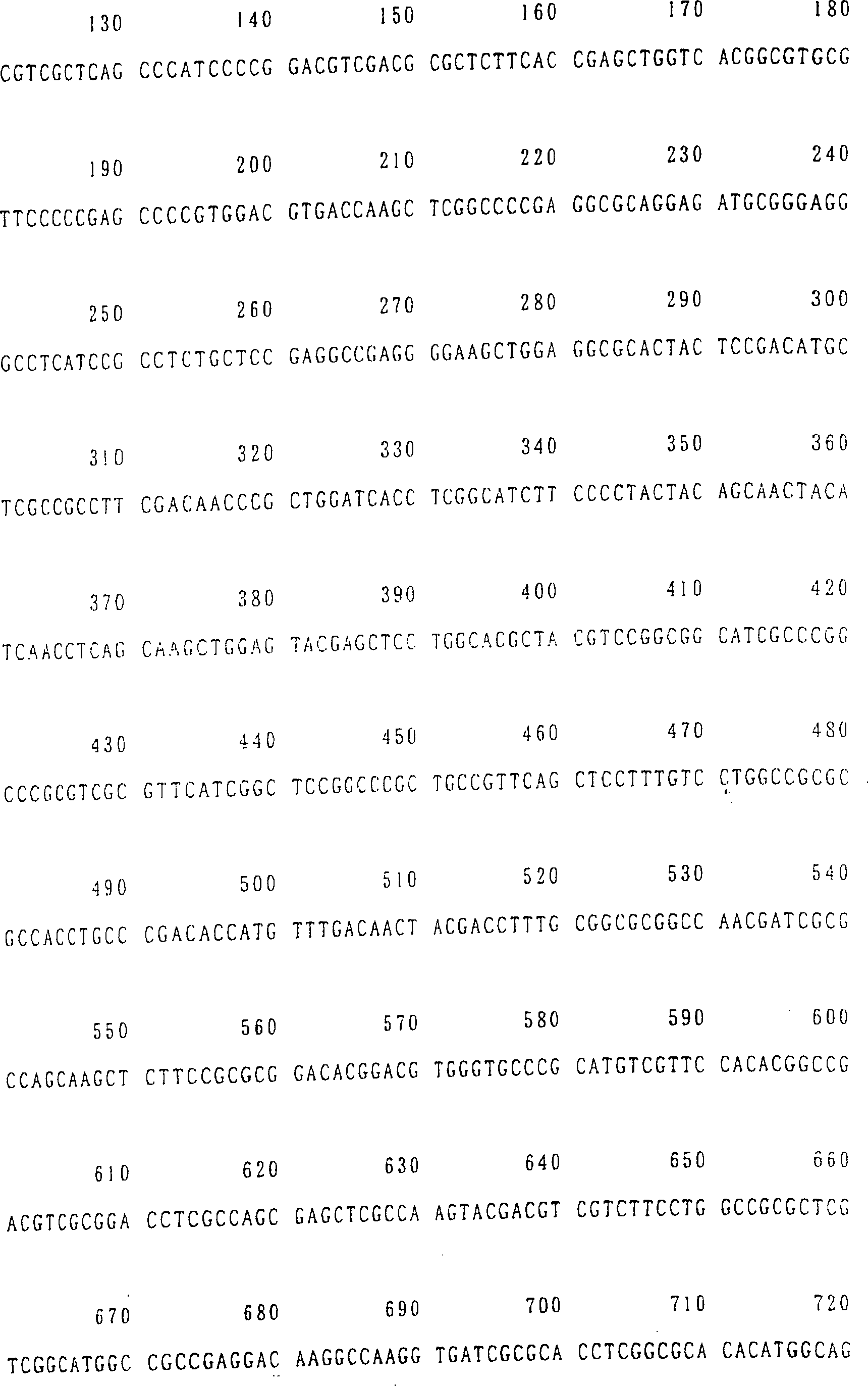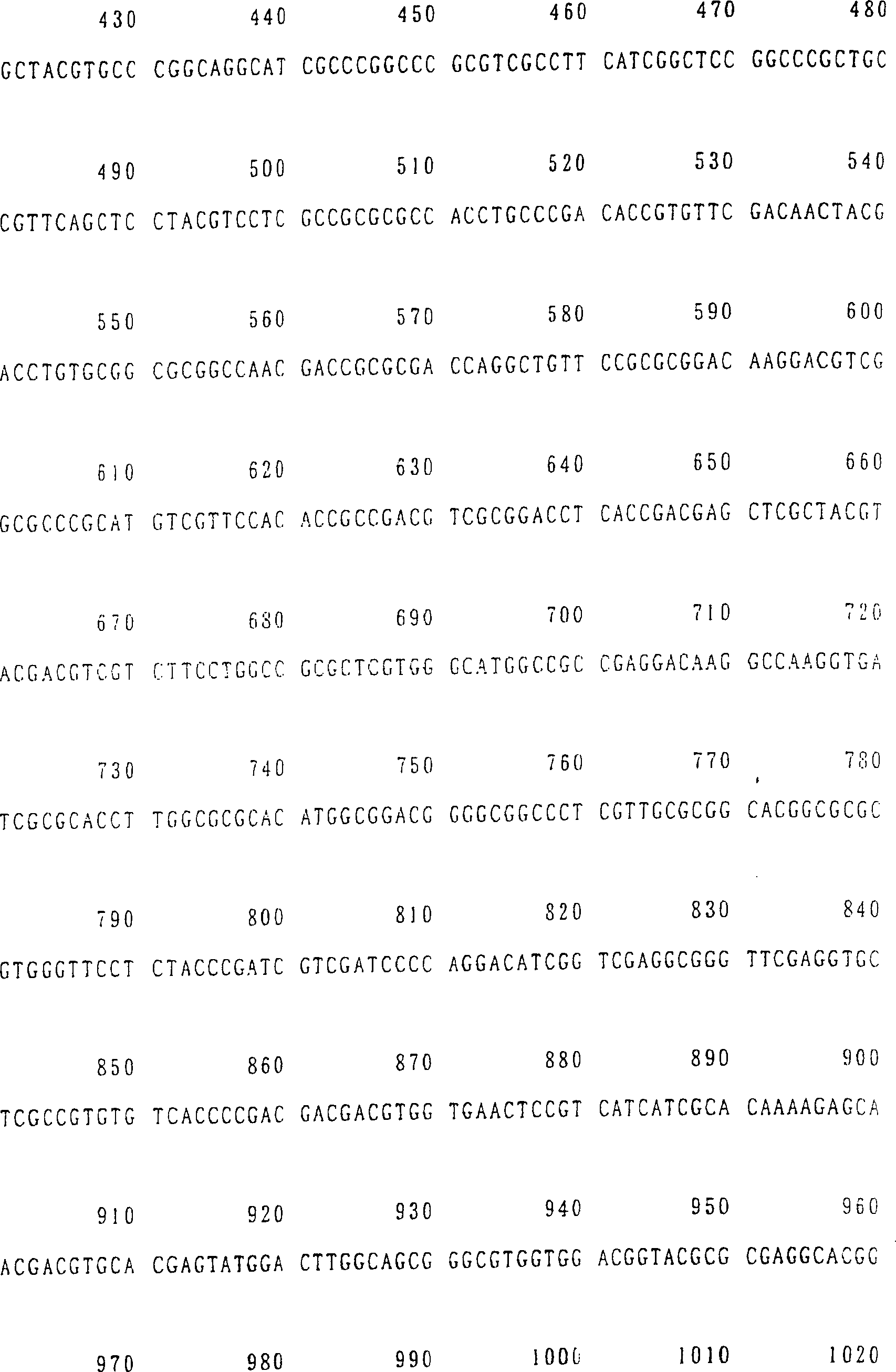-
Fachgebiet
-
Die
vorliegende Erfindung betrifft eine Nicotianaminsynthase, die am
Biosyntheseweg von Muginsäure beteiligt
ist, außerdem
deren Aminosäuresequenz,
ein Gen, das dieselbe codiert, einen Vektor, einen Prozess zur Herstellung
des Nicotianamins unter Verwendung derselben, Transformanden, wie
z.B. Pflanzen, die durch das Gen transformiert sind, das die Nicotianaminsynthase
codiert, als auch einen Antikörper
gegen die Nicotianaminsynthase.
-
Hintergrund
-
Gramineen-Pflanzen,
die durch Chelatieren des Fe(III) im unlöslichen Zustand im Boden mithilfe
der Muginsäure
absorbieren und den sogenannten Mechanismus der 2. Strategie zur
Fe-Anreicherung anwenden, sekretieren Fe-Chelatoren (Phytosiderophore)
aus ihren Wurzeln, um das kaum lösliche
Fe in der Rhizosphäre anzulösen (Roemheld,
1987). Die Menge an sekretierten Phytosiderophoren steigt unter
Fe-Defizienz-Stress. Die
Muginsäure-Familie
stellt das einzige bisher bekannte Beispiel von Phytosiderophoren
dar (Takagi, 1976). Die Toleranz gegenüber einer Fe-Defizienz in Gramineen
wird als von einer bestimmten Menge der Muginsäure-Familie, die von Pflanzen
sekretiert wird, abhängig
erachtet (Takagi et al. 1984, Roemheld und Marschner 1986, Marschner
et al. 1987, Mori et a. 1987, Kawai et al. 1988, Mori et al. 1988,
Mihasi und Mori 1989 und Shingh et al. 1993).
-
Der
Biosyntheseweg der Muginsäure
in Pflanzen ist in 1 gezeigt. S-Adenosylmethionin
wird aus Methionin durch die S-Adenosylmethioninsynthase synthetisiert.
Daraufhin werden drei Moleküle
des S-Adenosylmethionins zur Bildung eines Moleküls des Nicotianamins durch
die Nicotianaminsynthase kombiniert. Das erzeugte Nicotianamin wird
dann zu 3''-Ketosäure durch
Nicotianaminaminotransferase umgewandelt und dann 2'-Desoxymuginsäure durch
die anschließende
Wirkung einer Reduktase synthetisiert. Eine weitere Reihe von Hydroxylierungsschritten
erzeugt die anderen Muginsäure-Derivate
einschließlich
der Muginsäure
aus der Desoxymuginsäure
(Mori und Nishizawa 1987, Shojima et al. 1989, Shojima et al. 1990
und Ma und Nomoto 1993).
-
Eine
Verbindung in 1, die Verbindung in der unteren
rechten Hälfte,
worin R1 und R2 Wasserstoff sind
und R3 Hydroxyl ist, ist Muginsäure. Eine
Verbindung, worin R1 Wasserstoff ist und
R2 und R3 Hydroxyl sind,
ist 3-Hydroxymuginsäure.
Außerdem
ist eine Verbindung, worin R2 Wasserstoff
ist und R1 und R3 Hydroxyl sind,
3-Epihydroxymuginsäure.
-
Drei
S-Adenosylmethioninsynthase-Gene wurden aus Gerstenwurzeln isoliert,
doch wurden diese Gene durch eine Fe-Defizienz nicht induziert (Takizawa
et al. 1996). Ein Gen, Ids3, welches aus der Gerste durch Differentialscreening
erhalten wird, wird als ein Gen vermutet, welches Desoxymuginsäure durch
Hydroxylierung zu Muginsäure
umwandelt und das durch eine Fe-Defizienz stark induziert wird (Nakanishi
et al. 1993). Weiterhin wurde Nicotianaminaminotransferase aus Fe-defizienten
Gerstenwurzeln ausgereinigt und isoliert, und es wurden zwei Nicotianaminaminotransferase-Gene,
Naat-A und Naat-B, isoliert (Takahashi et al. 1997). Die Naat-A-Expression
wurde in Fe-defizienten Wurzeln induziert.
-
Die
Synthese von Nicotianamin aus S-Adenosylmethionin ist ähnlich der
Polyamin-Synthese
aus Decarboxy-S-Adenosylmethionin. Im Gegensatz zur Polyaminsynthase
katalysiert die Nicotianaminsynthase jedoch eine Kombination aus
drei S-Adenosylmethionin-Molekülen
und zugleich die Azetidin-Ringbildung (1). Daher
handelt es sich bei der Nicotianaminsynthase um einen neuartigen
Typ von Enzym. Zuvor haben wir von der teilweisen Ausreinigung von
Nicotianaminsynthase aus den Wurzeln der Fe-defizienten Gerste und
dem Expressionsmustern der Aktivität berichtet (Higuchi et al.
1994, Higuchi et al. 1995, Kanazawa et al. 1995, Higuchi et al.
1996a und Higuchi et al. 1996b). Da sich Nicotianaminsynthase während der
Extraktion und Ausreinigung leicht zersetzt, war es schwierig, ausreichende
Mengen für
die Aminosäure-Sequenzierung zu
reinigen.
-
Die
vorliegende Erfindung ermöglicht
die Erzeugung einer Pflanze, insbesondere einer Gramineen-Pflanze,
mit einer hochgradigen Toleranz gegenüber einem Fe-Defizienz, als
Ergebnis der Isolierung und Aufreinigung einer Nicotianaminsynthase,
das Klonieren des Gens dieses Enzyms, das Bestimmen seiner Basensequenz
und Aminosäuresequenz
und das Verwenden des Enzyms.
-
Beschreibung der Erfindung
-
Die
vorliegende Erfindung betrifft ein isoliertes Enzym, das eine Nicotianaminsynthase-Aktivität aufweist
und eine Aminosäuresequenz
umfasst, ausgewählt
aus folgendem:
- (a) SEQ ID NR. 1, oder
- (b) einer Sequenz, die homolog zu SEQ ID NR. 1 ist, vorzugsweise
ein Enzym, das aus Gerste stammt (und bevorzugter ein Enzym mit
der Aminosäuresequenz
der SEQ ID NR. 1, 3, 5, 7, 9, 11 oder 13) oder ein Enzym, das aus
Oryza sativa stammt (und bevorzugter ein Enzym mit der Aminosäuresequenz
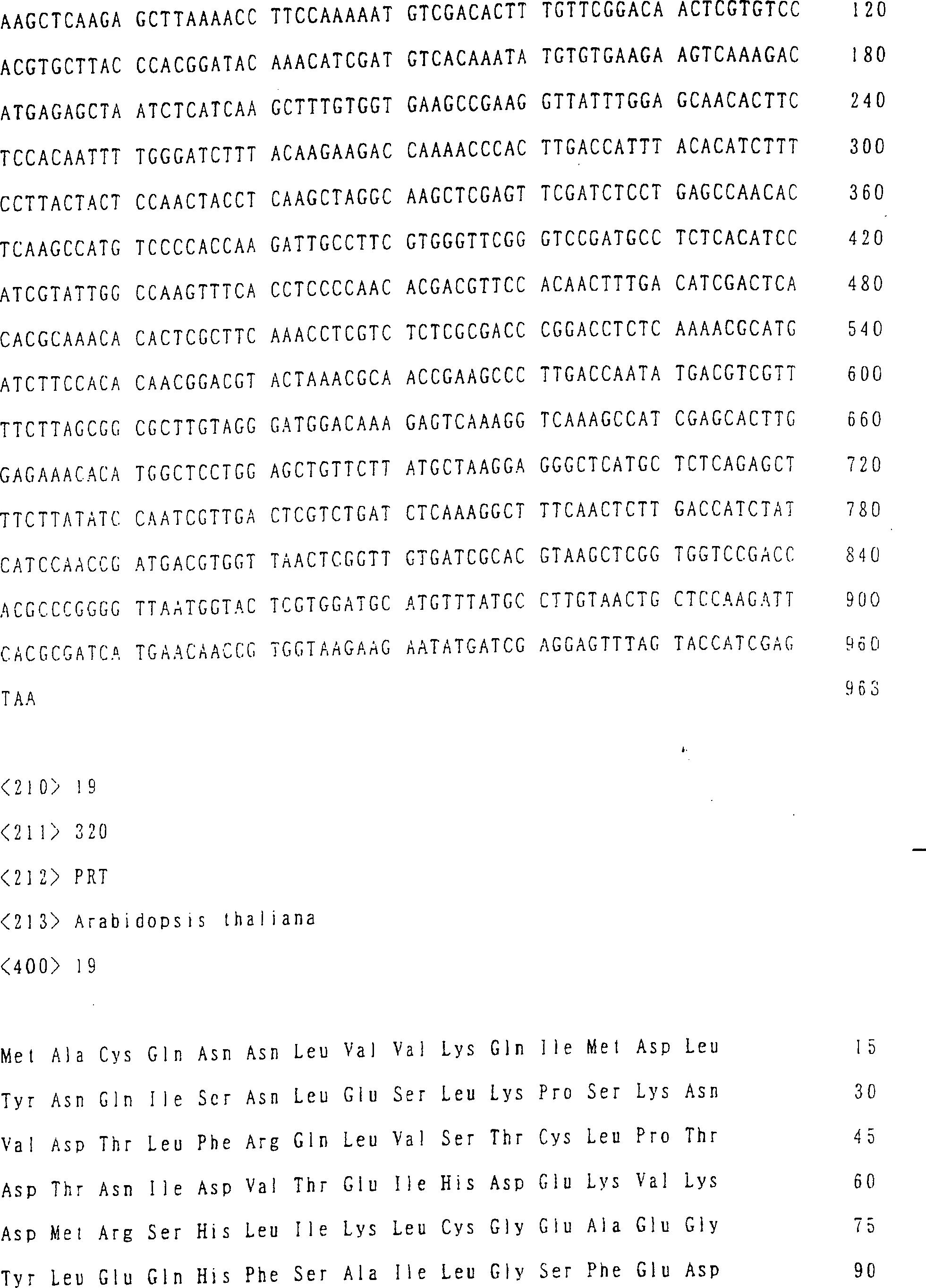
der SEQ ID NR. 15) oder ein Enzym, das aus Arabidopsis stammt (und
bevorzugter ein Enzym der Aminosäuresequenz der
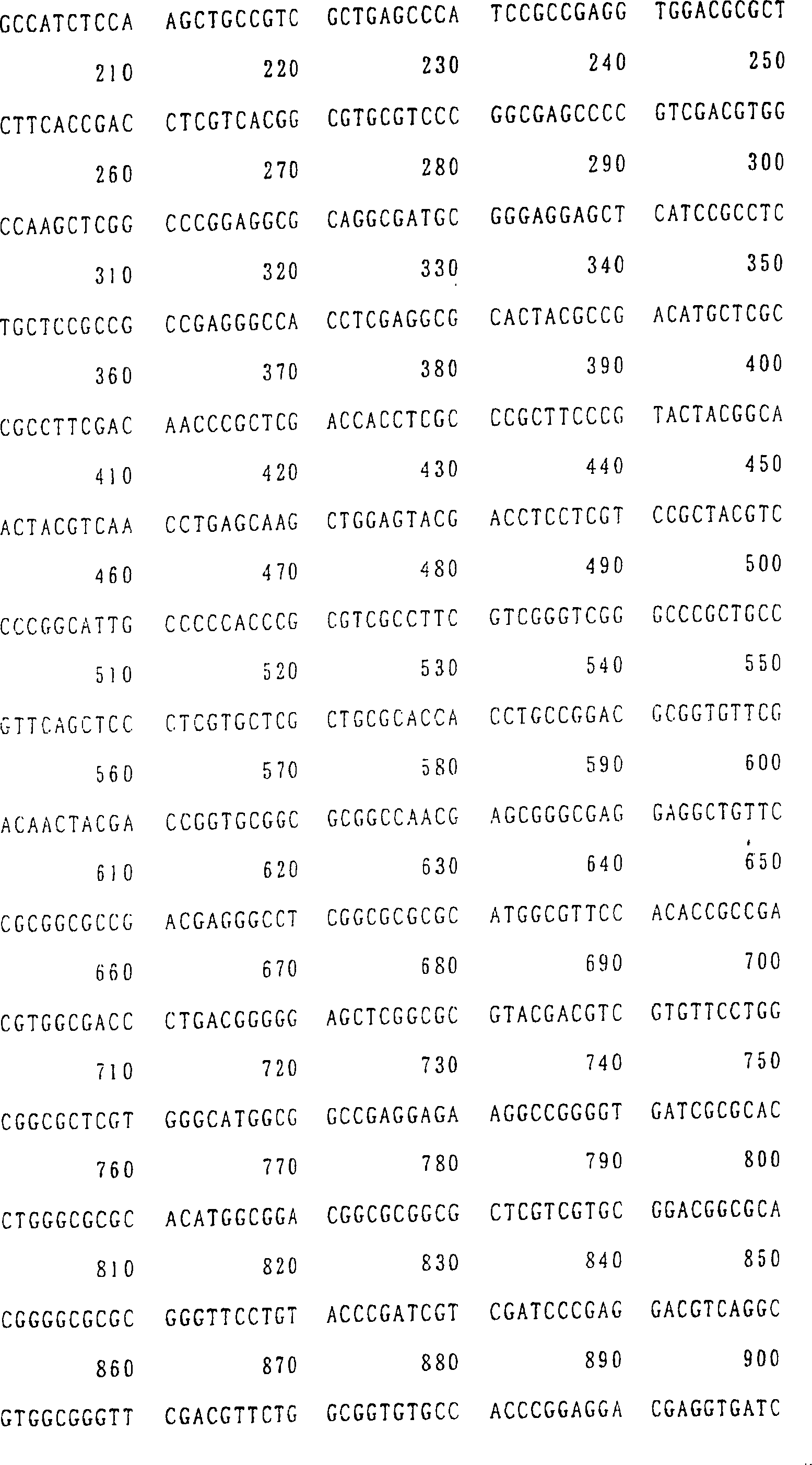
SEQ ID NR. 17, 19 oder 21).
-
Die
vorliegende Erfindung stellt außerdem
ein Gen bereit, das die Aminosäuresequenz
des Enzyms codiert, oder eine Nukleinsäure, die dieses Enzym codiert,
welches Nicotianaminsynthase-Aktivität aufweist, wobei die Nukleinsäure eine
Sequenz umfasst, ausgewählt
aus folgendem:
- (A) SEQ ID NR. 2
- (B) einer Sequenz, die homolog zu SEQ ID NR. 2 ist, vorzugsweise
eine Nukleinsäure,
die aus Gerste stammt (bevorzugter eine Nukleinsäure mit der Basensequenz der
SEQ ID NR. 2, 4, 6, 8, 10, 12 oder 14); oder eine Nukleinsäure, die
aus Oryza sativa stammt (bevorzugter eine Nukleinsäure mit
der Basensequenz der SEQ ID NR. 16) oder eine Nukleinsäure, die
aus Arabidopsis stammt (bevorzugter eine Nukleinsäure mit
der Basensequenz der SEQ ID NR. 18, 20 oder 22) und nicht bevorzugt,
dass die Nukleinsäure DNA
ist.
-
Die
vorliegende Erfindung stellt außerdem
einen Vektor bereit, der eine Nukleinsäure umfasst, wie oben definiert,
vorzugsweise eine Expressionsvektor, und einen nichthumanen Transformanden,
der durch die Vektoren transformiert ist, worin das Fremdgen vorzugsweise
ein Gen mit der Basensequenz der SEQ ID NR. 2, 4, 6, 8, 10, 12,
14, 16, 18, 20 oder 22 ist.
-
Bei
anderen Aspekten stellt die Erfindung ein Verfahren zur Erzeugung
eines Nicotianamins bereit, welches die Verwendung des obigen Transformanden
umfasst, eines Antikörpers
gegen die Enzyme, wie oben angegeben, vorzugsweise eines polyklonalen
Antikörpers
oder eines monoklonalen Antikörpers,
und ein Verfahren zur Aufreinigung der Nicotianaminsynthase, welches
umfasst: die Verwendung eines Thiolprotease-Hemmers, die Verwendung von CHAPS-Puffer,
die Verwendung der Ionenaustausch-Chromatographieträger DEAE-Sepharose FF® oder
DEAE Sephacel®,
die Verwendung des hydrophoben Chromatographieträgers TSK-Gel Butyl Toyopearl® und
die Verwendung des hydrophoben Chromatographieträgers TSK-Gel Ether Toyopearl®,
wobein der Thiolprotease-Hemmer vorzugsweise E-64 ist.
-
Kurze Beschreibung der
Zeichnungen
-
1 zeigt
den Biosyntheseweg der Muginsäure-Familie.
-
2 zeigt
einen Vergleich der Ausreinigung der Nicotianaminsynthase aus Feabhängigen und
aus Kontroll-Gerstenwurzeln.
-
3 zeigt
die präparative
SDS-PAGE (Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese, im folgenden bezeichnet
als SDS-PAGE) um 30 – 35
kDa. Die horizontalen Balken zeigen die relative Enzymaktivität, wie aus
den Gelen nachgewiesen.
-
4 zeigt
das Elutionsmuster der Nicotianaminsynthase-Aktivität aus der
Gelfiltrationssäule.
-
Die
großen
ausgefüllten
Kreise (•)
zeigen die Enzymaktivität.
-
5 zeigt
einen Vergleich mit einer Teilsequenz von sechs Aminosäuren, wie
bestimmt anhand von Nicotianaminsynthase, die aus Gerste stammt,
und einer ähnlichen
Sequenz von Gramineen, wie durch Computerdurchsuche der Datenbank
erhalten. Ein identischer Aminosäure-Rest
ist in ":" gezeigt.
-
6 zeigt
eine HvNAS1-cDNA der vollen Länge
und die daraus abgeleitete Aminosäuresequenz. Die unterstrichenen
Sequenzen geben die identischen partiellen Amino säuresequenzen
der in obiger 5 gezeigten Fragmente an. Die
Zahlen der Nukleotidsequenzen sind rechts jeder Reihe angegeben.
Die Zahlen der Aminosäuren
sind links jeder Reihe angegeben.
-
7 zeigt
einen Vergleich der abgeleiteten Aminosäuresequenzen der obigen 7 cDNAs,
die aus Gerste erhalten wurden. Sternchen "*" zeigt
identische Aminosäure-Reste in allen Sequenzen
auf.
-
8 zeigt
die Ergebnisse der dünnschichtchromatographischen
(TLC) Analyse der Nicotianaminsynthase-Aktivität, wie aus E. coli-Rohextrakt
erhalten, die ein Fusionsprotein mit dem Maltose-Bindungsprotein-HvNAS1-exprimiert.
-
9 zeigt
die Northern-Hybridisationsanalyse von HvNAS1 als einer Sonde.
-
10 zeigt
die Southern-Hybridisationsanalyse von HvNAS1 als einer Sonde.
-
11 zeigt
die Western-Blot-Analyse des Rohenzyms, wie zum Nachweis der Nicotianaminsynthase-Aktivität verwendet.
-
12 zeigt
die Western-Blot-Analyse des durch Trichloressigsäure/Aceton
extrahierten Gesamtproteins.
-
13 zeigt
einen Vergleich der Ausreinigung von Nicotianaminsynthase aus Fedefizienter
Gerste und Kontroll-Gerste nach DEAE-Sepharose FF.
-
14 zeigt
einen Vergleich der Ausreinigung von Nicotianaminsynthase aus Fedefizienter
Gerste und Kontroll-Gerste nach Ether Toyopearl* 650M (* eingetragenes
Markenzeichen).
-
15 zeigt
die Ergebnisse der dünnschichtchromatographischen
(TLC) Analyse der Nicotianaminsynthase-Aktivität, wie aus E. coli-Rohextrakt
erhalten, die ein Fusionsprotein mit dem Maltose-Bindungsprotein-OsNAS1-exprimiert.
-
16 zeigt
die Northern-Hybridisationsanalyse von OsNAS1 als einer Sonde.
-
17 zeigt
die Ergebnisse der dünnschichtchromatographischen
(TLC) Analyse der Nicotianaminsynthase-Aktivität, wie aus E. coli-Rohextrakt
erhalten, die Fusionsproteine mit dem Maltose-Bindungsprotein-AtNAS1,
AtNAS2 oder AtNAS3-exprimiert.
-
18 zeigt
die Ergebnisse der RT-PCR der Gesamt-RNA, die aus den über dem
Boden befindlichen Teilen und den Wurzeln von Arabidopsis thaliana
extrahiert wurden. Die rechte Gruppe gibt die positive Kontrolle
wieder.
-
Beste Ausführungsform
der Erfindung
-
Wir
haben versucht, Nicotianaminsynthase zu isolieren (Higuchi et al.
Plant & Soil,
Bd. 165, S. 173–179,
1994), doch da Nicotianaminsynthase sich leicht zersetzte und schwer
zu isolieren und zu reinigen war, konnten wir keine ausreichenden
Mengen an Protein erhalten, um seine partielle Aminosäuresequenz
zu bestimmen. Später
wurde festgestellt, dass ein Thiolprotease-Hemmer E-64 (im folgenden
als E-64 bezeichnet) sehr wirksam im Unterdrücken des Abbaus von Nicotianaminsynthase
war (Higuchi et al. Plant & Soil,
Bd. 178, S. 171–177,
1996a).
-
Bei
der vorliegenden Erfindung konnte als Folge des Zerstoßens von
gefrorenen Wurzeln zu einem Feinpulver in Flüssig-N2 und
dann schnellen Homogenisierens mit Puffer, der 0,1 mM Thiolprotease-Hemmer E-64
enthielt, das Nicotianaminsynthase-Protein isoliert werden, ebenso wie
sein Gen.
-
Ferner
gewann das Enzym der vorliegenden Erfindung seine Aktivität durch
Entfernen des SDS nach der SDS-PAGE-Behandlung zurück, doch
war die Rückgewinnungsrate
sehr niedrig (Higuchi et al. Plant & Soil, Bd. 165, S. 173–179, 1994).
Folglich sollte der Grad der Ausreinigung vor der SDS-PAGE-Behandlung
erhöht werden.
Daraufhin wurden die säulenchromatographischen
Verfahrensweisen weiter verbessert.
-
Wir
haben außerdem
festgestellt, dass das Enzym der vorliegenden Erfindung relativ
hydrophob ist und ein Puffer, der ein mildes oberflächenaktives
Mittel, CHAPS, enthielt, die Rückgewinnungsrate
erhöhte. Verschiedene
Ionenaustauscher-chromatograpische Träger wurden getestet, und DEAE-Sepharose
FF und DEAE-Sephacel wurden als am wirksamsten befunden. Über TSK-Gel
Butyl Toyopearl* hinaus entfernte auch ein anderer hydrophober chromatographischer
Träger,
TSK-Gel Ether Toyopearl* 650M, die Unreinheiten des 30 – 35 kDa
wirksam.
-
Vom
Enzym der vorliegenden Erfindung wurde berichtet, dass es das Peptid
von 30 – 35
kDa war, dessen Aktivität
durch Entfernen des SDS nach der SDS-PAGE-Behandlung wiederhergestellt wurde,
wobei die Aktivität
als ein breiter Molekulargewichtsbereich von 30 – 35 kDa nachgewiesen wurde
(siehe 3). 3 zeigt ein Ergebnis der präparativen
SDS-PAGE in den Fraktionen, die eine Enzymaktivität zeigen.
SDS-PAGE wurde unter Verwendung von 11% Acrylamid-Flachgelen vorgenommen.
Eine Portion des Gels wurde mit Coomassie-Brilliantblau angefärbt und
der Rest des Gels mit Cu gefärbt.
Das Gel, das Proteine von 30 – 35
kDa Größe enthielt,
wurde in sieben Fragmente geschnitten (angegeben durch die kurzen
Linien). Die dicken Balken in 3 zeigen
die relativen Enzymaktivitäten
an, die aus jedem Gelfragment bestimmt wurden.
-
Um
das Nicotianaminsynthase-Peptid aus den Proteinen mit diesen Molekulargewichten
zu identifizieren, wurden die Peptide, die in den Nicotianaminsynthase-Fraktionen,
ausgereinigt aus Fe-defizienten und Kontroll-Gerstenwurzeln, enthalten
waren, unter Anwendung von SDS-PAGE verglichen. Aus jeder Gerstenwurzel
von 200 g wurde das vorliegende Enzym gemäß der in nachstehendem Beispiel
3 beschriebenen Methode ausgereinigt.
-
Die
Enzymaktivität
der Kontrolle betrug ein Viertel der bei den Fe-defizienten Wurzeln.
-
Die
Peptidzusammensetzung der aktiven Enzymfraktion aus jedem Reinigungsschritt
des vorliegenden Enzyms wurde analysiert und mittels SDS-PAGE verglichen,
wobei die Ergebnisse in 2, 13 und 14 gezeigt
sind. 2, 13 und 14 zeigen
einen Vergleich mit der aktiven Fraktion aus dem Reinigungsschritt
der Fe-defizienten Gerstenwurzeln von 200 g [in der Figur mit (–) gezeigt]
und der aktiven Fraktion aus dem Reinigungsschritt der Kontroll-Gerstenwurzeln
von 200 g [in der Figur mit (+) gezeigt]. SDS-PAGE wurde unter Verwendung
von 12,5% Acrylamid-Flachgelen durchgeführt (Laemmli, Nature, Bd. 227,
S. 680–685,
1970). Die Gele wurden mit Coomassie-Brilliantblau angefärbt. 2 zeigt
einen Schritt vor der DEAE-Sepharose. Die obere Reihe zeigt das
Enzym aus Fe-defizienten Gerstenwurzeln, und die untere Reihe zeigt
das Enzym aus den Kontrollwurzeln. In jeder Bahn sind Bahnen 1,
Rohextrakt, 200 μg
Protein; Bahnen 2, nach Butyl Toyopearl 650M, 100 μg
Protein; Bahnen 3, nach Hydroxyapatit, 20 μg Protein; und Bahnen 4, nach
Butyl Toyopearl 650M, 15 μg
Protein, gezeigt.
-
13 zeigt
jede Bahn nach DEAE-Sepharose FF, 25 μg Protein. 14 zeigt
nach Ether Toyopearl* 650M, wobei im linken Teil die inaktive Fraktion
gezeigt ist und im rechten Teil die aktive Fraktion gezeigt ist, wobei
1/25 jeder Fraktion elektrophoretisch behandelt ist.
-
Als
Ergebnis wurde nahezu kein Unterschied sowohl bei Fe-defizienten
als auch Kontrollwurzeln vor dem DEAE-Sepharose-Schritt beobachtet
(siehe 2). Nach dem DEAE-Sepharose-Schritt wurde klar,
dass die 30- und 31-kDa-Peptide durch Fe-Defizienzl induziert wurden (siehe 13).
Nach dem Ether Toyopearl*-Schritt wurde das 31-kDa-Peptid aus der
aktiven Fraktion eliminiert. Die 32- und 33-kDa-Peptide wurden durch
Fe-Defizienz als neu induziert festgestellt (siehe 14).
Aktivitäten
wurden bei den 32- und 33-kDa-Peptiden nachgewiesen, doch wurde
keine Aktivität
beim 30-kDa-Peptid festgestellt (siehe 3).
-
Das
Molekulargewicht des Enzyms der vorliegenden Erfindung wurde durch
Gelfiltration bestimmt.
-
Das
mittels Gelfiltration beurteilte Molekulargewicht der Nicotianaminsynthase
betrug berichtetermaßen
40.000 – 50.000
(Higuchi et al. Plant & Soil,
Bd. 165, S 173–179,
1994). Dies stimmte allerdings nicht dem durch SDS-PAGE bestimmten
Wert überein.
-
In
der vorliegenden Studie erhöhte
der Puffer, der CHAPS enthielt, die Auflösung effektiv, und das Molekulargewicht
des vorliegenden Enzyms wurde mit 35.000 bewertet (siehe 4).
Dies entspricht dem mittels SDS-PAGE erhaltenen Wert.
-
4 zeigt
das Elutionsmuster der Nicotianaminsynthase aus der Gelfiltrationssäule. Die
schwarzen Kreise (•)
geben die Enzymaktivität
an, und die durchgezogene Linie gibt die Extinktion bei 280 nm an.
Die aktive Fraktion wurde nach der Hydroxyapatit-Chromatographie auf eine Sephacryl S300HR-(Pharmacia)-Säule (1,5
cm × 71
cm, 125 ml) aufgebraucht, äquilibriert
mit Entwicklerpuffer (50 mM Tris, 1 mM EDTA, 0,1 M KCl, 0,05% CHAPS,
0,1 mM p-APMSF und 3 mM DTT, pH 8,0). Die verwendeten Molekulargewichts-Marker
waren Thyroglobulin (MR 670.000), γ-Globulin (MR 158.000), Oval bumin
(MR 44.000) und Myoglobin (MR 17.000). Der lineare Fluss betrug
10 cm/Stunde.
-
Die
partielle Aminosäuresequenz
wurde aus gereinigter Nicotianaminsynthase bestimmt.
-
Die
oben erläuterten
30-kDa-, 32-kDa- und 33-kDa-Peptide wurden aus 1 kg Fedefizienten
Gerstenwurzeln unter Anwendung der hierin in Beispiel 3 beschriebenen
Methode ausgereinigt. Diese wurden unter Anwendung der hierin in
Beispiel 4 beschriebenen Methode teilweise zerlegt. Obschon 32-
und 33-kDa-Peptide nicht vollständig
voneinander getrennt werden konnten, könnten diese eine ähnliche
Sequenz aufweisen, oder war das 32-kDa-Peptid vermutlich das Abbauprodukt
des 33-kDa-Peptids,
und beide wurden gemeinsam zerlegt.
-
Die
bestimmten partiellen Aminosäuresequenzen
zeigten, dass diese Peptide sehr ähnlich zueinander waren (5).
Da außerdem
die Molekulargewichte der 33-kDa- und 32-kDa-(1)-Fragmente nahezu
unverändert
im Vergleich zu vor der Zerlegung waren, könnte diese Sequenz die N-terminale
Region des vorliegenden Enzyms sein. Als Ergebnis der Computerdurchsuche
der Datenbank wurde ein Gen von unbekannter Funktion mit sehr ähnlicher
Sequenz zu diesen Sequenzen gefunden, dass in Oryza sativa und Arabidogsis
thaliana vorliegt. Insbesondere die EST-cDNA-Klone D23792 und D24790
von Oryza sativa waren sehr ähnlich
bei 80,0% Identität
bei einer 33-Aminosäuren-Überlappung
bei ersterem und 68,4% Identität
bei einer 19-Aminosäuren-Überlappung
bei letzterem (5).
-
5 zeigt
einen Vergleich mit einer Teilsequenz aus sechs Aminosäuren, wie
bei aus Gerste stammender Nicotianaminsynthase bestimmt, und einer ähnlichen
Sequenz von Gramineen, wie durch Computerdurchsuche der Datenbank
erhalten. Ein identischer Aminosäure-Rest
ist in ":" gezeigt. Der durch
die Pfeile angegebene Teil der Nukleotidsequenzen wurde für die Sequenzen
des bei der PCR verwendeten Primers verwendet.
-
Das
Klonieren der Nukleotidsequenzen der cDNA-Klone, die Nicotianaminsynthase
codieren, wurde vorgenommen und bestimmt.
-
Die
PCR-Amplifikation der Gesamt-cDNA, die aus Fe-defizienten Gerstenwurzeln
unter Verwendung degenerierter Primer erhalten wurde, wie aus der
partiellen Aminosäuresequenz
konstruiert, die mittels der zuvor erörterten Methode erhalten worden
war, wurde durchgeführt,
doch konnte die Ziel-DNA nicht amplifiziert werden. Daraufhin wurden
die Primer mit einer einzelnen Nukleotidsequenz (durch Pfeile in 5 gezeigt)
aus Sequenzen von Oryza sativa, D23792 und D24790, synthetisiert
und die PCR-Amplifikation vorgenommen. Das 205-bp-Fragment wurde
mittels PCR unter Verwendung von NF- und NR-Primern amplifiziert
und das 274-bp-Fragment mittels PCR unter Verwendung von IF- und
IR-Primern amplifiziert, wobei diese die Zielsequenzen enthielten.
Eine unter Verwendung von Poly(A)+ RNA aus
Fe-defizienten Gerstenwurzeln hergestellte cDNA-Bank wurde gescreent
und 19 positive Klone unter Verwendung der 205-bp-Fragment-Sonde und 88 positive Klone
unter Verwendung der 274-bp-Fragment-Sonde erhalten.
-
Unter
den derart erhaltenen Klonen enthielt der als HvNAS1 bezeichnete
Klon eine translatierte Region von 985 bp, wobei die davon abgeleitete
Aminosäuresequenz
ein Rest aus 328 Aminosäuren
war mit einem abgeleiteten Molekulargewicht von 35.144. Dies entsprach
dem mittels SDS-PAGE und Gelfiltration bestimmten Wert gut. Die
partiellen Aminosäuresequenzen
der 32-kDA- und 33-kDa-Peptide waren vollständig in HvNAS1 enthalten (6).
-
6 zeigt
die HvNAS1-cDNA der vollen Länge
und die davon abgeleitete Aminosäuresequenz.
Die unterstrichenen Sequenzen zeigen die identischen partiellen
Aminosäuresequenzen
der in obiger 5 gezeigten Fragmente. Die Zahlen
der Nukleotidsequenzen sind rechts jeder Reihe angegeben. Die Zahlen
der Aminosäuren
sind links jeder Reihe angegeben.
-
Der
vorhergesagte pI von 5.2 entsprach dem durch native isoelektrische
Fokussierungs-Elektrophorese geschätzten Wert gut. Die sechs anderen
Klone als HvNAS1, nämlich
HvNAS2, HvNAS3, HvNAS4, HvNAS5, HvNAS6 und HvNAS7, mit sehr ähnlicher
Sequenz wurden ebenfalls erhalten (Tabelle 1, 7).
-
7 zeigt
einen Vergleich der abgeleiteten Aminosäuresequenzen der obigen 7 aus
Gerste erhaltenen cDNA. Sternchen, "*" weist
auf identische Aminosäure-Reste
in allen Sequenzen hin.
-
Die
Nukleotidsequenzen dieser Klone sind in SEQ ID NR. 2 (HvNAS1), SEQ
ID NR. 4 (HvNAS2), SEQ ID NR. 6 (HvNAS3), SEQ ID NR. 8 (HvNAS4),
SEQ ID NR. 10 (HvNAS5), SEQ ID NR. 12 (HvNAS6) bzw. SEQ ID NR. 14
(HvNAS7) gezeigt. Die Aminosäuresequenzen
dieser Aminosäuresequenzen
sind in SEQ ID NR. 1 (HvNAS1), SEQ ID NR. 3 (HvNAS12, SEQ ID NR.
5 (HvNAS3), SEQ ID NR. 7 (HvNAS4), SEQ ID NR. 9 (HvNAS5), SEQ ID
NR. 11 (HvNAS6) bzw. SEQ ID NR. 13 (HvNAS7) gezeigt. Tabelle
1 Eigenschaften der nas-Klone
-
Die
aus dem 30-kDa-Peptid bestimmten partiellen Aminosäuresequenzen
waren alle in HvNAS5 enthalten. Die 5'- und 3'-nicht-translatierten Regionen dieser
Klone waren nicht ähnlich
zueinander.
-
D23792
und D24790 ähnlich
der Nicotianaminsynthase von Oryzae sativa wurden von etwa 80% Identität zu HvNAS1
festgestellt. AC003114 und AB005248 von Arabidopsis thaliana wurden
von etwa 45% Identität
zu HvNAS1 festgestellt.
-
Das
erhaltene HvNAS1-Protein wurde in E. coli exprimiert.
-
Die
PCR-Amplifikation von HvNAS1 ORF wurde mit Vektor pMAL-c2 kloniert,
um C-terminal mit
dem Maltose-Bindungsprotein fusioniertes HvNAS1 zu exprimieren.
Die Expression des Fusionsproteins wird durch IPTG stark induziert.
-
Der
Rohextrakt wurde aus dem transformierten E. coli erhalten und die
Nicotianaminsynthase-Aktivität im
Zustand des Fusionsproteins untersucht. Im Rohextrakt aus dem mit
lediglich dem Vektor transformierten Stamm konnte die Aktivität nicht
nachgewiesen werden, wohingegen im Fall der Insertion von HvNAS1
ORF die Aktivität
nachgewiesen wurde. Das Ergebnis ist in 8 gezeigt.
-
8 zeigt
die Ergebnisse der dünnschichtchromatographischen
(TLC) Analyse der Nicotianaminsynthase, die aus E. coli-Rohextrakt
erhalten wurde, welche ein Fusionsprotein mit dem Maltose-Bindungsprotein-HvNAS1-exprimiert.
In 8, Bahn 1: eine standardmäßige Nicotianaminsynthase;
Bahn 2: E. coli, exprimierend Maltose-Bindungsprotein (SAM); und Bahn 3: E.
coli, exprimierend mit dem Maltose-Bindungsprotein fusioniertes Protein-HvNAS1.
-
Die
mittels der in untenstehendem Beispiel 7 beschriebenen Methode durchgeführte Northern-Hybridisationsanalyse
zeigte, dass dieses Gen in Fe-defizienten Wurzeln stark induziert
wurde (9). Dies stimmt mit dem Expressionsmuster der
vorliegenden Enzymaktivität überein (Higuchi
et al. 1994). 9 zeigt ein Ergebnis der Northern-Hybridisationsanalyse
unter Verwendung von HvNAS1 als einer Sonde. Die Gesamt-RNA wurde
aus Gerstenblätter
und -Wurzeln nach einwöchiger
Fe-defizienter Behandlung und Kontroll-Gerstenblätter und -Wurzeln extrahiert,
und in jeder Bahn wurden 5 μg
RNA elektrophoretisch behandelt.
-
Die
Southern-Hybridisationsanalyse der Gersten-Genom-DNA wurde gemäß der in
untenstehendem Beispiel 8 beschriebenen Methode vorgenommen. Das
Schneiden der DNA mit BamHI, EcoRI oder HindIII erzeugte eine Vielzahl
von Fragmenten, wobei allerdings keiner der derzeit erhaltenen Klone
mit BamHI und EcoRI verdaut werden konnte, weshalb folglich das
Nicotianaminsynthase-Gen in multipler Kopienzahl in den Genomen
von Gerste und Reis vorliegen könnte
(10).
-
10 zeigt
die Southern-Hybridisationsanalyse von HvNAS1 als einer Sonde. Genomische
DNAs von Gerste und Reis wurden mit BamHI (Bahnen B), EcoRI (Bahnen
R) und HindIII (Bahnen H) verdaut, und 10 μg davon wurden in jeder Bahn
elektrophoretisch behandelt.
-
Weiterhin
wurden unter Verwendung von Antigen, das mittels der in nachstehendem
Beispiel 9 beschriebenen Methode erhalten worden war, eine Western-Blot-Analyse
gemäß der in
Beispiel 10 beschriebenen Methode vorgenommen. Es wurde festgestellt,
dass das vorliegende Enzymprotein während des Vorgangs im Rohextrakt
schnell zersetzt wurde, welcher zum Nachweis der vorliegenden Enzymaktivität zubereitet
worden war (11). Das Färbungsmuster stimmte mit der
Tatsache überein,
dass die vorliegende Enzymaktivität in dem breiten Bereich von
30 – 35
kDa gemäß SDS-PAGE
nachgewiesen wurde (siehe 3).
-
11 zeigt
die Western-Blot-Analyse des zum Nachweis der Aktivität verwendeten
Rohenzyms. SDS-PAGE wurde unter Verwendung von 125% Acrylamid-Flachgel
durchgeführt.
100 μg des
Proteins wurden elektrophoretisch behandelt.
-
Der
aus denaturiertem Protein gemäß der in
nachstehendem Beispiel 10 beschriebenen Methode erhaltene Rohextrakt
wurde als nahezu einzige Bande mit 35 – 36 kDa nachgewiesen (12).
Dieser Wert stimmt mit dem hergeleiteten Wert aus der Aminosäuresequenz überein.
-
12 zeigt
die Western-Blot-Analyse des durch Trichloressigsäure/Aceton
extrahierten Gesamtproteins. SDS-PAGE wurde unter Verwendung von
12,5% Acrylamid-Flachgel
durchgeführt.
100 μg Protein
wurden elektrophoretisch behandelt. 200 μg aus Wurzeln extrahierte Proteine
und 500 μg
aus Blättern
extrahierte Proteine wurden elektrophoretisch behandelt.
-
Die
Western-Blot-Analyse nach zweidimensionaler Elektrophorese ergibt
den Nachweis mehrerer Flecken. Dies stimmt mit der Tatsache überein,
dass eine Vielzahl von Nicotianaminsynthase-Genen erhalten wird.
Alle Flecken waren in Fe-defizienten Wurzeln induziert.
-
Als
Ergebnis dessen, dass die cDNA-Bank aus Fe-defizienter Reiswurzel-Poly(A)
+ RNA unter Verwendung von Sonden gescreent wurde, die durch Schneiden
von HvNAS1 mit Restriktionsenzymen ApaLI und XhoI hergestellt wurden,
wurden 20 Klone erhalten. Diese Klone wurden in 3 Typen von Klonen
entsprechend ihren Sequenzen eingeteilt, von denen lediglich ein
Typ ORF der vollen Länge
enthielt, welcher als OsNAS1 bezeichnet wurde. Die Nukleotidsequenz
von OsNAS ist in SEQ ID NR. 16 gezeigt, und die Aminosäuresequenz
ist in SEQ ID NR. 15 gezeigt.
-
Die
PCR-Amplifikation von OsNAS1 ORF wurde mit einem Vektor pMAL-c2
kloniert, um eine C-terminal mit dem Maltose-Bindungsprotein fusionierte
Form zu exprimieren. Das Fusionsprotein wird durch IPTG in seiner
Expression stark induziert.
-
Ein
Rohextrakt des transformierten E. coli mit dem Fusionsprotein wurde
erhalten und die Nicotianaminsynthase-Aktivität im Zustand des Fusionsproteins
untersucht. Dieselbe Aktivität
wie bei HvNAS1 wurde nachgewiesen. Das Ergebnis ist in 15 gezeigt. 15 zeigt
die Ergebnisse der dünnschichtchromatographischen
(TLC) Analyse der aus E. coli-Rohextrakt erhaltenen Nicotianaminsynthase,
die ein Fusionsprotein mit dem Maltose-Bindungsprotein-OsNAS1-exprimiert.
In 15, Bahn 1: ein standardmäßiges Nicotianamin (NA); Bahn
2: ein Extrakt aus E. coli, das das Fusionsprotein mit dem Maltose-Bindungsprotein-OsNAS1-exprimiert;
und Bahn 3: ein Extrakt aus E. coli, das das Fusionsprotein mit
dem Maltose-Bindungsprotein-HvNAS1-exprimiert.
-
Die
mittels der in nachstehendem Beispiel 7 beschriebenen Methode durchgeführte Northern-Hybridisationsanalyse
ergab, dass im Gegensatz zu Gerste die Expression in Reis durch
eine Fe-defiziente Behandlung nicht nur in den Wurzeln, sondern
auch in den Blättern
induziert wurde (16). 16 zeigt
das Ergebnis der Northern-Hybridisationsanalyse
unter Verwendung von OsNAS1 ORF als einer Sonde. Die Ge samt-RNA
wurde nach zwei Wochen der Fe-defizienten Behandlung und Kontroll-Reisblättern und
-Wurzeln extrahiert, und in jeder Bahn wurden 5 μg RNA elektrophoretisch behandelt.
-
Die
Nukleotidsequenz von Arabidopsis thaliana ähnlich der von HvNAS1, wie
durch Computerdurchsuche der Datenbank erhalten, wurde als ein Primer
verwendet. Die PCR-Amplifikation der genomischen DNA von Arabidopsis
thaliana führte
zum Erhalt dreier Nicotianaminsynthase-Gene. Diese wurden als AtNAS1, AtNAS2
und AtNAS3 bezeichnet.
-
Die
Nukleotidsequenzen dieser Gene sind in SEQ ID NR. 18 (AtNAS1), SEQ
ID NR. 20 (AtNAS2) und SEQ ID NR. 22 (AtNAS3) gezeigt. Diese Aminosäuresequenzen
sind in SEQ ID NR. 17 (AtNAS1), SEQ ID NR. 19 (AtNAS2) und SEQ ID
NR. 21 (AtNAS3) gezeigt.
-
AtNAS1,
AtNAS2 und AtNAS3 ORF wurden mit PCR amplifiziert und mit dem Vektor
pMAL-c2 kloniert. Es wurde versucht, jeden von ihnen in der C-terminal
mit dem Maltose-Bindungsprotein fusionierten Form zu exprimieren.
Die Expression des Fusionsproteins wurde durch IPTG stark induziert.
-
Ein
Rohextrakt aus dem transformierten E. coli mit dem Fusionsprotein
wurde erhalten und die Nicotianaminsynthase-Aktivität im Zustand
des Fusionsproteins untersucht. Die Aktivität wurde nachgewiesen. Das Ergebnis
ist in 17 gezeigt. 17 zeigt
die Ergebnisse der TLC-Analyse der Nicotianaminsynthase-Aktivität, wie aus
E. coli-Rohextrakt erhalten, das ein Fusionsprotein mit dem Maltose-Bindungsprotein-AtNAS1-exprimierte. In 17,
Bahnen 1: ein standardmäßiges Nicotianamin
(NA) und S-Adenosylmethionin;
Bahnen 2: ein Extrakt aus E. coli, das lediglich Maltose-Bindungsprotein
exprimiert; Bahnen 3: ein Extrakt aus E. coli, das das Fusionsprotein
mit dem Maltose-Bindungsprotein-AtNAS1-exprimiert; Bahnen 4: ein
Extrakt aus E. coli, das das Fusionsprotein mit dem Maltose-Bindungsprotein-AtNAS2-exprimiert;
und Bahnen 5: ein Extrakt aus E. coli, das das Fusionsprotein mit
dem Maltose-Bindungsprotein-AtNAS3-exprimiert.
-
Eine
RT-PCR wurde gemäß der in
nachstehendem Beispiel 11 beschriebenen Methode durchgeführt. Es
wurde festgestellt, dass AtNAS1 in den Wurzeln und den über dem
Boden befindlichen Teilen von Arabidopsis thaliana exprimiert wurde,
wohingegen AtNAS2 weder in den Wurzeln noch in den über den
Boden gelegenen Teilen exprimiert wurde, und AtNAS3 wurde lediglich
in den Wurzeln exprimiert (18). In 18 zeigt
Bahn M den Molekulargewichts-Marker. Die Genexpression erfolgte
in den über
dem Boden gelegenen Teilen, den Wurzeln und den positiven Kontrollen
In der Figur, Bahnen C: AtNAS1 und AtNAS2 ORF der vollen Länge wurden
amplifiziert; Bahnen 1: AtNAS1-spezifische Amplifikationsfragmente;
Bahnen 2: AtNAS2-spezifische Amplifikationsfragmente; und Bahnen
3: AtNAS3-spezifische Amplifikationsfragmente.
-
Die
Menge an sekretierter Muginsäure
wurde auf bis zu 20 mg Muginsäure/g
Wurzeln Trockengewicht/Tag erhöht
berichtet (Takagi, 1993). Die von den vorliegenden Erfindern nachgewiesene
Roh-Nicotianaminsynthase-Aktivität
war ausreichend, um dies zu erfüllen.
Da die vorliegenden Enzymproteine in mehr als einigen Typen vorhanden
sind und ein 30-kDa-Peptid ohne Aktivität vorliegt, kann gemutmaßt werden,
dass als Folge der Aggregation dieser Peptide die konstruierte Struktur,
die zum Binden mit 3 Molekülen
S-Adenosylmethionin bevorzugt ist, eine maximale Aktivität zeigt.
Das mittels Gelfiltration bewertete Molekulargewicht betrug 35.000
(4).
-
Ein
Anstieg der Aktivität
durch Reaggregation der Untereinheiten wurde bisher nicht beobachtet.
Da das Fusionsprotein mit Maltose-Bindungsprotein und Untereinheiten
eine Aktivität
zeigte, verfolgen wir derzeit den Gedanken, dass das vorliegende
Enzym ein Monomer sein könnte.
Die Möglichkeit
jedoch, dass eine große
Aktivität
durch Konstruieren eines Multimers entfaltet werden kann, ist nicht
komplett von der Hand zu weisen.
-
Der
Reaktionsmechanismus bei der Synthese von Nicotianamin aus S-Adenosylmethionin
kann ähnlich
einer Methyltransfer-Reaktion unter Verwendung von S-Adenosylmethionin
als einem Methyl-Donor und einer Reaktion zur Synthese von Spermidin
und Spermin aus decarboxyliertem S-Adenosylmethionin sein. Die gemeinsame
katalytische Domäne
dieser Enzyme wurde bezüglich
der äquivalenten
Aminosäure- Konfiguration erörtert, bei
der ähnliche
Positionen in Strukturen einer höheren
Ordnung besetzt sind (Hashimoto et al. 1998 und Schluckebier et
al. 1995).
-
In
der Zukunft könnte
die katalytische Domäne
als Ergebnis eines Vergleichs mit der Nicotianaminsynthase von anderen
Pflanzenarten oder der Röntgenkristallographie
geklärt
werden.
-
Die
Induktion der Nicotianaminsynthase-Aktivität durch Fe-Defizienz stellt
ein spezifisches Phänomen bei
Gramineen dar und ist für
die Massenproduktion der Muginsäure-Familie wesentlich.
Bei Oryza sativa handelt es sich um eine Pflanze, in der die mengenmäßig geringste
Sekretion der Muginsäure-Familie
von den wichtigeren Gramineen erfolgt, weshalb sie sehr wenig ausgeprägt hinsichtlich
der Fe-Defizienz in Kalkboden ist.
-
Folglich
kann als Ergebnis der Schaffung von Oryza sativa-Transformanden
mit einer Toleranz gegenüber
einer Fe-Defizienz, indem das Nicotianaminsynthase-Gen der vorliegenden
Erfindung in Gramineen, insbesondere Oryza sativa, eingeführt wird
und diese bei Fe-Defizienz eine große Menge davon exprimieren,
die Kultivierung von Reis in Kalkboden möglich werden.
-
Hiervor
stellte man sich von Nicotianamin in Gramineen vor, dass es lediglich
eine Rolle als ein Vorläufer
für die
Synthese der Muginsäure-Familie
spielt. Da jedoch die vorliegende Erfindung die Erkenntnis gebracht
hat, dass das Nicotianaminsynthase-Gen die multiple Genfamilie darstellte,
kann es weitere wichtige Rollen in Gramineen spielen.
-
Bei
Pflanzen, denen die Sekretionsfähigkeit
der Muginsäure-Familie
fehlt, ausgenommen der Gramineen, wurde vorgeschlagen, dass Nicotianamin
eine Schlüsselrolle
als ein endogener Chelator von zweiwertigen Metallkationen wie Fe2+, Cu2+, Zn2+ und Mn2+ spielt
und dass es zur Homöostasie
dieser Metalle beiträgt (Stephan
et al. 1994). Folglich könnte
es dieselbe Rolle in Gramineen spielen.
-
Die
Nicotianaminsynthase-Aktivität
wird in Dikotyledonen nicht induziert, weshalb wohl die Expression des
Gens der vorliegenden Erfindung durch Fe-Defizienz nicht induziert
wird. Wir haben Nicotianaminsynthase-Gene von Arabidogsis thaliana
kloniert. Die Zusammensetzung der Promoter-Regionen in diesen Genen kann
den Mechanismus der Genexpression, wie durch Fe-Defizienz bewirkt,
erleuchten, und das Gen der vorliegenden Erfindung kann eine wichtige
Funktion nicht nur in Gramineen, sondern auch in Dikotyledonen spielen.
-
SEQ
ID NR. 1 zeigt die Aminosäuresequenz
der Nicotianaminsynthase der vorliegenden Erfindung.
-
Die
vorliegende Erfindung umfasst eine Nicotianaminsynthase mit der
in SEQ ID NR. 1 gezeigten Aminosäuresequenz.
Allerdings ist die vorliegende Erfindung nicht auf die obige Nicotianaminsynthase
beschränkt.
Die Nicotianaminsynthase der vorliegenden Erfindung enthält, sofern
sie nicht ihre Nicotianaminsynthase-Aktivität verliert, die Peptide, in
denen ein Teil der Aminosäuresequenz
des Peptids, vorzugsweise 50% oder weniger, bevorzugter 30% oder
weniger, oder sogar noch bevorzugter 10% oder weniger in den gesamten Aminosäuren, deletiert
ist oder durch andere Aminosäuren
substituiert ist, oder zu denen weitere Aminosäuren addiert sind oder in denen
diese Deletion, Substitution und Addition kombiniert sein kann.
-
Die
für die
Nicotianaminsynthase der vorliegenden Erfindung codierende Nukleotidsequenz
ist in SEQ ID NR. 2 gezeigt.
-
Die
vorliegende Erfindung umfasst nicht nur ein für Nicotianaminsynthase codierendes
Gen, wie in SEQ ID NR. 2 gezeigt, sondern auch die für Nicotianaminsynthase
codierenden Gene, die im folgenden genannt werden.
-
Der
Vektor der vorliegenden Erfindung zur Einführung des obigen Gens unterliegt
keiner speziellen Beschränkung,
weshalb verschiedene Vektoren verwendet werden können. Ein bevorzugter Vektor
ist der Expressionsvektor.
-
Verschiedene
Zellen können
in bequemer Weise unter Verwendung des rekombinanten Vektors der vorliegenden
Erfindung transformiert werden. Die Massenproduktion von Nicotianamid
kann unter Verwendung der derart erhaltenen Transformanden vorgenommen
werden. Diese Merhoden sind einem Fachmann des Gebiets wohlbekannt.
-
Beispiele
für die
Wirte zur Einführung
des Gens der vorliegenden Erfindung sind Bakterien, Hefen und Zellen.
Ein bevorzugter Wirt sind Pflanzen, insbesondere Gramineen.
-
Die
Methode zur Einführung
des Gens unterliegt keiner Beschränkung. Diese kann unter Verwendung eines
Vektors erfolgen, oder das Gen kann direkt in das Genom eingeführt werden.
-
Ein
Antikörper
der vorliegenden Erfindung gegen Nicotianaminsynthase kann in herkömmlicher
Weise unter Verwendung der Nicotianaminsynthase der vorliegenden
Erfindung erzeugt werden. Der Antikörper kann ein polyklonaler
Antikörper
oder, wenn erforderlich, ein monoklonaler Antikörper sein.
-
Weiterhin
kann eine selektive Zucht von Pflanzen, vorzugsweise Gramineen,
unter Verwendung des Gens der vorliegenden Erfindung vorgenommen
werden. Insbesondere kann das Gen der vorliegenden Erfindung zur
Verbesserung der Varietäten,
die selbst in Fe-defizientem Boden wachsen können, angewendet werden.
-
Beispiele
-
Die
folgenden Beispiele sollen zur Veranschaulichung der vorliegenden
Erfindung dienen, sind aber nicht als Beschränkung der vorliegenden Erfindung
gedacht.
-
Beispiel 1. (Präparierung
des Pflanzenmaterials)
-
Gerstensamen
(Hordeum vulgare L. cv Ehimehadakamugi Nr. 1) wurden auf feuchtem
Filterpapier auskeimen gelassen und in eine standardmäßige Wassernährlösung (Mori
und Nishizawa, 1987) in einem Glashaus bei Umgebungstemperatur unter
natürlichem
Licht übertragen.
Der pH-Wert der Wassernährlösung wurde
auf 5,5 mittels 0,5 N HCl täglich
eingestellt. Als sich die dritten Blätter entwickelten, wurden die
Pflanzen in eine Wasserkulturlösung
ohne Fe-Gehalt übertragen.
Der pH-Wert wurde auf 7,0 durch 0,5 N NaOH täglich gehalten. Die Kontrollpflanzen
wurden ebenfalls in der standardmäßigen Kulturlösung kontinuierlich
kultiviert. Die Kulturlösung
wurde einmal pro Woche erneuert. Zwei Wochen nach Beginn der Fe-defizienten
Behandlung, als sich eine schwere Eisenchlorose auf den 4. und 5.
Blättern
signifikant zeigte, wurden die Wurzeln geerntet und in Flüssig-N2 eingefroren und bei –80°C bis zur Verwendung gelagert.
-
Beispiel 2. (Assay der
Nicotianaminsynthase-Aktivität)
-
Es
wurde die zuvor von den vorliegenden Erfindern berichtete modifizierte
Assay-Methode (Higuchi
et al. 1996a) angewendet. Die Enzymlösungen wurden mit Reaktionspuffer
[50 mM Tris 1 mM EDTA, 3 mM Dithiothreitol (im folgenden bezeichnet
als DTT), 10 μM
(p-Amidinophenyl)methansulfonylfluorid (im folgenden bezeichnet
als p-APMSF) und
10 μM trans-Epoxysuccinylleucylamido-(4-guanidino)butan
(im folgenden bezeichnet als E-64), pH 8,7] äquilibriert. Der Pufferaustausch
wurde unter Verwendung einer Ultrafiltrationseinheit, Ultrafree
C3LGC NMWL10000 (Millipore Co.) vorgenommen. Mit14C
in der Carboxylgruppe markiertes S-Adenosylmethionin (Amsersham
Inc.) wurde der Enzymlösung
bei einer Endkonzentration von 20 μM zugegeben und dies bei 25°C für 15 Minuten
gehalten. Die Reaktionsprodukte wurden durch Dünnschichtchromatographie auf
Kieselgel LK6 (Whatman Inc.) unter Verwendung von Entwickler (Phenol
: Butanol : Ameisensäure
: Wasser = 12 : 3 : 2 : 3) aufgetrennt. Die Radioaktivität der Reaktionsprodukte
wurde mittels des Bildanalysegerätes BAAS-2000
(Fuji Film Co.) bestimmt. Der Proteingehalt wurde mittels der Bradford-Methode
unter Verwendung des Protein Assay Kit (Bio Rad Inc.) untersucht.
-
Beispiel 3 (Ausreinigung
der Nicotianaminsynthase)
-
Die
folgenden Vorgänge
wurden bei 4°C
vorgenommen; E-64 wurde den Nicotianaminsynthase-enthaltenden Fraktionen
bei einer Endkonzentration von 10 μM zugegeben.
-
Die
gefrorenen Wurzeln wurden zu einem Feinpulver in Flüssig-N2 zerstoßen
und in einem Haushalts-Entsafter mit 200 ml Extraktionspuffer [0,2
M Tris, 10 mM EDTA, 5 (Vol/Vol) Glycerol, 10 mM DTT, 0,1 mM E-64,
0,1 mM p-APMSF und 5% (Gew/Vol) unlösliches Polyvinylpyrrolidon
(PVP), pH 8,0] pro 100 g Wurzeln homogenisiert. Das Homogenat wurde
30 Minuten lang bei 22.500 × g
zum Erhalt des Überstands
zentrifugiert. Ammoniumsulfat wurde dem Überstand zu einer Endkonzentration
von 0,4 M zu gegeben und dies 1 Stunde lang stehengelassen. Das Gemisch
wurde nochmals 30 Minuten lang bei 22.500 × g zum Erhalt des Überstands
zentrifugiert.
-
Der Überstand
wurde auf eine TSK Gel* Butyl Toyopearl 650M-Säule aufgebracht
(10 ml Bettvolumen pro 100 g Wurzeln), mit dem Adsorptionspuffer äquilibriert
[20 mM Tris, 1 mM EDTA, 3 mM DTT, 0,4 M (NH4)2SO4 und 0,1 mM p-APMSF,
pH 8,0] und mit Elutionspuffer eluiert [10 mM Tris, 1 mM EDTA, 3
mM DTT, 0,1 mM p-APMSF, 5% Glycerol und 0,05% 3-[(3-Chloramidopropyl)dimethylammonio]propansulfonsäure (im
folgenden bezeichnet als CHAPS), pH 8,0].
-
KCl
wurde der aktiven Fraktion zu einer Endkonzentration von 0,4 M zugegeben,
und 1 M Kaliumphosphatpuffer (pH 8,0) wurde zu einer Endkonzentration
von 1 mM KCl zugegeben. Ein Hydroxyapatit-100-350-Mesh (Nacalai
Tesque), äquilibriert
mit dem Adsorptionspuffer (1 mM K-P, 10 mM KCl, 3 mM DTT und 0,1
mM p-APMSF, pH 8,0) wurde zu 10 ml pro 100 mg Protein zubereitet
und die die Nicotianaminsynthase enthaltenden Fraktionen aufgebracht.
Die Nicotianaminsynthase wurde ohne Adsorption durchgeleitet. Die durchgeleitete
Fraktion wurde auf eine TSK-Gel* Butyl Toyopearl 650M-Säule aufgebracht
(1 ml Bettvolumen pro 10 mg Protein), und die Nicotianaminsynthase
wurde in der oben beschriebenen Weise eluiert.
-
Die
aktive Fraktion wurde auf eine DEAE-Sepharose-FF-Säule aufgeladen
(5 ml Bettvolumen pro 25 mg Protein, Pharmacia), mit dem Adsorptionspuffer äquilibriert
(20 mM Tris, 1 mM EDTA, 3mM DTT, 0,1 mM p-APMSF und 0,05% CHAPS,
pH 8,0) und bei schrittweiser Gradientenelution einer Kaliumchloridkonzentration
von 0,05 M, 0,1 M, 0,15 M und 0,2 M eluiert. Die Nicotianaminsynthase
eluierte bei 0,15 M der KCl-Konzentration.
-
Die
aktive Fraktion wurde auf eine Ether Toyopearl* 650M-Säule aufgebracht
(10 ml Bettvolumen pro 100 Wurzeln), mit Adsorptionspuffer äquilibriert
[20 mM Tris, 1 mN EDTA, 3 mM DTT, 1,2 M (NH4)2SO4 und 0,1 mM p-APMSF,
pH 8,0]. Die Nicotianaminsynthase wurde nicht adsorbiert und wanderte
durch die Säule.
Die durchgewanderte Fraktion wurde auf eine TSK-Gel* Butyl Toyopearl*
650M-Säule
aufgebracht und die die Nicotianaminsynthase enthaltenden Fraktionen
eluiert. Die Peptide in der die Nicotianaminsynthase enthaltenden
aktiven Fraktionen, welche mittels den obigen säulenchromatographischen Behandlungen
gereinigt worden war, wurden durch Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese
(im folgenden bezeichnet als SDS-PAGE)
unter Verwendung von 11% Acrylamid-Flachgelen aufgetrennt. Nach
der SDS-PAGE wurde
das Gel mit 0,3 M Kupferchlorid angefärbt (Dzandu et al. 1988) und
die aufgetrennten Bande ausgeschnitten. Die Gelfragmente wurden
mit 0,25 M EDTA/0,25 M Tris (pH 9,0) entfärbt und mit dem Extraktionspuffer
(1% SDS, 25 mM Tris und 192 mM Glycin) homogenisiert. Jedes Homogenat
wurde mit SDS-freiem Puffer (25 mM Tris und 192 mM Glycin) elektroeluiert
und das Peptid rückgewonnen.
-
Beispiel 4. (Bestimmung
der partiellen Aminosäuresequenz)
-
Die
isolierte Nicotianaminsynthase wurde mit Cyanogenbromid chemisch
verdaut (Gross 1967).
-
Nach
der SDS-PAGE-Behandlung wurde ein 10-faches Volumen an 70% (Vol/Vol)
Ameisensäure
und 1% (Gew/Vol) Cyanogenbromid den die Nicotianaminsynthase enthaltenden
Gelfragmenten hinzugefügt
und dies bei 4°C über Nacht
zersetzt. Nach Beendigung des Verdaus wurde der flüssige Teil
entnommen und in vacuo getrocknet. Die getrocknete Substanz wurde
in SDS-PAGE-Probenpuffer gelöst
und bei Raumtemperatur über
Nacht stehengelassen, dann das verdaute Produkt mittels SDS-PAGE
unter Verwendung von 16,5% Acrylamidgel, das Tricin enthielt (Schagger
und Jagow, 1987) aufgetrennt. Die Peptide wurden auf eine PVDF-Membran
mittels Elektroblotting (Towbin et al. 1979) übertragen und mit Amidoschwarz
angefärbt.
Die gefärbten
Bande wurden ausgeschnitten und die Aminosäuresequenz von der N-terminalen
Seite jedes Peptids durch Edman-Abbau im Gasphasen-Sequenzierer
(Modell 492A-Proteinsequenzierer, Applied Biosystems Inc.) bestimmt.
-
Beispiel 5. (Klonierung
der Nicotianaminsynthase-Gene)
-
Die
PCR-Amplifikation wurde für
aus Fe-defizienten Gerstenwurzeln stammender cDNA unter Verwendung
von Primern vorgenommen, die auf der Basis der erhaltenen partiellen
Aminosäuresequenz
synthetisiert wurden. Eine aus der Poly(A)+RNA
der Fe defizienten Gerstenwurzeln hergestellte pYH23-cDNA-Bank wurde
mit den derart erhaltenen DNA-Fragmenten des PCR-Produkts gescreent,
welches mit [α-32P]dATP unter Verwendung des Random Primer
Kit (Takara Shuzo Co.) als den Primern markiert war. Die isolierten
cDNA-Klone wurden mittels des Cycle Sequencing Kits (Shimadzu Bunko
Co.) unter Verwendung des Shimadzu DNA-Sequenzierers DSQ-2000L sequenziert.
-
Die
PCR-Amplifikation wurde für
die genomische DNA von Arabidopsis thaliana unter Verwendung von
Primern durchgeführt,
die auf der Basis der Nukleotidsequenzen von AC003114 und AB005245
von Arabidopsis thaliana synthetisiert wurden. Die derart erhaltenen
DNA-Fragmente wurden mit dem Cycle Sequencing Kit (Shimadzu Bunko
Co.) unter Verwendung des Shimadzu DNA-Sequenzierers DSQ-1000L sequenziert.
-
Die
bestimmte Nukleotidsequenz ist in SEQ ID NR. 2 gezeigt.
-
Beispiel 6. (Expression
des NAS1-Proteins in E. coli)
-
Ein
Fragment, in dem die EcoRI-Stelle flussaufwärts des ersten ATG der HvNAS1-cDNA eingeführt wurde
und die PstI- und BamHI-Stellen flussabwärts des Stoppcodons der HvNAS1-cDNA
eingeführt
wurden, wurde mittels PCR amplifiziert. Das derart erhaltene amplifizierte
Produkt wurde in das pBluescriptII SK- unter Verwendung der EcoRI-Stelle und der BamHI-Stelle
subkloniert und die korrekte Nukleotidsequenz bestätigt. Das
Fragment zwischen der EcoRI-Stelle und der PstI-Stelle wurde in
pMAL-c2 kloniert, um eine Expression in Form des Fusionierens des
HvNAS1 an den C-Terminus des Maltose-Bindungsproteins zu erreichen.
-
Ein
Fragment, in dem die EcoRI-Stelle flussaufwärts des ersten ATG des OsNAS1
eingeführt
wurde und die HIndIII-Stelle flussabwärts des Stoppcodons des OsNAS1
eingeführt
wurde, wurde mittels PCR amplifiziert. Das derart erhaltene amplifizierte
Produkt wurde in das pBluescriptII SK- unter Verwendung der EcoRI-Stelle
und der HindIII-Stelle
subkloniert und die korrekte Nukleotidsequenz bestimmt. Das Fragment
zwischen der EcoRI-Stelle und der HindIII-Stelle wurde in pMAL-c2
kloniert, um die Expression in Form des Fusionierens des OsNAS1
an den C-Terminus des Maltose-Bindungsproteins
zu erreichen.
-
Ein
Fragment, in dem die EcoRI-Stelle flussaufwärts des ersten ATG von AtNAS1,
AtNAS2 und AtNAS3 eingeführt
wurde und die XbaI-Stelle flussabwärts des Stoppcodons von AtNAS1,
AtNAS2 und AtNAS3 eingeführt
wurde, wurde mittels PCR amplifiziert. Die derart erhaltenen amplifizierten
Produkte wurden in das pBLuescriptII SK- subkloniert, und die korrekten
Nukleotidsequenzen wurden bestätigt.
Das Fragment zwischen der EcoRI-Stelle und der XbaI-Stelle wurde
in pMAL-c2 kloniert, um die Expression in Form des Fusionierens
von AtNAS1, AtNAS2 und AtNAS3 jeweils an den C-Terminus der Maltose-Bindungsproteine zu
erreichen.
-
Der
E. coli-Stamm XL1-Blue wurde als ein Wirt zum Exprimieren des Fusionsproteins
verwendet. pMAL-c2-HvNAS bzw. pMAL-c2 wurden in XL1-Blue eingeführt. Die
derart erhaltenen rekombinanten Bakterien wurden in LB-Medium, das
Ampicillin und Tetracyclin zu jeweils 50 μg/ml enthielt, bei 37°C kultiviert,
bis die OD 600 der Kultur 0,5 erreicht hatte. Isopropyl-β-D-thiogalactopyranosid
(IPTG) wurde zu einer Endkonzentration von 0,3 mM hinzugefügt und kontinuierlich
bei 37°C
für 3 Stunden
gezüchtet
und die Bakterienzellen abgesammelt. Die Zellen wurden in 10 mM
Tris-Puffer suspendiert, der 0,2 M NaCl, 1 mM EDTA, 3 mM DTT und
0,1 mM E-64, pH 7,4, enthielt, und mit Flüssigstickstoff eingefroren.
Dies wurde in Eiswasser angetaut, und eine Ultrabeschallung für 15 Sekunden
wurde 10-mal wiederholt. Die Nicotianaminsynthase-Aktivität des derart
erhaltenen Rohextrakts wurde gemäß der in
Beispiel 2 beschriebenen Methode untersucht und die Enzymaktivität bestätigt.
-
Beispiel 7. (Northern-Hybridisation)
-
Die
Northern-Hybridisation der Gersten-RNA wurde unter Verwendung des
DNA-Fragments vorgenommen,
welches durch Ausschneiden von HvNAS1-cDNA mit HindIII und NotI
präpariert
und mit [α-32P]dATP als einer Sonde markiert wurde.
Die Gesamt-RNA wurde
aus Gerste extrahiert (Naito et al. 1988). Die extrahierte RNA wurde
mittels 1,4% Agarosegelelektrophorese aufgetrennt und auf Hybond-N–-Membranen (Amersham)
aufgeblottet. Die Northern-Hybridisation der Reis-RNA wurde unter
Verwendung von OsNAS1 ORF vorgenommen, die mit [α-32P]dATP
als einer Sonde markiert wurde. Die Gesamt-RNA wurde aus Reis extrahiert.
Die extrahierte RNA wurde mittels 1,4 Agarosegelelektrophorese aufgetrennt
und auf Hybond-N+-Membranen (Amersham) aufgeblottet.
Die Membran wurde mit der Sonde in 0,5 M Church-Phosphatpuffer (Church
und Gilbert 1984), 1 mM EDTA, 7% (Gew/Vol) SDS mit 100 μg/ml Lachssperma-DNA
bei 65°C über Nacht
hybridisiert. Die Membran wurde mit Puffer, der 40 mM Church-Phosphatpuffer
und 1% (Gew/Vol) SDS enthielt, bei 65°C für 10 Minuten gewaschen. Anschließend wurde
das Waschen nochmals wiederholt und die Membran mit Puffer, der
0,2 × SSPE
und 0,1% (Gew/Vol) SDS enthielt, bei 65°C für 10 Minuten gewaschen. Die
Radioaktivität
wurde unter Verwendung des Bildanalysegerätes BAS-2000 bestimmt.
-
Die
Ergebnisse sind in 9 und 16 gezeigt.
-
Beispiel 8. (Southern-Hybridisation)
-
Die
genomische DNA wurde aus den Blättern
von Gerste und Reis extrahiert. Der Extrakt wurde mit BamHI, EcoRI
oder HindIII verdaut, mit 0,8% (Gew/Vol) Agarose-Gelelektrophorese aufgetrennt und auf
Hybond-N+-Membranen (Amersham) übertragen.
Die Hybridisation wurde gemäß der in
Beispiel 7 beschriebenen Methode vorgenommen und die Radioaktivität bestimmt.
Das Ergebnis ist in 10 gezeigt.
-
Beispiel 9. (Herstellung
des polyklonalen Antikörpers)
-
Zwei
Ratten wurden unter Verwendung des Antigens immunisiert, das etwa
100 μg isolierte
Nicotianaminsynthase enthielt. Das Antigen stellte dieselbe Probe
dar, wie zur Bestimmung der partiellen Aminosäuresequenz verwendet. Komplettes
Freund-Adjuvans
wurde für
die erste Immunisierung verwendet, und für die zweite Immunisierung
wurde inkomplettes Freund-Adjuvans verwendet. Alle Bestandteile
des Bluts wurden gesammelt, nachdem die Ratten viermal immunisiert
worden waren, und das erhaltene Serum wurde bei –80°C konserviert.
-
Beispiel 10. (Western-Blotting-Analyse)
-
Das
Gesamtprotein wurde unter Verwendung von Trichloressigsäure und
Aceton extrahiert (Damerval et al. 1986). Die Pflanzen wurden in
Flüssigstickstoff
zerstoßen,
bis ein Pulver erhalten war, und mit Aceton vermischt, das 0,1%
(Vol/Vol) 2-Mercaptoethanol enthielt. Das Protein wurde ausgefällt, indem
es bei –20°C für 1 Stunde
stehen durfte, und das Präzipitat
wurde durch Zentrifugation bei 16.000 × g für 30 Minuten abgesammelt. Das
Präzipitat
wurde in Aceton suspendiert, das 0,1% (Vol/Vol) 2-Mercapto ethanol
enthielt, und bei –20°C für 1 Stunde
stehengelassen, woraufhin das Präzipitat
durch Zentrifugation bei 16.000 × g für 30 Minuten gesammelt wurde.
Das Präzipitat
wurde in vacuo getrocknet und in Probenpuffer [9,5 M Harnstoff,
2% (Gew/Vol) Triton X-100 und 5% (Vol/Vol)-ME] gelöst, dann
bei 16.000 × g
für 10
Minuten zum Erhalt des Überstands
zentrifugiert. Die im Überstand
enthaltenen Proteine wurden mittels SDS-PAGE oder der denaturierenden zweidimensionalen
Elektrophorese (O'Farrell
1975) aufgetrennt und auf eine PVDF-Membran übertragen. Die Western-Blotting-Analyse
wurde durch Aufbringen des primären
Antikörpers,
der Anti-Nicotianaminsnythase-Antikörper enthielt,
wie in Beispiel 9 präpariert,
und des sekundären
Antikörpers,
der Meerrettich-bindenden Ziege-Anti-Maus-IgG-(H + L)-Antikörper (Wako
Pure Chemicals Co.) enthielt, auf die Membran und Färben mit Diaminobenzidin
vorgenommen.
-
Das
Ergebnis ist in 12 gezeigt. Die SDS-PAGE wurde
unter Verwendung von 12,5 Acrylamid-Flachgel vorgenommen. 100 μg Protein
wurden elektrophoretisch behandelt. Die Proteine aus 200 μg Wurzeln
und 500 μg
Blättern
wurden elektrophoretisch behandelt.
-
Beispiel 11. (RT-PCR)
-
Die
Gesamt-RNA wurde aus Arabidogsis thaliana extrahiert. Die RT-PCR
wurde mit 1 μg
RNA als einer Template unter Verwendung des EZ rTth RNA PCR-Kits
(Parkin Elmer Inc.) vorgenommen. Spezifische Primer für AtNAS1,
AtNAS2 bzw. AtNAS3 wurden verwendet.
-
Das
Ergebnis ist in 18 gezeigt.
-
Industrielle Anwendbarkeit
-
Verschiedene
Zellen werden gemäß der herkömmlichen
Methode unter Verwendung der rekombinanten Vektoren der vorliegenden
Erfindung transformiert. Die Massenproduktion von Nicotianamin kann
unter Verwendung des erhaltenen Transformanden vorgenommen werden.
Diese Methoden können
gemäß der einem
Fachmann des Gebiets bekannten Methode durchgeführt werden.
-
Eine
selektive Züchtung
von Pflanzen, vorzugsweise Gramineen, kann ebenfalls unter Verwendung der
Gene der vorliegenden Erfindung durchgeführt werden. Insbesondere können die
Gene der vorliegenden Erfindung zur Verbesserung der Varietäten, die
auf Fe-defizientem Boden wachsen können, angewendet werden. SEQUENZLISTE