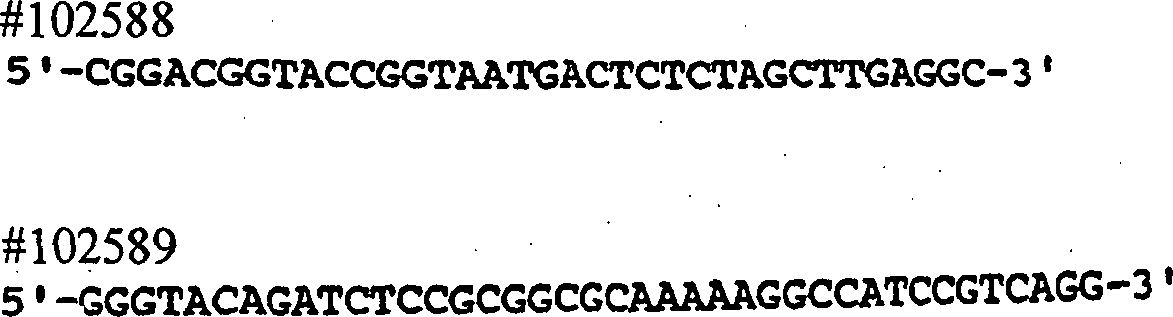-
Gebiet der
Erfindung
-
Die
vorliegende Erfindung bezieht sich auf ein Verfahren zur In-Vivo-Rekombination
homologer DNA-Sequenzen. Dieses Verfahren ist eine erzwungene, künstliche
Evolution, die zu einer DNA-Sequenz führt, die ein Polypeptid kodiert,
das eine vorteilhafte Eigenschaft hat.
-
Hintergrund
der Erfindung
-
Die
homologe Rekombination zwischen DNA-Sequenzen, die auf demselben
Plasmid angeordnet sind, ist dem Fachmann wohl bekannt (Weber und
Weissmann, Nucl. Acid. Res. (1983) 11, 5661–5669).
EP 252666 B1 (Novo Nordisk
A/S), WO 95/22625 A1 (Affymax Technologies N. V.) und WO 9101087
offenbaren die In-Vivo-Rekombination von Genen, die auf demselben
Plasmid angeordnet sind.
-
EP 449923 B1 (Setratech)
offenbart ein Verfahren zur intergenischen Rekombination in vivo
von teilweise homologen DNA-Sequenzen. Die Rekombination findet
in Zellen statt, deren enzymatisches Fehlpaarungs-Reparatursystem
gestört
ist.
-
J.
Biotechnol. (1991) 19, 221–240
offenbart ein Verfahren zur Inaktivierung eines Gens in dem Genom von
B. amyloliquefaciens. Das Verfahren umfasst die Integration und
Exzision einer pE194-Mutante.
-
In
Chemical Abstracts 118: 161925 (1993) wird die für die Rekombination erforderliche
minimale Sequenzlänge
der Homologie bestimmt. Das Verfahren zur Bestimmung schließt die Integration
eines pE194-Derivats, das ein Gluconasegen trägt, ein.
-
In
J. Bacteriol. (1982) 152, 524–526
wird ein Plasmid pBD9 offenbart, das die beiden Plasmide pE194 und
pUB110 von Staphylococcus aureus umfasst. Andere auf pE194 oder
pUB110 basierende Plasmide sind in Molecular and General Genetics
(1988), 213, 465–70,
Molecular and General Genetics (1984), 195, 374–7, Plasmid (1981), 6, 67–77 und
Genetika, (1986), 22, 2750–7
offenbart.
-
In
Yichuan Xuebao (1993), 20, 272–8
(siehe Chemical Abstracts 120: 1670 (1994)) offenbaren Chen et.
al. das Plasmid pNW102, ein pE194-Derivat, in dem das thermostabile α-Amylasegen
aus Bacillus licheniformis insertiert ist. pNW102 wurde in den Bacillus
subtilis-Stamm BF 7658 transformiert, gefolgt von Inkubation bei
nicht-permissiver Temperatur, was die Rekombination zwischen den α-Amylasegenen
von pNW102 und BF 7658 erlaubte. Die von den rekombinanten Stämmen produzierte α-Amylase
zeigt dieselben Charakteristika wie die α-Amylase von Bacillus licheniformis.
-
Jedoch
gibt es in dem verwandten Stand der Technik keinen Hinweis, dass
DNA-Sequenzen durch Wiederholen
von Integration und Auskreuzen in vivo rekombiniert werden können, um
ein Gen zu erhalten, das ein Polypeptid mit verbesserten Merkmalen,
wie z.B. verbesserter Stabilität,
verbesserter Spezifität
oder höherer
Aktivität,
kodiert.
-
Somit
ist es eine Aufgabe der Erfindung, ein verbessertes In-Vivo-Verfahren
zur Herstellung neuer DNA-Sequenzen bereitzustellen.
-
Kurze Beschreibung der
Erfindung
-
Die
vorliegende Erfindung bezieht sich auf ein In-Vivo-Verfahren zur
Herstellung neuer DNA-Sequenzen. Es hat sich herausgestellt, dass
neue DNA-Sequenzen mit verbesserten Eigenschaften auf eine schnelle und
effiziente Weise aus wenigstens zwei homologen DNA-Sequenzen durch
ein Verfahren hergestellt werden können, das den In-Vivo-Austausch von DNA
zwischen wenigstens zwei Vektoren, von denen jeder eine homologe
DNA-Sequenz und einen Replikationsursprung enthält, einschließt.
-
Das
erfindungsgemäße Verfahren
ist dadurch gekennzeichnet, dass der Austausch der homologen DNA-Sequenzen
durch wenigstens einmaliges Wiederholen in vivo der Integration
und des Ausschneidens eines Vektors in den anderen durchgeführt wird.
-
Somit
bezieht sich ein erster Aspekt der Erfindung auf ein Verfahren zur
In-Vivo-Rekombination
homologer DNA-Sequenzen, umfassend die folgenden Schritte:
- a) Inkubieren einer Zelle, die wengstens zwei
DNA-Strukturen enthält,
die jede eine DNA-Sequenz und einen Replikationsursprung enthalten,
unter Bedingungen, wobei i) wenigstens ein Replikationsursprung nicht-funktionell
ist und ii) die Zelle unter einem Selektionsdruck steht, der ein
Wachstum der Zelle nur erlaubt, falls sie die DNA-Struktur mit dem
nicht-funktionellen Ursprung enthält, wodurch eine Integration
einer der DNA-Strukturen in eine der anderen und dadurch die Bildung
einer hybriden DNA-Struktur begünstigt
wird;
- b) Verändern
der Bedingungen zu Bedingungen, unter denen weniger Replikationsursprünge nicht-funktionell
sind als in Schritt a), wodurch ein Auskreuzen aus der hybriden
DNA-Struktur und die Bildung von DNA-Strukturen begünstigt wird,
die eine rekombinierte DNA-Sequenz und einen Replikationsursprung
umfassen; und
- c) wenigstens einmaliges Wiederholen der Schritte a)–b).
-
Einer
der Vorteile des erfindungsgemäßen In-Vivo-Rekombinationsverfahrens
ist, dass es durch Wiederholen des In-Vivo-Austausches zwischen
den DNA-Sequenzen möglich
ist, zahlreiche verschiedene Rekombinationsmuster zwischen den DNA-Sequenzen
zu erhalten.
-
Dies
ist in der 1 veranschaulicht, die das Ergebnis
der wie in Beispiel 1 beschrieben durchgeführten In-Vivo-Rekombination
zwischen den zwei DNA-Sequenzen Savinase und Savisyn zeigt.
-
In 1 sind
Savinase/Savisyn-rekombinierte Klone, wie z.B. die Klone Nr. 3,
8, 12, von mehr als einem In-Vivo-Rekombinationsereignis zwischen
den beiden DNA-Sequenzen abgeleitet.
-
Kurze Beschreibung
der Zeichnung
-
1 veranschaulicht
die Rekombination von zwei DNA-Sequenzen, die auf den Plasmiden
pTR, temperaturempfindlich, da der Replikationsursprung bei 50°C funktionell
ist, und pTS, temperatursensitiv, da der Replikationsursprung über 45°C nicht-funktionell
ist, angeordnet sind. Unter nicht-permissiven Bedingungen (d.h.
bei 50°C)
integriert pTS in pTR, wobei ein Hybridplasmid gebildet wird.
-
2 veranschaulicht
das Auskreuzen aus dem Hybridplasmid, wobei zwei neue DNA-Sequenzen gebildet
werden, die jede auf einem Plasmid angeordnet sind. Ein wiederholtes
Ein- und Auskreuzen kann z.B. durch zyklischen Temperaturwechsel
oder einfach durch Aufrechterhalten des Temperaturdrucks und Selektieren
auf die Resistenz von pTS (d.h. Erm®) durchgeführt werden.
-
3 veranschaulicht
die Plasmide, die nach wiederholten Integrations- und Auskreuzungsereignissen
erhalten werden.
-
Außerdem veranschaulichen
die 1 und 2 ein DNA-Struktursystem, das
für die
In-Vivo-Messung
der Hybridstrukturbildung geeignet ist und für die Durchführung eines
erzwungenen Auskreuzens der DNA-Hybridstruktur geeignet ist.
-
1:
DNA-Struktur pTS:
Geeigneter aktiver Promotor, der die Expression
eines aktiven Proteins steuert
Erm®: Antibiotikum-Resistenzgen
Die
DNA-Struktur pTR umfasst:
Keinerlei aktiven Promotor für die Steuerung
der Transkription von GFP
GFP: Grün-fluoreszierendes Protein
-
Das
Erm®-Antibiotikum-Resistenzgen
in pTS ist wegen des aktiven Promotors aktiv.
-
Das
GFP-Gen in pTR ist inaktiv (d.h. das Gen ist nicht-transkribiert),
weil es keinen aktiven Promotor gibt, der die Expression des Gens
antreibt.
-
2:
Nach der Bildung der erfindungsgemäßen Hybridstruktur ist GFP
nun aktiv (angetrieben von dem Promotor). Dies ermöglicht es,
die Hybridstrukturbildung in vivo zu messen (siehe unten wegen weiterer Einzelheiten).
-
Im
Gegensatz dazu ist das Erm®-Antibiotikum-Resistenzgen
nun inaktiv (d.h. das Gen ist nicht-transkribiert), weil es keinen
aktiven Promotor gibt, der die Expression des Gens antreibt. Das
erzwungene Auskreuzen aus der DNA-Hybridstruktur wird durch Inkubieren
der Zellen unter antibiotischem Druck durch Erm® durchgeführt, wodurch
ein Auskreuzen aus der DNA-Hybridstruktur erzwungen wird, weil die
Zelle nur in der Lage ist, das Erm®-Antibiotikum-Resistenzgen
zu exprimieren, nachdem das Auskreuzen aus der DNA-Struktur stattgefunden
hat.
-
4 und 5:
Sie illustrieren die zwei Vektoren pSX120 (4) und pMB430
(5), die, wie in Beispiel 1 beschrieben, verwendet
werden, um die zwei DNA-Sequenzen Savinase und Savisyn in vivo zu
rekombinieren.
-
pSX120
umfasst i) einen offenen Leserahmen, der die Savinase kodiert, ii)
einen aktiven Promotor, der die Transkription der Savinase steuert,
und iii) ein Gen, das Resistenz gegen Chloramphenicol (Cam®)
verleiht.
-
pMB430
umfasst i) ein Savizyn-DNA-Fragment, ii) einen Terminator (der dazu
führt,
dass keine Transkription durch die auf ihn folgenden Regionen erfolgt),
iii) ein Gen, das Resistenz gegenüber Erythromycin (Erm®)
verleiht, und iv) einen offenen Leserahmen, der das Grün-fluoreszierende
Protein (GFP) kodiert.
-
6:
Illustriert die Ergebnisse der wie in Beispiel 1 beschrieben durchgeführten In-Vivo-Rekombination von
Savinase/Savisyn.
-
Die
schematische Darstellung zeigt 12 der gemischten/rekombinierten
Klone.
-
In
horizontaler Richtung sind die Positionen der Restriktionsstellen
angegeben, die nur in Savizyn vorkommen.
-
Der
Buchstabe „S" gibt an, dass die
Sequenz an dieser Position identisch zu der Savinase-Sequenz ist, und
der Buchstabe „Z" gibt an, dass die
Sequenz von Savizyn abstammt. Alle sequenzierten Klone sind in ihren
Rekombinationsmustern unterschiedlich.
-
Definitionen
-
Wie
hierin verwendet, haben die folgenden Ausdrücke die folgenden Bedeutungen:
Der
Ausdruck „DNA-Sequenz" schließt eine
beliebige DNA-Sequenz ein, bei der es wünschenswert ist, sie gemäß der Erfindung
zu modifizieren, z.B. eine DNA-Sequenz, die ein Polypeptid, zum
Beispiel ein Enzym, pharmakologisch aktive Polypeptide, z.B. Insulin,
Wachstumshormon, humane Hormone oder Wachstumsregulatoren, kodiert,
und Sequenzen, wie z.B. Promotoren, Transkriptions- oder Translationsregulatoren,
und andere intrazelluläre
Polynukleotide und Polypeptide.
-
Der
Begriff „homolog" bedeutet, dass die
DNA-Sequenzen einige Abschnitte identischer Nukleotide haben, die
die Rekombination in vivo ermöglichen.
Die besagten Abschnitte innerhalb der DNA-Sequenzen sind wengstens
15 Basenpaare lang, mehr bevorzugt wenigstens 25 Basenpaare lang,
noch mehr bevorzugt wenigstens 50 Basen lang und am meisten bevorzugt
wenigstens 150 Basenpaare oder mehr.
-
Der
Ausdruck „homolog" umfasst Abschnitte
von DNA-Sequenzen, die ab 60% identisch mit DNA-Sequenzen sind,
die sich nur in einem Nukleotid unterscheiden. Es ist bevorzugt,
dass die Abschnitte von DNA-Sequenzen zu mehr als 70% identisch
sind, mehr bevorzugt wenigstens 80%, speziell mehr als 90% identisch
und mehr bevorzugt, dass die Abschnitte von DNA-Sequenzen zu 95%,
am meisten bevorzugt zu wenigstens 97% identisch sind.
-
Innerhalb
der interessierenden Abschnitte der DNA-Sequenzen wird die oben
erwähnte
DNA-Sequenzhomologie als der Grad der Identität zwischen zwei Sequenzen bestimmt,
der eine Ableitung der ersten Sequenz von der zweiten anzeigt. Die
Homologie kann geeigneter Weise mittels im Stand der Technik bekannter
Computerprogramme bestimmt werden, wie z.B. dem in dem GCG-Programmpaket
bereitgestellten GAP (Programm Manual for the Wisconsin Package,
Version 8, August 1994, Genetics Computer Group, 575 Science Drive,
Madison, Wisconsin, USA 53711) (Needleman, S. B. und Wunsch, C.
D., (1970), Journal of Molecular Biology, 48, 443–453). Bei
der Verwendung von GAP mit den folgenden Einstellungen für den DNA-Sequenzvergleich:
GAP-Creation-Penalty von 5,0 und GAP-Extension-Penalty von 0,3,
zeigen die interessierenden homologen DNA-Sequenzen einen vorzugsweise
wie oben beschriebenen Grad der Identität.
-
Der
Ausdruck „Rekombinierte
DNA-Sequenz" bezeichnet
die DNA-Sequenz, die ein Ergebnis eines Rekombinationsereignisses
zwischen wenigstens zwei. DNA-Sequenzen ist. Das Rekombinationsereignis kann
ein einzelnes Rekombinationsereignis sein oder kann zuerst ein Rekombinationsereignis
zwischen wenigstens zwei DNA-Sequenzen, gefolgt von einem Auskreuzen
an einer anderen Stelle als das erste Rekombinationsereignis sein.
-
Der
Ausdruck „DNA-Struktur" bezeichnet ein DNA-Molekül, z.B.
einen Vektor, ein Plasmid, ein Chromosom (z.B. ein Bacillus-subtilis-Chromosom),
ein Virus, einen Phagen, ein Transposon oder ein Genom.
-
Der
Ausdruck „Vektor" bezeichnet ein beliebiges
lineares oder zirkuläres
DNA-Molekül,
das fähig
ist, eine DNA-Sequenz, die rekombiniert werden soll, und einen Replikationsursprung
zu enthalten. Der Ausdruck schließt einen beliebigen, dem Fachmann
bekannten Vektor, z.B. ein Plasmid oder einen Phagen, ein. Die DNA-Sequenz
befindet sich wahlweise, aber nicht notwendigerweise, unter der
Kontrolle eines Promotors.
-
Der
Ausdruck „eine
Zelle", der hierin
im Zusammenhang mit einem erfindungsgemäßen In-Vivo-Rekombinationsverfahren
verwendet wird, soll sowohl „eine
Zelle" als auch „eine Zellpopulation" umfassen.
-
Der
Ausdruck „Einführen" bedeutet, dass die
DNA-Struktur in eine Zelle eingeführt wird. Die DNA-Struktur
kann unter Verwendung von im Stand der Technik bekannten Techniken
für die
direkte Einführung
von DNA, z.B. mittels Elektroporation, Transformation kompetenter
Zellen, Protoplastentransformation, Transfektion, Transduktion,
Konjugation oder ballistischer Transformation, eingeführt werden.
-
Der
Ausdruck „Inkubieren" (oder Kultivieren,
Wachsenlassen) bedeutet, dass die Zelle in einer Weise behandelt
wird, so dass die normalen Zellfunktionen aktiv sind, speziell die
für die
Rekombination notwendigen Funktionen. Die Zelle kann auf einem festen
Substrat inkubiert werden (Agar-Petrischale), oder die Zelle kann unter
Submersbedingungen inkubiert werden. Es ist bevorzugt, dass die
Zelle in einem Teströhrchen,
z.B. einem Eppendorf-Teströhrchen,
inkubiert wird. Im Falle eines temperaturempfindlichen Replikationsursprungs wird
das Teströhrchen
zweckmäßiger Weise
in einen automatischen Thermozykler gestellt. Außerdem kann die Zelle in Standard-Schüttelflaschen
inkubiert werden.
-
Der
Ausdruck „Replikationsursprung" (ori) bezeichnet
eine Region, in der die Replikation einer DNA-Struktur initiiert
wird. Der Ausdruck funktionell im Zusammenhang mit Replikationsursprung
bedeutet, dass die Replikation an dem Ursprung initiiert werden
kann. Der Ausdruck nicht-funktionell bedeutet, dass keine Replikation
unter den gegebenen Bedingungen initiiert wird oder dass die Initiation
der Replikation verringert ist, z.B. auf ein Niveau, das für das Überleben
einer Zelle, die eine DNA-Struktur mit einem Markergen und dem Ori
enthält,
unter angemessenem Selektionsdruck nicht ausreicht.
-
Der
Ausdruck „temperaturempfindlich" bezeichnet im Zusammenhang
mit einem Replikationsursprung oder Vektor, dass der Vektor nicht
fähig ist,
unter z.B. erhöhten
Temperaturen, d.h. nicht-permissiven Bedingungen, die dennoch ein
Wachstum der Elternzelle erlauben, zu replizieren.
-
Der
Ausdruck „temperaturresistent" im Zusammenhang
mit Replikationsursprung oder Vektor bedeutet, dass der Vektor fähig ist,
bei erhöhten
Temperaturen zu replizieren.
-
Der
Ausdruck „DNA-Bibliothek" ist eine Bibliothek,
die zahlreiche verschiedene DNA-Sequenzen
umfasst. Eine DNA-Bibliothek kann gemäß im Stand der Technik bekannter
Standardverfahren hergestellt werden, wie z.B. durch In-vitro-DNA-Mischen/DNA-Rekombination (WO
95/17413, WO 95/22625) und/oder fehleranfällige PCR und Kassettenmutagenese.
-
Eine
DNA-Bibliothek kann erfindungsgemäß so wenige wie 2–20 verschiedene
Sequenzen umfassen, wie z.B. eine Bibliothek, in der nur eine Aminosäure variiert
ist. Außerdem
kann eine DNA-Bibliothek zahlreiche verschiedene DNA-Sequenzen,
z.B. bis zu 1010 verschiedene Sequenzen,
umfassen.
-
Detaillierte
Beschreibung der Erfindung
-
Die
vorliegende Erfindung umfasst ein Verfahren zur In-Vivo-Rekombination
homologer DNA-Sequenzen, umfassend die Schritte:
- a)
Inkubieren einer Zelle, die wenigstens zwei DNA-Strukturen enthält, die
jede eine DNA-Sequenz und einen Replikationsursprung umfassen, unter
Bedingungen, wobei i) wenigstens ein Replikationsursprung nicht-funktionell
ist und ii) die Zelle unter einem Selektionsdruck steht, der ein
Wachstum der Zelle nur erlaubt, falls sie die DNA-Struktur mit dem
nicht-funktionellen Ursprung enthält (d.h. wodurch eine Integration einer
der DNA-Strukturen in eine der anderen und dadurch die Bildung einer
hybriden DNA-Struktur begünstigt
wird);
- b) Verändern
der Bedingungen zu Bedingungen, unter denen weniger Replikationsursprünge nicht-funktionell
sind als in Schritt a) (d.h wodurch ein Auskreuzen aus der hybriden
DNA-Struktur begünstigt
wird); und
- c) wenigstens einmaliges Wiederholen der Schritte a)–b).
-
Die
rekombinierte DNA-Sequenz wird neue Polypeptide kodieren, die neue
Eigenschaften haben. Es ist dann möglich, in das erfindungsgemäße Verfahren
ein Selektions- oder Screeningsystem einzufügen, um gewünschte Eigenschaften, die von
oder mehreren der rekombinierten DNA-Sequenz(en) kodiert sind, zu
selektieren und/oder darauf zu screenen.
-
Die
Anzahl der Wiederholung(en) in Schritt (c) oben kann eine Wiederholung
sein, oder sie kann mehr sein, wie z.B. 5–10 Wiederholungen oder 50–100 oder
sogar eine noch höhere
Anzahl von Wiederholungen.
-
Es
ist bevorzugt, dass die Integration durch Inkubation der Zelle unter
Bedingungen, bei denen der Replikationsursprung einer DNA-Struktur
nicht-funktionell ist, erzwungen wird (oder für das Überleben der DNA-Struktur vorteilhaft
gemacht wird), und dass das Auskreuzen durch Inkubation der Zelle
unter Bedingungen, bei denen der Replikationsursprung einer DNA-Struktur
wieder funktionell ist, erzwungen wird.
-
In
einem anderen bevorzugten Ausführungsbeispiel
des oben in den Schritten (a) bis (c) dargestellten Ablaufs ist
es bevorzugt, dass die Zellen unter konstanten Bedingungen gehalten
werden (z.B. konstante Temperatur und Selektionsdruck); bei denen
sich ein spontanes Einkreuzen und Auskreuzen ereignet. Das heißt, das
Mischungsverfahren in den Zellen wird wiederholt, ohne die Bedingungen
wiederholt zu ändern.
-
In
einer Anzahl von DNA-Strukturen, wie z.B. in vielen Bacillus-DNA-Strukturen
(Vektoren) (Bacillus subtilis and Other Gram-Positive Bacteria,
Sonensheim et al., 1993, American Society for Microbiology, Washington
D.C.), umfasst der Replikationsursprung einen Ori(+) und einen Ori(–). Der
Ori(+) steuert, wo die Replikation des ersten DNA-Strangs beginnt,
und der Ori(–)
steuert den Initiationspunkt für
die Replikation auf dem zweiten Strang (Bacillus subtilis and Other
Gram-Positive Bacteria, Sonensheim et al., 1993, American Society
for Microbiology, Washington D.C.).
-
In
einem Ausführungsbeispiel
des erfindungsgemäßen Verfahrens
umfasst wenigstens eine DNA-Struktur nicht Ori(–) (d.h. sie umfasst nur Ori(+)).
Vorzugsweise ist die DNA-Struktur,
die Ori(–)
nicht umfasst, eine DNA-Struktur, deren Ursprung (d.h. hierin Ori(+))
während
der erfindungsgemäß durchgeführten Bildung
der hybriden DNA-Struktur funktionell ist.
-
Ein
Vorteil dieses erfindungsgemäßen Ausführungsbeispiels
ist, dass, wenn nur Ori(+) in der besagten DNA-Struktur funktionell
ist, diese DNA-Struktur für
eine vergleichsweise längere
Dauer in einem einzelsträngigen
Zustand ist als im Vergleich eine DNA-Struktur, die sowohl Ori(+)
als auch Ori(–)
umfasst. Dieser verlängerte
einzelsträngige
Zustand erleichtert die Bildung der hybriden DNA-Struktur gemäß dem erfindungsgemäßen Verfahren.
-
Vorzugsweise
ist die DNA-Struktur, die wie oben beschrieben Ori(–) nicht
umfasst, eine Bacillus-Struktur, mehr bevorzugt ein Bacillus-Vektor.
-
Um
dieses Konzept zu veranschaulichen, ist ein Beispiel ein modifizierter
pTR- (TR: temperaturresistent) Bacillus-Vektor, der Ori(–) nicht
umfasst (siehe 1). Falls die Hybridbildung
bei 50°C
durchgeführt
wird, ist nur der Ursprung von pTR funktionell (der Ursprung von
pTS (TS: temperatursensitiv) ist nicht funktionell) (siehe 1, 2).
Die DNA-Struktur pTR umfasst nur Ori(+) und ist deshalb für eine vergleichsweise
längere
Dauer in einem einzelsträngigen
Zustand, was das Rekombinationsereignis und folglich die Hybridbildung mit
der pTS-DNA-Struktur erleichtert (siehe 1, 2).
-
Es
ist mehr bevorzugt, dass eine DNA-Struktur ein Vektor ist, der fähig ist,
unter bestimmten (permissiven) Bedingungen zu replizieren und nicht
fähig ist,
unter anderen (nicht-permissiven) Bedingungen zu replizieren. Der
Vektor kann zum Beispiel einer sein, der bezüglich der Replikation temperaturempfindlich
ist. Somit kann der Vektor einer sein, der nicht fähig ist,
bei erhöhten
Temperaturen zu replizieren, die noch ein Wachstum der Elternzelle
erlauben. Die Zelle wird anfänglich
bei einer Temperatur inkubiert, die die Vektorreplikation erlaubt,
und wird nachfolgend, nachdem die Integration in die andere DNA-Struktur
stattgefunden haben mag, bei einer Temperatur inkubiert, die die
Vektorreplikation nicht erlaubt, sodass der Vektor aus den Zellen
verloren geht, sofern er nicht integriert wurde.
-
Der
Vektor kann außerdem
einen selektierbaren Marker umfassen. In diesem Fall kann die Inkubation bei
der nicht-permissiven Temperatur unter selektiven Bedingungen durchgeführt werden,
um sicherzustellen, dass nur Zellen, die den integrierten Vektor
(der die DNA-Sequenz und den selektierbaren Marker einschließt) enthalten, überleben
werden.
-
Außerdem ist
es bevorzugt, ein System zu konstruieren, in dem es möglich ist,
die Bildung der Hybridstruktur in vivo zu messen.
-
Vorzugsweise
wird dies durchgeführt,
indem ein System konstruiert wird, worin ein in-vivo-screenbares oder -selektierbares
Protein nur exprimiert wird, nachdem die Hybridstruktur gebildet
wurde.
-
Vorzugsweise
ist das screenbare Protein ein Protein, das unter geeigneter Exponierung
fluoreszierend ist, und insbesondere ist das fluoreszierende Protein
das Grünfluoreszierende
Protein (GFP) oder Varianten davon.
-
Das
Grün-fluoreszierende
Protein (GFP) oder Varianten davon, und wie GFPs aktiviert werden,
um zu fluoreszieren, sind im Stand der Technik beschrieben, und
es wird wegen weiterer Einzelheiten auf (Crameri et al., Nature
Biotechnology 14: 315–319
(1996); und Cromack et al., GENE 173: 33–38 (1996)) verwiesen.
-
Einer
der Vorteile der Verwendung solch eines solchen screenbaren Proteins,
das unter geeigneter Lichtexponierung fluoreszierend ist, und insbesondere
von GFP oder Varianten davon, besteht darin, dass es dann durch
z.B. die Fluoreszens-aktivierte Zellsortierungstechnologie (FACS)
möglich
ist, selektiv Zellen auszusortieren, die die besagte Hybridstruktur
umfassen. Dies kann den Hintergrund an Zellen eliminieren, die die Hybridstruktur
nach der Durchführung
von Schritt a) gemäß dem ersten
Aspekt der Erfindung nicht umfassen.
-
FACS
ist eine weithin bekannte Screeningtechnologie und es wird auf (Cormack
et al. „FACS-optimized
mutants of the green fluorescent protein (GFP)" GENE 173: 33–38 (1996)) wegen weiterer
Einzelheiten verwiesen.
-
Um
ein geeignetes System zu veranschaulichen, das die In-Vivo-Messung
der Bildung der Hybridstruktur ermöglicht, ist ein Beispiel eines
geeigneten Systems in 1 und 2 gezeigt.
-
Alternativ
zu der wie oben beschriebenen Verwendung eines screenbaren Proteins
kann es vorteilhaft sein, einen selektierbaren Marker zu verwenden,
wobei der selektierbare Marker nur transkriptionell aktiv ist, wenn
die Hybridstruktur gebildet wird.
-
Außerdem ist
es bevorzugt, dass das Auskreuzen aus der hybriden DNA-Struktur
in Schritt b) des ersten Aspekts der Erfindung ein erzwungenes Auskreuzungsereignis
ist.
-
Das
erzwungene Auskreuzen aus der Hybridstruktur kann durchgeführt werden
mittels:
- i) Herstellen eines Systems, worin
die DNA-Hybridstruktur keinen selektierbaren Marker exprimiert und
wenigstens eine der wenigstens zwei DNA-Strukturen in Schritt a)
des ersten Aspekts fähig
ist, den selektierbaren Marker zu exprimieren;
- ii) Inkubieren der Zelle unter selektierbaren Bedingungen, wodurch
ein Auskreuzen der DNA-Hybridstruktur erzwungen wird, da die Zelle
nur fähig
ist, das Markergen zu exprimieren, nachdem das Auskreuzen der DNA-Hybridstruktur
stattgefunden hat.
-
Vorzugsweise
wird das oben erwähnte
selektierbare Markergen auf einer DNA-Struktur exprimiert, in der
der Replikationsursprung während
einer wie oben beschrieben durchgeführten, erzwungenen Integration der
DNA-Strukturen in eine DNA-Hybridstruktur
nicht-funktionell ist, und vorzugsweise ist das selektierbare Markergen
ein Antibiotika-Resistenzgen. Siehe unten für die weitere Beschreibung
geeigneter selektierbarer Markergene.
-
Um
das Konzept des erzwungenen Auskreuzens aus der hybriden DNA-Struktur
zu veranschaulichen, ist ein Beispiel eines geeigneten Systems in
den 1 und 2 gezeigt.
-
Der
selektierbare Marker kann ein beliebiger im Stand der Technik bekannter
Marker sein, zum Beispiel ein Gen, das für ein Produkt kodiert, das
der Zelle eine Antibiotika-Resistenz verleiht, das einem auxotrophen
Stamm Prototrophie verleiht oder das einen Defekt des Wirts komplementiert
(z.B. in einen dal–-Stamm eingeführte dal-Gene;
vgl. B. Diderichsen (1986), Bacillus: Molecular Genetics and Biotechnology
Applications, A. T. Ganesan und J. A. Hoch, Hrsg., Academic Press,
Seiten 35–46).
Der selektierbare Marker kann z.B. aus einer bekannten Quelle ausgeschnitten
oder auf einem Vektor vorhanden sein, z.B. einem Plasmid, das für die Konstruktion
der DNA-Struktur verwendet wird, die in dem erfindungsgemäßen Verfahren
verwendet werden soll.
-
Diese
Selektion kann durch Wachsenlassen der Zellen unter Selektionsdruck
für den
von dem Markergen kodierten Selektionsmarker durchgeführt werden.
-
Die
DNA-Struktur (in die der Vektor integriert werden soll) kann ein
für einen
beliebigen selektierbaren Marker kodierendes Gen enthalten, z.B.
von einem beliebigen Typ, wie er oben im Zusammenhang mit dem Marker
beschrieben wurde, den der in dem erfindungsgemäßen Verfahren zu verwendende
Vektor wahlweise trägt.
Somit kann das Markergen eine Antibiotikaresistenz, wie z.B. eine
Resistenz gegen Kanamycin, Tetracyclin; Ampicillin, Erythromycin,
Chloramphenicol, oder eine Resistenz gegen verschiedene Schwermetalle, wie
z.B. Selenat, Antimon oder Arsenat, kodieren.
-
In
einem bevorzugten Ausführungsbeispiel
ist die in dem Verfahren verwendete Zelle bezüglich des enzymatischen Fehlpaarungs-Reparatursystems
gestört
oder vorübergehend
inaktiviert oder weist eine verringerte Aktivität der Fehlpaarungsreparatur
auf. Bei der Synthese von DNA können
Fehler vorkommen, und die resultierenden nicht-komplementären Basenpaare werden Fehlpaarungen
genannt. Es gibt ein Verfahren zur Korrektur von Fehlern (Fehlpaarungen)
in DNA. In E. coli werden Fehler sehr schnell und genau von zwei
Enzymen (MutS und MutL) entdeckt, die ein drittes Enzym (MutU) in
die Lage versetzen, die beiden DNA-Stränge zu entwinden, und ein viertes
Enzym (MutH) in die Lage versetzen, die neu synthetisierten Stränge an einer DNA-Sequenz
(GATC) zu schneiden, die ihrerseits später methyliert wird. Für eine genauere
Erklärung
wird auf
EP 449923 (Setratech)
verwiesen, deren Inhalt hierdurch mittels Verweis aufgenommen wird.
-
In
einem anderen bevorzugten Ausführungsbeispiel
ist die in dem Verfahren verwendete Zelle in den Haupt-Rekombinationsenzymen,
wie z.B. AddAB, beeinträchtigt
oder behindert. Es wurde gezeigt, dass ein in den AddAB-Genen beeinträchtigter
Stamm eine höhere
Häufigkeit
der homologen Rekombination zeigt. Sowohl die legitime als auch
die illegitime Rekombinationshäufigkeit
sind erhöht.
Jedoch ist speziell die Rekombinationshäufigkeit für die illegitime Rekombination
bevorzugt, wenn mit einem Stamm gearbeitet wird, der bezüglich der
ATP-abhängigen
Nuklease AddAB beeinträchtigt
oder behindert ist (Meima, R; Haijema, BJ; Dijkstra, H; Haan, GJ;
Venema, G; Bron S (1997) Role of enzymes of homologous recombination
in illegitimate plasmid recombination in Bacillus-subtilis. JOURNAL
OF BACTERIOLOGY Bd. 179, Nr. 4, Seiten 1219–1229).
-
In
einem anderen bevorzugten Ausführungsbeispiel überexprimiert
die in dem Verfahren verwendete Zelle eines der Schlüssel-Rekombinationsenzyme,
recA. Das recA-Enzym ist an der homologen Rekombination beteiligt,
indem es die Renaturierung von ssDNA fördert, um die Heteroduplex-Moleküle zu bilden,
die für das
Rekombinationsereignis notwendig sind, und initiiert den Austausch
von DNA-Strängen.
(PROGRESS IN NUCLEIC ACID RESEARCH AND MOLECULAR BIOLOGY Bd. 56,
Seiten 129–223
(1997)). Die recA-Überexpression
kann entweder durch klassische Mutagenese des Wirtsstamms oder durch
Klonieren eines hoch exprimierten recA-Gens erreicht werden.
-
In
einem anderen bevorzugten Ausführungsbeispiel
wird die in dem Verfahren verwendete Zelle UV-, Röntgenstrahlen
oder DNA-schädigenden
Wirkstoffen ausgesetzt, um die Rekombinationshäufigkeit zwischen den zwei
homologen Genen zu erhöhen,
die gemischt werden sollen. Stress, wie z.B. UV-, Röntgenstrahlen oder
DNA-schädigende
Wirkstoffe, induzieren bekanntlich die Rekombinationsenzyme über die
SOS-Antwort und aktivieren an Rekombinationsprozessen beteiligte
Enzyme wie recA. (BACILLUS SUBTILIS AND OTHER GRAM-POSITIVE BACTERIA,
Seiten 529–537
(1993)).
-
In
einem anderen bevorzugten Ausführungsbeispiel
wird das pTR-basierte Plasmid, das als ein Träger eines der zu mischenden
Gene benutzt wird, durch entweder eine Niedrigkopie- Version von pTR oder
ein anderes mit B. subtilis kompatibles Niedrigkopie-Plasmid, wie
z.B. pWVO1 (MOLECULAR AND GENERAL GENETICS, Bd. 249, Nr. 1, Seiten
43–50
(1995)), pTA1060 (JOURNAL OF BACTERIOLOGY, Bd. 142, Nr. 1, Seiten
315–8
(1980)) oder pAMb1 (JOURNAL OF BACTERIOLOGY, Bd. 157, Nr. 2, Seiten
445–53
(1984)), ersetzt.
-
In
einem weiteren bevorzugten Ausführungsbeispiel
ist das eine DNA-Konstrukt, das eines der zu mischenden Gene umfasst,
auf dem Chromosom der Zelle angeordnet und ein anderes DNA-Konstrukt,
das ein anderes zu rekombinierendes Gen von Interesse umfasst, ist
ein Plasmid. Bei der Durchführung
eines erfindungsgemäßen In-Vivo-Rekombinationsverfahrens
wird das Plasmid mit dem DNA-Konstrukt auf dem Chromosom rekombiniert.
Das Plasmid kann z.B. ein temperaturempfindliches Plasmid sein.
-
Die
Vorteile der Verwendung eines Niedrigkopie-pTR oder eines Systems,
bei dem eines der DNA-Konstrukte wie oben beschrieben auf dem Chromosom
lokalisiert ist, sind, dass das hybride DNA-Konstrukt nach der Rekombination
der beiden homologen Gene die vorherrschende Form der Gene in der
Zelle sein wird. Ein Hochkopie-Hintergrund nicht-rekombinierter pTR-Plasmide wird unausweichlich
die in der ersten Runde gebildeten Genhybride nach der zweiten Rekombinationsrunde
zurück
zum Wildtyp revertieren und deshalb jegliches Mischen der beiden
Gene verhindern. Wenn das Chromosom als ein Träger für eines der Gene verwendet
wird, kann kein Wildtyp-Hintergrund koexistieren, wenn das pTS über die
homologen Gene integriert hat.
-
Es
ist bevorzugt, dass die in dem Verfahren verwendete Zelle eine für die Produktion
des Produkts der rekombinierten DNA-Sequenz in großem Maßstab geeignete
Zelle ist. Es ist bevorzugt, dass die Zelle eine bakterielle Zelle
ist, mehr bevorzugt eine Bacillus-Zelle, am meisten bevorzugt eine
Bacillus-subtilis-Zelle.
-
Die
zu rekombinierenden DNA-Sequenzen können aus Organismen, die die
Sequenzen enthalten, in einer an sich bekannten Weise isoliert/kloniert
werden. Zum Beispiel kann eine geeignete Oligonukleotid-Sonde auf
der Basis einer ersten DNA-Sequenz von Interesse hergestellt werden,
um eine zweite DNA-Sequenz zu erhalten, die homolog zu der ersten
Sequenz ist. Alternativ kann die DNA-Sequenz mittels PCR hergestellt werden,
die auf Primern basiert, die aufgrund der Kenntnis konservierter
Regionen entworfen wurden.
-
In
verschiedenen Ausführungsbeispielen
des vorliegenden Inhalts wird der Ausdruck „ein Satz homologer DNA-Sequenzen
(„Satz
A", „Satz B", „Satz C" etc.)" verwendet, z.B.
im Zusammenhang mit einem Ausführungsbeispiel
der Erfindung, das sich auf ein erfindungsgemäßes Verfahren bezieht, wobei
die homologen DNA-Sequenzen aus zwei oder mehr verschiedenen Sätzen homologer
DNA-Sequenzen („Satz
A", „Satz B", „Satz C" etc.) bestehen und
wobei jeder der Sätze
homologer Sequenzen („Satz
A", „Satz B", „Satz C" etc.) in ein anderes
erfindungsgemäßes DNA-Konstrukt
(„DNA-Konstrukt
A", „DNA-Konstrukt
B", „DNA-Konstrukt
C" etc.) insertiert
wird.
-
Dieser
Ausdruck wird verwendet, um zu veranschaulichen, dass ein erfindungsgemäßes In-Vivo-Rekombinationsverfahren
verwendet werden kann, um zahlreiche „Sätze" homologer DNA-Sequenzen zu rekombinieren.
Jeder einzelne „Satz" homologer DNA-Sequenz(en)
kann jeweils von nur einer bis zu zahlreichen (z.B. ein „Satz", der eine DNA-Bibliothek
umfasst) einzelnen homologen DNA-Sequenz(en) umfassen.
-
Jeder
der „Sätze" homologer DNA-Sequenzen
(„Satz
A", „Satz B", „Satz C" etc.) kann dann
in ein anderes erfindungsgemäßes DNA-Konstrukt
(„DNA-Konstrukt
A", „DNA-Konstrukt B", „DNA-Konstrukt
C" etc.) insertiert
werden. Jedes der verschiedenen DNA-Konstrukte unterscheidet sich vorzugsweise
erfindungsgemäß in wichtigen
Parametern, wie z.B. einem verschiedenen Replikationsursprung, verschiedenen
selektierbaren Markern etc..
-
Alle
besagten DNA-Konstrukte („DNA-Konstrukt
A", „DNA-Konstrukt
B", „DNA-Konstrukt C" etc.) können dann
erfindungsgemäß in eine
Zelle eingeführt
werden, und wenn dann ein erfindungsgemäßes Verfahren zur In-Vivo-Rekombination
durchgeführt
wird, kann jedes einzelne DNA-Konstrukt („DNA-Konstrukt A", „DNA-Konstrukt
B", „DNA-Konstrukt
C" etc.) erfindungsgemäß zufällig rekombinieren.
Nach einigen Wiederholungen eines erfindungsgemäßen Verfahrens wird dies zu
einer DNA-Population in der Zellpopulation führen, die zahlreiche verschiedene
Kombinationen verschiedener rekombinierter DNA-Sequenzen umfasst,
die zufällig
zwischen allen in dem Verfahren verwendeten „Sätzen" homologer DNA-Sequenzen rekombiniert
sind.
-
Außerdem wird
der Ausdruck „ein
Satz homologer DNA-Sequenzen („Satz
A", „Satz B", „Satz C" etc.)" hierin verwendet,
um einen der Vorteile eines erfindungsgemäßen In-Vivo-Rekombinationsverfahrens zu veranschaulichen,
der die Flexibilität
des Systems ausmacht, beide Sätze
homologer DNA-Sequenzen, die eine, wenige und/oder zahlreiche (z.B.
große
DNA-Bibliotheken) einzelne homologe DNA-Sequenzen umfassen, zu rekombinieren.
-
Demgemäß bezieht
sich ein Ausführungsbeispiel
der Erfindung auf ein erfindungsgemäßes Verfahren, wobei die besagten
Sätze homologer
DNA-Sequenzen jeweils nur eine DNA-Sequenz (d.h. die Rekombination von
zwei oder mehr homologen Sequenzen) enthalten.
-
Des
Weiteren sind in erfindungsgemäßen Auführungsbeispielen
die zu rekombinierenden homologen DNA-Sequenzen von einer vorgefertigten
DNA-Bibliothek abgeleitet und/oder von einzelnen, verschiedenen, vorgefertigten
Bibliotheken abgeleitet.
-
Von
solchen DNA-Bibliotheken abgeleitete homologe DNAs werden in einen
erfindungsgemäßen Vektor
insertiert (d.h. DNA, die die DNA-Bibliothek repräsentiert,
wird in einen Vektor ligiert (insertiert)). Wie hierin beschrieben,
umfasst das erfindungsgemäße Verfahren
wenigstens zwei DNA-Strukturen (z.B. Vektoren), die sich in einer
Weise unterscheiden, die die erzwungene In-Vivo-Rekombination der
Vektoren erlaubt.
-
Homologe
DNA, die DNA-Bibliotheken repräsentiert,
kann in nur einen und/oder beide der besagten unterschiedlichen
Vektoren insertiert werden.
-
D.h.,
falls zum Beispiel homologe DNA, die eine DNA-Bibliothek repräsentiert,
in einen der unterschiedlichen Vektoren insertiert (ligiert) wird
und homologe DNA, die eine andere DNA-Bibliothek repräsentiert, in
den anderen Vektor insertiert wird, wird dies eine schnelle und
effiziente Rekombination zwischen zwei verschiedenen Bibliotheken
durch ein erfindungsgemäßes In-Vivo-Rekombinationsverfahren
erlauben. Um dieses Konzept zu veranschaulichen, kann DNA, die eine
Bibliothek repräsentiert,
in einen pTR-Vektor insertiert werden (siehe 1), und
DNA, die eine andere Bibliothek repräsentiert, kann in einen pTS-Vektor
insertiert werden (siehe 1). Schon wenige wiederholte
In-Vivo-Rekombinationsereignisse,
die mittels eines erfindungsgemäßen Verfahrens
durchgeführt
werden, werden dann eine Zellpopulation ergeben, die eine neue Bibliothek
repräsentiert,
die eine Kombination der beiden oben erwähnten Bibliotheken ist.
-
Somit
beziehen sich Ausführungsbeispiele
der Erfindung auf ein erfindungsgemäßes Verfahren, wobei wenigstens
einer der Sätze
homologer DNA-Sequenzen von einer vorgefertigten DNA-Bibliothek
abgeleitet ist und wenigstens einer der Sätze homologer DNA-Sequenzen
eine DNA-Sequenz ist, die homolog zu der DNA-Bibliothek ist (d.h.
was zu einem Verfahren zur In-Vivo-Rekombination DNA-Bibliothek
zu einer DNA-Sequenz führt,
die homolog zu der DNA-Bibliothek sind); oder
ein erfindungsgemäßes Verfahren,
wobei die Sätze
homologer DNA-Sequenzen von derselben vorgefertigen DNA-Bibliothek
abgeleitet sind (d.h. was zu einem Verfahren zur In-Vivo-Rekombination
der DNA-Bibliothek führt);
oder
ein erfindungsgemäßes Verfahren,
wobei die Sätze
homologer DNA-Sequenzen von den wenigstens zwei verschiedenen vorgefertigten
DNA-Bibliotheken abgeleitet sind (d.h. was zu einem Verfahren zur
In-Vivo-Rekombination von wenigstens zwei verschiedenen DNA-Bibliotheken
führt).
-
Um
noch weitere Rekombination zwischen den DNA-Sequenzen zu erhalten,
kann es bevorzugt sein, eine Isolierung der DNA-Strukturen nach
der ersten Runde der erfindungsgemäß durchgeführten In-Vivo-Rekombination
der DNA-Sequenzen durchzuführen.
Die isolierten Strukturen werden dann in die Zellen retransformiert,
und eine weitere Verfahrensrunde der In-Vivo-Rekombination wird
dann durchgeführt.
-
Die
Vorteile dieses Schritts werden durch das folgende Beispiel veranschaulicht:
Satz
A ist ein Vektor A, wobei die DNA-Sequenzen a, b einzeln in den
Vektor A eingeführt
werden (d.h. die Vektor-A-Population umfasst einen Vektor mit der
DNA-Sequenz a und
einen Vektor mit der Sequenz b); und
Satz B ist ein Vektor
B, wobei die DNA-Sequenzen c, d einzeln in den Vektor B eingeführt werden
(d.h, die Vektor-B-Population umfasst einen Vektor mit der DNA-Sequenz
c und einen Vektor mit der Sequenz d).
-
Die
Vektoren A und B werden dann in eine Zellpopulation transformiert
und erfindungsgemäß in vivo rekombiniert.
Dies wird zur Rekombination zwischen den DNA-Sequenzen ac, ad, bc, bd führen. Die
Vektoren A und B, die die rekombinierten Sequenzen umfassen, werden
isoliert und in die besagte Zellpopulation retransformiert. Die
erfindungsgemäße In-Vivo-Rekombination
wird dann alle möglichen
Rekombination der vier (a, b, c, d) DNA-Sequenzen ergeben, wie z.B.
die Rekombination von abc, abdc, etc.
-
In
noch weiteren Ausführungsbeispielen
bezieht sich die Erfindung auf ein erfindungsgemäßes Verfahren, wobei zwei Sätze homologer
DNA-Sequenzen („Satz
A" und „Satz B") in vivo rekombiniert
werden, umfassend die Schritte:
- a) Insertieren
einer(/von) Sequenz(en) aus „Satz
A" in einen Vektor,
der einen temperaturresistenten Replikationsursprung und ein Gen
für einen
ersten Marker enthält;
- b) Insertieren einer(/von) Sequenz(en) aus „Satz B" in einen Vektor, der einen temperaturempfindlichen
Replikationsursprung und ein Gen für einen zweiten Marker enthält;
- c) Einführen
der Vektoren in eine Zelle;
- d) Inkubieren der Zelle bei einer Temperatur, bei der beide
Replikationsursprünge
funktionell sind, wahlweise unter einem Selektionsdruck, der Zellen
mit beiden Markergenen begünstigt;
- e) Verändern
der Temperatur zu einer Temperatur, bei der nur der temperaturresistente
Replikationsursprung funktionell ist;
- f) wenigstens einmaliges Wiederholen der Schritte d)–e);
oder
ein
erfindungsgemäßes Verfahren,
wobei zwei Sätze
homologer DNA-Sequenzen („Satz
A" und „Satz B") in vivo rekombiniert
werden, umfassend die Schritte: - a) Insertieren
einer(/von) Sequenz(en) aus „Satz
A" in einen Vektor,
der einen temperaturresistenten Replikationsursprung und ein Gen
für einen
ersten Marker enthält;
- b) Insertieren einer(/von) Sequenz(en) aus „Satz B" in einen Vektor, der einen temperaturempfindlichen
Replikationsursprung und ein Gen für einen zweiten Marker enthält;
- c) Einführen
der Vektoren in eine Zelle;
- d) Inkubieren der Zelle bei einer Temperatur, bei der nur der
temperaturresistente Replikationsursprung funktionell ist, wahlweise
unter einem Selektionsdruck, der Zellen mit beiden Markergenen begünstigt;
- e) wahlweise Verändern
der Temperatur zu einer Temperatur, bei der beide Replikationsursprünge funktionell
sind;
- f) wenigstens einmaliges Wiederholen der Schritte d)–e).
-
Ein
weiterer Aspekt der Erfindung bezieht sich auf ein Verfahren zur
Herstellung eines oder mehrerer rekombinanten/r Proteins/Proteine
mit einer gewünschten
biologischen Aktivität,
umfassend:
- a) Durchführen eines Verfahrens zur In-Vivo-Rekombination
homologer DNA-Sequenzen
(das Verfahren wird gemäß der Erfindung
wie hierin beschrieben durchgeführt),
wobei wenigstens eine (der) DNA-Struktur(en) ein Expressionsvektor
ist;
- b) Exprimieren der zahlreichen verschiedenen rekombinanten Polypeptide,
die von den zahlreichen verschiedenen rekombinierten DNA-Sequenzen
aus Schritt a) kodiert werden; und
- c) Screenen oder Selektieren der zahlreichen verschiedenen rekombinanten
Proteine aus Schritt b) in einem geeigneten Screening- oder Selektionssystem
für ein
oder mehrere rekombinantes(e) Polypeptid(e), das/die eine gewünschte Aktivität besitzt/en.
-
Vorzugsweise
ist der Expressionsvektor in Schritt a) oben eine DNA-Struktur,
die während
einer erzwungenen Bildung einer hybriden DNA-Struktur (d.h. Integration
der Strukturen ineinander), die nach einem erfindungsgemäßen Verfahren
zur In-Vivo-Rekombination homologer DNA-Sequenzen durchgeführt wird, nicht
fähig ist
zu replizieren (z.B. da sie einen nicht-funktionellen Replikationsursprung
hat). Dies wird die Wahrscheinlichkeit verbessern, dass der Expressionsvektor
nach dem Auskreuzungsereignis der erfindungsgemäßen Hybridstruktur rekombinierte,
homologe DNA umfasst, weil die einzige Weise, auf die der Expressionsvektor
das Verfahren der erzwungenen Hybridbildung überlebt haben kann, darin besteht,
dass er mit einer anderen DNA-Struktur rekombiniert hat, die fähig ist,
unter den für
das Erzwingen der Hybridstruktur-Bildung verwendeten Bedingungen
zu replizieren.
-
Nach
der Durchführung
des Verfahrens zur In-Vivo-Rekombination (Schritt a) oben) und vor
den Expressions- und den Screening-Schritten b) und c) oben, kann
es vorteilhaft sein, eine Zellpopulation herzustellen, die hauptsächlich nur
den Expressionsvektor enthält
(d.h. andere(s) DNA-Konstrukt(e) wird/werden zum Teil oder beinahe
vollständig
aus der Zellpopulation entfernt). Dies ist besonders vorteilhaft,
wenn der Expressionsvektor während
der wie unmittelbar vorstehend beschriebenen, erzwungenen Bildung
der hybriden DNA-Struktur nicht fähig ist zu replizieren, da
eine solche Zellpopulation einen sehr begrenzten Hintergrund an nicht-rekombinierten
DNA-Sequenzen von Interesse haben wird, was in dem nachfolgenden
Screening-Schritt (Schritt c) oben) vorteilhaft ist.
-
Das
Herstellen einer Zellpopulation, die hauptsächlich nur den Expressionsvektor
umfasst, kann gemäß im Stand
der Technik bekannter Standardverfahren durchgeführt werden, wie z.B. Wachsenlassen
der Zellpopulation unter Bedingungen, die die Replikation des Expressionsvektors
im Vergleich zu den anderen DNA-Strukturen selektiv begünstigen;
oder selektives Reinigen des Expressionsvektors aus den Zellen und nachfolgendes
Einführen
von diesem in einen neue Zellpopulation.
-
Die
Expression der von der rekombinierten Sequenz kodierten rekombinanten
Polypeptide in Schritt a) oben kann unter Verwendung eines Standard-Expressionsvektors
durchgeführt
werden, und/oder ein Standard-Expressionsvektor kann modifiziert
werden, um Elemente zu umfassen, die für ein erfindungsgemäßes In-Vivo-Rekombinationsverfahren
speziell geeignet sind.
-
Ein
geeignetes Screening- oder Selektionssystem, um die zahlreichen
verschiedenen rekombinanten Proteine aus Schritt b) oben zu screenen
oder selektieren, wird von der gewünschten biologischen Aktivität abhängen.
-
Eine
Anzahl geeigneter Screening- oder Selektionssysteme für das Screenen
oder Selektieren auf eine gewünschte
biologische Aktivität
ist im Stand der Technik beschrieben. Beispiele sind:
Strauberg
et al. (Biotechnology 13: 669–673
(1995)), die ein Screeningsystem für das Screenen von Subtilisin-Varianten,
die eine Kalzium-unabhängige
Stabilität
aufweisen, beschreiben;
Bryan et al. (Proteins 1: 326–334 (1986)),
die einen Screening-Test für
das Screenen auf Protease, die eine verbesserte Hitzestabilität hat, beschreiben;
und
PCT-DK96/00322, die einen Screening-Test für das Screenen
auf Lipasen beschreibt, die eine verbesserte Waschleistung in Waschmitteln
haben.
-
Ein
bevorzugtes Ausführungsbeispiel
der Erfindung umfasst das Screenen oder die Selektion von rekombinantem/n
Protein(en), wobei die gewünschte
biologische Aktivität
eine verbesserte Leistung in Geschirrspülmitteln oder Waschmitteln
für Wäsche ist.
Beispiele geeigneter Geschirrspülmittel
oder Waschmittel für Wäsche sind
in PCT/DK96/00322 und WO 95/30011 offenbart.
-
Die
Erfindung umfasst auch einen Satz (wenigstens zwei) Vektoren, die
wenigstens zwei verschiedene Replikationsursprünge umfassen, die fähig sind,
in derselben Zelle unter permissiven Bedingungen zu funktionieren,
und wenigstens zwei homologe DNA- Sequenzen,
die Polypeptide kodieren. In einem bevorzugten Ausführungsbeispiel
ist wenigstens ein Replikationsursprung temperaturempfindlich. Der
Vektor kann vorzugsweise ein Plasmid sein.
-
Die
Erfindung umfasst außerdem
ein Vektorsystem, das wenigstens
- a) einen Vektor
A, der eine DNA-Sequenz, die ein Polypeptid kodiert, und einen Replikatonsursprung
enthält,
und
- b) einen Vektor B, der eine DNA-Sequenz, die homolog zu der
DNA-Sequenz in Vektor A ist, und einen Replikatonsursprung, der
sich von dem Replikationsursprung in Vektor A unterscheidet, enthält,
umfasst.
-
Die
Vektoren können
vorzugsweise Plasmide sein.
-
Es
ist bevorzugt, dass die DNA-Sequenz Polypeptide mit einer biologischen
Aktivität
kodiert, speziell Enzyme und insbesondere Enzyme, wie z.B. eine
Amylase, Lipase, Cutinase, Cellulase, Oxidase, Phytase und eine
Protease.
-
Die
Erfindung umfasst außerdem
eine neue rekombinierte DNA-Sequenz, dadurch gekennzeichnet, dass
sie durch ein erfindungsgemäßes Verfahren
hergestellt wurde oder ein rekombinantes Protein kodiert, das eine
gewünschte
biologische Aktivität
aufweist, wobei das rekombinante Protein, das eine gewünschte biologische
Aktivität
aufweist, durch das Verfahren gemäß dem Verfahren zur Herstellung
eines oder mehrerer rekombinanten/r Proteins/e, die eine erfindungsgemäße gewünschte biologische
Aktivität
aufweisen, hergestellt ist.
-
Die
Erfindung umfasst außerdem
ein Verfahren zur Herstellung eines Polypeptids, speziell eines
Enzyms, umfassend i) Wachsenlassen einer Zelle, die ein Polypeptid
exprimiert, das von einer rekombinierten DNA-Sequenz nach Anspruch
38 kodiert ist, und ii) Reinigen des exprimierten Polypeptids.
-
Die
Erfindung umfasst außerdem
ein neues Polypeptid, speziell ein Enzym, dadurch gekennzeichnet, dass
es durch ein erfindungsgemäßes Verfahren
hergestellt ist.
-
Die
Erfindung umfasst außerdem
ein Polypeptid, speziell ein Enzym, dadurch gekennzeichnet, dass es
von einer erfindungsgemäßen rekombinierten
DNA-Sequenz kodiert ist.
-
Erfindungsgemäße Polypeptide
schließen
Peptide und Proteine und wahlweise glycosylierte Formen davon ein.
Beispiele sind Enzyme, pharmazeutisch aktive Polypeptide, wie z.B.
Analoga von humanen oder von Säugerpolypeptiden,
z.B. Insulin, Wachstumshormon, menschliche Hormone oder Wachstumsregulatoren.
-
Die
Erfindung ist in den folgenden Beispielen, die in keiner Weise den
Umfang der Erfindung begrenzen sollen, mit weiteren Einzelheiten
beschrieben.
-
Beispiele
-
Materialien und Methoden
-
Stämme
-
E.
coli: SJ2 (Diderichsen, B., Wedsted, U., Hedegaard, L., Jensen,
B. R., Sjøholm,
C. (1990) Cloning of aldB, which encodes alpha-acetolactate decarboxylase,
an exoenzyme from Bacillus brevis. J. Bacteriol., 172, 4315–4321).
Elektrokompetente Zellen, hergestellt und transformiert unter Verwendung
eines Bio-Rad GenePulsersTM, wie von dem
Hersteller empfohlen.
-
B.
subtilis DN1885 (Diderichsen, B., Wedsted, U., Hedegaard, L., Jensen,
B. R., Sjøholm,
C. (1990) Cloning of aldB, which encodes alpha-acetolactate decarboxylase,
an exoenzyme from Bacillus brevis. J. Bacteriol., 172, 4315–4321).
Kompetente Zellen wurden wie von Yasbin, R. E., Wilson, G. A. und
Young, F. E. (1975) Transformation and transfection in lysogenic
strains of Bacillus subtilis: evidence for selective induction of
prophage in competent cells. J. Bacteriol., 121: 296–304, beschrieben
hergestellt und transformiert.
-
B.
subtilis PL1801. Dieser Stamm ist B. subtilis DN1885 mit disruptierten
apr- und npr-Genen
(Diderichsen, B., Wedsted, U., Hedegaard, L., Jensen, B. R., Sjøholm,
C. (1990) Cloning of aldB, which encodes alpha-acetolactate decarboxylase,
an exoenzyme from Bacillus brevis. J. Bacteriol., 172, 4315–4321).
-
Plasmide
-
pSX120:
B. subtilis-Expressionsvektor (beschrieben in
EP 405901 und WO 91100345). Dieser
Vektor umfasst das Wildtyp-Savinasegen.
-
pE194:
(Horinouchi, S. und Weisblum, B., 1982, J. Bacteriol. 150: 804–814).
-
pSX222:
B. subtilis-Expressionsvektor (beschrieben in WO 96/34946 und WO
92/19729). Dieser Vektor umfasst das synthetische Savinasegen, das
hierin Savisyn genannt wird. In das Savisyngen ist eine große Anzahl
von Restriktionsstellen im Vergleichen zu dem in pSX120 enthaltenen
Wildtyp-Savinasegen eingeführt.
-
pUC19:
Yanisch-Perron, C., Vieria, J., und Messing, J. (1985) Gene 33,
103–119.
-
pF64L-S65T-GFP:
ist das E. coli-Plasmid, das das DNA-Fragment kodiert, das das in
WO 97111094 beschriebene F64L-S65T-GFP kodiert.
-
pMUTIN-4-MCS:
Das Plasmid kann von dem Laboratoire de Genetique Microbienne, Institut
National de la Recherche Agronomique, 78352 Jouy en Josas – CEDEX,
Frankreich, erhalten werden.
-
Lösungen/Medien
-
TY-
und LB-Agar (wie beschrieben in Ausubel, F. M. et al. (Hrsg.) „Current
protocols in Molecular Biology".
John Wiley and Sons, 1995).
-
LBPG
ist LB-Agar, der mit 0,5% Glucose und 0,05 M Kaliumphosphat, pH
7,0, supplementiert ist.
-
LBPGSK-Platten
sind LBPG-Medium mit Agar und 1% Magermilch.
-
Antibiotika
werden in verschiedenen Medien in Konzentrationen von 6 μg/ml Chloramphenicol,
10 μg/ml
Kanamycin und 5 μg/ml
Erythromycin verwendet.
-
TE-Puffer
(wie beschrieben in Ausubel, F. M. et al. (Hrsg.) „Current
protocols in Molecular Biology". John
Wiley and Sons, 1995).
-
Allgemeine molekularbiologische
Verfahren
-
DNA-Manipulationen
und -Transformationen wurden unter Verwendung von Standardverfahren
der Molekularbiologie durchgeführt
(Sambrook et al. (1989) Molecular cloning: A laboratory manual,
Cold Spring Harbor Lab., Cold Spring Harbor, NY; Ausubel, F. M.
et al. (Hrsg.) "Current
protocols in Molecular Biology". John
Wiley and Sons, 1995; Harwood, C. R., und Cutting, S. M. (Hrsg.) "Molecular Biological
Methods for Bacillus".
John Wiley and Sons, 1990).
-
Enzyme
für DNA-Manipulationen
wurden gemäß den Angaben
der Lieferanten verwendet.
-
Enzyme für DNA-Manipulationen
-
Soweit
nicht anders angegeben, wurden alle Enzyme für DNA-Manipulationen, wie z.B.
Restriktionsendonukleasen, Ligasen, Polymerasen, PCR-Polymerasen
etc., von New England Biolabs, Inc., erhalten.
-
Konstruktion
des temperaturempfindlichen Mischungs-Plasmids
-
pMB430
wurde in einem Klonierungsverfahren mit fünf Schritten, wie im Folgenden
dargestellt, konstruiert, die gesamte DNA-Sequenz befindet sich
in SEQ ID Nr. 1.
-
1)
Das temperaturempfindliche Plasmid pE194 wurde durch Einführen multipler
Klonierungsstellen rund um die einzige ClaI-Stelle des Plasmids
pE194 und somit Zerstören
der ClaI-Stelle modifiziert. Dieses Einführen wurde mittels PCR unter
Verwendung der Primer #22899 und #24039 zur Amplifizierung des gesamten
Plasmids und Einführen
mehrerer nur einmal vorkommender Klonierungsstellen durchgeführt.
-
Die
PCR wurde wie folgt durchgeführt:
1 μl einer pE194-Qiaquick-Plasmidpräparation
wurde für
eine 50-μl-PCR-Amplifikation
in PCR-Puffer (10 mM Tris-HCl, pH 8,3, 50 mM KCl, 1,5 mM MgCl2, 0,01% (Gew/Vol) Gelatine) verwendet, der
200 μM jedes
dNTPs, 2,5 Units AmpliTaq-Polymexase (Perkin-Elmer, Cetus, USA) und 100 pmol jedes
Primers enthielt:
-
-
Die
PCR-Reaktionen wurden unter Verwendung eines DNA-Thermozyklers (Landgraf,
Deutschland) durchgeführt.
Eine Inkubation bei 94°C
für 1 min,
gefolgt von vierzig PCR-Zyklen,
die durchgeführt
wurden, indem ein Zyklusprofil von Denaturierung von 94°C für 1 min,
Annealing bei 55°C
für 1 min
und Verlängerung bei
72°C für 2 min
durchgeführt
wurden. Fünf-μl-Aliquots
des Amplifikationsprodukts wurden durch Elektrophorese in 0,7-%-Agarosegelen
(NuSieve, FMC) analysiert.
-
Subklonierung
von PCR-Fragment
-
Fünfundvierzig-μl-Aliquots
der wie oben beschrieben hergestellten PCR-Produkte wurden unter
Verwendung des QIAquick-PCR-Purification-Kits (Qiagen, USA) gemäß den Anweisungen
des Herstellers gereinigt. Die gereinigte DNA wurde in 50 μl 10 mM Tris-HCl, pH 8,5 eluiert.
Fünfzig μl des gereinigten
PCR Fragments wurden mit BamHI gespalten, in Gelen mit 0,8% Agarose
mit niedriger Gelierungstemperatur (SeaPlaque GTG, FMC) elektrophoretisiert,
das betreffende Fragment wurde aus den Gelen ausgeschnitten und
unter Verwendung des QIAquick-Gel-Extraction-Kits (Qiagen, USA)
gemäß den Anweisungen
des Herstellers gereinigt. Das isolierte DNA-Fragment wurde dann
an mit BamHI-gespaltenen pUC19 ligiert, und das Ligationsgemisch
wurde verwendet, um E. coli SJ2 zu transformieren.
-
Die
Zellen wurden auf LB-Agarplatten plattiert, die Erythromycin (200 μg/ml), enthielten
und mit X-gal (SIGMA, USA) (5-Brom-4-chlor-3-indolyl-alpha-D-galactopyranosid,
50 μg/ml)
supplementiert waren.
-
Identifizierung
und Charakterisierung positiver Klone
-
Die
transformierten Zellen wurden auf Erythromycin (200 μg/ml) enthaltende,
mit X-gal (50 μg/ml)
supplementierte LB-Agarplatten plattiert und bei 37°C über Nacht
inkubiert. Weiße
Kolonien wurden am nächsten Tag
durch Ausstreichen auf frische LB-Erythromycin-Agarplatten gerettet und
bei 37°C über Nacht
inkubiert. Die Einzelkolonien des nächsten Tages jedes Klons wurden
in flüssiges
LB-Medium transferiert, das Erythromycin (200 μg/ml) enthielt, und über Nacht
bei 37°C
unter Schütteln
bei 250 upm inkubiert.
-
Plasmide
wurden aus den Flüssigkulturen
unter Verwendung des QIAgen-Plasmid-Purification-Mini-Kits (Qiagen, USA)
nach den Anweisungen des Herstellers extrahiert. Fünf-μl-Proben
der Plasmide wurden mit BamHI gespalten. Die Spaltungen wurden durch
Gelelektrophorese auf einem 0,7-%-Agarosegel (NuSieve, FMC) überprüft. Das
Erscheinen von zwei DNA-Fragmenten von in etwa 2,7 kb und 3,7 zeigte
einen positiven Klon an. Der Klon wurde MB293 genannt und das Plasmid
pMB293.
-
2)
PMB293 wurde dann mit EcoRI gespalten und das 3,8-kb-Fragment wurde
auf einem 0,7--Agarosegel
und unter Verwendung des QiaQuick-Gelpurification-Kits von Qiagen
gemäß dem Hersteller
Gel-gereinigt. Dieses Fragment, das pE194 mit insertierten multiplen
Klonierungsstellen (die von den PCR-Primem und von der MCS von pUC19
stammen) darstellt, wurde durch Ligation zirkularisiert und verwendet,
um B. subtilis DN1885 zu transformieren, transformierte Zellen wurden
auf LBPG-Platten, die 5 μg/ml
Erythromycin enthielten, selektiert, und die Inkubation wurde bei
33°C über Nacht
durchgeführt.
Klone aus dieser Transformation wurden erneut ausgestrichen und über Nacht
in TY mit 5 μg/ml
Erythromycin kultiviert. Aus der Brühe der Übernachtkultur wurden Zellen
isoliert, und Plasmide wurden unter Verwendung des Qiaquick-Plasmid-Kits gereinigt. Die
Restriktionsenzym-Spaltung der Plasmide bestätigte die Korrektheit der Klone.
Ein solcher/s Klon und Plasmid wurde MB333 und pMB333 genannt.
-
3)
pMB333 wurde dann als das temperaturempfindliche Plasmid für die Konstruktion
eines Plasmids verwendet, das das Savizyngen ohne irgendeinen Promotor
enthielt. Dieses Konstrukt wurde wie folgt hergestellt.
-
Eine
PCR unter Verwendung der Primer #21547 und #21548 und von pSX222
als Vorlage wurde gemäß diesen
Bedingungen durchgeführt:
1 μl pSX222-Qiaquick-Plasmidpräparation
wurde für
eine 50 μl-PCR-Amplifizierung
in PCR-Puffer (10 mM Tris-HCl, pH 8,3, 50 mM KCl, 1,5 mM MgCl2, 0,01% (Gew/Vol) Gelatine) verwendet, der
200 μM jedes
dNTPs, 2,5 Units AmpliTaq-Polymerase (Perkin-Elmer, Cetus, USA) und 100 pmol jedes
Primers enthielt:
-
-
-
Die
PCR-Reaktionen wurden unter Verwendung eines DNA-Thermozyklers (Landgraf,
Deutschland) durchgeführt.
Eine Inkubation bei 94°C
für 1 min,
gefolgt von vierzig PCR-Zyklen,
die unter Verwendung eines Zyklusprofils von Denaturierung bei 94°C für 1 min,
Annealing bei 55°C
für 1 min
und Verlängerung
bei 72°C für 2 min,
durchgeführt
wurden. Fünf-μl-Aliquots
des Amplifikationsproduktes wurden durch Elektrophorese in 0,7-%-Agarosegelen
(NuSieve, FMC) analysiert.
-
Subklonierung von PCR-Fragment
-
Fünfundvierzig-μl-Aliquots
des wie oben beschrieben hergestellten PCR-Produkts wurden unter
Verwendung des QIAquick-PCR-Purification-Kits (Qiagen, USA) nach
den Anweisungen des Herstellers gereinigt. Die gereinigte DNA wurde
in 50 μl
10 mM Tris-HCl,
pH 8,5, eluiert. Fünfzig μl des gereinigten
PCR-Fragments wurden mit BamHI und ApaI gespalten, in Gelen mit
0,8% Agarose mit niedriger Gelierungstemperatur (SeaPlaque GTG,
FMC) elektrophoretisiert, das betreffende Fragment wurde aus den
Gelen ausgeschnitten und unter Verwendung des QIAquick-Gel-Extraction-Kits
(Qiagen, USA) nach den Anweisungen des Herstellers gereinigt. Gereinigtes
Fragment wurde an den Gel-gereinigten, BamHI- und ApaI-gespaltenen
pMB333 ligiert. Die Ligation wurde verwendet, um B. subtilis DN1885
zu transformieren. Transformierte Zellen wurden auf LBPG-Agarplatten
plattiert, die 5 μg/ml
Erythromycin enthielten, und über
Nacht bei 33°C
inkubiert. Klone wurden erneut auf ähnlichen Platten ausgestrichen,
und Plasmide wurden aus Übernachtkulturen
von Klonen gereinigt, die bei 33°C
in TY mit 5 μg/ml
Erythromycin inkubiert waren. Plasmide wurden unter Verwendung des
Qiaquick-Plasmid-Kits,
wie von dem Hersteller beschrieben, isoliert. Die Plasmide wurden
durch enzymatische Spaltung unter Verwendung von ApaI und BamHI überprüft. Ein
korrekter Klon wurde MB339 genannt und das Plasmid pMB339.
-
4)
Wir wollten einen einfachen Weg testen, um zu überwachen, dass sich Hybridplasmide
gebildet hatten. Deshalb wollten wir die erzwungene Rekombination
zwischen den beiden homologen Genen auf den beiden getrennten Plasmiden überwachen.
Dies wurde durchgeführt,
indem ein pE194-Derivat konstruiert wurde, das ein promotorloses
Savizyn aufwies, das transkriptionell an einen ORF fusioniert war,
der GFP kodierte. Auf diese Weise sollte es keine Expression von
GFP geben (wegen des Fehlens eines Promotors stromaufwärts des
Savizyns und damit des GFP-Gens), außer die beiden homologen Gene
hatten rekombiniert, wodurch das GFP unter die Kontrolle des sonst
auf pSX120 vorhandenen Promotors geriet. Das Plasmid wurde durch PCR-Amplifikation
des GFP-Gens und
dessen Einfügen
unmittelbar stromabwärts
des Savizyngens auf dem pMB339 konstruiert.
-
Die
hierin verwendete GFP-kodierende DNA ist ein Derivat des ursprünglichen
Wildtyp-Gens, das
Derivat von GFP wurde aus dem DNA-Konstrukt der Mutante F64L-S65T-GFP
kloniert, die wie in der internationalen Patentanmeldung WO 97/11094
beschrieben konstruiert wurde.
-
Die
Konstruktion des Vorlage-Plasmids wurde in WO 97/11094 beschrieben.
Das für
das F64L-S65T-GFP kodierende DNA-Fragment wurde mittels PCR aus
dem dieses Gen kodierenden E. coli-Plasmid wie folgt amplifiziert:
1 μl einer Qiaquick-Plasmidpräparation
von pF64L-S65T-GFP wurde für
eine 50-μl-PCR-Amplifizierung in FCR-Puffer
(10 mM Tris-HCl, pH 8,3, 50 mM KCl, 1,5 mM MgCl2,
0,01% (Gew/Vol) Gelatine) verwendet, der 200 μM jedes dNTPs, 2,5 Units AmpliTaq-Polymerase (Perkin-Elmer,
Cetus, USA) und 100 pmol jedes Primers enthielt:
-
-
Die
PCR-Reaktionen wurden unter Verwendung eines DNA-Thermozyklers (Landgraf,
Deutschland) durchgeführt.
Eine Inkubation bei 94°C
für 1 min,
gefolgt von vierzig PCR-Zyklen,
die unter Verwendung eines Zyklusprofils von Denaturierung bei 94°C für 1 min,
Annealing bei 55°C
für 1 min
und Verlängerung
bei 72°C für 2 min,
durchgeführt
wurden. Fünf-μl-Aliquots
des Amplifikationsproduktes wurden durch Elektrophorese in 0,7-%-Agarosegelen
(NuSieve, FMC) analysiert.
-
Subklonieren
des PCR-Fragments
-
Fünfundvierzig-μl-Aliquots
des wie oben beschrieben hergestellten PCR-Produkte wurden unter
Verwendung des QIAquick-PCR-Purification-Kits (Qiagen, USA) nach
den Anweisungen des Herstellers gereinigt. Die gereinigte DNA wurde
in 50 μl
10 mM Tris-HCl,
pH 8,5 eluiert. Das PCR-Fragment und pMB339 wurden mit ClaI und
NsiI gespalten, wie oben Gel-gereinigt und miteinander ligiert.
Das Ligationsgemisch wurde verwendet, um DN1885 zu transformieren,
transformierte Zellen wurden auf LBPG 5 μg/ml Erythromycin plattiert. Nach
18 Stunden der Inkubation wurden grün fluoreszierende Zellen als
einzelne Kolonien sichtbar. Eine von diesen wurde analysiert und
als MB406, sowie das entsprechende Plasmid als pMB406, gerettet.
Da der GFP-kodierende ORF keinen stromaufwärts gelegenen Promotor besaß, der die
Transkription antrieb, konnte eine möglich – Erklärung für die GFP-Expression ein Durchlesen
von einem weiter stromaufwärts
gelegenen Promotor der Savizyn-GFP-Transkriptionsfusion sein, nämlich der
Promotor des repF-Gens.
-
5)
Um jegliches Durchlesen in den ORF von GFP zu beseitigen, wurde
ein Transkriptionsterminator zwischen das repF-Gen und die Savizyn-DNA
insertiert. Der Terminator wurde aus dem pMUTIN-4-MCS-Plasmid unter
Verwendung der folgenden Primer und Bedingungen amplifiziert:
1 μl einer Qiaquick-Plasmidpräparation
von pMUTIN-4-MCS wurde für
eine 50-μl-PCR-Amplifizierung in PCR-Puffer
(10 mM Tris-HCl, pH 8,3, 50 mM KCl, 1,5 mM MgCl2,
0,01% (Gew/Vol) Gelatine) verwendet, der 200 μM jedes dNTPs, 2,5 Units AmpliTaq-Polymerase (Perkin-Elmer,
Cetus, USA) und 100 pmol jedes Primers enthielt:
-
-
Die
PCR-Reaktionen wurden unter Verwendung eines DNA-Thermozyklers (Landgraf,
Deutschland) durchgeführt.
Eine Inkubation bei 94°C
für 60
sec gefolgt von vierzig PCR-Zyklen,
die unter Verwendung eines Zyklusprofils von Denaturierung bei 94°C für 30 sec,
Annealing bei 55°C
für 30
min und Verlängerung
bei 72°C für 1 min,
durchgeführt
wurden. Fünf-μl-Aliquots
des Amplifikationsproduktes wurden durch Elektrophorese in 0,7-%-Agarosegelen
(NuSieve, FMC) analysiert.
-
Die
amplifizierte DNA wurde wie oben gereinigt und als ein KpnI-BglII-Fragment
in pMB406 kloniert, der ebenfalls mit BglII und KpnI gespalten war
(das gleiche Verfahren bezüglich
der Gel-Reinigung etc., wie oben beschrieben). Das Ligationsgemisch
wurde verwendet, um DN1885 zu transformieren, transformierte Zellen
wurden auf LBPG-5-μg/ml-Erythromycin
plattiert. Nach 18 Stunden der Inkubation wurden Zellen als einzelne
Kolonien sichtbar. Eine von diesen wurde analysiert und als MB430,
und entsprechend das Plasmid pMB430, gerettet. Dieser Klon wies
absolut keine Expression von GFP auf was darauf hindeutete, dass
der klonierte Terminator tatsächlich
wie erwartet funktionierte.
-
Die
resultierende Sequenz des gesamten Plasmids pMB430 ist als SEQ Nr.
1 aufgelistet.
-
Herstellen des Stamms
MB432
-
Bacillus
subtilis PL1801 wurde mit den zwei Plasmiden pMB430 und pSX120 transformiert,
wobei in etwa 1 μg
jedes Plasmids pro 100 μl
kompetenter PL1801 verwendet wurden. Die transfarmierten Zellen
wurden auf LBPG-5 μg/ml
Erythromycin plattiert, über
Nacht bei 33°C
inkubiert. Kolonien des nächsten
Tages wurde erneut auf LBPG-5 μg/ml
Erythromycin + 6 μg/ml
Chloramphenicol ausgestrichen und über Nacht bei 33°C inkubiert.
Klone wurden auch in Flüssigkultur
in TY 5 μg/ml
Erythromycin + 6 μg/ml
Chloramphenicol über Nacht
bei 33°C
wachsen gelassen, Plasmide wurde aus Kulturbrühe gereinigt, und das Vorhandensein
von sowohl pMB430 als auch pSX120 in dem PL1801-Stamm wurde verifiziert.
Dieser Klon wurde gerettet und MB432 genannt.
-
Beispiel 1
-
Sequenzmischen von zwei
homologen Sequenzen, die beide für
das Subtilisingen der Savinase kodieren
-
Das
Savinase-wt-Gen (in pSX120 enthalten), das für eine Protease des Subtilisintyps
kodiert, wurde als das Ziel für
das Mischen hoch homologer Gene ausgewählt. Eine ortsspezifische Variante
des Savinasegens (hierin im Folgenden Savizyn genannt, in pSX222
enthalten, worin eine große
Zahl stummer Mutationen, die zu neuen Restriktionsstellen führen, eingefügt ist),
wurde als der Mischungspartner für
die Savinase verwendet. Die beiden Gene wurden erfindungsgemäß auf zwei
getrennte, kompatible Plasmide kloniert. Ein transkriptionell aktives
Savinasegen wurde in ein auf dem Ursprung von pUB110 basierendes
Plasmid kloniert, das für
Resistenz gegenüber
Chloramphenicol kodiert (pSX120, 4). Ein
promotorloses Savizyngen wurde in ein Derivat des temperatur empfindlichen
Plasmids pE194 kloniert, das auch für eine Resistenz gegen Erythromycin
kodiert (pMB430). Zwei Transkriptionsterminatoren wurden stromaufwärts des
Savizyngens kloniert, um jegliches Durchlesen zu verhindern.
-
In
pMB430 war das Grün-fluoreszierende
Protein (GFP) transkriptionell an das Savizyngen fusioniert, um
ein visuelles Verfolgen der Einkreuzungs- und Auskreuzungsereignisse
zwischen den beiden Plasmiden zu ermöglichen.
-
Der
B. subtilis-Stamm MB432, pL1801, transformiert mit den zwei Plasmiden
pSX120 und pMB430, wurde zwei Zyklen von Temperaturverschiebungen
unterzogen, um zwei aufeinander folgende Doppel-Crossoverereignisse
zwischen den homologen Genen zu induzieren. Auf LBPG-Platten wurde
bei der restriktiven Temperatur (47–50°C) in der Gegenwart von Erythromycin
auf homologe Rekombination und Plasmid-Hybridbildung selektiert.
Unter diesen restriktiven Bedingungen wird das temperaturempfindliche
Plasmid pMB430 durch homologe Rekombination in der Savinase/Savizyn-Region
in das stabile pSX120-Plasmid gezwungen. Diese Kolonien, in denen
die Rekombination stattgefunden hat, werden außerdem als ein Ergebnis der
Rekombination zwischen den beiden Plasmiden und der gleichzeitigen
Aktivierung des Savizyn-GFP-Operons (2) fluoreszierend.
Auf Auskreuzungsereignisse wurde durch Wachsenlassen der Klone in
LB-Brühe bei der
permissiven Temperatur von 30°C
für zwei
Tage und Plattieren der Zellen auf LBPG-Platten mit Chloramphenicol
und Erythromycin gescreent. Die nicht-fluoreszierenden Kolonien wurden erneut
auf Platten ausgestrichen und einer zweiten, wie oben beschriebenen
Runde des Temperaturzyklus unterzogen.
-
Nach
der zweiten Mischungsrunde wurden Plasmidpräparationen aus 20 verschiedenen
Klonen hergestellt. Kompetenter PL1801 wurde mit jeder der 20 Plasmidpräparationen
transformiert und auf mit Erythromycin supplementiertes LBPG plattiert.
Die Platten wurden auf LBPG-Platten mit Chloramphenicol und Erythromycin
repliziert, um unerwünschte
Klone zu identifizieren, in denen sowohl pSX120 als auch pMB430
transformiert worden waren. Klone, in denen nur das pMB430-Plasmid
transformiert worden waren, wurden für das Sequenzieren der Savizyn-Region
ausgewählt,
um zu identifizieren, ob eine Mischung stattgefunden hatte. Es ist
aus der 6 ersichtlich, dass eine Rekombination
zwischen den beiden Genen Savinase und Savizyn stattgefunden hatte.
Alle 12 sequenzierten Klone zeigten einen gewissen Grad der Rekombination,
und 4 von diesen können
positiv als das Ergebnis von zwei aufeinander folgenden doppelten
Rekombinationsereignissen identifiziert werden. Es ist wichtig zu
bemerken, dass ein Austausch der Savinase- und Savizyn-Sequenz nur beobachtet
werden kann, falls das gemischte Fragment eine Restriktionsstelle überlappt,
die nur in Savizyn vorkommt. Die vier Klone mit einem positiven
Hinweis auf zwei doppelte Rekombinationen entsprechen deshalb der
Mindestanzahl, da die Rekombination sich zwischen zwei benachbarten
Restriktionsstellen ereignet haben könnte.
-
-
-
-
-
-