EP2098592A2 - RNA-abhängige RNA-Polymerase, Verfahren und Kits zur Amplifikation und/oder Markierung von RNA - Google Patents
RNA-abhängige RNA-Polymerase, Verfahren und Kits zur Amplifikation und/oder Markierung von RNA Download PDFInfo
- Publication number
- EP2098592A2 EP2098592A2 EP09155984A EP09155984A EP2098592A2 EP 2098592 A2 EP2098592 A2 EP 2098592A2 EP 09155984 A EP09155984 A EP 09155984A EP 09155984 A EP09155984 A EP 09155984A EP 2098592 A2 EP2098592 A2 EP 2098592A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- rna
- pol
- norovirus
- dependent
- primer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images





























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/12—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
- C12N9/1241—Nucleotidyltransferases (2.7.7)
- C12N9/127—RNA-directed RNA polymerase (2.7.7.48), i.e. RNA replicase
Definitions
- the invention relates to an RNA-dependent RNA polymerase as well as methods and kits for labeling and / or for primer-dependent and independent amplification of ribonucleic acid (RNA), in particular of viral, eukaryotic and prokaryotic and double-stranded ribonucleic acid (RNA) for use in biotechnology and the medicine.
- RNA ribonucleic acid
- the amplification method according to the invention is particularly suitable for microarray technology, for the production of siRNA and for the diagnosis of viral infections by the detection of viral RNA in patient material.
- the labeling method according to the invention is particularly suitable for the purification of RNA by affinity binding, as well as for the labeling of RNA for use in molecular biological methods that are used in the characterization of the function and / or structure of viral, eukaryotic and prokaryotic and double-stranded ribonucleic acid (RNA). be used.
- RNA ribonucleic acid
- RNA is an important component of the eukaryotic and prokaryotic cells. It is involved in several vital functions: transcription, i. the description of the information of the genetic material (deoxyribonucleic acid, DNA), the transmission of this information from the nucleus to the cytoplasm (mRNA), and the translation, i. the translation of the rewritten information into amino acids or proteins.
- the RNA also forms the so-called transfer RNA, an important building block for translation.
- RNA is also an important component of ribosomes. These are structural units of the cytoplasm, at which the translation of the information into protein takes place.
- RNA Ribonucleic acid
- miRNA miRNA
- siRNA small interfering RNA
- siPNA in vitro has hitherto been carried out by chemical synthesis or by DNA-dependent RNA polymerases such as T7 RNA polymerase.
- DNA-dependent RNA polymerases such as T7 RNA polymerase.
- two complementary DNA strands are synthesized by PCR using a primer that also contains the sequence for a T7 promoter.
- the resulting complementary RNA strands are hybridized and digested by RNAse 1.
- RNA amplification plays an important role in the detection of viral infections in patient material.
- RNA viruses ie viral pathogens with an RNA-derived genetic material.
- Important pathogens from this group of viruses include the human immunodeficiency virus (HIV), the hepatitis C virus (HCV), the hepatitis A virus, the flu virus (influenza A, B and C) but also the Avian influenza virus, the recently re-identified SARS virus, the polio virus (poliovirus), the measles virus, the mumps virus, the rubella virus and the diarrhea viruses (rotavirus, sapovirus and norovirus).
- RNA for example for the expression detection or for the production of siRNA, the transcription of the PCR product into RNA by DNA-dependent RNA polymerases, such as T7 polymerase, additionally takes place.
- DNA-dependent RNA polymerases such as T7 polymerase
- the implementation of these steps usually takes 18 to 24 hours. Also required is a certain "know-how", which is not accessible to all research institutions.
- RNA-dependent RNA polymerases e.g. the bacteriophage Q ⁇ RNA polymerase (also called Q ⁇ replicase), poliovirus or hepatitis C virus RNA polymerase.
- the genome of bacteriophage Q ⁇ consists of a single-stranded (+) RNA that is replicated by the RNA-dependent RNA polymerase.
- the Q ⁇ replicase produces a (-) strand RNA copy of the (+) strand, which as well as (+) strands can function as a template. In this way an exponential duplication can be achieved.
- the Q ⁇ system is already used to detect analytes, such as nucleic acids ( Chu et al. (1986), Nucleic Acids Research 14, 5591-5603 ; Lizardi et al (1988), Biotechnology 6, 1197-1202 ).
- RNA-dependent RNA polymerases require specific RNA sequences for the initiation of RNA replication. In addition, they are also dependent on certain auxiliary proteins for the amplification of RNA. Therefore, these RNA-dependent RNA polymerases are unable to amplify heterologous RNA.
- RNA at the 3'-terminus is possible in the prior art only with ATP by the use of E. coli poly (A) - transferase. Methods for labeling RNA with CTP, UTP and GTP are not known.
- RNA-dependent RNA polymerase and methods and kits for labeling and / or amplifying viral, eukaryotic and prokaryotic single and double-stranded ribonucleic acid (RNA) that are more efficient and faster.
- a so-called "right-hand conformation” is understood to mean that the tertiary structure (conformation) of the protein has a folding according to a right hand with finger (finger), palm (palm) and thumb (thumb).
- the sequence section "XXDYS” is the so-called A motif.
- the A motif is responsible for the discrimination between ribonucleosides and deoxyribonucleosides.
- the sequence section GXPSG is the so-called B motif, and the B motif is conserved in all members of the family Caliciviridae .
- the sequence section "YGDD” is the so-called C motif.
- the C motif represents the active site of the enzyme. This motif plays an important role in the coordination of metal ions.
- the sequence section "XXYGL” is the so-called D motif.
- the D motif is a feature of the template-dependent polymerases.
- the sequence section "XXXXFLXRXX” is the so-called E-motif.
- the E motif is a feature of RNA-dependent RNA polymerases, and does not occur in DNA-dependent RNA polymerases.
- the primer-dependent amplification of the RNA takes place only in the presence of a sequence-specific primer.
- the amplification is sequence-dependent and occurs by incorporation of AMP, GMP, CMP or UMP.
- the primer-independent amplification of the RNA takes place in the absence of a sequence-specific primer.
- the amplification is sequence-dependent and takes place by incorporation of AMP, GMP, CMP or UMP.
- RNA strand e.g., ATP or UTP or CTP or GTP
- RNA strand e.g., ATP or UTP or CTP or GTP
- the RNA-dependent RNA polymerase according to the invention is able to use in vitro heterologous viral, eukaryotic and prokaryotic RNA as a template for the amplification reaction. Both positive-stranded and negative-stranded single-stranded and double-stranded RNA can be used as a template for amplification.
- the RNA-dependent RNA polymerase according to the invention is preferably an RNA-dependent RNA polymerase of a virus of the family Caliciviridae.
- the genome of Caliciviridae consists of a polyadenylated (+) - stranded RNA single strand.
- the RNA-dependent RNA polymerase of calicivirus in viral replication writes the genomic calicivirus RNA into a (-) - stranded antisense RNA (aRNA) to. This results in an RNA aRNA hybrid.
- the (-) - stranded aRNA then serves as a template for the synthesis of new (+) - stranded genomic virus RNA.
- the RNA-dependent RNA polymerase is an RNA-dependent RNA polymerase of a human and / or non-human pathogenic calicivirus. Most preferably, it is an RNA-dependent RNA polymerase of a norovirus, sapovirus, vesivirus or a lagovirus, e.g. the RNA-dependent RNA polymerase of the norovirus strain HuCV / NL / Dresden174 / 1997 / GE or the sapovirus strain pJG-Sap01 or the virus strain Vesivirus FCV / Dresden / 2006 / GE.
- RNA-dependent RNA polymerases of the Norovirus, the sapovirus and the Vesivivirus can be produced recombinantly by cloning the Norovirus, Sapovirus or Vesivirus-RdRP-encoding nucleic acid into a suitable expression vector, for example pET-28 (Novagen).
- a suitable expression vector for example pET-28 (Novagen).
- the expression vector with the gene sequence coding for the virus RdRP is introduced into a suitable host organism for expression.
- the host organism is preferably selected from the prokaryotic, preferably Escherichia coli, or eukaryotic host organisms, preferably Sacharomyces cerevisae or insect cells (preferably Sf9 cells) which have been infected with recombinant baculoviruses, which is usually used for protein expression.
- This host organism contains at least one expression vector with one Gene sequence coding for the virus RdRP,
- the norovirus, sapovirus, or vesivirus calicivirus RdRP is fused at the N- or C-terminus to a protein sequence that facilitates purification of the RdRP fusion protein upon expression in a host organism.
- this sequence is a so-called histidine marker consisting of a sequence of at least 6 consecutive histidines (His or H).
- Histidine marker allows in a known manner the purification of the protein by affinity chromatography on a nickel or cobalt column.
- RNA-dependent RNA polymerase for the amplification and / or labeling of RNA.
- the RNA template is preferably used in amounts of from 1 ⁇ g to 4 ⁇ g per 50 ⁇ l reaction mixture.
- concentration of the ribonucleotides ATP, CTP, GTP and UTP (NTPs) used is preferably between 0.1 ⁇ mol / l and 1 ⁇ mol / l, particularly preferably 0.4 ⁇ mol / l.
- NTP analogues can also be used in the RNA amplification method according to the invention, for example fluorescently labeled NTPs, biotin-labeled NTPs or radioactively-labeled NTPs such as [P 32 ] -labeled NTPs.
- concentration of the RdRP according to the invention is preferably between 1 ⁇ mol / l and 3 ⁇ mol / l.
- the process is preferably carried out at a pH between 6.5 and 8.5 and at a temperature between 20 ° C and 40 ° C.
- a preferred embodiment of the method for RNA amplification is carried out at 30 ° C with norovirus RdRP or at 37 ° C with sapovirus RdRP under the following buffer conditions: 50 mmol / l HEPES, pH 8.0, 3 mmol / l magnesium acetate or manganese chloride, 4 mmol / l DTT.
- the RNA / antisense RNA duplex is separated into individual RNA strands by heat denaturation, by chemical denaturation and / or enzymatically, eg, by an enzyme capable of resolving double-stranded RNA into single-stranded RNA, such as a helicase ,
- Part of the invention is a corresponding kit for the amplification of RNA, which in addition to the kit according to the invention an enzyme with the ability to separate double-stranded RNA into single-stranded RNA, such as. a helicase containing.
- One embodiment of the invention is a method for primer-dependent amplification of RNA, wherein at least one primer which hybridizes with a section of the RNA template is used, and after the attachment of the RNA-dependent RNA polymerase according to the invention the primer by the RNA-dependent RNA Polymerase is extended according to the sequence of the RNA template.
- an RNA primer or a DNA primer e.g. with a length of 20 to 25 bases, preferably used in a concentration of 0.1 to 1 ⁇ mol / l.
- an RNA template e.g. viral, prokaryotic and eukaryotic RNA can be used.
- FIG. 2 The course of primer-dependent RNA amplification of a single-stranded RNA template using an RNA primer is in FIG. 2 shown schematically.
- polyadenylated RNA, polyuridylated RNA, polyguanylated RNA or polycytidylylated RNA are used as primers, a poly-U-RNA, poly-A-RNA, poly-C-RNA or poly-G-RNA primer.
- a poly-U-RNA primer With the help of the poly-U-RNA primer, a specific amplification of the entire cellular mRNA is possible.
- a poly-U primer of 20 to 24 bases in length is used for this purpose.
- the amplified cellular mRNA can be examined casually with the help of the so-called microarray method.
- the method according to the invention for sequence-specific RNA amplification is also advantageously used for detecting viral RNA in patient material.
- RNA primer that specifically hybridizes to a particular portion of viral RNA
- CSF CSF
- blood plasma or body fluids are used as patient material.
- plasma CSF
- body fluids are used as patient material.
- RNA viruses having a poly A tail at the 3 'end of the genome are also suitable. Also suitable is the method of detecting RNA viruses having a poly A tail at the 3 'end of the genome, DNA viruses and viral mRNA transcripts.
- the attachment of the RNA-dependent RNA polymerase to the RNA template is primer-independent.
- the sequence of sequence-independent RNA amplification of a single-stranded RNA template without the use of an RNA primer is in FIG. 1 shown schematically.
- a further embodiment of the invention is a method for the primer-independent amplification of poly (C) -RNA, characterized in that no primer which hybridizes with a section of the RNA template is used, which uses GTP, preferably 50 ⁇ M, as sole nucleotide is extended by the RNA-dependent RNA polymerase according to the sequence of the RNA template.
- GTP preferably 50 ⁇ M
- RNA-primer-independent amplification of the RNA The possible applications of the method according to the invention for the amplification of RNA are manifold.
- the direct detection of viral genome in patient material is possible.
- the entire cellular RNA is recovered and amplified using an RNA primer specific RNA sequence.
- the detection of the viral nucleic acid is then carried out by hybridization to specific probes.
- Another application concerns the non-specific (RNA-primer-independent) amplification of the RNA, which can make it possible to identify previously unidentified viruses or new viral variants.
- Another application concerns microarray technology. This has the differential detection of cellular expression as the goal. This is done by detecting the so-called cellular transcripts, i. the mRNA.
- the invention makes it possible to specifically amplify the mRNA by the use of a poly (U) -oligonucleotide in order subsequently to detect microarrays by hybridization.
- the invention enables the direct, efficient and simple production of double-stranded RNA in 1.5 hours and starting from an RNA sequence, without the need for a PCR step, in vitro transcription and hybridization of the RNA (which may be sub-optimal can).
- the procedure of labeling the 3 'end of the RNA template is schematically in FIG. 3 shown.
- the norovirus RdRP cDNA was obtained by PCR from the norovirus clone pUS-NorII (Genbank accession number: AY741811). It was cloned into the pET-28b (+) vector (Novagen), the expression vector was sequenced and transformed into E. coli CL21 (DE3) plysS. The cells were at 37 ° C in Luria-Bertani medium with kanamycin (50 mg / L). At an optical density of 0.6 (OD600), protein expression was induced by the addition of isopropyl- ⁇ -D-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. The cultures were then incubated at 25 ° C overnight.
- IPTG isopropyl- ⁇ -D-thiogalactopyranoside
- the cell pellets obtained from 250 ml of culture were washed once in 4 ml of phosphate buffered saline (PBS) and 1% Triton X 100 (Sigma).
- the cells were treated with DNase (10 U / ml) at 37 ° C for 15 minutes, sonicated on ice and resuspended in 40 ml of binding buffer (20 mM Tris / HCl, pH 7.9, 500 mM NaCl, 5 mM imidazole). After centrifugation at 4300 rpm at 4 ° C for 40 minutes, the clarified lysate was obtained.
- His6-tagged norovirus RdRP was bound to a Ni-nitrilotriacetacetic acid (NTA) -sepharose matrix (Novagen) preequilibrated with the binding buffer.
- NTA Ni-nitrilotriacetacetic acid
- the bound protein was washed with the binding buffer containing 60 mM imidazole and eluted with binding buffer containing 1 M imidazole.
- the eluted protein was dialyzed against buffer A (25 mM Tris-HCl, pH 8.0, 1 mM ⁇ -mercaptoethanol, 100 mM NaCl, 5 mM MgCl 2, 10% glycerol, 0.1% Triton X).
- the protein concentration was determined with the BCA protein assay kit (Pierce) based on the biuret reaction.
- the enzyme was resuspended in a final volume of 50% glycerol and stored at -20 ° C.
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g RNA template, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 0.4 each mM of ATP, CTP, GTP, UTP, 3 ⁇ M of norovirus RdRP prepared according to Example 1.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of stop solution (4 M ammonium acetate, 100 mM EDTA). Purification is by phenol-chloroform extraction or by MEGAclear kit (Ambion) according to the manufacturer's instructions.
- the transcription products are visualized after ethidium bromide staining by UV transillumination on TBE-buffered agarose gels. Formamide agarose gels can also be used.
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g RNA template, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 0.4 each mM of ATP, CTP, GTP, UTP, 0.1 to 1 ⁇ M gene-specific RNA primer, 3 ⁇ M of norovirus RdRP prepared according to Example 1.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of the stop solution (4 M ammonium acetate, 100 mM EDTA).
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g RNA template, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 0.4 each mM of ATP, CTP, GTP UTP, 0.1 to 1 ⁇ M poly (U) 20 primer, 3 ⁇ M of Norovirus RdRP prepared according to Example 1.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of stop solution (4 M ammonium acetate, 100 mM EDTA).
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g RNA template, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 0.4 each mM of ATP, CTP, GTP, UTP, 3 ⁇ M of norovirus RdRP prepared according to Example 1.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of stop solution (4 M ammonium acetate, 100 mM EDTA). Purification is by phenol-chloroform extraction or by MEGAclear kit (Ambion) according to the manufacturer's instructions.
- the transcription products are visualized after ethidium bromide staining by UV transillumination on TBE-buffered 10% polyacrylamide gels. Formamide agarose gels can also be used. Alternatively, armored RNA can be used.
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g RNA template, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 0.4 each mM of ATP, or CTP, or GTP, or UTP, 3 ⁇ M of the according to embodiment 1 Norovirus RdRP.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of stop solution (4 M ammonium acetate, 100 mM EDTA). Purification is by phenol-chloroform extraction or by MEGAclear kit (Ambion) according to the manufacturer's instructions.
- the transcription products are visualized after ethidium bromide staining by UV transillumination on TBE-puffed 10% polyacrylamide gels. Formamide agarose gels can also be used. Alternatively, armored RNA can
- the reaction mixture (50 ⁇ l) consists of 0.5 to 1 ⁇ g poly (C) RNA template, or an RNA with poly (C) RNA at the 3 'end, 10 ⁇ l of the reaction buffer (250 mM HEPES, pH 8.0, 15 mM magnesium acetate, 20 mM DTT), 50 U RNase inhibitor (RNAsin, Promega), 50 ⁇ M of GTP, and 3 ⁇ M of Norovirus RdRP prepared according to Example 1.
- the reaction is carried out at 30 ° C for 2 hours.
- the reaction is stopped by the addition of 50 ⁇ l of stop solution (4 M ammonium acetate, 100 mM EDTA).
- the present invention comprises an RNA-dependent RNA polymerase for amplification and / or labeling of RNA, the RNA-dependent RNA polymerase having a "right-hand conformation" and the amino acid sequence of the RNA-dependent RNA polymerase having the following Sequence sections comprises: a. XXDYS, b. GXPSG, c. YGDD, d. XXYGL, e. XXXXFLXRXX meaning: D: aspartate, Y: tyrosine, S: serine, G: glycine, P: proline, L: leucine, F: phenylalanine, R: arginine, X: any amino acid.
- RNA-dependent RNA polymerase is an RNA-dependent RNA polymerase of a virus of the family Caliciviridae.
- the RNA-dependent RNA polymerase is an RNA-dependent RNA polymerase of a human and / or non-human pathogenic virus of the family Caliciviridae.
- the RNA-dependent RNA polymerase is an RNA-dependent RNA polymerase of a norovirus, sapovirus, vesivirus or a lagovirus.
- the RNA-dependent RNA polymerase is the RNA-dependent RNA polymerase of a Norovirus strain such as the norovirus strain HuCV / NL / Dresden174 / 1997 / GE or one of a sapovirus strain such as the sapovirus strain pJG-SapO1, or a Vesivirus strain such as the Vesivirus strain FCV / Dresden / 2006 / GE.
- a Norovirus strain such as the norovirus strain HuCV / NL / Dresden174 / 1997 / GE or one of a sapovirus strain such as the sapovirus strain pJG-SapO1, or a Vesivirus strain such as the Vesivirus strain FCV / Dresden / 2006 / GE.
- the RNA-dependent RNA polymerase is a protein according to SEQ ID NO 1, SEQ ID NO 2, SEQ ID NO 3, SEQ ID NO 4, SEQ ID NO 5 or SEQ ID NO 6.
- the present invention further comprises a method for amplifying RNA by means of an RNA-dependent RNA polymerase with the steps a. Attachment of the RNA-dependent RNA polymerase to the RNA template in the presence or absence of an oligonucleotide primer, b. Rewriting of the RNA template into antisense RNA by the RNA-dependent RNA polymerase c. Separation of the RNA / antisense RNA duplex into individual RNA strands.
- the RNA / antisense RNA duplex is separated into individual RNA strands by heat denaturation, by chemical denaturation and / or enzymatically.
- the enzymatic separation of the RNA / antisense RNA duplex into individual RNA strands is performed by an enzyme having the ability to separate double-stranded RNA into single-stranded RNA.
- the present invention encompasses a method for primer-dependent amplification of RNA in which at least one primer which hybridizes with a section of the RNA template is used and after the attachment of the RNA-dependent RNA polymerase the primer is replaced by the RNA-dependent RNA Polymerase is extended according to the sequence of the RNA template.
- RNA primer or DNA primer is used as the primer.
- the primer employed is a poly-U-RNA, poly-A-RNA, poly-C-RNA or poly-G-RNA primer or any random oligo-RNA primer.
- RNA-dependent RNA polymerase In another embodiment of the method for primer-independent amplification of poly (C) RNA, no primer that hybridizes to a portion of the RNA template is used, GTP is used as a single nucleotide, and by the RNA-dependent RNA polymerase corresponding to the sequence of RNA template lengthened.
- the present invention further includes a method for labeling RNA using an RNA-dependent RNA polymerase, comprising the steps of: a. Attachment of the RNA-dependent RNA polymerase to the RNA to be labeled, b. Attaching at least one nucleotide to the 3 'end of the RNA to be labeled.
- the present invention also relates to the use of an RNA-dependent RNA polymerase for the amplification and / or labeling of RNA.
- the present invention comprises a kit for the amplification of RNA, at least consisting of: a. an RNA-dependent RNA polymerase according to any one of claims 1 to 6, b. a suitable reaction buffer, c. NTPs, d. optionally RNase inhibitor, e. if necessary stop solution.
- Another object of the invention is a kit for the amplification of RNA, at least consisting of a. an RNA-dependent RNA polymerase according to any one of claims 1 to 6, b. a suitable reaction buffer, c. NTPs, d. optionally RNase inhibitor, e. if necessary stop solution, f. a helicase.
- Another object of the invention is a kit for primer-dependent amplification of RNA, at least consisting of: a. an RNA-dependent RNA polymerase according to any one of claims 1 to 6, b. a suitable reaction buffer, c. NTPs, d. optionally RNase inhibitor, e. if necessary stop solution, f. Primers.
- the present invention also includes a kit for primer-independent amplification of RNA, at least consisting of: a. an RNA-dependent RNA polymerase according to any one of claims 1 to 6, b. a suitable reaction buffer, c. NTPs, d. optionally RNase inhibitor, e. if necessary stop solution.
- the present invention comprises a kit for labeling RNA, at least consisting of a. an RNA-dependent RNA polymerase according to any one of claims 1 to 6, b. a suitable reaction buffer, c. CTP or UTP or ATP or GTP or any nucleotide, d. optionally RNase inhibitor, e. if necessary stop solution.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
- Saccharide Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description
- Die Erfindung betrifft eine RNA-abhängige RNA-Polymerase sowie Verfahren und Kits zur Markierung und / oder zur primerabhängigen und -unabhängigen Amplifikation von Ribonukleinsäure (RNA), insbesondere von viraler, eukaryontischer und prokaryontischer und doppeisträngiger Ribonukleinsäure (RNA) zur Anwendung in der Biotechnologie und der Medizin. Das erfindungsgemäße Amplifikationsverfahren eignet sich insbesondere für die Microarray-Technologie, zur Herstellung von siRNA und zur Diagnose viraler Infektionen durch den Nachweis viraler RNA in Patientenmaterial. Das erfindungsgemäße Markierungsverfahren eignet sich insbesondere für die Aufreinigung von RNA mittels Affinitäts-Bindung, sowie für die Markierung von RNA für die Anwendung in molekularbiologischen Verfahren, die bei der Charakterisierung der Funktion und/oder Struktur viraler, eukaryontischer und prokaryontischer und doppelsträngiger Ribonukleinsäure (RNA) verwendet werden.
- Die RNA ist ein wichtiger Bestandteil der eukaryontischen und der prokaryontischen Zelle. Sie ist an mehreren vitalen Funktionen beteiligt: der Transkription, d.h. der Umschreibung der Information des Erbgutes (Desoxyribonukleinsäure, DNA), der Übertragung dieser Information vom Zellkern in das Zytoplasma (mRNA), und der Translation, d.h. die Übersetzung der umgeschriebenen Information in Aminosäuren bzw. Proteine. Die RNA bildet auch die sog. Transfer-RNA, einen wichtigen Baustein für die Translation. Die RNA ist auch ein wichtiger Bestandteil der Ribosomen. Dies sind strukturelle Einheiten des Zytoplasmas, an denen die Übersetzung der Information in Eiweiß stattfindet.
- Die Bedeutung der RNA im Bereich der Biotechnologie hat in den letzten Jahrzehnten zugenommen. Neue Entwicklungen wie z.B. die Microarray-Technologie, die miRNA (micro-RNA) oder die siRNA Technologie (small interfering RNA) haben stattgefunden, deren Relevanz im Bereich der Grundlagenforschung als auch der angewandten Forschung zunimmt. All diese Technologien beruhen auf der synthetischen Generierung von RNA durch Amplifikation in vitro.
- Die Herstellung von siPNA in vitro erfolgt bislang über chemische Synthese oder mit Hilfe von DNA abhängigen RNA-Polymerasen wie der T7-RNA-Polyrmerase. Dafür werden zwei komplementäre DNA-Stränge mittels PCR unter Einsatz eines Primers, der auch die Sequenz für einen T7-Promotor enthält synthetisiert. Nach Umschreiben der DNA mit Hilfe der T7 RNA-Polymerase werden die resultierenden komplementären RNA-Stränge hybridisiert und mittels RNAse 1 verdaut.
- Im medizinischen Bereich spielt die RNA-Amplifikation eine wichtige Rolle beim Nachweis viraler Infektionen im Patientenmaterial. Weltweit werden 80% aller viralen Infektionen durch RNA-Viren, d.h. virale Erreger mit einem aus RNA bestehenden Erbgut, verursacht. Wichtige Erreger aus dieser Gruppe von Viren sind u.a. das Humane Immundefizeinz-Virus (HIV), das Hepatitis-C-Virus (HCV), das Hepatitis-A-Virus, das Grippe-Virus (Influenza A, B und C) aber auch das Vogelpest-Virus, das vor kurzem neu identifizierte SARS-Virus, das Virus der Kinderlähmung (Poliovirus), das Masern-Virus, das Mumps-Virus, das Röteln-Virus und die Brechdurchfall-Viren (Rotavirus, Sapovirus und Norovirus).
- Der Nachweis von RNA im Patientenmaterial findet bis dato in vier Schritten statt. Nach der Gewinnung der RNA aus dem Untersuchungsmaterial folgen die Schritte:
- 1. Umschreibung der RNA in copy-SNA (cDNA) durch RNA-abhängige DNA-Polymerasen, die sog. reversen Transkriptasen. Dafür bedarf es auch des Einsatzes eines Primers. Primer sind Oligonukleotide, die spezifisch für die zu amplifizierende Sequenz sind. Primer und die RNA-abhängige DNA-Polymerase ermöglichen die gezielte Amplifikation vom Erbgut, allerdings in DNA-Form.
- 2. Amplifikation der cDNA durch Polymerasekettenreaktion (PCR),
- 3. Aufreinigung des PCR-Produkts,
- 4. Sequenzierung des PCR-Produkts.
- Für die Herstellung von RNA, beispielsweise zum Expressionsnachweis oder zur Herstellung von siRNA, erfolgt zusätzlich die Umschreibung des PCR-Produkts in RNA durch DNA-abhängige RNA-Polymerasen, wie z.B. die T7-Polymerase.
Die Durchführung dieser Schritte dauert in der Regel 18 bis 24 Stunden. Ebenfalls dazu benötigt wird auch ein gewisses "Know-How", was nicht allen Forschungseinrichtungen zugänglich ist. - Eine weitere Methode zur Amplifikation von RNA ist die Replikation durch RNA-abhängige RNA-Polymerasen wie z.B. die RNA-Polymerase des Bakteriophagen Qβ (auch Qβ - Replikase genannt), die RNA-Polymerase des Poliovirus oder des Hepatitis-C-Virus.
- Das Genom des Bakteriophagen Qβ besteht aus einer einzelsträngigen (+)-RNA, die durch die RNA-abhängige RNA-Polymerase repliziert wird. Die Qβ - Replikase stellt eine (-)-Strang-RNA-Kopie des (+)-Stranges her, welche ebenso wie (+)-Stränge als Matrize fungieren kann. Auf diese Weise kann eine exponentielle Vervielfältigung erreicht werden. Das Qβ -System wird bereits zum Nachweis von Analyten, wie etwa Nukleinsäuren, verwendet (Chu et al. (1986), Nucleic Acids Research 14, 5591 - 5603; Lizardi et al (1988), Biotechnology 6, 1197 - 1202).
- Diese RNA-abhängigen RNA-Polymerasen benötigen jedoch bestimmte RNA-Sequenzen zur Initiation der RNA-Replikation. Zudem sind sie für die Amplikation der RNA auch auf bestimmte Hilfsproteine angewiesen. Daher sind diese RNA-abhängigen RNA-Polymerasen nicht dazu in der Lage, heterologe RNA zu amplifizierten.
- Die Markierung von RNA am 3'-Terminus ist nach dem Stand der Technik nur mit ATP durch die Verwendung von E.coli poly(A)- Transferase möglich. Verfahren zur Markierung von RNA mit CTP, UTP und GTP sind nicht bekannt.
- Der Erfindung liegt daher die Aufgabe zugrunde, eine RNA-abhängige RNA-Polymerase sowie Verfahren und Kits zur Markierung und / oder zur Amplifikation von viraler, eukaryontischer und prokaryontischer einzel- und doppelsträngiger Ribonukleinsäure (RNA) bereitzustellen, die effizienter und schneller sind.
- Erfindungsgemäß wird die Aufgabe gelöst durch eine RNA-abhängige RNA-Polymerase (im folgenden auch als RdRP oder 3Dpol bezeichnet) zur Amplifikation und / oder Markierung von RNA, wobei die RNA-abhängige RNA-Polymerase zu den Viren der Familie der Caliciviridae gehört und eine "Rechte-Hand-KonformaHon" aufweist und die Aminosäuresequenz der RNA-abhängigen RNA-Polymerase die folgenden Sequenzabschnitte umfasst:
- a. XXDYS
- b. GXPSG
- c. YGDD
- d. XXYGL
- e. XXXXFLXRXX
- D: Aspartat
- Y: Tyrosin
- S: Serin
- G: Glycin
- P: Prolin
- L: Leucin
- F: Phenylalanin
- R: Arginin
- X: eine beliebige Aminosäure.
- Unter einer sog. "Rechte-Hand-Konformation" ist dabei zu verstehen, dass die tertiäre Struktur (Konformation) des Proteins eine Faltung nach einer rechten Hand mit Finger (finger), Handfläche (palm) und Daumen (thumb), aufweist.
- Bei dem Sequenzabschnitt "XXDYS" handelt es sich um das sog. A-Motiv. Das A-Motiv ist für die Diskriminierng zwischen Ribonukleosiden und Deoxyribonukleosiden zuständig.
Bei dem Sequenzabschnitt GXPSG" handelt es sich um das sog. B-Motiv. Das B-Motiv ist in allen Vertretern der Familie Caliciviridae konserviert.
Bei dem Sequenzabschnitt "YGDD" handelt es sich um das sog. C-Motiv. Das C-Motiv stellt die aktive Stelle des Enzyms dar. Dieses Motiv spielt bei der Koordinierung der Metallionen eine wichtige Rolle.
Bei dem Sequenzabschnitt "XXYGL" handelt es sich um das sog. D-Motiv. Das D- Motiv ist ein Merkmal der Template-abhängigen Polymerasen. - Bei dem Sequenzabschnitt "XXXXFLXRXX" handelt es sich um das sog. E-Motiv. Das E-Motiv ist ein Merkmal der RNA-abhängigen RNA Polymerasen, und kommt bei DNA-abhängigen RNA-Polymerasen nicht vor.
- Die erfindungsgemäße RNA-abhängige RNA-Polymerase verfügt über die folgenden Funktionen:
- Mg2+- oder Mn2+-abhängige RNA-abhängige RNA-Polymeraseaktivität, keine DNA-Abhängigkeit
- Tomplate-Abhängigkelt
- primer-abhängige RNA-Synthese mit homopolymerer RNA (poly(A)-RNA, poly(C)-RNA, poly(G)-RNA bzw. poly(U)-RNA) als Matrize und bei Anwesenheit der entsprechenden Primer (d.h. oligo(U)-RNA, oligo(G)-RNA, oligo-(C)-RNA, bzw. oligo(A)-RNA)
- primer-unabhängige RNA-Synthese mit poly(C)-RNA als Matrize in Anwesenheit von erhöhten GTP-Konzentrationen
- Primer-abhängige oder Primer-unabhängige RNA-Synthese mit heteropolymerer RNA als Matrize
- Terminale Transferase-Aktivität mit einer Präferenz für UTP und CTP, was zum "Labelling" des 3'-Endes eines einzelsträngigen RNA-Templates führt.
- Die Primer-abhängige Amplifikation der RNA erfolgt nur in der Anwesenheit eines Sequenzspezifischen Primers. Die Amplifikation ist Sequenz-abhängig und erfolgt durch Einbau von AMP, GMP, CMP oder UMP.
Die Primer-unabhängige Amplifikation der RNA erfolgt in der Abwesenheit eines Sequenzspezifischen Primers, Die Amplifikation ist Sequenz-abhängig und erfolgt durch Einbau von AMP, GMP, CMP oder UMP. - Durch die Terminale Transferase-Aktivität werden am 3'-Ende eines RNA-Stranges mehrere Nucleotide gleicher Art angehängt (z.B. ATP oder UTP oder CTP oder GTP), unabhängig von der Sequenz des RNA-Stranges.
- Überraschenderweise ist die erfindungsgemäße RNA-abhängige RNA-Polymerase in der Lage, in vitro heterologe virale, eukaryontische und prokaryontische RNA als Template für die Amplifikationsreaktion zu nutzen. Sowohl positiv-strängige und negativ-strängige einzelsträngige als auch doppelsträngige RNA- ist als Template zur Amplifikation einsetzbar.
- Vorzugsweise ist die erfindungsgemäße RNA-abhängige RNA-Polymerase eine RNA-abhängige RNA-Polymerase eines Virus aus der Familie der Caliciviridae. Das Genom der Caliciviridae besteht aus einem polyadenylierten (+)-strängigen RNA-Einzeistrang. In vivo schreibt die RNA-abhängige RNA-Polymerase des Calicivirus bei der viralen Replikation die genomische Calicivirus-RNA in eine (-) - strängige antisense-RNA (aRNA) um. Dabei entsteht ein RNA-aRNA-Hybrid. Die (-)-strängige aRNA dient im Anschluß als Templat zur Synthese neuer (+)-strängiger genomischer Virus-RNA.
- In weiteren bevorzugten Ausführungsformen der Erfindung ist die RNA-abhängige RNA-Polymerase eine RNA-abhängige RNA-Polymerase eines humanen und / oder nicht-human pathogenen Calicivirus. Besonders bevorzugt handelt es sich um eine RNA-abhängige RNA-Polymerase eines Norovirus, Sapovirus, Vesivirus oder eines Lagovirus, z.B. die RNA-abhängige RNA-Polymerase des Norovirus-Stammes HuCV/NL/Dresden174/1997/GE oder des Sapovirus-Stammes pJG-Sap01 oder des Vesivirus-Stammes FCV/Dresden/2006/GE.
- In besonders bevorzugten Ausführungsformen der Erfindung ist die RNA-abhängige RNA-Polymerase ein Protein mit einer Aminosäuresequenz gemäß SEQ ID NO 1, SEQ ID NO 2 oder SEQ ID NO 3,
- SEQ ID NO 1 ist die Aminosäuresequenz einer Norovirus-RdRP.
- SEQ ID NO 2 ist die Aminosäuresequenz einer Sapovirus-RdRP.
- SEQ ID NO 3 ist die Aminosäuresequenz einer Vesivirus-RdRP.



- Die RNA-abhängigen RNA-Polymerasen des Norovirus, des Sapovirus und des Vesivivirus sind rekombinant herstellbar durch Klonierung der die Norovirus-, Sapovirus bzw. Vesivirus-RdRP kodierenden Nukleinsäure in einen geeigneten Expressionsvektor, beispielsweise pET-28 (Novagen). Der Expressionsvektor mit der Gensequenz, die für die Virus-RdRP kodiert, wird zur Expression in einen geeigneten Wirtsorganismus eingebracht. Der Wirtsorganismus wird bevorzugt aus den üblicherweise für die Proteinexpression verwendeten prokaryontischen, bevorzugt Eschericha coli, oder eukaryontischen Wirtsorganismen, bevorzugt Sacharomyces cerevisae oder Insektenzellen (bevorzugt Sf9-Zellen), die mit rekombinanten Baculoviren infiziert worden sind, gewählt Dieser Wirtsorganismus enthält mindestens einen Expressionsvektor mit einer Gensequenz, die für die Virus-RdRP kodiert,
- In bevorzugten Ausführungsformen wird die Norovirus-, Sapovirus- oder Vesivirus- Calicivirus-RdRP am N- oder C-Terminus mit einer Proteinsequenz fusioniert, welche die Aufreinigung des RdRP-Fusionsproteins nach der Expression in einem Wirtsorganismus erleichtert. Bevorzugt ist diese Sequenz ein sogenannter Histidin-Marker, bestehend aus einer Sequenz von mindestens 6 aufeinanderfolgenden Histidinen (His oder H). Ein solcher Histidin-Marker ermöglicht in bekannter Weise die Aufreinigung des Proteins affinitätschromatographisch auf einer Nickel- oder Kobalt-Säule.
- Beispiele für mit einem Histidin-Marker fusionierte Ausführungsformen der RNA-abhängigen RNA-Polymerase sind die Proteine mit einer Aminosäuresequenz gemäß SEQ ID NO 4, SEQ ID NO 5 oder SEQ ID NO 6.
- SEQ ID NO 4 ist die Aminosäuresequenz einer Norovirus-RdRP mit einem Histidin-Marker (His-tag).
- SEQ ID NO 5 ist die Aminosäuresequenz einer Sapovirus-RdRP mit einem Histidin-Marker (His-tag).
- SEQ ID NO 6 ist die Aminosäuresequenz einer Vesivirus-RdRP mit einem Histidin-Marker (His-tag).




- Bestandteil der Erfindung ist auch die Verwendung einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase zur Amplifikation und / oder Markierung von RNA.
- Bestandteil der Erfindung ist auch ein Verfahren zur Amplifikation von RNA mittels einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase mit den Schritten
- a. Anlagerung der RNA-abhängigen RNA-Polymerese an die RNA-Matrize in der Anwesenheit oder Anwesenheit eines Oligonukleotid-Primers, z.B. eines Oligo-RNA-Primers oder Oligo-DNA-Primers,
- b. Umschreiben der RNA-Matrize in antisense-RNA durch die RNA-abhängige RNA-Polymerase,
- c. Trennung des RNA/antisense-RNA-Duplexes in einzelne RNA-Stränge
- a. einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase
- b. einem geeigneten Reaktions-Puffer
- c. NTPs
- d. ggf. RNase-Inhibitor
- e. ggf. Stoplösung
- Die RNA-Matrize wird beim erfindungsgemäßen Verfahren vorzugsweise in Mengen von 1 µg bis 4 µg pro 50 µl-Reaktionsansatz eingesetzt. Die Konzentration der eingesetzten Ribonukleotide ATP, CTP, GTP und UTP (NTPs) beträgt vorzugsweise zwischen 0,1 µmol/l und 1 µmol/l, besonders bevorzugt 0,4 µmol/l. Auch NTP-Analoga sind beim erfindungsgemäßen Verfahren zur RNA-Amplifikation einsetzbar, beispielsweise fluoreszenzmarkierte NTPs, Biotin-markierte NTPs oder radioaktiv-markierte NTPs Wie [P32]-markierte NTPs. Die Konzentration der erfindungsgemäßen RdRP beträgt bevorzugt zwischen 1 µmol/l und 3 µmol/l.
- In einer besonders bevorzugten Ausführungsform enthält das Kit
- a. 150 µmol/l rekombinant hergestellter Norovirus-RdRP gemäß SEQ ID NO 1 oder SEQ ID NO 4 und/oder Sapovirus-RdRP gemäß SEQ ID NO 2 oder SEQ ID NO 5 und/oder Vesivirus-RdRP gemäß SEQ ID NO 3 oder SEQ ID NO 6
- b. als Puffer: 50 mmol/l HEPES, pH 8.0, 3 mmol/l Magnesiumacetat oder Manganchlorid (MnCl2), 4 mM DTT
- c. 10 mmol/l ATP, 10 mmol/l CTP, 10 mmol/l GTP, 10 mmol/l UTP
- d. RNese-Inhibitor
- e. als Stoplösung: 4 mol/l Ammoniumacetat, 100 mmol/l EDTA
- Das Verfahren wird vorzugsweise bei einem pH-Wert zwischen 6,5 und 8,5 und bei einer Temperatur zwischen 20°C und 40°C durchgeführt. Eine bevorzugte Ausführungsform des Verfahrens zur RNA-Amplifikation wird bei 30°C mit Norovirus-RdRP oder bei 37°C mit Sapovirus-RdRP unter folgenden Pufferbedingungen durchgeführt: 50 mmol/l HEPES, pH 8.0, 3 mmol/l Magnesiumacetat oder Manganchlorid, 4 mmol/l DTT.
- In einer Ausführungsform der Erfindung erfolgt die Trennung des RNA / antisense-RNA-Duplexes in einzelne RNA-Stränge durch Hitzedenaturierung, durch chemische Denaturierung und / oder enzymatisch, z.B. mit einem Enzym mit der Fähigkeit, doppelsträngige RNA in einzelsträngige RNA aufzutrennen wie z.B. eine Helikase.
- Durch die Strangtrennung liegt wieder einzelsträngiges Template vor und eine weitere Amplifikationsrunde kann eingeleitet werden.
Durch den Einsatz eines Enzyms mit der Fähigkeit, doppelsträngige RNA in einzelsträngige RNA aufzutrennen, kann die Reaktion isotherm gehalten werden. - Bestandteil der Erfindung ist ein entsprechendes Kit zur Amplifikation von RNA, das zusätzlich zum erfindungsgemäßen Kit ein Enzym mit der Fähigkeit, doppelsträngige RNA in einzelsträngige RNA, aufzutrennen wie z.B. eine Helikase, enthält.
- Eine Ausführungsform der Erfindung ist ein Verfahren zur primerabhängigen Amplifikation von RNA, wobei mindestens ein Primer, der mit einem Abschnitt der RNA-Matrize hybridisiert, eingesetzt wird und nach der Anlagerung der erfindungsgemäßen RNA-abhängigen RNA-Polymerase der Primer durch die RNA-abhängige RNA-Polymerase entsprechend der Sequenz der RNA-Matrize verlängert wird.
- Als Primer wird bevorzugt ein RNA-Primer oder ein DNA-Primer, z.B. mit einer Länge von 20 bis 25 Basen, vorzugsweise in einer Konzentration von 0,1 bis 1 µmol/l eingesetzt. Als RNA-Matrize ist dabei z.B. virale, prokaryontische und eukaryontische RNA einsetzbar.
- Der Ablauf der primerabhängigen RNA-Amplifikation einer einzelsträngigen RNA-Matrize unter Einsatz eines RNA-Primers ist in
Figur 2 schematisch dargestellt. - Vorzugsweise wird bei polyadenylylierter RNA, polyurydylitierter RNA, polyguanylylierter RNA oder polycytidylylierter RNA als Primer, ein poly-U-RNA, poly-A-RNA, poly-C-RNA oder poly-G-RNA-Primer eingesetzt. Mit Hilfe des poly-U-RNA-Primers ist eine spezifische Amplifikation der gesamten zellulären mRNA möglich. Vorzugsweise wird dafür ein Poly-U-Primer von 20 bis 24 Basen Länge eingesetzt. Die amplifizierte zelluläre mRNA kann Anschluss beilspielsweise mit Hilfe der sog. Microarray-Verfahren untersucht werden.
- Das erfindungsgemäße Verfahren zur sequenzspezifischen RNA-Amplifikation wird vorteilhaft auch zum Nachweis viraler RNA in Patientenmaterial verwendet.
- Hierfür wird die gesamte zelluläre RNA aus dem Patientenmaterial gewonnen und unter Einsatz eines RNA-Primers, der spezifisch mit einem bestimmten Abschnitt viraler RNA hybridisiert, wird im Patientenmaterial vorhandene virale RNA amplifiziert.
- Als Patientenmaterial wird beispielsweise Liquor, Blut, Plasma oder Körperflüssigkeiten eingesetzt.
- Auch geeignet ist das Verfahren zum Nachweis von RNA-Viren mit einem poly-A-Schwanz am 3'-Ende des Genoms, DNA-Viren und viralen mRNA-Transkripten.
- Bestandteil der Erfindung ist ebenfalls ein Kit zur Durchführung des Verfahrens zur primerabhängigen Amplifikation von RNA, mindestens bestehend aus
- a. einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase
- b. einem geeigneten Reaktions-Puffer
- c. NTPs
- d. ggf. RNase-Inhibitor
- e. ggf. Stoplösung
- f. Primer
- a. 150 rekombinant hergestellter µmol/l Norovirus-RdRP gemäß SEQ ID NO 1 oder SEQ ID NO 4 und/oder Sapovirus-RdRP gemäß SEQ ID NO 2 oder SEQ ID NO 5 und/oder Vesivirus-RdRP gemäß SEQ ID NO 3 oder SEQ ID NO 6
- b. als Puffer: 50 mmol/l HEPES, pH 8.0, 3 mmol/l Magnesiumacetat oder Manganchlorid (MnCl2), 4 mM DTT
- c. 10 mmol/l ATP, 10 mmol/l CTP, 10 mmol/l GTP, 10 mmol/l UTP
- d. RNase-Inhibitor
- e. als Stoplösung: 4 mol/l Ammoniumacetat, 100 mmol/l EDTA
- f. 0,1 bis 3 µM Primer (homopolymeres oder heteropolymeres RNA- oder DNA-Oligonukleotid mit einer Länge von 15 bis 20 Basen)
- Ebenfalls Teil der Erfindung ist ein Verfahren zur primerunabhängigen) Amplifikation von RNA, wobei kein Primer, der mit einem Abschnitt der RNA-Matrize hybridisiert, eingesetzt und durch die RNA-abhängige RNA-Polymerase entsprechend der Sequenz der RNA-Matrize verlängert wird, sowie ein Kit zur primerunabhängigen Amplifikation von RNA, mindestens bestehend aus
- a. einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase
- b. einem geeigneten Reaktions-Puffer
- c. NTPs
- d. ggf. RNase-Inhibitor
- e. ggf. Stoplösung.
- In dieser Ausgestaltung des erfindungsgemäßen Verfahrens erfolgt die Anlagerung der RNA-abhängigen RNA-Polymerase an die RNA-Matrize primerunabhängig. Der Ablauf der sequenzunabhängigen RNA-Amplifikation einer einzelsträngigen RNA-Matrize ohne Einsatz eines RNA-Primers ist in
Figur 1 schematisch dargestellt. - Eine weitere Ausführungsform der Erfindung ist ein Verfahren zur primerunabhängigen Amplifikation von poly(C)-RNA, dadurch gekennzeichnet, dass kein Primer, der mit einem Abschnitt der RNA-Matrize hybridisiert, eingesetzt wird, dass GTP, vorzugsweise 50 µM, als einziger Nukleotid verwendet wird und durch die RNA-abhängige RNA-Polymerase entsprechend der Sequenz der RNA-Matrize verlängert wird.
Der Schematische Ablauf der sequenz-unabhängigen RNA-Synthese ausgehend von einer poly(C)-RNA in Anwesenheit von 50 µM GTP ist inFigur 4 dargestellt. - Die Anwendungsmöglichkeiten der erfindungsgemäßen Verfahren zur Amplifkation von RNA sind vielfältig, Zum einem ist der direkte Nachweis viralen Erbgutes In Patientenmaterial möglich. Hiefür wird die gesamte zelluläre RNA gewonnen und unter Einsatz eines RNA-Primers eine beliebige RNA-Sequenz spezifisch amplifiziert. Der Nachweis der viralen Nukleinsäure erfolgt dann durch Hybridisierung an spezifischen Sonden. Eine andere Anwendung betrifft die unspezifische (RNA-Primer-unabhängige) Amplifikation der RNA, die ermöglichen kann, bisher nicht identifizierte Viren bzw. neue virale Varianten zu identifizieren.
- Eine weitere Anwendung betrifft die Microarray-Technologie. Diese hat den differenziellen Nachweis der zellulären Expression zum Ziel. Dies geschieht durch den Nachweis der sog. zellulärer Transkripte, d.h. der mRNA. Die Erfindung ermöglicht ausgehend aus einem zellulären Gemisch, die mRNA spezifisch durch den Einsatz eines poly(U)-Oligonukleotid zu amplifizieren, um anschließend auf Microarrays durch Hybridisierung den Nachweis zu erbringen.
- Ein anderes Anwendungsgebiet der Erfindung ist die Herstellung von siRNA in vitro. Dies erfolgt bis dato mit Hilfe der T7-Polymerase. Es wird dafür
- 1. zwei komplementäre DNA-doppelstränge mittels PCR unter dem Einsatz eines T7-Promotor-Primer synthetisiert,
- 2. nach Umschreibung der DNA mit Hilfe der T7-Polymerase, werden beide resultierende komplementäre RNA-Stränge hybridisiert,
- 3. dann werden mittels Nase I beide Stränge verdaut.
- Diese Schritte dauern nach dem Stand der Technik in der Regel 24 bis 48 Stunden, und benötigen ebenso das notwendige "Know-How". Die Erfindung ermöglicht demgegenüber in 1,5 Stunden und ausgehend von einer RNA-Sequenz die direkte, effiziente und einfache Herstellung doppelsträngiger RNA, ohne die Notwendigkeit eines PCR-Schrittes, der in vitro Transkription und der Hybridisierung der RNA (die eventuell sub-optimal verlaufen kann).
- Bestandteil der Erfindung ist auch ein Verfahren zur Markierung von RNA mittels einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase mit den Schritten
- a. Anlagerung der RNA-abhängigen RNA-Polymerase an die zu markierende RNA,
- b. Anhängen mindestens eines Nukleotids an das 3'-Ende der zu markierenden RNA.
- a. einer erfindungsgemäßen RNA-abhängigen RNA-Polymerase
- b. einem geeigneten Reaktions-Puffer
- c. CTP oder UTP oder ATP oder GTP
- d. ggf. RNase-Inhibitor
- e. ggf. Stoplösung.
- Der Ablauf des Labelling des 3'-Endes des RNA-Templates ist schematisch in
Figur 3 dargestellt. - In einer besonders bevorzugten Ausführungsform enthält das Kit
- g. 150 rekombinant hergestellter µmol/l Norovirus-RdRP gemäß SEQ ID NO 1 oder SEQ ID NO 4 und/oder Sapovirus-RdRP gemäß SEQ ID NO 2 oder SEQ ID NO 5 und/oder Vesivirus-RdRP gemäß SEQ ID NO 3 oder SEQ ID NO 6
- h. als Puffer: 50 mmol/l HEPES, pH 8.0, 3 mmol/l Magnesiumacetat oder Manganchlorid (MnCl2), 4 mM DTT
- i. 10 mmol/l ATP, und 10 mmol/l CTP, und 10 mmol/l GTP, und 10 mmol/l UTP,
- j. RNase-Inhibitor
- k. als Stoplösung.: 4 mol/l Ammoniumacetat, 100 mmol/l EDTA
- Anhand der folgenden Figuren und Ausführungsbeispiele wird die Erfindung zäher erläutert. Dabei zeigen
- Figur 1:
- Schematische Ablauf der sequenz-abhängigen RNA-Amplifikation
- Figur 2:
- Schematischer Ablauf der sequenz-unabhängigen RNA-Ampliflkation
- Figur 3:
- Schematischer Ablauf des Labelling des 3'-Endes des RNA-Templates
- Figur 4:
- Schematischer Ablauf der sequenz-unabhängigen RNA- Synthese ausgehend von einer poly(C)-RNA In Anwesenheit von 50 µM GTP
- Figur 5:
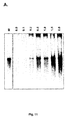
- Expression und Reinigung von Norovirus 3Dpol in E. coli.
- (A) SDS-PAGE-Anadyse von in E. coli exprimierter, gereinigter, rekombinanter Norovirus 3Dpol mit einem C-terminalen His-tag. 3Dpol, Wildtyp- Norovirus 3Dpol. m3Dpol, Norovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G). M, Molekulargewichtsmarker (KDa).
- (B) Western-Blot-Analyse von gereinigter rekombinanter Norovirus 3Dpol unter Verwendung eines Peffia-Histidin-Antikörpers (monoklonaler Maus-Antikörper, Qiagen) zur Detektion von Proteinen mit einem C-terminalen His-tag. 3Dpol, Wildtyp-Norovirus 3Dpol. Norovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G)- M, Molekulargewichtsmarker (KDa).
- Figur 6:
- RNA Synthese mit Norovirus 3D pol.
- (A) Die RNA-Synthese wurde in Anwesenheit einer synthetischen subgenomischen RNA (sG-RNA) als Template sowie in Anwesenheit von Wildtyp-Norovirus 3Dpol oder einer Norovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G) wie angegeben untersucht. Die Reaktionsprodukte wurden auf einem nicht-denaturierenden Agarosegel analysiert und mittels Autoradiographie visualisiert. T7, synthetische subgenomische RNA, die durch In vitro Transkription mit T7 hergestellt wurde.
- (B) Ethidiumbromid-Färbung desselben Gels und Visualisierung der Reaktionsprodukte durch UV-Transillumination. Die bei der Reaktion mit m3Dpol anstelle von Wildtyp 3Dpol sichtbare verbleibende Bande stammt von dem in der Reaktion eingesetzten synthetischen subgenomischen RNA-Template, M, RNA Molekulargewichtsmarker.
- (C) Strangseparationsanalyse des Reaktionsprodukts der in vitro RNA-Synthese mit Norovirus 3Dpol Das Reaktionsprodukt wurde durch RNA-Synthese mit Norovirus 3Dpol (3Dpol) mit einer synthetischen subgenomischen RNA (sG-RNA) als Template hergestellt. Die Reaktionsprodukte wurden auf nicht-denaturierenden (0.5 M; links) und denaturierenden (1.25 M; rechts) Formaldehyde-Agarosegelen sichtbar gemacht. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA Molekutargewichtsmarker.
- Figur 7:
- Northern-Blot Analyse der Norovirus 3Dpol Syntheseprodukte.
- (A) Die RNA-Synthese wurde in Anwesenheit einer synthetischen subgenomischen RNA (sG-RNA) als Template sowie in Anwesenheit von Wildtyp-Norovirus 3Dpol oder einer Norovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G) wie angegeben durchgeführt. Die Reaktionsprodukte wurden auf einem nicht-denaturierenden Agarosegel analysiert und nach Ethidiumbromidfärbung mittels UV-Transillumination visualiert. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA Molekulargewichtsmarker. Die bei der Reaktion mit m3Dpol anstelle von Wildtyp 3Dpol sichtbare verbleibende Bande stammt von dem in der Reaktion eingesetzten Template.
- (B) Hybridisierung einer (-)-strängigen RNA-Probe mit den Syntheseprodukten der Norovirus 3Dpol. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. sG-RNA, als Template eingesetzte synthetische subgenomische RNA. 3Dpol, Wildtyp-Norovirus 3Dpol. Norovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G).
- Figur 8:
- Analyse der Konzentrationsabhängigkeit, der Temperaturabhängigkeit und des Zeitverlaufs der Norovirus 3Dpol-Aktivität. Bei allen Reaktionen wurde subgenomische RNA als Template eingesetzt
- (A) Konzentrationsabhängige Aktivität der Norovirus 3Dpol. Norovirus 3Dpol Konzentration (µM) wie angegeben. Die Reaktion wurde für 2 h bei 30°C durchgeführt. Für jede Konzentration sind Mittelwert und Standardabweichung dreier unabhängiger Experimente gezeigt.
- (B) Zeitverlaufsanalyse der Norovirus 3Dpol-Aktivität. Einbau von [α-32P]UMP wie angegeben. Die Reaktion wurde bei 30°C durchgeführt und 3 µM Norovirus 3Dpol wurden eingesetzt. Für jeden Zeitpunkt sind Mittelwert und Standardabweichung dreier unabhängiger Experimente Experimenten gezeigt.
- (C) Temperaturabhängige Aktivität der Norovirus 3Dpol. Einbau von [α-32P]UMP wie angegeben. Die Reaktionszeit war 2 h. Für jede Konzentration von Norovirus 3Dpol sind Mittelwert und Standardabweichung dreier unabhängiger Experimente gezeigt.
- (D) Metallionen-Abhängigkeit der Norovirus 3Dpol-Aktivität. Die RNA-Synthese wurde über 2 h bei 30°C in Gegenwart von 3 µM Norovirus 3Dpol, subgenomischer RNA als Template (0.024 µM) und steigender Konzentrationen (0.5 bis 3 mM) von MgAcO, MnCl2 oder FeSO4 durchgeführt Einbau von [α-32P]UMP wie angegeben. Für jede Konzentration sind Mittelwert und Standardabweichung dreier unabhängiger Experimente gezeigt.
- Figur 9:
- Primer-abhänglge initiation der RNA Synthese auf hompolymeren Templaten. Die RNA-Synthese wurde in Anwesenheit (schwarze Balken) oder in Abwesenheit (graue Balken) eines komplementären RNA-Oligonukleotid-Primers durchgeführt. Der Einbau von [α-32P]CMP, [α-32P]AMP, [α-32P]GMP, oder [α-32P]UMP wurde nach TCA-Präzipitation und Sammeln von G/C-Glassfasefiltern gemessen. Die Einbauwerte sind angegeben,
- Figur 10:
- Terminale Transferase - Aktivität der Norovirus 3D pol
- (A) Zur Untersuchung der Terminalen Transferase - Aktivität wurde 3 µM Norovirus 3Dpol über 2 h bei 30°C in Gegenwart von subgenomischer RNA (sG-RNA) als Template (0.024 µM) und [α-32P]ATP, [α-32P]GTP, [α-32P]CTP oder [α-32P]UTP wie angegeben inkubiert. Die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen analysiert und mittels Autoradiographie visualisiert,
- (B) Zeitverlaufsexperiment der Norovirus 3Dpol Terminalen Transferase - Aktivität an subgenomischer Norovirus-RNA als Template, Die Reaktion wurde wie unter (A) beschrieben, jedoch unter Verwendung von [α-32P]UTP als gelabeltern Nukleotid durchgeführt, zu den angegebenen Zeiten (sec) gestoppt, und die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen mittels Autoradiographie visualisiert. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde.
- (C) Zeitverlaufsexperiment der RNA-Synthese mit 3 µM Norovirus 3Dpol unter Verwendung von subgenomischer RNA als Template. Die Reaktion wurde bei 30°C inkubiert, zu den angegebenen Zeiten (sec) gestoppt und die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen mittels Autoradiographie visualisiert T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. P, Norovirus 3Dpol Syntheseprodukt.
- Figur 11:
- Primer-abhängige Replikation von full-length subgenomischer polyadenylierter RNA durch Norovirus 3D pol. Bei allen Reaktionen wurde synthetische subgenomische polyadenylierte RNA als Template eingesetzt. Die Reaktionsprodukte wurden auf Formaldehyd-Agarosegelen analysiert und durch Autoradiographie visualisiert.
- (A) Die Reaktion wurde in Gegenwart eines oligo(U)20-RNA-Primers wie angegeben (mM) durchgeführt. M, Marker (in vitro transkribierte subgenomische polyadenylierte RNA).
- (B) Die Reaktion wurde in Gegenwart eines RNA-Oligonukleotids durchgeführt, das komplementär zu der dem poly(A)-Schwanz benachbarten Sequenz ist. RNA-Oligonukleotid Primerkonzentration (mM) wie angegeben. M, Marker (In vitro transkribierte subgenomische polyadenylierte RNA).
- Figur 12:
- De novo Initiation der RNA-Synthese an Norovirus anti-subgenomischer RNA. Bei allen Reaktionen wurde synthetische anti-subgenomische RNA als Template für die Replikationsreakton eingesetzt. Die RNA-Reaktionsprodukte wurden auf nativen Agarosegelen analysiert und nach Ethidiumbromidfärbung durch UV-Transillumination visualisiert.
- (A) Bahnen 1 bis 3: RNA-Synthese in Gegenwart von Wildtyp 3Dpol, mutierter 3Dpol bzw. in Abwesenheit von 3Dpol. Bahnen 4 bis 6: RNA-Synthese in Gegenwart von Wildtyp 3Dpol und einem RNA-Oligonukleotid komplementär zum 3'-Terminus, mutierter 3Dpol und einem RNA-Oligonukleotid komplementär zum 3'-Terminus bzw. mit einem RNA-Oligonukleotid komplementär zum 3'-Terminus, aber ohne Norovirus 3Dpol. Bahnen 7 bis 9, RNA-Synthese in Gegenwart von Wildtyp 3Dpol und einem DNA-Oligonukleotid komplementär zum 3'-Terminus, mutierter 3Dpol und einem DNA-Oligonukleotid komplementär zum 3'-Terminus, bzw. mit einem DNA-Oligonukleotid komplementär zum 3'-Terminus, aber ohne Norovirus 3Dpol. T, Template-RNA. R, Replikationsprodukt. M, RNA-Molekulargewichtsmarker (Kb).
- (B) Strangseparationsanalyse der Replikationsprodukte. Die Reaktionsprodukte wurden auf Formaldehyd-Agarosegelen analysiert und nach Ethidiumbromidfärbung durch UV-Transillumitiation visualisiert. Bahnen 1 und 2, Template (anti-subgenomische RNA) bzw. Norovirus 3Dpol Replikationsprodukt. M, RNA Molekulargewichtsmarker (Kb).
- Figur 13:
- Primer-unabhängige de novo Initiation der RNA Synthese an homopolymeren Templaten. Die RNA-Synthese wurde in Anwesenheit (schwarze Balken) oder in Abwesenheit (graue Balken) von kaltem CTP, ATP, GTP bzw. UTP für poly(rG), poly(rU), poly(rC) bzw. poly(rA)-Temptates durchgeführt. Der Einbau von [α-32P]CMP, [α-32P]AMP, [α-32P]GMP, oder [α-32P]UMP wurde nach TCA-Präzipitation und Sammeln von G/C-Glassfaserfiltern gemessen. Die Einbauwerte sind angegeben.
- Figur 14:
- Amplifikation viraler und eukaryontischer RNA mit dem Norovirus RNA-Polymerase-Enzym.
- (A) Autoradiogramm der inkorporation von [α32P]UMP. Reaktion 1, Subgenomische Norovirus RNA; Reaktion 2, Subgenomische Norovirus RNA mit einer 60 Nukleotid Deletion am 5'-Ende der RNA; Reaktion 3, Subgenomische Norovirus RNA mit einer 60 Nukleotid Deletion am 3'-Ende der RNA; Reaktion 4, Subgenomische Norovirus RNA mit einer 60 Nukleotid Deletion am 5'-Ende und 3'-Ende der RNA; Reaktion 5 u. 6, negative Kontrolle: Reaktion 7; Subgenomische Astrovirus RNA (Serotyp 1); Reaktion 8; Subgenomische Astrovirus RNA (Serotyp 2); Reaktion 9, eukaryontische RNA des elF4-Gens von Xenopus Laevi; Reaktion 10, Subgenomische Norovirus DNA; Reaktion 11; T7-transkriberte RNA des elF4-Gens von Xenopus Laevi; Reaktion 12, negative Kontrolle.
- (B) Quantifizierung der Inkorporation von [α-32P]UMP, Die Nummerierung der Reaktionen bezieht sich auf die Proben der
Figur 14 (A) .
- Figur 15:
- Expression und Reinigung von Sapovirus 3Dpol in E. coli.
- (A) SDS-PAGE Analyse von gereinigter rekombinanter Sapovirus 3Dpol die in E. coli exprimiert wurde und einen C-terminalen His-tag trägt. 3Dpol, Wildtyp-Sapovirus 3Dpol. m3Dpol, Sapovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G). M, Molekulargewichtsmarker (KDa).
- (B) Western-Blot Analyse von gereinigter rekombinanter Sapovirus 3Dpol unter Verwendung eines Penta-Histidin-Antikörpers (monoklonaler Maus-Antikörper, Qiagen) zur Detektion von Proteinen mit einem C-terminalen His-tag, 3Dpol, Wildtyp- Sapovirus 3Dpol. m3Dpol, Sapovirus 3Dpol mit einer Mutation in der 'active site' (YGD343GD344G). M, Molekulargewichtsmarker (KDa).
- Figur 16:
- Konzentrationsabhängigkeit, Substratabhängigkeit, Temperaturabhängigkeit und Metallionenabhängigkeit der Sapovirus 3Dpol Aktivität. Bei allen Reaktionen wurde subgenomische RNA als Template eingesetzt.
- (A) Konzentrationsabhängige Aktivität der Sapovirus 3Dpol. Die RNA-Synthese wurde in Gegenwart von 0.024 µM RNA und ansteigenden Konzentrationen von Sapovirus 3Dpol (0.0 bis 5.0 µM) durchgeführt. Einbau von [α-32P]UMP wie angegeben. Die Reaktion wurde 2 h bei 37°C durchgeführt.
- (B) Substratabhängigkeit der Sapovirus 3Dpol Aktivität. Die RNA-Synthese wurde in Gegenwart von 3 µM Sapovirus 3Dpol und ansteigenden Konzentrationen von subgenomischer RNA als Template (0.00, bis 0.100 µM) Für 2h bei 37°C durchgeführt. Einbau von [α-32P]UMP wie angegeben.
- (C) Temperaturabhängige Activität von Sapovirus 3Dpol. Die Reaktion wurde bei 30°C oder 37°C durchgeführt und zu den angegebenen Zeiten gestoppt. Bei allen Reaktionen wurden 3 µM Sapovirus 3Dpol eingesetzt. Einbau von [α-32P]UMP wie angegeben.
- (D) Metallionenabhängigkeit der Sapovirus 3Dpol-Aktivität. Die RNA-Synthese wurde in Gegenwart von 3 µM Sapovirus 3Dpol mit subgenomischer RNA als Template (0.024 µM) und steigenden Konzentrationen (0.5 bis 5 mM) von MgAcO oder MnCl2 bei 37°C für 2 h durchgeführt. Einbau von [α-32P]UMP wie angegeben.
- Figur 17:
- RNA-Synthese mit Sapovirus 3Dpol.
- (A) Die RNA-Synthese in Gegenwart einer synthetischen subgenomischen polyadenylierten RNA (sG-poly(A)-RNA) als Template wurde mit und ohne Zugabe eines 3 µM oligo(U)20 RNA-Primers und in Anwesenheit von Wildtyp Sapovirus 3Dpol (3Dpol) oder wie angegeben einer in der 'acitve site' mutierten 3Dpol (m3Dpol, YGD343GD344G) untersucht. Die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen analysiert und nach Ethidiumbromidfärbung mit UV-Transillumination visualisiert. Die sichtbare verbleibende Bande in den Reaktionen, bei denen m3Dpol anstelle von Wildtyp 3Dpol eingesetzt wurden oder bei denen kein Oligo(U)20 RNA-Primer zugegben wurde, resultiert vom in der Reaktion eingesetzten synthetischen subgenomischen RNA Template. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA-Molekulargewichtsmarker.
- (B) Strangseparationsanalyse des Reaktionsprodukts der in vitro RNA-Synthese mit Sapovirus 3Dpol. Das Reaktionsprodukt wurde wie angegeben durch RNA-Synthese mit Sapovirus 3Dpol (3Dpol) unter Verwendung einer subgenomeischen polyadenylierten RNA (sG-Po)y(A)-RNA) als Template erhalten. Die Reaktionsprodukte wurden auf denaturierenden Formaldehyd-Agarosegelen visualisiert. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA-Molekulargewichtsmarker.
- Figur 18:
- De novo Initiation der RNA-Synthese an Sapovirus anti-subgenomischer RNA.
- (A) Die RNA-Synthese wurde in Gegenwart einer synthetischen anti-subgenomische RNA (Anti-sG-RNA) als Template und Wildtyp-Sapovirus 3Dpol (3Dpol) oder einer in der 'active site' mutierten 3Dpol (m3Dpol, YGD343GD344G), wie angegeben. Die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen analysiert und nach Ethidiumbromidtärbung durch UV Trensilluminafion visualisiert. Die sichtbare verbleibende Bande bei der Reaktion, in der m3Dpol anstelle von Wildtyp 3Dpol eingesetzt wurde, resultiert aus dem in der Reaktion eingesetzten synthetischen anti-subgenomischen RNA-Template. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA-Molekulargewichtsmarker.
- (B) Strangseparationsanalyse des Reaktionsprodukts der in vitro RNA-synthese mit Sapovirus 3Dpol. Das Reaktionsprodukt wurde wie angegeben durch RNA-Synthese mit Sapovirus 3Dpol unter Verwendung einer synthetischen anti-subgenomische RNA (Anti-sG-RNA) als Template erhalten. Die Reaktionsprodukte wurden auf denaturierenden Formaldehyd-Agarosegelen visualisiert. T7, synthetische anti-subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA-Molekulargewichtsmarker.
- Figur 19:
- Terminale Transferase-Aktivität von Sapovirus 3Dpol.
Die Terminale Transferase-Aktivität wurde anhand einer Reaktion aus 3 µM Sapovirus 3Dpol oder einer in der 'active site' mutierten 3Dpol (m3Dpol) untersucht, die in Gegenwart von subgenomischer RNA (sG-RNA) als Template (0.024 µM) und [α-32P]ATP, [α-32P]GTP, [α-32P]CTP oder [α-32P]UTP für 2 h bei 37°C (0.024 µM) wie angegeben inkubiert wurde. Die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen analysiert und durch Autoradiographie visualisiert. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. - Figur 20:
- De novo Initiation der RNA-Synthese durch Sapovirus 3Dpol an homopolymeren Templates.
Die RNA-Synthese wurde durchgeführt, indem poly(G)-RNA, poly(U)-RNA, poly(C)-RNA und poly(A)-RNA Templates mit [α-32P]CTP, [α-32P]ATP, [α-32P]GTP, bzw. [α-32P]UTP und mit oder ohne 3 µM eines Oligo(C)20, Oligo(A)20, Oligo (G)20 bzw. Oligo(U)20-RNA-Primers inkubiert wurden. Parallel dazu wurde die RNA-Synthese in Abwesenheit eines Oligo(RNA)-Primers, aber in Anwesenheit von 50 µM kaltem CTP, ATP, GTP bzw. UTP für poly(G)-RNA, poly(U)-RNA, poly(C)-RNA bzw. poly(A)-RNA Templates. Einbau von [α-32P]CMP, [α-32P]AMP, [α-32P]GMP, oder [α-32P]UMP as wie angegeben. - Figur 21:
- Expression und Reinigung vom Vesivirus 3Dpol in E. coli.
- (A) SDS-PAGE Analyse von In E. coli exprimierter, gereinigter, rekombinanter Vesivirus 3Dpol mit einem C-terminalen His-tag. Gezeigt sind die Fractionen 11 bis 19 nach Affinitätsreinigung an einer Ni-NTA-Säule sowie die Konzentration von Fraktion 14. M, Molekulargewichtsmarker (KDa).
- Figur 22:
- RNA-Synthese mit Vesivirus 3Dpol.
- (A) Die RNA-Synthese in Gegenwart einer synthetischen subgenomischen polyadenylierten RNA (sG-poly(A)-RNA) als Template wurde mit und ohne Zugabe eines 3 µM oligo(U)20 RNA-Primers und in Anwesenheit von Wildtyp Vesivirus 3Dpol (3Dpol) untersucht. Die Reaktionsprodukte wurden auf nicht-denaturierenden Agarosegelen analysiert und nach Autoradiographie vizualisiert. T7, synthetische subgenomische RNA, die mittels in vitro Transkription mit T7 hergestellt wurde. M, RNA-Molekulargewichtsmarker.
- Die cDNA der Norovirus-RdRP wurde durch PCR aus dem Norovirus-Klon pUS-Norll (Genbank accession number: AY741811) erhalten. Sie wurde in den pET-28b(+)-Vektor (Novagen) kloniert, der Expressionsvektor wurde sequenziert und in E.coli CL21(DE3)plysS transformiert. Die Zellen wurden bei 37°C in Luria-Bertani-Medium mit Kanamycin (50 mg/L) kultiviert. Bei einer optischen Dichte von 0.6 (OD600) wurde die Proteinexpression durch den Zusatz von lsopropyl-β-D-thiogalactopyranosid (IPTG) mit einer Endkonzentration von 1 mM induziert. Die Kulturen wurden dann bei 25°C über Nacht inkubiert. Die aus 250 ml Kultur erhaltenen Zellpellets wurden einmal in 4 ml phosphate buffered saline (PBS) und 1 % Triton X 100 (Sigma) gewaschen. Die Zellen wurden bei 37°C für 15 Minuten mit DNase (10 U/ml) behandelt, auf Eis sonifiziert und in 40 ml Bindungspuffer (20 mM Tris/HCl, pH 7.9, 500 mM NaCl, 5 mM Imidazol) resuspendiert. Nach der Zentrifugation bei 4300 rpm bei 4 °C für 40 Minuten wurde das geklärte Lysat erhalten. Die mit einem His6-tag versehene Norovirus-RdRP wurde an eine Ni-Nitrilotriacetessigsäure (NTA)-Sepharosematrix (Novagen) gebunden, die mit dem Bindungspuffer prääquilibriert worden war. Das gebundene Protein wurde mit dem Bindungspuffer, der 60 mM Imidazol enthielt, gewaschen und mit Bindungspuffer, der 1 M Imidazol enthielt, eluiert. Das eluierte Protein wurde gegen Puffer A dialysiert (25 mM Tris-hCl, pH 8.0, 1 mM β-Mercaptoethanol, 100 mM NaCl, 5 mM MgCl2, 10 % Glycerin, 0.1 % Triton X). Die Proteinkonzentration wurde mit dem BCA proteinassay Kit (Pierce) auf der Grundlage der Biuret-Reaktion bestimmt. Das Enzym wurde in einem Endvolumen von 50 % Glycerin resuspendiert und bei -20°C aufbewahrt.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg RNA-Matrize, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-inhibitor (RNAsin, Promega), je 0.4 mM von ATP, CTP, GTP, UTP, 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Ethidiumbromid-Färbung durch UV-Transillumination auf TBE-gepufferten Agarosegelen sichtbar gemacht. Formamid-Agarosegele können ebenfalls verwendet werden.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg RNA-Matrize, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-Inhibitor (RNAsin, Promega), je 0.4 mM von ATP, CTP, GTP, UTP, 0.1 bis 1 µM genspezifischer RNA-Primer, 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Ethidiumbromid-Färbung durch UV-Transillumination auf TBE-gepufferten Agarosegelen sichtbar gemacht. Formamid-Agarosegele oder Harnstoff/Polyacrylamidgele können ebenfalls verwendet werden.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg RNA-Matrize, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-Inhibitor (RNAsin, Promega), je 0.4 mM von ATP, CTP, GTP UTP, 0.1 bis 1 µM poly-(U)20-Primer, 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Ethidiumbromid-Färbung durch UV-Transillumination auf TBE-gepufferten Agarosegelen sichtbar gemacht. Formamid-Agarosegele können ebenfalls verwendet werden.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg RNA-Matrize, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-Inhibitor (RNAsin, Promega), je 0.4 mM von ATP, CTP, GTP, UTP, 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Ethidiumbromid-Färbung durch UV-Transillumination auf TBE-gepufferten 10 %igen Polyacrylamidgelen sichtbar gemacht. Formamid-Agarosegele können ebenfalls verwendet werden. Alternativ kann armored-RNA verwendet werden.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg RNA-Matrize, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-Inhibitor (RNAsin, Promega), je 0.4 mM von ATP, oder CTP, oder GTP, oder UTP, 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Ethidiumbromid-Färbung durch UV-Transillumination auf TBE-gepufierten 10 %igen Polyacrylamidgelen sichtbar gemacht. Formamid-Agarosegele können ebenfalls verwendet werden. Alternativ kann armored-RNA verwendet werden.
- Die Reaktionsmischung (50 µl) besteht aus 0.5 bis 1 µg poly(C)-RNA-Matrize, oder eine RNA mit poly(C)-RNA am 3'-Ende, 10 µl des Reaktionspuffer (250 mM HEPES, pH 8.0, 15 mM Magnesiumacetat, 20 mM DTT), 50 U RNase-Inhibitor (RNAsin, Promega), 50 µM von GTP, und 3 µM der nach Ausführungsbeispiel 1 hergestellten Norovirus-RdRP. Die Reaktion wird bei 30 °C für 2 Stunden durchgeführt. Die Reaktion wird durch die Zugabe von 50 µl der Stoplösung (4 M Ammoniumacetat, 100 mM EDTA) gestoppt. Die Reinigung erfolgt durch Phenol-Chloroform-Extraktion oder mit dem MEGAclear Kit (Ambion) nach den Herstelleranweisungen. Die Transkriptionsprodukte werden nach Emidiumbromid-Färbung durch UV-Ttansillumination auf TBE-gepufferten 10 %igen Polyacrylamidgelen sichtbar gemacht. Formamid-Agarosegele können ebenfalls verwendet werden. Alternativ kann armored-RNA verwendet werden.
- Weiterhin umfasst die vorliegende Erfindung eine RNA-abhängige RNA-Polymerase zur Amplifikation und/oder Markierung von RNA, wobei die RNA-abhängige RNA-Polymerase eine "Rechte-Hand-Konformation" aufweist und die Aminosäuresequenz der RNA-abhängigen RNA-Polymerase die folgenden Sequenzabschnitte umfasst: a. XXDYS, b. GXPSG, c. YGDD, d. XXYGL, e. XXXXFLXRXX mit der Bedeutung: D: Aspartat, Y: Tyrosin, S: Serin, G: Glycin, P: Prolin, L: Leucin, F: Phenylalanin, R: Arginin, X: eine beliebige Aminosäure.
- Eine bevorzugte Ausführungsform der RNA-abhängigen RNA-Polymerase ist eine RNA-abhängige RNA-Polymerase eines Virus aus der Familie der Caliciviridae.
- Bei einer weiteren bevorzugten Ausführungsform ist die RNA-abhängige RNA-Polymerase eine RNA-abhängige RNA-Polymerase eines humanen und/oder nicht-humanen pathogenen Virus der Familie der Caliciviridae.
- Bei einer weiteren bevorzugten Ausführungsform ist die RNA-abhängige RNA-Polymerase eine RNA-abhängige RNA-Polymerase eines Norovirus, Sapovirus, Vesivirus oder eines Lagovirus.
- Bei einer weiteren bevorzugten Ausführungsform ist die RNA-abhängige RNA-Polymerase die RNA-abhängige RNA-Polymerase eines Norovirus-Stammes wie z.B. des Norovirus-Stammes HuCV/NL/Dresden174/1997/GE oder eines eines SapovirusStammes wie z.B. des Sapovirus-Stammes pJG-SapO1, oder eines Vesivirus-Stammes wie z.B. des Vesivirus-Stammes FCV/Dresden/2006/GE.
- Bei einer weiteren bevorzugten Ausführungsform ist die RNA-abhängige RNA-Polymerase ein Protein gemäß SEQ ID NO 1, SEQ ID NO 2, SEQ ID NO 3, SEQ ID NO 4, SEQ ID NO 5 oder SEQ ID NO 6.
- Die vorliegende Erfindung umfasst weiterhin ein Verfahren zur Amplifikation von RNA mittels einer RNA-abhängigen RNA-Polymerase mit den Schritten a. Anlagerung der RNA-abhängigen RNA-Polymerase an die RNA-Matrize in der Anwesenheit oder Abwesenheit eines Oligonukleotid-Primers, b. Umschreiben der RNA-Matrize in antisense-RNA durch die RNA-abhängige RNA-Polymerase c. Trennung des RNA/antisense-RNA-Duplexes in einzelne RNA-Stränge.
- Bei einer bevorzugten Ausführungsform des Verfahrens zur Amplifikation von RNA erfolgt die Trennung des RNA/antisense-RNA-Duplexes in einzelne RNA-Stränge durch Hitzedenaturierung, durch chemische Denaturierung und/oder enzymatisch.
- Bei einer weiteren bevorzugten Ausführungsform erfolgt die enzymatische Trennung des RNA/antisense-RNA-Duplexes in einzelne RNA-Stränge durch ein Enzym mit der Fähigkeit, doppelsträngige RNA in einzelsträngige RNA aufzutrennen.
- Weiterhin umfasst die vorliegende Erfindung ein Verfahren zur primerabhängigen Amplifikation von RNA bei dem mindestens ein Primer, der mit einem Abschnitt der RNA-Matrize hybridisiert, eingesetzt wird und nach der Anlagerung der RNA-abhängigen RNA-Polymerase der Primer durch die RNA-abhängige RNA-Polymerase entsprechend der Sequenz der RNA-Matrize verlängert wird.
- Bei einer bevorzugten Ausführungsform dieses Verfahrens wird als Primer ein heteropolymeres oder homopolymeres RNA-Primer oder DNA-Primer eingesetzt.
- Bei einer weiteren bevorzugten Ausführungsform wird als Primer ein poly-U-RNA-, poly-A-RNA-, poly-C-RNA- oder poly-G-RNA-Primer oder jedes beliebiges hompolymeres Oligo-RNA-Primer eingesetzt.
- Bei einer weiteren Ausführungsform des Verfahrens zur primerunabhängigen Amplifikation von poly(C)-RNA wird kein Primer, der mit einem Abschnitt der RNA-Matrize hybridisiert, eingesetzt, GTP als einziges Nukleotid verwendet und durch die RNA-abhängige RNA-Polymerase entsprechend der Sequenz der RNA-Matrize verlängert.
- Die vorliegende Erfindung umfasst weiterhin ein Verfahren zur Markierung von RNA mittels einer RNA-abhängigen RNA-Polymerase, mit den Schritten: a. Anlagerung der RNA-abhängigen RNA-Polymerase an die zu markierende RNA, b. Anhängen mindestens eines Nukleotids an das 3'-Ende der zu markierenden RNA.
- Die vorliegende Erfindung betrifft ebenfalls die Verwendung einer RNA-abhängigen RNA-Polymerase zur Amplifikation und/oder Markierung von RNA.
- Weiterhin umfasst die vorliegende Erfindung ein Kit zur Amplifikation von RNA, mindestens bestehend aus: a. einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 6, b. einem geeigneten Reaktions-Puffer, c. NTPs, d. ggf. RNase-Inhibitor, e. ggf. Stoplösung.
- Ein weiterer Gegenstand der Erfindung ist ein Kit zur Amplifikation von RNA, mindestens bestehend aus a. einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 6, b. einem geeigneten Reaktions-Puffer, c. NTPs, d. ggf. RNase-Inhibitor, e. ggf. Stoplösung, f. einer Helicase.
- Ein weiterer Gegenstand der Erfindung ist ein Kit zur primerabhängigen Amplifikation von RNA, mindestens bestehend aus: a. einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 6, b. einem geeigneten Reaktions-Puffer, c. NTPs, d. ggf. RNase-Inhibitor, e. ggf. Stoplösung, f. Primer.
- Die vorliegende Erfindung umfasst zudem ein Kit zur primerunabhängigen Amplifikation von RNA, mindestens bestehend aus: a. einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 6, b. einem geeigneten Reaktions-Puffer, c. NTPs, d. ggf. RNase-Inhibitor, e. ggf. Stoplösung.
- Weiterhin umfasst die vorliegende Erfindung ein Kit zur Markierung von RNA, mindestens bestehend aus a. einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 6, b. einem geeigneten Reaktions-Puffer, c. CTP oder UTP oder ATP oder GTP oder jeden beliebigen Nukleotid, d. ggf. RNase-Inhibitor, e. ggf. Stoplösung.












Claims (12)
- RNA-abhängige RNA-Polymerase mit einer Aminosäuresequenz gemäß SEQ ID NO: 2.
- RNA-abhängige RNA-Polymerase nach Anspruch 1, die am N- oder C-Terminus mit einer Sequenz fusioniert ist, welche die Aufreinigung des Fusionsproteins nach Expression in einem Wirtsorganismus erleichtert.
- RNA-abhängige RNA-Polymerase nach Anspruch 2, wobei die Sequenz ein Histidin-Marker ist.
- RNA-abhängige RNA-Polymerase nach Anspruch 3, die eine Aminosäuresequenz gemäß SEQ ID NO: 5 aufweist.
- Nukleinsäure, die für eine RNA-abhängige RNA-Polymerase nach einem der vorhergehenden Ansprüche kodiert.
- Expressionsvektor, der eine Nukleinsäure nach Anspruch 5 enthält.
- Wirtsorganismus, der einen Expressionsvektor nach Anspruch 6 enthält.
- Verfahren zur Herstellung der RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 4 mit den Schritteni. Klonierung der die RNA-abhängige RNA-Polymerase kodierenden Nukleinsäure in einen Expressionsvektor undii. Einbringen des Expressionsvektors in einen Wirtsorganismus.
- Kit zur Amplifikation von RNA, mindestens bestehend aus- einer RNA-abhängigen RNA-Polymerase nach einem der Ansprüche 1 bis 4- einem geeigneten Reaktionspuffer- Ribonukleotide ATP, UTP, GTP und CTP;- ggf. Stoplösung;- ggf. RNAse-Inhibitor.
- Kit nach Anspruch 9, das mindestens einen poly-U-RNA-Primer enthält.
- Kit nach Anspruch 9 oder 10, das mindestens einen Primer enthält, der spezifisch mit einem Abschnitt viraler RNA hybridisiert.
- Kit zur Markierung von RNA, mindestens bestehend aus- einer RNA-abhängigen RNA Polymerase nach einem der Ansprüche 1 bis 4;- einem geeigneten Reaktionspuffer;- ATP, GTP, UTP oder CTP;- ggf. Stoplösung;- ggf. RNAse-Inhibitor.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005036085 | 2005-07-25 | ||
| DE102006008219 | 2006-02-13 | ||
| EP06761834A EP1910528A2 (de) | 2005-07-25 | 2006-07-25 | Rna-abhängige rna-polymerase, verfahren und kits zur amplifikation und / oder markierung von rna |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP06761834A Division EP1910528A2 (de) | 2005-07-25 | 2006-07-25 | Rna-abhängige rna-polymerase, verfahren und kits zur amplifikation und / oder markierung von rna |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2098592A2 true EP2098592A2 (de) | 2009-09-09 |
| EP2098592A3 EP2098592A3 (de) | 2009-09-30 |
Family
ID=37682762
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP06761834A Withdrawn EP1910528A2 (de) | 2005-07-25 | 2006-07-25 | Rna-abhängige rna-polymerase, verfahren und kits zur amplifikation und / oder markierung von rna |
| EP09155974A Not-in-force EP2077322B1 (de) | 2005-07-25 | 2006-07-25 | Verfahren und Kit zur Amplifikation von heteropolymerer oder poly(C)- RNA |
| EP09155984A Withdrawn EP2098592A3 (de) | 2005-07-25 | 2006-07-25 | RNA-abhängige RNA-Polymerase, Verfahren und Kits zur Amplifikation und/oder Markierung von RNA |
| EP10158113A Not-in-force EP2204445B1 (de) | 2005-07-25 | 2006-07-25 | Verfahren und Kit zur primer-abhängigen Amplifikation heterologer viraler, eukaryontischer oder prokaryontischer RNA |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP06761834A Withdrawn EP1910528A2 (de) | 2005-07-25 | 2006-07-25 | Rna-abhängige rna-polymerase, verfahren und kits zur amplifikation und / oder markierung von rna |
| EP09155974A Not-in-force EP2077322B1 (de) | 2005-07-25 | 2006-07-25 | Verfahren und Kit zur Amplifikation von heteropolymerer oder poly(C)- RNA |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP10158113A Not-in-force EP2204445B1 (de) | 2005-07-25 | 2006-07-25 | Verfahren und Kit zur primer-abhängigen Amplifikation heterologer viraler, eukaryontischer oder prokaryontischer RNA |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20080176293A1 (de) |
| EP (4) | EP1910528A2 (de) |
| JP (1) | JP5383189B2 (de) |
| CN (1) | CN101228268A (de) |
| AT (1) | ATE478946T1 (de) |
| DE (2) | DE112006002611A5 (de) |
| WO (1) | WO2007012329A2 (de) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003279624A1 (en) | 2002-11-08 | 2004-06-07 | Pantarhei Bioscience B.V. | Synthesis of estetrol via estrone derived steroids |
| US20110053226A1 (en) * | 2008-06-13 | 2011-03-03 | Riboxx Gmbh | Method for enzymatic synthesis of chemically modified rna |
| JP2012508571A (ja) | 2008-11-13 | 2012-04-12 | リボックス・ゲーエムベーハー | Rna検出法 |
| US8409805B2 (en) * | 2009-02-13 | 2013-04-02 | Asuragen, Inc. | Method of amplification of GC-rich DNA templates |
| EP2401375B1 (de) * | 2009-02-24 | 2017-08-23 | RiboxX GmbH | Verbesserte konstruktion von small-interfering-rna |
| US20120208242A1 (en) * | 2009-10-21 | 2012-08-16 | Riboxx Gmbh | Method and RNA Reactor for Exponential Amplification of RNA |
| JP2013507942A (ja) * | 2009-10-21 | 2013-03-07 | リボックス・ゲーエムベーハー | 熱安定性rna依存性rnaポリメラーゼを用いる、rnaの指数関数的増幅のための方法 |
| EP2619318B1 (de) * | 2010-09-21 | 2014-12-24 | RiboxX GmbH | Durch mikrowellen induzierte rna polymerisation mittels rna polymerasen von caliciviren |
| US20130183718A1 (en) | 2010-09-21 | 2013-07-18 | RibpxX GmbH | Method for Synthesizing RNA using DNA Template |
| EP2673376A1 (de) | 2011-02-09 | 2013-12-18 | RiboxX GmbH | Verfahren für den nachweis von polynukleotidsequenzen |
| WO2012107538A1 (en) | 2011-02-09 | 2012-08-16 | Riboxx Gmbh | Method for labelling double-stranded dna or dna/rna hybrids |
| SG195118A1 (en) | 2011-06-01 | 2013-12-30 | Estetra S A | Process for the production of estetrol intermediates |
| PT2714712T (pt) | 2011-06-01 | 2016-11-08 | Estetra Sprl | Processo para a produção de intermediários de estetrol |
| EP2383279A1 (de) | 2011-07-19 | 2011-11-02 | Pantarhei Bioscience B.V. | Herstellungsverfahren für Esterol |
| WO2013064584A1 (en) | 2011-10-31 | 2013-05-10 | Riboxx Gmbh | Double-stranded RNA for immunostimulation |
| WO2013097965A1 (en) | 2011-12-30 | 2013-07-04 | Riboxx Gmbh | Triphosphate-containing double-stranded rna for immunostimulation |
| ES2759023T3 (es) | 2012-07-20 | 2020-05-07 | Asuragen Inc | Genotipificación completa de FMR1 |
| EP3126501A1 (de) | 2014-03-24 | 2017-02-08 | RiboxX GmbH | Doppelsträngige rna-konjugate und deren verwendung |
| WO2016112963A1 (en) | 2015-01-13 | 2016-07-21 | Riboxx Gmbh | Delivery of biomolecules into cells |
| WO2017121494A1 (en) | 2016-01-15 | 2017-07-20 | Riboxx Gmbh | 5'-triphosphated short immunostimulatory nucleotides, oligonucleotides and polynucleotides |
| JP6792847B2 (ja) * | 2016-12-27 | 2020-12-02 | 国立大学法人 東京大学 | mRNAの機能化方法 |
| US10870891B2 (en) * | 2017-01-05 | 2020-12-22 | Biodesix, Inc. | Diagnostic test system for specific, sensitive and reproducible detection of circulating nucleic acids in whole blood |
| CN108893467B (zh) * | 2018-06-21 | 2022-02-15 | 广东省微生物研究所(广东省微生物分析检测中心) | 一种gi.1型札幌病毒基因组扩增引物和扩增方法 |
| WO2021231891A1 (en) * | 2020-05-15 | 2021-11-18 | Quidel Corporation | Method for direct amplification and detection of rna |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2429814C (en) * | 2000-12-01 | 2014-02-18 | Thomas Tuschl | Rna interference mediating small rna molecules |
| WO2002092774A2 (en) * | 2001-05-14 | 2002-11-21 | Shi-Lung Lin | Replicase cycling reaction amplification |
| JP2005110517A (ja) * | 2003-10-03 | 2005-04-28 | Bml Inc | Rna依存性rnaポリメラーゼ蛋白質の製造 |
-
2006
- 2006-07-25 EP EP06761834A patent/EP1910528A2/de not_active Withdrawn
- 2006-07-25 WO PCT/DE2006/001326 patent/WO2007012329A2/de not_active Ceased
- 2006-07-25 DE DE112006002611T patent/DE112006002611A5/de not_active Withdrawn
- 2006-07-25 EP EP09155974A patent/EP2077322B1/de not_active Not-in-force
- 2006-07-25 DE DE502006007749T patent/DE502006007749D1/de active Active
- 2006-07-25 EP EP09155984A patent/EP2098592A3/de not_active Withdrawn
- 2006-07-25 JP JP2008523122A patent/JP5383189B2/ja not_active Expired - Fee Related
- 2006-07-25 EP EP10158113A patent/EP2204445B1/de not_active Not-in-force
- 2006-07-25 CN CNA2006800270288A patent/CN101228268A/zh active Pending
- 2006-07-25 AT AT09155974T patent/ATE478946T1/de active
-
2008
- 2008-01-24 US US12/019,206 patent/US20080176293A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007012329A3 (de) | 2007-08-23 |
| EP2077322A3 (de) | 2009-09-30 |
| CN101228268A (zh) | 2008-07-23 |
| WO2007012329A2 (de) | 2007-02-01 |
| EP2077322B1 (de) | 2010-08-25 |
| DE112006002611A5 (de) | 2008-07-03 |
| ATE478946T1 (de) | 2010-09-15 |
| EP1910528A2 (de) | 2008-04-16 |
| DE502006007749D1 (de) | 2010-10-07 |
| JP2009502145A (ja) | 2009-01-29 |
| EP2077322A2 (de) | 2009-07-08 |
| EP2098592A3 (de) | 2009-09-30 |
| US20080176293A1 (en) | 2008-07-24 |
| EP2204445B1 (de) | 2013-01-02 |
| JP5383189B2 (ja) | 2014-01-08 |
| EP2204445A1 (de) | 2010-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2077322B1 (de) | Verfahren und Kit zur Amplifikation von heteropolymerer oder poly(C)- RNA | |
| DE69730926T2 (de) | Modifizierte thermostabile DNA Polymerase | |
| US7662594B2 (en) | Helicase-dependent amplification of RNA | |
| US20210139884A1 (en) | Reverse transcriptase for nucleic acid sequencing | |
| JP6587544B2 (ja) | 熱安定なTthPrimPolを用いた増幅及び配列決定方法 | |
| DE69133204T2 (de) | Gereinigtes thermostabiles nukleinsäure-polymeraseenzym aus thermotoga maritima | |
| US20220348892A1 (en) | Modified heat-resistant dna polymerase | |
| DE60112746T2 (de) | Kälteempfindliche dna-polymerasemutanten | |
| EP2697372B1 (de) | Dna-polymerasen mit verbesserter aktivität | |
| US20150210989A1 (en) | Novel reverse transcriptases for use in high temperature nucleic acid synthesis | |
| KR20230013167A (ko) | 핵산 샘플을 프로세싱하는 방법 | |
| JP2003519488A (ja) | 活性部位に1又は複数の変異を有するdnaポリメラーゼ変異体 | |
| CN113801917B (zh) | 基于crispr技术进行多重核酸检测的方法 | |
| KR20220038668A (ko) | 유형 III CRISPR/Cas 기반 진단 | |
| ES2553400T3 (es) | ADN polimerasas con actividad mejorada | |
| US6867027B1 (en) | RNA polymerase | |
| US5516633A (en) | DNA sequencing with a T7-type gene 6 exonuclease | |
| ES2285766T3 (es) | Arn-polimerasa dependiente de arn que funciona preferentemente sobre molde de arn y procedimiento de transcripcion de arn bajo la dependencia de un promotor con dicha arn polimerasa dependiente de arn. | |
| CN114592043A (zh) | 一种等温核酸检测酶组合物、试剂盒及其用途和检测方法 | |
| JP7160142B2 (ja) | 核酸増幅用試薬及び核酸増幅法 | |
| EP1100923A2 (de) | Thermostabiler in vitro-komplex mit polymeraseaktivität | |
| JP4453115B2 (ja) | 標識されたdnaの調製方法 | |
| JP4329365B2 (ja) | 単一成分から成るトリ骨芽球症ウイルス逆転写酵素とその用途 | |
| KR20240141182A (ko) | 열안정성 rna 중합효소 | |
| EA004630B1 (ru) | Способ образования комплекса |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AC | Divisional application: reference to earlier application |
Ref document number: 1910528 Country of ref document: EP Kind code of ref document: P |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA HR MK RS |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA HR MK RS |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: RIBOXX GMBH |
|
| AKX | Designation fees paid | ||
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 20100401 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: 8566 |