
wurde zweifach mit 50 mM Tris-HCI, pH 8,0 und 0,1 m M DTT verdünnt und
48 h lang bei 4 °C
stehengelassen. Die Probe wurde danach nacheinander gegen 150 ml,
500 ml und 210,1 M Natriumphosphat, pH 6,0, dialysiert. Die dialysierte
Probe wurde auf eine DEAE-Sepharose-Säule (1,4 × 18 cm) aufgebracht, die mit
0,1 M Phosphat, pH 6,0, equilibriert war. Nachdem die Säule gewaschen
worden war, wurden die Proteine eluiert mittels eines linearen Gradienten,
der gebildet wurde durch jeweils 100 ml 0 M und 0,5 M NaCl in Phosphat-Puffer.
Der Haupt-Protein-Peak, der eine antikoagulierende Aktivität enthielt,
wurde gepoolt, und Ammoniumsulfat wurde bis zu einer 80 %igen Sättigung
zugesetzt. Die Niederschläge,
die durch Zentrifugieren gesammelt wurden, wurden in 5 ml 50 mM
Natriumacetat, pH 5,6, 150 mM NaCl und 10 mM DTT gelöst und auf
eine Sephadex G-75-Säule
(2,5 × 90
cm) aufgebracht, die mit demselben Puffer mit Ausnahme von DTT equilibriert
war. Das Annexin V-N6 eluierte als einzelner Peak und es war homogen
an SDS-PAGE mit einer Molekularmasse von 36 kDa. Die Ausbeute betrug
35 mg Protein aus 3 1 Kulturmedium. Das modifizierte Annexin, das
wie oben beschrieben hergestellt worden war, wurde danach in Übereinstimmung
mit der Erfindung verwendet.
-
BEISPIEL XI
-
Herstellung
eines modifizierten Annexins V und seines Dimers
-
Durch
Zusatz zugänglicher
Sulfhydrylgruppen modifiziertes Annexin V und sein Dimer wurden
wie folgt hergestellt: Das existierende Plasmid pJ110 (auch bekannt
als pANXVC-S-N6) wurde in den E. coli-Stamm BL21 (DE3) transfektiert.
Eine cyctoplasmatische Expression erfolgte durch Wachstum über Nacht bis
zur Sättigung
in 4 × 1
1 einer T-Brühe.
Die Zellen wurden durch Zentrifugieren gewonnen. Eine Ultraschall-Behandlung
in einem Puffer wurde durchgeführt,
der aus 50 mM Tris-HCl, pH 7,2, 10 mM CaCl2 bestand. Es
wurde 20 Minuten lang bei 23.000 × g zentrifugiert, und der Überstand
wurde verworfen. Modifiziertes Annexin wurde aus dem Pellet freigesetzt
durch Suspendieren in 50 mM Tris-HCI, pH 7,2, 20 mM EDTA. Es wurde bei
23.000 × g
20 Minuten lang zentrifugiert, und das Pellet wurde verworfen. Die überstehende
Flüssigkeit wurde
mit RNase A (3 μg/ml)
für 2 h
bei Raumtemperatur behandelt. Es erfolgte eine Dialyse in Schläuchen mit
einem Molekulargewicht-Cutt off von 25.000 gegen 3 × 2120mM
Tris-HCI, pH 8,0 (gut durchlüftet).
Eine Teilmenge des vorhandenen modifizierten Annexins wurde in das
A5-Dimer durch spontane Disulfid-Bindungs-Bildung
aufgrund der Luftoxidation umgewandelt. Eine Reinigung erfolgte
durch Ionenaustausch-FPLC auf einer MonoQ HR10/10-Säule (Firma
Pharmacia) mit einem linearen Gradienten von 0 bis 0,5 M NaCl in
20 mM Tris-HCl (pH 8,0). Es wurde 1/30 Volumenteil 1 M 2-[N-Morpholino-]ethansulfonsäure (Firma
Sigma Chemical) zu den Fraktionen zugesetzt, um den pH-Wert bei
5,8 zu halten und eine spontane Disulfid-Bindungs-Bildung zu inhibieren.
Das Monomer wurde bei 0,22 M NaCl eluiert und das Dimer wurde bei
0,28 M NaCl eluiert. Diese Verfahrensweise ergibt etwa 5 mg sowohl
des reinen Monomers als auch des reinen Dimers pro Liter an Ausgangs-Kulturmaterial.
-
Sobald
das über
Disulfid-Bindungen gebundene Dimer von modifiziertem Annexin V produziert
ist, kann es im Hinblick auf seine Membran- und Thrombus-Bindungs-Aktivität sowie
im Hinblick auf pharmakokinetische Daten und Organaufnahme in Mäusen bewertet
werden. Seine Affinität
für Zellmembranen
wurde bestimmt durch direkte Titration (siehe nachfolgend (1)),
seine Kurve des Verschwindens aus dem Blut und seine Bioverteilung
wurde in Mäusen
bestimmt (siehe nachfolgend (2)), und das in vivo bestimmte Verhältnis der
Präsenz
in Thrombus : in Blut im Schweizeherz-Vorhof-Thrombus-Modell 30
Minuten und 120 Minuten nach der Injektion und im Vergleich mit
dem entsprechenden Verhältnis
für monomeres
Annexin V wurde bestimmt (siehe nachfolgend (3)).
-
- 1. Binden von modifiziertem Annexin V und Annexin
V-Dimer an Phosphatidylserin enthaltende Membranen: Sofern Derivate
mit bereits Tc-99m markiert waren, wurde jedes Derivat nicht mit
I-125 durch das Iodo-Gen-Verfahren markiert und durch Gelfiltration
gereinigt, wie dies früher
für Annexin
V beschrieben wurde (siehe Tait et al., J. Lab. Clin. Med., 123,
741 – 748
(1994)). Die Affinität
jedes Derivats gegenüber
Zellmembranen wurde gemessen durch direkte Titration von aufbewahrten
Erythrocyten mit freigesetztem Phosphatidylserin, wie dies früher beschrieben
wurde in „Taft
et al., J. Lab. Clin. Med., 123, 741 – 748 (1994)". Gebundener und
freier Ligand wurden durch Zentrifugation von Zellen durch eine
Siliconöl-Barriere getrennt.
Die Dissoziationskonstante wurde bestimmt durch Einpassen der Daten
in ein einfaches Modell des Bindens an homogene Stellen, wie dies
von Tait beschrieben wurde. Die Verwendung von künstlich behandelten Erythrocyten
mit hohen Konzentrationen an freigesetztem PS ist eine experimentelle
Erleichterung, die Ergebnisse liefert, die equivalent denjenigen
sind, die erhalten wurden mit aktivierten Blutplättchen mit freigesetztem PS.
Normale Erythrocyten haben in vivo kein extracellulär freigesetztes
PS (Taft et al., J. Lab. Clin. Med., 123, 741 – 748 (1994)).
- 2. Freisetzung aus dem Blut, Bioverteilung und Exkrektion in
Mäusen:
Ein intravenöser
Bolus von 10 μg/kg (0,3 μg pro 30
Gramm Maus) an jeweils mit I-125 markiertem oder mit Tc-99m-markiertem
Derivat (Dimer; Galactose-modifiziertes Annexin; Tc-99m-modifiziertes
Annexin; Tc-99m-Serylsuccinat-markiertes Annexin V) wurde injiziert.
Blutproben wurden zum Zeitpunkt von 1, 2, 3, 5, 7, 10, 15, 20, 30,
45, 60, 90 und 120 Minuten hergestellt. Die folgenden Organe wurden
von den Mäusen
jeweils zum Zeitpunkt von 30, 60 und 120 Minuten nach der Injektion
gewonnen: Gehirn, Herz, Aorta, Vena cava, Lunge, Milz, Leber, Magen,
Gedärme,
Niere, Blase, Skelettmuskel, Knochen und Blut. Die Ergebnisse sind
ausgedrückt
als % ID/g Gewebe und % ID/Organ.
- 3. Verhältnis
Thrombus/Blut in vivo in Schweinen: Es wurden etablierte Verfahrensweisen
angewendet, wie sie für
akute Vorhof-Thromben beschrieben sind (Stratton et al., Circulation,
92, 3113 – 3121
(1995)), und zwar für
das I-125 markierte Dimer, für
Galactose-modifiziertes Annexin und für Annexin V als Kontrollprobe.
Auf der Basis früherer
Erfahrungen mit nativem Annexin V in diesem Modell kann ein zweifacher
Unterschied in den Thrombus/Blut-Verhältnissen zwischen modifiziertem
und nativem Annexin V bei einem P-Wert von ≤ 0,05 im zweigängigen t-Test
mit einem Wert n von 5 Tieren dargelegt werden.
-
Sobald
ein Molekül
mit wünschenswerten
Eigenschaften identifiziert wurde, kann das Annexin V-Dimer direkt
als Fusionsprotein unter Verwendung eines existierenden Expressions-Systems
produziert werden, wie dies beschrieben wurde in Tait et al., J.
Biol. Chem., 270, 21594 – 21599,
wobei die beiden Annexin V-Einheiten über einen Peptid-Linker verbunden
sind. Dieses dimere Molekül
kann auch zusätzliche
funktionelle Stellen enthalten, wie beispielsweise eine endogene
Tc-99m-Chelatisierungsstelle
oder eine freie Sulfhydrylgruppe, an der die Bindung von Galactose
möglich
werden kann.
-
BEISPIEL XII
-
Endogene Radiomarkierung
von modifiziertem Annexin
-
Es
wurde eine Untersuchung des Radiomarkierens von modifziertem Annexin
(Annexin V-N-6 von Beispiel X) mit Tc-99m durchgeführt, wobei
man der Verfahrensweise von George et al., Proc. Natl. Acad. Sci. USA,
92, 8358 – 8362
(1995) folgte. Das modifizierte Annexin (160 μg) wurde mit 2,5 mCi Tc-99m-Pertechnetat zusammengegeben,
und zwar zusammen mit Methylendiphosphonat als Austausch-Ligand und Zinnionen
als Reduktionsmittel. Die Endkonzentration des Proteins, wie sie
verfügbar
war, betrug 200 μg/ml.
Jedoch führte eine
Inkubation bei einem pH-Wert von 10 für 30 Minuten bei 37 °C zu einer
Radiochemie-Ausbeute von 52 %. Diese offensichtliche Ausbeute war
konsistent mit den Ergebnissen, die nach Reinigung durch Gelfiltration erhalten
wurden, die eine Ausbeute von 56 % der eingesetzten Radioaktivität in 89
%iger radiochemischer Reinheit zeigten. Die Lagerung des gereinigten
Materials zeigte keine Änderung
der radiochemischen Reinheit bei Messung mittels TLC- und Zorbax-HPLC-Chromatographie
nach 18 h in PBS bei Raumtemperatur. Im Gegensatz dazu zeigte natives
Annexin V nur eine Markierung von 2 % unter denselben Bedingungen.
So zeigten die Ergebnisse, daß das
modifizierte Annexin V-Molekül
direkt mit Tc-99m markiert werden kann. Ein Fachmann mit üblichem
Sachverstand in diesem technischen Bereich kann das modifizierte
Annexin V-Molekül
für eine
Optimierung der Tc-99m-Radiomarkierungs-Ausbeute (Proteinkonzentration,
Austausch-Chelate, pH-Wert-Abhängigkeit,
Temperatur und Zeit) bewerten. Die Radiomarkierungs-Stabilität (Behandlung
mit Serum und unterschiedlichen Konzentrationen Cystein), die Zell-
und Thrombus-Bindung, die Freisetzung aus dem Blut und die Bioverteilungs-Eigenschaften,
sowie Katabolit-Formen
von ausgeschiedener Radioaktivität (HPLC
von biologischen Proben und Vergleich mit synthetischen Peptid-Tc-99m-Chelat-Standards
zur Indetifizierung). Geeignete Methoden schließen diejenigen ein, die zur
Charakterisierung verschiedener N,S-Amid-Thiolat-Chelatisierungsmittel
für Tc-99m-bifunktionelle
Protein- Radiomarkierungs-Mittel
verwendet wurden (Kasina et al., J. Nucl. Med., 32, 1445 – 1451 (1991);
Fritzberg et al., Proc. Natl. Acad. Sic. USA, 95, 4025 – 4029 (1988).
-
BEISPIEL XIII
-
Herstellung
eines radiomarkierten Anneacin-Konjugats mit einem Esteraseempfindlichen
Chelat
-
-
Experimentelle
Vorgehensweise: Annexin V wurde mit Tc-99m-N3S-Serylsuccinat über den
Weg mit vorgeformtem Chelat radiomarkiert, wie dies für Antikörper Fab-Fragmente
beschrieben ist (Srinivasan et al., J. Nucl. Med., 32, 1017 (Zusammenfassung)
(1991)). Die Anwendung dieses Ansatzes auf dem C5N2S2-Aktivester war ähnlich für beide
Antikörper-Proteine
(Kasina et al., J. Nucl. Med., 32, 1445 – 1441 (1991)) und Annexin
V (Stratton et al., Circulation, 92, 3113 – 3121 (1995)). In der vorliegenden
Verfahrensweise wurde der Serylsuccinat-Ester in dem Radiomarkierungs-Verfahren
ersetzt. Dieses Derivat wurde in vitro im Hinblick auf Markierungsstabilität, Membranbindung
und Gerinselbindung bewertet. Es kann in Mäuse injiziert werden, und seine
Aufnahme in Organen, einschließlich
Leber und Niere über
den Zeitraum von 10 Minuten bis 2 Stunden kann gemessen werden.
Dies zeigt die Geschwindigkeit der Organ-Aufnahme sowie die Rückhaltung
an, und ermöglicht
einen Vergleich des Zeitablaufs mit früheren Daten für das Amid-verknüpfte Tc-99m-Chelat.
-
Ein
Erfolg wird angezeigt durch eine signifikante Reduktion der Rückhaltung
von Radioaktivität
in der Leber. Eine erhöhte
Rate des Verschwindens aus den Organen im Anschluß an das
Aufnehmen von Tc-99m-Annexin V zeigt das Potential von verbesserten
Thrombus : Organ-Verhältnissen
innerhalb der Zeit im Anschluß an
die Bildgebung auf. Bildgebungs-Experimente können mit dem Schweine-Modell
durchgeführt werden,
um vergleichbare Verbesserungen unter klinischen Bedingungen vorherzusagen.
-
Markieren
von Annexin V mit Tc-99m-Serylsuccinat-Ester: Annexin V, das mit
dem Serylsuccinat-Ester konjugiert war, wurde mit Tc-99m nach der
Onco Trac-Kit-Verfahrensweise
markiert (Kasina et al., J. Nucl. Med., 32, 1445 – 1451 (1991))
und wurde dann an Annexin V unter Verwendung des vorher beschriebenen Verfahrens
zur Konjugation Tc-99m-N2S2-TFP
an Annexin V konjugiert (Stratton et al., Circulation, 92, 3113 – 3121 (1995)).
-
BEISPIEL XIV
-
Herstellung eines radiomarkierten
modifizierten Annexin V-Galactose-Cluster-Chelat-Konjugats
-
Ein
Maleimid-verknüpfter
Galactose-Cluster gemäß der vorliegenden
Erfindung kann spezifisch an die Sulfhydrylgruppe des modifizierten
Annexins V konjugiert werden. Nach Verifizierung der Stöchiometrie
der Galactose-Markierung wurde das Galactose-modifizierte Annexin
radioiodiert, und das Produkt wurde hinsichtlich seiner Bindung
in vitro an die Membran und einen Thrombus bewertet, und es wurden
pharmako-kinetische Daten und Organ-Aufnahme-Daten in Mäusen ermittelt.
Einer Aufrechterhaltung der Bindung und einem schnelleren Verschwinden
aus dem Blut folgte die in vivo-Bestimmung des Thrombus : Blut-Verhältnisses
im Schweine-Modell zum Zeitpunkt von 30 und 120 Minuten nach der
Injektion und im Vergleich zu entsprechenden Verhältnissen
für nicht-modifiziertes
Annexin V.
-
Die
Geschwindigkeit des Verschwindens des modifizierten Annexins V aus
dem Blut ohne Kompromisse beim Vermögen des modifizierten Annexins
V, sich an Zellmembranen oder Thromben in vitro zu binden, wird
verglichen mit den Werten von unmodifiziertem Annexin V. Das in
vivo-Verhältnis
Thrombus : Blut zum Zeitpunkt von 30 Minuten nach der Injektion
sollte höher
sein verglichen mit nativem Annexin V, und sollte genauso hoch oder
höher sein
zum Zeitpunkt von 120 Minuten nach der Injektion. Wenn jedoch die
Anwendung dieses Mechanismus auch das Hintergrundsignal von der
Leber zu speziellen Zeiten erhöht,
die für
eine Bildgebung ausgewählt
werden, kann der Einsatz des Mechanismus über einen Esteraseempfindlichen
Linker zur Beschleunigung der Ausscheidung von Radiomarkierung aus
der Leber verwendet werden, um das Hintergrundsignal zu senken.
Die weiteren Beispiele beziehen sich auf weitere Ausführungsformen
der vorliegenden Erfindung.
-
-
-
-
-
Herstellung von Methyl-6-bromhexanoat
(1)
-
In
einen 2 Liter-Rundkolben, der mit 99,7 g (0,511 Mol) 6-Bromhexanoat
(Aldrich Chemical Co., Milwaukee, Wisconsin) und 1 Liter Methanol
befüllt
war, wurde 1 bis 2 Minuten lang Chlorwasserstoffgas eingeblasen.
Die Mischung wurde 18 h lang bei 20 bis 30 °C gerührt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde mit 500 ml Diethylether verdünnt und mit 150 ml entionisiertem
Wasser, 200 ml gesättigtem
Natriumbicarbonat und danach nochmals mit 200 ml entionisiertem
Wasser gewaschen. Die organische Phase wurde über wasserfreiem Magnesiumsulfat
getrocknet, filtriert und mittels Rotationsverdampfung konzentriert.
Der Rückstand
wurde unter Vakuum verdampft und dies ergab 99,6 g des Produktes (1)
in Form eines farblosen Öls
(93 %): Siedepunkt = 93 bis 96 °C
bei 3 mm Hg: 1H-NMR (d6-DMSO);
d 3,57 (3H, s), 3,51 (2H, t), 2,30 (2H, t), 1,78 (2H, Pentett) und
1,62 bis 1,28 (4H, m) ppm.
-
Herstellung von Methyl-6-aminohexanoat-hydrochlorid
(2)
-
In
einen 2 Liter-Rundkolben, der mit 101,3 g (0,722 Mol) 6-Aminohexanoat
(Aldrich Chemical Co.) in 1 Liter Methanol befällt war, wurde 3 bis 4 Minuten
lang Chlorwasserstoffgas eingeblasen. Die Mischung wurde 16 h lang
bei 20 bis 30 °C
gerührt
und danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde zweimal mit 500 ml Methanol verdünnt und erneut konzentriert
(< 0,5 mm Hg).
Dies ergab 140,1 g des Produktes (2) in Form eines weißen Feststoffes
(100 %): 1H-NMR (d6-DMSO);
d 9,40 (1H, breites Triplett), 3,57 (3H, s), 3,15 (2H, Quartett),
2,29 (2H, t), 1,60 bis 1,38 (4H, m) und 1,32 bis 1,19 (2H, m) ppm.
-
Herstellung von Methyl-6-(trifluoracetamido-)hexanoat
(3)
-
Einem
2 Liter-Rundkolben, der mit 100,2 g (0,552 Mol) Aminhydrochlorid
(2) und 1 Liter Methanol befällt
war, wurden 100 g (0,703 Mol) Ethyltrifluoracetat und anschließend 120
ml (0,861 Mol) Triethylamin zugesetzt. Die Mischung wurde 19 h lang
bei 20 bis 30 °C
gerührt
und danach mittels Rotationverdampfung konzentriert. Der Rückstand
wurde mit 500 ml Diethylether verdünnt und danach filtriert. Das
Filtrat wurde mit 3 × 300
ml Aliquota 1 N wässriger
HCl, 200 ml entionisiertem Wasser, 2 × 200 ml Aliquot gesättigter
wäßriger Natriumbicarbonat-Lösung und
letztendlich mit 200 ml entionisiertem Wasser gewaschen. Die organische
Phase wurde über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wurde unter Vakuum destilliert und ergab 115,8 g des Produktes (3)
in Form eines farblosen Öls:
Siedepunkt = 113 bis 116 °C
bei 120 mm Hg: 1H-NMR (d6-DMSO);
d 3,57 (3H, s), 2,75 (2H, m), 2,29 (2H, t), 1,60 bis 1,40 (4H, m)
und 1,37 bis 1,19 (2H, m) ppm.
-
Herstellung von N,N-Bis-(5-methoxycarbonylpentyl-)amin-hydrochlorid
(4)
-
Einem
5 Liter-Dreihalskolben, der mit einem Rückflußkühler ausgestattet war, der
mit einem Gasblasenzähler
verbunden war, und der mit 20,9 g 60 % Natriumhydroxid (0,523 Mol)
in 1 Liter wasserfreiem Dioxan befällt war, wurden 100 g (0,416
Mol) sekundäres
Amid 3 in 200 ml trockenem Dioxan über eine Zeitdauer von 20 Minuten
zugesetzt. Die Mischung wurde 1 h lang bei 20 bis 30 °C gerührt, und
danach wurden 130 g (0,622 Mol) Bromid 1 in 100 ml Dioxan zugesetzt.
Die Mischung wurde unter Rückfluß erhitzt
und 7 h lang gerührt. Zusätzlich wurden
10 g der Verbindung 1 zugesetzt, und die resultierende Mischung
wurde für
weitere 15 h gerührt.
Die Mischung wurde abgekühlt
und mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 600 ml 1 N wäßriger HCl
verdünnt
und mit 1 Liter Ethylacetat extrahiert. Die organische Phase wurde
danach mit 250 ml entionisiertem Wasser, 250 ml 5 %iger wäßriger Natriumbisulfit-Lösung und
letztendlich mit 250 ml entionisiertem Wasser gewaschen. Die organische
Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und konzentriert.
-
Der
Rückstand
wurde mit 300 ml entionisiertem Wasser und 500 ml Methanol verdünnt und
mit 200 ml 10 N wäßrigem Natriumhydroxid
behandelt. Die Mischung wurde 16 h lang bei 20 bis 30 °C gerührt und
mittels Rotationsverdampfung zu einem dicken Syrup konzentriert.
Der Rückstand
wurde mit 800 ml entionisiertem Wasser verdünnt und mit 200 ml konzentrierter
HCl auf einen pH-Wert von 1 bis 2 angesäuert. Die Mischung wurde danach
mit 3 × 300
ml Aliquots Diethylether gewaschen und die wäßrige Phase wurde danach mittels Rotationsverdampfung
zu einem dicken Sirup konzentriert. Der Rückstand wurde mit 1 Liter trockenem
Methanol verdünnt
und mittels Rotationsverdampfung erneut konzentriert. Der Rückstand
wurde mit 1 Liter trockenem Methanol verdünnt und danach wurde Chlorwasserstoffgas
2 bis 3 Minuten lang in die Mischung eingeblasen. Die Mischung wurde
18 h lang bei 20 bis 30 °C
gerührt,
und danach durch ein Celite-Filter (hergestellt durch die Firma
J.T. Baker) vakuumfiltriert. Die Feststoffe wurden mit 200 ml Methanol
gespült.
Die vereinigten Filtrate wurden konzentriert. Der Rückstand
wurde mit 1 Liter Methanol verdünnt
und Chlorwasserstoffgas wurde erneut 2 bis 3 Minuten lang in die
Mischung eingeblasen. Die Mischung wurde 3 h lang gerührt und
danach konzentriert. Der Rückstand
wurde mit 1 Liter Methanol verdünnt
und 10 g Aktivkohle wurden zugesetzt. Die Mischung wurde 30 Minuten
lang gerührt
und danach durch ein Celite-Filter vakuumfiltriert. Die Feststoffe
wurden mit 100 ml Methanol gewaschen und die vereinigten Filtrate
wurden konzentriert. Der Rückstand
wurde in heißem
2-Propanol gelöst
und man ließ ihn
danach erneut, zuerst bei Raumtemperatur und dann mittels Verwendung
eines Eisbades, kristallisieren. Die Feststoffe wurden filtriert
und mit 3 × 75
ml Aliquots kaltem 2-Propanol gespült. Die Feststoffe wurden an
der Luft getrocknet und dies ergab 70,5 g des Produktes (4) in Form eines
weißen
Feststoffes. Die Filtrate wurden vereinigt und konzentriert. Der
Rückstand
wurde erneut in 200 ml 2-Propanol kristallisiert und ergab zusätzlich 15,3
g des Produktes von insgesamt 85,8 g (67 %): 1H-NMR (d6-DMSO);
d 8,69 (2H, breit), 3,57 (6H, s), 2,82 (4H, m), 2,30 (4H, t), 1,67
bis 1,43 (8H, m) und 1,28 bis 1,19 (4H, m) ppm; 1H-NMR
(CD3OD); d 3,66 (6H, s), 3,42 (4H, t), 2,34
(4H, t), 1,75 bis 1,55 (8H, m) und 1,45 bis 1,25 (4H, m) ppm.
-
Herstellung von N-BOC-S-Aminopentanol
(5)
-
Einem
2 Liter-Dreihals-Rundkolben, der an dem mittleren Hals mit einem
500 ml Zugabe-Trichter und an einem Seitenhals mit einem Übergangsstück zum Entlüften eines
Gasblasenzählers
ausgestattet war, wurden 40 g (0,388 Mol) 5-Aminopentanol in 500
ml trockenem Acetonitril zugesetzt. Danach wurden 84,5 g (0,387 Mol)
Di-t-butyldicarbonat
in 400 ml trockenem Acetonitril über
einer Zeitdauer von 50 Minuten zugesetzt. Die Mischung wurde 15
h lang bei 20 bis 30 °C
gerührt
und danach konzentriert. Der Rückstand
wurde mit 600 ml Ethylacetat verdünnt und mit 2 x 200 ml Aliquots
0,5 N wäßriger HCl
und 2 × 200
ml Aliquot entionisiertem Wasser gewaschen. Die organische Phase
wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und zunächst mittels
Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 74,5 g des Produktes (5) in Form eines nahezu farblosen Öls (88 %): 1H-NMR (d6-DMSO);
d 6,72 (1H, breites Triplett), 4,31 (1H, t), 3,43 bis 3,27 (2H,
m), 2,87 (2H, Quartett) und 1,45 bis 1,10 (15H, s und Multiplett)
ppm; 1H-NMR (CDCl3);
d 4,58 (1H, breites s), 3,65 (2H, t), 3,13 (2H, Quartett) und 1,70
bis 1,30 (15H, Singlett und Multiplett) ppm: Dünnschichtchromatographie (Sichtbarmachung
mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf =
0,28 (Methylenchlorid/Methanol (95:5)).
-
Herstellung von N-BOC-5-Aminopentyltoluolsulfonat
(6)
-
Einem
1 Liter-Rundkolben, der mit 74,5 g (0,366 Mol) N-BOC-Aminopentanol
(6) in 400 ml Methylenchlorid befüllt war, wurden 45 ml wasserfreies
Pyridin und anschließend
74,1 g (0,389 Mol) p-Toluolsulfonylchlorid zugesetzt. Die Mischung
wurde 17 h lang bei Raumtemperatur gerührt, mit 200 ml Methylenchlorid
verdünnt
und mit 400 ml 0,5 N HCl, 2 × 200
ml Aliquots 0,5 N HCl und 2 × 100
ml Aliquots entionisiertem Wasser gewaschen. Die organische Phase
wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und konzentriert.
Der Rückstand
wurde an einer Silica-Gel-Säule
(11 × 23
cm) chromatographiert, zunächst
mit Methylenchlorid und danach mit Ethylacetat/Methylenchlorid (3
: 97) eluiert. Die Fraktionen, die das Produkt enthielten, wurden
vereinigt und zunächst
mittels Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 82,13 g des Produktes (6) in Form eines weißen Feststoffes: 1H-NMR (CDCl3); d
7,77 (2H, d), 7,31 (2H, d), 4,45 (1H, breites s), 3,98 (2H, t),
3,03 (2H, t), 2,41 (3H, s) und 1,80 bis 1,20 (15H, Singlett und
Multiplett) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf
= 0,50 (Ethylacetat/Methylenchlorid (3 : 97)).
-
Herstellung von 1-b, 3,4,6-Tetra-O-acetyl-N-acetyl-galactosamin
(7)
-
Einem
500 ml-Rundkolben, der mit 25,0 g (116 mMol) Galactosaminhydrochlorid
(Sigma Chemical Co., St. Louis, Missouri) befüllt war, wurden 180 ml wasserfreies
Pyridin und anschließend
115 ml Essigsäureanhydrid
(1,22 Mol) zugesetzt. Die Mischung wurde 44 h lang bei 20 bis 30 °C gerührt und
danach in einen 2 Liter-Becher
gegossen, der 600 g Eis und 200 ml entionisiertes Wasser enthielt.
Die Mischung wurde 10 bis 15 Minuten lang bei Raumtemperatur gerührt und
danach vakuumfiltriert. Die sammelten Feststoffe wurden mit 4 × 100 ml
Aliquotsentionisiertem Wasser gewaschen, 2 h lang an der Luft getrocknet
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) 14 h lang getrocknet; dies
ergab 39,8 g des Produktes in Form eines weißen Feststoffes (88 %): 1H-NMR (d6-DMSO);
d 7,89 (1H, d), 5,63 (1H, d), 5,16 (1H, d); 5,07 (1H, dd), 4,28 bis
3,92 (4H, m), 2,11 (3H, s), 2,02 (3H, s), 1,99 (3H, s), 1,90 (3H,
s) und 1,88 (3H, s) ppm.
-
Herstellung von 3,4,6-Tri-O-acetvlgalactosamin-1-b-pseudothioharnstoff-hydrochlorid
-
Einem
1 Liter-Rundkolben, der mit 39,8 g (102 mMol) der Verbindung 7 befüllt war,
wurden 400 ml Acetylchlorid zugesetzt. Die Mischung wurde 64 h lang
bei 47 bis 48 °C
gerührt.
Die Mischung wurde konzentriert und danach zweimal mit 200 ml Methylenchlorid
verdünnt
und erneut zunächst
mittels Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) erneut
konzentriert; dies ergab 40,2 g des Rohproduktes (8) in Form eines
dunkel-bernsteinfarbenen schaumigen Feststoffes: 1H-NMR
(CDCl3); d 6,24 (1H, d), 5,61 (1H d), 5,43
(1H, dd), 5,27 (1H, dd), 4,83 bis 4,71 (1H, m), 4,48 (1H, t), 4,22
bis 4,01 (2H, 2 dd), 2,15 (3H, s), 2,02 (3H, s), 2,00 (3H, s) und
1,98 (3H, s) ppm. Dem rohen Chlorid (8) in einem 1 Liter-Rundkolben wurden
9,3 g (122 mMol) Thioharnstoff und 150 ml Aceton zugesetzt. Die
Mischung wurde unter Rückfluß 40 Minuten
lang gerührt
und danach in einem Eisbad 30 Minuten lang abgekühlt und danach vakuumfiltriert.
Die gesammelten Feststoffe wurden mit 2 × 75 ml Aliquots Aceton gespült. Die
Feststoffe wurden danach 45 Minuten lang an der Luft getrocknet
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) 2 h lang getrocknet; dies
ergab 33,0 g des Produktes (9) in Form eines hellbeigen Feststoffes
(74 % Gesamtausbeute aus 7): 'H-NMR
(d6-DMSO); d 9,38 und 9,12 (2 breite Singletts,
3H), 8,36 (1H, d), 5,56 (1H, d), 5,34 (1H, d), 5,01 (1H, dd), 4,38
(1H, t), 4,22 bis 4,00 (3H, m), 2,11 (3H, s), 2,01 (3H, s), 1,92
(3H, s) und 1,81 (3H, s) PPm.
-
Herstellung von 1-b-Mercapto-3,4,6-tri-O-acetyl-N-acetylgalactosamin
(10)
-
Einem
1 Liter-Rundkolben, der mit 30,0 g (67,9 mMol) des Pseudothioharnstoffes
(9) in 175 ml Methylenchlorid und 175 ml entionisiertem Wasser befüllt war,
wurden 7,08 g (37,24 mMol) Natriummetabisulfit zugesetzt und anschließend vorsichtig
10,2 g (74,5 mMol) Kaliumcarbonat zugesetzt. Die Mischung wurde
40 Minuten lang bei Raumtemperatur gerührt und die Mischung wurde
danach in einen 500 ml-Scheidetrichter überführt. Die Schichten wurden getrennt
und die wäßrige Phase
wurde danach mit 2 × 125
ml Aliquotss Methylenchlorid gewaschen. Die organischen Extrakte
wurden vereinigt, über
wasserfreiem Natriumsulfat getrocknet, filtriert und konzentriert.
Dies ergab 24,2 g des Produktes (10) in Form eines sehr blassgelben (cremefarbenen)
Feststoffes (98 %):1H-NMR (CDCl3);
d 6,24 (1H, d), 5,61 (1H, d), 5,43 (1H, dd), 5,27 (1H, dd), 4,83
bis 4,71 (1H, m), 4,48 (1H, t), 4,22 bis 4,01 (2H, 2 dd), 2,15 (3H,
s), 2,02 (3H, s), 2,00 (3H, s) und 1,98 (3H, s) ppm.
-
Herstellung von 3,4,6-Tri-O-acetyl-N-acetylgalactosamin-1-a-S-[5'-thiopentyl-N-BOC-amin] (11)
-
Einem
1 Liter-Rundkolben, der mit 24,2 g (66,6 mMol) des Thiols (10) unter
Stickstoffatmosphäre
befüllt
war, wurden 350 ml trockenes Acetonitril zugesetzt. Die Mischung
wurde über
eine Zeitdauer von 20 Minuten auf 40 bis 42 °C erhitzt, bis sich letztendlich
die Feststoffe gelöst
hatten. 1,8-Diazabicyclo[5.4.0]undec-7en (DBU; im Handel erhältlich von
der Firma Aldrich Chemical Company; 10,5 ml, 70,2 mMol) wurden danach
zugesetzt und die Mischung wurde 20 Minuten lang gerührt. Danach wurden
24,0 g (67,1 mMol) des Tosylats 6 in 75 ml Acetonitril über eine
Zeitdauer von 3 bis 4 Minuten zugesetzt. Die resultierende Mischung wurde
1,5 h lang bei 40 bis 25°C
gerührt
und danach konzentriert. Der Rückstand
wurde mit 400 ml Methylenchlorid verdünnt und zuerst mit 250 ml 0,5
N wäßriger HCl
und danach mit 250 ml einer 5 %igen wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesium getrocknet, vakuumfiltriert und mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde an einer Silica-Gel-Säule
(21 × 7
cm) (hergestellt von der Firma E.M. Merck) chromatographiert und
mit Ethylacetat/Hexan/Ethanol (55 : 42,5 : 2,5) eluiert. Die Fraktionen,
die das Produkt enthielten, wurden vereinigt, konzentriert und erneut
an einer RP-18-Silica-Gel-Säule
(21 × 7
cm) (hergestellt von der Firma J.T. Baker) chromatographiert und
mit jeweils 500 ml Methanol/Wasser im Verhältnis 50 : 50; 55 : 45; 60
: 40; 65 : 35 und 70 : 30 und anschließend mit Methanol/Wasser (75
: 25) eluiert, bis das gesamte Produkt von der Säule eluiert war. Die Fraktionen,
die das Produkt enthielten, wurden vereinigt und konzentriert. Der
Rückstand
wurde mit 500 ml Methylenchlorid verdünnt und mit wasserfreiem Magnesiumsulfat
behandelt. Die Mischung wurde vakuumfiltiert und das Filtrat wurde
zuerst mittels Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 17,9 g des Produktes (11) in Form eines schaumigen weißen Feststoffes: 1H-NMR (d6-DMSO);
d 9,38 und 9,12 (2 breite Singletts, 3H), 8,36 (1H, d), 5,56 (1
H, d), 5,34 (1H, d), 5,01 (1H, dd), 4,38 (1H, t), 4,22 bis 4,00
(3H, m), 2,11 (3H, s), 2,01 (3H, s), 1,92 (3H, s) und 1,81 (3H,
s) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit p-Anisaldehyd-Spray und Hitze); Silica-Gel,
Rf = 0,50 (Ethylacetat/Hexan/Ethanol (57 : 40,5 : 2,5)); RP-18-Silica-Gel, Rf= 0,21
(Methanol/Wasser (65 : 35)).
-
Herstellung von Methyl-6-methylaminohexanoat-hydrochlorid
(12)
-
Ein
2 Liter-Dreihals-Rundkolben, der mit 8,77 g 60 % NaH in Mineralöl (219 mMol;
1,1 Equivalente) in 500 ml wasserfreiem Tetrahydrofuran befüllt war,
wurde mit einem 500 ml Zugabetrichter am mittleren Hals ausgestattet.
Danach wurden 34,5 g (144 mMol) des sekundären Amids 3 in 300 ml wasserfreiem
Tetrahydrofuran über
eine Zeitdauer von 30 Minuten zugesetzt. Die Mischung wurde weitere
50 Minuten lang gerührt
und anschließend
wurden 22,6 ml (363 mMol) Iodmethan zugesetzt. Die Mischung wurde
23 h lang bei Raumtemperatur gerührt
und danach in einen 2 Liter-Rundkolben überführt und
mittels Rotationsverdampfung konzentriert. Der Rückstand wurde mit 400 ml 1
N wäßriger HCl
behandelt und anschließend
mit 300 ml Ethylacetat und danach mit 2 × 200 ml Aliquotss Ethylacetat
extrahiert. Die organischen Extrakte wurden vereinigt und zuerst
mit 3 × 125
ml Aliquotss 5 %iger wäßriger Natriumthiosulfat-Lösung und
anschließend
mit 100 ml entionisiertem Wasser gewaschen. Die organische Phase
wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und konzentriert.
Der Rückstand
wurde in 250 ml Methanol gelöst
und erneut konzentriert. Der Rückstand
wurde mit 250 ml Methanol verdünnt
und mit 50 ml 10 N wäßrigem Natriumhydroxid
und anschließend
mit 100 ml entionisiertem Wasser behandelt. Die Mischung wurde bei
Raumtemperatur 17 h lang gerührt, mit
weiteren 50 ml entionisiertem Wasser verdünnt und danach mit 3 × 200 ml
Aliquotss Hexan gewaschen. Die wäßrige Phase
wurde mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 500 ml Methanol verdünnt
und Chlorwasserstoffgas wurde 2 bis 3 Minuten lang in die Mischung
eingeblasen (10 g). Die Mischung wurde 3 h lang bei Raumtemperatur
gerührt
und danach vakuumfiltriert und mittels Rotationsverdampfung konzentriert.
Dem Rückstand
wurden 500 ml Methanol zugesetzt und anschließend wurde nochmals Chlorwasserstoffgas
2 bis 3 Minuten lang in die Mischung eingeblasen (9,2 g). Die Mischung
wurde 18 h lang bei Raumtemperatur gerührt. Die Mischung wurde in
einem Eisbad abgekühlt
und danach vakuumfiltriert. Das Filtrat wurde mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde zweimal mit 250 ml Methanol verdünnt und erneut konzentriert.
Der Rückstand
wurde mit 300 ml 2-Propanol verdünnt
und mit 4 g Aktivkohle 30 Minuten lang behandelt. Die Mischung wurde
durch ein Celite-Filter vakuumfiltriert und die Feststoffe wurden
mit 2 × 75
ml Aliquotss 2-Propanol gespült.
Die Filtrate wurden vereinigt und zuerst mittels Rotationsverdampfung
und anschließend
unter vollem Vakuumpumpen-Druck konzentriert. Der Rückstand
wurde mit 250 ml Methanol verdünnt
und erneut mittels Rotationsverdampfung und anschließend unter
vollem Vakuumpumpen-Druck konzentriert. Der Rückstand wurde mit 250 ml Methanol verdünnt und
Chlorwasserstoffgas wurde 1 bis 2 Minuten lang in die Mischung eingeblasen
(5,0 g). Die Mischung wurde bei Raumtemperatur 2 h lang gerührt und
danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde zweimal mit 250 ml Methanol verdünnt und zuerst mittels Rotationsverdampfung
und letztendlich unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert;
dies ergab das Produkt (12) in Form eines sehr hellgelben, schaumigen
Feststoffes (85 %): 1H-NMR (d6-DMSO);
d 8,72 (2H, breites s), 3,58 (3H, s), 2,82 (2H, m), 2,49 (3H, s),
2,32 (2H, t), 1,68 bis 1,45 (4H, m) und 1,39 bis 1,21 (2H, m) ppm; 1H-NMR (CD3OD); d
3,63 (3H, s), 2,97 (2H, t), 2,34 (2H, t), 1,75 bis 1,56 (4H, m)
und 1,49 bis 1,31 (2H, m) ppm.
-
Herstellung von N-BOC-N,N-Bis-(5-methoxycarbonylpentyl-)amin
(15)
-
Einem
500 ml-Rundkolben, der mit 6,34 g (29,1 mMol) Di-t-butyldicarbonat
und 9,00 g (29,1 mMol) N,N-Bis-(5-methoxycarbonylpentyl-)amin-hydrochlorid
4 befüllt
war, wurden 125 ml wasserfreies Acetonitril und anschließend 7,5
ml Triethylamin zugesetzt. Die Mischung wurde 22 h lang bei Raumtemperatur
gerührt und
anschließend
mittels Rotationsverdampfung konzentriert. Der Rückstand wurde mit 300 ml Ethylacetat verdünnt und
mit 2 × 100
ml Aliquotss 0,1 N wäßrige HCl,
100 ml entionisiertem Wasser und 100 ml einer 5 %igen wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
Magnesiumsulfat getrocknet, vakuumfiltriert und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert; dies ergab
10,5 g des Produktes (15) als nahezu farbloses Öl (97 %): 1H-NMR
(d6-DMSO); d 3,57 (6H,s), 3,07 (4H, t),
228 (4H, t), 1,60 bis 1,10 und 1,37 (21H, m und s) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf
= 0,33 (Ethylacetat/Hexan (20 : 80)); RP-18-Silica-Gel, Rf= 0,17
(Methanol/Wasser (70 : 30)).
-
Herstellung von N-BOC-N,N-Bis-(5-hydroxycarbonylpentyl-)amin
(16)
-
Einem
500 ml-Rundkolben, der mit 10,5 g Bis-methylester 15 befällt war,
wurden 75 ml Methanol und anschließend 75 ml 1 N wäßriges Natriumhydroxid zugesetzt.
Die Mischung wurde 16 h lang bei Raumtemperatur gerührt und
danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 75 ml entionisiertem Wasser verdünnt und der pH-Wert der resultierenden
Lösung
wurde durch langsame Zugabe von etwa 75 ml 1 N wäßriger HCl auf 2,0 bis 2,5
eingestellt. Danach wurden 200 ml Ethylacetat zugesetzt und die
Mischung wurde 3 Minuten lang heftig gerührt. Die Mischung wurde in
einen Scheidetrichter überführt und
die Schichten wurden getrennt. Die wäßrige Phase wurde mit 2 × 150 ml
Aliquotss Ethylacetat extrahiert. Die organischen Extrakte wurden
vereinigt, über
wasserfreiem Magnesiumsulfat getrocknet und vakuumfiltriert. Die Filtrate
wurden zuerst mittels Rotationsverdampfung und anschließend unter
vollem Vakuumpumpen-Druck (< 0,5
mm Hg) konzentriert; dies ergab 9,52 g des Produktes in Form eines
viskosen, nahezu farblosen Öls
(98 %): 1H-NMR (d6-DMSO);
d 3,07 (4H, t), 2,28 (4H, t), 1,58 bis 1,10 und 1,37 (21H, m und
s) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); RP-18-Silica-Gel,
Rf = 0,44 (Methanol/UWasser (70 : 30)).
-
Herstellung von N-BOC-N,N-Bis-(N',N'-bis-(5-methoxycarbonylpentyl-)5-carbamylpentyl-)amin
(17)
-
Einem
1 Liter-Rundkolben, der mit 9,52 g (27,6 mMol) Bis-Säure (16)
in 250 ml wasserfreiem Dimethylformamid befüllt war, wurden 19,0 g 861,3
mMol) N,N-Bis-(5-methoxycarbonylpentyl-)amin-hydrochlorid (4)
und anschließend
30 ml Triethylamin zugesetzt. Während
die Mischung gerührt
wurde, wurden 25,7 g (58,1 mMol) BOP zugesetzt. Die resultierende
Mischung wurde 14 h lang bei Raumtemperatur gerührt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde mit 750 ml Ethylacetat verdünnt und mit 250 ml 0,2 N wäßriger HCl,
100 ml 0,1 N wäßriger HCl,
100 ml entionisiertem Wasser und 2 × 100 ml Aliquots 5 %iger wäßriger Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und mittels
Rotationsverdampfung konzentriert. Der Rückstand wurde an einer Silica-Gel-Säule (9 × 21 cm)
chromatographiert und zuerst mit 70 % Ethylacetat/Hexan und danach mit
100 % Ethylacetat eluiert. Die Fraktionen, die das Produkt (17)
enthielten, wurden vereinigt und mittels Rotationsverdampfung konzentriert.
Der Rückstand
wurde an einer RP-18-Silica-Gel-Säule (7 × 23 cm)
chromatographiert und zuerst mit Methanol/Wasser (75 25), danach
mit Methanol/Wasser (80 : 20) und letztendlich mit Methanol/Wasser
(85 15) eluiert. Die Fraktionen, die das Produkt enthielten, wurden
vereinigt und zuerst mittels Rotationsverdampfung und anschließend unter
vollem Vakuumpumpen-Druck konzentriert. Der Rückstand wurde mit 500 ml Diethylether
verdünnt
und die resultierende Lösung
wurde mit wasserfreiem Magnesiumsulfat getrocknet. Die Mischung
wurde vakuumfiltriert und das Filtrat wurde zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert; dies ergab
17,80 g des Produktes (17) in Form eines nahezu farblosen, viskosen Öls (75 %): 1H-NMR (d6-DMSO);
d 3,57 (12H, s), 3,18 und 3,07 (12H, 2 t), 2,32 bis 2,16 (12H, m),
1,61 bis 1,09 und 1,37 (45H, m und s) ppm: Dünnschichtchromatographie (Sichtbarmachung
mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf = 0,50 (Ethylacetat);
RP-18-Silica-Gel, Rf = 0,30 (Methanol/Wasser (85 15)).
-
Herstellung von N-BOC-N,N-Bis-(N',N'-bis-(5-hydroxycarbonylpentyl-)5-carbamyl-pentyl-)amin (18)
-
Einem
500 ml-Rundkolben, der mit 7,88 g (9,20 mMol) Tetramethylester (18)
in 75 ml Methanol befällt war,
wurden 70 ml 1 N wäßriges Natriumhydroxid
zugesetzt. Die Mischung wurde 16 h lang bei Raumtemperatur gerührt und
danach mittels Rotationsverdampfung zu einem dicken Sirup konzentriert.
Der Rückstand wurde
mit 50 ml entionisiertem Wasser verdünnt und unter heftigem Rühren wurde
der pH-Wert der Lösung durch
langsame Zugabe von etwa 70 ml 1 N wäßriger HCl auf 2 bis 2,5 eingestellt,
wobei das Produkt (18) in dem Verfahren als Öl austrat (eine flüssige Phase
trennte sich von einer anderen flüssigen Phase). Die Mischung
wurde mit 200 ml 2-Propanol/Methylenchlorid
(3 : 1) und anschließend
mit 3 × 100
ml Aliquots 2-Propanol/Methylenchlorid
(3 : 1) extrahiert. Die organischen Extrakte wurden vereinigt, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert. Dies ergab
7,70 g eines nahezu farblosen, dicken Sirups, der aus 6,93 g des
erwünschten
Produktes (18; 94 %) und 0,77 g 2-Propanol bestand (durch NMR-Integration): 1H-NMR (d6-DMSO);
d 3,18 und 3,07 (12H, 2 t), 2,37 bis 2,12 (12H, m), 1,60 bis 1,10
und 1,37 (45H, m und s) ppm; Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); RP-18-Silica-Gel,
Rf = 0,50 (Methanol/Wasser (70 : 30)).
-
Herstellung eines N-BOC-Tet-Gal-Nac-1-a-S-C5-Zweiges
(20)
-
Einem
250 ml-Rundkolben, der mit 4,05 g (7,38 mMol) 3,4,6-Tri-O-acetyl-N-acetyl-galactosamin-1-a-S-[5'-thiopentyl-N-BOC-amin]
(11) befällt
war, wurden 20 ml Methylenchlorid und anschließend 20 ml Trifluoressigsäure zugesetzt.
Die Mischung wurde 15 Minuten lang bei Raumtemperatur gerührt. Die
Mischung wurde mittels Rotationsverdampfung konzentriert und der
Rückstand
wurde dreimal mit 75 ml Methylenchlorid verdünnt und erneut konzentriert;
dies ergab 6,27 g Rückstand,
nämlich
eine Mischung aus dem erwünschten Produkt
(19) und restlicher Trifluoressigsäure: 1H-NMR
(CD3OD); d 5,61 (1H, d), 5,41 (1H, dd),
5,01 (1H, dd), 4,62 bis 4,47 (2H, m), 4,11 (2H, d), 2,91 (2H, t),
2,74 bis 2,48 (2H, m), 2,11 (3H, 2 s), 2,00 (3H, s), 1,93 und 1,91
(6H, 2s) und 1,37 bis 1,10 (6H, m) ppm. Einem separaten 250 ml-Rundkolben,
der mit 1,33 g des Syrups, der 90 Gew.-% der Verbindung 18 enthielt
(netto 1,20 g; 1,50 mMol) befüllt
war, wurden 50 ml wasserfreies Dimethylformamid zugesetzt. Um das
restliche 2-Propanol zu entfernen, wurde die Mischung zuerst mittels Rotationsverdampfung
und anschließend
unter vollem Vakuumpumpen-Druck (< 0,5
mm Hg) konzentriert. Dem Rückstand
wurden 20 ml wasserfreies Dimethylformamid und 10 ml trockenes Triethylamin
zugesetzt. Der resultierenden gerührten Lösung wurde eine Dimethylformamid-Lösung des
Rohproduktes 19 (insgesamt 30 ml wasserfreies Dimethylformamid)
zugesetzt und die resultierende Mischung wurde 2 h lang bei Raumtemperatur
gerührt.
Die Mischung wurde danach mittels Rotationsverdampfung konzentriert.
Der Rückstand wurde
danach mit 250 ml Methylenchlorid vedünnt und mit 2 × 100 ml
Aliquots 1 N wäßriger HCl,
100 ml entionisiertem Wasser und danach mit 100 ml gesättigter
wäßriger Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltiert und mittels
Rotationsverdampfung konzentriert. Der Rückstand wurde an einer RP-18-Silica-Gel-Säule (5,5 × 19 cm)
chromatographiert und mit jeweils 250 ml Methanol/Wasser (65 : 35),
Methanol/Wasser (70 : 30), Methanol/Wasser (75 : 25) und anschließend mit
800 ml Methanol/Wasser (80 : 20) eluiert. Die Fraktionen, die das
Produkt enthielten, wurden vereinigt und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck konzentriert. Dies ergab
3,55 g eines schaumigen weißen
Feststoffes (94 %). Dieses Material wurde danach an einer Silica-Gel-Säule (5,5 × 20 cm)
chromatographiert und mit Ethylacetat/Methanol (80 : 20) eluiert.
Die Fraktionen, die nur das erwünschte
Produkt (20) enthielten, wurden vereinigt und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert. Dies ergab
2,83 g des erwünschten
Produktes in Form eines rein weißen schaumigen Feststoffes
(75 %): 1H-NMR (CD3OD);
d 5,58 (4H, d), 5,42 (4H, dd), 5,01 (4H, dd), 4,63 bis 4,51 (8H,
m), 4,20 bis 4,00 (8H, m), 3,35 bis 3,10 (20H, m), 2,73 bis 2,47
(8H, m), 2,32 (4H, t), 2,25 bis 2,08 (20H, m und s), 2,00 (12H,
s), 1,93 und 1,91 (24H, 2 s), 1,71 bis 1,20 (69H, m und s) ppm;
Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray
und Hitze); Silica-Gel, Rf = 0,47 (Ethylacetat/Methanol (75 : 25);
RP-18-Silica-Gel,
Rg = 0,33 (Methanol/Wasser (80 : 20)).
-
Herstellung von N,N-Bis-(N',N'-his-(5-methoxycarbonylpentyl-)5-carbamylpentyl-)
amin TFA (21)
-
Einem
250 ml-Rundkolben, der mit 1,50 g (1,75 mMol) N-BOC-N,N-Bis-(N',N'-bis-(5-methoxycarbonylpentyl-)5-carbamylpentyl-)amin
(17) in 15 ml Methylenchlorid befüllt war, wurden 15 ml Trifluoressigsäure zugesetzt.
Die Mischung wurde 15 Minuten lang bei Raumtemperatur gerührt und
danach konzentriert. Der Rückstand
wurde mit 50 ml Methylenchlorid verdünnt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde danach mit 50 ml Methanol verdünnt und erneut mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde nochmals mit 50 ml Methylenchlorid verdünnt und erneut mittels Rotationsverdampfung und
danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert.
-
Herstellung von TFA-Salz
eines Tet-Gal-Nac-1-a-S-Pentylamin-Zweiges (22)
-
Einem
100 ml-Rundkolben, der mit 790 mg (0,313 mMol) eines N-BOC-Tet-Gal-Nac-1-a-S-C5-Zweiges
(20) befüllt
war, wurden 10 ml Methylenchlorid und anschließend 10 ml Trifluoressigsäure zugesetzt.
Die Mischung wurde 15 Minuten lang bei Raumtemperatur gerührt und
danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 150 ml Methylenchlorid verdünnt und mit 2 × 100 ml
Aliquots einer gesättigten
wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
Magnesiumsulfat getrocknet, vakuumfiltriert und danach zuerst mittels
Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 690 mg des Produktes (22) in Form eines schaumigen cremefarbenen
Feststoffes (91 %).
-
Herstellung eines Tet-Gal-Nac-1-a-S-Pentylamin-Konjugats
von modifiziertem Annexin V mittels SMCC-Derivatisierung (25)
-
Das
Tetragalactosyl-N-AC-1-a-S-pentylamin (22) wurde mit SMCC-Reagens
in Triethylamin und DMF bei Raumtemperatur umgesetzt. Das Maleimidyl-Derivat
(23) wird mit einem Anionenaustausch-Harz des Typs AG-1 X8 (BioRad;
Hydroxid-Form; 2 bis 6 m Equivalente/g) in wäßrigem Methanol trituriert.
Die Mischung wurde 15 Stunden lang bei Raumtemperatur gerührt und
danach vakuumfiltriert. Das Harz wurde mit 50 ml entionisiertem
Wasser und danach mit 50 ml Methanol gespült. Die Filtrate wurden vereinigt
und mittels Rotationsverdampfung und anschließend mit einer Vakuumpumpe
bei vollem Druck (< 0,5
mm Hg) konzentriert; dies ergab das Produkt 24 in Form eines trockenen
Feststoffes. Die Konjugation von modifiziertem Annexin V-SH mit einer mit
einem Maleimidyl-Rest derivatisierten gesäuertem Galactose wird durchgeführt in 5
bis 15 % DMSO-Lösungsmittel
mit einem pH-Wert im Bereich von 5 bis 8 in Borat-Puffer. Der Maleimidyl-Galactose-Cluster wird
dem Annexin V in molaren Verhältnissen
von 1 : 1; 5 : 1 und 10 : 1 angeboten. Das monomere Galactose-Cluster-Annexin V-Konstrukt
wird danach mittels Gelfiltrations-Methoden gereinigt. Das Produkt
wird danach auf seinen Sulfhydrylgehalt untersucht, um den Derivatisierungsgrad
zu bestimmen, um das Konjugat 25 in guter Ausbeute zu erhalten.
-
Herstellung von 99mTc-Galactose-Cluster-Annexin V-Konjugat
(26)
-
Der 99mTc-N2S2-TFP-Ester wird hergestellt mittels des
VerlumaTM-Kit-Markierungsverfahrens (Kasina, S., Rao,
T.N., Srinivasan, A., et al., Development and Biologic Evaluation
of a Kit for Performed Chelate Technetium-99m Radiolabeling of an
Antibody Fab Fragment Using a Diamide Dimercaptide Chelating Agent,
J. Nucl. Med., 32, 1445-1451 (1991). Das 99mTc
wurde in den N2S2-Liganden
mittels Transchelatisierung von 99mTc-Gluconat
durch Erhitzen eingearbeitet. Der 99mTc-N2S2-TFP-Ester wird danach
konjugiert mit dem Galactose-Cluster-Annexin V-Konstrukt in einem
0,2 m Bicarbonat-Puffer, pH 10,0, für eine Zeitdauer von 20 bis
30 Minuten bei Raumtemperatur. Das 99mTc-N2S2-Galactose-Cluster-Annexin
V-Konjugat wird durch Gelchromatographie gereinigt; dies ergab 30
bis 40 % einer radiochemischen Ausbeute mit einer radiochemischen
Reinheit von ≥85
% des Produktes 26.
-
BEISPIEL XVI
-
Herstellung von Annexin-S-Galactose-Chelat-Verbindung
-
-
Herstellung
von modifiziertem Annexin V-Monomer-Konjugat von trifunktionellem
Lysin-Addukt, welches die Galactose-Cluster-Chelat-Verbindung-Einheit
enthält
(36)
-
Die
epsilon-Amino-BOC-Schutzgruppe des trifunktionellen Lysin-Addukts
des Galactose-Clusters mit der Chelat-Verbindung 31 wird umgesetzt
mit Trifluoressigsäure
in Methylenchlorid. Die Reaktionsmischung wurde bei Raumtemperatur
gerührt.
Das Lösungsmittel
wurde unter verringertem Druck entfernt und getrocknet und ergab
ein epsilon-Amin ohne BOC-Schutzgruppe in guter Ausbeute. Das Rohprodukt
wurde ohne weitere Reinigung mit einem AG-1 X8-Anionenaustausch-Harz
in wäßrigem Methanol
20 Stunden lang bei Raumtemperatur trituriert. Die Mischung wurde
filtriert, und das Harz wurde mit Methanol gespült. Die Filtrate wurden vereinigt
und konzentriert und dies ergab den O-deacetylierten N-Acetylgalactose-Cluster-Chelat-Verbindung-Liganden des trifunktionellen
Lysins als epsilon-Amin in guter Ausbeute. Die epsilon-Amin-Funktionalität wurde
mit 1,2 Equivalenten SMCC-Reagens in DMF-Lösungsmittel
in Gegenwart von 10 Equivalenten Triethylamin bei Raumtemperatur
umgesetzt. Das Lösungsmittel
wurde von der Reaktionsmischung entfernt, und das Rohprodukt wurde
an einer Silica-Gel-Säule
chromatographiert. Dies ergab das mit Maleimidyl derivatisierte
trifunktionelle Galactose-Cluster-Chelat-Verbindung-Ligand-SMCC-Addukt des trifunktionellen
Lysin-Produktes 36 in einer guten Ausbeute.
-
BEISPIEL XVII
-
Herstellung von Annexin-S-Galactose-(Peptid)-Chelat-Verbindung
-
-
-
-
N,N1-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin
13 (auch 5, 3) wurde
in DMF gelöst.
Der Lösung
wurden 0,5 Equivalente N-Hydroxysuccinimid zugesetzt und die Lösung wurde
mit einem Magneten gerührt.
Der Reaktionsmischung wurden 0,6 Equivalente 10-(3-Dimethylaminopropyl-)3-ethylcarbodiimid
(CDI) in Form seines Hydrochlorid-Salzes zugesetzt. Der pH-Wert
der Reaktionsmischung wurde mit Phosphatpuffer, 1,0 M, pH 7,0, auf
einen Wert zwischen 5 und 6 eingestellt. Der Fortschritt der Reaktion
wurde mittels Dünnschichtchromatographie
an Silica-Gel-Platten
aufgezeichnet. Das Lösungsmittel
wurde unter Vakuum von der Reaktionsmischung entfernt und getrocknet.
Das Rohprodukt wurde mittels Silica-Gel-Säulenchromatographie
gereinigt. Die Fraktionen, die den Mono-NHS-Ester und die Mono-Säure enthielten,
wurden vereinigt und das Lösungsmittel
wurde unter verringertem Druck entfernt und getrocknet. Dies ergab
die erwünschte
Verbindung 14 in niedriger Ausbeute.
-
Herstellung des Annexin
V-Monomer-S-Konjugat des Galactose-Cluster-Liganden des trifunktionellen
Lysins (43)
-
Der
N,N'-Bis-(2-disulfidyl-6-hydroxycarbonylphenyl-)1,3-propyldiamin-mono-NHS-Ester (14) wurde mit
1,2 Equivalenten t-Butyltetraglycincarboxylat in Form seines Hydrochlorid-Salzes,
im Handel erhältlich
von der Firma Aldrich Chemical Company, in DMF-Lösungsmittel unter Verwendung
von 10 Equivalenten von Triethylamin als Base umgesetzt. Das Lösungsmittel
aus der Reaktionsmischung wurde unter Vakuum entfernt und das Rohprodukt
wurde mittels Silica-Gel-Säulenchromatographie
gereinigt. Dies ergab das Peptid-Addukt 38 in guter Ausbeute. Der
tertiäre
Butylester der Verbindung 38 wurde in einer TFA-CH2Cl2-Mischung entfernt. Die Mischung wurde 1
Stunde lang gerührt
und das Lösungsmittel
entfernt und getrocknet und ergab die Carbonsäure des Ligand-Tetrapeptid-Addukts
39 in guter Ausbeute.
-
Die
Carboxyl-Funktionalität
des Tetraglycin-Ligand-Addukts 39 wurde mit 1,2 Equivaltenten 3,5,6-Tri-O-acetyl-NH-acetyl-tetra-galactosyl-∈-HNBOC-α-Aminolysin
29a in DMF-Lösungsmittel
und 5 Equivalenten Triethylamin in Gegenwart von 1,2 Equivalenten
BOP-Reagens konjugiert. Das Lösungsmittel
aus der Reaktionsmischung wurde unter Vakuum entfernt und getrocknet.
Das Rohprodukt wurde mittels Silica-Gel-Säulenchromatographie gereinigt.
Dies ergab Verbindung 40 des trifunktionellen Lysis-epsilon-Amins, geschützt mit
BOC.
-
Die
BOC-Gruppe von Verbindung 40 wird in einer TFA-CH2Cl2-Mischung entfernt. Die Reaktionsmischung
wird 1 bis 2 Stunden lang bei Raumtemperatur gerührt. Das Lösungsmittel aus der Reaktionsmischung wurde
entfernt und getrocknet. Das Rohprodukt wurde danach mit AG-1 X8-Anionenaustausch-Harz
in wäßrigem Methanol
bei Raumtemperatur trituriert. Die Mischung wurde filtriert, und
das Harz wurde mit Methanol gespült.
Die Filtrate wurden vereinigt und konzentriert und dies ergab o-Deacetyl-N-acetyltetra-galactosyl-Ligand-Chelat-Verbindung
des trifunktionellen Lysins in Form eines epsilon-Amins 41 in guter
Ausbeute. Die Aminoverbindung 41 wurde mit 1,2 Equivalenten SMCC-Reagens
in 10 Equivalenten Triethylamin und DMF-Lösungsmittel derivatisiert.
Die Reaktionsmischung wurde 1 bis 2 Stunden lang bei Raumtemperatur
gerührt.
Das Lösungsmittel
wurde unter verringertem Druck entfernt und das Rohprodukt wurde
an einer Silica-Gel-Säule
chromatographiert und ergab Maleimidyl-tetra-galactose-tetra-glycyl-peptidyl-Ligand-Chelat-Verbindung
des trifunktionellen Lysins 42 in guter Ausbeute. Das Maleimidyl-Derivat
42 wurde danach konjugiert mit modifiziertem Annexin V-SH bei einem
pH-Wert von 5 bis 9 in Boratpuffer und/oder Phosphatpuffer. Die
Reaktionsmischung wurde 2 Stunden lang bei 25 bis 37 °C inkubiert.
Das Schwefel-Addukt des modifizierten Annexin-Konjugats wurde danach
mittels Gelfiltration gereinigt und ergab das Konjugat 43 in guter
Ausbeute.
-
BEISPIEL XVIII
-
Verfahren
zur Herstellung von Dimer-N-Galactose-Chelat-Verbindung oder S-Annexin-N-Galactose (endogene
Chelatisierung, kein Dimer) oder S-Annexin-N-Galactose-Chelat-Verbindung
-
-
-
-
Herstellung von N-∈-(tert-Butoxycarbonyl-1N'-α-trifluoracetyl-L-Lysin (28)
-
Einem
250 ml-Rundkolben, der mit 10,0 g (0,041 Mol) N-∈-(tert-Butoxycarbonyl-)L-Lysin 27, im Handel erhältlich von
der Firma BACHEM Bioscience Inc. (Tochtergesellschaft der BACHEM
Switzerland, 3700 Horizon Drive, Renaissance at Gulph Mills, King
of Prussio, PA, 19406, USA) und 100 ml Methanol befüllt war,
wurden 10,0 g (0,07 Mol) Ethyltrifluoracetat und anschließend 20,0
ml (0,144 Mol) Triethylamin zugesetzt. Die Mischung wurde 20 Stunden
lang bei 20 bis 30°C
gerührt
und danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 50 ml Diethylether verdünnt und danach filtriert. Das
Filtrat wurde danach mit 3 × 30 ml
Aliquots einer 1 N wäßrigen HCl,
20 ml entionisiertem Wasser und letztendlich mit 20 ml entionisiertem
Wasser gewaschen. Die organische Phase wurde über wasserfreiem Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wurde unter Vakuum
destilliert und dies ergab Produkt 28 in guter Ausbeute. N-∈-(tert-Butoxycarbonyl-)N'-α-trifluoracetyl-L-Lysin wurde
mit Tri-o-acetyl-tet-Gal-Nac-1-a-S-Pentylamin in Gegenwart von BOP-Reagens
umgesetzt. Die aktivierte Carbonsäwe reagierte mit dem Galasctosamin-Cluster
in DMF und Triethylamin und ergab das Galactose-Cluster-Lysin-Addukt
29 in guter Ausbeute. Der N-α-Trifluoracetyl-Schutz
des Lysins wurde entfernt und ergab den primären α-Amino-Lysin-Galactose-Cluster 29a
in mäßiger Ausbeute.
-
Konjugation von Galactose-Cluster
mit einer Chelat-Verbindung und Annexin-Komponenten (34)
-
Tetragalactosyl-Chelat-Verbindung-Konstrukt
(29a–34)
-
Einer
Mischung aus mit Amin verlängertem
Tetra-Galactosylamin 29a und 1,2 Equivalenten N,N'-Bis-(2-disulfidyl-4-hydroxycarbonylphenyl-)1,3-propyldiamin-mono-NHS-Ester in
Dimethylformamid wurden 5 Equivalente Triethylamin zugesetzt. Die
Mischung wurde 2 bis 24 Stunden lang bei 15 bis 30 °C gerührt und
danach konzentriert. Der Rückstand
wurde mit entionisiertem Wasser verdünnt und der pH-Wert wurde durch
Zugabe von 1 N wäßriger Chlorwasserstoffsäure auf
2,5 eingestellt. Die Mischung wurde mit Ethylacetat gewaschen. Die
wäßrige Phase
wurde konzentriert. Der Rückstand
wurde an einem Phasenumkehr-C-18-Silica-Gel chromatographiert. Die
Fraktionen, die das Produkt enthielten, wurden vereinigt und konzentriert,
und dies ergab so das Produkt 30 in guter Ausbeute. Die mit BOC
geschützte
epsiolon-Amin-Funktionalität
von Verbindung 30 wurde entfernt durch Rühren bei Raumtemperatur in
einer Trifluoressigsäure-Methylenchlorid-Mischung.
Die resultierende primäre
epsilon-Amin-Verbindung 31 wurde mit AG-1 X8-Anionenaustausch-Harz (Hydroxid-Form)
in einer Methanol/Wasser-Mischung 15 Stunden lang bei Raumtemperatur
trituriert. Die Mischung wurde filtriert, und das Harz wurde mit
Methanol gespült.
Die Filtrate wurden vereinigt und konzentriert und dies ergab den
o-deacetylierten N-Acetyl-Galactose-Cluster, der nach Umsetzung
mit Bernsteinsäureanhydrid
in Triethylamin und DMF bei Raumtemperatur das Hemibernsteinsäure-Addukt
32 in guter Ausbeute ergab. Die Hemibernsteinsäure-Funktionalität der Verbindung 32 wurde aktiviert
durch Umsetzung mit 1,1 Equivalenten N-Hydroxysuccinimid in Gegenwart
von 1,2 Equivalenten eines DCC-Kupplungsmittels.
Das Reaktionsprodukt wurde unter Vakuum konzentriert, und ergab
ein Rohprodukt, das nach Reinigung mittels Silica-Gel-Säulenchromatogaphie
den NHS-Ester 33 in mäßiger Ausbeute
ergab. Der NHS-Ester des Galactose-Cluster-Chelat-Verbindung-Addukts des trifunktionellen
Lysins wurde mit Annexin V bei einem pH-Wert von 6 bis 10 in Gegenwart
von DMSO (0 bis 15 %) umgesetzt. Die Reaktion wurde durchgeführt bei
25 bis 37 °C für eine Zeitdauer
von 1 bis 2 Stunden. Das rohe Konjugat wurde gereinigt durch Gelchromatographie
und dies ergab das Galactose-Cluster-Chelat-Verbindung-Annexin V-Lysin-Konjugat
34 in guter Ausbeute.
-
BEISPIEL XIX
-
Radiomarkierungs-Verfahren
für die
Beispiele XVI, XVII und XVIII
-
Charakterisierung der Galactose-Cluster-Chelat-Annexin
V-Konjugate
-
Die
Proteinkonzentration wurde bestimmt unter Verwendung von A280 von 0,6 für 1 mg/ml Lösung von Annexin V. Die Anzahl
der Galactose-Reste pro Molekül
Annexin V wurde bestimmt durch Messen der Gesamtzahl reaktiver Amine
am Annexin V vor und nach der Umsetzung mit dem Galactosyl-Cluster-Chelat-Konjugats
unter Verwendung von Trinitrobenzolsulfonsäure, wie dies beschrieben wurde
von Habeeb, Analytical Biochemistry, 14, 328-336 (1966).
-
Auch
die Anzahl der Galactose-Reste pro Molekül modifizierten Annexins V,
die mittels Sulfhydryl konjugiert waren, wurde bestimmt durch Messen
des reaktiven Sulfhydryls an Annexin V vor und nach der Umsetzung
mit dem Galactosyl-Cluster-Chelat-Konjugat
unter Verwendung von 3-Carboxy-4-nitrophenyldisulfid, DTNB, Ellman's Reagens, wie dies
beschrieben wurde von Deakin, et al., Biochem. J., 89, 296 (1963).
Die Fähigkeit
des Galactose-Chelat-Annexins V an aktivierte Blutplättchen zu
binden wurde bewertet, indem man seine Fähigkeit bestimmte, die Bindung
von unmodifiziertem, I-125 radiomarkiertem Annexin V an frisch isolierte menschliche
Blutplättchen
zu binden in Anlehnung an das Verfahren von Thiagarajan und Tait,
J. Biol. Chem., 265, 17, 240-243 (1990).
-
Radiomarkierungs-Verfahren
zur Verwendung in nachgelagerten Chelat-Konjugations-Verfahren
-
Verfahren
A: Zinn-Gluconat-Kits wurden hergestellt, die 5 mg Natriumgluconat,
100 Mikrogram Zinnchlorid, 1,0 mg (1 mg/ml) Galactose-Cluster-Chelat-Annexin
V und 0,1 bis 1,0 mg Lactose enthielten. Der pH-Wert wurde unter
Verwendung von HCl, Essigsäure
und NaOH zwischen 5 und 7 gehalten. Dem Zinn-Gluconat-Kit wurden
1,0 ml Natriumpertechnetat (Tc-99m) mit einer spezifischen Aktivität von etwa
50 mCi zugesetzt. Die Phiole wurde 5 bis 30 Minuten lang bei 25
bis 30 °C
inkubiert. Die prozentuale Bildung von markiertem Konjugat, verbleibendem
Pertecnetat und hydrolysiertem/reduziertem Technetium wurde mittels
ITLC in 12 % TCA als Entwicklungs-Lösungsmittel bestimmt.
-
Verfahren
B: Zinn-Tartrat-Kits, die 0,5 ml Dinatriumtartrat (10 mg/ml) und
0,1 ml Zinnchlorid (1,0 mg/ml Ethanol) enthielten, wurden in einer
Vakuum-Phiole unter Stickstoff hergestellt. Der pH-Wert der Lösung wurde zwischen
5 und 7, vorzugsweise bei 6,0, gehalten. Dieser Zinn-Tartrat-Lösung wurden
1,0 ml Natriumpertechnetat (50 mCi) zugesetzt, und man ließ die Lösung bei
Raumtemperatur stehen. Einer evakuierten Phiole wurden nacheinander
200 μl Natriumphosphat
(0,5 M, pH 8 oder 10) und 1,0 ml Galactose-Cluster-Chelat-Verbindung-Annexin
V-Konjugat (1,0 mg/lml) zugesetzt. Anschließend wurde das Tc-99m-tartrat
(50 mCi) zugesetzt, und die Phiole wurde 5 bis 30 Minuten lang bei
25 bis 37 °C
inkubiert. Die prozentuale Bildung von markiertem Konjugat, verbleibendem
Pertechnetat und hydrolysiertemireduziertem Technetium wurde mittels
ITLC in 12 % TCA als Entwicklungs-Lösungsmittel bestimmt.
-
Radiomarkierte
Verbindung von Beispiel XVI
-
Radiomarkierte
Verbindung von Beispiel XVII
-
Radiomarkierte
Verbindung von Beispiel XVIII
-
BEISPIEL XX
-
Herstellung von modifiziertem
Annexin V-Dimer durch rekombinante Verfahren
-
Ein
Dimer aus zwei modifizierten Annexin V-Molekülen kann hergestellt werden
durch rekombinante DMS-Verfahren in Anlehnung an die Verfahren,
die beschrieben wurden von Tait et al., J. Biol. Chem., 270, 21594 – 21599
(1995), zur Konstruktion und Expression von chimären Molekülen, die Annexin V enthalten. Zuerst
wurde an dem Annexin V-cDNS-Template-pPPAP-I-1 eine PCR durchgeführt mit
Oligonukleotid-Primern, die an der Ndel-Stelle an dem 5'-Ende und an der
BamHI- Stelle an
dem 3'-Ende der
Annexin V-Kodierungssequenz (Aminosäuren 1-320 und Stop-Codon)
eingeführt
wurden. Dieses PCR-Produkt wurde durch Standardverfahren an den
NdeI- und BamHI-Stellen des Plasmids pEP-12a (Novagen Corp., Madison,
WI) kloniert, um das Plasmid pET-12a-Axl zu erzeugen. PCR wurde
danach nochmals an dem Annexin V-cDNS-Template-pPAP-1-1,6 mit Oligonukleotid-Primern,
die an der NdeI-Stelle an dem 5'-Ende
eingeführt
wurden und einer Sequenz, die die Aminosäuren Gly-Gly-Gly-Gly-Gly-Gly
codiert, und anschließend
an der NdeI-Stelle an dem 3'-Ende
der Annexin V-Kodierungssequenz (Aminosäuren 1-3210) durchgeführt. Dieses
pCR-Produkt wurde
danach durch Standardverfahren an der NdeI-Stelle des Plasmids pET-12a-Axl kloniert,
um das Plasmid PET-12a-Ax2 zu erzeugen. Die Herstellung des dimeren
Annexin V-Moleküls
durch Cytoplasma-Expression in E. coli aus Plasmid pET-12a-Ax2 wurde
danach durch Standardverfahren durchgeführt, wie dies beispielsweise
gezeigt wurde durch Tait et al., 7. Biol. Chem., 270, 21594 – 21599
(1995).
-
BEISPIEL XXI
-
Durchführung der Galactosylierung
(4-Mer-Zucker-Cluster) von modifziertem Annexin
-
A. Synthese von Zucker mit
Linker
-
-
-
-
-
-
E.
Konjugation von modifiziertem Annexin V (Monomer) mit Verbindung
28
-
Herstellung von 1-b 3 4
6-Tetra-O-Acetyl-N-acetyl-galactosamin (2)
-
Einem
500 ml-Rundkolben, der mit 25,0 g (116 mMol) Galactosaminhydrochlorid
(1, Sigma Chemical Co., St. Louis, Missouri) befüllt war, wurden 180 ml wasserfreies
Pyridin und danach 115 ml Essigsäureanhydrid
(1,22 Mol) zugesetzt. Die Mischung wurde 44 h lang bei 20 bis 30 °C gerührt und
danach in einen 2 Liter-Becher, der 600 g Eis und 600 ml entionisiertes
Wasser enthielt, gegossen. Die Mischung wurde 10 bis 15 Minuten
lang bei Raumtemperatur gerührt
und danach vakuumfiltriert. Die gesammelten Feststoffe wurden mit 4 × 100 ml
Aliquots entionisiertem Wasser gespült, 2 h lang an der Luft getrocknet
und danach 14 h lang unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg} getrocknet. Dies ergab
39,8 g des Produktes in Form eines weißen Feststoffes (88 %): 1H-NMR (d6-DMSO);
d 7,89 (1H, d), 5,63 (1H, d), 5,16 (1H, d), 5,07 (1H, dd), 4,28
bis 3,92 (4H, m), 2,11 (3H, s), 2,02 (3H, s), 1,99 (3H, s), 1,90
(3H, s) und 1,88 (3H, s) ppm.
-
Herstellung von 3,4,6,-Tri-O-Acetyl-N-acetyl-Galactosamin-l-b-nseudothioharnstoffhydrochlorid
(4)
-
Einem
1 Liter-Rundkolben, der mit 39,8 g (102 mMol) 2 befüllt war,
wurden 400 ml Acetylchlorid zugesetzt. Die Mischung wurde 64 h lang
bei 47 bis 48 °C
gerührt.
Die Mischung wurde konzentriert und danach zweimal mit 200 ml Methylenchlorid
verdünnt
und zuerst mittels Rotationsverdampfung und danach unter vollem
Vakuumpumpen-Druck (< 0,5
mm Hg) erneut konzentriert. Dies ergab 40,2 g des Rohproduktes (3)
in Form eines dunkel-bernsteinfarbenen schaumigen Feststoffes: 1H-NMR(CDCl3); d
6,24 (1H, d), 5,61 (1H d), 5,43 (1H, dd), 5,27 (1H, dd), 4,83 bis
4,71 (1H, m), 4,48 (1H, t), 4,22 bis 4,01 (2H, 2 dd), 2,15 (3H,
s), 2,02 (3H, s), 2,00 (3H, s) und 1,98 (3H, s) ppm. Dem rohen Chlorid
(3) in einem 1 Liter-Rundkolben wurden 9,3 g (122 mMol) Thioharnstoff
und 150 ml Aceton zugesetzt. Die Mischung wurde unter Rückfluß 40 Minuten
lang gerührt
und danach in einem Eisbad 30 Minuten lang abgekühlt und danach vakuumfiltriert.
Die gesammelten Feststoffe wurden mit 2 × 75 ml Aliquots Aceton gespült. Die
Feststoffe wurden danach 45 Minuten lang an der Luft getrocknet
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) 2 h lang getrocknet; dies
ergab 33,0 g des Produktes (4) in Form eines hellbeigen Feststoffes
(74% Gesamtausbeute aus 7): 1H-NMR (d6-DMSO);
d 9,38 und 9,12 (2 breite Singletts, 3H), 8,36 (1H, d), 5,56 (1H,
d), 5,34 (1H, d), 5,01 (1H, dd), 4,38 (1H, t), 4,22 bis 4,00 (3H,
m), 2,11 (3H, s), 2,01 (3H, s), 1,92 (3H, s) und 1,81 (3H, s) ppm.
-
Herstellung von 1-b-Mercapto-3,4
6-tri-O-acetyl-N-acetyl-N-acetylgalactosamin (5)
-
Einem
1 Liter-Rundkolben, der mit 30,0 g (67,9 mMol) des Pseudothioharnstoffes
(4) in 175 ml Methylenchlorid und 175 ml entionisiertem Wasser befüllt war,
wurden 7,08 g (37,24 mMol) Natriummetabisulfit zugesetzt und anschließend vorsichtig
10,2 g (74,5 mMol) Kaliumcarbonat zugesetzt. Die Mischung wurde
40 Minuten lang bei Raumtemperatur gerührt und die Mischung wurde
danach in einen 500 ml-Scheidetrichter überführt. Die Schichten wurden getrennt
und die wäßrige Phase
wurde danach mit 2 × 125
ml Aliquots Methylenchlorid gewaschen. Die organischen Extrakte
wurden vereinigt, über
wasserfreiem Natriumsulfat getrocknet, filtriert und konzentriert.
Dies ergab 24,2 g (5) in Form eines sehr blassgelben (cremefarbenen)
Feststoffes (98 %): 1H-NMR (CDCl3); d 6,24 (1H, d), 5,61 (1H, d), 5,43 (1H,
dd), 5,27 (1H, dd), 4,83 bis 4,71 (1H, m), 4,48 (1H, t), 4,22 bis
4,01 (2H, 2 dd), 2,15 (3H, s), 2,02 (3H, s), 2,00 (3H, s) und 1,98
(3H, s) ppm.
-
Herstellung von N-BOC-S-Aminopentanol
(7)
-
Einem
2 Liter-Dreihals-Rundkolben, der an dem mittleren Hals mit einem
500 ml Zugabe-Trichter und an einem Seitenhals mit einem Übergangsstück zum Entlüften eines
Gasblasenzählers
ausgestattet war, wurden 40 g (0,388 Mol) 5-Aminopentanol (6) in
500 ml trockenem Acetonitril zugesetzt. Danach wurden 84,5 g (0,387
Mol) Di-t-butyldicarbonat
in 400 ml trockenem Acetonitril über
einer Zeitdauer von 50 Minuten zugesetzt. Die Mischung wurde 15
h lang bei 20 bis 30 °C
gerührt
und danach konzentriert. Der Rückstand
wurde mit 600 ml Ethylacetat verdünnt und mit 2 × 200 ml
Aliquots 0,5 N wäßriger HCl
und 2 × 200
ml Aliquots entionisiertem Wasser gewaschen. Die organische Phase
wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und zunächst mittels
Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 74,5 g des Produktes (7) in Form eines nahezu farblosen Öls (88 %): 1H-NMR (d6-DMSO);
d 6,72 (1H, breites Triplett), 4,31 (1H, t), 3,43 bis 3,27 (2H,
m), 2,87 (2H, Quartett), und 1,45 bis 1,10 (15H, s und Multiplett)
ppm; 1H-NMR (CDCl3);
d 4,58 (1H, breites Singlett), 3,65 (2H, t), 3,13 (2H, Quartett) und
1,70 bis 1,30 (15H, Singlett und Multiplett) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf = 0,28 (Methylenchlorid/Methanol (95:5)).
-
Herstellung von N-BOC-5-Aminopentyltoluolsulfonat
(8)
-
Einem
1 Liter-Rundkolben, der mit 74,5 g (0,366 mol) N-BOC-Aminopentanol
(7) in 400 ml Methylenchlorid befüllt war, wurden 45 ml wasserfreies
Pyridin und anschließend
74,1 g (0,389 Mol) p-Toluolsulfonylchlorid zugesetzt. Die Mischung wurde
17 h lang bei Raumtemperatur gerührt,
mit 200 ml Methylenchlorid verdünnt
und mit 400 ml 0,5 N HCl, 2 × 200
ml Aliquots 0,5 N HCl und 2 × 100
ml Aliquots entionisiertem Wasser gewaschen. Die organische Phase
wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und konzentriert.
Der Rückstand
wurde an einer Silica-Gel-Säule
(11 × 23
cm) chromatographiert, zunächst
mit Methylenchlorid und danach mit Ethylacetat/Methylenchlorid (3
: 97) eluiert. Die Fraktionen, die das Produkt enthielten, wurden
vereinigt und zunächst
mittels Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 82,13 g des Produktes (8) in Form eines weißen Feststoffes: 1H-NMR (CDCl3); d
7,77 (2H, d), 7,31 (2H, d), 4,45 (1H, breites Singlett), 3,98 (2H,
t), 3,03 (2H, t), 2,41 (3H, s) und 1,80 bis 1,20 (15H, Singlett
und Multiplett) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf
= 0,50 (Ethylacetat/Methylenchlorid (3 : 97)).
-
Herstellung von 3,4,6-Tri-O-acetyl-N-acetylgalactosamin-1-a-S-[5'-thiopentyl-N-BOC-amin] (9)
-
Einem
1 Liter-Rundkolben, der mit 24,2 g (66,6 mMol) des Thiols (5) unter
Stickstoffatmosphäre
befüllt war,
wurden 350 ml trockenes Acetonitril zugesetzt. Die Mischung wurde über eine
Zeitdauer von 20 Minuten auf 40 bis 42 °C erhitzt, bis sich letztendlich
die Feststoffe gelöst
hatten. 1,8-Diazabicyclo[5.4.0]undec-7-en (DBU; im Handel erhältlich von
der Firma Aldrich Chemical Company; 10,5 ml, 70,2 mMol) wurden danach
zugesetzt und die Mischung wurde 20 Minuten lang gerührt. Danach
wurden 24,0 g (67,1 mMol) des Tosylats 6 in 75 ml Acetonitril über eine
Zeitdauer von 3 bis 4 Minuten zugesetzt. Die resultierende Mischung
wurde 1,5 h lang bei 40 bis 25°C
gerührt
und danach konzentriert. Der Rückstand
wurde mit 400 ml Methylenchlorid verdünnt und zuerst mit 250 ml 0,5
N wäßriger HCl
und danach mit 250 ml einer 5% igen wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesium getrocknet, vakuumfiltriert und mittels eines
Rotationsverdampfers konzentriert. Der Rückstand wurde an einer Silica-Gel-Säule (21 × 7 cm)
(hergestellt von der Firma E.M. Merck) chromatographiert und mit Ethylacetat/Hexan/Ethanol
(55 : 42,5 : 2,5) eluiert. Die Fraktionen, die das Produkt enthielten,
wurden vereinigt, konzentriert und erneut chromatographiert an einer
RP-18-Silica-Gel-Säule (21 × 7 cm)
(hergestellt von der Firma J.T. Baker) und mit jeweils 500 ml Methanol/Wasser
im Verhältnis
50 : 50; 55 : 45; 60 : 40; 65 : 35 und 70 : 30 und anschließend mit Methanol/Wasser
(75 : 25) eluiert, bis das gesamte Produkt von der Säule eluiert
war. Die Fraktionen, die das Produkt enthielten, wurden vereinigt
und konzentriert. Der Rückstand
wurde mit 500 ml Methylenchlorid verdünnt und mit wasserfreiem Magnesiumsulfat
behandelt. Die Mischung wurde vakuumfiltiert und das Filtrat wurde
zuerst mittels Rotationsverdampfung und danach unter vollem Vakuumpumpen-Druck
(< 0,5 mm Hg) konzentriert;
dies ergab 17,9 g des Produktes (9) in Form eines schaumigen weißen Feststoffes: 1H-NMR (d6-DMSO);
d 9,38 und 9,12 (2 breite Singletts, 3H), 8,36 (1H, d), 5,56 (1
H, d), 5,34 (1H, d), 5,01 (1H, dd), 4,38 (1H, t), 4,22 bis 4,00
(3H, m), 2,11 (3H, s), 2,01 (3H, s), 1,92 (3H, s) und 1,81 (3H,
s) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit p-Anisaldehyd-Spray und Hitze); Silica-Gel,
Rf = 0,50 (Ethylacetat/Hexan/Ethanol (57 : 40,5 : 2,5)); RP-18-Silica-Gel, Rf= 0,21
(Methanol/Wasser (65 : 35)).
-
Herstellung von Methyl-6-bromhexanoat
(12)
-
In
einen 2 Liter-Rundkolben, der mit 99,7 g (0,511 Mol) 6-Bromhexanoat
(Aldrich Chemical Co., Milwaukee, Wisconsin) und 1 Liter Methanol
befüllt
war, wurde 1 bis 2 Minuten lang Chlorwasserstoffgas eingeblasen.
Die Mischung wurde 18 h lang bei 20 bis 30 °C gerührt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde mit 500 ml Diethylether verdünnt und mit 150 ml entionisiertem
Wasser, 200 ml gesättigtem
Natriumbicarbonat und danach nochmals mit 200 ml entionisiertem
Wasser gewaschen. Die organische Phase wurde über wasserfreiem Magnesiumsulfat
getrocknet, filtriert und mittels Rotationsverdampfung konzentriert.
Der Rückstand
wurde unter Vakuum destilliert und dies ergab 99,6 g des Produktes (12)
in Form eines farblosen Öls
(93 %): Siedepunkt = 93 bis 96 °C
bei 3 mm Hg: 1H-NMR (d6-DMSO);
d 3,57 (3H, s), 3,51 (2H, t), 2,30 (2H, t), 1,78 (2H, Pentett und
1,62 bis 1,28 (4H, m) ppm.
-
Herstellung von Methvl-6-aminohexanoat-hydrochlorid
(14)
-
In
einen 2 Liter-Rundkolben, der mit 101,3 g (0,722 Mol) 6-Aminohexanoat
(13) (Aldrich Chemical Co.) in 1 Liter Methanol befüllt war,
wurde 3 bis 4 Minuten lang Chlorwasserstoffgas eingeblasen. Die
Mischung wurde 16 h lang bei 20 bis 30 °C gerührt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde zweimal mit 500 ml Methanol verdünnt und erneut konzentriert
(< 0,5 mm Hg).
Man erhielt 140,1 g des Produktes (14) in Form eines weißen Feststoffes
(100 %): 1H-NMR (d6-DMSO);
d 9,40 (1H, breites Triplett), 3,57 (3H, s), 3,15 (2H, Quartett),
2,29 (2H, t), 1,60 bis 1,38 (4H, m) und 1,32 bis 1,19 (2H, m) ppm.
-
Herstellung von Methyl-6-trifluoracetamido-)hexanoat
(15)
-
Einen
2 Liter-Rundkolben, der mit 100,2 g (0,552 Mol) Aminhydrochlorid
(14) und 1 Liter Methanol befüllt
war, wurden 100 g (0,703 Mol) Ethyltrifluoracetat und anschließend 120
ml (0,861 Mol) Triethylamin zugesetzt. Die Mischung wurde 19 h lang
bei 20 bis 30 °C
gerührt
und danach mittels Rotationverdampfung konzentriert. Der Rückstand
wurde mit 500 ml Diethylether verdünnt und danach filtriert. Das
Filtrat wurde mit 3 × 300
ml Aliquots 1 N wässriger
HCl, 200 ml entionisiertem Wasser, 2 × 200 ml Aliquots gesättigter
wäßriger Natriumbicarbonat-Lösung und
letztendlich mit 200 ml entionisiertem Wasser gewaschen. Die organische
Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und konzentriert.
Der Rückstand
wurde unter Vakuum destilliert und ergab 115,8 g des Produktes (15)
in Form eines farblosen Öls:
Siedepunkt = 113 bis 116 °C
bei 120 mm Hg: 1H-NMR (d6-DMSO);
d 3,57 (3H, s), 2,75 (2H, m), 2,29 (2H, t), 1,60 bis 1,40 (4H, m)
und 1,37 bis 1,19 (2H, m) ppm.
-
Herstellung von N,N-Bis-(5-methox
carbonvlpentyl-) amin-hydrochlorid (18)
-
Einem
5 Liter-Dreihalskolben, der mit einem Rückflußkühler ausgestattet war, der
mit einem Gasblaszähler
verbunden war, und der mit 20,9 g 60 % Natriumhydroxid (0,523 Mol)
in 1 Liter wasserfreiem Dioxan befüllt war, wurden 100 g (0,416
Mol) sekundäres
Amid 15 in 200 ml trockenem Dioxan über eine Zeitdauer von 20 Minuten
zugesetzt. Die Mischung wurde 1 h lang bei 20 bis 30 °C gerührt, und
danach wurden 130 g (0,622 mol) Bromid 12 in 100 ml Dioxan zugesetzt.
Die Mischung wurde unter Rückfluß erhitzt
und 7 h lang gerüht.
Zusätzlich
wurden 10 g der Verbindung 12 zugesetzt, und die resultierende Mischung
wurde für
weitere 15 h gerührt.
Die Mischung wurde abgekühlt
und mittels Rotationsverdampfung konzentriert. Der Rückstand wurde
mit 600 ml 1 N wäßriger HCl
verdünnt
und mit 1 Liter Ethylacetat extrahiert. Die organische Phase wurde danach
mit 250 ml entionisiertem Wasser, 250 ml 5 % wäßriger Natriumbisulfit-Lösung und
letztendlich mit 250 ml entionisiertem Wasser gewaschen. Die organische
Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und konzentriert.
-
Der
Rückstand
wurde mit 300 ml entionisiertem Wasser und 500 ml Methanol verdünnt und
mit 200 ml 10 N wäßrigem Natriumhydroxid
behandelt. Die Mischung wurde 16 h lang bei 20 bis 30 °C gerührt und
mittels Rotationsverdampfung zu einem dicken Sirup konzentriert.
Der Rückstand
wurde mit 800 ml entionisiertem Wasser verdünnt und mit 200 ml konzentrierter
HCl auf einen pH-Wert von 1 bis 2 angesäuert. Die Mischung wurde danach
mit 3 × 300
ml Aliquots Diethylether gewaschen und die wäßrige Phase wurde danach mittels Rotationsverdampfung
zu einem dicken Sirup konzentriert. Der Rückstand wurde mit 1 Liter trockenem
Methanol verdünnt
und mittels Rotationsverdampfung erneut konzentriert. Der Rückstand
wurde mit 1 Liter trockenem Methanol verdünnt und danach wurde Chlorwasserstoffgas
2 bis 3 Minuten lang in die Mischung eingeblasen. Die Mischung wurde
18 h lang bei 20 bis 30 °C
gerührt,
und danach durch eine Celite-Filter (hergestellt durch die Firma
J.T. Baker) vakuumfiltriert. Die Feststoffe wurden mit 200 ml Methanol
gespült.
Die vereinigten Filtrate wurden konzentriert. Der Rückstand
wurde mit 1 Liter Methanol verdünnt
und Chlorwasserstoffgas wurde erneut 2 bis 3 Minuten lang in die
Mischung eingeblasen. Die Mischung wurde 3 h lang gerührt und
danach konzentriert. Der Rückstand
wurde mit 1 Liter Methanol verdünnt
und 10 g Aktivkohle wurden zugesetzt. Die Mischung wurde 30 Minuten
lang gerührt
und danach durch eine Celite-Filter vakuumfiltriert. Die Feststoffe wurden
mit 100 ml Methanol gewaschen und die vereinigten Filtrate wurden
konzentriert. Der Rückstand
wurde in heißem
2-Propanol gelöst
und man ließ ihn
danach erneut, zuerst bei Raumtemperatur und dann mittels Verwendung
eines Eisbades, kristallisieren. Die Feststoffe wurden filtriert
und mit 3 × 75
ml Aliquots kaltem 2-Propanol gespült. Die Feststoffe wurden an
der Luft getrocknet; dies ergab 70,5 g des Produktes (18) in Form
eines weißen
Feststoffes. Die Filtrate wurden vereinigt und konzentriert. Der
Rückstand
wurde erneut in 200 ml 2-Propanol kristallisiert und ergab zusätzlich 15,3
g des Produktes von insgesamt 85,8 g (67 %): 1H-NMR (d6-DMSO); d 8,69 (2H, breit), 3,57 (6H, s),
2,82 (4H, m), 2,30 (4H, t), 1,67 bis 1,43 (8H, m) und 1,28 bis 1,19 (4H,
m) ppm; 1H-NMR (CD3OD); d 3,66 (6H, s),
3,42 (4H, t), 2,34 (4H, t), 1,75 bis 1,55 (8H, m) und 1,45 bis 1,25
(4H, m) ppm.
-
Herstellung von N-BOC-N,N-Bis-(5-methoxycarbonylphentyl-)amin
(19)
-
Einem
500 ml-Rundkolben, der mit 6,34 g (29,1 mMol) Di-t-butyldicarbonat
und 9,00g (29,1 mMol) N,N-Bis-(5-methoxycarbonylpentyl-)amin-hydrochlorid
(18) befüllt
war, wurden 125 ml wasserfreies Acetonitril und anschließend 7,5
ml Triethylamin zugesetzt. Die Mischung wurde 22 h lang bei Raumtemperatur
gerührt und
anschließend
mittels Rotationsverdampfung konzentriert. Der Rückstand wurde mit 300 ml Ethylacetat verdünnt und
mit 2 × 100
ml Aliquots 0,1 N wäßrige HCl,
100 ml entionisiertem Wasser und 100 ml einer 5 %igen wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
Magnesiumsulfat getrocknet, vakuumfiltriert und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert; dies ergab
10,5 g des Produktes (19) als nahezu farbloses Öl (97 %): 1H-NMR
(d6-DMSO); d 3,57 (6H, s), 3,07 (4H, t),
2,28 (4H, t), 160 bis 1,10 und 1,37 (21H, m und s) ppm: Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf
= 0,33 (Ethylacetat/Hexan (20 : 80)); RP-18-Silica-Gel, Rf = 0,17
(Methanol/Wasser (70 : 30)).
-
Herstellung von N-BOC-N,N-Bis-(5-hydroxycarbonylphentyl-)amin
(20)
-
Einem
500 ml-Rundkolben, der mit 10,5 g Bis-methylester 19 befüllt war,
wurden 75 ml Methanol und anschließend 75 ml 1 N wäßriges Natriumhydroxid
zugesetzt. Die Mischung wurde 16 h lang bei Raumtemperatur gerührt und
danach mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde mit 75 ml entionisiertem Wasser verdünnt und der pH-Wert der resultierenden
Lösung
wurde durch langsame Zugabe von etwa 75 ml 1 N wäßriger HCl auf 2 bis 2,5 eingestellt.
Danach wurden 200 ml Ethylacetat zugesetzt und die Mischung wurde
3 Minuten lang heftig gerührt.
Die Mischung wurde in einen Scheidetrichter überführt und die Schichten wurden
getrennt. Die wäßrige Phase
wurde mit 2 × 150
ml Aliquots Ethylacetat extrahiert. Die organischen Extrakte wurden
vereinigt, über
wasserfreiem Magnesiumsulfat getrocknet und vakuumfiltriert. Die
Filtrate wurden zuerst mittels Rotationsverdampfung und anschließend unter
vollem Vakuumpumpen-Druck (< 0,5
mm Hg) konzentriert; dies ergab 9,52 g des Produktes in Form eines
viskosen, nahezu farblosen Öls
(98 %): 'H-NMR (d6-DMSO); d 3,07 (4H, t), 2,28 (4H, t), 1,58
bis 1,10 und 1,37 (21H, m und s) ppm: Dünnschichtchromatographie (Sichtbarmachung
mit Ninhydrin-Spray und Hitze); RP-18-Silica-Gel, Rf= 0,44 (Methanol/Wasser
(70 : 30)).
-
Herstellung von N-BOC-N
N-Bis-(N',N'-bis-(5-methoxycarbonylpentyl-)5-carbamylpentyl-)amin
(21)
-
Einem
1 Liter-Rundkolben, der mit 9,52 g (27,6 mMol) Bis-Säure (16)
in 250 ml wasserfreiem Dimethylformamid befüllt war, wurden 19,0 g (61,3
mMol) N,N-Bis-(5-methoxycarbonylpentyl-)amin-hydrochlorid (18)
und anschließend
30 ml Triethylamin zugesetzt. Während
die Mischung gerührt
wurde, wurden 25,7 g (58,1 mMol) BOP zugesetzt. Die resultierende
Mischung wurde 14 h lang bei Raumtemperatur gerührt und danach mittels Rotationsverdampfung
konzentriert. Der Rückstand
wurde mit 750 ml Ethylacetat verdünnt und mit 250 ml 0,2 N wäßriger HCl,
100 ml 0,1 N wäßriger HCl,
100 ml entionisiertem Wasser und 2 × 100 ml Aliquots 5 %iger wäßriger Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltriert und mittels
Rotationsverdampfung konzentriert. Der Rückstand wurde an einer Silica-Gel-Säule (9 × 21 cm) chromatogaphiert
und zuerst mit 70 % Ethylacetat/Hexan und danach mit 100 % Ethylacetat
eluiert. Die Fraktionen, die das Produkt (21) enthielten, wurden
vereinigt und mittels Rotationsverdampfung konzentriert. Der Rückstand
wurde an einer RP-18-Silica-Gel-Säule (7 × 23 cm)
chromatogaphiert und zuerst mit Methanol/Wasser (75 25), danach
mit Methanol/Wasser (80 : 20) und letztendlich mit Methanol/Wasser
(85 15) eluiert. Die Fraktionen, die das Produkt enthielten, wurden
vereinigt und zuerst mittels Rotationsverdampfung und anschließend unter
vollem Vakuumpumpen-Druck konzentriert. Der Rückstand wurde mit 500 ml Diethylether
verdünnt
und die resultierende Lösung
wurde mit wasserfreiem Magnesiumsulfat getrocknet. Die Mischung
wurde vakuumfiltriert und das Filtrat wurde zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert; dies ergab 17,80
g des Produktes (21) in Form eines nahezu farblosen, viskosen Öls (75 %): 1H-NMR (d6-DMSO);
d 3,57 (12H, s), 3,18 und 3,07 (12H, 2t), 2,32 bis 2,16 (12H, m),
1,61 bis 1,09 und 1,37 (45H, m und s) ppm: Dünnschichtchromatogaphie (Sichtbarmachung
mit Ninhydrin-Spray und Hitze); Silica-Gel, Rf = 0,50 (Ethylacetat); RP-18-Silica-Gel,
Rf = 0,30 (Methanol/Wasser (85 15)).
-
Herstellung von N-BOC-N,N-Bis-(N',N'-bis-(5-hydroxycarbonylpentyl-)5-carbamyl-pentyl)amin (22)
-
Einem
500 ml-Rundkolben, der mit 7,88 g (9,20 mMol) Tetramethylester (21)
in 75 ml Methanol befüllt war,
wurden 70 ml 1 N wäßriges Natriumhydroxid
zugesetzt. Die Mischung wurde 16 h lang bei Raumtemperatur gerührt und
danach mittels Rotationsverdampfung zu einem dicken Sirup konzentriert.
Der Rückstand wurde
mit 50 ml entionisiertem Wasser verdünnt und unter heftigem Rühren wurde
der pH-Wert der Lösung durch
langsame Zugabe von etwa 70 ml 1 N wäßriger HCl auf 2 bis 2,5 eingestellt,
wobei das Produkt (22) in dem Verfahren als Öl austrat (eine flüssige Phase
trennte sich von einer anderen flüssigen Phase). Die Mischung
wurde mit 200 ml 2-Propanol/Methylenchlorid
(3 : 1) und anschließend
mit 3 × 100
ml Aliquots 2-Propanol/Methylenchlorid
(3 : 1) extrahiert. Die organischen Extrakte wurden vereinigt, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 mm Hg) konzentriert. Dies ergab
7,70 g eines nahezu farblosen dicken Sirups, der aus 6,93 g des
erwünschten
Produktes (22; 94 %) und 0,77 g 2-Propanol bestand (durch NMR-Integration): 1H-NMR (d6-DMSO);
d 3,18 und 3,07 (12H, 2 t), 2,37 bis 2,12 (12H, m), 1,60 bis 1,10
und 1,37 (45H, m und s) ppm; Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray und Hitze); RP-18-Silica-Gel,
Rf = 0,50 (Methanol/Wasser (70 : 30)).
-
Herstellung eines N-BOC-Tet-Gal-Nac-1-a-S-C5-Zweiges
(23)
-
Einem
250 ml-Rundkolben, der mit 4,05 g (7,38 mMol) 3,4,6-Tri-O-acetyl-N-acetyl-galactosamin-1-a-S-[5'-thiopentyl-N-BOC-amin]
(9) befüllt
war, wurden 20 ml Methylenchlorid und anschließend 20 ml Trifluoressigsäre zugesetzt.
Die Mischung wurde 15 Minuten lang bei Raumtemperatw gerührt. Die
Mischung wurde mittels Rotationsverdampfung konzentriert und der
Rückstand
wurde dreimal mit 75 ml Methylenchlorid verdünnt und erneut konzentriert;
dies ergab 6,27 g Rückstand,
nämlich
eine Mischung aus dem erwünschten Produkt
(10) und restlicher Trifluoressigsäure: 1H-NMR
(CD3OD); d 5,61 (1H, d), 5,41 (1H, dd),
5,01 (1H, dd), 4,62 bis 4,47 (2H, m), 4,11 (2H, d), 2,91 (2H, t),
2,74 bis 2,48 (2H, m), 2,11 (3H, 2 s), 2,00 (3H, s), 1,93 und 1,91
(6H, 2 s) und 1,37 bis 1,10 (6H, m) ppm. Einem separaten 250 ml-Rundkolben,
der mit 1,33 g des Sirups, der 90 Gew.-% der Verbindung 20 enthielt
(netto 1,20 g; 1,50 mMol) befüllt
war, wurden 50 ml wasserfreies Dimethylformamid zugesetzt. Um das
restliche 2-Propanol zu entfernen, wurde die Mischung zuerst mittels Rotationsverdampfung
und anschließend
unter vollem Vakuumpumpen-Druck (< 0,5
mm Hg) konzentriert. Dem Rückstand
wurden 20 ml wasserfreies Dimethylformamid und 10 ml trockenes Triethylamin
zugesetzt. Der resultierenden gerührten Lösung wurde eine Dimethylformamid-Lösung des
Rohproduktes 10 (insgesamt 30 ml wasserfreies Dimethylformamid)
zugesetzt und die resultierende Mischung wurde 2 h lang bei Raumtemperatur
gerührt.
Die Mischung wurde danach mittels Rotationsverdampfung konzentriert.
Der Rückstand wurde
danach mit 250 ml Methylenchlorid verdünnt und mit 2 × 100 ml
Aliquots 1 N wäßriger HCl,
100 ml entionisiertem Wasser und danach mit 100 ml gesättigter
wäßriger Natriumbicarbonat-Lösung gewaschen.
Die organische Phase wurde über
wasserfreiem Magnesiumsulfat getrocknet, vakuumfiltiert und mittels
Rotationsverdampfung konzentriert. Der Rückstand wurde an einer RP-18-Silica-Gel-Säule (5,5 × 19 cm)
chromatographiert und mit jeweils 250 ml Methanol/Wasser (65 : 35),
Methanol/Wasser (70 : 30), Methanol/Wasser (75 : 25) und anschließend mit
800 ml Methanol/Wasser (80 : 20) eluiert. Die Fraktionen, die das
Produkt enthielten, wurden vereinigt und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck konzentriert. Dies ergab
3,55 g eines schaumigen weißen
Feststoffes (94 %). Dieses Material wurde danach an einer Silica-Gel-Säule (5,5 × 20 cm)
chromatographiert und mit Ethylacetat/Methanol (80 : 20) eluiert.
Die Fraktionen, die nur das erwünschte
Produkt (23) enthielten, wurden vereinigt und zuerst mittels Rotationsverdampfung
und danach unter vollem Vakuumpumpen-Druck (< 0,5 nun Hg) konzentriert. Dies ergab
2,83 g des erwünschten
Produktes in Form eines rein weißen schaumigen Feststoffes
(75 %): 1H-NMR (CD3OD);
d 5,58 (4H, d), 5,42 (4H, dd), 5,01 (4H, dd), 4,63 bis 4,51 (8H,
m), 4,20 bis 4,00 (8H, m), 3,35 bis 3,10 (20H, m), 2,73 bis 2,47
(8H, m), 2,32 (4H, t), 2,25 bis 2,08 (20H, m und s), 2,00 (12H,
s), 1,93 und 1,91 (24H, 2s), 1,71 bis 1,20 (69H, m und s) ppm; Dünnschichtchromatographie
(Sichtbarmachung mit Ninhydrin-Spray
und Hitze); Silica-Gel, Rf = 0,47 (Ethylacetat/Methanol (75 : 25)).
-
Einer
Lösung
aus 155 mg 23 in 1,0 ml CH2Cl2 wurden
1,0 ml Trifluoressigsäure
zugesetzt. Die Mischung wurde 15 Minuten lang bei Raumtemperatur
gerührt
und danach konzentriert. Der Rückstand
wurde mit 5 ml CH2Cl2 verdünnt und
erneut konzentriert. Der Rückstand
wurde in 5 ml CH2Cl2 gelöst und mit
10 ml gesättigter
wäßriger Natriumbicarbonat-Lösung gewaschen.
Die wäßrige Phase
wurde zurückextrahiert
mit 3 × 5
ml CH2Cl2/2-Propanol
(3 : 1). Die organischen Phasen wurden vereinigt, über Natriumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wurde dreimal in 5
ml CH2Cl2 gelöst und erneut
konzentriert; dies ergab 153 mg des Rohproduktes.
-
Einer
Lösung
aus 145 mg des Rohproduktes 24 in 2 ml Methanol wurden 1,0 g AG1-X8-Anionenaustausch-Harz
(Biorad; Hydroxid-Form; 2,3 mequiv/g) und anschließend 3 ml
entionisiertes Wasser zugesetzt. Die Mischung wurde 16 h lang bei
Raumtemperatur gerührt
und danach filtriert. Das Harz wurde mit 10 ml Methanol/Wasser (1
: 1) gespült
und die vereinigten Filtrate wurden danach konzentriert. Der Rückstand
wurde an einer RP-18-Silica-Gel-Säule (3 × 11 cm) chromatographiert
und mit Methanol/Wasser/1 N wäßrige HCl
(50 : 50 : 0,2) eluiert. Dies ergab 112 mg Produkt (96 %): NMR (D2O); 6 5,48 (4H, d), 4,37 bis 4,20 (8H, m),
3,98 (4H, d), 3,87 bis 3,70 (12H, m), 3,40 bis 3,21 (8H, m), 3,15
(8H, t), 3,03 (4H, t), 2,61 (8H, m), 2,40 (4H, t), 2,21 (8H, t),
2,02 (12H, s) und 1,78 bis 1,15 (60H, m) ppm. TLC, Rf= 0,59 (Methanol/Wasser/1
N wäßrige HCl
(70 : 30 : 0,2)).
-
1,01
g Maleimidopropionsäure-NHS-Ester
26 in 10 ml trockenem DMF wurden 750 mg 6-Aminocapronsäure und
anschließend
1,5 ml trockenes Diisopropylethylamin zugesetzt. Die Mischung wurde
3,5 h lang bei Raumtemperatur gerührt und danach konzentriert.
Der Rückstand
wurde an einer RP-18-Silica-Gel-Säule (3,5 × 18 cm) chromatographiert
und mit Methanol/Wasser/1 N wäßrige HCl
(40 : 60 : 0,1) und danach mit Methanol/Wasser (50 : 50) eluiert.
Die Fraktionen, die das Produkt enthielten, wurden vereinigt und
konzentriert. Dies ergab 956 mg des Produktes 27 in Form eines weißen Feststoffes
(89 %): NMR (DMSO – 200
MHz); 6 7,90 (1H, t), 7,01 (2H, s), 3,60 (2H, t), 2,98 (2H, Quartett),
2,31 (2H, t), 2,19 (2H, t) und 1,58 bis 1,13 (5H, m) ppm. TLC, Rf=
0,55 (Methanol/Wasser (50 : 50)).
-
10
ml Maleimid-Carbonsäure
27 in 1,0 ml trocknem DMF wurden 100 μl trockenes Diisopropylethylamin
und 15 mg BOP zugesetzt. Die Mischung wurde 15 Minuten lang bei
Raumtemperatur gerührt
und danach wurden 17 mg 25 zugesetzt. Die Mischung wurde zwei Stunden
lang bei Raumtemperatur gerührt
und danach konzentriert. Der Rückstand
wurde an einer RP-18-Silica-Gel-Säule (2,5 × 16 cm) chromatographiert
und mit Methanol/Wasser (60 : 40 : 0,2) eluiert. Dies ergab 6,5
mg des Produktes 28 (34 %): NMR (CD3OD – 200 MHz); 6
6,83 (2H, s), 5,53 (4H, d), 4,41 (4H, dd), 4,18 (4H, t), 3,90 (4H,
d), 3,81 bis 3,67 (12H, m), 3,33 (12H, m), 3,15 (10H, t), 2,60 (8H,
m), 2,38 (6H, m), 2,19 (8H, m), 1,97 (12H, s) und 1,75 bis 1,20
(66H, m) ppm.
-
C. Konjugation von Annexin
V-SH mit Verbindung 28
-
Die
Konjugation von Annexin an den Maleimidyl-4-mer-Zucker-Cluster bedingt
die Inkubation von Annexin V-SH mit Dithiothreitol (DTT), um sicherzustellen,
daß die
Sulfhydryl-Einheit des Annexins vollständig reduziert wird, die Reinigung
durch Größen-Ausschlußchromatographie,
um überschüssiges DTT
zu entfernen, und anschließend
die sofortige Umsetzung mit dem Maleimidyl-Agens.
-
Insbesondere
wurden 2,25 ml Annexin V-SH (4,1 mg pro ml 10 mMol MES-Puffer, pH 6,0, der
150 mMol Natriumchlorid enthielt) (d.h. 0,26 μMol) wurden 45 μMol DTT in
0,05 ml Wasser zugesetzt, um eine Endlösung von 20 mMol DTT zu erhalten.
Nach Inkubation bei Raumtemperatur für eine Zeitdauer von 30 Minuten wurden
1,1 ml Aliquots der Reaktionslösung
auf PD-10-SephadexR-Größen-Ausschlußchromatophraphie-Säulen aufgetragen,
die zuvor mit einem mit Stickstoff gesättigten 30 mMol Natriumphosphat-Puffer, pH-Wert:
6,75, der 150 mMol Natriumchlorid enthielt, equilibriert worden
waren. Die 2,4 bis 4,8 ml-Fraktionen aus jeder Säule wurden danach gepoolt und
sofort danach wurden 2,6 μMol
(5,7 mg) der festen Verbindung 26 zugesetzt. Nach dem Rühren, um
alles aufzulösen,
wurde die Reaktionsmischung 2 h lang bei Raumtemperatur inkubiert
und danach erschöpfend
gegen eine mit Phosphat gepufferte Kochsalzlösung dialysiert.
-
Um
die Derivatisierung zu bestätigen,
wurde eine Matrix-unterstützte
Laser-Desorptions-Ionisation (MALDI)
an mit destilliertem Wasser dialysiertem Konjugat durchgeführt, die
eine Spezies mit einem MW von 38.548 (m/e) anzeigte, was auf ein
1 : 1-Konjugat hinwies.