EP0486900B1 - Procédé pour la préparation d'indoles - Google Patents
Procédé pour la préparation d'indoles Download PDFInfo
- Publication number
- EP0486900B1 EP0486900B1 EP91119053A EP91119053A EP0486900B1 EP 0486900 B1 EP0486900 B1 EP 0486900B1 EP 91119053 A EP91119053 A EP 91119053A EP 91119053 A EP91119053 A EP 91119053A EP 0486900 B1 EP0486900 B1 EP 0486900B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- hydrogen
- phenylhydrazine
- process according
- sodium
- branched
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 *C(*1)=C(*)C2=C1C/C(/*)=C/C=*(\*)/C2 Chemical compound *C(*1)=C(*)C2=C1C/C(/*)=C/C=*(\*)/C2 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/86—Carbazoles; Hydrogenated carbazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the ring system
Definitions
- the present invention relates to a process for the preparation of indoles by cyclization of phenylhydrazones, which are formed from phenylhydrazine and ketones, in the presence of less than 5 equivalents of sodium hydrogen sulfate.
- Indoles are important intermediates in the dye industry. It is known to cyclize phenylhydrazones in organic media using Lewis or protonic acids, for example in ethanol in the presence of zinc chloride (Chem. Zeitg. 22 (1898), 38), in glacial acetic acid as the reaction medium (J. Org. Chem. 42 (1977), 2474), in chloroform in the presence of polyphosphoric acid ester (Chem. And Ind. 1965 , 473). Furthermore, such a reaction is described in polyphosphoric acid without further solvent addition (J. Am. Chem. Soc. 74 (1952), 3948).
- indole synthesis in aqueous reaction medium can also be carried out with sodium hydrogen sulfate in short reaction times with good yields, using less than 5 equivalents of sodium hydrogen sulfate, based on the phenylhydrazine.
- This is all the more surprising since it is known from DE-OS 19 06 832 that when using substituted aliphatic or aromatic acids, the pK value of which is close to 1.3, large amounts of acid and long reaction times are required in order to obtain satisfactory yields achieve.
- Suitable phenylhydrazines for the process according to the invention are: Phenylhydrazine, 4-methylphenylhydrazine, 4-dodecylphenylhydrazine, 2,5-dimethylphenylhydrazine, 3,4-dimethylphenylhydrazine, 4-methoxyphenylhydrazine, 4-hydroxyphenylhydrazine, 4-chlorophenylhydrazine, 5-chloro-2-methylphenylhydrazine, 2,4-dichlorophenylhydrazine.
- Suitable ketones for the process according to the invention are: 2-butanone, 3-methyl-2-butanone, 3-pentanone, 4-methyl-2-pentanone, cyclohexanone, 2-methylcyclohexanone, 2-dodecanone.
- Sodium hydrogen sulfate is used in an amount of less than 5 equivalents per 1 mol of the phenylhydrazine used, preferably in an amount of 1-4.9 equivalents, particularly preferably 1.5-4.5 equivalents, very particularly preferably 2-4 equivalents.
- the process according to the invention is carried out at a temperature of 50-150 ° C., preferably 80-110 ° C.
- both the phenylhydrazine and the ketone can be initially introduced together with sodium hydrogen sulfate in an aqueous medium, brought to the reaction temperature and then the reaction component which is still missing is added. It is often advantageous to present the phenylhydrazine. It is also possible to add the phenylhydrazine and the ketone simultaneously to the aqueous medium at the reaction temperature with sodium bisulfate; it may also be advantageous here to provide some phenylhydrazine and then to add both components simultaneously. The simultaneous addition is suitable also for a continuous way of working. In all cases, the formation of the phenylhydrazone and its cyclization to the indole take place in succession with NH 3 elimination.
- the reaction mixture is neutralized.
- the indole is obtained as an organic phase; if necessary, the indole can be further purified in a manner familiar to the person skilled in the art.
- the saline aqueous phase which is obtained during the neutralization, is disposed of in a known manner. This salt load requiring disposal is smaller in the process according to the invention than in conventional indole syntheses.
- a phenylhydrazine is used which is prepared from the associated sodium phenylhydrazodisulfonate and / or the associated sodium phenylhydrazo- ⁇ -sulfonate by adding sulfuric acid. This preparation generally takes place in the presence of residual sodium sulfite and / or sodium hydrogen sulfite, which had been used to form the hydrazosulfonates.
- the amount of sulfuric acid required is set for the use of such a phenylhydrazine such that the remaining sodium sulfite, sodium bisulfite and the sulfate split off from the hydrazosulfonate are used less than 5 equivalents of sodium hydrogen sulfate per mole of phenylhydrazine.
- the ketone is then added to such a phenylhydrazine, which contains the stated amount of sodium hydrogen sulfate, and the process according to the invention is carried out as described in more detail above.
- aqueous benzene diazonium chloride solution prepared by conventional methods from 2 moles of aniline, 4.2 moles of hydrochloric acid and 2.02 moles of sodium nitrite was reacted according to known methods with 5.2 moles of a mixture of sodium sulfite and sodium bisulfite to give a phenylhydrazodisulfonate solution.
- 80 ° C were too 150 ml of 48% sulfuric acid (1 mol) were added dropwise to this solution. The mixture was stirred at 80 ° C. for 2 h, then 225 ml (2.1 mol) of methyl isopropyl ketone were metered in over 30 min.
- aniline was diazotized with hydrochloric acid and sodium nitrite in a molar ratio of 1: 2.3: 1.07.
- This diazonium salt solution was also continuously reacted with 2.55 mol of sodium sulfite / bisulfite mixture per mol of aniline to give a phenylhydrazodisulfonate solution.
- a portion of this mixture, corresponding to 1.91 mol of aniline, was added dropwise at 80 ° C. to 200 ml of a 48% sulfuric acid (1.35 mol). After a reaction time of 2 h at 80 ° C., 225 ml (2.1 mol) of methyl isopropyl ketone were metered in in 30 min. After a reaction time of 3 h at 95 to 100 ° C., the mixture was cooled. The mixture was neutralized, the organic phase was separated off and distilled.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Indole Compounds (AREA)
Claims (8)
- Procédé de préparation des 1-H- et 3-H-indoles de formules respectives

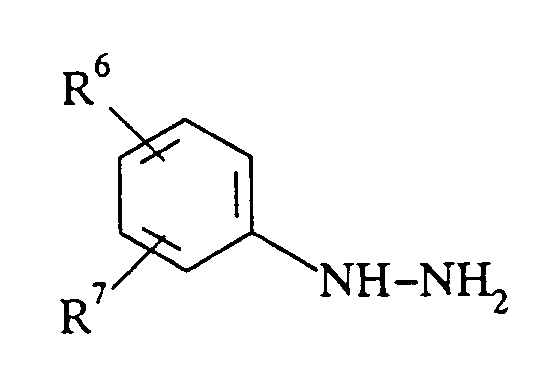
 R1 représente l'hydrogène,R2 représente un groupe méthyle,R3 représente l'hydrogène ou un groupe méthyle,R1 pouvant en outre représenter un groupe méthyle lorsque R3 représente l'hydrogène,R1 et R2 peuvent également former ensemble un groupe triméthylène etR2 peut en outre représenter un groupe isopropyle, n-butyle ou n-nonyle lorsqueR1 et R3 représentent l'hydrogène,R6 et R7 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe alkyle à chaîne droite ou ramifiée en C1-C12, un groupe alcoxy à chaîne droite ou ramifiée en C1-C4, un groupe phényle, phénoxy, un halogène, un groupe hydroxy, nitro, cyano ou N(R8,R9),R6 pouvant en outre représenter CON(R8R9), COR8 ou COOR8 etR8 et R9 représentent chacun, indépendamment l'un de l'autre, l'hydrogène ou un groupe alkyle à chaîne droite ou ramifiée en C1-C4,caractérisé en ce que l'on met en oeuvre du bisulfate de sodium en quantité inférieure à cinq équivalents pour une mole de phénylhydrazine.
R1 représente l'hydrogène,R2 représente un groupe méthyle,R3 représente l'hydrogène ou un groupe méthyle,R1 pouvant en outre représenter un groupe méthyle lorsque R3 représente l'hydrogène,R1 et R2 peuvent également former ensemble un groupe triméthylène etR2 peut en outre représenter un groupe isopropyle, n-butyle ou n-nonyle lorsqueR1 et R3 représentent l'hydrogène,R6 et R7 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe alkyle à chaîne droite ou ramifiée en C1-C12, un groupe alcoxy à chaîne droite ou ramifiée en C1-C4, un groupe phényle, phénoxy, un halogène, un groupe hydroxy, nitro, cyano ou N(R8,R9),R6 pouvant en outre représenter CON(R8R9), COR8 ou COOR8 etR8 et R9 représentent chacun, indépendamment l'un de l'autre, l'hydrogène ou un groupe alkyle à chaîne droite ou ramifiée en C1-C4,caractérisé en ce que l'on met en oeuvre du bisulfate de sodium en quantité inférieure à cinq équivalents pour une mole de phénylhydrazine. - Procédé selon la revendication 1, caractérisé en ce que l'on met en oeuvre une phénylhydrazine ayant au moins une position ortho non substituée et répondant à la formule
 R16 et R17 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe alkyle à chaîne droite ou ramifiée en C1-C12, un groupe alcoxy à chaîne droite ou ramifiée en C1-C4, un groupe phényle, phénoxy, un halogène, un groupe hydroxy, cyano ou N(R18,R19),R18 et R19 représentant chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe méthyle ou éthyle,de préférence une phénylhydrazine ayant au moins une position ortho non substituée et répondant à la formule
R16 et R17 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe alkyle à chaîne droite ou ramifiée en C1-C12, un groupe alcoxy à chaîne droite ou ramifiée en C1-C4, un groupe phényle, phénoxy, un halogène, un groupe hydroxy, cyano ou N(R18,R19),R18 et R19 représentant chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe méthyle ou éthyle,de préférence une phénylhydrazine ayant au moins une position ortho non substituée et répondant à la formule R26 et R27 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe méthyle, éthyle, méthoxy, éthoxy ou un halogène, R26 pouvant en outre représenter un groupe hydroxy ou N(R18,R19).
R26 et R27 représentent chacun, indépendamment l'un de l'autre, l'hydrogène, un groupe méthyle, éthyle, méthoxy, éthoxy ou un halogène, R26 pouvant en outre représenter un groupe hydroxy ou N(R18,R19). - Procédé selon la revendication 1, caractérisé en ce que l'on met en oeuvre de 1 à 4,9 équivalents, de préférence de 1,5 à 4,5 équivalents et tout spécialement de 2 à 4 équivalents de bisulfate de sodium par mole de phénylhydrazine.
- Procédé selon la revendication 1, caractérisé en ce que l'on opère à des températures de 50 à 150°C, de préférence de 80 à 110°C.
- Procédé selon la revendication 1, caractérisé en ce que l'on met en oeuvre une phénylhydrazine qui a été préparée à partir du phénylhydrazodisulfonate alcalin correspondant et/ou du phénylhydrazo-β-sulfonate alcalin correspondant en présence du sulfite de sodium et/ou du bisulfite de sodium résiduels par addition d'acide sulfurique.
- Procédé selon la revendication 5, caractérisé en ce que l'on règle la quantité d'acide sulfurique en sorte que le sulfate libéré à partir du sulfite et du bisulfite de sodium résiduel et de l'hydrazosulfonate, représente moins de 5 équivalents de bisulfate de sodium par mole d'hydrazine.
- Procédé selon la revendication 5, caractérisé en ce que la scission du groupe sulfonate à partir du phénylhydrazodisulfonate de sodium et/ou du phénylhydrazo-β-sulfonate de sodium est réalisée en présence de la cétone et en ce que la synthèse de l'indole est réalisée dans le même milieu sans isolement préalable de la phénylhydrazine.
- Procédé selon la revendication 7, caractérisé en ce que, pour la préparation de l'indole, on part d'une aniline non substituée dans au moins une position ortho et répondant à la formule
 R6 pouvant en outre représenter CON(R8,R9), COR8 ou COOR8, et on procède aux opérations de diazotation, de réduction du sel de diazonium à l'aide d'un sulfite, de scission du groupe sulfonate et finalement de réaction avec la cétone sans isoler les produits intermédiaires.
R6 pouvant en outre représenter CON(R8,R9), COR8 ou COOR8, et on procède aux opérations de diazotation, de réduction du sel de diazonium à l'aide d'un sulfite, de scission du groupe sulfonate et finalement de réaction avec la cétone sans isoler les produits intermédiaires.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4037004A DE4037004A1 (de) | 1990-11-21 | 1990-11-21 | Verfahren zur herstellung von indolen |
| DE4037004 | 1990-11-21 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0486900A2 EP0486900A2 (fr) | 1992-05-27 |
| EP0486900A3 EP0486900A3 (en) | 1992-08-19 |
| EP0486900B1 true EP0486900B1 (fr) | 1996-09-04 |
Family
ID=6418634
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP91119053A Expired - Lifetime EP0486900B1 (fr) | 1990-11-21 | 1991-11-08 | Procédé pour la préparation d'indoles |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US5179211A (fr) |
| EP (1) | EP0486900B1 (fr) |
| JP (1) | JPH04266871A (fr) |
| DE (2) | DE4037004A1 (fr) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6133453A (en) * | 1998-05-15 | 2000-10-17 | Pharm-Eco Laboratories, Inc. | Method for making substituted indoles |
| MX2011006564A (es) | 2008-12-19 | 2011-09-26 | Amgen Inc | Procedimiento mejorado para la preparacion de 1-acetil-6-amino-3,3-dimetil-2,3-dihidroindol. |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE574840C (de) * | 1931-11-01 | 1933-04-20 | I G Farbenindustrie Akt Ges | Verfahren zur Darstellung von Indolen |
| US3639420A (en) * | 1968-02-21 | 1972-02-01 | Toms River Chemical Corp | Process for the preparation of 2 3 3-trimethyl indolenines |
| IT1015908B (it) * | 1974-04-05 | 1977-05-20 | Snam Progetti | Procedimento per la sintesi di indolenine alchil sostituite |
| FR2275461A1 (fr) * | 1974-06-18 | 1976-01-16 | Labaz | Nouveaux stabilisants des polymeres et copolymeres du chlorure de vinyle |
-
1990
- 1990-11-21 DE DE4037004A patent/DE4037004A1/de not_active Withdrawn
-
1991
- 1991-11-08 DE DE59108138T patent/DE59108138D1/de not_active Expired - Fee Related
- 1991-11-08 EP EP91119053A patent/EP0486900B1/fr not_active Expired - Lifetime
- 1991-11-13 US US07/792,317 patent/US5179211A/en not_active Expired - Fee Related
- 1991-11-18 JP JP3328296A patent/JPH04266871A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JPH04266871A (ja) | 1992-09-22 |
| EP0486900A3 (en) | 1992-08-19 |
| DE4037004A1 (de) | 1992-05-27 |
| EP0486900A2 (fr) | 1992-05-27 |
| US5179211A (en) | 1993-01-12 |
| DE59108138D1 (de) | 1996-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69123336T2 (de) | Neue Synthese für Pyrazolfarbstoffe | |
| DE1443877C3 (fr) | ||
| DE2842263A1 (de) | Herstellung chromogener pyridinverbindungen | |
| EP0486900B1 (fr) | Procédé pour la préparation d'indoles | |
| DE1237580B (de) | Verfahren zur Herstellung von in 1-Stellung substituierten 3-Phenylamino-2-pyrazolin-5-onen | |
| DE1670914A1 (de) | Verfahren zur Herstellung von 2-Aryl-v-triazolen | |
| DE2948142C2 (de) | Verfahren zur Herstellung von 21-Acylthio-Steroiden | |
| EP0368085B1 (fr) | Procédé de préparation d'esters d'acide de 1,2-naphtoquinone-(2)-diazide-4-sulfoniques substitués et leur emploi dans des mélanges photosensibles | |
| EP1001929B1 (fr) | Procede pour la preparation d'acide 3-cyano-2, 4-diahalogene-5-fluor-benzoique | |
| EP0008323B1 (fr) | Procédé de préparation d'aldéhydes de pyridine et leurs dérivés et composés préparés selon ce procédé | |
| DE3035056C2 (de) | Verfahren zur Herstellung von 1,3,4-Thiadiazolazofarbstoffen | |
| EP0283898A2 (fr) | Procédé de préparation d'acides aryldiazidesulfoniques | |
| DE2300221A1 (de) | Verfahren zur herstellung von 3anilino-pyrazolonen-(5) | |
| DE3241886A1 (de) | Verfahren zur herstellung eines 2-aequivalent-purpurrotkupplers | |
| DE1643463C3 (de) | Verfahren zur Herstellung von l-(p-Chlorbenzoyl>2-methyl-3-indolylessigsäuren | |
| EP0014454A2 (fr) | Procédé et intermédiaires pour la préparation de dérivés de 2-benzazépine | |
| DE69728703T2 (de) | Verfahren zur Herstellung von 1,1-disubstituierten-1H-Benz[e]indol Derivaten und Hydroxyl-substituierte 1,1-disubstituierte-1H-Benz[e]indol Derivate | |
| EP0342532B1 (fr) | Procédé pour la préparation du 4-chloro-2-méthyl-5-nitro-phénol | |
| DE2546657A1 (de) | Verfahren zur herstellung von benzidinpigmenten | |
| DE1620014C3 (de) | Verfahren zur Herstellung von (2-Methyl-3-indolyl)-essigsäure-tertbutylestern | |
| EP0095638A1 (fr) | 2,6-Dicyanoanilines | |
| AT274800B (de) | Verfahren zur Herstellung von Indolderivaten | |
| EP0125621B1 (fr) | Procédé dans un seul réacteur pour la préparation de N-alkylanilines substituées dans le noyau | |
| DE2054564A1 (de) | Verfahren zur Herstellung wasser löslicher kationischer Farbstoffe | |
| EP0072508A2 (fr) | Dérivés de pyrazolone, leur préparation et leur utilisation comme copulants |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): CH DE FR GB IT LI |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): CH DE FR GB IT LI |
|
| 17P | Request for examination filed |
Effective date: 19920907 |
|
| 17Q | First examination report despatched |
Effective date: 19950216 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): CH DE FR GB IT LI |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: E. BLUM & CO. PATENTANWAELTE |
|
| REF | Corresponds to: |
Ref document number: 59108138 Country of ref document: DE Date of ref document: 19961010 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19961014 Year of fee payment: 6 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 19960923 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19961021 Year of fee payment: 6 |
|
| ITF | It: translation for a ep patent filed | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19961030 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19961121 Year of fee payment: 6 |
|
| ET | Fr: translation filed | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19971108 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19971130 Ref country code: FR Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 19971130 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19971130 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19971108 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980801 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20051108 |