DE69920508T2 - Quaternäre phosponiumsalz-katalysatoren in der katalytischen hydrolyse der alkylenoxide - Google Patents
Quaternäre phosponiumsalz-katalysatoren in der katalytischen hydrolyse der alkylenoxide Download PDFInfo
- Publication number
- DE69920508T2 DE69920508T2 DE69920508T DE69920508T DE69920508T2 DE 69920508 T2 DE69920508 T2 DE 69920508T2 DE 69920508 T DE69920508 T DE 69920508T DE 69920508 T DE69920508 T DE 69920508T DE 69920508 T2 DE69920508 T2 DE 69920508T2
- Authority
- DE
- Germany
- Prior art keywords
- water
- quaternary
- alkylene
- catalyst
- anion
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003054 catalyst Substances 0.000 title description 41
- 238000006460 hydrolysis reaction Methods 0.000 title description 14
- 230000003197 catalytic effect Effects 0.000 title description 12
- 230000007062 hydrolysis Effects 0.000 title description 12
- 150000003839 salts Chemical class 0.000 title description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 32
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 25
- 238000000034 method Methods 0.000 claims abstract description 25
- 150000001450 anions Chemical class 0.000 claims abstract description 21
- -1 alkylene glycols Chemical class 0.000 claims abstract description 17
- 239000000203 mixture Substances 0.000 claims abstract description 16
- XYFCBTPGUUZFHI-UHFFFAOYSA-O phosphonium Chemical group [PH4+] XYFCBTPGUUZFHI-UHFFFAOYSA-O 0.000 claims abstract description 9
- 229920000642 polymer Polymers 0.000 claims abstract description 6
- 238000002360 preparation method Methods 0.000 claims abstract description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 5
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 4
- 125000002877 alkyl aryl group Chemical group 0.000 claims abstract description 3
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 3
- 125000003118 aryl group Chemical group 0.000 claims abstract description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 3
- 239000007787 solid Substances 0.000 claims description 15
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims description 14
- 239000003957 anion exchange resin Substances 0.000 claims description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims description 5
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 4
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 claims description 2
- 150000001735 carboxylic acids Chemical class 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 abstract description 3
- 229910052736 halogen Inorganic materials 0.000 abstract description 2
- 150000002367 halogens Chemical class 0.000 abstract 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 31
- 239000011347 resin Substances 0.000 description 21
- 229920005989 resin Polymers 0.000 description 21
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 17
- 238000006243 chemical reaction Methods 0.000 description 16
- 239000003456 ion exchange resin Substances 0.000 description 14
- 229920003303 ion-exchange polymer Polymers 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 10
- 230000002378 acidificating effect Effects 0.000 description 9
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 8
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 8
- 125000005496 phosphonium group Chemical group 0.000 description 8
- 125000001453 quaternary ammonium group Chemical group 0.000 description 7
- 239000001569 carbon dioxide Substances 0.000 description 6
- 229910002092 carbon dioxide Inorganic materials 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 6
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000000536 complexating effect Effects 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 239000004793 Polystyrene Substances 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 150000003003 phosphines Chemical class 0.000 description 4
- 229920002223 polystyrene Polymers 0.000 description 4
- 150000004023 quaternary phosphonium compounds Chemical class 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 101710134784 Agnoprotein Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 150000004714 phosphonium salts Chemical group 0.000 description 3
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 3
- DFQPZDGUFQJANM-UHFFFAOYSA-M tetrabutylphosphanium;hydroxide Chemical compound [OH-].CCCC[P+](CCCC)(CCCC)CCCC DFQPZDGUFQJANM-UHFFFAOYSA-M 0.000 description 3
- WPWHSFAFEBZWBB-UHFFFAOYSA-N 1-butyl radical Chemical compound [CH2]CCC WPWHSFAFEBZWBB-UHFFFAOYSA-N 0.000 description 2
- MNZAKDODWSQONA-UHFFFAOYSA-N 1-dibutylphosphorylbutane Chemical compound CCCCP(=O)(CCCC)CCCC MNZAKDODWSQONA-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 2
- 239000004280 Sodium formate Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 150000004679 hydroxides Chemical class 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- MEFBJEMVZONFCJ-UHFFFAOYSA-N molybdate Chemical compound [O-][Mo]([O-])(=O)=O MEFBJEMVZONFCJ-UHFFFAOYSA-N 0.000 description 2
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000010517 secondary reaction Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 2
- 235000019254 sodium formate Nutrition 0.000 description 2
- 239000011343 solid material Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- 238000009283 thermal hydrolysis Methods 0.000 description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- RBACIKXCRWGCBB-UHFFFAOYSA-N 1,2-Epoxybutane Chemical compound CCC1CO1 RBACIKXCRWGCBB-UHFFFAOYSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- PQXKWPLDPFFDJP-UHFFFAOYSA-N 2,3-dimethyloxirane Chemical compound CC1OC1C PQXKWPLDPFFDJP-UHFFFAOYSA-N 0.000 description 1
- TYDSIOSLHQWFOU-UHFFFAOYSA-N 2-cyclohexylidenecyclohexan-1-one Chemical compound O=C1CCCCC1=C1CCCCC1 TYDSIOSLHQWFOU-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-OUBTZVSYSA-N Carbon-13 Chemical compound [13C] OKTJSMMVPCPJKN-OUBTZVSYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- CTKINSOISVBQLD-UHFFFAOYSA-N Glycidol Chemical compound OCC1CO1 CTKINSOISVBQLD-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920001283 Polyalkylene terephthalate Polymers 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 150000003868 ammonium compounds Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000002528 anti-freeze Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- GDVKFRBCXAPAQJ-UHFFFAOYSA-A dialuminum;hexamagnesium;carbonate;hexadecahydroxide Chemical compound [OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Al+3].[Al+3].[O-]C([O-])=O GDVKFRBCXAPAQJ-UHFFFAOYSA-A 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 125000003700 epoxy group Chemical group 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 230000000887 hydrating effect Effects 0.000 description 1
- 229960001545 hydrotalcite Drugs 0.000 description 1
- 229910001701 hydrotalcite Inorganic materials 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- ALTWGIIQPLQAAM-UHFFFAOYSA-N metavanadate Chemical compound [O-][V](=O)=O ALTWGIIQPLQAAM-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- DJFBJKSMACBYBD-UHFFFAOYSA-N phosphane;hydrate Chemical group O.P DJFBJKSMACBYBD-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001281 polyalkylene Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- WQDSRJBTLILEEK-UHFFFAOYSA-N sulfurous acid Chemical compound OS(O)=O.OS(O)=O WQDSRJBTLILEEK-UHFFFAOYSA-N 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- BJQWBACJIAKDTJ-UHFFFAOYSA-N tetrabutylphosphanium Chemical compound CCCC[P+](CCCC)(CCCC)CCCC BJQWBACJIAKDTJ-UHFFFAOYSA-N 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- XKFPGUWSSPXXMF-UHFFFAOYSA-N tributyl(methyl)phosphanium Chemical compound CCCC[P+](C)(CCCC)CCCC XKFPGUWSSPXXMF-UHFFFAOYSA-N 0.000 description 1
- PBYZMCDFOULPGH-UHFFFAOYSA-N tungstate Chemical compound [O-][W]([O-])(=O)=O PBYZMCDFOULPGH-UHFFFAOYSA-N 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/09—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis
- C07C29/10—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis of ethers, including cyclic ethers, e.g. oxiranes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/09—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis
- C07C29/10—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis of ethers, including cyclic ethers, e.g. oxiranes
- C07C29/103—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis of ethers, including cyclic ethers, e.g. oxiranes of cyclic ethers
- C07C29/106—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrolysis of ethers, including cyclic ethers, e.g. oxiranes of cyclic ethers of oxiranes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
- Die vorliegende Erfindung betrifft ein Verfahren zur Herstellung von Alkylenglycolen durch Umsetzung eines Alkylenoxids mit Wasser in Gegenwart einer katalytischen Zusammensetzung.
- Hintergrund der Erfindung
- Alkylenglycolen, insbesondere Monoalkylenglycolen, kommt ein etabliertes kommerzielles Interesse zu. Beispielsweise werden Monoalkylenglycole als Gefrierschutzmittelzusammensetzungen, als Lösungsmittel und als Grundmaterialien in der Herstellung von Polyalkylenterephthalaten, beispielsweise für Fasern oder Flaschen, verwendet.
- Die Herstellung von Alkylenglycolen durch Flüssigphasenhydrolyse von Alkylenoxid ist bekannt. Die Hydrolyse wird ohne Katalysator durch Zusetzen eines großen Überschusses an Wasser, beispielsweise 20 bis 25 Mol Wasser pro Mol Alkylenoxid, vorgenommen, oder sie wird mit einem kleineren Überschuß an Wasser in einem katalytischen System ausgeführt. Die Reaktion wird als eine nukleophile Substitutionsreaktion betrachtet, wobei ein Öffnen des Alkylenoxidringes stattfindet, worin Wasser als das nukleophile Agens wirkt. Da das zunächst gebildete Monoalkylenglycol ebenfalls als ein Nukleophil wirkt, wird in der Regel ein Gemisch aus Monoalkylenglycol, Dialkylenglycol und höheren Alkylenglycolen ausgebildet. Um die Selektivität in Richtung Monoalkylenglycol zu erhöhen, ist es erforderlich, die Sekundärreaktion zwischen dem Primärprodukt und dem Alkylenglycol zu unterdrücken, welche mit der Hydrolyse des Alkylenoxids konkurriert.
- Ein wirksames Mittel zur Unterdrückung der Sekundärreaktion besteht in der Erhöhung der Relativmenge an Wasser, die im Reaktionsgemisch vorhanden ist. Obwohl diese Maßnahme die Selek tivität in Richtung der Ausbildung des Monoalkylenglycols verbessert, stellt sie ein Problem dar, da große Mengen an Wasser zur Gewinnung des Produktes entfernt werden müssen.
- Es wurden beträchtliche Anstrengungen unternommen, um eine Alternative zur Erhöhung der Reaktionsselektivität aufzufinden, ohne einen großen Wasserüberschuß anwenden zu müssen. Üblicherweise haben sich diese Bemühungen auf die Auswahl von wirksameren Hydrolysekatalysatoren konzentriert, und es sind verschiedene Katalysatoren beschrieben worden.
- Es wurden sowohl saure als auch alkalische Hydrolysekatalysatoren untersucht, wobei es scheint, daß die Verwendung von sauren Katalysatoren die Reaktionsgeschwindigkeit erhöht, ohne die Selektivität signifikant zu beeinträchtigen, wogegen bei Anwendung von alkalischen Katalysatoren im allgemeinen geringere Selektivitäten im Hinblick auf das Monoalkylenglycol erhalten werden.
- Bestimmte Anionen, beispielsweise Bicarbonat (Hydrogencarbonat), Bisulfit (Hydrogensulfit), Formiat und Molybdat sind dafür bekannt, eine gute katalytische Aktivität in Bezug auf Alkylenoxidumwandlung und Selektivität in Richtung Monoalkylenglycol zu zeigen. Wenn die Salze dieser Anionen als Katalysator in einem homogenen System verwendet werden, wird jedoch die Aufarbeitung des Reaktionsproduktes durch Destillation ein Problem darstellen, da die Salze im Glycol schlecht löslich sind und dazu neigen, dieses halbfest zu machen. Quaternäre Ammoniumsalze bleiben in dem Glycolreaktionsprodukt löslich.
- Hohe Umwandlungen, eine gute Selektivität und ein niedriges Wasser/Alkylenoxid-Verhältnis können mit dem Verfahren erhalten werden, das in EP-A 0 156 449 und EP-A 0 160 330 (beide von Union Carbide) beschrieben wird. Gemäß diesen Dokumenten wird die Hydrolyse von Alkylenoxiden in Gegenwart eines die Selektivität erhöhenden, ein Metalatanion enthaltenden Materials, vorzugsweise eines Feststoffes mit elektropositiven Komplexierungsstellen mit einer Affinität für Metalatanionen, ausgeführt. Der Feststoff ist vorzugsweise ein Anionenaustauscherharz, insbesondere ein Styrol-Divinylbenzol-Copolymer. Die elektropositiven Komplexierungsstellen sind insbesondere quaternäres Ammonium, protoniertes tertiäres Amin oder quaternäres Phosphonium. Dem quaternären Phosphonium wird kein spezifischer Vorteil zugeschrieben. Die Metalatanionen werden als Molybdat-, Wolframat-, Metavanadat-, Hydrogenpyrovanadat- und Pyrovanadatanionen beschrieben. Eine Komplizierung dieses Verfahrens besteht darin, daß der Alkylenglycol enthaltende Produktstrom auch eine erhebliche Menge an Metalatanionen umfaßt, welche aus den elektropositiven Komplexierungsstellen des festen, das Metalatanion enthaltenden Materials verdrängt wurden. Zur Verringerung der Menge an Metalatanionen im Alkylenglycolproduktstrom wird dieser Strom mit einem Feststoff in Kontakt gebracht, der elektropositive Komplexierungsstellen enthält, die mit Anionen assoziiert sind, die durch die genannten Metalatanion ersetzbar sind.
- In WO 95/20559 (Shell) wird ein Verfahren zur Herstellung von Alkylenglycolen beschrieben, worin ein Alkylenoxid mit Wasser in Gegenwart einer Katalysatorzusammensetzung, die ein festes Material mit einer oder mit mehreren elektropositiven Stellen umfaßt, dessen elektropositiven Stellen mit einem oder mit mehreren Anionen, welche keine Metalat- oder Halogenanionen sind, beispielsweise Bicarbonat, Bisulfit und Carboxylat, koordiniert sind, umgesetzt wird, mit der Maßgabe, daß dann, wenn es sich bei dem festen Material um ein Anionenaustauscherharz vom quaternären Ammoniumtyp handelt und das Anion Bicarbonat ist, das Verfahren im wesentlichen in Abwesenheit von Kohlendioxid ausgeführt wird. Gemäß diesem Dokument ist das Vorliegen von Kohlendioxid in dem Einsatzmaterial für die katalytische Wirkung von Bicarbonat-ausgetauschten Harzen vom quaternären Ammoniumtyp schädlich.
- Wie oben ausgeführt, können aus katalytischen Anionen und quaternären Ammoniumkationen zusammengesetzte Salze sowohl in einem homogenen System als auch in einem heterogenen System verwendet werden. In der Tat sind in heterogenen Systemen derartige quaternäre Ammoniumionen die traditionellerweise verwendeten Kationen der meisten Anionenaustauscherharze. Ein von diesen quaternären Ammoniumverbindungen geteilter Nachteil ist ihre beschränkte Wärmebeständigkeit. Bei Ausführung des Verfahrens der Alkylenoxidhydrolyse gemäß der WO 95/20559 mit Katalysatorzusammensetzungen auf der Basis von konventionellen organischen quaternären Ammoniumionaustauschern wurde gefunden, daß unter strengen Alkylenoxidhydrolysereaktionsbedingungen (hohe Temperatur und/oder lange Betriebszeiten) die katalytische Aktivität (Selektivität und/oder Umwandlung) und/oder das Quellverhalten der konventionellen Katalysatoren auf Harzbasis zu einer Verschlechterung neigen.
- In der US-A-4,160,116 (Showa Denko) wird ein Verfahren zur Herstellung eines Alkylenglycols durch Hydratisieren eines Alkylenoxids in Gegenwart einer erheblichen Menge von Kohlendioxid unter Anwendung eines quaternären Phosphoniumsalzes von Iod, Brom oder Chlor als Katalysator beschrieben.
- Zusammenfassung der Erfindung
- Die vorliegende Erfindung bezieht sich auf ein Verfahren zur Herstellung von Alkylenglycolen durch Umsetzung eines Alkylenoxids mit Wasser in Gegenwart wenigstens einer ionischen Zusammensetzung aus einem quaternären Phosphoniumkation mit der allgemeinen Formel
- Im allgemeinen wird Kohlendioxid nicht benötigt.
- In einer bevorzugten Ausführungsform der vorliegenden Erfindung ist das quaternäre Phosphoniumkation auf einem festen Träger immobilisiert, wie auf einem Anionenaustauscherharz.
- Eingehende Beschreibung der Erfindung
- Die quaternären Phosphoniumverbindungen, wie sie hier definiert werden, sind als solche als Alkylenoxidhydrolysekatalysatoren in einem homogenen flüssigen Reaktionssystem wirksam. Ein spezieller Vorteil dieser quaternären Phosphoniumverbindungen tritt jedoch dann zutage, wenn sie in einem heterogenen Reaktionssystem verwendet werden, worin die quaternären Phosphoniumkationen die elektropositiven Stellen eines festen Trägers ausmachen, wie in WO 95/20559 definiert. Insbesondere dann, wenn der feste Träger ein stark basisches Anionenaustauscherharz ist, dessen Basis ein quaternäres Phosphoniumkation gemäß der vorliegenden Erfindung ist, wird eine katalytische Zusammensetzung ausgebildet – mit dem Anion gemäß der Erfindung –, die beständig ist und die ihre Selektivität und Stabilität unter strengen Reaktionsbedingungen beibehält und auch gegenüber einem Quellen beständiger ist.
- Aus der großen Anzahl von Ionenaustauschharztypen kann ein beliebiges Harz als der feste Träger verwendet werden, insbeson dere die stark basischen (anionischen) Ionenaustauscherharze, worin die basischen Gruppen quaternäre Phosphoniumgruppen sind, die an ein polymeres Rückgrat gebunden (d.h. adsorbiert, damit umgesetzt oder aufgepfropft) sind. Geeignete polymere Rückgrate umfassen hochmolekulare Polymere und Copolymere, beispielsweise Additions- und Kondensationspolymere, einschließlich Polyalkylen, Polyester, Polycarbonat, Polyurethan, Formaldehydharze usw.. Zu im Handel erhältlichen Ionenaustauscherharzen zählen Harze auf der Basis von Polyacrylat oder Styrol-Divinylbenzol-Copolymeren. Viele dieser Ionenaustauscherharze sind rein organische Polymere, es können aber auch Harze auf Siliciumoxidbasis, wie Polysiloxane, in bequemer Weise verwendet werden. Alternative Materialien, bei denen die elektropositive Komplexierungsstelle vom quaternären Phosphoniumtyp durch Adsorption, Reaktion oder Pfropfen gebunden ist, umfassen solche Materialien mit anorganischer Natur, wie Kohlenstoff, Siliciumoxid, Siliciumoxid-Aluminiumoxid, Zeolithe, Glas und Tone, wie Hydrotalcit.
- Die Katalysatorzusammensetzung gemäß der Erfindung kann durch Immobilisieren des katalytisch aktiven Anions auf dem festen Träger vervollständigt werden, indem es in wäßriger Lösung zu einer Suspension des festen Trägers zugesetzt wird, der in einer vorangehenden präparativen Stufe adaptiert werden kann oder auch nicht. Wenn beispielsweise der feste Träger ein anionisches Austauscherharz ist, kann die Immobilisierung in einem einzigen Schritt durch Vermischen des Harzes mit dem Katalysator in einem wäßrigen Medium und durch anschließendes Waschen mit Wasser vorgenommen werden, oder alternativ in zwei Schritten, indem zunächst das Harz mit einem Hydroxid wie wäßrigem Natriumhydroxid in seine Hydroxylform umgewandelt wird und darauf der Katalysator zugesetzt wird.
- Die als Ausgangsmaterialien im Verfahren der Erfindung verwendeten Alkylenoxide entsprechen ihrer konventionellen Definiti on, d.h., sie sind Verbindungen mit einer vicinalen Oxidgruppe (Epoxygruppe) in ihren Molekülen.
- Besonders geeignet sind Alkylenoxide mit der allgemeinen Formelworin R1 bis R4 unabhängig ein Wasserstoffatom oder eine gegebenenfalls substituierte Alkylgruppe mit 1 bis 6 Kohlenstoffatomen bedeuten. Jede beliebige, durch R1, R2, R3 und/oder R4 dargestellte Alkylgruppe weist vorzugsweise 1 bis 3 Kohlenstoffatome auf. Als Substituenten können inaktive Reste wie Hydroxygruppen zugegen sein. Vorzugsweise stellen R1, R2 und R3 Wasserstoffatome dar und R4 steht für eine unsubstituierte C1-C3-Alkylgruppe, und stärker bevorzugt stellen alle Reste R1, R2, R3 und R4 Wasserstoffatome dar.
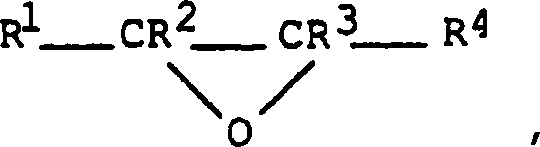
- Beispiele für geeignete Alkylenoxide umfassen daher Ethylenoxid, Propylenoxid, 1,2-Epoxybutan, 2,3-Epoxybutan und Glycidol. Ethylenoxid und Propylenoxid sind von besonderer kommerzieller Bedeutung.
- Wie zuvor erwähnt, ist es vorteilhaft, die Hydrolyse der Alkylenoxide ohne Anwendung überschüssiger Mengen an Wasser vorzunehmen. Im Verfahren gemäß der Erfindung sind Wassermengen im Bereich von 1 bis 15 Mol pro Mol Alkylenoxid durchaus geeignet, wobei Mengen im Bereich von 1 bis 6, auf die gleiche Basis bezogen, bevorzugt werden. Im Verfahren der Erfindung werden hohe Selektivitäten hinsichtlich des Monoalkylenglycols häufig bereits dann erzielt, wenn nur 4 oder 5 Mol Wasser pro Mol Alkylenoxid zugeführt werden.
- Das erfindungsgemäße Verfahren kann im ansatzweisen Betrieb ausgeführt werden. Insbesondere für großtechnische Anlagen wird es jedoch bevorzugt, das Verfahren kontinuierlich zu betreiben.
- Ein derartiges kontinuierliches Verfahren kann in einem Festbettreaktor durchgeführt werden, der im Aufstrom oder im Abstrom betrieben wird. Ein Abstrombetrieb wird bevorzugt.
- Der Reaktor kann unter isothermischen, adiabatischen oder unter Hybridbedingungen gehalten werden. Isothermische Reaktoren sind im allgemeinen Rohr-und-Mantel-Reaktoren, meistens vom Mehrrohrtyp, worin die Rohre den Katalysator enthalten und ein Kühlmittel die Rohre außen umstreicht. Adiabatische Reaktoren werden nicht gekühlt, und der aus ihnen austretende Produktstrom kann in einem gesonderten Wärmeaustauscher gekühlt werden.
- Unter bestimmten gewählten Umständen kann die katalytische Umwandlung von Ethylenoxid unvollständig sein, in welcher Situation restliches Ethylenoxid im Totraum des Reaktors unterhalb des Katalysatorbettes thermisch hydrolysiert werden kann. Da diese thermische Hydrolyse weniger spezifisch in Richtung Monoethylenglycol verläuft, wird es empfohlen, die Flüssigkeitsverweilmenge im Reaktor zu minimieren. Dies kann durch Füllen des Rektorauslaßteils mit Einbauten oder inertem Packungsmaterial zur Verringerung seines Volumens erreicht werden, und/oder durch Zusetzen eines Inertgases, wie Stickstoff, zu dem Reaktoreinspeisungsgemisch und durch Betreiben des Reaktors unter sogenannten Tropfenströmungsbedingungen.
- Zur Erzielung adäquater Zeit-Ausbeute-Werte wird es empfohlen, das Verfahren unter Bedingungen einer erhöhter Temperatur und eines erhöhten Drucks auszuführen.
- Geeignete Reaktionstemperaturen liegen generell im Bereich von 80 bis 200°C, wobei Temperaturen im Bereich von 90 bis 150°C bevorzugt werden. Der Reaktionsdruck wird üblicherweise im Bereich von 200 bis 3.000, vorzugsweise 200 bis 2.000 kPa gewählt. Für einen ansatzweisen Betrieb des Verfahrens wird der ausgewählte Reaktionsdruck vorteilhaft durch Aufdrücken eines Inertgases, wie Stickstoff, erreicht. Gewünschtenfalls können Gemische von Gasen eingesetzt werden, beispielsweise ist ein Gemisch aus Kohlendioxid und Stickstoff in bestimmten Fällen von Vorteil.
- Um einem etwaigen Quellen des Katalysators während des Betriebes Rechnung zu tragen, kann das Reaktorvolumen vorteilhaft größer sein als das vom Katalysator darin eingenommene volumen, beispielsweise um 10 bis 70 Vol.-% größer.
- In bestimmten Situationen, insbesondere dann, wenn im kontinuierlichen Strömungsmodus gearbeitet wird, hat es sich als vorteilhaft erwiesen, wenigstens einen Teil, wie etwa 30 bis 60 Gew.-% des Alkylenoxideinspeisestroms, in Abwesenheit eines Katalysators einer partiellen thermischen Hydrolyse zu unterziehen, bevor die Hydrolyse katalytisch vervollständigt wird. Es hat sich gezeigt, daß eine partielle Hydrolyse, selbst in Abwesenheit eines Katalysators, noch immer ausreichend selektiv hinsichtlich des Monoalkylenglycols verläuft, während anderseits diese Maßnahme zur Katalysatoreinsparung wirksam ist.
- Ein Problem, das gelegentlich im jedem Verfahren auftreten kann, worin Ethylenoxid hydrolysiert wird, ist das Auftreten kleiner Mengen von Aminen und/oder Phosphinen als Verunreinigungen im Produktstrom. Wenn ein stark basisches Anionenaustauscherharz gemäß der vorliegenden Erfindung als fester Träger für das katalytische Anion eingesetzt wird, sind dessen basische Gruppen quaternäre Phosphoniumgruppen. Es hat sich gezeigt, daß im Betrieb kleine Mengen an Phosphinen aus dem Harz in den Produktstrom auslaugen können. Darüber hinaus kann der Produktstrom kleine Mengen an Aminen enthalten, die aus Korrosionsinhibitoren stammen, die dem im Verfahren eingesetzten Wasser zugesetzt werden. Obwohl die Mengen derartigen Amin- und/oder Phosphinverunreinigungen, die in das Endprodukt gelangen, im allgemeinen sehr klein sind, können sie die Qualität des Endproduktes derart beeinträchtigen, daß es wünschenswert sein kann, sie unter der Erfassungsgrenze zu halten. Beispielsweise können Trimethylamin (TMA) und/oder Dimethylamin (DMR) in einer Menge von bis zu 10 ppm in das Endprodukt gelangen, wogegen der fischige Geruch von TMA in einer so geringen Menge wie 1 ppb detektiert werden kann.
- Eine wirksame Maßnahme zur Beseitigung von Aminen und/oder Phosphinen, die im Produktstrom von im allgemeinen jedem beliebigen Verfahren zugegen sein können, worin Ethylenoxid hydrolysiert wird, einschließlich des Verfahrens der vorliegenden Erfindung, ist, wie sich gezeigt hat, die Anwendung eines Schutzbettes, das ein stark saures Ionenaustauscherharz enthält, das wirksam die Amine oder Phosphine abfängt. Stark saure Ionenaustauscherharze entsprechen dem Sulfonsäuretyp. Im Handel verfügbare Beispiele davon sind die unter den Handelsmarken AMBERLYST 15, AMBERJET 1500H, AMBERJET 1200H, DOWEX MSC-1, DOWEX 50W, DIANON SK1B, LEWATIT VP OC 1812, LEWATIT S 100 MB und LEWATIT S 100 G1 bekannten Austauscherharze. Diese stark sauren Ionenaustauscherharze sind in der H+-Form und in der Salzform erhältlich, wie der Na+-Form. Wenn im Schutzbett nur die H+-Form des stark sauren Harzes verwendet wird, kann der Produktstrom nach dem Passieren des Harzes sauer werden. Die Verwendung eines Gemisches des stark sauren Ionenaustauscherharzes in seiner H+-Form und in der Salzform hat den Vorteil, daß der pH-Wert des Produktstroms nahe beim Neutralwert bleibt.
- Ein zusätzlicher Vorteil des stark sauren Schutzbettes liegt darin, daß etwaiges restliches Alkylenoxid, das noch im Produktstrom zugegen sein könnte, zu Alkylenglycol hydrolysiert wird, wenngleich mit einer geringeren Selektivität hinsichtlich Monoalkylenglycol.
- Um einer Erschöpfung des stark sauren Ionenaustauscherharzes während des Betriebes Rechnung zu tragen, ist es vorteilhaft, das Schutzbett in zwei oder mehreren getrennten Gefäßen zu betreiben.
- Erschöpftes stark saures Ionenaustauscherharz kann durch Behandlung mit eines Säure regeneriert werden, die stärker ist als die Sulfonsäuregruppen in der Harzmatrix, wie HCl und H2SO4. Heiße Schwefelsäure mit einer Normalität von 0,1 bis 2 hat sich als wirksam erwiesen.
- Die nachfolgenden Beispiele werden die Erfindung erläutern.
- Beispiele
- 1. Herstellung von Katalysatoren
- 1.1 Homogene Bicarbonatkatalysatoren
- Ein quaternäres Phosphoniumsalz und (zum Vergleich) ein ähnliches quaternäres Ammoniumsalz in Hydroxidform wurden als Vorläufer für die zu untersuchenden Bicarbonatkatalysatoren verwendet:
- – Tetra-n-butylphosphoniumhydroxid: (n-C4H9)4P+OH–
- – Tetra-n-butylammoniumhydroxid: (n-C4H9)4N+OH–
- Diese Basen wurden vor Gebrauch durch Rühren über Nacht unter 1.000 kPa Kohlendioxid in die Bicarbonatsalze umgewandelt:
- 1.2 Katalysatoren auf der Basis von stark basischen Ionenaustauscherharzen
-
- • Ein stark basisches Ionenaustauscherharz vom quaternären Phosphoniumtyp (Tributylmethylphosphoniumbromid auf einem Polystyrol/1% Divinylbenzol-Polymerträger von der Firma Fluka, Chloridform, Austauschkapazität 0,9 mÄq/g) wurde in folgender weise behandelt, um den Bicarbonatkatalysator zu bereiten: – 25 g trockenes Harz wurden 20 Stunden mit 250 ml entmineralisiertem Wasser und mit 18,9 g (10 molarer Überschuß) Natriumbicarbonat (NaHCO3) gerührt. Nach einem Filtrieren wurde diese Vorgangsweise dreimal wiederholt – das ausgetauschte Harz wurde mit 1.200 ml Wasser 2 Stunden lang gewaschen, bis im Waschwasser kein Chlorid mehr festgestellt werden konnte (AgNO3).
- • Ein stark basisches Ionenaustauscherharz vom quaternären Phosphoniumtyp (EGL-660, monodisperses vernetztes Polystyrol/Divinylbenzolharz, Firma Rohm & Haas, Chloridform, Austauschkapazität 1,7 mÄq/g) wurde in folgender Weise behandelt, um den Formiatkatalysator zu bereiten: – 100 g nasses (50 Gew.-%) Harz wurde in einem mit Wasser gefüllten Glasrohr (60 × 2,5 cm) aufgeschlämmt – das Chlorid wurde durch Behandlung mit 122,4 g Natriumformiat in wäßriger Lösung (10 molarer Überschuß, in 2.500 g Wasser) etwa 5 Stunden lang ausgetauscht (LHSV: 4 l/h) – das ausgetauschte Harz wurde 2 Stunden lang mit 1.200 ml Wasser gewaschen (LHSV: 4 l/h), bis in dem Waschwasser kein Chlorid mehr festgestellt werden konnte (AgNO3).
- • Ein stark basisches Ionenaustauscherharz vom quaternären Ammoniumtyp (AMBERJET 4200 (Handelsmarke), monodisperses vernetztes Polystyrol/Divinylbenzolharz von Rohm & Haas, Chloridform, Austauschkapazität 1,4 mÄq/ml) wurde in folgender Weise behandelt, um den Bicarbonat- oder Formiat-Vergleichskatalysator zu bereiten: – 150 ml nasses Harz wurden in einem mit Wasser gefüllten Glasrohr (60 × 2,5 cm) aufgeschlämmt – das Chlorid wurde durch Behandeln mit 176,4 g Natriumbicarbonat oder 151,2 g Natriumformiat in wäßriger Lösung (10 molarer Überschuß, in 2.500 g Wasser) während etwa 5 Stunden ausgetauscht (LHSV: 4 l/h) – das ausgetauschte Harz wurde mit 1.200 ml Wasser 2 Stunden lang gewaschen (LHSV: 4 l/h), bis in dem Waschwasser kein Chlorid mehr festgestellt werden konnte (AgNO3).
- 2. Ansatzweise Ethylenoxid-Hydrolysereaktion bei 100°C
- Ein 250 ml Autoklav wurde mit dem jeweiligen Katalysator (30 mMol) und mit Wasser (100 g; 5,55 Mol) gefüllt. Die Gaskappe wurde dreimal mit Stickstoff gespült, und es wurde ein Ausgangsdruck von 1.000 kPa Stickstoff angelegt. In den Versuchen 2.3 und 2.4 wurde das Gemisch über Nacht bei Raumtemperatur unter CO2 gerührt. In allen Fällen wurde das Gemisch auf 100°C erhitzt. Das Ethylenoxid (44 g; 1 Mol) wurde langsam unter Rühren (500 UpM) zugeführt. Das Reaktionsgemisch wurde 6 Stunden lang unter fortgesetztem Rühren auf der Reaktionstemperatur gehalten. Nach einem Abkühlen auf Raumtemperatur (20°C) wurde das Rühren über Nacht fortgesetzt und am Ende des Versuchsdurchganges wurde für die GLC-Analyse eine Probe genommen.
- Die Ergebnisse der katalytischen ansatzweisen Ethylenoxid-Hydrolyseversuche, ausgedrückt in Ethylenoxidumwandlung und Selektivität zu Monoethylenglycol, unter Anwendung der Katalysatoren vom Phosphoniumtyp in Bicarbonatform, sowie die Ergebnisse von Vergleichsversuchen (kein Katalysator, NaHCO3, AMBERJET 4200/Bicarbonat und zwei Tetraalkylammoniumbicarbonatkatalysatoren) sind in Tabelle 1 zusammengestellt.
- Tabelle 1. Ansatzweise EO-Hydrolyse bei 100°C mit Phosphoniumkatalysatoren und Vergleich mit ähnlichen Ammoniumkatalysatoren

- Die Ergebnisse zeigen, daß die Phosphonium/Bicarbonatkatalysatoren (sowohl homogen als auch auf der Polystyrol/Divinylbenzolmatrix) ein sehr attraktives katalytisches Verhalten, ausgedrückt als Selektivität zu MEG (83,8 bzw. 87,1%), aufweisen. Die Leistung ist sehr ähnlich zu derjenigen von anderen Katalysatoren vom Bicarbonattyp.
- 3. Katalysatorstabilitätstest
- Zum Vergleich der thermischen Stabilität einer quaternären Phosphoniumverbindung mit einer ähnlichen quaternären Ammoniumverbindung wurden beide in ihrer Hydroxidform überprüft, weil derartige Hydroxide gegenüber einem thermischen Abbau empfindlicher sind als die entsprechenden Bicarbonatformen.
- Die thermische Stabilität von Tetrabutylphosphoniumhydroxid (TBPH) wurde bewertet und mit der thermischen Stabilität von Tetrabutylammoniumhydroxid (TBAH) verglichen. Die Hydroxide (jeweils eine 40%-ige wäßrige Lösung) wurden mehrere Tage lang bei 100°C in einem Autoklaven gehalten. Für die Analyse wurden in. zeitlichen Intervallen Proben gezogen. Die Zersetzung der quaternären Basen wurde durch Kernmagnetresonanzspektroskopie (NMR) bestimmt, unter Heranziehung der Kohlenstoff-13 (13C)-NMR für die Ammoniumverbindungen und von Phosphor-31 (31P)-NMR für die Phosphoniumverbindungen.
- Die NMR-Analyse ergab, daß das thermische Abbauprodukt des quaternären Ammoniumhydroxids TBAH Tri-n-butylamin (TBA) war, und daß das thermische Abbauprodukt des quaternären Phosphoniumhydroxids TBPH Tri-n-butylphosphinoxid (TBPO) war.
- Die Ergebnisse dieser Stabilitätsuntersuchungen sind in Tabelle 2 zusammengefaßt.
- Tabelle 2. Thermische Stabilität eines quaternären Phosphoniumkatalysators im Vergleich mit einem ähnlichen quaternären Ammoniumkatalysator

- Diese Ergebnisse zeigen, daß die thermische Stabilität einer quaternären Phosphoniumverbindung signifikant besser ist als die thermische Stabilität einer ähnlichen quaternären Ammoniumverbindung.
Claims (4)
- Verfahren zur Herstellung von Alkylenglycolen durch Umsetzung eines Alkylenoxids mit Wasser in Gegenwart wenigstens einer ionischen Zusammensetzung aus einem quaternären Phosphoniumkation mit der allgemeinen Formel
- Verfahren nach Anspruch 1, worin das Anion aus der Gruppe von Formiat und Citrat ausgewählt ist.
- Verfahren nach einem der Ansprüche 1 bis 2, worin das quaternäre Phosphoniumkation auf einem festen Träger immobilisiert ist.
- Verfahren nach Anspruch 3, worin der feste Träger ein Anionenaustauschharz ist.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98204232 | 1998-12-14 | ||
| EP98204232 | 1998-12-14 | ||
| EP99201349 | 1999-04-29 | ||
| EP99201349 | 1999-04-29 | ||
| PCT/EP1999/010047 WO2000035840A1 (en) | 1998-12-14 | 1999-12-13 | Quaternary phosphonium salt catalysts in catalytic hydrolysis of alkylene oxides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69920508D1 DE69920508D1 (de) | 2004-10-28 |
| DE69920508T2 true DE69920508T2 (de) | 2005-10-20 |
Family
ID=26150967
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69920508T Expired - Lifetime DE69920508T2 (de) | 1998-12-14 | 1999-12-13 | Quaternäre phosponiumsalz-katalysatoren in der katalytischen hydrolyse der alkylenoxide |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP1140748B1 (de) |
| JP (1) | JP4323099B2 (de) |
| KR (1) | KR100649295B1 (de) |
| CN (1) | CN1149186C (de) |
| AT (1) | ATE276987T1 (de) |
| AU (1) | AU761984B2 (de) |
| BR (1) | BR9916254B1 (de) |
| CA (1) | CA2354775C (de) |
| DE (1) | DE69920508T2 (de) |
| ES (1) | ES2224725T3 (de) |
| MY (1) | MY120595A (de) |
| PE (1) | PE20001100A1 (de) |
| WO (1) | WO2000035840A1 (de) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GC0000169A (en) * | 2000-09-28 | 2005-06-29 | Shell Int Research | Catalytic process for producing an alkylene glycolwith reactor-output recycle. |
| US20040220433A1 (en) * | 2003-03-28 | 2004-11-04 | Evert Van Der Heide | Process for the preparation of propylene glycol |
| WO2004089866A1 (en) * | 2003-04-09 | 2004-10-21 | Shell Internationale Research Maatschappij B.V. | Process for the preparation of alkanediol |
| CA2469696A1 (en) | 2003-06-06 | 2004-12-06 | Shell Internationale Research Maatschappij B.V. | Process for the production of alkylene glycols using homogeneous catalysts |
| US7435858B2 (en) | 2004-12-23 | 2008-10-14 | Shell Oil Company | Process for the preparation of alkylene glycols |
| US8053609B2 (en) * | 2007-10-15 | 2011-11-08 | Sd Lizenzverwertungsgesellschaft Mbh & Co. Kg | Solid catalyst useful for converting an alkylene oxide to an alkylene glycol |
| TWI422429B (zh) | 2010-12-22 | 2014-01-11 | Ind Tech Res Inst | 奈米碳材承載型觸媒及碳酸酯的製造方法 |
| JP2013129613A (ja) * | 2011-12-20 | 2013-07-04 | Mitsubishi Chemicals Corp | トリアルキレングリコールの製造方法 |
| CN106478375A (zh) * | 2015-08-25 | 2017-03-08 | 中国石油化工股份有限公司 | 一种以阴离子交换树脂为催化剂合成邻二醇类化合物的方法 |
| EP3788029B1 (de) | 2018-04-30 | 2025-08-20 | Scientific Design LLC | Verfahren zur verbesserung der herstellung von ethylenglykol |
| US10807929B2 (en) | 2018-04-30 | 2020-10-20 | Scientific Design Company, Inc. | Process for preparing ethylene glycol |
| CN112041292A (zh) | 2018-04-30 | 2020-12-04 | 科学设计有限公司 | 用于制备乙二醇的再循环方法 |
| WO2019213034A1 (en) | 2018-04-30 | 2019-11-07 | Scientific Design Company, Inc. | Epoxidation process with concentrated ethylene oxide solutions |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4160116A (en) * | 1978-08-28 | 1979-07-03 | Showa Denko K.K. | Process for the production of alkylene glycols |
| GB2086894B (en) * | 1980-11-10 | 1984-06-20 | Ici Plc | Production of alkylene glycols |
| CA1314566C (en) * | 1984-03-28 | 1993-03-16 | John Howard Robson | Process for the production of alkylene glycols with metalate-containing solid |
| JPH09508136A (ja) * | 1994-01-31 | 1997-08-19 | シエル・インターナシヨネイル・リサーチ・マーチヤツピイ・ベー・ウイ | アルキレングリコールの製造方法 |
-
1999
- 1999-12-10 PE PE1999001221A patent/PE20001100A1/es not_active Application Discontinuation
- 1999-12-10 MY MYPI99005371A patent/MY120595A/en unknown
- 1999-12-13 AT AT99959416T patent/ATE276987T1/de not_active IP Right Cessation
- 1999-12-13 ES ES99959416T patent/ES2224725T3/es not_active Expired - Lifetime
- 1999-12-13 EP EP99959416A patent/EP1140748B1/de not_active Expired - Lifetime
- 1999-12-13 WO PCT/EP1999/010047 patent/WO2000035840A1/en not_active Ceased
- 1999-12-13 JP JP2000588104A patent/JP4323099B2/ja not_active Expired - Fee Related
- 1999-12-13 DE DE69920508T patent/DE69920508T2/de not_active Expired - Lifetime
- 1999-12-13 BR BRPI9916254-7A patent/BR9916254B1/pt not_active IP Right Cessation
- 1999-12-13 KR KR1020017007323A patent/KR100649295B1/ko not_active Expired - Fee Related
- 1999-12-13 CA CA002354775A patent/CA2354775C/en not_active Expired - Fee Related
- 1999-12-13 CN CNB998156736A patent/CN1149186C/zh not_active Expired - Fee Related
- 1999-12-13 AU AU16591/00A patent/AU761984B2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| CN1149186C (zh) | 2004-05-12 |
| PE20001100A1 (es) | 2000-11-16 |
| WO2000035840A1 (en) | 2000-06-22 |
| CN1333740A (zh) | 2002-01-30 |
| BR9916254A (pt) | 2001-10-02 |
| EP1140748B1 (de) | 2004-09-22 |
| JP2002532446A (ja) | 2002-10-02 |
| ATE276987T1 (de) | 2004-10-15 |
| EP1140748A1 (de) | 2001-10-10 |
| DE69920508D1 (de) | 2004-10-28 |
| CA2354775C (en) | 2009-03-24 |
| CA2354775A1 (en) | 2000-06-22 |
| KR20010082337A (ko) | 2001-08-29 |
| KR100649295B1 (ko) | 2006-11-24 |
| ES2224725T3 (es) | 2005-03-01 |
| JP4323099B2 (ja) | 2009-09-02 |
| AU1659100A (en) | 2000-07-03 |
| AU761984B2 (en) | 2003-06-12 |
| MY120595A (en) | 2005-11-30 |
| BR9916254B1 (pt) | 2010-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69505641T2 (de) | Verfahren zur herstellung von alkylenglykolen | |
| DE69920508T2 (de) | Quaternäre phosponiumsalz-katalysatoren in der katalytischen hydrolyse der alkylenoxide | |
| RU2222522C2 (ru) | Способ получения алкиленгликолей | |
| EP0523089B1 (de) | Verwendung von hydrophobierten hydrotalciten als katalysatoren für die ethoxylierung bzw. propoxylierung | |
| EP0857709B1 (de) | Verfahren zur Herstellung von 1,3-Propandiol | |
| DE69815154T2 (de) | Verfahren zur herstellung von ethylenglykol | |
| WO2001087809A1 (de) | Verfahren zur herstellung von gesättigten c3-c20-alkoholen | |
| DE69606723T2 (de) | Verfahren zur herstellung von alkylenglykolen | |
| DE60104870T2 (de) | Katalytische verfahren zur herstellung von alkylenglykol mit reaktorausgangsleistungskreislauf | |
| DE69817392T2 (de) | Katalytische hydrolyse von alkylenoxiden | |
| DE102010013992A1 (de) | Verfahren zur Herstellung eines Carbamats, ein in dem Verfahren eingesetzter Katalysator, ein Verfahren zur Herstellung des Katalysators und seine Verwendung | |
| DE19928549A1 (de) | Bisphenolethoxylat-Syntheseverfahren | |
| DE69911249T2 (de) | Stabilisierender zusatz des katalysators in der hydrolyse von alkylenoxiden | |
| DE69408813T2 (de) | Verfahren zur Herstellung von Diol-Verbindungen | |
| EP1123912B1 (de) | Verfahren zur Entfernung von gelöstem Sauerstoff aus Phenol | |
| DE3236765C2 (de) | Verfahren zur Abtrennung von Indol durch Destillation | |
| DE602004005702T2 (de) | Verfahren zur entfernung von organischen halogenhaltigen verbindungen | |
| DE4213870C2 (de) | Bisphenol-Synthese an modifizierten Ionenaustauscherharzen unter Verwendung speziell gereinigter Carbonylverbindungen | |
| WO1999033772A1 (de) | Verfahren zur herstellung von alkylenglykol | |
| DE68915694T2 (de) | Verfahren zur Herstellung von verzweigten 1,3-Glykolen und ihren Monoestern. | |
| DE10031187A1 (de) | Verfahren zur Herstellung von Monoalkylenglykol aus Alkylenoxid und Wasser | |
| DE69502002T2 (de) | Verfahren zur Herstellung von Hydroxyaldehyden | |
| DE2431034A1 (de) | Verfahren und katalysator zur oxidation von olefinen | |
| DE69706514T2 (de) | Methode zur Wiedergewinnung und Gebrauch von Wärme durch Anwendung chemischer Energie bei der Synthese und Spaltung von Methylformiat | |
| DE3338882C2 (de) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8328 | Change in the person/name/address of the agent |
Representative=s name: JUNG, SCHIRDEWAHN, GRUENBERG, SCHNEIDER PATENTANWAELTE |
|
| 8328 | Change in the person/name/address of the agent |
Representative=s name: ADVOTEC. PATENT- UND RECHTSANWAELTE, 80538 MUENCHE |