-
TECHNISCHES
GEBIET
-
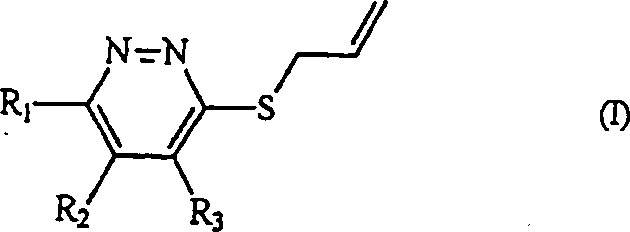
Die vorliegende Erfindung betrifft
ein neues Allylthiopyridazinderivat, dargestellt durch die Formel
(I), welches eine überlegene
Wirkung für
die Vorbeugung und Behandlung von hepatitischen Krankheiten aufzeigt, die
durch toxische Substanzen induziert sind und für den Schutz von humanen Geweben
vor Strahlung:
oder ein pharmazeutisch
akzeptables Salz davon, wobei R
1 ein Halogenatom,
C
1-C
4 Alkoxy, n-Pentyloxy, i-Pentyloxy, Dialkylaminoalkoxy,
Hydroxyalkoxy, Phenoxy substituiert oder nicht substituiert mit
Methyl, Benzyloxy oder Phenyl darstellt, und R
2 und
R
3 unabhängig
voneinander Wasserstoff oder Methyl oder R
2 und
R
3 gemeinsam in einem Kohlenstoffatom mit
welchem sie verbunden sind, einen gesättigten oder ungesättigten 6-gliedrigen
Ring bilden, darstellen, vorausgesetzt, daß R
2 und
R
3 nicht Wasserstoff sind, wenn R
1 Chlor ist.
-
Zusätzlich betrifft die Erfindung
ein Verfahren zum Herstellen der Verbindung der Formel (I) wie oben, ein
neues Intermediat, das bei diesen Verfahren verwendet wird und eine
pharmazeutische Zusammensetzung, die die Verbindung der Formel (I)
als effektiven Bestandteil enthält.
-
HINTERGRUND
UND STAND DER TECHNIK
-
Oltipraz erhöht die Expressionsrate von
mikrosomaler Epoxidhydrolase (mEH) und Glutathione S-Transferasen
(GST), intrazellulären
Oxidoreduktasen, und erhöht
so deren Spiegel in Zellen. Oltipiraz war bekannt dafür, das zelluläre Gewebe
vor Strahlung zu schützen
(siehe S. G. Kim, et al., Molekular Pharmacology, 51, 225–233, 1997;
S. Y. Nam, et al., Radiation Research, 147, 613–620, 1997]. D. h., es kann
bemerkt werden, daß der
Anstieg der intrazellulären
Expressionsrate von mEH und GST eng mit dem Schutz von Zellen vor
Strahlung in Verbindung steht. Weiterhin wurde allgemein erkannt,
daß der
Anstieg der intrazellulären
Expressionsrate von mEH und GST ebenso mit dem Schutz des menschlichen
Körpers
vor toxischen Substanzen in Verbindung steht [siehe Ansher et al.,
Hepatology, 3, 932–935,
1983; Lu & Miwa,
Annual Review of Pharmacology and Toxicology, 20, 513–531, 1980].
-
Es wurde berichtet, daß der Gewebeschaden,
der durch toxische Substanzen, so wie Acetaminophen oder Kohlenstofftetrachlorid
induziert wurde, eine enge Beziehung zu der Aktivität von Cytochrom
P450 2E1 hat, welches solche toxische Substanzen metabolisiert [siehe
S. K. Kim et al., J. Pharmacol. Exp. Therap. 277, 1058, 1996]. Daher
tritt ernsthafter hepatitischer Schaden sogar bei niedriger Konzentration
von Kohlenstofftetrachlorid auf, wenn Laborratten mit Pyridin, einem
Cytochrom P450 2E1-Derivat behandelt werden. Daher wurde erwartet,
daß wenn
die Aktivität
von Cytochrom P450 2E1 effektiv inhibiert werden kann, daß dann der Gewebeschaden
an menschlichen Organen (insbesondere Leber) aufgrund von toxischen
Substanzen vermieden werden könnte
und aus dem selben Grund könnte
der menschliche Körper
ebenso von oncogenen Substanzen, Strahlung, Antitumorchemotherapeutika,
usw. gegenüber
des Dünndarms,
des Dickdarms, der Gallenblase, der Bronchien, der Bauchspeicheldrüse, der
Brustdrüse
und der Haut geschützt
werden.
-
Als eines der therapeutischen Mittel,
die gegenwärtig
auf dem klinischen Gebiet für
die Vorbeugung und Behandlung von hepatitischen Krankheiten, induziert
durch toxische Substanzen, verwendet werden, wurde für Malotilat
gezeigt, daß es
eine gute Wirkung hinsichtlich des Vorbeugens und des Behandelns
von hepatitischen Krankheiten, die durch Kohlenstofftetrachlorid
und Acetaminophen induziert wurden, besitzt. Zusätzlich wurde ebenfalls erkannt,
daß Diallylsulfid
und Allicin als eine der aromatischen Substanzen in Knoblauchöl den Effekt
des Inhibierens von Oncogenese aufgrund von 1,2-Dimethylhydrazin haben und die Leber vor
der Hepatotoxizität
des 1,2-Dimethylhydrazins schützen.
-
Da die vorliegenden Erfinder viel
technisches Wissen über
den konventionellen Stand der Technik haben, haben sie die Möglichkeit
in Erwägung
gezogen, daß die
Allylthiogruppe eine wichtige Rolle beim Schützen von humanen Organen und
vor toxischen Substanzen oder vor Strahlung spielen könnte, und
daß durch Erhöhen der
intrazellulären
Expression von mEH und GST gleichzeitig die Aktivität von Cytochrom
P450 2E1 wie oben erwähnt
effektiv inhibiert wird. Daher haben wir ausführlich eine Studie ausgeführt, um
eine Vielzahl von Verbindungen, die eine Allylthiogruppe aufweisen,
neu zu systhetisieren und deren pharmakologische Aktivitäten zu untersuchen.
Als ein Ergebnis haben wir identifiziert, daß die Verbindung der Formel
(I), wie oben definiert, in welcher eine Allylthiogruppe als eine
pharmakologisch aktive Gruppe in den Pyridazinnucleus eingeführt wird
und ein Substituent, so wie Halogen, Alkoxy, usw. in die Para-Position
der Allylthiogruppe eingeführt
wird, menschliche Gewebe vor Strahlung und aktiven toxischen Substanzen
durch Erhöhen
der Expression von mEH und GST und gleichzeitig durch Inhibieren
der Expression von methabolischen Enzymen schützen kann.
-
OFFENBARUNG
DER ERFINDUNG
-
Dementsprechend betrifft die vorliegende
Erfindung eine neue Allylthiopyridazinverbindung der Formel (I)
wie oben definiert oder ein pharmazeutisch akzeptables Salz davon.
-
Weiterhin betrifft die vorliegende
Erfindung ein Verfahren zur Herstellung der Verbindung der Formel (I).
-
Zusätzlich betrifft die vorliegende
Erfindung eine pharmazeutische Zusammensetzung für die Vorbeugung und Behandlung
von hepatitischen Krankheiten, welche eine effektive Menge der Verbindung
der Formel (I) oder dessen Salz gemeinsam mit einem pharmazeutisch
akzeptablen Träger
umfaßt.
-
Auch betrifft die vorliegende Erfindung
eine pharmazeutische Zusammensetzung zum Schützen von humanen Geweben vor
Strahlung, welche eine effektive Menge der Formel (I) oder deren
Salz gemeinsam mit einem pharmazeutisch akzeptablen Träger umfaßt.
-
KURZE BESCHREIBUNG
DER ZEICHNUNG
-
Für
ein gründliches
Verständnis
der Natur und der Gegenstände
der vorliegenden Erfindung sollte Bezug auf die folgende Beschreibung
gemeinsam mit der begleitenden Zeichnung genommen werden, in welcher:
-
1 das
Ergebnis einer Northern Blot Analyse zum Messen eines Anstiegs der
mRNA von mikrosomaler Epoxydhydrolase in Lebergewebe 24 Stunden
nach der Verabreichung der erfindungsgemäßen Verbindung zeigt;
-
2 das
Ergebnis einer Northern Blot Analyse zum Messen eines Anstiegs der
mRNA von Glutation S-Transferase
A2 in Lebergewebe 24 Stunden nach der Verabreichung der erfindungsgemäßen Verbindung zeigt;
und
-
3 die Überlebensrate
von Mäusen
während
30 Tagen mit 8 Gy Bestrahlung zeigt, nachdem ihnen die Verbindung
von Beispiel 4 (K6) oder Beispiel 8 (K16) gemäß der vorliegenden Erfindung
verabreicht wurde.
-
BESTES VERFAHREN
ZUM AUSFÜHREN
DER ERFINDUNG
-
Hiernach wird die vorliegende Erfindung
genauer erklärt
werden.
-
Die vorliegende Erfindung betrifft
eine neue Allythiopyridazinverbindung, dargestellt durch die Formel (I),
welche eine überlegene
Wirkung bei der Vorbeugung und bei der Behandlung von hepatitischen
Krankheiten aufzeigt, die durch toxische Substanzen induziert werden,
und für
den Schutz von humanen Geweben vor Strahlung aufzeigt:
oder ein pharmazeutisch
akzeptables Salz davon, in welchem R
1 ein
Halogenatom, C
1-C
4 Alkoxy,
n-Pentyloxy, i-Pentyloxy,
Dialkyaminoalkoxy, Hydroxyalkoxy, Phenoxy substituiert oder nicht
substituiert mit Methyl, Benzyloxy oder Phenyl darstellt, und R
2 und R
3 unabhängig voneinander
Wasserstoff oder Methyl darstellen oder R
2 und
R
3 gemeinsam mit einem Kohlenstoffatom,
mit welchen sie verbunden sind, einen gesättigten oder ungesättigten
6-gliedrigen Ring bilden, vorausgesetzt, daß R
2 und
R
3 anders als Wasserstoff sind, wenn er
R
1 ein Chlor ist.
-
Unter den Verbindungen der Formel
(I), die eine überlegene
pharmakologische Wirkung haben, beinhalten die bevorzugten Verbindungen
diejenigen, bei denen R1 Chlor, Metoxy,
Etoxy, n-Propoxy, 2-Propoxy, n-Butoxy, 2-Butoxy, t-Butoxy, n-Pentyloxy,
i-Pentyloxy, 2-(N,N-Dimethylamino)ethoxy,
2-Hydroxyethoxy, Phenoxy, Benzyloxy, 4-Methylphenoxy, 3-Methylphenoxy,
2-Methylphenoxy
oder Phenyl darstellt und R2 und R3 unabhängig
voneinander Wasserstoff oder Methyl darstellen oder R2 und
R3 gemeinsam mit dem Kohlenstoffatom mit
welchem sie verbunden sind, einen gesättigten oder ungesättigten
6-gliedrigen Ring bilden.
-
Die pharmazeutisch akzeptablen Salze
der Verbindung (I) gemäß der vorliegenden
Erfindung können pharmazeutisch
akzeptable Säureadditionsalze,
so wie Asparaginat, Glukanat, Glutamat, p-Toluolsulfonat oder Citrat
beinhalten, Salze mit Alkalimetallen, so wie Natrium, Kalium oder
Lithium beinhalten oder Salze mit anderen Säuren oder Basen beinhalten,
von welchen bekannt ist, daß sie
konventionell für
Verbindungen, so wie Dialylsulfid, Allicin, usw. verwendet werden.
Diese Salze können
leicht durch konventionelle Transformationsverfahren aus der freien
Verbindung der Formel (I) hergestellt werden.
-
Zusätzlich kann die Verbindung
der Formel (I) gemäß der vorliegenden
Erfindung durch ein Verfahren hergestellt werden, das dadurch gekennzeichnet
ist, daß (A)
eine Verbindung der Formel (II):
wobei R
1,
R
2 und R
3 definiert
sind wie oben und X ein Chlor repräsentiert, mit einer Verbindung
der Formel (III):
HS-CH2-CH=CH2 (III) in
einem Lösungsmittel
in der Gegenwart einer Base umgesetzt wird, um die Verbindung der
Formel (I) herzustellen; oder (b) eine Verbindung der Formel (IV):
wobei X Chlor repräsentiert,
mit einer Verbindung der Formel (V):
R1'ONa (V) umgesetzt
wird, wobei R
1' C
1-C
4 Alkyl, n-Pentyl, i-Pentyl, Dialkylaminoalkyl, Hydroxyalkyl,
Phenyl substituiert oder nicht substituiert mit Methyl oder Benzyl
darstellt, in einem Lösungsmittel
um eine Verbindung der Formel (Ia):
herzustellen, wobei R
1' wie
oben definiert ist.
-
Dementsprechend ist ein anderer Zweck
der vorliegenden Erfindung, solch ein Verfahren zum Herstellen der
Verbindung der Formel (I) zur Verfügung zu stellen.
-
Bei der Variante (a) kann das Lösungsmittel
aus Methanol, Ethanol, Aceton, Methylethylketon oder einer Mischung
daraus ausgewählt
werden; und die Base kann ausgewählt
werden aus der Gruppe bestehend aus Alkalimetallen so wie Natrium,
Kalium, Lithium, usw. vorzugsweise wird die Umsetzung für ein bis
24 Stunden unter Rückfluß unter
Erhitzung ausgeführt.
-
Das Lösungsmittel, welches bei der
Variante (b) verwendet werden kann, kann das selbe sein, wie diejenigen,
die bei der Variante (a) oben erwähnt sind. Die Verbindung der
Formel (V) verwendet als Reaktant bei diesem Verfahren, wird hergestellt
durch Umsetzen von Natrium mit einer Verbindung der Formel R1'OH
in dem Umsetzungsgefäß und wird
dann augenblicklich für
die Umsetzung mit der Verbindung der Formel (IV) verwendet. Die
Umsetzung wird 24 Stunden lang unter Rückfluß mit Erhitzen ausgeführt.
-
Zwischenzeitlich ist die Verbindung
der Formel (II), die als Ausgangsmaterial bei der Variante (a) verwendet
wird, eine neue Verbindung. Daher ist weiterhin ein anderer Zweck
der vorliegenden Erfindung, die neue Verbindung der Formel (III),
wie oben definiert zur Verfügung
zu stellen.
-
Die Verbindung der Formel (II) kann
durch ein Verfahren hergestellt werden, das in den folgenden Umsetzungsschemas
1 und 2 gezeigt ist. Das spezifische Verfahren kann in den Herstellungsbeispielen,
die hierunter beschrieben sind, gesehen werden.
-
-
Bei dem obigen Umsetzungsschema sind
R1, R2, R3 und X wie oben definiert.
-
Bei dem obigen Umsetzungsschema 1
wird die Verbindung der Formel R1'H, die zu der Umsetzungslösung hinzugefügt wird,
mit einem Alkalimetall umgesetzt, um das Alkalimetallsalz zu bilden,
und das resultierende Alkalimetallsalz wird mit dem Ausgangsmaterial
umgesetzt, um die Verbindung der Formel (II) herzustellen. Diese
Umsetzung wird im Allgemeinen bei Raumtemperatur oder unter Erwärmen ausgeführt. Die
Umsetzung kann innerhalb von 30 Minuten bis zu 12 Stunden abgeschlossen
werden.
-
Alternativ kann die Verbindung der
Formel (II), wobei die reaktive Abgangsgruppe Chlor ist und die
R1 Position ebenso mit Chlor substituiert
ist, durch Umsetzen der Verbindung, wobei beide der 3- und 6-Positionen des
Pyridazinrings mit Hydroxyl substituiert sind mit Phosphoroxychlorid
(POCl3) hergestellt werden, wie gezeigt
in dem folgenden Reaktionsschema 2. Bei diesem Verfahren wird die
Umsetzung 5–10
Stunden unter Rückfluß mit Erhitzen
ausgeführt.
-
-
Bei dem obigen Reaktionsschema sind
R2 und R3 wie oben
definiert.
-
Die Verbindung der Formel (IV) (X=Cl),
verwendet als Ausgangsmaterial bei der Umsetzungsvariante (b) zum
Herstellen der Verbindung der Formel (I), ist eine bekannte Verbindung
und kann hergestellt werden unter Bezugnahme auf das Verfahren,
daß in
der Literatur offenbart ist [siehe M. Kocevar et al., Croat. Chem. Acta.,
45, 457, 1973].
-
Um die Wirkung der Allylthiopyridazinderivate
gemäß der vorliegenden
Erfindung auf die Expressionsrate von mEH und GST zu bestätigen, haben
die vorliegenden Erfinder die Menge der mRNA von mEH und GSTA2,
die im Lebergewebe nach einer gewissen Zeitperiode ab der oralen
Verabreichung der entsprechenden Verbindung an Mäuse gemäß Northern Blot Analyse (siehe 1 und 2) quantifiziert. Als ein Ergebnis konnten
wir einen signifikanten Anstieg bei der Expressionsrate der mRNA
beobachten. Solch eine mRNA erhöhende
Wirkung der Verbindung der Formel (I) wird betrachtet als einen
direkten Bezug zu der Wirkung für die
Vorbeugung und Behandlung von hepatitischen Krankheiten, die durch
toxische Substanzen induziert sind, und zu dem Schutz von humanem
Gewebe vor Bestrahlung habend. Daher haben die vorliegenden Erfinder das
folgende Experiment ausgeführt,
um die Wirkungen der Verbindung der Formel (I) für dei Vorbeugung und für die Behandlung
von hepatitischen Krankheiten und zum Schutz von humanem Gewebe
vor Bestrahlung zu bestimmen.
-
Zunächst wurden die Wirkungen des
Allylthiopyridazinderivates der Formel (I) zur Vorbeugung und bei der
Behandlung von hepatitischen Krankheiten unter Verwendung des Kohlenstofftetrachloridmodells
und des Acetaminophenmodells untersucht.
-
Das Kohlenstofftetrachloridmodell
[siehe Philippe Letteron et al., Biochemical Pharmacology, 39, 12, 2027– 2034,
1990] wird am häufigsten
als ein experimentelles Modell für
hepatitische Fehlzustände
verwendet und wurde auf der Basis der Tatsache etabliert, daß Kohlenstofftetrachlorid
in freie Trichlormethylradikale (CCl3 · ), ein
sehr toxisches Metabolit, durch die Wirkung von Cytochrom P-450
im Körper
umgewandelt wird. Dieser Metabolit bindet stark an Thiolgruppen
von microsomalem Membranprotein in der Leber, um ein Lipidradikal zu
produzieren, welches dann in ein Peroxidradikal in Gegenwart von
Sauerstoff umgewandelt wird und dabei die Peroxidierungsumsetzung
der Membranlipide stimuliert. D. h. Kohlenstofftetrachlorid inhibiert
Proteinsynthese in der Leber, erhöht Blut ALT (Alaninaminotransferase)
und verursacht zentrilobulare Nekrose der Leberzellen.
-
Zusätzlich können die leberschützenden
Wirkungen der Verbindung der vorliegenden Erfindung ebenso durch
das Acetaminophenmodell bestätigt
werden [siehe Wang et al., Toxicology and Applied Pharmacology,
136, 146–154,
1996]. Acetaminophen wird leicht durch CYP2E1 metabolisiert, um
hepatitische Toxizität
zu verursachen, welche in struktureller und funktionalen Veränderungen
in der zellulären
Membran der Leber resultiert, was Nekrose in hepatitischen Lobule,
einen Anstieg an Blut ALT und des LDH (Lactatdehydrogenase) Spiegels
verursacht.
-
In der vorliegenden Erfindung wurde
der inhibierende Effekt der Verbindung der Formel (I) auf hepatitische
Fehlzustände,
induziert durch Kohlentetrachlorid und Acetaminophen, nach dreitägiger oraler
Verabreichung der neuen Verbindung gemäß der vorliegenden Erfindung
an Mäuse
als Versuchstiere bestimmt. Der Grad der Leberschädigung in
dem entsprechenden Experimentiertier wurde bestimmt durch Messen
der ALT und LDH Spiegel im Blut [siehe Biol. Pharm. Bull., 20, 4,
381–385,
1997; Toxicology and Applied Pharmacology, 95, 1–11, 1988] und weiterhin durch
Entfernen der Leber, Färben
(× 100)
der Leber mit Hematoxylin und Eosin und dann Beobachten der Leber
mit einem Mikroskop um den Grad der Hepatozyten Nekrose zu untersuchen.
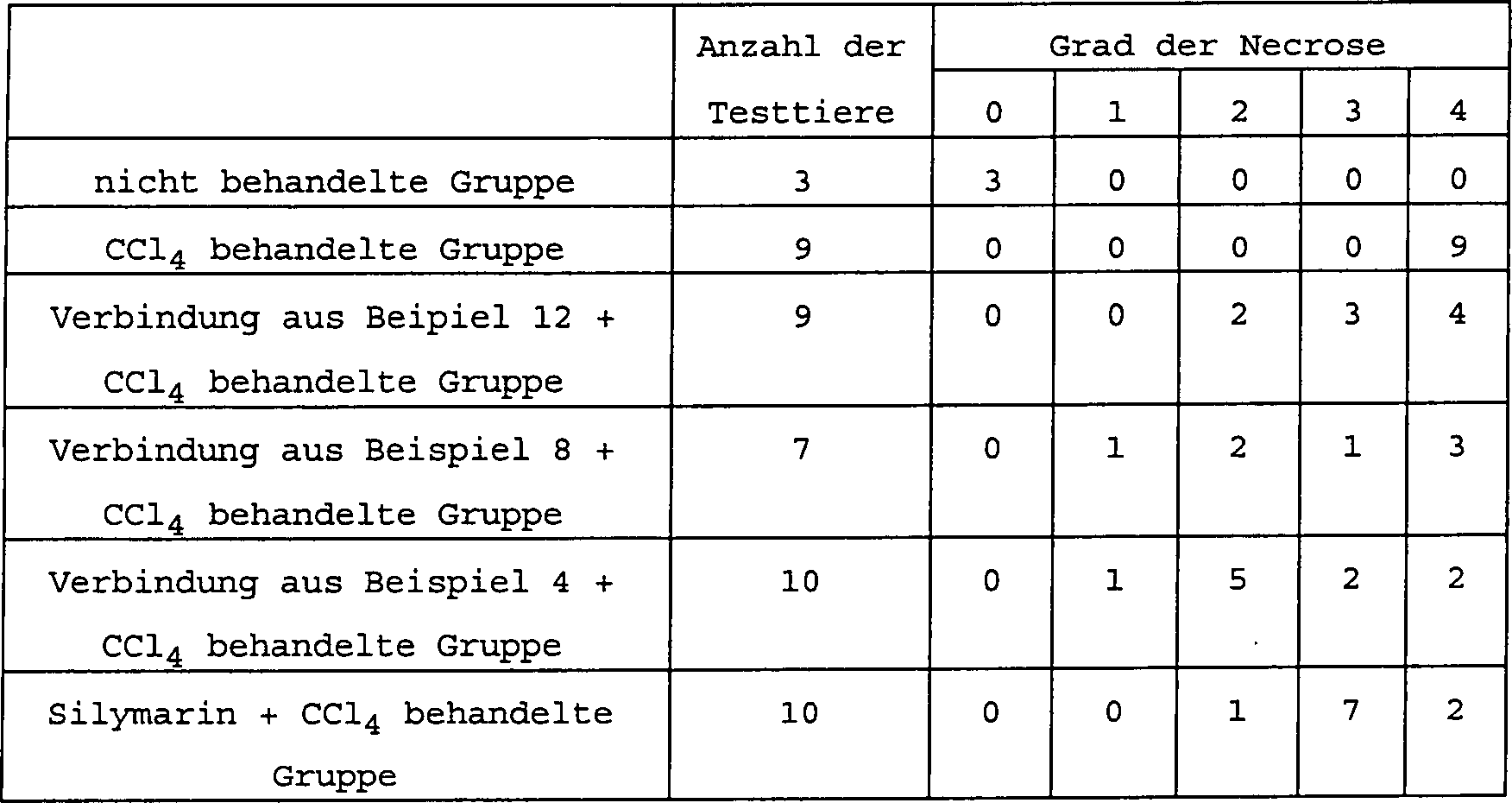
Als ein Ergebnis dieses Experimentes wurde identifiziert, daß die Verbindung
der Formel (I) gemäß der vorliegenden
Erfindung eine überlegene
leberschützende
Wirkung sogar im Vergleich mit Silymarin hat, welches weithin als
leberschützendes
Mittel bekannt ist.
-
Weiterhin haben die vorliegenden
Erfinder die schützende
Wirkung des erfindungsgemäßen Allylthiopyridazinderivats
gegen Strahlung durch Verabreichen der aktiven Verbindung an Mäuse, Bestrahlen
mit der Strahlung von 8 oder 9 Gy und nach einer gewissen Zeitperiode
Messen der Überlebensrate
der Mäuse
bestätigt.
Beispielsweise kann die Überlebensrate
während
30-tägiger
Bestrahlung mit 8 Gy aus 3 ersehen werden,
und das spezifische experimentelle Ergebnis ist in der folgenden
Tabelle 7 beschrieben. Spezifisch, wenn 8 Gy Strahlung verwendet
wird, beträgt
die Überlebensrate
während
30 Tagen in den Gruppen, bei denen nur Strahlung verwendet wurde,
lediglich 48%, während
die Überlebensrate
während
30 Tagen in den Gruppen, an welche die Verbindungen der Beispiele
4 und 8 vorher verabreicht wurden 67% und 70% betrug. Daher kann
bemerkt werden, daß der
schützende
Effekt der Verbindung gemäß der vorliegenden
Erfindung gegen Strahlung bewiesen worden ist, eine sehr signifikante
neue Entdeckung zu sein.
-
Darüberhinaus, als ein Ergebnis
eines akuten Toxizitätstest
unter Verwendung von Mäusen,
um die generelle Toxizität
der Verbindung gemäß der vorliegenden
Erfindung zu evaluieren, wenn die Verbindung alleine auf oralem
Wege verabreicht wurde, betrug der LD50 Wert
der entsprechenden Verbindung 3,5 g/kg oder mehr. Daher wurde bestimmt,
daß die
Verbindung der vorliegenden Erfindung sehr sicher ist.
-
Aus den obigen experimentellen Ergebnissen
kann identifiziert werden, daß die
Verbindung der Formel (I) gemäß der vorliegenden
Erfindung sicher ist und eine ausgezeichnete Wirkung für die Vorbeugung
und die Behandlung von hepatitischen Krankheiten hat. Daher ist
es ein weiterer Zweck der vorliegenden Erfindung, eine pharmazeutische
Zusammensetzung für
die Vorbeugung und Behandlung von hepatitischen Krankheiten zur
Verfügung
zu stellen, welcher als einen effektiven Bestandteil die Verbindung
der Formel (I) oder dessen pharmazeutisch azteptables Salz enthält. Zusätzlich,
da die Verbindung der vorliegenden Erfindung und dessen Salz als
eine schützende
Wirkung gegen Strahlung habend identifiziert wurden, ist die Zusammensetzung
zum Schutz von humanen Geweben vor Strahlung, welche die Verbindung
als einen effektiven Bestandteil umfaßt ebenso ein Gegenstand der
vorliegenden Erfindung. Solche Wirkungen der pharmazeutischen Zusammensetzungen
gemäß der vorliegenden
Erfindung stehen mit dem Wirkmechanismus der Formel (I) in Verbindung,
welcher die Expressionsrate von mEH und GST als Oxidoreduktasen
erhöht
und ebenso die Aktivität
von Cytochrom P450 2E1 inhibiert. Wie oben erwähnt, da der Wirkmechanismus,
welcher die Expressionsrate von mEH und GST erhöht und die Aktivität von Cytochrom
P450 2E1 reduziert, mit den Wirkungen des Reduzierens der Toxizität von Antitumorchemotherapeutika
und des Inhibierens der Oncogenese und von chemischen Substanzen,
ebenso wie die Wirkungen des Vorbeugens und des Behandelns von hepatitischen Krankheiten
und des Schützens
von humanem Gewebe vor Strahlung in Verbindung steht, könnte verstanden werden,
daß die
Verbindung der vorliegenden Erfindung ebenso solche Wirkungen aufzeigen
könnte.
-
Bei der Verwendung der erfindungsgemäßen pharmazeutischen
Zusammensetzung zu einem klinischen Zweck kann die Verbindung der
Formel (I) in Kombination mit einem pharmazeutisch akzeptablen inerten
Träger
in eine feste, halbfeste oder flüssige
pharmazeutische Präparation
formuliert werden, welche für orale
oder parenterale Verabreichung geeignet ist.
-
Pharmazeutisch akzeptable inerte
Träger,
welche zu diesem Zweck verwendet werden können, können fest oder flüssig sein
und beinhalten ein Glied oder mehrere ausgewählt aus einer Gruppe bestehend
aus Verdünnungsmitteln,
Geschmacksmitteln, Solubilisierungsmitteln, Befeuchtungsmitteln, Suspensionsmitteln, Bindemitteln,
Tablettenanschwellmitteln, usw. Als ein spezifisches Beispiel für den geeigneten
festen oder flüssigen
Träger,
welcher in der vorliegenden Erfindung verwendet werden kann, können Stärke, Lactose,
Zellulosederivate (Avicel®),
Zucker, usw. erwähnt
werden.
-
Wenn die pharmazeutische Zusammensetzung
gemäß der vorliegenden
Erfindung zum Zwecke des Vorbeugens und des Behandelns von hepatitischen
Krankheiten und zum Schützen
der Leber vor Bestrahlung verwendet wird, beträgt die tägliche Dosis der aktiven Verbindung
gemäß der vorliegenden
Erfindung vorzugsweise 1 bis 500 mg/kg Körpergewicht, im initialen Stadium.
Jedoch sollte verstanden werden, daß die Dosierung variiert werden
kann, abhängig
vom Bedarf des Patienten, der Schwere der Krankheit, die behandelt
wird, der Art der verwendeten Verbindung, usw., und die optimale
Dosierung kann gemäß dem bekannten
Verfahren durch einen Fachmann bestimmt werden. Im Allgemeinen wird
die therapeutische Prozedur bei einem Dosierungsniveau initiiert,
welches niedriger ist, als die optimale Dosierung der Verbindung,
und dann wird die Dosierung graduell erhöht, abhängig vom Zustand, bis die optimale
Wirkung erhalten wird. Wenn angemessen kann die tägliche Dosierung
in einige Teile unterteilt werden und dann über einer eintägige Zeitperiode
verabreicht werden.
-
Die vorliegende Erfindung wird genauer
durch die folgenden Herstellungen und Beispiele beschrieben werden.
Jedoch sollte verstanden werden, daß diese Herstellungen und Beispiele
nur dafür
vorgesehen sind, beim Verständnis
der vorliegenden Erfindung zu helfen, aber die vorliegende Erfindung
wird nicht durch diese Beispiele in irgendeiner Art und Weise limitiert.
-
Herstellung 1
-
Synthese von 3,6-Dihydroxy-4,5-dimethylpyridazin
-
50 ml auf gereinigtes Wasser wurde
zu 3,4 ml (0,07 mol) Hydrazinmonohydrat hinzugefügt, und dann wurden 14 ml (0,14
mol), konzentrierte Salzsäure
tropfenweise hinzugefügt.
Dann wurde die Umsetzungslösung
bis zum Kochen erwärmt.
Als die Umsetzungsmischung begann, unter Rückfluß zu sieden, wurden 8,83 g
(0,07 mol) 2,3-Dimethylmaleinsäureanhydrid
hinzugegeben. Die Umsetzungslösung
wurde kontinuierlich unter Rückfluß 3 Stunden
lang erhitzt und dann abgekühlt.
Die präzipitierten
weißen
Kristalle wurden gefiltert, mit auf gereinigtem Wasser gewaschen,
gelöst
in aufgereinigtem kochenden Wasser um unlösliches Material zu entfernen
und dann unkristallisiert, um die Titelverbindung als einen amorphen
weißen
Kristall zu erhalten.
Ausbeute: 9,10 g (92,8%)
Schmelzpunkt:
298–300°C (Zersetzung)
Umkristallisationslösungsmittel:
Wasser
NMR (CDCl3 + DMSO-d6, δ): 2,00 (s,
3H × 2,
CH3), 3,30 (s, 1H × 2, OH)
-
Herstellung 2
-
Synthese von 1,4-Dihydroxy-5,6,7,8-tetrahydophthalazin
-
25 ml aufgereinigtes Wasser wurde
zu 1,7 ml (0,35 mol) Hydrazinmonohydrat hinzugegeben, und dann wurden
7 ml (0,07 mol) konzentrierte Salzsäure tropfenweise hinzugegeben.
Dann wurde die Umsetzungslösung
zum Kochen erwärmt.
Als die Umsetzungslösung
begann, unter Rückfluß zu sieden,
wurden 5,33 g (0,035 mol) 3,4,5,6-Tetrahydrophthalsäureanhydird hinzugegeben. Die
Umsetzungslösung
wurde kontinuierlich unter Rückfluß 3 Stunden
lang erhitzt und dann abgekühlt.
Dann wurde die Umsetzungslösung
auf die selbe Art und Weise wie Herstellung 1 behandelt, um die
Titelverbindung als einen weißen
Nadelkristall zu erhalten.
Ausbeute: 5,47 g (94,0%)
Schmelzpunkt:
298–300°C
Umkristallisationslösungsmittel:
Wasser
NMR (CDCl3 + DMSO-d6, δ): 1,65 (s,
2H × 2,
CH2), 2,35 (s 2H × 2, CH2),
3,30 (s, 1H × 2,
OH)
-
Herstellung 3
-
Synthese von 3,6-Dichlor-4,5-dimethylpyridazin
-
2,80 g (0,02 mol) vollständig getrocknetes
3,6-Dihydroxy-4,5-dimethylpyridazin
(erhalten aus Herstellung 1) wurden zu 20 ml Phosphoroxichlorid
(POCl3) hinzugegeben und die Umsetzungsmischung
wurde unter Rückfluß 7 Stunden
lang erhitzt. Zu diesem Zeitpunkt wurde der Punkt, bei dem die farblose
Raktionslösung sich
lila färbte
als der Endpunkt der Umsetzung erwogen. Die Umsetzungslösung wurde
unter reduziertem Druck konzentriert, um überschüssiges Phosphoroxidchlorid
zu entfernen. Die kleine Menge Eiswasser wurde zu dem Rückstand
hinzugegeben und dann wurde gerührt,
um eine Suspension zu bilden. 28%-ige wäßrige Ammoniumhydroxidlösung wurde
zu der Suspension hinzugegeben, bis die Suspension alkalisch wurde,
um braunes Präzipitat
zu produzieren. Das Präzipitat
wurde gefiltert und dann in heißem
Ethanol gelöst,
um unlösliches
Material zu entfernen. Aktivkohle wurde zu der resultierenden Lösung hinzugefügt, und
die Mischung wurde 5 Minuten lang unter Rückfluß erhitzt, entfärbt durch
Laufenlassen durch Siliziumdioxid und dann destilliert unter reduziertem
Druck, um überschüssiges Ethanol
zu entfernen. Auf diese Art und Weise wurde die Titelverbindung
als weiße
nadelförmige
Kristalle erhalten.
Ausbeute: 2,45 g (69,2%)
Schmelzpunkt:
109–111°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 2,05 (s,
3H × 2,
CH3)
-
Herstellung 4
-
Synthese von 1,4-Dichlor-5,6,7,8-tetrahydrophtalazin
-
3,32 g (0,02 mol) vollständig getrocknetes
1,4-Dihydroxy-5,6,7,8-tetrahydrophalazin
(erhalten aus Herstellung 2) wurde zu 20 ml Phosphoroxidchlorid
hinzugegeben. Dann wurde die Umsetzung auf die selbe Art und Weise
ausgeführt
wie Herstellung 3, um die Titelverbindung als weiße nadelförmige Kristalle
zu erhalten.
Ausbeute: 2,68 g (66,0%)
Schmelzpunkt: 148–150°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 1,85 (s,
2H × 2,
CH2), 2,75 (s, 2H × 2, CH2)
-
Herstellung 5
-
Synthese von 3-Methoxy-4,5-dimethyl-6-chlorpyridazin
-
0,46 g (0,02 mol) metallisches Natrium
wurden in 30 ml absolutem Ethanol gelöst und dann wurden 3,54 g (0,02
mol) 3,6-Dichlor-4,5-dimethylpyrazin, erhalten aus Herstellung 3,
hinzugegeben und vollständig gelöst. Die
Umsetzungslösung
wurde eine Stunde bei Raumtemperatur gerührt und unter reduziertem Druck filtriert,
um überschüssiges Methanol
zu entfernen. 100 ml Diethylether wurde zu dem Rückstand hinzugegeben, und dann
wurde 10 Minuten lang heftig gerührt.
Die diethyletherunlöslichen
Materialien wurden entfernt und die verbleibende Lösung wurde
zweimal mit 50 ml gereinigtem Wasser gewaschen, getrocknet über wasserfreiem
Natriumsulfat und dann unter reduziertem Druck konzentriert, um
Diethylether zu entfernen. Auf diese Art und weise wurde die Titelverbindung
als weiße
nadelförmige
Kristalle erhalten.
Ausbeute: 2,63 g (76,2%)
Schmelzpunkt:
80–82°C
Umkristallisationslösungsmittel:
Ethanol
-
Herstellung 6
-
Synthese von 1-Methoxy-4-chlor-5,6,7,8-tetrahydrophthalazin
-
0,34 g (0,015 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann wurden 3,05 g (0,015
mol) 1,4-Dichlor-5,6,7,8-tetrahydrophthalazin, erhalten aus Herstellung
4, hinzugefügt
und vollständig
gelöst.
Die Umsetzungslösung
wurde eine Stunde lang bei Raumtemperatur gelöst und dann auf die selbe Art
und weise wie bei Herstellung 5 behandelt, um die Titelverbindung
als weiße
nadelförmige
Kristalle zu erhalten.
Ausbeute: 1,78 g (59,7%)
Schmelzpunkt:
117–119°C
Umkristallisationslösungsmittel:
Ethanol
-
Herstellung 7
-
Synthese von 1-Methoxy-4-chlorphthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 50 ml absolutem Methanol gelöst, und dann wurden 1,99 g
(0,01 mol) 1,4-Dichlorphtalazin hizugegeben und vollständig gelöst. Die
Umsetzungslösung
wurde eine Stunde lang bei Raumtemperatur gelöst und dann auf die selbe Art
und Weise behandelt wie bei Herstellung 5, um die Titelverbindung
als amorphe weiße
Kristalle zu erhalten.
Ausbeute: 1,81 g (92,8%)
Schmelzpunkt:
92–94°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 4,28 (s,
3H, OCH3), 7,90–8,25 (m, 1H × 4, aromatisch)
-
Herstellung 8
-
Synthese von 3-Ethoxy-4,5-dimethyl-6-chlorpyridazin
-
0,69 g (0,03 mol) metallisches Natrium
wurden in 100 ml absolutem Methanol gelöst und dann wurden 5,31 g (0,03
mol) 3,6-Dichlor-4,5-dimethylpyridazin zugegeben und vollständig gelöst. Die
Umsetzungslösung wurde
4 Stunden lang bei Raumtemperatur gerührt und dann auf die selbe
Art und weise behandelt wie in Herstellung 5, um die Titelverbindung
als einen blaß-weißen Kristall
zu erhalten.
Ausbeute: 4,22 g (75,4%)
Schmelzpunkt: 60–62°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 1,42 (t,
3H, CH3), 2,20 (s, 3H, CH3),
2,32 (s, 3H, CH3), 4,52 (q, 2H, OCH3)
-
Herstellung 9
-
Synthese von 1-Ethoxy-4-chlor-5,6,7,8-tetrahydrophthalazin
-
0,69 g (0,03 mol) metallisches Natrium
wurden in 100 ml absolutem Ethanol gelöst und dann wurden 6,09 g (0,03
mol) 1,4-Dichlor-5,6,7,8-tetrahydrophtlazin, erhalten aus Herstellung
4, zugegeben und vollständig
gelöst.
Die Umsetzungslösung
wurde 4 Stunden bei Raumtemperatur gerührt und dann auf die selbe
Art und Weise, wie in Herstellung 5, behandelt, um die Titelverbindung
als einen blaß-weißen Kristall
zu erhalten.
Ausbeute: 5,98 g (93,7%)
Schmelzpunkt: 105–107°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 1,40 (t,
3H, CH3), 1,80–2,53 (m, 2H × 4, aromatisch),
4,52 (q, 2H, OCH2)
-
Herstellung 10
-
Synthese von 1-Ethoxy-4-chlorphthalazin
-
0,69 g (0,03 mol) metallisches Natrium
wurden in 100 ml absolutem Methanol gelöst, und dann wurden 5,97 g
(0,03 mol) 1,4-Dichlorphthalazin hinzugegeben und vollständig gelöst. Die
Umsetzungslösung
wurde eine Stunde lang bei 50 ± 10°C gerührt und
dann auf die selbe Art und Weise wie bei Herstellung 5 behandelt, um
die Titelverbindung als weiße
nadelförmige
Kristalle zu erhalten.
Ausbeute: 5,02 g (80,2%)
Schmelzpunkt:
76–78°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 1,55 (t,
3H, CH3), 4,70 (q, 2H, OCH2,
7,92–8,20
(m, 1H × 4,
aromatisch)
-
Beispiel 1
-
Synthese von 3-Chlor-4,5-Dimethyl-6-Allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 40 ml absolutem Methanol gelöst, und die resultierende Lösung wurde
mit 0,93 ml (0,01 mol) Allylmercaptan (CH2=CH-CH2-SH) gemischt. Zu dieser Mischung wurden
1,77 g (0,01 mol) 3,6- Dichlor-4,5-dimethylpyridazin,
erhalten aus Herstellung 3, hinzugefügt, und die Umsetzungslösung wurde
unter Rückfluß eine Stunde
lang erhitzt und dann unter reduziertem Druck konzentriert, um Methanol
zu entfernen. 50 ml Diethylether wurden zu dem Rückstand hinzugegeben, und es
wurde kräftig
10 Minuten lang gerührt.
Diethyletherunlösliche
Materialien wurden entfernt, und die verbleibende Lösung wurde
zweimal mit 30 ml gereinigtem Wasser gewaschen, getrocknet über wasserfreiem
Natriumsulfat und unter reduziertem Druck konzentriert, um den Rückstand
als ein gelbes Öl
zu erhalten. Der erhaltene Rückstand
wurde TLC ausgesetzt, um 5 Punkte (Rf = 0,8, 0,6, 0,5, 0,2, 0,1)
zu erhalten. Unter ihnen wurde die Titelverbindung mit einem Rf-Wert
von 0,5 durch Silikagel-Säulenchromatographie
(Eluent: n-Hexan-Ethylacetat
= 5/1, v/v) abgetrennt. Der Eluent wurde unter reduziertem Druck
entfernt und der Rückstand
wurde konzentriert, um ein weißes Öl zu erhalten,
welches zwei Stunden lang unter Hochvakuum getrocknet wurde, um die
Titelverbindung als einen gefrorenen weißen Kristall zu erhalten.
Ausbeute:
1,16 g (54,0%)
Schmelzpunkt: 51–53°C
NMR (CDCl3, δ): 2,25 (s,
3H, CH3) 2,35 (s, 3H, CH3),
3,95 (d, 2H, SCH2), 5,25 (dd, 2H, CH2), 6,00 (m, 1H, CH)
-
Beispiel 2
-
Synthese von 1-Chlor-4-allylthio-5,6,7,8-tetrahydrophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurde in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 2,0 g (0,01
mol) 1,4-Dichlor-5,6,7,8-tetrahydrophthalazin, erhalten aus Herstellung
4, hinzugefügt.
Die Umsetzungslösung
wurde eine Stunde lang unter Rückfluß erhitzt
und dann auf die selbe Art und Weise wie Beispiel 1 behandelt, um
die Titelverbindung als einen gefrorenen weißen Kristall zu erhalten.
Ausbeute:
1,81 g (75%)
Schmelzpunkt: 43–45°C
NMR (CDCl3, δ): 1,85–2,65 (m,
2H × 4,
aromatisch), 3,95 (d, 2H, SCH2), 5,25 (dd,
2H, CH2), 6,00 (m, 1H, CH)
-
Beispiel 3
-
Synthese von 1-Chlor-4-allylthiophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,99 g (0,01
mol) 1,4-Dichlorphthalazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als blasse weiße nadelförmige Verbindung
zu erhalten.
Ausbeute: 1,27 g (53,6%)
Schmelzpunkt: 76–78°C
NMR
(CDCl3, δ):
4,15 (d, 2H, SCH2), 5,30 (dd, 2H, CH2), 6,10 (m, 1H, CH), 8,10 (m, 1H × 4, aromatisch)
-
Beispiel 4
-
Synthese von 3-Methoxy-6-Allylthiopyridazin
-
1,15 g (0,05 mol) metallisches Natrium
wurden in 50 ml absolutem Methanol gelöst und dann mit 4,98 ml (0,05
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 7,23 g (0,05
mol) 3-Methoxy-6-chlorpyridazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen blassen
weißen
Kristall zu erhalten.
Ausbeute: 3,99 g (53,7%)
Schmelzpunkt:
25–27°C
NMR
(CDCl3, δ):
3,95 (d, 2H, SCH2), 4,08 (s, 3H, OCH3), 5,25 (dd, 2H, CH2),
6,00 (m, 1H, CH), 6,80–7,30
(dd, 1H × 2,
CH)
-
Beispiel 5,
-
Synthese von 3-Methoxy-4,5-dimethyl-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
lmol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,73 g
(0,01 mol) 3-Methoxy-4,5-dimethyl-6-chlorpyridazin erhalten aus
Herstellung 5 hinzugefügt.
Die Umsetzungslösung
wurde unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als gefrorene blasse weiße Kristalle
zu erhalten.
Ausbeute: 0,67 g (31,9%)
Schmelzpunkt: 51–53°C
-
Beispiel 6
-
Synthese von 1-Methoxy-4-allylthio-5,6,7,8-tetrahydrophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,99 g (0,01
mol) 1-Methoxy-4-Chlor-5,6,7,8-Tetrahydrophthalazin, erhalten aus
Beispiel 6, hinzugefügt.
Die Umsetzungslösung
wurde unter Rückfluß 1 Stunde
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen blassen
weißen
Kristall zu erhalten.
Ausbeute: 1,37 g (58,1%)
Schmelzpunkt:
46–48°C
-
Beispiel 7
-
Synthese von 1-Methoxy-4-allylthiophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,95 g (0,01
mol) 1-Methoxy-4-chlorphthalazin, erhalten aus Herstellung 7 hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 1 Stunde
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen blassen
weißen
Kristall zu erhalten.
Ausbeute: 1,58 g (68,1%)
Schmelzpunkt:
32–34°C
NMR
(CDCl3, δ):
4,08 (d, 2H, SCH2), 4,20 (s, 3H, OCH3) 5, 26 (dd, 2H, CH2),
6,10 (m, 1H, CH), 7,82–8,17
(m, 1H × 4,
aromatisch)
-
Beispiel 8
-
Synthese von 3-Ethoxy-6-allylthipyridazin
-
0,57 g (0,025 mol) metallisches Natrium
wurden in 40 ml absolutem Methanol gelöst und dann mit 2,49 ml (0,025
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 3,96 g (0,025
mol) 3-Methoxy-6-chlorpyridazin, hinzugegeben. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die gleiche Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen blassen
weißen Kristall
zu erhalten.
Ausbeute: 1,85 g (37,7%)
Schmelzpunkt: 28–30°C
NMR
(CDCl3, δ):
1,35 (t, 3H, CH3), 4,45 (q, 2H, OCH2) 5, 15 (dd, 2H, CH2),
5,95 (m, 1H, CH), 6,70–7,15
(dd, 1H × 2,
CH)
-
Beispiel 9
-
Synthese von 3-Ethoxy-4,5-dimethyl-6-allylthipyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 100 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,87 g (0,01
mol) 3-Ethoxy-4,5-dimethyl-6-chlorpyridazin, erhalten aus Beispiel
8, hinzugefügt.
Die Umsetzungslösung
wurde unter Rückfluß 3 Stunden
lang erhitzt und dann auf die gleiche Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen weißen Kristall
zu erhalten.
Ausbeute: 0,15 g (6,7%)
Schmelzpunkt: 29–31°C
-
Beispiel 10
-
Synthese von 1-Ethoxy-4-allylthio-5,6,7,8-tetrahydrophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 100 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 2,13 g (0,01
mol) 1-Ethoxy-4-chlor-5,6,7,8-tetrahydrophthalazin, erhalten aus
Herstellung 9 hinzugefügt.
Die Umsetzungslösung
wurde unter Rückfluß 3 Stunden
lang erhitzt und dann auf die gleiche Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen gefrorenen weißen Kristall
zu erhalten.
Ausbeute: 0,36 g (14,4%)
Schmelzpunkt: 39–41°C
-
Beispiel 11
-
Synthese von 1-Ethoxy-4-allylthiophthalazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 100 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 2,09 g (0,01
mol) 1-Ethoxy-4-chlorphthalazin, erhalten aus Herstellung 10, hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 3 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als gefrorene weiße Kristalle
zu erhalten.
Ausbeute: 0,79 g (32,1%)
Schmelzpunkt: 44–46°C
-
Beispiel 12
-
Synthese von 3-(n-Propoxy)-6-allylthiopyridazin
-
1,15 g (0,05 mol) metallisches Natrium
wurden in 75 ml absolutem Methanol gelöst und dann mit 4,98 ml (0,05
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 8,63 g (0,05
mol) 3-(n-Propoxy)-6-chlorpyridazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als einen blaß-gelben öligen Rückstand zu erhalten.
Ausbeute:
3,11 g (29,6%)
NMR (CDCl3, δ): 1,10 (t,
3H, CH3), 1,82 (q, 2H, CH2)
3,95 (d, 2H, SCH2), 4,40 (q, 2H, OCH2), 5,25 (dd, 2H, CH2),
6,00 (m, 1H, CH), 6,75–7,30
(dd, 1H × 2,
CH)
-
Beispiel 13
-
Synthese von 3-(2-Propoxy)-6-allylthiopyridazin
-
2,30 g (0,1 mol) metallisches Natrium
wurden in 150 ml absolutem Methanol gelöst und dann mit 9,28 ml (0,1
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 17,26 g
(0,1 mol) 3-(2-Propoxy)-6-chlorpyridazin hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blaß-gelbes Öl zu erhalten.
Ausbeute:
4,39 g (20,9%)
NMR (CDCl3, δ): 1,35 (d,
3H × 2,
CH(CH3)2), 3,90
(d, 2H, SCH2) 5,20 (dd, 2H, CH2),
5,45 (m, 1H, OCH), 5,95 (m, 1H, CH), 6,70–7,20 (dd, 1H × 2, CH)
-
Beispiel 14
-
Synthese von 3-(n-Butoxy)-6-allylthiopyridazin
-
1,15 g (0,05 mol) metallisches Natrium
wurden in 75 ml absolutem Methanol gelöst und dann mit 4,98 ml (0,05
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 9,33 g (0,05
mol) 3-(n-Butoxy)-6-chlorpyridazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blaß-gelbes Öl zu erhalten.
Ausbeute:
2,24 g (20,2%)
NMR (CDCl3, δ): 0,90 (t,
3H, CH3), 1,42 (q, 2H, CH2)
1,75 (q, 2H, CH2), 3,85 (d, 1H, SCH2), 4,40 (t, 2H, OCH2), 5,18
(dd, 2H, CH2), 5,18 (dd, 2H, CH2),
6,00 (m, 1H, CH), 6,70–7,20
(dd, 1H × 2,
CH)
-
Beispiel 15
-
Synthese von 3-(2-Butoxy)-6-allylthiopyridazin
-
0,92 g (0,04 mol) metallisches Natrium
wurden in 120 ml absolutem Methanol gelöst und dann mit 3,98 ml (0,04
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 7,47 g (0,04
mol) 3-(2-Butoxy)-6-chlorpyridazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blaß-gelbes Öl zu erhalten.
Ausbeute:
1,19 g (13,3%)
NMR (CDCl3, δ): 0,90 (t,
3H, CH3), 1,30 (d, 3H, CH3)
1,70 (m, 2H, CH2), 3,90 (d, 2H, SCH2), 5,15 (dd, 2H, CH2), 5,30
(m, 1H, OCH2), 5,98 (m, 1H, CH), 6,70–7,15 (dd,
1H × 2,
CH)
-
Beispiel 16
-
Synthese von 3-(t-Butoxy)-6-allylthiopyridazin
-
0,46 g (0,02 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 1,99 ml (0,02
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 3,57 g (0,02
mol) 3-(t-Butoxy)-6-chlorpyridazin hinzugefügt. Die Umsetzungslösung wurde
unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blaß-gelbes Öl zu erhalten.
Ausbeute:
1,13 g (25,1%)
NMR (CDCl3, δ): 1,60 (s,
3H × 3,
C (CH3)3), 3,92
(d, 2H, SCH2) 5,20 (dd, 2H, CH2),
6,00 (m, 1H, CH), 6,65–7,20 (dd,
1H × 2,
CH)
-
Beispiel 17
-
Synthese von 3-(n-Pentyloxy)-6-allylthiopyridazin
-
1,15 g (0,05 mol) metallisches Natrium
wurden in 75 ml absolutem Methanol gelöst und dann mit 4,98 ml (0,05
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 10,03 g
(0,05 mol) 3-(n-Pentyloxy)-6-chlorpyridazin hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als blaß-gelbe nadelförmige Kristalle
zu erhalten.
Ausbeute: 4,21 g (35,3%)
Schmelzpunkt: 39–42°C
NMR
(CDCl3, δ):
0,95 (t, 3H, CH3), 1,40 (t, 2H × 2, CH2) 1,80 (q, 2H, CH2),
3,95 (d, 2H, SCH2), 4,45 (m, 2H, OCH2), 5,22 (dd, 2H, CH2)
6,00 (m, 1H, CH), 6,75–7,30
(m, 1H × 2,
CH)
-
Beispiel 18
-
Synthese von 3-Isopentaloxy-6-allylthiopyridazin
-
1,15 g (0,05 mol) metallisches Natrium
wurden in 150 ml absolutem Methanol gelöst und dann mit 4,98 ml (0,05
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 10,03 g
(0,05 mol) 3-Isopentyloxy-6-chlorpyridazin hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blaß-gelbes Öl zu erhalten.
Ausbeute:
1,13 g (25,1%)
Schmelzpunkt: 39–42°C
NMR (CDCl3, δ): 0,92 (d,
3H × 2,
C(CH3)2), 1,70 (m,
1H&2H, CHCH2) 3,90 (q, 2H, SCH2),
4,45 (t, 2H, OCH2), 5,15 (dd, 2H, CH2), 5,95 (m, 1H, CH), 6,70–7,20 (dd,
1H × 2,
CH)
-
Beispiel 19
-
Synthese von 3-(2-N,N-dimethylaminoethoxy)-6-allylthiopyridazin
-
0,46 g (0,02 mol) metallisches Natrium
wurden in 30 ml absolutem 2-Dimethylaminoethanol gelöst. Zu der
resultierenden Lösung
wurden 3,73 g (0,02 mol) 3-Chlorallylthiopyridazin hinzugefügt. Die
Umsetzungslösung
wurde 2 Stunden bei Raumtemperatur gerührt und auf die selbe Art und
Weise, wie Beispiel 1, behandelt, um die Titelverbindung als gelbe
nadelförmige
Kristalle zu erhalten.
Ausbeute: 0,25 g (5,2%)
Schmelzpunkt:
36–38°C NMR (CDCl3, δ):
1,88 (d, 3H × 2,
N(CH3)2), 1,95 (d,
2H&2, OCH2CH2) 4,00 (d, 2H, SCH2), 5,25 (dd, 2H, CH2),
6,10 (m, 1H, CH), 6,85–7,20
(m, 1H × 2,
CH)
-
Beispiel 20
-
Synthese von 3-(2-Hydroxyethoxy)-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 1,00 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,75 g (0,01
mol) 3-(2-Hydroxyethoxy)-6-chlorpyridazin hinzugefügt. Die
Umsetzungslösung
wurde unter Rückfluß 24 Stunden
lang erhitzt und dann auf die selbe Art und Weise behandelt, wie
Beispiel 1, um die Titelverbindung als ein blasse nadelförmige Kristalle
zu erhalten.
Ausbeute: 1,24 g (58,5%)
Schmelzpunkt: 38–40°C
NMR
(CDCl3, δ):
3,92 (d, 2H, SCH2), 4,10 (s, 2H&2, OCH2CH2O) 5,22 (dd,
2H, CH2), 6,00 (m, 1H, CH), 6,80– 7,25 (dd,
1H × 2,
CH)
-
Beispiel 21
-
Synthese von 3-Phenoxy-6-allylthiopyridazin
-
0,11 g (0,005 mol) metallisches Natrium
wurden in 30 g trockenem Phenol gelöst. Zu der resultierenden Lösung wurden
0,93 g (0,005 mol) 3-Chlor-6-allylthiopyridazin hinzugefügt, und
dann wurde die Umsetzungslösung
3 Stunden lang bei 160 ± 5°C gerührt. Nachdem
es der Reaktion erlaubt wurde zu stoppen, wurde die Reaktion abgekühlt und
auf einen alkalischen pH-Wert (pH 14) mit 2-N-NaOH eingestellt,
wobei die Kristalle präzipitierten.
Die präzipitierten
Kristalle wurden mit Diethylether extrahiert und dann auf die selbe
Art und Weise wie Beispiel 1, behandelt, um die Titelverbindung
als Kristall zu erhalten.
Ausbeute: 0,42 g (34,5%)
Schmelzpunkt:
71–73°C
NMR
(CDCl3, δ):
3,95 (d, 2H, SCH2), 5,22 (dd, 2H, CH2), 6,00 (m, 1H, CH), 6,80–7,10 (m,
1H × 2,
CH), 7,15–7,45
(m, 1H × 5,
aromatisch)
-
Beispiel 22
-
Synthese von 3-Benzyloxy-6-allylthiopyridazin
-
0,11 g (0,005 mol) metallisches Natrium
wurden in 20 ml trockenem Benzylalkohol gelöst. Zu der resultierenden Lösung wurden
0,93 g (0,005 mol) 3-Chlor-6-Allylthiopyridazin
hinzugefügt,
und dann wurde die Umsetzungslösung
2 Stunden lang bei 80 ± 5°C gerührt. Um überschüssigen Benzylalkohol
und das unlösliche Material (NaCl)
zu entfernen, wurde die Umsetzungsmischung mit Silikagel Säulenchromatographie
auf gereinigt (Eluent: n-Hexan-Ethylacetat
= 40/1, v/v), um die Titelverbindung als nadelförmige Kristalle zu erhalten.
Ausbeute:
0,92 g (71,3%)
Schmelzpunkt: 56–58°C
Umkristallisationslösungsmittel:
Ethanol
NMR (CDCl3, δ): 4,00 (d,
2H, SCH2), 5,25 (dd, 2H, CH2),
5,55 (s, 2H, OCH2), 6,05 (m, 1H, CH), 6,85–7,30 (dd, 1H × 2, CH),
7,35–7,65
(m, 1H × 5,
aromatisch)
-
Beispiel 23
-
Synthese von 3-(4-Methylphenoxy)-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml trockenem p-Cresol gelöst. Zu der resultierenden Lösung wurden
1,87 g (0,01 mol) 3-Chlor-6-Allylthiopyridazin hinzugefügt, und
dann wurde die Umsetzungslösung
5 Stunden lang bei 100 ± 5°C gerührt. Nachdem
es der Umsetzung erlaubt wurde zu stoppen, wurde die Umsetzungslösung abgekühlt und
auf einen alkalischen pH-Wert
(pH 14) mit 150 ml 2-N-NaOH eingestellt, wobei die Kristalle präzipitierten.
Die präzipitierten
Kristalle wurden mit Diethylether extrahiert und zweimal mit aufgereinigtem
Wasser gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet und dann konzentriert, um
Diethylether zu entfernen. Der resultierende ölige Rückstand wurde langsam abgekühlt, um die
Titelverbindung als einen blaß-weißen amorphen
Kristall zu erhalten.
Ausbeute: 1,07 g (41,5%)
Schmelzpunkt:
115–117°C
NMR
(CDCl3, δ):
2,35 (s, 3H, CH3), 3,95 (d, 2H, SCH2), 5,20 (dd, 2H, CH2),
6,00 (m, 1H, CH), 6,98–7,10
(dd, 1H × 2,
CH), 7,18–7,35
(m, 1H × 4,
aromatisch)
-
Beispiel 24
-
Synthese von 3-(3-Methylphenoxy)-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml trockenem m-Cresol gelöst. Zu der resultierenden Lösung wurden
1,87 g (0,01 mol) 3-Chlor-6-allylthiopyridazin hinzugefügt. Die
Reaktionslösung
wurde 5 Stunden lang bei 100 ± 5°C gerührt und
dann auf die selbe Art und Weise, wie Beispiel 23 behandelt, um
die Titelverbindung als blaß-weiße nadelförmige Kristalle
zu erhalten.
Ausbeute: 0,94 g (36,4%)
Schmelzpunkt: 48–50°C
NMR
(CDCl3, δ):
2,30 (s, 3H, CH3), 3,90 (d, 2H, SCH2), 5,15 (dd, 2H, CH2),
5,95 (m, 1H, CH), 6,95 (m, 1H × 2, CH),
7,22 (m, 1H × 4,
aromatisch)
-
Beispiel 25
-
Synthese von 3-(2-Methylphenoxy)-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml trockenem o-Cresol gelöst. Zu der resultierenden Lösung wurden
1,87 g (0,01 mol) 3-Chlor-6-Allylthiopyridazin hinzugefügt. Die
Umsetzungslösung
wurde 5 Stunden lang bei 100 ± 5°C gerührt und
dann auf die selbe Art und Weise, wie Beispiel 23, behandelt, um die
Titelverbindung als blaß-weiße nadelförmige Kristalle
zu erhalten.
Ausbeute: 0,48 g (18,6%)
Schmelzpunkt: 78–80°C
NMR
(CDCl3, δ):
2,20 (s, 3H, CH3), 3,92 (d, 2H, SCH2), 5,20 (dd, 2H, CH2),
6,00 (m, 1H, CH), 7,00–7,35
(m, 1H × 2, & 1H × 4, CH & aromatisch)
-
Beispiel 26
-
Synthese von 3-Phenyl-6-allylthiopyridazin
-
0,23 g (0,01 mol) metallisches Natrium
wurden in 30 ml absolutem Methanol gelöst und dann mit 0,93 ml (0,01
mol) Allylmercaptan gemischt. Zu dieser Mischung wurden 1,90 g (0,01
mol) 6-Chlor-3-phenylpyridazin hinzugefügt. Die Umsetzungslösung wurde
5 Stunden lang unter Rückfluß erhitzt
und unter reduzierten Druck konzentriert, um Methanol zu entfernen.
100 ml Diethylether wurde zu dem Rückstand hinzugegeben und es
wurde 10 Minuten lang heftig gerührt.
Die in Diethylether unlöslichen
Materialien wurden entfernt und die verbleibende Lösung wurde
zweimal mit 50 ml auf gereinigtem Wasser gewaschen, getrocknet über wasserfreiem
Magnesiumsulfat und dann unter reduziertem Druck konzentriert, um
Diethylether zu entfernen. Auf diese Weise wurde die Titelverbindung
als ein weißer
Kristall erhalten.
Ausbeute: 2,18 g (95,6%)
Schmelzpunkt:
96–98°C
NMR
(CDCl3, δ):
4,08 (d, 2H, SCH2), 5,28 (dd, 2H, CH2), 6,08 (m, 1H, CH), 7,48 (m, 1H × 2, CH)
-
Experiment 1
-
Akuter Toxizitätstest
-
Der akute Toxizitätstest wurde ausgeführt, um
die Toxizität,
welche innerhalb einer kurzen Zeitperiode nach einer einfachen Verabreichung
der Testverbindung am Experimentiertier auftritt, qualitativ und
quantitativ zu untersuchen.
-
Bei diesem Experiment wurde der akute
Toxizitätstest
ausgeführt
unter Verwendung von 5–6
Wochen alten ICR männlichen
Mäusen,
erhalten vom Korea Experimental Animal Center. Die Mäuse fasteten
von 6 Uhr abends am Tag vor der Verabreichung der Testverbindung
bis 9 Uhr morgens am Tag, an dem die Testverbindung verabreicht
wird. Die Testverbindung wurde gemahlen und in Maisöl suspendiert,
und dann in der Dosis von 1000–6000
mg/kg dem Experimentiertier verabreicht. Für jede Dosis wurden 6 Tiere
verwendet. Die Testverbindung wurde an einem Tag hergestellt und
verwendet und wurde einmal auf oralem Wege mittels einer Nadel verabreicht.
Nachdem die Verbindung von Beispiel 4 als typisches Beispiel der
erfindungsgemäßen Verbindung
verabreicht wurde, wurden die überlebenden
und gestorbenen Tiere 14 Tage beobachtet. Das Resultat davon ist
in der folgenden Tabelle 1 gezeigt. Entsprechend der Analyse dieses
Ergebnisses durch das Litchfield-Wilcoxon-Verfahren wurde gezeigt,
daß der
LD50-Wert der Verbindung 3,95 g/kg beträgt (95%
Vertrauensintervall: 3,26–4,78
g/kg).
-
-
Experiment 2
-
Wirkung der erfindungsgemäßen Verbindung
auf die Expressionsrate von mEH und GST
-
Um die Wirkung des erfindungsgemäßen Allylthiopyridazinderivates
auf die Eypressionsrate von mEH und GST zu bestätigen, wurde die entsprechende
Verbindung oral 5–6
Wochen an SD männliche
Ratte in der Dosis von 100 mg/kg verabreicht. Nachdem 24 Stunden
seit der Verabreichung vergangen waren, wurden die mRNA Mengen für mEH und
GSTA2, die im Lebergewebe produziert wurden gemäß der Northern Blot Analyse quantifiziert.
Die erhaltenen Resultate sind in 1 und 2 hinsichtlich des relativen
Anstiegs um ein Mehrfaches im Vergleich zu der nicht behandelten
Gruppe dargestellt. Aus 1 und 2 kann geschlossen werden, daß alle Testverbindungen
zweifache oder größere Anstiege
in der Expressionsrate zeigen, und einige Verbindungen zeigen einen
außergewöhnlichen
Anstieg in der Expressionsrate.
-
Experiment 3
-
Wirkung des Inhibierens
von hepatitischen
-
Fehlzuständen, induziert durch Kohlenstofftetrachlorid
Der schützende
Effekt für
die Leber der erfindungsgemäßen Verbindung
wurde untersucht nach dem bekannten Verfahren, wie spezifisch hierunter
beschrieben [siehe Philippe Letteron et al., Biochemical Pharmacology,
39, 12, 2027–2034,
1990].
-
Die erfindungsgemäße Verbindung wurde in Maisöl suspendiert,
und die resultierende Suspension wurde in einer Menge von 1,0 mg/kg
verabreicht. Jede Gruppe war aus 3–10 männlichen ICR-Mäusen (5–10 Wochen)
zusammengesetzt, und die Testverbindung wurde oral 3 Tage lang in
der Dosis von 50 mg/kg pro Tag verabreicht. Nachdem die Testverbindung
verabreicht wurde, wurde Kohlenstofftetrachlorid intraperitoneal
in einer Menge von 100 μl/kg
injiziert. Bei diesem Experiment wurde Silymarin als Vergleichsverbindung
verwendet. Nach 24 Stunden seit der Verabreichung von Kohlenstofftetrachlorid
wurde Blut aus dem Testtier durch Kardialperforation entnommen und
ein ALT-Wert im Serum wurde bestimmt. Das erhaltene Ergebnis ist
in der folgenden Tabelle 2 beschrieben (in welcher der ALT-Wert durch
den Mittelwert ± Standartabweichung
dargestellt ist). Die ALT-Einheit im Serum wurde bestimmt durch
Zentrifugieren des Blutes, um Serum als Überstand abzutrennen, und dann
wurde das abgetrennte Serum dem Reitman-Frankel-Verfahren mittels
eines kommerziell erhältlichen
Kits (BC101-O,P, Youngdong Pharm.) ausgesetzt. D. h. DL-Aalanin
und α-Ketoglutarat
wurden als Substratlösung
verwendet, zu welcher 2,4-Dinitrophenylhydrazin
hinzugegeben wurde, um Hydrazon herzustellen. Dann wurde Natriumhydroxyd
zum Hydrazon zur Farbgebung hinzugegeben und die Absorbtion wurde
bei 505 nm gemessen.
-
Zusätzlich wurde die Leber aus
den Testtieren entfernt, gefärbt
(× 100)
mit Hematoxylin und Eosin und dann mittels eines Mikroskops beobachtet.
Der Grad der Hepatozytennekrose wurde gemäß den folgenden 5 Graden evaluiert:
0 keine Läsion,
1 milde Läsion,
2 moderate Läsion,
3 schwere Läsion
und 4 diffuse Läsion. Das
Resultat, wie erhalten, ist beschrieben in der folgenden Tabelle
3. Gemäß der statistischen
Analyse der Resultate, beschrieben in Tabellen 2 und 3 kann erkannt
werden, daß nur
die nicht-behandelte Gruppe und die Gruppe, die mit der Verbindung
aus Beispiel 4 behandelt wurden, signifikant von mit der CCl4 behandelten Gruppe abweicht (p < 0,01).
-
-
-
-
Experiment 4
-
Wirkung des Inhibierens
von hepatitischen Fehlzuständen,
induziert durch Acetaminophen (APAP)
-
Der leberschützende Effekt der Verbindung
gemäß der vorliegenden
Erfindung wurde untersucht entsprechend bekannter Verfahren, wie
spezifisch hierunter beschrieben [siehe Wang et al., Toxicology
and applied Pharmacology, 136, 146–154, 1996].
-
Die erfindungsgemäße Verbindung wurde in Maisöl suspendiert
und die resultierende Suspension wurde in einer Menge von 1,0 mg/kg
verabreicht. Jede Gruppe war aus 3–10 männlichen ICR-Mäusen (5–6 Wochen)
zusammengesetzt, und die Testverbindung wurde oral 3 Tage lang in
der Dosis von 50 mg/kg pro Tag verabreicht. Nach 2 Stunden ab der
letzten Verabreichung der Testverbindung wurde Acetaminophen oral
in einer Menge von 0,5 g/kg verabreicht. Bei diesem Experiment wurde
Silymarin als Vergleichsverbindung verwendet. Nach 22 Stunden ab
der Verabreichung von Acetaminophen wurde Blut aus dem individuellen
Testtier entnommen und ALT- und LDH-Werte im Serum wurden bestimmt.
Die Resultate, wie erhalten, sind in den folgenden Tabellen 4 und
5 beschrieben (in welchen ALT und LDH-Werte dargestellt sind als
Mittelwert ± Standartabweichung).
Zusätzlich
wurde die Leber aus den Testtieren entfernt und dann auf die selbe
Art und Weise, wie in Experiment 3 beobachtet, um dem Grad der Hepatozytennekrose
zu evaluieren. Das Resultat, wie erhalten ist beschrieben in der
folgenden Tabelle 6. Gemäß der statistischen
Analyse der Resultate, die in Tabellen 4–6 beschrieben sind, konnte
erkannt werden, daß nur
die nicht behandelte Gruppe und die Gruppen, die mit den Verbindungen
der Beispiele 4 und 8 behandelt wurden, signifikant von der APAP
behandelten Gruppe abwich (p < 0,01).
-
-
-
-
-
Experiment 5
-
Schützender Effekt gegen Strahlungsschaden
-
Um den schützenden Effekt der erfindungsgemäßen Verbindung
gegen Strahlungsschaden zu bestätigen,
wurde der folgende Test ausgeführt.
-
Männliche
ICR-Mäuse
wurden in einer Kammer gezogen, in der eine sterile Atmosphäre zur Verfügung gestellt
wird, bei einer inneren Temperatur von 20–23°C und relativer Luftfeuchtigkeit
von 50% mit ausreichend Wasser und Futtermitteln. Als das Gewicht
der Maus 20–25
g erreichte, wurde die erfindungsgemäße Verbindung in Maisöl suspendiert
und dann 2 Tage lang in der Dosis von 100 mg/kg (1 mg/kg) pro Tag
verabreicht. Nach 3 Stunden seit der letzten Verabreichung der Testverbindung
wurde die Maus mit 8 oder 9 Gy Strahlung bestrahlt. Spezifisch wurde
die Maus in eine Kiste eingeführt,
die aus Acryl gefertigt war, welche ihre Bewegung limitierte; und
dann wurde der gesamte Körper
mit Strahlung, die mit 60CO radioaktivem
Material produziert wurde, bei einer Bestrahlungsrate von 115,8
cGy/min bestrahlt. Die Dimension der Acrylbox war 31 × 31 cm2, welches der mit der Strahlung betstrahlten
Fläche
entspricht. 7 Tage nach der Bestrahlung mit γ-Strahlen wurde die Maus geopfert,
um histopathologische Beobachtungen auszuführen. Die Resultate des Untersuchens
der Überlebensrate
während
30-tägiger Bestrahlung
mit 8 Gy Bestrahlung ist in 3 gezeigt, und
das Ergebnis des Bestimmens der Überlebensrate
nach einer gewissen Zeitperiode (10, 13 oder 30 Tage) durch die
Bestrahlung mit 8 oder 9 Gy Strahlung ist in der folgenden Tabelle
7 beschrieben.
-
-

 wobei X Chlor repräsentiert, mit einer Verbindung der Formel (V):
wobei X Chlor repräsentiert, mit einer Verbindung der Formel (V): herzustellen, wobei R1' wie oben definiert ist.
herzustellen, wobei R1' wie oben definiert ist.












 wobei R1, R2 und R3 wie oben definiert sind und X für Chlor steht, umgesetzt wird mit einer Verbindung der Formel (III)
wobei R1, R2 und R3 wie oben definiert sind und X für Chlor steht, umgesetzt wird mit einer Verbindung der Formel (III) wobei X für Chlor steht, umgesetzt wird mit einer Verbindung der Formel (V):
wobei X für Chlor steht, umgesetzt wird mit einer Verbindung der Formel (V): wobei R1' wie oben definiert ist.
wobei R1' wie oben definiert ist.
