ES2704525T3 - Derivados de naftiridina como antagonistas de integrina AlfaVBeta6 para el tratamiento de, por ejemplo, enfermedades fibróticas - Google Patents
Derivados de naftiridina como antagonistas de integrina AlfaVBeta6 para el tratamiento de, por ejemplo, enfermedades fibróticas Download PDFInfo
- Publication number
- ES2704525T3 ES2704525T3 ES15767476T ES15767476T ES2704525T3 ES 2704525 T3 ES2704525 T3 ES 2704525T3 ES 15767476 T ES15767476 T ES 15767476T ES 15767476 T ES15767476 T ES 15767476T ES 2704525 T3 ES2704525 T3 ES 2704525T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- fluoro
- acid
- tetrahydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims description 34
- 201000010099 disease Diseases 0.000 title claims description 30
- 238000011282 treatment Methods 0.000 title claims description 30
- 102000006495 integrins Human genes 0.000 title description 38
- 108010044426 integrins Proteins 0.000 title description 38
- 239000005557 antagonist Substances 0.000 title description 27
- 230000003176 fibrotic effect Effects 0.000 title description 9
- 150000005054 naphthyridines Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 243
- 150000003839 salts Chemical class 0.000 claims abstract description 77
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 9
- -1 3-fluoro-3- (2- (5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl) ethyl) pyrrolidin-1-yl Chemical group 0.000 claims description 114
- 239000008194 pharmaceutical composition Substances 0.000 claims description 32
- 238000002560 therapeutic procedure Methods 0.000 claims description 18
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 10
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 9
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 5
- 229940044551 receptor antagonist Drugs 0.000 claims description 3
- 239000002464 receptor antagonist Substances 0.000 claims description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 101
- 239000000243 solution Substances 0.000 description 99
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 89
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- 239000000203 mixture Substances 0.000 description 87
- 235000002639 sodium chloride Nutrition 0.000 description 71
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 68
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 59
- 239000000543 intermediate Substances 0.000 description 53
- 238000002360 preparation method Methods 0.000 description 45
- 238000006243 chemical reaction Methods 0.000 description 43
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 42
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 41
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 40
- 239000002253 acid Substances 0.000 description 40
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 33
- 239000007787 solid Substances 0.000 description 28
- 230000002829 reductive effect Effects 0.000 description 27
- 239000011541 reaction mixture Substances 0.000 description 26
- 235000019439 ethyl acetate Nutrition 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- IMNFDUFMRHMDMM-UHFFFAOYSA-N anhydrous n-heptane Natural products CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 22
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 21
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 20
- 229910052757 nitrogen Inorganic materials 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 19
- 238000000034 method Methods 0.000 description 19
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 18
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 18
- 238000005481 NMR spectroscopy Methods 0.000 description 18
- 235000019441 ethanol Nutrition 0.000 description 18
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 18
- 239000000725 suspension Substances 0.000 description 18
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 16
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 16
- 239000003814 drug Substances 0.000 description 16
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 14
- 239000001099 ammonium carbonate Substances 0.000 description 14
- 238000004296 chiral HPLC Methods 0.000 description 14
- 239000003921 oil Substances 0.000 description 14
- 239000010948 rhodium Substances 0.000 description 14
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 238000004587 chromatography analysis Methods 0.000 description 13
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- 239000002585 base Substances 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 11
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 11
- 210000004027 cell Anatomy 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 10
- 206010016654 Fibrosis Diseases 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000012267 brine Substances 0.000 description 10
- 238000003352 cell adhesion assay Methods 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- 239000003446 ligand Substances 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 10
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 10
- 239000012453 solvate Substances 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 9
- 229910021529 ammonia Inorganic materials 0.000 description 9
- MDSGTKLDIYJRNX-UHFFFAOYSA-N benzyl 3-fluoro-3-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound OCC1(F)CCN(C1)C(=O)OCc1ccccc1 MDSGTKLDIYJRNX-UHFFFAOYSA-N 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 230000004761 fibrosis Effects 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 239000013543 active substance Substances 0.000 description 8
- 238000002425 crystallisation Methods 0.000 description 8
- 230000008025 crystallization Effects 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 208000005069 pulmonary fibrosis Diseases 0.000 description 8
- 239000000377 silicon dioxide Substances 0.000 description 8
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 7
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 7
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000006071 cream Substances 0.000 description 7
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 7
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- OHGOMHOEZUWGOH-UHFFFAOYSA-N (3-morpholin-4-ylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(N2CCOCC2)=C1 OHGOMHOEZUWGOH-UHFFFAOYSA-N 0.000 description 6
- YVMHYDQOUZWQQA-UHFFFAOYSA-N 3-fluoro-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-3-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC(F)(C(O)=O)C1 YVMHYDQOUZWQQA-UHFFFAOYSA-N 0.000 description 6
- BMGSGGYIUOQZBZ-UHFFFAOYSA-N 3-morpholin-4-ylphenol Chemical compound OC1=CC=CC(N2CCOCC2)=C1 BMGSGGYIUOQZBZ-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 229940123038 Integrin antagonist Drugs 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- 206010028980 Neoplasm Diseases 0.000 description 6
- 235000019502 Orange oil Nutrition 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 6
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 6
- 238000000113 differential scanning calorimetry Methods 0.000 description 6
- 239000000499 gel Substances 0.000 description 6
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 150000002688 maleic acid derivatives Chemical class 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 239000010502 orange oil Substances 0.000 description 6
- 210000000056 organ Anatomy 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 235000019698 starch Nutrition 0.000 description 6
- 239000008107 starch Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- CMSYDJVRTHCWFP-UHFFFAOYSA-N triphenylphosphane;hydrobromide Chemical compound Br.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 CMSYDJVRTHCWFP-UHFFFAOYSA-N 0.000 description 6
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- RWOGZQDBFUFMSJ-RXMQYKEDSA-N [(3R)-3-fluoropyrrolidin-3-yl]methanol Chemical compound F[C@]1(CNCC1)CO RWOGZQDBFUFMSJ-RXMQYKEDSA-N 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 150000001860 citric acid derivatives Chemical class 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000001530 fumaric acid Substances 0.000 description 5
- 235000011087 fumaric acid Nutrition 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 235000011167 hydrochloric acid Nutrition 0.000 description 5
- 238000011065 in-situ storage Methods 0.000 description 5
- 239000011976 maleic acid Substances 0.000 description 5
- 238000010899 nucleation Methods 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 235000011181 potassium carbonates Nutrition 0.000 description 5
- 239000001632 sodium acetate Substances 0.000 description 5
- 235000017281 sodium acetate Nutrition 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 238000002411 thermogravimetry Methods 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 208000028006 Corneal injury Diseases 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 235000015165 citric acid Nutrition 0.000 description 4
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 208000017169 kidney disease Diseases 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- RWIKCBHOVNDESJ-NSCUHMNNSA-N methyl (e)-4-bromobut-2-enoate Chemical compound COC(=O)\C=C\CBr RWIKCBHOVNDESJ-NSCUHMNNSA-N 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 4
- 230000035699 permeability Effects 0.000 description 4
- 235000011007 phosphoric acid Nutrition 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 238000000634 powder X-ray diffraction Methods 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- NYCVCXMSZNOGDH-UHFFFAOYSA-N pyrrolidine-1-carboxylic acid Chemical compound OC(=O)N1CCCC1 NYCVCXMSZNOGDH-UHFFFAOYSA-N 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 230000037390 scarring Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 4
- ZCZVGQCBSJLDDS-UHFFFAOYSA-N 1,2,3,4-tetrahydro-1,8-naphthyridine Chemical compound C1=CC=C2CCCNC2=N1 ZCZVGQCBSJLDDS-UHFFFAOYSA-N 0.000 description 3
- NRMBSORJNKJEKR-AWEZNQCLSA-N 7-[2-[(3S)-3-fluoropyrrolidin-3-yl]ethyl]-1,2,3,4-tetrahydro-1,8-naphthyridine Chemical compound F[C@@]1(CCC2=CC=C3CCCNC3=N2)CCNC1 NRMBSORJNKJEKR-AWEZNQCLSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 229920000856 Amylose Polymers 0.000 description 3
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 3
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 3
- NRMBSORJNKJEKR-UHFFFAOYSA-N FC1(CCC2=CC=C3CCCNC3=N2)CCNC1 Chemical compound FC1(CCC2=CC=C3CCCNC3=N2)CCNC1 NRMBSORJNKJEKR-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 241001629494 Nephelium Species 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 238000002441 X-ray diffraction Methods 0.000 description 3
- OTQMNVRNDGSBDR-UHFFFAOYSA-M [Br-].C(C1=CC=C2CCCNC2=N1)[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound [Br-].C(C1=CC=C2CCCNC2=N1)[P+](C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 OTQMNVRNDGSBDR-UHFFFAOYSA-M 0.000 description 3
- 235000011054 acetic acid Nutrition 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- NTECHUXHORNEGZ-UHFFFAOYSA-N acetyloxymethyl 3',6'-bis(acetyloxymethoxy)-2',7'-bis[3-(acetyloxymethoxy)-3-oxopropyl]-3-oxospiro[2-benzofuran-1,9'-xanthene]-5-carboxylate Chemical compound O1C(=O)C2=CC(C(=O)OCOC(C)=O)=CC=C2C21C1=CC(CCC(=O)OCOC(C)=O)=C(OCOC(C)=O)C=C1OC1=C2C=C(CCC(=O)OCOC(=O)C)C(OCOC(C)=O)=C1 NTECHUXHORNEGZ-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 235000012501 ammonium carbonate Nutrition 0.000 description 3
- 239000000427 antigen Substances 0.000 description 3
- 102000036639 antigens Human genes 0.000 description 3
- 108091007433 antigens Proteins 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- VJRITMATACIYAF-UHFFFAOYSA-N benzenesulfonohydrazide Chemical compound NNS(=O)(=O)C1=CC=CC=C1 VJRITMATACIYAF-UHFFFAOYSA-N 0.000 description 3
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 3
- 229920002988 biodegradable polymer Polymers 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000005038 ethylene vinyl acetate Substances 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 210000002744 extracellular matrix Anatomy 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 238000009169 immunotherapy Methods 0.000 description 3
- 239000012442 inert solvent Substances 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- 235000010981 methylcellulose Nutrition 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 3
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 239000006072 paste Substances 0.000 description 3
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- DQAKJEWZWDQURW-UHFFFAOYSA-N pyrrolidonecarboxylic acid Chemical compound OC(=O)N1CCCC1=O DQAKJEWZWDQURW-UHFFFAOYSA-N 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 238000003828 vacuum filtration Methods 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- SBTVLCPCSXMWIQ-UHFFFAOYSA-N (3,5-dimethylphenyl) carbamate Chemical compound CC1=CC(C)=CC(OC(N)=O)=C1 SBTVLCPCSXMWIQ-UHFFFAOYSA-N 0.000 description 2
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 2
- FBPINGSGHKXIQA-UHFFFAOYSA-N 2-amino-3-(2-carboxyethylsulfanyl)propanoic acid Chemical compound OC(=O)C(N)CSCCC(O)=O FBPINGSGHKXIQA-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- CDDGNGVFPQRJJM-UHFFFAOYSA-N 3-fluoropyrrolidine Chemical compound FC1CCNC1 CDDGNGVFPQRJJM-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- RGXYNUKDIDIYHV-UHFFFAOYSA-N 7-(bromomethyl)-1,2,3,4-tetrahydro-1,8-naphthyridine Chemical compound BrCC1=CC=C2CCCNC2=N1 RGXYNUKDIDIYHV-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- PVARBNSRKJYNSU-UHFFFAOYSA-N CC1(C)OB(OC1(C)C)C1=C(F)C=CC(=C1)N1CCOCC1 Chemical compound CC1(C)OB(OC1(C)C)C1=C(F)C=CC(=C1)N1CCOCC1 PVARBNSRKJYNSU-UHFFFAOYSA-N 0.000 description 2
- PLVOIZFSQZJALP-UHFFFAOYSA-N CCCCCCC.C(C)(C)N Chemical compound CCCCCCC.C(C)(C)N PLVOIZFSQZJALP-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282465 Canis Species 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 2
- 208000010837 Diabetic eye disease Diseases 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 239000001692 EU approved anti-caking agent Substances 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 2
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 2
- YGDPENGSJXXWLU-UHFFFAOYSA-N FC1(CCN(C1)C(=O)OCC1=CC=CC=C1)C=CC1=CC=C2CCCNC2=N1 Chemical compound FC1(CCN(C1)C(=O)OCC1=CC=CC=C1)C=CC1=CC=C2CCCNC2=N1 YGDPENGSJXXWLU-UHFFFAOYSA-N 0.000 description 2
- AKEKWOVOTCBVRG-UHFFFAOYSA-N FC1=C(C=C(C=C1)N1CCOCC1)B(O)O Chemical compound FC1=C(C=C(C=C1)N1CCOCC1)B(O)O AKEKWOVOTCBVRG-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 201000003838 Idiopathic interstitial pneumonia Diseases 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 2
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 102000011016 Type 5 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 description 2
- 108010037581 Type 5 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 description 2
- 108010031318 Vitronectin Proteins 0.000 description 2
- 102100035140 Vitronectin Human genes 0.000 description 2
- PBHQURAZXUZRPX-UHFFFAOYSA-N [Rh+].C12=CC=C(CC1)C2.C21=CC=C(CC2)C1 Chemical compound [Rh+].C12=CC=C(CC1)C2.C21=CC=C(CC2)C1 PBHQURAZXUZRPX-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- 229960004308 acetylcysteine Drugs 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 206010064930 age-related macular degeneration Diseases 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000003510 anti-fibrotic effect Effects 0.000 description 2
- 238000011861 anti-inflammatory therapy Methods 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000000305 astragalus gummifer gum Substances 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001743 benzylic group Chemical group 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000002843 carboxylic acid group Chemical group 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 2
- 229950009003 cilengitide Drugs 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- LDHQCZJRKDOVOX-NSCUHMNNSA-M crotonate Chemical compound C\C=C\C([O-])=O LDHQCZJRKDOVOX-NSCUHMNNSA-M 0.000 description 2
- WACQKHWOTAEEFS-UHFFFAOYSA-N cyclohexane;ethyl acetate Chemical compound CCOC(C)=O.C1CCCCC1 WACQKHWOTAEEFS-UHFFFAOYSA-N 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 210000000981 epithelium Anatomy 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 2
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000002329 infrared spectrum Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 229950001014 intetumumab Drugs 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 229940041616 menthol Drugs 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- CLHZAAANKSXSEL-ONEGZZNKSA-N methyl (e)-4-acetyloxybut-2-enoate Chemical compound COC(=O)\C=C\COC(C)=O CLHZAAANKSXSEL-ONEGZZNKSA-N 0.000 description 2
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 description 2
- 201000004071 non-specific interstitial pneumonia Diseases 0.000 description 2
- 230000000414 obstructive effect Effects 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 238000012856 packing Methods 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 235000010413 sodium alginate Nutrition 0.000 description 2
- 239000000661 sodium alginate Substances 0.000 description 2
- 229940005550 sodium alginate Drugs 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000011121 sodium hydroxide Nutrition 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000010922 spray-dried dispersion Methods 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 229960005294 triamcinolone Drugs 0.000 description 2
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- RQEUFEKYXDPUSK-SSDOTTSWSA-N (1R)-1-phenylethanamine Chemical compound C[C@@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-SSDOTTSWSA-N 0.000 description 1
- TWDOPJXHIBEHIL-YMQJAAJZSA-N (1r,2s,5r)-5-methyl-2-propan-2-ylcyclohexan-1-ol;5-methyl-2-propan-2-ylcyclohexan-1-ol Chemical compound CC(C)C1CCC(C)CC1O.CC(C)[C@@H]1CC[C@@H](C)C[C@H]1O TWDOPJXHIBEHIL-YMQJAAJZSA-N 0.000 description 1
- SHXGGYLLZCCNAX-UHFFFAOYSA-N (3-chlorophenyl) carbamate Chemical compound NC(=O)OC1=CC=CC(Cl)=C1 SHXGGYLLZCCNAX-UHFFFAOYSA-N 0.000 description 1
- PRRIGGBFRPGBRY-UHFFFAOYSA-N (3-diphenylphosphanyl-1-naphthalen-1-ylnaphthalen-2-yl)-diphenylphosphane Chemical group C1=CC=CC=C1P(C=1C(=C(C=2C3=CC=CC=C3C=CC=2)C2=CC=CC=C2C=1)P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 PRRIGGBFRPGBRY-UHFFFAOYSA-N 0.000 description 1
- PZBWVCPGDUUTHD-ZCFIWIBFSA-N (3R)-3-fluoro-3-(hydroxymethyl)pyrrolidine-1-carboxylic acid Chemical compound OC[C@@]1(F)CCN(C1)C(O)=O PZBWVCPGDUUTHD-ZCFIWIBFSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- YIBDLWLJOYCFLR-GFCCVEGCSA-N (4S)-4-(3-morpholin-4-ylphenyl)oxolan-2-one Chemical compound O1CCN(CC1)C=1C=C(C=CC=1)[C@@H]1CC(OC1)=O YIBDLWLJOYCFLR-GFCCVEGCSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical compound O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- CDQVVPUXSPZONN-WPPLYIOHSA-N 1-[6-amino-9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]purin-2-yl]-n-methylpyrazole-4-carboxamide;hydrate Chemical compound O.C1=C(C(=O)NC)C=NN1C1=NC(N)=C(N=CN2[C@H]3[C@@H]([C@H](O)[C@@H](CO)O3)O)C2=N1 CDQVVPUXSPZONN-WPPLYIOHSA-N 0.000 description 1
- ZPHKNDPUMBNUAX-UHFFFAOYSA-N 1-chlorocycloocta-1,5-diene rhodium Chemical class [Rh].ClC1=CCCC=CCC1 ZPHKNDPUMBNUAX-UHFFFAOYSA-N 0.000 description 1
- PVDGZHKJXXVONO-INIZCTEOSA-N 2-[(4s)-3-oxo-8-[3-(pyridin-2-ylamino)propoxy]-2-(2,2,2-trifluoroethyl)-4,5-dihydro-1h-2-benzazepin-4-yl]acetic acid Chemical compound C([C@H](C(N(CC(F)(F)F)CC1=C2)=O)CC(=O)O)C1=CC=C2OCCCNC1=CC=CC=N1 PVDGZHKJXXVONO-INIZCTEOSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000000143 2-carboxyethyl group Chemical group [H]OC(=O)C([H])([H])C([H])([H])* 0.000 description 1
- BMIBJCFFZPYJHF-UHFFFAOYSA-N 2-methoxy-5-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound COC1=NC=C(C)C=C1B1OC(C)(C)C(C)(C)O1 BMIBJCFFZPYJHF-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- UCFSYHMCKWNKAH-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC1(C)OBOC1(C)C UCFSYHMCKWNKAH-UHFFFAOYSA-N 0.000 description 1
- WTJXVDPDEQKTCV-UHFFFAOYSA-N 4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide;hydron;chloride Chemical compound Cl.C1C2=C(N(C)C)C=CC(O)=C2C(O)=C2C1CC1C(N(C)C)C(=O)C(C(N)=O)=C(O)C1(O)C2=O WTJXVDPDEQKTCV-UHFFFAOYSA-N 0.000 description 1
- NSTVGWPAPZDCDY-UHFFFAOYSA-N 4-(4-fluorophenyl)morpholine Chemical compound C1=CC(F)=CC=C1N1CCOCC1 NSTVGWPAPZDCDY-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- WZRJTRPJURQBRM-UHFFFAOYSA-N 4-amino-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide;5-[(3,4,5-trimethoxyphenyl)methyl]pyrimidine-2,4-diamine Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1.COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 WZRJTRPJURQBRM-UHFFFAOYSA-N 0.000 description 1
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical compound CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 1
- WCZIBQKXBMHRLJ-UHFFFAOYSA-N 5,6,7,8-tetrahydro-1,8-naphthyridin-2-ylmethanol Chemical compound C1CCNC2=NC(CO)=CC=C21 WCZIBQKXBMHRLJ-UHFFFAOYSA-N 0.000 description 1
- 102000004023 5-Lipoxygenase-Activating Proteins Human genes 0.000 description 1
- 108090000411 5-Lipoxygenase-Activating Proteins Proteins 0.000 description 1
- 239000003406 5-lipoxygenase-activating protein inhibitor Substances 0.000 description 1
- KKYABQBFGDZVNQ-UHFFFAOYSA-N 6-[5-[(cyclopropylamino)-oxomethyl]-3-fluoro-2-methylphenyl]-N-(2,2-dimethylpropyl)-3-pyridinecarboxamide Chemical compound CC1=C(F)C=C(C(=O)NC2CC2)C=C1C1=CC=C(C(=O)NCC(C)(C)C)C=N1 KKYABQBFGDZVNQ-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- ORVNHOYNEHYKJG-UHFFFAOYSA-N 8-(2,6-difluorophenyl)-2-(1,3-dihydroxypropan-2-ylamino)-4-(4-fluoro-2-methylphenyl)pyrido[2,3-d]pyrimidin-7-one Chemical compound CC1=CC(F)=CC=C1C1=NC(NC(CO)CO)=NC2=C1C=CC(=O)N2C1=C(F)C=CC=C1F ORVNHOYNEHYKJG-UHFFFAOYSA-N 0.000 description 1
- JRLTTZUODKEYDH-UHFFFAOYSA-N 8-methylquinoline Chemical group C1=CN=C2C(C)=CC=CC2=C1 JRLTTZUODKEYDH-UHFFFAOYSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-UHFFFAOYSA-N 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid Chemical compound FC1=CC(C(C(C(O)=O)=C2)=O)=C3N2C(C)COC3=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-UHFFFAOYSA-N 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 208000022309 Alcoholic Liver disease Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- YXHZRHLLCYJTFM-UHFFFAOYSA-N C(c1nc(NCCC2)c2cc1)P(c1ccccc1)(c1ccccc1)c1ccccc1 Chemical compound C(c1nc(NCCC2)c2cc1)P(c1ccccc1)(c1ccccc1)c1ccccc1 YXHZRHLLCYJTFM-UHFFFAOYSA-N 0.000 description 1
- 102100024167 C-C chemokine receptor type 3 Human genes 0.000 description 1
- 101710149862 C-C chemokine receptor type 3 Proteins 0.000 description 1
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- HWINQKZLNKBASI-UHFFFAOYSA-N C1CNc2nc(C=P(c3ccccc3)(c3ccccc3)c3ccccc3)ccc2C1 Chemical compound C1CNc2nc(C=P(c3ccccc3)(c3ccccc3)c3ccccc3)ccc2C1 HWINQKZLNKBASI-UHFFFAOYSA-N 0.000 description 1
- 102100032976 CCR4-NOT transcription complex subunit 6 Human genes 0.000 description 1
- KKTPPUNNCIHOFA-UHFFFAOYSA-N COC(c1nc(NCCC2)c2cc1)OC Chemical compound COC(c1nc(NCCC2)c2cc1)OC KKTPPUNNCIHOFA-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- ZKLPARSLTMPFCP-UHFFFAOYSA-N Cetirizine Chemical compound C1CN(CCOCC(=O)O)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZKLPARSLTMPFCP-UHFFFAOYSA-N 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- LUKZNWIVRBCLON-GXOBDPJESA-N Ciclesonide Chemical compound C1([C@H]2O[C@@]3([C@H](O2)C[C@@H]2[C@@]3(C[C@H](O)[C@@H]3[C@@]4(C)C=CC(=O)C=C4CC[C@H]32)C)C(=O)COC(=O)C(C)C)CCCCC1 LUKZNWIVRBCLON-GXOBDPJESA-N 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 206010056533 Congenital hepatic fibrosis Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 208000001708 Dupuytren contracture Diseases 0.000 description 1
- 208000010975 Dystrophic epidermolysis bullosa Diseases 0.000 description 1
- 208000002197 Ehlers-Danlos syndrome Diseases 0.000 description 1
- 229940118365 Endothelin receptor antagonist Drugs 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 206010014954 Eosinophilic fasciitis Diseases 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- KEKFBOHNNGLSRN-UHFFFAOYSA-N FC1(CCC2=CC=C3CCCNC3=N2)CCN(C1)C(=O)OCC1=CC=CC=C1 Chemical compound FC1(CCC2=CC=C3CCCNC3=N2)CCN(C1)C(=O)OCC1=CC=CC=C1 KEKFBOHNNGLSRN-UHFFFAOYSA-N 0.000 description 1
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 1
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000006933 Hermanski-Pudlak Syndrome Diseases 0.000 description 1
- 206010071775 Hermansky-Pudlak syndrome Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- 208000024934 IgG4-related mediastinitis Diseases 0.000 description 1
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 description 1
- 102100021854 Inhibitor of nuclear factor kappa-B kinase subunit beta Human genes 0.000 description 1
- 101710205525 Inhibitor of nuclear factor kappa-B kinase subunit beta Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 208000029523 Interstitial Lung disease Diseases 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- 229940110339 Long-acting muscarinic antagonist Drugs 0.000 description 1
- 208000005777 Lupus Nephritis Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000002805 Mediastinal fibrosis Diseases 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 208000003510 Nephrogenic Fibrosing Dermopathy Diseases 0.000 description 1
- 206010067467 Nephrogenic systemic fibrosis Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102100029438 Nitric oxide synthase, inducible Human genes 0.000 description 1
- 101710089543 Nitric oxide synthase, inducible Proteins 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- YIBDLWLJOYCFLR-LBPRGKRZSA-N O1CCN(CC1)C=1C=C(C=CC=1)[C@H]1CC(OC1)=O Chemical compound O1CCN(CC1)C=1C=C(C=CC=1)[C@H]1CC(OC1)=O YIBDLWLJOYCFLR-LBPRGKRZSA-N 0.000 description 1
- YGDPENGSJXXWLU-ZHACJKMWSA-N O=C(N(CC1)CC1(/C=C/c1nc(NCCC2)c2cc1)F)OCc1ccccc1 Chemical compound O=C(N(CC1)CC1(/C=C/c1nc(NCCC2)c2cc1)F)OCc1ccccc1 YGDPENGSJXXWLU-ZHACJKMWSA-N 0.000 description 1
- HKDUWRRLEDNORE-UHFFFAOYSA-N OC(=O)CC(CN1CCC(F)(CCC2=CC=C3CCCNC3=N2)C1)C1=CC(=CC=C1)N1CCOCC1 Chemical compound OC(=O)CC(CN1CCC(F)(CCC2=CC=C3CCCNC3=N2)C1)C1=CC(=CC=C1)N1CCOCC1 HKDUWRRLEDNORE-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 206010031023 Oral submucosal fibrosis Diseases 0.000 description 1
- 102000004264 Osteopontin Human genes 0.000 description 1
- 108010081689 Osteopontin Proteins 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229940124780 PI3K delta inhibitor Drugs 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 208000004362 Penile Induration Diseases 0.000 description 1
- 229940083963 Peptide antagonist Drugs 0.000 description 1
- 208000020758 Peyronie disease Diseases 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001244 Poly(D,L-lactide) Polymers 0.000 description 1
- 229920002319 Poly(methyl acrylate) Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920000331 Polyhydroxybutyrate Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 206010036805 Progressive massive fibrosis Diseases 0.000 description 1
- 208000032056 Radiation Fibrosis Syndrome Diseases 0.000 description 1
- 238000001237 Raman spectrum Methods 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038748 Restrictive cardiomyopathy Diseases 0.000 description 1
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 229940122605 Short-acting muscarinic antagonist Drugs 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 206010046798 Uterine leiomyoma Diseases 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- GRFUMPFWJKGLQC-ZETCQYMHSA-N [(1s)-1-phenylethyl]carbamic acid Chemical compound OC(=O)N[C@@H](C)C1=CC=CC=C1 GRFUMPFWJKGLQC-ZETCQYMHSA-N 0.000 description 1
- LQIWYVNQUXQNQQ-YFKPBYRVSA-N [(3S)-3-fluoropyrrolidin-3-yl]methanamine Chemical class NC[C@@]1(F)CCNC1 LQIWYVNQUXQNQQ-YFKPBYRVSA-N 0.000 description 1
- 229950005008 abituzumab Drugs 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- TVWAEQRFKRTYIG-JIDHJSLPSA-N acetic acid;4-[(1r)-2-[6-[2-[(2,6-dichlorophenyl)methoxy]ethoxy]hexylamino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol Chemical compound CC(O)=O.C1=C(O)C(CO)=CC([C@@H](O)CNCCCCCCOCCOCC=2C(=CC=CC=2Cl)Cl)=C1 TVWAEQRFKRTYIG-JIDHJSLPSA-N 0.000 description 1
- YDCNECXDBVLHLS-UHFFFAOYSA-N acetonitrile;azane Chemical compound N.CC#N YDCNECXDBVLHLS-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- PWACSDKDOHSSQD-IUTFFREVSA-N acrivastine Chemical compound C1=CC(C)=CC=C1C(\C=1N=C(\C=C\C(O)=O)C=CC=1)=C/CN1CCCC1 PWACSDKDOHSSQD-IUTFFREVSA-N 0.000 description 1
- 229960003792 acrivastine Drugs 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000005910 alkyl carbonate group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- OUJTZYPIHDYQMC-LJQANCHMSA-N ambrisentan Chemical compound O([C@@H](C(OC)(C=1C=CC=CC=1)C=1C=CC=CC=1)C(O)=O)C1=NC(C)=CC(C)=N1 OUJTZYPIHDYQMC-LJQANCHMSA-N 0.000 description 1
- 229960002414 ambrisentan Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 230000003266 anti-allergic effect Effects 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 210000000270 basal cell Anatomy 0.000 description 1
- 238000010945 base-catalyzed hydrolysis reactiony Methods 0.000 description 1
- 229950000210 beclometasone dipropionate Drugs 0.000 description 1
- 239000003659 bee venom Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- YGDPENGSJXXWLU-NEQMZLFVSA-N benzyl (3R)-3-fluoro-3-[(E)-2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethenyl]pyrrolidine-1-carboxylate Chemical compound F[C@]1(CCN(C1)C(=O)OCC1=CC=CC=C1)\C=C\C1=CC=C2CCCNC2=N1 YGDPENGSJXXWLU-NEQMZLFVSA-N 0.000 description 1
- KEKFBOHNNGLSRN-JOCHJYFZSA-N benzyl (3R)-3-fluoro-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]pyrrolidine-1-carboxylate Chemical compound F[C@]1(CN(CC1)C(=O)OCC1=CC=CC=C1)CCC1=NC=2NCCCC=2C=C1 KEKFBOHNNGLSRN-JOCHJYFZSA-N 0.000 description 1
- VALPXRNQZYGXPS-UHFFFAOYSA-N benzyl pyrrolidine-1-carboxylate Chemical compound C1CCCN1C(=O)OCC1=CC=CC=C1 VALPXRNQZYGXPS-UHFFFAOYSA-N 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- 229940124748 beta 2 agonist Drugs 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 229920013641 bioerodible polymer Polymers 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 208000002352 blister Diseases 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000003918 blood extract Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 235000010338 boric acid Nutrition 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- VIHAEDVKXSOUAT-UHFFFAOYSA-N but-2-en-4-olide Chemical compound O=C1OCC=C1 VIHAEDVKXSOUAT-UHFFFAOYSA-N 0.000 description 1
- PVEOYINWKBTPIZ-UHFFFAOYSA-M but-3-enoate Chemical compound [O-]C(=O)CC=C PVEOYINWKBTPIZ-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 210000001054 cardiac fibroblast Anatomy 0.000 description 1
- 230000009787 cardiac fibrosis Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 229960001803 cetirizine Drugs 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- SOYKEARSMXGVTM-UHFFFAOYSA-N chlorphenamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 SOYKEARSMXGVTM-UHFFFAOYSA-N 0.000 description 1
- 229960003291 chlorphenamine Drugs 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229960003728 ciclesonide Drugs 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 229940047766 co-trimoxazole Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 208000018631 connective tissue disease Diseases 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- RXPAYNWWOUGGHI-UHFFFAOYSA-K cycloocta-1,5-diene;rhodium(3+);trichloride Chemical compound Cl[Rh](Cl)Cl.C1CC=CCCC=C1 RXPAYNWWOUGGHI-UHFFFAOYSA-K 0.000 description 1
- GCUVBACNBHGZRS-UHFFFAOYSA-N cyclopenta-1,3-diene cyclopenta-2,4-dien-1-yl(diphenyl)phosphane iron(2+) Chemical compound [Fe++].c1cc[cH-]c1.c1cc[c-](c1)P(c1ccccc1)c1ccccc1 GCUVBACNBHGZRS-UHFFFAOYSA-N 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 210000005220 cytoplasmic tail Anatomy 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 201000011190 diabetic macular edema Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 description 1
- 229910000071 diazene Inorganic materials 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 229950009763 dilmapimod Drugs 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- FVTWTVQXNAJTQP-UHFFFAOYSA-N diphenyl-[1-(2-phenylmethoxyethyl)-1-azoniabicyclo[2.2.2]octan-4-yl]methanol Chemical compound C=1C=CC=CC=1C(C12CC[N+](CCOCC=3C=CC=CC=3)(CC1)CC2)(O)C1=CC=CC=C1 FVTWTVQXNAJTQP-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003602 elastase inhibitor Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 201000010048 endomyocardial fibrosis Diseases 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 208000004298 epidermolysis bullosa dystrophica Diseases 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- 229950009569 etaracizumab Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- UMYZHWLYICNGRQ-UHFFFAOYSA-N ethanol;heptane Chemical compound CCO.CCCCCCC UMYZHWLYICNGRQ-UHFFFAOYSA-N 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 229940126864 fibroblast growth factor Drugs 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000005454 flavour additive Substances 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 229960001347 fluocinolone acetonide Drugs 0.000 description 1
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229960001469 fluticasone furoate Drugs 0.000 description 1
- XTULMSXFIHGYFS-VLSRWLAYSA-N fluticasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(F)[C@@]4(C)C=CC(=O)C=C4[C@@H](F)C[C@H]3[C@@H]2C[C@H]1C)C(=O)SCF)C(=O)C1=CC=CO1 XTULMSXFIHGYFS-VLSRWLAYSA-N 0.000 description 1
- 229960000289 fluticasone propionate Drugs 0.000 description 1
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- XLYMOEINVGRTEX-UHFFFAOYSA-N fumaric acid monoethyl ester Natural products CCOC(=O)C=CC(O)=O XLYMOEINVGRTEX-UHFFFAOYSA-N 0.000 description 1
- 150000002238 fumaric acids Chemical class 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000011141 high resolution liquid chromatography Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 1
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000008611 intercellular interaction Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- 208000014861 isolated congenital hepatic fibrosis Diseases 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 201000010666 keratoconjunctivitis Diseases 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 229960000681 leflunomide Drugs 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 150000002617 leukotrienes Chemical class 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- JCCNYMKQOSZNPW-UHFFFAOYSA-N loratadine Chemical compound C1CN(C(=O)OCC)CCC1=C1C2=NC=CC=C2CCC2=CC(Cl)=CC=C21 JCCNYMKQOSZNPW-UHFFFAOYSA-N 0.000 description 1
- 229960003088 loratadine Drugs 0.000 description 1
- 229950003265 losmapimod Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229940076783 lucentis Drugs 0.000 description 1
- JGCMEBMXRHSZKX-UHFFFAOYSA-N macitentan Chemical compound C=1C=C(Br)C=CC=1C=1C(NS(=O)(=O)NCCC)=NC=NC=1OCCOC1=NC=C(Br)C=N1 JGCMEBMXRHSZKX-UHFFFAOYSA-N 0.000 description 1
- 229960001039 macitentan Drugs 0.000 description 1
- 208000002780 macular degeneration Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 150000002689 maleic acids Chemical class 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- BUTPBERGMJVRBM-UHFFFAOYSA-N methanol;methylsulfinylmethane Chemical compound OC.CS(C)=O BUTPBERGMJVRBM-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 1
- CPMDPSXJELVGJG-UHFFFAOYSA-N methyl 2-hydroxy-3-[N-[4-[methyl-[2-(4-methylpiperazin-1-yl)acetyl]amino]phenyl]-C-phenylcarbonimidoyl]-1H-indole-6-carboxylate Chemical compound OC=1NC2=CC(=CC=C2C=1C(=NC1=CC=C(C=C1)N(C(CN1CCN(CC1)C)=O)C)C1=CC=CC=C1)C(=O)OC CPMDPSXJELVGJG-UHFFFAOYSA-N 0.000 description 1
- 238000004452 microanalysis Methods 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960002421 minocycline hydrochloride Drugs 0.000 description 1
- 230000003990 molecular pathway Effects 0.000 description 1
- 229960002744 mometasone furoate Drugs 0.000 description 1
- WOFMFGQZHJDGCX-ZULDAHANSA-N mometasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(Cl)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)C(=O)CCl)C(=O)C1=CC=CO1 WOFMFGQZHJDGCX-ZULDAHANSA-N 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- UUIQMZJEGPQKFD-UHFFFAOYSA-N n-butyric acid methyl ester Natural products CCCC(=O)OC UUIQMZJEGPQKFD-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 239000010434 nepheline Substances 0.000 description 1
- 229910052664 nepheline Inorganic materials 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 229960001699 ofloxacin Drugs 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 208000005207 oral submucous fibrosis Diseases 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960003073 pirfenidone Drugs 0.000 description 1
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 1
- 206010035653 pneumoconiosis Diseases 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 239000004632 polycaprolactone Substances 0.000 description 1
- 201000006038 polycystic kidney disease 4 Diseases 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- UAJUXJSXCLUTNU-UHFFFAOYSA-N pranlukast Chemical compound C=1C=C(OCCCCC=2C=CC=CC=2)C=CC=1C(=O)NC(C=1)=CC=C(C(C=2)=O)C=1OC=2C=1N=NNN=1 UAJUXJSXCLUTNU-UHFFFAOYSA-N 0.000 description 1
- 229960004583 pranlukast Drugs 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- XYKIUTSFQGXHOW-UHFFFAOYSA-N propan-2-one;toluene Chemical compound CC(C)=O.CC1=CC=CC=C1 XYKIUTSFQGXHOW-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000011253 protective coating Substances 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000003379 purinergic P1 receptor agonist Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003235 pyrrolidines Chemical class 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 229960003614 regadenoson Drugs 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 201000004193 respiratory failure Diseases 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 150000003284 rhodium compounds Chemical class 0.000 description 1
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 239000000565 sealant Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 229940040944 tetracyclines Drugs 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- 239000004634 thermosetting polymer Substances 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-L thiosulfate(2-) Chemical compound [O-]S([S-])(=O)=O DHCDFWKWKRSZHF-UHFFFAOYSA-L 0.000 description 1
- 230000009974 thixotropic effect Effects 0.000 description 1
- LERNTVKEWCAPOY-DZZGSBJMSA-N tiotropium Chemical compound O([C@H]1C[C@@H]2[N+]([C@H](C1)[C@@H]1[C@H]2O1)(C)C)C(=O)C(O)(C=1SC=CC=1)C1=CC=CS1 LERNTVKEWCAPOY-DZZGSBJMSA-N 0.000 description 1
- 229940110309 tiotropium Drugs 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 239000002750 tryptase inhibitor Substances 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 229960004258 umeclidinium Drugs 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 229960004026 vilanterol Drugs 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pyrrole Compounds (AREA)
Abstract
Un compuesto de fórmula (I): **Fórmula** en donde R representa H o F, o una sal del mismo.
Description
DESCRIPCIÓN
Derivados de naftiridina como antagonistas de integrina avp6 para el tratamiento de, por ejemplo, enfermedades fibróticas
Campo de la invención
La presente invención se refiere a compuestos de pirrolidina que son antagonistas de integrina avp6, a composiciones farmacéuticas que comprenden dichos compuestos y a su uso en terapia, en especial en el tratamiento de afecciones para las que está indicado un antagonista de integrina avp6, para el uso de un compuesto en la fabricación de un medicamento para el tratamiento de afecciones en las que está indicado un antagonista de integrina avp6 y un método para el tratamiento o la profilaxis de trastornos en los que está indicado el antagonismo de integrina avp6 en un ser humano.
Antecedentes de la invención
Las proteínas de la superfamilia de las integrinas son receptores de superficie celular heterodímeros, compuestos de subunidades alfa y beta. Se ha informado de al menos 18 subunidades alfa y 8 subunidades beta, que se ha demostrado que forman 24 heterodímeros alfa/beta distintos. Cada cadena comprende un dominio extracelular grande (>640 aminoácidos para la subunidad beta, >940 aminoácidos para la subunidad alfa), con una región que abarca la transmembrana de alrededor de 20 aminoácidos por cadena, y en general una cola citoplásmica corta de 30 a 50 aminoácidos por cadena. Se ha demostrado que diferentes integrinas participan en una plétora de biologías celulares, incluyendo la adhesión celular a la matriz extracelular, interacciones célula-célula y efectos sobre la migración, proliferación, diferenciación y supervivencia celulares (Barczyk et al., Cell and Tissue Research, 2010, 339, 269).
Los receptores de integrinas interaccionan con las proteínas de unión por medio de las interfases de unión proteína-proteína cortas. La familia de las integrinas se puede agrupar en subfamilias que comparten motivos de reconocimiento de unión similares en dichos ligandos. Una subfamilia principal son las integrinas RGD, que reconocen los ligandos que contienen un motivo RGD (arginina-glicina-ácido aspártico) dentro de la secuencia de proteína. Existen 8 integrinas en esta subfamilia, a saber, avpi , avp3 , avp5 , avp6, avpa, a iibp3 , a5pi , aapi , donde la nomenclatura demuestra que avpi , avp3 , avp5 , avp6, y avpa comparten una subunidad av común con una subunidad p divergente, y avpi , a5pi y aspi comparten una subunidad pi común con una subunidad a divergente. Se ha demostrado que la subunidad pi se empareja con i i subunidades a diferentes, de las que solo las 3 enumeradas anteriormente reconocen comúnmente el motivo peptídico RGD (Humphries et al, Journal of Cell Science, 2006, 119, 390i).
Las 8 integrinas de unión a RGD tienen diferentes afinidades y especificidades de unión para diferentes ligandos que contienen RGD. Los ligandos incluyen proteínas tales como fibronectina, vitronectina, osteopontina y los péptidos asociados a la latencia (LAP) del factor de crecimiento transformante pi y p3 (TGFpi y TGFp3). Se ha demostrado que la unión de integrinas a los LAP de TGFpi y TGFp3 en varios sistemas permite la activación de las actividades biológicas de TGFpi y TGFp3, y las posteriores biologías activadas por TGFp (Worthington et al., Trends in Biochemical Sciences, 2011, 36, 47). La diversidad de dichos ligandos, acoplados con los patrones de expresión de las integrinas de unión a RGD, genera múltiples oportunidades para la intervención de la enfermedad. Dichas enfermedades incluyen enfermedades fibróticas (Margadant et al, EMBO reports, 2010, 11, 97), trastornos inflamatorios, cáncer (Desgrosellier et al, Nature Reviews Cancer, 2010, 10, 9), reestenosis y otras enfermedades con un componente angiogénico (Weis et al, Cold Spring. Harb. Perspect. Med. 2011, 1, a 006478).
Se ha descrito en la bibliografía un número significativo de antagonistas de integrina av (Goodman et al, Trends in Pharmacological Sciences, 2012, 33, 405) incluyendo anticuerpos inhibidores, péptidos y moléculas pequeñas. Para los anticuerpos, estos incluyen los antagonistas de pan-av intetumumab y abituzumab (Gras, Drugs of the Future, 2015, 40, 97), el antagonista selectivo de avp3 etaracizumab, y los antagonistas selectivos de avp6 STX-i00. La cilengitida es un antagonista peptídico cíclico que inhibe tanto avp3 como avp5 y el SB-267268 es un ejemplo de un compuesto (Wilkinson-Berka et al, Invest. Ophthalmol. Vis. Sci., 2006, 47, i600) que inhibe tanto avp3 como avp5. La invención de compuestos que actúan como antagonistas de diferentes combinaciones de integrinas av permite que se generen agentes novedosos personalizados para indicaciones de enfermedades específicas.
La fibrosis pulmonar representa la fase terminal de varias enfermedades pulmonares intersticiales, incluyendo neumonías intersticiales idiopáticas, y se caracteriza por el depósito excesivo de matriz extracelular dentro del intersticio pulmonar. Entre las neumonías intersticiales idiopáticas, la fibrosis pulmonar idiopática (IPF, en inglés) representa la afección más común y más mortal con una supervivencia típica de 3 a 5 años después del diagnóstico. La fibrosis en IPF es, en general, progresiva, resistente a la intervención farmacológica actual y lleva inexorablemente a una insuficiencia respiratoria debida a la obstrucción de las unidades alveolares funcionales. La IPF afecta aproximadamente a 500000 personas en EE. UU. y Europa.
Existen datos inmunohistoquímicos experimentales in vitro en animales y pacientes con IPF, que apoyan un papel clave para la integrina restringida epitelialmente, avp6, en la activación de TGFpi. La expresión de esta integrina es baja en los tejidos epiteliales normales y se regula por incremento significativamente en epitelios lesionados e inflamados incluyendo el epitelio activado en la IPF. Seleccionar esta integrina como objetivo, por tanto, reduce la posibilidad teórica de interferencia con papeles homeostáticos más amplios del TGFp. Se ha demostrado que la
inhibición parcial de la integrina avp6 por bloqueo de anticuerpos evita la fibrosis pulmonar sin empeorar la inflamación (Horan GS et al., «Partial inhibition of integrin avp6 prevents pulmonary fibrosis without exacerbating inflammation». Am J Respir Crit Care Med 2008, 177: 56-65). Aparte de la fibrosis pulmonar, también se considera la avp6 un promotor importante de enfermedad fibrótica de otros órganos, incluyendo el hígado y el riñón (revisado en Henderson NC et al., «Integrin-mediated regulation of TGFp in Fibrosis», Biochimica et Biophysica Acta - Molecular Basis of Disease 2013, 1832:891-896), lo que sugiere que un antagonista de avp6 podría ser eficaz en el tratamiento de enfermedades fibróticas en múltiples órganos.
Acorde con la observación de que varias integrinas de unión a RGD pueden unirse a TGFp y activarse, recientemente se han implicado diferentes integrinas av en la enfermedad fibrótica (Henderson NC et al. «Targeting of av integrin identifies a core molecular pathway that regulates fibrosis in several organs», Nature Medicine 2013, vol. 19, número 12: 1617-1627; Sarrazy V et al., «Integrins avp5 and avp3 promote latent TGF-p1 activation by human cardiac fibroblast contraction», Cardiovasc Res 2014, 102:407-417; Minagawa S et al., «Selective targeting of TGF-p activation to treat fibroinflammatory airway disease», Sci Transl Med 2014, vol. 6, edición 241: 1-14; Reed NI et al. «The avp1 integrin plays a critical in vivo role in tissue fibrosis», Sci Transl Med 2015, vol. 7, edición 288: 1-8). Por lo tanto, los inhibidores frente a miembros específicos de la familia de las integrinas de unión a RGD, o con huellas identificativas con selectividad específica dentro de la familia de las integrinas de unión a RGD, pueden ser eficaces en el tratamiento de enfermedades fibróticas en múltiples órganos.
Se han descrito relaciones SAR (relaciones estructura-actividad, en inglés) de una serie de antagonistas de integrina frente a avp3 avp5, avp6 y avpa (Macdonald, SJF et al. «Structure activity relationships of av integrin antagonists for pulmonary fibrosis by variation in aryl substituents». ACS Med Chem Lett 2014, 5, 1207-1212. 19 de septiembre de 2014).
Es deseable proporcionar antagonistas de avp6 que puedan tener también actividades frente a otras integrinas av, tales como avp-i, avp5 o avpa.
Breve compendio de la invención
En un primer aspecto de la presente invención, se proporciona un compuesto de fórmula (I) o una sal del mismo, más en particular un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo:

en donde R representa H o F.
Los compuestos de fórmula (I) y sus sales tienen actividad antagonista de avp6 y se cree que tienen uso potencial para el tratamiento o la profilaxis de ciertos trastornos. El término actividad antagonista de avp6 incluye actividad inhibidora de avp6 en la presente memoria.
En un segundo aspecto de la presente invención, se proporciona una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables.
En un tercer aspecto de la presente invención, se proporciona un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, para su uso en la terapia, en particular en el tratamiento de una enfermedad o afección para la que está indicado un antagonista del receptor de integrina avp6.
En un cuarto aspecto de la presente invención, se proporciona el uso de un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, en la fabricación de un medicamento para el tratamiento de una enfermedad o afección para la que está indicado un antagonista del receptor de integrina avp6.
Descripción detallada de la invención
En un primer aspecto de la invención, se proporciona un compuesto de fórmula (I) o una sal del mismo:

en donde R representa H o F.
Un compuesto de fórmula (I) es el ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (IA) o una sal del mismo:
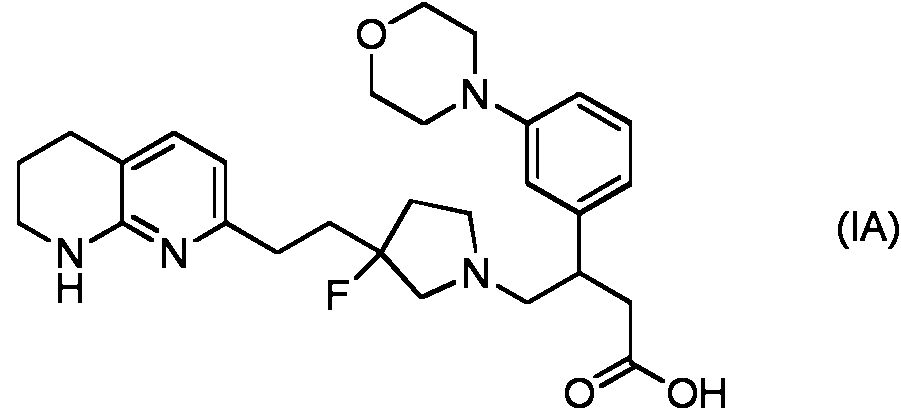
En una realización, el compuesto de fórmula (IA) es una sal farmacéuticamente aceptable del ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico.
En una realización, el compuesto de fórmula (IA) tiene la fórmula (IA1):

ácido (R)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o una sal farmacéuticamente aceptable del mismo.
En otra realización, el compuesto de fórmula (I) tiene la fórmula (IA2):

ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o una sal farmacéuticamente aceptable del mismo.
En otra realización, el compuesto de fórmula (I) tiene la fórmula (IA3):

ácido (S)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(3-morfol¡nofen¡l)butano¡co o una sal farmacéut¡camente aceptable del m¡smo.
En otra real¡zac¡ón, el compuesto de fórmula (I) t¡ene la fórmula (IA4):

ác¡do (R)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(3-morfol¡nofen¡l)butano¡co o una sal farmacéut¡camente aceptable del m¡smo.
Otro compuesto de fórmula (I) es el ác¡do 4-(3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(2-fluoro-5-morfol¡nofen¡l)butano¡co (IB) o una sal del m¡smo:

En una real¡zac¡ón, el compuesto de fórmula (IB) es una sal farmacéut¡camente aceptable del ác¡do 4-(3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(2-fluoro-5-morfol¡nofen¡l)butano¡co.
En una real¡zac¡ón, el compuesto de fórmula (IB) t¡ene la fórmula de (IB1):

ác¡do (R)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l) p¡rrol¡d¡n-1-¡l)-3-(2-fluoro-5-morfol¡nofen¡l) butano¡co o una sal farmacéut¡camente aceptable del m¡smo.
En otra real¡zac¡ón, el compuesto de fórmula (IB) t¡ene la fórmula (IB2):
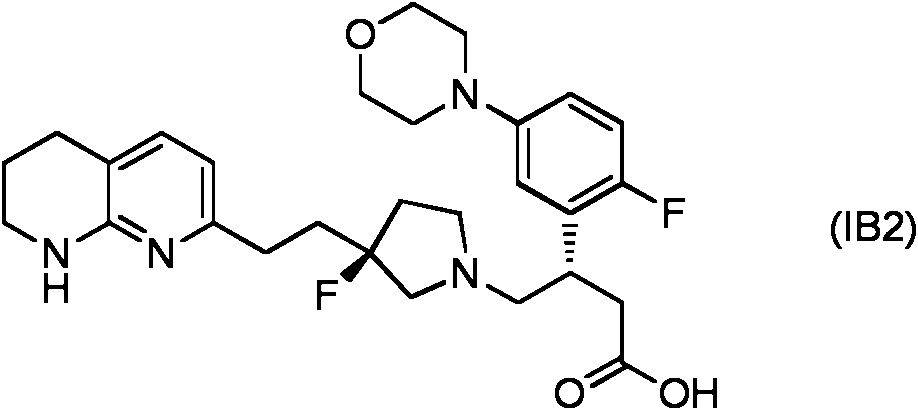
ác¡do (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l) p¡rrol¡d¡n-1-¡l)-3-(2-fluoro-5-morfol¡nofen¡l) butano¡co o una sal farmacéut¡camente aceptable del m¡smo.
En otra realización, el compuesto de fórmula (IB) tiene la fórmula (IB3):

ácido (S)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil) pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil) butanoico o una sal farmacéuticamente aceptable del mismo.
En otra realización, el compuesto de fórmula (IB) tiene la fórmula (IB4):

ácido (R)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil) pirrolidin-1 -il)-3-(2-fluoro-5-morfolinofenil) butanoico o una sal farmacéuticamente aceptable del mismo.
Los compuestos de fórmula (I) tienen tanto un grupo amino básico como un grupo ácido carboxílico y, por consiguiente, pueden formar una sal interna, es decir, un zwitterión o sales internas. Por lo tanto, en una realización, el compuesto de fórmula (I) es el ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o el ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico o uno cualquiera de los compuestos IA1, IA2, IA3, IA4, IB1, IB2, IB3 o IB4 en forma de sal zwitteriónica. En otra realización, el compuesto de fórmula (I) es el ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o el ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico o uno cualquiera de los compuestos IA1, IA2, IA3, IA4, IB1, IB2, IB3 o IB4 en forma no zwitteriónica.
Se apreciará que la presente invención cubre los compuestos de fórmula (I) como compuesto precursor, como zwitterión (el compuesto precursor está protonado internamente por su grupo ácido carboxílico y existe normalmente como zwitterión) y como sales de los mismos, por ejemplo como una sal farmacéuticamente aceptable de los mismos. En una realización, la invención se refiere a compuestos de fórmula (I) o una sal farmacéuticamente aceptable de los mismos.
Para una revisión sobre sales adecuadas, véase Berge et al., J. Pharm. Sci., 66:1-19, (1977). Las sales farmacéuticamente aceptables adecuadas se enumeran en P H Stahl y C G Wermuth, editores, Handbook of Pharmaceutical Salts; Properties, Selection and Use, Weinheim/Zurich: Wiley- VCH/VHCA, 2002. Las sales farmacéuticamente aceptables adecuadas pueden incluir sales de adición de ácido con ácidos inorgánicos tales como, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido ortofosfórico, ácido nítrico, ácido fosfórico o ácido sulfúrico, o con ácidos orgánicos tales como, por ejemplo, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido acético, ácido propiónico, ácido láctico, ácido cítrico, ácido fumárico, ácido málico, ácido succínico, ácido salicílico, ácido maleico, ácido glicerofosfórico, tartárico, benzoico, glutámico, aspártico, bencenosulfónico, naftalenosulfónico tales como ácido 2-naftalenosulfónico, ácido hexanoico o ácido acetilsalicílico. Los ácidos particularmente adecuados son los ácidos fumárico y maleico. Típicamente, se puede preparar fácilmente una sal farmacéuticamente aceptable usando un ácido o una base deseados según sea apropiado. La sal resultante puede precipitar de la solución y recogerse por filtración o puede recuperarse por evaporación del disolvente.
Se pueden usar otras sales no farmacéuticamente aceptables, por ejemplo, formiatos, oxalatos o trifluoroacetatos, por ejemplo en el aislamiento de los compuestos de fórmula (I), y están incluidas dentro del alcance de esta invención.
Se puede formar una sal de adición de base farmacéuticamente aceptable por reacción de un compuesto de fórmula (I) con una base orgánica adecuada, (por ejemplo, trietilamina, etanolamina, trietanolamina, colina, arginina, lisina o histidina), opcionalmente en un disolvente adecuado, para dar la sal de adición de base que usualmente se aísla, por ejemplo, por cristalización y filtración. Las sales básicas farmacéuticamente aceptables incluyen sales de amonio,
sales de metales alcalinos tales como las de sodio y potasio, sales de metales alcalinotérreos tales como las de calcio y magnesio, y sales con bases orgánicas, incluyendo sales de aminas primarias, secundarias y terciarias, tales como isopropilamina, dietilamina, etanolamina, trimetilamina, diciclohexilamina y N-metil-D-glucamina.
La invención incluye dentro de su alcance todas las formas estequiométricas y no estequiométricas posibles de las sales de los compuestos de fórmula (I).
Los compuestos de fórmula (I) pueden estar en forma cristalina o amorfa. Además, algunas de las formas cristalinas de los compuestos de fórmula (I) pueden existir como polimorfos, que están incluidos dentro del alcance de la presente invención. Las formas polimórficas de los compuestos de fórmula (I) pueden caracterizarse y diferenciarse usando varias técnicas analíticas convencionales, incluyendo patrones de difracción en polvo de rayos X (XRPD), espectros de infrarrojo (IR), espectros Raman, calorimetría diferencial de barrido (DSC), análisis termogravimétrico (TGA) y resonancia magnética nuclear de estado sólido (SSNMR) (todas por sus siglas en inglés), pero sin limitarse a estas.
Los compuestos de fórmula (I) también se pueden preparar como una dispersión molecular amorfa en una matriz polimérica, tal como acetato-succinato de hidroxipropilmetilcelulosa, usando un procedimiento de dispersión secada por pulverización (SDD) para mejorar la estabilidad y la solubilidad de la sustancia farmacológica.
Se apreciará que muchos compuestos orgánicos pueden formar complejos con disolventes en los que reaccionen o en los que precipiten o cristalicen. Estos complejos se conocen como «solvatos». Por ejemplo, un complejo con agua se conoce como un «hidrato». Pueden usarse para formar solvatos disolventes con altos puntos de ebullición y/o capaces de formar enlaces de hidrógeno, tales como agua, xileno, N-metilpirrolidinona, metanol y etanol. Los métodos para la identificación de solvatos incluyen, RMN y microanálisis, pero no se limitan a estos. Se apreciará que las formas cristalinas pueden estar opcionalmente solvatadas para formar, por ejemplo, solvatos farmacéuticamente aceptables, tales como hidratos que pueden ser hidratos estequiométricos así como compuestos que contienen cantidades variables de agua. Los solvatos incluyen solvatos estequiométricos y solvatos no estequiométricos. Los compuestos de fórmula (I ) pueden existir en forma solvatada o no solvatada.
Los compuestos descritos en la presente memoria contienen dos centros asimétricos de modo que se pueden formar isómeros ópticos, por ejemplo, diastereoisómeros y enantiómeros. De acuerdo con esto, la presente invención abarca isómeros de los compuestos de fórmula (I), ya sea como isómeros individuales aislados tales como para estar sustancialmente exentos del otro isómero (es decir, puros) o como mezclas. Se puede aislar un isómero individual aislado para que esté sustancialmente exento del otro isómero (es decir, puro) de modo que esté presente menos de un 10 %, en particular menos de aproximadamente un 1 %, por ejemplo menos de aproximadamente un 0,1 % del otro isómero.
Los expertos en la materia entenderán que ciertos diastereoisómeros pueden ser menos activos que otros y que la actividad de un diastereoisómero individual puede estar por debajo de un límite seleccionado.
La separación de los isómeros se puede lograr por técnicas convencionales conocidas para los expertos en la materia, por ejemplo, por cristalización fraccionada, cromatografía, HPLC o una combinación de estas técnicas.
Los compuestos de fórmula (I) pueden existir en una de varias formas tautómeras. Se entenderá que la presente invención abarca todos los tautómeros de los compuestos de fórmula (I) ya sea como tautómeros individuales o como mezclas de los mismos.
Se apreciará de lo anterior que dentro del alcance de la invención están incluidos solvatos, isómeros y formas polimórficas de los compuestos de fórmula (I) y sales de los mismos.
Preparación de compuestos
Los compuestos de la invención se pueden fabricar por varios métodos, incluyendo química estándar. Cualquier variable definida previamente seguirá teniendo el significado definido previamente a menos que se indique de otro modo. Se exponen a continuación métodos sintéticos generales ilustrativos y después se preparan compuestos específicos de la invención en los ejemplos de trabajo.
Los expertos en la materia apreciarán que la descripción de (E) o (Z) de algunos compuestos intermedios que pueden existir como dos isómeros geométricos, puede contener el otro isómero geométrico como componente secundario.
Los compuestos de fórmula estructural (I) se pueden preparar por un procedimiento que implica en primer lugar la desprotección de un compuesto de fórmula estructural (II), es decir, escisión del grupo éster, seguido opcionalmente de la conversión en una sal:

donde R2 es un grupo alquilo C1-C6, por ejemplo un grupo terc-butilo, etilo o metilo. De forma alternativa, R2 es un alcohol quiral, por ejemplo (-)-mentol [(1R,2S,5R)-2-isopropil-5-metilciclohexanol].
Un sexto aspecto de la invención proporciona un compuesto de fórmula (II).
La desprotección del compuesto de fórmula estructural (II), donde R2 es metilo, etilo, un alcohol quiral tal como mentol o ferc-butilo, puede llevarse a cabo por hidrólisis ácida usando, por ejemplo, ácido clorhídrico, bromhídrico, sulfúrico o trifluoroacético, en un disolvente inerte, tal como diclorometano, 2-metiltetrahidrofurano, tetrahidrofurano, 1,4-dioxano o éter ciclopentilmetílico o agua.
De forma alternativa, la desprotección del compuesto de fórmula estructural (II), donde R2 es metilo, etilo o un alcohol quiral tal como mentol, se puede llevar a cabo por hidrólisis básica usando, por ejemplo, hidróxido de litio, hidróxido de sodio, hidróxido de potasio en un disolvente adecuado, por ejemplo, un disolvente acuoso tal como metanol acuoso.
Después de la escisión del grupo éster, el producto resultante se puede convertir en la sal requerida por métodos conocidos para los expertos en la materia.
En una realización, la conversión del zwitterión en la sal fumarato se logra por tratamiento de una solución etanólica del zwitterión con una solución etanólica de ácido fumárico, calentando la solución salina resultante a 40 °C y dejando que se enfríe a 5 °C para que se produzca la cristalización.
En otra realización, la conversión del zwitterión en la sal maleato se logra por tratamiento de una solución de acetonitrilo del zwitterión con una solución acuosa de ácido maleico, calentando la solución resultante a 40 °C y dejando que se enfríe a 5 °C para que se produzca la cristalización.
Los compuestos de fórmula estructural (II) se pueden obtener a partir de los compuestos de fórmula estructural (III):

donde R2 es como se definió anteriormente, por reacción con un compuesto de ácido borónico de fórmula estructural (IV), donde R es H o F:

De forma alternativa, se puede usar un éster de boronato, tal como éster de pinacol, lo que proporciona el ácido borónico precursor in situ. Los compuestos de fórmula estructural (IV) están comercialmente disponibles, por ejemplo, de Enamina LLC, Princeton Corporate Plaza, 7 Deer Park Drive Ste. 17-3, Monmouth Jct. NJ (EE. UU.) 08852, Manchester Organics o Fluorochem. La reacción entre el compuesto de las fórmulas estructurales (III) y (IV) puede realizarse en presencia de un catalizador adecuado, tal como un catalizador de rodio, por ejemplo el dímero de (1,5-ciclooctadieno)cloruro de rodio, [Rh(COD)Cl]2 y un aditivo tal como un ligando de fosfina, por ejemplo bis(difenilfosfino)-1,1'-binaftilo (BINAP), preferentemente en presencia de una base, tal como hidróxido de potasio acuoso, a temperatura elevada, tal como de 5o °C a 95 °C, y en un disolvente miscible en agua, tal como 1,4-dioxano. Preferentemente, se lleva a cabo la reacción en condiciones estrictamente anaerobias, donde se purga la mezcla de reacción con un gas inerte tal como nitrógeno, y se evacúa a presión reducida, repitiendo este procedimiento de evacuación y purgando con nitrógeno tres veces. De forma alternativa, se puede llevar a cabo la reacción en un vial de microondas y se calienta la mezcla en un reactor de microondas a temperatura elevada. Esta reacción produce una mezcla de isómeros, normalmente en la proporción 1 : 1. Puede separarse la mezcla de isómeros producidos por cromatografía, HPLC o por cristalización. Se puede lograr una síntesis asimétrica por la inclusión de un enantiómero del ligando quiral, por ejemplo (R)-(+)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (R-BINAP) en presencia de un catalizador basado en un compuesto de rodio. La geometría del doble enlace en el compuesto de fórmula estructural (III) puede ser isómero (E) o mezcla
de isómeros (E) y (Z), preferentemente isómero (E) puro.
La reacción entre un enantiómero de un compuesto de fórmula (III) con un compuesto de fórmula (IV) produce dos diastereoisómeros, en proporción aproximadamente 1 : 1, que se pueden separar por cristalización, cromatografía o por HPLC. El método preferente de separación es HPLC quiral en un soporte quiral, tal como columnas Chiralpak o Chiralcel. La proporción de los diastereoisómeros formados se puede incrementar sustancialmente a, por ejemplo, aproximadamente 80 : 20 o mayor en presencia de aproximadamente un 10% de aditivos, tales como (R)-(+)-2,2'-bis(difenilfosfino)-1,1-binaftaleno [(R)-BINAP], lo que proporciona como isómero principal el diastereoisómero biológicamente más activo.
De forma alternativa, varias combinaciones del compuesto (III) con diferentes grupos R2 quirales, ligando, ácido borónico (IV), catalizador y disolvente seleccionados por los expertos en la materia o por examen de un gran número de combinaciones, pueden proporcionar una mayor proporción de diastereoisómeros.
La proporción diastereoisómera se puede incrementar además a, por ejemplo, más de 99 : 1, por HPLC quiral o por cristalización.
Los compuestos de fórmula estructural (III) se pueden obtener a partir de los compuestos de fórmula estructural (V):

por reacción con un compuesto de fórmula estructural (VI)

donde R2 es como se definió anteriormente, en presencia de una base orgánica tal como N,N-diisopropiletilamina («DIPEA») y un catalizador a base de paladio adecuado, por ejemplo PdCh(dppf)-CH2Cl2 [1,1-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejado con diclorometano, en un disolvente tal como diclorometano. El compuesto de fórmula (V) se puede usar como compuesto precursor, o se puede generar in situ a partir de una sal, tal como la sal diclorhidrato, en presencia de una base de amina terciaria.
Los compuestos de fórmula estructural (VI) se pueden preparar por métodos descritos en la presente memoria. A modo de ilustración, el compuesto de fórmula estructural (VI), donde R2 es metilo, y teniendo el doble enlace la geometría (E), se puede preparar por el método mostrado a continuación, partiendo del 4-bromocrotonato de metilo comercialmente disponible y acetato de sodio o potasio en acetonitrilo a temperatura elevada, por ejemplo, 50 °C:

Los compuestos de fórmula estructural (V) se pueden preparar a partir de los compuestos de fórmula estructural (VII):

por hidrogenolisis catalítica usando, por ejemplo, un catalizador de paladio depositado sobre carbón, en un disolvente inerte, tal como etanol o acetato de etilo.
Los compuestos de fórmula estructural (VII) se pueden obtener a partir de compuestos de fórmula estructural (VIII):

por reducción de diimida, generada, por ejemplo, a partir de bencenosulfonilhidrazida en presencia de una base, tal como carbonato de potasio, en un disolvente adecuado, tal como DMF, y a temperatura elevada, tal como 130 °C.
Los compuestos de fórmula estructural (VIII) existen como isómeros geométricos, por ejemplo, forma (E) o (Z) y se pueden usar como isómeros puros o bien como mezclas. Los compuestos de fórmula estructural (VIII) se pueden obtener partiendo de compuestos comercialmente disponibles conocidos (por ejemplo, de Wuxi App Tec, 288 Fute Zhong Road, Waigaoquiao Free Trade, Shanghai 200131, China) de fórmula estructural (IX):

que se puede oxidar, por ejemplo, con trióxido de azufre en piridina al correspondiente aldehído de fórmula estructural (X):

Este compuesto de fórmula estructural (X) se puede hacer reaccionar a continuación, lo que puede realizarse sin aislamiento del compuesto de fórmula (X), con un iluro de fórmula estructural (XI):

para formar de este modo el compuesto de fórmula (VIII) que existe como una mezcla de isómeros geométricos (E) y (Z). Los expertos en la materia apreciarán que existen otros métodos para formar el compuesto de fórmula (VIII) a partir del aldehído (X). Los isómeros geométricos se pueden separar por cromatografía o se pueden usar en la siguiente etapa como una mezcla. Este esquema global para la preparación de compuestos de fórmula estructural (I) se resume a continuación como el esquema (I):
Esquema (I):

Los iluros de fórmula estructural (XI) se pueden preparar partiendo de los compuestos de fórmula (XII) (disponibles de Fluorochem):
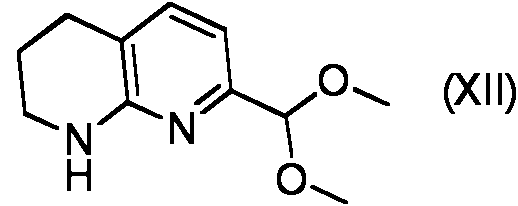
que por reacción con ácido clorhídrico en primer lugar, seguido de neutralización con bicarbonato de sodio, se pueden convertir a continuación en un aldehído de fórmula estructural (XIII):

que se puede reducir, por ejemplo, usando borohidruro de sodio al correspondiente alcohol de fórmula estructural (XIV):

(Véanse también las rutas descritas en la Patente de EE. UU. A-20040092538 para la preparación del alcohol de fórmula (XIV)) que se puede bromar a continuación, por ejemplo, usando tribromuro de fósforo para producir el correspondiente compuesto de bromo de fórmula estructural (XV):
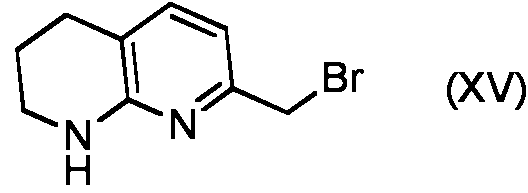
que se puede convertir en el bromuro de trifenilfosfonio (XVI) haciéndolo reaccionar con trifenilfosfina en un disolvente tal como acetonitrilo.

El compuesto de iluro mencionado anteriormente de fórmula estructural (XI) se puede obtener por reacción del compuesto de fórmula estructural (XVI) con una base, tal como una solución de terc-butóxido de potasio en un disolvente inerte, tal como THF. El iluro de fórmula estructural (XI) se puede aislar o preferentemente se puede formar in situ y hacerse reaccionar en el mismo recipiente con un aldehído de fórmula estructural (X) sin aislamiento previo.
El esquema global para la preparación del iluro de fórmula estructural (XI) se resume a continuación como el esquema (II):
Esquema (II)

Cada uno de los dos enantiómeros comercialmente disponibles del compuesto de fórmula (IX) proporciona un diastereoisómero de compuesto de fórmula (I) que es más potente que el otro.
Se apreciará que cualquiera de las rutas descritas anteriormente puede ser ventajosa para proteger uno o más grupos funcionales. Se pueden encontrar ejemplos de grupos protectores y de los medios para su retirada en T. W. Greene, Protective groups in organic synthesis (3a edición, J. Wiley and Sons, 1999). Los grupos protectores de amina adecuados incluyen acilo (por ejemplo, acetilo), carbamato (por ejemplo, 2',2',2'-tricloroetoxicarbonilo, benciloxicarbonilo o t-butoxicarbonilo) y arilalquilo (por ejemplo, bencilo), que se pueden retirar por hidrólisis (por ejemplo, usando un ácido tal como ácido clorhídrico en dioxano o ácido trifluoroacético en diclorometano) o de forma reductora (por ejemplo, hidrogenolisis de un grupo bencilo o benciloxicarbonilo o retirada reductora de un grupo 2',2',2'-tricloroetoxicarbonilo usando cinc en ácido acético) según sea apropiado. Otros grupos protectores de amina adecuados incluyen trifluoroacetilo (-COCF3) que se puede retirar por hidrólisis catalizada por bases.
Se apreciará que en cualquiera de las rutas descritas anteriormente, se puede variar el orden preciso de las etapas sintéticas por las que se introducen en la molécula varios grupos y restos. Estará dentro de la habilidad del experto en la materia asegurar que los grupos o restos introducidos en una fase del procedimiento no se vean afectados por las posteriores transformaciones y reacciones, y seleccionar el orden de las etapas sintéticas de acuerdo con esto.
También se cree que determinados compuestos de fórmulas (III), (V) a (VIII), (X), (XI), (XV) y (XVI) son novedosos y, por lo tanto, forman otro aspecto adicional de la invención.
La configuración absoluta de los compuestos de fórmula (I) se puede obtener siguiendo una síntesis enantioselectiva independiente a partir de un compuesto intermedio de configuración absoluta conocida. De forma alternativa, un compuesto enantioméricamente puro de fórmula (I) se puede convertir en un compuesto con una configuración absoluta que sea conocida. En cualquier caso, se puede usar una comparación de los datos espectroscópicos, rotación
óptica y tiempos de retención en una columna de HPLC analítica para confirmar la configuración absoluta. Cuando sea posible, una tercera opción es la determinación de la configuración absoluta a través de cristalografía de rayos X.
Métodos de uso
Se cree que los compuestos de fórmula (I) y las sales de los mismos tienen actividad antagonista de integrina av, en particular actividad del receptor de avp6, y por tanto tienen una utilidad potencial en el tratamiento de enfermedades o afecciones para las que está indicado un antagonista de avp6.
Por tanto, la presente invención proporciona un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo para su uso en la terapia. El compuesto de fórmula (I) o la sal farmacéuticamente aceptable del mismo puede ser para su uso en el tratamiento de una enfermedad o afección para la que está indicado un antagonista de integrina avp6.
Por tanto, la presente invención proporciona un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de una enfermedad o afección para la que está indicado un antagonista de integrina avp6.
También se proporciona el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo en la fabricación de un medicamento para el tratamiento de una enfermedad o afección para la que está indicado un antagonista de integrina avp6.
De forma adecuada, el sujeto que lo necesita es un mamífero, en particular un ser humano.
Como se usa en la presente memoria, el término «cantidad eficaz» quiere decir una cantidad de un fármaco o agente farmacéutico que provocará la respuesta biológica o médica de un tejido, sistema, animal o ser humano que está buscando, por ejemplo, un investigador o un médico. Además, el término «cantidad terapéuticamente eficaz» quiere decir cualquier cantidad que, en comparación con un sujeto correspondiente que no ha recibido dicha cantidad, da como resultado una mejora en el tratamiento, curación, prevención o mejoría de una enfermedad, trastorno o efecto secundario, o una disminución en la velocidad de avance de una enfermedad o trastorno. El término también incluye dentro de su alcance cantidades eficaces para potenciar la función fisiológica normal.
Las enfermedades fibróticas implican la formación de un exceso de tejido conjuntivo fibroso en un órgano o tejido en un proceso reparador o reactivo. Se cree que los antagonistas de avp6 son útiles en el tratamiento de varias de dichas enfermedades o afecciones incluyendo las dependientes de la función de integrina avp6 y de la activación del factor de crecimiento transformante beta por medio de integrinas alfa v. Las enfermedades pueden incluir: fibrosis pulmonar (por ejemplo, fibrosis pulmonar idiopática, neumonía intersticial no específica (NINE), neumonía intersticial usual (NIU), síndrome de Hermansky-Pudlak, fibrosis masiva progresiva (una complicación de la neumoconiosis de los mineros), fibrosis pulmonar relacionada con enfermedades del tejido conjuntivo, fibrosis de vías respiratorias en asma y EPOC, fibrosis asociada al SDRA (síndrome de dificultad respiratoria aguda), lesión pulmonar aguda, fibrosis inducida por radiación, fibrosis pulmonar familiar, hipertensión pulmonar); fibrosis renal (nefropatía diabética, nefropatía de IgA, nefritis lúpica, glomeruloesclerosis focal segmentaria (GEFS), nefropatía de trasplante, nefropatía autoinmunitaria, nefropatía farmacógena, nefropatía relacionada con la hipertensión, fibrosis sistémica nefrogénica); fibrosis hepáticas (fibrosis inducida víricamente (por ejemplo, hepatitis C o B), hepatitis autoinmunitaria, cirrosis biliar primaria, hepatopatía alcohólica, esteatosis hepática no alcohólica incluyendo esteatohepatitis no alcohólica (EHNA), fibrosis hepática congénita, colangitis esclerosante primaria, hepatitis farmacógena, cirrosis hepática); fibrosis cutánea (cicatriz hipertrófica, esclerodermia, queloides, dermatomiositis, fascitis eosinofílica, contractura de Dupuytren, síndrome de Ehlers-Danlos, enfermedad de La Peyronie, epidermólisis ampollosa distrófica, fibrosis submucosa oral); fibrosis ocular (degeneración macular relacionada con la edad (DMRE), edema macular diabético, queratoconjuntivitis seca, glaucoma), nefelio, lesión corneal y cicatrización corneal, prevención de cicatrización de ampollas filtrantes después de cirugía de trabeculectomía; fibrosis cardiaca (insuficiencia cardiaca congestiva, ateroesclerosis, infarto de miocardio, fibrosis endomiocárdica, miocardiopatía hipertrófica (MCH)) y otras afecciones fibróticas diversas (fibrosis mediastínica, mielofibrosis, fibrosis retroperitoneal, enfermedad de Crohn, neurofibromatosis, liomiomas uterinos (miomas uterinos), rechazo crónico del órgano trasplantado. Existen beneficios adicionales para la inhibición adicional de las integrinas avpi, avp5 o avp8.
Además, también se pueden tratar lesiones precancerosas o tumores malignos asociados a integrinas avp6 (estos pueden incluir tumores malignos endometriales, basocelulares, hepáticos, de colon, cervicouterinos, orales, de páncreas, de mama y de ovario, sarcoma de Kaposi, tumores de células gigantes y estroma asociado a cáncer, pero sin limitarse a estos). También se pueden beneficiar las afecciones que pueden beneficiarse de los efectos sobre la angiogénesis (por ejemplo, tumores sólidos).
El término «enfermedad o afección para la que está indicado un antagonista de avp6», pretende incluir cualquiera o todas las condiciones patológicas anteriores.
En una realización, la enfermedad o afección para la que está indicado un antagonista de avp6 es la fibrosis pulmonar idiopática.
En otra realización, la enfermedad o afección para la que está indicado un antagonista de avp6 se selecciona de nefelio, lesión corneal y cicatrización corneal.
Composiciones
Si bien es posible que, para su uso en la terapia, se pueda administrar un compuesto de fórmula (I), así como sales farmacéuticamente aceptables del mismo, como producto químico en bruto, es común presentar el principio activo como una composición farmacéutica.
Por lo tanto, la presente invención proporciona en otro aspecto una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable y uno o más vehículos, diluyentes y/o excipientes farmacéuticamente aceptables. Los compuestos de la fórmula (I) y las sales farmacéuticamente aceptables son como se describieron anteriormente. El (los) vehículo(s), diluyente(s) o excipiente(s) debe(n) ser aceptable(s) en el sentido de ser compatible(s) con los otros principios activos de la composición y no perjudicial(es) para el receptor de los mismos.
De acuerdo con otro aspecto de la invención, también se proporciona un procedimiento para la preparación de una composición farmacéutica que incluye mezclar un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, con uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables. La composición farmacéutica puede ser para su uso en el tratamiento de cualquiera de las afecciones descritas en la presente memoria.
Se proporciona además una composición farmacéutica para el tratamiento de enfermedades o afecciones para las que está indicado un antagonista de integrina avp6 que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo.
Se proporciona además una composición farmacéutica que comprende de 0,01 mg a 3000 mg de un compuesto de fórmula (I) o una sal farmacéutica del mismo y de 0,1 g a 2 g de uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables.
Ya que los compuestos de fórmula (I) están destinados a su uso en composiciones farmacéuticas, se entenderá fácilmente que cada uno se proporciona preferentemente en forma sustancialmente pura, por ejemplo, pura al menos en un 60 %, más adecuadamente pura al menos en un 75 % y preferentemente pura al menos en un 85 %, en especial pura al menos en un 98 % (% en una base de peso por peso).
Las composiciones farmacéuticas se pueden presentar en forma de dosis unitarias que contienen una cantidad predeterminada de principio activo por dosis unitaria. Las composiciones de dosificación unitaria preferentes son las que contienen una dosis diaria o una subdosis, o una fracción apropiada de las mismas, de un principio activo. Dichas dosis unitarias se pueden administrar, por tanto, más de una vez al día. Las composiciones de dosificación unitaria preferentes son las que contienen una dosis diaria o subdosis (para su administración más de una vez al día), como se indicó anteriormente en la presente memoria, o una fracción apropiada de las mismas, de un principio activo.
Las composiciones farmacéuticas se pueden adaptar para su administración por cualquier vía apropiada, por ejemplo, por la vía oral (incluyendo bucal o sublingual), rectal, inhalatoria, intranasal, tópica (incluyendo bucal, sublingual o transdérmica), vaginal, ocular o parenteral (incluyendo subcutánea, intramuscular, intravenosa o intradérmica). Dichas composiciones se pueden preparar por cualquier método conocido en la técnica de farmacia, por ejemplo, mezclando el principio activo con el (los) vehículo(s) o excipiente(s).
En una realización, la composición farmacéutica está adaptada para su administración oral.
Las composiciones farmacéuticas adaptadas para su administración oral se pueden presentar como unidades discretas tales como cápsulas o comprimidos; polvos o gránulos; soluciones o suspensiones en líquidos acuosos o no acuosos; espumas o batidos comestibles o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
Por ejemplo, para la administración oral en forma de un comprimido o una cápsula, el componente farmacológico activo se puede asociar con un vehículo inerte farmacéuticamente aceptable no tóxico, oral, tal como etanol, glicerol, agua y similares. Se pueden preparar polvos adecuados para su incorporación en comprimidos o cápsulas reduciendo el compuesto a un tamaño de partícula fino adecuado (por ejemplo, por micronización) y mezclando con un vehículo farmacéutico preparado de forma similar tal como un carbohidrato comestible, como por ejemplo, almidón o manitol. También pueden estar presentes un agente saborizante, conservante, dispersante y colorante.
Las cápsulas se pueden fabricar preparando una mezcla en polvo, como se describió anteriormente y rellenando las cubiertas de gelatina formadas. Se pueden añadir agentes antiapelmazantes y lubricantes tales como sílice coloidal, talco, estearato de magnesio, estearato de calcio o polietilenglicol sólido, a la mezcla en polvo antes de la operación de llenado. También se puede añadir un agente disgregante o solubilizante tal como agar-agar, carbonato de calcio o carbonato de sodio para mejorar la disponibilidad del medicamento cuando se ingiere la cápsula.
Además, cuando se desee o sea necesario, también se pueden incorporar a la mezcla aglutinantes, agentes antiapelmazantes, lubricantes, agentes edulcorantes, saborizantes, agentes disgregantes y agentes colorantes adecuados. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, goma de tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares.
Los lubricantes usados en estas formas farmacéuticas incluyen: oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares.
Los disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares. Los comprimidos se formulan, por ejemplo, preparando una mezcla en polvo, granulando o formando lingotes, añadiendo un lubricante y disgregante y prensando en comprimidos. Se prepara una mezcla en polvo mezclando el compuesto, molido adecuadamente, con un diluyente o base como se describió anteriormente, y opcionalmente, con un aglutinante tal como carboximetilcelulosa, un alginato, gelatina o polivinilpirrolidona, un retardante de solución tal como parafina, un acelerador de resorción tal como una sal cuaternaria y/o un agente de absorción tal como bentonita, caolín o fosfato de dicalcio. La mezcla en polvo se puede granular por humectación con un aglutinante tal como jarabe, pasta de almidón, mucílago de Acadia o soluciones de materiales celulósicos o poliméricos y forzándola a pasar a través de un tamiz. Como alternativa a la granulación, se puede hacer pasar la mezcla en polvo a través de una máquina para formar comprimidos y el resultado son lingotes formados imperfectamente que se rompen en gránulos. Los gránulos se pueden lubricar para evitar que se adhieran a los moldes que forman los comprimidos por medio de la adición de ácido esteárico, una sal de estearato, talco o aceite mineral. La mezcla lubricada se comprime a continuación en comprimidos. Los compuestos de la presente invención también se pueden asociar con un vehículo inerte fluido y comprimirse en comprimidos directamente sin pasar a través de las etapas de granulación o pegado. Se puede proporcionar un recubrimiento protector transparente u opaco que consiste en un revestimiento sellante de goma laca, un recubrimiento de azúcar o material polimérico y un recubrimiento pulido de cera. Se pueden añadir tintes a estos recubrimientos para distinguir dosificaciones unitarias diferentes.
Se pueden preparar fluidos orales tales como solución, jarabes y elixires en forma farmacéutica unitaria de modo que una cantidad dada contenga una cantidad predeterminada del compuesto. Se pueden preparar jarabes disolviendo el compuesto en una solución acuosa adecuadamente saborizada, mientras que se preparan elixires a través del uso de un vehículo alcohólico no tóxico. Se pueden formular suspensiones dispersando el compuesto en un vehículo no tóxico. También se pueden añadir solubilizantes y emulsionantes, tales como alcoholes isoestearílicos etoxilados y éteres de polioxietilensorbitol, conservantes, aditivos saborizantes tales como aceite de menta o edulcorantes naturales o sacarina u otros edulcorantes artificiales y similares.
Cuando sea apropiado, se pueden microencapsular composiciones de unidad de dosificación para administración oral. También se puede preparar la formulación para prolongar o mantener la liberación como, por ejemplo, recubriendo o incrustando material particulado en polímeros, cera o similares.
Los compuestos de la invención también se pueden administrar en forma de sistemas de suministro de liposomas, tales como vesículas unilaminares pequeñas, vesículas unilaminares grandes y vesículas multilaminares. Se pueden formar liposomas a partir de varios fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
Las composiciones farmacéuticas adaptadas para administración transdérmica se pueden presentar como parches discretos destinados a permanecer en contacto íntimo con la epidermis del receptor durante un periodo de tiempo prolongado.
Las composiciones farmacéuticas adaptadas para administración tópica se pueden formular como pomadas, cremas, suspensiones, lociones, polvos, soluciones, pastas, geles, pulverizaciones, aerosoles o aceites.
Para tratamientos de los ojos u otros tejidos externos, por ejemplo la boca y la piel, las composiciones se aplican preferentemente como pomada o crema tópica. Cuando se formula en una pomada, el principio activo se puede emplear con una base de pomada parafínica o una miscible con agua. De forma alternativa, el principio activo se puede formular en una crema con una base de crema de aceite en agua o una base de agua en aceite. Los compuestos de esta invención se pueden administrar como colirios tópicos. Los compuestos de esta invención se pueden administrar por medio de vías subconjuntival, intracameral o intravítrea, lo que necesitaría periodos de administración que fueran mayores que a diario.
Las formulaciones farmacéuticas adaptadas para administraciones tópicas para los ojos incluyen colirios en donde el principio activo se disuelve o suspende en un vehículo adecuado, en especial un disolvente acuoso. Las formulaciones que se van a administrar en los ojos tendrán pH y osmolalidad compatibles oftálmicamente. En una composición de la invención se pueden incluir uno o más agentes tamponadores y/o agentes de ajuste del pH oftálmicamente aceptables, incluyendo ácidos tales como ácidos acético, bórico, cítrico, láctico, fosfórico y clorhídrico; bases tales como hidróxido de sodio, fosfato de sodio, borato de sodio, citrato de sodio, acetato de sodio y lactato de sodio y tampones tales como citrato/dextrosa, bicarbonato de sodio y cloruro de amonio. Dichos ácidos, bases y tampones se pueden incluir en una cantidad requerida para mantener el pH de la composición en un intervalo oftálmicamente aceptable. En la composición se pueden incluir una o más sales oftálmicamente aceptables en una cantidad suficiente para llevar la
osmolalidad de la composición a un intervalo oftálmicamente aceptable. Dichas sales incluyen las que tienen cationes sodio, potasio o amonio y aniones cloruro, citrato, ascorbato, borato, fosfato, bicarbonato, sulfato, tiosulfato o bisulfito.
El dispositivo de suministro ocular se puede diseñar para la liberación controlada de uno o más agentes terapéuticos con múltiples velocidades de liberación y cinéticas de dosificación sostenida y permeabilidad definidas. La liberación controlada se puede obtener a través del diseño de matrices poliméricas que incorporan diferentes elecciones y propiedades de polímeros biodegradables/bioerosionables (por ejemplo, poli(etileno-acetato de vinilo) (EVA), PVA superhidrolizado), hidroxialquilcelulosa (HPC), metilcelulosa (MC), hidroxipropilmetilcelulosa (HPMC) (todos por sus siglas en inglés), policaprolactona, poli(ácido glicólico), poli(ácido láctico), polianhídrido, de pesos moleculares poliméricos, cristalinidad polimérica, proporciones copoliméricas, condiciones de procesamiento, acabado superficial, geometría, adición de excipientes y recubrimientos poliméricos que potenciarán la difusión, erosión, disolución y ósmosis del fármaco.
Las formulaciones para el suministro de fármaco usando dispositivos oculares pueden asociar uno o más agentes activos y adyuvantes apropiados para la vía de administración indicada. Por ejemplo, los agentes activos se pueden mezclar con cualquier excipiente farmacéuticamente aceptable, lactosa, sacarosa, almidón en polvo, ésteres celulósicos de ácidos alcanoicos, ácido esteárico, talco, estearato de magnesio, óxido de magnesio, sales de sodio y calcio de ácidos fosfórico y sulfúrico, goma arábiga, gelatina, alginato de sodio, polivinilpirrolidina y/o alcohol polivinílico, comprimidos o encapsulados para su administración convencional. De forma alternativa, los compuestos se pueden disolver en polietilenglicol, propilenglicol, soluciones coloidales de carboximetilcelulosa, etanol, aceite de maíz, aceite de cacahuete, aceite de semilla de algodón, aceite de sésamo, goma de tragacanto y/o varios tampones. Los compuestos también se pueden mezclar con composiciones de polímeros tanto biodegradables como no biodegradables y un vehículo o diluyente que tenga una propiedad de retraso temporal. Los ejemplos representativos de composiciones biodegradables pueden incluir albúmina, gelatina, almidón, celulosa, dextranos, polisacáridos, poli(D, L-lactida), poli(D, L-lactida-co-glicolida), poli(glicolida), poli(hidroxibutirato), poli(alquilcarbonato) y poli(ortoésteres) y mezclas de los mismos. Los ejemplos representativos de polímeros no biodegradables pueden incluir copolímeros de EVA, caucho de silicona y poli(metilacrilato) y mezclas de los mismos.
Las composiciones farmacéuticas para su suministro ocular también incluyen composición acuosa gelificable in situ. Una composición de este tipo comprende un agente gelificante en una concentración eficaz para promover la gelificación cuando se ponen en contacto con el ojo o con líquido lagrimal. Los agentes gelificantes adecuados incluyen polímeros termoendurecibles, pero no se limitan a estos. El término «gelificable in situ», como se usa en la presente memoria, incluye no solo líquidos de viscosidad baja que forman geles cuando se ponen en contacto con el ojo o con líquido lagrimal, sino que también incluye líquidos más viscosos tales como geles semifluidos y tixotrópicos que presentan un incremento sustancial en la viscosidad o la rigidez del gel después de la administración en el ojo. Véase, por ejemplo, Ludwig (2005) Adv. Drug Deliv. Rev. 3; 57:1595-639, incorporado en la presente memoria por referencia con el propósito de la explicación de los ejemplos de polímeros para su uso en el suministro ocular de fármaco.
Las composiciones farmacéuticas adaptadas para su administración tópica en la boca incluyen pastillas para chupar, pastillas y enjuagues bucales.
Las composiciones farmacéuticas adaptadas para su administración rectal se pueden presentar como supositorios o como enemas.
Las formas farmacéuticas para su administración nasal o inhalatoria se pueden formular convenientemente como aerosoles, soluciones, suspensiones, geles o polvos secos.
Las composiciones farmacéuticas adaptadas para su administración vaginal se pueden presentar como óvulos vaginales, tampones, cremas, geles, pastas, espumas o formulaciones de pulverización.
Las composiciones farmacéuticas adaptadas para su administración parenteral incluyen soluciones de inyección estériles acuosas y no acuosas que pueden contener antioxidantes, tampones, bacteriostáticos y solutos que hacen que la composición sea isotónica con la sangre del receptor destinado y suspensiones estériles acuosas y no acuosas que pueden incluir agentes de suspensión y agentes espesantes. Las composiciones se pueden presentar en recipientes de dosis unitaria o de múltiples dosis, por ejemplo ampollas y viales sellados, y se pueden almacenar en una condición secada por congelación (liofilizada) que requiere solo la adición del vehículo líquido estéril, por ejemplo, agua para inyecciones, inmediatamente antes de su uso. Las soluciones y suspensiones de inyección sin preparación previa se pueden preparar a partir de polvos estériles, gránulos y comprimidos.
Las composiciones farmacéuticas adaptadas para su administración subcutánea o intramuscular incluyen el copolímero poli(ácido láctico-co-glicólico) (PLGA) para formar micropartículas que contienen el principio activo farmacéutico para proporcionar la liberación mantenida.
Una cantidad terapéuticamente eficaz de un compuesto de la presente invención dependerá de varios factores incluyendo, por ejemplo, la edad y el peso del sujeto, la afección precisa que requiere tratamiento y su gravedad, la naturaleza de la formulación, y la vía de administración, y en última instancia, dependerá del juicio del médico o veterinario. En la composición farmacéutica, cada unidad de dosificación para su administración oral o parenteral contiene preferentemente de 0,01 mg a 3000 mg, más preferentemente de 0,1 mg a 2000 mg, de un compuesto de la
invención, calculado como el compuesto precursor zwitteriónico.
Los compuestos farmacéuticamente aceptables de la invención se pueden administrar en una dosis diaria (para un paciente adulto) de, por ejemplo, una dosis oral o parenteral de 0,01 mg a 3000 mg al día o de 0,5 mg a 1000 mg al día del compuesto de la fórmula (I) o una sal farmacéuticamente aceptable del mismo, calculada como zwitterión. Esta cantidad se puede dar en una única dosis al día o más usualmente en varias (tales como dos, tres, cuatro, cinco o seis) subdosis al día de modo que la dosis diaria total sea la misma. Una cantidad eficaz de una sal de los mismos se puede determinar como una relación de la cantidad eficaz del compuesto de fórmula (I) per se.
Los compuestos de la invención se pueden emplear solos o en combinación con otros agentes terapéuticos. Las terapias asociadas de acuerdo con la presente invención comprenden, por tanto, la administración de al menos un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, y el uso de al menos otro agente farmacéuticamente activo. Preferentemente, las terapias asociadas de acuerdo con la presente invención comprenden la administración de al menos un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, y al menos otro agente farmacéuticamente activo. El(los) compuesto(s) de la invención y el(los) otro(s) agente(s) farmacéuticamente activo(s) se pueden administrar juntos en una única composición farmacéutica o por separado y, cuando se administran por separado, esto se puede producir simultánea o secuencialmente en cualquier orden. Las cantidades del(de los) compuesto(s) de la invención y el(los) otro(s) agente(s) farmacéuticamente activo(s) y los momentos de administración relativos se seleccionarán con el fin de lograr el efecto terapéutico asociado deseado.
Por tanto, en otro aspecto, se proporciona una asociación que comprende un compuesto de la invención y al menos otro agente farmacéuticamente activo.
Por tanto, en un aspecto, el compuesto y las composiciones farmacéuticas de acuerdo con la invención se pueden usar en asociación con otro u otros agentes terapéuticos más, o incluirlos, incluyendo terapias para enfermedad alérgica, enfermedad inflamatoria, enfermedad autoinmunitaria, terapias antifibróticas y terapias para enfermedad obstructiva de las vías respiratorias, terapias para enfermedades oculares diabéticas y terapias para nefelio, lesión corneal y cicatrización corneal.
Las terapias antialérgicas incluyen inmunoterapia con antígenos (tal como componentes y fragmentos de veneno de abeja, polen, leche, cacahuete, motivos de CpG, colágeno, otros componentes de la matriz extracelular que se pueden administrar como antígenos orales o sublinguales), antihistaminas (tales como cetirizina, loratidina, acrivastina, fexofenidina, clorfenamina) y corticosteroides (tales como propionato de fluticasona, furoato de fluticasona, dipropionato de beclometasona, budesonida, ciclesonida, furoato de mometasona, triamcinolona, flunisolida, prednisolona, hidrocortisona).
Las terapias antiinflamatorias incluyen los AINE (tales como aspirina, ibuprofeno, naproxeno), moduladores de leucotrieno (tales como montelukast, zafirlukast, pranlukast), y otras terapias antiinflamatorias (tales como inhibidores de iNOS, inhibidores de triptasa, inhibidores de IKK2, inhibidores de p38 (losmapimod, dilmapimod), inhibidores de elastasa, agonistas de beta2, antagonistas de DP1, antagonistas de DP2, inhibidores de pI3K delta, inhibidores de ITK, inhibidores de LP (lisofosfatídicos) o inhibidores de FLAP (proteína activadora de 5-lipoxigenasa) (tales como 3-(3-(terc-butiltio)-1-(4-(6-etoxipiridin-3-il)bencil)-5-((5-metilpiridin-2-il)metoxi)-1H-indol-2-il)-2,2-dimetilpropanoato de sodio); agonistas de adenosina a2a (tales como adenosina y regadenosón), antagonistas de quimiocinas (tales como antagonistas de CCR3 o antagonistas de CCR4), inhibidores de la liberación de mediadores.
Las terapias para enfermedad autoinmunitaria incluyen los FARME (tales como metotrexato, leflunomida, azatioprina), terapias biofarmacéuticas (tales como anti-IgE, anti-TNF, antiinterleucinas (tales como anti-IL-1, anti-IL-6, anti-IL-12, anti-IL-17, anti-IL-18)), terapias con receptores (tales como etanercept y agentes similares); inmunoterapias no específicas de antígenos (tales como interferón u otras citocinas/quimiocinas, moduladores de receptores de citocina/quimiocina, agonistas o antagonistas de citocina, agonistas de TLR y agentes similares).
Otras terapias antifibróticas incluyen inhibidores de la síntesis de TGFp (tales como pirfenidona), inhibidores de tirosina cinasa que seleccionan como objetivo el factor de crecimiento de células endoteliales vasculares (VEGF), factor de crecimiento derivado de plaquetas (PDGF) y factor de crecimiento fibroblástico (FGF) receptores de cinasas (tales como Nintedanib (BIBF-1120) y mesilato de imatinib (Gleevec)), antagonistas del receptor de endotelina (tales como ambrisentán o macitentán), antioxidantes (tales como N-acetilcisteína (NAC); antibióticos de amplio espectro (tales como cotrimoxazol, tetraciclinas (clorhidrato de minociclina)), inhibidores de fosfodiesterasa 5 (PDE5) (tales como sildenafilo), anticuerpos y fármacos anti-avpx (tales como anticuerpos monoclonales anti-avp6 (tales como los descritos en la Patente Internacional W02003100033A2); intetumumab; cilengitida) se pueden usar en asociación.
Las terapias para enfermedades obstructivas de las vías respiratorias incluyen broncodilatadores tales como agonistas de p2 de acción breve, tales como salbutamol), agonistas de p2 de acción prolongada (tales como salmeterol, formoterol y vilanterol), antagonistas muscarínicos de acción breve (tales como bromuro de ipratropio), antagonistas muscarínicos de acción prolongada, (tales como tiotropio, umeclidinio).
En algunas realizaciones, el tratamiento también puede implicar la asociación de un compuesto de esta invención con otros modos de tratamiento existentes, por ejemplo agentes existentes para el tratamiento de enfermedades oculares diabéticas, tales como tratamientos anti-VEGF, por ejemplo, Lucentis®, Avastin® y Aflibercept y esteroides, por
ejemplo, triamcinolona e implantes esteroideos que contienen acetónido de fluocinolona.
En algunas realizaciones, el tratamiento también puede implicar la asociación de un compuesto de esta invención con otros modos de tratamiento existentes, por ejemplo, agentes existentes para el tratamiento de nefelio, lesión corneal o cicatrización corneal, tales como Gentel®, extracto de sangre de ternera, Levofloxacin® y Ofloxacin®.
Los compuestos y las composiciones de la invención se pueden usar para tratar tumores malignos solos o en asociación con terapias para el cáncer que incluyen quimioterapia, radioterapia, agentes seleccionados como objetivo, inmunoterapia y terapia celular o génica.
Quedará claro para un experto en la materia, cuando sea apropiado, que se puede(n) usar el(los) otro(s) ingrediente(s) terapéutico(s) en forma de sales, por ejemplo como sales de metal alcalino o amina o como sales de adición de ácido, o profármacos, o como ésteres, por ejemplo ésteres de alquilos inferiores, o como solvatos, por ejemplo hidratos, para optimizar la actividad y/o estabilidad y/o características físicas, tales como solubilidad, del ingrediente terapéutico. También quedará claro, cuando sea apropiado, que los ingredientes terapéuticos se pueden usar en forma ópticamente pura.
Las asociaciones referidas anteriormente se pueden presentar convenientemente para su uso en forma de una composición farmacéutica y, por tanto, las composiciones farmacéuticas que comprenden una asociación como se definió anteriormente junto con un diluyente o vehículo farmacéuticamente aceptable representan otro aspecto de la invención. Los compuestos individuales de dichas combinaciones se pueden administrar secuencialmente o bien simultáneamente en composiciones farmacéuticas separadas o asociadas. Preferentemente, los compuestos individuales se administrarán simultáneamente en una composición farmacéutica asociada. Las dosis apropiadas de agentes terapéuticos conocidos serán fácilmente apreciadas por los expertos en la materia.
Se apreciará que, cuando el compuesto de la presente invención se administra en asociación con otro u otros agentes terapéuticamente activos más administrados normalmente por vía inhalatoria, intravenosa, oral, intranasal, ocular, tópica u otra vía, la composición farmacéutica resultante se puede administrar por la misma vía. De forma alternativa, los componentes individuales de la composición se pueden administrar por vías diferentes.
Las presentes invenciones se ilustrarán ahora solo a modo de ejemplo.
Abreviaturas
La siguiente lista proporciona definiciones de ciertas abreviaturas como se usan en la presente memoria. Se apreciará que la lista no es exhaustiva, pero el significado de las abreviaturas no definidas a continuación en la presente memoria será evidente para los expertos en la materia.
Ac (acetilo)
BCECF-AM (éster acetoximetílico de 2',7'-bis-(2-carboxietil)-5-(y-6)-carboxifluoresceína)
BEH (Ethylene Bridged Hybrid Technology, tecnología de partículas híbridas con puentes de etileno)
Bu (butilo)
CBZ (carboxibencilo)
CHAPS (3-[(3-colamidopropil)dimetilamonio]-1-propanosulfonato)
Chiralcel OD-H (tris(3,5-dimetilfenilcarbamato) de celulosa recubierto en gel de sílice, 5 pm)
Chiralpak AD-H (tris(3,5-dimetilfenilcarbamato) de amilosa recubierto en gel de sílice, 5 pm)
Chiralpak ID (tris(3-clorofenilcarbamato) de amilosa inmovilizado en gel de sílice, 5 pm)
Chiralpak AS (tris((S)-alfa-metilbencilcarbamato) de amilosa recubierto en gel de sílice, 5 pm)
CDI (carbonildiimidazol)
CSH (Charged Surface Hybrid Technology, tecnología de partículas híbridas de superficie cargada)
VC (volumen de columna)
DCM (diclorometano)
DIPEA (diisopropiletilamina)
DMF (N, N-dimetilformamida)
DMSO (dimetilsulfóxido)
DSC (calorimetría diferencial de barrido)
Et (etilo)
EtOH (etanol)
EtOAc (acetato de etilo)
h (hora/horas)
HCl (ácido clorhídrico)
HEPES (ácido 4-(2-hidroxietil)-1-piperazinetanosulfónico)
CLEM (cromatografía líquida-espectrometría de masas)
M (molar)
MDAP (HPLC autopreparativa dirigida por masa)
MDCK (riñón canino Madin-Darby)
Me (metilo)
MeCN (acetonitrilo)
MeI (yoduro de metilo)
MeOH (metanol)
min (minuto/minutos)
EM (espectro de masas)
PdCl2(dppf)-CH2Cl2 [1,1LBis(difenilfosfino)ferroceno]dicloropaladio(N), complejo con diclorometano Ph (fenilo)
iPr (isopropilo)
(R)-BINAP (R)-(+)-2,2,-Bis(difenilfosfino)-1,1,-binaftaleno
[Rh(COD)Cl]2 (dímero cloro(1,5-ciclooctadieno)rodio(I))
SPE (extracción en fase sólida)
TBME (éter metil-terc-butílico)
TEA (trietilamina)
TFA (ácido trifluoroacético)
TGA (análisis termogravimétrico)
TGA-IR (analizador termogravimétrico interconectado con infrarrojo)
THF (tetrahidrofurano)
TLC (cromatografía en capa fina)
UPLC (cromatografía líquida de alta resolución)
XRPD (difracción en polvo de rayos X)
Todas las referencias a salmuera se refieren a una solución acuosa saturada de cloruro de sodio.
Detalles experimentales
CLEM analítica
La CLEM analítica se realizó en uno de los siguientes sistemas A o B.
La detección UV en todos los sistemas fue una señal promedio de una longitud de onda de 220 nm a 350 nm y los espectros de masas se registraron en un espectrómetro de masas usando ionización por electropulverización en modo positivo y negativo con barrido alterno.
Los detalles experimentales de los sistemas A-C de CLEM como se hace referencia en la presente memoria son como sigue:
Sistema A
Columna: columna Acquity UPLC BEH C18 de 1,7 pm, 50 mm x 2,1 mm de DI
Caudal: 1 ml/min
Temp.: 40 °C
Disolventes: A: bicarbonato de amonio 10 mM en agua ajustado a pH 10 con solución de amoníaco
B: acetonitrilo
Gradiente: Tiempo (min)___________ % de A_________% de B
0 99 1
1,5 3 97
1,9 3 97
2,0 99 1
Sistema B
Columna: columna Acquity UPLC BEH C18de 1,7 pm, 50 mm x 2,1 mm de DI
Caudal: 1 ml/min
Temp.: 40 °C
Disolventes: A: solución de ácido fórmico en agua al 0,1 % v/v
B: solución de ácido fórmico en acetonitrilo al 0,1 % v/v
Gradiente: Tiempo (min) % de A % de B
0 97 3
1,5 0 100
1,9 0 100
2,0 97 3
Sistema C
Columna: columna Acquity UPLC CSH C18de 1,7 pm, 50 mm x 2,1 mm de DI
Caudal: 1 ml/min.
Temp.: 40 °C
Disolventes: A: bicarbonato de amonio 10 mM en agua ajustado a pH 10 con solución de amoníaco B: acetonitrilo
Gradiente: Tiempo (min) % de A % de B
0 97 3
1,5 5 95
1,9 5 95
2,0 97 3
Compuesto intermedio 1: 7-(bromometil)-1,2,3,4-tetrahidro-1,8-naftiridina (compuesto (XV))

Se añadió gota a gota tribromuro de fósforo (0,565 ml, 5,99 mmol) a una suspensión de (5,6,7,8-tetrahidro-1,8-naftiridin-2-il)metanol (compuesto (XIV)): véase la Patente de EE. UU. 20040092538, página 80, [0844]) (820 mg, 4,99 mmol) en acetonitrilo anhidro (50 ml) a 0 °C en nitrógeno. Tras la adición, se formó un precipitado de color naranja oscuro, que se volvió naranja pálido. La mezcla de reacción se agitó a 0 °C durante 1 h, tiempo en que se completó la reacción. La mezcla se concentró al vacío y el residuo se dividió entre acetato de etilo (250 ml) y una solución acuosa saturada de NaHCO3 (250 ml). La fase acuosa se extrajo adicionalmente con acetato de etilo (250 ml). Las soluciones orgánicas combinadas se hicieron pasar a través de una frita hidrófoba y, a continuación, se concentraron al vacío para dar el compuesto del título (1,05 g, 93 %) como un sólido cremoso esponjoso: CLEM (sistema C) TR=0,95 min, ES+vo m/z 227, 229 (M+H)+.
Compuesto intermedio 2: bromuro de trifenil((5,6,7,8-tetrahidro-1,8-naftiridin-2-il)metil)fosfonio (compuesto (XVI))

Se trató una solución de 7-(bromometil)-1,2,3,4-tetrahidro-1,8-naftiridina (compuesto (XV), para una preparación véase el compuesto intermedio 1) (1,00 g, 4,40 mmol) en acetonitrilo (98 ml) con trifenilfosfina (1,270 g, 4,84 mmol) y la solución se agitó a temperatura ambiente en nitrógeno durante la noche. La mezcla se concentró al vacío para dar un sólido crema oscuro, que se trituró a continuación con éter dietílico para dar el compuesto del título (2,139 g, 99 %) como un sólido crema pálido: CLEM (sistema C) TR= 1,23 min, ES+vo m/z 409 (M+H)+.
Compuesto intermedio 3: (E, Z) 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo (compuesto (VIII))

Se trató una solución agitada de (+) 3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo (compuesto (IX): disponible de Wuxi App Tec) (260 mg, 1,03 mmol) en DCM (3 ml) y DMSO (0,3 ml), en nitrógeno, con DIPEA (0,896 ml, 5,13 mmol). Después de enfriar a (0-5) °C (baño de hielo), se añadió piridina-trióxido de azufre (327 mg, 2,05 mmol) en porciones durante aprox. 5 min para oxidar el compuesto alcohólico (IX) al correspondiente compuesto aldehido (X) que no fue aislado. Se retiró el baño de enfriamiento y se continuó la agitación durante 0,5 h. Mientras tanto, se trató gota a gota una solución de bromuro de trifenil((5,6,7,8-tetrahidro-1,8-naftiridin-2-il) metil)fosfonio (compuesto (XVI), para una preparación véase el compuesto intermedio 2) (553 mg, 1,13 mmol) en DCM anhidro (10 ml), en nitrógeno, con terc-butóxido de potasio (1 M en THF) (1,232 ml, 1,232 mmol) durante aprox. 5 min dando como resultado una solución de color naranja. Se continuó la agitación durante 10 min y, a continuación, se añadió la solución de aldehido (fórmula (X)) a la solución de iluro de una vez y la mezcla se agitó a temperatura ambiente durante 22 h. La mezcla de reacción se diluyó con DCM (20 ml), se lavó con bicarbonato de sodio acuoso saturado (20 ml) y salmuera (20 ml), se secó (Na2SO4), a continuación, se evaporó al vacío. El residuo marrón oscuro se purificó mediante cromatografía en un cartucho de SPE de 20 g de sílice y se eluyó con un gradiente de 0 % a 100 % de acetato de etilo-ciclohexano durante 30 min para obtener el compuesto del título como dos isómeros geométricos:
isómero 1: una goma de color pajizo (123,4 mg, 31 %); CLEM (sistema A) TR=1,28 min, 95 %, ES+vo m/z 382 (M+H)+
e
isómero 2 : una goma de color pajizo (121,5 mg, 31 %); CLEM (sistema A) TR=1,22 min, 91 %, ES+vo m/z 382 (M+H)+
Rendimiento global = 244,9 mg, 62,5 %.
Posteriormente, la configuración del compuesto intermedio 3 mostró que era (R) y los dos isómeros geométricos eran: (R,E) 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo y (R,Z) 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo.
Compuesto intermedio 4: 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-carboxilato de bencilo (compuesto (VII))

Se trató una solución de (E/Z) 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo (compuesto VIII, para una preparación véase el compuesto intermedio 3) (1:1, E:Z) (244 mg, 0,640 mmol) en DMF (2 ml) con bencenosulfonilhidrazida (disponible de Alfa Aesar) (275 mg, 1,60 mmol) y carbonato de potasio (354 mg, 2,56 mmol). La mezcla de reacción se calentó a 130 °C durante 1 h, a continuación, se dejó enfriar y se dividió entre DCM y agua. La fase orgánica se lavó con agua y se secó por medio de una frita hidrófoba. La solución orgánica se evaporó al vacío y el aceite naranja residual se purificó mediante cromatografía en un cartucho de sílice (20 g) eluyendo con un gradiente de 0 % a 50 % [(EtOAc: EtOH 3:1) - EtOAc] durante 20 min. Las fracciones apropiadas se combinaron y evaporaron al vacío para dar el compuesto del título (150 mg, 61 %) como una goma amarillo pálido: CLEM (sistema A) TR=1,24min, 90%, ES+vo m/z 384 (M+H)+. Posteriormente, se mostró que la configuración absoluta del compuesto intermedio 4 era (S), por ende el compuesto es (S) 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-carboxilato de bencilo. El cambio de (R) en el compuesto intermedio 3 a (S) en el compuesto intermedio 4 se debe al cambio en la prioridad de retirar el doble enlace.
Compuesto intermedio 5: 7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina (compuesto (V))

Se agitó una solución agitada de 3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-carboxilato de bencilo (compuesto (VII, para una preparación véase el compuesto intermedio 4) (4,67 g, 12,2 mmol) en etanol (70 ml) que contenía un 10 % de paladio sobre carbón (0,50 g) en una atmósfera de hidrógeno durante 7 h. La CLEM mostró desprotección incompleta y se añadió un 10 % de paladio sobre carbón adicional (0,25 g) y la mezcla se agitó en una atmósfera de hidrógeno durante la noche. La mezcla de reacción se presentó como una suspensión gris oscuro, por lo que se le añadió DCM para disolver el material hasta que la mezcla llegó a ser negra. Se retiró el catalizador mediante filtración a través de una almohadilla de celite y el líquido filtrado y los lavados se evaporaron al vacío. El residuo se evaporó de DCM para obtener el compuesto del título como un aceite naranja (3,28 g): CLEM (sistema A) TR = 0,79 min, 90 %, ES+vo m/z 250 (M+H)+. Posteriormente, se estableció por inferencia la configuración del compuesto intermedio 5 como (S) y el nombre del compuesto es (S)-7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina.
Compuesto intermedio 6. Sal de ácido metanosulfónico de [7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina, (compuesto (V))

Esta sal del compuesto (V) se puede preparar y cristalizar como un método de purificación del compuesto (V) anterior.
Se añadió 2-butanol (5 ml) a 7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina (compuesto (V), para una preparación véase el compuesto intermedio 5) (1,0 g, 4,0 mmol) y la mezcla se calentó hasta que se alcanzó la disolución completa. Se añadió ácido metanosulfónico (0,260 ml, 4,01 mmol) a la solución caliente y la mezcla se calentó a 80 °C con agitación. A continuación, la solución se dejó enfriar a temperatura ambiente. De forma inmediata no fue evidente ninguna precipitación, por lo que la solución se enfrió adicionalmente en un refrigerador (ca. 4 °C). Después de 3 días, se observó una cantidad significativa de sólido. El sólido se aisló mediante filtración y se lavó con 2-butanol frío y se secó adicionalmente al vacío para proporcionar el compuesto del título (600 mg, 43 %) como un sólido amarillo pálido: CLEM (sistema A) TR = 0,80 min, 100 %, ES+vo m/z 250 (M+H)+; Hp Lc quiral analítica en una columna Chiralpak AD (250 mm x 4,6 mm) TR = 8,41 min, 99,6 % y TR = 12,03 min, 0,4 %, eluyendo con un 40 % de EtOH-heptano (que contenía un 0,2 % de isopropilamina), caudal de 1 ml/min, detectándose a 235 nm. Posteriormente, se estableció por inferencia la configuración del compuesto intermedio 6 como (S) y el nombre del compuesto es sal de ácido metanosulfónico de (S)-7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina.
Compuesto intermedio 7: (E)-4-acetoxibut-2-enoato de metilo (compuesto (VI))

Se trató una suspensión de acetato de sodio (3,5 g, 42 mmol) en MeCN (30 ml) con 4-bromocrotonato de metilo (disponible de Aldrich) (3,33 ml, 5 g, 28 mmol) y la mezcla se calentó a 50 °C durante 3 d. La mezcla se diluyó con éter y, a continuación, se filtró. El sólido se lavó con éter y el líquido filtrado combinado y los lavados se evaporaron a presión reducida. Después de la evaporación, el residuo se dividió entre éter y agua. La fase orgánica se lavó con bicarbonato de sodio acuoso, se secó sobre MgSO4 y se evaporó a presión reducida para dar un aceite naranja pálido. La RMN indicó una mezcla de producto y material de partida, por lo tanto, se añadió acetato de sodio (3,44 g, 42 mmol) al aceite residual, seguido de MeCN (10 ml) y la mezcla se calentó a 70 °C durante el fin de semana. La mezcla se concentró a presión reducida y el residuo se dividió entre éter y agua. La solución orgánica se lavó con agua, salmuera, se secó (MgSO4) y se filtró. El líquido filtrado se evaporó a presión reducida para dar el compuesto del título (3,55 g, 80 %) como un aceite naranja: RMN 6 (CDCla) 6,92 (1H, td, J 16,5 Hz), 6,01 (1H, td, J 16,2 Hz), 4,72 (2H, dd, J 5,2 Hz), 3,73 (3H, s), 2,10 (3H, s).
Compuesto intermedio 8: (E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-2-enoato de metilo (compuesto (III)

Se agitó una mezcla de (E)-4-acetoxibut-2-enoato de metilo (compuesto (VI), para una preparación véase el compuesto intermedio 7) (127 mg, 0,802 mmol), 7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina (compuesto (V), para una preparación véase el compuesto intermedio 5) (200 mg, 0,802 mmol) y aducto de PdCl2(dppf)-CH2^2 (65,7 mg, 0,080 mmol) en DCM (2 ml) a temperatura ambiente durante 2 h. La CLEM mostró alrededor de un 50 % de conversión y se añadió DIPEA (0,279 ml, 1,60 mmol) y la solución se agitó durante 2 h a temperatura ambiente. La CLEM mostró prácticamente una conversión completa en el producto. El material se cargó directamente en una columna y purificó mediante cromatografía (cartucho de 20 g de aminopropilo) eluyendo con un gradiente de 0 % a 100% de EtOAc en ciclohexano durante 20 min. Las fracciones apropiadas se combinaron y evaporaron para dar el compuesto del título (101,4 mg, 36 % de rendimiento): CLEM (sistema C) TR = 1,08 min, 95 %,
ES+vo m/z 348 (M+H)+. Se estableció por inferencia la configuración del compuesto intermedio 8 como (S) y el nombre como (S,E) 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-3-enoato de metilo.
Compuesto intermedio 9: 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoato de metilo. Isómero A e isómero B)

Se disolvieron (E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-2-enoato de metilo (compuesto (III), para una preparación véase el compuesto intermedio 8) (145 mg, 0,334 mmol), (R)-BINAP (31 mg, 0,05 mmol), [Rh(COD)Cl]2 (10 mg, 0,020 mmol), ácido (3-morfolinofenil)borónico (disponible de, por ejemplo, CombiBlocks, Manchester Organics o Fluorochem) (259 mg, 1,251 mmol) y KOH 3,8 M (0,22 ml, 0,836 mmol) en 1,4-dioxano (2 ml) en un vial de microondas y la solución se calentó en un horno de microondas (100 min, 95 °C). La mezcla de reacción se filtró a través de celite, se lavó con EtOAc (10 ml) y se concentró. La mezcla de reacción se suspendió en MeOH (300 ^l) y se purificó mediante cromatografía de fase inversa (C18, 40 g, MeCN al (5-95) % (que contenía amoníaco al 0,1 %) en bicarbonato de amonio 10 mM, 20 VC). Las fracciones apropiadas se combinaron y evaporaron para dar una mezcla diastereómera del compuesto del título (II) (99 mg, 58 %) como una goma.
La mezcla se disolvió en EtOH (2 ml) y heptano (1 ml) y los diastereoisómeros se separaron mediante HPLC quiral en una columna Chiralcel OD-H (3 cm * 25 cm) eluyendo con EtOH al 30 % (que contenía isopropilamina al 0,2 %) -heptano al 70 % (caudal = 30 ml/min, detectándose a 215 nm) para dar los dos diastereoisómeros del compuesto (II).
Isómero A (17 mg, 10 %): HPLC quiral analítica TR = 8,0 min, >99,5 % en una columna Chiralcel OD-H (4,6 mm de DI * 25 cm) eluyendo con (EtOH al 30 % (que contenía isopropilamina al 0,2 %) - heptano, caudal = 1,0 ml/min, detectándose a 215 nm; CLEM (sistema A) TR = 1,21 min, 99 %, ES+vo m/z 511 (M+H)+; RMN de 1H (400 MHz, CD3OD) 57,17 (t, J = 7,5 Hz, 1H), 7,13 (d, J = 7,5 Hz, 1H), 6,88-6,84 (m, 1H), 6,76 (d, J = 7,5 Hz, 1H), 6,38 (d, J = 7,5 Hz, 1H), 3,87-3,81 (m, 4H), 3,58 (s, 3H), 3,42-3,36 (m, 2H), 3,17-3,10 (m, 4H), 2,90-2,49 (m, 12H), 2,11-1,84 (m, 6H), 1,38-1,28 (m, 2H).
Isómero B (77 mg, 45 %): HPLC quiral analítica TR = 17,2 min, >99,5 % en una columna Chiralcel OD-H (4,6 mm de DI * 25 cm) eluyendo con (EtOH al 30% (que contenía isopropilamina al 0,2%) - heptano, caudal = 1,0 ml/min, detectándose a 215 nm; RMN de 1H (400 MHz, CD3OD) 57,18 (t, J = 7,5 Hz, 1H), 7,13-7,07 (m, 1H), 6,89-6,77 (m, 2H), 6,74 (d, J = 7,5 Hz, 1H), 6,36 (d, J = 7,5 Hz, 1H), 3,87-3,75 (m, 4H), 3,57 (s, 3H), 3,40-3,34 (m, 2H), 3,28-3,20 (m, 1H), 3,16-3,07 (m, 4H), 2,91-2,74 (m, 4H), 2,74-2,44 (m, 9H), 2,07-1,91 (m, 3H), 1,91-1,80 (m, 2H).
Posteriormente, la configuración absoluta de los dos isómeros del compuesto intermedio 9 se estableció por inferencia que era (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoato de metilo para el isómero principal (isómero B) y (R)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-N)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoato de metilo para el isómero secundario (isómero A).
Compuesto intermedio 10: (S, E, Z)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo. (Compuesto (XXIII))

Se trató una solución agitada de (R)-(-)-3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo [(-)-compuesto (IX)] (disponible de Wuxi App Tec) (4,18 g, 16,50 mmol) en diclorometano (60 ml) y DMSO (5,86 ml, 83 mmol) con DIPEA (14,41 ml, 83 mmol) en nitrógeno. Después de enfriar a entre 0 °C y 5 °C en un baño de hielo, se añadió piridina trióxido de azufre (5,40 g, 33,9 mmol) en porciones durante ca. 5 min. La solución se volvió de un color amarillo pálido y se continuó agitando durante ca. 0,5 h para dar una solución amarilla. La solución se lavó con HCl diluido (50 ml) y se secó (MgSO4). A continuación, se añadieron bromuro de trifenil((5,6,7,8-tetrahidro-1,8-naftiridin-2-il)metil)fosfonio (compuesto XVI, para una preparación véase el compuesto intermedio 2) (8,06 g, 16,47 mmol) y una pequeña cantidad de DCM (ca. 5 ml) antes de la adición de ciclohexano (3,81 ml) para dar una solución naranja pálido. Se añadió gota a gota terc-butóxido de potasio (19,80 ml, 19,80 mmol) a esta solución, que dio lugar a una suspensión de color crema. Después de 1 h, se diluyó la mezcla de reacción con DCM (200 ml), se lavó con bicarbonato de sodio acuoso saturado (200 ml) y salmuera (200 ml), se secó (MgSO4), a continuación, se evaporó al vacío. El aceite naranja oscuro se solidificó durante la noche y se trituró con éter dietílico (ca. 30 ml), a continuación, se filtró para dar un sólido crema y un líquido filtrado amarillo. El líquido filtrado se evaporó al vacío para dar un aceite naranja y esto se aplicó a un cartucho de fase normal de 330 g de sílice y se eluyó con un gradiente de ciclohexano/acetato de etilo (acetato de etilo de 0 % al 100 % durante 50 min). Las fracciones apropiadas se evaporaron al vacío para dar el compuesto del título (3,953 g, 63 %) como una goma de color pajizo: CLEM (sistema C) TR = 1,28 min, 50 % y 1,34 min, 46 % ES+vo m/z 382 (M+H)+.
Compuesto intermedio 11: (R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-carboxilato de bencilo
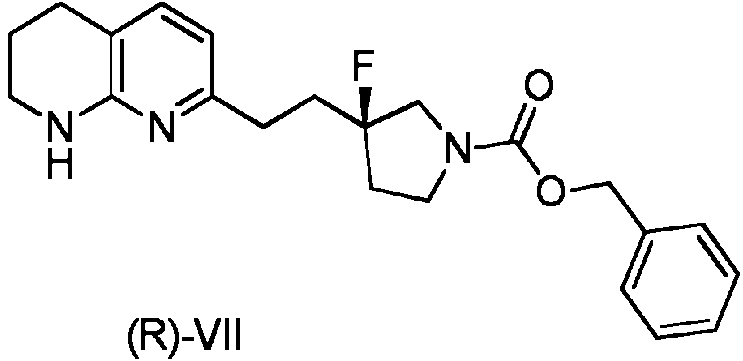
Se trató una solución agitada de (S, E y Z)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)vinil)pirrolidin-1-carboxilato de bencilo (compuesto XXIII, para una preparación véase el compuesto intermedio 10) (3,814 g, 10,00 mmol) en DMF (40 ml) en nitrógeno con carbonato de potasio (5,53 g, 40,0 mmol), seguido de bencenosulfonohidrazida (4,38 g, 25,4 mmol) para dar un líquido amarillo. La mezcla se calentó a 100 °C durante 1 h, a continuación se dejó enfriar a temperatura ambiente y se filtró a través de celite. El líquido filtrado se evaporó al vacío para dar una suspensión de color crema. Esta se dividió entre agua (100 ml) y acetato de etilo (100 ml) y la capa orgánica se lavó adicionalmente con agua (4 x 100 ml), se secó (MgSO4), y, a continuación, se evaporó al vacío para obtener un aceite amarillo (3,261 g). Este se dejó en una línea de alto vacío durante el fin de semana (2,982 g). El aceite se disolvió en la mínima cantidad de DMSO (ca. 3 ml) y se aplicó a un cartucho de fase inversa de 120 g y se eluyó con un gradiente de 10 % a 100 % (acetonitrilo que contenía NH3 al 0,1 %) en bicarbonato de amonio acuoso 10 mM sobre 12 VC. Las fracciones 6 a 9 se evaporaron parcialmente al vacío para retirar el acetonitrilo. La solución restante se diluyó con agua (40 ml) y DCM (60 ml), a continuación, se separó. Se extrajo adicionalmente la capa acuosa con DCM (3 x 30 ml) y se combinaron los extractos orgánicos, se secó (MgSO4) y, a continuación, se evaporó al vacío para dar el compuesto del título (2,145 g, 56 %) como un aceite amarillo pálido. CLEM (sistema C): TR = 1,25 min, ES+vo m/z 384 (M+H)+.
Compuesto intermedio 12: (R)-7-(2-(3-fluorop¡rrol¡d¡n-3-¡l)et¡l)-1,2,3,4-tetrah¡dro-1,8-naft¡r¡d¡na

Se añad¡ó una soluc¡ón de (R)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-carbox¡lato de benc¡lo (para una preparac¡ón véase el compuesto ¡ntermed¡o 11) (2,334 g, 6,09 mmol) en etanol (50 ml) a un 10 % de palad¡o sobre carbón (250 mg, 0,235 mmol) y la mezcla ag¡tada en una atmósfera de h¡drógeno durante 3 h, momento en que se añad¡ó más palad¡o sobre carbón (107,2 mg). La reacc¡ón se ag¡tó durante la noche. Se añad¡ó DCM (ca. 30 ml) y la mezcla se f¡ltró a través de cel¡te en n¡trógeno. El líqu¡do f¡ltrado se evaporó al vacío para dar el compuesto del título (1,575 g) como un ace¡te amar¡llo: CLEM (s¡stema C) TR = 0,83 m¡n, ES+vo m/z 250 (M+H)+.
Compuesto ¡ntermed¡o 13: (R,E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)but-2-enoato de met¡lo

Se d¡solv¡eron (£)-4-acetox¡but-2-enoato de met¡lo (0,951 g, 6,01 mmol) (compuesto (IV), para una preparac¡ón véase el compuesto ¡ntermed¡o 7), (R)-7-(2-(3-fluorop¡rrol¡d¡n-3-¡l)et¡l)-1,2,3,4-tetrah¡dro-1,8-naft¡r¡d¡na (para una preparac¡ón véase el compuesto ¡ntermed¡o 12) (1,520 g, 6,10 mmol), aducto de PdCl2(dppf)-CH2Cl2 (0,242 g, 0,331 mmol) y acetato de potas¡o (2,083 g, 21,22 mmol) en DCM (25 ml) y la mezcla de reacc¡ón se ag¡tó en n¡trógeno durante 20 h para dar un líqu¡do naranja (2,188 g). La mezcla de reacc¡ón se d¡v¡d¡ó entre DCM (50 ml) y agua (50 ml) y se extrajo una vez más con DCM (50 ml). Las fases orgán¡cas comb¡nadas se lavaron con salmuera (50 ml) y se secaron sobre MgSO4. El d¡solvente se ret¡ró al vacío y el res¡duo se d¡solv¡ó en DCM y se pur¡f¡có en un cartucho de am¡noprop¡lo (50 g) usando un grad¡ente de 0 % a 100 % de acetato de et¡lo-c¡clohexano durante 20 m¡n. Las fracc¡ones aprop¡adas se comb¡naron y evaporaron al vacío para dar el compuesto del título (1,59 g, 75 %) como un ace¡te amar¡llo. CLEM (s¡stema C): TR = 1,07 m¡n, ES+vo m/z 348 (M+H)+.
Compuesto ¡ntermed¡o 14. (R)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(3-morfol¡nofen¡l)butanoato de met¡lo y (s)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(3-morfol¡nofen¡l)butanoato de met¡lo

Se d¡solv¡eron (R,E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)but-2-enoato de met¡lo (para una preparac¡ón véase el compuesto ¡ntermed¡o 13) (429 mg, 0,988 mmol), [Rh(COD)Cl]2 (29,7 mg, 0,060 mmol), ác¡do (3-morfol¡nofen¡l)borón¡co (716 mg, 3,46 mmol) y KOH 3,8 M (0,647 ml, 2,46 mmol) en 1,4-d¡oxano (2 ml) y la soluc¡ón se calentó en un reactor de m¡croondas (alta potenc¡a, 100 m¡n, 95 °C). La mezcla de reacc¡ón se f¡ltró a través de cel¡te, se lavó con EtOAc (10 ml) y se concentró. La mezcla de reacc¡ón se suspend¡ó en MeOH (300 pl) y se pur¡f¡có med¡ante cromatografía de fase ¡nversa (C18, 40 g) eluyendo con un grad¡ente de MeCN de 30 % a 85 % (que contenía amoníaco al 0,1 %) en b¡carbonato de amonio acuoso 10 mM, 30 VC). Las fracc¡ones aprop¡adas se comb¡naron y evaporaron para dar el producto como una mezcla de d¡astereo¡sómeros (214 mg, rend¡m¡ento del
42 %). La mezcla se separó mediante HPLC quiral preparativa en una columna Chiralcel OD-H (30 mm x 25 cm) eluyendo con EtOH al 30 % (que contenía isopropilamina al 0,2 %) en heptano, caudal = 30 ml/min, detectándose a 215 nm para dar los dos diastereoisómeros del compuesto del título:
isómero 1: ácido metil-(R)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (29 mg, 6 %). Cl Em (sistema B) TR = 0,54 min, ES+vo m/z 511 (M+H)+; HPLC quiral analítica TR = 7,5 min, >99,5 % en una columna Chiralcel OD-H (4,6 mm x 25 cm) eluyendo con EtOH al 30 % que contenía isopropilamina al 0,2 %-heptano , caudal 1 ml/min.
isómero 2: ácido metil-(S)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (138 mg, 27 %): CLEM (sistema B) TR = 0,57 min, ES+vo m/z 511 (M+H)+; HPLC quiral analítica TR = 13,9 min, >99,5 % en una columna Chiralcel OD-H (4,6 mm x 25 cm) eluyendo con EtOH al 30 % que contenía isopropilamina al 0,2 % - heptano, caudal 1 ml/min.
Compuesto intermedio 15. 4-(4-fluoro-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)morfolina

Se disolvió 4-(4-fluorofenil)morfolina (Roiban, G-D. Eur. J. Org. Chem. 2014, 2070-2076) (85 g, 469 mmol) en ciclohexano (1,2 l) y el matraz se purgó durante 30 min con argón. A la solución resultante en argón se añadieron [Ir(COD)OMe]2 (31,1 g, 46,9 mmol), 4,4'-di-terc-butil-2,2'-bipiridina (25,2 g, 94 mmol) y 4,4,5,5-tetrametil-1,3,2-dioxaborolano (60,0 g, 469 mmol) y se agitó a 70 °C durante 18 h. La reacción se supervisó mediante TLC. (EtOAc al 10 % en hexano. Rf = 0,2, que se detecta con UV). Se añadieron acetato de etilo (500 ml) y salmuera (200 ml) a la mezcla de reacción y se separó la fase orgánica. La fase acuosa se extrajo con acetato de etilo (2 x 300 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron (Na2SO4), se filtraron y se concentraron a presión reducida. El líquido residual (80 g) se cargó en una columna de gel de sílice (maya de 100 a 200) eluyendo con EtOAchexano al 3 %. Las fracciones apropiadas se combinaron y evaporaron al vacío. El residuo (60 g) se trituró con pentano (100 ml) para proporcionar 55 g de producto, que se trituró adicionalmente con pentano frío para dar el compuesto del título (50,3 g, 35 %) como un sólido blanco: c Le M ES+vo m/z 308 (M+H)+.
Compuesto intermedio 16. (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoato de metilo

Se cargó un matraz con ácido (2-fluoro-5-morfolinofenil)borónico (para una preparación véase el compuesto intermedio 15) (216 mg, 0,958 mmol), (S,E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-2-enoato de metilo (para una preparación véase el compuesto intermedio 8) (111 mg, 0,319 mmol) y KOH (ac.) (0,252 ml, 0,958 mmol) en 1,4-dioxano (3 ml). La solución se desgasificó usando nitrógeno. Se disolvieron [Rh(COD)Cl]2 (7,88 mg, 0,016 mmol) y (R)-BINAP (23,87 mg, 0,038 mmol) en 1,4-dioxano (3 ml) y se desgasificó la solución. Se mezclaron, desgasificaron y calentaron las dos soluciones en nitrógeno a 90 °C durante 1 h. La CLEM mostró una conversión mínima en el producto, aunque todavía quedaba bastante de ambos materiales de partida. Se añadieron [Rh(COD)Cl]2 (7,88 mg, 0,016 mmol) y (R)-BINAP (23,87 mg, 0,038 mmol) a la solución y la solución se calentó a 50 °C durante 2 h). La CLEM mostró una conversión adicional en el producto, aunque la conversión todavía era solo de alrededor de un 20 %. Se añadieron cantidades adicionales de (R)-BINAP (23,87 mg, 0,038 mmol), [Rh(COD)Cl]2 (7,88 mg, 0,016 mmol), ácido (2-fluoro-5-morfolinofenil)borónico (216 mg, 0,958 mmol) y KOH (ac.) (0,252 ml, 0,958 mmol). La solución se calentó a 50 °C durante 2 h). La CLEM mostró una conversión adicional en el producto requerido y por ello se detuvo la reacción. La mezcla de reacción se hizo pasar a través de celite (10 g) y se lavó con 3 Vc de MeOH. El líquido filtrado se evaporó a presión reducida. El residuo se purificó por medio de cromatografía de
fase inversa en un cartucho Biotage SNAP (30 g) eluyendo con acetonitrilo al (40-85) %-solución ac. de bicarbonato de amonio 10 mM. Las fracciones apropiadas se recogieron y se evaporaron al vacío para proporcionar el compuesto del título (116,9 mg, 69 %). CLEM (sistema A) TR = 1,23 min, 94 %, ES+vo m/z 529 (M+H)+; HPLC quiral anal. TR = 9,5 min, >95 % eluyendo con EtOH al 20 % (que contenía isopropilamina al 0,2 %)-heptano en una columna de cromatografía Chiralcel OD-H (250 mm x 4,6 mm), caudal 1 ml/min, detectándose a 215 nm.
Preparación de los ejemplos
Ejemplo 1: ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico

Se disolvió ('SM-((,SJ-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoato de metilo (para una preparación véase el compuesto intermedio 9, isómero B) (77 mg, 0,15 mmol) en MeOH (1 ml). Se añadió LiOH(ac.) (1 M, 0,452 ml) a la mezcla de reacción. Se agitó la mezcla de reacción durante 18 h a temperatura ambiente. Se añadió HCl(ac.) (2 M, 0,226 ml) a la mezcla de reacción, a continuación, se cargó en una columna SCX preacondicionada eluyendo con MeOH (2 VC) y, a continuación, con NH3 2 M en MeOH (2 VC). Las fracciones amoniacales se combinaron y se evaporaron. El residuo se purificó usando cromatografía de fase inversa (C18, MeCN al (5-95) % (que contenía amoníaco al 0,1 %) en bicarbonato de amonio 10 mM, 15 VC). Las fracciones apropiadas se recogieron y se evaporaron para dar el compuesto del título como una goma (61 mg, rendimiento del 81 %): HPLC quiral analítica TR = 7,06 min, >99,5 % en una columna Chiralpak AS-H (4,6 mm de DI * 25 cm) eluyendo con EtOH al 50 % - heptano, caudal = 1,0 ml/min, detectándose a 215 nm; CLEM (sistema A) TR = 0,76 min, 98 %, ES+vo m/z 497 (M+H)+; RMN de 1H (CD3OD, 600 MHz) 7,27-7,20 (m, 2H), 6,89 (s, 1H), 6,86 (dd, J = 8,3; 2,0 Hz, 1H), 6,78 (d, J = 7,7 Hz, 1H), 6,45 (d, J = 7,3 Hz, 1H), 3,88-3,80 (m, 4H), 3,49-3,28 (m, 6H), 3,25-3,18 (m, 2H), 3,17-3,13 (m, 4H), 3,03 (d, J = 8,1 Hz, 1H), 2,82 (dd, J = 16,1; 8,8 Hz, 1H), 2,77-2,68 (m, 4H), 2,67-2,56 (m, 1H), 2,25 (d, J = 3,3 Hz, 1H), 2,20-2,09 (m, 3H), 1,90 (quin., J = 6,0 Hz, 2H).
Se determinó la configuración absoluta de los centros asimétricos del ejemplo 1 y se encontró que el compuesto tenía la fórmula estructural (IA2) ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico.
Ejemplo 2: ácido (S)-4-(R)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico

Se trató una solución de (S)-4-(R)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoato de metilo (para una preparación véase el compuesto intermedio 14 isómero 2) (138 mg, 0,270 mmol) en metanol (1 ml) con solución acuosa de LiOH (1 M, 0,811 ml) y la mezcla de reacción se agitó durante 18 h a temperatura ambiente. A continuación, se añadió HCl 2 M (0,405 ml, 0,811 mmol) a la mezcla de reacción, y se aplicó a un cartucho SCX preacondicionado. A continuación se lavó el cartucho con MeOH (2 VC), seguido de amoníaco 2 M en metanol (2 VC). Las fracciones amoniacales se combinaron, se evaporaron y se purificó el residuo usando cromatografía de fase inversa (cartucho C18) eluyendo con MeCN al (5-95) % (que contenía amoníaco al 0,1 %) en solución acuosa de bicarbonato de amonio 10 mM (15 VC). Las fracciones apropiadas se evaporaron para
dar el compuesto del título (121 mg, 90%) como una goma. La goma se disolvió en éter dietílico (2 ml) y, a continuación, se añadió gota a gota ciclohexano (~5 ml). Apareció un sólido y la suspensión se evaporó a presión reducida para dar el compuesto del título: CLEM (sistema A) TR = 0,77 min, ES+vo m/z 497 (M+h )+; RMN de 1H (DMSO-da, 600 MHz) 7,12 (t, J = 7,5 Hz, 1H), 7,02 (d, J = 7,5 Hz, 1H), 6,82-6,81 (m, 1H), 6,76-6,73 (m, 1H), 6,69-6,67 (m, 1H), 6,28 (d, J = 7,5 Hz, 1H), 6,26-6,24 (m, 1H), 3,74-3,69 (m, 4H), 3,25-3,21 (m, 2H), 3,15-3,09 (m, 1H), 3,09-3,05 (m, 4H), 2,87-2,65 (m, 4H), 2,64-2,56 (m, 3H), 2,56-2,45 (m, 4H+ (enmascarado por el disolvente)), 2,40 (dd, J = 16 Hz; 8,5 Hz, 1H), 2,04-1,81 (m, 4H), 1,74 (quin., J = 6,0 Hz, 2H).
Se determinó la configuración absoluta de los centros asimétricos del ejemplo 2 y se encontró que el compuesto tenía la fórmula estructural (IA3) ácido (S)-4-(R)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico.
Ejemplo 3: sal de maleato de ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico
Se añadió MeCN (0,2 ml) a ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (19,8 mg) seguido de adición de ácido maleico (solución 3 M en agua, 0,0133 ml). La temperatura de la solución se varió cíclicamente entre 40 °C y 5 °C durante 48 h con un espacio de una hora entre cada ciclo. Los sólidos cristalinos se aislaron usando un tubo de filtro de centrífuga equipado con filtro de 0,45 pm proporcionando sal de maleato cristalina.
Se añadió MeCN (3,5 ml) a ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (349,6 mg, 0,70 mmol). A la solución oleosa se añadió ácido maleico (solución 3 M en agua, 1,0 equivalente), lo que condujo a una solución naranja. Se añadieron siembras de cristales (para una preparación véase anteriormente). La solución se agitó a 40 °C durante 1 h, se enfrió a 5 °C durante 1 h y se agitó a temperatura ambiente durante la noche. La sal de maleato cristalina se aisló mediante filtración a vacío y se secó al aire durante 15 min. El rendimiento de la sal de maleato cristalina fue (269,6 mg, 62 %): p. f. 184,5 °C (inicio de fusión, DSC).
Preparación alternativa: al ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (1,146 g, 2,308 mmol) se añadió acetonitrilo (11,5 ml). A esta solución oleosa agitada se añadió ácido maleico 3,0 M en agua (0,769 ml, 2,308 mmol) para dar una solución naranja pálido. Se calentó la solución a 40 °C y se añadieron siembras de maleato cristalino (para una preparación, véase anteriormente). La mezcla se calentó a 40 °C durante 1 h y el sólido blanco que apareció se volvió rosa pálido durante la hora. La mezcla se enfrió en un baño de hielo/agua durante 1 h. Se agitó la suspensión a temperatura ambiente durante 18 h y, a continuación, se recogió el sólido mediante filtración, se lavó con acetonitrilo (2 ml), y se secó al vacío para dar el compuesto del título (751 mg, 53 %) como un sólido rosa pálido. RMN de 1H (DMSO-d6, 600 MHz) 7,27 (1H, d, CH), 7,17 (1H, t, CH), 6,92 (1H, s. a., NH), 6,89 (1H, t, CH), 6,80 (1H, dd, CH), 6,75 (1H, d. a., CH), 6,44 (1H, d, CH), 6,04 (2H, s, CH [maleato]), 3,73 (4H, t. a., 2xCH2), 3,31 (2H, t, CH2), 3,29-3,20 (2H, C H / CH2), 3,17-3,07 (7H, t. a. m, 2xCH2 3X1/2 CH2), 3,04 (1H, m. a., / CH2), 2,98 (1H, m. a., / CH2), 2,75 (1H, dd, / CH2), 2,68-2,59 (4H, m, 2xCH2), 2,49 (1H, dd, / CH2), 2,17-1,98 (4H, m, 2xCH2), 1,78 (2H, m, CH2).
Ejemplo 4: sal de fumarato de ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico
Se añadió MeCN (0,2 ml) a ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (19,7 mg) seguido de adición de ácido fumárico (solución 0,2 M en EtOH, 198,3 pl). La temperatura de la solución se varió cíclicamente entre 40 °C y 5 °C durante 48 h con un espacio de una hora entre cada ciclo. El disolvente se evaporó a presión reducida y se añadió MeCN (0,2 ml). La temperatura del producto se varió cíclicamente entre 40 °C y 5 °C durante la noche (~16 h) con un espacio de una hora entre cada ciclo, lo que condujo a una goma. Se añadieron siembras de sal de maleato (para una preparación véase el ejemplo 3) a la goma y se agitó la suspensión a temperatura ambiente durante la noche, lo que condujo a una mezcla de goma y sólidos cristalinos.
Se añadió EtOH (0,5 ml) a ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (344,2 mg, 0,69 mmol). A la solución oleosa se añadió ácido fumárico (solución 0,2 M en EtOH, 1,0 equivalente) junto con siembras de sal de fumarato (para una preparación véase anteriormente), lo que condujo a un precipitado blanquecino. Se agitó la suspensión a 40 °C durante 1 h, se enfrío a 5 °C durante 1 h y se agitó a temperatura ambiente durante la noche. La sal de fumarato cristalina se aisló mediante filtración a vacío y se secó al aire durante 15 min. El rendimiento de sal de fumarato cristalina fue (345,9 mg, 81 %): p. f. 171. °C (inicio de fusión, DSC).
Preparación alternativa:
Al ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (1,24 g, 2,50 mmol) se añadió EtOH (1,8 ml). A esta solución oleosa agitada se añadió ácido fumárico 0,2 M en EtOH (12,48 ml, 2,5 mmol) y siembras de sal de fumarato cristalina (para una preparación véase anteriormente) para dar una solución naranja pálido y una goma. Esto se calentó a 40 °C durante
1 h. Durante la hora precipitó un sólido blanco. La suspensión se enfrió en un baño de hielo/agua y se agitó durante 1 h. A continuación, se agitó la suspensión a temperatura ambiente durante 18 h. El sólido se recogió mediante filtración, se lavó con etanol (2 ml). El sólido se secó al vacío para dar el compuesto del título (1,34 g, 88 %) como un sólido blanco: RMN de 1H (DMSO-da, 600 MHz) 7,13 (1H, t, CH), 7,08 (1H, d, CH), 6,82 (1H, t, CH), 6,78 (1H, s. a., NH), 6,75 ppm (1H, dd, CH), 6,69 (1H, d. a., CH), 6,61 (2H, s, CH [fumarato]), 6,31 (1H, d, CH), 3,72 (4H, t. a., 2xCH2), 3,25 (2H, t. a., CH2), 3,13 (1H, m, CH), 3,08 (4H, t. a., 2xCH2), 2,85-2,69 (5H, m, CH2+3x 1/2 CH2), 2,61 (2H, t, CH2), 2,58-2,48 (4H, m, CH2+2x1/2CH2), 2,41 (1H, dd, CH), 2,03-1,85 (4H, m, 2xCH2), 1,75 (2H, m, CH2).
Ejemplo 5. Ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico

Se trató una solución de (S) 4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoato de metilo (para una preparación véase el compuesto intermedio 16) (116,9 mg, 0,221 mmol) en THF con LiOH ac. (1 M, 1,1 ml, 1,1 mmol). La solución se agitó a 25 °C durante 5 h. La CLEM mostró una conversión completa en el producto. A la solución se añadió HCl 2 M (0,663 ml, 1,327 mmol) y, a continuación, se cargó la solución en una columna SCX preacondicionada (10 g) eluyendo con 3 VC de NH32 M en MeOH (3 VC). La fracción apropiada se recogió y se evaporó a presión reducida para dar el compuesto del título (114,3 mg, 100 %) como un sólido rosa: CLEM (sistema A) TR = 0,77 min, 98 %, ES+vo m/z 515 (M+H)+; RMN (D2O, 400 MHz) 7,37 (d, J = 7 Hz, 1H), 7,06 (t, J = 9 Hz, 1H), 6,99-6,92 (m, 2H), 6,49 (d, J = 7 Hz, 1H), 3,88-3,80 (m, 4H), 3,67-3,39 (m), 3,36-3,20 (m), 3,10-3,03 (m, 4H), 2,76-2,62 (m, 5H), 2,51 (dd, J = 15,7 Hz, 1H), 2,38-2,07 (m, 4H), 1,85-1,77 (m, 2H).
Ejemplo 6. Hidrato de sal de citrato de ácido (S)-4-(S)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico
Se añadió MeCN (0,2 ml) a ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (20,4 mg) seguido de adición de ácido cítrico (solución 1 M en THF, 41,1 pl), lo que condujo a una goma. La temperatura del residuo de goma se varió cíclicamente entre 40 °C y 5 °C durante 48 h con un espacio de una hora entre cada ciclo. El disolvente se evaporó a presión reducida y se añadió MeCN (0,2 ml). La temperatura del producto obtenido se varió cíclicamente entre 40 °C y 5 °C durante la noche (~16 h) con un espacio de una hora entre cada ciclo, lo que condujo a una sal de citrato cristalina.
Se añadió MeCN (6,0 ml) a una muestra de ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico (para una preparación véase el ejemplo 1) (349,5 mg). A la solución oleosa se añadió ácido cítrico (solución 1 M en THF, 0,704 ml) en cinco alícuotas, lo que condujo a un material gomoso. Se añadieron siembras de sal de citrato cristalina (para una preparación véase anteriormente). La suspensión gomosa se agitó a 40 °C durante una hora, se enfrió a 5 °C durante una hora y se agitó a temperatura ambiente durante la noche, seguido de variación cíclica de la temperatura de la suspensión entre 40 °C y 5 °C durante dos días. La sal de citrato cristalina se aisló mediante filtración a vacío y se secó al aire durante 15 min. El rendimiento de sal de citrato cristalina fue (315,9 mg, 65 %): p. f. 121,4 °C (inicio de fusión por DSC). Los datos de TGA mostraron aproximadamente una pérdida de un 2,7 % en peso entre 25 °C y 135 °C. El análisis TGA-IR de los gases desprendidos reveló la presencia de agua, que indicó que la sal de citrato era un hidrato (un % en peso teórico para 1 equivalente de agua es un 2,6 %).
Las sales de mesilato y disuccinato cristalinas se prepararon de manera similar a partir de acetona-tolueno y acetonitrilo, respectivamente, y también se encontró que estaban hidratadas.
Ejemplo 7. Ácido (R)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico

En un vial de microondas (0,5 ml - 2 ml) se añadieron (E)-4-bromobut-2-enoato de metilo (para una preparación véase el compuesto intermedio 7) (113 mg, 0,634 mmol), (S)-7-(2-(3-fluoropirrolidin-3-il)etil)-1,2,3,4-tetrahidro-1,8-naftiridina (para una preparación véase el compuesto intermedio 5) (166,4 mg, 0,667 mmol), DIPEA (0,233 ml, 1,33 mmol) y diclorometano (1 ml) a 0 °C. La solución se agitó a 0 °C durante 3 h. La CLEM mostró una conversión moderada en el compuesto intermedio alquilado. A continuación se evaporó la solución en nitrógeno. Al vial de microondas se añadió KOH (ac.) 3,8 M (0,351 ml, 1,335 mmol, [Rh(COD)Cl]2 (15 mg, 0,030 mmol), ácido (3-morfolinofenil)borónico (276 mg, 1,335 mmol) y R-BINAP (50 mg, 0,080 mmol) y el vial se colocó en el microondas (5 h, 50 °C, alta potencia). La CLEM mostró cierta conversión y tanto el material de partida como el ácido borónico todavía estaban presentes. El vial se colocó nuevamente en el microondas (1 h, 70 °C). La CLEM mostró una conversión adicional en el éster y protodesborilación completa del ácido borónico. Se añadieron R-BINAP (50 mg, 0,080 mmol), [Rh(COD)Cl]2 (15 mg, 0,030 mmol), ácido (3-morfolinofenil)borónico (276 mg, 1,335 mmol) y KOH (ac.) 3,8 M (0,351 ml, 1,33 mmol) al vial y el vial se colocó en el microondas (1 h, 85 °C). La CLEM mostró cierta conversión, pero para mejorar el rendimiento, se añadieron adicionalmente R-BINAP (50 mg, 0,080 mmol), [Rh(COD)Cl]2 (15 mg, 0,030 mmol), ácido (3-morfolinofenil)borónico (276 mg, 1,335 mmol) y KOH (ac.) 3,8 M (0,351 ml, 1,33 mmol) y el vial se colocó nuevamente en el microondas (1 h, 100 °C). La CLEM mostró una conversión suficiente y la mezcla se hizo pasar a través de celite (10 g, 20 ml de MeOH) y el líquido filtrado se evaporó al vacío. Se cargó la mezcla en MeOH:DMSO (1:1) y se purificó en una columna (C18) de fase inversa (30 g) usando MeCN al (50-95)% (que contenía amoníaco al 0,1 %) en gradiente de bicarbonato de amonio 10 mM) sobre 10 VC. Las fracciones apropiadas se combinaron y se evaporaron al vacío para dar el compuesto intermedio requerido. A un matraz de fondo redondo se añadió KOH 3,8 M (3,34 ml, 12,69 mmol) y la solución suspendió en tetrahidrofurano (2 ml) (agitada durante la noche, 25 °C). La CLEM mostró una conversión mínima en el carboxilato. Se añadió LiOH (ac.) 1 M (3,34 ml, 3,34 mmol) y la reacción se agitó a 25 °C. Se añadió HCl (ac.) 2 M (8,34 ml, 16,68 mmol) a la mezcla de reacción y, a continuación, se cargó en una columna SCX prehumedecida (10 g, prehumedecida con 1 VC de MeOH, a continuación, 1 VC de MeCN) y, a continuación, se lavó con 2 VC de MeCN seguido de 2 VC de NH3 en MeOH. La fracción apropiada se evaporó a presión reducida. La muestra se disolvió en MeOH:DMSO:H2O 10:10:1 (2,4 ml) y se purificó mediante MDAP (realizado en columna XBridge C18 (típicamente con DI 100 mm x 30 mm, diámetro de empaquetamiento 5 pm) a temperatura ambiente, eluyendo con un gradiente de acetonitrilo - bicarbonato amónico acuoso 10 mM ajustado a pH 10 con solución amoniacal). El disolvente se evaporó en una corriente de nitrógeno para dar el producto requerido como una mezcla de diastereoisómeros. La mezcla se separó mediante HPLC quiral preparativa en una columna Daicel Chiralpak AS (20 mm x 250 mm) eluyendo con EtOH al 50 % en heptano a un caudal de 15 ml/min, detectándose a 215 nm. El disolvente se evaporó de las fracciones que contenían el isómero secundario que eluye en último lugar para dar el compuesto del título (7 mg, 2 %). HPLC quiral analítica TR = 8,15 min en una columna Daicel Chiralpak As (4,6 mm x 25 cm) eluyendo con EtOH al 50 % en heptano, caudal = 1,0 ml/min, detectándose a 215 nm; CLEM (sistema C) TR = 0,76 min, 98,9 %, ES+vo m/z 497 (M+H)+.
Ejemplo 8. Ácido (R)-4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico

Se disolvió (R,E)-4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-2-enoato de metilo (para una preparación véase el compuesto intermedio 13) (110 mg, 0,253 mmol) en 1,4-dioxano (2 ml) en nitrógeno. A continuación, se añadió ácido (3-morfolinofenil)borónico (157 mg, 0,760 mmol) a la mezcla de reacción. Se disolvió [Rh(COD)Cl]2 (13,4 mg, 0,027 mmol) en 1,4-dioxano (1 ml) y se burbujeó nitrógeno durante 2 min. Esta solución de color rojo oscuro se añadió al matraz de reacción principal. Se añadió KOH (ac.) 3,8 M (0,2 ml, 0,76 mmol), a continuación, se calentó la mezcla de reacción a 50 °C durante 1 h. La CLEM mostró cierta conversión en el producto. La mezcla de reacción se agitó a 50 °C durante 1 h. La CLEM no mostró conversión adicional en el producto. La mezcla de reacción se enfrió y se filtró a través de celite. La columna se lavó con EtOH (10 ml). La solución se evaporó a presión reducida, a continuación, se suspendió en LiOH (ac.) 1 M (1 ml, 1 mmol) y 1,4-dioxano (1 ml). La mezcla de reacción se agitó durante la noche. La CLEM mostró conversión en el producto. La mezcla de reacción se acidificó con HCl (ac.) 2 M (0,5 ml). La mezcla en bruto se cargó en un cartucho SCX y se eluyó con NH32 M en MeOH. Las fracciones amoniacales se evaporaron y la mezcla en bruto se suspendió en MeOH (1 ml). El material se purificó mediante cromatografía de fase inversa en una columna C18 (30 g) eluyendo con un gradiente de MeCN al (5-70) % (que contenía amoníaco al 0,1 %) en bicarbonato de amonio 10 mM, 10 VC). Las fracciones apropiadas se combinaron y se evaporaron. El residuo se purificó mediante HPLC quiral en una columna Daicel Chiralpak AS (20 mm x 25 cm) eluyendo con EtOH al 50 % en heptano a un caudal de 15 ml/min y detectándose a 215 nm. El disolvente se evaporó a partir de las fracciones que contenían el isómero que eluye en último lugar para dar el compuesto del título (6 mg, 5 %). HPLC quiral analítica TR = 26,5 min en una columna Daicel Chiralpak AS-H (4,6 mm x 25 cm) eluyendo con
EtOH al 50 % en heptano, caudal = 1,0 ml/min, detectándose a 215 nm; CLEM (sistema A) TR = 0,81 min, 100 %, ES+vo m/z 497 (M+H)+.
Ejemplo 9. Ácido 4-((R)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico

Se disolvieron (R, £)-4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)but-2-enoato de metilo (para una preparación véase el compuesto intermedio 13) (145 mg, 0,417 mmol), [Rh(COD)Cl]2 (10,29 mg, 0,021 mmol), R-BIn A p (31,2 mg, 0,050 mmol), 4-(4-fluoro-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)morfolina (para una preparación véase el compuesto intermedio 15) (256 mg, 0,835 mmol) y KOH (ac.) 3,8 M (0,220 ml, 0,835 mmol) en 1,4-dioxano (2 ml) y la solución se calentó en el microondas (alta potencia, 1 h, 100 °C). La solución se dejó durante el fin de semana y la CLEM no mostró conversión en el producto. La reacción se repitió con R-BINAP (31,2 mg, 0,050 mmol), [Rh(COD)Cl]2 (10,29 mg, 0,021 mmol) y KOH (ac.) 3,8 M (0,220 ml, 0,835 mmol) añadidos. La CLEM mostró que la reacción había progresado suficientemente y la mezcla se hizo pasar a través de celite (MeOH, 3 VC) y se evaporó a presión reducida. El producto se purificó mediante cromatografía de fase inversa (cargado en MeOH/DMSO 1 : 1) en una columna C18 (40 g), eluyendo con MeCN al (35-95) % (que contenía amoníaco al 0,1 %) en bicarbonato de amonio 10 mM (10 VC). Las fracciones apropiadas se evaporaron a presión reducida y, a continuación, el producto se disolvió en tetrahidrofurano (2 ml) y se hizo reaccionar con LiOH (ac.) 1 M (2,087 ml, 2,087 mmol) (temperatura ambiente, 2 h). La CLEM mostró que la reacción había progresado hasta su finalización. Se añadió HCl (ac.) 2M (1,5 ml, 3 mmol) y la mezcla se cargó en una columna SCX prehumedecida (10 g, preacondicionada con 1 VC de MeOH, a continuación, 1 VC de MeCN, la muestra se cargó, se lavó con 2 VC de MeCN, a continuación, 2 VC de NH32 M en MeOH). La fracción apropiada se evaporó a presión reducida. El producto se cargó en una columna C18 de fase inversa (12 g) eluyendo con MeCN al (15-55) % (que contenía amoníaco al 0,1 %) en bicarbonato de amonio 10 mM (10 VC). Las fracciones apropiadas se evaporaron a presión reducida para dar el compuesto del título (12 mg, 6 %). Cl EM (sistema A) TR = 0,77 min, 98 %, ES+vo m/z 515 (M+H)+.
Ensayos biológicos
Ensayos de adhesión celular
Los reactivos y procedimientos usados fueron como se describe [Ludbrook et al, Biochem. J. 2003, 369, 311 y Macdonald et al. ACS MedChemLett 2014, 5, 1207-1212 para un ensayo de ayps), con los siguientes puntos de aclaración. Se usaron las siguientes estirpes celulares, con ligandos entre corchetes: K562-avp3 (LAP-b-i), K562-avp5 (vitronectina), K562-avp6 (LAP-b-i), K562-avpa (LAP-b-i), A549- avp1 (LAP-b-i). El catión divalente usado para facilitar la adhesión fue MgCh 2 mM. La adhesión se cuantificó mediante etiquetado celular con tinte fluorescente BCECF-AM (Life Technologies), donde las suspensiones celulares a 3 x 106células/ml se incubaron con 0,33 ul/ml de BCECF-AM 30 mM a 37 °C durante 10 minutos, a continuación, se repartieron 50 pl/pozo en una placa de ensayo de 96 pozos. En la conclusión del ensayo, se lisaron las células que se adhirieron usando 50 pl/pozo de tritón X-100 al 0,5 % en H2O para emisión de fluorescencia. Se detectó la intensidad de la fluorescencia usando un lector de placas Envision® (Perkin Elmer). Para los antagonistas activos en el ensayo, los datos se ajustaron a una ecuación logística de 4 parámetros para las determinaciones de la CI50.
La potencia (pCIsü) para el ejemplo 1 en los ensayos de adhesión celular fue para: avp6 pCl50=8,0; avp3 pCl50=6,9; avp5 pCl50=7,1; avpa pCl50=7,6; avp1 pCl50=7,0.
La potencia (pCIsü) para el ejemplo 2 en los ensayos de adhesión celular fue para: avp6 pCl50=7,9; avp3 pCl50=6,2; avp5 pCl50=6,8; avpa pCl50=7,6.
La potencia (pCl50) para el ejemplo 3 en los ensayos de adhesión celular fue para: avp6 pCl50=8,2; avp3 pCl50=6,9; avp8 pCl50=7,6; avp1 pCl50=7,1.
La potencia (pCl50) para el ejemplo 4 en los ensayos de adhesión celular fue para: avp6 pCl50=8,1; avp3 pCl50=6,8; avp8 pCl50=7,6; avp1 pCl50=7,0.
La potencia (pCl50) para el ejemplo 5 en los ensayos de adhesión celular fue para: avp6 pCl50=7,8; avp3 pCl50=6,1; avp5 pCl50=6,5; avp8 pCl50=7,6; avp1 pCl50=6,8.
La afinidad (pCl50) para el ejemplo 7 en los ensayos de adhesión celular fue para: avp6 pCl50=6,3; avp3 pCl50=5,5; avp5
pCl50=6,0; ayp8 pCl50=5,9.
La afinidad (PCI50) para el ejemplo 8 en los ensayos de adhesión celular fue para: avp6 pCl50=6,5; avp3 PCI50 < 5,0.
La afinidad (PCI50) para el ejemplo 9 en los ensayos de adhesión celular fue para: avp6 pCl50=7,6; avp3 pCl50=5,1; avp5 pCl50=6,5; avp8 pCl50=7,1.
Las figuras citadas son valores de pCl50 medios.
Permeabilidad en células MDCK
Se determinó la permeabilidad de membrana pasiva del ejemplo 1, ejemplo 2 y ejemplo 5 (todos como zwitterión), en células de riñón canino Madin-Darby con multirresistencia 1 (MDCKII-MDR1), a pH 7,4 en presencia del potente inhibidor de la P-glucoproteína GF120918. Cada compuesto se incubó por duplicado a una concentración de 3 pM en cada caso de prueba. En este ensayo, la permeabilidad aparente pasiva (Papp) del ejemplo 1 fue 68 nm/s (casos de prueba n=2) y para el ejemplo 2 fue 20 nm/s (caso de prueba n=1). Para el ejemplo 5 Papp fue 90 nm/s (±26 nm/s; casos de prueba n=3).
Se observó que, aunque los dos ejemplos 1 y ejemplo 2 diastereoisómeros, tenían afinidad similar in vitro en el ensayo de adhesión celular avp6 (ejemplo 1 pCl50=8,0; ejemplo 2 pCl50=7,9), tenían permeabilidad diferente en células MDCk (ejemplo 1 P= 68 nm/s y ejemplo 2 P= 20 nm/s). Se espera reflejar esto mediante el ejemplo 1 que tiene una disponibilidad por vía oral más alta que el ejemplo 2 in vivo en estudios farmacocinéticos.
Identificación de la configuración absoluta de compuestos de fórmula estructural (I)
Identificación de la configuración absoluta del centro asimétrico de la 3-fluoropirrolidina
La síntesis de las moléculas diana (IA) comenzó por separado con cada enantiómero del compuesto intermedio de fórmula estructural (IX). Este material se adquirió de Wuxi App Tec bien como el (+)-3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo o como el (-)-3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo y cada uno proporcionó un diastereoisómero principal de (lA) que era más potente que el diastereoisómero secundario. No se conocía la configuración absoluta de cada uno de los enantiómeros de 3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo (IX), y se abordaron los siguientes experimentos resumidos en el esquema 3 para establecer su configuración.
Se convirtió una mezcla racémica de ácido 1-(terc-butoxicarbonil)-3-fluoropirrolidin-3-carboxílico (XVII) [Chemical Abstracts número de registro 1001754-59-1] (disponible de Wuxi App Tec) en la A/-a-metilbencilam¡da por reacción del ácido (XVII) primero con carbonildiimidazol (CDI), seguido de (+)-(R)-a-metilbencilamina. Esto proporcionó una mezcla diastereoisómera de amidas, separable mediante cromatografía sobre gel de sílice (P. K. Mykhailiuk et. al. «Convenient synthesis of enantiopure (R) - and (S)-3-fluoro-3-aminomethylpyrrolidines», Tetrahedron 2014, 70, 3011 -3017). Se estableció la configuración del isómero más polar de manera independiente mediante tanto los estudios de Mykhailiuk como los nuestros por difracción de rayos X y se mostró que era (S)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1 -carboxilato de terc-butilo [compuesto (XVIII)] (figura 1), y por ende para el isómero menos polar como (R)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de terc-butilo [compuesto (XIX)]. Además, esto proporcionó materiales de referencia para la comparación con el compuesto obtenido por la secuencia muestra en el esquema III. Aunque nuestros datos de rayos X sobre el isómero polar [compuesto (XVIII)] estaban de acuerdo con la estructura de cristal de rayos X indicada por Mykhailiuk et. al., el espectro de RMN de 1H difería de nuestro espectro obtenido. Los espectros para los dos diastereoisómeros [compuestos (XVIII) y (XIX)] eran muy similares; sin embargo, había una pequeña diferencia de diagnóstico para el protón C4 de la pirrolidina. Se observó a 2,22 ppm. Mykhailiuk indicó que estaba a 2,15 ppm.
El (-)-enantiómero del compuesto de fórmula estructural (IX) [(-)-3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de bencilo], que proporcionó el diastereómero de (IA) ejemplo 2, se hidrogenó sobre Pd/C al 10 % en etanol para retirar el grupo protector CBZ, y la amina resultante (XX) se protegió con dicarbonato de di-terc-butilo para dar (-)-3-fluoro-3-(hidroximetil)pirrolidin-1 -carboxilato de terc-butilo (XXI). Se oxidó el último con tricloruro de rutenio y peryodato de sodio en acetonitrilo-agua. A continuación, se convirtió el ácido carboxílico (XXII) resultante en la amida como antes usando CDI y (+)-(R)-a-metilbencilamina. Esta amida se comparó con las muestras de amida (XVIII) y (XIX) de referencia y se encontró que era idéntica, mediante espectroscopía de RMN, rotación óptica y HPLC quiral, a (R)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de terc-butilo (XIX). El (-)-enantiómero de (IX) es el isómero que proporciona los diastereoisómeros (IA3) y (IA4) y estos tienen la configuración (R) en el centro asimétrico de la pirrolidina. El (+)-enantiómero de (IX) proporcionó (IA1) y (IA2), que tienen la configuración absoluta (S) en el centro asimétrico de la pirrolidina.

Esquema III. Identificación de la configuración absoluta del centro asimétrico de la 3-fluoropirrolidina
Identificación de la configuración absoluta del centro asimétrico bencílico
La configuración absoluta del centro asimétrico bencílico del ejemplo 1 se obtuvo mediante el experimento de degradación mostrado en el esquema IV. Por lo tanto, se trató el ejemplo 1 con yoduro de metilo en DCM a temperatura ambiente durante la noche para cuaternizar el nitrógeno de la pirrolidina y, a continuación, se añadió carbonato de potasio, se calentó a 120 °C durante 1 h en un reactor de microondas para dar (S)-4-(3-morfolinofenil)dihidrofuran-2(3H)-ona (compuesto XXIII). Se comparó el producto de degradación con (R)-4-(3-morfolinofenil)dihidrofuran-2(3H)-ona (compuesto XXIV) auténtica preparada mediante adición de ácido (3-morfolinofenil)borónico a furan-2(5H)-ona usando tetrafluoroborato de bis(norbornadieno)rodio (I) como el catalizador y (R)-BINAP como el ligando quiral usando la clásica reacción asimétrica de Hayashi (T. Hayashi et. al. Tetrahedron Asymmetry, 1999, 10, 4047-4056) y se demostró que era el enantiómero del producto de degradación, estableciendo así la configuración del ejemplo 1 en su centro bencílico como (S).

Esquema IV. Reactivos y condiciones: i) MeI, DCM, temperatura ambiente, 18 h; ii) K2CO3, 120 °C, 1 h; iii) tetrafluoroborato de bis(norbornadieno)rodio(I) y (R)-BlNAP, Ko H, 1,4-dioxano, 100 °C, 1 h.
Adicionalmente, se confirmó de forma independiente la configuración del compuesto XXIII por un estudio de difracción de rayos X y se demostró que era (S) (figura 2).
Basándose en los experimentos anteriores para identificar la configuración absoluta de cada centro asimétrico en el compuesto de fórmula estructural (I) la configuración absoluta de los ejemplos se resume como sigue:
El ejemplo 1 es el compuesto de fórmula estructural (IA2) ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico. El ejemplo 2 es el compuesto de fórmula estructural (IA3) ácido (S)-4-(R)-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico.
Parte experimental
(S)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de ferc-butilo (compuesto XVIII) y (R)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de ferc-butilo (compuesto XIX)
Se trató una solución de ácido (±)-1-(ferc-butoxicarbonil)-3-fluoropirrolidin-3-carboxílico (compuesto XVII) [número de registro de Chemical Abstracts 1001754-59-1] (disponible de Wuxi App Tec) (3,00 g, 12,9 mmol) en THF (70 ml) a temperatura ambiente con CDI sólido (2,5 g, 15,4 mmol) y, a continuación, se calentó la mezcla a 80 °C durante 1,5 h. Se añadió (R)-(+)-a-metilbencilamina (disponible de Fluka) (1,6 g, 13,2 mmol) a esta temperatura y, a continuación, se calentó la mezcla durante 1,5 h adicional a 80 °C. Se diluyó la mezcla con acetato de etilo y se lavó con HCl diluido, NaHCO3, salmuera, se secó (MgSO4), se filtró y se dejó que se evaporara lentamente a temperatura ambiente. Finalmente se concentró la mezcla a presión reducida ya que no se separó mediante cristalización ningún sólido. Se purificó el residuo por cromatografía en cartuchos de sílice (2 x 100 g) eluyendo con EtOAc al (0-25) % - ciclohexano durante 40 min. Se obtuvo el compuesto que se eluyó en primer lugar como una espuma blanca (1,54 g, 36 %): CLEM (sistema A) TR = 1,17 min, ES+vo m/z 337 (M+H)+; RMN de 1H (500 MHz, CDCls) 1,43-1,49 (m, 9H), 1,54 (d, J = 7,0 Hz, 3H), 2,08-2,19 (m, 1H), 2,37-2,62 (m, 1H), 3,43-3,56 (m, 1H), 3,61-3,93 (m, 3H), 5,14 (quin., J = 7,1 Hz, 1H), 6,71 6,76 (m, 1H), 7,27-7,39 (m, 5H) contenía aproximadamente un 10 % del diastereómero más polar; [a]D20 61 (c = 1,27 en MeOH); HPLC quiral analítica TR = 7,58 min, 90 %, y TR = 9,53 min, 10 % en una columna Chiralpak AD (250 mm x 4,6 mm), eluyendo con EtOH al 10% - heptano, caudal = 1 ml/min, detectándose a 215 nm. Se purificó adicionalmente una porción de 50 mg de esta muestra en un cartucho de sílice (20 g) eluyendo con EtOAc al (0-25) % - ciclohexano durante 20 min. Se evaporó la fracción apropiada a presión reducida para dar una muestra analíticamente pura (30 mg) de (R)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de ferc-butilo (compuesto XIX) CLEM (sistema C) TR = 1,16 min, ES+vo m/z 337 (M+H)+ y 354 (M+NH4)+ y ES-vo m/z 335 (M-H)-;
[a]D 20 63 (c=0,933 en MeOH).
Se cristalizó el segundo compuesto que eluyó de la columna (diastereómero más polar) (1,2 g, 28 %) a partir de éter para dar cristales blancos de (S)-3-fluoro-3-(((R)-1-feniletil)carbamoil)pirrolidin-1-carboxilato de terc-butilo (compuesto XVIII): p. f. = 113 °C a 115 °C; CLEM (sistema C) TR = 1,16 min, ES+vo m /z337 (M+H)+; RMN de 1H (500 MHz, CDCls) 1,43-1,48 (m, 9H), 1,54 (d, J = 7,0 Hz, 3H), 2,14-2,26 (m, 1H), 2,44-2,70 (m, 1H), 3,46-3,55 (m, 1H), 3,56-3,87 (m, 3H), 5,14 (quin., J = 7,1 Hz, 1H), 6,73 (s. a., 1H), 7,27-7,40 (m, 5H); [a]D20 73 (c = 0,876 en MeOH); HPLC quiral analítica TR = 9,50 min, 100 % en una columna Chiralpak AD (250 mm x 4,6 mm) eluyendo con EtOH al 10 % - heptano, caudal = 1 ml/min, detectándose a 215 nm. Se estableció la configuración absoluta de este diastereómero a partir de un estudio de difracción de rayos X.
(R)-(-)-(3-fluoropirrolidin-3-il)metanol (compuesto XX)
Se hidrogenó una solución de (-)-A/-CBZ-3-fluoro-3-(hidroximetil)pirrolidina, (-)-isómero de compuesto (IX), (disponible de Wuxi App Tec) (4,0 g, 15,8 mmol) sobre Pd/C al 10 % (400 mg) en etanol (150 ml) durante la noche. Se retiró el catalizador mediante filtración a través de celite y se lavó con etanol. Se evaporaron el líquido filtrado y los lavados a presión reducida para dar el compuesfo del fífulo (2,0 g, 106 %, contenía parte de etanol por RMN) como un aceite amarillo, que solidificó en un sólido ceroso: CLEM (sistema C) TR = 0,22 min, ES+vo m/z 120 (M+H)+ y ES-vo m/z 118 (M-H)'. Se secó adicionalmente el producto en un concentrador con nitrógeno a 40 °C. RMN de 1H (500 MHz, CDCla) 3,82 (dd, J = 18,7; 12,5 Hz, 1H), 3,73 (dd, J = 22,0; 12,2 Hz, 1H), 3,22-3,15 (m, 1H), 3,23-3,14 (m, 1H), 2,99-2,92 (m, 1H), 2,91 (dd, J = 29,1; 13,2 Hz, 1H), 2,66 (s. a., 2H), 2,10-1,98 (m, 1H), 1,94-1,81 (m, 1H); [a]D 20= -4 (c = 1,19 en EtOH).
(R)-(-)-3-fluoro-3-(hidroximetil)pirrolidin-1-carboxilato de ferc-butilo (compuesto XXI)
Se trató una solución de (R)-(3-fluoropirrolidin-3-il)metanol (compuesto XX) (1,88 g, 15,8 mmol) en DCM (15 ml) y diisopropiletilamina (4,13 ml, 23,7 mmol) con dicarbonato de di-ferc-butilo (3,79 g, 17 mmol) y se agitó la mezcla a 20 °C durante 3 h. Se dividió la mezcla entre HCl 2M y DCM y se separó en un cartucho separador de fases. Se concentró la capa orgánica a presión reducida y se purificó el residuo por cromatografía en un cartucho de sílice (70 g) eluyendo con un gradiente de EtOAc al (0-50) %-ciclohexano durante 40 min. Se comprobaron las fracciones por TLC en sílice (EtOAc al 50 %-ciclohexano) y se tiñó con solución de KMnO4. Se combinaron las fracciones apropiadas y se evaporó a presión reducida para dar el compuesfo del fífulo (2,73 g, 79 %) como un aceite incoloro: CLEm (sistema C) TR = 0,79 min, ES+vo m/z 220 (M+H)+ y 439 (2M+H)+; RMN de 1H (400 MHz, DMSO-d6) 81,42 (s, 9H), 1,96-2,14 (m, 2H), 3,32-3,41 (m, 2H), 3,42-3,50 (m, 2H), 3,54-3,61 (m, 1H), 3,62-3,69 (m, H), 4,90 (t, J = 5,8 Hz, 1H); [a]D20 = 28
(c = 3,51 en CHCla).
(R)-3-fluoro-3-(((R)-1-fen¡let¡l)carbamo¡l)p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (compuesto XIX)
Se trató una solución de (-)-3-fluoro-3-(h¡drox¡met¡l)p¡rrolid¡n-1-carbox¡lato de ferc-butilo (compuesto XXI) (200 mg, 0,9 mmol) en MeCN (1 ml) y agua (1 ml) con RuCh (9,5 mg, 0,05 mmol) y peryodato de sod¡o (976 mg, 4,5 mmol) y se ag¡tó la mezcla a 20 °C durante 16 h. Se ac¡d¡f¡có la mezcla con HCl 1 M (5 ml) y se d¡v¡d¡ó en DCM. Se volv¡ó a extraer dos veces la fase acuosa con DCM y se separaron las fases en un cartucho de separac¡ón de fases. Se evaporó la soluc¡ón orgán¡ca en un concentrador para dar ác¡do (R)-1-(terc-butox¡carbon¡l)-3-fluorop¡rrol¡d¡n-3-carboxíl¡co (compuesto XXII) (125 mg, 59 %): EM ES-vo m/z 232 (M-H)x Se d¡solv¡ó el ác¡do (125 mg, 0,54 mmol) en acetato de et¡lo (10 ml) y se trató con CDI (360 mg, 2,2 mmol) y se ag¡tó la mezcla a temperatura amb¡ente durante 1 h y, a cont¡nuac¡ón, se calentó a 50 °C durante 0,5 h. Se concentró la mezcla en un concentrador, se d¡solv¡ó el res¡duo en THF (6 ml) y se trató con (R)-(+)-a-met¡lbenc¡lam¡na (200 mg, 1,9 mmol) y se ag¡tó a 20 °C durante 1,5 h. Se d¡luyó la mezcla con acetato de et¡lo y se lavó con soluc¡ón de HCl 2 M dos veces, segu¡do de salmuera. Se secó la soluc¡ón orgán¡ca (MgSO4) y se evaporó a pres¡ón reduc¡da para dar un sól¡do gr¡s (290 mg). Se d¡solv¡ó el res¡duo en MeOH-DMSO (1:1; 3 ml) y se pur¡f¡có por MDAP en una columna XSELECT Cs H C18 (150 mm x 30 mm D. I., d¡ámetro de empaquetado de 5 pm) a temperatura amb¡ente, eluyendo con un grad¡ente del (30-85) % (b¡carbonato de amonio 10 mM en agua ajustado a pH 10 con soluc¡ón ac. de amoníaco-aceton¡tr¡lo) en func¡onam¡ento durante 30 m¡n, detectándose a 254 nm y recog¡endo el máx¡mo con TR = 17,4 m¡n, ES+vo m/z 337 (M+H)+. Se concentró la fracc¡ón en un concentrador a 45 °C en n¡trógeno y se extrajo la suspens¡ón res¡dual con EtOAc. Se lavó la soluc¡ón orgán¡ca con HCl 2 M dos veces y, a cont¡nuac¡ón, con salmuera, se secó (MgSO4) y se evaporó a pres¡ón reduc¡da para dar una goma amar¡lla (35 mg). Se repur¡f¡có la goma por MDAP en una columna XBr¡dge C18 (100 mm x 19 mm D. I. d¡ámetro de empaquetam¡ento de 5 pm) a temperatura amb¡ente eluyendo con un grad¡ente de (b¡carbonato de amonio 10 mM en agua ajustado a pH 10 con soluc¡ón ac. de amoníaco-aceton¡tr¡lo) en func¡onam¡ento durante 25 m¡n, detectándose a 254 nm) recog¡endo la pr¡mera fracc¡ón (TR = 10 m¡n). Se ret¡ró el d¡solvente en un concentrador con n¡trógeno a 45 °C para dar el compuesto del título (16 mg, 5 %) como una goma ¡ncolora: CLEM (s¡stema C) TR = 1,16 m¡n, ES+vo m/z 337 (M+H)+, 354 (M+NH4)+; HPLC qu¡ral analít¡ca TR = 7,58 m¡n, 97,7% en una columna Ch¡ralpak AD (250 mm x 4,6 mm) eluyendo con EtOH al 10 %-heptano, caudal = 1 ml/m¡n, detectándose a 215 nm;
[a]o 20 63 (c = 1,15 en MeOH). El espectro de RMN de 1H (500 MHz, CDCh) así como la rotac¡ón ópt¡ca y el TR de HPLC qu¡ral co¡nc¡den todos con el de (R)-3-fluoro-3-(((R)-1-fen¡let¡l)carbamo¡l)p¡rrol¡d¡n-1-carbox¡lato de ferc-but¡lo (compuesto XIX).
Determ¡nac¡ón de la conf¡gurac¡ón absoluta del centro as¡métr¡co bencíl¡co del ejemplo 1 por degradac¡ón a (+)-(S)-4-(3-morfol¡nofen¡l)d¡h¡drofuran-2(3H)-ona (compuesto XXIII)
Se trató una soluc¡ón de ác¡do (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrah¡dro-1,8-naft¡r¡d¡n-2-¡l)et¡l)p¡rrol¡d¡n-1-¡l)-3-(3-morfol¡nofen¡l)butano¡co (ejemplo 1) (100 mg, 0,201 mmol) en DCM (8 ml) a temperatura amb¡ente con yodometano (0,195 ml, 3,13 mmol) y se ag¡tó durante 18 h. Se concentró la reacc¡ón al vacío (para ret¡rar el exceso de yodometano). Se red¡solv¡ó el res¡duo en DCM (5 ml) y se añad¡ó carbonato de potas¡o (122 mg, 0,884 mmol). Se calentó la reacc¡ón en un reactor de m¡croondas a 120 °C durante 1 h. Se f¡ltró la soluc¡ón y se concentró al vacío. Se pur¡f¡có el ace¡te res¡dual por cromatografía en columna sobre síl¡ce (10 g) eluyendo con un grad¡ente de TBME al (0-100)% en c¡clohexano. Se concentraron las fracc¡ones relevantes al vacío para dar (S)-4-(3-morfol¡nofen¡l)d¡h¡drofuran-2(3H)-ona (compuesto XXIII) (32 mg, 64 %) como un sól¡do blanco: CLEM (s¡stema C) TR = 0,82 m¡n, 100%, ES+vo m/z 247 (M+H)+; RMN de 1H (400 MHz, CDCla) 7,32-7,26 (m, 1H), 6,89-6,84 (m, 1H), 6,79 6,73 (m, 2H), 4,67 (dd, J = 9,8 Hz, 1H), 4,30 (dd, J = 9,06; 7,5 Hz, 1H), 3,92-3,85 (m, 4H), 3,76 (qu¡n., J = 8,31 Hz, 1H), 3,21-3,17 (m, 4H), 2,93 (dd, J = 17,5 Hz; 8,7 Hz, 1H), 2,93 (dd, J = 17,5 Hz; 8,7 Hz, 1H), 2,70 (dd, J = 17,5 Hz; 8,7 Hz, 1H); [ao22 = 37,1 (c = 1,40 en CHCh); HPLC qu¡ral TR = 25,4 m¡n en una columna Ch¡ralpak ID (25 cm x 4,6 mm) eluyendo con ¡sopropanol al 20 %-heptano, caudal 1 ml/m¡n, detectándose a 215 nm. Se recr¡stal¡zó una porc¡ón de compuesto XXIII a part¡r de cloroformo por cr¡stal¡zac¡ón lenta para proporc¡onar cr¡stales que fueran adecuados para un estud¡o de d¡fracc¡ón de rayos X.
Síntes¡s de (R)-4-(3-morfol¡nofen¡l)d¡h¡drofuran-2(3H)-ona (compuesto XXIV) autént¡ca para la comparac¡ón con el compuesto (XXIII)
Se trató una soluc¡ón de tetrafluoroborato de b¡s(norbornad¡eno)rod¡o(I) (d¡spon¡ble de Aldr¡ch) (18,70 mg, 0,05 mmol) y ác¡do (3-morfol¡nofen¡l)borón¡co (1035 mg, 5,00 mmol) en 1,4-d¡oxano (10 ml) con furan-2(5H)-ona (0,142 ml, 2,0 mmol) y soluc¡ón de KOH (3,8 M, 1,053 ml, 4,00 mmol). Se calentó la soluc¡ón resultante a 100 °C durante 1 h en un reactor de m¡croondas. Se dejó enfr¡ar la reacc¡ón y se concentró al vacío para dar un ace¡te marrón. Se pur¡f¡có el res¡duo por cromatografía (cartucho KPNH, 50 g) eluyendo con un grad¡ente de EtOAc al (0-50) % en c¡clohexano durante 45 m¡n. Se concentraron las fracc¡ones relevantes al vacío para dar (R)-4-(3-morfol¡nofen¡l)d¡h¡drofuran-2(3H)-ona (compuesto XXIV) (132 mg, 27 %) como un sól¡do blanco: CLEM (s¡stema C) TR = 0,82 m¡n, 100 %, ES+vo m/z 248 (M+H)+; [aD22= -28,3 (c = 1,70 en CHCla); HPLC qu¡ral TR = 23,4 m¡n, 94 %, TR = 25,4 m¡n 6 % en una columna Ch¡ralpak ID (25 cm x 4,6 mm) eluyendo con ¡sopropanol al 20 %-heptano, caudal 1 ml/m¡n, detectándose a 215 nm.
Claims (10)
1. Un compuesto de fórmula (I):

en donde R representa H o F, o una sal del mismo.
2. Un compuesto de acuerdo con la reivindicación 1, de fórmula (IA):

que es ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o una sal del mismo.
3. Un compuesto de acuerdo con la reivindicación 1 o con la reivindicación 2, que tiene la fórmula (IA2):

ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(3-morfolinofenil)butanoico o una sal farmacéuticamente aceptable del mismo.
4. Un compuesto de acuerdo con la reivindicación 1, de fórmula (IB)

ácido 4-(3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico o una sal del mismo.
5. Un compuesto de acuerdo con la reivindicación 1 o la reivindicación 4, que tiene la fórmula (IB2):

ácido (S)-4-((S)-3-fluoro-3-(2-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)etil)pirrolidin-1-il)-3-(2-fluoro-5-morfolinofenil)butanoico o una sal farmacéuticamente aceptable del mismo.
6. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo, para su uso en terapia.
7. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad o una afección para la que está indicado un antagonista del receptor ayp6.
8. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de fibrosis pulmonar idiopática.
9. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo y uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables.
10. Una composición farmacéutica de acuerdo con la reivindicación 9, en una forma adaptada para su administración oral.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1417011.2A GB201417011D0 (en) | 2014-09-26 | 2014-09-26 | Novel compounds |
| PCT/EP2015/071777 WO2016046226A1 (en) | 2014-09-26 | 2015-09-22 | Novel compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2704525T3 true ES2704525T3 (es) | 2019-03-18 |
Family
ID=51901167
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15767476T Active ES2704525T3 (es) | 2014-09-26 | 2015-09-22 | Derivados de naftiridina como antagonistas de integrina AlfaVBeta6 para el tratamiento de, por ejemplo, enfermedades fibróticas |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US10000489B2 (es) |
| EP (1) | EP3197893B1 (es) |
| JP (1) | JP6665169B2 (es) |
| KR (1) | KR20170063590A (es) |
| CN (1) | CN107074849A (es) |
| AR (1) | AR101995A1 (es) |
| AU (1) | AU2015320859A1 (es) |
| BR (1) | BR112017006253A2 (es) |
| CA (1) | CA2962326A1 (es) |
| ES (1) | ES2704525T3 (es) |
| GB (1) | GB201417011D0 (es) |
| RU (1) | RU2017114346A (es) |
| TW (1) | TW201629057A (es) |
| UY (1) | UY36315A (es) |
| WO (1) | WO2016046226A1 (es) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201305668D0 (en) | 2013-03-28 | 2013-05-15 | Glaxosmithkline Ip Dev Ltd | Avs6 Integrin Antagonists |
| GB201417002D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| GB201417094D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201417018D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201604681D0 (en) * | 2016-03-21 | 2016-05-04 | Glaxosmithkline Ip Dev Ltd | Chemical Compounds |
| GB201604680D0 (en) | 2016-03-21 | 2016-05-04 | Glaxosmithkline Ip Dev Ltd | Chemical Compounds |
| BR112019009245A2 (pt) | 2016-11-08 | 2019-07-16 | Bristol-Myers Squibb Company | azol amidas e aminas como inibidores de alfav integrina |
| AU2017359023B2 (en) | 2016-11-08 | 2021-12-09 | Bristol-Myers Squibb Company | 3-substituted propionic acids as alpha v integrin inhibitors |
| WO2018089357A1 (en) | 2016-11-08 | 2018-05-17 | Bristol-Myers Squibb Company | INDAZOLE DERIVATIVES AS αV INTEGRIN ANTAGONISTS |
| HRP20210228T1 (hr) * | 2016-11-08 | 2021-03-19 | Bristol-Myers Squibb Company | Amidi pirola kao inhibitori alfa v integrina |
| ES2984394T3 (es) | 2016-11-08 | 2024-10-29 | Bristol Myers Squibb Co | Compuestos mono y espirocíclicos que contienen ciclobutano y azetidina como inhibidores de la integrina alfa v |
| US10696672B2 (en) | 2016-12-23 | 2020-06-30 | Pliant Therapeutics, Inc. | Amino acid compounds and methods of use |
| MA52117A (fr) | 2017-02-28 | 2022-04-06 | Morphic Therapeutic Inc | Inhibiteurs de l'intégrine (alpha-v) (bêta-6) |
| MX394210B (es) | 2017-02-28 | 2025-03-04 | Morphic Therapeutic Inc | INHIBIDORES DE INTEGRINA AVß6. |
| CN107823208A (zh) * | 2017-10-25 | 2018-03-23 | 南京多宝生物科技有限公司 | 吗啉类化合物在制备治疗和预防肾纤维化药物中的应用 |
| KR102700471B1 (ko) | 2017-11-07 | 2024-08-28 | 브리스톨-마이어스 스큅 컴퍼니 | 알파 v 인테그린 억제제로서의 피롤로피라진 유도체 |
| CN108208177A (zh) * | 2017-11-28 | 2018-06-29 | 上海交通大学医学院附属新华医院 | 含丁酸类化合物的组合物及其应用 |
| SI3844162T1 (sl) * | 2018-08-29 | 2025-06-30 | Morphic Therapeutic, Inc. | Zaviralci integrina alfa v beta6 |
| WO2020047239A1 (en) | 2018-08-29 | 2020-03-05 | Morphic Therapeutic, Inc. | INHIBITING αV β6 INTEGRIN |
| WO2020047208A1 (en) | 2018-08-29 | 2020-03-05 | Morphic Therapeutic, Inc. | Inhibitors of (alpha-v)(beta-6) integrin |
| TW202028179A (zh) | 2018-10-08 | 2020-08-01 | 美商普萊恩醫療公司 | 胺基酸化合物及使用方法 |
| US20240165195A1 (en) * | 2018-10-09 | 2024-05-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of alpha-v-integrin (cd51) inhibitors for the treatment of cardiac fibrosis |
| JP2022512648A (ja) * | 2018-10-09 | 2022-02-07 | アンスティチュ ナショナル ドゥ ラ サンテ エ ドゥ ラ ルシェルシュ メディカル | 心筋線維化の処置のためのαV-インテグリン(CD51)阻害剤の使用 |
| US11034710B2 (en) * | 2018-12-04 | 2021-06-15 | Sumitomo Dainippon Pharma Oncology, Inc. | CDK9 inhibitors and polymorphs thereof for use as agents for treatment of cancer |
| JP2025518533A (ja) * | 2022-05-18 | 2025-06-17 | プライアント・セラピューティクス・インコーポレイテッド | インテグリン阻害剤の安定化 |
| EP4605008A1 (en) | 2022-12-14 | 2025-08-27 | Alnylam Pharmaceuticals, Inc. | Alpha-v beta-6 integrin ligands for extrahepatic delivery |
| WO2025259743A1 (en) | 2024-06-12 | 2025-12-18 | Alnylam Pharmaceuticals, Inc. | Dual conjugate compounds for extrahepatic delivery |
| WO2025259747A2 (en) | 2024-06-12 | 2025-12-18 | Alnylam Pharmaceuticals, Inc. | Dystrophy myotonic protein kinase (dmpk) irna compositions and methods of use thereof |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2315370A1 (en) | 1997-12-17 | 1999-06-24 | Merck & Co., Inc. | Integrin receptor antagonists |
| KR20010033248A (ko) | 1997-12-17 | 2001-04-25 | 폴락 돈나 엘. | 인테그린 수용체 길항제 |
| CN1589145A (zh) | 1999-06-02 | 2005-03-02 | 麦克公司 | αv整联蛋白受体拮抗剂 |
| WO2000078317A1 (en) | 1999-06-23 | 2000-12-28 | Merck & Co., Inc. | Integrin receptor antagonists |
| EP1229910A4 (en) * | 1999-10-04 | 2003-10-01 | Merck & Co Inc | integrin |
| PL355011A1 (en) | 1999-11-08 | 2004-03-22 | Merck & Co, Inc. | Process and intermediates for the preparation of imidazolidinone alpha v integrin antagonists |
| US7119098B2 (en) | 2000-06-15 | 2006-10-10 | Pharmacia Corporation | Heteroarylakanoic acids as intergrin receptor antagonists |
| AU7793501A (en) | 2000-07-26 | 2002-02-05 | Merck & Co Inc | Alpha v integrin receptor antagonists |
| WO2002022616A2 (en) | 2000-09-14 | 2002-03-21 | Merck & Co., Inc. | Alpha v integrin receptor antagonists |
| CA2432504A1 (en) | 2001-01-03 | 2002-07-11 | Merck & Co., Inc. | Methods and compositions for treating periodontal disease |
| DE10112771A1 (de) | 2001-03-16 | 2002-09-26 | Merck Patent Gmbh | Inhibitoren des Integrins alpha¶v¶beta¶6¶ |
| US20040224986A1 (en) * | 2002-08-16 | 2004-11-11 | Bart De Corte | Piperidinyl targeting compounds that selectively bind integrins |
| EP1592421A1 (en) | 2002-12-20 | 2005-11-09 | Pharmacia Corporation | Heteroarylalkanoic acids as integrin receptor antagonists |
| US6932865B2 (en) | 2003-04-11 | 2005-08-23 | Lockheed Martin Corporation | System and method of making single-crystal structures through free-form fabrication techniques |
| EP2139882B1 (en) | 2007-03-23 | 2013-12-25 | Amgen Inc. | 3- substituted quinoline or quinoxaline derivatives and their use as phosphatidylinositol 3-kinase (pi3k) inhibitors |
| EP2211615A4 (en) | 2007-10-22 | 2010-10-13 | Glaxosmithkline Llc | PYRIDOSULFONAMIDE DERIVATIVES AS PI3 KINASE INHIBITORS |
| WO2011111880A1 (ko) | 2010-03-08 | 2011-09-15 | 주식회사 메디젠텍 | 세포핵에서 세포질로의 gsk3의 이동을 억제하는 화합물을 함유하는 세포핵에서 세포질로의 gsk3 이동에 의해 발생되는 질환의 치료 또는 예방용 약학적 조성물 |
| GB201305668D0 (en) * | 2013-03-28 | 2013-05-15 | Glaxosmithkline Ip Dev Ltd | Avs6 Integrin Antagonists |
| WO2015048819A1 (en) | 2013-09-30 | 2015-04-02 | The Regents Of The University Of California | Anti-alphavbeta1 integrin compounds and methods |
| GB201417002D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| GB201417018D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201417094D0 (en) | 2014-09-26 | 2014-11-12 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| US9790222B2 (en) | 2015-02-19 | 2017-10-17 | Scifluor Life Sciences, Inc. | Fluorinated tetrahydronaphthyridinyl nonanoic acid derivatives and uses thereof |
| WO2016145258A1 (en) | 2015-03-10 | 2016-09-15 | The Regents Of The University Of California | Anti-alphavbeta1 integrin inhibitors and methods of use |
| GB201604589D0 (en) | 2016-03-18 | 2016-05-04 | Glaxosmithkline Ip Dev Ltd | Chemical compound |
| GB201604680D0 (en) | 2016-03-21 | 2016-05-04 | Glaxosmithkline Ip Dev Ltd | Chemical Compounds |
| GB201604681D0 (en) | 2016-03-21 | 2016-05-04 | Glaxosmithkline Ip Dev Ltd | Chemical Compounds |
-
2014
- 2014-09-26 GB GBGB1417011.2A patent/GB201417011D0/en not_active Ceased
-
2015
- 2015-09-22 UY UY0001036315A patent/UY36315A/es not_active Application Discontinuation
- 2015-09-22 ES ES15767476T patent/ES2704525T3/es active Active
- 2015-09-22 AR ARP150103048A patent/AR101995A1/es unknown
- 2015-09-22 CN CN201580051539.2A patent/CN107074849A/zh active Pending
- 2015-09-22 US US15/514,407 patent/US10000489B2/en not_active Expired - Fee Related
- 2015-09-22 RU RU2017114346A patent/RU2017114346A/ru unknown
- 2015-09-22 JP JP2017516365A patent/JP6665169B2/ja not_active Expired - Fee Related
- 2015-09-22 KR KR1020177007734A patent/KR20170063590A/ko not_active Withdrawn
- 2015-09-22 WO PCT/EP2015/071777 patent/WO2016046226A1/en not_active Ceased
- 2015-09-22 TW TW104131367A patent/TW201629057A/zh unknown
- 2015-09-22 BR BR112017006253A patent/BR112017006253A2/pt not_active Application Discontinuation
- 2015-09-22 EP EP15767476.3A patent/EP3197893B1/en active Active
- 2015-09-22 CA CA2962326A patent/CA2962326A1/en active Pending
- 2015-09-22 AU AU2015320859A patent/AU2015320859A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016046226A1 (en) | 2016-03-31 |
| CN107074849A (zh) | 2017-08-18 |
| RU2017114346A (ru) | 2018-11-02 |
| US20170298063A1 (en) | 2017-10-19 |
| AR101995A1 (es) | 2017-01-25 |
| TW201629057A (zh) | 2016-08-16 |
| JP6665169B2 (ja) | 2020-03-13 |
| JP2017528503A (ja) | 2017-09-28 |
| CA2962326A1 (en) | 2016-03-31 |
| EP3197893A1 (en) | 2017-08-02 |
| BR112017006253A2 (pt) | 2017-12-12 |
| GB201417011D0 (en) | 2014-11-12 |
| KR20170063590A (ko) | 2017-06-08 |
| AU2015320859A1 (en) | 2017-03-09 |
| UY36315A (es) | 2016-04-29 |
| EP3197893B1 (en) | 2018-10-24 |
| US10000489B2 (en) | 2018-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2704525T3 (es) | Derivados de naftiridina como antagonistas de integrina AlfaVBeta6 para el tratamiento de, por ejemplo, enfermedades fibróticas | |
| JP6611799B2 (ja) | 新規化合物 | |
| ES2796235T3 (es) | Derivados de naftiridina como antagonistas de la integrina alfa V beta 6 para el tratamiento de enfermedades fibróticas | |
| CN104341451B (zh) | 新磷酸酯化合物、其制备方法及含有它们的药物组合物 | |
| ES2814000T3 (es) | Naftiridinas como antagonistas de integrinas | |
| CA2962321A1 (en) | Naphthyridine derivatives as alpha v beta 6 integrin antagonists for the treatment of e.g. fibrotic diseases | |
| CA3018014A1 (en) | Naphthyridines as integrin antagonists | |
| JP2019508475A (ja) | 化合物(s)−4−((s)−3−フルオロ−3−(2−(5,6,7,8−テトラヒドロ−1,8−ナフチリジン−2−イル)エチル)ピロリジン−1−イル)−3−(3−(2−メトキシエトキシ)フェニル)ブタン酸のクエン酸塩 | |
| WO2018028491A1 (zh) | 吲哚胺2,3-双加氧酶抑制剂及其在药学中的用途 | |
| HK1234041B (en) | Naphthyridine derivatives as alpha v beta 6 integrin antagonists for the treatment of e.g. fibrotic diseases | |
| HK1234041A1 (en) | Naphthyridine derivatives as alpha v beta 6 integrin antagonists for the treatment of e.g. fibrotic diseases |